
IMRALDI injektioneste, liuos, esitäytetty kynä 40 mg/0,4 ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Vaikuttavat aineet ja niiden määrät
Imraldi 40 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty 0,4 ml:n kerta-annoskynä sisältää 40 mg adalimumabia.
Adalimumabi on rekombinantti ihmisen monoklonaalinen vasta-aine, joka on tuotettu kiinanhamsterin munasarjasoluissa (CHO).
Apuaine(et), joiden vaikutus tunnetaan:
Tämä lääkevalmiste ei sisällä apuainetta (apuaineita), joiden vaikutus tunnetaan.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Nivelreuma
Imraldi-valmisteen ja metotreksaatin yhdistelmä on tarkoitettu:
- keskivaikeaa tai vaikeaa, aktiivista nivelreumaa sairastavien aikuispotilaiden hoitoon silloin, kun varsinaisilla taudin kulkuun vaikuttavilla reumalääkkeillä (DMARD = disease-modifying anti-rheumatic drugs), kuten metotreksaatilla, ei ole saatu riittävää vastetta
- vaikean, aktiivisen ja progressiivisen nivelreuman hoitoon aikuisilla, jotka eivät ole aiemmin saaneet metotreksaattihoitoa.
Imraldi-valmistetta voidaan antaa myös yksinään, jos potilas ei siedä metotreksaattia tai metotreksaattihoidon jatkaminen ei ole tarkoituksenmukaista.
Adalimumabin on metotreksaattiin yhdistettynä osoitettu vähentävän nivelvaurion etenemistä röntgenkuvista mitattuna ja parantavan fyysistä toimintakykyä.
Idiopaattinen juveniili artriitti
Idiopaattinen juveniili polyartriitti
Imraldi yhdessä metotreksaatin kanssa on tarkoitettu käytettäväksi aktiivisen idiopaattisen juveniilin polyartriitin hoitoon vähintään 2-vuotiailla potilailla, kun yhdellä tai useammalla DMARD-lääkkeellä ei ole saatu riittävää vastetta. Imraldi-valmistetta voidaan antaa myös yksinään, jos potilas ei siedä metotreksaattia tai metotreksaattihoidon jatkaminen ei ole tarkoituksenmukaista (monoterapian teho, ks. kohta Farmakodynamiikka). Adalimumabia ei ole tutkittu alle 2-vuotiailla potilailla.
Entesiitteihin liittyvä artriitti
Imraldi on tarkoitettu aktiivisen entesiitteihin liittyvän artriitin hoitoon 6-vuotiailla ja sitä vanhemmilla potilailla, kun tavanomaisilla hoidoilla ei ole saatu riittävää vastetta tai ne ovat olleet huonosti siedettyjä (ks. kohta Farmakodynamiikka).
Aksiaalinen spondylartriitti
Selkärankareuma
Imraldi on tarkoitettu vaikean aktiivisen selkärankareuman hoitoon aikuisilla, kun potilaan vaste tavanomaisille hoidoille on ollut riittämätön.
Aksiaalinen spondylartriitti (ilman radiografista näyttöä selkärankareumasta)
Imraldi on tarkoitettu vaikean aksiaalisen spondylartriitin (ilman radiografista näyttöä selkärankareumasta, mutta selvät merkit tulehduksesta magneettikuvissa ja/tai kohonnut CRP) hoitoon aikuisilla, jotka eivät ole saaneet riittävää vastetta steroideihin kuulumattomille tulehduskipulääkkeille NSAID‑lääkkeille tai jotka eivät siedä NSAID-hoitoa.
Nivelpsoriaasi
Imraldi on tarkoitettu aktiivisen ja progressiivisen nivelpsoriaasin hoitoon aikuisilla, kun potilaan vaste aiemmalle DMARD-hoidolle on ollut riittämätön.
Adalimumabin on osoitettu hidastavan röntgenkuvissa todettavien perifeeristen nivelvaurioiden etenemistä symmetristä polyartikulaarista nivelpsoriaasia sairastavilla potilailla (ks. kohta Farmakodynamiikka) ja parantavan potilaiden fyysistä toimintakykyä.
Psoriaasi
Imraldi on tarkoitettu keskivaikean tai vaikean läiskäpsoriaasin hoitoon aikuispotilailla, joille harkitaan systeemistä hoitoa.
Läiskäpsoriaasi lapsilla
Imraldi on tarkoitettu vaikean, kroonisen läiskäpsoriaasin hoitoon vähintään 4-vuotiailla lapsilla ja nuorilla, joilla paikallishoito ja valohoidot eivät ole tuottaneet riittävää vastetta tai joille ne eivät sovellu.
Hidradenitis suppurativa (HS-tauti)
Imraldi on tarkoitettu keskivaikean ja vaikean aktiivisen hidradenitis suppurativan (taiveaknen) hoitoon aikuisille ja nuorille 12 vuoden iästä alkaen, kun tavanomaisella systeemisellä HS-hoidolla ei ole saatu riittävää vastetta (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Crohnin tauti
Imraldi on tarkoitettu keskivaikean tai vaikean aktiivisen Crohnin taudin hoitoon aikuisille potilaille, joilla täysimääräinen ja riittävä kortikosteroidihoito ja/tai immunosuppressanttihoito ei ole saanut aikaan hoitovastetta, sekä potilaille, jotka eivät siedä tällaisia hoitoja tai joilla on tällaisille hoidoille jokin lääketieteellinen vasta-aihe.
Crohnin tauti lapsilla
Imraldi on tarkoitettu keskivaikean tai vaikean, aktiivisen Crohnin taudin hoitoon vähintään 6-vuotiailla lapsipotilailla, kun tavanomaisilla hoidoilla (mm. primaarinen ravitsemushoito ja kortikosteroidi ja/tai immunomodulantti) ei ole saavutettu riittävää vastetta tai ne ovat olleet huonosti siedettyjä tai kyseiset hoidot ovat vasta-aiheisia.
Ulseratiivinen koliitti
Imraldi on tarkoitettu keskivaikean tai vaikean aktiivisen ulseratiivisen koliitin hoitoon aikuispotilaille, joilla vaste tavanomaisille hoidoille mukaan lukien kortikosteroidi ja 6‑merkaptopuriini (6-MP) tai atsatiopriini on ollut riittämätön, tai potilaille, jotka eivät siedä tällaisia hoitoja tai joilla on tällaisille hoidoille jokin lääketieteellinen vasta-aihe.
Ulseratiivinen koliitti lapsilla
Imraldi on tarkoitettu keskivaikean tai vaikean, aktiivisen ulseratiivisen koliitin hoitoon lapsille 6 vuoden iästä alkaen, kun tavanomaisilla hoidoilla (mm. kortikosteroidit ja/tai 6-merkaptopuriini (6-MP) tai atsatiopriini (AZA)) ei ole saavutettu riittävää vastetta tai ne ovat olleet huonosti siedettyjä tai kyseiset hoidot ovat vasta-aiheisia.
Uveiitti
Imraldi on tarkoitettu ei-infektioperäisen intermediaarisen, posteriorisen ja panuveiitin hoitoon aikuispotilaille, joilla vaste kortikosteroideille on riittämätön, joiden kortikosteroidien käyttöä täytyy rajoittaa, tai joille kortikosteroidihoito ei sovi.
Uveiitti lapsilla
Imraldi on tarkoitettu pediatrisen kroonisen ei-infektioperäisen anteriorisen uveiitin hoitoon 2 vuoden iästä alkaen, kun tavanomaiselle hoidolle ei ole saatu riittävää vastetta tai tavanomainen hoito on ollut huonosti siedettyä, tai se ei ole tarkoituksenmukaista.
Ehto
Hoito tulee toteuttaa indikaation mukaiseen hoitoon perehtyneen lääkärin aloittamana ja valvonnassa. Hoitoa saaville potilaille tulee antaa erityinen potilaskortti.
Annostus ja antotapa
Imraldi-hoito tulee toteuttaa indikaation mukaiseen diagnosointiin ja hoitoon perehtyneen lääkärin aloittamana ja valvonnassa. Silmätautien erikoislääkäreitä kehotetaan konsultoimaan asianmukaista erikoislääkäriä ennen Imraldi-hoidon aloitusta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Imraldi-hoitoa saaville potilaille on annettava Potilaskortti.
Kun potilas hallitsee pistämistekniikan kunnolla, hän voi pistää Imraldi-annoksensa itse, jos lääkäri pitää tätä soveliaana ja seuraa tarvittaessa potilaan tilaa.
Muut samanaikaiset hoidot (esim. kortikosteroidit ja/tai immunomoduloivat hoidot) tulee optimoida Imraldi-hoidon aikana.
Annostus
Nivelreuma
Imraldi-valmisteen annossuositus aikuisille nivelreumapotilaille on 40 mg adalimumabia kerta-annoksena joka toinen viikko pistoksena ihon alle. Metotreksaattihoitoa tulee jatkaa Imraldi-hoidon aikana.
Glukokortikoidien, salisylaattien, steroideihin kuulumattomien tulehduskipulääkkeiden (NSAID‑lääkkeiden) tai kipulääkkeiden käyttöä voidaan jatkaa Imraldi-hoidon aikana. Yhdistäminen muihin taudin kulkuun vaikuttaviin lääkkeisiin metotreksaattia lukuun ottamatta, ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka.
Jos vaste Imraldi-annostukselle 40 mg joka toinen viikko heikkenee monoterapiahoidon aikana, voi annoksen nostaminen tasolle 40 mg adalimumabia kerran viikossa tai 80 mg joka toinen viikko hyödyttää osaa potilaista.
Saatavilla olevien tietojen perusteella hoitovaste saavutetaan yleensä 12 viikon kuluessa. Jos hoitovastetta ei saavuteta tämän ajan kuluessa, hoidon jatkamista tulee harkita uudelleen.
Käytön keskeyttäminen
Imraldi-valmisteen käyttö voi olla tarpeen keskeyttää esimerkiksi leikkauksen tai vakavan infektion takia.
Saatavilla olevien tietojen perusteella adalimumabin uudelleen aloittaminen 70 päivän tai pidemmän tauon jälkeen johti samansuuruiseen kliiniseen vasteeseen ja samanlaiseen turvallisuusprofiiliin kuin ennen taukoa.
Selkärankareuma, aksiaalinen spondylartriitti (ilman radiografista näyttöä selkärankareumasta) ja nivelpsoriaasi
Imraldi-valmisteen suositusannos selkärankareumaa, aksiaalista spondylartriittia (ilman radiografista näyttöä selkärankareumasta) ja nivelpsoriaasia sairastaville potilaille on 40 mg adalimumabia kerta-annoksena joka toinen viikko pistoksena ihon alle.
Saatavilla olevien tietojen perusteella hoitovaste saavutetaan yleensä 12 viikon kuluessa. Jos hoitovastetta ei saavuteta tämän ajan kuluessa, hoidon jatkamista tulee harkita uudelleen.
Psoriaasi
Aikuispotilailla suositellaan Imraldi-hoidon aloittamista aloitusannoksella 80 mg ihon alle, minkä jälkeen potilas jatkaa annoksella 40 mg ihon alle joka toinen viikko. Ensimmäinen 40 mg:n annos otetaan viikon kuluttua aloitusannoksesta.
Jos potilaalla ei saavuteta vastetta 16 viikon hoidon aikana, hoidon jatkamista tulee harkita tarkoin.
Potilaat, joilla vaste ei ole riittävä Imraldi-annostuksella 40 mg joka toinen viikko 16 viikon hoidon jälkeen, voivat hyötyä Imraldi-annoksen nostamisesta 40 milligrammaan kerran viikossa tai 80 milligrammaan joka toinen viikko. Jos potilaalla ei saavuteta riittävää vastetta annoksen nostamisen jälkeen, viikoittaisen 40 mg annoksen tai joka toinen viikko otettavan 80 mg annoksen jatkamisen hyödyt ja riskit tulee harkita tarkoin (ks. kohta Farmakodynamiikka). Jos riittävä vaste saavutetaan annostuksella 40 mg kerran viikossa tai 80 mg joka toinen viikko, voidaan annos myöhemmin laskea takaisin 40 milligrammaan joka toinen viikko.
Hidradenitis suppurativa
Hidradenitis suppurativaa (HS-tautia) sairastaville aikuispotilaille suositeltava Imraldi-annostus on ensin 160 mg päivänä 1 (neljä 40 mg:n injektiota samana päivänä tai kaksi 40 mg:n injektiota vuorokaudessa kahtena peräkkäisenä päivänä) ja tämän jälkeen 80 mg kaksi viikkoa myöhemmin päivänä 15 (kaksi 40 mg:n injektiota samana päivänä). Kaksi viikkoa myöhemmin (päivänä 29) siirrytään käyttämään 40 mg:n annosta kerran viikossa tai 80 mg annosta joka toinen viikko (kaksi 40 mg injektiota samana päivänä). Antibioottihoitoa voidaan tarvittaessa jatkaa Imraldi-hoidon aikana. On suositeltavaa, että potilas käsittelee HS-leesiot paikallisesti käytettävällä antiseptisella ihohuuhteella päivittäin Imraldi-hoidon aikana.
Jos oireet eivät lievity 12 viikon kuluessa, hoidon jatkamista on harkittava tarkoin.
Jos hoito on keskeytettävä, hoito voidaan aloittaa uudelleen Imraldi-annostuksella 40 mg kerran viikossa tai 80 mg joka toinen viikko (ks. kohta Farmakodynamiikka).
Pitkäaikaishoidon jatkamisen hyödyt ja riskit on arvioitava säännöllisin väliajoin (ks. kohta Farmakodynamiikka).
Crohnin tauti
Keskivaikeaa tai vaikeaa aktiivista Crohnin tautia sairastavien aikuispotilaiden Imraldi-hoito tulisi aloittaa annostuksella 80 mg viikolla 0 ja 40 mg viikolla 2. Jos hoitovaste on saavutettava nopeammin, voidaan hoito aloittaa antamalla potilaalle 160 mg Imraldi-valmistetta viikolla 0 (joko neljä 40 mg injektiota samana päivänä tai kaksi 40 mg injektiota päivässä kahtena peräkkäisenä päivänä) ja sen jälkeen 80 mg viikolla 2 (kaksi 40 mg injektiota samana päivänä). Haittatapahtumariskin suureneminen hoidon aloitusvaiheessa on kuitenkin otettava huomioon.
Aloitusvaiheen jälkeen suositusannos on 40 mg injektiona ihon alle joka toinen viikko. Mikäli potilas on keskeyttänyt Imraldi-hoidon ja taudin merkit ja oireet uusiutuvat, Imraldi voidaan aloittaa uudelleen. Hoidon uudelleenaloittamisesta tilanteessa, jossa edellisen annoksen antamisesta on kulunut yli 8 viikkoa, on vain vähän kokemusta.
Kortikosteroidiannosta voidaan pienentää ylläpitohoidon aikana hoitosuositusten mukaisesti.
Imraldi-annoksen nostamisesta 40 milligrammaan kerran viikossa tai 80 milligrammaan joka toinen viikko voi olla hyötyä joillekin potilaille, joiden hoitovaste annostuksella 40 mg joka toinen viikko osoittaa heikkenemisen merkkejä.
Myös sellaiset potilaat, jotka eivät ole saaneet vastetta viikkoon 4 mennessä, voivat hyötyä ylläpitohoidon jatkamisesta 12 viikkoon asti. Hoidon jatkamista on harkittava huolellisesti uudelleen, jos potilaalla ei saavuteta 12 viikossa hoitovastetta.
Ulseratiivinen koliitti
Keskivaikeaa tai vaikeaa ulseratiivista koliittia sairastavien aikuispotilaiden suositettu aloitusannos on 160 mg viikolla 0 (joko neljä 40 mg injektiota samana päivänä tai kaksi 40 mg injektiota päivässä kahtena peräkkäisenä päivänä) ja 80 mg viikolla 2 (kaksi 40 mg injektiota samana päivänä). Aloitusvaiheen jälkeen suositusannos on 40 mg injektiona ihon alle joka toinen viikko.
Kortikosteroidiannosta voidaan pienentää ylläpitohoidon aikana hoitosuositusten mukaisesti.
Imraldi-annoksen nostamisesta 40 milligrammaan kerran viikossa tai 80 milligrammaan joka toinen viikko voi olla hyötyä joillekin potilaille, joiden hoitovaste annostuksella 40 mg joka toinen viikko osoittaa heikkenemisen merkkejä.
Saatavilla olevien tietojen perusteella hoitovaste saavutetaan yleensä 2–8 viikon kuluessa. Imraldi-hoitoa ei tule jatkaa potilailla, jotka eivät saa vastetta tässä ajassa.
Uveiitti
Uveiittia sairastaville aikuispotilaille suositeltava aloitusannos on 80 mg, jonka jälkeen annetaan 40 mg joka toinen viikko alkaen viikon kuluttua aloitusannoksesta. Kokemusta hoidon aloituksesta yksinään Imraldi-valmisteella on rajallisesti. Imraldi-hoidon voi aloittaa yhdessä kortikosteroidin ja/tai toisen ei-biologisen immunomoduloivan hoidon kanssa. Samanaikaista kortikosteroidiannosta voidaan pienentää hoitosuositusten mukaisesti kahden viikon kuluttua Imraldi-hoidon aloituksesta.
On suositeltavaa, että jatkuvan, pitkäaikaisen hoidon hyödyt ja riskit arvioidaan vuosittain (ks. kohta Farmakodynamiikka).
Erityisryhmät
Iäkkäät potilaat
Annoksen muuttaminen ei ole tarpeen.
Munuaisten ja/tai maksan vajaatoiminta
Adalimumabia ei ole tutkittu näissä potilasryhmissä. Annossuosituksia ei voida antaa.
Pediatriset potilaat
Esitäytetty Imraldi‑kynä on saatavana vain 40 mg:n annoksena. Tämän vuoksi esitäytettyä Imraldi‑kynää ei voida käyttää pediatrisille potilaille, joille pitää antaa kokonaista 40 mg:n annosta pienempi annos. Jos tarvitaan vaihtoehtoinen annos, on käytettävä muita pakkausmuotoja, joista on tarjolla sopiva vaihtoehto.
Idiopaattinen juveniili artriitti
Idiopaattinen juveniili polyartriitti 2 vuoden iästä alkaen
Idiopaattista juveniilia polyartriittia sairastavien potilaiden Imraldi suositeltu kerta-annos 2 vuoden iästä alkaen lasketaan potilaan painon perusteella (taulukko 1) Imraldi annostellaan joka toinen viikko injektiona ihon alle.
Taulukko 1. Imraldi-annos idiopaattista juveniilia polyartriittia sairastaville potilaille
Potilaan paino | Annos |
10 kg‑< 30 kg | 20 mg joka toinen viikko |
≥ 30 kg | 40 mg joka toinen viikko |
Kliininen vaste saavutetaan yleensä 12 hoitoviikossa. Jos potilas ei saavuta vastetta tämän ajan kuluessa, hoidon jatkamista on harkittava tarkoin.
Ei ole asianmukaista käyttää adalimumabia alle 2 vuoden ikäisille tämän käyttöaiheen hoidossa.
Entesiitteihin liittyvä artriitti
Entesiitteihin liittyvää artriittia sairastavilla potilailla Imraldi-valmisteen suositeltu kerta-annos 6 vuoden iästä alkaen lasketaan potilaan painon perusteella (taulukko 2). Imraldi annostellaan joka toinen viikko injektiona ihon alle.
Taulukko 2. Imraldi-annos entesiitteihin liittyvää artriittia sairastaville potilaille
Potilaan paino | Annos |
15 kg‑< 30 kg | 20 mg joka toinen viikko |
≥ 30 kg | 40 mg joka toinen viikko |
Adalimumabia ei ole tutkittu entesiitteihin liittyvää artriittia sairastavilla alle 6-vuotiailla potilailla.
Läiskäpsoriaasi lapsilla
Läiskäpsoriaasia sairastavien 4–17-vuotiaiden potilaiden suositeltu kerta-annos lasketaan painon perusteella (taulukko 3). Imraldi annostellaan injektiona ihon alle.
Taulukko 3. Imraldi-annos läiskäpsoriaasia sairastaville lapsipotilaille
Potilaan paino | Annos |
15 kg‑< 30 kg | Aloitusannos 20 mg, jonka jälkeen 20 mg joka toinen viikko alkaen viikon kuluttua aloitusannoksesta. |
≥ 30 kg | Aloitusannos 40 mg, jonka jälkeen 40 mg joka toinen viikko alkaen viikon kuluttua aloitusannoksesta. |
Jos potilas ei saavuta vastetta 16 viikon kuluessa, hoidon jatkamista on harkittava tarkoin.
Jos uusintahoito Imraldi-valmisteella on aiheellista, noudatetaan edellä annettuja ohjeita annoksesta ja hoidon kestosta.
Adalimumabin turvallisuutta on arvioitu pediatrisilla läiskäpsoriaasipotilailla keskimäärin 13 kuukauden ajan.
Ei ole asianmukaista käyttää adalimumabia alle 4 vuoden ikäisille lapsille tämän käyttöaiheen hoidossa.
Hidradenitis suppurativa nuorilla (12 vuoden iästä alkaen, kun paino on vähintään 30 kg)
Nuorilla HS-tautia sairastavilla potilailla ei ole tehty kliinisiä adalimumabitutkimuksia. Adalimumabin annostus näillä potilailla on määritetty farmakologisen mallinnuksen ja simulaation avulla (ks. kohta Farmakokinetiikka).
Suositeltu Imraldi-annos on 80 mg ihonalaisena injektiona viikolla 0, jonka jälkeen 40 mg joka toinen viikko alkaen viikosta 1.
Nuorille potilaille, joilla vaste 40 mg:n Imraldi-annokselle joka toinen viikko on riittämätön, voidaan harkita annoksen nostamista 40 mg:aan joka viikko tai 80 mg:aan joka toinen viikko.
Antibioottihoitoa voidaan tarvittaessa jatkaa Imraldi-hoidon aikana. On suositeltavaa, että potilas käsittelee HS-leesiot paikallisesti käytettävällä antiseptisella ihohuuhteella päivittäin Imraldi-hoidon aikana.
Jos oireet eivät lievity 12 viikon kuluessa, hoidon jatkamista on harkittava tarkoin.
Jos hoito on keskeytettävä, Imraldi voidaan aloittaa uudelleen tarvittaessa.
Pitkäaikaishoidon jatkamisen hyödyt ja riskit on arvioitava säännöllisin väliajoin (ks. aikuisia koskeva tieto kohdasta Farmakodynamiikka).
Ei ole asianmukaista käyttää adalimumabia alle 12 vuoden ikäisille tämän käyttöaiheen hoidossa.
Crohnin tauti lapsilla
Crohnin tautia sairastavien 6–17-vuotiaiden potilaiden suositeltu kerta-annos lasketaan painon perusteella (taulukko 4). Imraldi annostellaan injektiona ihon alle.
Taulukko 4. Imraldi-annos Crohnin tautia sairastavilla pediatrisilla potilailla
Potilaan paino | Aloitusannos | Ylläpito annos alkaen viikolta 4 |
< 40 kg |
Jos nopeampi hoitovaste on tarpeen, voidaan antaa
On kuitenkin muistettava, että suurempia induktioannoksia käytettäessä haittatapahtumariski voi olla suurempi. | 20 mg joka toinen viikko |
≥ 40 kg |
Jos nopeampi hoitovaste on tarpeen, voidaan antaa
On kuitenkin muistettava, että suurempia induktioannoksia käytettäessä haittatapahtumariski voi olla suurempi. | 40 mg joka toinen viikko |
Potilaat, joilla vaste on riittämätön, saattavat hyötyä annoksen nostamisesta:
- < 40 kg: 20 mg joka viikko
- ≥ 40 kg: 40 mg joka viikko tai 80 mg joka toinen viikko.
Hoidon jatkamista on harkittava tarkoin, jos potilas ei saavuta vastetta viikkoon 12 mennessä.
Ei ole asianmukaista käyttää adalimumabia < 6 vuoden ikäisille tämän käyttöaiheen hoidossa.
Ulseratiivinen koliitti lapsilla
Ulseratiivista koliittia sairastavien 6–17-vuotiaiden potilaiden suositeltu Imraldi-annos lasketaan painon perusteella (taulukko 5). Imraldi annostellaan injektiona ihon alle.
Taulukko 5. Imraldi-annos ulseratiivista koliittia sairastavilla pediatrisilla potilailla
Potilaan paino | Aloitusannos | Ylläpitoannos alkaen viikolta 4* |
< 40 kg |
|
|
≥ 40 kg |
|
|
* Pediatristen potilaiden, jotka täyttävät 18 vuotta Imraldi-hoidon aikana, tulee jatkaa hoitoa heille määrätyllä ylläpitoannoksella.
Hoidon jatkamista on harkittava tarkoin, jos potilas ei saavuta vastetta viikkoon 8 mennessä.
Ei ole asianmukaista käyttää Imraldia alle 6 vuoden ikäisille tämän käyttöaiheen hoidossa.
Imraldia voi olla saatavilla myös muita vahvuuksia ja/tai annostelumuotoja riippuen yksilöllisestä hoidon tarpeesta.
Nivelpsoriaasi ja aksiaalinen spondylartriitti (myös selkärankareuma)
Ei ole asianmukaista käyttää adalimumabia pediatrisille potilaille selkärankareuman ja nivelpsoriaasin hoidossa.
Uveiitti lapsilla
Uveiittia sairastavien lapsipotilaiden suositeltu Imraldi‑kerta-annos 2 vuoden iästä alkaen lasketaan painon perusteella (taulukko 6). Imraldi pistetään injektiona ihon alle.
Ei ole olemassa kokemuksia lasten uveiitin hoidosta adalimumabilla ilman samanaikaista metotreksaattihoitoa.
Taulukko 6. Imraldi-annos uveiittia sairastavilla pediatrisilla potilailla
Potilaan paino | Annos |
< 30 kg | 20 mg joka toinen viikko yhdessä metotreksaatin kanssa |
≥ 30 kg | 40 mg joka toinen viikko yhdessä metotreksaatin kanssa |
Kun Imraldi-hoito aloitetaan, voidaan viikkoa ennen ylläpitohoidon aloitusta antaa 40 mg latausannos < 30 kg potilaille tai 80 mg latausannos ≥ 30 kg potilaille. Kliinistä tutkimustietoa adalimumabi-latausannoksen käytöstä alle 6-vuotiailla lapsilla ei ole (ks. kohta Farmakokinetiikka).
Ei ole asianmukaista käyttää adalimumabia alle 2 vuoden ikäisille tämän käyttöaiheen hoidossa.
Pitkäaikaisen hoidon hyödyt ja riskit on suositeltavaa arvioida vuosittain (ks. kohta Farmakodynamiikka).
Antotapa
Imraldi pistetään ihon alle (subkutaanisesti). Katso käyttöohjeet pakkausselosteesta.
