
ASPAVELI infusionsvätska, lösning 1080 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Terveydenhuollon ammattilainen
Yleinen
Observera
▼ Detta läkemedel är föremål för utökad övervakning. Detta kommer att göra det möjligt att snabbt identifiera ny säkerhetsinformation. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning. Se avsnitt Biverkningar om hur man rapporterar biverkningar.
Kvalitativ och kvantitativ sammansättning
Varje 20 ml injektionsflaska innehåller 1 080 mg pegcetakoplan.
Varje ml innehåller 54 mg pegcetakoplan.
Hjälpämne(n) med känd effekt
Varje ml innehåller 41 mg sorbitol.
Varje injektionsflaska innehåller 820 mg sorbitol.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Infusionsvätska, lösning.
Kliniska uppgifter
Terapeutiska indikationer
ASPAVELI är avsett som monoterapi för behandling av vuxna patienter med paroxysmal nokturn hemoglobinuri (PNH) som har hemolytisk anemi.
ASPAVELI är avsett för behandling av vuxna och ungdomar i åldern 12 till 17 år med C3-glomerulopati (C3G) eller primär immunkomplexmedierad membranoproliferativ glomerulonefrit (IC-MPGN) i kombination med en renin-angiotensinsystemblockerare (RAS-blockerare), såvida inte behandling med RAS-blockerare är kontraindicerad eller inte kan tolereras.
Villkor
Hoito on aloitettava hematologisiin sairauksiin perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Dosering och administreringssätt
Behandling ska inledas under överinseende av hälso- och sjukvårdspersonal med erfarenhet av behandling av patienter med hematologiska eller renala sjukdomar. Självadministrering och infusion i hemmet ska övervägas för patienter som har tolererat behandling väl på mottagning med erfarenhet av sådan behandling. Beslut om eventuell självadministrering och infusion i hemmet ska tas efter bedömning och rekommendation från behandlande läkare.
Dosering
Pegcetakoplan kan ges av hälso- och sjukvårdspersonal eller administreras av patienten eller vårdgivare efter noggranna anvisningar.
PNH
Vuxna patienter med PNH
Pegcetakoplan administreras två gånger per vecka som en 1 080 mg subkutan infusion via en infusionspump med ett sprutsystem eller ett kroppsburet infusionssystem (OBDS, on-body delivery system) som kan administrera doser upp till 20 ml. Dosen administreras två gånger per vecka, dag 1 och dag 4 per behandlingsvecka.
PNH är en kronisk sjukdom och behandling med ASPAVELI bör fortsätta livslångt, såvida inte utsättning av detta läkemedel är kliniskt indicerad (se avsnitt Varningar och försiktighet).
Patienter med PNH som byter till ASPAVELIfrån en C5‑hämmare
Under de första 4 veckorna administreras pegcetakoplan som subkutana doser om 1 080 mg två gånger per vecka i tillägg till patientens aktuella dos av C5‑hämmare för att minska risken för hemolys vid abrupt behandlingsutsättning. Efter 4 veckor ska behandlingen med C5‑hämmaren vara avslutad. Därefter fortsätter behandlingen med ASPAVELI i monoterapi.
Byte från komplementhämmare andra än ekulizumab har inte studerats. Utsättning av andra komplementhämmare innan steady-state för pegcetakoplan har uppnåtts ska göras med försiktighet (se avsnitt Farmakokinetiska egenskaper).
Dosjustering vid PNH
Doseringsregimen kan ändras till 1 080 mg var tredje dag (t.ex. dag 1, dag 4, dag 7, dag 10, dag 13 osv.) om patientens nivå av laktatdehydrogenas (LDH) överstiger 2 x den övre normalgränsen (ULN). Vid en dosökning ska LDH kontrolleras två gånger per vecka under minst 4 veckor (se avsnitt Varningar och försiktighet).
C3G och primär IC-MPGN
Pegcetakoplan administreras två gånger per vecka som en subkutan infusion via en infusionspump med ett sprutsystem eller ett kroppsburet infusionssystem (OBDS) som kan administrera doser upp till 20 ml. Dosen administreras två gånger per vecka, dag 1 och dag 4 per behandlingsvecka.
C3G och primär IC-MPGN är kroniska sjukdomar. Utsättning av detta läkemedel rekommenderas inte såvida det inte är kliniskt indicerat.
Vuxna patienter med C3G eller primär IC-MPGN
Pegcetakoplan administreras två gånger per vecka som en 1 080 mg subkutan infusion.
Ungdomar med C3G eller primär IC-MPGN Comment by SE Linguistic comment: Revised in line with common text and how it is named in the indication in 4.1 in the sentence below.
Doseringsregimen för ungdomar baseras på patientens kroppsvikt och består av följande: Comment by SE Linguistic comment: Proposed to change to ”baseras”.
Kroppsvikt | Första dosen (infusionsvolym) | Andra dosen (infusionsvolym) | Underhållsdos (infusionsvolym) |
≥ 50 kg | 1 080 mg två gånger i veckan (20 ml) | ||
35 till < 50 kg | 648 mg (12 ml) | 810 mg (15 ml) | 810 mg två gånger i veckan (15 ml) |
30 till < 35 kg | 540 mg (10 ml) | 540 mg (10 ml) | 648 mg två gånger i veckan (12 ml) |
Glömd dos
Om en dos av pegcetakoplan för behandling av PNH, C3G eller primär IC-MPGN glöms bort ska den administreras så snart som möjligt och därefter ska det vanliga behandlingsschemat återupptas. Detta gäller även om det blir mindre än 3 dagar mellan ersättningsdosen och nästa dos.
Patienter med recidiverande C3G eller primär IC-MPGN efter transplantation
Diagnosen recidiverande C3G eller primär IC-MPGN efter transplantation ska ställas genom transplantatbiopsi. Recidiv av C3G eller primär IC-MPGN kan upptäckas vid rutinmässig posttransplantationsbiopsi, i övrigt ska en biopsi göras vid kliniska tecken på att sjukdomen återkommit. Liksom i studien APL2-C3G-204 (se avsnitt Farmakodynamiska egenskaper) kan behandling med pegcetakoplan påbörjas innan kliniska tecken uppstått, såsom minskad beräknad glomerulär filtrationshastighet (eGFR) eller ökad protein-kreatininkvot i urin. Erfarenheten från användningen av pegcetakoplan till patienter med recidiverande C3G eller primär IC-MPGN efter transplantation i kliniska studier är begränsad.
Särskilda populationer
Äldre
Även om inga tydliga åldersrelaterade skillnader observerades i kliniska studier var antalet patienter i åldern 65 år och äldre inte tillräckligt för att fastställa om de svarar på ett annat sätt än yngre patienter. Det finns inga data som tyder på att någon särskild försiktighet behöver vidtas vid behandling av en äldre population.
Nedsatt njurfunktion
Svårt nedsatt njurfunktion (kreatininclearance < 30 ml/min) hade ingen effekt på farmakokinetiken för pegcetakoplan. Således är ingen dosjustering av pegcetakoplan nödvändig hos patienter med nedsatt njurfunktion. Inga data finns tillgängliga om användning av pegcetakoplan hos patienter med njursjukdom i terminalfas (ESRD) som kräver dialys (se avsnitt Farmakokinetiska egenskaper).
Nedsatt leverfunktion
Säkerhet och effekt för pegcetakoplan har inte studerats hos patienter med nedsatt leverfunktion men ingen dosjustering rekommenderas eftersom nedsatt leverfunktion inte förväntas påverka clearance av pegcetakoplan.
Pediatrisk population
Säkerhet och effekt för ASPAVELI för barn i åldern 0 till < 18 år med PNH har ännu inte fastställts. Inga data finns tillgängliga.
Säkerhet och effekt för ASPAVELI för barn under 12 år med C3G eller primär IC-MPGN har ännu inte fastställts. Inga data finns tillgängliga.
Detta läkemedel ska inte ges till barn < 12 års ålder eftersom icke‑kliniska säkerhetsdata inte är tillgängliga för denna åldersgrupp.
Administreringssätt
ASPAVELI ska bara administreras som en subkutan infusion med en kommersiellt tillgänglig infusionspump med ett sprutsystem eller ett kroppsburet infusionssystem.
Detta läkemedel kan självadministreras. När självadministrering påbörjas ska patienten få utbildning av kvalificerad hälso- och sjukvårdspersonal i infusionstekniker, användning av en infusionspump med ett sprutsystem eller ett kroppsburet infusionssystem, registrering av behandlingen, hur eventuella biverkningar känns igen och vilka åtgärder som kan vidtas om dessa uppkommer.
- Vid användning av en infusionspump med ett sprutsystem ska ASPAVELI infunderas i buken, låren, höfterna eller överarmarna. Det måste vara minst 7,5 cm mellan infusionsställena. Infusionsställen ska bytas mellan administreringarna. Infusionstiden är cirka 30 minuter (vid användning av två ställen) eller cirka 60 minuter (vid användning av ett ställe).
- Vid användning av ett kroppsburet infusionssystem ska ASPAVELI infunderas på ett ställe i buken. Infusionsstället ska bytas mellan administreringarna i enlighet med anvisningarna för utrustningen från tillverkaren. Infusionstiden varierar mellan patienter och är vanligtvis mellan 30 och 60 minuter.
Infusion i områden med hud som är öm, har blåmärken, har rodnad eller är hård ska undvikas. Infusion i tatueringar, ärr eller hudbristningar ska undvikas. Infusionen måste påbörjas omedelbart efter att detta läkemedel har dragits upp i sprutan. Administrering måste avslutas inom 2 timmar efter beredning av sprutan. Anvisningar om beredning och infusion av läkemedlet finns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Kontraindikationer
Överkänslighet mot pegcetakoplan eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Behandling med pegcetakoplan får inte sättas in för patienter:
- med kvarvarande infektion orsakad av kapslade bakterier, inklusive Neisseria meningitidis, Streptococcus pneumoniae och Haemophilus influenzae (se avsnitt Varningar och försiktighet).
