
KISQALI tabletti, kalvopäällysteinen 200 mg
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää ribosiklibisuksinaattia vastaten 200 mg ribosiklibia (ribociclib.).
Apuaine, jonka vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 0,344 mg soijalesitiiniä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Varhaisvaiheen rintasyöpä
Kisqali on tarkoitettu käytettäväksi yhdessä aromataasinestäjän kanssa adjuvanttihoitona potilaille, joilla on suuren uusiutumisriskin hormonireseptoripositiivinen, HER2-negatiivinen varhaisvaiheen rintasyöpä (soveltuvuuskriteerit, ks. kohta Farmakodynamiikka).
Miehillä ja pre- tai perimenopausaalisilla naisilla aromataasinestäjähoitoon on yhdistettävä LHRH-agonisti.
Edennyt tai etäpesäkkeinen rintasyöpä
Kisqali on tarkoitettu käytettäväksi naisille, joilla on hormonireseptoripositiivinen, HER2-negatiivinen paikallisesti edennyt tai etäpesäkkeinen rintasyöpä, yhdessä aromataasinestäjän tai fulvestrantin kanssa ensimmäisenä endokriinisena hoitona, tai naisille, jotka ovat saaneet aiempaa endokriinista hoitoa.
Pre‑ tai perimenopausaalisilla naisilla endokriiniseen hoitoon on yhdistettävä LHRH-agonisti.
Ehto
Hoito tulee aloittaa syövän hoitoon tarkoitettujen lääkevalmisteiden antamiseen perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Kisqali-hoidon aloittaa lääkäri, jolla on kokemusta syöpähoitojen toteuttamisesta.
Hormonireseptoripositiivisuuden, HER2-negatiivisuuden testaus
Potilaiden valinta Kisqali-hoitoon kasvaimen hormonireseptoreiden ja HER2:n ilmentymisen perusteella on arvioitava CE-merkityllä in vitro -diagnostiikkaan (IVD) tarkoitetulla lääkinnällisellä laitteella, jolla on vastaava käyttötarkoitus. Jos CE-merkittyä IVD-laitetta ei ole saatavilla, on käytettävä vaihtoehtoista validoitua testiä.
Annostus
Varhaisvaiheen rintasyöpä
Suositeltu annos on 400 mg (kaksi 200 mg:n kalvopäällysteistä tablettia) ribosiklibia kerran vuorokaudessa 21 perättäisenä päivänä, minkä jälkeen pidetään 7 päivän hoitotauko. Hoitojakson kokonaispituus on siis 28 päivää. Varhaisvaiheen rintasyöpää sairastavilla potilailla Kisqali-hoitoa jatketaan 3 vuoden ajan tai taudin uusiutumiseen asti tai kunnes ilmenee sietämätöntä toksisuutta.
Kun Kisqali-valmistetta käytetään yhdessä aromataasinestäjän kanssa, aromataasinestäjä otetaan suun kautta kerran vuorokaudessa jatkuvasti koko 28 päivän hoitojakson ajan. Lisätiedot, ks. kyseisen aromataasinestäjän valmisteyhteenveto.
Miehillä ja pre- tai perimenopausaalisilla naisilla aromataasinestäjähoitoon on yhdistettävä LHRH-agonisti.
Edennyt tai etäpesäkkeinen rintasyöpä
Suositeltu annos on 600 mg (kolme 200 mg:n kalvopäällysteistä tablettia) ribosiklibia kerran vuorokaudessa 21 perättäisenä päivänä, minkä jälkeen pidetään 7 päivän hoitotauko. Hoitojakson kokonaispituus on siis 28 päivää. Edenneen tai etäpesäkkeisen rintasyövän hoitoa jatketaan niin kauan kuin potilas hyötyy hoidosta tai kunnes ilmenee sietämätöntä toksisuutta.
Kun Kisqali-valmistetta käytetään yhdessä aromataasinestäjän kanssa, aromataasinestäjä otetaan suun kautta kerran vuorokaudessa jatkuvasti koko 28 päivän hoitojakson ajan. Lisätiedot, ks. kyseisen aromataasinestäjän valmisteyhteenveto.
Kun Kisqali-valmistetta käytetään yhdessä fulvestrantin kanssa, fulvestrantti annetaan lihakseen päivinä 1, 15 ja 29 ja tämän jälkeen kerran kuukaudessa. Lisätiedot, ks. fulvestrantin valmisteyhteenveto.
Pre‑ ja perimenopausaalisten naisten hoitoon on hyväksyttyjen Kisqali-yhdistelmähoitojen lisäksi kuuluttava myös LHRH-agonisti paikallisen kliinisen hoitokäytännön mukaan.
Annosmuutokset
Vaikeiden tai sietämättömien haittavaikutusten hoito voi edellyttää Kisqali-hoidon tauottamista, Kisqali-annoksen pienentämistä tai Kisqali-hoidon lopettamista. Jos annosta on pienennettävä, annoksen pienennyssuositukset esitetään taulukossa 1.
Taulukko 1 Annosmuutossuositukset
| Kisqali | ||
| Annos | 200 mg:n tablettien määrä | |
| Varhaisvaiheen rintasyöpä | ||
| Aloitusannos | 400 mg/vrk | 2 |
| Pienennetty annos | 200 mg*/vrk | 1 |
| Edennyt tai etäpesäkkeinen rintasyöpä | ||
| Aloitusannos | 600 mg/vrk | 3 |
| 1. pienennetty annos | 400 mg/vrk | 2 |
| 2. pienennetty annos | 200 mg*/vrk | 1 |
| * Jos annosta on tarpeen pienentää edelleen alle tason 200 mg/vrk, hoito on lopetettava pysyvästi. | ||
Taulukoissa 2, 3, 4, 5 ja 6 esitetään yhteenvedot Kisqali-hoidon tauottamista, annoksen pienentämistä ja hoidon lopettamista koskevista suosituksista tiettyjen haittavaikutusten hoidossa. Kunkin potilaan hoitosuunnitelman tulee perustua hoitavan lääkärin kliiniseen arvioon ja potilaskohtaiseen hyöty-riskiarvioon (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen verenkuva on määritettävä ennen Kisqali-hoidon aloittamista. Hoidon aloittamisen jälkeen täydellistä verenkuvaa seurataan 2 viikon välein ensimmäisten 2 hoitojakson ajan, aina hoitojakson alussa seuraavien 4 hoitojakson ajan ja tämän jälkeen kliinisen tarpeen mukaan.
Taulukko 2 Annosmuutokset ja hoito – Neutropenia
Aste 1 tai 2* (absoluuttinen neutrofiilimäärä 1,0–1,5 x 109/l) | Aste 3* (absoluuttinen neutrofiilimäärä 0,5- <1,0 x 109/l) | Aste 3* kuumeinen neutropenia** | Aste 4* (absoluuttinen neutrofiilimäärä < 0,5 x 109/l) | |
| Neutropenia | Annosta ei tarvitse muuttaa. | Hoito tauotetaan, kunnes vaikeusaste lievittyy tasolle ≤ 2. Kisqali-hoito aloitetaan uudelleen samalla annostasolla. Jos toksisuus uusiutuu asteen 3 tasoisena: hoito tauotetaan, kunnes toksisuus korjautuu tasolle ≤ 2, minkä jälkeen Kisqali-hoito aloitetaan uudelleen 1 annostasoa pienemmällä annoksella. | Hoito tauotetaan, kunnes vaikeusaste lievittyy tasolle ≤ 2. Kisqali-hoito aloitetaan uudelleen 1 annostasoa pienemmällä annoksella. | Hoito tauotetaan, kunnes vaikeusaste lievittyy tasolle ≤ 2. Kisqali-hoito aloitetaan uudelleen 1 annostasoa pienemmällä annoksella. |
* Vaikeusasteet perustuvat CTCAE-kriteerien (CTCAE = Common Terminology Criteria for Adverse Events) versioon 4.03. ** Asteen 3 neutropenia ja yhden kerran kuumetta > 38,3 °C (tai ≥ 38 °C yli yhden tunnin ajan ja/tai samanaikainen infektio) | ||||
Maksan toimintakokeet on tehtävä ennen Kisqali-hoidon aloittamista. Hoidon aloittamisen jälkeen maksan toimintakokeita tehdään 2 viikon välein ensimmäisten 2 hoitojakson ajan, aina hoitojakson alussa seuraavien 4 hoitojakson ajan ja tämän jälkeen kliinisen tarpeen mukaan. Jos asteen ≥ 2 poikkeavuuksia todetaan, suositellaan tiiviimpää seurantaa.