Saatavana on 40 mg:n esitäytetty kynä potilaskäyttöön täyden 40 mg:n annoksen pistämistä varten.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiivinen tuberkuloosi tai jokin muu vakava infektio, kuten sepsis, sekä opportunistiset infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Keskivaikea tai vaikea sydämen vajaatoiminta (NYHA III/IV) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Potilaat, jotka käyttävät TNF-antagonisteja ovat herkempiä vakaville infektioille. Heikentynyt keuhkojen toiminta voi lisätä infektioiden kehittymisen riskiä. Potilaita tulee täten seurata huolellisesti infektioiden (mm. tuberkuloosin) varalta ennen Imraldi-hoidon aloittamista, hoidon aikana ja sen jälkeen. Koska adalimumabin eliminoituminen voi viedä neljä kuukautta, seurantaa tulee jatkaa siihen saakka.
Imraldi-hoitoa ei tule aloittaa potilailla, joilla on aktiivinen infektio (krooninen tai paikallinen), ennen kuin infektio on hallinnassa. Potilailla, jotka ovat altistunet tuberkuloosille sekä potilailla, jotka ovat matkustaneet korkean tuberkuloosin tai endeemisen mykoosin (kuten histoplasmoosi, koksidioidomykoosi tai blastomykoosi) riskialueilla, Imraldi-hoidon hyödyt ja riskit on huomioitava ennen hoidon aloittamista (ks. Muut opportunistiset infektiot).
Potilaita, joille kehittyy uusi infektio Imraldi-hoidon aikana, tulee seurata huolellisesti ja heille on tehtävä täydellinen diagnostinen arvio. Jos potilaalle kehittyy uusi, vakava infektio tai sepsis, tarvittava antimikrobinen tai antimykoottinen hoito on aloitettava ja Imraldi-valmisteen antaminen on keskeytettävä välittömästi, kunnes infektio saadaan hallintaan. Lääkärien tulee noudattaa varovaisuutta harkitessaan Imraldi-valmisteen käyttöä potilailla, joilla on ollut toistuvia infektioita tai perussairaus, joka voi altistaa infektioille mukaan lukien samanaikainen immunosuppressiivisen lääkityksen käyttö.
Vakavat infektiot
Vakavia infektioita, mukaan lukien sepsis, joka johtuu bakteereista, mykobakteereista, invasiivisista sieni-infektioista, parasiiteista, viruksista tai muista opportunistisista infektioista, kuten listerioosista, legionelloosista ja pneumokystoosista, on raportoitu adalimumabia käyttävillä potilailla.
Muita vakavia infektioita, joita on ilmennyt kliinisissä tutkimuksissa, sisältävät pneumonian, pyelonefriitin, septisen niveltulehduksen ja sepsiksen kaltaisia infektioita. Infektioihin liittyvää sairaalahoitoa tai kuolemaan johtavia tapauksia on raportoitu.
Tuberkuloosi
Adalimumabihoitoa saaneilla potilailla on raportoit tuberkuloosia, mukaan lukien tuberkuloosin reaktivaatiota ja uusia tapauksia. Raportit sisälsivät sekä keuhko- että keuhkojen ulkopuolista (eli disseminoitunutta) tuberkuloosia.
Ennen Imraldi-hoidon aloittamista tulee kaikki potilaat tutkia sekä aktiivisen että inaktiivisen (”latentin”) tuberkuloosin varalta. Tähän tutkimukseen tulee kuulua huolellinen potilaan arviointi, jotta saadaan selville potilaan aikaisemmin sairastama tuberkuloosi ja aiemmat kontaktit aktiivista tuberkuloosia sairastavan henkilön kanssa sekä aikaisemmat ja/tai käynnissä olevat immunosuppressiiviset hoidot. Asianmukaisia seulontatestejä (tuberkuliinikoe ja keuhkoröntgen) voidaan joutua tekemään kaikille potilaille (paikallisten vaatimusten mukaisesti). Nämä kokeet ja niiden tulokset on suositeltavaa merkitä Potilaskorttiin. Lääkäriä muistutetaan, että tuberkuliinikokeessa saatetaan saada väärä negatiivinen tulos, jos kyseessä on vakavasti sairas tai immuunivajavuudesta kärsivä potilas.
Jos todetaan aktiivinen tuberkuloosi, Imraldi-hoitoa ei saa aloittaa (ks. kohta Vasta-aiheet).
Alla kuvatuissa tilanteissa Imraldi-hoidon hyötyjen ja riskien suhdetta on harkittava hyvin huolellisesti.
Jos potilaalla epäillään latenttia tuberkuloosia, on tuberkuloosin hoitoon perehtynyttä lääkäriä konsultoitava.
Jos potilaalla todetaan latentti tuberkuloosi, sen hoito profylaktisella tuberkuloosihoidolla on käynnistettävä ennen Imraldi-hoidon aloittamista ja paikallisten suositusten mukaisesti.
Profylaktista tuberkuloosihoitoa on harkittava ennen Imraldi-hoidon aloittamista myös siinä tapauksessa, että potilaalla on useita tai merkittäviä tuberkuloosin riskitekijöitä, mutta latentin tuberkuloosin osoituskoe on negatiivinen, ja potilaalla, jolla on aiemmin ollut latentti tai aktiivinen tuberkuloosi, eikä hoidon asianmukaisesta toteutuksesta ole varmuutta.
Adalimumabihoitoa saaneilla potilailla on ilmennyt tuberkuloosin reaktivaatiotapauksia tuberkuloosin profylaktisesta hoidosta huolimatta. Joillekin potilaille, joita on aiemmin hoidettu menestyksekkäästi aktiivisen tuberkuloosin vuoksi, on uudelleen kehittynyt tuberkuloosi adalimumabihoidon aikana.
Potilaita tulee myös kehottaa kääntymään lääkärin puoleen, jos heillä esiintyy tuberkuloosi-infektioon viittaavia merkkejä/oireita (esim. sitkeää yskää, laihtumista/painon laskua, lämpöilyä, voimattomuutta) Imraldi-hoidon aikana tai sen jälkeen.
Muut opportunistiset infektiot
Adalimumabia saavilla potilailla on tavattu opportunistisia infektioita, kuten invasiivisia sieni-infektioita. TNF-antagonisteja saavilla potilailla näitä infektioita ei ole välttämättä tunnistettu ja tämä johti asianmukaisen hoidon viivästymiseen ja joskus kuolemaan.
Jos potilaalle kehittyy sellaisia merkkejä ja oireita kuten kuume, huonovointisuus, painonlasku, hikoilu, yskä, hengenahdistus, ja/tai keuhkoinfiltraatteja tai muita vakavia systeemisiä sairauksia, liittyi niihin sokki tai ei, invasiivista sieni-infektiota tulisi epäillä ja Imraldi-valmisteen anto lopettaa viipymättä. Diagnoosi ja empiirisen antifungaalisen hoidon aloitus tulisi tehdä yhteistyössä lääkärin kanssa, jolla on kokemusta invasiivisten sieni-infektioiden hoidosta.
B-hepatiitin uudelleenaktivoituminen
B-hepatiittiviruksen kroonisilla kantajilla (eli pinta-antigeenipositiivisilla) on todettu B-hepatiitin uudelleenaktivoitumista TNF-salpaajahoidon, myös adalimumabihoidon, aikana. Jotkin tapaukset ovat johtaneet kuolemaan. Potilaat on tutkittava HBV-infektion varalta ennen Imraldi-hoidon aloittamista. Jos potilas osoittautuu HBV-tartunnan kantajaksi, suositellaan hepatiitti B-infektion hoitoon perehtyneen lääkärin konsultoimista.
Jos HBV-kantaja tarvitsee ehdottomasti Imraldi-hoitoa, häntä on seurattava tarkasti aktiiviseen HBV-infektioon viittaavien merkkien ja oireiden varalta koko hoitojakson ajan ja useita kuukausia hoidon päättymisen jälkeen. TNF-salpaajahoitoa saavien HBV-kantajien hoitamisesta samanaikaisesti viruslääkkeillä B-hepatiitin uudelleenaktivoitumisen estämiseksi ei ole riittävästi tietoa. Jos potilaan B-hepatiitti aktivoituu uudelleen, on Imraldi-hoito lopetettava ja tehokas viruslääkitys ja asianmukainen tukihoito aloitettava.
Neurologiset tapahtumat
TNF-salpaajiin, kuten adalimumabiin, on harvinaisissa tapauksissa liittynyt keskushermoston demyelinoivan sairauden (mukaan lukien MS-tauti ja optikusneuriitti) ja ääreishermoston demyelinoivan sairauden (mukaan lukien Guillain-Barrén oireyhtymä) kliinisten oireiden ja/tai röntgenlöydösten ilmeneminen tai paheneminen. Varovaisuutta tulee siis noudattaa määrättäessä Imraldi-valmistetta potilaille, joilla on joko aiemmin tai hiljattain alkanut keskus- tai ääreishermoston myeliinikatosairaus. Imraldi-hoidon keskeytystä on harkittava, jos potilaalle kehittyy jokin näistä sairauksista. Intermediaarisen uveiitin ja keskushermoston demyelinoivien sairauksien välillä on tunnettu yhteys. Potilaille, joilla on ei-infektioperäinen intermediaarinen uveiitti, on ennen Imraldi-hoidon aloitusta sekä säännöllisesti hoidon aikana tehtävä neurologinen arvio, jossa arvioidaan aiemmin alkaneita tai kehittymässä olevia keskushermoston demyelinoivia sairauksia.
Allergiset reaktiot
Kliinisissä tutkimuksissa adalimumabihoitoon liittyvät vakavat allergiset reaktiot olivat harvinaisia. Adalimumabiin liitetyt lievät allergiset reaktiot olivat kliinisissä tutkimuksissa melko harvinaisia. Adalimumabin annon yhteydessä on raportoitu vakavia allergisia reaktioita, mukaan lukien anafylaksia. Jos anafylaktinen tai muu vakava allerginen reaktio ilmenee, tulee Imraldi-valmisteen antaminen keskeyttää välittömästi ja aloittaa asianmukainen hoito.
Immunosuppressio
Tutkimuksessa, jossa 64 nivelreumapotilasta sai adalimumabihoitoa, ei havaittu viivästyneen yliherkkyysreaktion heikkenemistä, immunoglobuliinipitoisuuden pienenemistä eikä muutoksia efektori T- tai B-solujen, luonnollisten tappajasolujen, monosyyttien/makrofagien eikä neutrofiilien määrässä.
Maligniteetit ja lymfoproliferatiiviset häiriöt
TNF-antagonisteilla tehtyjen kliinisten tutkimusten kontrolloiduissa osioissa TNF-antagonistia saaneilla potilailla on havaittu enemmän maligniteetteja, mukaan lukien lymfoomia, kuin verrokeilla. Niitä esiintyi kuitenkin harvoin. Leukemiaa on raportoitu markkinoilletulon jälkeen potilailla, joita hoidettiin TNF-antagonisteilla. Lymfooman ja leukemian taustariski on suurentunut nivelreumapotilailla, joilla on hyvin aktiivinen, pitkään kestänyt tulehduksellinen tauti, mikä vaikeuttaa riskin arviointia. Tämänhetkisten tietojen perusteella lymfoomien, leukemian ja muiden maligniteettien kehittymisriskiä ei voida sulkea pois TNF-antagonistihoitoa saavien potilaiden kohdalla.
Maligniteetteja, myös kuolemaan johtaneita, on ilmoitettu lapsilla, nuorilla ja nuorilla aikuisilla (alle 22-vuotiailla), jotka ovat saaneet TNF-antagonistihoitoa (hoidon aloitusikä ≤ 18 vuotta), markkinoilletulon jälkeinen adalimumabihoito mukaan lukien. Noin puolet tapauksista oli lymfoomia. Muut tapaukset olivat erilaisia maligniteetteja, ja mukana oli harvinaisia, yleensä immunosuppressioon liittyviä muotoja. Maligniteettiriskiä ei voida sulkea pois lapsilla ja nuorilla, jotka saavat TNF-antagonistihoitoa.
Adalimumabilla hoidetuilla potilailla on markkinoilletulon jälkeen raportoitu harvoin hepatospleenistä T-solulymfoomaa. Tämä harvinainen T-solulymfooma on taudinkuvaltaan aggressiivinen ja tavallisesti fataali. Osa adalimumabihoidon aikana ilmenneistä hepatospleenisistä T-solulymfoomista on ilmennyt nuorilla aikuisilla, joilla on hoidettu tulehduksellista suolistotautia samanaikaisesti atsatiopriinilla tai 6-merkaptopuriinilla. Atsatiopriinin tai 6-merkaptopuriinin ja adalimumabin yhdistelmähoitoon liittyvä mahdollinen riski on huomioitava. Hepatospleenisen T-solulymfooman kehittymisen riskiä ei voida poissulkea potilailla, joita hoidetaan Imraldi-valmisteella (ks. kohta Haittavaikutukset).
Tutkimuksia ei ole tehty potilailla, joilla on ollut jokin maligniteetti ennen adalimumabihoitoa, tai joilla adalimumabihoitoa olisi jatkettu maligniteetin kehittymisen jälkeen. Siksi erityistä varovaisuutta tulee noudattaa harkittaessa adalimumabihoitoa näille potilaille (ks. kohta Haittavaikutukset).
Kaikki potilaat tulee tutkia muiden ihosyöpien kuin melanooman varalta ennen Imraldi-hoitoa ja sen aikana. Tämä koskee etenkin potilaita, jotka ovat käyttäneet runsaasti immunosuppressiivisia hoitoja, sekä PUVA-hoitoa saaneita psoriaasipotilaita. TNF-salpaajia, kuten adalimumabia, saaneilla potilailla on ilmoitettu esiintyneen myös melanoomaa ja merkelinsolukarsinoomaa (ks. kohta Haittavaikutukset).
Eksploratiivisessa kliinisessä tutkimuksessa, jossa arvioitiin erään toisen TNF-antagonistin, infliksimabin, käyttöä potilailla, joilla oli keskivaikea tai vaikea keuhkoahtaumatauti, infliksimabihoitoa saaneilla potilailla ilmoitettiin enemmän maligniteetteja etenkin keuhkojen, pään ja kaulan alueella kuin verrokkipotilailla. Kaikki potilaat olivat aiemmin tupakoineet runsaasti. Tämän vuoksi varovaisuutta on noudatettava, kun keuhkoahtaumatautipotilaita hoidetaan millä tahansa TNF-antagonistilla, samoin kuin potilailla, joiden maligniteettiriski on suurentunut runsaan tupakoinnin vuoksi.
Tämänhetkisen tiedon perusteella ei tiedetä vaikuttaako adalimumabihoito dysplasian tai paksusuolen syövän kehittymisen riskiin. Kaikki ulseratiivista koliittia sairastavat potilaat, joilla on lisääntynyt riski dysplasiaan tai paksusuolen syöpään (esimerkiksi potilaat, joilla on pitkäaikainen ulseratiivinen koliitti tai primaarinen sklerosoiva kolangiitti), tai potilaat, joilla on aiemmin ollut dysplasia tai paksusuolen syöpä, tulee seuloa dysplasian varalta säännöllisin väliajoin ennen hoidon aloitusta ja hoidon aikana. Seulonnan tulisi sisältää kolonoskopia ja biopsia paikallisten suositusten mukaisesti.
Verenkuvamuutokset
Pansytopeniaa, mukaan lukien aplastista anemiaa, on ilmoitettu harvinaisissa tapauksissa TNF-salpaajien käytön yhteydessä. Hematologiseen järjestelmään kohdistuvia haittavaikutuksia, kuten lääketieteellisesti merkitsevää sytopeniaa (esim. trombosytopenia, leukopenia) on ilmoitettu adalimumabihoidon yhteydessä. Kaikkia potilaita tulee kehottaa hakeutumaan välittömästi lääkärin hoitoon, jos heille kehittyy Imraldi-hoidon aikana verenkuvamuutoksiin viittaavia merkkejä ja oireita (esim. sitkeä kuume, mustelmanmuodostus, verenvuoto, kalpeus). Imraldi-hoidon keskeyttämistä tulee harkita, jos potilaalla todetaan merkitseviä hematologisia poikkeavuuksia.
Rokotukset
Kun 226 aikuista nivelreumapotilasta sai adalimumabi- tai lumelääkehoitoa, havaitut vasta-ainevasteet tavanomaiselle 23-valentille pneumokokkirokotteelle ja trivalentille influenssavirusrokotteelle olivat samankaltaiset. Elävien rokotteiden aiheuttamista infektioiden sekundaarisista siirtymistä ei ole tietoa adalimumabihoitoa saaneilla potilailla.
Kaikkien lapsipotilaiden kohdalla on suositeltavaa varmistaa mahdollisuuksien mukaan, että potilas saa kaikki ajankohtaisten rokotussuositusten mukaiset rokotukset ennen adalimumabihoidon aloittamista.
Potilaille voidaan antaa rokotuksia adalimumabihoidon aikana eläviä rokotteita lukuun ottamatta. Elävien rokotteiden (esim. BCG-rokotteen) antamista vauvoille, jotka ovat altistuneet adalimumabille kohdussa, ei suositella 5 kuukauteen äidin viimeisestä raskaudenaikaisesta adalimumabi-injektiosta.
Kongestiivinen sydämen vajaatoiminta
Erästä toista TNF-salpaajaa koskevassa kliinisessä tutkimuksessa havaittiin kongestiivisen sydämen vajaatoiminnan pahenemista ja kongestiivisesta sydämen vajaatoiminnasta johtuvan kuolleisuuden lisääntymistä. Myös adalimumabihoidon aikana on raportoitu kongestiivisen sydämen vajaatoiminnan pahenemista. Imraldi-valmisteen käytössä on noudatettava varovaisuutta potilailla, joilla on lievä sydämen vajaatoiminta (NYHA I/II). Keskivaikea ja vaikea sydämen vajaatoiminta ovat Imraldi-valmisteen käytön vasta-aiheita (ks. kohta Vasta-aiheet). Imraldi-hoito tulee keskeyttää, jos potilaalle tulee kongestiivisen sydämen vajaatoiminnan oireita tai jos oireet pahenevat.
Autoimmuuniprosessit
Imraldi-hoito voi johtaa autovasta-aineiden muodostukseen. Pitkäaikaisen adalimumabihoidon vaikutusta autoimmuunisairauksien kehittymiseen ei tunneta. Jos potilaalle kehittyy lupuksen kaltaiseen oireyhtymään viittaavia oireita Imraldi-hoidon jälkeen ja hänellä todetaan vasta-aineita kaksijuosteiselle DNA:lle, Imraldi-hoitoa ei tule jatkaa (ks. kohta Haittavaikutukset).
Samanaikainen biologisten DMARDien tai TNFα-salpaajan antaminen
Kliinisissä tutkimuksissa anakinran ja toisen TNFα-salpaajan, etanerseptin, yhteiskäytössä on todettu vakavia infektioita, eikä yhteiskäytöstä ollut lisääntynyttä kliinistä hyötyä verrattuna etanerseptin antamiseen yksinään. Samantyyppiset haittavaikutukset ovat mahdollisia anakinran ja adalimumabin yhteiskäytössä. Siksi adalimumabin ja anakinran yhteiskäyttöä ei suositella (ks. kohta Yhteisvaikutukset).
Adalimumabin ja muiden biologisten DMARDien (kuten anakinra ja abatasepti) tai muiden TNFα-salpaajien samanaikaista antoa ei suositella perustuen infektioiden mahdollisesti lisääntyneeseen riskiin, mukaan lukien vakavat infektiot ja muut farmakologiset yhteisvaikutukset (ks. kohta Yhteisvaikutukset).
Leikkaus
Turvallisuustiedot adalimumabilla hoidettujen potilaiden leikkaushoidoista ovat rajalliset. Leikkausta suunniteltaessa on otettava huomioon adalimumabin pitkä puoliintumisaika. Leikkaushoitoa vaatineen Imraldi-potilaan infektioita on tarkkaan seurattava ja asianmukaisiin toimenpiteisiin on tarvittaessa ryhdyttävä. Turvallisuustiedot atroplastiahoitoa vaativien adalimumabipotilaiden hoidosta ovat rajalliset.
Ohutsuolitukos
Jos Crohnin tauti ei reagoi hoitoon, potilaalla saattaa olla kiinteä fibroottinen striktuura, joka saattaa vaatia leikkaushoitoa. Nykyisten tietojen perusteella adalimumabi ei pahenna striktuuroita eikä aiheuta niitä.
Iäkkäät potilaat
Vakavien infektioiden esiintyvyys oli korkeampi yli 65-vuotiailla potilailla (3,7 %), joita hoidettiin adalimumabilla, kuin alle 65-vuotiailla (1,5 %). Jotkin näistä infektioista olivat fataaleja. Vanhuksia hoidettaessa tulee erityisesti ottaa huomioon infektioriski.
Pediatriset potilaat
Katso Rokotukset yllä.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 0,4 ml:n annos, eli se on olennaisesti natriumiton.
Yhteisvaikutukset
Adalimumabia on tutkittu nivelreumaa, idiopaattista juveniilia polyartriittia ja nivelpsoriaasia sairastavilla potilailla sekä yksinään että metotreksaattiin yhdistettynä. Vasta-ainemuodostus oli vähäisempää metotreksaatin kanssa annettuna kuin yksinään. Adalimumabin käyttö ilman metotreksaattia lisäsi vasta-ainemuodostusta, tehosti adalimumabin puhdistumaa ja heikensi sen tehoa (ks. kohta Farmakodynamiikka).
Imraldi-valmisteen ja anakinran yhdistämistä ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet Samanaikainen biologisten DMARDien tai TNFα-salpaajan antaminen).
Imraldi-valmisteen ja abataseptin yhdistämistä ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet Samanaikainen biologisten DMARDien tai TNFα-salpaajan antaminen).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on harkittava riittävää raskauden ehkäisyä Imraldi-hoidon aikana ja vähintään viiden kuukauden ajan viimeisen Imraldi-annoksen jälkeen.
Raskaus
Laajat tiedot eivät viittaa epämuodostumien lisääntymiseen vastasyntyneillä. Tiedot kerättiin prospektiivisesti noin 2 100 raskaudesta, joiden aikana oli tapahtunut adalimumabialtistus ja jotka olivat johtaneet elävän lapsen syntymään tiedossa olevin lopputuloksin (mukaan lukien tiedot yli 1 500:sta ensimmäisen raskauskolmanneksen aikana tapahtuneesta altistuksesta).
Prospektiivisessa kohorttirekisterissä oli mukana 257 naista, joilla oli nivelreuma tai Crohnin tauti ja jotka saivat adalimumabia vähintään ensimmäisellä raskauskolmanneksella, ja 120 naista, joilla oli nivelreuma tai Crohnin tauti ja jotka eivät saaneet adalimumabia. Ensisijainen päätetapahtuma oli merkittävien synnynnäisten kehityshäiriöiden esiintyvyys syntymähetkellä. Vähintään yhden elävän, merkittävästi kehityshäiriöisen lapsen syntymään johtaneiden raskauksien osuus oli adalimumabihoitoa saaneilla nivelreumaa sairastavilla naisilla 6/69 (8,7 %) ja hoitamattomilla nivelreumaa sairastavilla naisilla 5/74 (6,8 %) (korjaamaton vetosuhde 1,31; 95 % luottamusväli [lv] 0,38–4,52). Adalimumabihoitoa saaneilla Crohnin tautia sairastavilla naisilla vastaava osuus oli 16/152 (10,5 %) ja hoitamattomilla Crohnin tautia sairastavilla naisilla 3/32 (9,4 %) (korjaamaton vetosuhde 1,14; 95 % lv 0,31–4,16). Korjattu vetosuhde (lähtötilanteen eroja koskeva) oli 1,10 (95 % lv 0,45–2,73), kun nivelreumaa ja Crohnin tautia koskevat tiedot yhdistettiin. Selviä eroja adalimumabihoitoa saaneiden ja hoitamattomien naisten välillä ei todettu toissijaisten
Apinoilla tehdyssä sikiökehityksen toksisuustutkimuksessa ei havaittu merkkejä emoon eikä alkioon kohdistuvasta toksisuudesta eikä teratogeenisuudesta. Adalimumabin postnataalitoksisista vaikutuksista ei prekliinistä ole tietoa (ks. kohta Prekliiniset tiedot turvallisuudesta).
Koska adalimumabi estää TNFα:aa, sen antaminen raskausaikana voi vaikuttaa vastasyntyneen normaaliin immuunivasteeseen. Adalimumabia saa käyttää raskausaikana vain, jos se on selvästi tarpeellista.
Adalimumabi voi läpäistä adalimumabihoitoa saavan naisen istukan ja päätyä sikiön verenkiertoon raskauden aikana. Tästä johtuen näillä imeväisillä voi olla syntymän jälkeen kohonnut infektioriski. Elävien rokotteiden (esim. BCG-rokotteen) antamista imeväisille, jotka ovat altistuneet adalimumabille in utero, ei suositella 5 kuukauteen äidin viimeisestä raskaudenaikaisesta adalimumabi-injektiosta.
Imetys
Julkaistusta kirjallisuudesta saadut suppeat tiedot viittaavat siihen, että adalimumabi erittyy hyvin pieninä pitoisuuksina ihmisen rintamaitoon ja sen pitoisuus ihmisen rintamaidossa on 0,1–1 % äidin seerumin adalimumabipitoisuudesta. Suun kautta annettuna immunoglobuliini G ‑proteiinit hajoavat suolistossa proteolyysin kautta ja niiden biologinen hyötyosuus on pieni. Imetettävään vastasyntyneeseen / vauvaan kohdistuvia vaikutuksia ei ole odotettavissa. Näin ollen adalimumabia voi käyttää imetyksen aikana.
Hedelmällisyys
Prekliinistä tietoa adalimumabin vaikutuksista hedelmällisyyteen ei ole saatavilla.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Imraldi voi vaikuttaa vähäisessä määrin ajokykyyn ja kykyyn käyttää koneita. Imraldi-valmisteen ottamisen jälkeen saattaa esiintyä kiertohuimausta ja näön heikkenemistä (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Adalimumabia on tutkittu tärkeimmissä kontrolloiduissa ja avoimissa tutkimuksissa 9 506 potilaalla jopa 60 kuukauden ajan tai yli. Näihin tutkimuksiin osallistui nivelreumapotilaita, joiden tauti oli kestänyt lyhyen tai pitkän aikaa, sekä idiopaattista juveniilia artriittia (idiopaattista juveniilia polyartriittia ja entesiitteihin liittyvää artriittia), aksiaalista spondylartriittia (selkärankareumaa ja aksiaalista spondylartriittia (ilman radiografista näyttöä selkärankareumasta)), nivelpsoriaasia, Crohnin tautia, ulseratiivista koliittia, psoriaasia, hidradenitis suppurativaa ja uveiittia sairastavia potilaita. Tärkeimmissä kontrolloiduissa tutkimuksissa 6 089 potilasta sai adalimumabia ja 3 801 potilasta sai plaseboa tai vaikuttavaa vertailuvalmistetta kontrolloidun vaiheen aikana.