- som i nuläget inte är vaccinerade mot Neisseria meningitidis, Streptococcus pneumoniae och Haemophilus influenzae såvida de inte får profylaktisk behandling med lämpliga antibiotika till 2 veckor efter vaccination (se avsnitt Varningar och försiktighet).
Varningar och försiktighet
Allvarliga infektioner orsakade av kapslade bakterier
Användning av pegcetakoplan kan predisponera individer för allvarliga infektioner orsakade av kapslade bakterier, inklusive Neisseria meningitidis, Streptococcus pneumoniae och Haemophilus influenzae. För att minska risken för infektion måste alla patienter vaccineras mot dessa bakterier enligt gällande lokala riktlinjer minst 2 veckor innan de får pegcetakoplan, såvida inte risken för att behandlingen blir fördröjd uppväger risken för att utveckla en infektion.
Patienter med känd vaccinationsanamnes
Innan behandling med pegcetakoplan ges till patienter med känd vaccinationsanamnes måste det säkerställas att patienterna har fått vacciner mot kapslade bakterier, inklusive Streptococcus pneumoniae, Neisseria meningitidis serogrupp A, C, W, Y och B och Haemophilus influenzae typ B, inom 2 år innan behandling med pegcetakoplan påbörjas.
Patienter utan känd vaccinationsanamnes
För patienter utan känd vaccinationsanamnes ska de vacciner som krävs administreras minst 2 veckor innan den första dosen av pegcetakoplan ges. Om omedelbar behandling är indicerad ska de vacciner som krävs administreras så snart som möjligt och patienterna behandlas med lämpliga antibiotika till 2 veckor efter vaccination.
Övervakning av patienter för allvarliga infektioner
Vaccination kanske inte räcker för att förhindra allvarlig infektion. Hänsyn ska tas till officiella riktlinjer om lämplig användning av antibakteriella medel. Alla patienter ska kontrolleras för tidiga tecken på infektioner orsakade av kapslade bakterier, inklusive Neisseria meningitidis, Streptococcus pneumoniae och Haemophilus influenzaee, och utvärderas omedelbart vid misstanke om infektion och vid behov behandlas med lämpliga antibiotika. Patienterna ska informeras om dessa tecken och symtom och omedelbart uppsöka läkare. Läkaren måste diskutera fördelarna och riskerna med behandling med pegcetakoplan med patienten.
Överkänslighet
Överkänslighetsreaktioner har rapporterats. Om en svår överkänslighetsreaktion (inklusive anafylaxi) uppkommer måste infusionen med pegcetakoplan omedelbart avbrytas och lämplig behandling sättas in.
Reaktioner vid injektionsstället
Reaktioner vid injektionsstället har rapporterats vid subkutan användning av pegcetakoplan (se avsnitt Biverkningar). Patienterna ska få utbildning i korrekt injektionsteknik.
Laboratoriekontroller av PNH
Patienter med PNH som får pegcetakoplan måste regelbundet kontrolleras för tecken och symtom på hemolys, inklusive mätning av LDH‑nivåer, och dosjustering av det rekommenderade doseringsschemat kan krävas (se avsnitt Dosering och administreringssätt).
Effekter på laboratorieprover
Interferens kan uppkomma mellan reagens av kiseldioxid vid koagulationsscreening och pegcetakoplan som leder till artificiell förlängning av aktiverad partiell tromboplastintid (aPTT). Användning av reagens av kiseldioxid vid koagulationsscreening måste således undvikas.
Behandlingsutsättning vid PNH
Om patienter med PNH sätter ut behandlingen med pegcetakoplan måste de kontrolleras noggrant för tecken och symtom på allvarlig intravaskulär hemolys. Allvarlig intravaskulär hemolys identifieras med höjda nivåer av LDH tillsammans med en plötslig minskning av PNH:s klonstorlek eller hemoglobin (Hb) eller återkomst av symtom såsom trötthet, hemoglobinuri, buksmärta, dyspné, svår vaskulär biverkning (inklusive trombos), dysfagi eller erektil dysfunktion. Om utsättning av detta läkemedel är nödvändig ska alternativ behandling övervägas. Om allvarlig hemolys uppkommer efter utsättning av ASPAVELI ska följande ingrepp/behandlingar övervägas: blodtransfusion (packade erytrocyter), utbytestransfusion, antikoagulation och kortikosteroider. Patienterna måste kontrolleras noggrant under minst 8 veckor från den senaste dosen, motsvarande mer än 5 halveringstider för detta läkemedel, för att medge washout av läkemedel (se avsnitt Farmakokinetiska egenskaper) för att detektera allvarlig hemolys och andra reaktioner. Dessutom ska långsam avvänjning övervägas.
Preventivmedel hos fertila kvinnor
Fertila kvinnor bör använda effektiva preventivmetoder för att förhindra graviditet under behandling med pegcetakoplan och under minst 8 veckor efter den sista dosen av pegcetakoplan (se avsnitt Fertilitet, graviditet och amning).
Ackumulering av polyetylenglykol (PEG)
ASPAVELI är ett pegylerat läkemedel. De eventuella långtidseffekterna av PEG-ackumulering i njurarna, hjärnans plexus choroideus och andra organ är okänd (se avsnitt Prekliniska säkerhetsuppgifter). Njurfunktionen bör kontrolleras regelbundet med laboratorietester.
Utbildningsmaterial
Alla läkare som avser att förskriva ASPAVELI måste säkerställa att de har fått och känner till utbildningsmaterialet för läkare. Läkarna måste förklara och diskutera fördelarna och riskerna avseende behandling med ASPAVELI med patienten och ge honom/henne patientinformationspaketet och patientkortet. Patienten ska instrueras att omedelbart uppsöka hälso- och sjukvården vid tecken eller symtom på allvarlig infektion eller överkänslighet under behandling med ASPAVELI, särskilt om de tyder på infektion med kapslade bakterier.
Hjälpämne(n) med känd effekt
Sorbitolinnehåll
ASPAVELI 1 080 mg innehåller 820 mg sorbitol per injektionsflaska.
Patienter med hereditär fruktosintolerans ska inte ta/använda detta läkemedel.
Natriuminnehåll
Detta läkemedel innehåller mindre än 1 mmol (23 mg) natrium per dos, d.v.s. är näst intill ”natriumfritt”.
Interaktioner
Inga interaktionsstudier har utförts. Baserat på in vitro‑data har pegcetakoplan låg potential för läkemedelsinteraktioner.
Fertilitet, graviditet och amning
Fertila kvinnor
Fertila kvinnor bör använda effektiva preventivmetoder för att förhindra graviditet under behandling med pegcetakoplan och under minst 8 veckor efter den sista dosen av pegcetakoplan. För kvinnor som planerar att bli gravida kan användning av pegcetakoplan övervägas efter en bedömning av riskerna och nyttan (se Graviditet).
Graviditet
Det finns begränsad mängd data från användningen av pegcetakoplan hos gravida kvinnor. Data från djurstudier har visat reproduktionstoxikologiska effekter (se avsnitt Prekliniska säkerhetsuppgifter).
Pegcetakoplan rekommenderas inte under graviditet eller till fertila kvinnor som inte använder preventivmedel.
Amning
Det är okänt om pegcetakoplan utsöndras i bröstmjölk. Risken för absorption och skada hos det ammade barnet är okänd. Djurstudier tyder på en låg utsöndring (mindre än 1 %, inte farmakologiskt signifikant) av pegcetakoplan i mjölk hos apor (se avsnitt Prekliniska säkerhetsuppgifter). Det är inte troligt att ett ammat barn utsätts för en kliniskt relevant exponering.
Amning bör avbrytas under behandling med pegcetakoplan.
Fertilitet
Det saknas djur- eller humandata om effekten av pegcetakoplan på fertilitet. I toxicitetsstudier sågs inga mikroskopiska avvikelser i reproduktionsorgan hos apor av han- och honkön (se avsnitt Prekliniska säkerhetsuppgifter).
Effekter på förmågan att framföra fordon och använda maskiner
ASPAVELI har ingen eller försumbar effekt på förmågan att framföra fordon och använda maskiner.
Biverkningar
Sammanfattning av säkerhetsprofilen
PNH
De vanligaste rapporterade biverkningarna hos patienter med PNH behandlade med pegcetakoplan var reaktioner vid injektionsstället: erytem vid injektionsstället, hudklåda vid injektionsstället, svullnad vid injektionsstället, smärta vid injektionsstället, blåmärken vid injektionsstället. Andra biverkningar som rapporterats hos mer än 10 % av patienter under kliniska studier var övre luftvägsinfektion, diarré, hemolys, buksmärta, huvudvärk, trötthet, feber, hosta, urinvägsinfektion, vaccinationskomplikation, extremitetssmärta, yrsel, artralgi och ryggvärk. De vanligaste rapporterade allvarliga biverkningarna var hemolys och sepsis.
C3G och primär IC-MPGN
De vanligaste rapporterade biverkningarna hos patienter med C3G eller primär IC-MPGN behandlade med pegcetakoplan var reaktioner vid injektionsstället och övre luftvägsinfektioner. De vanligaste rapporterade allvarliga biverkningarna var akut njurskada och lunginflammation.
