
XENPOZYME kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 20 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Terveydenhuollon ammattilainen
Koti-infuusiohoitoa koskeva opas
Yleinen
Mitä Xenpozyme on, Merkittävät haittavaikutukset, Naiset ja raskaus
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Xenpozyme 4 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo sisältää 4 mg olipudaasi alfaa*.
Apuaine, jonka vaikutus tunnetaan
Yksi injektiopullo sisältää 0,60 mg natriumia.
Xenpozyme 20 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo sisältää 20 mg olipudaasi alfaa*.
Apuaine, jonka vaikutus tunnetaan
Yksi injektiopullo sisältää 3,02 mg natriumia.
Käyttökuntoon saattamisen jälkeen yksi injektiopullo sisältää 4 mg olipudaasi alfaa per millilitra. Jokaisen injektiopullon sisältö on laimennettava edelleen ennen käyttöä (ks. kohta Käyttö- ja käsittelyohjeet).
*Olipudaasi alfa on rekombinantti ihmisen hapan sfingomyelinaasi, ja se tuotetaan kiinanhamsterin munasolulinjassa (CHO-solulinjassa) yhdistelmä-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos (kuiva-aine välikonsentraattia varten).
Kliiniset tiedot
Käyttöaiheet
Xenpozyme-valmistetta käytetään entsyymikorvaushoitoon happaman sfingomyelinaasin puutoksen (Acid Sphingomyelinase Deficiency, ASMD) muualla kuin keskushermostossa todettavien ilmentymien hoidossa pediatrisille potilaille ja aikuispotilaille, joiden tauti on tyyppiä A/B tai tyyppiä B.
Ehto
Hoito on toteutettava perinnöllisten aineenvaihduntahäiriöiden hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Annostus ja antotapa
Xenpozyme-hoito on toteutettava happaman sfingomyelinaasin puutoksen tai muiden perinnöllisten aineenvaihduntahäiriöiden hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa. Xenpozyme-infuusion antaa terveydenhuollon ammattilainen, jolla on käytettävissä asianmukaiset lääketieteelliset tukitoimet mahdollisten vaikeiden reaktioiden, kuten vakavien systeemisten yliherkkyysreaktioiden, hoitoon.
Annostus
Elimistöön kertyneen sfingomyeliinin nopea metaboloituminen olipudaasi alfan vaikutuksesta johtaa proinflammatoristen hajoamistuotteiden muodostumiseen, mikä voi aiheuttaa infuusioon liittyviä reaktioita ja/tai ohimenevää maksaentsyymiarvojen suurenemista.
Xenpozyme-hoito täytyy aina aloittaa annoksen suurentamisen ohjelmalla (ks. jäljempänä), jolla pyritään minimoimaan infuusioon liittyvien reaktioiden (akuutin vaiheen reaktiot mukaan lukien) riski ja maksan transaminaasiarvojen suurenemisen riski. Yliannostuksen (ks. kohta Yliannostus) riskin välttämiseksi on noudatettava kaikkia annostus- ja anto-ohjeita (ks. jäljempänä) sekä kaikkia valmistelu- ja käsittelyohjeita (ks. kohta Käyttö- ja käsittelyohjeet). On huomattava, että annoksen suurentaminen toteutetaan pediatrisilla potilailla eri tavalla kuin aikuisilla. Annoksen suurentamisen ohjelman noudattamisen lisäksi jokainen annos täytyy antaa porrastetulla infuusionopeudella (ks. taulukot 3 ja 4).
Antamatta jääneet annokset, ks. myös jäljempänä. Koti-infuusiohoitoa voi harkita vasta annoksen suurentamisen vaiheen jälkeen.
Xenpozyme-annos perustuu potilaan todelliseen painoon, jos hänen painoindeksinsä on ≤ 30, ja optimaaliseen painoon, jos painoindeksi on > 30 (ks. kohta, jossa kerrotaan potilaista, joiden painoindeksi on > 30).
Aikuiset
Annoksen suurentamisen vaihe
Xenpozyme-valmisteen suositeltu aloitusannos on 0,1 mg/kg* aikuisille (ks. myös lisäohjeet kohdassa, jossa kerrotaan antamatta jääneistä annoksista), ja tämän jälkeen annosta suurennetaan taulukon 1 mukaista annoksen suurentamisen ohjelmaa noudattaen:
Taulukko 1: Annoksen suurentamisen ohjelma aikuisilla
| Aikuiset potilaat (≥ 18-vuotiaat) | |
| Annos 1 (päivä 1 / viikko 0) | 0,1 mg/kg* |
| Annos 2 (viikko 2) | 0,3 mg/kg* |
| Annos 3 (viikko 4) | 0,3 mg/kg* |
| Annos 4 (viikko 6) | 0,6 mg/kg* |
| Annos 5 (viikko 8) | 0,6 mg/kg* |
| Annos 6 (viikko 10) | 1 mg/kg* |
| Annos 7 (viikko 12) | 2 mg/kg* |
| Annos 8 (viikko 14) | 3 mg/kg* (suositeltu ylläpitoannos) |
*Jos potilaan painoindeksi on ≤ 30, käytetään todellista painoa. Jos potilaan painoindeksi on > 30, käytetään optimaalista painoa, kuten jäljempänä on kuvattu.
Ylläpitovaihe
Xenpozyme-valmisteen suositeltu ylläpitoannos on 3 mg/kg* 2 viikon välein.
*Jos potilaan painoindeksi on ≤ 30, käytetään todellista painoa. Jos potilaan painoindeksi on > 30, käytetään optimaalista painoa, kuten jäljempänä on kuvattu.
Pediatriset potilaat
Annoksen suurentamisen vaihe
Xenpozyme-valmisteen suositeltu aloitusannos pediatrisille potilaille on 0,03 mg/kg*, ja tämän jälkeen annosta suurennetaan taulukossa 2A esitetyn annoksen suurentamisen ohjelman mukaisesti:
Taulukko 2A: Annoksen suurentamisen ohjelma pediatrisilla potilailla
| Pediatriset potilaat (0 – < 18-vuotiaat) | |
| Annos 1 (päivä 1 / viikko 0) | 0,03 mg/kg* |
| Annos 2 (viikko 2) | 0,1 mg/kg* |
| Annos 3 (viikko 4) | 0,3 mg/kg* |
| Annos 4 (viikko 6) | 0,3 mg/kg* |
| Annos 5 (viikko 8) | 0,6 mg/kg* |
| Annos 6 (viikko 10) | 0,6 mg/kg* |
| Annos 7 (viikko 12) | 1 mg/kg* |
| Annos 8 (viikko 14) | 2 mg/kg* |
| Annos 9 (viikko 16) | 3 mg/kg* (suositeltu ylläpitoannos) |
*Jos potilaan painoindeksi on ≤ 30, käytetään todellista painoa. Jos potilaan painoindeksi on > 30, käytetään optimaalista painoa, kuten jäljempänä on kuvattu.
Ylläpitovaihe
Xenpozyme-valmisteen suositeltu ylläpitoannos on 3 mg/kg* 2 viikon välein.
*Jos potilaan painoindeksi on ≤ 30, käytetään todellista painoa. Jos potilaan painoindeksi on > 30, käytetään optimaalista painoa, kuten jäljempänä on kuvattu.
Potilaat, joiden painoindeksi on > 30
Jos aikuisen tai pediatrisen potilaan painoindeksi on > 30, hänelle arvioidaan Xenpozyme-annoksen laskemiseen käytettävä paino seuraavalla menetelmällä (annoksen suurentamisen vaihetta ja ylläpitovaihetta varten).
Annoksen laskemiseen käytettävä paino (kg) = 30 × (todellinen pituus metreinä)2
Esimerkki:
Oletetaan, että potilaan ominaisuudet ovat seuraavat:
painoindeksi 38
paino 110 kg
pituus 1,7 m.
Annettavan annoksen laskennassa käytetään painoa 30 × 1,72 = 86,7 kg.
Antamatta jääneet annokset
Annoksen katsotaan jääneen antamatta, jos sitä ei anneta 3 vuorokauden kuluessa suunnitellusta päivämäärästä. Jos Xenpozyme-annos jää antamatta, seuraava annos on annettava mahdollisimman pian, kuten jäljempänä on kuvattu. Myöhemmät annokset sovitaan annettavaksi 2 viikon välein viimeisimmän antopäivän jälkeen.
Xenpozyme-valmisteen antamisessa käytettävä annoksen suurentamisen ohjelma estää kataboliittien nopeaa vapautumista, joka voisi johtaa vakaviin toksisiin vaikutuksiin, kuten maksatulehdukseen tai transaminaasiarvojen suurenemiseen, vakaviin ja hengenvaarallisiin infuusioon liittyviin reaktioihin tai jopa kuolemaan (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset ja Yliannostus). Jos potilaan elimistössä on edelleen runsaasti sfingomyeliinikertymiä tai epäillään sfingomyeliinin uudelleenkertymistä antamatta jääneiden annosten vuoksi, hoito on aloitettava uudelleen pienemmällä annoksella.
Taulukko 2B: Xenpozyme-valmisteen annostussuositukset aikuisilla ja pediatrisilla potilailla, joille on jäänyt antamatta yksi tai useampia annoksia
| Peräkkäiset antamatta jääneet annokset | Annoksen suurentamisen vaihe | Ylläpitovaihe |
| Jos 1 infuusio on jäänyt antamatta*: | Annetaan viimeisin siedetty annos ja sen jälkeen jatketaan annoksen suurentamista aikuisille tarkoitetun ohjelman (taulukko 1) tai pediatrisille potilaille tarkoitetun ohjelman (taulukko 2A) mukaisesti. | Annetaan ylläpitoannos ja muutetaan hoidon aikataulua asianmukaisesti. |
| Jos 2 peräkkäistä infuusiota on jäänyt antamatta*: | Annetaan annos, joka on yhden annostason verran pienempi kuin viimeisin siedetty annos (vähimmäisannos on 0,3 mg/kg), ja sen jälkeen jatketaan annoksen suurentamista taulukon 1 tai taulukon 2A mukaisesti. | Annetaan annos, joka on yhden annostason verran pienempi kuin ylläpitoannos (eli 2 mg/kg). Myöhempien infuusioiden yhteydessä annetaan ylläpitoannos (3 mg/kg) 2 viikon välein. |
| Jos vähintään 3 peräkkäistä infuusiota on jäänyt antamatta: | Aikuisilla potilailla, joilla ei ole toteutettu annoksen suurentamisen ohjelmaa kokonaan, annoksen suurentamisen ohjelma aloitetaan uudelleen antamalla ensimmäinen taulukossa 1 kuvattu suurennettu annos.
