
ENJAYMO infuusioneste, liuos 50 mg/ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Lääkäri
Potilas
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi millilitra infuusionestettä sisältää 50 mg sutimlimabia*.
Yksi injektiopullo sisältää 1 100 mg sutimlimabia 22 ml:ssa.
* Sutimlimabi on immunoglobuliini G4:n (IgG4:n) monoklonaalinen vasta-aine, joka on tuotettu kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-tekniikalla.
Apuaine, jonka vaikutus tunnetaan
Yksi millilitra infuusionestettä sisältää 3,5 mg natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusioneste, liuos (infuusio)
Kliiniset tiedot
Käyttöaiheet
Enjaymo on tarkoitettu hemolyyttisen anemian hoitoon aikuispotilaille, joilla on kylmäagglutiniinisairaus (CAD, cold agglutinin disease).
Ehto
Valmistetta saa antaa vain terveydenhoitoalan ammattilainen hyväksytyn käyttöaiheen hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Enjaymo-valmisteen antaa terveydenhuollon ammattilainen veritauteja sairastavien potilaiden hoitoon perehtyneen lääkärin valvonnassa.
Annostus
Potilaat on rokotettava viimeisten voimassa olevien paikallisten pitkäkestoista komplementtipuutosta sairastaville potilaille annettujen suositusten mukaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Valmisteen suositeltu annos perustuu painoon. Suositeltu annos potilaille, jotka painavat 39 kg – alle 75 kg, on 6 500 mg, ja suositeltu annos potilaille, jotka painavat vähintään 75 kg, on 7 500 mg. Enjaymo annetaan laskimoon kerran viikossa ensimmäisten kahden viikon ajan ja tämän jälkeen kahden viikon välein. Enjaymo on annettava annostusohjelmassa suositeltuina ajankohtina tai kahden vuorokauden sisällä näistä ajankohdista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Enjaymo on tarkoitettu vain jatkuvaan käyttöön pitkäaikaishoitona, ellei Enjaymo-hoidon lopettaminen ole kliinisesti aiheellista.
Annoksen jääminen väliin
Jos annos jää väliin, se on annettava mahdollisimman pian. Jos viimeisen annoksen antamisesta on kulunut yli 17 päivää, hoito on aloitettava uudelleen siten, että valmiste annetaan laskimoon kerran viikossa ensimmäisten kahden viikon ajan ja tämän jälkeen kahden viikon välein.
Erityisryhmät
Iäkkäät
Annoksen muuttaminen ei ole tarpeen vähintään 65-vuotiailla potilailla, joilla on kylmäagglutiniinisairaus (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen maksan vajaatoimintaa sairastavilla potilailla.
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen munuaisten vajaatoimintaa sairastavilla potilailla.
Pediatriset potilaat
Enjaymo-valmistetta ei ole tarkoitettu kätettäväksi alle 18-vuotiaille lapsille kylmäagglutiniinisairauden hoitoon.
Antotapa
Enjaymo on tarkoitettu annettavaksi vain infuusiona laskimoon. Ei saa antaa laskimoinjektiona tai -boluksena. Ks. kohdasta Käyttö- ja käsittelyohjeet valmistelu ja anto-ohjeet.
Valmistelun jälkeen Enjaymo-infuusioliuos annetaan laskimoon taulukossa 1 esitetyllä infuusionopeudella.
Taulukko 1 – Infuusion antonopeus
| Painon vaihteluväli | Annos (mg) | Tarvittava injektiopullojen määrä | Tilavuus (ml) | Infuusion maksiminopeus |
| Vähintään 39 kg – alle 75 kg | 6 500 | 6 | 130 | 130 ml/tunti |
| Vähintään 75 kg | 7 500 | 7 | 150 | 150 ml/tunti |
Potilaille, joilla on kardiopulmonaalinen sairaus, valmiste voidaan antaa 120 minuuttia kestävänä infuusiona.
Jos Enjaymo-valmisteen annon aikana ilmenee jokin haittavaikutus, infuusio voidaan antaa hitaammin tai infuusion anto voidaan lopettaa lääkärin harkinnan mukaisesti. Jos potilaalla ilmenee yliherkkyysreaktio, Enjaymo-infuusio on keskeytettävä ja aloitettava asianmukainen hoito. Potilasta on tarkkailtava vähintään kahden tunnin ajan ensimmäisen infuusion päättymisen jälkeen infuusioreaktion ja/tai yliherkkyysreaktion merkkien ja oireiden varalta. Potilasta on tarkkailtava yhden tunnin ajan seuraavien infuusioiden päättymisen jälkeen infuusioreaktion merkkien ja oireiden varalta.
Koti-infuusio
Koti-infuusiot antaa terveydenhuollon ammattilainen.
Jos infuusion antoa kotona harkitaan, päätöksen on perustuttava potilaan yksilöllisiin kliinisiin ominaisuuksiin ja yksilöllisiin tarpeisiin. Jos infuusioiden antaminen siirretään hoitoyksiköstä potilaan kotiin, on varmistettava, että käytettävissä on hoitavan lääkärin määräysten mukainen asianmukainen infrastruktuuri ja riittävät resurssit. Enjaymo-infuusioiden antamista kotona voidaan harkita potilaille, jotka ovat sietäneet hoitoyksikössä annetun infuusion hyvin ja joilla ei ole ilmennyt infuusioon liittyviä reaktioita. Kun arvioidaan potilaan soveltuvuutta infuusioiden antoon kotona, potilaan perussairaudet ja kyky sitoutua kotona annettaviin infuusioihin liittyviin vaatimuksiin on otettava huomioon. Lisäksi on otettava huomioon seuraavat kriteerit:
- Potilaalla ei saa olla parhaillaan sellaista samanaikaista terveydentilaa, joka lääkärin mielestä saattaa suurentaa potilaalle koituvia riskejä, jos infuusio annetaan kotona hoitoyksikön sijaan. Ennen koti-infuusioiden aloittamista on tehtävä kattava arviointi, jotta varmistetaan potilaan vakaa terveydentila.
- Enjaymo-infuusion annon on täytynyt onnistua hoitoyksikössä (sairaalassa tai avoterveydenhuollossa) vähintään kolmen kuukauden ajan kylmäagglutiniinisairauden hoitoon perehtyneen lääkärin tai muun terveydenhuollon ammattilaisen valvonnassa.
- Potilaan on oltava halukas ja kykenevä noudattamaan kotona annettaviin infuusioihin liittyviä menettelyjä ja hoitavan lääkärin tai muun terveydenhuollon ammattilaisen antamia suosituksia.