Lista över biverkningar i tabellform
I tabell 1 visas biverkningar som observerats i kliniska studier och erfarenhet efter marknadsintroduktionen av pegcetakoplan hos patienter med PNH, C3G och primär IC-MPGN. Biverkningarna är listade enligt MedDRA:s klassificering av organsystem och frekvens med följande konvention: mycket vanliga (≥1/10), vanliga (≥1/100, <1/10), mindre vanliga (≥1/1 000, <1/100), sällsynta (≥1/10 000, <1/1 000), mycket sällsynta (<1/10 000) och ingen känd frekvens (kan inte beräknas från tillgängliga data). Inom varje frekvensgrupp presenteras biverkningarna i fallande svårighetsgrad.
Tabell 1: Biverkningar från kliniska studier1 och erfarenhet efter marknadsintroduktionen
| Biverkning enligt MedDRA:s klassificering av organsystem | Frekvens vid PNH | Frekvens vid C3G eller primär IC-MPGN |
| Infektioner och infestationer | ||
| Influensa | Mycket vanliga | |
| Övre luftvägsinfektion2 | Mycket vanliga | Mycket vanliga |
| Urinvägsinfektion | Mycket vanliga | Vanliga |
| Sepsis | Vanliga3 | |
| Opportunistiska infektioner | Vanliga4 | |
| Covid-19, infektion i magtarmkanalen, svampinfektion, hudinfektion, oral infektion | Vanliga | |
| Öroninfektion | Vanliga | Vanliga |
| Infektion, luftvägsinfektion5, virusinfektion, bakterieinfektion, vaginal infektion, ögoninfektion | Vanliga | |
| Cervicit, ljumskinfektion | Mindre vanliga | |
| Lunginflammation | Mindre vanliga | Vanliga |
| Näsabscess, tuberkulos, esofageal kandidos, Covid-19-pneumoni, anal abscess | Mindre vanliga | |
| Immunsystemet | ||
| Överkänslighetsreaktion | Mycket vanliga6 | |
| Blodet och lymfsystemet | ||
| Hemolys | Mycket vanliga | |
| Trombocytopeni | Vanliga | Vanliga7 |
| Neutropeni | Vanliga | Vanliga |
| Metabolism och nutrition | ||
| Hypokalemi | Vanliga | Vanliga |
| Centrala och perifera nervsystemet | ||
| Huvudvärk | Mycket vanliga | Mycket vanliga |
| Yrsel | Mycket vanliga | |
| Blodkärl | ||
| Hypertoni | Vanliga | |
| Andningsvägar, bröstkorg och mediastinum | ||
| Hosta | Mycket vanliga | Vanliga |
| Dyspné, orofaryngeal smärta, nästäppa | Vanliga | |
| Näsblod | Vanliga | Vanliga |
| Magtarmkanalen | ||
| Buksmärta | Mycket vanliga | |
| Diarré | Mycket vanliga | Mycket vanliga |
| Illamående | Vanliga | Mycket vanliga |
| Hud och subkutan vävnad | ||
| Erytem, utslag, urtikaria | Vanliga | |
| Muskuloskeletala systemet och bindväv | ||
| Artralgi, ryggvärk | Mycket vanliga | |
| Extremitetssmärta | Mycket vanliga | Vanliga |
| Myalgi | Vanliga | Vanliga |
| Muskelspasmer | Vanliga | |
| Njurar och urinvägar | ||
| Akut njurskada | Vanliga | Mycket vanliga |
| Kromaturi | Vanliga | |
| Allmänna symtom och/eller symtom vid administreringsstället | ||
| Feber | Mycket vanliga | Mycket vanliga |
| Utmattning | Mycket vanliga | Vanliga |
| Reaktion vid infusionsstället8 | Mycket vanliga | Mycket vanliga |
| Undersökningar och provtagningar | ||
| Förhöjt alaninaminotransferas, förhöjt bilirubin | Vanliga | |
| Skador, förgiftningar och behandlingskomplikationer | ||
| Vaccinationskomplikationer | Mycket vanliga | |
1Studierna APL2‑308, APL2‑302, APL2‑202, APL2‑CO‑PNH‑204 och APL‑CP0514 på PNH-patienter, och studierna APL2-C3G-310, APL2-C3G-314, APL2-201 och APL2-C3G-204 på patienter med C3G och primär IC-MPGN.
Medicinska likartade termer är, om tillämpligt, grupperade på basis av likartat medicinskt begrepp.
2Omfattar nasofaryngit, övre luftvägsinfektion, faryngit, rinit och sinuit.
3Sepsis inkluderar ett fall av septisk chock och ett fall av icke kapslad Neisseria meningitidis.
4Herpes zoster (inklusive Herpes zoster-meningoencefalit) och Pneumocystis jirovecii-infektion.
5Omfattar luftvägsinfektion och virusinfektion i luftvägar.
6Omfattar hudutslag och eksem.
7Omfattar minskat trombocytantal.
8Rekommenderade termer inkluderade i reaktion vid infusionsstället: erytem vid infusionsställe, klåda vid infusionsstället, svullnad vid infusionsstället, blåmärke vid infusionsstället, smärta vid infusionsstället, förhårdnad vid infusionsstället.
Beskrivning av utvalda biverkningar
Infektioner
Ingen allvarlig infektion orsakad av kapslade bakterier rapporterades under PNH-studie APL2‑302. Fyrtioåtta patienter fick en infektion under studien. De vanligaste infektionerna hos patienter behandlade med pegcetakoplan under PNH-studien APL2‑302 var övre luftvägsinfektion (28 fall, 35 %). De flesta infektioner som rapporterades hos patienter behandlade med pegcetakoplan under PNH-studie APL2‑302 var icke‑allvarliga och deras intensitet var i huvudsak lätt. Tio patienter utvecklade infektioner rapporterade som allvarliga, däribland en patient som avled på grund av covid‑19. De vanligaste allvarliga infektionerna var sespis (3 fall) (som ledde till utsättning av pegcetakoplan hos en patient) och gastroenterit (3 fall); varav alla gick över.
I kliniska studier av C3G och primär IC‑MPGN rapporterades fyra allvarliga luftvägsinfektioner orsakade av kapslade bakterier hos patienter som behandlades med pegcetakoplan: en epiglottit, en pneumoni orsakad av pneumokocker och en atypisk pneumoni som ledde till behandlingsavbrott, samt en pneumoni orsakad av Haemophilus utan dosjustering. Tillstånden gick tillbaka och patienterna återhämtade sig, utom i fallen med Haemophilus-pneumoni och atypisk pneumoni som gick tillbaka med följdtillstånd. Ett allvarligt fall av urinvägsinfektion orsakad av Escherichia rapporterades också, vilket gick tillbaka med återhämtning utan dosjustering.
Hemolys
Nitton patienter som behandlades med pegcetakoplan rapporterade hemolys under PNH-studie APL2‑302. Sju fall rapporterades som allvarliga och 5 fall ledde till utsättning av pegcetakoplan och dosen av pegcetakoplan höjdes hos 10 patienter. Under PNH-studien APL2-308 förekom 3 fall av hemolys hos patienter som behandlades med pegcetakoplan. Inget av dessa fall rapporterades som allvarligt eller ledde till utsättning av pegcetakoplan. Dosen av pegcetakoplan höjdes hos alla 3 patienter.
Akut njurskada
I kliniska studier av C3G och primär IC-MPGN rapporterades 10 allvarliga fall av akut njurskada hos 8 patienter (5,7 %) som behandlades med pegcetakoplan, varav 5 händelser observerades hos 4 patienter efter transplantation. Av dessa allvarliga händelser ledde endast 1 till läkemedelsutsättning och 1 till behandlingsavbrott. Alla händelser gick tillbaka och patienterna återhämtade sig, utom i det enda fall som ledde till läkemedelsutsättning.
Patienter med recidiverande C3G eller primär IC-MPGN efter transplantation
Säkerhetsprofilen för patienterna med recidiverande C3G eller primär IC-MPGN efter transplantation (N=22) som ingick i studierna APL2-C3G-310 och APL2-C3G-204 liknade säkerhetsprofilen för hela populationen, men med högre frekvenser av svåra och allvarliga biverkningar, vilket förväntas i denna patientpopulation.
Pediatrisk population
Hos ungdomar med C3G eller primär IC-MPGN (N=28, i åldern 12 till 17 år) som deltog i studien APL2‑C3G-310 stämde säkerhetsprofilen med de övergripande resultaten. Den vanligast rapporterade biverkningen i denna patientpopulation var reaktioner vid infusionsstället.
Säkerheten för pegcetakoplan har inte studerats hos pediatriska patienter under 12 år.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via
Suomi/Finland
[Finnish]
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
[Swedish]
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Fall av överdosering har rapporterats efter marknadsintroduktionen. Inga nya säkerhetsrelaterade händelser har observerats. Vid överdosering bör patienten övervakas för tecken eller symtom på biverkningar och lämplig symtomatisk behandling sättas in.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Immunsuppressiva medel, komplementhämmare, ATC-kod: L04AJ03
Verkningsmekanism
Pegcetakoplan är en symmetrisk molekyl som består av två identiska pentadekapeptider som är kovalent bundna till ändarna på en linjär 40 kDa PEG-molekyl. Peptiddelarna binder till komplement C3 och C3b och utövar en omfattande hämning av komplementkaskaden. PEG‑delen på 40 kDa ger bättre löslighet och längre residenstider i kroppen efter administrering av läkemedlet.
Pegcetakoplan binder till komplementprotein C3 och dess aktiveringsfragment C3b med hög affinitet och reglerar därmed klyvningen av C3 och bildandet av effektorer nedströms i komplementaktiveringen. Vid PNH underlättas extravaskulär hemolys (EVH) av C3b‑opsonisering medan intravaskulär hemolys (IVH) medieras av membranattackkomplex (MAC) nedströms. Pegcetakoplan utövar omfattande reglering av komplementkaskaden genom att agera proximalt vid bildande av både C3b och MAC och därmed kontrollera de mekanismer som leder till EVH och IVH.