Taulukko 3 Annosmuutokset ja hoito – Maksa- ja sappitoksisuus
Aste 1* (> ULN – 3 x ULN) | Aste 2* (> 3 – 5 x ULN) | Aste 3* (> 5 – 20 x ULN) | Aste 4* (> 20 x ULN) | |
| ASAT- ja/tai ALAT-arvojen suureneminen lähtötasolta**, ei kokonaisbilirubiiniarvon suurenemista yli tason 2 x ULN | Annosta ei tarvitse muuttaa. | Lähtötason aste < 2: Hoito tauotetaan, kunnes tilanne korjautuu lähtötason vaikeusastetta vastaavaksi tai sitä lievemmäksi; tämän jälkeen Kisqali-hoito aloitetaan uudelleen samalla annostasolla. Jos asteen 2 toksisuus uusiutuu, Kisqali-hoito aloitetaan uudelleen yhtä annostasoa pienemmällä annoksella. | Hoito tauotetaan, kunnes tilanne korjautuu lähtötason vaikeusastetta vastaavaksi tai sitä lievemmäksi; tämän jälkeen Kisqali-hoito aloitetaan uudelleen yhtä annostasoa pienemmällä annoksella. Jos asteen 3 toksisuus uusiutuu, Kisqali-hoito lopetetaan. | Kisqali-hoito lopetetaan. |
Lähtötason aste = 2: Hoitoa ei tauoteta. | ||||
| ASAT- ja/tai ALAT-arvon suureneminen ja samanaikainen kokonaisbilirubiiniarvon suureneminen, ei kolestaasia | Jos potilaan ALAT- ja/tai ASAT-arvo suurenee tasolle > 3 x ULN ja kokonaisbilirubiiniarvo tasolle > 2 x ULN, Kisqali-hoito lopetetaan lähtötason vaikeusasteesta riippumatta. | |||
* Vaikeusasteet perustuvat CTCAE-kriteerien (CTCAE = Common Terminology Criteria for Adverse Events) versioon 4.03. ** Lähtötaso = ennen hoidon aloitusta ULN = viitearvojen yläraja | ||||
EKG-löydökset on arvioitava jokaiselta potilaalta ennen Kisqali-hoidon aloittamista.
Kisqali-hoidon saa aloittaa vain, jos potilaan QTcF-arvo on alle 450 ms. Hoidon aloittamisen jälkeen EKG-tutkimus toistetaan suunnilleen ensimmäisen hoitojakson päivänä 14, sekä tämän jälkeen kliinisen tarpeen mukaan.
Jos hoidon aikana todetaan QTcF-ajan pitenemistä, suositellaan sekä varhaisvaiheen rintasyöpää sairastavien että edennyttä tai etäpesäkkeistä rintasyöpää sairastavien potilaiden kohdalla tiiviimpää EKG-seurantaa.
Taulukko 4 Annosmuutokset ja hoito – QT-ajan piteneminen
| QTcF*-ajan piteneminen | Varhaisvaiheen rintasyöpä | Edennyt tai etäpesäkkeinen rintasyöpä |
| > 480 – ≤ 500 ms | Kisqali-hoito tauotetaan, kunnes QTcF-aika korjautuu < 481 ms:n pituiseksi. | |
| Hoito aloitetaan uudelleen samalla annostasolla. | Hoitoa jatketaan yhtä annostasoa pienemmällä annoksella. | |
| Jos QTcF-aika pitenee uudelleen ≥ 481 ms:n pituiseksi, Kisqali-hoito tauotetaan, kunnes QTcF-aika korjautuu < 481 ms:n pituiseksi; tämän jälkeen hoito aloitetaan uudelleen yhtä annostasoa pienemmällä annoksella. | ||
| > 500 ms | Kisqali-hoito tauotetaan, kunnes QTcF-aika korjautuu < 481 ms:n pituiseksi; tämän jälkeen Kisqali-hoito aloitetaan uudelleen yhtä annostasoa pienemmällä annoksella. Jos QTcF-aika pitenee uudestaan yli 500 ms:n pituiseksi, Kisqali-hoito lopetetaan. | |
| Jos QTcF-aika pitenee yli 500 ms:n pituiseksi tai muuttuu lähtötilanteesta yli 60 ms ja potilaalla on samanaikaisesti kääntyvien kärkien takykardiaa, polymorfista kammiotakykardiaa tai vakavien rytmihäiriöiden oireita/löydöksiä, Kisqali-hoito lopetetaan pysyvästi. | ||
Huom. Jos annosta on tarpeen pienentää edelleen alle tason 200 mg/vrk, hoito on lopetettava. *QTcF = Friderician kaavan mukaan korjattu QT-aika | ||
Taulukko 5 Annosmuutokset ja hoito – interstitiaalinen keuhkosairaus (ILD)/keuhkotulehdus
Aste 1* (oireeton) | Aste 2* (oireellinen) | Aste 3 tai 4* (vaikea) | |
| ILD/keuhkotulehdus | Annosta ei tarvitse muuttaa. Aloitetaan asianmukainen lääketieteellinen hoito ja seurataan kliinisen tarpeen mukaan. | Hoito tauotetaan, kunnes tilanne korjautuu vaikeusasteen ≤ 1 tasolle, tämän jälkeen Kisqali-hoito aloitetaan uudelleen yhtä annostasoa pienemmällä annoksella**. | Kisqali-hoito lopetetaan. |
* Vaikeusasteet perustuvat CTCAE-kriteerien (CTCAE = Common Terminology Criteria for Adverse Events) versioon 4.03. **Harkittaessa Kisqali-hoidon aloittamista uudelleen, on tehtävä yksilöllinen hyöty-riski-arvio. ILD = interstitiaalinen keuhkosairaus | |||
Taulukko 6 Annosmuutokset ja hoito – Muu toksisuus*
| Muu toksisuus | Aste 1 tai 2** | Aste 3** | Aste 4** |
| Annosta ei tarvitse muuttaa. Aloitetaan asianmukainen lääketieteellinen hoito ja seurataan kliinisen tarpeen mukaan. | Hoito tauotetaan, kunnes tilanne korjautuu vaikeusasteen ≤ 1 tasolle; tämän jälkeen Kisqali-hoito aloitetaan uudelleen samalla annostasolla. Jos asteen 3 toksisuus uusiutuu, Kisqali-hoito aloitetaan uudelleen yhtä annostasoa pienemmällä annoksella. | Kisqali-hoito lopetetaan. | |
* Neutropeniaa, maksatoksisuutta, QT-ajan pitenemistä ja interstitiaalista keuhkosairautta/keuhkotulehdusta lukuun ottamatta. ** Vaikeusasteiden määritys perustuu CTCAE-kriteerien (CTCAE= Common Terminology Criteria for Adverse Events) versioon 4.03. | |||
Katso annosmuutosohjeet ja muut oleelliset turvallisuustiedot samanaikaisesti käytettävän aromataasinestäjän, fulvestrantin tai LHRH-agonistin valmisteyhteenvedosta, mikäli ilmenee toksisuutta.
Annosmuutokset, kun Kisqali-valmistetta käytetään yhdessä vahvojen CYP3A4:n estäjien kanssa
Vahvojen CYP3A4:n estäjien samanaikaista käyttöä on vältettävä, ja samanaikaiseen käyttöön on harkittava jotakin toista lääkevalmistetta, jolla on pienempi CYP3A4:n estopotentiaali. Jos potilaalle on annettava vahvaa CYP3A4:n estäjää samanaikaisesti ribosiklibin kanssa, Kisqali-annos pienennetään (ks. kohta Yhteisvaikutukset).
Potilailla, joiden käyttämä ribosiklibiannos on 600 mg vuorokaudessa ja joiden kohdalla vahvan CYP3A4:n estäjän aloittamista ei voida välttää, tulee ribosiklibiannosta pienentää 400 mg:aan.
Potilailla, joiden käyttämä ribosiklibiannos on 400 mg vuorokaudessa ja joiden kohdalla vahvan CYP3A4:n estäjän aloittamista ei voida välttää, tulee ribosiklibiannosta pienentää edelleen 200 mg:aan.
Potilailla, joiden ribosiklibiannos on pienennetty 200 mg:aan vuorokaudessa ja joiden kohdalla vahvan CYP3A4:n estäjän aloittamista ei voida välttää, Kisqali-hoito tauotetaan.
Potilaiden välisen vaihtelun vuoksi suositeltavat annosmuutokset eivät välttämättä ole optimaalisia kaikkien potilaiden kohdalla, joten on suositeltavaa seurata potilaita tarkoin toksisuuden merkkien varalta. Jos vahvan estäjän käyttö lopetetaan, Kisqali-annos muutetaan kyseisen vahvan CYP3A4:n estäjän käyttöönottoa edeltäneeksi annokseksi, kun on kulunut vähintään 5 kyseisen vahvan CYP3A4:n estäjän puoliintumisaikaa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakokinetiikka).
Erityisryhmät
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta. Potilaille, joilla on vaikea munuaisten vajaatoiminta, suositeltava aloitusannos on 200 mg. Kisqali-valmistetta ei ole tutkittu rintasyöpäpotilailla, joilla on vaikea munuaisten vajaatoiminta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Farmakodynamiikka ja Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa, jos varhaisvaiheen rintasyöpää sairastavalla potilaalla on maksan vajaatoiminta (ks. kohta Farmakokinetiikka). Jos edennyttä tai etäpesäkkeistä rintasyöpää sairastavalla potilaalla on lievä maksan vajaatoiminta (Child–Pugh-luokka A), annosta ei tarvitse muuttaa, mutta jos tällaisella potilaalla on keskivaikea (Child–Pugh-luokka B) tai vaikea maksan vajaatoiminta (Child–Pugh-luokka C), ribosiklibialtistus voi suurentua (alle 2-kertaiseksi). Tällöin Kisqali-hoidon suositeltava aloitusannos on 400 mg kerran vuorokaudessa (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Kisqali-hoidon turvallisuutta ja tehoa lasten ja alle 18-vuotiaiden nuorten hoidossa ei ole varmistettu. Saatavissa olevat tiedot Kisqali-valmisteen käytöstä yhdessä topotekaanin ja temotsolomidin kanssa pediatrisilla potilailla on kuvattu kohdissa Farmakodynamiikka ja Farmakokinetiikka, mutta suosituksia annostuksesta ei voida antaa.