Tärkeimmissä kontrolloiduissa kaksoissokkotutkimuksissa hoidon keskeytti haittatapahtumien vuoksi 5,9 % adalimumabia ja 5,4 % vertailuhoitoa saaneista potilaista.
Yleisimmin ilmoitettuja haittavaikutuksia ovat infektiot (esim. nenänielutulehdus, ylähengitystieinfektiot ja sinuiitti), pistoskohdan reaktiot (punoitus, kutina, verenvuoto, kipu tai turvotus), päänsärky ja tuki- ja liikuntaelimistön kipu.
Adalimumabihoidon yhteydessä on ilmoitettu vakavia haittavaikutuksia. TNF-antagonistit, kuten adalimumabi, vaikuttavat immuunijärjestelmään, ja niiden käyttö voi vaikuttaa elimistön kykyyn torjua infektioita ja syöpää.
Adalimumabin käytön yhteydessä on ilmoitettu kuolemaan johtaneita ja henkeä uhanneita infektioita (mm. sepsistä, opportunistisia infektioita ja tuberkuloosia), HBV-infektion reaktivaatiota ja eri syöpätauteja (mm. leukemiaa, lymfoomia ja hepatospleenistä T-solulymfoomaa).
Myös vakavia hematologisia, neurologisia ja autoimmuunireaktioita on ilmoitettu. Näistä pansytopeniaa, aplastista anemiaa ja keskus- ja ääreishermoston myeliinikatotapauksia on ilmoitettu harvoin. Myös lupusta, lupuksen kaltaisia oireistoja ja Stevens–Johnsonin oireyhtymää on ilmoitettu.
Pediatriset potilaat
Lapsipotilailla todetut haittatapahtumat olivat yleisesti ottaen yleisyydeltään ja luonteeltaan samanlaisia kuin aikuispotilailla todetut haitat.
Haittavaikutustaulukko
Alla olevassa taulukossa 7 on lueteltu kliinisissä tutkimuksissa ja markkinoilletulon jälkeen esiintulleet haittatapahtumat, ja ne on jaoteltu elinryhmittäin ja esiintyvyyden perusteella: hyvin yleiset (≥ 1/10); yleiset (≥ 1/100 ja < 1/10); melko harvinaiset (≥ 1/1 000 ja < 1/100); harvinaiset (≥ 1/10 000 ja < 1/1 000); sekä tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Kunkin yleisyysluokan haittavaikutukset on esitetty vakavuuden mukaisessa järjestyksessä vakavimmasta haittavaikutuksesta alkaen. Eri käyttöaiheiden tiedoista on valittu suurin yleisyysluku. Elinjärjestelmäsarakkeessa oleva tähti (*) tarkoittaa, että kohdissa Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset on lisätietoa aiheesta.
Taulukko 7
Haittavaikutukset
| Elinjärjestelmä | Yleisyys | Haittavaikutus |
| Infektiot* | Hyvin yleiset | Hengitystieinfektiot (mm. ylä- ja alahengitystieinfektiot, keuhkokuume, sinuiitti, nielutulehdus, nenänielutulehdus ja herpesviruspneumonia) |
| Yleiset | Systeemiset infektiot (mm. sepsis, kandidiaasi ja influenssa), suolistoinfektiot (mm. virusperäinen gastroenteriitti), iho- ja pehmytkudosinfektiot (mm. paronykia, selluliitti, märkärupi, nekrotisoiva faskiitti ja vyöruusu), korvatulehdukset, suutulehdukset (mm. herpes simplex, huuliherpes ja hammasinfektiot), sukuelininfektiot (mm. vulvovaginan sieni-infektiot), virtsatieinfektiot (mm. pyelonefriitti), sieni-infektiot, nivelinfektiot | |
| Melko harvinaiset | Hermostoinfektiot (mm. virusmeningiitti), opportunistiset infektiot ja tuberkuloosi (mm. Coccidioides-infektiot, histoplasmoosi ja Mycobacterium avium ‑infektiot), bakteeriperäiset infektiot, silmäinfektiot, divertikuliitti1) | |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit)* | Yleiset | Ei-melanoomatyyppiset ihosyövät (mm. tyvisolusyöpä ja okasolusyöpä), hyvänlaatuiset kasvaimet |
| Melko harvinaiset | Lymfooma**, kiinteät kasvaimet (mm. rintasyöpä, keuhkokasvaimet ja kilpirauhaskasvaimet), melanooma** | |
| Harvinaiset | Leukemia1) | |
| Tuntematon | Hepatospleeninen T-solulymfooma1), merkelinsolukarsinooma (ihon neuroendokriininen karsinooma)1), Kaposin sarkooma | |
| Veri ja imukudos* | Hyvin yleiset | Leukopenia (mm. neutropenia ja agranulosytoosi), anemia |
| Yleiset | Leukosytoosi, trombosytopenia | |
| Melko harvinaiset | Idiopaattinen trombosytopeeninen purppura | |
| Harvinaiset | Pansytopenia | |
| Immuunijärjestelmä* | Yleiset | Yliherkkyys, allergiat (myös kausiallergiat) |
| Melko harvinaiset | Sarkoidoosi1), vaskuliitti | |
| Harvinaiset | Anafylaksia1) | |
| Aineenvaihdunta ja ravitsemus | Hyvin yleiset | Kohonneet rasva-arvot |
| Yleiset | Hypokalemia, kohonneet virtsahappoarvot, poikkeavat veren natriumarvot, hypokalsemia, hyperglykemia, hypofosfatemia, nestehukka | |
| Psyykkiset häiriöt | Yleiset | Mielialanvaihtelut (mm. masennus), ahdistuneisuus, unettomuus |
| Hermosto* | Hyvin yleiset | Päänsärky |
| Yleiset | Parestesiat (mm. hypestesia), migreeni, hermojuuren kompressio | |
| Melko harvinaiset | Aivohalvaus1), vapina, neuropatia | |
| Harvinaiset | MS-tauti, myeliinikatohäiriöt (esim. optikusneuriitti, Guillain-Barrén oireyhtymä)1) | |
| Silmät | Yleiset | Näköhäiriöt, sidekalvotulehdus, luomitulehdus, silmän turvotus |
| Melko harvinaiset | Kaksoiskuvat | |
| Kuulo ja tasapainoelin | Yleiset | Kiertohuimaus |
| Melko harvinaiset | Kuurous, tinnitus | |
| Sydän* | Yleiset | Takykardia |
| Melko harvinaiset | Sydänkohtaus1), rytmihäiriöt, kongestiivinen sydämen vajaatoiminta | |
| Harvinaiset | Sydämenpysähdys | |
| Verisuonisto | Yleiset | Hypertensio, kuumat aallot, hematooma |
| Melko harvinaiset | Aortan aneurysma, valtimotukos, tromboflebiitti | |
| Hengityselimet, rintakehä ja välikarsina* | Yleiset | Astma, hengenahdistus, yskä |
| Melko harvinaiset | Keuhkoembolia1), interstitiaalinen keuhkosairaus, keuhkoahtaumatauti, pneumoniitti, pleuraeffuusio1) | |
| Harvinaiset | Keuhkofibroosi1) | |
| Ruoansulatuselimistö | Hyvin yleiset | Vatsakipu, pahoinvointi ja oksentelu |
| Yleiset | Ruoansulatuskanavan verenvuoto, dyspepsia, refluksitauti, Sjögrenin oireyhtymä | |
| Melko harvinaiset | Haimatulehdus, dysfagia, kasvojen turvotus | |
| Harvinaiset | Suolen puhkeama1) | |
| Maksa ja sappi* | Hyvin yleiset | Kohonneet maksaentsyymiarvot |
| Melko harvinaiset | Kolekystiitti ja sappikivet, maksan rasvoittuminen, kohonneet bilirubiiniarvot | |
| Harvinaiset | Hepatiitti, B-hepatiitin uudelleenaktivoituminen1), autoimmuunihepatiitti1) | |
| Tuntematon | Maksan vajaatoiminta1) | |
| Iho ja ihonalainen kudos | Hyvin yleiset | Ihottuma (mm. eksfoliatiivinen ihottuma) |
| Yleiset | Psoriaasin puhkeaminen tai paheneminen (mukaan lukien palmoplantaarinen pustuloosi psoriaasi)1), nokkosihottuma, mustelmat (mm. purppura), ihotulehdus (mm. ekseema), kynsien murtuminen, voimakas hikoilu, alopesia1), kutina | |
| Melko harvinaiset | Öinen hikoilu, arpimuodostus | |
| Harvinaiset | Erythema multiforme1), Stevens‑Johnsonin oireyhtymä1), angioedeema1), ihon vaskuliitti1), likenoidi (punajäkälää muistuttava) ihoreaktio1) | |
| Tuntematon | Dermatomyosiitin oireiden paheneminen1) | |
| Luusto, lihakset ja sidekudos | Hyvin yleiset | Luusto- ja lihaskipu |
| Yleiset | Lihasspasmit (mm. kohonneet veren kreatiinifosfokinaasiarvot) | |
| Melko harvinaiset | Rabdomyolyysi, systeeminen lupus erythematosus (SLE/LED) | |
| Harvinaiset | Lupuksen kaltainen oireyhtymä1) | |
| Munuaiset ja virtsatiet | Yleiset | Munuaisten vajaatoiminta, hematuria |
| Melko harvinaiset | Nokturia | |
| Sukupuolielimet ja rinnat | Melko harvinaiset | Erektiohäiriöt |
| Yleisoireet ja antopaikassa todettavat haitat* | Hyvin yleiset | Pistoskohdan reaktiot (mm. pistoskohdan punoitus) |
| Yleiset | Rintakipu, turvotus, kuume1) | |
| Melko harvinaiset | Inflammaatio | |
| Tutkimukset* | Yleiset | Hyytymis- ja verenvuotohäiriöt (mm. APTT-ajan piteneminen), positiivinen tulos autovasta-ainetestissä (mm. kaksijuosteisen DNA:n vasta-aineet), kohonneet veren laktaattidehydrogenaasiarvot |
| Tuntematon | Painonnousu2) | |
| Vammat, myrkytykset ja hoitokomplikaatiot | Yleiset | Hidas paraneminen |
* lisätietoa kohdissa Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset
** myös avoimissa jatkotutkimuksissa
1) mukaan lukien spontaaniraportit
2) Adalimumabin yhteydessä keskimääräinen painon muutos lähtötilanteesta oli 0,3–1,0 kg aikuisten käyttöaiheissa, kun taas lumelääkettä käytettäessä paino laski tai nousi keskimäärin 0,4 kg 4–6 kuukauden pituisen hoitojakson aikana. Pitkäaikaisissa jatkotutkimuksissa, joissa ei ollut vertailuryhmää, on havaittu myös 5–6 kg:n painonnousua, kun potilaat ovat altistuneet lääkevalmisteelle noin 1–2 vuoden ajan. Tämä koskee erityisesti potilaita, joilla on Crohnin tauti ja haavainen paksusuolitulehdus. Mekanismi, johon tämä vaikutus perustuu, on epäselvä, mutta se voi liittyä adalimumabin tulehduksia estävään ja lievittävään vaikutukseen.
Hidradenitis suppurativa
Turvallisuusprofiili adalimumabia viikoittain saaneilla HS-potilailla oli yhdenmukainen adalimumabin tunnetun turvallisuusprofiilin kanssa.
Uveiitti
Turvallisuusprofiili adalimumabia joka toinen viikko saaneilla uveiittipotilailla oli yhdenmukainen adalimumabin tunnetun turvallisuusprofiilin kanssa.
Tiettyjen haittavaikutusten kuvaus
Pistoskohdan reaktiot
Tärkeimmissä kontrolloiduissa tutkimuksissa aikuisilla ja lapsilla pistoskohdan reaktioita (punoitus ja/tai kutina, verenvuoto, kipu tai turvotus) ilmeni 12,9 %:lla adalimumabia ja 7,2 %:lla plaseboa tai vaikuttavaa vertailuvalmistetta saaneista potilaista. Pistoskohdan reaktioiden yhteydessä ei lääkityksen keskeyttäminen yleensä ollut tarpeen.
Infektiot
Tärkeimmissä kontrolloiduissa tutkimuksissa aikuisilla ja lapsilla infektioiden esiintyvyys oli adalimumabia saaneilla 1,51 tapausta ja plaseboa tai vaikuttavaa vertailuvalmistetta saaneilla 1,46 tapausta potilasvuotta kohti. Valtaosa infektioista oli nenänielutulehduksia, ylähengitystieinfektioita ja nenän sivuontelotulehduksia (sinuiitti). Useimmat potilaat jatkoivat adalimumabilääkitystä infektion parannuttua.
Vakavien infektioiden esiintyvyys oli adalimumabia saaneilla 0,04 tapausta ja plaseboa tai vaikuttavaa vertailuvalmistetta saaneilla 0,03 tapausta potilasvuotta kohti.
Adalimumabilla tehdyissä kontrolloiduissa ja avoimissa tutkimuksissa aikuisilla ja lapsilla on ilmoitettu vakavia infektioita (myös kuolemaan johtaneita infektioita, joita esiintyi harvoin), esim. tuberkuloosia (esim. miliaarituberkuloosia ja keuhkojen ulkopuolella esiintyvää tuberkuloosia) ja invasiivisia opportunistisia infektioita (esim. disseminoitunutta tai extrapulmonaalista histoplasmoosia, blastomykoosia, koksidioidomykoosia, pneumokystoosia, kandidiaasia, aspergilloosia ja listerioosia). Valtaosa tuberkuloositapauksista kehittyi kahdeksan kuukauden kuluessa hoidon aloittamisesta, mikä saattaa viitata latentin sairauden uudelleenaktivoitumiseen.
Maligniteetit ja lymfoproliferatiiviset häiriöt
Idiopaattista juveniilia artriittia (idiopaattista juveniilia polyartriittia ja entesiitteihin liittyvää artriittia) koskeneissa adalimumabitutkimuksissa 249:llä pediatrisella potilaalla, joiden kokonaisaltistus oli 655,6 potilasvuotta, ei havaittu maligniteetteja. Maligniteetteja ei havaittu myöskään lasten Crohnin tautia koskeneissa adalimumabitutkimuksissa 192 lapsipotilaalla, joiden kokonaisaltistus oli 498,1 potilasvuotta. Maligniteetteja ei havaittu 77:llä pediatrisella potilaalla lasten kroonista läiskäpsoriaasia koskeneissa adalimumabitutkimuksissa, joiden kokonaisaltistus oli 80,0 potilasvuotta. Ulseratiivista koliittia koskeneessa adalimumabitutkimuksessa 93:lla pediatrisella potilaalla, joiden kokonaisaltistus oli 65,3 potilasvuotta, ei havaittu maligniteetteja. Lasten uveiittia koskeneessa adalimumabitutkimuksessa 60:llä lapsipotilaalla, joiden kokonaisaltistus oli 58,4 potilasvuotta, ei havaittu maligniteetteja.
Tärkeimpiin vähintään 12 viikkoa kestäneisiin aikuisten adalimumabitutkimuksiin osallistui potilaita, joilla oli keskivaikea tai vaikea aktiivinen nivelreuma, selkärankareuma, aksiaalinen spondylartriitti (ilman radiografista näyttöä selkärankareumasta), nivelpsoriaasi, psoriaasi, Crohnin tauti, ulseratiivinen koliitti tai uveiitti. Näiden tutkimusten kontrolloiduissa osissa 5 291 adalimumabihoitoa saaneella potilaalla havaittiin 6,8 (95 % luottamusväli 4,4–10,5) maligniteettia (lukuun ottamatta lymfoomia ja muita ihosyöpiä kuin melanoomia) 1 000 potilasvuotta kohti, kun taas 3 444 verrokkipotilaalla todettiin 6,3 (95 % luottamusväli 3,4–11,8) tapausta 1 000 potilasvuotta kohti. Hoidon mediaanikesto oli 4,0 kk adalimumabiryhmässä ja 3,8 kk verrokkipotilailla. Muiden ihosyöpien kuin melanooman esiintymistiheys oli adalimumabihoitoa saaneilla potilailla 8,8 (95 % luottamusväli 6,0–13,0) tapausta 1 000 potilasvuotta kohti ja verrokkipotilailla 3,2 (95 % luottamusväli 1,3–7,6) tapausta 1 000 potilasvuotta kohti. Näistä ihosyövistä levyepiteelikarsinoomien esiintymistiheys oli adalimumabihoitoa saaneilla potilailla 2,7 (95 % luottamusväli 1,4–5,4) tapausta 1 000 potilasvuotta kohti ja verrokkipotilailla 0,6 (95 % luottamusväli 0,1–4,5) tapausta 1 000 potilasvuotta kohti. Lymfoomien esiintymistiheys oli adalimumabihoitoa saaneilla potilailla 0,7 (95 % luottamusväli 0,2–2,7) tapausta 1 000 potilasvuotta kohti ja verrokkiryhmässä 0,6 (95 % luottamusväli 0,1–4,5) tapausta 1 000 potilasvuotta kohti.
Näiden tutkimusten kontrolloitujen osien ja parhaillaan tehtävien sekä lopetettujen avointen jatkotutkimusten mediaanikesto on noin 3,3 vuotta, ja niihin on osallistunut 6 427 potilasta yhteensä yli 26 439 potilashoitovuoden ajan. Kun näiden tutkimusten tulokset yhdistetään, havaittujen maligniteettien esiintymistiheydeksi saadaan noin 8,5 tapausta 1 000 potilasvuotta kohti (lymfoomia ja muita ihosyöpiä kuin melanoomaa lukuun ottamatta). Muiden ihosyöpien kuin melanooman havaittu esiintymistiheys on noin 9,6 tapausta 1 000 potilasvuotta kohti, ja lymfoomien havaittu esiintymistiheys noin 1,3 tapausta 1 000 potilasvuotta kohti.
Markkinoille tulon jälkeen tammikuusta 2003 joulukuuhun 2010 pääasiassa nivelreumapotilailla spontaanisti ilmoitettujen maligniteettien esiintymistiheys on noin 2,7 tapausta 1 000 potilashoitovuotta kohti. Muiden ihosyöpien kuin melanooman spontaanisti ilmoitettu esiintymistiheys on noin 0,2 tapausta 1 000 potilashoitovuotta kohti ja lymfoomien spontaanisti ilmoitettu esiintymistiheys noin 0,3 tapausta 1 000 potilashoitovuotta kohti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Adalimumabilla hoidetuilla potilailla on markkinoilletulon jälkeen raportoitu harvoin hepatospleenistä T-solulymfoomaa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Autovasta-aineet
Potilaiden seeruminäytteistä määritettiin nivelreumatutkimuksissa I–V autovasta-aineet useana ajankohtana. Näissä tutkimuksissa 11,9 %:lla adalimumabia ja 8,1 %:lla plaseboa tai vaikuttavaa vertailuvalmistetta saaneista potilaista lähtötason negatiivinen tumavasta-ainetitteri muuttui positiiviseksi viikolla 24. Kaikissa nivelreuma- ja nivelpsoriaasitutkimuksissa kahdella 3 441:stä adalimumabia saaneesta potilaasta havaittiin kliinisiä merkkejä lupuksen kaltaisen oireiston kehittymisestä. Tila korjaantui hoidon keskeyttämisen jälkeen. Yhdellekään potilaalle ei kuitenkaan tullut lupuksesta johtuvaa nefriittiä eikä keskushermosto-oireita.
Maksa- ja sappitapahtumat
Nivelreuma- ja nivelpsoriaasipotilailla tehdyissä kontrolloiduissa vaiheen 3 adalimumabitutkimuksissa, joiden kontrolloidun osan kesto oli 4–104 viikkoa, ALAT-arvojen suurenemista ≥ 3 kertaa viitevälin ylärajan (ULN) suuruisiksi esiintyi 3,7 %:lla adalimumabihoitoa saaneista ja 1,6 %:lla vertailuhoitoa saaneista.
Idiopaattista juveniilia polyartriittia sairastavilla 4–17-vuotiailla potilailla ja entesiitteihin liittyvää artriittia sairastavilla 6–17-vuotiailla potilailla tehdyissä kontrolloiduissa vaiheen 3 adalimumabitutkimuksissa ALAT-tason suurenemista tasolle ≥ 3 × ULN esiintyi 6,1 %:lla adalimumabihoitoa saaneista ja 1,3 %:lla vertailuhoitoa saaneista. ALAT-tason suurenemisista useimmat havaittiin potilailla, jotka käyttivät samanaikaisesti myös metotreksaattia. Kenelläkään vaiheen 3 adalimumabitutkimukseen osallistuneista idiopaattista juveniilia polyartriittia sairastavista 2–<4-vuotiaista potilaista ei esiintynyt ALAT-tason suurenemista ≥ 3 × ULN.
Crohnin tautia tai ulseratiivista koliittia sairastavilla potilailla tehdyissä kontrolloiduissa vaiheen 3 adalimumabitutkimuksissa, joiden kontrolloidun osan kesto oli 4–52 viikkoa, ALAT-arvojen suurenemista tasolle ≥ 3 × ULN esiintyi 0,9 %:lla adalimumabihoitoa saaneista ja 0,9 %:lla vertailuhoitoa saaneista.
Crohnin tautia sairastavilla lapsipotilailla tehdyssä vaiheen 3 kliinisessä tutkimuksessa, jossa selvitettiin adalimumabin tehoa ja turvallisuutta painonmukaisen induktiohoidon jälkeisessä ylläpitohoidossa lapsipotilailla, jotka saivat adalimumabia painonmukaisella annoksella (2 painoluokkaa) 52 viikon ajan, ALAT-arvojen suurenemista tasolle ≥ 3 × ULN esiintyi 2,6 %:lla (5/192) potilaista. Näistä potilaista 4 oli saanut immonosuppressiivista hoitoa lähtötilanteessa.
Läiskäpsoriaasipotilailla tehdyissä kontrolloiduissa vaiheen 3 adalimumabitutkimuksissa, joiden kontrolloidun osan kesto oli 12–24 viikkoa, ALAT-arvojen suurenemista tasolle ≥ 3 ×ULN esiintyi 1,8 %:lla adalimumabihoitoa saaneista ja 1,8 %:lla vertailuhoitoa saaneista.
Kenelläkään vaiheen 3 adalimumabitutkimukseen osallistuneista läiskäpsoriaasia sairastaneista lapsipotilaista ei havaittu ALAT-tason suurenemista ≥ 3 × ULN.
Hidradenitis suppurativaa sairastavilla potilailla tehdyissä kontrolloiduissa adalimumabitutkimuksissa (aloitusannos 160 mg viikolla 0 ja 80 mg viikolla 2, minkä jälkeen 40 mg kerran viikossa alkaen viikolta 4), joiden kontrolloidun osan kesto oli 12–16 viikkoa, ALAT-arvojen kohoamista ≥ 3 × ULN esiintyi 0,3 %:lla adalimumabihoitoa saaneista ja 0,6 %:lla vertailuhoitoa saaneista.
Uveiittia sairastavilla potilailla tehdyissä kontrolloiduissa 80 viikkoa kestäneissä adalimumabitutkimuksissa (aloitusannos 80 mg viikolla 0, jonka jälkeen 40 mg joka toinen viikko alkaen viikolta 1), joissa altistumisajan mediaani adalimumabihoitoa saaneilla potilailla oli 166,5 vuorokautta ja vertailuhoitoa saaneilla potilailla 105,0 vuorokautta, ALAT-arvojen kohoamista ≥ 3 × ULN esiintyi 2,4 %:lla adalimumabihoitoa saaneista ja 2,4 %:lla vertailuhoitoa saaneista.
Ulseratiivista koliittia sairastavilla pediatrisilla potilailla (N = 93) tehdyssä kontrolloidussa vaiheen 3 tutkimuksessa, jossa arvioitiin adalimumabin tehoa ja turvallisuutta ylläpitoannoksella 0,6 mg/kg (enintään 40 mg) joka toinen viikko (N = 31) ja ylläpitoannoksella 0,6 mg/kg (enintään 40 mg) viikossa (N = 32) sen jälkeen, kun potilaat olivat saaneet painonmukaista induktiohoitoa annoksella 2,4 mg/kg (enintään 160 mg) viikolla 0 ja viikolla 1 sekä annoksella 1,2 mg/kg (enintään 80 mg) viikolla 2 (N = 63) tai induktiohoitoa annoksella 2,4 mg/kg (enintään 160 mg) viikolla 0, lumelääkettä viikolla 1 ja induktiohoitoa annoksella 1,2 mg/kg (enintään 80 mg) viikolla 2 (N = 30), ALAT-arvojen suurenemista tasolle ≥ 3 x ULN esiintyi 1,1 %:lla (1/93) potilaista.
Potilaat, joilla esiintyi ALAT-arvojen suurenemista, olivat kaikkien käyttöaiheiden kliinisissä tutkimuksissa oireettomia. Arvojen suureneminen oli useimmiten ohimenevää ja korjautui, kun hoitoa jatkettiin. Adalimumabilla hoidetuilla potilailla on kuitenkin markkinoilletulon jälkeen raportoitu myös maksan vajaatoimintaa ja lievempiä maksan häiriöitä, jotka saattavat edeltää maksan vajaatoimintaa, mm. hepatiittia (myös autoimmuunihepatiittia).
Samanaikainen hoito atsatiopriinilla/6-merkaptopuriinilla
Aikuisten Crohnin tautia käsittelevissä tutkimuksissa pahanlaatuisten ja vakavien infektioihin liittyvien haittatapahtumien ilmaantuvuus oli suurempi käytettäessä adalimumabin ja atsatiopriinin/6‑merkaptopuriinin yhdistelmähoitoa verrattuna pelkkään adalimumabiin.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Annosta rajoittavaa toksisuutta ei havaittu kliinisissä tutkimuksissa. Suurin tutkittu annos on 10 mg/kg laskimonsisäisesti useana annoksena, mikä on noin 15 kertaa suositusannoksen verran.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, Tuumorinekroositekijä alfan (TNF-α) estäjät, ATC-koodi: L04AB04
Imraldi on ns. biosimilaari lääkevalmiste. Yksityiskohtaisempaa tietoa on saatavilla Euroopan lääkeviraston kotisivulta: http://www.ema.europa.eu.
Vaikutusmekanismi
Adalimumabi sitoutuu spesifisesti tuumorinekroositekijään (TNF) ja neutraloi TNF:n biologisen toiminnan estämällä sen vaikutuksen solukalvon TNF-reseptoreihin (p55 ja p75).
Adalimumabi säätelee myös TNF:n indusoimia tai säätelemiä biologisia vasteita, mm. leukosyyttien migraatiosta vastaavien adheesiomolekyylien määrän muutoksia (ELAM-1, VCAM-1 ja ICAM-1, joilla IC50 on 0,1–0,2 nM).