Vid C3G och primär IC-MPGN sker en kraftig ansamling av nedbrytningsprodukter från C3 i njurarnas glomeruli, vilket leder till skada på njurparenkymet och nedsatt njurfunktion. Pegcetakoplan verkar på uppströms effektorer på komplementaktivering (C3 och C3b) och hämmar därmed aktivering initierad av alla komplementvägar (alternativa, klassiska och lektinvägen). Genom att hämma C3 avhjälper pegcetakoplan den felaktiga C3-aktiveringen direkt och modifierar den underliggande sjukdomen genom att minska den kraftiga ansamlingen av nedbrytningsprodukter från C3 i njurarnas glomeruli. Genom att verka på C3b hämmar pegcetakoplan också aktiviteten hos C3-konvertas i den alternativa vägen genom en ytterligare verkningsmekanism i komplementkaskaden. Detta förebygger ansamlingen av nedbrytningsprodukter av C3 i glomeruli ytterligare.
Farmakodynamisk effekt
PNH
I studie APL2‑302 ökade den genomsnittliga C3‑koncentrationen i serum från 0,94 g/l vid baslinjen till 3,83 g/l vecka 16 i pegcetakoplangruppen och bibehölls på den nivån t.o.m. vecka 48. I studie APL2‑308 ökade genomsnittlig C3‑koncentration i serum från 0,95 g/l vid baslinjen till 3,56 g/l vecka 26.
I studie APL2‑302 ökade den genomsnittliga procentandelen av PNH typ II + III erytrocyter från 66,80 % vid baslinjen till 93,85 % vecka 16 och bibehölls på den nivån t.o.m. vecka 48. I studie APL2‑308 ökade den genomsnittliga procentandelen av PNH typ II + III erytrocyter från 42,4 % vid baslinjen till 90,0 % vecka 26.
I studie APL2‑302 minskade den genomsnittliga procentandelen av PNH typ II + III erytrocyter med deponering av C3 från 17,73 % vid baslinjen till 0,20 % vecka 16 och bibehölls på den nivå t.o.m. vecka 48. I studie APL2‑308 minskade den genomsnittliga procentandelen av PNH typ II + III erytrocyter med deponering av C3 från 2,85 % vid baslinjen till 0,09 % vecka 26.
C3G och primär IC-MPGN
I studien APL2-C3G-310 ökade den genomsnittliga C3-koncentrationen i serum från 0,62 g/l vid baslinjen till 3,71 g/l vecka 26 i pegcetakoplangruppen, och effekten bibehölls fram till vecka 52. I placebogruppen förblev C3-koncentrationerna stabila fram till vecka 26 (0,57 g/l vid baslinjen; 0,58 g/l vecka 26) och ökade vid byte till pegcetakoplan till 3,59 g/l vecka 52.
Den genomsnittliga serumkoncentrationen av sC5b-9 minskade från 902,5 ng/ml vid baslinjen till 290,2 ng/ml vecka 26 i pegcetakoplangruppen, och effekten bibehölls fram till vecka 52. I placebogruppen förblev koncentrationerna av sC5b-9 stabila (768,3 ng/ml vid baslinjen; 759,9 ng/ml vecka 26) och minskade vid byte till pegcetakoplan till 272,9 ng/ml vecka 52.
Clearance av glomerulära C3-ansamlingar efter 6 månader observerades baserat på en större andel patienter som fick infärgningsgrad noll i pegcetakoplangruppen (71,4 %) jämfört med placebogruppen (8,8 %).
Klinisk effekt och säkerhet
PNH
Effekt och säkerhet för pegcetakoplan hos patienter med PNH bedömdes i två öppna, randomiserade, kontrollerade fas 3‑studier: på patienter som tidigare behandlats med komplementhämmare i studie APL2‑302 och på patienter som inte tidigare behandlats med komplementhämmare i studie APL2-308. I båda studierna var dosen pegcetakoplan 1 080 mg två gånger i veckan. Vid behov kunde dosen justeras till 1 080 mg var tredje dag.
Studie på vuxna patienter som tidigare behandlats med komplementhämmare (APL2-302)
Studie APL2-302 var en öppen, randomiserad studie med en aktiv komparatorkontrollerad period på 16 veckor som följdes av en 32‑veckors öppen period (open labelled period, OLP). Till studien rekryterades patienter med PNH som hade fått behandling med en stabil dos av ekulizumab under minst de föregående 3 månaderna och med Hb-nivåer < 10,5 g/dl. Lämpliga patienter började i en run‑in‑fas på 4 veckor under vilken de fick pegcetakoplan 1 080 mg subkutant två gånger per vecka i tillägg till deras aktuella dos av ekulizumab. Patienterna randomiserades i kvoten 1:1 till att få antingen 1 080 mg pegcetakoplan två gånger per vecka eller deras aktuella dos av ekulizumab under den randomiserade kontrollerade perioden (randomized controlled period, RCP) på 16 veckor. Randomiseringen stratifierades till antalet transfusioner av packade erytrocyter (PRBC) inom 12 månader före dag ‑28 (< 4; ≥ 4) och antalet trombocyter vid screening (< 100 000/mm3; ≥ 100 000/mm3). Patienter som slutförde RCP gick in i OLP under vilken alla patienter fick pegcetakoplan i upp till 32 veckor (patienter som fick ekulizumab under RCP gick in i en 4‑veckors run‑in‑period innan de gick över till monoterapi med pegcetakoplan).
De primära och sekundära effektmåtten bedömdes vecka 16. Det primära effektmåttet var förändring av Hb-nivå från baslinjen till vecka 16 (under RCP). Baslinjen definierades som de genomsnittliga mätningarna före den första dosen av pegcetakoplan (i början av run‑in‑perioden). Viktiga sekundära effektmått var undvikande av transfusion, definierat som andelen patienter som inte krävde en transfusion under RCP, och förändring från baslinjen till vecka 16 av absolut retikulocytantal, LDH‑nivå och poängskala på FACIT-(Functional Assessment of Chronic Illness Therapy)‑Fatigue.
Totalt 80 patienter deltog i run‑in‑perioden. I slutet av run‑in‑perioden randomiserades alla 80, 41 till pegcetakoplan och 39 till ekulizumab. Demografiska egenskaper och sjukdomsegenskaper vid baslinjen var generellt väl balanserade mellan behandlingsgrupperna (se tabell 2). Totalt 38 patienter i gruppen som fick behandling med pegcetakoplan och 39 patienter i ekulizumabgruppen avslutade 16 veckors RCP och fortsatte i den öppna perioden på 32 veckor. Totalt 12 av 80 (15 %) patienter som fick pegcetakoplan avslutade studien på grund av biverkningar. 15 patienter fick sin dos justerad till 1 080 mg var tredje dag i enlighet med protokollet. Tolv patienter utvärderades för nytta och 8 av de 12 patienterna uppvisade nytta av dosjusteringen.
Tabell 2: Patientdemografi och -egenskaper vid baslinjen i studie APL2‑302
| Parameter | Statistik | Pegcetakoplan (N = 41) | Ekulizumab (N = 39) |
Ålder (år) 18–64 år ≥ 65 år | Medelvärde (SD) n (%) n (%) | 50,2 (16,3) 31 (75,6) 10 (24,4) | 47,3 (15,8) 32 (82,1) 7 (17,9) |
Dosnivå av ekulizumab vid baslinjen Varannan vecka 900 mg i.v. Var 11:e dag 900 mg i.v. Varannan vecka 1 200 mg i.v. Varannan vecka 1 500 mg i.v. | n (%) n (%) n (%) n (%) | 26 (63,4) 1 (2,4) 12 (29,3) 2 (4,9) | 29 (74,4) 1 (2,6) 9 (23,1) 0 |
| Kvinnor | n (%) | 27 (65,9) | 22 (56,4) |
| Tid sedan PNH‑diagnos (år) till dag ‑28 | Medelvärde (SD) | 8,7 (7,4) | 11,4 (9,7) |
| Hemoglobinnivå (g/dl) | Medelvärde (SD) | 8,7 (1,1) | 8,7 (0,9) |
| Retikulocytantal (109/l) | Medelvärde (SD) | 218 (75,0) | 216 (69,1) |
| LDH‑nivå (U/l) | Medelvärde (SD) | 257,5 (97,6) | 308,6 (284,8) |
| Total FACIT‑Fatigue* | Medelvärde (SD) | 32,2 (11,4) | 31,6 (12,5) |
| Antal transfusioner under de senaste 12 månaderna före dag ‑28 | Medelvärde (SD) | 6,1 (7,3) | 6,9 (7,7) |
| < 4 | n (%) | 20 (48,8) | 16 (41,0) |
| ≥ 4 | n (%) | 21 (51,2) | 23 (59,0) |
| Trombocytantal vid screening (109/l) | Medelvärde (SD) | 167 (98,3) | 147 (68,8) |
| Trombocytantal vid screening < 100 000/mm3 | n (%) | 12 (29,3) | 9 (23,1) |
| Trombocytantal vid screening ≥ 100 000/mm3 | n (%) | 29 (70,7) | 30 (76,9) |
| Anamnes på aplastisk anemi | n (%) | 11 (26,8) | 9 (23,1) |
| Anamnes på myelodysplastiskt syndrom | n (%) | 1 (2,4) | 2 (5,1) |
*FACIT‑Fatigue är mätt på skalan 0–52 och högre värden anger mindre trötthet.