Pediatrisilla potilailla, joilla ei ole toteutettu annoksen suurentamisen ohjelmaa kokonaan, annoksen suurentamisen ohjelma aloitetaan uudelleen antamalla ensimmäinen taulukossa 2A kuvattu suurennettu annos. | Jos aikuiselle potilaalle on jäänyt antamatta vähintään kolme peräkkäistä ylläpitoannosta, sfingomyeliiniä on voinut kertyä uudelleen. Tässä tapauksessa hoitavaa lääkäriä kehotetaan aloittamaan annoksen suurentamisen ohjelma uudelleen antamalla ensimmäinen taulukossa 1 kuvattu suurennettu annos.
Jos pediatriselle potilaalle on jäänyt antamatta vähintään kolme peräkkäistä ylläpitoannosta, sfingomyeliiniä on voinut kertyä uudelleen. Tässä tapauksessa hoitavaa lääkäriä kehotetaan aloittamaan annoksen suurentamisen ohjelma uudelleen antamalla ensimmäinen taulukossa 2A kuvattu suurennettu annos. |
* Jos annos on jäänyt antamatta ja seuraavan suunnitellun infuusion yhteydessä annettava annos on 0,3 tai 0,6 mg/kg, tämä annos on annettava kahdesti taulukon 1 tai taulukon 2A mukaisesti.
Transaminaasiarvojen seuranta
Transaminaasiarvot (alaniiniaminotransferaasi [ALAT] ja aspartaattiaminotransferaasi [ASAT]) on määritettävä ennen hoidon aloittamista, ja niitä on seurattava aina annoksen suurentamisen vaiheiden aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Jos ennen infuusiota määritetyt transaminaasiarvot ovat lähtötasoa suuremmat ja > 2-kertaiset viitealueen ylärajaan (ULN) nähden, Xenpozyme-annosta voidaan muuttaa (voidaan antaa sama annos kuin aiemmin tai aiempaa annosta voidaan pienentää) tai hoito voidaan tilapäisesti keskeyttää sen mukaan, miten voimakkaasti transaminaasiarvot ovat suurentuneet. Jos potilaan annosta on muutettava tai hoito on keskeytettävä, hoidon uudelleen aloittamisen yhteydessä on noudatettava annoksen suurentamisen ohjelmaa, joka on kuvattu aikuisille potilaille taulukossa 1 ja pediatrisille potilaille taulukossa 2A, sekä ohjeita, jotka koskevat antamatta jääneitä annoksia (ks. antamatta jääneitä annoksia koskeva kohta).
Erityisryhmät
Iäkkäät potilaat
Annoksen muuttamista ei suositella yli 65-vuotiaiden potilaiden kohdalla (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttamista ei suositella maksan vajaatoimintaa sairastavien potilaiden kohdalla (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttamista ei suositella munuaisten vajaatoimintaa sairastavien potilaiden kohdalla (ks. kohta Farmakokinetiikka).
Antotapa
Xenpozyme annetaan vain laskimoon. Infuusiot on annettava vaiheittain, mieluiten infuusiopumpulla.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Käyttökuntoon saattamisen ja laimentamisen jälkeen liuos annetaan infuusiona laskimoon. Infuusionopeuksia suurennetaan vähitellen infuusion aikana, mutta vain, jos infuusioon liittyviä reaktioita ei ilmene (toimintaohjeet infuusioon liittyvien reaktioiden yhteydessä, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Infuusionopeus ja infuusion kesto (+/- 5 min) infuusion kussakin vaiheessa on esitetty yksityiskohtaisesti taulukossa 3 ja taulukossa 4.
Kun infuusionopeus määritetään taulukoiden 3 ja 4 perusteella, käytetään joko taulukossa 1 (aikuiset) tai taulukossa 2A (pediatriset potilaat) esitetyn annoksen suurentamisen ohjelman mukaista annostasoa.
Taulukko 3: Infuusionopeudet ja infuusion kesto aikuisilla potilailla
| Annos* (mg/kg) | Infuusionopeus Infuusion kesto | Infuusion kesto suunnilleen | |||
| vaihe 1 | vaihe 2 | vaihe 3 | vaihe 4 | ||
| 0,1 | 20 ml/h 20 min:n ajan | 60 ml/h 15 min:n ajan | ei oleellinen | ei oleellinen | 35 min |
| 0,3–3 | 3,33 ml/h 20 min:n ajan | 10 ml/h 20 min:n ajan | 20 ml/h 20 min:n ajan | 33,33 ml/h 160 min:n ajan | 220 min |
h: tunti; min: minuutti
*Taulukossa 1 esitetyn annoksen suurentamisen ohjelman mukainen annostaso
Taulukko 4: Infuusionopeudet ja infuusion kesto pediatrisilla potilailla
| Annos* (mg/kg) | Infuusionopeus Infuusion kesto | Infuusion kesto suunnilleen | |||
| vaihe 1 | vaihe 2 | vaihe 3 | vaihe 4 | ||
| 0,03 | 0,1 mg/kg/h koko infuusion ajan | ei oleellinen | ei oleellinen | ei oleellinen | 18 min |
| 0,1 | 0,1 mg/kg/h 20 min:n ajan | 0,3 mg/kg/h infuusion loppuun asti | ei oleellinen | ei oleellinen | 35 min |
| 0,3 | 0,1 mg/kg/h 20 min:n ajan | 0,3 mg/kg/h 20 min:n ajan | 0,6 mg/kg/h infuusion loppuun asti | ei oleellinen | 60 min |
| 0,6 | 0,1 mg/kg/h 20 min:n ajan | 0,3 mg/kg/h 20 min:n ajan | 0,6 mg/kg/h 20 min:n ajan | 1 mg/kg/h infuusion loppuun asti | 80 min |
| 1 | 100 min | ||||
| 2 | 160 min | ||||
| 3 | 220 min | ||||
h: tunti; min: minuutti
*Taulukossa 2A esitetyn annoksen suurentamisen ohjelman mukainen annostaso
Potilaan vointia on seurattava infuusioon liittyvien reaktioiden oireiden ja merkkien, kuten päänsäryn, nokkosihottuman, kuumeen, pahoinvoinnin ja oksentelun, sekä muiden yliherkkyyden oireiden tai merkkien varalta infuusion aikana. Oireiden vaikeusasteen mukaan infuusiota voidaan hidastaa tai se voidaan keskeyttää tai lopettaa, ja asianmukainen hoito voidaan aloittaa tarvittaessa.
Jos potilaalla ilmenee vaikea yliherkkyysreaktio ja/tai anafylaktinen reaktio, Xenpozyme-hoito on keskeytettävä heti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infuusion lopussa (kun ruisku tai infuusiopussi on tyhjä) infuusiolinja huuhdellaan natriumkloridi-injektioliuoksella (9 mg/ml, 0,9 %) käyttäen samaa infuusionopeutta kuin infuusion viimeisessä vaiheessa.
Koti-infuusiohoito ylläpitovaiheessa
Terveydenhuollon ammattilaisen valvonnassa toteutettavaa koti-infuusiohoitoa voidaan harkita, jos potilas saa ylläpitoannosta ja sietää infuusioita hyvin. Päätös potilaan siirtämisestä koti-infuusiohoitoon on tehtävä lääkkeen määränneen lääkärin arvion ja suosituksen mukaan.
Asianmukaisten lääketieteellisten tukitoimien, hätätoimenpiteisiin koulutettu henkilöstö mukaan lukien, on oltava helposti saatavilla Xenpozyme-valmistetta annettaessa. Jos potilaalla ilmenee anafylaktinen reaktio tai muu akuutti reaktio, on välittömästi keskeytettävä Xenpozyme-infuusio, aloitettava asianmukainen hoito ja otettava yhteys lääkäriin. Jos potilaalla ilmenee vaikea yliherkkyysreaktio, myöhemmät infuusiot on aina annettava ympäristössä, jossa on elvytysvalmius. Koti-infuusiohoidossa on käytettävä edelleen samaa annosta ja infuusionopeutta, joita valvotussa hoitoympäristössä käytettiin, eikä niitä saa muuttaa muutoin kuin lääkkeen määränneen lääkärin valvonnassa. Jos annos jää antamatta tai infuusio viivästyy, on otettava yhteys lääkkeen määränneeseen lääkäriin, sillä myöhemmät infuusiot on mahdollisesti annettava valvotussa hoitoympäristössä.
Vasta-aiheet
Hengenvaarallinen yliherkkyys (anafylaktinen reaktio) olipudaasi alfalle tai kohdassa Apuaineet mainituille apuaineille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Ei kulkeutumista veri-aivoesteen läpi
Xenpozyme-valmisteen ei odoteta läpäisevän veri-aivoestettä eikä vaikuttavan taudin ilmentymiin keskushermostossa.
Infuusioon liittyvät reaktiot
Infuusioon liittyviä reaktioita ilmeni noin 58 %:lla potilaista, jotka saivat Xenpozyme-valmistetta kliinisissä tutkimuksissa. Tällaisiin infuusioon liittyviin reaktioihin kuului yliherkkyysreaktioita ja akuutin vaiheen reaktioita (ks. kohta Haittavaikutukset). Yleisimpiä infuusioon liittyviä reaktioita olivat päänsärky, nokkosihottuma, kuume, pahoinvointi ja oksentelu (ks. kohta Haittavaikutukset). Infuusioon liittyviä reaktioita ilmeni tyypillisesti infuusion aikana ja enintään 24 tunnin kuluessa infuusion päättymisen jälkeen.
Annoksen suurentamisen vaiheen aikana tapahtuneen yliannostuksen jälkeen on ilmennyt vakavia haittavaikutuksia, mukaan lukien kuolema (ks. kohdat Annostus ja antotapa ja Yliannostus).
Yliherkkyys/anafylaksi
Xenpozyme-valmistetta saaneilla potilailla on ilmoitettu yliherkkyysreaktioita, anafylaksi mukaan lukien (ks. kohta Haittavaikutukset). Kliinisissä tutkimuksissa yliherkkyysreaktioita ilmeni 7 aikuisella potilaalla (17,5 %) ja 9 pediatrisella potilaalla (45 %), mukaan lukien anafylaksi yhdellä pediatrisella potilaalla.
Hoito
Potilaiden vointia on seurattava tarkoin infuusion aikana ja asianmukainen aika infuusion jälkeen kliinisen arvion mukaisesti. Potilaille on kerrottava yliherkkyyden/anafylaksin mahdollisista oireista, ja heitä on kehotettava hakeutumaan välittömästi lääkärin hoitoon, jos oireita ilmenee. Infuusioon liittyvien reaktioiden hoidon on perustuttava oireiden ja merkkien vaikeusasteeseen, ja hoitoon voi kuulua Xenpozyme-infuusion tilapäinen keskeyttäminen, infuusionopeuden hidastaminen ja/tai asianmukainen hoito.
