
LUPKYNIS kapseli, pehmeä 7,9 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi pehmeä kapseli sisältää 7,9 mg voklosporiinia (voclosporin).
Apuaineet, joiden vaikutus tunnetaan
Yksi pehmeä kapseli sisältää 21,6 mg etanolia ja 28,7 mg sorbitolia.
Lupkynis saattaa sisältää soijalesitiinin jäämiä, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, pehmeä (kapseli)
Kliiniset tiedot
Käyttöaiheet
Lupkynis on tarkoitettu aikuispotilaiden aktiivisen luokan III, IV tai V (myös sekamuotoisen, luokan III/V tai IV/V) SLE-nefriitin hoitoon yhdistelmänä mykofenolaattimofetiilin kanssa.
Ehto
SLE-nefriitin diagnosointiin ja hoitoon perehtyneen lääkärin on aloitettava hoito ja valvottava sen toteuttamista.
Annostus ja antotapa
SLE -nefriitin diagnosointiin ja hoitoon perehtyneen lääkärin on aloitettava Lupkynis-hoito ja valvottava sen toteuttamista.
Annostus
Suositeltu annos on 23,7 mg (kolme 7,9 mg:n pehmeää kapselia) kahdesti vuorokaudessa.
Lupkynis-annokset on suositeltavaa ottaa samaan aikaan vuorokaudesta siten, että annosten väli on mahdollisimman lähellä 12 tuntia. Annosten minimiväli on 8 tuntia. Unohtunut annos on otettava mahdollisimman pian 4 tunnin kuluessa unohtumisesta. Jos 4 tuntia ylittyy, on odotettava seuraavaan tavanomaisen lääkkeenottoaikataulun mukaiseen annokseen. Seuraavaa annosta ei pidä ottaa kaksinkertaisena.
Lupkynis-valmistetta on käytettävä yhdessä mykofenolaattimofetiilin kanssa.
Lääkärin on arvioitava hoidon teho vähintään 24 viikon jälkeen ja punnittava asianmukaisesti hoidon jatkamisen riskit ja hyödyt.
Annoksen muuttaminen eGFR:n perusteella
Ennen voklosporiinihoidon aloittamista on suositeltavaa määrittää lähtötilanteen glomerulusten laskennallinen suodatusnopeus (eGFR), minkä jälkeen arvoa seurataan kahden viikon välein ensimmäisenä hoitokuukautena ja sen jälkeen neljän viikon välein.
Annoksen muuttaminen on tarpeen potilaille, joiden eGFR on vahvistetusti (kaksi peräkkäistä arviota 48 tunnin sisällä) laskenut alle 60 ml/min/1,73 m2. Jos eGFR pysyy tasolla ≥ 60 ml/min/1,7 m2, annosta ei tarvitse muuttaa (ks. taulukko 1).
Taulukko 1: Suositellut eGFR-arvoon perustuvat annosmuutokset
Vahvistettu eGFR:n lasku lähtötilanteesta1 | Suositus |
≥ 30 %:n lasku | Lopeta voklosporiinin anto. eGFR:n korjauduttua aloita hoito uudelleen 7,9 mg:lla (1 kapselilla) kahdesti vuorokaudessa ja suurenna annosta potilaan sietokyvyn mukaan munuaistoiminnan perusteella. |
> 20 %:n – < 30 %:n lasku | Pienennä voklosporiiniannosta 7,9 mg:lla (1 kapselilla) kahdesti vuorokaudessa. Arvioi tilanne uudelleen kahden viikon kuluessa; jos eGFR:n lasku ei ole korjautunut, pienennä annosta jälleen 7,9 mg:lla (1 kapselilla) kahdesti vuorokaudessa. |
≤ 20 %:n lasku | Jatka nykyisellä annoksella ja seuraa. |
1 Jos eGFR pysyy tasolla ≥ 60 ml/min/1,7 m2, annosta ei tarvitse muuttaa.
Jos potilaan annosta on pienennettävä, eGFR on suositeltavaa arvioida uudelleen kahden viikon kuluessa annoksen pienentämisestä. Potilaille, joiden annosta on pienennetty eGFR:n laskun vuoksi, on harkittava annoksen suurentamista 7,9 mg:lla kahdesti vuorokaudessa jokaista arviointia kohden, jossa eGFR on ≥ 80 % lähtötilanteen arvosta. Aloitusannosta ei saa kuitenkaan ylittää.
Samanaikainen anto kohtalaisten CYP3A4:n estäjien kanssa
Jos Lupkynis-valmistetta annetaan samanaikaisesti kohtalaisten sytokromi P450 (CYP)3A4:n estäjien kanssa (esim. verapamiili, flukonatsoli, diltiatseemi), vuorokausiannos on pienennettävä 15,8 mg:aan aamulla ja 7,9 mg:aan illalla (ks. kohta Yhteisvaikutukset).
Maksan vajaatoiminta
Potilaille, joilla on lievä tai keskivaikea maksan vajaatoiminta (Child-Pugh-luokka A tai B), suositeltu aloitusannos on 15,8 mg kahdesti vuorokaudessa. Voklosporiinin vaikusta ei ole arvioitu vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavilla potilailla, eikä voklosporiinia suositella tälle potilasryhmälle (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Munuaistoiminnan huolellinen seuranta on suositeltavaa (ks. taulukko 1 ja kohta Varoitukset ja käyttöön liittyvät varotoimet). Lupkynis-valmisteen käytöstä potilaille, joiden lähtötilanteen eGFR on 30 – < 45 ml/min/1,73 m2, on vain vähän tietoja. Lupkynis-valmisteen käyttöä näille potilaille suositellaan vain siinä tapauksessa, että hyödyt ovat riskejä suuremmat, ja tällöin aloitusannos on 23,7 mg kahdesti vuorokaudessa.
Lupkynis-valmistetta ei ole tutkittu vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min/1,73 m2) sairastavilla potilailla, eikä käyttöä tällaisille potilaille suositella, elleivät hyödyt ole riskejä suuremmat. Jos sitä käytetään, suositeltu aloitusannos on 15,8 mg kahdesti vuorokaudessa (ks. kohta Farmakokinetiikka).
Iäkkäät potilaat
SLE-nefriittiä sairastavista yli 65-vuotiaista potilaista on vain vähän tietoja, eikä yli 75-vuotiaista potilaista ole lainkaan tietoja saatavilla. Lupkynis-valmistetta ei suositella yli 75-vuotiaille potilaille (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Lupkynis-valmisteen turvallisuutta ja tehoa 5–18 vuoden ikäisten lasten ja nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla. Ei ole asianmukaista käyttää Lupkynis-valmistetta alle 5 vuoden ikäisille lapsille SLE-nefriitin hoitoon.
Antotapa
Suun kautta.
Pehmeät kapselit on nieltävä kokonaisina, ja ne voidaan ottaa ruoan kanssa tai ilman ruokaa.
Lupkynis-valmisteen ottamista greipin tai greippimehun kanssa ei suositella (ks. kohta Yhteisvaikutukset).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Voklosporiinin samanaikainen käyttö voimakkaiden CYP3A4:n estäjien kanssa (esim. ketokonatsoli, itrakonatsoli, klaritromysiini) (ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Lymfoomat ja muut maligniteetit
Immunosuppressantit lisäävät lymfoomien ja muiden maligniteettien muodostumisen riskiä, etenkin ihossa. On suositeltavaa, että potilaita neuvotaan välttämään tai rajoittamaan suojaamatonta altistumista auringolle tai UV-valolle.