- Kotona infuusiota antavan terveydenhuollon ammattilaisen on oltava koko ajan paikalla infuusion aikana ja vähintään 1 tunnin ajan sen jälkeen.
Jos potilaalla ilmenee haittavaikutuksia kotona annettavan infuusion aikana, infuusion anto on keskeytettävä välittömästi, asianmukainen hoito on aloitettava (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) ja asiasta on ilmoitettava hoitavalle lääkärille. Tällaisissa tapauksissa hoitavan lääkärin on päätettävä, annetaanko infuusioita myöhemmin ja jos annetaan, annetaanko ne sairaalassa vai valvotusti avoterveydenhuollossa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Enjaymo-valmisteen vaikutus kohdistuu komplementin klassiseen tiehen; lääkeaine sitoutuu spesifisesti komplementtiproteiinikomponentti 1:n s-osaan (C1s) ja estää komplementtiproteiini C4:n pilkkoutumisen. Vaikka valmiste ei vaikutakaan lektiinitiehen eikä oikotiehen, potilaiden alttius vakaville infektioille saattaa suurentua. Tämä koskee erityisesti kapselillisten bakteerien, kuten Neisseria meningitidis, Streptococcus pneumoniae ja Haemophilus influenzae, aiheuttamia infektioita. Potilaat on rokotettava kapselillisia bakteereja vastaan ennen Enjaymo-hoidon aloittamista, ks. ”Rokotukset” jäljempänä.
Enjaymo-hoitoa saaneilla potilailla on kylmäagglutiniinisairautta arvioineissa kliinisissä tutkimuksissa ilmoitettu vakavia infektioita, myös sepsistä (ks. kohta Haittavaikutukset). Enjaymo-hoitoa ei pidä aloittaa potilaille, joilla on aktiivinen, vakava infektio. Potilaita on tarkkailtava infektioiden varhaisten merkkien ja oireiden varalta, ja heitä on kehotettava hakeutumaan välittömästi lääkärin hoitoon, jos tällaisia oireita ilmenee.
Potilaat, joilla oli virushepatiitti tai HIV, suljettiin pois kliinisistä tutkimuksista. Jos potilaalla todetaan B- tai C-hepatiitti tai HIV-infektio ennen hoitoa tai hoidon aikana, hänen on kerrottava asiasta lääkärille. Varovaisuutta on noudatettava hoidettaessa potilaita, joilla on anamneesissa B- tai C-hepatiitti tai HIV-infektio.
Rokotukset
Potilaat rokotetaan viimeisten voimassa olevien paikallisten pitkäkestoista komplementtipuutosta sairastaville potilaille annettujen suositusten mukaisesti. Lisäksi annetaan meningokokki- ja streptokokkirokotteet. Uusintarokotukset annetaan paikallisten suositusten mukaisesti.
Potilaat, joita ei ole aiemmin rokotettu kapselillisia bakteereja vastaan, rokotetaan vähintään 2 viikkoa ennen ensimmäisen Enjaymo-annoksen antamista. Jos kiireellinen Enjaymo-hoito on tarpeen rokottamattomalle potilaalle, rokote tai rokotteet on annettava mahdollisimman pian. Antibioottiprofylaksin hyötyjä ja riskejä infektioiden ehkäisyssä ei ole varmistettu Enjaymo-hoitoa saavilla potilailla.
Yliherkkyysreaktiot
Kuten muidenkin proteiinivalmisteiden, myös Enjaymo-valmisteen anto saattaa aiheuttaa yliherkkyysreaktioita, myös anafylaksin. Kliinisissä tutkimuksissa Enjaymo-hoitoon ei todettu liittyvän vakavia yliherkkyysreaktioita. Jos potilaalla ilmenee yliherkkyysreaktio, Enjaymo-infuusio on keskeytettävä ja aloitettava asianmukainen hoito.
Infuusioon liittyvät reaktiot
Enjaymo-valmisteen anto saattaa aiheuttaa infuusioon liittyviä reaktioita infuusion aikana tai välittömästi infuusion jälkeen (ks. kohta Haittavaikutukset). Potilasta on tarkkailtava infuusioon liittyvien reaktioiden varalta, ja jos reaktio ilmenee, infuusio on keskeytettävä ja aloitettava asianmukainen hoito.
Systeeminen lupus erythematosus (SLE)
Perinnöllistä komplementin klassisen tien puutosta sairastavilla henkilöillä on suurentunut SLE:n kehittymisen riski. SLE-potilaat suljettiin pois kliinisistä Enjaymo-tutkimuksista. Enjaymo-hoitoa saavia potilaita on tarkkailtava SLE:n merkkien ja oireiden varalta ja arvioitava asianmukaisesti. Enjaymo-valmisteen käytössä on noudatettava varovaisuutta, jos potilaalla on SLE tai hänelle kehittyy SLE:n merkkejä ja oireita.
Kylmäagglutiniinisairauden oireiden seuranta Enjaymo-hoidon lopettamisen jälkeen
Hemolyysiin kohdistuvat vaikutukset heikkenevät hoidon päätyttyä. Näin ollen potilaita on tarkkailtava hemolyysin merkkien ja oireiden varalta, jos hoito lopetetaan. Jos hoidon lopettamisen jälkeen ilmenee hemolyysin merkkejä ja oireita, on tarvittaessa harkittava Enjaymo-hoidon aloittamista uudelleen.
Natrium
Tämä lääkevalmiste sisältää 3,5 mg natriumia per millilitra eli 77 mg natriumia per injektiopullo, mikä vastaa 3,85 %:a WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Enjaymo ei todennäköisesti aiheuta sytokromi P450 välitteisiä yhteisvaikutuksia muiden lääkkeiden kanssa, koska se on rekombinantti ihmisen proteiini. Sutimlimabin yhteisvaikutuksia CYP-entsyymien substraattien kanssa ei ole tutkittu. Sutimlimabi kuitenkin pienentää proinflammatoristen sytokiinien pitoisuuksia potilailla, ja yksi tällaisista sytokiineista on IL‑6, jonka tiedetään vähentävän tiettyjen maksan CYP450-entsyymien (CYP1A2, CYP2C9, CYP2C19 ja CYP3A4) ilmentymistä. Sutimlimabihoitoa aloitettaessa tai lopetettaessa on tämän vuoksi noudatettava varovaisuutta potilailla, jotka saavat myös CYP3A4:n, CYP1A2:n, CYP2C9:n tai CYP2C19:n substraatteja, etenkin jos niiden terapeuttinen indeksi on kapea (kuten varfariinilla, karbamatsepiinilla, fenytoiinilla ja teofylliinillä), ja annoksia on tarvittaessa muutettava.