Pegcetakoplan var överlägset ekulizumab för det primära effektmåttet hemoglobinförändring från baslinjen (P < 0,0001).
Figur 1. Justerad genomsnittlig förändring av hemoglobin (g/dl) från baslinjen till vecka 16 i APL2-302
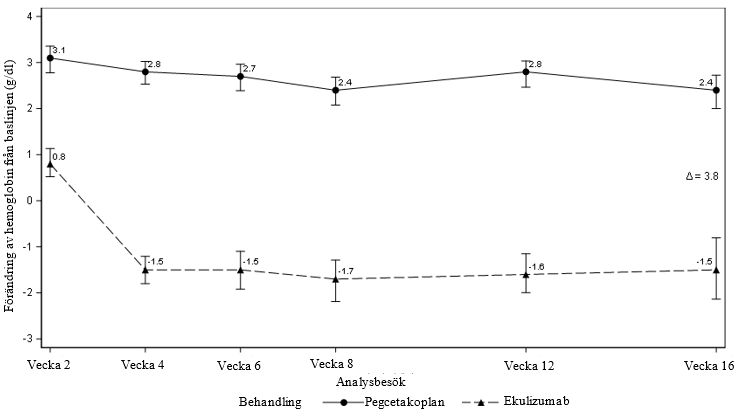
Non‑inferiority påvisades för viktiga sekundära effektmått som undvikande av transfusion och förändring av ARC från baslinjen.
Non‑inferiority påvisades inte för förändring av LDH från baslinjen.
På grund av hierarkisk testning utfördes ingen statistisk formell testning för förändring från baslinjen för poäng på FACIT‑Fatigue.
Justerade medelvärden, behandlingsskillnad, konfidensintervall och statistiska analyser som utfördes för viktiga sekundära effektmått visas i figur 2.
Figur 2. Analys av viktiga sekundära effektmått i APL2-302
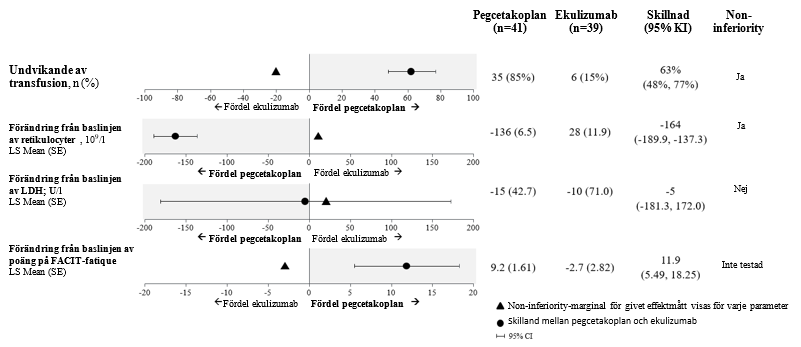
Resultaten överensstämde över alla stödjande analyser för primära och viktiga sekundära effektmått, inklusive alla observerade data med data efter transfusion inkluderade.
Normalisering av Hb uppnåddes hos 34 % av patienterna i gruppen som fick pegcetakoplan jämfört med 0 % i ekulizumabgruppen vecka 16. Normalisering av LDH uppnåddes hos 71 % av patienterna i gruppen som fick pegcetakoplan jämfört med 15 % i ekulizumabgruppen.
Totalt 77 patienter gick in i 32‑veckors OLP, under vilken alla patienter fick pegcetakoplan, vilken gav en total exponering på upp till 48 veckor. Resultaten vecka 48 överensstämde generellt med resultaten vecka 16 och stödjer ihållande effekt.
Studie på vuxna patienter som inte tidigare behandlats med komplementhämmare (APL2‑308)
Studie APL2‑308 var en öppen, randomiserad, kontrollerad studie som rekryterade patienter med PNH som inte behandlats med komplementhämmare inom 3 månader före rekrytering och som hade Hb-nivåer under den nedre normalgränsen (LLN). Lämpliga patienter randomiserades i förhållandet 2:1 till att få pegcetakoplan eller understödjande vård (t.ex. transfusioner, kortikosteroider, tillskott av järn, folsyra och vitamin B12), nedan kallad kontrollarmen, under hela 26-veckorsperioden.
Randomiseringen stratifierades på basis av antalet transfusioner av PRBC inom 12-månadersperioden före dag -28 (<4; ≥4). En patient som placerats i kontrollarmen och som någon gång under studien hade Hb-nivåer ≥2 g/dl under baslinjen eller drabbades av en PNH-associerad tromboembolisk händelse kunde enligt protokollet gå över till pegcetakoplan under återstoden av studien.
Totalt 53 patienter randomiserades, 35 till pegcetakoplan och 18 till kontrollarmen. Demografi och sjukdomskarakteristika var generellt välbalanserade mellan behandlingsgrupperna. Genomsnittsåldern var 42,2 år i pegcetakoplanarmen och 49,1 år i kontrollarmen. Genomsnittligt antal PRBC-transfusioner under de 12 månaderna före screening var 3,9 i pegcetakoplanarmen och 5,1 i kontrollarmen. Fem patienter i vardera arm (14,3 % i pegcetakoplanarmen och 27,8 % i kontrollarmen) hade anamnes på aplastisk anemi. Ytterligare baslinjevärden var följande: genomsnittliga Hb-nivåer vid baslinjen (pegcetakoplanarmen: 9,4 g/dl jämfört med kontrollarmen: 8,7 g/dl) ARC (pegcetakoplanarmen: 230,2 × 109/l, jämfört med kontrollarmen: 180,3 × 109/l), LDH (pegcetakoplanarmen:2 151,0 >U/l jämfört med kontrollarmen: 1 945,9 U/l och trombocytantal (pegcetakoplanarmen: 191,4 × 109/l jämfört med kontrollarmen: 125,5 × 109/l). Elva av 18 patienter randomiserade till kontrollarmen gick över till pegcetakoplan på grund av Hb-nivåer som sjönk med ≥2 g/dl under baslinjen. Av de 53 randomiserade patienterna fick 52 (97,8 %) profylaktisk antibiotikabehandling enligt lokala förskrivningsriktlinjer.
De primära och sekundära effektmåtten bedömdes vecka 26. De två ko-primära effektmåtten var Hb-stabilisering, definierad som undvikande av en >1 g/dl sänkning av Hb-koncentrationen från baslinjen utan transfusion, och förändring av LDH-koncentration från baslinjen.
I gruppen som behandlades med pegcetakoplan uppnådde 30 av 35 patienter (85,7 %) Hb-stabilisering jämfört med 0 patienter i kontrollarmen. Den justerade skillnaden mellan pegcetakoplan och kontrollarmen var 73,1 % (95 % KI, 57,2 % till 89,0 %; p<0,0001).
Minsta kvadrat-(LS)-medelvärde (SE) för förändringar från baslinjen av LDH-koncentration vecka 26 var -1 870 U/l i gruppen behandlad med pegcetakoplan jämfört med -400 U/l i kontrollarmen (p<0,0001). Skillnaden mellan pegcetakoplan och kontrollarmen var -1 470 (95 % KI, -2 113 till ‑827). Behandlingsskillnader mellan pegcetakoplan och kontrollarmen var tydliga vecka 2 och kvarstod t.o.m. vecka 26 (figur 3). LDH-koncentrationerna i kontrollarmen förblev förhöjda.
Figur 3. Genomsnittlig (±SE) LDH-koncentration (U/l) över tid per behandlingsgrupp i studie APL2‑308
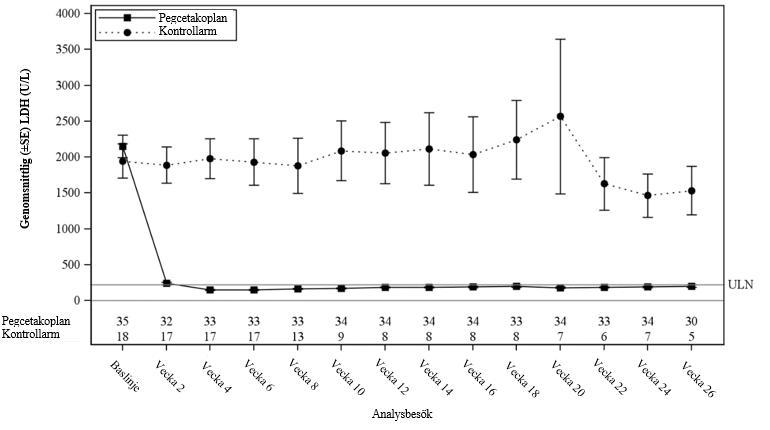
För de sekundära effektmåtten hemoglobinsvar i avsaknad av transfusioner, förändring av hemoglobinnivå och ARC-förändring uppvisade gruppen behandlad med pegcetakoplan en signifikant behandlingsskillnad jämfört med kontrollarmen (tabell 3).
Tabell 3: Analys av viktiga sekundära effektmått i studie APL2‑308
| Parameter | Pegcetakoplan (N=35) | Kontrollarm (N=18) | Skillnad (95 % KI) p-värde |
Hemoglobinsvar i avsaknad av transfusionera n (%) | 25 (71%) | 1 (6%) | 54% (34%, 74%) p < 0,0001) |
Förändring av hemoglobinnivå (g/dl) från baslinjen till vecka 26 LS-medelvärde (SE) | 2,9 (0,38) | 0,3 (0,76) | 2,7 (1,0; 4,4) |
Förändring av ARC (109/l) från baslinjen till vecka 26 LS-medelvärde (SE) | -123 (9,2) | -19 (25,2) | -104 (-159, -49) |
a Hemoglobinsvar definierades som en ökning på ≥ 1 g/dl av hemoglobin från baslinjen vid vecka 26.