Vakavat infektiot
Immunosuppressantit, myös voklosporiini, saattavat lisätä bakteeri-, virus-, sieni- ja alkueläininfektioiden riskiä ja myös mahdollisesti vakavien tai kuolemaan johtavien opportunististen infektioiden riskiä (ks. kohta Haittavaikutukset). Potilaita on seurattava huolellisesti infektioiden varalta voklosporiinihoidon aikana. Infektion ilmaantuessa voklosporiinihoidon jatkamisen hyödyt ja riskit on arvioitava.
Munuaistoksisuus
Kuten muidenkin kalsineuriinin estäjien käytön yhteydessä, voklosporiinihoitoa saavilla potilailla on havaittu haittavaikutuksina munuaistoiminnan akuuttia heikentymistä tai eGFR:n laskua. Hemodynaamista eGFR:n laskua on havaittu voklosporiinihoidon ensimmäisten neljän viikon aikana (ks. kohta Haittavaikutukset). Lasku on korjattavissa annosmuutoksilla. Säännöllinen eGFR:n seuranta on suositeltavaa (ks. kohta Annostus ja antotapa).
Punasoluaplasia
Potilailla, jotka ovat saaneet hoitoa eräällä toisella kalsineuriinin estäjällä, on raportoitu punasoluaplasiatapauksia. Kaikilla näillä potilailla oli punasoluaplasian riskitekijöitä, kuten parvovirus B19 -infektio, jokin perussairaus tai punasoluaplasiaan yhdistettyjä muita samanaikaisia hoitoja. Mekanismia, jolla kalsineuriinin estäjät aiheuttavat punasoluaplasian, ei tunneta. Jos potilaalla todetaan punasoluaplasia, Lupkynis-hoidon lopettamista on harkittava.
Hyperkalemia
Kalsineuriinin estäjien, myös voklosporiinin, käytössä on raportoitu hyperkalemiaa, joka saattaa olla vakavaa ja vaatia hoitoa (ks. kohta Haittavaikutukset). Samanaikainen käyttö hyperkalemiaan yhdistettyjen lääkevalmisteiden kanssa (esim. kaliumia säästävät diureetit, angiotensiinikonvertaasin [ACE:n] estäjät, angiotensiinireseptorin salpaajat) saattaa lisätä hyperkalemian riskiä. Potilaiden seerumin kaliumpitoisuuden säännöllinen seuranta on suositeltavaa hoidon aikana.
Hypertensio
Voklosporiini voi aiheuttaa tai pahentaa systeemistä hypertensiota (ks. kohta Haittavaikutukset). Verenpainetta on seurattava kahden viikon välein voklosporiinihoidon ensimmäisenä kuukautena ja sen jälkeen kliinisen tarpeen mukaan. Jos verenpaine kohoaa kliinisesti merkittävästi, on noudatettava taulukon 2 suosituksia.
Taulukko 2: Suositukset hypertension hallintaan
Verenpaine | Suositus |
Systolinen paine > 130 ja ≤ 165 mmHg ja diastolinen paine > 80 ja ≤ 105 mmHg | Hypertensiolääkitys voidaan aloittaa tai sen annostusta muuttaa |
Verenpaine > 165/105 mmHg, johon liittyy hypertension oireita | Lopeta voklosporiinin anto ja aloita hypertensiolääkitys tai muuta sen annostusta |
QT-ajan pidentyminen
Voklosporiinin samanaikainen käyttö muiden lääkevalmisteiden kanssa, joiden tiedetään pidentävän QTc-aikaa, saattaa aiheuttaa kliinisesti merkittävää QT-ajan pidentymistä. Tietyt tilanteet saattavat lisätä kääntyvien kärkien takykardian ja/tai äkkikuoleman riskiä QTc-aikaa pidentävien lääkevalmisteiden käytön yhteydessä; tällaisia ovat esimerkiksi bradykardia, hypokalemia tai hypomagnesemia, muiden QTc-aikaa pidentävien lääkevalmisteiden samanaikainen käyttö ja synnynnäinen QT-ajan pidentyminen.
Neurotoksisuus
Immunosuppressiiviseen hoitoon, myös voklosporiiniin, liittyy lisääntynyt neurotoksisuuden riski (ks. kohta Haittavaikutukset). Potilaita on seurattava uusien tai pahenevien neurologisten oireiden varalta. Niitä ovat muun muassa kouristuskohtaukset, vapina tai posterioriseen reversiibeliin enkefalopatiaoireyhtymään (PRES) viittaavat merkit ja oireet. Jos tällaisia ilmenee, on harkittava voklosporiinin annoksen pienentämistä tai hoidon lopettamista.
Maksan vajaatoiminta
Voklosporiinia ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla (Child-Pugh-luokka C), eikä sen käyttöä siksi suositella tässä potilasryhmässä.
Rokotteet
Immunosuppressantit saattavat heikentää immuunivastetta rokotteille. Voklosporiinihoidon aikana annettujen rokotteiden teho saattaa olla tavallista heikompi. Elävien, heikennettyjä taudinaiheuttajia sisältävien rokotteiden käyttöä on vältettävä.
Samanaikainen käyttö muiden lääkevalmisteiden kanssa
Voklosporiinin samanaikaista antoa kohtalaisten tai voimakkaiden CYP3A4:n indusoijien kanssa ei suositella (ks. kohta Yhteisvaikutukset).
Voklosporiinin turvallisuutta ja tehoa yhteiskäytössä syklofosfamidin kanssa ei ole varmistettu.
Apuaineet
Etanoli
Tämä lääkevalmiste sisältää 21,6 mg alkoholia (etanolia) per pehmeä kapseli. Näin ollen 23,7 mg:n annos Lupkynis-valmistetta sisältää 64,8 mg etanolia. Alkoholimäärä 23,7 mg:ssa tätä lääkevalmistetta vastaa alle 2 ml:aa olutta tai 1 ml:aa viiniä. Tämän lääkevalmisteen sisältämä pieni määrä alkoholia ei aiheuta havaittavia vaikutuksia.
Sorbitoli
Tämä lääkevalmiste sisältää 28,7 mg sorbitolia per pehmeä kapseli. Sorbitolia (tai fruktoosia) sisältävien muiden valmisteiden samanaikaisen annon sekä ravinnosta saatavan sorbitolin (tai fruktoosin) additiivinen vaikutus on huomioitava. Suun kautta otettavien lääkevalmisteiden sorbitoli saattaa vaikuttaa muiden suun kautta otettavien lääkkeiden biologiseen hyötyosuuteen.
Soijalesitiini (mahdolliset jäämät valmistusprosessista)
Tämä lääkevalmiste saattaa sisältää soijalesitiinin jäämiä. Potilaat, jotka ovat saaneet anafylaktisen reaktion soijasta tai maapähkinästä, eivät saa käyttää tätä lääkevalmistetta.
Yhteisvaikutukset
Voklosporiini metaboloituu CYP3A4:n välityksellä, ja se on P-glykoproteiinin (P-gp:n) sekä orgaanisten anionien kuljettajapolypeptidi (OATP)1B1:n ja OATP1B3:n estäjä.
Muiden lääkevalmisteiden mahdolliset vaikutukset voklosporiinialtistukseen
Voklosporiini metaboloituu CYP3A4:n välityksellä. Samanaikainen käyttö sellaisten lääkevalmisteiden tai kasvirohdosvalmisteiden kanssa, joiden tiedetään estävän tai indusoivan CYP3A4:ää, saattaa vaikuttaa voklosporiinin metaboliaan ja siten suurentaa tai pienentää voklosporiinin pitoisuutta veressä.