Farmakodynaamiset vaikutukset
Adalimumabihoidon jälkeen todettiin, että nivelreumapotilailla tulehduksen akuutin vaiheen osoittajien (C-reaktiivinen proteiini ja lasko) ja seerumin sytokiinien (IL-6) määrät pienenivät lähtötasoon verrattuna nopeasti. Myös rustotuhoa aiheuttavaa kudosten uudismuodostusta tuottavien matriksin metalloproteinaasientsyymien (MMP-1 ja MMP-3) pitoisuudet seerumissa pienenivät adalimumabin antamisen jälkeen. Adalimumabia saaneilla potilailla havaittiin usein kroonisen tulehduksen hematologisten merkkien parantumista.
Adalimumabia käyttävillä idiopaattista juveniilia polyartriittia, Crohnin tautia, ulseratiivista koliittia ja hidradenitis suppurativaa sairastavilla potilailla todettiin myös CRP-arvojen pienenevän nopeasti. Crohnin tauti -potilailla havaittiin tulehdusmarkkereita ilmentävien solujen määrän väheneminen paksusuolessa, mukaan lukien TNFα:n ilmentymisen merkittävä väheneminen. Suolen limakalvon endoskooppiset tutkimukset ovat osoittaneet limakalvon paranemista tapahtuvan adalimumabilla hoidetuissa potilaissa.
Kliininen teho ja turvallisuus
Nivelreuma
Adalimumabia on kliinisissä tutkimuksissa arvioitu yhteensä yli 3 000:lla nivelreumapotilaalla. Adalimumabin tehokkuutta ja turvallisuutta nivelreuman hoidossa on arvioitu viidessä satunnaistetussa, hyvin kontrolloidussa kaksoissokkotutkimuksessa. Osa potilaista sai hoitoa 120 kuukauden ajan. Adalimumabi 40 mg/0,4 ml -valmisteen aiheuttamaa pistoskohdan kipua arvioitiin kahdessa satunnaistetussa, yksöissokkoutetussa kaksijaksoisessa vaihtovuoroisessa tutkimuksessa, jossa käytettiin verrokkina vaikuttavaa valmistetta.
Nivelreumatutkimuksessa I arvioitiin 271 vähintään 18-vuotiasta kohtalaisen aktiivista tai hyvin aktiivista nivelreumaa sairastavaa potilasta, joilla hoito vähintään yhdellä taudin kulkuun vaikuttavalla reumalääkkeillä oli epäonnistunut ja joilla metotreksaatti ei ollut riittävän tehokas annostasolla 12,5‑25 mg/vko (10 mg/vko jos potilas ei siedä metotreksaattia) annoksen pysyessä vakiona 10‑25 mg/vko. Adalimumabia tai plaseboa annettiin 20, 40 tai 80 mg:n annoksina joka toinen viikko 24 viikon ajan.
Nivelreumatutkimuksessa II arvioitiin 544 vähintään 18-vuotiasta kohtalaisen aktiivista tai hyvin aktiivista nivelreumaa sairastavaa potilasta, joilla hoito vähintään yhdellä taudin kulkuun vaikuttavalla reumalääkkeellä oli epäonnistunut. Adalimumabia annettiin ihonalaisina injektioina 20 tai 40 mg:n annoksina siten, että adalimumabia annettiin yksinään joko viikoittain tai vuoroviikoin plasebon kanssa 26 viikon ajan; plaseboa annettiin viikoittain samanmittaisen ajanjakson verran. Muiden taudin kulkuun vaikuttavien reumalääkkeiden käyttö ei ollut sallittua.
Nivelreumatutkimuksessa III arvioitiin 619 vähintään 18-vuotiasta kohtalaisen aktiivista tai hyvin aktiivista nivelreumaa sairastavaa potilasta, joilla metotreksaattihoito ei ollut riittävän tehokas annostasolla 12,5–25 mg/vko tai jotka eivät sietäneet 10 mg/vko annoksia metotreksaattia. Tutkimuksessa oli kolme ryhmää. Ensimmäinen ryhmä sai plaseboa viikoittain pistoksena 52 viikon ajan. Toinen ryhmä sai 20 mg adalimumabia viikoittain 52 viikon ajan. Kolmas ryhmä sai vuoroviikoin 40 mg adalimumabia ja vuoroviikoin plaseboa. Ensimmäisen 52 viikon jälkeen 457 potilasta siirrettiin avoimeen jatkovaiheeseen, jossa annettiin 40 mg adalimumabia/metotreksaattia joka toinen viikko 10 vuoden ajan.
Nivelreumatutkimuksessa IV arvioitiin lähinnä turvallisuutta 636 vähintään 18-vuotiaalla kohtalaisen aktiivista tai hyvin aktiivista nivelreumaa sairastavalla potilaalla. Potilailla ei ollut aikaisempaa reumalääkitystä tai he saivat jatkaa nykyistä reumalääkitystään, kunhan hoito pysyi samana vähintään 28 päivän ajan. Lääkitys oli metotreksaatti, leflunomidi, hydroksiklorokiini, sulfasalatsiini ja/tai kultasuolat. Potilaat satunnaistettiin saamaan joko 40 mg adalimumabia tai plaseboa joka toinen viikko 24 viikon ajan.
Nivelreumatutkimuksessa V arvioitiin 799 aikuispotilasta, joilla oli keskivaikea tai vaikea aktiivinen varhaisvaiheen nivelreuma (taudin kesto keskimäärin alle 9 kuukautta) ja jotka eivät olleet saaneet aiemmin metotreksaattihoitoa. Tässä tutkimuksessa arvioitiin adalimumabi 40 mg joka toinen viikko / metotreksaatti -kombinaatiohoitoa, adalimumabi 40 mg joka toinen viikko -monoterapiaa sekä metotreksaatti-monoterapiaa nivelreuman oireiden ja löydösten sekä nivelvaurioiden etenemisen estämisen suhteen nivelreumapotilailla 104 viikon ajan. Ensimmäisen 104 viikon jälkeen 497 potilasta siirrettiin avoimeen jatkovaiheeseen, jossa annettiin 40 mg adalimumabia joka toinen viikko 10 vuoden ajan.
Nivelreumatutkimuksissa VI ja VII tarkasteltiin molemmissa 60 keskivaikeaa tai vaikeaa aktiivista nivelreumaa sairastavaa ≥ 18-vuotiasta potilasta. Tutkimukseen osallistuneet potilaat olivat joko adalimumabi 40 mg/0,8 ml -valmisteen käyttäjiä ja he olivat arvioineet keskimääräisen pistoskohdan kivun ainakin 3 cm (0–10 cm VAS-mittarilla) tai he eivät olleet aiemmin saaneet biologista lääkitystä ja olivat aloittamassa adalimumabi 40 mg/0,8 ml -valmisteen käytön. Potilaat satunnaistettiin saamaan ensin kerta-annos joko adalimumabi 40 mg/0,8 ml- tai adalimumabi 40 mg/0,4 ml -valmistetta. Seuraavalla pistoskerralla potilaalle annettiin toista valmistetta kuin mitä hän oli saanut ensimmäisellä kerralla.
Ensisijainen päätetapahtuma nivelreumatutkimuksissa I, II ja III ja toissijainen päätetapahtuma nivelreumatutkimuksessa IV oli niiden potilaiden prosentuaalinen osuus, jotka saavuttivat ACR 20-vasteen viikolla 24 tai 26. Ensisijainen päätetapahtuma nivelreumatutkimuksessa V oli niiden potilaiden prosentuaalinen osuus, jotka saavuttivat ACR 50-vasteen viikolla 52. Ensisijaisena päätetapahtumana nivelreumatutkimuksissa III ja V oli lisäksi taudin etenemisen hidastuminen (röntgentutkimuksella todettuna) viikolla 52. Ensisijaisena päätetapahtumana nivelreuma-tutkimuksessa III oli lisäksi elämänlaadussa tapahtunut muutos. Ensisijainen päätetapahtuma nivelreumatutkimuksissa VI ja VII oli pistoskohdan kipu välittömästi lääkkeenannon jälkeen VASasteikolla (0–10 cm) mitattuna.
ACR-vaste
Prosentuaaliset osuudet niistä adalimumabia saaneista potilasta, jotka saavuttivat ACR 20, 50 ja 70 ‑vasteet, olivat yhdenmukaiset nivelreumatutkimuksissa I, II ja III. Yhteenveto annostasolla 40 mg joka toinen viikko saaduista tuloksista on esitetty taulukossa 8.
Taulukko 8
ACR-vasteet plasebokontrolloiduissa tutkimuksissa (prosentuaalinen osuus potilaista)
Vaste | Nivelreumatutkimus Ia** | Nivelreumatutkimus IIa** | Nivelreumatutkimus IIIa** | |||||
Plasebo/ MTXc | Adalimumabib/ MTXc | Plasebo | Adalimumabib | Plasebo/ MTXc | Adalimumabib/ MTXc | |||
ACR 20 | ||||||||
6 kk:n kuluttua | 13,3 % | 65,1 % | 19,1 % | 46,0 % | 29,5 % | 63,3 % | ||
12 kk:n kuluttua | - | - | - | - | 24,0 % | 58,9 % | ||
ACR 50 | ||||||||
6 kk:n kuluttua | 6,7 % | 52,4 % | 8,2 % | 22,1 % | 9,5 % | 39,1 % | ||
12 kk:n kuluttua | - | - | - | - | 9,5 % | 41,5 % | ||
ACR 70 | ||||||||
6 kk:n kuluttua | 3.3 % | 23,8 % | 1,8 % | 12,4 % | 2,5 % | 20,8 % | ||
12 kk:n kuluttua | - | - | - | - | 4,5 % | 23,2 % | ||
a Nivelreumatutkimus I viikolla 24, nivelreumatutkimus II viikolla 26 ja nivelreumatutkimus III viikoilla 24 & 52
b 40 mg adalimumabia joka toinen viikko
c MTX = metotreksaatti
** p < 0,01, adalimumabi vs. plasebo
- Ei käytettävissä
Nivelreumatutkimuksissa I-IV viikon 24 tai 26 kohdalla mitattu paraneminen kaikkien yksittäisten ACR-vastekriteerien osalta [aristavien ja turvonneiden nivelten lukumäärä, lääkärin ja potilaan arvio sairauden aktiivisuudesta ja kivusta, toimintakykyindeksin (HAQ) pisteet ja CRP (mg/dl)] oli huomattavampaa kuin plaseboryhmässä. Nivelreumatutkimuksessa III nämä paranemat säilyivät viikolle 52 asti.
Nivelreumatutkimuksen III avoimessa jatkovaiheessa vaste säilyi useimmilla ACR-vasteen saavuttaneilla potilailla enintään 10 vuoden seurannan ajan. 207 potilaasta, jotka oli satunnaistettu käyttämään 40 mg:n adalimumabiannostusta joka toinen viikko, 114 jatkoi 40 mg:n adalimumabiannosten käyttöä joka toinen viikko 5 vuoden ajan. Näistä potilaista 86 (75,4 %) saavutti ACR 20 -vasteen. 72 potilasta (63,2 %) saavutti ACR 50 -vasteen ja 41 potilasta (36 %) ACR 70 -vasteen. 207 potilaasta 81 jatkoi 40 mg:n adalimumabiannosten käyttöä joka toinen viikko 10 vuoden ajan. Näistä potilaista 64 (79,0 %) saavutti ACR 20 -vasteen. 56 potilasta (69,1 %) saavutti ACR 50 -vasteen ja 43 potilasta (53,1 %) ACR 70 -vasteen.
Nivelreumatutkimuksessa IV adalimumabia ja standardihoitoa saaneiden potilaiden ACR 20 -vaste oli tilastollisesti merkitsevästi parempi kuin plaseboa ja standardihoitoa saaneiden (p < 0,001).
Nivelreumatutkimuksissa I–IV adalimumabia saaneet potilaat saavuttivat plaseboon verrattuna tilastollisesti merkitsevät ACR 20 ja 50-vasteet jopa 1–2 viikon kuluttua hoidon aloittamisesta.
Nivelreumatutkimuksessa V potilailla, joilla oli varhaisvaiheen nivelreuma ja jotka eivät olleet aiemmin saaneet metotreksaattihoitoa, adalimumabin ja metotreksaatin yhdistelmä sai aikaan nopeamman ja merkitsevästi paremman ACR-vasteen viikolla 52 kuin pelkkä metotreksaatti tai pelkkä adalimumabi, ja vaste säilyi viikkoon 104 saakka (ks. taulukko 9).
Taulukko 9
ACR-vasteet nivelreumatutkimuksessa V (potilaiden prosentuaalinen osuus)
Vaste | MTX | Adali-mumabi | Adalimumabi/MTX | p-arvoa | p-arvob | p-arvoc |
ACR 20 | ||||||
Viikko 52 | 62,6 % | 54,4 % | 72,8 % | 0,013 | < 0,001 | 0,043 |
Viikko 104 | 56,0 % | 49,3 % | 69,4 % | 0,002 | < 0,001 | 0,140 |
ACR 50 | ||||||
Viikko 52 | 45,9 % | 41,2 % | 61,6 % | < 0,001 | < 0,001 | 0,317 |
Viikko 104 | 42,8 % | 36,9 % | 59,0 % | < 0,001 | < 0,001 | 0,162 |
ACR 70 | ||||||
Viikko 52 | 27,2 % | 25,9 % | 45,5 % | < 0,001 | < 0,001 | 0,656 |
Viikko 104 | 28,4 % | 28,1 % | 46,6 % | < 0,001 | < 0,001 | 0,864 |
a p-arvo on saatu metotreksaattimonoterapian ja adalimumabi/metotreksaattiyhdistelmähoidon parivertailusta (Mann-Whitneyn U -testillä).
b p-arvo on saatu adalimumabimonoterapian ja adalimumabi/metotreksaattiyhdistelmähoidon parivertailusta (Mann-Whitneyn U -testillä).
c p-arvo on saatu adalimumabimonoterapian ja metotreksaattimonoterapian parivertailusta (Mann-Whitneyn U -testillä).
Nivelreumatutkimuksen V avoimessa jatkovaiheessa ACR-vasteet säilyivät enintään 10 vuoden seurannassa. 542:sta potilaasta, jotka oli satunnaistettu saamaan 40 mg adalimumabia joka toinen viikko, 170 jatkoi 40 mg:n adalimumabiannosten käyttöä joka toinen viikko 10 vuoden ajan. Näistä potilaista 154 (90,6 %) saavutti ACR 20 -vasteen, 127 potilasta (74,7 %) saavutti ACR 50 -vasteen ja 102 potilasta (60,0 %) ACR 70 -vasteen.
Viikolla 52 adalimumabi/metotreksaattiyhdistelmähoitoa saaneista potilaista 42,9 %:llä saavutettiin kliininen remissio (DAS28 < 2,6), kun vastaava luku oli pelkkää metotreksaattia saaneilla 20,6 % ja pelkkää adalimumabia saaneilla 23,4 %. Adalimumabi/metotreksaattiyhdistelmähoito oli kliinisesti ja tilastollisesti parempi kuin pelkkä metotreksaatti (p < 0,001) tai pelkkä adalimumabi (p < 0,001) taudin lievittämisessä potilailla, joilla oli äskettäin diagnosoitu keskivaikea tai vaikea nivelreuma. Monoterapiaryhmissä vasteet olivat samankaltaiset (p = 0,447). Niistä 342:sta potilaasta, jotka oli alunperin satunnaistettu saamaan adalimumabia monoterapiana tai adalimumabi/metotreksaattiyhdistelmähoitoa ja jotka jatkoivat avoimessa jatkotutkimuksessa, 171 jatkoi adalimumabihoitoa 10 vuotta. Heistä 109:n (63,7 %) raportoitiin olevan remissiossa 10 vuoden kohdalla.
Radiologinen vaste
Nivelreumatutkimuksessa III, jossa adalimumabia saavilla potilailla oli ollut nivelreuma noin 11 vuotta, rakenteelliset nivelvauriot arvioitiin röntgenkuvista ja ilmaistiin muutoksina modifioiduissa Sharpin (TSS) kokonaispisteissä ja sen komponenteissa, eroosiopisteissä ja nivelraon madaltumispisteissä. Adalimumabi/metotreksaattipotilailla todettiin 6 ja 12 kuukauden kohdalla merkitsevästi vähemmän etenemistä kuin pelkkää metotreksaattia saavilla potilailla (ks. taulukko 10).
Avoimen nivelreumatutkimuksen III jatkotutkimuksessa rakenteellisten vaurioiden etenemisnopeuden hidastuminen säilyy 8 ja 10 vuotta osalla potilaista. 8 vuoden kohdalla arvioitiin röntgenkuvista 81 potilasta 207 potilaasta, joita alun perin hoidettiin adalimumabilla (40 mg joka toinen viikko). 48 potilaalla näistä ei havaittu rakenteellisten vaurioiden etenemistä (määritelmä: mTSS‑arvon muutokset lähtötilanteesta 0,5 tai vähemmän). 10 vuoden kohdalla tehtiin radiologiset arvioinnit 79:lle 207:sta potilaasta, joita alun perin hoidettiin adalimumabilla (40 mg joka toinen viikko). 40:llä näistä potilaista ei todettu rakenteellisten vaurioiden etenemistä (määritelmä: mTSS-arvon muutos lähtötilanteesta enintään 0,5 pistettä).
Taulukko 10
Radiologiset keskiarvomuutokset 12 kuukauden aikana nivelreumatutkimuksessa III
Plasebo/ | Adalimumabi/MTX | Plasebo/MTX-Adalimumabi/MTX (95 % luottamusvälib) | p-arvo | |
TSS | 2,7 | 0,1 | 2,6 (1,4, 3,8) | < 0,001c |
Eroosioaste | 1,6 | 0,0 | 1,6 (0,9, 2,2) | < 0,001 |
Nivelraon madaltuma | 1,0 | 0,1 | 0,9 (0,3, 1,4) | 0,002 |
a metoreksaatti
b 95 % luottamusväli muutosasteen eroissa metotreksaatin ja adalimumabin välillä.
c perustuu ranking-analyysiin
d Joint Space Narrowing
Nivelreumatutkimuksessa V rakenteelliset nivelvauriot arvioitiin röntgenkuvista ja ilmaistiin muutoksina modifioiduissa Sharpin kokonaispisteissä (ks. taulukko 11).
Taulukko 11
Radiologiset keskiarvomuutokset viikolla 52 nivelreumatutkimuksessa V
MTX | Adalimumabi | Adalimumabi/MTX | p-arvoa | p-arvob | p-arvoc | |
TSS | 5,7 (4,2–7,3) | 3,0 (1,7–4,3) | 1,3 (0,5–2,1) | < 0,001 | 0,0020 | < 0,001 |
Eroosioaste | 3,7 (2,7–4,7) | 1,7 (1,0–2,4) | 0,8 (0,4–1,2) | < 0,001 | 0,0082 | < 0,001 |
Nivelraon madaltuma | 2,0 (1,2–2,8) | 1,3 (0,5–2,1) | 0,5 (0–1,0) | < 0,001 | 0,0037 | 0,151 |
a p-arvo on saatu metotreksaattimonoterapian ja adalimumabi/metotreksaattiyhdistelmähoidon parivertailusta (Mann-Whitney U -testillä).
b p-arvo on saatu adalimumabimonoterapian ja adalimumabi/metotreksaattiyhdistelmähoidon parivertailusta (Mann-Whitney U -testillä).
c p-arvo on saatu adalimumabimonoterapian ja metotreksaattimonoterapian parivertailusta (Mann-Whitney U -testillä).
52 ja 104 viikon hoidon jälkeen niiden potilaiden prosentuaalinen osuus, joilla tauti ei ollut edennyt (muutos lähtötilanteesta modifioiduissa Sharpin kokonaispisteissä ≤ 0,5) oli merkitsevästi suurempi potilailla, jotka olivat saaneet adalimumabi/metotreksaattiyhdistelmähoitoa (63,8 % ja 61,2 %) kuin potilailla, jotka olivat saaneet pelkkää metotreksaattia (37,4 % ja 33,5 %, p < 0,001) tai pelkkää adalimumabia (50,7 %, p < 0,002 ja 44,5 %, p < 0,001).
Avoimessa nivelreuman jatkotutkimuksessa V keskimääräinen muutos lähtötilanteesta 10 vuoden kohdalla modifioiduissa Sharpin kokonaispisteissä (TSS) oli alun perin metotreksaattimonoterapiaan satunnaistetuilla potilailla 10,8, adalimumabimonoterapiaan satunnaistetuilla potilailla 9,2 ja adalimumabi/metoreksaattiyhdistelmähoitoon satunnaistetuilla potilailla 3,9. Vastaavat osuudet potilaista, joilla ei tapahtunut radiologista etenemistä, olivat 31,3 %, 23,7 % ja 36,7 %.
Elämänlaatu ja fyysinen toimintakyky
Terveyteen liittyvää elämänlaatua ja fyysistä toimintakykyä arvioitiin kaikissa neljässä asianmukaisessa ja hyvin kontrolloidussa tutkimuksessa toimintakykyindeksillä (HAQ), joka oli nivelreumatutkimuksessa III ennalta määritelty ensisijainen päätetapahtuma viikolla 52. Kaikki adalimumabiannokset/annostusohjelmat kaikissa neljässä tutkimuksessa osoittivat, että paraneminen mitattuna HAQ-toimintakykyindeksillä lähtötasolta kuukaudelle 6 oli plaseboon verrattuna tilastollisesti merkitsevästi parempaa, ja nivelreumatutkimuksessa III havaittiin samaa viikolla 52. Short Form Health Survey (SF 36) ‑kyselyn tulokset adalimumabin kaikkien annosten/annostusohjelmien osalta tukevat näitä löydöksiä (mm. PCS-pisteet ja kipu- ja vitaliteettiosioiden pisteet annokselle 40 mg joka toinen viikko olivat tilastollisesti merkitseviä). FACIT-pisteet osoittavat, että väsymys väheni tilastollisesti merkitsevästi kaikissa niissä kolmessa tutkimuksessa, joissa sitä arvioitiin (nivelreumatutkimukset I, II, III).
Nivelreumatutkimuksessa III fyysisessä toimintakyvyssä havaitut paranemiset säilyivät useimmilla potilailla, joiden fyysinen toimintakyky parani ja jotka jatkoivat hoitoa vielä avoimen tutkimuksen viikolla 520 (120 kuukautta). Elämänlaadun paraneminen mitattiin viikolle 156 (36 kuukautta) asti. Elämänlaadun paraneminen pysyi samalla tasolla koko tämän ajan.
Nivelreumatutkimuksessa V toimintakykyindeksin (HAQ) ja SF 36 -kyselyn fyysisen komponentin paraneminen oli merkittävämpää (p < 0,001) adalimumabi/metotreksaattiyhdistelmähoidon kuin pelkän metotreksaattihoidon tai pelkän adalimumabihoidon yhteydessä viikolla 52, ja vaikutus säilyi viikolle 104. Niillä 250:llä potilaalla, jotka olivat avoimessa jatkotutkimuksessa loppuun asti, fyysisessä toimintakyvyssä havaitut paranemiset säilyivät 10 hoitovuoden ajan.
Pistoskohdan kipu
Nivelreuman yhdistetyissä vaihtovuoroisissa tutkimuksissa VI ja VII havaittiin tilastollisesti merkitsevä ero välittömästi lääkkeenannon jälkeen havaitussa pistoskohdan kivussa valmisteiden adalimumabi 40 mg/0,8 ml ja adalimumabi 40 mg/0,4 ml välillä (VAS keskiarvo 3,7 cm vs. 1,2 cm asteikolla 0-10 cm, p < 0,001). Tämä vastasi 84 % mediaanivähenemää pistoskohdan kivussa.
Aksiaalinen spondylartriitti
Selkärankareuma
Adalimumabia (40 mg joka toinen viikko) arvioitiin kahdessa satunnaistetussa, 24 viikkoa kestäneessä, kaksoissokkoutetussa plasebokontrolloidussa tutkimuksessa 393 potilaalla, joilla oli aktiivinen selkärankareuma (taudin aktiivisuutta kuvaavat pisteet [Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)] olivat lähtötilanteessa 6,3 kaikissa ryhmissä) ja joiden vaste tavanomaisille hoidoille oli ollut riittämätön. 79 potilasta (20,1 %) sai samanaikaisesti DMARD-hoitoa ja 37 potilasta (9,4 %) glukokortikoidihoitoa. Sokkoutettua vaihetta seurasi avoin vaihe, jonka aikana potilaat saivat adalimumabia 40 mg ihon alle joka toinen viikko vielä enintään 28 viikon ajan. Potilaat, joilla ASAS 20 jäi saavuttamatta viikkojen 12, 16 tai 20 kohdalla (n = 215; 54,7 %), siirtyivät avoimelle adalimumabi 40 mg ihon alle joka toinen viikko -hoidolle. Näitä potilaita pidettiin tämän jälkeen hoidolle vasteettomina potilaina kaksoissokkoutetuissa tilastollisissa analyyseissä.
Suuremmassa selkärankareumatutkimuksessa I, johon osallistui 315 potilasta, adalimumabia saaneiden potilaiden selkärankareuman merkeissä ja oireissa todettiin merkitsevää paranemista plaseboa saaneisiin potilaisiin verrattuna. Merkitsevä vaste havaittiin viikon 2 kohdalla, ja se säilyi viikolle 24 asti (taulukko 12).
Taulukko 12
Tehokkuusvaste plasebokontrolloidussa selkärankareumatutkimuksessa (selkärankareumatutkimus I)
Merkkien ja oireiden väheneminen
Vaste | Plasebo | Adalimumabi |
ASASa 20 | ||
Viikko 2 | 16 % | 42 %*** |
Viikko 12 | 21 % | 58 %*** |
Viikko 24 | 19 % | 51 %*** |
ASAS 50 | ||
Viikko 2 | 3 % | 16 %*** |
Viikko 12 | 10 % | 38 %*** |
Viikko 24 | 11 % | 35 %*** |
ASAS 70 | ||
Viikko 2 | 0 % | 7 %** |
Viikko 12 | 5 % | 23 %*** |
Viikko 24 | 8 % | 24 %*** |
BASDAIb 50 | ||
Viikko 2 | 4 % | 20 %*** |
Viikko 12 | 16 % | 45 %*** |
Viikko 24 | 15 % | 42 %*** |
***, ** Tilastollisesti merkitsevä p < 0,001; < 0,01 kaikissa adalimumabi/plasebovertailuissa viikkojen 2, 12 ja 24 kohdalla
a Assessments in Ankylosing Spondylitis
b Bath Ankylosing Spondylitis Disease Activity Index
Adalimumabihoitoa saaneilla potilailla oli merkitsevästi paremmat vasteet viikon 12 kohdalla, ja ne säilyivät viikolle 24 asti sekä SF36-kyselyllä että selkärankareumaa koskevalla elämänlaatukyselyllä (ASQoL) mitattuna.