Iäkkäät
Yli 65-vuotiaiden potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Antotapa
Kisqali otetaan suun kautta kerran vuorokaudessa ruoan kanssa tai ilman ruokaa (ks. kohta Yhteisvaikutukset). Potilaita kehotetaan ottamaan annos suunnilleen samaan aikaan joka päivä, mieluiten aamuisin. Jos potilas oksentaa annoksen ottamisen jälkeen tai annos jää väliin, kyseisenä päivänä ei pidä ottaa uutta annosta. Seuraava lääkemääräyksen mukainen annos otetaan tavanomaiseen aikaan. Tabletit niellään kokonaisina, eikä niitä saa pureskella, murskata eikä pilkkoa ennen nielemistä. Tablettia ei saa niellä, jos se on rikkoutunut, halkeillut tai muutoin rikki.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai maapähkinälle, soijalle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Kriittinen viskeraalinen tauti
Ribosiklibin tehoa ja turvallisuutta ei ole tutkittu potilailla, joilla on kriittinen viskeraalinen tauti.
Neutropenia
Neutropenian vaikeusasteesta riippuen voi olla tarpeellista tauottaa Kisqali-hoito, pienentää Kisqali-annosta tai lopettaa Kisqali-hoito taulukossa 2 kuvattavaan tapaan (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Maksa- ja sappitoksisuus
Maksan toimintakokeet on tehtävä ennen Kisqali-hoidon aloittamista. Hoidon aloittamisen jälkeen maksan toimintaa on seurattava (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Transaminaasiarvojen nousun vaikeusasteesta riippuen voi olla tarpeellista tauottaa Kisqali-hoito, pienentää Kisqali-annosta tai lopettaa Kisqali-hoito taulukossa 3 kuvattavaan tapaan (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Potilaille, joiden ASAT/ALAT-arvot ovat lähtötilanteessa astetta ≥ 3 vastaavasti koholla, ei ole laadittu suosituksia.
QT-ajan piteneminen
Kisqali-hoitoa on vältettävä, jos potilaalla on jo entuudestaan pitkä QTc-aika tai merkittävä QTc-ajan pitenemisriski. Tämä koskee seuraavia potilasryhmiä:
- potilaat, joilla on pitkä QT ‑oireyhtymä;
- potilaat, joilla on huonossa hoitotasapainossa oleva tai merkittävä sydäntauti, esim. äskettäinen sydäninfarkti, kongestiivinen sydämen vajaatoiminta, epästabiili angina pectoris tai bradyarytmiaa;
- potilaat, joilla on elektrolyyttihäiriöitä.
Kisqalin käyttöä yhdessä tunnetusti QTc-aikaa pidentävien lääkevalmisteiden ja/tai vahvojen CYP3A4:n estäjien kanssa on vältettävä, sillä se voi johtaa QTcF-ajan kliinisesti merkittävään pitenemiseen (ks. kohdat Annostus ja antotapa, Yhteisvaikutukset ja Farmakodynamiikka). Jos Kisqalin ja vahvan CYP3A4:n estäjän samanaikainen käyttö on välttämätöntä, Kisqali-annosta on muutettava kohdassa Annostus ja antotapa kuvattavaan tapaan.
E2301-tutkimuksen (MONALEESA‑7) löydösten perusteella Kisqali-valmisteen käyttö yhdessä tamoksifeenin kanssa ei ole suositeltavaa (ks. kohdat Haittavaikutukset ja Farmakodynamiikka).
Varhaisvaiheen rintasyöpä
O12301C-tutkimuksessa (NATALEE) QTcF-ajan pitenemistä > 60 ms lähtötilanteesta todettiin Kisqali-valmisteen ja aromataasinestäjän yhdistelmää saaneessa ryhmässä 19 potilaalla (0,8 %).
EKG-löydökset on arvioitava ennen hoidon aloittamista. Kisqali-hoidon saa aloittaa vain, jos potilaan QTcF-arvo on alle 450 ms. EKG-tutkimus toistetaan suunnilleen ensimmäisen hoitojakson päivänä 14, tämän jälkeen kliinisen tarpeen mukaan (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Varhaisvaiheen rintasyöpää sairastavilla potilailla seerumin elektrolyyttiarvoja (mm. kalium, kalsium, fosfori ja magnesium) on seurattava asianmukaisesti ennen hoidon aloittamista, ensimmäisten 6 hoitojakson alussa ja tämän jälkeen kliinisen tarpeen mukaan. Mahdolliset poikkeavuudet on korjattava ennen Kisqali-hoidon aloittamista ja Kisqali-hoidon aikana.
Hoidon aikana todetusta QT-ajan pitenemisestä riippuen voi olla tarpeellista tauottaa Kisqali-hoito, pienentää Kisqali-annosta tai lopettaa Kisqali-hoito taulukossa 4 kuvattavaan tapaan (ks. kohdat Annostus ja antotapa, Haittavaikutukset ja Farmakokinetiikka).
Edennyt tai etäpesäkkeinen rintasyöpä
E2301-tutkimuksessa (MONALEESA-7) QTcF-ajan pitenemistä > 60 ms lähtötilanteesta todettiin Kisqali- ja tamoksifeenihoitoa saaneessa ryhmässä 14/87 potilaalla (16,1 %) ja Kisqali‑valmistetta ja ei-steroidista aromataasinestäjää saaneessa ryhmässä 18/245 potilaalla (7,3 %).
EKG-löydökset on arvioitava ennen hoidon aloittamista. Kisqali-hoidon saa aloittaa vain, jos potilaan QTcF-arvo on alle 450 ms. EKG-tutkimus toistetaan suunnilleen ensimmäisen hoitojakson päivänä 14, tämän jälkeen kliinisen tarpeen mukaan (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla seerumin elektrolyyttiarvoja (mm. kalium, kalsium, fosfori ja magnesium) on seurattava asianmukaisesti ennen hoidon aloittamista, ensimmäisten 6 hoitojakson alussa ja tämän jälkeen kliinisen tarpeen mukaan. Mahdolliset poikkeavuudet on korjattava ennen Kisqali-hoidon aloittamista ja Kisqali-hoidon aikana.
Hoidon aikana todetusta QT-ajan pitenemisestä riippuen voi olla tarpeellista tauottaa Kisqali-hoito, pienentää Kisqali-annosta tai lopettaa Kisqali-hoito taulukossa 4 kuvattavaan tapaan (ks. kohdat Annostus ja antotapa, Haittavaikutukset ja Farmakokinetiikka).
Vaikeat ihoreaktiot
Kisqali-hoidon yhteydessä on raportoitu toksista epidermaalista nekrolyysiä (TEN). Jos potilaalla esiintyy vaikeisiin ihoreaktioihin viittaavia oireita tai löydöksiä (esim. etenevä laaja ihottuma, johon usein liittyy rakkuloita tai limakalvovaurioita), Kisqali-hoito täytyy keskeyttää välittömästi.
Interstitiaalinen keuhkosairaus/keuhkotulehdus
Interstitiaalista keuhkosairautta (ILD)/keuhkotulehdusta on raportoitu Kisqalin käytön yhteydessä. Potilaita täytyy seurata ILD:hen/keuhkotulehdukseen viittaavien keuhko-oireiden varalta. Oireisiin voi kuulua hypoksiaa, yskää ja hengenahdistusta. Muutokset annostuksessa täytyy tehdä taulukon 5 mukaisesti (ks. kohta Annostus ja antotapa).
ILD:n/keuhkotulehduksen, jotka voivat johtaa kuolemaan, vakavuudesta riippuen Kisqali-hoito voidaan joutua tauottamaan, annosta pienentämään tai hoito voidaan joutua lopettamaan, kuten taulukossa 5 on kuvattu (ks. kohta Annostus ja antotapa).
Suurentunut veren kreatiniinipitoisuus
Ribosiklibi voi suurentaa veren kreatiniinipitoisuutta, sillä se estää munuaisten kuljettajaproteiineja OCT2:ta (orgaanisten kationien kuljettajaproteiini 2) ja MATE1:tä (multidrug and toxin extrusion protein 1), jotka osallistuvat kreatiniinin aktiiviseen eritykseen proksimaalisista tubuluksista (ks. kohta Yhteisvaikutukset). Jos veren kreatiniinipitoisuus suurenee hoidon aikana, munuaisten toiminnan lisätutkimuksia suositellaan munuaisten vajaatoiminnan poissulkemiseksi.
CYP3A4:n substraatit
Ribosiklibi on 600 mg:n annoksilla vahva CYP3A4:n estäjä ja 400 mg:n annoksilla keskivahva CYP3A4:n estäjä. Ribosiklibilla voi siis olla yhteisvaikutuksia CYP3A4-välitteisesti metaboloituvien lääkevalmisteiden kanssa, mikä voi suurentaa CYP3A4:n substraattien pitoisuuksia seerumissa (ks. kohta Yhteisvaikutukset). Varovaisuus on suositeltavaa, jos samanaikaisesti käytetään herkkiä, terapeuttiselta leveydeltään kapeita CYP3A4:n substraatteja. CYP3A4:n estäjien samanaikaista käyttöä koskevat suositukset on tarkistettava toisen valmisteen valmisteyhteenvedosta.