CYP3A4:n estäjät
Voklosporiinialtistus oli 18,6-kertainen, kun sitä annettiin samanaikaisesti voimakkaan CYP3A4:n estäjän ketokonatsolin kanssa, verrattuna siihen, kun voklosporiinia annettiin yksinään. Voklosporiinin samanaikainen anto voimakkaiden CYP3A4:n estäjien kanssa (esim. ketokonatsoli, itrakonatsoli, klaritromysiini) on vasta-aiheista (ks. kohta Vasta-aiheet).
Voklosporiinialtistus oli 2,71-kertainen, kun sitä annettiin samanaikaisesti kohtalaisen CYP3A4:n estäjän verapamiilin kanssa, verrattuna siihen, kun voklosporiinia annettiin yksinään. Jos voklosporiinia annetaan samanaikaisesti kohtalaisten CYP3A4:n estäjien kanssa (esim. verapamiili, flukonatsoli, erytromysiini, diltiatseemi, greippi ja greippimehu), annos on pienennettävä 15,8 mg:aan aamulla ja 7,9 mg:aan illalla (ks. kohta Annostus ja antotapa).
Heikot CYP3A4:n estäjät saattavat lisätä voklosporiinialtistusta, mutta in vivo -tutkimuksia ei ole tehty. Voklosporiiniannosta ei tarvitse muuttaa, jos sitä annetaan samanaikaisesti heikkojen CYP3A4:n estäjien kanssa, mutta eGFR:n lisäseuranta on suositeltavaa, kun hoito heikolla CYP3A4:n estäjällä aloitetaan.
CYP3A4:n indusoijat
Voklosporiinialtistus oli 87 % vähäisempi ja voklosporiinin huippupitoisuus (Cmax) 68 % pienempi, kun sitä annettiin samanaikaisesti voimakkaan CYP3A4:n indusoijan rifampisiinin kanssa (600 mg kerran vuorokaudessa 10 peräkkäisen päivän ajan), verrattuna siihen, kun voklosporiinia annettiin yksinään. Myös useiden samanaikaisesti annettujen kohtalaisen CYP3A4:n indusoijan annosten odotetaan vähentävän voklosporiinialtistusta kliinisesti merkittävällä tavalla.
Voimakkaiden ja kohtalaisten CYP3A4:n indusoijien (esim. karbamatsepiini, fenobarbitaali, rifampisiini, mäkikuisma, efavirentsi) samanaikaista antoa voklosporiinin kanssa ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Heikot CYP3A4:n indusoijat saattavat myös pienentää altistusta ja mahdollisesti heikentää voklosporiinin vaikutusta, mutta tämän kliinistä merkitystä ei tunneta.
Voklosporiinin mahdolliset vaikutukset muille lääkevalmisteille altistukseen
P-gp:n substraatit
Voklosporiini on P-glykoproteiinin (P-gp:n) estäjä. Voklosporiinin samanaikainen anto useiden digoksiiniannosten kanssa suurensi digoksiinin Cmax-arvoa 1,51-kertaisesti ja käyrän alle jäävää pinta-alaa (AUC) 1,25-kertaisesti. Varovaisuutta on noudatettava, jos voklosporiinia annetaan samanaikaisesti herkkien P-gp:n substraattien kanssa, etenkin sellaisten, jotka ovat terapeuttiselta indeksiltään kapeita (esim. digoksiini, dabigatraanieteksilaatti, feksofenadiini), ja potilaita on tällöin seurattava asianmukaisesti kunkin valmisteen valmistetietojen mukaan.
OATP1B1:n ja OATP1B3:n substraatit
Voklosporiini on kuljettajaproteiinien OATP1B1 ja OATP1B3 estäjä. Eräässä kliinisessä tutkimuksessa simvastatiinin 40 mg:n kerta-annoksen samanaikainen anto kahdesti vuorokaudessa annettujen 23,7 mg:n voklosporiiniannosten kanssa suurensi aktiivisen metaboliitin simvastatiinihapon (joka on herkkä OATP1B1:n ja OATP1B3:n substraatti) Cmax-arvoa 3,1-kertaisesti ja AUC-arvoa 1,8-kertaisesti. Samassa tutkimuksessa altistus kanta-aine simvastatiinille (joka on myös rintasyöpäresistenssiproteiinin [BCRP] substraatti) ei muuttunut AUC-arvon osalta, mutta Cmax-arvo suureni 1,6-kertaisesti, mikä voi mahdollisesti johtua suoliston BCRP:n ja voklosporiinin välisestä yhteisvaikutuksesta. Potilaita on seurattava haittavaikutusten, kuten myopatian ja rabdomyolyysin, varalta, kun OATP1B1:n ja OATP1B3:n substraatteja (esim. simvastatiini, atorvastatiini, pravastatiini, rosuvastatiini) käytetään samanaikaisesti voklosporiinin kanssa.
BCRP:n substraatit
Voklosporiini estää BCRP:tä in vitro. Kliinisesti oleellista suoliston BCRP:n estoa ei voida poissulkea, ja voklosporiini saattaa suurentaa näiden substraattien pitoisuutta in vivo. BCRP:n substraattien käyttöä on seurattava samanaikaisessa käytössä voklosporiinin kanssa, sillä pienetkin pitoisuuden muutokset saattavat aiheuttaa vakavaa toksisuutta (esim. rosuvastatiini).
Mykofenolaattimofetiili
Voklosporiinin samanaikainen anto mykofenolaattimofetiilin kanssa ei vaikuttanut kliinisesti merkittävällä tavalla mykofenolihapon pitoisuuteen veressä.
CYP3A4:n substraatit
Useiden voklosporiiniannosten (0,4 mg/kg kahdesti vuorokaudessa) antaminen suun kautta ei vaikuttanut kliinisesti merkittävästi herkän CYP3A4:n substraatin midatsolaamin farmakokinetiikkaan.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja (alle 300 raskaudesta) voklosporiinin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Lupkynis-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
12 imettävällä tutkittavalla tehdyssä tutkimuksessa suurin arvioitu täysimetetyn vauvan saama voklosporiiniannos oli 1,4 % painon mukaan korjatusta äidin annoksesta (ks. kohta Farmakokinetiikka). Voklosporiinin vaikutuksia imetettävään vauvaan ei tunneta.
On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Lupkynis-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Ei ole olemassa tietoja voklosporiinin vaikutuksesta ihmisen hedelmällisyyteen. Eläinkokeissa havaittiin voklosporiiniin liittyviä muutoksia urosten sukupuolielimissä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Lupkynis-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitetut haittavaikutukset voklosporiinin käytössä ovat pienentynyt eGFR (26,2 %) ja hypertensio (19,1 %).
Yleisimmin ilmoitetut vakavat haittavaikutukset voklosporiinin käytöstä olivat infektiot (10,1 %), akuutti munuaisvaurio (3 %) ja hypertensio (1,9 %).
Voklosporiinihoidon ensimmäisten neljän viikon aikana esiintyy yleisesti hemodynaamista eGFR:n laskua, joka tasaantuu sen jälkeen, vaikka hoitoa jatkettaisiin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutusten taulukkomuotoinen luettelo
Taulukossa 3 on yhteenveto haittavaikutuksista SLE-nefriittiä sairastavilla potilailla, jotka saivat voklosporiinia suositellulla annoksella kahdessa lumekontrolloidussa tutkimuksessa, joissa hoidon mediaanikesto oli 1 vuosi, ja/tai markkinoille saattamisen jälkeisessä käytössä.