Raskaus ja imetys
Raskaus
Ei ole saatavilla tietoja sutimlimabin käytöstä raskaana oleville naisille. Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kappale Prekliiniset tiedot turvallisuudesta).
Ihmisen IgG-vasta-aineet läpäisevät istukan, joten sutimlimabi saattaa siirtyä äidistä sikiöön.
Varmuuden vuoksi sutimlimabin käyttöä on suositeltavaa välttää raskauden aikana. Sutimlimabia saa antaa raskauden aikana vain, jos se on selvästi aiheellista.
Imetys
Ihmisen IgG-vasta-aineiden tiedetään erittyvän rintamaitoon muutamien vuorokausien ajan synnytyksen jälkeen, minkä jälkeen niiden pitoisuus pienenee nopeasti. Imetettävään lapseen tällä lyhyellä ajanjaksolla kohdistuvia riskejä ei siis voida sulkea pois. Ei tiedetä, erittyvätkö sutimlimabi tai sen metaboliitit ihmisen rintamaitoon. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko sutimlimabihoito, ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Sutimlimabin vaikutuksia urosten ja naaraiden hedelmällisyyteen ei ole arvioitu eläintutkimuksissa. Toistuvilla annoksilla tehdyissä tutkimuksissa, joissa sutimlimabia annettiin jaavanmakakeille annoksilla, joilla saavutettu altistus vastasi enimmillään noin 4-kertaisesti ihmiselle suositeltua annosta, ei havaittu lisääntymiselimiin kohdistuvia vaikutuksia.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Enjaymo-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kliinisissä CADENZA ja CARDINAL-tutkimuksissa Enjaymo-valmisteen yleisimmin ilmoitetut haittavaikutukset olivat päänsärky, hypertensio, virtsatieinfektio, ylähengitystieinfektio, nasofaryngiitti, pahoinvointi, vatsakipu, infuusioon liittyvät reaktiot ja syanoosi (jota on ilmoitettu akrosyanoosina).
Haittavaikutustaulukko
Enjaymo-valmisteen turvallisuusarviointi potilailla, joilla on kylmäagglutiniinisairaus, perustui ensisijaisesti vaiheen 3 satunnaistettuun, lumekontrolloituun tutkimukseen (CADENZA) ja avoimeen yksihaaraiseen tutkimukseen (CARDINAL) osallistuneista 66 potilaasta saatuihin tietoihin.
Taulukossa 2 on lueteltu CADENZA ja CARDINAL-tutkimuksissa havaitut haittavaikutukset elinjärjestelmän ja esiintymistiheyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000). Lääkkeen haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2 Luettelo CADENZA ja CARDINAL-tutkimuksissa todetuista haittavaikutuksista
MedDRA-elinjärjestelmäluokka | Hyvin yleiset | Yleiset |
Infektiot | Virtsatieinfektio | Alahengitystieinfektiotc |
Yleisoireet ja antopaikassa todettavat haitat | Kuumef | |
Hermosto | Päänsärky | Auraf |
Verisuonisto | Hypertensiod | Hypotensiof* |
Ruoansulatuselimistö | Vatsakipue | Ripulif |
Hengityselimet, rintakehä ja välikarsina | Epämukava tunne rintakehälläf* | |
Iho ja ihonalainen kudos | Kutinaf* |
aYlähengitystieinfektiot: ylähengitystieinfektio, keuhkoputkitulehdus ja virusperäinen ylähengitystieinfektio
bNasofaryngiitti: nasofaryngiitti, nielutulehdus
cAlahengitystieinfektiot: Klebsiella pneumoniaen aiheuttama keuhkokuume, COVID-19-keuhkokuume, alahengitystieinfektio, virusperäinen hengitystieinfektio, hengitystieinfektio, keuhkokuume
dHypertensio: hypertensio, kohonnut verenpaine, essentiaalinen hypertensio, hypertensiivinen kriisi, valkotakkiverenpaine
eVatsakipu: vatsakipu, alavatsakipu, ylävatsakipu, vatsan aristus
fInfuusioon liittyvä reaktio: Kaikki infuusioon liittyvät reaktiot ilmenivät 24 tunnin kuluessa Enjaymo-infuusion aloittamisesta. *Taulukossa ovat mukana yliherkkyysreaktioihin viittaavat tapahtumat.
Vakavat infektiot
CADENZA ja CARDINAL-tutkimuksiin osallistuneista 66 potilaasta vakavia infektioita ilmoitettiin 10 potilaalla (15,2 %:lla). Haittavaikutustaulukossa lueteltuja vakavia infektioita ovat hengitystieinfektio [Klebsiella pneumoniaen aiheuttama keuhkokuume (n = 1), hengitystieinfektio (n = 1) ja COVID‑19-keuhkokuume (n = 1)], virtsatieinfektio [urosepsis (n = 1), virtsatieinfektio (n = 1) ja bakteeriperäinen virtsatieinfektio (n = 1)] ja herpes zoster (n = 1). Yhdellä potilaalla sutimlimabihoito lopetettiin vakavan infektion vuoksi. Kyseessä oli kuolemaan johtanut Klebsiella pneumoniaen aiheuttama keuhkokuume. Muita kuolemaan johtaneita infektiotapahtumia ei ilmoitettu. Ks. kohdasta Varoitukset ja käyttöön liittyvät varotoimet tietoa rokotussuosituksista vakavien infektioiden ehkäisemiseksi ja potilaan tarkkailusta infektioiden varhaisten merkkien ja oireiden varalta.