Jos potilaalla ilmenee vaikea yliherkkyys tai anafylaksi, Xenpozyme-valmisteen anto on keskeytettävä välittömästi ja asianmukainen hoito on aloitettava. Potilaalle, jolle kehittyi anafylaksi kliinisessä tutkimuksessa, järjestettiin yksilöllinen siedätysohjelma, jonka ansiosta hän pystyi aloittamaan uudelleen pitkäkestoisen Xenpozyme-hoidon suositellulla ylläpitoannoksella. Lääkkeen määränneen lääkärin on arvioitava riskit ja hyödyt, jotka liittyvät Xenpozyme-valmisteen antamiseen uudelleen anafylaksin tai vaikean yliherkkyysreaktion jälkeen. Jos harkitaan Xenpozyme-valmisteen antamista uudelleen anafylaksin jälkeen, lääkkeen määränneen lääkärin on otettava yhteys Sanofi‑yhtiön paikalliseen edustajaan, jolta saa ohjeita lääkkeen antamisesta uudelleen. Tällaisten potilaiden hoidossa on noudatettava äärimmäistä varovaisuutta, ja asianmukaisen elvytysvalmiuden on oltava käytettävissä, kun Xenpozyme-valmistetta annetaan uudelleen.
Jos potilaalla ilmenee lieviä tai keskivaikeita infuusioon liittyviä reaktioita, infuusionopeutta voidaan hidastaa tai infuusion anto voidaan tilapäisesti keskeyttää, yksittäisen infuusion kunkin vaiheen kestoa voidaan pidentää ja/tai Xenpozyme-annosta voidaan pienentää. Jos potilaan annosta on pienennettävä, annoksen uudelleen suurentamisen yhteydessä on noudatettava annoksen suurentamisen ohjelmaa, joka on kuvattu aikuisille potilaille taulukossa 1 ja pediatrisille potilaille taulukossa 2A (ks. kohta Annostus ja antotapa).
Potilaille voidaan antaa esilääkityksenä antihistamiineja, antipyreettejä ja/tai glukokortikoideja allergisten reaktioiden ennaltaehkäisemiseksi tai vähentämiseksi.
Immunogeenisuus
Kliinisissä tutkimuksissa aikuisilla ja pediatrisilla potilailla ilmoitettiin kehittyneen hoidon aikana lääkevasta-aineita (antidrug antibodies, ADA) (ks. kohta Haittavaikutukset). Infuusioon liittyviä reaktioita ja yliherkkyysreaktioita saattaa ilmetä lääkevasta-aineiden kehittymisestä riippumatta. Suurin osa infuusioon liittyvistä reaktioista ja yliherkkyysreaktioista oli lieviä tai keskivaikeita ja ne voitiin hoitaa tavanomaisin kliinisin keinoin.
IgE-tyypin lääkevasta-aineiden tutkimista voidaan harkita, jos potilaalla on ilmennyt vaikea yliherkkyysreaktio olipudaasi alfalle.
Kliinisissä tutkimuksissa ei ilmoitettu tehon häviämistä. IgG-tyypin lääkevasta-aineiden tutkimista voidaan kuitenkin harkita, jos hoitovaste häviää.
Ohimenevä transaminaasiarvojen suureneminen
Transaminaasiarvojen (ALAT tai ASAT) ohimenevää suurenemista 24–48 tunnin kuluessa infuusioiden antamisesta ilmoitettiin Xenpozyme-valmisteen annoksen suurentamisen vaiheen aikana kliinisissä tutkimuksissa (ks. kohta Haittavaikutukset). Seuraavan suunnitellun infuusion ajankohtana nämä suurentuneet transaminaasiarvot yleensä palasivat tasolle, joka oli havaittu ennen Xenpozyme-infuusiota.
Transaminaasiarvot (ALAT ja ASAT) on mitattava yhden kuukauden kuluessa ennen Xenpozyme-hoidon aloittamista (ks. kohta Annostus ja antotapa). Annoksen suurentamisen aikana tai tilanteessa, jossa hoito aloitetaan uudelleen annosten jäätyä antamatta, transaminaasiarvot on mitattava 72 tunnin kuluessa ennen seuraavaa suunniteltua Xenpozyme-infuusiota. Jos joko lähtötilanteessa tai ennen infuusiota mitattu transaminaasiarvo on > 2-kertainen viitealueen ylärajaan nähden annoksen suurentamisen aikana, transaminaasiarvot on mitattava uudelleen 72 tunnin kuluessa infuusion päättymisestä. Jos ennen infuusiota määritetyt transaminaasiarvot ovat lähtötasoa suuremmat ja > 2-kertaiset viitealueen ylärajaan nähden, Xenpozyme-annosta voidaan muuttaa (voidaan antaa sama annos kuin aiemmin tai aiempaa annosta voidaan pienentää) tai hoito voidaan tilapäisesti keskeyttää sen mukaan, miten voimakkaasti transaminaasiarvot ovat suurentuneet (ks. kohta Annostus ja antotapa).
Kun suositeltu ylläpitoannos on saavutettu, transaminaasiarvot voidaan määrittää osana happaman sfingomyelinaasin puutoksen tavanomaista kliinistä hoitoa.
Natriumin määrä
Tämä lääkevalmiste sisältää 0,60 mg natriumia per 4 mg:n injektiopullo tai 3,02 mg natriumia per 20 mg:n injektiopullo, mikä vastaa 0,03 %:a tai 0,15 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille tai nuorille ja ≤ 0,08 %:a tai ≤ 0,38 %:a natriumin hyväksyttävästä päivittäisestä enimmäissaannista alle 16 vuoden ikäisille lapsille.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Olipudaasi alfa on rekombinantti ihmisen proteiini, joten sytokromi P450 -välitteisiä yhteisvaikutuksia muiden lääkkeiden kanssa ei ole odotettavissa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisia, jotka voivat tulla raskaaksi, neuvotaan käyttämään tehokasta ehkäisyä hoidon aikana ja 14 päivän ajan viimeisen annoksen saamisen jälkeen, jos Xenpozyme-hoito lopetetaan.
Raskaus
Ei ole olemassa tietoja olipudaasi alfan käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Xenpozyme-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä tehokasta ehkäisyä, elleivät hoidon mahdolliset hyödyt äidille ole suurempia kuin mahdolliset riskit, mukaan lukien sikiöön kohdistuvat riskit.
Imetys
Ei tiedetä, erittyykö olipudaasi alfa ihmisen rintamaitoon. Olipudaasi alfaa on todettu imettävien hiirten maidossa (ks. kohta Prekliiniset tiedot turvallisuudesta). Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Xenpozyme-hoito, ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Saatavilla ei ole ihmisiin perustuvaa tietoa olipudaasi alfan vaikutuksista miesten ja naisten hedelmällisyyteen. Eläinkokeissa ei ole havaittu hedelmällisyyteen kohdistuvia suoria tai epäsuoria haitallisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kliinisissä tutkimuksissa on ilmoitettu hypotensiota, joten Xenpozyme-valmisteella saattaa olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Xenpozyme-hoitoa saaneilla potilailla ilmoitettuja vakavia haittavaikutuksia olivat aiemmin kardiomyopatiaa sairastaneella henkilöllä todettu ekstrasystoletapahtuma, joka ilmeni yhdellä aikuisella potilaalla (2,5 %), sekä anafylaktinen reaktio, nokkosihottuma, ihottuma, yliherkkyys ja alaniiniaminotransferaasiarvon suureneminen, joista kutakin esiintyi yhdellä pediatrisella potilaalla (5 %). Vakavien infuusioon liittyvien yliherkkyysreaktioiden ilmaantuvuus oli suurempi pediatrisilla potilailla kuin aikuisilla.
Yleisimmin ilmoitettuja haittavaikutuksia olivat päänsärky (31,7 %), kuume (25 %), nokkosihottuma (21,7 %), pahoinvointi (20 %), oksentelu (16,7 %), vatsakipu (15 %), lihaskipu (11,7 %), kutina (10 %) ja suurentunut C-reaktiivisen proteiinin pitoisuus (10 %).
Haittavaikutustaulukko
Neljän kliinisen tutkimuksen (siedettävyystutkimus aikuisilla potilailla, ASCEND, ASCEND-Peds ja jatkotutkimus aikuisilla ja pediatrisilla potilailla) yhdistettyyn turvallisuusanalyysiin sisältyi yhteensä 60 potilasta (40 aikuista ja 20 pediatrista potilasta), jotka saivat Xenpozyme-valmistetta enintään annoksella 3 mg/kg 2 viikon välein.
Kliinisten tutkimusten yhdistetyssä turvallisuusanalyysissä ilmoitetut haittavaikutukset on lueteltu taulukossa 5 elinjärjestelmäluokittain ja esiintymistiheyksien mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1 000), hyvin harvinainen (< 1 / 10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 5: Xenpozyme-valmistetta saaneilla potilailla todetut haittavaikutukset kliinisten tutkimusten yhdistetyssä analyysissä
Elinjärjestelmäluokka | Esiintymistiheys | |
Hyvin yleinen | Yleinen | |
Immuunijärjestelmä | Anafylaksi ja yliherkkyys | |
Hermosto | Päänsärky | |
Silmät | Silmän verekkyys, epämiellyttävä tuntemus silmässä, silmän kutina | |
Sydän | Sydämentykytys, takykardia | |
Verisuonisto | Hypotensio, kuumat aallot, lehahdusoire | |
Hengityselimet, rintakehä ja välikarsina | Nielun edeema, nielun turvotus, kiristävä tuntemus kurkussa, hengityksen vinkuminen, kurkunpään ärsytys, hengenahdistus, kurkun ärsytys | |
Ruoansulatuselimistö | Pahoinvointi, vatsakipu, oksentelu | Ripuli, ylävatsakipu, vatsavaivat, ruoansulatuskanavan kipu |
Maksa ja sappi | Kipu maksassa | |
Iho ja ihonalainen kudos | Nokkosihottuma, kutina | Angioedeema, toistopunoittuma, ihottuma, papulaarinen ihottuma, makulaarinen ihottuma, makulopapulaarinen ihottuma, punoittava ihottuma, kutiava ihottuma, tuhkarokkomainen ihottuma, papulat, makulat, punoitus |
Luusto, lihakset ja sidekudos | Lihaskipu | Luukipu, nivelkipu, selkäkipu |
Yleisoireet ja antopaikassa todettavat haitat | Kuume | Kipu, vilunväristykset, kanyylikohdan kipu, kanyylikohtaan liittyvä reaktio, kanyylikohdan kutina, kanyylikohdan turvotus, uupumus, voimattomuus |
Tutkimukset | Suurentunut C-reaktiivisen proteiinin pitoisuus | Suurentunut alaniiniaminotransferaasiarvo, suurentunut aspartaattiaminotransferaasiarvo, suurentunut seerumin ferritiinipitoisuus, poikkeava C‑reaktiivisen proteiinin pitoisuus, kohonnut ruumiinlämpö |
Valikoitujen haittavaikutusten kuvaus
Infuusioon liittyvät reaktiot, mukaan lukien yliherkkyysreaktiot ja anafylaktiset reaktiot
Infuusioon liittyviä reaktioita ilmoitettiin 55 %:lla aikuisista potilaista ja 65 %:lla pediatrisista potilaista. Infuusioon liittyvien reaktioiden yleisimmin ilmoitettuja oireita aikuisilla potilailla olivat päänsärky (22,5 %), pahoinvointi (15 %), nokkosihottuma (12,5 %), nivelkipu (10 %), lihaskipu (10 %), kuume (10 %), kutina (7,5 %), oksentelu (7,5 %) ja vatsakipu (7,5 %). Infuusioon liittyvien reaktioiden yleisimmin ilmoitettuja oireita pediatrisilla potilailla olivat kuume (40 %), nokkosihottuma (35 %), oksentelu (30 %), päänsärky (20 %), pahoinvointi (20 %) ja ihottuma (15 %). Infuusioon liittyviä reaktioita ilmeni tyypillisesti infuusion aikana ja 24 tunnin kuluessa infuusion päättymisen jälkeen.