Kaikki haittavaikutukset on lueteltu elinjärjestelmäluokan ja seuraavan yleisyysluokituksen mukaan: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), erittäin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 3: Haittavaikutukset
| Elinjärjestelmäluokka | Hyvin yleinen | Yleinen | Tuntematon |
| Infektiot | Ylähengitystieinfektio1 | Pneumonia Influenssa Vyöruusu Maha-suolitulehdus Virtsatieinfektio | |
| Veri ja imukudos | Anemia | ||
| Immuunijärjestelmä | Yliherkkyys | ||
| Aineenvaihdunta ja ravitsemus | Hyperkalemia Heikentynyt ruokahalu | ||
| Hermosto | Päänsärky | Kouristuskohtaus Vapina | |
| Verisuonisto | Hypertensio2 | ||
| Hengityselimet, rintakehä ja välikarsina | Yskä | ||
| Ruoansulatuselimistö | Ripuli Vatsakipu3 | Pahoinvointi Ienhyperplasia4 Dyspepsia Suun haavauma | |
| Iho ja ihonalainen kudos | Alopesia Hypertrikoosi5 | ||
| Munuaiset ja virtsatiet | Pienentynyt glomerulusten suodatusnopeus6, 7 | Akuutti munuaissairaus6 Akuutti munuaisvaurio6 | |
| Yleisoireet ja antopaikassa todettavat haita | Uupumus |
1 Sisältää seuraavat Preferred Term (PT) -termit: ylähengitysteiden virusinfektio ja ylähengitysteiden bakteeri-infektio
2 Sisältää seuraavat PT-termit: kohonnut verenpaine, kohonnut diastolinen verenpaine, diastolinen hypertensio
3 Sisältää seuraavat PT-termit: ylävatsakipu, epämiellyttävät tuntemukset vatsassa
4 Sisältää seuraavat PT-termit: ientulehdus, ienverenvuoto, ienhypertrofia, ienturvotus
5 Sisältää seuraavat PT-termit: hypertrikoosi, hirsutismi
6 Sisältää seuraavan PT-termin: munuaisten vajaatoiminta
7 Sisältää seuraavan PT-termin: kohonnut veren kreatiniinipitoisuus
Valikoitujen haittavaikutusten kuvaus
Infektiot
Infektioiden kokonaisilmaantuvuus oli 62,2 % voklosporiinia saavilla potilailla ja 54,9 % lumelääkettä saavilla potilailla. Infektioita, joita esiintyi vähintään 5 %:lla voklosporiinia saaneista potilaista ja joiden esiintyvyys oli vähintään 1 % suurempi kuin lumelääkettä saavilla potilailla, olivat virtsatieinfektio, ylähengitysteiden virusinfektio, vyöruusu ja maha-suolitulehdus. Vakavia infektioita esiintyi 10,1 %:lla voklosporiinia saaneista ja 10,2 %:lla lumelääkettä saaneista potilaista. Yleisimmät niistä olivat keuhkokuume (voklosporiini 4,1 %, lumelääke 3,8 %), maha-suolitulehdus (voklosporiini 1,5 %, lumelääke 0,4 %) ja virtsatieinfektio (voklosporiini 1,1 %, lumelääke 0,4 %). Vakavia opportunistisia infektioita esiintyi 1,1 %:lla voklosporiinia saaneista potilaista ja 0,8 %:lla lumelääkettä saaneista potilaista. Kuolemaan johtaneita infektioita esiintyi 0,7 %:lla voklosporiinia saaneista potilaista ja 0,8 %:lla lumelääkettä saaneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Munuaistoksisuus
Munuaistoksisuuteen viittaavia haittavaikutuksia, joiden esiintyvyys oli ≥ 1 % suurempi voklosporiinia saavilla potilailla kuin lumelääkettä saavilla potilailla, olivat pienentynyt eGFR (26,2 % vs. 9,4 %), munuaisten vajaatoiminta (5,6 % vs. 2,6 %), akuutti munuaisvaurio (3,4 % vs. 0,8 %) ja hyperkalemia (1,9 % vs. 0,8 %). Vakavia haittavaikutuksia raportoitiin 5,2 %:lla voklosporiinia saaneista potilaista ja 3,4 %:lla lumelääkettä saaneista potilaista. Yleisimmät haittavaikutukset, jotka johtivat annoksen muutokseen (annoksen pienentämiseen tai hoidon tilapäiseen keskeyttämiseen), olivat pienentynyt eGFR (voklosporiini 23,6 %, lumelääke 6,8 %), munuaisten vajaatoiminta (voklosporiini 3,0 %, lumelääke 0,8 %) ja akuutti munuaisvaurio (voklosporiini 0,7 %, lumelääke 0 %). Yleisimmät haittavaikutukset, jotka johtivat lääkevalmisteen käytön pysyvään lopettamiseen, olivat eGFR:n lasku (voklosporiini 3,7 %, lumelääke 1,9 %) ja munuaisten vajaatoiminta (voklosporiini 1,9 %, lumelääke 1,5 %). Mediaaniaika eGFR:n laskun korjautumiseen, kun eGFR:n lasku oli ≥ 20 %, oli 49 päivää voklosporiinihoitoa saavilla potilailla. Kun eGFR:n lasku oli ≥ 30 %, mediaaniaika korjautumiseen oli 102 päivää voklosporiinia saavilla potilailla.
Hypertensio
Hypertensiota ilmoitettiin 19,1 %:lla voklosporiinia saaneista potilaista ja 8,6 %:lla lumelääkettä saaneista potilaista. Hypertension ilmaantuvuus oli suurimmillaan voklosporiinihoidon ensimmäisen 4 viikon aikana, minkä jälkeen se väheni. Hypertensio oli vaikeaa 1,1 %:lla voklosporiinia saaneista potilaista ja 0,8 %:lla lumelääkettä saaneista potilaista. Vakavaa hypertensiota esiintyi 1,9 %:lla voklosporiinia saaneista potilaista ja 0,4 %:lla lumelääkettä saaneista potilaista.
Pitkäaikainen altistus (enintään 36 kuukautta)
Jatkohoidon (kuukaudet 12–36) aikana esiintyneet haittavaikutukset olivat yhdenmukaisia ensimmäisen hoitovuoden aikana esiintyneiden haittavaikutusten kanssa, mutta useimpien tapahtumien ilmaantuvuus oli pienempi kuin ensimmäisenä hoitovuoden aikana. Infektioiden kokonaisilmaantuvuus oli 49,1 % voklosporiinia saavilla potilailla ja 43,0 % lumelääkettä saavilla potilailla. Infektioita, joita esiintyi vähintään 5 %:lla voklosporiinia saaneista potilaista ja joiden esiintyvyys oli vähintään 1 % suurempi kuin lumelääkettä saavilla potilailla, olivat virtsatieinfektio, ylähengitystieinfektio, ylähengitysteiden virusinfektio ja maha-suolitulehdus. Vakavia infektioita esiintyi 6,9 %:lla voklosporiinia saaneista ja 8,0 %:lla lumelääkettä saaneista potilaista. Yleisimmät niistä olivat koronavirusinfektio (voklosporiini 1,7 %, lumelääke 5,0 %) ja viruksen aiheuttama keuhkokuume (voklosporiini 1,7 %, lumelääke 0 %). Munuaistoksisuuteen viittaavia haittavaikutuksia, joiden esiintyvyys oli suurempi voklosporiinia saavilla potilailla kuin lumelääkettä saavilla potilailla, olivat pienentynyt eGFR (10,3 % vs. 5,0 %) ja munuaisten vajaatoiminta (3,4 % vs. 2,0 %). Hypertensiota ilmoitettiin 8,6 %:lla voklosporiinia saaneista potilaista ja 7,0 %:lla lumelääkettä saaneista potilaista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Voklosporiinin käytöstä on ilmoitettu vahingossa tapahtuneita yliannostustapauksia. Oireita olivat vapina ja takykardia. Terveillä tutkittavilla tehdyssä yhteisvaikutustutkimuksessa ketokonatsolin ja voklosporiinin samanaikainen anto suurensi voklosporiinialtistusta 18,6-kertaisesti. Lisäksi havaittiin seerumin kreatiniinipitoisuuden suurenemista, seerumin magnesiumpitoisuuden pienenemistä ja verenpaineen kohoamista. Muiden kalsineuriinin estäjien yliannostuksen oireita (ei havaittu voklosporiinilla) ovat päänsärky, pahoinvointi ja oksentelu, infektiot, nokkosihottuma, letargia, muutokset elektrolyyttien pitoisuuksissa sekä veren ureatyppipitoisuuden ja alaniiniaminotransferaasin (ALAT) suureneminen.