Immunogeenisuus
Sutimlimabin immunogeenisuutta arvioitiin CARDINAL- ja CADENZA-tutkimuksissa kylmäagglutiniinisairautta sairastavilla potilailla lähtötilanteessa, hoitojakson aikana ja hoidon lopussa (viikko 26). Kahdelle 24:stä CARDINAL-tutkimukseen otetusta potilaasta (8,3 %:lle), jotka olivat saaneet vähintään yhden sutimlimabiannoksen, kehittyi hoidon aikana lääkevasta-aineita. CADENZA-tutkimuksessa kuudelle 42:sta sutimlimabia saaneesta potilaasta (14,3 %:lle) kehittyi hoidon aikana lääkevasta-aineita. Näitä lääkevasta-aineita ilmeni ohimenevästi ja niiden titteri oli pieni, eikä niihin liittynyt muutoksia farmakokineettisessä profiilissa, kliinisessä vasteessa tai haittatapahtumissa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksessa suositellaan infuusion välitöntä keskeyttämistä ja potilaan tarkkaa seurantaa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, komplementin estäjät, ATC-koodi: L04AJ04
Vaikutusmekanismi
Sutimlimabi on immunoglobuliini G:n alaluokka 4:n (IgG4:n) monoklonaalinen vasta-aine, joka estää klassisen tien aktivaatiota; lääkeaine sitoutuu spesifisesti komplementtiproteiinikomponentti 1:n s-osaan (C1s), joka on C4:ää pilkkova seriiniproteaasi. Sutimlimabi ei estä lektiinitien eikä oikotien aktivaatiota. Komplementin klassisen tien esto C1s-alakomponentin tasolla estää komplementtiopsoniinien kertymistä punasolujen pinnalle, mikä estää hemolyysia potilailla, joilla on kylmäagglutiniinisairaus. Lisäksi se estää proinflammatoristen C3a ja C5a-anafylatoksiinien ja alavirran puoleisen terminaalisen C5b-9-komplementtikompleksin muodostumista.
Kliininen teho ja turvallisuus
Potilailla, joilla on kylmäagglutiniinisairaus, klassisen tien aktivaation havaittiin estyvän yli 90-prosenttisesti ensimmäisen Enjaymo-infuusion jälkeen, ja C4-pitoisuudet palautuivat normaaleiksi (0,2 g/l) viikon kuluessa ensimmäisen Enjaymo-annoksen saamisesta.
Enjaymo-valmisteen turvallisuutta ja tehoa kylmäagglutiniinisairautta sairastavien potilaiden hoidossa arvioitiin vaiheen 3 satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa (CADENZA) 42 potilaalla (n = 22 Enjaymo-ryhmässä ja n = 20 lumeryhmässä) sekä vaiheen 3 avoimessa, yksihaaraisessa tutkimuksessa (CARDINAL) 24 potilaalla 26 viikon ajan. Kuuden kuukauden hoitojaksojen (osa A) päätyttyä kummankin tutkimuksen potilaat saivat edelleen Enjaymo-valmistetta jatkovaiheessa (osa B), jossa arvioitiin pitkän aikavälin turvallisuutta ja vasteen kestoa vielä 12 kuukauden ajan (CADENZA) tai 24 kuukauden ajan (CARDINAL) sen jälkeen kun viimeinen potilas oli saanut osan A päätökseen. Molemmissa tutkimuksissa oli 9 viikon seurantajakso viimeisen Enjaymo-annoksen jälkeen. Tärkeimmät soveltuvuuskriteerit olivat lähtötilanteen hemoglobiiniarvo (Hb) ≤ 10 g/dl ja aktiivinen hemolyysi, johon liittyi viitealueen ylärajaa suurempi bilirubiinipitoisuus. Potilaat, joilla oli kylmäagglutiniinioireyhtymä (CAS), suljettiin pois tutkimuksista. CADENZA-tutkimuksen potilailla ei ollut anamneesissa verensiirtoa 6 kuukauden sisällä eikä enempää kuin yksi verensiirto tutkimukseen ottoa edeltäneiden 12 kuukauden aikana, kun taas CARDINAL-tutkimukseen otetuilla potilailla oli anamneesissa vähintään yksi dokumentoitu verensiirto tutkimukseen ottoa edeltäneiden 6 kuukauden aikana. 39 – < 75 kg:n painoisille potilaille annettiin 6 500 mg ja ≥ 75 kg:n painoisille potilaille 7 500 mg Enjaymo-valmistetta laskimoon noin 60 minuutin aikana päivänä 0, päivänä 7 ja sen jälkeen 14 päivän välein. Yhteenveto tutkimuspopulaation tärkeimmistä ominaisuuksista lähtötilanteessa on esitetty alla taulukossa 3.
Taulukko 3 – Kliinisiin tutkimuksiin osallistuneiden potilaiden ominaisuudet lähtötilanteessa
| Parametri | Tilastotieto | CADENZA | CARDINAL | |
|---|---|---|---|---|
Lumelääke N = 20 | Enjaymo N = 22 | Enjaymo N = 24 | ||
| Ikä | Keskiarvo Minimi, maksimi | 68,2 51, 83 | 65,3 46, 88 | 71,3 55, 85 |
Sukupuoli Miehiä | n (%) | 4 (20,0) 16 (80,0) | 5 (22,7) 17 (77,3) | 9 (37,5) 15 (62,5) |
| Paino | Keskiarvo, kg Minimi, maksimi | 64,9 48, 95 | 66,8 39, 100 | 67,8 40, 112 |
| Hemoglobiini | Keskiarvo, g/dl | 9,33 | 9,15 | 8,59 |
| (Kokonais)bilirubiini* | μmol/l | 35,77 (1,75 x ULN) | 41,17 (2 x ULN) | 53,26 (2,6 × ULN†) |
Anamneesissa verensiirto Viimeisten 6 kuukauden aikana Viimeisten 12 kuukauden aikana | Verensiirtojen määrän keskiarvo (vaihteluväli) |
0
0 |
0
0,14 (0, 1) |
3,2 (1, 19)
4,8 (1, 23) |
| FACIT†-F-pistemäärä | Keskiarvo | 32,99 | 31,67 | 32,5 |
*Potilaita, joista saatavilla bilirubiinia koskevat tiedot: N = 21 CARDINAL-tutkimuksessa; N = 18 (lumeryhmä) ja N = 20 (Enjaymo-ryhmä) CADENZA-tutkimuksessa pois lukien potilaat, joilla oli joko positiivinen Gilbertin oireyhtymän testitulos tai ei testitulosta saatavilla.
†ULN: viitealueen yläraja, FACIT: Functional Assessment of Chronic Illness Therapy; FACIT-Fatigue-mittarilla (FACIT-F) arvioidaan väsymystä asteikolla 0 (pahin mahdollinen väsymys) – 52 (ei väsymystä)
CADENZA-tutkimus
42 potilasta satunnaistettiin saamaan Enjaymo-valmistetta (n = 22) tai lumelääkettä (n = 20) viikolle 25.