Kliinisissä tutkimuksissa ilmeni infuusioon liittyviä yliherkkyysreaktioita (mukaan lukien anafylaksi) 26,7 %:lla potilaista: 17,5 %:lla aikuisista potilaista ja 45 %:lla pediatrisista potilaista. Infuusioon liittyvien yliherkkyysreaktioiden yleisimmin ilmoitettuja oireita olivat nokkosihottuma (20 %), kutina (6,7 %), punoitus (6,7 %) ja ihottuma (5 %).
Kliinisissä tutkimuksissa yhdellä pediatrisella potilaalla todettiin vaikea anafylaktinen reaktio. Lisäksi 16 kuukauden ikäisellä potilaalla, jolla happaman sfingomyelinaasin puutos oli tyyppiä A ja joka sai Xenpozyme-hoitoa kliinisen tutkimusohjelman ulkopuolella, ilmeni kaksi anafylaktista reaktiota. Molemmilla potilailla todettiin IgE-vasta-aineita olipudaasi alfaa vastaan.
Kahdella aikuisella ja kolmella pediatrisella potilaalla infuusioon liittyvien reaktioiden oireiden yhteydessä todettiin laboratorioarvojen (esim. C-reaktiivisen proteiinin pitoisuuden ja ferritiinipitoisuuden) muutoksia, jotka viittasivat akuutin vaiheen reaktioon.
Transaminaasiarvojen suureneminen
Transaminaasiarvojen (ALAT tai ASAT) ohimenevää suurenemista 24–48 tunnin kuluessa infuusion antamisesta todettiin kliinisissä tutkimuksissa joillakin Xenpozyme-valmistetta saaneilla potilailla annoksen suurentamisen vaiheen aikana. Nämä suurentuneet arvot korjautuivat yleensä aiempien, ennen infuusiota määritettyjen transaminaasiarvojen tasolle seuraavaan suunniteltuun infuusioon mennessä.
52 viikon Xenpozyme-hoidon jälkeen ALAT-arvon keskiarvo pieneni kaiken kaikkiaan 45,9 % ja ASAT-arvon keskiarvo pieneni 40,2 % verrattuna lähtötilanteeseen. Kun tarkasteltiin aikuisia potilaita, ALAT-arvo oli viitealueella kaikilla niillä 16 potilaalla, joilla ALAT-arvo oli ollut suurentunut lähtötilanteessa, ja ASAT-arvo oli viitealueella 10:llä niistä 12 potilaasta, joilla ASAT-arvo oli ollut suurentunut lähtötilanteessa.
Immunogeenisuus
Yhteensä 16 aikuiselle potilaalle 40:stä (40 %) ja 13 pediatriselle potilaalle 20:stä (65 %), jotka saivat Xenpozyme-valmistetta, kehittyi hoidon aikana lääkevasta-aineita. Mediaaniaika ensimmäisen Xenpozyme-infuusion antamisesta serokonversioon oli aikuisilla noin 33 viikkoa ja pediatrisilla potilailla noin 10 viikkoa. Useimmilla lääkevasta-ainepositiivisilla potilailla (11 aikuisella potilaalla 16:sta ja 8 pediatrisella potilaalla 13:sta) lääkevasta-ainevaste oli heikko (≤ 400) tai lääkevasta-ainetestin tulos muuttui myöhemmin negatiiviseksi. Neljällä 16:sta lääkevasta-ainepositiivisesta aikuisesta potilaasta ja viidellä 13:sta lääkevasta-ainepositiivisesta pediatrisesta potilaasta todettiin neutraloivia vasta-aineita (NAb), jotka estivät olipudaasi alfan vaikutusta. Kuudelle potilaalle kehittyi neutraloivia vasta-aineita kerran ja 3 potilaalla todettiin niitä ajoittain. Yhdellä pediatrisella potilaalla hoito voimisti lääkevasta-ainevastetta. Yhdellä pediatrisella potilaalla ilmeni anafylaktinen reaktio ja kehittyi IgE-tyypin lääkevasta-aineita sekä IgG-tyypin lääkevasta-aineita, joiden titteri oli suurimmillaan 1 600.
Lääkevasta-aineiden ei todettu vaikuttavan Xenpozyme-valmisteen farmakokinetiikkaan eikä tehoon aikuisilla eikä pediatrisilla potilailla. Niiden potilaiden prosenttiosuus, joilla ilmeni hoidon aikana infuusioon liittyviä reaktioita (mukaan lukien yliherkkyysreaktiot), oli suurempi potilailla, joille kehittyi hoidon aikana lääkevasta-aineita (75,9 %), kuin niillä, joille ei kehittynyt lääkevasta-aineita (41,9 %).
Pediatriset potilaat
Xenpozyme-valmisteen turvallisuusprofiili oli samanlainen pediatrisilla ja aikuisilla potilailla lukuun ottamatta sitä, että infuusioon liittyvien yliherkkyysreaktioiden ilmaantuvuus oli pediatrisilla potilailla suurempi kuin aikuisilla.
Pitkäaikainen käyttö
Aikuisilla ja pediatrisilla potilailla pitkäaikaisessa käytössä havaittu haittatapahtumaprofiili vastasi yleisesti ensimmäisen hoitovuoden aikana havaittua haittatapahtumaprofiilia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Xenpozyme-valmisteen yliannostustapauksia on ilmoitettu pediatrisilla potilailla annoksen suurentamisen aikana. Osalla näistä potilaista ilmeni 24 tunnin sisällä hoidon aloittamisesta vakavia haittavaikutuksia, mukaan lukien kuolema. Merkittävimpiä kliinisiä löydöksiä olivat hengitysvajaus, hypotensio, huomattavasti suurentuneet arvot maksan toimintakokeissa ja ruoansulatuskanavan verenvuoto.
Xenpozyme-valmisteen yliannostukseen ei tunneta spesifistä vastalääkettä. Yliannostustapauksessa infuusion anto on lopetettava välittömästi ja potilasta on seurattava tarkasti sairaalassa infuusioon liittyvien reaktioiden, myös akuutin vaiheen reaktioiden, varalta. Haittavaikutusten hoito, ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut ruuansulatuselimistön sairauksien ja aineenvaihduntasairauksien lääkkeet, Entsyymit, ATC-koodi: A16AB25
Vaikutusmekanismi
Olipudaasi alfa on rekombinantti ihmisen hapan sfingomyelinaasi, joka vähentää sfingomyeliinin kertymistä elimiin potilailla, joilla on happaman sfingomyelinaasin puutos (ASMD).
Kliininen teho ja turvallisuus
Xenpozyme-valmisteen tehoa on arvioitu kolmessa kliinisessä tutkimuksessa (ASCEND-tutkimuksessa aikuisilla potilailla, ASCEND-Peds-tutkimuksessa pediatrisilla potilailla ja jatkotutkimuksessa sekä aikuisilla että pediatrisilla potilailla), joihin osallistuneilla yhteensä 61 potilaalla oli happaman sfingomyelinaasin puutos.
Kliininen tutkimus aikuisilla potilailla
ASCEND-tutkimus on satunnaistettu, kaksoissokkoutettu, lumekontrolloitu, toistuvilla annoksilla toteutettava vaiheen II/III monikeskustutkimus aikuisilla potilailla, joilla on happaman sfingomyelinaasin puutos (tyyppi A/B tai tyyppi B). Yhteensä 36 potilasta satunnaistettiin suhteessa 1:1 saamaan joko Xenpozyme-valmistetta tai lumelääkettä. Molempien ryhmien potilaat saivat hoitoa infuusiona laskimoon 2 viikon välein. Xenpozyme-valmistetta saaneiden potilaiden annos suurennettiin vähitellen annoksesta 0,1 mg/kg tavoiteannokseen 3 mg/kg. Tutkimukseen kuului kaksi peräkkäistä jaksoa: satunnaistettu, lumekontrolloitu, kaksoissokkoutettu primaarianalyysin jakso, joka jatkui viikkoon 52 asti, ja sen jälkeen enintään 4 vuoden pituinen jatkohoitojakso.
Potilaat, jotka satunnaistettiin primaarianalyysin jakson ajaksi lumelääkeryhmään, siirtyivät jatkohoitojaksolla saamaan Xenpozyme-valmistetta, ja heidän hoidossaan pyrittiin tavoiteannokseen 3 mg/kg; alkuperäiseen Xenpozyme-ryhmään satunnaistetut potilaat taas jatkoivat aiempaa hoitoaan.
Tutkimukseen otetuilla potilailla keuhkojen hiilimonoksidin diffuusiokapasiteetti (DLco) oli ≤ 70 % normaalista odotusarvosta, pernan tilavuus oli ≥ 6-kertainen normaaliin nähden (normaalin kerrannaisia ≥ 6) magneettikuvauksella mitattuna ja splenomegaliaan liittyvä pistemäärä (Splenomegaly Related Score, SRS) oli ≥ 5. Demografiset tiedot ja taudin ominaisuudet lähtötilanteessa olivat yleisesti samankaltaiset molemmissa hoitoryhmissä. Potilaiden mediaani-ikä oli 30 vuotta (vaihteluväli 18–66 vuotta). Iän keskiarvo (keskihajonta) happaman sfingomyelinaasin puutoksen toteamishetkellä oli 18 (18,4) vuotta. Lähtötilanteessa 9 aikuisella potilaalla 36:sta (25 %) oli neurologisia ilmentymiä, jotka vastasivat happaman sfingomyelinaasin puutoksen tyypin A/B kliinistä diagnoosia. Lopuilla 27 potilaalla oli kliininen diagnoosi, joka vastasi happaman sfingomyelinaasin puutoksen tyyppiä B.