Voklosporiinille ei ole saatavilla erityistä vastalääkettä. Yliannostuksen yhteydessä on tuettava elintoimintoja ja annettava oireenmukaista hoitoa. Voklosporiinihoito on keskeytettävä tilapäisesti ja veren ureatyppipitoisuutta, seerumin kreatiniinipitoisuutta, eGFR-arvoa ja ALAT-arvoa on seurattava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, kalsineuriinin estäjät, ATC-koodi: L04AD03
Vaikutusmekanismi
Voklosporiini on kalsineuriinin estäjiin kuuluva immunosuppressantti, jonka kalsineuriinia estävä vaikutus on annosriippuvainen enintään 1,0 mg/kg:n annokseen asti. Lymfosyyttien aktivoitumiseen liittyy solunsisäisen kalsiumpitoisuuden suureneminen. Kalsineuriini on kalsium-/kalmoduliiniriippuvainen fosfataasi, jota tarvitaan T-solujen lymfokiinituotannon ja proliferaation käynnistämiseen. Immunosuppressanttivaikutus estää lymfosyyttien proliferaatiota, T-solujen sytokiinituotantoa ja T-soluja aktivoivien pinta-antigeenien ilmentymistä.
Farmakodynaamiset vaikutukset
Sydämen sähköfysiologia
Satunnaistetussa, lumelääke- ja aktiivikontrolloidussa (moksifloksasiini 400 mg) kerta-annoksen vaikutusta arvioivassa rinnakkaisryhmätutkimuksessa havaittiin annosriippuvaista QT-ajan pidentymistä voklosporiinin annosalueella 0,5–4,5 mg/kg (enintään 9-kertainen terapeuttinen altistus). Tällöin aika QTc-ajan pitenemisen huippuun oli kaikilla annostasoilla 4–6 tuntia. QTcF-ajan suurimmat keskimääräiset lumelääkekorjatut muutokset lähtötilanteesta voklosporiinin eri annoksilla olivat 0,5 mg/kg:n annoksella 6,4 msek, 1,5 mg/kg:n annoksella 17,5 msek, 3,0 mg/kg:n annoksella 25,7 msek ja 4,5 mg/kg:n annoksella 34,6 msek.
Erillisessä satunnaistetussa, lumekontrolloidussa vaihtovuorotutkimuksessa, johon osallistui 31 tervettä tutkittavaa, suurta keskimääräistä pidentymistä (> 20 msek) ei havaittu 7 päivän voklosporiinihoidon jälkeen annoksilla 0,3 mg/kg, 0,5 mg/kg ja 1,5 mg/kg kahdesti vuorokaudessa (enintään noin 6-kertainen terapeuttinen altistus). Kerta-annosta ja useita annoksia arvioivissa tutkimuksissa havaitun QT-aikaa pidentävän vaikutuksen taustamekanismia ei tunneta.
Lumelääkekorjattua lähtötilanteen jälkeistä QTcF-ajan muutosta koskevassa regressioanalyysissa, joka perustui voklosporiinia 23,7 mg tai 39,5 mg kahdesti vuorokaudessa saaneiden SLE-nefriittiä sairastavien potilaiden tietoihin, muutoksen kulmakerroin oli minimaalisen negatiivinen (−0,065344 msek/ng/ml) eikä eronnut tilastollisesti merkitsevästi kulmakertoimesta 0 (p = 0,1042).
Kliininen teho ja turvallisuus
Voklosporiinin turvallisuutta ja tehoa arvioitiin kahdessa lumekontrolloidussa kliinisessä tutkimuksessa (AURORA 1 ja AURA-LV) SLE-nefriittiä sairastavilla potilailla, joiden sairaus oli luokaltaan III (puhdas tai sekamuoto III/V), IV (puhdas tai sekamuoto IV/V) tai V. Kaikki potilaat saivat taustahoitona mykofenolaattimofetiilia (MMF) (2 g/vrk) ja kortikosteroideja (yhteensä enintään 1 g metyyliprednisolonia laskimoon päivien 1 ja 2 aikana, minkä jälkeen suun kautta annettavia kortikosteroideja aloitusannoksella 25 mg/vrk [tai 20 mg/vrk, jos potilaan paino < 45 kg], joka pienennettiin asteittain 2,5 mg:aan/vrk viikkoon 16 mennessä).
Potilailla, jotka olivat mukana AURORA 1 -tutkimuksessa sen päättymiseen asti, oli mahdollisuus siirtyä kahden vuoden pituiseen jatkotutkimukseen (AURORA 2).
Vaiheen 3 tutkimus AURORA 1
AURORA 1 -tutkimus oli vaiheen 3 prospektiivinen, satunnaistettu, kaksoissokkoutettu tutkimus, jossa verrattiin voklosporiinia annoksella 23,7 mg kahdesti vuorokaudessa (vastaa annosta 0,37 mg/kg) (n = 179) ja lumelääkettä (n = 178) 52 viikon hoitojakson ajan. Potilaiden demografiset ominaisuudet oli hyvin tasapainotettu tutkimuksen hoitoryhmien välillä. Keski-ikä oli 33 vuotta (vaihteluväli 18–72 vuotta), ja suurin osa potilaista oli naisia (87,7 %), joista 81,8 % oli naisia, jotka voivat tulla raskaaksi.
Useimmat potilaat olivat valkoihoisia (36,1 %) tai aasialaisia (30,5 %), ja noin kolmannes tutkimuspopulaatiosta oli latinalaisamerikkalaisia. Keskipaino oli 66,5 kg (vaihteluväli 36–142 kg). Mediaani-aika systeemisen lupus erythematosuksen (SLE) toteamisesta oli 5,0 vuotta ja mediaani-aika SLE-nefriitin toteamisesta 2,0 vuotta.
Ennen AURORA 1 -tutkimukseen osallistumista suurin osa potilaista (98 %) oli saanut aiempaa hoitoa SLE-nefriittiin, ja noin 55 % käytti seulonnan aikaan mykofenolaattimofetiilia. Niiden potilaiden osuus, jotka eivät olleet aiemmin saaneet hoitoa SLE-nefriittiin, oli hyvin pieni (2 %).
Useampi potilas voklosporiiniryhmässä kuin lumelääkeryhmässä saavutti ensisijaisen päätetapahtuman eli munuaisvasteen (taulukko 4).
Taulukko 4: AURORA 1 – Yhteenveto tärkeimmistä tehon päätetapahtumista
Voklosporiini (n = 179) n (%) | Lumelääke (n = 178) n (%) | Vetosuhde vs. lumelääke (95 %:n CI) | p-arvo | |
Munuaisvaste 52 viikon aikapisteessä | 73 (40,8) | 40 (22,5) | 2,65 (1,64, 4,27) | < 0,001 |
Munuaisvaste 24 viikon aikapisteessä | 58 (32,4) | 35 (19,7) | 2,23 (1,34, 3,72) | = 0,002 |
Osittainen munuaisvaste* 24 viikon aikapisteessä | 126 (70,4) | 89 (50,0) | 2,43 (1,56, 3,79) | < 0,001 |
Osittainen munuaisvaste* 52 viikon aikapisteessä | 125 (69,8) | 92 (51,7) | 2,26 (1,45, 3,51) | < 0,001 |
* Osittaisen munuaisvasteen määritelmä: UPCR-arvo pieneni 50 %.