ARC = absolut retikulocytantal, KI = konfidensintervall, LS = minsta kvadrat, SE = standardfel
C3G och primär IC-MPGN
Effekt och säkerhet för pegcetakoplan hos patienter med C3G eller primär IC-MPGN bedömdes i den randomiserade, placebokontrollerade, dubbelblinda fas 3-studien APL2-C3G-310, som inkluderade vuxna och ungdomar med C3G eller primär IC-MPGN i ursprunglig njure eller recidiverande efter transplantation.
Dosen pegcetakoplan var 1 080 mg två gånger i veckan för vuxna och ungdomar som vägde ≥ 50 kg, eller viktbaserad för ungdomar som vägde < 50 kg.
Studie på vuxna patienter och ungdomar med C3G eller primär IC-MPGN (APL2-C3G-310)
Studien APL2-C3G-310 var en randomiserad, dubbelblind studie med en placebokontrollerad period på 26 veckor som följdes av en 26 veckor lång öppen period. Denna studie rekryterade ungdomar mellan 12 och 17 år och vuxna med C3G eller primär IC-MPGN. Studien rekryterade patienter med sjukdom i ursprunglig njure eller recidiverande sjukdom efter transplantation som hade proteinuri ≥ 1 g/dag och eGFR ≥ 30 ml/min/1,73 m2. Patienterna stod på en stabil och optimerad dosregim för behandling av C3G/primär IC‑MPGN (t.ex. RAS-blockerare, natriumglukostransportör 2-hämmare [SGLT2-hämmare], immunsupprimerande medel, systemiska kortikosteroider som ej överskred en prednisonekvivalent om 20 mg/dag) i minst 12 veckor före randomisering.
Lämpliga patienter randomiserades i förhållandet 1:1 till att få pegcetakoplan eller placebo subkutant två gånger i veckan under den 26 veckor långa randomiserade kontrollerade perioden. Två stratifieringsfaktorer applicerades på randomiseringen: patienter med recidiv efter transplantation mot patienter med sjukdom i ursprunglig njure, och patienter med njurbiopsi vid baslinjen (antingen tagen under screeningen eller inom 28 veckor före randomiseringen) mot patienter utan njurbiopsi vid baslinjen. Under den randomiserade kontrollerade perioden hölls förändringar av baslinjebehandlingarna för C3G/primär IC-MPGN till ett minimum och gjordes endast när det krävdes för patientens hälsas skull. Patienter som slutförde den randomiserade kontrollerade perioden gick vidare till den 26 veckor långa öppna perioden, där alla deltagare behandlades med pegcetakoplan två gånger i veckan.
Totalt 124 patienter randomiserades; 63 till pegcetakoplan och 61 till placebo. Demografiska och sjukdomsrelaterade egenskaper vid baslinjen var generellt balanserade mellan de två grupperna (se tabell 4). Totalt 118 patienter slutförde de 26 veckornas randomiserad kontrollerad period, varav 114 patienter slutförde den öppna behandlingsperioden med pegcetakoplan (N=59 pegcetakoplan-till-pegcetakoplan; N=55 placebo-till-pegcetakoplan).
Tabell 4: Patienternas demografiska och sjukdomsrelaterade egenskaper vid baslinjen i studien APL2-C3G-310
| Parameter | Statistik | Pegcetakoplan (N=63) | Placebo (N=61) |
| Ålder (år) | Medelvärde (SD) | 28,2 (17,1) | 23,6 (14,3) |
| Ungdomar (12–17 år) | n (%) | 28 (44,4) | 27 (44,3) |
| Vuxna ≥ 18 år | n (%) | 35 (55,6) | 34 (55,7) |
Kön Man Kvinna | n (%) n (%) | 26 (41,3) 37 (58,7) | 28 (45,9) 33 (54,1) |
| Sjukdomstyp vid screening | |||
| C3G | n (%) | 51 (81,0) | 45 (73,8) |
| C3GN | n (%) | 45 (71,4) | 41 (67,2) |
| DDD | n (%) | 4 (6,3) | 4 (6,6) |
| Obestämd | n (%) | 2 (3,2) | 0 |
| IC-MPGN | n (%) | 12 (19,0) | 16 (26,2) |
| Tid sedan diagnos på C3G/IC-MPGN (år) | Medelvärde (SD) | 3.64 (3,47) | 3,76 (3,62) |
| Tidigare njurtransplantation | n (%) | 5 (7,9) | 4 (6,6) |
| Tid sedan senaste njurtransplantation (år) | Medelvärde (SD) | 11,4 (6,7) | 5,8 (6,4) |
| Tid sedan senaste recidiv efter transplantation (år) | Medelvärde (SD) | 1,47 (1,49) | 1,38 (1,64) |
| Baslinjevärde FMU uPCR (mg/g) i triplikat | Medelvärde (SD) | 3 124 (2 408) | 2 541 (2 015) |
| Baslinjevärde eGFR (ml/min/1,73 m2) | Medelvärde (SD) | 78,5 (34,1) | 87,2 (37,2) |
| C3c-infärgning vid baslinjebiopsi | |||
| 3+ | n (%) | 51 (81,0) | 51 (83,6) |
| 2+ | n (%) | 12 (19,0) | 10 (16,4) |
| Baslinjevärde serumalbumin (g/dl) | Medelvärde (SD) | 3,31 (0,61) | 3,39 (0,70) |
| Baslinjevärde C3 i serum (mg/dl) | Medelvärde (SD) | 60,6 (45,7) | 56,3 (35,6) |
| Sjukdomsmanifestationer | |||
| Ödem | n (%) | 45 (71,4) | 32 (52,5) |
| Utmattning | n (%) | 16 (25,4) | 8 (13,1) |
| Hematuri | n (%) | 37 (58,7) | 39 (63,9) |
| Högt blodtryck | n (%) | 35 (55,6) | 29 (47,5) |
| Nefrotiskt syndrom | n (%) | 32 (50,8) | 27 (44,3) |
| Användning av andra behandlingar vid baslinjen* | |||
| Medel som verkar på renin-angiotensinsystemet | n (%) | 59 (93,7) | 54 (88,5) |
| Immunsupprimerande medel | n (%) | 49 (77,8) | 45 (73,8) |
| Glukokortikoider | n (%) | 29 (46,0) | 27 (44,3) |
*Inom 12 veckor före inskrivning i studien.
C3G = C3-glomerulopati, C3GN = C3-glomerulonefrit, DDD = dense deposit disease, IC MPGN = immunkomplexmedierad membranoproliferativ glomerulonefrit, FMU = första morgonurinen, uPCR = protein-kreatininkvot i urin, eGFR = beräknad glomerulär filtrationshastighet, SD = standardavvikelse
Det primära och de huvudsakliga sekundära effektmåtten bedömdes vecka 26. Det primära effektmåttet var log-transformerad protein-kreatininkvot i första morgonurin (FMU) vecka 26 jämfört med vid baslinjen.
Pegcetakoplan var överlägset placebo. Det gav en statistiskt signifikant minskning om 68,1 % (95 % KI: 57,3 % till 76,2 %, p < 0,0001) av uPCR från baslinjen jämfört med placebo efter 26 veckors behandling (‑67,2 % [95 % KI: -74,9 % till -57,2 %] och + 2,9 % [95 % KI: -8,6 % till 15,9 %] för pegcetakoplan respektive placebo). Effekt av liknande storlek observerades i undergrupper oavsett ålder (ungdomar mot vuxna), sjukdomstyp (C3G mot primär IC-MPGN), sjukdomsstatus (ursprunglig njure mot recidiv efter transplantation), och samtidig användning av immunsupprimerande medel/glukokortikoider (ja mot nej). Effekten av pegcetakoplan på uPCR bibehölls till och med vecka 52 (-67,2 % från baslinjen). Patienter som bytte från placebo till pegcetakoplan vecka 26 (figur 4) upplevde en liknande minskning (‑51,3 %) vecka 52.
Figur 4. Geometrisk medelkvot (95 % CI) för FMU uPCR jämfört med baslinjevärdena över tid enligt behandlingsgrupp från MMRM-modell i studien APL2-C3G-310
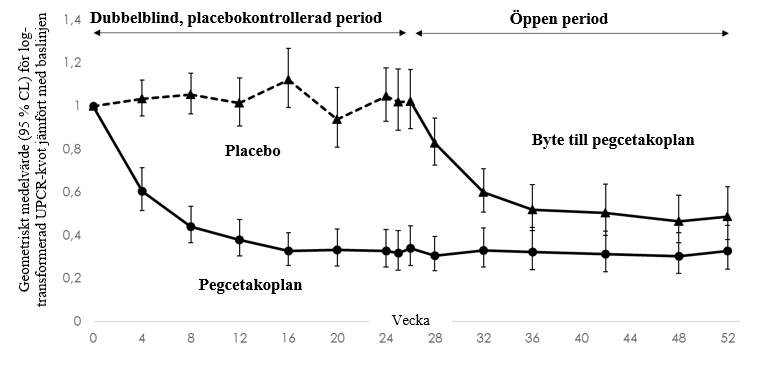
Obs: Geometrisk medelkvot beräknad från återexponentierade minsta kvadrat-medelvärden.
KI = konfidensintervall, FMU = första morgonurin, uPCR = protein-kreatininkvot i urin, MMRM = blandad modell med upprepade mätningar (mixed model of repeated measure)
Pegcetakoplanbehandling i 26 veckor uppvisade en statistiskt signifikant förbättring av det huvudsakliga sekundära effektmåttet relaterat till minskad proteinuri: 60,3 % av patienterna behandlade med pegcetakoplan uppnådde en ≥ 50 % minskning av uPCR jämfört med 4,9 % i placebogruppen, en skillnad på 52,7 % (95 % KI: 29,2 %‑76,2 %; p < 0,0001).