Tässä tutkimuksessa oli kaksi erillistä ensisijaista tehoa koskevaa päätemuuttujaa: DLco-arvon (% normaalista odotusarvosta) prosentuaalinen muutos ja pernan tilavuuden (normaalin kerrannaiset) prosentuaalinen muutos magneettikuvauksella mitattuna lähtötilanteesta viikkoon 52 mennessä. Toissijaisiin tehoa koskeviin päätemuuttujiin kuuluivat maksan tilavuuden prosentuaalinen muutos (normaalin kerrannaiset) ja verihiutalemäärien prosentuaalinen muutos lähtötilanteesta viikkoon 52 mennessä. Lisäksi arvioitiin farmakodynaamisia parametreja (keramidipitoisuuksia ja lysosfingomyeliinin [eli sfingomyeliinin deasyloituneen muodon] pitoisuuksia).
52 viikon pituisella primaarianalyysin jaksolla keskimääräisten prosentuaalisten muutosten todettiin olevan parempia Xenpozyme-ryhmässä kuin lumelääkeryhmässä, kun arvioitiin DLco-arvoa (% odotusarvosta; p = 0,0004) ja pernan tilavuutta (p < 0,0001) sekä keskimääräistä maksan tilavuutta (p < 0,0001) ja verihiutalemäärää (p = 0,0185). Hoidon viikolla 26 eli ensimmäisessä lääkkeenannon jälkeen toteutetussa päätemuuttujien arvioinnissa DLco-arvon (% odotusarvosta), pernan tilavuuden, maksan tilavuuden ja verihiutalemäärän keskimääräisten prosentuaalisten muutosten todettiin olevan merkitsevästi paremmat.
Primaarianalyysin jakson tulokset viikolta 52 on esitetty taulukossa 6.
Taulukko 6: Tehoa koskevien päätemuuttujien keskiarvot (keskihajonta) lähtötilanteessa ja pienimmän neliösumman keskiarvon prosentuaalinen muutos (keskivirhe) lähtötilanteesta viikkoon 52 mennessä
Lumelääke (n = 18) | Xenpozyme (n = 18) | Erotus [95 %:n luottamusväli] | p-arvo* | |
Ensisijaiset päätemuuttujat | ||||
DLco-arvon (% odotusarvosta) keskiarvo lähtötilanteessa DLco-arvon (% odotusarvosta) prosentuaalinen muutos lähtötilanteesta viikkoon 52 mennessä | 48,5 (10,8) 3 (3,4) | 49,4 (11,0) 22 (3,3) | ei oleellinen 19 (4,8) [9,3; 28,7] | ei oleellinen 0,0004 |
Pernan tilavuuden (normaalin kerrannaiset) keskiarvo lähtötilanteessa Pernan tilavuuden prosentuaalinen muutos lähtötilanteesta viikkoon 52 mennessä | 11,2 (3,8) 0,5 (2,5) | 11,7 (4,9) -39,4 (2,4) | ei oleellinen ‑39,9 (3,5) [‑47,1; ‑32,8] | ei oleellinen < 0,0001 |
Toissijaiset päätemuuttujat | ||||
Maksan tilavuuden (normaalin kerrannaiset) keskiarvo lähtötilanteessa Maksan tilavuuden prosentuaalinen muutos lähtötilanteesta viikkoon 52 mennessä | 1,6 (0,5) ‑1,5 (2,5) | 1,4 (0,3) -28,1 (2,5) | ei oleellinen ‑26,6 (3,6) [‑33,9; ‑19,3] | ei oleellinen < 0,0001 |
Verihiutalemäärän (109/l) keskiarvo lähtötilanteessa Verihiutalemäärän prosentuaalinen muutos lähtötilanteesta viikkoon 52 mennessä | 115,6 (36,3) 2,5 (4,2) | 107,2 (26,9) 16,8 (4,0) | ei oleellinen +14,3 (5,8) [2,6; 26,1] | ei oleellinen 0,0185 |
*Tilastollisesti merkitsevä, kun tiedot korjattiin kerrannaisuuden suhteen
Lisäksi lysosfingomyeliinin pitoisuudet, jotka ovat happaman sfingomyelinaasin puutosta sairastavien potilaiden plasmassa huomattavan suuret, pienenivät merkitsevästi. Tämä kuvastaa kudosten sfingomyeliinimäärän pienenemistä. Pienimmän neliösumman keskiarvon prosentuaalinen muutos (keskivirhe) lähtötilanteesta viikkoon 52 mennessä oli plasmasta ennen infuusiota mitattujen lysosfingomyeliinipitoisuuksien kohdalla 77,7 % (3,9) Xenpozyme-hoitoryhmässä ja 5,0 % (4,2) lumelääkeryhmässä. Maksan sfingomyeliinimäärä, joka arvioitiin histopatologisella tutkimuksella, pieneni 92,0 % (keskivirhe 8,1) lähtötilanteesta viikkoon 52 mennessä Xenpozyme-hoitoryhmässä (lumelääkeryhmässä muutos oli +10,3 % [keskivirhe 7,8]).
17 potilasta niistä 18:sta, jotka olivat saaneet aiemmin lumelääkettä 52 viikon ajan (primaarianalyysin jakso), aloitti Xenpozyme-hoidon ja jatkoi sitä enintään 4 vuoden ajan, ja 18 potilasta niistä 18:sta, jotka olivat saaneet aiemmin primaarianalyysin jakson aikana Xenpozyme-hoitoa, jatkoi Xenpozyme-hoitoa enintään 4 vuoden ajan. Xenpozyme-valmisteen pitkäkestoiset vaikutukset tehoa koskeviin päätemuuttujiin viikkoon 104 mennessä on esitetty kuvissa 1 ja 2 sekä taulukossa 7.
Kuva 1: Kuvaaja DLco-arvojen (% odotusarvosta) lähtötilanteesta viikkoon 104 arvioidun prosentuaalisen muutoksen pienimmän neliösumman keskiarvoista (95 %:n luottamusväli) – modifioitu hoitoaiepopulaatio (mITT-populaatio)
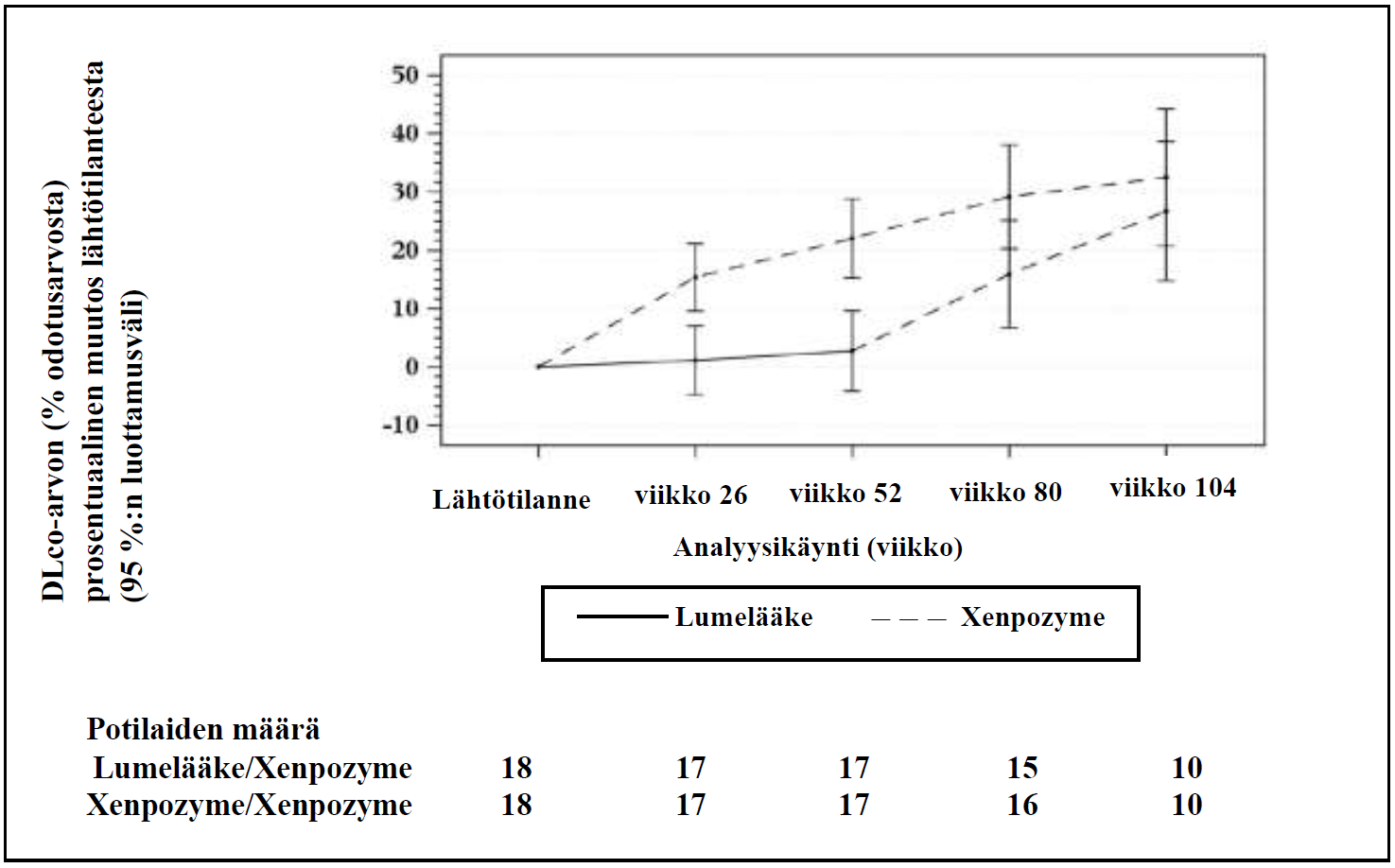
Pystyviivat kuvaavat pienimmän neliösumman keskiarvojen 95 %:n luottamusvälejä.
Pienimmän neliösumman keskiarvot ja 95 %:n luottamusvälit perustuvat toistomittausten sekamalliin ja viikkoon 104 mennessä kertyneisiin tietoihin.