Teho perustui niiden potilaiden osuuteen, jotka täyttivät ensisijaiset päätemuuttujakriteerit: hemoglobiiniarvon suureneminen lähtötilanteeseen verrattuna vähintään 1,5 g/dl hoidon arviointiajankohtana (keskiarvo viikoilta 23, 25 ja 26), ei verensiirtoa viikolta 5 viikolle 26 eikä muuta kylmäagglutiniinisairauden hoitoa kuin mikä oli sallittua tutkimussuunnitelman mukaan viikolta 5 viikolle 26. Potilaalle annettiin verensiirto, jos hemoglobiini laski seuraavien raja-arvojen mukaiseksi: Hb < 7 g/dl tai Hb < 9 g/dl, johon liittyi oireita. Kiellettyjä hoitoja olivat rituksimabi ainoana hoitona tai yhdessä solunsalpaajien kanssa.
Tehoa arvioitiin lisäksi seuraavilla kahdella keskeisellä toissijaisella päätemuuttujalla: Enjaymo-hoidon vaikutus Hb-arvon keskimuutokseen lähtötilanteesta sekä vaikutus FACIT-F-pistemäärän keskimuutokseen lähtötilanteesta elämänlaadun muutoksen arvioimiseksi. Muita toissijaisia päätemuuttujia olivat: hemolyysin laboratoriomittaukset, mukaan lukien kokonaisbilirubiinin keskimuutos lähtötilanteesta. Tutkimusta varten kerättyihin, täydentäviin tehoa koskeviin tietoihin sisältyi annettujen verensiirtojen määrä viiden hoitoviikon kuluttua.
Tehotulokset on kuvattu alla taulukoissa 4 ja 5.
Taulukko 4 – Tehotulokset kylmäagglutiniinisairautta sairastavilla potilailla CADENZA-tutkimuksessa – osa A
| Parametri | Tilastotieto | Lumelääke N = 20 | Enjaymo N = 22 | Hoidon teho |
|---|---|---|---|---|
| Vasteen saaneeta | % (95 %:n luottamusväli)
Kerroinsuhde (95 %:n luottamusväli) p-arvo | 3 (15,0 (3,2, 37,9)
| 16 (72,7) (49,8, 89,3)
|
15,94 < 0,001 |
| Hemoglobiini | Keskimuutos lähtötilanteesta (LS†-keskiarvo), g/dl | 0,09 | 2,66 | 2,56 |
| LS-keskiarvon 95 %:n luottamusväli | (-0,5, 0,68) | (2,09, 3,22) | (1,75, 3,38) | |
| p-arvo | < 0,001 | |||
| Verensiirtojen määrän keskiarvo (viikolta 5 viikolle 26) | n (keskihajonta) | 0,5 (1,1) | 0,05 (0,2) | Ei laskettu |
| FACIT†-F-pistemäärä | Keskiarvo | 33,66 | 43,15 | |
| Keskimuutos lähtötilanteesta (LS†-keskiarvo) | 1,91 | 10,83 | 8,93 | |
| LS-keskiarvon 95 %:n luottamusväli | (-1,65, 5,46) | (7,45, 14,22) | (4, 13,85) | |
| p-arvo | < 0,001 | |||
| Kokonaisbilirubiini* | Keskiarvo, μmol/l | 33,95 | 12,12 | |
| Keskimuutos lähtötilanteesta | -1,83 | -22,13 | Ei laskettu | |
| Niiden potilaiden määrä, joilla arvo normalisoitui (%) | 4 (22,2 %) | 15 (88,2) |
aVasteen saanut määriteltiin potilaaksi, jonka hemoglobiiniarvo oli suurentunut lähtötilanteesta vähintään 1,5 g/dl hoidon arviointiajankohtana (keskiarvo viikoilta 23, 25 ja 26) ja joka ei ollut saanut verensiirtoa viikolta 5 viikolle 26 eikä muuta kylmäagglutiniinisairauden hoitoa kuin mikä oli sallittua tutkimussuunnitelman mukaan viikolta 5 viikolle 26.
*Bilirubiinia koskevien tietojen N = 18 lumeryhmässä ja N = 17 Enjaymo-ryhmässä pois lukien potilaat, joilla oli joko positiivinen Gilbertin oireyhtymän testitulos tai ei testitulosta saatavilla
†LS: pienin neliösumma, FACIT: Functional Assessment of Chronic Illness Therapy
Hemoglobiiniarvon (Hb) keskimuutosta lähtötilanteesta kuvaava käyrä on esitetty alla kuvassa 1.
Kuva 1 CADENZA-tutkimus, osa A: Hemoglobiiniarvon (g/dl) keskimääräinen muutos lähtötilanteesta (+/-keskivirhe) käynnin mukaan
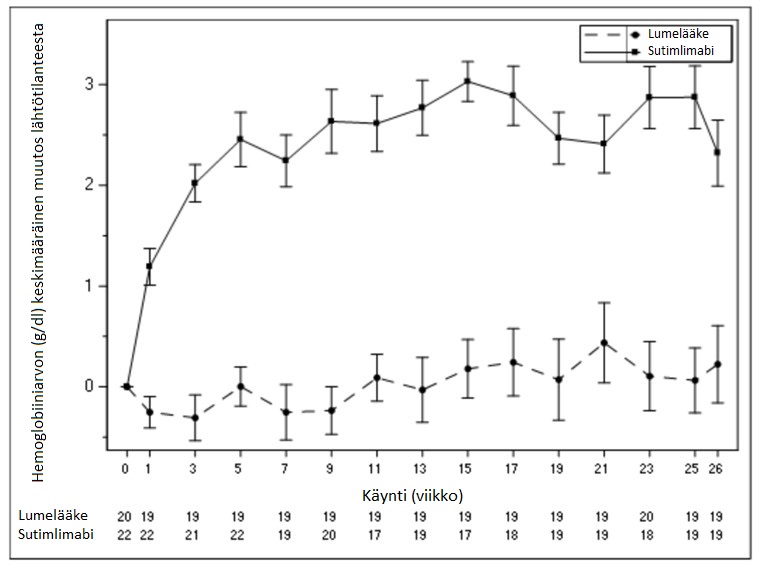
Bilirubiinipitoisuuksien keskiarvoja kuvaava käyrä käynnin mukaan on esitetty alla kuvassa 2.