Selitteet: CI = luottamusväli; UPCR = virtsan proteiinin ja kreatiniinin suhde
Ensisijaisen päätetapahtuman kunkin arvioidun osan saavuttaneiden potilaiden osuudet voklosporiiniryhmässä vs. lumelääkeryhmässä olivat seuraavat:
| 45,3 % vs. 23,0 % |
| 82,1 % vs. 75,8 % |
| 87,2 % vs. 85,4 % |
| 91,1 % vs. 86,5 % |
Useampi potilas voklosporiiniryhmässä kuin lumelääkeryhmässä saavutti UPCR-arvon ≤ 0,5 mg/mg (64,8 % vs. 43,8 %), ja aika UPCR-arvon ≤ 0,5 mg/mg saavuttamiseen oli merkitsevästi lyhyempi voklosporiiniryhmässä kuin lumelääkeryhmässä (mediaaniaika 169 päivää vs. 372 päivää); riskisuhde: 2,02; 95 %:n luottamusväli: 1,51, 2,70; p < 0,001).
Kuva 1: Kaplan-Meierin käyrä ajalle (päivää) UPCR-arvon ≤ 0,5 mg/mg saavuttamiseen
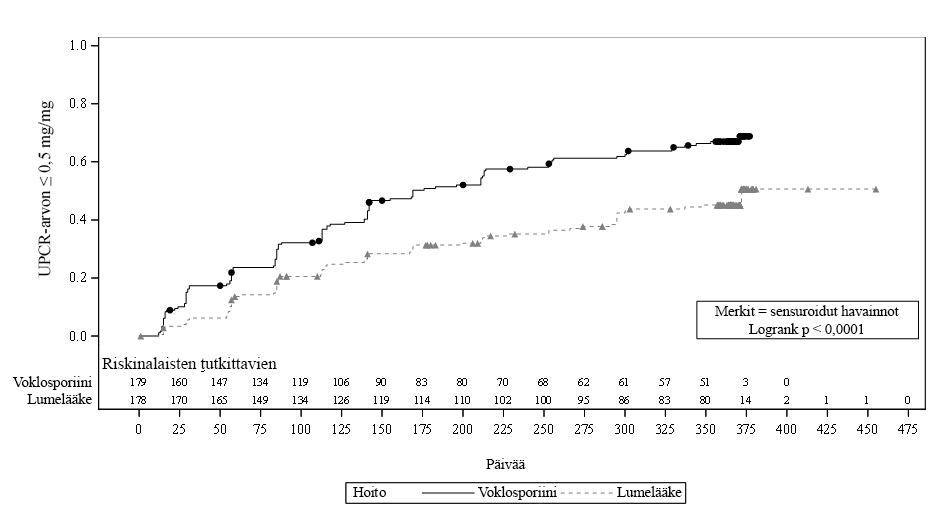
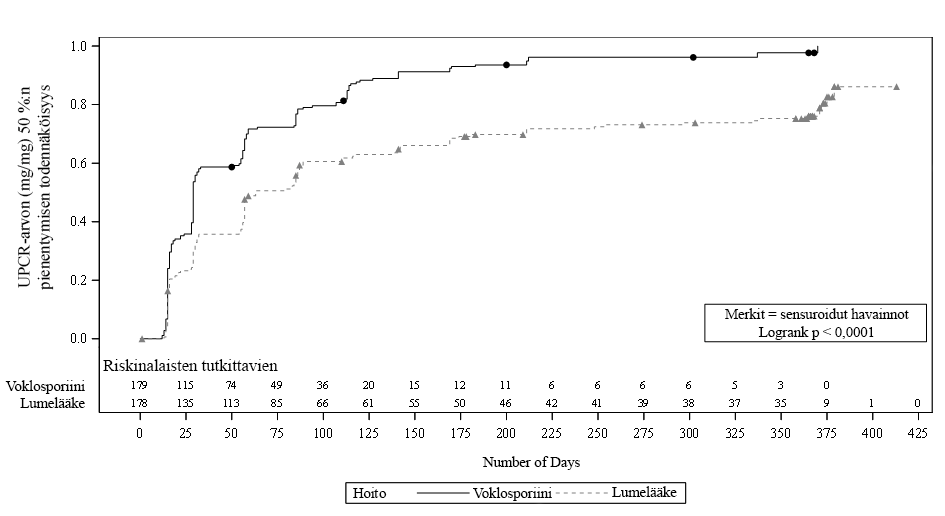
Aika UPCR-arvon 50 %:n pienentymisen saavuttamiseen oli merkitsevästi lyhyempi voklosporiiniryhmässä kuin lumelääkeryhmässä (riskisuhde: 2,05; 95 %:n luottamusväli: 1,62, 2,60; p < 0,001). Mediaaniaika siihen, että UPCR-arvo oli pienentynyt 50 %, oli 29 päivää voklosporiiniryhmässä vs. 63 päivää lumelääkeryhmässä (kuva 2).
Kuva 2: Kaplan-Meierin käyrä ajalle (päivää) UPCR-arvon pienentymiseen 50 %:lla lähtötilanteesta
Yli 80 prosentilla AURORA 1 -tutkimuksen potilaista suun kautta annettavien kortikosteroidien annos oli viikkoon 24 mennessä pienennetty ≤ 2,5 mg:aan/vrk, ja 52 viikon aikapisteessä annos oli säilynyt tällä tasolla yli 75 %:lla potilaista.
Faasin 3 tutkimus AURORA 2
AURORA 2 -tutkimus oli jatkotutkimus, jossa arvioitiin voklosporiinin pitkäaikaista turvallisuutta ja tehoa potilailla, jotka olivat mukana AURORA 1 -tutkimuksessa sen päättymiseen asti. Potilaat jatkoivat samaa voklosporiinihoitoa (n = 116) tai lumelääkehoitoa (n = 100), jota he olivat saaneet AURORA 1 -tutkimuksen päättyessä, myös annoksen osalta, ja jatkoivat siinä enintään kahden vuoden ajan. Yli 85 % potilaista (87,1 % voklosporiiniryhmästä ja 85,0 % lumelääkeryhmästä) oli mukana tutkimuksen päättymiseen asti, ja 79,3 % voklosporiiniryhmän potilaista ja 73 % lumelääkeryhmän potilaista sai edelleen tutkimushoitoa tutkimuksen päättyessä.
Munuaisvasteen saavuttaneiden potilaiden osuus 36 kuukauden aikapisteessä oli 33 % (59/179) voklosporiiniryhmässä ja 22 % (39/178) lumelääkeryhmässä (hoitoaikeen mukainen populaatio, AURORA 1) sekä 51 % (59/116) voklosporiiniryhmässä ja 39 % (39/100) lumelääkeryhmässä (hoitoaikeen mukainen populaatio, AURORA 2).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Lupkynis-valmisteen käytöstä SLE-nefriitin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Suun kautta annon jälkeen (23,7 mg voklosporiinia kahdesti vuorokaudessa) aika (mediaani) huippupitoisuuden (Cmax) saavuttamiseen kokoveressä on noin 1,5 tuntia (vaihteluväli: 0,75–2 tuntia). Kahdesti vuorokaudessa annettaessa voklosporiinin vakaan tilan pitoisuus saavutetaan 6 vuorokaudessa. Voklosporiinin kumuloituminen on noin kaksinkertaista suhteessa kerta-annokseen. Vakaassa tilassa voklosporiinin keskimääräinen Cmax kokoveressä oli 120 ng/ml (CV: 32 %) ja annosta edeltävä aallonpohjapitoisuus 15,0 ng/ml (CV: 49 %). In vitro -tiedot, joiden avulla on arvioitu, onko voklosporiini effluksikuljettajaproteiinien P-gp tai BCRP substraatti, eivät ole vakuuttavia, mutta P-gp:n/BCRP:n estäjien ei odoteta vaikuttavan kliinisesti merkittävällä tavalla.