Lumelääke/Xenpozyme-ryhmässä potilaat saivat lumelääkettä viikkoon 52 asti, minkä jälkeen he siirtyivät saamaan Xenpozyme-valmistetta.
Kuva 2: Kuvaaja pernan tilavuuden (normaalin kerrannaiset) lähtötilanteesta viikkoon 104 arvioidun prosentuaalisen muutoksen pienimmän neliösumman keskiarvoista (95 %:n luottamusväli) – modifioitu hoitoaiepopulaatio
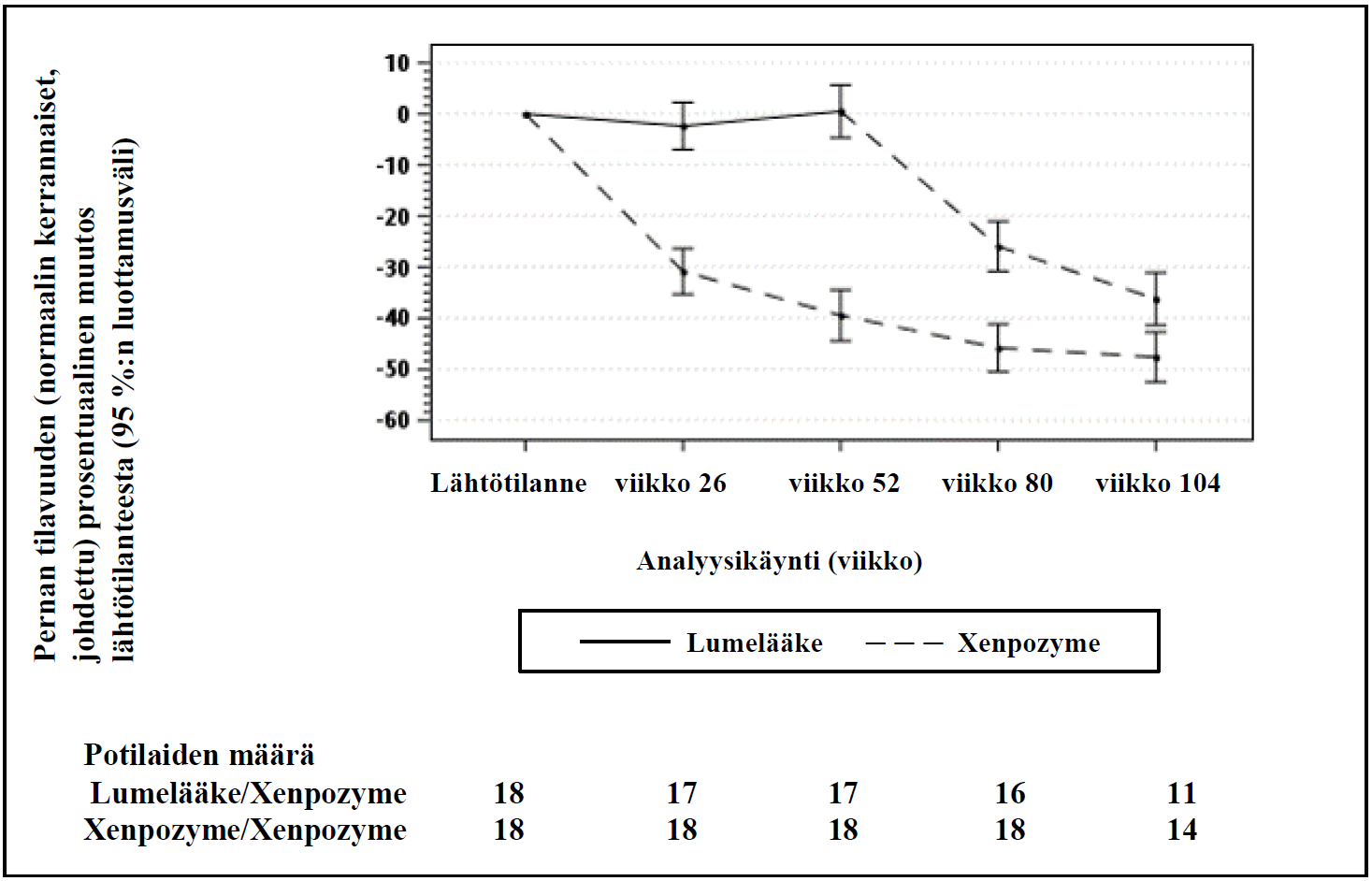
Pystyviivat kuvaavat pienimmän neliösumman keskiarvojen 95 %:n luottamusvälejä.
Pienimmän neliösumman keskiarvot ja 95 %:n luottamusvälit perustuvat toistomittausten sekamalliin ja viikkoon 104 mennessä kertyneisiin tietoihin.
Lumelääke/Xenpozyme-ryhmässä potilaat saivat lumelääkettä viikkoon 52 asti, minkä jälkeen he siirtyivät saamaan Xenpozyme-valmistetta.
Taulukko 7: Maksan tilavuuden (normaalin kerrannaiset) ja verihiutalemäärän (109/l) pienimmän neliösumman keskiarvon prosentuaalinen muutos (keskivirhe) lähtötilanteesta viikkoon 104 mennessä potilailla, jotka saivat Xenpozyme-valmistetta 104 viikon ajan
Aiemmin olipudaasi alfaa saaneiden ryhmä | ||
viikko 52 (jatkohoitojakson alku) | viikko 104 | |
N Maksan tilavuuden prosentuaalinen muutos (keskihajonta) | 17 ‑27,8 (2,5) | 14 ‑33,4 (2,2) |
N Verihiutalemäärän prosentuaalinen muutos (keskihajonta) | 18 16,6 (4,0) | 13 24,9 (6,9) |
N: potilaiden lukumäärä
Jatkotutkimus aikuisilla potilailla
Viisi aikuista potilasta, jotka osallistuivat happaman sfingomyelinaasin puutosta sairastavilla potilailla tehtyyn, suurenevilla lääkeannoksilla toteutettuun avoimeen tutkimukseen, jatkoi hoitoa avoimessa jatkotutkimuksessa ja sai Xenpozyme-valmistetta pisimmillään yli 7 vuoden ajan.
Aikuisilla todettiin tutkimuksen aikana pitkäkestoisia parannuksia DLco-arvossa (% odotusarvosta), pernan ja maksan tilavuuksissa ja verihiutalemäärässä verrattuna lähtötilanteeseen (ks. taulukko 8).
Taulukko 8: Tehoa koskevien parametrien keskimääräiset prosentuaaliset muutokset (keskihajonta) lähtötilanteesta kuukauteen 78 mennessä
Kuukausi 78 (N = 5) | |
DLco-arvon (% odotusarvosta) prosentuaalinen muutos (keskihajonta) | 55,3 % (48,1) |
Pernan tilavuuden prosentuaalinen muutos (keskihajonta) | ‑59,5 % (4,7) |
Maksan tilavuuden prosentuaalinen muutos (keskihajonta) | ‑43,7 % (16,7) |
Verihiutalemäärän prosentuaalinen muutos (keskihajonta) | 38,5 % (14,7) |
N: potilaiden lukumäärä
Pediatriset potilaat
ASCEND-Peds-tutkimus (vaiheen 1/2 kliininen tutkimus) on avoin, toistuvilla annoksilla toteutettava monikeskustutkimus, jossa arvioidaan Xenpozyme-valmisteen turvallisuutta ja siedettävyyttä 64 viikon aikana alle 18‑vuotiailla pediatrisilla potilailla, joilla on happaman sfingomyelinaasin puutos (tyyppi A/B tai tyyppi B). Lisäksi viikolla 52 arvioitiin tehoa koskevia eksploratiivisia päätemuuttujia, jotka liittyivät organomegaliaan, keuhkojen ja maksan toimintaan ja pituuskasvuun.
Yhteensä 20 potilasta (4 nuorta, joiden ikä oli 12 vuotta – < 18 vuotta; 9 lasta, joiden ikä oli 6 vuotta – < 12 vuotta; ja 7 imeväistä/lasta, joiden ikä oli < 6 vuotta) sai Xenpozyme-valmistetta, jonka annos suurennettiin annoksesta 0,03 mg/kg tavoiteannokseen 3 mg/kg annoksen suurentamisen ohjelman mukaisesti. Hoitoa annettiin infuusiona laskimoon 2 viikon välein enintään 64 viikon ajan. Tutkimukseen osallistuneilla potilailla pernan tilavuus oli ≥ 5‑kertainen normaaliin nähden magneettikuvauksella mitattuna. Potilaat jakautuivat kaikkiin ikäryhmiin 1,5‑vuotiaista 17,5‑vuotiaisiin, ja molemmat sukupuolet olivat yhtäläisesti edustettuina. Iän keskiarvo (keskihajonta) happaman sfingomyelinaasin puutoksen toteamishetkellä oli 2,5 (2,5) vuotta. Lähtötilanteessa 8 pediatrisella potilaalla 20:stä (40 %) oli neurologisia ilmentymiä, jotka vastasivat happaman sfingomyelinaasin puutoksen tyypin A/B kliinistä diagnoosia. Lopuilla 12 potilaalla oli kliininen diagnoosi, joka vastasi happaman sfingomyelinaasin puutoksen tyyppiä B.
Xenpozyme-hoidolla oli saavutettu viikolla 52 paremmat keskimääräiset prosentuaaliset muutokset DLco-arvojen (% odotusarvosta), pernan ja maksan tilavuuksien, verihiutalemäärien ja pituuskasvun etenemisen (pituuden Z-luvuilla mitattuna) kohdalla verrattuna lähtötilanteeseen (ks. taulukko 9).