Kuva 2 – CADENZA-tutkimus, osa A: Bilirubiinipitoisuuden keskiarvo (μmol/l) (+/-keskivirhe) käynnin mukaan (pois lukien tutkittavat, joilla Gilbertin oireyhtymän testitulos oli positiivinen tai tuntematon)
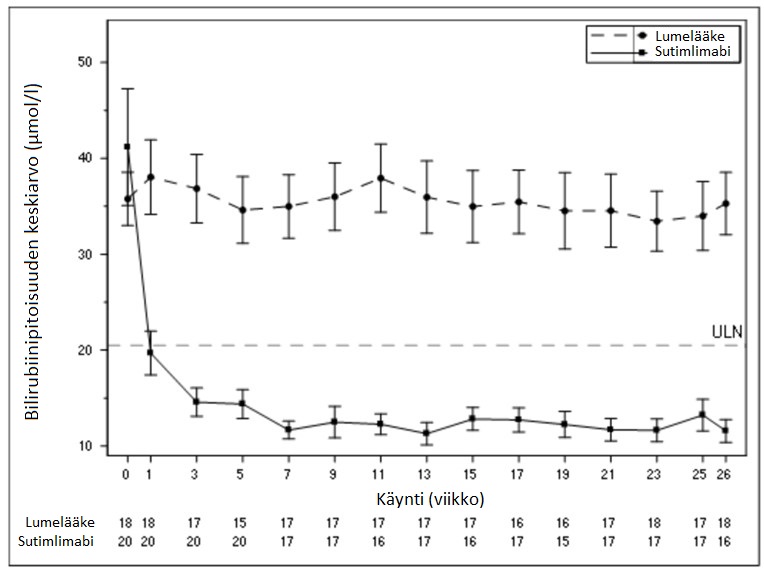
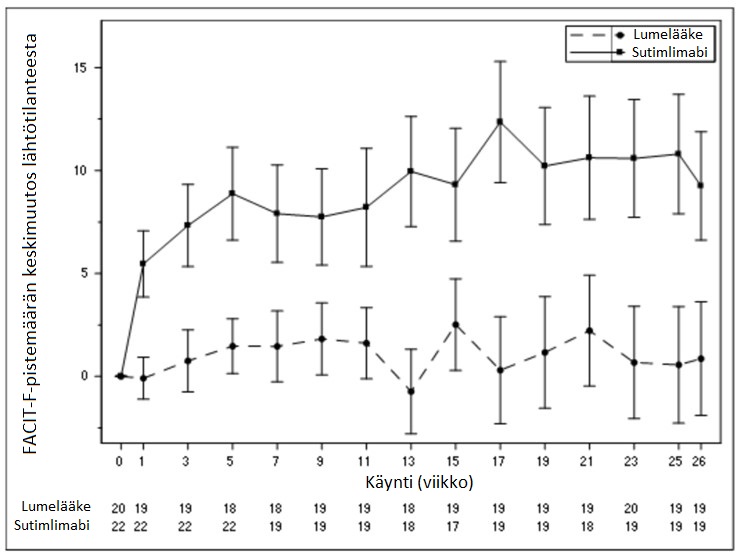
Terveyteen liittyvä elämänlaatu
FACIT-pistemäärien keskiarvon suureneminen osassa A on esitetty alla olevassa kuvassa 3.
Kuva 3 – CADENZA-tutkimus, osa A: FACIT-F-pistemäärän keskimuutos (keskivirhe) käynnin mukaan – Havainnot – Täydellinen analyysijoukko
Osassa B hemoglobiinipitoisuuksien keskiarvo pysyi suurempana kuin 11 g/dl, ja bilirubiinipitoisuuksien keskiarvo normalisoitui pitkäaikaisesti, mikä viittasi pysyvään hemolyysin vähenemiseen. Osassa A havaitut suurentuneet FACIT-F-pistemäärät säilyivät.
Kun tutkimuksen viimeinen Enjaymo-annos oli annettu, havaittiin hemolyysin uusiutumisen merkkejä ja oireita. 9 viikon kuluttua viimeisen annoksen antamisesta osassa B hemoglobiinipitoisuuden keskiarvo oli pienentynyt 2,41 g/dl (keskihajonta: 2,21) ja bilirubiinipitoisuuden keskiarvo oli suurentunut 21,80 µmol/l (keskihajonta: 18,14) viimeisistä hoidon aikana saatavilla olleista arvoista. FACIT-F-pistemäärän keskiarvo (31,29) palautui lähes lähtötasolle, ja pistemäärän muutoksen keskiarvo (keskihajonta) lähtötilanteeseen verrattuna oli ‑1,40 (11,48).
CARDINAL-tutkimus
24 potilaalle annettiin Enjaymo-valmistetta viikolle 25.
Teho perustui niiden potilaiden osuuteen, jotka täyttivät ensisijaiset päätemuuttujakriteerit: hemoglobiiniarvon suureneminen lähtötilanteeseen verrattuna vähintään 2 g/dl tai hemoglobiini vähintään 12 g/dl hoidon arviointiajankohtana (keskiarvo viikoilta 23, 25 ja 26), ei verensiirtoa viikolta 5 viikolle 26 eikä muuta kylmäagglutiniinisairauden hoitoa kuin mikä oli sallittua tutkimussuunnitelman mukaan viikolta 5 viikolle 26. Potilaalle annettiin verensiirto, jos hemoglobiini laski seuraavien raja-arvojen mukaiseksi: Hb < 7 g/dl tai Hb < 9 g/dl, johon liittyi oireita. Kiellettyjä hoitoja olivat rituksimabi ainoana hoitona tai yhdessä solunsalpaajien kanssa.
Tehoa arvioitiin lisäksi seuraavilla toissijaisilla päätemuuttujilla: Enjaymo-valmisteen vaikutus hemoglobiiniarvoon ja hemolyysia mittaaviin laboratorioarvoihin, mukaan lukien kokonaisbilirubiinin keskimuutos lähtötilanteesta. Elämänlaadun muutosta arvioitiin toissijaisena päätemuuttujana; mittarina oli FACIT-F-pistemäärän keskimuutos lähtötilanteesta. Tutkimusta varten kerättyihin, täydentäviin tehoa koskeviin tietoihin sisältyi annettujen verensiirtojen määrä viiden hoitoviikon kuluttua.
Taulukossa 5 esitetään tehotulokset kylmäagglutiniinisairautta sairastavilla potilailla CARDINAL-tutkimuksessa.