Voklosporiinin anto ruoan kanssa pienensi sekä imeytymisnopeutta että imeytyvää osuutta. Voklosporiinin Cmax ja AUC pienenivät 53 % ja 25 % runsasrasvaisen aterian kanssa annettaessa ja 29 % ja 15 % vähärasvaisen aterian kanssa annettaessa. Näitä muutoksia ei pidetty kliinisesti merkittävinä. Näin ollen voklosporiinia voidaan ottaa ruoan kanssa tai ilman ruokaa.
Jakautuminen
Voklosporiini sitoutuu 97-prosenttisesti plasman proteiineihin. Voklosporiini jakautuu laajasti punasoluihin. Jakautuminen kokoveren ja plasman välillä on pitoisuus- ja lämpötilariippuvaista. Populaatiofarmakokineettisessä analyysissa näennäinen jakautumistilavuus (Vss/F) oli 2 154 l.
Biotransformaatio
Voklosporiini metaboloituu laajasti, pääasiassa CYP3A4:n välityksellä muodostaen oksidatiivisia metaboliitteja. Voklosporiini on tärkein kiertävä komponentti [14C]-voklosporiinin kerta-annoksen jälkeen. Ihmisen kokoveressä havaittiin yksi päämetaboliitti, jonka osuus kokonaisaltistuksesta oli 16,7 %. Päämetaboliitin ei odoteta osallistuvan voklosporiinin farmakologiseen vaikutukseen, koska sen tehon ilmoitettiin olevan kahdeksan kertaa voklosporiinia heikompi lymfosyyttiproliferaatiotestin perusteella ja koska sille altistus on vähäisempää kuin voklosporiinille.
Eliminaatio
Keskimääräinen vakaan tilan näennäinen puhdistuma (CLss/F), kun voklosporiinia annetaan 23,7 mg kahdesti vuorokaudessa, on 63,6 l/h (CV: 37,5 %). Keskimääräinen terminaalinen puoliintumisaika (t½) vakaassa tilassa on noin 30 tuntia (vaihteluväli: 24,9–36,5 tuntia).
Suun kautta annetun 70 mg:n [14C]-voklosporiinikerta-annoksen radioaktiivisuudesta 94,8 % poistui 168 tunnin kuluessa annosta: 92,7 % erittyi ulosteeseen (siitä 5 % oli muuttumatonta voklosporiinia) ja 2,1 % virtsaan (siitä 0,25 % oli muuttumatonta voklosporiinia).
Lineaarisuus/ei-lineaarisuus
Terveillä tutkittavilla havaittiin ei-lineaarisuutta annoksen ja altistuksen välillä tutkitun annosalueen (0,25–1,5 mg/kg kahdesti vuorokaudessa) alapäässä. Tämän vaikutus farmakokinetiikkaan oli melko vähäinen. Annosriippuvaisuuden kerroin oli jatkuvasti alle 1,5. Tätä ei-lineaarisuutta ei ole havaittu annosalueella, jota on tutkittu SLE-nefriittiä sairastavilla potilailla.
Farmakokinetiikka erityisryhmissä
Munuaisten vajaatoiminta
Kliinisissä tutkimuksissa munuaistoiminnan seurantaan käytettiin eGFR-arvoa, ja potilaiden annoksia muutettiin etukäteen määritetyn suunnitelman mukaisesti. Tutkimukseen otettujen SLE-nefriittiä sairastavien potilaiden lähtötilanteen eGFR oli > 45 ml/min/1,73 m2. Annoksen muuttaminen on tehtävä taulukon 1 suositusten mukaisesti.
Erillisessä munuaisten vajaatoimintaa koskevassa tutkimuksessa voklosporiinin kerta-annoksen ja toistuvien annosten jälkeiset Cmax- ja AUC-arvot olivat samankaltaisia lievää munuaisten vajaatoimintaa sairastavilla (kreatiniinipuhdistuma [CLCr] 60–89 ml/min Cockcroft-Gaultin kaavalla laskettuna) ja keskivaikeaa munuaisten vajaatoimintaa sairastavilla (CLCr 30–59 ml/min) verrattuna munuaistoiminnaltaan normaaleihin (CLCr ≥ 90 ml/min) tutkittaviin. Vaikeaa munuaisten vajaatoimintaa sairastavilla tutkittavilla (CLCr < 30 ml/min) voklosporiinikerta-annoksen jälkeinen Cmax oli 1,5 kertaa suurempi ja AUC 1,7 kertaa suurempi. Hemodialyysilla tai ilman hemodialyysia hoidettavan loppuvaiheen munuaissairauden vaikutusta voklosporiinin farmakokinetiikkaan ei tunneta (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Erillisessä maksan vajaatoimintaa koskevassa tutkimuksessa verrattiin systeemistä voklosporiinialtistusta lievää ja keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden (Child-Pugh-luokat A ja B) ja niiden verrokkien välillä, joiden maksa toiminta oli normaalia. Lievää ja keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden voklosporiinin Cmax oli 1,5-kertainen ja AUC0-48 noin kaksinkertainen verrokkeihin nähden (ks. kohta Annostus ja antotapa). Voklosporiinia ei ole arvioitu vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavilla potilailla, eikä sen käyttöä näille potilaille suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ikä, sukupuoli, rotu ja paino
Populaatiofarmakokineettinen analyysi, jossa arvioitiin iän, sukupuolen, rodun ja painon vaikutuksia, ei osoittanut näillä kovariaateilla olevan kliinisesti merkittävää vaikutusta voklosporiinialtistukseen.
Imetys
Imettävillä vapaaehtoisilla (ks. kohta Raskaus ja imetys) erittyi voklosporiinia rintamaitoon keskimäärin 0,00472 mg 48 tunnin aikana voklosporiinin 23,7 mg:n kerta-annoksen jälkeen. Tästä määrästä 80 % erittyi 12 tunnin kuluessa. Tiedot osoittivat, että maidon ja äidin veren välinen voklosporiinialtistuksen suhde oli 0,42–0,95. Kun rintamaidon saanti oli 200 ml/kg/vrk, suurin suhteellinen vauvan saama annos oli 1,4 % painon mukaan korjatusta äidin annoksesta.
Prekliiniset tiedot turvallisuudesta
Jäljempänä mainittuja haittavaikutuksia ei ole todettu kliinisissä tutkimuksissa, mutta niitä on todettu koe-eläimillä, jotka ovat saaneet hoitoannoksia vastaavia määriä lääkeainetta. Siksi haitoilla voi olla kliinistä merkitystä.
Toistuvaa altistusta koskevissa eläinkokeissa on havaittu neurohistologisina löydöksinä aivojen ja selkäytimen glioosia ja perivaskulaarisia infiltraatteja rotilla, mutta ei koirilla ja apinoilla. Näitä löydöksiä ei havaittu annoksilla, jotka vastaavat noin 0,3-kertaista suurinta ihmiselle suositeltua annosta (MRHD), 23,7 mg voklosporiinia kahdesti vuorokaudessa, lääkevalmisteelle altistuksen (AUC) perusteella.