Samantyyppiset vasteet nähtiin pienemmässä satunnaistetussa, kaksoissokkoutetussa plasebokontrolloidussa selkärankareumatutkimuksessa II, johon osallistui 82 aktiivista selkärankareumaa sairastavaa aikuispotilasta (mutta ne eivät aina olleet tilastollisesti merkitseviä).
Aksiaalinen spondylartriitti (ilman radiografista näyttöä selkärankareumasta)
Adalimumabin tehoa ja turvallisuutta arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa potilailla, joilla oli röntgennegatiivinen aksiaalinen spondylartriitti. Tutkimuksessa nr-axSpA I arvioitiin potilaita, joilla oli aktiivinen röntgennegatiivinen aksiaalinen spondylartriitti. Tutkimus nr-axSpA II oli hoidon keskeyttämistutkimus aktiivista röntgennegatiivista aksiaalista spondylartriittia sairastavilla potilailla, jotka saavuttivat remission avoimen adalimumab-hoidon aikana.
Tutkimus nr-axSpA I
Tutkimuksessa nr-axSpA I, adalimumabihoitoa (40 mg joka toinen viikko) arvioitiin 185 potilaalla satunnaistetussa, 12 viikon pituisessa lumekontrolloidussa kaksoissokkotutkimuksessa, johon osallistuneilla potilailla oli aktiivinen, röntgennegatiivinen aksiaalinen spondylartriitti (tautiaktiivisuuspisteiden [Bath Ankylosing Spondylitis Disease Activity Index, BASDAI] lähtötilanteen keskiarvo adalimumabiryhmässä 6,4 ja lumeryhmässä 6,5) ja ≥ 1 tulehduskipulääke oli ollut teholtaan riittämätön tai huonosti siedetty tai tulehduskipulääkkeiden käyttö oli vasta-aiheista.
33 potilasta (18 %) käytti samanaikaisesti tautiprosessiin vaikuttavia reumalääkkeitä, ja 146 potilasta (79 %) käytti lähtötilanteessa tulehduskipulääkettä. Kaksoissokkovaiheen jälkeen seurasi avoin hoitovaihe, jonka aikana kaikki potilaat saivat adalimumabihoitoa (40 mg ihon alle joka toinen viikko) vielä enintään 144 viikon ajan. Viikon 12 tulokset osoittivat, että aktiivisen röntgennegatiivisen spondylartriitin oireet ja löydökset lievittyivät adalimumabiryhmässä tilastollisesti merkitsevästi verrattuna lumeryhmään (taulukko 13).
Taulukko 13
Teho lumekontrolloidussa nr-axSpA I tutkimuksessa
Kaksoissokkovaihe | Lume | Adalimumabi |
ASASa 40 | 15 % | 36 %*** |
ASAS 20 | 31 % | 52 %** |
ASAS 5/6 | 6 % | 31 %*** |
ASAS PR (osittainen vaste) | 5 % | 16 %*** |
BASDAIb 50 | 15 % | 35 %** |
ASDASc,d,e | -0,3 | -1,0*** |
ASDAS ID (inaktiivinen tauti) | 4 % | 24 %*** |
hs-CRPd,f,g | -0,3 | -4,7*** |
SPARCCh MRI sakroiliaaliset niveletd,i | -0,6 | -3,2** |
SPARCC MRI selkärankad,j | -0,2 | -1,8** |
a Assessments of SpondyloArthritis international Society
b Bath Ankylosing Spondylitis Disease Activity Index
c Ankylosing Spondylitis Disease Activity Score
d keskimääräinen muutos lähtöarvoihin nähden
e N = 91 lume ja N = 87 adalimumabi
f herkkä CRP-määritys (mg/l)
g N = 73 lume ja N = 70 adalimumabi
h Spondyloarthritis Research Consortium of Canada
i N = 84 lume ja adalimumabi
j N = 82 lume ja N = 85 adalimumabi
***, **, *: tilastollisesti merkitsevä, p-arvot < 0,001, < 0,01 ja < 0,05. Kaikki vertailut adalimumabin ja lumeryhmän välillä.
Avoimessa jatkotutkimuksessa adalimumabihoitoa saaneilla potilailla paranema havaituissa oireissa ja löydöksissä säilyi 156 viikon ajan.
Inflammaation esto
Herkällä CRP-määrityksellä (hs-CRP) mitattuna tulehdusarvossa havaittiin merkitsevä paranema, joka säilyi 156 viikon ajan. Sakroiliaalinivelten ja selkärangan MRI:n tulehduslöydöksissä havaittiin merkitsevä paranema 104 viikon ajan.
Elämänlaatu ja fyysinen toimintakyky
Terveyteen liittyvää elämänlaatua ja fyysistä toimintakykyä arvioitiin HAQ-S- ja SF-36-kyselylomakkeilla. Adalimumabi paransi HAQ-S-kokonaispisteitä ja SF-36-mittarin fyysisen osion pisteitä (Physical Component Score, PCS) tilastollisesti merkitsevästi enemmän kuin lumelääke, kun viikon 12 arvoja verrattiin lähtöarvoihin. Terveyteen liittyvän elämänlaadun ja fyysisen toimintakyvyn paranema säilyi avoimen jatkotutkimuksen ajan viikolle 156.
Tutkimus nr-axSpA II
Tutkimuksen nr-axSpA II avoimeen vaiheeseen otettiin 673 potilasta, joilla oli aktiivinen röntgennegatiivinen aksiaalinen spondylartriitti (tautiaktiivisuuspisteiden [BASDAI] lähtötilanteen keskiarvo 7,0) ja joilla vaste ≥ 2 tulehduskipulääkkeeseen oli riittämätön tai jotka eivät sietäneet tulehduskipulääkkeitä tai joilla tulehduskipulääkkeiden käyttö oli vasta-aiheista. Avoimen vaiheen aikana potilaat saivat adalimumab 40 mg -hoitoa joka toinen viikko 28 viikon ajan. Potilailla oli myös objektiivista näyttöä tulehduksesta (magneettikuvauksessa todettu sakroiliaalinivelen tai selkärangan tulehdus tai erittäin herkkä CRP koholla). Potilaat, jotka saavuttivat avoimessa vaiheessa pitkäkestoisen, vähintään 12 viikon pituisen remission (N = 305) (ASDAS < 1,3 viikoilla 16, 20, 24 ja 28), satunnaistettiin tämän jälkeen joko jatkamaan adalimumab -hoitoa (40 mg joka toinen viikko; N = 152) tai saamaan lumehoitoa (N = 153) vielä 40 viikon ajan kaksoissokkoutetussa, lumekontrolloidussa vaiheessa (tutkimuksen kokonaiskesto 68 viikkoa). Jos tutkittavalle kehittyi pahenemisvaihe kaksoissokkoutetun vaiheen aikana, hänelle voitiin antaa varahoitona adalimumab (40 mg joka toinen viikko) vähintään 12 viikon ajan.
Ensisijainen tehon päätetapahtuma oli niiden potilaiden osuus, joille ei kehittynyt pahenemisvaihetta tutkimuksen viikkoon 68 mennessä. Pahenemisvaiheeksi määriteltiin ASDAS-pistemäärä ≥ 2,1 kahdella peräkkäisellä käynnillä, joiden välillä kului neljä viikkoa. Potilaita, joille ei kehittynyt pahenemisvaihetta kaksoissokkoutetun vaiheen aikana, oli adalimumab-ryhmässä enemmän kuin lumeryhmässä (70,4 % vs. 47,1 %, p < 0,001) (kuva 1).
Kuva 1: Pahenemisvaiheeseen kulunut aika Kaplan–Meier-yhteenvetokäyrinä
tutkimuksessa nr-axSpA II
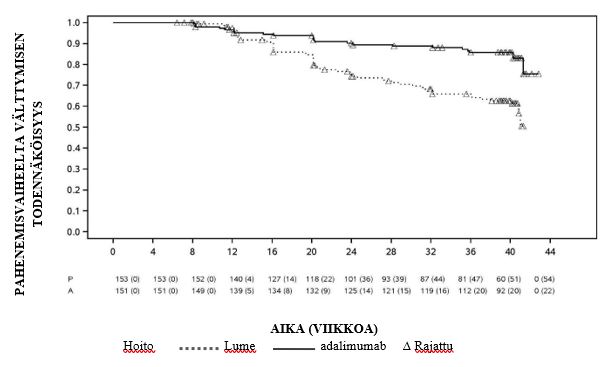
Huom. P = lume [riskille alttiina (pahenemisvaiheen kokeneet)]; A = ADALIMUMAB [riskille alttiina (pahenemisvaiheen kokeneet)].
Hoidon keskeytysryhmässä 68 potilaalle kehittyi pahenemisvaihe. Heistä 65 käytti adalimumab-varahoitoa 12 viikon ajan. Näistä potilaista 37 potilasta (56,9 %) saavutti uudelleen remission (ASDAS < 1,3) 12 viikon kuluessa avoimen hoidon uudelleenaloittamisesta.
Tutkimuksen kaksoissokkoutetun vaiheen aikana todettiin, että aktiivisen röntgennegatiivisen aksiaalisen spondylartriitin oireet ja löydökset lievittyivät viikkoon 68 mennessä tilastollisesti merkitsevästi useammin potilailla, jotka saivat jatkuvaa adalimumab-hoitoa, kuin hoidon keskeytysryhmään määrätyillä potilailla (taulukko 14).
Taulukko 14
Teho nr-axSpA II -tutkimuksen lumekontrolloidussa vaiheessa
Kaksoissokkoutettu | Lume | Adalimumab |
ASASa,b 20 | 47,1 % | 70,4 %*** |
ASASa,b 40 | 45,8 % | 65,8 %*** |
ASASa, osittainen remissio | 26,8 % | 42,1 %** |
ASDASc, inaktiivinen tauti | 33,3 % | 57,2 %*** |
Osittainen pahenemisvaihed | 64,1 % | 40,8 %*** |
a Assessment of SpondyloArthritis international Society | ||
Nivelpsoriaasi
Adalimumabia (40 mg joka toinen viikko) tutkittiin kahdessa plasebokontrolloidussa tutkimuksessa (nivelpsoriaasitutkimukset I ja II) potilailla, joilla oli keskivaikea tai vaikea aktiivinen nivelpsoriaasi. Nivelpsoriaasitutkimus I kesti 24 viikkoa, ja siihen osallistui 313 aikuispotilasta, joilla steroideihin kuulumattomat tulehduskipulääkkeet eivät olleet saaneet aikaan tyydyttävää vastetta. Noin 50 % näistä potilaista käytti metotreksaattia. Nivelpsoriaasitutkimus II kesti 12 viikkoa, ja siihen osallistui 100 potilasta, joilla DMARD-hoito ei ollut saanut aikaan tyydyttävää vastetta. Näiden tutkimusten päätyttyä 383 potilasta siirtyi avoimeen jatkotutkimukseen, jossa he jatkoivat adalimumabia annoksella 40 mg joka toinen viikko.
Adalimumabin tehokkuudesta ei ole riittävästi näyttöä potilailla, joilla on selkärankareuman kaltainen psoriaattinen artropatia, sillä tutkittujen potilaiden lukumäärä on pieni.
Taulukko 15
ACR-vaste plasebokontrolloiduissa nivelpsoriaasitutkimuksissa (potilaiden prosentuaalinen osuus)
Nivelpsoriaasitutkimus I | Nivelpsoriaasitutkimus II | |||
Vaste | Plasebo | Adalimumabi | Plasebo | Adalimumabi |
ACR 20 | ||||
Viikko 12 | 14 % | 58 %*** | 16 % | 39 %* |
Viikko 24 | 15 % | 57 %*** | - | - |
ACR 50 | ||||
Viikko 12 | 4 % | 36 %*** | 2 % | 25 %*** |
Viikko 24 | 6 % | 39 %*** | - | - |
ACR 70 | ||||
Viikko 12 | 1 % | 20 %*** | 0 % | 14 % * |
Viikko 24 | 1 % | 23 %*** | - | - |
*** p < 0,001 kaikissa adalimumabi/plasebovertailuissa
* p < 0,05 kaikissa adalimumabi/plasebovertailuissa
- ei käytettävissä
Nivelpsoriaasitutkimuksessa I ACR-vasteet olivat samankaltaiset riippumatta siitä, saivatko potilaat samanaikaisesti metotreksaattihoitoa vai eivät. ACR-vasteet säilyivät avoimessa jatkotutkimuksessa jopa 136 viikon ajan.
Nivelpsoriaasitutkimuksissa arvioitiin radiologisesti todettavia muutoksia. Kädet, ranteet ja jalkaterät kuvannettiin lähtötilanteessa viikolla 24, kun potilaat saivat kaksoissokkoutetusti joko adalimumabia tai lumelääkettä, sekä viikolla 48, jolloin kaikki potilaat saivat avoimesti adalimumabia. Arvioinnissa käytettiin modifioitua TSS-pisteytystä (mTSS), jossa sormien ja varpaiden kärkinivelet otettiin huomioon (ts. pisteytys erosi nivelreuman arviointiin käytetystä TSS-pisteytyksestä).
Adalimumabihoito hidasti perifeeristen nivelvaurioiden etenemistä verrattuna lumehoitoon. Lumeryhmässä mTSS-arvon muutos lähtötilanteeseen nähden (keskiarvo ± keskihajonta) oli 0,8 ± 2,5 viikolla 24, kun taas adalimumabiryhmässä se oli 0,0 ± 1,9 viikolla 48 (p < 0,001).
Taudin radiologinen eteneminen pysähtyi hoitoviikkoon 144 asti 84 prosentilla niistä adalimumabihoitoa saaneista potilaista, joiden tauti ei ollut radiologisesti edennyt lähtötilanteen ja viikon 48 välillä (n = 102). Adalimumabihoitoa saaneiden potilaiden fyysinen toimintakyky parani tilastollisesti merkitsevässä määrin, kun sitä arvioitiin HAQ-indeksillä ja SF 36 -kyselyllä (Short Form Health Survey) ja verrattiin lumehoitoon viikolla 24. Fyysinen toimintakyky pysyi lähtötilannetta parempana avoimen jatkotutkimuksen viikkoon 136 asti.
Psoriaasi
Adalimumabin tehoa ja turvallisuutta tutkittiin satunnaistetuissa, kaksoissokkoutetuissa tutkimuksissa aikuispotilailla, joilla oli krooninen läiskäpsoriaasi (≥ 10 % kehon pinta-alasta ja PASI-indeksi ≥ 12 tai ≥ 10) ja joille harkittiin systeemistä hoitoa tai valohoitoa. 73 % psoriaasitutkimuksiin I ja II osallistuneista potilaista oli käyttänyt aiemmin systeemistä hoitoa tai valohoitoa. Adalimumabin tehoa ja turvallisuutta tutkittiin satunnaistetussa kaksoissokkoutetussa tutkimuksessa (Psoriaasitutkimus III) myös aikuispotilailla, jotka sairastivat keskivaikeaa tai vaikeaa kroonista läiskäpsoriaasia ja siihen liittyvää käsi- ja/tai jalkapsoriaasia ja joille harkittiin systeemistä hoitoa.
Psoriaasitutkimuksessa I (REVEAL) arvioitiin 1 212 potilasta kolmen hoitojakson puitteissa. Hoitojaksolla A potilaat saivat joko lumehoitoa tai adalimumabihoitoa (80 mg:n aloitusannos, jonka jälkeen 40 mg joka toinen viikko; ensimmäinen 40 mg:n annos otettiin viikon kuluttua aloitusannoksesta). 16 hoitoviikon jälkeen ne potilaat, joilla saavutettiin vähintään PASI 75 -vaste (PASI-indeksi parani vähintään 75 % lähtötilanteeseen nähden), siirtyivät hoitojaksoon B ja saivat avointa adalimumabihoitoa annoksella 40 mg joka toinen viikko. Potilaat, joilla oli edelleen ≥ PASI 75 -vaste viikolla 33 ja jotka oli alunperin satunnaistettu hoitojaksolla A saamaan vaikuttavaa hoitoa, satunnaistettiin uudelleen hoitojaksolla C saamaan joko lumehoitoa tai 40 mg adalimumabia joka toinen viikko vielä 19 viikon ajan. Kaikissa hoitoryhmissä lähtötilanteen PASI-indeksi oli keskimäärin 18,9 ja lääkärin yleisarvio potilaan sairaudesta oli lähtötilanteessa ”keskivaikea” (53 % tutkimushenkilöistä), ”vaikea” (41 %) tai ”hyvin vaikea” (6 %).
Psoriaasitutkimuksessa II (CHAMPION) adalimumabin tehoa ja turvallisuutta verrattiin metotreksaattiin ja lumehoitoon 271 potilaalla. Potilaat saivat 16 viikon ajan joko lumehoitoa, metotreksaattia (aloitusannos 7,5 mg, jonka jälkeen annosta nostettiin viikolle 12 asti; maksimiannos 25 mg) tai adalimumabihoitoa (80 mg:n aloitusannos, jonka jälkeen 40 mg joka toinen viikko; ensimmäinen 40 mg annos viikon kuluttua aloitusannoksesta). Adalimumabia ja metotreksaattia vertailevia tietoja 16 hoitoviikkoa pidemmältä ajalta ei ole. Metotreksaattiryhmässä potilaiden annosta ei enää nostettu, jos heillä saavutettiin ≥ PASI 50 -vaste viikolla 8 ja/tai viikolla 12. Kaikissa hoitoryhmissä lähtötilanteen PASI-indeksi oli keskimäärin 19,7 ja lääkärin yleisarvio potilaan sairaudesta oli lähtötilanteessa ”lievä” (< 1 % tutkimushenkilöistä), ”keskivaikea” (48 %), ”vaikea” (46 %) tai ”hyvin vaikea” (6 %).
Kaikkiin vaiheen 2 ja vaiheen 3 psoriaasitutkimuksiin osallistuneet potilaat soveltuivat avoimeen jatkotutkimukseen, jossa adalimumabia annettiin vielä vähintään 108 viikon ajan.
Psoriaasitutkimusten I ja II ensisijainen päätetapahtuma oli niiden potilaiden osuus, jotka saavuttivat viikolla 16 PASI 75 -vasteen lähtötilanteeseen nähden (ks. taulukot 16 ja 17).
Taulukko 16
Psoriaasitutkimus I (REVEAL) - Tehotulokset 16 viikon kohdalla
Lumehoito | Adalimumabi (40 mg joka 2. viikko) | |
≥ PASI 75a | 26 (6,5) | 578 (70,9)b |
PASI 100 | 3 (0,8) | 163 (20,0)b |
Lääkärin yleisarvio: puhdas/melkein puhdas | 17 (4,3) | 506 (62,2)b |
a PASI 75 -vasteen saavuttaneiden potilaiden prosenttiosuus laskettiin keskusten mukaan vakioituna
b p < 0,001, adalimumabi vs. lumehoito
Taulukko 17
Psoriaasitutkimus II (CHAMPION) - Tehotulokset 16 viikon kohdalla
Lumehoito | Metotreksaatti | Adalimumabi (40 mg joka 2. viikko) | |
≥ PASI 75 | 10 (18,9) | 39 (35,5) | 86 (79,6)a, b |
PASI 100 | 1 (1,9) | 8 (7,3) | 18 (16,7)c, d |
Lääkärin yleisarvio: puhdas/melkein puhdas | 6 (11,3) | 33 (30,0) | 79 (73,1)a, b |
a p < 0,001 adalimumabi vs. lumehoito
b p < 0,001 adalimumabi vs. metotreksaatti
c p < 0,01 adalimumabi vs. lumehoito
d p < 0,05 adalimumabi vs. metotreksaatti
Psoriaasitutkimuksessa I 28 % potilaista, joilla saavutettiin PASI 75 -vaste ja jotka satunnaistettiin uudelleen saamaan lumehoitoa viikolla 33, menetti riittävän vasteen (PASI-indeksi viikon 33 jälkeen ja viikolla 52 tai sitä ennen oli sellainen, että PASI-vaste lähtötilanteeseen nähden oli < 50, ja indeksi suureni vähintään 6 pistettä viikkoon 33 nähden). Adalimumabihoitoa jatkaneessa ryhmässä näin tapahtui vain 5 %:lle potilaista (p < 0,001). 38 % (25/66) niistä potilaista, jotka menettivät riittävän vasteen lumehoitoon satunnaistamisen jälkeen ja siirtyivät myöhemmin avoimeen jatkotutkimukseen, saavutti jälleen PASI 75 -vasteen 12 hoitoviikon jälkeen ja 55 % (36/66) 24 hoitoviikon jälkeen.
Yhteensä 233 PASI 75 -vasteen viikolla 16 ja viikolla 33 saavuttanutta potilasta sai jatkuvaa adalimumabihoitoa 52 viikon ajan psoriaasitutkimuksessa I ja jatkoi adalimumabihoitoa avoimessa jatkotutkimuksessa. 108 viikon avoimen lisähoidon jälkeen (tutkimuksen alusta yhteensä 160 hoitoviikkoa) 74,7 % potilaista sai PASI 75 -vasteen ja 59,0 %:lla lääkärin yleisarvio oli puhdas tai melkein puhdas. Analyysissä, jossa kaikki potilaat, jotka keskeyttivät tutkimuksen haittatapahtumien tai riittämättömän tehon vuoksi tai joiden annosta suurennettiin, luokiteltiin ilman vastetta jääneiksi potilaiksi, PASI 75 -vasteen sai 69,6 % potilaista ja 55,7 %:lla lääkärin yleisarvio oli puhdas tai melkein puhdas 108 viikon avoimen lisähoidon jälkeen (yhteensä 160 viikkoa).
Yhteensä 347 pysyvän vasteen saanutta potilasta osallistui hoidon keskeyttämistä ja uusintahoitoa arvioivaan avoimeen jatkotutkimukseen. Keskeyttämisjakson aikana psoriaasioireet uusiutuivat ajan myötä, ja relapsiin (lääkärin yleisarvio huononi ”keskivaikeaksi” tai huonommaksi) kuluneen ajan mediaani oli noin 5 kuukautta. Yhdelläkään potilaista ei esiintynyt rebound-ilmiötä keskeyttämisjakson aikana. Yhteensä 76,5 %:lla (218/285) uusintahoitojakson aloittaneista potilaista lääkärin yleisarvio oli ”puhdas” tai ”melkein puhdas” 16 uusintahoitoviikon kuluttua riippumatta siitä, esiintyikö relapseja keskeyttämisjakson aikana (69,1 % [123/178] potilaista, joilla oli relapsi keskeyttämisjakson aikana, ja 88,8 % [95/107] potilaista, joilla ei ollut relapsia keskeyttämisjakson aikana). Uusintahoidon aikana turvallisuusprofiili oli samankaltainen kuin ennen hoidon keskeyttämistä.
DLQI-elämänlaatumittarilla (Dermatology Life Quality Index) mitattuna potilaiden vointi koheni merkitsevästi viikkoon 16 mennessä lähtötilanteeseen nähden, kun tuloksia verrattiin lumehoitoon (tutkimukset I ja II) ja metotreksaattiin (tutkimus II). Tutkimuksessa I myös SF-36-mittarin fyysisten ja psyykkisten osioiden yhteispisteet paranivat merkitsevästi verrattuna lumehoitoon.
Avoimessa jatkotutkimuksessa osa potilaista siirtyi riittämättömän PASI-vasteen (< 50 %) vuoksi käyttämään 40 mg annosta joka viikko joka toisen viikon sijasta. Näistä potilaista 26,4 % (92/349) saavutti PASI 75 -vasteen 12 viikon kuluttua ja 37,8 % (132/349) 24 viikon kuluttua tehostettuun hoitoon siirtymisestä.
Psoriaasitutkimus III (REACH) vertasi adalimumabi- ja lumehoidon tehoa ja turvallisuutta 72 potilaalla, joilla oli keskivaikea tai vaikea krooninen läiskäpsoriaasi ja käsi- ja/tai jalkapsoriaasi. Potilaat saivat 16 viikon ajan joko lumehoitoa tai adalimumabia aloitusannoksen 80 mg ja tämän jälkeen 40 mg joka toinen viikko (alkaen yksi viikko aloitusannoksen jälkeen). 16 viikon kohdalla tilastollisesti merkitsevästi suurempi osa potilaista, jotka saivat adalimumabihoitoa (30,6 %), saavutti lääkärin yleisarvioksi puhdas tai melkein puhdas käsien ja/tai jalkojen osalta verrattuna niihin potilaisiin, jotka saivat lumehoitoa (4,3 %, [p = 0,014]).
Psoriaasitutkimus IV vertasi adalimumabi- ja lumehoidon tehoa ja turvallisuutta 217 aikuispotilaalla, joilla oli keskivaikea tai vaikea kynsipsoriaasi. Potilaat saivat 26 viikon ajan joko lumehoitoa tai adalimumabia aloitusannoksen 80 mg ja tämän jälkeen 40 mg joka toinen viikko (alkaen yksi viikko aloitusannoksen jälkeen), minkä jälkeen he saivat avointa adalimumabihoitoa vielä toisten 26 viikon ajan. Kynsipsoriaasin arvioinnissa käytettyjä mittareita olivat mm. Modified Nail Psoriasis Severity Index (mNAPSI), Physician’s Global Assessment of FingernailPsoriasis (PGA-F) ja Nail Psoriasis Severity Index (NAPSI) (ks. taulukko 18). Adalimumabi osoitti hoitohyötyä kynsipsoriaasipotilailla vaihtelevan kokoisilla iho-alueilla (BSA ≥ 10 % (60 % potilaista) ja BSA < 10 % ja ≥5 % (40 % potilaista)).
Taulukko 18
Psoriaasitutkimus IV - Tehotulokset 16, 26 ja 52 viikon kohdalla
Päätetapahtuma | Viikko 16 | Viikko 26 | Viikko 52 | ||
Lumelääke | Adalimumabi | Lumelääke | Adalimumabi | Adalimumabi | |
≥ mNAPSI 75 (%) | 2,9 | 26,0a | 3,4 | 46,6a | 65,0 |
PGA-F puhdas/hyvin vähäinen ja ≥ 2-asteen paraneminen (%) | 2,9 | 29,7a | 6,9 | 48,9a | 61,3 |
Prosentuaalinen muutos kynnen kokonais-NAPSI-mittarilla (%) | -7,8 | -44,2a | -11,5 | -56,2a | -72,2 |
a p < 0,001, adalimumabi vs. lumehoito
Adalimumabilla hoidetuilla potilailla todettiin merkitsevää paranemista viikon 26 kohdalla verrattuna lumevalmistetta saaneisiin potilaisiin DLQI-elämänlaatumittarilla mitattuna.