Pegcetakoplanbehandling i 26 veckor resulterade i en större andel patienter som uppnådde en minst tvåfaldig minskning, på en skala mellan 0 och 3, av intensiteten av infärgning av C3 i njurvävnad: 26 (74,3 %) patienter på pegcetakoplan jämfört med 4 (11,8 %) på placebo och en skillnad på 64,3 % (95 % KI: 41,4 %‑87,2 %, nominellt p < 0,0001).
Pegcetakoplanbehandling i 26 veckor visade stabilisering av eGFR med en förändring från baslinjen på -1,497 (2,242) på pegcetakoplan jämfört med -7,808 (1,919) på placebo, och en behandlingsskillnad på 6,312 ml/min/1,73m2 (95 % KI: 0,501; 12,122, nominellt p = 0,0333). Effekten av pegcetakoplan på eGFR bibehölls till och med vecka 52. Patienter som bytte från placebo till pegcetakoplan vecka 26 upplevde en liknande stabilisering vecka 52.
Effekt av liknande storlek observerades i allmänhet gällande ≥ 50 % minskning av proteinuri, clearance av infärgning av C3 och stabilisering av eGFR i undergrupper oavsett ålder (ungdomar mot vuxna), sjukdomstyp (C3G mot primär IC-MPGN), sjukdomsstatus (sjukdom i ursprunglig njure mot recidiv efter transplantat) och samtidig användning av immunsupprimerande medel/glukokortikoider (ja mot nej) vecka 26.
Studie på vuxna patienter med recidiverande C3G eller primär IC-MPGN efter transplantation (APL2-C3G-204)
Studien APL2-C3G-204 var en öppen, randomiserad fas 2-studie på 13 vuxna patienter med recidiverande C3G (N=10) eller primär IC-MPGN (N=3) efter transplantation som pågick i 52 veckor.
Under studiens första 12 veckor fick 10 patienter pegcetakoplan utöver standardbehandling, och 3 fick endast standardbehandling. Alla patienterna fick pegcetakoplan från vecka 13 till vecka 52.
Det primära effektmåttet, minskning av intensiteten av C3-infärgning på njurbiopsi vecka 12, observerades hos 50 % av patienterna som behandlades med pegcetakoplan (5 av 10 patienter, varav 4 fick infärgningsgrad noll), och 33,3 % av patienterna i kontrollgruppen (1 av 3 patienter; denna patient fick infärgningsgrad 1).
Generellt var förändringarna och de procentuella förändringarna från baslinjen vad gäller eGFR (sekundärt effektmått) små. Genomsnittlig (SD) eGFR förändrades från 52,3 (12,11) ml/min/1,73 m2 vid baslinjen till 57,3 (25,12) ml/min/1,73 m2 vecka 52, och medianvärdet för eGFR förändrades från 50,5 ml/min/1,73 m2 vid baslinjen till 58,5 ml/min/1,73 m2 vecka 52. De flesta patienterna (9 av 13 patienter [69,2 %]) i båda grupperna hade uppnått stabilisering eller förbättring av eGFR vid vecka 52.
Immungenicitet
Två olika analyser för detektion av anti‑läkemedelsantikroppar (ADA) i form av anti‑pegcetakoplan‑peptidantikroppar användes i kliniska studier av PNH respektive C3G och primär IC-MPGN. Analysen som användes för C3G eller primär IC-MPGN hade högre sensitivitet. Skillnaderna mellan analyserna förhindrar meningsfull jämförelse mellan studierna som beskrivs nedan vad gäller incidensen av ADA.
I kliniska studier av PNH var incidensen av ADA (behandlingsutlöst ADA eller boostrad ADA från befintlig nivå) låg och om de förekom hade de ingen noterbar påverkan på farmakodynamik/farmakokinetik, effekt eller säkerhetsprofil för pegcetakoplan. Under hela studierna APL2‑302 och APL2-308 utvecklade 3 av 126 patienter som exponerades för pegcetakoplan positiva anti‑pegcetakoplan‑peptidantikroppar. Alla 3 patienter testade också positivt för neutraliserande antikropp (NAb). NAb‑svar hade ingen tydlig påverkan på farmakokinetik eller klinisk effekt. Arton av 126 patienter utvecklade anti‑PEG‑antikroppar: 9 var behandlingsutlösta och 9 var behandlingsboostrade.
I kliniska studier av C3G och primär IC-MPGN var incidensen av ADA (behandlingsutlöst ADA eller boostrad ADA från befintlig nivå) i studien APL2-C3G-310 23,6 % för anti-PEG och 16,3% för anti‑pegcetakoplan‑peptid. Baserat på populationsfarmakokinetisk och -farmakodynamisk analys hade ADA ingen kliniskt meningsfull påverkan på effekt eller farmakokinetik/farmakodynamik i en poolad analyspopulation. Fem patienter testade också positivt för NAb. NAb-svar hade ingen synbar påverkan på farmakokinetik eller klinisk effekt. Tjugonio av 123 patienter utvecklade anti-PEG-antikroppar; 14 var behandlingsutlösta och 15 var behandlingsboostrade. Bland patienter med recidiverande sjukdom efter transplantation i studien APL2-C3G-204 var det ingen som utvecklade ett positivt ADA-svar (behandlingsutlöst ADA eller boostrad ADA från befintlig nivå) mot pegcetakoplan-peptid eller PEG. Under den 26 veckor långa placebokontrollerade perioden i studien APL2‑C3G-310 sågs ingen påvisbar påverkan av ADA på säkerheten hos pegcetakoplanbehandling.
Pediatrisk population
Europeiska läkemedelsmyndigheten har senarelagt kravet att skicka in studieresultat för ASPAVELI för en eller flera grupper av den pediatriska populationen för PNH respektive C3G eller primär IC-MPGN (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Absorption
Pegcetakoplan administreras som en subkutan infusion och absorberas successivt till den systemiska cirkulationen med median Tmax mellan 108 och 144 timmar (4,5 till 6,0 dagar) efter en subkutan engångsdos till friska frivilliga.
Serumkoncentrationer i steady‑state efter dosering två gånger per vecka av 1 080 mg till patienter med PNH uppnåddes cirka 4 till 6 veckor efter den första dosen. Hos patienter som tidigare behandlats med komplementhämmare (studie APL2-302) var det geometriska medelvärdet (%CV) för serumkoncentrationer i steady‑state mellan 655 (18,6 %) och 706 (15,1 %) µg/ml hos patienter behandlade under 16 veckor. Steady‑state‑koncentrationerna hos patienterna (n=22) som fortsatte att få pegcetakoplan fram till vecka 48 var 623 µg/ml (39,7 %), vilket tyder på att terapeutiska koncentrationer av pegcetakoplan bibehölls till och med vecka 48. Hos patienter som inte tidigare behandlats med komplementhämmare (studie APL2‑308) var det geometriska medelvärdet (%CV) för serumkoncentrationer i steady‑state vecka 26 744 µg/ml (25,5 %) vid dosering två gånger i veckan. Biotillgängligheten för en subkutan dos av pegcetakoplan uppskattas vara 76 % baserat på en farmakokinetisk populationsanalys.
Serumkoncentrationer i steady-state efter dosering två gånger per vecka av 1 080 mg till patienter med C3G eller primär IC-MPGN uppnåddes cirka 4 till 8 veckor efter den första dosen, och terapeutiska koncentrationer av pegcetakoplan bibehölls till och med vecka 52. Hos patienterna i studien APL2-C3G-310 varierade de genomsnittliga serumkoncentrationerna i steady-state (%CV) mellan 715,8 (31,2 %) och 765,7 (23,2 %) μg/ml fram till vecka 26 och kvarstod mellan 670,1 (30,1 %) och 726,6 (30,5 %) μg/ml fram till vecka 52.
Distribution
Genomsnittlig (%CV) distributionsvolym för pegcetakoplan är cirka 3,98 l (32 %) hos patienter med PNH baserat på en farmakokinetisk populationsanalys.
Genomsnittlig (%CV) central distributionsvolym för pegcetakoplan är cirka 4,31 l (32,1 %) hos vuxna patienter med C3G eller primär IC-MPGN.
Metabolism/eliminering
Baserat på dess pegylerade peptidstruktur förväntas metabolismen för pegcetakoplan uppkomma via katabola vägar och brytas ned till små peptider, aminosyror och PEG. Resultat av en radioaktivt märkt studie på cynomolgusapor tyder på att den preliminära elimineringsvägen för den radioaktivt märkta peptiden är via urinutsöndring. Även om elimineringen av PEG inte har studerats är det känt för att genomgå renal utsöndring.
Pegcetakoplan visade ingen hämning eller induktion av isoformerna för det CYP‑enzym som testats vilket resultat av in vitro-studier visar. Pegcetakoplan är varken ett substrat eller en hämmare av humana upptags- eller effluxtransportörer.
Efter flera subkutana doser av pegcetacoplan till patienter med PNH var genomsnittlig (%CV) clearance 0,015 l/timme (30 %) och median effektiv elimineringshalveringstid (t1/2) var 8,6 dagar enligt beräkning i den farmakokinetiska populationsanalysen.
Beräknat genomsnitt (CV%) för clearance är 0,012 l/timme (43 %) hos vuxna patienter med C3G eller primär IC-MPGN. Medianvärdet för terminal halveringstid t1/2 är 10,1 dagar för vuxna patienter med C3G eller primär IC-MPGN.