Jaavanmakakeilla tehdyssä 39 viikon pituisessa suun kautta annetun voklosporiinin toksisuutta koskevassa tutkimuksessa esiintyi pahanlaatuisia lymfoomia annoksella 150 mg/kg/vrk (uroksilla noin 4-kertainen ja naarailla noin 7-kertainen MRHD-annos lääkevalmisteelle altistuksen [AUC] perusteella). Tämä annos aiheutti apinoille voimakasta immunosuppressiota, mikä ilmeni kalsineuriinin eston maksimitason (Emax) kohoamisena yli 80 prosenttiin. Suurin annos, jolla tätä löydöstä ei havaittu (NOAEL-arvo), oli 75 mg/kg/vrk (sekä uroksilla että naarailla noin 4-kertainen MRHD-annos lääkevalmisteelle altistuksen [AUC] perusteella).
Genotoksisuutta koskevissa konventionaalisissa tutkimuksissa voklosporiinilla ei havaittu mutageenisia tai genotoksisia vaikutuksia.
Hiirillä tehdyssä kahden vuoden pituisessa suun kautta annetun voklosporiinin karsinogeenisuutta koskevassa tutkimuksessa havaittiin pahanlaatuisten lymfoomien esiintyvyyden kasvua suurimmalla testatulla annoksella, 30 mg/kg/vrk (noin 7,5-kertainen MRHD-annos lääkevalmisteelle altistuksen [AUC] perusteella). Tämän löydöksen katsottiin johtuvan voklosporiiniin liittyvästä immunosuppressiosta. NOAEL-arvo oli 10 mg/kg/vrk (noin 1-kertainen MRHD-annos lääkevalmisteelle altistuksen [AUC] perusteella).
Rotilla tehdyssä hedelmällisyystutkimuksessa, jossa eläimille annettiin voklosporiinia ja sen cis-isomeeria 50:50-yhdistelmänä, annoksella 25 mg/kg/vrk havaittiin urosten sukupuolielinten, myös lisäkivesten hännän (cauda epidymis), rakkularauhasten, eturauhasen ja kivesten, painon laskua. Näiden löydösten NOAEL-arvo oli 10 mg/kg/vrk (noin 5-kertainen MRHD-annos lääkevalmisteelle altistuksen [AUC] perusteella). Vaikutuksia parittelu- ja hedelmällisyysparametreihin, siittiöiden liikkuvuuteen, määrään ja tiheyteen, kiimavaiheiden lukumäärään per 14 vuorokautta ja sektioparametreihin ei havaittu. Eturauhasen ja kivesten painon laskua havaittiin myös toistuvan altistuksen aiheuttamaa toksisuutta koskevissa 13 viikon ja 26 viikon pituisissa tutkimuksissa, joissa eläimille annettiin voklosporiinia ja sen cis-isomeeria 50:50-seoksena suun kautta annoksilla 25 mg/kg/vrk ja 10 mg/kg/vrk (18-kertainen ja 7-kertainen MRHD-annos lääkevalmisteelle altistuksen [AUC] perusteella). Näiden vaikutusten NOAEL-arvo toistuvaa altistusta koskevassa 26 viikon pituisessa tutkimuksessa oli 2,5 mg/kg/vrk (noin 1-kertainen MRHD-annos lääkevalmisteelle altistuksen [AUC] perusteella).
Alkioiden ja sikiöiden kehitystä tutkittiin voklosporiinin ja sen cis-isomeerin 50:50-yhdistelmällä, jota annettiin rotille ja kaniineille, sekä voklosporiinilla, jota annettiin kaniineille. Alkio- ja sikiötoksisuutta havaittiin emolle toksisilla annoksilla (rotilla noin 15-kertainen ja kaniineilla noin 1-kertainen MRHD-annos lääkevalmisteelle altistuksen [AUC] perusteella). Emoon kohdistuvia vaikutuksia olivat painon muutokset ja/tai maitorauhasten turvotus ja sikiöihin kohdistuvia vaikutuksia lievä painonlasku ja siihen liittyvät luuston kehityksen vaihtelut. Epämuodostumia aiheuttavia vaikutuksia ei havaittu tutkimuksissa. NOAEL-arvo oli rotilla 10 mg/kg/vrk ja kaniineilla 1 mg/kg/vrk (rotilla noin 7-kertainen ja kaniineilla noin 0,01-kertainen MRHD-annos lääkevalmisteelle altistuksen [AUC] perusteella).
Rotilla tehdyssä pre- ja postnataalista kehitystä koskevassa tutkimuksessa emoon kohdistuva toksisuus voklosporiinin ja sen cis-isomeerin 50:50-seoksenannoksella 25 mg/kg/vrk (noin 17-kertainen MRHD-annos lääkevalmisteelle altistuksen [AUC] perusteella) viivästytti synnytystä (dystokia), mikä johti synnytettyjen poikasten sekä poikueiden eloon jäävien poikasten keskilukumäärien pienenemiseen. Tätä annosta pidettiin emolle toksisena painonnousun pienentymisen vuoksi. Emoon tai poikasiin kohdistuvia haittavaikutuksia ei havaittu enintään noin 3-kertaisilla MRHD-annoksilla (perustuen lääkevalmisteelle altistukseen [AUC], ja emolle suun kautta annettu NOAEL-annos oli 10 mg/kg/vrk). Vaikutuksia uros- tai naaraspoikasten käyttäytymiseen, fyysiseen kehitykseen tai lisääntymiskykyyn ei havaittu. Suurin annos, joka ei vaikuttanut synnytykseen ja poikasten eloonjääntiin, oli 10 mg/kg/vrk.
Imettäville rotille suun kautta annetun [14C]-voklosporiinin radioaktiivisuus jakautui nopeasti eläinten maitoon. Kun lääkevalmistetta erittyy eläinten maitoon, on todennäköistä, että sitä erittyy myös ihmisen rintamaitoon.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Etanoli
E-vitamiini (E307) -polyeteeniglykolisuksinaatti (tokofersolaani)
Polysorbaatti 40
Keskipitkäketjuiset triglyseridit
Kapselin kuori
Gelatiini
Sorbitoli
Glyseriini
Puhdistettu vesi
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Apuaineet valmistuksessa
Soijalesitiini
Yhteensopimattomuudet
Ei oleellinen
Kestoaika
4 vuotta
Säilytys
Säilytä alkuperäisessä läpipainopakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LUPKYNIS kapseli, pehmeä
7,9 mg (L:ei) 180 fol (899,83 €)
PF-selosteen tieto
Pehmeät kapselit on pakattu kylmämuovattuihin alumiiniläpipainopakkauksiin, joiden laminoidut kansi- ja alusmateriaalit on kuumasaumattu yhteen. Yksi läpipainopakkaus sisältää 18 pehmeää kapselia. Yksi kotelo sisältää 180 tai 576 pehmeää kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Vaaleanpunaiset/oranssit, soikeat, pehmeät kapselit, joiden koko on noin 13 mm × 6 mm.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
LUPKYNIS kapseli, pehmeä
7,9 mg 180 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Voklosporiini: Aikuisten SLE-nefriitin hoito yhdistelmänä mykofenolaattimofetiilin kanssa erityisin edellytyksin (3083).
ATC-koodi
L04AD03
Valmisteyhteenvedon muuttamispäivämäärä
17.11.2025
Yhteystiedot
Sveavägen 151
113 46 Stockholm
Sweden
+46 8 545 28660
www.otsuka.se
info@otsuka.se