Taulukko 9: Tehoa koskevien parametrien pienimmän neliösumman keskiarvojen prosentuaalinen muutos (keskivirhe) tai muutos (keskihajonta) lähtötilanteesta viikkoon 52 mennessä (kaikki ikäkohortit)
| Arvo lähtötilanteessa (n = 20) | Viikko 52 (n = 20) |
DLco-arvon (% odotusarvosta) keskiarvo (keskihajonta) DLco-arvon (% odotusarvosta) prosentuaalinen muutos* 95 %:n luottamusväli | 54,8 (14,2) | 71,7 (14,8) 32,9 (8,3) 13,4; 52,5 |
Pernan tilavuuden (normaalin kerrannaiset) keskiarvo (keskihajonta) Pernan tilavuuden (normaalin kerrannaiset) prosentuaalinen muutos 95 %:n luottamusväli | 19,0 (8,8) | 9,3 (3,9) ‑49,2 (2,0) ‑53,4; ‑45,0 |
Maksan tilavuuden (normaalin kerrannaiset) keskiarvo (keskihajonta) Maksan tilavuuden (normaalin kerrannaiset) prosentuaalinen muutos 95 %:n luottamusväli | 2,7 (0,7) | 1,5 (0,3) ‑40,6 (1,7) ‑44,1; ‑37,1 |
Verihiutalemäärän (109/l) keskiarvo (keskihajonta) Verihiutalemäärän prosentuaalinen muutos 95 %:n luottamusväli | 137,7 (62,3) | 173,6 (60,5) 34,0 (7,6) 17,9; 50,1 |
Pituuden Z-lukujen keskiarvo (keskihajonta) Pituuden Z-lukujen muutos* 95 %:n luottamusväli | -2,1 (0,8) | ‑1,6 (0,8) 0,6 (0,4) (0,38; 0,73) |
* DLco-arvo arvioitiin 9 pediatrisella potilaalla, joiden ikä oli ≥ 5 vuotta ja jotka kykenivät suoriutumaan DLco-arvon tutkimuksesta, ja pituuden Z-lukujen muutos arvioitiin 19 pediatrisella potilaalla.
Lisäksi plasmasta ennen infuusiota mitattujen keramidi- ja lysosfingomyeliinipitoisuuksien pienimmän neliösumman keskiarvot olivat pienentyneet keramidien kohdalla 57 % (keskivirhe 5,1) ja lysosfingomyeliinin kohdalla 87,2 % (keskivirhe 1,3) 52 viikon hoidon jälkeen verrattuna lähtötilanteeseen.
Xenpozyme-valmisteen todettiin vaikuttavan pernan ja maksan tilavuuksiin, verihiutalemääriin ja pituuden Z-lukuihin kaikissa tutkimukseen sisältyneissä pediatrisissa ikäkohorteissa.
Jatkotutkimus pediatrisilla potilailla
20 pediatrista potilasta, jotka osallistuivat ASCEND-Peds-tutkimukseen, jatkoi hoitoa avoimessa jatkotutkimuksessa ja sai Xenpozyme-valmistetta pisimmillään yli 5 vuoden ajan.
Pediatrisilla potilailla todettiin pitkäkestoisia parannuksia tehoa koskevissa parametreissa (DLco-arvo [% odotusarvosta], pernan ja maksan tilavuudet, verihiutalemäärät, pituuden Z-luvut ja luustoikä) tutkimuksen aikana kuukauteen 48 mennessä (ks. taulukko 10).
Taulukko 10: Tehoa koskevien parametrien keskimääräiset prosentuaaliset muutokset tai muutokset (keskihajonta) lähtötilanteesta kuukauteen 48 mennessä (kaikki ikäkohortit)
Kuukausi 48 | |
N DLco-arvon (% odotusarvosta) prosentuaalinen muutos (keskihajonta) | 5 60,3 (58,5) |
N Pernan tilavuuden prosentuaalinen muutos (keskihajonta) | 7 -69,1 (4,1) |
N Maksan tilavuuden prosentuaalinen muutos (keskihajonta) | 7 -55,4 (11,0) |
N Verihiutalemäärän prosentuaalinen muutos (keskihajonta) | 5 35,8 (42,4) |
N Pituuden Z-lukujen muutos (keskihajonta) | 5 2,3 (0,8) |
N Luustoiän muutos (kuukausina) (keskihajonta) | 7 18,5 (19,0) |
N: potilaiden lukumäärä
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Xenpozyme-valmisteen käytöstä happaman sfingomyelinaasin puutoksen hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Olipudaasi alfan farmakokinetiikkaa arvioitiin 49 aikuisella potilaalla, joilla oli happaman sfingomyelinaasin puutos ja jotka osallistuivat johonkin kaikista happaman sfingomyelinaasin puutosta sairastavilla potilailla tehdyistä kliinisistä tutkimuksista ja saivat lääkettä kerran tai useita kertoja. Annoksella 3 mg/kg 2 viikon välein huippupitoisuuden (Cmax) keskiarvo (variaatiokerroin prosentteina, CV%) vakaassa tilassa oli 30,2 µg/ml (17 %) ja pitoisuus-aikakuvaajan alle annosvälillä jäävän pinta-alan (AUC0–τ) keskiarvo (CV%) vakaassa tilassa oli 607 µg·h/ml (20 %).
Imeytyminen
Imeytymistä ei tapahdu, sillä Xenpozyme annetaan laskimoon.
Jakautuminen
Olipudaasi alfan arvioidun jakautumistilavuuden keskiarvo (variaatiokerroin, CV%) on 13,1 l (18 %).
Biotransformaatio
Olipudaasi alfa on rekombinantti ihmisen entsyymi, ja sen odotetaan poistuvan elimistöstä hajoamalla proteolyyttisesti pieniksi peptideiksi ja aminohapoiksi.
Eliminaatio
Olipudaasi alfan puhdistuman keskiarvo (CV%) on 0,331 l/h (22 %). Terminaalisen puoliintumisajan (t1/2) keskiarvo oli 31,9–37,6 tuntia.
Lineaarisuus/ei-lineaarisuus
Olipudaasi alfan farmakokinetiikka oli lineaarinen annosalueella 0,03–3 mg/kg. Kun annoksen suurentamisen ohjelman mukaisesti siirryttiin vaiheittain annoksesta 0,1 mg/kg ylläpitoannokseen 3 mg/kg 2 viikon välein, plasmasta mitatuissa olipudaasi alfan pitoisuuksissa todettiin erittäin vähäistä kumuloitumista.
Erityisryhmät
Olipudaasi alfan farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja sukupuolten välillä.
Populaatiofarmakokineettisen analyysin mukaan altistukset aasialaisilla potilailla (n = 2) ja muita etnisiä taustoja edustavilla potilailla (n = 2) olivat valkoihoisilla potilailla todettujen altistusten vaihteluvälillä.
Iäkkäät (vähintään 65‑vuotiaat) potilaat
Populaatiofarmakokineettinen analyysi ei viitannut muista potilaista poikkeavaan altistukseen iäkkäillä potilailla. (Xenpozyme-valmisteen kliinisiin tutkimuksiin osallistui vain kaksi potilasta, joiden ikä oli 65 vuoden ja 75 vuoden välillä.)
Pediatriset potilaat
Olipudaasi alfan farmakokinetiikkaa arvioitiin 20 pediatrisella potilaalla, joihin kuului 4 nuorta potilasta, 9 lapsipotilasta ja 7 lapsipotilasta / imeväisikäistä potilasta (taulukko 11). Altistukset olipudaasi alfalle olivat pediatrisilla potilailla pienemmät kuin aikuisilla potilailla. Näitä eroja ei kuitenkaan pidetty kliinisesti merkittävinä.
Taulukko 11: Olipudaasi alfan farmakokineettisten parametrien keskiarvot (CV%) annoksella 3 mg/kg 2 viikon välein nuorilla potilailla, lapsipotilailla ja lapsipotilailla / imeväisikäisillä potilailla, joilla oli happaman sfingomyelinaasin puutos
Ikäryhmä | Ikä (vuosina) | Cmax (µg/ml) | AUC0–τ (µg·h/ml) | |
Nuoret (n = 4) | 12 – < 18 | 27,5 (8) | 529 (7) | |
Lapset (n = 9) | 6 – < 12 | 24,0 (10) | 450 (15) | |
Lapset/imeväisikäiset (n = 7) | < 6 | 22,8 (8) | 403 (11) | |
Kuvailevat tilastotiedot edustavat populaatiofarmakokineettiseen analyysiin perustuvia vakaan tilan altistuksen post hoc ‑estimaatteja. | ||||
Maksan vajaatoiminta
Olipudaasi alfa on rekombinantti proteiini, ja sen odotetaan poistuvan elimistöstä proteolyyttisesti hajoamalla. Maksan vajaatoiminnan ei siis odoteta vaikuttavan olipudaasi alfan farmakokinetiikkaan.
Munuaisten vajaatoiminta
ASCEND-tutkimukseen osallistui neljä potilasta (11,1 %), joilla oli lievä munuaisten vajaatoiminta (kreatiniinipuhdistuma 60 ml/min – < 90 ml/min). Olipudaasi alfan farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja potilailla, joilla oli lievä munuaisten vajaatoiminta. Kohtalaisen tai vaikean munuaisten vajaatoiminnan vaikutuksia olipudaasi alfan farmakokinetiikkaan ei tunneta. Olipudaasi alfan ei odoteta poistuvan elimistöstä erittymällä munuaisten kautta. Munuaisten vajaatoiminnan ei siis odoteta vaikuttavan olipudaasi alfan farmakokinetiikkaan.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, kerta-annoksen aiheuttamaa toksisuutta ja toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Kyseiset tutkimukset toteutettiin villin tyypin eläimillä (hiirillä, rotilla, kaniineilla, koirilla ja apinoilla), ja annokset olivat 10‑kertaisia ihmiselle suositeltuun enimmäisannokseen nähden. Ei ole tehty tutkimuksia, joissa arvioitaisiin olipudaasi alfan mutageenisuutta ja karsinogeenisuutta.
Poistogeenisillä ASMKO-hiirillä, joilta puuttui hapanta sfingomyelinaasia koodaava geeni (kyseessä on happaman sfingomyelinaasin puutosta kuvaava tautimalli), havaittiin kuolleisuutta hiirten saatua olipudaasi alfaa bolusinjektiona laskimoon kerta-annoksina, jotka olivat ≥ 3,3‑kertaisia ihmiselle suositeltuun enimmäisannokseen nähden. Toistuvilla annoksilla toteutetuissa tutkimuksissa kuitenkin osoitettiin, että olipudaasi alfan antaminen annoksen suurentamisen ohjelman mukaisesti ei johtanut valmisteeseen liittyvään kuolleisuuteen ja että muiden havaittujen toksisten vaikutusten vaikeusaste pieneni, kun suurin tutkittu annos oli 10‑kertainen ihmiselle suositeltuun enimmäisannokseen nähden.
Eksenkefalian ilmaantuvuuden havaittiin suurentuneen, kun tiineille hiirille annettiin päivittäin olipudaasi alfaa ja altistukset olivat pienemmät kuin ihmisen altistus käytettäessä suositeltua ylläpitohoitoannosta ja antotiheyttä. Ilmaantuvuus oli hieman suurempi kuin historiallisissa vertailutiedoissa. Tämän havainnon merkitystä ihmisille ei tunneta. Olipudaasi alfan päivittäinen anto laskimoon tiineille kaniineille ei johtanut sikiöiden epämuodostumiin eikä poikkeavuuksiin, kun altistukset olivat merkittävästi suurempia kuin ihmisen altistus käytettäessä suositeltua ylläpitohoitoannosta ja antotiheyttä.