Hidradenitis suppurativa
Adalimumabin turvallisuutta ja tehoa arvioitiin satunnaistetuissa, kaksoissokkoutetuissa, lumekontrolloiduissa tutkimuksissa ja avoimessa jatkotutkimuksessa keskivaikeaa ja vaikeaa hidradenitis suppurativaa (HS-tautia) sairastavilla aikuispotilailla, kun potilas ei sietänyt systeemistä antibioottihoitoa, kyseinen hoito oli vasta-aiheinen tai kyseisellä hoidolla ei saatu riittävää vastetta vähintään 3 kuukaudessa. HS-I- ja HS-II-tutkimuksien potilailla oli Hurley-asteen II tai III tauti, johon liittyi vähintään 3 absessia tai tulehduksellista kyhmyä.
HS-I-tutkimuksessa (PIONEER I) arvioitiin 307 potilasta kahden hoitojakson puitteissa. Hoitojaksolla A potilaat saivat lumehoitoa tai adalimumabia (aloitusannos 160 mg viikolla 0, 80 mg viikolla 2 ja 40 mg kerran viikossa viikolta 4 viikolle 11). Samanaikainen antibioottien käyttö ei ollut sallittua tutkimuksen aikana. 12 hoitoviikon jälkeen hoitojaksolla A adalimumabia saaneet potilaat satunnaistettiin uudelleen hoitojaksolla B yhteen kolmesta hoitoryhmästä (adalimumabi 40 mg kerran viikossa, adalimumabi 40 mg joka toinen viikko tai lumehoito viikolta 12 viikolle 35). Hoitojaksolla A lumehoitoryhmään satunnaistetut potilaat saivat hoitojaksolla B 40 mg adalimumabia kerran viikossa.
HS-II-tutkimuksessa (PIONEER II) arvioitiin 326 potilasta kahden hoitojakson puitteissa. Hoitojaksolla A potilaat saivat lumehoitoa tai adalimumabia (aloitusannos 160 mg viikolla 0, 80 mg viikolla 2 ja 40 mg kerran viikossa viikolta 4 viikolle 11). 19,3 % potilaista jatkoi lähtötilanteen oraalista antibioottihoitoa tutkimuksen aikana. 12 hoitoviikon jälkeen hoitojaksolla A adalimumabia saaneet potilaat satunnaistettiin uudelleen hoitojaksolla B yhteen kolmesta hoitoryhmästä (adalimumabi 40 mg kerran viikossa, adalimumabi 40 mg joka toinen viikko tai lumehoito viikolta 12 viikolle 35). Hoitojaksolla A lumehoitoryhmään satunnaistetut potilaat saivat lumehoitoa hoitojaksolla B.
HS-I- ja HS-II-tutkimuksiin osallistuneet potilaat soveltuivat avoimeen jatkotutkimukseen, jossa adalimumabia annettiin 40 mg kerran viikossa. Keskimääräinen altistus kaikissa adalimumabiryhmissä oli 762 päivää. Potilaat käyttivät paikallisesti antiseptista ihohuuhdetta päivittäin kaikkien kolmen tutkimuksen ajan.
Kliininen vaste
Tulehduksellisten leesioiden vähenemistä ja absessien ja vuotavien fistelien pahenemisen ehkäisemistä arvioitiin HiSCR-vasteen avulla (Hidradenitis Suppurativa Clinical Response; absessien ja tulehduksellisten kyhmyjen kokonaismäärän pieneneminen vähintään 50 %:lla ilman absessien ja vuotavien fistelien määrän lisääntymistä suhteessa lähtötilanteeseen). HS-tautiin liittyvän ihokivun vähenemistä arvioitiin numeerisella arviointiasteikolla potilailla, joilla lähtötilanteen aloituspistemäärä oli tutkimukseenottohetkellä vähintään kolme 11-portaisella asteikolla.
Merkitsevästi suurempi osuus adalimumabia saaneista potilaista saavutti HiSCR-vasteen viikon 12 kohdalla verrattuna lumehoitoa saaneisiin. HS-II-tutkimuksessa merkitsevästi suuremmalla osalla potilaista HS-tautiin liittyvä ihokipu väheni kliinisesti merkittävästi viikon 12 kohdalla (ks. taulukko 19). Adalimumabia saaneilla potilailla taudin pahenemisvaiheen riski oli merkitsevästi pienempi ensimmäisten 12 hoitoviikon aikana.
Taulukko 19
Tehotulokset 12 viikon kohdalla (HS-tutkimukset I ja II)
HS-tutkimus I | HS-tutkimus II | |||
Lumehoito | 40 mg adalimumabia kerran viikossa | Lumehoito | 40 mg adalimumabia kerran viikossa | |
HiSCR-vaste (Hidradenitis Suppurativa clinical response)a | N = 154 | N = 153 | N = 163 | N = 163 |
≥ 30 % ihokivun vähenemäb | N = 109 | N = 122 | N = 111 | N = 105 |
* p < 0,05, ***p < 0,001, adalimumabi vs. lumehoito
a Kaikkien satunnaistettujen potilaiden keskuudessa.
b Potilailla, joilla HS-tautiin liittyvän ihokivun arvioitu pistemäärä lähtötilanteessa ≥ 3 numeerisella arviointiasteikolla 0–10; 0 = ei ihokipua, 10 = pahin kuviteltavissa oleva ihokipu.
Adalimumabihoito (40 mg kerran viikossa) pienensi merkitsevästi absessien ja vuotavien fistelien pahenemisen riskiä. Lumeryhmässä adalimumabiryhmään verrattuna noin kaksinkertaisella määrällä potilaita esiintyi absessien pahenemista (23,0 % vs. 11,4 %) ja vuotavien fistelien pahenemista (30,0 % vs. 13,9 %) HS-I- ja HS-II-tutkimuksen ensimmäisten 12 viikon aikana.
Viikon 12 kohdalla seuraavilla osa-alueilla tapahtui lähtötilanteeseen verrattuna enemmän kohentumista kuin lumehoidossa: ihospesifinen terveyteen liittyvä elämänlaatu DLQI-elämänlaatumittarilla mitattuna (HS-I- ja HS-II-tutkimus), potilaan yleinen tyytyväisyys lääkehoitoon TSQM-kyselylomakkeella mitattuna (HS-I- ja HS-II-tutkimus) ja fyysinen terveys SF-36-mittarin fyysisen osion yhteispisteillä mitattuna (HS-I-tutkimus).
Vähintään osittaisen vasteen adalimumabihoitoon (40 mg kerran viikossa) viikon 12 kohdalla saaneilla potilailla HiSCR-prosentti viikon 36 kohdalla oli adalimumabihoitoa viikoittain jatkaneilla potilailla suurempi kuin potilailla, joilla annostelutiheyttä harvennettiin joka toiseen viikkoon tai joilla hoito keskeytettiin (ks. taulukko 20).
Taulukko 20
Osuus potilaistaa, jotka saavuttivat HiSCR-vasteenb 24 ja 36 viikon kuluttua siitä, kun heidät viikoittaisen adalimumabihoidon jälkeen uudelleensatunnaistettiin (viikko 12)
Lumehoito(hoidon keskeyttäminen) | 40 mg adalimumabiajoka toinen viikko | 40 mg adalimumabiakerran viikossa | |
Viikko 24 | 24 (32,9 %) | 36 (51,4 %) | 40 (57,1 %) |
Viikko 36 | 22 (30,1 %) | 28 (40,0 %) | 39 (55,7 %) |
- Potilaat, jotka saivat vähintään osittaisen vasteen adalimumabihoidolle (40 mg kerran viikossa) 12 hoitoviikon jälkeen.
- Jos tutkimussuunnitelmassa nimetyt kriteerit täyttyivät eli vaste menetettiin tai tila ei kohentunut, potilaan oli keskeytettävä osallistuminen tutkimukseen ja potilas tulkittiin ei vastetta saaneeksi.
68,3 % potilaista, jotka saivat vähintään osittaisen vasteen viikolla 12 ja jotka saivat jatkuvaa viikoittaista adalimumabihoitoa, saavutti HiSCR-vasteen viikolla 48; viikolla 96 sen saavutti 65,1 % potilaista. Kun adalimumabihoitoa annettiin pitempikestoisesti 40 mg 96 viikon ajan, uusia turvallisuuteen liittyviä löydöksiä ei havaittu.
Potilailla, joilla adalimumabihoito keskeytettiin HS-I- ja HS-II-tutkimuksissa viikon 12 kohdalla, HiSCR-prosentti palautui 12 viikon kuluttua adalimumabihoidon (40 mg kerran viikossa) uudelleenaloittamisesta samalle tasolle kuin ennen keskeyttämistä (56,0 %).
Crohnin tauti
Adalimumabin tehoa ja turvallisuutta arvioitiin satunnaistetuissa, kaksoissokkoutetuissa ja lume-kontrolloiduissa tutkimuksissa yli 1 500 potilaalla, joilla oli keskivaikea tai vaikea aktiivinen Crohnin tauti (pisteet taudin aktiivisuutta mittaavalla CDAI-indeksillä ≥ 220 ja ≤ 450). Aminosalisylaatit, kortikosteroidit ja/tai immunomodulaattorit muuttumattomina annoksina olivat tutkimuksen aikana sallittuja, ja 80 % potilaista sai ainakin yhtä näistä lääkityksistä.
Kliinisen remission (määritelmän mukaan CDAI < 150) induktiota arvioitiin Crohn-tutkimuksessa I (CLASSIC I) ja Crohn-tutkimuksessa II (GAIN). Crohn-tutkimuksessa I 299 potilasta, jotka eivät olleet aiemmin saaneet TNF-salpaajia, satunnaistettiin johonkin neljästä tutkimusryhmästä. Ryhmille annettiin joko lumelääkettä viikoilla 0 ja 2, 160 mg adalimumabia viikolla 0 ja 80 mg adalimumabia viikolla 2, 80 mg adalimumabia viikolla 0 ja 40 mg adalimumabia viikolla 2, tai 40 mg adalimumabia viikolla 0 ja 20 mg adalimumabia viikolla 2. Crohn-tutkimukseen II otettiin 325 potilasta, jotka olivat joko menettäneet hoitovasteensa tai eivät sietäneet infliksimabia. Heidät satunnaistettiin saamaan joko 160 mg adalimumabia viikolla 0 ja 80 mg adalimumabia viikolla 2 tai lumelääkettä viikoilla 0 ja 2. Potilaat, joilla ei saavutettu primaarista hoitovastetta, suljettiin pois tutkimuksista, eikä heitä arvioitu pidempään.
Crohn-tutkimuksessa III (CHARM) arvioitiin kliinisen remission pysyvyyttä. Crohn-tutkimuksessa III 854 potilasta sai avoimesti 80 mg adalimumabia viikolla 0 ja 40 mg adalimumabia viikolla 2. Viikolla 4 potilaat satunnaistettiin saamaan joko 40 mg adalimumabia joka toinen viikko, 40 mg adalimumabia kerran viikossa tai lumelääkettä. Tutkimuksen kokonaiskesto oli 56 viikkoa. Potilaat, joilla todettiin kliininen hoitovaste (CDAI-pisteiden väheneminen ≥ 70:lla) viikolla 4, stratifioitiin ja analysoitiin erillään potilaista, joilla ei saavutettu kliinistä hoitovastetta viikkoon 4 mennessä. Kortikosteroidiannoksen pienentäminen sallittiin viikon 8 jälkeen.
Crohn-tutkimuksissa I ja II saavutetut remissio- ja vasteprosentit on esitetty taulukossa 21.
Taulukko 21
Kliininen remissio ja vasteet (% potilaista)
Crohn-tutkimus I: potilaat, jotka eivät olleet aiemmin saaneet infliksimabia | Crohn-tutkimus II: aiemmin infliksimabia saaneet potilaat | ||||
Lumelääke | Adalimumabi | Adalimumabi | Lumelääke | Adalimumabi | |
Viikko 4 | |||||
Kliininen remissio | 12 % | 24 % | 36 %* | 7 % | 21 %* |
Kliininen vaste (CR-100) | 24 % | 37 % | 49 %** | 25 % | 38 %** |
Kaikki p-arvot on saatu adalimumabin ja lumelääkkeen osuuksien parivertailuista.
* p < 0,001
** p < 0,01
Remissioprosentit olivat viikon 8 kohdalla samankaltaiset riippumatta siitä, saivatko potilaat hoidon alussa 160/80 mg vai 80/40 mg adalimumabia, mutta 160/80 mg saaneilla potilailla haittatapahtumien esiintymistiheys oli suurempi.
Crohn-tutkimuksessa III 58 %:lla potilaista (499/854) todettiin kliininen vaste viikolla 4, ja nämä potilaat otettiin mukaan päävasteanalyysiin. Viikkoon 4 mennessä kliinisen hoitovasteen saavuttaneista potilaista 48 % oli saanut aiemmin jotain muuta TNF-antagonistihoitoa. Remission ja vasteiden pysyvyys on esitetty taulukossa 22. Kliinisen remission ylläpidon suhteen tulokset pysyivät melko muuttumattomina aiemmista TNF-antagonistihoidoista riippumatta.
Sairauteen liittyvien sairaalahoitojaksojen ja leikkausten määrä väheni adalimumabihoidolla lumehoitoon verrattuna tilastollisesti merkitsevästi viikolla 56.
Taulukko 22
Kliinisen remission ja vasteiden ylläpito (% potilaista)
Lumelääke | 40 mg adalimumabia joka toinen viikko | 40 mg adalimumabia kerran viikossa | |
Viikko 26 | N = 170 | N = 172 | N = 157 |
Kliininen remissio | 17 % | 40 %* | 47 %* |
Kliininen vaste (CR-100) | 27 % | 52 %* | 52 %* |
Potilaat, joiden remissio ilman steroideja kesti ≥ 90 päivääa | 3 % (2/66) | 19 % (11/58)** | 15 % (11/74)** |
Viikko 56 | N = 170 | N = 172 | N = 157 |
Kliininen remissio | 12 % | 36 %* | 41 %* |
Kliininen vaste (CR-100) | 17 % | 41 %* | 48 %* |
Potilaat, joiden remissio ilman steroideja kesti ≥ 90 päivääa | 5 % (3/66) | 29 % (17/58)* | 20 % (15/74)** |
* p < 0,001 saatu adalimumabin ja lumelääkkeen osuuksien parivertailuista
** p < 0,02 saatu adalimumabin ja lumelääkkeen osuuksien parivertailuista
a Potilaista, jotka käyttivät lähtötilanteessa kortikosteroideja
Potilaista, joilla ei todettu hoitovastetta viikolla 4, 43 % adalimumabia ylläpitohoitona saaneista ja 30 % lumelääkettä ylläpitohoitona saaneista potilaista saavuttivat hoitovasteen viikkoon 12 mennessä. Näiden tulosten perusteella ylläpitohoidon jatkamisesta viikolle 12 voi olla hyötyä joillekin potilaille, joilla ei saavuteta hoitovastetta viikkoon 4 mennessä. Hoidon jatkaminen yli 12 viikon ajan ei lisännyt merkitsevästi vasteen saaneita (ks. kohta Annostus ja antotapa).
117/276 potilasta Crohn-tutkimuksessa I ja 272/777 potilasta Crohn-tutkimuksista II ja III seurattiin 3 vuoden ajan avoimessa adalimumabihoidossa. 88 (tutkimuksessa I) ja 189 (tutkimuksissa II ja III) potilasta pysyi kliinisessä remissiossa. Kliininen vaste (CR-100) säilyi 102 (tutkimuksessa I) ja 233 potilaalla (tutkimuksissa II ja III).
Elämänlaatu
Crohn-tutkimuksissa I ja II potilailla, jotka oli satunnaistettu saamaan adalimumabia joko annoksena 80/40 mg tai 160/80 mg, todettiin lumelääkettä saaneisiin potilaisiin verrattuna tilastollisesti merkitsevää paranemista tulehduksellisia suolistosairauksia spesifisesti arvioivan IBDQ-kyselylomakkeen kokonaispisteissä viikolla 4. Samansuuntaista paranemista todettiin myös Crohn-tutkimuksessa III viikolla 26 ja viikolla 56 adalimumabia saaneissa hoitoryhmissä lumelääkkeeseen verrattuna.
Ulseratiivinen koliitti
Toistuvien adalimumabiannosten turvallisuutta ja tehoa arvioitiin satunnaistetuissa, kaksoissokkoutetuissa, lumekontrolloiduissa tutkimuksissa aikuispotilailla, joiden ulseratiivinen koliitti oli aktiivisuudeltaan keskivaikea tai vaikea (Mayo-pisteet 6–12, endoskopiaosion pisteet 2–3).
UC-I-tutkimuksessa 390 potilasta, jotka eivät olleet saaneet aiemmin TNF-antagonisteja, satunnaistettiin saamaan joko lumelääkettä viikkojen 0 ja 2 kohdalla tai 160 mg adalimumabia viikon 0 kohdalla ja tämän jälkeen 80 mg viikon 2 kohdalla tai 80 mg adalimumabia viikon 0 kohdalla ja tämän jälkeen 40 mg viikon 2 kohdalla. Viikon 2 jälkeen kummankin adalimumabiryhmän potilaat saivat 40 mg 2 viikon välein. Kliinisen remission saavuttaminen (määritelmänä Mayo-pisteet ≤ 2, kunkin osion pisteet ≤ 1) arvioitiin viikon 8 kohdalla.
UC-II-tutkimuksessa 248 potilasta sai 160 mg adalimumabia viikon 0 kohdalla, 80 mg viikon 2 kohdalla ja tämän jälkeen 40 mg 2 viikon välein, ja 246 potilasta sai lumelääkettä. Kliinisiä tuloksia arvioitiin remission induktion osalta viikon 8 kohdalla ja remission ylläpidon osalta viikon 52 kohdalla.
Kliinisen remission saavuttaminen viikon 8 kohdalla oli tilastollisesti merkitsevästi yleisempää alussa 160/80 mg adalimumabia saaneilla potilailla kuin lumelääkettä saaneilla sekä UC-I-tutkimuksessa (adalimumabi 18 % vs. lume 9 %, p = 0,031) että UC-II-tutkimuksessa (adalimumabi 17 % vs. lume 9 %, p = 0,019). UC-II-tutkimuksessa niistä adalimumabia saaneista potilaista, jotka saavuttivat remission viikon 8 kohdalla, 21/41 (51 %) oli edelleen remissiossa viikon 52 kohdalla.
Taulukossa 23 esitetään koko UC-II-tutkimuspopulaation tulokset.
Taulukko 23
Hoitovaste, remissio ja limakalvon paraneminen UC-II-tutkimuksessa (Prosenttia potilaista)
Lume | Adalimumabi 40 mg joka toinen viikko | |
Viikko 52 | N = 246 | N = 248 |
Kliininen vaste | 18 % | 30 %* |
Kliininen remissio | 9 % | 17 %* |
Limakalvon paraneminen | 15 % | 25 %* |
Remissiossa ilman steroidihoitoa ≥ 90 vrk ajana | 6 % | 13 %* |
Viikot 8 ja 52 | ||
Pitkäkestoinen vaste | 12 % | 24 %** |
Pitkäkestoinen remissio | 4 % | 8 %* |
Pitkäkestoinen limakalvon paraneminen | 11 % | 19 %* |
Kliininen remissio: Mayo-pisteet ≤ 2, kunkin osion pisteet > 1;
Kliininen vaste: Mayo-pisteiden lasku ≥ 3 pistettä ja ≥ 30 % lähtötilanteesta; lisäksi rektaaliverenvuotoa kuvaavan RBS-pistearvon tulee laskea ≥ 1 tai olla 0 tai 1.
* p < 0,05, adalimumabi vs. lume, prosenttiosuuksien pareittainen vertailu
** p < 0,001, adalimumabi vs. lume, prosenttiosuuksien pareittainen vertailu
a Potilaista, jotka saivat kortikosteroidihoitoa lähtötilanteessa
Niistä potilaista, jotka saivat vasteen viikolla 8, viikon 52 kohdalla 47 % oli säilyttänyt vasteen, 29 % oli remissiossa, 41 %:lla havaittiin limakalvon paraneminen ja 20 % oli ollut steroidivapaassa remissiossa ≥ 90 vuorokautta.
Aiempi infliksimabilla toteutettu TNF-antagonistihoito oli epäonnistunut noin 40 prosentilla UC-II-tutkimuksen potilaista. Näillä potilailla adalimumabin teho oli heikompi kuin potilailla, jotka eivät olleet saaneet aiempaa TNF-antagonistihoitoa. Potilaista, joiden aiempi TNF-antagonistihoito oli epäonnistunut, viikon 52 kohdalla remission saavutti 3 % lumehoitoa saaneista ja 10 % adalimumabihoitoa saaneista.
UC-I ja -II -tutkimusten potilaat saivat siirtyä avoimeen, pitkäkestoiseen jatkotutkimukseen (UC-III). Seuraavan kolmen vuoden adalimumabihoidon jälkeen 75 % (301/402) oli edelleen kliinisessä remissiossa osittaisen Mayo-pisteytyksen mukaan.
Sairaalahoitoon joutuminen
Adalimumabitutkimushaarassa hoidetut potilaat tarvitsivat vähemmän sekä kaikista syistä johtuvaa että ulseratiiviseen koliittiin liittyvää sairaalahoitoa 52 viikkoa kestäneiden UC-I ja UC-II -tutkimusten aikana kuin lumelääketutkimushaarassa hoidetut potilaat. Kun huomioon otettiin kaikki sairaalahoitoon johtaneet syyt, Adalimumabihoitoa saaneista 0,18 ja lumehoitoa saaneista 0,26 tapausta/potilasvuosi tarvitsi sairaalahoitoa. Vastaavasti ulseratiivisen koliitin vuoksi sairaalahoitoon joutui adalimumabihoitoa saaneista 0,12 ja lumehoitoa saaneista 0,22 tapausta/potilasvuosi.
Elämänlaatu
UC-II –tutkimuksessa Inflammatory Bowel Disease Questionaire (IBDQ)-pisteet paranivat adalimumabihoitoa saaneilla.
Uveiitti
Adalimumabin turvallisuutta ja tehoa arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa (UV I ja II) aikuispotilailla, joilla oli ei-infektioperäinen intermediaarinen uveiitti, posteriorinen uveiitti tai panuveiitti. Pois suljettiin potilaat, joilla oli isoloitu anteriorinen uveiitti. Potilaat saivat joko lumehoitoa tai adalimumabihoitoa (80 mg aloitusannos, jonka jälkeen 40 mg joka toinen viikko; ensimmäinen 40 mg:n annos otettiin viikon kuluttua aloitusannoksesta). Samanaikainen hoito yhdellä vakaa-annoksisella ei-biologisella immunosuppressantilla sallittiin.
UV I -tutkimuksessa arvioitiin 217 potilasta, joilla oli aktiivinen uveiitti kortikosteroidihoidosta huolimatta (peroraalinen prednisoni annoksella 10–60 mg/vrk). Kaikki potilaat saivat 2 viikon ajan prednisonihoitoa standardiannoksella 60 mg/vrk tutkimukseenottovaiheessa, minkä jälkeen oli pakollinen annoksen pienentämisvaihe, ja kortikosteroidihoito lopetettiin kokonaan viikkoon 15 mennessä.
UV II -tutkimuksessa arvioitiin 226 potilasta, joiden inaktiivinen uveiitti vaati lähtötilanteessa pitkäaikaista kortikosteroidihoitoa (peroraalinen prednisoni 10–35 mg/vrk) taudin hallinnassa pysymiseksi. Tämän jälkeen oli pakollinen annoksen pienentämisvaihe, ja kortikosteroidihoito lopetettiin kokonaan viikkoon 19 mennessä.
Ensisijainen tehon päätetapahtuma oli kummassakin tutkimuksessa ”hoidon epäonnistumiseen kuluva aika”. Hoidon epäonnistumisen määritelmänä oli monitekijäinen vastemuuttuja, joka perustui inflammatorisiin suoni- ja verkkokalvon ja/tai inflammatorisiin verkkokalvon verisuonien leesioihin, etukammion solujen määrään, lasiaissamentumien asteeseen ja parhaaseen lasikorjattuun näöntarkkuuteen (BCVA).
Potilaat jotka olivat mukana UV I ja UV II -tutkimuksissa olivat oikeutettuja osallistumaan kontrolloimattomaan pitkäkestoiseen jatkotutkimukseen, jonka alkuperäinen suunniteltu kesto oli 78 viikkoa. Potilaat saivat jatkaa tutkimuslääkkeellä viikon 78 jälkeen, siihen saakka kun he saivat adalimumabia.
Kliininen vaste
Kummassakin tutkimuksessa havaittiin adalimumabiryhmässä tilastollisesti merkitsevä hoidon epäonnistumisriskin pienentyminen vs. lumeryhmä (ks. taulukko 24). Kummassakin tutkimuksessa adalimumabihoidolla havaittiin varhainen ja pitkäkestoinen vaikutus hoidon epäonnistumisosuuteen vs. lume (ks. kuva 2).
Taulukko 24
Hoidon epäonnistumiseen kulunut aika UV I- ja UV II -tutkimuksissa
Analyysi Hoito | N | Epäonnistuminen N (%) | Mediaaniaika epäonnistumiseen (kk) | HRa | 95 % lv, HR | P-arvob |
Hoidon epäonnistumiseen kulunut aika viikon 6 kohdalla tai sen jälkeen UV I-tutkimuksessa | ||||||
Lume | 107 | 84 (78,5) | 3,0 | - | - | - |
Adalimumabi | 110 | 60 (54,5) | 5,6 | 0,50 | 0,36 –0,70 | < 0,001 |
Hoidon epäonnistumiseen kulunut aika viikon 2 kohdalla tai sen jälkeen tutkimuksessa UV II | ||||||
Lume | 111 | 61 (55,0) | 8,3 | - | - | - |
Adalimumabi | 115 | 45 (39,1) | EAc | 0,57 | 0,39–0,84 | 0,004 |
Huom. Hoidon epäonnistuminen viikon 6 kohdalla tai sen jälkeen (UV I -tutkimus) tai viikon 2 kohdalla tai sen jälkeen (UV II -tutkimus) laskettiin tapahtumaksi. Muusta syystä kuin hoidon epäonnistumisesta johtuneet keskeytykset tutkimuksessa rajattiin keskeytyshetken kohdalta.
a Adalimumabin vs. lumeen riskisuhde (HR) laskettiin suhteellisen riskin regressiolla, jossa tekijänä oli hoito.
b 2-tahoinen P-arvo laskettiin log-rank-testillä.
c EA = ei arvioitavissa. Alle puolella riskille alttiista potilaista esiintyi tapahtuma.
Kuva 2: Hoidon epäonnistumiseen kulunut aika yhteenvetona Kaplan–Meier-käyrinä viikon 6 kohdalla tai sen jälkeen (UV I -tutkimus) tai viikon 2 kohdalla tai sen jälkeen (UV II ‑tutkimus)
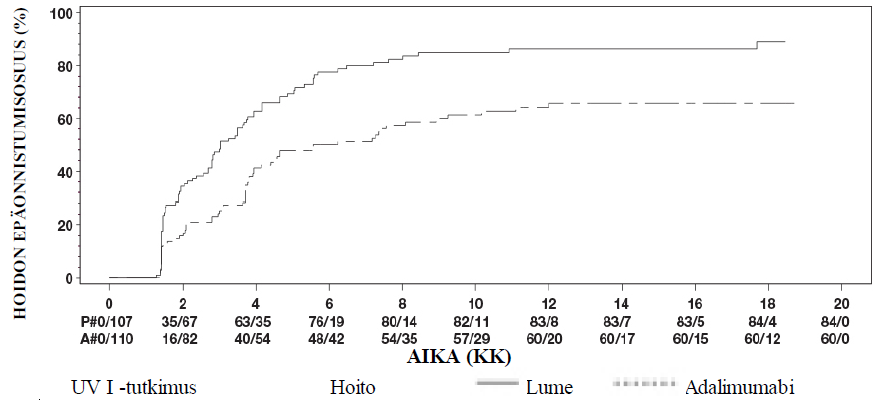
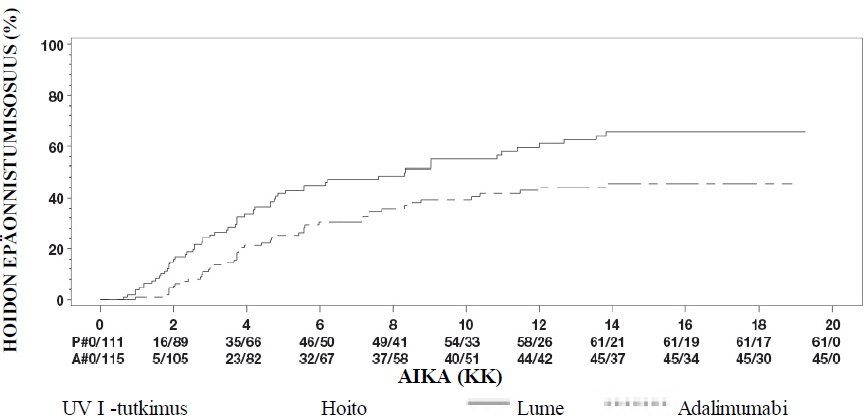
Huom. P# = Lume (tapahtumien määrä/riskille alttiiden määrä); A# = Adalimumabi (tapahtumien määrä/riskille alttiiden määrä).
UV I -tutkimuksessa havaittiin tilastollisesti merkitseviä eroja adalimumabin hyväksi vs. lume kunkin hoidon epäonnistumisen osatekijän osalta. UV II -tutkimuksessa havaittiin tilastollisesti merkitseviä eroja vain näöntarkkuuden osalta, mutta muutkin osatekijät olivat adalimumabilla numeerisesti paremmat.
Tutkimusten UV I ja UV II kontrolloimattomaan pitkäkestoiseen jatkotutkimukseen otettiin 424 tutkittavaa. Heistä 60 tutkittavan ei katsottu soveltuvan analyysiin (esim. poikkeamien, diabeettisen retinopatian komplikaatioiden, kaihileikkauksen tai vitrektomian vuoksi), ja heidät suljettiin pois ensisijaisesta tehoanalyysistä. Jäljelle jääneistä 364 tutkittavasta 269 arviointikelpoista tutkittavaa (74 %) sai avointa adalimumabihoitoa 78 viikon ajan. Havaintotietojen perusteella 216 tutkittavan (80,3 %) tilanne oli rauhallinen (ei aktiivisia tulehdusmuutoksia, etukammion solujen määrä ≤ 0,5+, lasiaissamentumien aste ≤ 0,5+) ja samanaikaisesti käytössä ollut steroidiannos oli ≤ 7,5 mg/vrk. 178 tutkittavalla (66,2 %) tilanne oli rauhallinen eikä käytössä ollut steroidilääkitystä. Paras lasikorjattu näöntarkkuus (BCVA) oli joko aiempaa parempi tai ennallaan (huonontunut < 5 kirjaimen verran) 88,6 %:ssa silmistä viikolla 78. Viikon 78 jälkeiset tiedot olivat yleisesti yhdenmukaisia näiden tulosten kanssa, mutta (tutkimukseen) osallistuneiden tutkittavien määrä laski tämän ajan jälkeen. Kaiken kaikkiaan tutkimuksen keskeyttäneistä tutkittavista 18 % keskeytti osallistumisensa haittatapahtumien vuoksi ja 8 % siksi, että adalimumabihoidolla saavutettu vaste oli riittämätön.
Elämänlaatu
Kummassakin kliinisessä tutkimuksessa arvioitiin potilaiden ilmoittamia lopputulemia näköön liittyvästä toimintakyvystä NEI VFQ-25-kyselyllä. Adalimumabi oli numeerisesti parempi valtaosassa alaosioista, ja tilastollisesti merkitsevät keskimääräiset erot saavutettiin UV I -tutkimuksessa yleisen näkökyvyn, silmäkivun, lähinäön, mielenterveyden ja kokonaispisteiden alaosioissa sekä UV II ‑tutkimuksessa yleisen näkökyvyn ja mielenterveyden alaosioissa. Näkökykyyn liittyvät vaikutukset eivät olleet adalimumabihoidolla numeerisesti parempia UV I -tutkimuksessa värinäön osalta ja UV II -tutkimuksessa värinäön, perifeerisen näön ja lähinäön osalta.
Immunogeenisuus
Adalimumabihoidon aikana saattaa kehittyä anti-adalimumabivasta-aineita. Anti-adalimumabivasta-aineiden muodostus tehostaa adalimumabin puhdistumaa ja heikentää sen tehoa. Anti-adalimumabivasta-aineiden muodostuksen ja haittatapahtumien esiintymistiheyden välillä ei ole mitään selkeää yhteyttä.
Pediatriset potilaat
Idiopaattinen juveniili artriitti (JIA)
Idiopaattinen juveniili polyartriitti (pJIA)
Adalimumabin tehoa ja turvallisuutta arvioitiin kahdessa tutkimuksessa (pJIA-tutkimukset I ja II) lapsilla, joilla oli aktiivinen idiopaattinen juveniili polyartriitti tai taudinkulultaan polyartikulaarinen idiopaattinen juveniili artriitti; idiopaattisen juveniilin artriitin alkuvaihe vaihteli, mutta useimmiten kyseessä oli aluksi reumatekijänegatiivinen tai -positiivinen polyartriitti tai leviävä oligoartriitti.
pJIA-I
Adalimumabin tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa, rinnakkaisryhmissä toteutetussa monikeskustutkimuksessa, johon osallistui 171 lasta, joilla oli idiopaattinen juveniili polyartriitti (ikä 4–17 v). Avoimessa aloitusvaiheessa potilaat stratifioitiin kahteen ryhmään, metotreksaattia käyttäviin ja ei metotreksaattia käyttäviin. Potilaat, jotka eivät käyttäneet metotreksaattia, eivät joko olleet käyttäneet sitä koskaan tai olivat lopettaneet sen käytön viimeistään kaksi viikkoa ennen tutkimuslääkkeen antamista. Potilaat käyttivät edelleen vakaina annoksina steroideihin kuulumattomia tulehduskipulääkkeitä (NSAID-lääkkeitä) ja/tai prednisonia (≤ 0,2 mg/kg/vrk tai enintään 10 mg/vrk). Avoimessa aloitusvaiheessa kaikki potilaat saivat 24 mg/m2 adalimumabihoitoa (enintään 40 mg) joka toinen viikko 16 viikon ajan. Potilaiden jakaumat iän ja avoimessa aloitusvaiheessa käytetyn minimi-, mediaani- ja maksimiannoksen mukaan esitetään taulukossa 25.
Taulukko 25
Potilaiden jakauma iän ja avoimessa aloitusvaiheessa käytetyn adalimumabiannoksen mukaan
Ikäryhmä | Potilasmäärä lähtötilanteessa, N (%) | Minimi-, mediaani- ja maksimiannos |
4–7 v | 31 (18,1) | 10, 20 ja 25 mg |
8–12 v | 71 (41,5) | 20, 25 ja 40 mg |
13–17 v | 69 (40,4) | 25, 40 ja 40 mg |
Potilaat, joilla saavutettiin Paediatric ACR 30 -vaste viikolla 16, voitiin satunnaistaa kaksoissokkoutettuun vaiheeseen, jossa he saivat joko adalimumabia (24 mg/m2, enintään 40 mg) tai lumelääkettä joka toinen viikko vielä 32 viikon ajan tai taudin pahenemisvaiheeseen saakka. Pahenemisvaiheen kriteerit määriteltiin seuraavasti: lähtötasoon verrattuna ≥ 30 % huonompi tulos vähintään 3:ssa Paediatric ACR -kriteerien 6 keskeisestä kriteeristä, tautiaktiivisuutta ≥ 2 nivelessä ja > 30 % parempi tulos enintään 1 kriteerin kohdalla 6:sta. 32 viikon kuluttua tai taudin pahenemisvaiheessa potilaat saivat siirtyä avoimeen jatkovaiheeseen.
Taulukko 26
Idiopaattinen juveniili polyartriitti ‑tutkimuksen PaedACR 30 ‑vasteprosentit
Ryhmä | Metotreksaatti | Ei metotreksaattia | ||
Vaihe | ||||
Avoin aloitusvaihe, 16 viikkoa | ||||
PaedACR 30 ‑vaste (n/N) | 94,1 % (80/85) | 74,4 % (64/86) | ||
Tehokkuustulokset | ||||
Kaksoissokkoutettu vaihe, 32 viikkoa | Adalimumabi/MTX (N = 38) | Lume/MTX (N = 37) | Adalimumabi (N = 30) | Lume (N = 28) |
Potilaat, joilla esiintyi taudin pahenemisvaiheita 32 viikon aikanaa (n/N) | 36,8 % (14/38) | 64,9 % (24/37)b | 43,3 % (13/30) | 71,4 % (20/28)c |
Pahenemisvaiheen alkamiseen kuluneen ajan mediaani | > 32 viikkoa | 20 viikkoa | > 32 viikkoa | 14 viikkoa |
a PaedACR 30/50/70 -vasteprosentit viikolla 48 merkitsevästi suuremmat kuin lumeryhmässä
b p = 0,015
c p = 0,031
Jos potilas saavutti vasteen viikolla 16 (n = 144), Paediatric ACR 30/50/70/90 -vaste säilyi jopa 6 vuoden ajan avoimessa jatkovaiheessa niillä potilailla, jotka saivat adalimumabia koko tutkimuksen ajan. Yhteensä 19 tutkimushenkilöä sai hoitoa vähintään 6 vuoden ajan. 11 heistä kuului lähtötilanteessa 4–12-vuotiaiden ikäryhmään ja 8 taas lähtötilanteessa 13–17-vuotiaiden ikäryhmään.
Kokonaisvasteprosentit olivat yleisesti ottaen paremmat ja harvemmille potilaille kehittyi vasta-aineita adalimumabin ja metotreksaatin yhdistelmää käytettäessä kuin pelkän adalimumabihoidon aikana. Kun nämä tulokset otetaan huomioon, adalimumabia suositellaan käytettäväksi yhdessä metotreksaatin kanssa ja käytettäväksi ainoana lääkkeenä niillä potilailla, joille metotreksaatin käyttö ei sovi (ks. kohta Annostus ja antotapa).
pJIA-II
Adalimumabin turvallisuutta ja tehoa arvioitiin avoimessa monikeskustutkimuksessa, johon osallistui 32 lasta (ikä 2 – < 4 v tai ikä vähintään 4 v ja paino < 15 kg), joilla oli keskivaikea tai vaikea, aktiivinen idiopaattinen juveniili polyartriitti. Potilaat saivat 24 mg/m2 (enintään 20 mg) kerta-annoksen adalimumabia joka toinen viikko injektiona ihon alle vähintään 24 viikon ajan. Useimmat potilaat käyttivät tutkimuksen aikana samanaikaisesti myös metotreksaattia, kun taas kortikosteroidien tai tulehduskipulääkkeiden käyttöä ilmoitettiin harvemmin.
Viikolla 12 PaedACR 30-vasteprosentti oli 93,5 % ja viikolla 24 90,0 % (havaintotiedot). Viikolla 12 PaedACR 50/70/90-vasteen saavutti 90,3 %/61,3 %/38,7 % potilaista ja viikolla 24 83,3 %/73,3 %/36,7 % potilaista. Niistä potilaista, jotka saavuttivat Paediatric ACR 30 -vasteen viikolla 24 (n = 27 yhteensä 30 potilaasta), Paediatric ACR 30 -vaste säilyi jopa 60 viikon ajan avoimessa jatkovaiheessa, kun potilaat saivat adalimumabia koko tämän ajan. Yhteensä 20 potilasta sai hoitoa vähintään 60 viikon ajan.
Entesiitteihin liittyvä artriitti
Adalimumabin turvallisuutta ja tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa, johon osallistui 46 potilasta (6–17-vuotiaita), joilla oli keskivaikea entesiitteihin liittyvä artriitti. Potilaat satunnaistettiin saamaan joko adalimumabia 24 mg/m2 (korkeintaan 40 mg) tai lumelääkettä joka toinen viikko 12 viikon ajan. Tutkimus jatkuu kaksoissokkoutetun jakson jälkeen avoimena tutkimuksena vielä 192 viikon ajan. Avoimen tutkimusjakson aikana potilaat saivat adalimumabia 24 mg/m2 (korkeintaan 40 mg) joka toinen viikko injektiona ihon alle. Ensisijaisena päätetapahtuma pidettiin prosentuaalista muutosta lähtötilanteesta viikolle 12 aktiivien tulehtuneiden nivelten määrässä (nivelen turvotusta, joka ei johtunut epämuodostumasta, tai niveliä, joissa oli liikerajoitteita sekä kipua ja/tai arkuutta). Adalimumabiryhmässä saavutettiin keskimäärin -62,6 % (mediaani -88,9 %) muutos ja lumelääkeryhmässä -11,6 % (mediaani -50,0 %) muutos. Aktiivien tulehtuneiden nivelten määrässä havaittu positiivinen muutos säilyi samana avoimen tutkimusjakson viikolle 156 asti 26:lla tutkimuksessa jatkaneesta adalimabiryhmän 31:sta potilaasta (84 %). Enemmistöllä potilaista myös toissijaisten päätetapahtumien mittareissa, kuten entesiitin esiintymiskohtien määrä, arkojen nivelten määrä (tender joint count, TJC), turvonneiden nivelten määrä (swollen joint count, SJC), Paediatric ACR 50 -vaste ja Paediatric ACR 70 -vaste, havaittiin paranemista, vaikkakaan ei tilastollisesti merkitsevästi.
Läiskäpsoriaasi lapsilla
Adalimumabin tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa, kontrolloidussa tutkimuksessa 114:llä vähintään 4-vuotiaalla pediatrisella potilaalla, joilla oli vaikea krooninen läiskäpsoriaasi (määritelmänä lääkärin yleisarvio ≥ 4 tai > 20 % kehon pinta-alasta tai > 10 % kehon pinta-alasta sekä hyvin paksuja leesioita tai PASI ≥ 20 tai ≥ 10 sekä kliinisesti merkittäviä muutoksia kasvoissa, sukuelimissä tai käsissä/jalkaterissä) ja joilla paikallishoito ja auringonvalohoito tai valohoito eivät olleet tuottaneet riittävää vastetta.
Potilaat saivat adalimumabia 0,8 mg/kg joka toinen viikko (enintään 40 mg), 0,4 mg/kg joka toinen viikko (enintään 20 mg) tai metotreksaattia 0,1–0,4 mg/kg viikoittain (enintään 25 mg). Viikon 16 kohdalla positiivinen tehovaste (esim. PASI 75) havaittiin useammin adalimumabi 0,8 mg/kg -hoitoon satunnaistetuilla potilailla kuin potilailla, jotka oli satunnaistettu saamaan adalimumabia 0,4 mg/kg joka toinen viikko tai metotreksaattia.
Taulukko 27
Tehotulokset pediatrisilla läiskäpsoriaasipotilailla 16 viikon kohdalla
MTXa | Adalimumabi (0,8 mg/kg joka 2. viikko) | |
PASI 75b | 12 (32,4 %) | 22 (57,9 %) |
Lääkärin yleisarvio: puhdas/melkein puhdasc | 15 (40,5 %) | 23 (60,5 %) |
a MTX = metotreksaatti
b P = 0,027, adalimumabi 0,8 mg/kg vs. MTX
c P = 0,083, adalimumabi 0,8 mg/kg vs. MTX
Jos potilas saavutti PASI 75 -vasteen ja lääkärin yleisarvio oli puhdas tai melkein puhdas, hoito keskeytettiin enintään 36 viikoksi ja potilasta seurattiin taudin pahenemisen varalta (lääkärin yleisarvio huononee vähintään 2 luokkaa). Tämän jälkeen potilaat saivat uusintahoitona 0,8 mg/kg adalimumabia joka toinen viikko vielä 16 viikon ajan. Uusintahoidon aikana havaitut vasteet olivat samaa luokkaa kuin aiemman kaksoissokkovaiheen aikana: PASI 75 –vasteen saavutti 78,9 % tutkimushenkilöistä (15 tutkimushenkilöä 19:stä) ja lääkärin yleisarvion puhdas tai melkein puhdas 52,6 % tutkimushenkilöistä (10 tutkimushenkilöä 19:stä).
Tutkimuksen avoimessa vaiheessa PASI 75 -vaste ja lääkärin yleisarvio puhdas tai melkein puhdas säilyivät vielä enintään 52 viikon ajan ilman uusia turvallisuuslöydöksiä.
Hidradenitis suppurativa nuorilla
Nuorilla HS-tautia sairastavilla potilailla ei ole tehty kliinisiä adalimumabitutkimuksia. Adalimumabin teho nuorten HS-tautia sairastavien potilaiden hoidossa on arvioitu perustuen aikuisilla HS-tautia sairastavilla potilailla osoitettuun tehoon ja altistus-vastesuhteeseen sekä siihen todennäköisyyteen, että taudin kulku, patofysiologia ja lääkkeen vaikutukset ovat olennaisesti samanlaiset kuin aikuisilla samoilla altistumistasoilla. Suositellun adalimumabiannoksen turvallisuus nuorille HS-tautia sairastaville potilaille perustuu adalimumabin turvallisuusprofiiliin muissa käyttöaiheissa sekä aikuis- että lapsipotilailla samoilla tai tiheämmillä annoksilla (ks. kohta Farmakokinetiikka).
Crohnin tauti lapsilla
Adalimumabihoitoa arvioitiin satunnaistetussa, kaksoissokkoutetussa kliinisessä monikeskustutkimuksessa, jossa arvioitiin induktio- ja ylläpitohoidon tehoa ja turvallisuutta painon (< 40 kg tai ≥ 40 kg) mukaisina annoksina 192 lapsipotilaalla, joiden ikä oli 6–17 vuotta ja joilla oli keskivaikea tai vaikea Crohnin tauti (määritelmä: Paediatric Crohn’s Disease Activity Index -pisteet [PCDAI] > 30). Mukaan otettiin vain potilaita, joilla Crohnin taudin tavanomainen hoito (mm. kortikosteroidi- ja/tai immunomodulanttihoito) oli epäonnistunut. Myös aiemman infliksimabihoitovasteen menettäminen tai infliksimabihoitoon liittyneet siedettävyysongelmat olivat hyväksyttäviä mukaanottoperusteita.
Kaikki potilaat käyttivät avointa induktiohoitoa, jonka annos riippui heidän painostaan lähtötilanteessa. ≥ 40 kg:n painoiset potilaat saivat 160 mg:n annoksen viikolla 0 ja 80 mg:n annoksen viikolla 2. Alle 40 kg:n painoiset taas saivat 80 mg viikolla 0 ja 40 mg viikolla 2.
Viikolla 4 potilaat satunnaistettiin ajankohtaisen painon mukaan suhteessa 1:1 käyttämään joko pieniä tai tavanomaisia annoksia ylläpitohoitona (ks. taulukko 28).
Taulukko 28
Ylläpitohoito
Paino | Pieni annos | Tavanomainen annos |
< 40 kg | 10 mg joka 2. viikko | 20 mg joka 2. viikko |
≥ 40 kg | 20 mg joka 2. viikko | 40 mg joka 2. viikko |
Tehotulokset
Tutkimuksen ensisijainen päätetapahtuma oli kliininen remissio viikolla 26 (määritelmä: PCDAI-pisteet ≤ 10).
Kliiniset remissio- ja vasteprosentit esitetään taulukossa 29 (kliinisen vasteen määritelmä: PCDAI-pisteet pienenivät vähintään 15 pistettä lähtöarvoihin nähden). Kortikosteroidi- tai immunomodulanttihoidon lopettamisprosentit esitetään taulukossa 30.
Taulukko 29
Tutkimus lasten Crohnin taudista
Kliininen PCDAI-remissio ja vaste
Tavanomainen annos | Pieni annos | p-arvo* | |
Viikko 26 | |||
Kliininen remissio | 38,7 % | 28,4 % | 0,075 |
Kliininen vaste | 59,1 % | 48,4 % | 0,073 |
Viikko 52 | |||
Kliininen remissio | 33,3 % | 23,2 % | 0,100 |
Kliininen vaste | 41,9 % | 28,4 % | 0,038 |
* Tavanomaisen ja pienen annoksen vertailun p-arvo.
Taulukko 30
Tutkimus lasten Crohnin taudista
Kortikosteroidien tai immunomodulanttien lopettaminen ja fistelien remissio
Tavanomainen annos | Pieni annos | p-arvo1 | |
Lopetti kortikosteroidit | N = 33 | N = 38 | |
Viikko 26 | 84,8 % | 65,8 % | 0,066 |
Viikko 52 | 69,7 % | 60,5 % | 0,420 |
Lopetti immunomodulantit2 | N = 60 | N = 57 | |
Viikko 52 | 30,0 % | 29,8 % | 0,983 |
Fistelien remissio3 | N = 15 | N = 21 | |
Viikko 26 | 46,7 % | 38,1 % | 0,608 |
Viikko 52 | 40,0 % | 23,8 % | 0,303 |
1 Tavanomaisen ja pienen annoksen vertailun p-arvo.
2 Immunosuppressanttihoito voitiin lopettaa aikaisintaan viikolla 26 tutkijan arvion mukaan, jos kliinisen vasteen kriteerit täyttyivät.
3 Määritelmä: kaikki lähtötilanteessa vuotaneet fistelit sulkeutuneina vähintään kahdella peräkkäisellä käynnillä lähtötasokäynnin jälkeen.
Molemmissa hoitoryhmissä todettiin, että painoindeksi ja pituuskasvunopeus suurenivat (paranivat) tilastollisesti merkitsevästi lähtötilanteen ja viikkojen 26 ja 52 välillä.
Molemmissa hoitoryhmissä todettiin myös elämänlaatuparametrien (mm. IMPACT III) parantuneen tilastollisesti ja kliinisesti merkitsevässä määrin lähtötilanteeseen nähden.
Lasten Crohnin tauti -tutkimukseen osallistuneista potilaista sata (n = 100) jatkoi avoimeen pitkäkestoiseen jatkotutkimukseen. 5 vuoden adalimumabihoidon jälkeen 74,0 % (37/50) 50:stä tutkimuksessa jäljellä olevasta potilaasta pysyi kliinisessä remissiossa, ja 92,0 %:lla (46/50) potilaista säilyi kliininen vaste PCDAI:n mukaan.
Ulseratiivinen koliitti lapsilla
Adalimumabin turvallisuutta ja tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa, johon osallistui 93 pediatrista potilasta (5–17-vuotiaita), joilla oli keskivaikea tai vaikea ulseratiivinen koliitti (Mayo-pisteet 6–12, endoskopiaosion pisteet 2–3, vahvistettu keskitetysti arvioidulla endoskopialla) ja joiden kohdalla tavanomaisilla hoidoilla ei ollut saatu riittävää vastetta tai ne olivat olleet huonosti siedettyjä. Aiempi TNF-antagonistihoito oli epäonnistunut noin 16 %:lla tutkimukseen osallistuneista potilaista. Kortikosteroideja tutkimukseenottovaiheessa saaneiden potilaiden kortikosteroidiannoksen pienentäminen sallittiin viikon 4 jälkeen.
Tutkimuksen aloitusvaiheessa 77 potilasta satunnaistettiin suhteessa 3:2 saamaan kaksoissokkoutettua Adalimumabihoitoa induktioannoksella 2,4 mg/kg (enintään 160 mg) viikolla 0 ja viikolla 1 sekä 1,2 mg/kg (enintään 80 mg) viikolla 2 tai induktioannoksella 2,4 mg/kg (enintään 160 mg) viikolla 0, lumelääkettä viikolla 1 sekä annoksella 1,2 mg/kg (enintään 80 mg) viikolla 2. Molemmat ryhmät saivat annoksena 0,6 mg/kg (enintään 40 mg) viikolla 4 ja viikolla 6. Tutkimusasetelmaan tehdyn muutoksen jälkeen loput 16 aloitusvaiheeseen osallistunutta potilasta saivat avoimesti Adalimumabihoitoa induktioannoksella 2,4 mg/kg (enintään 160 mg) viikolla 0 ja viikolla 1 sekä annoksella 1,2 mg/kg (enintään 80 mg) viikolla 2.
Viikolla 8 62 potilasta, joilla todettiin kliininen vaste osittaisen Mayo-pisteytyksen mukaan (määritelmä: osittaisten Mayo-pisteiden lasku ≥ 2 pistettä ja ≥ 30 % lähtötilanteesta), satunnaistettiin yhtäläisessä suhteessa saamaan joko kaksoissokkoutettua adalimumabiylläpitohoitoa annoksella 0,6 mg/kg (enintään 40 mg) viikossa tai ylläpitoannoksella 0,6 mg/kg (enintään 40 mg) joka toinen viikko. Lisäksi 12 potilasta, joilla todettiin kliininen vaste osittaisen Mayo-pisteytyksen mukaan, satunnaistettiin saamaan lumelääkettä ennen tutkimusasetelman muuttamista, mutta heitä ei otettu mukaan tehon varmistamiseksi tehtyyn analyysiin.
Taudin pahenemisvaihe määriteltiin osittaisten Mayo-pisteiden nousuksi vähintään 3 pisteellä (potilaat, joiden osittaiset Mayo-pisteet olivat 0–2 viikolla 8), vähintään 2 pisteellä (potilaat, joiden osittaiset Mayo-pisteet olivat 3–4 viikolla 8) tai vähintään 1 pisteellä (potilaat, joiden osittaiset Mayopisteet olivat 5–6 viikolla 8).
Potilaat, jotka täyttivät taudin pahenemisvaiheen kriteerit viikolla 12 tai sen jälkeen, satunnaistettiin saamaan uutena induktioannoksena 2,4 mg/kg (enintään 160 mg) tai annoksena 0,6 mg/kg (enintään 40 mg), minkä jälkeen he jatkoivat hoitoa omalla ylläpitoannoksellaan.
Tehotulokset
Tutkimuksen ensisijaisia yhdistettyjä päätetapahtumia olivat kliininen remissio osittaisen Mayopisteytyksen mukaan (määritelmä: osittaiset Mayo-pisteet ≤ 2 eikä yhdenkään osion pisteet > 1) viikolla 8 ja kliininen remissio Mayo-kokonaispisteiden mukaan (määritelmä: Mayo-pisteet ≤ 2 eikä yhdenkään osion pisteet > 1) viikolla 52 niiden potilaiden osalta, jotka saavuttivat kliinisen vasteen osittaisen Mayo-pisteytyksen mukaan viikolla 8. Kliiniset remissioprosentit osittaisen Mayo-pisteytyksen mukaan viikolla 8 kaksoissokkoutettua adalimumabi-induktiohoitoa saaneisiin ryhmiin kuuluneilla potilailla esitetään taulukossa 31).
Taulukko 31
Kliininen remissio osittaisen Mayo-pisteytyksen mukaan viikolla 8
Adalimumabia | Adalimumabib, c | |
Kliininen remissio | 13/30 (43,3 %) | 28/47 (59,6 %) |
a Adalimumibabi 2,4 mg/kg (enintään 160 mg) viikolla 0, lumelääkettä viikolla 1 ja 1,2 mg/kg (enintään 80 mg) viikolla 2 | ||
Viikolla 8 hoitoon vastanneiden kliinistä remissiota Mayo-kokonaispisteiden mukaan, viikolla 8 hoitoon vastanneiden kliinistä vastetta Mayo-kokonaispisteiden mukaan (määritelmä: Mayo-pisteiden lasku ≥ 3 pistettä ja ≥ 30 % lähtötilanteesta), viikolla 8 hoitoon vastanneiden limakalvojen paranemista (määritelmä: Mayo-endoskopiaosion pisteet ≤ 1), viikolla 8 remissiossa olleiden kliinistä remissiota Mayo-kokonaispisteiden mukaan sekä niiden viikolla 8 hoitoon vastanneiden tutkittavien osuutta, jotka olivat remissiossa ilman kortikosteroidihoitoa Mayo-kokonaispisteiden mukaan, arvioitiin viikolla 52 niiden potilaiden osalta, jotka saivat kaksoissokkoutettua adalimumabiylläpitohoitoa enintään 40 mg joka toinen viikko (0,6 mg/kg) ja enintään 40 mg viikossa (0,6 mg/kg) (taulukko 32).
Taulukko 32
Tehotulokset viikolla 52
Adalimumabia | Adalimumabib | |
Kliininen remissio potilailla, jotka vastasivat hoitoon viikolla 8 osittaisen Mayo-pisteytyksen mukaan | 9/31 (29,0 %) | 14/31 (45,2 %) |
Kliininen vaste potilailla, jotka vastasivat hoitoon viikolla 8 osittaisen Mayopisteytyksen mukaan | 19/31 (61,3 %) | 21/31 (67,7 %) |
Limakalvojen paraneminen potilailla, jotka vastasivat hoitoon viikolla 8 osittaisen Mayo-pisteytyksen mukaan | 12/31 (38,7 %) | 16/31 (51,6 %) |
Kliininen remissio potilailla, joiden tauti oli remissiossa viikolla 8 osittaisen Mayopisteytyksen mukaan | 9/21 (42,9 %) | 10/22 (45,5 %) |
Remissio ilman kortikosteroideja potilailla, jotka vastasivat hoitoon viikolla 8 osittaisen Mayopisteytyksen mukaanc | 4/13 (30,8 %) | 5/16 (31,3 %) |
a Adalimumabi 0,6 mg/kg (enintään 40 mg) joka toinen viikko | ||
Muita eksploratiivisia tehon päätetapahtumia olivat kliininen vaste pediatrisen ulseratiivisen koliitin aktiivisuusindeksin (PUCAI) mukaan (määritelmä: PUCAI-pisteiden lasku ≥ 20 pistettä lähtötilanteesta) ja kliininen remissio PUCAI-pisteiden mukaan (määritelmä: PUCAI-pisteet < 10) viikolla 8 ja viikolla 52 (taulukko 33).
Taulukko 33
Eksploratiivisten päätetapahtumien tulokset PUCAI-pisteiden mukaan
Viikko 8 | ||
Adalimumabia | Adalimumabib,c | |
Kliininen remissio PUCAIpisteiden mukaan | 10/30 (33,3%) | 22/47 (46,8%) |
Kliininen vaste PUCAIpisteiden mukaan | 15/30 (50,0%) | 32/47 (68,1%) |
Viikko 52 | ||
Adalimumabid | Adalimumabie | |
Kliininen remissio PUCAIpisteiden mukaan potilailla, jotka vastasivat hoitoon viikolla 8 osittaisen Mayopisteytyksen mukaan | 14/31 (45,2%) | 18/31 (58,1%) |
Kliininen vaste PUCAIpisteiden mukaan potilailla, jotka vastasivat hoitoon viikolla 8 osittaisen Mayopisteytyksen mukaan | 18/31 (58,1%) | 16/31 (51,6%) |
a Adalimumabi 2,4 mg/kg (enintään 160 mg) viikolla 0, lumelääkettä viikolla 1 ja 1,2 mg/kg (enintään 80 mg) viikolla 2 | ||
Niistä adalimumabihoitoa saaneista potilaista, jotka saivat uuden induktiohoidon ylläpitovaiheessa, 2/6 (33 %) saavutti kliinisen vasteen Mayo-kokonaispisteiden mukaan viikolla 52.
Elämänlaatu
Adalimumabihoitoa saaneissa ryhmissä todettiin IMPACT III- pisteiden sekä huoltajan tuottavuuden ja aktiivisuuden heikentymistä mittaavien WPAI-pisteiden parantuneen kliinisesti merkitsevästi lähtötilanteeseen nähden. Pituuskasvunopeuden kliinisesti merkittävää lisääntymistä (paranemista) lähtötilanteeseen nähden todettiin adalimumabia saaneissa ryhmissä, ja painoindeksin kliinisesti merkittävää nousua (paranemista) lähtötilanteeseen nähden todettiin niillä tutkittavilla, jotka saivat suurena ylläpitoannoksena enintään 40 mg (0,6 mg/kg) viikossa.
Uveiitti lapsilla
Adalimumabin turvallisuutta ja tehoa arvioitiin satunnaistetussa, kaksoissokkoutetussa, kontrolloidussa tutkimuksessa, johon osallistui 90 2 – < 18-vuotiasta lapsipotilasta, joilla oli aktiivinen JIA:iin liittyvä ei-infektioperäinen anteriorinen uveiitti, ja joilla tauti oli refraktaarinen vähintään 12 viikkoa kestäneestä metotreksaattihoidosta huolimatta. Potilaat saivat joko lumelääkettä tai 20 mg adalimumabia (paino < 30 kg) tai 40 mg adalimumabia (paino ≥ 30 kg) joka toinen viikko yhdessä lähtötilanteen metotreksaattiannoksensa kanssa.
Ensisijainen päätetapahtuma oli "hoidon epäonnistumiseen kuluva aika". Hoidon epäonnistumisen määritelmänä oli silmän tulehduksen pahentuminen tai pysyvä ei-paraneminen, osittainen paraneminen, johon liittyi muiden samanaikaisten pysyvien silmäsairauksien kehittyminen tai paheneminen, muun samanaikaisen lääkityksen luvaton käyttö ja hoidon keskeytys pidemmäksi ajaksi.
Kliininen vaste
Adalimumabi viivästytti merkitsevästi hoidon epäonnistumiseen kuluvaa aikaa verrattuna lumelääkkeeseen (ks. kuva 3, p < 0,0001 log-rank-testillä). Mediaaniaika hoidon epäonnistumiseen oli 24,1 viikkoa lumelääkettä saaneilla potilailla. Adalimumabihoitoa saaneiden potilaiden mediaaniaikaa hoidon epäonnistumiseen ei voitu arvioida, koska alle puolella heistä hoito epäonnistui. Adalimumabi pienensi merkitsevästi hoidon epäonnistumisen riskiä 75 %:lla verrattuna lumelääkkeeseen, kuten riskisuhde osoittaa (HR = 0,25 [95 % luottamusväli 0,12‑0,49]).
Kuva 3: Hoidon epäonnistumiseen kulunut aika yhteenvetona Kaplan-Meier-käyrinä lasten uveiittitutkimuksessa
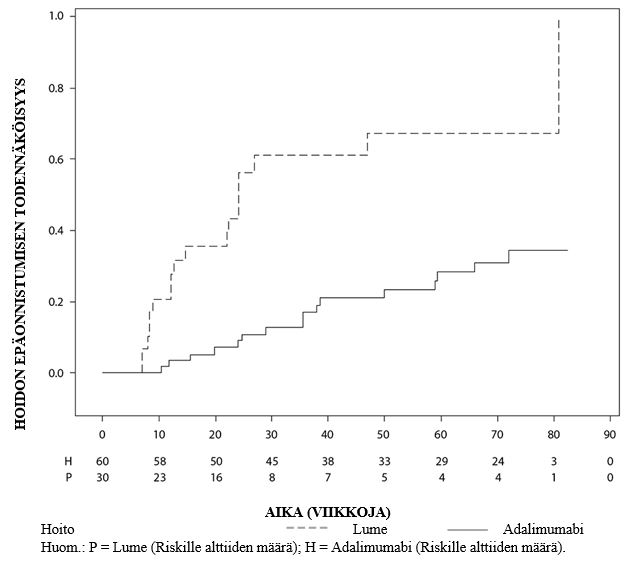
Farmakokinetiikka
Imeytyminen ja jakautuminen
Ihonalaisen 40 mg:n kerta-annoksen jälkeen adalimumabin imeytyminen ja jakautuminen oli hidasta, huippupitoisuus seerumissa saavutettiin n. 5 päivän kuluttua lääkkeen antamisesta. Kolmesta tutkimuksesta saatujen tulosten perusteella adalimumabin absoluuttisen biologisen hyötyosuuden arvioidaan olevan keskimäärin 64 % 40 mg:n ihonalaisen kerta-annoksen jälkeen. Laskimonsisäisten kerta-annosten (0,25–10 mg/kg) jälkeen pitoisuudet olivat annoksesta riippuvaisia. Puhdistuma 0,5 mg/kg:n annosten (~ 40 mg) jälkeen oli 11–15 ml/h, jakautumistilavuus (Vss) vaihteli välillä 5–6 litraa, ja terminaalivaiheen puoliintumisajan keskiarvo oli noin kaksi viikkoa. Useilla nivelreumapotilailla adalimumabin pitoisuudet nivelnesteessä olivat 31–96 % seerumista mitattaviin pitoisuuksiin verrattuna.
Ihonalaisen 40 mg:n adalimumabikerta-annoksen jälkeen vakaan tilan pienin pitoisuus aikuisilla nivelreumapotilailla oli keskimäärin n. 5 mikrog/ml (jos samanaikaisesti ei käytetty metotreksaattia) tai 8‑9 mikrog/ml (jos samanaikaisesti käytettiin metotreksaattia). Vakaassa tilassa seerumin pienin adalimumabipitoisuus suureni lähes annoksesta riippuvaisesti, kun valmistetta annettiin ihonalaisesti 20, 40 ja 80 mg joka toinen viikko tai viikoittain.
Kun idiopaattista juveniilia polyartriittia sairastavat 4–17-vuotiaat potilaat saivat adalimumabia 24 mg/m2 (enintään 40 mg) ihon alle joka toinen viikko, adalimumabin vakaan tilan minimipitoisuuksien keskiarvo seerumissa (arvot viikoilta 20–48) oli 5,6 ± 5,6 mikrog/ml (variaatiokerroin 102 %) käytettäessä adalimumabia ilman metotreksaattia ja 10,9 ± 5,2 mikrog/ml (variaatiokerroin 47,7 %) samanaikaisen metotreksaattihoidon aikana.
Kun idiopaattista juveniilia polyartriittia sairastaville potilaille, joiden ikä oli 2 – < 4 v tai ikä vähintään 4 v ja paino < 15 kg, annettiin adalimumabia 24 mg/m2, adalimumabin vakaan tilan minimipitoisuuksien keskiarvo seerumissa oli 6,0 ± 6,1 mikrog/ml (variaatiokerroin 101 %) käytettäessä adalimumabia ilman metotreksaattia ja 7,9 ± 5,6 mikrog/ml (variaatiokerroin 71,2 %) samanaikaisen metotreksaattihoidon aikana.
Kun entesiitteihin liittyvää artriittia sairastavat 6–17-vuotiaat potilaat saivat 24 mg/m2 (enintään 40 mg) ihon alle joka toinen viikko, adalimumabin vakaan tilan minimipitoisuuksien keskiarvo seerumissa (mitattu viikolla 24) oli 8,8 ± 6,6 mikrog/ml käytettäessä adalimumabia ilman metotreksaattia ja 11,8 ± 4,3 mikrog/ml samanaikaisen metotreksaattihoidon aikana.
Kun röntgennegatiivista aksiaalista spondylartriittia sairastaville aikuispotilaille annettiin 40 mg adalimumabia ihon alle joka toinen viikko, vakaan tilan minimipitoisuuksien keskiarvo (± keskihajonta) oli viikon 68 kohdalla 8,0 ± 4,6 µg/ml.
Aikuisilla psoriaasipotilailla vakaan tilan minimipitoisuuksien keskiarvo oli 5 mikrog/ml adalimumabimonoterapian aikana (40 mg joka toinen viikko).
Kun kroonista läiskäpsoriaasia sairastaville lapsipotilaille oli annettu 0,8 mg/kg (enintään 40 mg) adalimumabia ihon alle joka toinen viikko, adalimumabin vakaan tilan minimipitoisuuksien keskiarvo (± keskihajonta) oli noin 7,4 ± 5,8 mikrog/ml (variaatiokerroin 79 %).
Kun hidradenitis suppurativaa sairastaville aikuispotilaille annettiin 160 mg adalimumabiannos viikolla 0 ja sen jälkeen 80 mg viikolla 2, seerumin pienin adalimumabipitoisuus oli noin 7–8 mikrog/ml viikolla 2 ja viikolla 4. Vakaan tilan minimipitoisuuksien keskiarvo (arvot viikoilta 12–36) oli noin 8–10 mikrog/ml, kun adalimumabia annettiin 40 mg kerran viikossa.
Adalimumabialtistus nuorilla HS-potilailla ennustettiin käyttäen populaatiofarmakokineettistä mallinnusta ja simulaatiota, jotka perustuivat kaikkien käyttöaiheiden farmakokinetiikkaan muilla pediatrisilla potilailla (lasten psoriaasi, idiopaattinen juveniili nivelreuma, Crohnin tauti lapsilla ja entesiitteihin liittyvä artriitti). Suositeltu annostusohjelma HS-tautia sairastaville nuorille 40 mg joka toinen viikko. Koska adalimumabialtistukseen voivat vaikuttaa kehon koko, painavammat nuoret, joilla vaste on riittämätön, saattavat hyötyä siitä, että saavat aikuisille tarkoitetun annoksen eli 40 mg joka viikko.
Kun Crohnin tautia sairastaville potilaille annettiin 80 mg:n latausannos adalimumabia viikolla 0 ja sen jälkeen 40 mg viikolla 2, seerumin pienimmät adalimumabipitoisuudet olivat hoidon aloitusvaiheen aikana noin 5,5 mikrog/ml. Kun potilaille annettiin 160 mg:n latausannos adalimumabia viikolla 0 ja sen jälkeen 80 mg viikolla 2, seerumin pienimmät adalimumabipitoisuudet olivat hoidon aloitusvaiheen aikana noin 12 mikrog/ml. Vakaassa tilassa pienimmät pitoisuudet Crohnin tautia sairastavilla potilailla olivat keskimäärin noin 7 mikrog/ml, kun potilaat saivat 40 mg adalimumabia ylläpitoannoksena joka toinen viikko.
Keskivaikeaa tai vaikeaa Crohnin tautia sairastavien lapsipotilaiden avoin adalimumabiannos oli induktiohoidon viikoilla 0 ja 2 joko 160/80 mg (vähintään 40 kg painoiset) tai 80/40 mg (alle 40 kg painoiset). Viikolla 4 potilaat satunnaistettiin suhteessa 1:1 käyttämään ylläpitohoitoa joko tavanomaisina annoksina (40/20 mg joka toinen viikko painosta riippuen) tai pieninä annoksina (20/10 mg joka toinen viikko painosta riippuen). Seerumin adalimumabipitoisuuksien minimin keskiarvo (± keskihajonta) viikolla 4 oli 15,7 ± 6,6 mikrog/ml, jos potilas painoi ≥ 40 kg (160/80 mg), ja 10,6 ± 6,1 mikrog/ml, jos potilas painoi < 40 kg (80/40 mg).
Satunnaistettua hoitoaan jatkaneilla potilailla adalimumabin minimipitoisuuksien keskiarvo (± keskihajonta) viikolla 52 oli tavanomaisen annoksen ryhmässä 9,5 ± 5,6 mikrog/ml ja pienen annoksen ryhmässä 3,5 ± 2,2 mikrog/ml. Minimipitoisuuksien keskiarvo pysyi ennallaan, kun potilaat jatkoivat adalimumabihoidon käyttöä joka toinen viikko 52 viikon ajan. Jos hoitoa tehostettiin ottamalla lääke joka viikko, seerumin adalimumabipitoisuuksien keskiarvo (± keskihajonta) oli viikolla 52 joko 15,3 ± 11,4 mikrog/ml (40/20 mg joka viikko) tai 6,7 ± 3,5 mikrog/ml (20/10 mg joka viikko).
Kun ulseratiivista koliittia sairastaville potilaille annettiin 160 mg:n latausannos adalimumabia viikolla 0 ja sen jälkeen 80 mg viikolla 2, seerumin pienimmät adalimumabipitoisuudet olivat hoidon aloitusvaiheen aikana noin 12 mikrog/ml. Vakaassa tilassa pienimmät pitoisuudet ulseratiivista koliittia sairastavilla potilailla olivat keskimäärin noin 8 mikrog/ml, kun potilaat saivat 40 mg adalimumabia ylläpitoannoksena joka toinen viikko.
Kun ulseratiivista koliittia sairastaville pediatrisille potilaille annettiin painoon perustuva annos 0,6 mg/kg (enintään 40 mg) ihon alle joka toinen viikko, keskimääräinen pienin vakaan tilan adalimumabipitoisuus seerumissa oli 5,01 ± 3,28 µg/ml viikolla 52. Potilailla, jotka saivat annoksena 0,6 mg/kg (enintään 40 mg) viikossa, keskimääräinen (± keskihajonta) pienin vakaan tilan adalimumabipitoisuus seerumissa oli 15,7 ± 5,60 μg/ml viikolla 52.
Kun uveiittia sairastaville aikuispotilaille annettiin 80 mg:n latausannos adalimumabia viikolla 0 ja sen jälkeen 40 mg adalimumabia joka toinen viikko alkaen viikolla 1, vakaan tilan pitoisuuden keskiarvo oli noin 8–10 mikrog/ml.
Adalimumabialtistus uveiittia sairastavilla lapsilla arvioitiin käyttämällä populaatiofarmakokineettistä mallinnusta ja simulaatiota, jotka perustuivat muiden käyttöaiheiden farmakokinetiikkaan muilla lapsipotilailla (psoriaasi lapsilla, juveniili idiopaattinen artriitti, Crohnin tauti lapsilla ja entesiitteihin liittyvä artriitti). Kliinistä tietoa altistumisesta latausannokselle alle 6-vuotiailla lapsilla ei ole saatavilla. Ennustetut altistukset viittaavat siihen, että ilman metotreksaattia latausannos voi johtaa aluksi systeemisen altistuksen nousuun.
Populaatiofarmakokineettinen ja farmakokineettinen/farmakodynaaminen mallinnus ja simulaatio ennustivat adalimumabialtistuksen ja tehon olevan samaa luokkaa annoksella 40 mg kerran viikossa ja 80 mg joka toinen viikko (potilaina aikuisia, joilla oli nivelreuma, HS-tauti, ulseratiivinen koliitti, Crohnin tauti tai läiskäpsoriaasi, nuoria, joilla oli hidradenitis suppurativa, sekä ≥ 40 kg painavia lapsipotilaita, joilla oli Crohnin tauti ja ulseratiivinen koliitti).
Altistus-vastesuhde pediatrisilla potilailla
Plasmapitoisuuden ja PedACR 50 -vasteen välinen altistus-vastesuhde arvioitiin juveniilia idiopaattista artriittia (pJIA ja ERA) sairastavilla potilailla tehtyjen kliinisten tutkimusten perusteella. Havaittava adalimumabin pitoisuus plasmassa, joka aiheuttaa puolet PedACR 50 -vasteen maksimivaikutuksesta (EC50) oli 3 μg/ml (95 % luottamusväli 1‑6 μg/ml).
Adalimumabipitoisuuden ja tehon välinen altistus-vastesuhde pediatrisilla potilailla, joilla oli vaikea kroonien läiskäpsoriaasi, arvioitiin PASI 75-vasteelle ja PGA puhdas/melkein puhdas -vasteelle. PASI 75 ja PGA puhdas/melkein puhdas -vasteet paranivat, kun adalimumabipitoisuus nousi. Molemmilla havaittu EC50 oli sama, noin 4,5 μg/ml (95 % luottamusväli 0,4‑47,6 [PASI 75] ja 1,9‑10,5 [PGA])
Eliminaatio
Farmakokineettisissä yli 1 300 nivelreumapotilasta käsittävissä väestötutkimuksissa havaittiin adalimumabin näennäisen puhdistuman lisääntyvän ruumiinpainon mukaan. Kun tulokset vakioitiin painoerojen suhteen, havaittiin sukupuolella ja iällä olevan ainoastaan minimaalinen vaikutus adalimumabin puhdistumaan. Vapaan adalimumabin (adalimumabin, joka ei ole sitoutunut adalimumabin vasta-aineisiin) pitoisuudet seerumissa todettiin pienemmiksi niillä potilailla, joilla oli mitattavissa olevat määrät adalimumabin vasta-aineita.
Maksan tai munuaisten vajaatoiminta
Adalimumabia ei ole tutkittu maksan tai munuaisten vajaatoimintaa sairastavilla potilailla.
Prekliiniset tiedot turvallisuudesta
Kerta- ja toistuvan altistuksen aiheuttamaa toksisuutta ja genotoksisuutta koskevien tutkimusten tulokset eivät viittaa mihinkään erityiseen vaaraan ihmisille.
Makakiapinoilla tehdyssä alkion/sikiön kehitykseen kohdistuvaa toksisuutta/postnataalia kehitystä koskevassa tutkimuksessa ei havaittu adalimumabin aiheuttavan haittaa sikiölle annostasoilla 0, 30 ja 100 mg/kg (9-17 apinaa/ryhmä). Adalimumabilla ei ole tehty karsinogeenisuustutkimuksia eikä fertiliteettiä tai postnataalitoksisuutta koskevia standardiarviointeja, koska ei ole saatavilla sopivia malleja, joissa vasta-aine reagoisi rajoitetusti ristiin jyrsijän TNF:n kanssa, ja koska jyrsijöissä kehittyy neutraloivia vasta-aineita.
Farmaseuttiset tiedot
Apuaineet
Yksiemäksinen natriumfosfaatti, monohydraatti
Kaksiemäksinen natriumfosfaatti, heptahydraatti
Meripihkahappo
Kaksiemäksinen natriumsuksinaatti
Histidiini
Histidiinihydrokloridimonohydraatti
Mannitoli
Polysorbaatti 20
Injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
30 kuukautta
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä esitäytetty kynä ulkopakkauksessaan. Herkkä valolle.
Yksittäinen esitäytetty Imraldi-kynä voidaan säilyttää huoneenlämmössä enintään 25 °C:ssa 31 päivän ajan. Kynä tulee suojata valolta, ja se on hävitettävä, jos sitä ei käytetä 31 päivän aikana.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
IMRALDI injektioneste, liuos, esitäytetty kynä
40 mg (L:ei) 2 x 0,4 ml (40 mg/0,4 ml) (400,79 €), 6 x 0,4 ml (40 mg/0,4 ml) (246,89 €)
PF-selosteen tieto
Imraldi 40 mg injektioneste, liuos, esitäytetty kynä
0,4 ml injektioneste, liuos, esitäytetyssä kertakäyttökynässä, jonka sisällä on esitäytetty ruisku. Kynän sisällä oleva ruisku on valmistettu tyypin I lasista, jossa on ruostumattomasta teräksestä valmistettu neula, kova neulansuojus ja kumimäntä (klorobutyylia).
Pakkaukset:
- 1 esitäytetty kynä ja 2 puhdistuslappua
- 2 esitäytettyä kynää, kummassakin 1 puhdistuslappu
- 4 esitäytettyä kynää, kussakin 1 puhdistuslappu
- 6 esitäytettyä kynää, kussakin 1 puhdistuslappu
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai opaalinhohtoinen, väritön tai vaaleanruskea liuos.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
IMRALDI injektioneste, liuos, esitäytetty kynä
40 mg 2 x 0,4 ml, 6 x 0,4 ml
- Alempi erityiskorvaus (65 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli ja tosilitsumabi (tulehdukselliset reumasairaudet): Nivelreuman, juveniilin polyartriitin, psoriaasiin liittyvän niveltulehduksen, selkärankareuman tai edellä mainittuja niveltulehduksia läheisesti muistuttavan niveltulehduksen hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman hoito erityisin edellytyksin (281).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli, tosilitsumabi ja ustekinumabi (tulehdukselliset reumasairaudet): Eräiden reumasairauksien hoito erityisin edellytyksin / Adalimumabi: Uveiitin hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman ja jättisoluarteriitin hoito erityisin edellytyksin (313), Adalimumabi, bimekitsumabi, brodalumabi, etanersepti, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sekukinumabi, sertolitsumabipegoli, tildrakitsumabi ja ustekinumabi (ihopsoriaasi): Vaikean kroonisen ihopsoriaasin hoito erityisin edellytyksin (319), Adalimumabi, golimumabi, infliksimabi, risankitsumabi, ustekinumabi ja vedolitsumabi (tulehdukselliset suolistosairaudet): Eräiden suolistosairauksien hoito erityisin edellytyksin (326), Adalimumabi ja sekukinumabi: Keskivaikean ja vaikean aktiivisen hidradenitis suppurativan hoito erityisin edellytyksin (380).
- Oikeus käyttää samaa biologista lääkettä (sama kauppanimi) 6 kk ajan.
ATC-koodi
L04AB04
Valmisteyhteenvedon muuttamispäivämäärä
26.09.2024
Yhteystiedot
Bertel Jungin aukio 5 C
02600 Espoo
0207 401 200
www.biogen.fi
www.MS-nyt.fi