Linjäritet/icke-linjäritet
Exponering av pegcetakoplan ökar på ett dosproportionellt sätt från 45 till 1 440 mg.
Särskilda populationer
Ingen påverkan på farmakokinetiken för pegcetakoplan identifierades för ålder (12–81 år), etnicitet eller kön baserat på resultaten från den farmakokinetiska populationsanalysen av patienter med PNH, C3G eller primär IC-MPGN.
Jämfört med en referenspatient som väger 70 kg förutspås den genomsnittliga koncentrationen vid steady-state vara cirka 20 % högre hos patienter med en kroppsvikt på 50 kg. PNH-patienter som väger 40 kg förutspås ha 45 % högre genomsnittlig koncentration. Det finns mycket få data om säkerhetsprofilen för pegcetakoplan för PNH-patienter med en kroppsvikt under 50 kg.
Äldre
Även om inga tydliga åldersrelaterade skillnader observerades i dessa studier är antalet patienter över 65 år inte tillräckligt för att fastställa om de svarar på ett annat sätt än yngre patienter. Se avsnitt Dosering och administreringssätt.
Pediatrisk population
Baserat på farmakokinetisk populationsanalys påverkar kroppsvikten hos ungdomar (12-17 år) clearance och distributionsvolym. Doseringsregimen för ungdomar med C3G eller primär IC-MPGN bygger på patientens kroppsvikt. Se avsnitt Dosering och administreringssätt. Den enligt modell förutspådda exponeringen hos ungdomar med C3G eller primär IC-MPGN stämmer i tillräcklig grad med referensexponeringen för vuxna.
Nedsatt njurfunktion
I en studie med 8 patienter med svårt nedsatt njurfunktion, definierad som kreatininclearance (CrCl) under 30 ml/min med Cockcroft‑Gaults formel (med 4 patienter med värden under 20 ml/min), hade nedsatt njurfunktion ingen effekt på farmakokinetiken för en engångsdos om 270 mg pegcetakoplan. Det finns mycket få data om patienter med PNH med nedsatt njurfunktion som har fått den kliniska dosen 1 080 mg två gånger per vecka. Baserat på farmakokinetisk populationsanalys hade eGFR ingen kliniskt meningsfull påverkan på pegcetakoplanexponering i en poolad analyspopulation. Inga kliniska data finns tillgängliga för användning av pegcetakoplan hos patienter med ESRD som kräver dialys. Se avsnitt Dosering och administreringssätt.
Prekliniska säkerhetsuppgifter
Toxikologiska data in vitro och in vivo visade ingen toxicitet som är av särskild betydelse för människa. Effekter observerade på djur vid exponeringsnivåer som liknar kliniska exponeringsnivåer beskrivs nedan. Dessa effekter observerades inte i kliniska studier.
Reproduktion hos djur
Behandling med pegcetakoplan till dräktiga cynomolgusapor vid en subkutan dos på 28 mg/kg/dag (2,9 gånger human steady‑state Cmax) från dräktighetsperioden till och med nedkomst resulterade i en statistiskt signifikant ökning av missfall och dödfödslar. Ingen maternell toxicitet och inga teratogena effekter observerades hos avkomman efter fullgången period. Dessutom sågs inga effekter på utvecklingen hos avkomman upp till 6 månader postpartum. Systemisk exponering för pegcetakoplan detekterades hos foster från apor som behandlats med 28 mg/kg/dag från organogenesperioden till och med den andra trimestern men exponeringen var minimal (mindre än 1 %, inte farmakologiskt signifikant).
Karcinogenes
Långvariga karcinogenicitetsstudier på djur med pegcetakoplan har inte utförts.
Gentoxicitet
Pegcetakoplan var inte mutagent vid testning in vitro i en analys av bakteriell omvänd mutation (Ames) och var inte gentoxiskt i en analys in vitro på humana TK6‑celler eller i en mikrokärnanalys in vivo på möss.
Toxicitet hos djur
Allmänna dosstudier utfördes på kaniner och cynomolgusapor med dagliga subkutana doser av pegcetakoplan upp till 7 gånger den humana dosen (1 080 mg två gånger per vecka). Histologiska fynd hos båda arterna inkluderade dosberoende epitelvakuolisering och infiltrat av vakuoliserade makrofager i flera vävnader. Dessa resultat har associerats med stora kumulativa doser av långkedjig PEG i andra godkända pegylerade läkemedel, var utan kliniska effekter och betraktades inte som biverkningar. Reversibilitet påvisades inte i djurstudier med pegcetakoplan efter en månad och utvärderades inte under en längre tid. Data från litteraturen tyder på reversibilitet för PEG-vakuoler.
Renal tubulär degeneration observerades mikroskopiskt hos båda könen vid exponeringar (Cmax och AUC) under eller jämförbara med dosen till människa och var minimal och icke‑progressiv mellan 4 veckor och 9 månader av daglig administrering av pegcetakoplan. Även om inga tecken på renal dysfunktion observerades hos djur är den kliniska signifikansen och den funktionella effekten av dessa resultat okänd.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Sorbitol (E 420)
Ättiksyra, koncentrerad
Natriumacetattrihydrat
Natriumhydroxid (för pH‑justering)
Vatten för injektionsvätskor
Inkompatibiliteter
Då blandbarhetsstudier saknas får detta läkemedel inte blandas med andra läkemedel.
Hållbarhet
30 månader.
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 °C‑8 °C).
Förvaras i originalkartongen. Ljuskänsligt.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
ASPAVELI infuusioneste, liuos
1080 mg (L:ei) 1 kpl (20 ml (54 mg/ml)) (3596,95 €), 8 x 1 kpl (20 ml (54 mg/ml)) (27511,40 €)
PF-selosteen tieto
Injektionsflaska av typ I‑glas med en propp (klorbutyl eller brombutyl) och en försegling (aluminium) med ett flip‑off‑lock (polypropen) innehållande 54 mg/ml steril lösning.
Varje enskild förpackning innehåller 1 injektionsflaska.
Multipack innehållande 8 (8 förpackningar med 1) injektionsflaskor.
Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras.
Läkemedlets utseende:
Klar, färglös till lätt gulaktig vattenlösning med pH 5,0.
Särskilda anvisningar för destruktion och övrig hantering
ASPAVELI levereras i en bruksfärdig injektionsflaska för engångsbruk. Eftersom lösningen inte innehåller något konserveringsmedel ska detta läkemedel infunderas omedelbart efter beredning av sprutan.
ASPAVELI är en klar, färglös till lätt gulaktig vattenlösning. Den får inte användas om vätskan är grumlig, innehåller partiklar eller är mörkgul.
Låt alltid injektionsflaskan vara i rumstemperatur under cirka 30 minuter före användning.
Ta bort flip‑off‑locket på injektionsflaskan för att exponera den centrala delen av injektionsflaskans grå gummipropp. Rengör proppen med en ny alkoholservett och låt proppen torka. Använd inte om flip‑off‑locket saknas eller är skadat.
Förberedelse av sprutan
Alternativ 1: Vid användning av en nållös överföringsenhet (såsom en adapter för injektionsflaska), följ anvisningarna som tillhandahålls med utrustningen från tillverkaren.
Alternativ 2: Om överföring görs med en överföringsnål och en spruta, följ anvisningarna nedan:
- Montera en steril överföringsspruta till en steril nål.
- Dra tillbaka kolven för att fylla sprutan med luft, vilket ska vara cirka 20 ml.
- Kontrollera att injektionsflaskan är i upprätt läge. Vänd inte injektionsflaskan upp och ned.
- Tryck in den luftfyllda sprutan med den monterade överföringsnålen mitt på injektionsflaskans propp.
- Spetsen på överföringsnålen ska inte vara i lösningen för att förhindra att bubblor bildas.
- Tryck försiktigt ut luften från sprutan till injektionsflaskan. Detta injicerar luften från sprutan till injektionsflaskan.
- Vänd injektionsflaskan.
- Dra långsamt i kolven med överföringsnålen i lösningen för att fylla sprutan med den ordinerade dosen av ASPAVELI.
- Ta bort den fyllda sprutan och överföringsnålen från injektionsflaskan.
- Sätt inte på locket på överföringsnålen. Skruva loss nålen och kassera den i en behållare för vassa och skärande föremål.
Administrering
ASPAVELI ska bara administreras som en subkutan infusion med en infusionspump med ett sprutsystem eller ett kroppsburet infusionssystem (OBDS).
- Följ anvisningarna för utrustningen från tillverkaren för att förbereda infusionspumpen och slangen. Vid användning av en infusionspump är infusionsställena buken, låren, höfterna eller överarmarna. Byt infusionsställe mellan injektionerna. Vid flera infusionsställen måste det vara minst 7,5 cm mellan dessa. Infusionstiden är cirka 30 minuter (vid användning av två ställen) eller cirka 60 minuter (vid användning av ett ställe).
- Följ anvisningarna för utrustningen från tillverkaren för att förbereda det kroppsburna infusionssystemet. Vid användning av ett kroppsburet infusionssystem ska ASPAVELI administreras på ett ställe i buken. Byt infusionsställe mellan injektionerna. Infusionstiden varierar mellan patienter och är vanligtvis mellan 30 och 60 minuter.
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
ASPAVELI infuusioneste, liuos
1080 mg 1 kpl, 8 x 1 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Iptakopaani ja pegsetakoplaani: Aikuisten kohtauksittaisen yöllisen hemoglobiinivirtsaisuuden (PNH) hoito erityisin edellytyksin (3078).
Atc-kod
L04AJ03
Datum för översyn av produktresumén
15.01.2026
Yhteystiedot
Äyritie 18
01510 Vantaa
0201 558 840
www.sobi.fi
mail.fi@sobi.com