Kun hiirille annettiin 3 mg/kg olipudaasi alfaa päivänä 7 synnytyksen jälkeen, olipudaasi alfaa todettiin maidossa 2 päivän kuluttua valmisteen annosta.
Farmaseuttiset tiedot
Apuaineet
L-metioniini
Dinatriumfosfaattiheptahydraatti
Natriumdivetyfosfaattimonohydraatti
Sakkaroosi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamattomat injektiopullot
60 kuukautta.
Käyttökuntoon saatettu lääkevalmiste
Kun lääkevalmiste on saatettu käyttökuntoon injektionesteisiin käytettävällä steriilillä vedellä, sen kemiallisen, fysikaalisen ja mikrobiologisen käytönaikaisen säilyvyyden on osoitettu olevan enintään 24 tuntia 2–8 °C:ssa tai 6 tuntia huoneenlämmössä (enintään 25 °C:ssa).
Käyttökuntoon saatettu lääkevalmiste on mikrobiologisista syistä käytettävä välittömästi. Jos valmistetta ei laimenneta välittömästi, käytönaikaiset säilytysajat ja säilytysolosuhteet ennen laimentamista ovat käyttäjän vastuulla, eikä valmistetta yleensä säilytetä yli 24:ää tuntia 2–8 °C:ssa.
Laimennettu lääkevalmiste
Kun lääkevalmiste on laimennettu natriumkloridi-injektioliuoksella (9 mg/ml, 0,9 %), sen kemiallisen, fysikaalisen ja mikrobiologisen käytönaikaisen säilyvyyden on osoitettu olevan pitoisuusalueella 0,1–3,5 mg/ml 24 tuntia 2–8 °C:ssa ja enintään 12 tuntia (infuusioaika mukaan lukien) huoneenlämmössä (enintään 25 °C:ssa).
Laimennettu lääkevalmiste on mikrobiologisista syistä käytettävä välittömästi. Jos valmistetta ei käytetä välittömästi laimentamisen jälkeen, käytönaikaiset säilytysajat ja säilytysolosuhteet ovat käyttäjän vastuulla, eikä valmistetta saa yleensä säilyttää yli 24:ää tuntia 2–8 °C:ssa eikä tämän jälkeen yli 12:ta tuntia (infuusioaika mukaan lukien) huoneenlämmössä (enintään 25 °C:ssa).
Säilytys
Säilytä jääkaapissa (2–8 °C).
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
XENPOZYME kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
20 mg (L:ei) 1 kpl (4057,17 €)
PF-selosteen tieto
Xenpozyme 4 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
4 mg kuiva-ainetta välikonsentraatiksi infuusionestettä varten, liuos, 5 ml:n injektiopullossa (tyypin I lasia), jossa on silikonoitu klooributyylielastomeeristä valmistettu kylmäkuivaustulppa ja alumiinikiinnitys sekä irtinapsautettava muovikorkki.
Pakkauksessa on 1, 5 tai 10 injektiopulloa. Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Xenpozyme 20 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
20 mg kuiva-ainetta välikonsentraatiksi infuusionestettä varten, liuos, 20 ml:n injektiopullossa (tyypin I lasia), jossa on silikonoitu klooributyylielastomeeristä valmistettu kylmäkuivaustulppa ja alumiinikiinnitys sekä irtinapsautettava muovikorkki.
Pakkauksessa on 1, 5, 10 tai 25 injektiopulloa. Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen kylmäkuivattu jauhe.
Käyttö- ja käsittelyohjeet
Injektiopullot on tarkoitettu vain kerta-antoon.
Infuusiot on annettava vaiheittain, mieluiten infuusiopumpulla.
Annettavan liuoksen valmistelu
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos, on saatettava käyttökuntoon injektionesteisiin käytettävällä steriilillä vedellä, laimennettava natriumkloridi-injektioliuoksella (9 mg/ml, 0,9 %) ja annettava sitten infuusiona laskimoon.
Käyttökuntoon saattaminen ja laimentaminen on toteutettava aseptisissa olosuhteissa. Suodatuslaitteita ei saa käyttää missään vaiheessa infuusioliuoksen valmistelun aikana. Käyttökuntoon saattamisen ja laimentamisen aikana on vältettävä vaahdon muodostumista.
1) Määritä käyttökuntoon saatettavien injektiopullojen määrä kyseessä olevan potilaan painon ja hänelle määrätyn annoksen mukaan.
Potilaan paino (kg) × annos (mg/kg) = potilaan annos (mg). Esimerkiksi 20 mg:n injektiopulloja käytettäessä potilaan annos (mg) jaettuna 20 mg:lla/injektiopullo = käyttökuntoon saatettavien injektiopullojen määrä. Jos injektiopullojen määrä ei ole kokonaisluku, pyöristä seuraavaan kokonaislukuun.
2) Ota tarvittava määrä injektiopulloja jääkaapista ja anna niiden lämmetä huoneenlämpöön noin 20–30 minuutin aikana.
3) Saata kunkin injektiopullon sisältö käyttökuntoon ruiskuttamalla
- 4 mg:n injektiopulloon 1,1 ml injektionesteisiin käytettävää steriiliä vettä
- 20 mg:n injektiopulloon 5,1 ml injektionesteisiin käytettävää steriiliä vettä
hitaasti, pisara kerrallaan injektiopullon sisäseinämää pitkin.
4) Kallistele ja pyörittele jokaista injektiopulloa varovasti. Jokaiseen injektiopulloon muodostuu kirkas, väritön liuos, jonka pitoisuus on 4 mg/ml.
5) Tarkista injektiopulloissa oleva käyttökuntoon saatettu liuos silmämääräisesti hiukkasten ja värimuutosten varalta. Xenpozyme-liuoksen on oltava kirkasta ja väritöntä. Jos injektiopullossa on läpinäkymättömiä hiukkasia tai värimuutoksia, sitä ei saa käyttää.
6) Vedä asianmukaisesta määrästä injektiopulloja sellainen tilavuus käyttökuntoon saatettua liuosta, joka vastaa potilaalle määrättyä annosta, ja laimenna natriumkloridi-injektioliuoksella (9 mg/ml, 0,9 %) ruiskussa tai infuusiopussissa infuusionesteen tilavuuden mukaan (ks. taulukko 12, suositeltu infuusionesteen kokonaistilavuus potilaan iän ja/tai painon mukaan).
Taulukko 12: Suositellut infuusiotilavuudet
| Paino ≥ 3 kg – < 10 kg | Paino ≥ 10 kg – < 20 kg | Paino ≥ 20 kg (< 18‑vuotiaat pediatriset potilaat) | Aikuiset potilaat (≥ 18‑vuotiaat) |
Annos (mg/kg) | Infuusionesteen kokonaistilavuus (ml) | Infuusionesteen kokonaistilavuus (ml) | Infuusionesteen kokonaistilavuus (ml) | Infuusionesteen kokonaistilavuus (ml) |
0,03 | Tilavuus vaihtelee painon mukaan | Tilavuus vaihtelee painon mukaan | 5 | ei oleellinen |
0,1 | Tilavuus vaihtelee painon mukaan | 5 | 10 | 20 |
0,3 | 5 | 10 | 20 | 100 |
0,6 | 10 | 20 | 50 | 100 |
1 | 20 | 50 | 100 | 100 |
2 | 50 | 75 | 200 | 100 |
3 | 50 | 100 | 250 | 100 |
- Jos infuusionesteen lopullinen tilavuus vaihtelee pediatrisilla potilailla potilaan painon mukaan (ks. taulukko 12):
- Valmistele infuusioliuos, jonka pitoisuus on 0,1 mg/ml, ottamalla tyhjään 10 ml:n ruiskuun 0,25 ml (1 mg) vaiheessa 3) valmisteltua käyttökuntoon saatettua liuosta ja 9,75 ml natriumkloridi-injektioliuosta (9 mg/ml, 0,9 %).
- Laske tilavuus (ml), joka tarvitaan, jotta saadaan potilaan annos (mg).
Esimerkki: 0,3 mg ÷ 0,1 mg/ml = 3 ml
- Laimennusohjeet, kun kokonaistilavuus on 5–20 ml ja laimentamisessa käytetään ruiskua:
- Ruiskuta tarvittava tilavuus käyttökuntoon saatettua liuosta hitaasti tyhjän ruiskun sisäseinämää pitkin.
- Lisää hitaasti natriumkloridi-injektioliuosta (9 mg/ml, 0,9 %) määrä, jolla saavutetaan tarvittava infuusionesteen kokonaistilavuus (vältä vaahdon muodostumista ruiskuun).
- Laimennusohjeet, kun kokonaistilavuus on ≥ 50 ml ja laimentamisessa käytetään infuusiopussia:
- Tyhjä infuusiopussi:
- Ruiskuta tarvittava tilavuus vaiheessa 3) valmisteltua käyttökuntoon saatettua liuosta hitaasti steriiliin sopivan kokoiseen infuusiopussiin.
- Lisää hitaasti natriumkloridi-injektioliuosta (9 mg/ml, 0,9 %) määrä, jolla saavutetaan tarvittava infuusionesteen kokonaistilavuus (vältä vaahdon muodostumista pussiin).
- Esitäytetty infuusiopussi:
- Vedä natriumkloridi-injektioliuoksella (9 mg/ml, 0,9 %) esitäytetystä infuusiopussista fysiologista keittosuolaliuosta määrä, jolla saavutetaan taulukossa 12 ilmoitettu lopullinen tilavuus.
- Lisää tarvittava tilavuus vaiheessa 3) valmisteltua käyttökuntoon saatettua liuosta hitaasti infuusiopussiin (vältä vaahdon muodostumista pussiin).
- Tyhjä infuusiopussi:
7) Kääntele ruiskua tai infuusiopussia varovasti, jotta sen sisältö sekoittuu. Älä ravista. Kyseessä on proteiiniliuos, joten laimentamisen jälkeen esiintyy silloin tällöin lievää flokkulaatiota (joka on kuvattu ohuiden, läpikuultavien kuitujen muodostumiseksi).
8) Laimennettu liuos on suodatettava antamisen yhteydessä kiinteän (in-line), niukasti proteiineja sitovan 0,2 μm:n suodattimen läpi.
9) Infuusion päätyttyä infuusiolinja huuhdellaan natriumkloridi-injektioliuoksella (9 mg/ml, 0,9 %) käyttäen samaa infuusionopeutta kuin infuusion viimeisessä vaiheessa.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
XENPOZYME kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
20 mg 1 kpl
- Ei korvausta.
ATC-koodi
A16AB25
Valmisteyhteenvedon muuttamispäivämäärä
13.02.2025
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi