
LUTATHERA infuusioneste, liuos 370 MBq/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
LUTATHERA®-hoidon aloittaminen: opas potilaille
Vaikuttavat aineet ja niiden määrät
Yksi millilitra liuosta sisältää 370 MBq lutetium(177Lu)oksodotreotidia kalibroinnin päivämääränä ja kellonaikana.
Radioaktiivisuuden kokonaismäärä kerta-annospulloa kohden on 7 400 MBq infuusion päivämääränä ja kellonaikana. Huomioiden kiinteän 370 MBq/ml:n volumetrisen aktiivisuuden kalibroinnin päivämääränä ja kellonaikana liuoksen tilavuus infuusiopullossa vaihtelee välillä 20,5–25,0 ml tarvittavan radioaktiivisuuden määrän tuottamiseksi infuusion päivämäärän ja kellonajan kohdalla.
Fysikaaliset ominaisuudet
Lutetium‑177:n puoliintumisaika on 6,647 vuorokautta. Lutetium‑177 hajoaa β-‑säteilyn kautta vakaaksi hafnium-177:ksi. Kaikkein voimakkaimman β-‑säteilyn (79,3 %) maksimienergia on 0,498 MeV. Keskimääräinen beetaenergia on noin 0,13 MeV. Matalaa gammaenergiasäteilyä ilmenee myös, esimerkiksi tasoilla 113 keV (6,2 %) ja 208 keV (11 %).
Apuaine(et), joiden vaikutus tunnetaan
Yksi millilitra liuosta sisältää enintään 0,14 mmol (3,2 mg) natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
Lutathera on tarkoitettu leikkaushoitoon soveltumattomien tai etäpesäkkeisten, levinneiden, hyvin erilaistuneiden (G1 tai G2), somatostatiinireseptoripositiivisten gastroenteropankreaattisten neuroendokriinikasvainten (GEP‑NET‑kasvainten) hoitoon aikuisilla.
Ehto
Valmistetta saavat antaa vain radioaktiivisten aineiden käsittelyyn valtuutetut henkilöt tähän tarkoitukseen varatuissa tiloissa sen jälkeen, kun pätevä lääkäri on tutkinut potilaan.
Annostus ja antotapa
Tärkeät turvallisuusohjeet
Lutathera‑valmistetta saavat antaa vain radiofarmaseuttisten valmisteiden käsittelyyn valtuutetut henkilöt siihen tarkoitetussa hoitoympäristössä (ks. kohta Käyttö- ja käsittelyohjeet) ja sen jälkeen, kun pätevä lääkäri on arvioinut potilaan.
Potilasvalinta
Ennen Lutathera‑hoidon aloittamista somatostatiinireseptorien yliekspressio kasvainkudoksessa on vahvistettava näiden reseptorien kuvantamisella skintigrafialla tai positroniemissiotomografialla [PET]. Kasvaimeen oton on oltava vähintään yhtä suurta kuin normaali maksaan otto.
Annostus
Aikuiset
Suositeltava aikuisten Lutathera-hoito koostuu neljästä 7 400 MBq:n infuusiosta. Suositeltava aikaväli kunkin antokerran välillä on 8 viikkoa (± 1 viikko).
Tiedot annosmuutoksista vaikeiden tai sietämättömien haittavaikutusten hoitamiseksi esitetään asiaa koskevassa kohdassa jäljempänä.
Aminohappoliuos
Munuaisten suojaamiseen tarkoitettu L‑lysiiniä ja L‑arginiinia sisältävä aminohappoliuos on annettava laskimoon neljä tuntia kestävänä infuusiona (koostumus, ks. taulukot 1 ja 2). Aminohappoliuoksen laskimoinfuusio on aloitettava 30 minuuttia ennen Lutathera‑infuusion aloittamista. On suositeltavaa infusoida aminohappoliuos ja Lutathera eri käsivarsien laskimoon. Jos kahden laskimokanyylin käyttö ei kuitenkaan ole mahdollista huonon suoniyhteyden tai hoitolaitoksen linjauksen / kliinisen valinnan vuoksi, aminohappoliuos ja Lutathera voidaan infusoida saman kanyylin kautta kolmitiehanaa käyttäen, kunhan otetaan huomioon virtausnopeus ja suoniyhteyden hallinta. Aminohappoliuosannosta ei pidä pienentää, vaikka Lutathera-annosta pienennettäisiin.
Aminohappoliuosta, joka sisältää vain L‑lysiiniä ja L‑arginiinia taulukossa 1 esitettyinä määrinä, pidetään suositeltavana lääkevalmisteena johtuen pienemmästä infusoitavasta kokonaistilavuudesta ja pienemmästä osmolaalisuudesta.
Aminohappoliuos voidaan valmistaa sairaalassa steriilien lääkevalmisteiden hyviä valmistustapoja noudattaen. Koostumus määritellään taulukossa 1.
Taulukko 1 Valmistettavan aminohappoliuoksen koostumus
| Yhdiste | Määrä |
| L‑lysiinihydrokloridi | 25 g* |
| L‑arginiinihydrokloridi | 25 g** |
| Natriumkloridi 9 mg/ml (0,9 %) injektioneste, liuos, tai injektionesteisiin käytettävä vesi | 1 litra |
* vastaa 20,0 g L‑lysiiniä ** vastaa 20,7 g L‑arginiinia | |
Vaihtoehtoisesti voidaan käyttää kaupallisesti saatavilla olevia aminohappoliuoksia, jos ne vastaavat taulukossa 2 kuvattua spesifikaatiota.
Taulukko 2 Kaupallisesti saatavilla olevien aminohappoliuosten spesifikaatio
| Ominaisuus | Spesifikaatio |
| L‑lysiinihydrokloridi | 18–25 g* |
| L‑arginiinihydrokloridi | 18–25 g** |
| Tilavuus | 1–2 litraa |
| Osmolaalisuus | < 1 200 mosm/kg |
* vastaa 14,4–20 g L‑lysiiniä ** vastaa 14,9–20,7 g L‑arginiinia | |
Hoidon valvonta
Ennen jokaista antoa ja Lutathera‑hoidon aikana laboratoriotestit ovat tarpeen potilaan voinnin arvioimiseksi ja hoitoprotokollan muokkaamiseksi tarvittaessa (annos, infuusioväli, infuusioiden määrä) (ks. taulukko 3).
Ennen jokaista infuusiota tarvitaan vähintään seuraavat laboratoriotestit:
- Verenkuva (hemoglobiini [Hb], veren valkosolujen määrä ja erittelylaskenta, verihiutaleiden määrä)
- Munuaisten toiminta (seerumin kreatiniini ja kreatiniinipuhdistuma Cockcroft-Gaultin kaavan mukaan)
- Maksan toiminta (alaniiniaminotransferaasi [ALAT], aspartaattiaminotransferaasi [ASAT], seerumin albumiini, INR‑arvo ja bilirubiini).
Nämä laboratoriotestit on tehtävä vähintään kerran 2–4 viikkoa ennen antoa, ja hieman ennen antoa. On myös suositeltavaa tehdä nämä testit 4 viikon välein vähintään 3 kuukauden ajan viimeisen Lutathera‑infuusion jälkeen, ja tämän jälkeen 6 kuukauden välein, jotta mahdolliset viivästyneet haittavaikutukset pystytään havaitsemaan (ks. kohta Haittavaikutukset). Annostusta voi olla tarpeen muuttaa testitulosten perusteella (ks. taulukko 3).
Annoksen muuttaminen
Vaikeiden tai sietämättömien haittavaikutusten hoito voi edellyttää hoidon tauottamista (annosvälin pidentämistä 8 viikosta enintään 16 viikkoon), annoksen pienentämistä tai Lutathera‑hoidon pysyvää lopettamista (ks. taulukko 3 ja kuva 1).
Taulukko 3 Suositukset Lutathera‑annoksen muuttamisesta haittavaikutusten vuoksi
| Haittavaikutus | Haittavaikutuksen vaikeusaste | Annosmuutos |
| Trombosytopenia | Ensimmäinen ilmaantuminen: Aste 2 (trombosyytit < 75–50 x 109/l) Aste 3 (trombosyytit < 50–25 x 109/l) Aste 4 (trombosyytit < 25 x 109/l) | Lääke tauotetaan, kunnes tilanne korjautuu kokonaan tai osittain (aste 0 tai 1). Lutathera‑hoito aloitetaan uudelleen 3 700 MBq:n annoksella (100 mCi), jos tilanne on korjautunut kokonaan tai osittain. Jos pienennetty annos ei aiheuta asteen 2, 3 eikä 4 trombosytopeniaa, seuraava annettava Lutathera‑annos on 7 400 MBq (200 mCi). Lutathera‑hoito lopetetaan pysyvästi, jos asteen 2 tai vaikeampi trombosytopenia vaatii yli 16 viikon pituisen annosvälin. |
| Uusiutunut, aste 2, 3 tai 4 | Lutathera‑hoito lopetetaan pysyvästi. | |
| Anemia ja neutropenia | Anemian ensimmäinen ilmaantuminen: Aste 3 (Hb < 8,0 g/dl); verensiirto aiheellinen Aste 4 (henkeä uhkaavat seuraukset) Neutropenian ensimmäinen ilmaantuminen: Aste 3 (absoluuttinen neutrofiiliarvo [ANC] < 1,0–0,5 x 109/l) Aste 4 (ANC < 0,5 x 109/l) | Lääke tauotetaan, kunnes tilanne korjautuu kokonaan tai osittain (aste 0, 1 tai 2). Lutathera‑hoito aloitetaan uudelleen 3 700 MBq:n annoksella (100 mCi), jos tilanne on korjautunut kokonaan tai osittain. Jos pienennetty annos ei aiheuta asteen 3 eikä 4 anemiaa eikä neutropeniaa, seuraava annettava Lutathera‑annos on 7 400 MBq (200 mCi). Lutathera‑hoito lopetetaan pysyvästi, jos asteen 3 tai vaikeampi anemia tai neutropenia vaatii yli 16 viikon pituisen annosvälin. |
| Uusiutunut, aste 3 tai 4 | Lutathera‑hoito lopetetaan pysyvästi. | |
| Munuaistoksisuus | Ensimmäinen ilmaantuminen: • kreatiniinipuhdistuma alle 40 ml/min; laskettuna Cockcroft‑Gaultin kaavalla ja todellisen painon perusteella; tai • seerumin kreatiniinipitoisuus suurenee 40 % lähtötilanteesta, tai • kreatiniinipuhdistuma pienenee 40 % lähtötilanteesta; laskettuna Cockcroft‑Gaultin kaavalla ja todellisen painon perusteella. | Hoito tauotetaan, kunnes tilanne korjautuu tai palautuu lähtötasolle. Lutathera‑hoito aloitetaan uudelleen 3 700 MBq:n annoksella (100 mCi), jos tilanne on korjautunut tai palautunut lähtötasolle. Jos pienennetty annos ei aiheuta munuaistoksisuutta, seuraava annettava Lutathera‑annos on 7 400 MBq (200 mCi). Lutathera‑hoito lopetetaan pysyvästi, jos munuaistoksisuus vaatii yli 16 viikon pituisen annosvälin. |
| Uusiutunut munuaistoksisuus | Lutathera‑hoito lopetetaan pysyvästi. | |
| Maksatoksisuus | Ensimmäinen ilmaantuminen: • veren bilirubiinipitoisuus suurempi kuin 3 kertaa normaaliarvojen yläraja (aste 3 tai 4) tai • albuminemia alle arvon 30 g/l ja INR > 1,5 | Hoito tauotetaan, kunnes tilanne korjautuu tai palautuu lähtötasolle. Lutathera‑hoito aloitetaan uudelleen 3 700 MBq:n annoksella (100 mCi), jos tilanne on korjautunut tai palautunut lähtötasolle. Jos pienennetty Lutathera‑annos ei aiheuta maksatoksisuutta, seuraava annettava Lutathera‑annos on 7 400 MBq (200 mCi). Lutathera‑hoito lopetetaan pysyvästi, jos maksatoksisuus vaatii yli 16 viikon pituisen annosvälin. |
| Uusiutunut maksatoksisuus. | Lutathera‑hoito lopetetaan pysyvästi. | |
| Mikä tahansa muu CTCAE*‑asteen 3 tai 4 haittavaikutus1 | Ensimmäinen ilmaantuminen: Aste 3 tai 4 | Lääke tauotetaan, kunnes tilanne korjautuu kokonaan tai osittain (aste 0–2). Lutathera‑hoito aloitetaan uudelleen 3 700 MBq:n annoksella (100 mCi), jos tilanne on korjautunut kokonaan tai osittain. Jos pienennetty annos ei aiheuta asteen 3 eikä 4 toksisuutta, seuraava annettava Lutathera‑annos on 7 400 MBq (200 mCi). Lutathera‑hoito lopetetaan pysyvästi, jos asteen 3 tai vaikeampi haittavaikutus vaatii yli 16 viikon pituisen annosvälin. |
| Uusiutunut, aste 3 tai 4 | Lutathera‑hoito lopetetaan pysyvästi. | |
1 Annosta ei tarvitse muuttaa asteen 3 tai 4 hematologisen toksisuuden vuoksi, jos toksisuus johtuu pelkästään lymfopeniasta. * CTCAE: Common Terminology Criteria for Adverse Events ‑haittatapahtumaluokitus, Yhdysvaltain National Cancer Institute ‑organisaatio | ||
Kuva 1 Yhteenveto ohjeista annoksen muuttamiseen
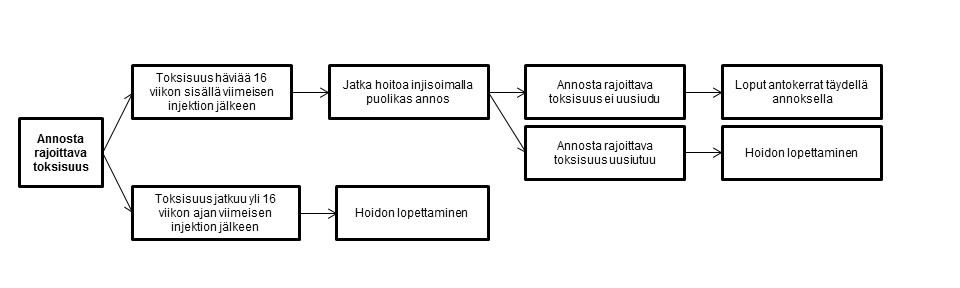
Muita tekijöitä, joiden yhteydessä on harkittava Lutathera‑hoidon tauottamista, ovat mm. sairastuminen muuhun samanaikaiseen sairauteen (esim. virtsatieinfektioon), joka lääkärin arvion mukaan voi suurentaa Lutathera‑valmisteen antoon liittyviä riskejä ja jonka on ehdittävä korjautua tai vakautua ennen hoidon jatkamista, sekä suuret leikkaukset. Suuren leikkauksen jälkeen hoidosta tulee pidättäytyä 12 viikon ajan leikkauspäivästä lukien.