Taulukko 5 – Tehotulokset kylmäagglutiniinisairautta sairastavilla potilailla CARDINAL-tutkimuksessa – osa A
| Parametri | Tilastotieto | ENJAYMO N = 24 |
| Vasteen saaneeta | n (%) | 13 (54) |
| Hemoglobiini | Keskimuutos lähtötilanteesta (LS†-keskiarvo), g/dl LS-keskiarvon 95 %:n luottamusväli | 2,60 (0,74, 4,46) |
| Verensiirtojen määrän keskiarvo (viikolta 5 viikolle 26) | n | 0,9 |
| Kokonaisbilirubiini* | Keskiarvo, μmol/l Keskimuutos lähtötilanteesta (LS†-keskiarvo) Niiden potilaiden määrä, joilla arvo normalisoitui (%) | 15,48 (0,76 × ULN†) -38,18 13 (54,2) |
| Pisteet FACIT†-väsymysasteikolla | Keskiarvo Keskimuutos lähtötilanteesta (LS†-keskiarvo) LS-keskiarvon 95 %:n luottamusväli | 44,26 10,85 (8,0, 13,7) |
aVasteen saanut määriteltiin potilaaksi, jonka hemoglobiiniarvo suureni lähtötilanteeseen verrattuna vähintään 2 g/dl tai hemoglobiiniarvo oli vähintään 12 g/dl hoidon arviointiajankohtana (keskiarvo viikoilta 23, 25 ja 26) ja joka ei ollut saanut verensiirtoa viikolta 5 viikolle 26 eikä muuta kylmäagglutiniinisairauden hoitoa kuin mikä oli sallittua tutkimussuunnitelman mukaan viikolta 5 viikolle 26.
*N = 21 bilirubiinitietojen osalta lukuun ottamatta potilaita, joilla oli Gilbertin oireyhtymä
†LS: pienin neliösumma, ULN: viitealueen yläraja, FACIT: Functional Assessment of Chronic Illness Therapy
Osassa B hemoglobiinipitoisuuksien keskiarvo pysyi suurempana kuin 11 g/dl, ja bilirubiinipitoisuuksien keskiarvo normalisoitui pitkäaikaisesti, mikä viittasi pysyvään hemolyysin vähenemiseen.
Kun tutkimuksen viimeinen Enjaymo-annos oli annettu, havaittiin hemolyysin uusiutumisen merkkejä ja oireita. 9 viikon kuluttua viimeisen annoksen antamisesta osassa B hemoglobiinipitoisuuden keskiarvo oli pienentynyt 2,28 g/dl (keskihajonta: 1,80) ja bilirubiinipitoisuuden keskiarvo oli suurentunut 24,27 µmol/l (keskihajonta: 13,51) viimeisistä hoidon aikana saatavilla olleista arvoista. FACIT-F-pistemäärien keskiarvo palautui kohti lähtötasoa, ja pistemäärän muutoksen keskiarvo (keskihajonta) verrattuna lähtötilanteessa ennen hoitoa todettuihin arvoihin oli 1,05 (8,15).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Enjaymo-valmisteen käytöstä primaarin kylmäagglutiniinisairauden hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Iäkkäät potilaat
Enjaymo-valmisteella tehtyihin, kylmäagglutiniinisairautta arvioineisiin kliinisiin tutkimuksiin osallistuneista potilaista suurin osa (43/66, 65 %) oli vähintään 65-vuotiaita. Raportoiduissa kliinisissä kokemuksissa ei tullut esiin eroja vasteissa yli 65-vuotiaiden ja sitä nuorempien potilaiden välillä.
Farmakokinetiikka
Sutimlimabin farmakokinetiikkaa tarkasteltiin 24 potilaalla (CARDINAL-tutkimuksessa) ja 42 potilaalla (CADENZA-tutkimuksessa). Näistä potilaista 51 sai hoitoa 6 500 mg:n annoksella ja 15 potilasta 7 500 mg:n annoksella annostussuositusten mukaisesti. Kokonaisaltistukset vakaassa tilassa ehdotetulla annostusohjelmalla on esitetty taulukossa 6.
Taulukko 6 – Vakaan tilan altistusparametrit, keskiarvo (keskihajonta)
CARDINAL ja CADENZA | Annos (mg) | Cmin (µg/ml)* | AUCSS (µg·h/ml)* |
|
|
|
|
* Lyhenteet: AUCss = kahden peräkkäisen annoksen annon välinen käyrän alle jäävä pinta-ala, kun vakaa tila on saavutettu; Cmin = minimipitoisuus vakaassa tilassa määritettynä 1 tunti ennen seuraavan annoksen antoa
Vakaa tila saavutettiin viikkoon 7 mennessä sutimlimabihoidon aloittamisen jälkeen, ja kumulaatiosuhde oli alle 2.
Jakautuminen
Sentraalinen ja perifeerinen jakautumistilavuus vakaassa tilassa oli noin 5,8 l potilailla, joilla on kylmäagglutiniinisairaus.
Biotransformaatio
Sutimlimabi on proteiini. Yleisesti tiedetään, että vasta-aineet metaboloituvat hajoamalla pieniksi peptideiksi ja yksittäisiksi aminohapoiksi.
Eliminaatio
Sutimlimabin puoliintumisaika riippuu sen pitoisuudesta plasmassa. Sutimlimabin vakaan tilan terminaalisen eliminaation puoliintumisaika on kokonaispuhdistuman (lineaarinen ja ei-lineaarinen puhdistuma) perusteella 16 päivää.
Lineaarisuus/ei-lineaarisuus
Kerta-annosten annon jälkeen sutimlimabin puhdistuma pieneni alkuvaiheessa voimakkaasti, kun annokset olivat alle 30 mg/kg (noin 2 g); 60–100 mg/kg:n sutimlimabiannoksilla puhdistuma ei enää ollut riippuvainen annoksesta.
Erityisryhmät
Sutimlimabin farmakokinetiikassa ei havaittu kliinisesti merkittäviä eroja sukupuolen, iän eikä maksan tai munuaisten vajaatoiminnan perusteella. Vakaan tilan altistustasojen (Cmax, Cmin ja AUC) arviointi perustui 6 500 mg:n (potilaan paino < 75 kg) ja 7 500 mg:n (potilaan paino ≥ 75 kg) annoksiin, jotka annettiin päivinä 0 ja 7 ja sen jälkeen 14 päivän välein. Populaatiofarmakokineettisessä analyysissä tarkasteltiin 101 miespuolista ja 95 naispuolista tutkittavaa, ja havaitut altistusparametrit olivat sukupuolten välillä samankaltaiset.
Populaatiofarmakokineettisessä analyysissä tarkasteltiin tutkittavien rotua (94 valkoihoista, 10 mustaihoista, 42 aasialaista) ja havaitut altistusparametrit olivat näiden tutkittavien välillä samankaltaiset.
Populaatiofarmakokineettisen analyysin mukaan paino ja etninen tausta (japanilaiset vs. ei-japanilaiset) vaikuttivat sutimlimabin farmakokinetiikkaan. Tutkittavilla, joiden paino oli suurempi, havaittiin altistuksen olevan pienempi. Tutkimusten välisen vertailun perusteella sutimlimabin AUC0–168 oli 30–100 mg/kg:n annosten jälkeen jopa 38 % suurempi japanilaisilla kuin ei-japanilaisilla tutkittavilla.
Farmakokineettiset/farmakodynaamiset suhteet
Yli 100 μg/ml:n sutimlimabipitoisuus esti klassisen tien aktivaatiota maksimaalisesti. Ehdotetulla annostusohjelmalla saatiin vakaassa tilassa riittävä sutimlimabialtistus, jotta vaikutukset hemoglobiini- ja bilirubiiniarvoihin ja C4-kokonaispitoisuuksiin olivat kliinisesti merkittävät.
Prekliiniset tiedot turvallisuudesta
Jaavanmakakeilla tehdyssä tehostetussa pre- ja postnataalista kehitystä (enhanced pre- and post-natal development, ePPND) koskeneessa tutkimuksessa ei havaittu merkkejä haitallisista vaikutuksista kehitykseen, kun sutimlimabia annettiin laskimoon organogeneesista synnytykseen asti annoksilla, joilla saavutettu altistus vastasi noin 2–3-kertaisesti ihmiselle suositellulla enimmäisannostuksella koituvia AUC-arvoja. Toistuvilla annoksilla tehdyissä tutkimuksissa, joissa sutimlimabia annettiin jaavanmakakeille annoksilla, joilla saavutettu altistus vastasi enimmillään noin 4-kertaisesti ihmiselle suositeltua annosta, ei havaittu lisääntymiselimiin kohdistuvia vaikutuksia.
Eläimillä ei ole tehty tutkimuksia, joissa olisi arvioitu sutimlimabin karsinogeenisuutta.
Jaavanmakakeilla tehtyjen konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
Polysorbaatti 80 (E 433)
Natriumkloridi
Dinatriumfosfaatti (E 339)
Natriumdivetyfosfaatti (E 339)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo
4 vuotta
Lääkevalmisteen säilytys avaamisen jälkeen:
Valmisteen kemiallisen ja fysikaalisen käytönaikaisen säilyvyyden on osoitettu olevan 16 tuntia 18–25 °C:ssa tai 72 tuntia 2–8 °C:ssa. Mikrobiologisista syistä valmiste on käytettävä välittömästi.
Jos sitä ei käytetä välittömästi, käytönaikaiset säilytysajat ja olosuhteet ennen käyttöä ovat käyttäjän vastuulla, eivätkä ne yleensä saa ylittää 24 tuntia 2–8°C:ssa tai 8 tuntia huoneenlämmössä, ellei injektiopullojen avaamista ja niiden sisällön yhdistämistä infuusiopussiin ole tehty valvotuissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Ei saa jäätyä.
Lääkevalmisteen säilytys injektiopullon ensimmäisen käyttökerran jälkeen, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ENJAYMO infuusioneste, liuos
50 mg/ml (L:ei) 1 kpl (22 ml) (1466,33 €)
PF-selosteen tieto
22 ml liuosta injektiopullossa (tyypin I lasia), jossa on tulppa (butyylikumia), suljin (alumiinia) ja irtinapsautettava korkki.
Pakkauksessa on 1 tai 6 injektiopulloa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Samea, väritön tai kellertävä liuos, jossa ei käytännössä ole näkyviä hiukkasia ja jonka pH on noin 6,1 ja osmolaalisuus 268–312 mOsm/kg.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Enjaymo toimitetaan liuoksena kerta-annosinjektiopullossa, ja terveydenhuollon ammattilaisen on valmisteltava se aseptista tekniikkaa noudattaen.
Valmistelu
- Ota Enjaymo jääkaapista. Jotta vaahtoaminen olisi mahdollisimman vähäistä, älä ravista.
- Tarkasta injektiopullot silmämääräisesti hiukkasten ja värimuutosten varalta ennen valmisteen antoa. Liuos on samea ja väritön tai kellertävä neste. Älä anna valmistetta, jos siinä on värimuutoksia tai hiukkasia.
- Vedä suositellun annoksen mukaan (ks. taulukko 1) laskettu tilavuus valmistetta tarvittavasta määrästä injektiopulloja ja lisää tyhjään infuusiopussiin. Hävitä injektiopulloon jäljelle jäänyt liuos.
- Valmisteltu liuos on annettava välittömästi. Säilytys, ks. kohta Kestoaika.
Anto
- Anna infuusioliuoksen lämmetä huoneenlämpöiseksi (18–25 °C) ennen antoa. Katso infuusionopeus taulukosta 1, ks. kohta Annostus ja antotapa. Infuusio annetaan 1–2 tunnin aikana potilaan painon mukaan. Anna infuusio aina 0,22 mikronin suodattimen kautta, jossa on polyeetterisulfonista (PES) valmistettu kalvo. Infuusionesteen lämmitintä voidaan käyttää; 40 °C:n lämpötilaa ei saa ylittää.
- Infuusiokatetri ja letkut on esitäytettävä annettavalla liuoksella juuri ennen infuusiota ja huuhdeltava välittömästi infuusion päättymisen jälkeen riittävällä määrällä (noin 20 ml) 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionestettä.
- Yhteensopimattomuuksia ei ole havaittu Enjaymo-infuusioliuoksen ja polyvinyylikloridista (PVC) valmistettujen di(2-etyyliheksyyli)ftalaatilla (DEHP) pehmennettyjen tai etyylivinyyliasetaatista (EVA) tai polyolefiinista (PO) valmistettujen infuusiopussien; DEHP:llä pehmennetystä PVC:stä, DEHP-vapaasta polypropeenista (PP) tai polyeteenistä (PE) valmistettujen infuusioletkustojen; eikä polykarbonaatista (PC) tai akryylinitriilibutadieenistyreenistä (ABS) valmistettujen injektiopulloliitinten välillä.
Korvattavuus
ENJAYMO infuusioneste, liuos
50 mg/ml 1 kpl
- Ei korvausta.
ATC-koodi
L04AJ04
Valmisteyhteenvedon muuttamispäivämäärä
01.08.2025
Yhteystiedot
Berzelius väg 8
171 65 Solna
Sverige
+46 8 545 802 30
www.recordati.com