Erityiset potilasryhmät
Iäkkäät
Annosta ei tarvitse muuttaa 65 vuotta täyttäneiden potilaiden kohdalla, sillä kliinisen kokemuksen perusteella vasteessa ei ole havaittu eroja iäkkäiden ja nuorten potilaiden välillä. Koska kohonnut hematotoksisuuden esiintymisen riski on kuitenkin havaittu iäkkäillä potilailla (≥ 70‑vuotiailla), tarkka seuranta, joka mahdollistaa nopean annoksen muuttamisen (annosta rajoittavan toksisuuden tilanteessa) on suositeltavaa tämän potilasryhmän kohdalla.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastaville potilaille annettava aktiivisuus on harkittava huolellisesti, koska suurentunut säteilyaltistus on mahdollista näillä potilailla. Lutetium(177Lu)oksodotreotidin farmakokineettistä profiilia ja turvallisuutta ei ole tutkittu lähtötilanteessa vaikea‑asteista munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min Cockcroft-Gaultin kaavalla) tai loppuvaiheen munuaistautia sairastavilla potilailla. Lutathera‑hoito on vasta‑aiheista potilailla, joilla on munuaisten vajaatoiminta ja joiden kreatiniinipuhdistuma on < 30 ml/min (ks. kohta Vasta-aiheet). Lutathera‑hoitoa ei suositella potilaille, joiden kreatiniinipuhdistuma on lähtötilanteessa < 40 ml/min (Cockcroft‑Gaultin kaavalla). Annosmuutoksia ei suositella, jos potilaalla on munuaisten vajaatoiminta ja kreatiniinipuhdistuma on lähtötilanteessa ≥ 40 ml/min. Koska tämän lääkevalmisteen kuitenkin tiedetään erittyvän merkittävästi munuaisten kautta, munuaistoimintaa on seurattava tavallista tiheämmin hoidon aikana, sillä toksisuusriski voi olla suurentunut tässä potilasryhmässä.
Lisätietoja sellaisten potilaiden hoidosta, joilla ilmenee munuaistoksisuutta, löytyy taulukosta 3 kohdasta Annostus ja antotapa ja kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastaville potilaille annettava aktiivisuus on harkittava huolellisesti, koska suurentunut säteilyaltistus on mahdollista näillä potilailla. Lutetium(177Lu)oksodotreotidin farmakokineettistä profiilia ja turvallisuutta ei ole tutkittu lähtötilanteessa vaikea‑asteista maksan vajaatoimintaa sairastavilla potilailla (kokonaisbilirubiini > 3 x normaaliarvojen yläraja ASAT‑arvosta riippumatta). Potilaille, joilla on lähtötilanteessa maksan vajaatoiminta ja joko kokonaisbilirubiini > 3 x normaaliarvojen yläraja tai albuminemia < 30 g/l ja INR-arvo > 1,5, tulisi antaa Lutathera‑hoitoa vain huolellisen hyöty‑riskiarvioinnin jälkeen. Annosmuutoksia ei suositella potilaille, joilla on lähtötilanteessa lievä tai keskivaikea maksan vajaatoiminta.
Lisätietoja sellaisten potilaiden hoidosta, joilla ilmenee maksatoksisuutta, löytyy taulukosta 3 kohdasta Annostus ja antotapa ja kohdasta Varoitukset ja käyttöön liittyvät varotoimet.
Pediatriset potilaat
Lutathera-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. 12-vuotiaiden ja sitä vanhempien lasten hoitoa koskevan saatavilla olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka, Farmakokinetiikka ja 11, ei voida antaa suosituksia annostuksesta.
Antotapa
Lutathera annetaan laskimoon. Se on käyttövalmis radioaktiivinen lääkevalmiste, joka on tarkoitettu vain kertakäyttöön.
Anto-ohjeet
Suositusannoksen antoon voidaan käyttää painovoimamenetelmää, peristalttista infuusiopumppua tai ruiskupumppua. Terveydenhuollon ammattilaiset voivat käyttää myös muita asianmukaisiksi ja turvallisiksi katsottuja menetelmiä etenkin, jos annoksen pienentäminen on tarpeen.
Jos käytetään painovoimamenetelmää tai peristalttista infuusiopumppua, Lutathera on infusoitava suoraan sen alkuperäisestä infuusiopullosta. Jos annosta muutetaan haittavaikutuksen vuoksi (ks. taulukko 3, kohta Annostus ja antotapa), pienennetyn Lutathera-annoksen antoon on käytettävä peristalttista infuusiopumppua tai ruiskupumppua. Jos pienennetyn Lutathera-annoksen antoon käytetään painovoimamenetelmää eikä annosta ole muutettu ennen antoa, potilas voi saada väärän tilavuuden Lutathera-valmistetta. Säteilyturvallisuusvarotoimet on huomioitava antomenetelmästä riippumatta (ks. kohta Käyttö- ja käsittelyohjeet).
Seuraavassa taulukossa on esitetty yhteenveto koko Lutathera‑hoidon antotoimenpiteestä:
Taulukko 4 Pahoinvointilääkkeen, aminohappoliuoksen ja Lutathera‑valmisteen antotoimenpide
| Annettavat aineet | Aloitusaika (min) | Infuusionopeus (ml/h) | Kesto |
| Pahoinvointilääke | Viimeistään 30 minuuttia ennen aminohappoliuosta | Valmisteyhteenvedon mukaisesti | Valmisteyhteenvedon mukaisesti |
| Aminohappoliuos, joko ex tempore ‑valmiste (1 litra) tai kaupallinen (1–2 litraa) | 0 | 250–500 tilavuudesta riippuen | 4 tuntia |
| Lutathera ja natriumkloridi 9 mg/ml (0,9 %) injektioneste, liuos | 30 | Enintään 400 | 30 ± 10 minuuttia |
Ks. kohdasta 12 ohjeet valmistelusta ja antotavoista laskimoon.
Ks. kohdasta Varoitukset ja käyttöön liittyvät varotoimet suositukset ekstravasaation varalta.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Raskaus tai raskausepäily tai kun raskauden mahdollisuutta ei ole poissuljettu (kohta Raskaus ja imetys).
- Munuaisten vajaatoiminta, kreatiniinipuhdistuma < 30 ml/min.
Varoitukset ja käyttöön liittyvät varotoimet
Yksilöllinen hyöty‑riskiperustelu
Säteilyaltistuksen pitää olla perusteltavissa todennäköisellä hyödyllä jokaisen potilaan kohdalla. Annetun aktiivisuuden on oltava jokaisessa tapauksessa niin vähäinen kuin on kohtuudella saavutettavissa tarvittavan terapeuttisen vaikutuksen aikaansaamiseksi.
Lutathera‑valmisteen vaikutusmekanismin ja siedettävyysprofiilin vuoksi on suositeltavaa, että Lutathera‑hoitoa ei aloiteta, jos potilaalla on somatostatiinireseptorikuvantamisen mukaan somatostatiinireseptorinegatiivisia tai sekamuotoisia sisäelinleesioita.
Myelosuppressio
Hematologisten haittavaikutusten mahdollisuuden vuoksi potilaan verenkuvaa on seurattava lähtötilanteessa ja hoidon aikana ennen jokaista Lutathera‑annosta ja kunnes mahdollinen toksisuus korjautuu (ks. kohta Annostus ja antotapa). Lutathera‑hoidon aikana ilmenevän hematologisen toksisuuden riski voi olla suurentunut, jos potilaan luuydintoiminta on heikentynyt tai hän on saanut aiemmin solunsalpaajahoitoa tai ulkoista sädehoitoa (joka on kattanut yli 25 % luuytimestä). Hoito ei ole suositeltavaa, jos potilaan hematologinen toiminta on merkittävästi heikentynyt lähtötilanteessa ja hoidon aikana (esim. Hb < 4,9 mmol/l tai 8 g/dl, trombosyytit < 75 x 109/l tai valkosolut < 2 x 109/l), ellei heikentyminen johdu pelkästään lymfopeniasta.
Myelodysplastinen oireyhtymä ja akuutti leukemia
Viivästynyttä myelodysplastista oireyhtymää (MDS) ja akuuttia leukemiaa (AL) on havaittu Lutathera‑hoidon jälkeen (ks. kohta Haittavaikutukset). MDS on alkanut noin 29 kuukautta (9–45) ja AL noin 55 kuukautta (32–125) ensimmäisen Luthathera‑infuusion jälkeen. Näiden hoitoon liittyvien toissijaisten myeloidisten sairauksien etiologia on epäselvä. Yli 70 vuoden ikää, heikentynyttä munuaisten toimintaa, lähtötilanteen sytopenioita, aiempien hoitojen lukumäärää, aiempaa altistusta solunsalpaajille (erityisesti alkyloiville aineille) ja aiempaa sädehoitoa pidetään mahdollisina MDS:n/AL:n riski‑ ja/tai niille altistavina tekijöinä.
Munuaistoksisuus
Koska lutetium(177Lu)oksodotreotidi eliminoituu lähes pelkästään munuaisjärjestelmän kautta, on pakollista antaa samanaikaisesti aminohappoliuosta, joka sisältää aminohappoja L‑lysiini ja L‑arginiini. Aminohappoliuos auttaa vähentämään lutetium(177Lu)oksodotreotidin takaisinimeytymistä proksimaalisten tiehyeiden kautta, mikä vähentää munuaisiin absorboituvaa annosta merkittävästi (ks. kohta Annostus ja antotapa). Kun suositeltava aminohappoliuosinfuusio annetaan 4 tunnin aikana, munuaisten säteilyannoksen on ilmoitettu vähenevän keskimäärin 47 %.
Potilasta on kehotettava ylläpitämään nesteytystä ja virtsaamaan tiheästi ennen Lutathera‑valmisteen antoa, antopäivänä ja antopäivän jälkeisenä päivänä (esim. 1 lasillinen vettä joka tunti).
Munuaisten toimintaa on arvioitava seerumin kreatiniinin ja Cockcroft-Gaultin kaavalla lasketun kreatiniinipuhdistuman perusteella lähtötilanteessa, hoidon aikana ja vähintään ensimmäisen hoidon jälkeisen vuoden aikana (ks. kohta Annostus ja antotapa).
Toksisuusriski voi olla säteilyaltistuksen suurenemisen vuoksi suurentunut, jos potilaalla on munuaisten vajaatoiminta lähtötilanteessa tai munuais- tai virtsatiepoikkeamia (ks. kohta Annostus ja antotapa).
Jos potilaan kreatiniinipuhdistuma on < 50 ml/min, on myös otettava huomioon, että aminohappoliuoksen antoon liittyvän ohimenevän hyperkalemian riski on suurentunut (ks. samanaikaisesti annettavaan munuaisia suojaavaan aminohappoliuokseen liittyvä varoitus ja varotoimi).
Maksatoksisuus
Koska monilla Lutathera‑hoitoon lähetetyillä potilailla on maksaetäpesäke, on tavallista kohdata potilaita, joilla maksan toiminta on muuttunutta lähtötilanteessa. Säteilyaltistuksen aiheuttaman maksatoksisuuden riski voi olla suurentunut, jos potilaalla on maksaetäpesäke tai jo entuudestaan pitkälle edennyt maksan vajaatoiminta. Näin ollen on suositeltavaa seurata ALAT‑, ASAT‑, bilirubiini‑, seerumin albumiini- ja INR‑arvoja hoidon aikana (ks. kohta Annostus ja antotapa).
Yliherkkyys
Yliherkkyysreaktioita (mukaan lukien yksittäiset angioedeematapahtumat) on raportoitu markkinoille tulon jälkeen potilailla, joita on hoidettu Lutathera-valmisteella (ks. kohta Haittavaikutukset). Mikäli vakavia yliherkkyysreaktioita esiintyy, meneillään oleva Lutathera-infuusion anto on lopetettava välittömästi. Asianmukainen lääkitys ja hoitovälineistö näiden reaktioiden hoitamiseksi tulee olla välittömästi saatavilla.
Pahoinvointi ja oksentelu
Hoitoon liittyvän pahoinvoinnin ja oksentelun ehkäisemiseksi on injisoitava pahoinvointilääkettä laskimoboluksena viimeistään 30 minuuttia ennen aminohappoliuosinfuusion aloittamista, jotta täysi pahoinvointia ehkäisevä teho saavutetaan (ks. kohta Annostus ja antotapa).
Samanaikainen somatostatiinianalogien käyttö
Somatostatiini ja sen analogit sitoutuvat kilpailevasti somatostatiinireseptoreihin ja saattavat heikentää Lutathera‑valmisteen tehoa (ks. kohta Yhteisvaikutukset).
Neuroendokriiniset hormonaaliset kriisit
Liiallisesta hormonien tai bioaktiivisten aineiden vapautumisesta johtuvia kriisejä voi ilmetä Lutathera‑hoidon jälkeen. Tämän vuoksi potilaiden tarkkailua yön yli tapahtuvan sairaalahoidon avulla on harkittava joissakin tapauksissa (esim. potilaiden kohdalla, joiden oireita on vaikea hoitaa lääkehoidolla). Hormonaalisen kriisin ilmetessä suositeltavia hoitoja ovat: suuriannoksiset laskimoon annettavat somatostatiinianalogit, laskimonsisäinen nesteytys, kortikosteroidit sekä oksentelevien ja/tai ripuloivien potilaiden elektrolyyttihäiriöiden korjaaminen.
Tuumorilyysioireyhtymä
Tuumorilyysioireyhtymää on raportoitu lutetium‑177:ää sisältävillä lääkevalmisteilla tapahtuneen hoidon jälkeen. Erityistä varovaisuutta on noudatettava sellaisten potilaiden hoidossa, joilla on ennestään munuaisten vajaatoimintaa ja suuri tuumoritaakka, ja siten mahdollisesti korkeampi tuumorilyysioireyhtymän todennäköisyys. Munuaisten toimintakykyä ja nestetasapainoa on arvioitava lähtötilanteessa ja hoidon aikana.
Säteilysuojauksen säännöt
Lutathera‑hoitoa saavat potilaat on pidettävä poissa muiden lähettyviltä annon aikana ja siihen saakka, kunnes lakisääteiset säteilyrajat täyttyvät, yleensä 4–5 tuntia lääkevalmisteen annon jälkeen. Terveydenhuollon ammattilaisen on määriteltävä, milloin potilas voi poistua sairaalan valvotulta alueelta, eli milloin säteilyaltistus kolmansille osapuolille ei ylitä valvontaviranomaisten kynnysarvoja.
Potilaita on kehotettava ylläpitämään nesteytystä ja virtsaamaan tiheästi ennen Lutathera‑valmisteen antoa, antopäivänä ja antopäivän jälkeisenä päivänä (esim. 1 lasillinen vettä joka tunti) eliminaation edistämiseksi. Potilaita on myös kehotettava ulostamaan joka päivä ja käyttämään tarvittaessa laksatiivia. Virtsa ja ulosteet on hävitettävä kansallisten säännösten mukaisesti.
Jos potilaan iho ei ole kontaminoitunut, esimerkiksi infuusiojärjestelmän vuodon tai virtsainkontinenssin takia, ihon tai oksennuksen aiheuttama radioaktiivinen kontaminaatio ei ole odotettavissa. On kuitenkin suositeltavaa, että kun tehdään hoitotoimenpiteitä tai tutkimuksia lääkinnällisillä laitteilla tai muilla instrumenteilla, jotka ovat kosketuksessa ihoon (esim. sydänsähkökäyrä [EKG]), huomioidaan perussuojaustoimenpiteet kuten käsineiden käyttäminen, materiaalin/elektrodin asettaminen paikoilleen ennen radioaktiivisen lääkevalmisteen infuusiota, materiaalin/elektrodin vaihtaminen mittauksen jälkeen sekä radioaktiivisuuden valvonta käytön jälkeen.
Ennen kotiuttamista potilaalle on selitettävä tarvittavat säteilysuojauksen säännöt ja toimintatavat, joita noudatetaan oltaessa tekemisissä samassa taloudessa asuvien henkilöiden ja yleisväestön kanssa, sekä yleiset varotoimet, joita potilaan on noudatettava päivittäisten toimintojen aikana hoidon jälkeen (löytyvät seuraavasta kappaleesta ja pakkausselosteesta), jotta muiden altistuminen säteilylle minimoidaan.
Kansallisten, paikallisten ja hoitolaitoskohtaisten toimenpiteiden ja ohjeiden lisäksi kunkin annon jälkeen voidaan harkita seuraavia yleissuosituksia:
- Läheistä (alle 1 metrin etäisyydellä tapahtuvaa) kanssakäymistä muiden henkilöiden kanssa on rajoitettava 7 vuorokauden ajan.
- Lasten ja/tai raskaana olevien naisten osalta lähikontakti (alle 1 metrin etäisyydellä) on rajoitettava alle 15 minuuttiin vuorokaudessa 7 vuorokauden ajan.
- Potilaan on nukuttava 7 vuorokauden ajan erillisessä makuuhuoneessa, jossa ei nuku muita ihmisiä.
- Potilas ei saa nukkua 15 vuorokauteen makuuhuoneessa, jossa nukkuu myös lapsia ja/tai raskaana oleva nainen.
Suositeltavat toimenpiteet ekstravasaation sattuessa
Kertakäyttöisiä vedenkestäviä käsineitä on käytettävä. Lääkevalmisteen infuusio on välittömästi keskeytettävä ja antolaite (katetri tms.) on poistettava. Asiasta on ilmoitettava isotooppilääketieteen erikoislääkärille ja radiofarmasian asiantuntijalle.
Kaikki antolaitteen materiaalit on säilytettävä, jotta voidaan mitata jäännösradioaktiivisuus ja todellinen annettu aktiivisuus ja arvioida absorboitu annos. Ekstravasaatioalue on rajattava häviämättömällä kynällä ja siitä on mahdollisuuksien mukaan otettava kuva. On myös suositeltavaa kirjata ekstravasaation aika ja arvioitu ekstravasoitunut tilavuus.
Lutathera‑valmisteen infuusion jatkamiseksi on pakollista käyttää uutta katetria, mahdollisesti asettaen se kontralateraaliseen laskimotiehen.
Muita lääkevalmisteita ei saa antaa samalle puolelle, jossa ekstravasaatio tapahtui.
Lääkevalmisteen dispersion nopeuttamiseksi ja sen kudoksiin patoutumisen estämiseksi on suositeltavaa lisätä veren virtausta nostamalla vaurioitunutta käsivartta ylöspäin. Tapauksesta riippuen on harkittava ekstravasaationesteen aspiraatiota, huuhteluinjektiota natriumkloridi 9 mg/ml (0,9 %) ‑injektionesteellä, tai lämpimien kääreiden tai lämpötyynyn asettamista infuusiokohtaan vasodilataation nopeuttamiseksi.
Oireita on hoidettava, erityisesti tulehdusta ja/tai kipua. Tilanteesta riippuen isotooppilääketieteen erikoislääkärin on kerrottava potilaalle ekstravasaatiovammaan liittyvistä riskeistä ja annettava neuvoja mahdollisesta hoidosta ja tarvittavasta seurannasta. Ekstravasaatioaluetta on seurattava siihen saakka, kunnes potilas kotiutetaan sairaalasta. Vaikeusasteesta riippuen tämä tapahtuma on ilmoitettava haittavaikutukseksi.
Potilaat, joilla on virtsainkontinenssi
Tämän lääkevalmisteen antoa seuraavien 2 vuorokauden aikana on suoritettava erityiset varotoimet radioaktiivisen kontaminaation leviämisen välttämiseksi, jos potilaalla on virtsainkontinenssi. Varotoimet sisältävät sellaisten materiaalien käsittelyn, jotka ovat mahdollisesti kontaminoituneet virtsasta.
Potilaat, joilla on aivoetäpesäkkeitä
Ei ole olemassa tietoja tehosta potilailla, joilla on todettu aivoetäpesäkkeitä. Näin ollen yksilöllinen hyöty‑riskisuhde on arvioitava näiden potilaiden kohdalla.
Sekundaariset pahanlaatuiset kasvaimet
Altistus ionisoivalle säteilylle on yhteydessä syövän syntyyn ja mahdollisten perimän virheiden kehittymiseen. Hoitoaltistuksesta aiheutuva säteilyannos voi saada aikaan korkeamman syövän ja mutaation insidenssin. Kaikissa tapauksissa on tarpeen varmistaa, että säteilyaltistuksen riskit ovat vähäisempiä kuin itse sairauden aiheuttamat riskit.
Muut potilaat, joilla on riskitekijöitä
Potilaat, joilla on jokin jäljempänä mainituista sairaustiloista, ovat alttiimpia haittavaikutusten kehittymiselle. Näin ollen tällaisten potilaiden tiheämpi seuranta on suositeltavaa hoidon aikana. Ks. taulukko 3 annosta rajoittavan toksisuuden tilanteessa.
- Luuetäpesäke;
- Aiempi radiometabolinen syöpähoito 131I‑yhdisteillä tai muu hoito, jossa käytetään suojaamattomia radioaktiivisuuden lähteitä;
- Aiemmat muut pahanlaatuiset kasvaimet, ellei potilaan katsotaan olleen remissiossa vähintään 5 vuoden ajan.
Raskauden ehkäisy miehillä ja naisilla
Naispotilaita, jotka voivat tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä hoidon aikana ja 7 kuukauden ajan viimeisen Lutathera‑annoksen jälkeen (ks. kohta Raskaus ja imetys).
Miespotilaita, joiden kumppani voi tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä hoidon aikana ja 4 kuukauden ajan viimeisen Lutathera‑annoksen jälkeen (ks. kohta Raskaus ja imetys).
Samanaikaisesti annettavaan munuaisia suojaavaan aminohappoliuokseen liittyvät erityiset varoitukset ja varotoimet
Hyperkalemia
Arginiinia ja lysiiniä saavilla potilailla voi esiintyä ohimenevää seerumin kaliumpitoisuuden suurenemista. Pitoisuus palaa yleensä normaalille tasolle 24 tunnin kuluessa aminohappoliuosinfuusion aloittamisesta. Potilailla, joiden kreatiniinipuhdistuma on pienentynyt, voi olla suurentunut ohimenevän hyperkalemian riski (ks. ”Munuaistoksisuus”, kohta Varoitukset ja käyttöön liittyvät varotoimet).
Seerumin kaliumpitoisuus on määritettävä ennen kutakin aminohappoliuosantokertaa. Jos hyperkalemiaa ilmenee, potilaan hyperkalemia‑anamneesi ja samanaikainen lääkitys on selvitettävä. Hyperkalemia on korjattava asianmukaisesti ennen infuusion aloittamista.
Jos potilaalla on entuudestaan kliinisesti merkittävä hyperkalemia, kaliumpitoisuus on määritettävä uudelleen ennen aminohappoliuosinfuusiota. Näin varmistetaan, että hyperkalemia on korjautunut. Potilasta on seurattava tarkoin hyperkalemian oireiden ja löydösten varalta. Näitä ovat esimerkiksi hengenahdistus, heikotus, tunnottomuus, rintakipu ja sydänlöydökset (johtumishäiriöt ja sydämen rytmihäiriöt). EKG on otettava ennen potilaan kotiuttamista.
Elintoimintoja on seurattava infuusion aikana seerumin kaliumpitoisuuden lähtötasosta riippumatta. Potilaita on kehotettava ylläpitämään nesteytystä ja virtsaamaan tiheästi ennen Lutathera‑valmisteen antoa, antopäivänä ja antopäivän jälkeisenä päivänä (esim. 1 lasillinen vettä joka tunti), jotta liiallisen kaliumin poistuminen seerumista tehostuu.
Jos aminohappoliuosinfuusion aikana kehittyy hyperkalemian oireita, on ryhdyttävä asianmukaisiin korjaaviin toimiin. Vaikean oireisen hyperkalemian yhteydessä on harkittava aminohappoliuosinfuusion lopettamista ja otettava huomioon munuaisten suojaamisen hyödyt ja riskit suhteessa akuuttiin hyperkalemiaan.
Sydämen vajaatoiminta
Volyymiylikuormitukseen liittyvien kliinisten komplikaatioiden mahdollisuuden vuoksi varovaisuus on tarpeen, jos arginiinia ja lysiiniä annetaan potilaalle, jolla on vaikea sydämen vajaatoiminta eli NYHA‑luokituksen (New York Heart Association) luokan III tai IV sydämen vajaatoiminta. Jos potilaalla on vaikea sydämen vajaatoiminta eli NYHA‑luokan III tai IV sydämen vajaatoiminta, hoitoa saa antaa vain huolellisen hyöty‑riskiarvioinnin jälkeen. Arvioinnissa on otettava huomioon aminohappoliuoksen tilavuus ja osmolaalisuus.
Metabolinen asidoosi
Täydellisen parenteraalisen ravitsemuksen (TPN) osana annettujen monimutkaisten aminohappoliuosten käytön yhteydessä on todettu metabolista asidoosia. Happo‑emästasapainon muuttuminen vaikuttaa solunulkoisen ja solunsisäisen kaliumin tasapainoon, ja asidoosin yhteydessä plasman kaliumpitoisuus voi suurentua nopeasti.
Erityisvaroitukset
Natriumpitoisuus
Tämä lääkevalmiste sisältää enintään 3,5 mmol (81,1 mg) natriumia per infuusiopullo, joka vastaa 4 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Ks. kohdasta Käyttö- ja käsittelyohjeet varotoimet ympäristöhaittoihin liittyen.
Yhteisvaikutukset
Somatostatiinianalogit
Somatostatiini ja sen analogit sitoutuvat kilpailevasti somatostatiinireseptoreihin ja saattavat huonontaa Lutatheran tehoa. Tämän vuoksi pitkävaikutteisten somatostatiinianalogien antoa on vältettävä 30 vuorokauden sisällä ennen tämän lääkevalmisteen antoa. Tarvittaessa potilasta voidaan hoitaa lyhytvaikutteisilla somatostatiinianalogeilla siihen asti, kunnes Lutathera‑valmisteen antoon on 24 tuntia.
Glukokortikoidit
On jonkin verran näyttöä siitä, että glukokortikoidit voivat aiheuttaa alatyypin 2 somatostatiinireseptoreiden (SSTR2) vaimennussäätelyä. Siksi toistuvaa glukokortikoidien antoa korkeilla annoksilla on varmuuden vuoksi vältettävä Lutathera‑hoidon aikana. Potilailta, joilla on aiempaa pitkäaikaista glukokortikoidien käyttöä, on arvioitava huolellisesti somatostatiinireseptoreiden riittävä ekspressio. Ei tiedetä, voiko glukokortikoidien jaksoittainen käyttö pahoinvoinnin ja oksentelun ehkäisyyn Lutathera‑valmisteen annon aikana aiheuttaa SSTR2‑reseptoreiden vaimennussäätelyä. Varmuuden vuoksi glukokortikoidien käyttöä myös pahoinvoinnin ja oksentelun ennaltaehkäisyyn on vältettävä. Jos pahoinvoinnin ja oksentelun ehkäisyyn ennen aminohappoliuoksen infusointia käytetyt lääkkeet osoittautuvat riittämättömiksi, voidaan käyttää yksittäistä glukokortikoidiannosta edellyttäen, että sitä ei anneta ennen Lutathera‑infuusion aloittamista eikä Lutathera‑infuusion päättymistä seuraavan tunnin aikana.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Jos radiofarmaseuttista valmistetta aiotaan antaa hedelmällisessä iässä olevalle naiselle, on tärkeää selvittää, onko nainen raskaana. Kuukautisten jäädessä väliin on aina oletettava, että nainen on raskaana, kunnes toisin todistetaan. Jos raskauden mahdollisuus on epäselvä (kuukautiset jääneet väliin, hyvin epäsäännöllinen kuukautiskierto jne.), potilaan kohdalla on käytettävä vaihtoehtoisia menetelmiä, joihin ei liity ionisoivaa säteilyä (mikäli tällaisia on saatavilla). Ennen Lutathera‑valmisteen käyttöä raskaus on poissuljettava asianmukaisella/validoidulla testillä.
Raskauden ehkäisy miehillä ja naisilla
Lutatheran anto raskaana olevalle naiselle voi vahingoittaa sikiötä.
Naispotilaita, jotka voivat tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä hoidon aikana ja 7 kuukauden ajan viimeisen Lutathera‑annoksen jälkeen.
Miespotilaita, joiden kumppani voi tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä hoidon aikana ja 4 kuukauden ajan viimeisen Lutathera‑annoksen jälkeen.
Raskaus
Lutetium(177Lu)oksodotreotidin vaikutuksesta lisääntymiseen ei ole tehty eläinkokeita.
Raskaana olevalle naiselle tehtävät radionukliditoimenpiteet altistavat myös sikiön säteilylle. Ionisoivaan säteilyyn liittyvän riskin vuoksi Lutathera‑valmisteen käyttö on vasta‑aiheista todetun tai epäillyn raskauden aikana ja silloin, kun raskautta ei ole poissuljettu (ks. kohta Vasta-aiheet). Raskaana oleville naisille on kerrottava sikiöön kohdistuvasta riskistä.
Imetys
Ei tiedetä, erittyykö lutetium(177Lu)oksodotreotidi ihmisen rintamaitoon. Ionisoivaan säteilyyn liittyvää riskiä imeväiselle ei voida poissulkea. Imettämistä on vältettävä tämän lääkehoidon aikana. Jos Lutathera‑hoito on tarpeen imetyksen aikana, lapsi on vieroitettava.
Hedelmällisyys
Lutetium(177Lu)oksodotreotidin aiheuttamia, miesten ja naisten hedelmällisyyteen kohdistuvia vaikutuksia ei ole tutkittu eläimillä. Lutetium(177Lu)oksodotreotidin ionisoivalla säteilyllä voi mahdollisesti olla tilapäisiä toksisia vaikutuksia naisten ja miesten sukurauhasiin. Geneettinen konsultaatio on suositeltavaa, jos potilas haluaa saada lapsia hoidon jälkeen. Siemennesteen ja munasolujen pakastuksen mahdollisuudesta voidaan keskustella potilaiden kanssa ennen hoitoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Lutathera‑valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Potilaan yleinen vointi ja mahdolliset hoidon haittavaikutukset on otettava huomioon ennen ajamista ja koneiden käyttöä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Lutathera‑valmisteen yleinen turvallisuusprofiili perustuu yhdistettyihin potilastietoihin, jotka ovat peräisin kliinisistä tutkimuksista (vaiheen III NETTER‑1‑tutkimus ja vaiheen I/II Erasmus‑tutkimuksen hollantilaiset potilaat), sekä tietoihin vaiheen II NETTER-P-tutkimuksesta ja ns. compassionate use ‑ohjelmista.
Yleisimmät haittavaikutukset Lutathera‑valmistetta saavilla potilailla olivat infuusion alussa esiintyvä pahoinvointi (58,9 %:lla potilaista) ja oksentelu (45,5 %:lla potilaista). Pahoinvoinnin/oksentelun syy‑yhteyden arviointia sekoittaa samanaikaisesti munuaisten suojaamiseksi annettavan aminohappoliuoksen emeettinen vaikutus.
Lutathera‑valmisteen luuydintoksisuuden vuoksi todennäköisimmin odotettavissa olevat haittavaikutukset liittyivät hematologiseen toksisuuteen: trombosytopenia (25 %), lymfopenia (22,3 %), anemia (13,4 %) ja pansytopenia (10,2 %).
Muita ilmoitettuja hyvin yleisiä haittavaikutuksia ovat uupumus (27,7 %) ja ruokahalun heikkeneminen (13,4 %).
NETTER‑1‑tutkimuksen loppuanalyysin hetkellä, kun seurannan mediaanikesto oli 76 kuukautta kummassakin tutkimusryhmässä, turvallisuusprofiili säilyi johdonmukaisena aiempaan verrattuna.
Taulukkomuotoinen luettelo haittavaikutuksista
Haittavaikutukset on lueteltu taulukossa 5 esiintymistiheyden ja MedDRA‑elinjärjestelmäluokan (System Organ Class, SOC) mukaisesti. Esiintymistiheydet on luokiteltu seuraavasti: hyvin yleinen (≥1/10), yleinen (≥1/100, <1/10), melko harvinainen (≥1/1 000, <1/100), harvinainen (≥1/10 000, <1/1 000), hyvin harvinainen (<1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 5 Kliinisissä tutkimuksissa ja markkinoille tulon jälkeisessä valvonnassa ilmoitettujen haittavaikutusten esiintymistiheys
| MedDRA‑elinjärjestelmäluokka (SOC) | Hyvin yleinen | Yleinen | Melko harvinainen | Tuntematon |
| Infektiot | Konjuktiviitti Hengitystieinfektio Kystiitti Keuhkokuume Vyöruusu Silmänseudun vyöruusu Influenssa Stafylokokki‑infektiot Streptokokkibakteremia | |||
| Hyvän‑ ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Refraktorinen sytopenia ja usean solulinjan dysplasia (myelodysplastinen oireyhtymä) | Akuutti myelooinen leukemia Akuutti leukemia Krooninen myelomonosyyttinen leukemia | ||
| Veri ja imukudos | Trombosytopenia2 Lymfopenia3 Anemia4 Pansytopenia | Leukopenia5 Neutropenia6 | Refraktorinen sytopenia ja yhden solulinjan dysplasia Nefrogeeninen anemia Luuytimen vajaatoiminta Trombosytopeeninen purppura | |
| Immuunijärjestelmä | Yliherkkyys | Angioedeema | ||
| Umpieritys | Sekundaarinen kilpirauhasen vajaatoiminta | Kilpirauhasen vajaatoiminta Diabetes mellitus Karsinoidikriisi Hyperparatyroidismi | ||
| Aineenvaihdunta ja ravitsemus | Ruokahalun heikkeneminen | Hyperglykemia Nestehukka Hypomagnesemia Hyponatremia | Hypoglykemia Hypernatremia Hypofosfatemia Tuumorilyysioireyhtymä Hyperkalsemia Hypokalsemia Hypoalbuminemia Metabolinen asidoosi | |
| Psyykkiset häiriöt | Unihäiriöt | Ahdistuneisuus Aistiharha Sekavuus | ||
| Hermosto | Heitehuimaus Makuhäiriö Päänsärky10 Letargia Pyörtyminen | Formikaatio Hepaattinen enkefalopatia Parestesia Parosmia Uneliaisuus Selkäytimen kompressio | ||
| Silmät | Silmien häiriöt | |||
| Kuulo ja tasapainoelin | Kiertohuimaus | |||
| Sydän | Sydänsähkökäyrän pidentynyt QT‑aika | Eteisvärinä Sydämentykytykset Sydäninfarkti Angina pectoris Kardiogeeninen shokki | ||
| Verisuonisto | Hypertensio7 Punastuminen Kuuma aalto Hypotensio | Vasodilataatio Perifeerinen kylmyys Kalpeus Ortostaattinen hypotensio Laskimotulehdus | ||
| Hengityselimet, rintakehä ja välikarsina | Hengenahdistus | Suunielun kipu Keuhkopussin nestekertymä Lisääntynyt limaneritys Tukehtumisen tunne | ||
| Ruoansulatuselimistö | Pahoinvointi Oksentelu | Vatsan turpoaminen Ripuli Vatsakipu Ummetus Ylävatsan kipu Dyspepsia Gastriitti | Suun kuivuminen Ilmavaivat Askites Ruoansulatuselimistön kipu Suutulehdus Veriuloste Vatsavaivat Suolitukos Koliitti Akuutti haimatulehdus Peräsuoliverenvuoto Mustat veriulosteet Alavatsan kipu Verioksennus Hemorraginen vesivatsa Ileus | |
| Maksa ja sappi | Hyperbilirubinemia9 | Haimaentsyymien väheneminen Maksasoluvaurio Sappitukos Maksakongestio Maksan vajaatoiminta | ||
| Iho ja ihonalainen kudos | Alopesia | Ihottuma Ihon kuivuminen Kasvojen turvotus Liikahikoilu Yleistynyt kutina | ||
| Luusto, lihakset ja sidekudos | Lihas‑ ja luukipu8 Lihaskouristukset | |||
| Munuaiset ja virtsatiet | Akuutti munuaisvaurio Verivirtsaisuus Munuaisten vajaatoiminta Proteinuria | Leukosyturia Virtsainkontinenssi Glomerulusfiltraation vähentyminen Munuaisten toimintahäiriö Akuutti prerenaalinen vajaatoiminta Munuaisten vajaatoiminta | ||
| Yleisoireet ja antopaikassa todettavat haitat | Uupumus1 | Injektiokohdan reaktio11 Perifeerinen turvotus Antokohdan kipu Vilunväristykset Influenssan kaltainen sairaus | Injektiokohdan kovettuma Epämukava tunne rinnassa Rintakipu Kuume Huonovointisuus Kipu Kuolema Epänormaali olo | |
| Tutkimukset | Veren kreatiniiniarvon kohoaminen GGT*‑arvon kohoaminen ALAT**‑arvon kohoaminen ASAT***‑arvon kohoaminen Veren AFOS****‑arvon kohoaminen | Veren kaliumarvon laskeminen Veren urea‑arvon kohoaminen Glykosyloidun hemoglobiinin kohoaminen Hematokriittiarvon laskeminen Proteiinia virtsassa Painon lasku Veren kreatiinifosfokinaasiarvon kohoaminen Veren laktaattidehydrogenaasiarvon kohoaminen Veren katekoliamiinit C‑reaktiivisen proteiinin kohoaminen | ||
| Vammat, myrkytykset ja hoitokomplikaatiot | Solisluun murtuma | |||
| Kirurgiset ja lääketieteelliset toimenpiteet | Verensiirto | Vatsaontelon tyhjennys Dialyysi Maha‑suolikanavan letkun asettaminen Stentin asettaminen Paiseen tyhjennys Luuytimen kerääminen Polypektomia | ||
| Sosiaaliset olosuhteet | Fyysinen toiminnanvajaus |
1 Sisältää astenian ja uupumuksen
2 Sisältää trombosytopenian ja verihiutaleiden määrän vähenemisen
3 Sisältää lymfopenian ja lymfosyyttien määrän vähenemisen
4 Sisältää anemian ja hemoglobiinin vähenemisen
5 Sisältää leukopenian ja veren valkosolujen määrän vähenemisen
6 Sisältää neutropenian ja neutrofiilien määrän vähenemisen
7 Sisältää hypertension ja hypertensiivisen kriisin
8 Sisältää nivelkivun, raajakivun, selkäkivun, luukivun, kylkikivun, muskuloskeletaalisen rintakivun ja niskakivun
9 Sisältää veren bilirubiinin lisääntymisen ja hyperbilirubinemian
10 Sisältää päänsäryn ja migreenin
11 Sisältää injektiokohdan reaktion, injektiokohdan yliherkkyyden, injektiokohdan kovettuman, injektiokohdan turvotuksen
*Gammaglutamyylitransferaasiarvo
**Alaniiniaminotransferaasi
***Aspartaattiaminotransferaasi
****Alkalinen fosfataasi
Valittujen haittavaikutusten kuvaus
Myelosuppressio
Pääasiassa lievä tai keskivaikea luuydintoksisuus (myelo‑/hematotoksisuus) ilmeni palautuvina/ohimenevinä verisolujen määrien vähenemisinä, jotka vaikuttavat kaikkiin solulinjoihin (sytopeniat kaikkina yhdistelminä: pansytopenia, bisytopeniat, isoloidut monosytopeniat – anemia, neutropenia, lymfosytopenia ja trombosytopenia). Havaitusta merkittävästä selektiivisestä B‑solukadosta huolimatta infektiokomplikaatioiden esiintymistiheys ei lisääntynyt pepitidireseptoriradionuklidihoidon (PRRT) jälkeen. Korjautumattomien hematologisten patologioiden, eli premalignien (myelodysplastinen oireyhtymä) ja malignien (akuutti myelooinen leukemia) veren neoplasmojen tapauksia on ilmoitettu Lutathera‑hoidon jälkeen.
NETTER‑1-tutkimuksessa verihiutalemäärän nadiirin mediaaniajankohta oli 5,1 kuukauden kuluttua ensimmäisestä annoksesta. Tutkimuksessa 59 potilaalle kehittyi trombosytopenia; heistä 68 %:lla verihiutalemäärä korjautui lähtötasolle tai viitealueelle. Mediaaniaika verihiutalemäärän korjautumiseen oli 2 kuukautta. 19 potilaalla ei dokumentoitu verihiutalemäärän korjautumista lähtötasolle tai viitealueelle; heistä 15:llä todettiin kuitenkin verihiutalemäärän suurenemista nadiirin jälkeen.
Munuaistoksisuus
Lutetium(177Lu)oksodotreotidi erittyy munuaisten kautta.
Kliinisissä tutkimuksissa havaittiin pitkäaikainen etenevä glomerulusfiltraatiotoiminnon heikkeneminen. Lutathera‑valmisteeseen liittyvä nefropatia on näin ollen krooninen munuaissairaus, joka kehittyy etenevästi kuukausien tai vuosien kuluessa altistuksen jälkeen. Yksilöllistä hyöty‑riskiarviointia suositellaan ennen Lutathera‑hoitoa potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta. Katso lisätietoja kohdasta Annostus ja antotapa (taulukko 3 ja alakohta ”Munuaisten vajaatoiminta”) ja kohdasta Varoitukset ja käyttöön liittyvät varotoimet. Lutathera on vasta‑aiheinen munuaisten vajaatoimintaa sairastavilla potilailla, joiden kreatiniinipuhdistuma on < 30 ml/min (ks. kohta Vasta-aiheet).
Neuroendokriiniset hormonaaliset kriisit
Bioaktiivisten aineiden vapautumiseen liittyviä hormonaalisia kriisejä (jotka johtuvat todennäköisesti neuroendokriinikasvainsolujen hajoamisesta) on havaittu harvoin ja ne ovat loppuneet asianmukaisen lääkehoidon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Julkaistun kirjallisuuden ja 11:n NETTER-P-tutkimukseen osallistuneen nuoren kliinisten tietojen perusteella ei raportoitu uusia turvallisuussignaaleja. Nuorilla oli somatostatiinireseptoripositiivinen GEP-NET, feokromosytooma tai paragangliooma (PPGL).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‑haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostus on epätodennäköistä Lutathera‑valmistetta käytettäessä, koska tämä lääkevalmiste toimitetaan kerta‑annoksina ja käyttövalmiina ennalta määritellyn radioaktiivisuuden sisältävänä valmisteena ja sen antavat radiofarmaseuttisten valmisteiden käsittelyyn valtuutetut henkilöt sen jälkeen, kun pätevä lääkäri on arvioinut potilaan. Yliannostuksen yhteydessä säteilytoksisuuteen liittyvien haittavaikutusten esiintymistiheyden odotetaan lisääntyvän.
Mikäli Lutathera‑valmistetta annettaessa tapahtuu säteily‑yliannostus, potilaaseen absorboitunutta annosta on vähennettävä mahdollisuuksien mukaan lisäämällä radionuklidin eliminaatiota kehosta tiheällä virtsaamisella tai pakotetulla diureesilla ja tiheällä virtsarakon tyhjennyksellä ensimmäisten 48 tunnin aikana infuusion jälkeen. Voi olla hyödyllistä arvioida käytetty efektiivinen annos.
Seuraavat laboratoriotutkimukset on suoritettava joka viikko seuraavien 10 viikon ajan:
- Hematologinen seuranta: veren valkosolujen määrä ja erittelylaskenta, verihiutaleet ja hemoglobiini
- Veren kemian seuranta: seerumin kreatiniini ja glykemia.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: terapeuttiset radioaktiiviset lääkevalmisteet, muut terapeuttiset radioaktiiviset lääkevalmisteet, ATC‑koodi: V10XX04
Vaikutusmekanismi
Lutetium(177Lu)oksodotreotidilla on korkea affiniteetti alatyypin 2 somatostatiinireseptoreille (SSTR2). Se sitoutuu maligneihin soluihin, joissa SSTR2‑reseptorit ovat yliekspressoituneita.
Lutetium‑177 on β-‑säteilevä radionuklidi, jonka maksimaalinen tunkeutumisalue kudoksissa on 2,2 mm (keskimääräinen tunkeutumisalue 0,67 mm). Se tappaa kohteena olevia kasvainsoluja vaikuttaen vain rajatusti läheisiin normaaleihin soluihin.
Farmakodynaamiset vaikutukset
Käytettävällä pitoisuudella (noin 10 μg/ml; vapaa ja radioleimattu muoto yhteensä), oksodotreotidipeptidillä ei ole mitään kliinisesti merkittävää farmakodynaamista vaikutusta.
Kliininen teho ja turvallisuus
NETTER‑1-tutkimus
Vaiheen III NETTER‑1‑tutkimus oli ositettu, avoin, satunnaistettu, vertailuvalmistekontrolloitu, rinnakkaisryhmissä toteutettu monikeskustutkimus. Lutathera‑valmistetta (neljä 7 400 MBq:n annosta, yksi annos 8 viikon [± 1 viikko] välein) annettiin aminohappoliuoksen ja parhaan mahdollisen tukihoidon (pitkävaikutteinen oktreotidi [LAR] 30 mg jokaisen Lutathera‑annoksen jälkeen ja 4 viikon välein Lutathera‑hoidon päättymisen jälkeen oireiden hallintaan, korvattiin lyhytvaikutteisella oktreotidilla Lutathera‑antoa edeltävällä 4 viikon aikajaksolla) kanssa. Lutathera‑hoitoa verrattiin suuriannoksiseen oktreotidiin (LAR, 60 mg 4 viikon välein) potilailla, joilla oli leikkaushoitoon soveltumattomia, levinneitä, somatostatiinireseptoripositiivisia keskisuolen karsinoidikasvaimia. Tutkimuksen ensisijainen päätetapahtuma oli etenemisvapaa elossaolo (progression‑free survival, PFS) Response Evaluation Criteria in Solid Tumours (RECIST, versio 1.1) ‑kriteerien perusteella arvioituna, sokkoutetun riippumattoman arviointitoimikunnan (BIRC, blinded independent review committee) arvioinnin pohjalta. Toissijaisia tehon päätetapahtumia olivat objektiivinen vaste (ORR, objective response rate), kokonaiselossaolo (OS, overall survival), aika kasvaimen etenemiseen (TTP, time to tumour progression), lääkevalmisteen turvallisuus ja siedettävyys sekä terveyteen liittyvä elämänlaatu (HRQoL, health-related quality of life).
Ensisijaisen analyysin hetkellä 229 potilasta satunnaistettiin saamaan joko Lutathera‑valmistetta (n = 116) tai oktreotidi LAR ‑valmistetta suurina annoksina (n = 113). Demografiset tiedot ja sairauksien lähtötilanteen ominaisuudet olivat hyvin tasapainossa hoitoryhmien välillä. Potilaiden mediaani‑ikä oli 64 vuotta, ja 82,1 % heistä oli valkoihoisia.
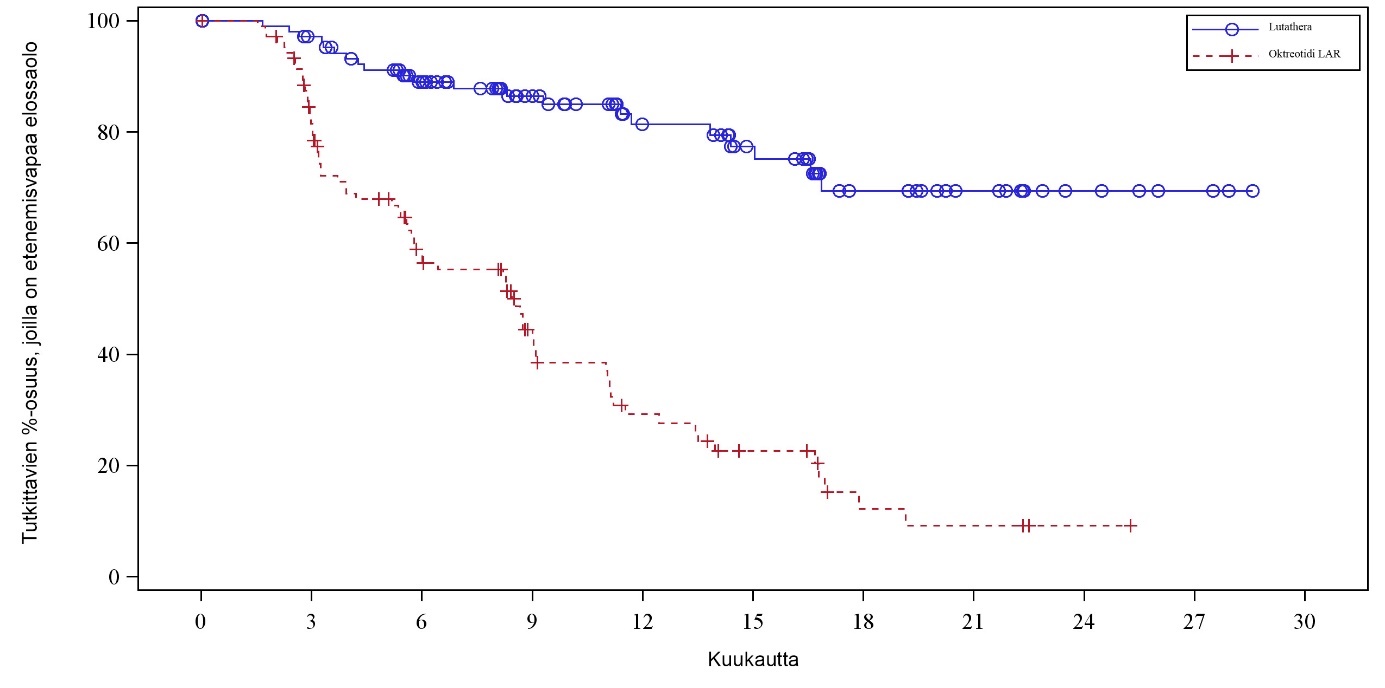
Etenemisvapaata elossaoloa (PFS) koskevan ensisijaisen analyysin hetkellä (katkaisupäivämäärä 24.7.2015) keskitetysti vahvistettujen taudin etenemisten tai kuolemien lukumäärä oli 21 tapahtumaa Lutathera‑ryhmässä ja 70 tapahtumaa suuriannoksisessa oktreotidi LAR ‑ryhmässä (taulukko 6). Etenemisvapaassa elossaolossa oli merkitsevä ero (p < 0,0001) hoitoryhmien välillä. Lutathera‑ryhmässä etenemisvapaan elossaolon mediaania ei ollut saavutettu katkaisupäivämäärään mennessä; suuriannoksisessa oktreotidi LAR ‑ryhmässä etenemisvapaan elossaolon mediaani oli 8,5 kuukautta. Riskitiheyssuhde (hazard ratio, HR) Lutathera‑ryhmässä verrattuna suuriannoksiseen oktreotidi LAR ‑ryhmään oli 0,18 (95 %:n lv: 0,11; 0,29), mikä merkitsee 82 % pienempää taudin etenemisen tai kuoleman riskiä Lutathera‑ryhmässä.
Taulukko 6 Vaiheen III NETTER‑1‑tutkimuksessa havaittu etenemisvapaa elossaolo (PFS) potilailla, joilla oli levinneitä keskisuolen karsinoidikasvaimia – katkaisupäivämäärä 24.7.2015 (täysi analyysijoukko [FAS], N = 229)
| Hoito | ||
| Lutathera ja oktreotidi LAR | Suuriannoksinen oktreotidi LAR | |
| N | 116 | 113 |
| Potilaita, joilla oli tapahtumia | 21 | 70 |
| Sensuroituja potilaita | 95 | 43 |
| Kuukausien mediaani (95 %:n lv) | Ei saavutettu | 8,5 (5,8; 9,1) |
| Log‑rank-testin p‑arvo | < 0,0001 | |
| Riskitiheyssuhde (95 %:n lv) | 0,177 (0,108; 0,289) | |
N: potilaiden lukumäärä, lv: luottamusväli.
Etenemisvapaan elossaolon Kaplan–Meierin käyrä täyden analyysijoukon (FAS) osalta katkaisupäivänä 24.7.2015 on esitetty kuvassa 2.
Kuva 2 Etenemisvapaan elossaolon (PFS) Kaplan–Meierin käyrät potilailla, joilla oli levinneitä keskisuolen karsinoidikasvaimia – katkaisupäivämäärä 24.7.2015 (vaiheen III NETTER‑1‑tutkimus; FAS, N = 229)
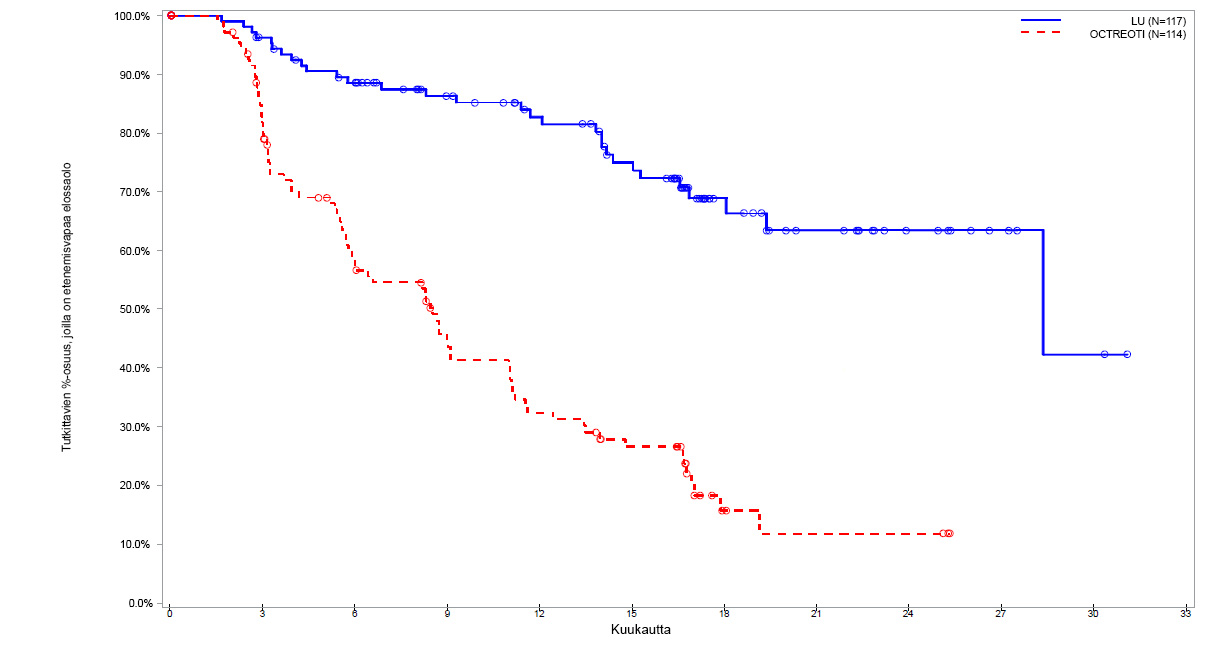
Tilastollisessa post hoc ‑analyysissa (katkaisupäivämäärä 30.6.2016), jossa mukana oli kaksi ylimääräistä satunnaistettua potilasta (N = 231), keskitetysti vahvistettujen taudin etenemisten tai kuolemien lukumäärä oli katkaisupäivämäärän hetkellä 30 tapahtumaa Lutathera‑ryhmässä ja 78 tapahtumaa suuriannoksisessa oktreotidi LAR ‑ryhmässä (taulukko 7). Etenemisvapaassa elossaolossa oli merkitsevä ero (p < 0,0001) hoitoryhmien välillä. Lutathera‑ryhmässä etenemisvapaan elossaolon mediaani oli 28,4 kuukautta, kun suuriannoksisessa oktreotidi LAR ‑ryhmässä etenemisvapaan elossaolon mediaani oli 8,5 kuukautta. Riskitiheyssuhde Lutathera‑ryhmässä verrattuna suuriannoksiseen oktreotidi LAR ‑ryhmään oli 0,21 (95 %:n lv: 0,14; 0,33), mikä merkitsee 79 % pienempää taudin etenemisen tai kuoleman riskiä Lutathera‑ryhmässä.
Taulukko 7 Vaiheen III NETTER‑1‑tutkimuksessa havaittu etenemisvapaa elossaolo (PFS) potilailla, joilla oli levinneitä keskisuolen karsinoidikasvaimia – katkaisupäivämäärä 30.6.2016 (FAS, N = 231)
| Hoito | ||
| Lutathera ja oktreotidi LAR | Suuriannoksinen oktreotidi LAR | |
| N | 117 | 114 |
| Potilaita, joilla oli tapahtumia | 30 | 78 |
| Sensuroidut potilaat | 87 | 36 |
| Kuukausien mediaani (95 %:n lv) | 28,4 (28,4; NE) | 8,5 (5,8; 11,0) |
| Log‑rank‑testin p‑arvo | < 0,0001 | |
| Riskitiheyssuhde (95 %:n lv) | 0,214 (0,139; 0,330) | |
N: potilaiden lukumäärä, lv: luottamusväli.
Etenemisvapaan elossaolon Kaplan–Meierin käyrä täyden analyysijoukon osalta katkaisupäivänä 30.6.2016 on esitetty kuvassa 3.
Kuva 3 Etenemisvapaan elossaolon Kaplan–Meierin käyrät potilailla, joilla oli levinneitä keskisuolen karsinoidikasvaimia – katkaisupäivämäärä 30.6.2016 (vaiheen III NETTER‑1‑tutkimus; FAS, N = 231)
Kokonaiselossaoloa (OS) koskevan välianalyysin hetkellä (katkaisupäivämäärä 24.7.2015) Lutathera‑ryhmässä oli esiintynyt 17 kuolemaa ja suuriannoksisessa oktreotidi LAR ‑ryhmässä 31 kuolemaa; riskitiheyssuhde oli 0,459 (99,9915 %:n lv: 0,140; 1,506) Lutathera‑ryhmän hyväksi. Kokonaiselossaolon mediaania ei ollut saavutettu Lutathera‑ryhmässä katkaisupäivämäärään mennessä; suuriannoksisessa oktreotidi LAR ‑ryhmässä se oli 27,4 kuukautta. Kokonaiselossaoloa koskevat välianalyysin tulokset eivät olleet tilastollisesti merkitseviä. Samanlainen suuntaus näkyi noin vuotta myöhemmin tehdyssä päivityksessä (katkaisupäivämäärä 30.6.2016), jossa oli mukana kaksi ylimääräistä satunnaistettua potilasta (N = 231). Tällöin Lutathera‑ryhmässä oli esiintynyt 28 kuolemaa ja suuriannoksisessa oktreotidi LAR ‑ryhmässä 43 kuolemaa; riskitiheyssuhde oli 0,536 Lutathera‑ryhmän hyväksi. Kokonaiselossaelon mediaania ei ollut edelleenkään saavutettu Lutathera‑ryhmässä katkaisupäivämäärään mennessä; suuriannoksisessa oktreotidi LAR ‑ryhmässä se oli 27,4 kuukautta.
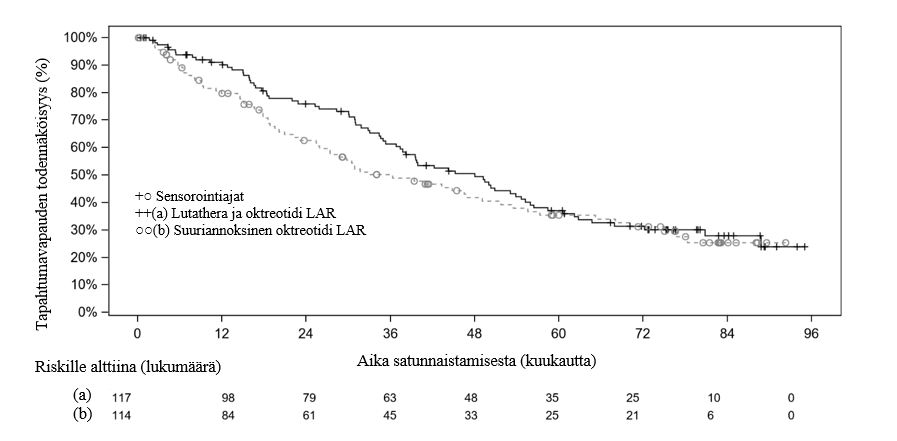
Kokonaiselossaoloa koskevan loppuanalyysin hetkellä viisi vuotta viimeisen potilaan satunnaistamisesta (N = 231, katkaisupäivämäärä 18.1.2021) seurannan mediaanikesto oli kummassakin hoitoryhmässä 76 kuukautta. Lutathera‑ryhmässä oli esiintynyt 73 kuolemaa (62,4 %) ja suuriannoksisessa oktreotidi LAR ‑ryhmässä 69 kuolemaa (60,5 %); riskitiheyssuhde oli 0,84 (95 %:n lv, 0,60; 1,17; kaksitahoinen stratifioimaton log‑rank‑testi, p = 0,3039) Lutathera‑ryhmän hyväksi. Kokonaiselossaolon mediaani oli kliinisesti merkittävästi 11,7 kuukautta pidempi Lutathera‑ryhmään satunnaistetuilla potilailla kuin suuriannoksiseen oktreotidi LAR ‑ryhmään satunnaistetuilla potilailla. Kokonaiselossaolon mediaani oli Lutathera‑ryhmässä 48,0 kuukautta (95 %:n lv: 37,4; 55,2) ja suuriannoksisessa oktreotidi LAR ‑ryhmässä 36,3 kuukautta (95 %:n lv: 25,9; 51,7). Lopulliset kokonaiselossaolon tulokset eivät olleet tilastollisesti merkitseviä. Suuriannoksisessa oktreotidi LAR ‑ryhmässä 22,8 % potilaista sai myöhempää radioligandihoitoa (mukaan lukien lutetium(177Lu)oksodotreotidia) 24 kuukauden kuluessa satunnaistamisesta, ja 36 % potilaista sai uudelleen radioligandihoitoa lopullisten kokonaiselossaolotietojen katkaisupäivämäärään mennessä. Tällä ja muilla tekijöillä saattoi olla vaikutusta tämän potilasalaryhmän kokonaiselossaoloon.
Kokonaiselossaolon Kaplan–Meierin käyrä täyden analyysijoukon osalta katkaisupäivänä 18.1.2021 on esitetty kuvassa 4.
Kuva 4 Kokonaiselossaolon Kaplan–Meierin käyrä potilailla, joilla oli levinneitä keskisuolen karsinoidikasvaimia – katkaisupäivämäärä 18.1.2021 (vaiheen III NETTER‑1‑tutkimus; FAS, N = 231)
Ei‑suhteellisten riskien vuoksi kokonaiselossaolon loppuanalyysin hetkellä tehtiin vielä ylimääräinen herkkyysanalyysi (rajattu keskimääräinen elossaolo [RMST, restricted mean survival time]), jotta saataisiin lisätietoa hoitovaikutuksesta (taulukko 8). Keskimääräinen kokonaiselossaolohyöty oli 60 kuukauden kohdalla satunnaistamisesta 5,1 kk pidempi (95 %:n lv: -0,5; 10,7) Lutathera‑ryhmässä kuin suuriannoksisessa oktreotidi LAR ‑ryhmässä.
Taulukko 8 Kokonaiselossaolo; rajattu keskimääräinen elossaolo (restricted mean survival time, RMST) vaiheen III NETTER‑1‑tutkimuksessa potilailla, joilla oli levinneitä keskisuolen karsinoidikasvaimia (FAS, N = 231)
| Lutathera ja oktreotidi LAR, N = 117 | Suuriannoksinen oktreotidi LAR, N = 114 | ||
| 24 kk | Kuolemat, n (%) | 26 (22,2) | 39 (34,2) |
| RMST (95 %:n lv) | 21,2 (20,2; 22,3) | 19,3 (18,0; 20,7) | |
| Ero (95 %:n lv) | 1,9 (0,1; 3,6) | ||
| 36 kk | Kuolemat, n (%) | 41 (35,0) | 51 (44,7) |
| RMST (95 %:n lv) | 29,7 (27,7; 31,6) | 26,0 (23,7; 28,3) | |
| Ero (95 %:n lv) | 3,7 (0,7; 6,7) | ||
| 48 kk | Kuolemat, n (%) | 53 (45,3) | 58 (50,9) |
| RMST (95 %:n lv) | 36,2 (33,4; 39,0) | 31,5 (28,3; 34,8) | |
| Ero (95 %:n lv) | 4,6 (0,3; 8,9) | ||
| 60 kk | Kuolemat, n (%) | 65 (55,6) | 63 (55,3) |
| RMST (95 %:n lv) | 41,2 (37,6; 44,9) | 36,1 (31,9; 40,4) | |
| Ero (95 %:n lv) | 5,1 (-0,5; 10,7) | ||
Terveyteen liittyvää elämänlaatua (HRQoL) arvioitiin käyttämällä EORTC‑järjestön (European Organisation for Research and Treatment of Cancer) elämänlaatukyselyä (EORTC QLQ‑C30) (geneerinen instrumentti) sekä sen neuroendokriinikasvainmoduulia (EORTC QLQ‑GI.NET‑21).
Tulokset viittaavat siihen, että yleisterveyteen liittyvä elämänlaatu paranee viikkoon 84 asti Lutathera‑hoitoa saavassa ryhmässä verrattuna suuriannoksista oktreotidi LAR ‑hoitoa saavaan ryhmään.
ERASMUS-tutkimus
Vaiheen I/II Erasmus‑tutkimus oli yksihaarainen avoin yksikeskustutkimus samanaikaisesti aminohappoliuoksen kanssa annetun Lutathera‑valmisteen (neljä 7 400 MBq:n annosta, yksi annos 8 viikon välein) tehosta potilailla, joilla oli somatostatiinireseptoripositiivisia kasvaimia. Tutkimukseen mukaan otettujen potilaiden mediaani‑ikä oli 59 vuotta. Useimmat potilaat olivat hollantilaisia (811). Loput potilaista (403) olivat eri Euroopan maiden ja muiden kuin Euroopan maiden kansalaisia. Pääanalyysi koski 811:tä hollantilaista potilasta, joilla oli erilaisia somatostatiinireseptoripositiivisia neuroendokriinikasvaintyyppejä (NET‑kasvaimia). Objektiivinen vaste (ORR) (mukaan lukien täydellinen vaste [CR, complete response] ja osittainen vaste [PR, partial response] RECIST‑kriteerien mukaisesti) ja vasteen kesto (DoR, duration of response) hollantilaisella FAS‑populaatiolla, jolla oli gastroenteropankreaattisia (GEP) ja bronkiaalisia NET‑kasvaimia (360 potilasta) on esitetty taulukossa 9, myös kasvaintyypin mukaan jaoteltuina.
Taulukko 9 Paras vaste, objektiivinen vaste (ORR) ja vasteen kesto (DoR) vaiheen I/II Erasmus‑tutkimuksessa hollantilaisilla potilailla, joilla oli GEP‑ ja bronkiaalisia NET‑kasvaimia – (FAS, N = 360)
| N | CR | PR | SD | ORR | DoR (kuukautta) | |||||||||
| Kasvaimen tyyppi | n | % | n | % | N | % | n | % | 95 %:n lv | Mediaani | 95 %:n lv | |||
| Kaikki NET-kasvaimet* | 360 | 11 | 3 % | 151 | 42 % | 183 | 51 % | 162 | 45 % | 40 % | 50 % | 16,3 | 12,2 | 17,8 |
| Keuhko | 19 | 0 | 0 % | 7 | 37 % | 11 | 58 % | 7 | 37 % | 16 % | 62 % | 23,9 | 1,7 | 30,0 |
| Haima | 133 | 7 | 5 % | 74 | 56 % | 47 | 35 % | 81 | 61 % | 52 % | 69 % | 16,3 | 12,1 | 21,8 |
| Etusuoli** | 12 | 1 | 8 % | 6 | 50 % | 4 | 33 % | 7 | 58 % | 28 % | 85 % | 22,3 | 0,0 | 38,0 |
| Keskisuoli | 183 | 3 | 2 % | 58 | 32 % | 115 | 63 % | 61 | 33 % | 27 % | 41 % | 15,3 | 10,5 | 17,7 |
| Takasuoli | 13 | 0 | 0 % | 6 | 46 % | 6 | 46 % | 6 | 46 % | 19 % | 75 % | 17,8 | 6,2 | 29,9 |
CR = Täydellinen vaste; PR = Osittainen vaste; SD = Vakaa tauti (stable disease); ORR = Objektiivinen vasteprosentti (CR+PR); DoR = Vasteen kesto
* Sisältää etusuolen, keskisuolen ja takasuolen; ** Etusuolen muut NET‑kasvaimet kuin bronkiaaliset ja haiman kasvaimet
Hollantilaisen FAS‑potilasjoukon, jolla oli GEP‑ ja bronkiaalisia NET‑kasvaimia, etenemisvapaan elossaolon ja kokonaiselossaolon mediaani sekä kasvaintyypin mukainen jaottelu on esitetty taulukossa 10.
Taulukko 10 Etenemisvapaa elossaolo ja kokonaiselossaolo vaiheen I/II Erasmus‑tutkimuksessa hollantilaisilla potilailla, joilla oli GEP‑ ja bronkiaalisia NET‑kasvaimia – (FAS, N = 360)
PFS Aika (kuukautta) | OS Aika (kuukautta) | ||||||
| Mediaani | 95 %:n lv | Mediaani | 95 %:n lv | ||||
| Kaikki NET-kasvaimet* | 360 | 28,5 | 24,8 | 31,4 | 61,2 | 54,8 | 67,4 |
| Keuhko | 19 | 18,4 | 10,4 | 25,5 | 50,6 | 31,3 | 85,4 |
| Haima | 133 | 30,3 | 24,3 | 36,3 | 66,4 | 57,2 | 80,9 |
| Etusuoli** | 12 | 43,9 | 10,9 | ND | NR | 21,3 | ND |
| Keskisuoli | 183 | 28,5 | 23,9 | 33,3 | 54,9 | 47,5 | 63,2 |
| Takasuoli | 13 | 29,4 | 18,9 | 35,0 | NR | ND | ND |
PFS = Etenemisvapaa elossaolo; OS = Kokonaiselossaolo; ND = Ei havaittu (not detected); NR = Ei saavutettu (not reached)
* Sisältää etusuolen, keskisuolen ja takasuolen; ** Etusuolen muut NET‑kasvaimet kuin bronkiaaliset ja haiman kasvaimet
Vaiheen I/II Erasmus‑tutkimuksessa 188 potilasta (52 %) sai ja 172 (48 %) potilasta ei saanut samanaikaisesti oktreotidi LAR ‑valmistetta Lutathera‑hoidon aikana. Etenemisvapaassa elossaolossa ei havaittu tilastollisesti merkitsevää eroa niiden potilaiden välillä, jotka eivät saaneet oktreotidi LAR ‑valmistetta (25,4 kuukautta [95 %:n lv: 22,8; 30,6]) ja jotka saivat samanaikaisesti oktreotidi LAR ‑hoitoa (30,9 kuukautta [95 %:n lv: 25,6; 34,8]) (p = 0,747).
Pediatriset potilaat
NETTER-P
NETTER-P oli vaiheen II monikeskuksinen, avoin, yksihaarainen tutkimus, jossa arvioitiin Lutatheran turvallisuutta ja dosimetriaa 12– < 18-vuotiailla nuorilla, joilla oli somatostatiinireseptoripositiivinen GEP-NET tai PPGL. Hoito koostui neljästä 8 viikon välein (± 1 viikko) annettavasta 7 400 MBq:n (200 mCi) suuruisesta Lutathera-annoksesta, jotka annettiin yhdessä 2,5 % arginiinia ja 2,5 % lysiiniä sisältävän aminohappoliuoksen kanssa.
Ensisijaiset päätetapahtumat olivat kohde-elimiin absorboituneiden säteilyannosten mittaaminen (ks. kohta 11) sekä haittatapahtumien ja laboratorioarvoissa ilmenevän toksisuuden esiintyvyys ensimmäisen Lutathera-annoksen jälkeen GEP-NET- ja PPGL-potilaiden yhdistetyssä populaatiossa. Kaikki tehoa koskevat päätetapahtumat olivat eksploratiivisia.
Ensisijaisen analyysin ajankohtana (katkaisupäivämäärä 12.3.2024) tutkimukseen osallistui 11 potilasta, joilla oli somatostatiinireseptoripositiivinen kasvain; neljällä oli GEP-NET ja seitsemällä PPGL. Potilaiden mediaani-ikä oli 15 vuotta (vaihteluväli: 13–17 vuotta) ja heistä kuusi oli naisia. Annettujen Lutathera-syklien keskimääräinen lukumäärä oli 3,6 (± 0,9). Yhdeksän potilasta (neljä GEP-NET- ja viisi PPGL-potilasta) oli saanut neljä sykliä Lutatheraa, yksi PPGL-potilas kolme sykliä ja yksi PPGL-potilas yhden syklin. NETTER-P-tutkimusprotokollan annosmuutoskriteerien mukaisesti kahden PPGL-potilaan annosta pienennettiin 50 % ensimmäisen syklin jälkeen, koska arvioitu kumulatiivinen munuaisiin absorboitunut annos ylitti 29 Gy.
Neljästä GEP-NET-potilaasta kahdella oli G1- ja kahdella G2-kasvain. Kaikilla GEP-NET-potilailla oli metastaattinen tauti. Kaikki neljä GEP-NET-potilasta oli leikattu ja he olivat aiemmin saaneet vähintään yhden linjan antineoplastista hoitoa. Kaikilla PPGL-potilailla oli metastaattinen tauti, mukaan lukien viidellä potilaalla, joilla oli lisämunuaisen ulkopuolinen paragangliooma ja kahdella potilaalla, joilla oli lisämunuaisen feokromosytooma. Kahdelle näistä potilaista oli aiemmin tehty toispuolinen nefrektomia. Kuusi PPGL-potilasta oli saanut aiemmin antineoplastista hoitoa.
Ensisijaisen analyysin ajankohtana (katkaisupäivämäärä 12.3.2024) oli käytettävissä yhdeksän potilaan (kolme GEP-NET- ja kuusi PPGL-potilasta) kokonaisvastetiedot. Näiden potilaiden saavuttama paras kokonaisvaste (BOR, best overall response) oli vakaa tautitilanne. Yhdellä kolmesta GEP-NET-potilaasta tauti eteni 3 kuukauden päästä viimeisestä annoksesta, ja hänen osallistumisensa tutkimukseen keskeytettiin. Neljännen GEP-NET-potilaan kasvainta ei voitu arvioida CT/MRI-kuvauksella. Yksi PPGL-potilas keskeytti hoidon lääkärin päätöksen perusteella ensimmäisen hoitojakson jälkeen ja jäi tutkimukseen pitkäaikaisseurantaa varten. Kymmenen yhdestätoista potilaasta jatkoi tutkimuksessa ja oli elossa ilman kasvaimen etenemistä.
Lutatheran yleinen turvallisuusprofiili NETTER-P-tutkimuksen nuorilla GEP-NET- tai PPGL-potilailla oli yhdenmukainen aikuisilla GEP-NET-potilailla raportoidun turvallisuusprofiilin kanssa. NETTER-P-tutkimuksesta ei kuitenkaan ole tällä hetkellä saatavilla nuoria potilaita koskevia pitkäaikaisia turvallisuustietoja.
Lutathera-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Lutathera‑valmisteen käytöstä GEP‑NET‑kasvainten hoidossa pediatrisissa potilasryhmissä syntymästä < 12 vuoden ikäisiin saakka (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Lääkevalmiste annetaan laskimoon ja se on välittömästi ja täysin biosaatava.
Jakautuminen
Ihmisen plasmalla tehty tutkimus ei‑radioaktiivisen yhdisteen (lutetium(175Lu)oksodotreotidi) plasman proteiineihin sitoutumisen määrän määrittämiseksi osoitti, että noin 50 % yhdisteestä sitoutuu plasman proteiineihin.
Lutetium‑177:n transkelaatiota lutetium(175Lu)oksodotreotidista seerumin proteiineihin ei ole havaittu.
Elimiin otto
4 tunnin kuluessa annosta lutetium(177Lu)oksodotreotidin jakautumisessa on havaittavissa nopea otto munuaisiin, kasvainleesioihin, maksaan ja pernaan, sekä joillakin potilailla aivolisäkkeeseen ja kilpirauhaseen. Samanaikainen aminohappoliuoksen anto vähentää munuaisiin ottoa, tehostaen radioaktiivisuuden eliminaatiota (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Biojakautumistutkimukset osoittavat, että lutetium(177Lu)oksodotreotidi puhdistuu nopeasti verestä.
Biotransformaatio
Vaiheen III NETTER‑1‑tutkimuksen dosimetria‑, farmakokinetiikka‑ ja EKG‑osatutkimukseen osallistuneiden 20 potilaan virtsanäytteiden analyysien perusteella lutetium(177Lu)oksodotreotidi metaboloituu vain niukasti ja erittyy pääasiassa muuttumattomana yhdisteenä munuaisten kautta.
Korkean suorituskyvyn nestekromatografia (HPLC) ‑analyysit, jotka tehtiin 48 tuntia infuusion jälkeen otetuista virtsanäytteistä, osoittivat muuttumattoman lutetium(177Lu)oksodotreotidin osuuden olevan lähes 100 % useimmissa analysoiduissa näytteissä (matalin arvo oli yli 92 %). Tämä merkitsee sitä, että yhdiste eliminoituu virtsaan pääasiassa muuttumattomana yhdisteenä.
Tämä näyttö vahvistaa vaiheen I/II Erasmus‑tutkimuksessa tehdyt havainnot. Kyseisessä tutkimuksessa 1 tunti lutetium(177Lu)oksodotreotidin annon jälkeen otettiin virtsanäyte potilaalta, joka sai 1,85 MBq lutetium(177Lu)oksodotreotidia. Virtsanäytteen HPLC-analyysi osoitti, että pääosa (91 %) erittyi muuttumattomana.
Näitä löydöksiä tukevat ihmisen maksasoluilla in vitro tehdyt metaboliatutkimukset, joissa ei havaittu lutetium(175Lu)oksodotreotidin metabolista hajoamista.
Eliminaatio
Vaiheen I/II Erasmus‑tutkimuksessa ja vaiheen III NETTER‑1‑tutkimuksessa kerättyjen tietojen perusteella lutetium(177Lu)oksodotreotidi eliminoituu pääasiassa munuaiserittymisen kautta: noin 60 % lääkevalmisteesta eliminoituu virtsaan 24 tunnin sisällä ja noin 65 % 48 tunnin sisällä annon jälkeen.
Iäkkäät potilaat
Farmakokineettista profiilia iäkkäillä potilailla (≥ 75‑vuotiailla) ei ole tutkittu. Tietoja ei ole saatavilla.
Pediatriset potilaat (12– < 18-vuotiaat)
Kerätyt farmakokineettiset tiedot koskivat 11:a vähintään 12-vuotiasta nuorta, joilla oli somatostatiinireseptoripositiivinen GEP-NET tai PPGL. Nuoret osallistuivat NETTER-P-tutkimukseen ja heille käytettiin aikuisten annostusta. Farmakokineettiset tiedot olivat aikuisilta mitattujen arvojen vaihteluvälin sisällä: keskimääräinen AUCinf 35,8 ng.h/ml (CV 12,5 %), keskimääräinen puhdistuma (CL, clearance) 6,0 l/h (CV 11,5 %) ja Lutathera-infuusion lopussa saavutettu keskimääräinen Cmax 10,3 ng/ml (CV 5,2 %).
Yhteisvaikutusten mahdollisuuden arviointi in vitro
Metaboliset ja kuljettajaproteiineihin liittyvät yhteisvaikutukset
Prekliinisissä tutkimuksissa ei havaittu ihmisen CYP450‑entsyymien inhibitiota eikä merkittävää induktiota eikä spesifisiä yhteisvaikutuksia P‑glykoproteiinin (ulosvirtauskuljettajaproteiini) eikä OAT1-, OAT3-, OCT1-, OCT2-, OATP1B1-, OATP1B3- eikä BCRP‑kuljettajaproteiinien kanssa. Tämä viittaa siihen, että Lutathera-valmisteella on vähäinen todennäköisyys aiheuttaa merkittäviä metabolismi- tai kuljettajaproteiinivälitteisiä yhteisvaikutuksia.
Prekliiniset tiedot turvallisuudesta
Toksikologiset rotilla tehdyt eläinkokeet osoittivat, että yksi enintään 4 550 MBq/kg:n laskimoinjektio oli hyvin siedetty eikä kuolemia havaittu. Tutkittaessa ”kylmää” yhdistettä (ei‑radioaktiivista lutetium(175Lu)oksodotreotidia) yhtenä laskimoinjektiona rotilla ja koirilla enintään 20 000 µg/kg:n annoksilla (rotilla) ja 3 200 µg/kg:n annoksilla (koirilla), ”kylmä” yhdiste (ei‑radioaktiivinen lutetium(175Lu)oksodotreotidi) oli hyvin siedetty molemmilla lajeilla eikä kuolemia havaittu. Toksisuutta ei havaittu toistetun annon tutkimuksessa (”kylmää” yhdistettä neljä kahden viikon välein toistuvaa antoa) annoksilla 1 250 µg/kg rotilla ja 80 µg/kg koirilla. Tämä lääkevalmiste ei ole tarkoitettu säännölliseen tai jatkuvaan antoon.
Mutageenisuustutkimuksia ja pitkäkestoisia karsinogeenisuustutkimuksia ei ole toteutettu.
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta ja genotoksisuutta koskevien konventionaalisten tutkimusten tulokset liittyen ”kylmään” yhdisteeseen (ei‑radioaktiivinen lutetium(175Lu)oksodotreotidi) eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
Etikkahappo
Natriumasetaatti
Gentisiinihappo
Askorbiinihappo
Dietyleeni‑triamiini‑pentaetikkahappo
Natriumkloridi
Natriumhydroksidi
Injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa 12.
Kestoaika
72 tuntia kalibroinnin päivämäärästä ja kellonajasta
Säilytys
Säilytä alle 25 °C.
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa ionisoivalta säteilyltä suojaamiseksi (lyijysuoja).
Radiofarmaseuttisten valmisteiden säilytyksessä on noudatettava radioaktiivisia materiaaleja koskevia kansallisia määräyksiä.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LUTATHERA infuusioneste, liuos
370 MBq/ml (L:ei) 1 kpl (20,5 - 25 ml) (24015,60 €)
PF-selosteen tieto
Kirkas, väritön, tyypin I lasista valmistettu infuusiopullo, joka on suljettu bromobutyylikumitulpalla ja alumiinisinetillä.
Yksi infuusiopullo sisältää tilavuuden, joka vaihtelee välillä 20,5–25,0 ml liuosta. Tämä vastaa 7 400 MBq:n aktiivisuutta infuusion päivämäärän ja kellonajan kohdalla.
Infuusiopullo on lyijysäiliössä suojausta varten.
Valmisteen kuvaus:
Kirkas, väritön tai hieman keltainen liuos.
Käyttö- ja käsittelyohjeet
Vain kertakäyttöön.
Yleinen varoitus
Radiofarmaseuttisia valmisteita saavat ottaa vastaan, käyttää ja antaa vain siihen valtuutetut henkilöt siihen tarkoitetussa hoitoympäristössä. Niiden vastaanoton, säilytyksen, käytön, siirtämisen ja hävittämisen tulee tapahtua määräysten ja/tai toimivaltaisen viranomaisen myöntämien asianmukaisten lupien mukaisesti.
Radiofarmaseuttisten valmisteiden valmistelussa on huomioitava sekä säteilyturvallisuus että farmaseuttiset laatuvaatimukset. Asianmukaisia aseptisia varotoimia on noudatettava.
Ks. kohdasta 12 ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen antoa.
Jos lyijysäiliö tai infuusiopullo vahingoittuu milloin tahansa tämän lääkevalmisteen käyttövalmiiksi saattamisen aikana, sitä ei pidä käyttää.
Antotoimenpiteet on toteutettava siten, että minimoidaan lääkevalmisteen kontaminaation ja käyttäjien säteilytyksen riski. Riittävä suojautuminen on pakollista.
Lääkevalmistetta käsiteltäessä on käytettävä vesitiiviitä käsineitä ja noudatettava asianmukaisia aseptisia tekniikoita.
Radioaktiivisten lääkevalmisteiden anto aiheuttaa riskejä muille henkilöille ulkoisen säteilyn tai virtsan, oksennuksen tms. roiskeesta johtuvan kontaminaation muodossa. Säteilysuojauksen varotoimet on sen vuoksi toteutettava kansallisten säännösten mukaisesti.
Tämä valmiste aiheuttaa todennäköisesti suhteellisen korkean säteilyannoksen useimmille potilaille. 7 400 MBq:n anto voi aiheuttaa merkittävän ympäristöhaitan.
Tämä voi olla huolenaiheena potilaan kanssa samassa taloudessa asuvien henkilöiden tai yleisväestön osalta annetun aktiivisuuden tasosta riippuen. Siksi on noudatettava säteilyltä suojautumista koskevia sääntöjä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Sopivat kansallisten säännösten mukaiset varotoimet on toteutettava potilaiden eliminoimaa aktiivisuutta koskien, kontaminaatioiden välttämiseksi.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Lutetium-177:ä Lutathera-valmistetta varten voidaan valmistaa käyttämällä kahta eri vakaan nuklidin lähdettä (lutetium-176 tai ytterbium-176), mikä vaikuttaa jätteenkäsittelytoimiin. Asianmukaisen jätteenkäsittelyn varmistamiseksi käyttäjän on ennen Lutathera-valmisteen käyttöä tutustuttava mukana toimitettavaan dokumentaatioon.
Korvattavuus
LUTATHERA infuusioneste, liuos
370 MBq/ml 1 kpl
- Ei korvausta.
ATC-koodi
V10XX04
Valmisteyhteenvedon muuttamispäivämäärä
15.07.2026
Dosimetria
Seuraavat Lutathera‑hoitoa koskevat johtopäätökset määriteltiin kliinisissä tutkimuksissa tehdyistä säteilydosimetria‑arvioinneista:
Kriittinen elin on luuydin. Suositellulla Lutathera‑valmisteen kumulatiivisella annoksella 29 600 MBq (neljä 7 400 MBq:n antokertaa) ei kuitenkaan ole havaittu korrelaatiota hematologisen toksisuuden ja annetun säteilyn kokonaismäärän tai luuytimeen absorboituneen annoksen välillä vaiheen I/II Erasmus‑tutkimuksessa eikä vaiheen III NETTER‑1‑tutkimuksessa.
Munuainen ei ole kriittinen elin, jos samanaikaisesti annetaan asianmukaista aminohappoliuosta (ks. kohta Annostus ja antotapa).
Vaiheen III NETTER‑1‑tutkimuksen osatutkimuksen ja vaiheen I/II Erasmus‑tutkimuksen dosimetrisen analyysin tulokset ovat kokonaisuutena samansuuntaisia. Ne osoittavat Lutathera‑valmisteen annostuksen (neljä 7 400 MBq:n antokertaa) olevan turvallista.
Taulukko 11 Lutetium(177Lu)oksodotreotidin arvioidut absorboituneet annokset vaiheen III NETTER‑1‑tutkimuksesta (Olinda output)
| Elin | Elimeen absorboitunut annos aktiivisuusyksikköä kohti (mGy/MBq) (n = 20) | |
| Keskiarvo | Keskihajonta | |
| Lisämunuaiset | 0,037 | 0,016 |
| Aivot | 0,027 | 0,016 |
| Rinnat | 0,027 | 0,015 |
| Sappirakon seinämä | 0,042 | 0,019 |
| Alempi paksusuolen seinämä | 0,029 | 0,016 |
| Ohutsuoli | 0,031 | 0,015 |
| Mahan seinämä | 0,032 | 0,015 |
| Ylempi paksusuolen seinämä | 0,032 | 0,015 |
| Sydämen seinämä | 0,032 | 0,015 |
| Munuaiset | 0,654 | 0,295 |
| Maksa* | 0,299 | 0,226 |
| Keuhkot | 0,031 | 0,015 |
| Lihas | 0,029 | 0,015 |
| Munasarjat*** | 0,031 | 0,013 |
| Haima | 0,038 | 0,016 |
| Punainen luuydin | 0,035 | 0,029 |
| Osteogeeniset solut | 0,151 | 0,268 |
| Iho | 0,027 | 0,015 |
| Perna | 0,846 | 0,804 |
| Kivekset** | 0,026 | 0,018 |
| Kateenkorva | 0,028 | 0,015 |
| Kilpirauhanen | 0,027 | 0,016 |
| Virtsarakon seinämä | 0,437 | 0,176 |
| Kohtu*** | 0,032 | 0,013 |
| Koko keho | 0,052 | 0,027 |
*n = 18 (kaksi potilasta suljettiin pois, koska otto maksan etäpesäkkeisiin vaikutti maksaan absorboituneeseen annokseen)
**n = 11 (vain miespotilaita)
***n = 9 (vain naispotilaita)
Sairauden kulun aiheuttamat patofysiologiset muutokset voivat vaikuttaa merkittävästi niiden eri elinten säteilyannokseen, jotka eivät ole hoidon kohde‑elimiä. Tämä on otettava huomioon seuraavia tietoja käytettäessä.
Pediatriset potilaat
Lutetium(177Lu)oksodotreotidin dosimetriaa nuorilla on tutkittu neljällä GEP-NET- ja kuudella PPGL-potilaalla (ikähaarukka: 12– < 18 vuotta), jotka osallistuivat vaiheen II NETTER-P-tutkimukseen. Dosimetrianäytteitä kerättiin lutetium(177Lu)oksodotreotidin biodistribuutioprofiilin määrittelemiseksi ja koko kehon ja elinten säteilyannosten laskemiseksi, kiinnittäen erityistä huomiota kriittisiin elimiin (esim. munuaisiin ja luuytimeen) absorboituneeseen säteilyannokseen.
Taulukossa 12 esitetään NETTER-P-tutkimukseen osallistuneiden nuorten arvioitujen absorboituneiden säteilyannosten keskiarvo ja keskihajonta.
Taulukko 12 Lutetium(177Lu)oksodotreotidin arvioidut absorboituneet annokset vähintään 12-vuotiailla lapsipotilailla (n = 10) vaiheen II NETTER-P-tutkimuksessa
| Elin | Elimeen absorboitunut annos aktiivisuusyksikköä kohti (mGy/MBq) | Laskennallinen absorboitunut annos, mikä saadaan 4 x 7,4 GBq:sta (29,6 GBq:n kumulatiivinen aktiivisuus) (Gy) | Teoreettinen kumulatiivinen absorboitunut annos (Gy) | ||
| Keskiarvo | Keskihajonta | Keskiarvo | Keski-hajonta | Min-Max | |
| Lisämunuaiset | 0,045 | 0,011 | 1,3 | 0,3 | 0,64–1,7 |
| Aivot | 0,020 | 0,006 | 0,6 | 0,2 | 0,38–0,86 |
| Rinnata | 0,018 | 0,005 | 0,5 | 0,2 | 0,37‑0,75 |
| Ruokatorvi | 0,023 | 0,006 | 0,7 | 0,2 | 0,40–0,93 |
| Silmät | 0,020 | 0,006 | 0,6 | 0,2 | 0,38–0,86 |
| Sappirakon seinämä | 0,030 | 0,010 | 0,9 | 0,3 | 0,48–1,5 |
| Sydämen seinämä | 0,023 | 0,006 | 0,7 | 0,2 | 0,40–0,92 |
| Munuaiset | 0,778 | 0,280 | 23,0 | 8,3 | 14–40 |
| Vasemmanpuoleinen paksusuoli | 0,273 | 0,074 | 8,1 | 2,2 | 4,6–12 |
| Maksa | 0,210 | 0,205 | 6,2 | 6,1 | 2,4–23 |
| Keuhkot | 0,023 | 0,006 | 0,7 | 0,2 | 0,40–0,91 |
| Osteogeeniset solut | 0,045 | 0,017 | 1,3 | 0,5 | 0,64–2,1 |
| Munasarjatb | 0,026 | 0,007 | 0,8 | 0,2 | 0,49–1,0 |
| Haima | 0,027 | 0,006 | 0,8 | 0,2 | 0,46–1,1 |
| Aivolisäkec | 1,114 | 0,425 | 33,0 | 12,6 | 18–56 |
| Eturauhanend | 0,025 | 0,006 | 0,7 | 0,2 | 0,62–0,98 |
| Peräsuoli | 0,277 | 0,076 | 8,2 | 2,2 | 4,8–12 |
| Punainen luuydin (veri)e | 0,026 | 0,005 | 0,8 | 0,1 | 0,55–1,0 |
| Punainen luuydin (kuvantaminen)e | 0,057 | 0,027 | 1,7 | 0,8 | 0,70–2,8 |
| Oikeanpuoleinen paksusuoli | 0,156 | 0,041 | 4,6 | 1,2 | 2,7–6,5 |
| Sylkirauhaset | 0,033 | 0,017 | 1,0 | 0,5 | 0,38–1,7 |
| Ohutsuoli | 0,045 | 0,011 | 1,3 | 0,3 | 0,85–1,9 |
| Perna | 0,742 | 0,275 | 22,0 | 8,1 | 8,3–34 |
| Mahan seinämä | 0,026 | 0,006 | 0,8 | 0,2 | 0,44–1,0 |
| Kiveksetd | 0,020 | 0,005 | 0,6 | 0,1 | 0,50–0,81 |
| Kateenkorva | 0,022 | 0,006 | 0,6 | 0,2 | 0,39–0,88 |
| Kilpirauhanen | 0,027 | 0,017 | 0,8 | 0,5 | 0,38–2,2 |
| Virtsarakon seinämä | 0,552 | 0,089 | 16,4 | 2,6 | 14–20 |
| Kohtub | 0,030 | 0,008 | 0,9 | 0,2 | 0,59–1,2 |
| Koko keho | 0,040 | 0,010 | 1,2 | 0,3 | 0,66–1,6 |
a n = 5 (vain naispotilaita).
b n = 6 (vain naispotilaita).
c n = 9 (3 GEP-NET- ja 6 PPGL-potilasta). Aivolisäkkeen dosimetria-arviot tehtiin vain silloin, kun aivolisäkkeeseen otto oli selvästi havaittavissa tasokuvissa. Arviot voivat olla hyvin epävarmoja, sillä aivolisäkkeen koko on pieni, kvantifiointi on mahdollista vain tasokuvista ja nenän limakalvon aktiivisuus aiheuttaa häiriöitä.
d n = 4 (vain miespotilaita).
e Punaista luuydintä koskevat dosimetria-arviot määritettiin joko veren radioaktiivisuudesta tai kuvantamalla ja skaalaamalla edustava alue lannerangasta.
Radiofarmaseuttisten valmisteiden valmistusohjeet
Ennen Lutathera-valmisteen käyttöä käyttäjän on tutustuttava mukana toimitettavaan dokumentaatioon asianmukaisen jätteenkäsittelyn varmistamiseksi (ks. kohta Käyttö- ja käsittelyohjeet).
Valmisteluohjeet
- Käytä Lutathera-liuoksen annon aikana aseptista tekniikkaa ja säteilysuojausta. Käytä infuusiopullon käsittelyyn pihtejä säteilyaltistuksen minimoimiseksi.
- Ennen antoa tarkasta valmiste suojaavan lasin takaa silmämääräisesti hiukkasten ja värimuutosten varalta. Hävitä infuusiopullo, jos havaitset hiukkasia ja/tai värimuutoksia.
- Tarkasta, ettei pakkaus ole vaurioitunut. Määritä kalibroidulla säteilymittarilla, onko radioaktiivista kontaminaatiota tapahtunut. Älä käytä valmistetta, jos infuusiopullo tai lyijysäiliö on vaurioitunut.
- Älä injisoi Lutathera-liuosta suoraan mihinkään toiseen laskimonsisäisesti annettavaan liuokseen.
- Määritä potilaan Lutathera-valmisteesta saaman radioaktiivisuuden määrä toteuttamalla mittaus kalibroidulla radioaktiivisuuden mittausjärjestelmällä ennen kutakin Lutathera-valmisteen antoa ja kunkin annon jälkeen. Varmista, että saadun radioaktiivisuuden määrä vastaa suunniteltua määrää.
- Älä anna Lutathera-valmistetta boluksena laskimoon.
- Aloita potilaasta tulevan radioaktiivisen säteilyn seuraaminen kalibroidulla säteilymittarilla pian infuusion alkamisen jälkeen. Säteilymäärän seurannalla varmistetaan, että potilas saa annoksen. Infuusion aikana potilaasta tulevan radioaktiivisen säteilyn pitäisi lisääntyä tasaisesti ja Lutathera-infuusiopullosta tulevan säteilyn pitäisi vähentyä.
- Infuusion aikana on suositeltavaa seurata potilaan elintoimintoja huolellisesti.
Antotavat laskimoon
Ohjeet antoon painovoimamenetelmällä (letkunpuristimen tai infuusiopumpun avulla)
1. Työnnä Lutathera‑infuusiopulloon 2,5 cm, 20 G neula (lyhyt neula) ja yhdistä infuusiopullo kanyylin kautta steriiliin 500 ml:n 0,9 % NaCl‑infuusionesteeseen (käytetään Lutathera‑liuoksen infusoimiseen). Varmista, ettei lyhyt neula kosketa infuusiopullossa olevaa Lutathera‑liuosta. Älä yhdistä lyhyttä neulaa suoraan potilaaseen. Älä päästä NaCl‑infuusionestettä virtaamaan Lutathera‑infuusiopulloon ennen Lutathera‑infuusion aloitusta. Älä myöskään injisoi Lutathera‑liuosta suoraan NaCl‑infuusionesteeseen.
2. Työnnä Lutathera‑infuusiopulloon toinen, 9 cm, 18 G neula (pitkä neula). Varmista, että pitkä neula on tiiviisti kiinni Lutathera‑infuusiopullon pohjassa koko infuusion ajan. Yhdistä pitkä neula potilaan laskimokanyyliin, joka on esitäytetty steriilillä 0,9 % NaCl‑infuusionesteellä ja jota käytetään Lutathera‑infuusion antoon potilaalle.
3. Käytä letkunpuristinta tai infuusiopumppua säätelemään NaCl‑infuusionesteen virtausta lyhyen neulan kautta Lutathera‑infuusiopulloon. NaCl‑infuusioneste virtaa infuusiopulloon lyhyen neulan kautta ja kuljettaa Lutathera‑liuoksen infuusiopullosta potilaalle laskimokanyyliin yhdistetyn pitkän neulan kautta; infuusion kokonaiskesto on 30 ± 10 minuuttia infuusionopeuden ollessa enintään 400 ml/h. Infuusion alussa käytetään ensimmäisten 5–10 minuutin ajan hitaampaa infuusionopeutta, < 100 ml/h, minkä jälkeen infuusionopeutta suurennetaan potilaan laskimostatuksesta riippuen. Infuusiopullon sisäinen paine on pidettävä tasaisena koko infuusion ajan.
4. Varmista, että Lutathera‑infuusiopullon sisällä olevan liuoksen määrä pysyy tasaisena infuusion aikana. Jos käytössä on läpinäkyvä suojasäiliö, määrä tarkastetaan toistuvasti silmämääräisellä tarkastuksella, tai jos käytössä on lyijyinen kuljetussäiliö, käyttämällä pihtejä infuusiopullon käsittelyyn.
5. Seuraa Lutathera‑valmisteen virtausta infuusiopullosta potilaalle koko infuusion ajan.
6. Kun radioaktiivisuuden taso on pysynyt vakaana vähintään viiden minuutin ajan, irrota pitkään neulaan yhdistetty letku infuusiopullosta ja sulje NaCl‑infuusionesteen letku puristimella.
7. Huuhtele infuusion jälkeen laskimokanyyli antamalla sen kautta potilaalle 25 ml steriiliä 0,9 % NaCl‑infuusionestettä.
Ohjeet antoon peristalttisen infuusiopumpun avulla
1. Työnnä Lutathera‑infuusiopulloon 2,5 cm, 20 G suodatinneula (lyhyt ilmausneula). Varmista, ettei lyhyt neula kosketa infuusiopullossa olevaa Lutathera‑liuosta. Älä yhdistä lyhyttä neulaa suoraan potilaaseen äläkä peristalttiseen infuusiopumppuun.
2. Työnnä Lutathera‑infuusiopulloon toinen, 9 cm, 18 G neula (pitkä neula). Varmista, että pitkä neula on tiiviisti kiinni Lutathera‑infuusiopullon pohjassa koko infuusion ajan. Yhdistä pitkä neula ja steriili 0,9 % NaCl‑infuusioneste asianmukaisilla letkuilla kolmitiehanaan.
3. Yhdistä kolmitiehanan porttiin peristalttisen infuusiopumpun sisäänmenopuoleen liitetty letku pumpun valmistajan ohjeiden mukaisesti.
4. Esitäytä letku avaamalla kolmitiehana ja pumppaamalla Lutathera‑liuosta letkuun, kunnes se saavuttaa kolmitiehanan ulosmenoaukon.
5. Esitäytä potilaaseen yhdistettävä laskimokanyylijärjestelmä avaamalla kolmitiehana steriilin 0,9 % NaCl‑infuusionesteen suuntaan ja pumppaamalla järjestelmään steriiliä 0,9 % NaCl‑infuusionestettä, kunnes sitä poistuu järjestelmän ulosmenoaukosta.
6. Yhdistä esitäytetty laskimokanyylijärjestelmä potilaaseen ja käännä kolmitiehana auki peristalttisen infuusiopumpun Lutathera‑liuoksen suuntaan.
7. Infusoi tarvittavan radioaktiivisuuden mukainen tilavuus Lutathera‑liuosta 30 ± 10 minuutin aikana.
8. Kun tarvittava radioaktiivisuusannos on annettu, pysäytä peristalttinen infuusiopumppu ja käännä kolmitiehanaa niin, että se on auki peristalttisen infuusiopumpun steriilin 0,9 % NaCl‑infuusionesteen suuntaan. Käynnistä peristalttinen infuusiopumppu uudelleen ja huuhtele laskimokanyyli antamalla sen kautta potilaalle 25 ml steriiliä 0,9 % NaCl‑infuusionestettä.
Ohjeet antoon ruiskupumpun avulla
1. Vedä tarvittavan radioaktiivisuuden mukainen tilavuus Lutathera‑liuosta kertakäyttöruiskuun, jossa on suojus ja johon on liitetty steriili 9 cm, 18 G kertakäyttöneula (pitkä neula). Liuoksen vetämistä ruiskuun voidaan helpottaa 2,5 cm, 20 G suodatinneulalla (lyhyt ilmausneula), joka vähentää infuusiopullon paineen aiheuttamaa vastusta. Varmista, ettei lyhyt neula kosketa infuusiopullossa olevaa Lutathera‑liuosta.
2. Aseta ruisku säteilysuojattuun ruiskupumppuun. Liitä kolmitiehana ruiskun ja Lutathera‑infuusion antoon käytettävän steriilillä 0,9 % NaCl‑infuusionesteellä esitäytetyn laskimokanyylin väliin.
3. Infusoi tarvittavan radioaktiivisuuden mukainen tilavuus Lutathera‑liuosta 30 ± 10 minuutin aikana.
4. Kun tarvittava radioaktiivisuusannos on annettu, pysäytä ruiskupumppu ja käännä kolmitiehanaa huuhdellaksesi ruiskun 25 ml:lla steriiliä 0,9 % NaCl‑infuusionestettä. Käynnistä ruiskupumppu uudelleen.
5. Kun ruisku on huuhdeltu, huuhtele laskimokanyyli antamalla sen kautta potilaalle 25 ml steriiliä 0,9 % NaCl‑infuusionestettä.
Kuva 5 Antotavat tiivistettynä
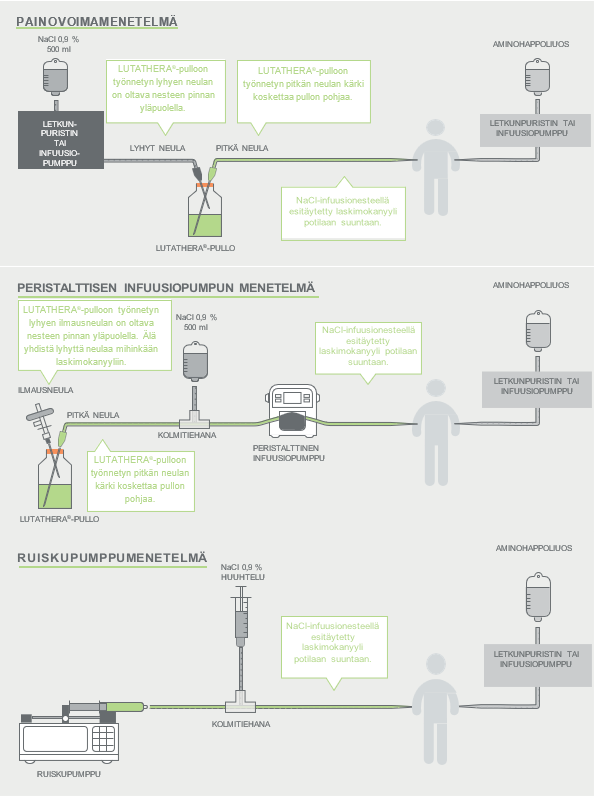
Lisätietoa tästä lääkevalmisteesta on Euroopan lääkeviraston verkkosivulla https://www.ema.europa.eu.
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com