Munuaisten vajaatoiminta
Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden 200 mg:n aloitusannoksen on arvioitu aiheuttavan 45 % alhaisemman altistuksen verrattuna tavanomaiseen 600 mg:n aloitusannokseen edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla, joilla on normaali munuaisten toiminta. Tämän aloitusannoksen tehoa ei ole vahvistettu. Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden hoidossa on noudatettava varovaisuutta, ja potilaita on seurattava tarkoin toksisuuden merkkien varalta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Naiset, jotka voivat tulla raskaaksi
Naisia, jotka voivat tulla raskaaksi, on kehotettava käyttämään tehokasta ehkäisymenetelmää Kisqali-hoidon aikana ja vähintään 21 päivän ajan viimeisen annoksen jälkeen (ks. kohta Raskaus ja imetys).
Soijalesitiini
Kisqali sisältää soijalesitiiniä. Potilaat, jotka ovat yliherkkiä maapähkinälle tai soijalle, eivät saa käyttää Kisqalia (ks. kohta Vasta-aiheet).
Yhteisvaikutukset
Aineet, jotka saattavat suurentaa plasman ribosiklibipitoisuuksia
Ribosiklibi metaboloituu lähinnä CYP3A4-välitteisesti. CYP3A4-entsyymitoimintaan vaikuttavat lääkevalmisteet saattavat siis vaikuttaa ribosiklibin farmakokinetiikkaan. Kun terveille henkilöille annettiin vahvaa CYP3A4:n estäjä ritonaviiria (100 mg kahdesti vuorokaudessa 14 päivän ajan) yhdessä 400 mg:n ribosiklibikerta-annoksen kanssa, suureni ribosiklibialtistus (AUCinf) 3,2-kertaiseksi ja ribosiklibin huippupitoisuus (Cmax) 1,7-kertaiseksi verrattuna pelkän 400 mg:n ribosiklibikerta-annoksen antoon. LEQ803-metaboliitin (ribosiklibin merkittävä metaboliitti, joka tuottaa alle 10 % kanta-aineen altistuksesta) Cmax-arvo pieneni 96 % ja AUClast-arvo 98 %. Fysiologiaan perustuvan farmakokineettisen (PBPK) simulaation perusteella arvioitiin, että annettaessa ritonaviiria (100 mg kahdesti vuorokaudessa) samanaikaisesti ribosiklibin (400 mg kerran vuorokaudessa) kanssa, ribosiklibin vakaan tilan Cmax-arvo suureni 1,5-kertaiseksi ja AUC0-24h-arvo 1,8-kertaiseksi.
Potilaiden hoidossa on vältettävä vahvojen CYP3A4:n estäjien samanaikaista käyttöä eli mm. seuraavien lääkkeiden samanaikaista käyttöä: klaritromysiini, indinaviiri, itrakonatsoli, ketokonatsoli, lopinaviiri, ritonaviiri, nefatsodoni, nelfinaviiri, posakonatsoli, sakinaviiri, telapreviiri, telitromysiini, verapamiili ja vorikonatsoli (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.) Samanaikaista käyttöä varten on harkittava muita lääkevalmisteita, joiden CYP3A4-toiminnan estopotentiaali on pienempi, ja potilaita on seurattava ribosiklibiin liittyvien haittavaikutusten varalta (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Jos Kisqalin ja vahvan CYP3A4:n estäjän samanaikainen käyttö on välttämätöntä, Kisqali-annosta on muutettava kohdassa Annostus ja antotapa kuvatun mukaisesti. Näistä annosmuutoksista ei kuitenkaan ole kliinistä tietoa. Potilaiden välisen vaihtelun vuoksi suositeltavat annosmuutokset eivät välttämättä ole optimaalisia kaikkien potilaiden kohdalla, joten on suositeltavaa seurata potilaita tarkoin ribosiklibiin liittyvien haittavaikutusten varalta. Ribosiklibihoitoon liittyvän toksisuuden yhteydessä annosta muutetaan tai hoito tauotetaan, kunnes toksisuus on korjautunut (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka). Jos vahvan CYP3A4:n estäjän käyttö lopetetaan ja vähintään 5 kyseisen CYP3A4:n estäjän puoliintumisaikaa on kulunut (katso kyseisen CYP3A4:n estäjän valmisteyhteenveto), Kisqali-annos palautetaan vahvan CYP3A4:n estäjän aloittamista edeltäneelle tasolle.
PBPK-simulaatiot viittaavat siihen, että 600 mg:n ribosiklibiannoksia käytettäessä keskivahva CYP3A4:n estäjä (erytromysiini) saattaa suurentaa ribosiklibin vakaan tilan Cmax-arvon 1,1-kertaiseksi ja AUC-arvon 1,1-kertaiseksi. PBPK-simulaatiot viittaavat siihen, että 400 mg:n ribosiklibiannoksia käytettäessä keskivahva CYP3A4:n estäjä saattaa suurentaa ribosiklibin vakaan tilan Cmax-arvon 1,1‑kertaiseksi ja AUC-arvon 1,2‑kertaiseksi. Annoksella 200 mg kerran vuorokaudessa vaikutuksen ennustettiin olevan vakaan tilan Cmax-arvon 1,3‑kertainen ja AUC-arvon 1,5‑kertainen suureneminen. Lievän tai keskivahvan CYP3A4:n estäjän käytön aloittaminen ei edellytä ribosiklibiannoksen muuttamista. On kuitenkin suositeltavaa seurata potilasta ribosiklibiin liittyvien haittavaikutusten varalta.
Potilaita on kehotettava välttämään greippiä ja greippimehua. Niiden tiedetään estävän sytokromi CYP3A4‑entsyymitoimintaa, ja ne saattavat suurentaa ribosiklibialtistusta.
Aineet, jotka saattavat pienentää plasman ribosiklibipitoisuuksia
Kun terveille henkilöille annettiin vahvaa CYP3A4:n indusori rifampisiinia (600 mg vuorokaudessa 14 päivän ajan) yhdessä 600 mg:n ribosiklibikerta-annoksen kanssa, pieneni ribosiklibialtistus (AUCinf) 89 % ja ribosiklibin Cmax-arvo 81 % verrattuna pelkän 600 mg:n ribosiklibikerta-annoksen antoon. LEQ803-metaboliitin Cmax-arvo suureni 1,7-kertaiseksi ja AUCinf-arvo pieneni 27 %. Vahvojen CYP3A4:n indusorien samanaikainen käyttö voi siis pienentää altistusta ja aiheuttaa siten tehon heikkenemisen riskin. Vahvojen CYP3A4:n indusorien kuten mm. fenytoiinin, rifampisiinin, karbamatsepiinin ja mäkikuisman (Hypericum perforatum) samanaikaista käyttöä on vältettävä. Samanaikaista käyttöä varten on harkittava muita lääkevalmisteita, joiden CYP3A4-toimintaa indusoiva vaikutus on minimaalinen tai puuttuu kokonaan.
Keskivahvan CYP3A4:n indusoijan vaikutusta ribosiklibialtistukseen ei ole tutkittu. PBPK-simulaatiot viittaavat siihen, että keskivahva CYP3A4:n indusori (efavirentsi) saattaa pienentää ribosiklibin vakaan tilan Cmax-arvoa 55 % ja AUC-arvoa 74 % ribosiklibiannoksen ollessa 400 mg ja vakaan tilan Cmax-arvoa 52 % ja AUC-arvoa 71 % ribosiklibiannoksen ollessa 600 mg. Keskivahvojen CYP3A4:n indusoijien samanaikainen käyttö voi siten johtaa pienempään altistukseen ja tämän seurauksena tehon heikkenemiseen, erityisesti potilailla, jotka saavat ribosiklibihoitoa annoksella 400 mg tai 200 mg kerran vuorokaudessa.
Aineet, joiden pitoisuuksia plasmassa Kisqali saattaa muuttaa
Ribosiklibi on keskivahva tai vahva CYP3A4:n estäjä, ja sillä voi olla yhteisvaikutuksia CYP3A4-välitteisesti metaboloituvien substraattilääkevalmisteiden kanssa. Tämä voi suurentaa samanaikaisesti käytettävän lääkevalmisteen pitoisuuksia seerumissa.
Kun terveille henkilöille annettiin samanaikaisesti midatsolaamia (CYP3A4:n substraatti) ja toistuvia Kisqali-annoksia (400 mg), midatsolaamialtistus suureni 280 % (3,80-kertaiseksi) verrattuna pelkän midatsolaamin antoon. PBPK-simulaatiot viittaavat siihen, että 600 mg:n Kisqali-annosten anto suurentaa oletettavasti midatsolaamin AUC-arvon 5,2-kertaiseksi. Kun ribosiklibia annetaan samanaikaisesti muiden lääkevalmisteiden kanssa, onkin yleensä tutustuttava toisen lääkevalmisteen valmisteyhteenvedon suosituksiin CYP3A4:n estäjien samanaikaisesta käytöstä. Varovaisuus on tarpeen, jos samanaikaisesti käytetään terapeuttiselta leveydeltään kapeita, herkkiä CYP3A4:n substraatteja (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Herkän ja terapeuttiselta leveydeltään kapean CYP3A4:n substraatin annosta on ehkä pienennettävä, sillä ribosiklibi saattaa suurentaa altistusta. Tällaisia lääkkeitä ovat esimerkiksi alfentaniili, siklosporiini, everolimuusi, fentanyyli, sirolimuusi ja takrolimuusi.
Ribosiklibin antoa samanaikaisesti seuraavien CYP3A4:n substraattien kanssa on vältettävä: alfutsosiini, amiodaroni, sisapridi, pimotsidi, kinidiini, ergotamiini, dihydroergotamiini, ketiapiini, lovastatiini, simvastatiini, sildenafiili, midatsolaami, triatsolaami.
Kun terveille henkilöille annettiin kofeiinia (CYP1A2:n substraatti) samanaikaisesti toistuvien Kisqali-annosten (400 mg) kanssa, kofeiinialtistus suureni 20 % (1,20-kertaiseksi) verrattuna pelkän kofeiinin antoon. Fysiologiaan pohjautuvia farmakokinetiikan malleja hyödyntäneet simulaatiot ennustavat ribosiklibin kliinisesti merkityksellisillä 600 mg:n annoksilla olevan vain heikkoa estovaikutusta CYP1A2:n substraatteihin (AUC-arvo suurenee < 2-kertaiseksi).
Kuljettajaproteiinien substraattiaineet
In vitro ‑arvioinnit viittaavat siihen, että ribosiklibi saattaa estää P-gp-, BRCP-, OATP1B1/1B3-, OCT1-, OCT2-, MATE1- ja BSEP-kuljettajaproteiinien toimintaa. Varovaisuus on tarpeen ja potilasta on seurattava toksisuuden varalta, jos hän käyttää samanaikaisesti näiden kuljettajaproteiinien herkkiä substraatteja, joiden terapeuttinen leveys on kapea, esim. digoksiinia, pitavastatiinia, pravastatiinia, rosuvastatiinia tai metformiinia.
Lääkkeiden ja ruoan yhteisvaikutukset
Kisqali voidaan antaa ruoan kanssa tai ilman ruokaa (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Mahan pH-arvoa suurentavat lääkevalmisteet
Ribosiklibi on erittäin liukoinen pH-arvon ollessa 4,5 tai sitä pienempi sekä biorelevanteissa väliaineissa (pH-arvon ollessa 5,0 ja 6,5). Ribosiklibin antoa samanaikaisesti mahan pH-arvoa suurentavien lääkevalmisteiden kanssa ei arvioitu kliinisessä tutkimuksessa. Populaatiofarmakokinetiikan analyysissä ja tilavapaissa farmakokinetiikan analyyseissä ei kuitenkaan todettu muutoksia ribosiklibin imeytymisessä.
Ribosiklibin ja letrotsolin väliset lääkeaineinteraktiot
Rintasyöpäpotilailla toteutetun kliinisen tutkimuksen ja populaatiofarmakokinetiikan analyysin tiedot eivät viittaa lääkeaineinteraktioihin samanaikaisesti annettavan ribosiklibin ja letrotsolin välillä.
Ribosiklibin ja anastrotsolin väliset lääkeaineinteraktiot
Rintasyöpäpotilailla tehdyn kliinisen tutkimuksen tiedot eivät viittaa siihen, että samanaikaisesti annettavan ribosiklibin ja anastrotsolin välillä olisi kliinisesti merkittäviä lääkeaineinteraktioita.
Ribosiklibin ja fulvestrantin väliset lääkeaineinteraktiot
Rintasyöpäpotilailla tehdyn kliinisen tutkimuksen tiedot eivät viittaa siihen, että fulvestrantti vaikuttaisi kliinisesti merkittävästi ribosiklibialtistukseen, kun näitä lääkevalmisteita annetaan samanaikaisesti.
Ribosiklibin ja tamoksifeenin väliset lääkeaineinteraktiot
Rintasyöpäpotilailla tehdyn kliinisen tutkimuksen tiedot viittaavat siihen, että tamoksifeenialtistus suurenee noin 2‑kertaiseksi, kun ribosiklibia ja tamoksifeenia annetaan samanaikaisesti.
Ribosiklibin ja ehkäisytablettien väliset lääkeaineinteraktiot
Ribosiklibin ja ehkäisytablettien välisiä yhteisvaikutuksia koskevia lääkeaineinteraktiotutkimuksia ei ole tehty (ks. kohta Raskaus ja imetys).
Odotettavissa olevat yhteisvaikutukset
Rytmihäiriölääkevalmisteet ja muut mahdollisesti QT-aikaa pidentävät lääkevalmisteet
On aiheellista välttää Kisqalin antoa samanaikaisesti sellaisten lääkevalmisteiden kanssa, joiden tiedetään voivan pidentää QT-aikaa, kuten rytmihäiriölääkevalmisteiden kanssa (mm. amiodaroni, disopyramidi, prokaiiniamidi, kinidiini ja sotaloli) ja muiden tunnetusti QT-aikaa pidentävien lääkevalmisteiden kanssa (mm. klorokiini, halofantriini, klaritromysiini, siprofloksasiini, levofloksasiini, atsitromysiini, haloperidoli, metadoni, moksifloksasiini, bepridiili, pimotsidi ja laskimoon annettu ondansetroni) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kisqalin käyttö yhdessä tamoksifeenin kanssa ei myöskään ole suositeltavaa (ks. kohdat Käyttöaiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy
Ennen Kisqali-hoidon aloittamista on tarkistettava, onko potilas raskaana.
Kisqali-valmistetta saavien naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä (esim. kahta ehkäisymenetelmää) Kisqali-hoidon aikana ja vähintään 21 päivän ajan sen lopettamisen jälkeen.
Raskaus
Raskaana oleville naisille ei ole tehty asianmukaisia ja hyvin kontrolloituja tutkimuksia. Eläintutkimuslöydösten perusteella ribosiklibin anto raskaana olevalle naiselle voi aiheuttaa haittaa sikiölle (ks. kohta Prekliiniset tiedot turvallisuudesta). Kisqali-valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä ehkäisyä.
Imetys
Ei tiedetä, esiintyykö ribosiklibia ihmisillä äidinmaidossa. Ribosiklibin vaikutuksista imetettävään vauvaan tai maidoneritykseen ei ole tietoa. Ribosiklibi ja sen metaboliitit erittyvät tehokkaasti imettävien rottien maitoon. Kisqali‑valmistetta saavien potilaiden ei tule imettää ennen kuin viimeisestä annoksesta on kulunut vähintään 21 päivää.
Hedelmällisyys
Ribosiklibin vaikutuksesta hedelmällisyyteen ei ole kliinistä tietoa. Eläintutkimusten perusteella ribosiklibi saattaa heikentää lisääntymiskykyisten urosten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kisqali-valmisteella voi olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaita on kehotettava olemaan varovaisia ajaessaan autoa tai käyttäessään koneita, jos heillä on uupumusta, heitehuimausta tai kiertohuimausta Kisqali-hoidon aikana (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Varhaisvaiheen rintasyöpä
Yleisimpiä haittavaikutuksia (ilmoitettu esiintymistiheys ≥ 20 %), joiden esiintymistiheys oli Kisqali-hoidon ja aromataasinestäjän yhdistelmää saaneilla suurempi kuin pelkkää aromataasinestäjää saaneilla, olivat neutropenia, infektiot, pahoinvointi, päänsärky, uupumus, leukopenia ja maksan toimintakokeiden poikkeavuudet.
Yleisimpiä asteen 3/4 haittavaikutuksia (ilmoitettu esiintymistiheys ≥ 2 %), joiden esiintymistiheys oli Kisqali-hoidon ja aromataasinestäjän yhdistelmää saaneilla suurempi kuin pelkkää aromataasinestäjää saaneilla, olivat neutropenia, maksan toimintakokeiden poikkeavuudet ja leukopenia.
Annosta pienennettiin haittatapahtumien vuoksi (syysuhteesta riippumatta) 22,8 %:lla Kisqali-valmisteen ja aromataasinestäjän yhdistelmää vaiheen III kliinisessä tutkimuksessa saaneista potilaista. Hoito lopetettiin pysyvästi 19,7 %:lla potilaista, jotka saivat Kisqali-valmisteen ja aromataasinestäjän yhdistelmää vaiheen III kliinisessä tutkimuksessa.
Edennyt tai etäpesäkkeinen rintasyöpä
Yleisimpiä haittavaikutuksia (ilmoitettu esiintymistiheys ≥ 20 %), joiden esiintymistiheys oli yhdistettyjen tietojen mukaan Kisqali-hoitoa saaneilla (kaikki lääkeyhdistelmät) suurempi kuin lumelääkettä saaneilla (kaikki lääkeyhdistelmät), olivat neutropenia, infektiot, pahoinvointi, uupumus, ripuli, leukopenia, oksentelu, päänsärky, ummetus, hiustenlähtö, yskä, ihottuma, selkäkipu, anemia ja maksan toimintakokeiden poikkeavuudet.
Yleisimpiä asteen 3/4 haittavaikutuksia (ilmoitettu esiintymistiheys ≥ 2 %), joiden esiintymistiheys oli yhdistettyjen tietojen mukaan Kisqali-hoitoa saaneilla (kaikki lääkeyhdistelmät) suurempi kuin lumelääkettä saaneilla (kaikki lääkeyhdistelmät), olivat neutropenia, leukopenia, maksan toimintakokeiden poikkeavuudet, lymfopenia, infektiot, selkäkipu, anemia, uupumus, hypofosfatemia ja oksentelu.
Annosta pienennettiin haittatapahtumien vuoksi (syysuhteesta riippumatta) 39,5 %:lla Kisqali-valmistetta vaiheen III kliinisissä tutkimuksissa saaneista potilaista käytetystä lääkeyhdistelmästä riippumatta. Hoito lopetettiin pysyvästi 8,7 %:lla potilaista, jotka saivat Kisqali-valmistetta minkä tahansa lääkeyhdistelmän osana vaiheen III kliinisissä tutkimuksissa.
Haittavaikutustaulukko
Varhaisvaiheen rintasyöpä
Kisqalin yleinen turvallisuusarviointi perustuu tietoihin 2 525 potilaasta, jotka saivat Kisqali-valmistetta yhdessä aromataasinestäjän kanssa. Kyseiset potilaat osallistuivat satunnaistettuun, avoimeen vaiheen III kliiniseen NATALEE-tutkimukseen.
Tutkimushoidolle altistumisen mediaanikesto oli 33,0 kk, ja 64,9 % potilaista altistui valmisteelle ≥ 24 kk ajan. 42,8 % potilaista sai ribosiklibia 36 kk:n pituisen hoito-ohjelman loppuun asti.
Edennyt tai etäpesäkkeinen rintasyöpä
Kisqalin yleinen turvallisuusarviointi perustuu yhdistettyihin tietoihin 1 065 potilaasta, jotka saivat Kisqali-valmistetta yhdessä endokriinisen hoidon kanssa (N = 582 yhdessä aromataasinestäjän kanssa ja N = 483 yhdessä fulvestrantin kanssa). Kyseiset potilaat osallistuivat satunnaistettuihin, kaksoissokkoutettuihin, lumekontrolloituihin vaiheen III kliinisiin tutkimuksiin MONALEESA-2, MONALEESA-7-tutkimuksen ei-steroidista aromataasinestäjää saanut alaryhmä ja MONALEESA-3.
Tutkimushoidolle altistumisen mediaanikesto oli vaiheen III tutkimusten yhdistetyissä tiedoissa 19,2 kk, ja 61,7 % potilaista altistui valmisteelle ≥ 12 kk ajan.
Vaiheen III kliinisissä tutkimuksissa ja myyntiluvan myöntämisen jälkeen varhaisvaiheen rintasyövän hoidossa ja edenneen tai etäpesäkkeisen rintasyövän hoidossa ilmoitetut haittavaikutukset (taulukko 7) esitetään MedDRA-elinjärjestelmäluokittain. Kunkin elinjärjestelmäluokan haittavaikutukset on esitetty yleisyysjärjestyksessä yleisimmästä alkaen. Kunkin yleisyysluokan haittavaikutukset on esitetty vakavuusjärjestyksessä vakavimmasta alkaen. Lisäksi kunkin haittavaikutuksen yleisyysluokka perustuu seuraavaan luokitukseen (CIOMS III): hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 7 Vaiheen III kliinisissä tutkimuksissa ja markkinoille tulon jälkeen raportoidut haittavaikutukset
| Esiintymistiheys | Varhaisvaiheen rintasyöpää sairastavat potilaat, joiden aloitusannos oli 400 mg ribosiklibia | Edennyttä tai etäpesäkkeistä rintasyöpää sairastavat potilaat, joiden aloitusannos oli 600 mg ribosiklibia |
| Infektiot | ||
| Hyvin yleinen | Infektiot1 | Infektiot1 |
| Veri ja imukudos | ||
| Hyvin yleinen | Neutropenia, leukopenia | Neutropenia, leukopenia, anemia, lymfopenia |
| Yleinen | Anemia, trombosytopenia, lymfopenia | Trombosytopenia, kuumeinen neutropenia |
| Melko harvinainen | Kuumeinen neutropenia | – |
| Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleinen | – | Ruokahalun heikkeneminen |
| Yleinen | Hypokalsemia, hypokalemia, ruokahalun heikkeneminen | Hypokalsemia, hypokalemia, hypofosfatemia |
| Hermosto | ||
| Hyvin yleinen | Päänsärky | Päänsärky, heitehuimaus |
| Yleinen | Heitehuimaus | Kiertohuimaus |
| Silmät | ||
| Yleinen | – | Lisääntynyt kyynelnesteen eritys, kuivasilmäisyys |
| Sydän | ||
| Yleinen | – | Pyörtyminen |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleinen | Yskä | Hengenahdistus, yskä |
| Yleinen | Hengenahdistus, interstitiaalinen keuhkosairaus (ILD) /keuhkotulehdus | Interstitiaalinen keuhkosairaus (ILD) /keuhkotulehdus |
| Ruoansulatuselimistö | ||
| Hyvin yleinen | Pahoinvointi, ripuli, ummetus, vatsakipu2 | Pahoinvointi, ripuli, oksentelu, ummetus, vatsakipu2, suutulehdus, dyspepsia |
| Yleinen | Oksentelu, suutulehdus3 | Makuaistin häiriöt |
| Maksa ja sappi | ||
| Yleinen | Maksatoksisuus4 | Maksatoksisuus4 |
| Iho ja ihonalainen kudos | ||
| Hyvin yleinen | Hiustenlähtö | Hiustenlähtö, ihottuma5, kutina |
| Yleinen | Ihottuma5, kutina | Ihon kuivuus, punoitus, valkopälvi |
| Harvinainen | – | Erythema multiforme |
| Tuntematon | – | Toksinen epidermaalinen nekrolyysi (TEN) |
| Luusto, lihakset ja sidekudos | ||
| Hyvin yleinen | – | Selkäkipu |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleinen | Uupumus, voimattomuus, kuume | Uupumus, ääreisosien turvotus, kuume, voimattomuus |
| Yleinen | Ääreisosien turvotus, suunielun kipu | Suunielun kipu, suun kuivuus |
| Tutkimukset | ||
| Hyvin yleinen | Poikkeavat maksan toimintakoearvot6 | Poikkeavat maksan toimintakoearvot6 |
| Yleinen | Veren kreatiniinipitoisuuden suureneminen, QT-ajan piteneminen EKG-tutkimuksessa | Veren kreatiniinipitoisuuden suureneminen, QT-ajan piteneminen EKG-tutkimuksessa |
1 Infektiot: virtsatieinfektiot, hengitystieinfektiot, gastroenteriitti (vain edenneen tai etäpesäkkeisen rintasyövän hoidossa), sepsis (< 1 %, vain edenneen tai etäpesäkkeisen rintasyövän hoidossa). 2 Vatsakipu: vatsakipu, ylävatsakipu. 3 Suutulehdus varhaisvaiheen rintasyövässä sisältää seuraavat: suutulehdus, mukosiitti. 4 Maksatoksisuus: maksan sytolyysi, maksasoluvaurio (vain edenneen tai etäpesäkkeisen rintasyövän hoidossa), lääkkeen aiheuttama maksavaurio (< 1 %, varhaisvaiheen rintasyövän hoidossa ja edenneen tai etäpesäkkeisen rintasyövän hoidossa), maksatoksisuus, maksan vajaatoiminta (vain edenneen tai etäpesäkkeisen rintasyövän hoidossa), autoimmuunihepatiitti. 5 Ihottuma: ihottuma, makulopapulaarinen ihottuma, kutiava ihottuma. 6 Poikkeavat maksan toimintakoearvot: ALAT-arvon nousu, ASAT-arvon nousu, veren bilirubiinipitoisuuden nousu. | ||
Tiettyjen haittavaikutusten kuvaus
Neutropenia
Vaiheen III tutkimuksessa varhaisvaiheen rintasyöpää sairastavilla potilailla neutropenia oli yleisin ilmoitettu haittavaikutus (62,5 %), ja asteen 3 tai 4 neutrofiiliarvojen laskua (laboratoriokokeiden perusteella) ilmoitettiin 45,1 %:lla potilaista, jotka saivat Kisqali-valmisteen ja aromataasinestäjän yhdistelmää.
Varhaisvaiheen rintasyöpää sairastavilla potilailla mediaaniaika asteen 2, 3 tai 4 neutropeniatapahtumien alkamiseen oli 0,6 kk. Kun hoito tauotettiin ja/tai annosta pienennettiin ja/tai hoito lopetettiin, mediaaniaika tilan korjautumiseen asteen ≥ 3 tasolta (normaaliarvoihin tai asteen < 3 tasolle) oli Kisqali-valmisteen ja aromataasinestäjän yhdistelmää saaneilla 0,3 kk. Kuumeista neutropeniaa ilmoitettiin noin 0,3 %:lla potilaista, jotka altistuivat Kisqali-valmisteen ja aromataasinestäjän yhdistelmälle. Hoidon lopettaminen neutropenian vuoksi oli Kisqali-valmisteen ja aromataasinestäjän yhdistelmää saaneilla potilailla vähäistä (1,1 %) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Vaiheen III tutkimuksissa edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla neutropenia oli yleisin ilmoitettu haittavaikutus (75,4 %), ja asteen 3 tai 4 neutrofiiliarvojen laskua (laboratoriokokeiden perusteella) ilmoitettiin 62,0 %:lla potilaista, jotka saivat Kisqali-valmistetta minkä tahansa lääkeyhdistelmän osana.
Edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla mediaaniaika asteen 2, 3 tai 4 neutropeniatapahtumien alkamiseen oli 17 vrk. Kun hoito tauotettiin ja/tai annosta pienennettiin ja/tai hoito lopetettiin, mediaaniaika tilan korjautumiseen asteen ≥ 3 tasolta (normaaliarvoihin tai asteen < 3 tasolle) oli Kisqali-valmistetta saaneilla 12 vrk (kaikki lääkeyhdistelmät). Kuumeista neutropeniaa ilmoitettiin noin 1,7 %:lla potilaista, jotka altistuivat Kisqali-valmisteelle vaiheen III tutkimuksissa. Hoidon lopettaminen neutropenian vuoksi oli vähäistä (0,8 %) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kaikille potilaille on kerrottava, että mahdollisesta kuumeesta on ilmoitettava ripeästi.
Maksa- ja sappitoksisuus
Vaiheen III tutkimuksissa varhaisvaiheen rintasyöpää sairastavilla potilailla ja edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla todettiin transaminaasiarvojen nousua.
Vaiheen III kliinisessä tutkimuksessa varhaisvaiheen rintasyöpää sairastavilla potilailla maksa- ja sappitoksisuustapahtumia esiintyi yleisemmin Kisqali-hoidon ja aromataasinestäjän yhdistelmää saaneilla (26,4 %) kuin pelkkää aromataasinestäjää saaneilla (11,2 %), ja Kisqali-hoidon ja aromataasinestäjän yhdistelmää saaneilla ilmoitettiin enemmän asteen 3/4 haittatapahtumia (8,6 % vs. 1,7 %). ALAT- tai ASAT-arvon nousua yli 3 kertaa viitealueen ylärajan suuruiseksi ja samanaikaista kokonaisbilirubiiniarvon nousua yli 2 kertaa viitealueen ylärajan suuruiseksi tilanteessa, jossa AFOS-arvo oli normaali, todettiin 8 potilaalla, jotka saivat Kisqali-hoidon ja aromataasinestäjän yhdistelmää (6 potilaalla ALAT- tai ASAT-arvo korjautui normaaliksi 65–303 päivän kuluessa Kisqali-hoidon lopettamisesta).
Maksa- ja sappitoksisuustapahtumien ilmoitettiin johtaneen hoidon tauottamiseen 12,4 %:lla Kisqali-valmisteen ja aromataasinestäjän yhdistelmää saaneista, varhaisvaiheen rintasyöpää sairastaneista potilaista. Syynä oli lähinnä ALAT-arvon nousu (10,1 %) ja/tai ASAT-arvon nousu (6,8 %). Maksa- ja sappitoksisuustapahtumien ilmoitettiin johtaneen annosmuutokseen 2,6 %:lla Kisqali-valmisteen ja aromataasinestäjän yhdistelmää saaneista potilaista. Syynä oli lähinnä ALAT-arvon nousu (1,9 %) ja/tai ASAT-arvon nousu (0,6 %). 8,9 % potilaista lopetti Kisqali-hoidon maksan toimintakoearvojen poikkeavuuksien vuoksi ja 0,1 % maksatoksisuuden vuoksi (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Vaiheen III kliinisessä tutkimuksessa varhaisvaiheen rintasyöpää sairastavilla potilailla 80,9 % (165/204) asteen 3 tai 4 ALAT- tai ASAT-nousutapahtumista tapahtui ensimmäisten 6 hoitokuukauden aikana. Mediaaniaika asteen 3 tai 4 ALAT/ASAT-arvojen nousun alkamiseen oli Kisqali-valmisteen ja aromataasinestäjän yhdistelmää saaneilla 2,8 kk. Mediaaniaika tilan korjautumiseen (normaaliksi tai asteen ≤ 2 tasolle) oli Kisqali-valmisteen ja aromataasinestäjän yhdistelmää saaneilla 0,7 kk.
Vaiheen III kliinisissä tutkimuksissa edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla maksa- ja sappitoksisuustapahtumia esiintyi yleisemmin Kisqali-hoitoa saaneilla lääkeyhdistelmästä riippumatta (27,3 %) kuin lumelääkettä saaneilla lääkeyhdistelmästä riippumatta (19,6 %), ja Kisqali-hoitoa saaneilla (kaikki lääkeyhdistelmät) ilmoitettiin enemmän asteen 3/4 haittatapahtumia (13,2 % vs. 6,1 %). Asteen 3 tai 4 ALAT-arvojen nousua todettiin Kisqali-ryhmässä 11,2 %:lla ja lumelääkeryhmässä 1,7 %:lla ja vastaavaa ASAT-arvojen nousua taas 7,8 %:lla ja 2,1 %:lla. ALAT- tai ASAT-arvon nousua yli 3 kertaa viitealueen ylärajan suuruiseksi ja samanaikaista kokonaisbilirubiiniarvon nousua yli 2 kertaa viitealueen ylärajan suuruiseksi tilanteessa, jossa AFOS-arvo oli normaali eikä potilaalla ollut kolestaasia, todettiin 6 potilaalla (4 potilaalla A2301-tutkimuksessa [MONALEESA-2]), joiden arvot korjautuivat normaaleiksi 154 päivän kuluessa Kisqali-hoidon lopettamisesta, ja F2301-tutkimuksessa [MONALEESA-3] 2 potilaalla, joiden arvot korjautuivat normaaleiksi 121 ja 532 päivän kuluessa Kisqali-hoidon lopettamisesta). E2301-tutkimuksessa (MONALEESA-7) ei ilmoitettu yhtään tällaista tapausta.
Maksa- ja sappitoksisuustapahtumien ilmoitettiin johtaneen hoidon tauottamiseen ja/tai annosmuutokseen 12,3 %:lla Kisqali-valmistetta minkä tahansa lääkeyhdistelmän osana saaneista, edennyttä tai etäpesäkkeistä rintasyöpää sairastaneista potilaista. Syynä oli lähinnä ALAT-arvon nousu (7,9 %) ja/tai ASAT-arvon nousu (7,3 %). 2,4 % potilaista lopetti Kisqali-hoidon (kaikki lääkeyhdistelmät) maksan toimintakoearvojen poikkeavuuksien vuoksi ja 0,3 % maksatoksisuuden vuoksi (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Vaiheen III kliinisissä tutkimuksissa edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla 70,9 % (90/127) asteen 3 tai 4 ALAT- tai ASAT-nousutapahtumista tapahtui ensimmäisten 6 hoitokuukauden aikana. Mediaaniaika asteen 3 tai 4 ALAT/ASAT-arvojen nousun alkamiseen oli Kisqali-hoitoa saaneilla (kaikki lääkeyhdistelmät) 92 vrk. Mediaaniaika tilan korjautumiseen (normaaliksi tai asteen ≤ 2 tasolle) oli Kisqali-hoitoa saaneilla (kaikki lääkeyhdistelmät) 21 vrk.
QT-ajan piteneminen
Vaiheen III kliinisessä tutkimuksessa varhaisvaiheen rintasyöpää sairastavilla potilailla 5,3 %:lla Kisqalia ja aromataasinestäjää saaneista potilaista ja 1,4 %:lla pelkkää aromataasinestäjää saaneista potilaista ilmoitettiin QT-ajan pitenemistapahtumia. Kisqalia ja aromataasinestäjää saaneiden potilaiden QT-ajan pitenemistapahtumat olivat pääasiassa QT-ajan pitenemisiä EKG-tutkimuksessa (4,3 %). QT-ajan piteneminen EKG-tutkimuksessa oli ainoa Kisqali-hoidon vahvistettu haittavaikutus. QT-ajan pitenemisen EKG-tutkimuksessa ja pyörtymisen ilmoitettiin johtaneen hoidon tauottamiseen 1,1 %:lla Kisqalia saaneista potilaista. QT-ajan pitenemisen EKG-tutkimuksessa ilmoitettiin johtaneen annosmuutokseen 0,1 %:lla Kisqalia saaneista potilaista.
EKG-löydösten keskitetyssä analyysissä todettiin, että 10 potilaalla (0,4 %) Kisqalia ja aromataasinestäjää saaneista ja 4 potilaalla (0,2 %) pelkkää aromataasinestäjää saaneista esiintyi QTcF-ajan pitenemistä > 480 ms:n pituiseksi vähintään kerran lähtötilanteen jälkeen. Kisqalia ja aromataasinestäjää saaneilla potilailla, joiden QTcF-aika piteni > 480 ms:n pituiseksi, mediaaniaika tilanteen alkamiseen oli 15 vrk. Muutokset korjautuivat, kun hoito tauotettiin ja/tai annosta pienennettiin. Kisqalia ja aromataasinestäjää saaneista 19 potilaalla (0,8 %) todettiin QTcF-ajan muutos > 60 ms lähtötilanteesta ja 3 potilaalla (0,1 %) lähtötilanteen jälkeinen QTcF-aika > 500 ms.
E2301-tutkimuksessa (MONALEESA‑7) edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla QTcF-ajan havaittu keskipitenemä lähtötilanteesta oli noin 10 ms pidempi tamoksifeeni- ja lumelääkeryhmässä kuin ei-steroidista aromataasinestäjää ja lumelääkettä saaneilla. Tämä viittaa siihen, että pelkällä tamoksifeenilla oli QTcF-aikaa pidentävä vaikutus. Tämä voi osaltaan selittää Kisqali- ja tamoksifeeniryhmässä todettuja QTcF-arvoja. Lumelääkeryhmässä QTcF-aika pitenemistä > 60 ms lähtötilanteesta todettiin tamoksifeenia saaneessa ryhmässä 6/90 potilaalla (6,7 %), mutta ei yhdelläkään ei-steroidista aromataasinestäjää saaneella potilaalla (ks. kohta Farmakokinetiikka). QTcF-välin pitenemistä > 60 ms lähtötilanteesta todettiin Kisqali- ja tamoksifeenihoitoa saaneessa ryhmässä 14/87 potilaalla (16,1 %) ja Kisqali‑valmistetta ja ei-steroidista aromataasinestäjää saaneessa ryhmässä 18/245 potilaalla (7,3 %). Kisqali-valmisteen ja tamoksifeenin yhteiskäyttö ei ole suositeltavaa (ks. kohta Farmakodynamiikka).
Vaiheen III kliinisissä tutkimuksissa edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla 9,3 %:lla Kisqalia ja aromataasinestäjää tai fulvestranttia saaneiden ryhmien potilaista ja 3,5 %:lla lumelääkettä ja aromataasinestäjää tai fulvestranttia saaneista potilaista oli vähintään yksi QT-ajan pitenemistapahtuma (joita olivat QT-ajan piteneminen EKG-tutkimuksessa sekä pyörtyminen). EKG-löydösten arvioinnissa todettiin, että 15 potilaalla (1,4 %) QTcF-aika piteni lähtötilanteen jälkeen > 500 ms:n pituiseksi ja 61 potilaan (5,8 %) QTcF-ajat pitenivät lähtötilanteesta> 60 ms. Yhtäkään kääntyvien kärkien takykardiatapausta ei ilmoitettu. QT-ajan pitenemisen EKG-tutkimuksessa tai pyörtymisen ilmoitettiin johtaneen hoidon tauottamiseen / annosmuutokseen 2,9 %:lla Kisqalia ja aromataasinestäjää tai fulvestranttia saaneista potilaista.
EKG-löydösten analyysissä todettiin, että 55 potilaalla (5,2 %) Kisqalia ja aromataasinestäjää tai fulvestranttia saaneista ja 12 potilaalla (1,5 %) lumelääkettä ja aromataasinestäjää tai fulvestranttia saaneista esiintyi QTcF-ajan pitenemistä > 480 ms:n pituiseksi vähintään kerran lähtötilanteen jälkeen. Potilailla, joiden QTcF-aika piteni > 480 ms:n pituiseksi, mediaaniaika tilanteen alkamiseen oli 15 vrk käytetystä lääkeyhdistelmästä riippumatta. Muutokset korjautuivat, kun hoito tauotettiin ja/tai annosta pienennettiin (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Munuaisten vajaatoimintapotilaat
Ribosiklibihoitoa annettiin varhaisvaiheen rintasyöpää koskeneessa vaiheen III tutkimuksessa 983 potilaalle, joilla oli lievä munuaisten vajaatoiminta, ja 71 potilaalle, joilla oli keskivaikea munuaisten vajaatoiminta. Kyseiseen tutkimukseen ei otettu yhtään vaikeaa munuaisten vajaatoimintaa sairastavaa potilasta (ks. kohta Farmakodynamiikka).
Ribosiklibihoitoa annettiin kolmessa edennyttä tai etäpesäkkeistä rintasyöpää koskeneessa avaintutkimuksessa 341 potilaalle, joilla oli lievä munuaisten vajaatoiminta, ja 97 potilaalle, joilla oli keskivaikea munuaisten vajaatoiminta. Kyseisiin tutkimuksiin ei otettu yhtään vaikeaa munuaisten vajaatoimintaa sairastavaa potilasta (ks. kohta Farmakodynamiikka). Munuaisten vajaatoiminnan lähtötilanteen vaikeusasteen ja hoidonaikaisten veren kreatiniinipitoisuuksien välillä havaittiin korrelaatio. Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla havaittiin QT-ajan pitenemisen ja trombosytopenian esiintymistiheyksien vähäistä lisääntymistä. Suositukset näihin haittoihin liittyvästä seurannasta ja annoksen muuttamisesta, ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Raportoiduista Kisqali-yliannostustapauksista on vain rajallisesti kokemusta. Yliannostuksen yhteydessä voi esiintyä oireita kuten pahoinvointia ja oksentelua. Lisäksi voi esiintyä hematologista toksisuutta (esim. neutropenia, trombosytopenia) ja mahdollista QTc-ajan pitenemistä. Kaikissa yliannostustapauksissa on ryhdyttävä yleisluontoisiin tukihoitoihin tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, proteiinikinaasin estäjät, ATC-koodi: L01EF02
Vaikutusmekanismi
Ribosiklibi on selektiivinen sykliinistä riippuvaisten kinaasien (CDK) 4 ja 6 estäjä, jonka CDK4:n 50 %:n estopitoisuus (IC50) on biokemiallisissa testeissä 0,01 μM (4,3 ng/ml) ja CKD6:n IC50-arvo 0,039 μM (16,9 ng/ml). Nämä kinaasit aktivoituvat D-sykliinien sitoutumisen yhteydessä, ja niillä on keskeisen tärkeä asema solusyklin etenemiseen ja solujen proliferaatioon johtavissa signalointireiteissä. Sykliini D-CDK4/6-kompleksi säätelee solusyklin etenemistä retinoblastoomaproteiinin (pRb) fosforylaation kautta.
In vitro ‑tutkimuksissa rintasyövästä johdetuilla malleilla ribosiklibi vähensi pRb:n fosforylaatiota, mikä johti solusyklin pysähtymiseen G1-vaiheessa, solujen proliferaation vähenemiseen ja senesenttiin fenotyyppiin. In vivo yksilääkehoito ribosiklibilla johti kasvaimen regressioon, joka korreloi pRb:n fosforylaation eston kanssa.
Potilasperäisen estrogeenireseptoripositiivisen rintasyövän ksenograftimallissa toteutetuissa in vivo ‑tutkimuksissa ribosiklibin ja antiestrogeenien (ts. letrotsolin) yhdistelmät estivät tehokkaammin kasvaimen kasvua ja johtivat kasvaimen pitkäkestoiseen regressioon sekä pidensivät kasvaimen uudelleenkasvuun kuluvaa aikaa lääkityksen päättymisen jälkeen, kun niitä verrattiin kummankin lääkkeen käyttöön erikseen. Potilaille annetulla ribosiklibilla voi olla myös immunomodulaatiovaikutus, jolloin se pienentää säätelijä-T‑solujen määrää ja CD3+‑T‑solujen suhteellista määrää. Ribosiklibin ja fulvestrantin yhdistelmän tehoa kasvaimiin arvioitiin myös in vivo immuunipuutteisilla hiirillä, joilla oli ihmisen estrogeenireseptoripositiivisen rintasyövän ZR751‑ksenografteja. Tämä fulvestranttia sisältänyt yhdistelmähoito esti kasvainten kasvun täysin.
Kun ribosiklibia testattiin useissa rintasyövän solulinjoissa, joiden ER-status tiedettiin, se osoittautui tehokkaammaksi ER-positiivisten kuin ER-negatiivisten rintasyöpäsolulinjojen hoidossa. Tähän mennessä prekliinisillä malleilla testattaessa, ribosiklibin aktiviteettiin vaadittiin intakti pRb.
Sydämen elektrofysiologia
Ribosiklibin vaikutusta QTc-aikaan arvioitiin ottamalla pitkälle edennyttä syöpää sairastavilta tutkittavilta kolme peräkkäistä EKG-käyrää kerta-annoksen jälkeen ja vakaassa tilassa. Farmakokinetiikan-farmakodynamiikan analyysiin otettiin yhteensä 997 potilasta, jotka saivat ribosiklibihoitoa 50–1 200 mg:n annoksilla. Analyysin tulokset viittasivat siihen, että ribosiklibi pidentää QTc-aikaa pitoisuuksista riippuvaisesti.
Edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla QTcF-ajan arvioitu keskimuutos lähtötilanteesta oli 600 mg:n Kisqali-annoksia käytettäessä ja vakaan tilan geometrisen Cmax-keskipitoisuuden yhteydessä 22,0 ms (90 % luottamusväli (lv) 20,56; 23,44), kun samanaikaisesti annettiin ei-steroidista aromataasinestäjää, ja 23,7 ms (90 % lv 22,31; 25,08), kun samanaikaisesti annettiin fulvestranttia; vastaavien Kisqali-annosten anto yhdessä tamoksifeenin kanssa johti 34,7 ms:n QTcF-ajan arvioituun keskimuutokseen lähtötilanteesta (90 % lv 31,64; 37,78) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varhaisvaiheen rintasyöpää sairastavilla potilailla QTc-aika pitenee samalla pitoisuudesta riippuvaisella tavalla. Arvioidun QTcF-ajan keskimääräisen muutoksen lähtötilanteesta arvioidaan olevan Kisqali 400 mg ‑hoitoa saavilla, varhaisvaiheen rintasyöpää sairastavilla potilailla pienempi kuin Kisqali 600 mg ‑hoitoa saavilla, edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla.
Kliininen teho ja turvallisuus
Varhaisvaiheen rintasyöpä
CLEE011O12301C-tutkimus (NATALEE)
Kisqali-valmisteen ja aromataasinestäjän (letrotsoli tai anastrotsoli) yhdistelmää verrattiin pelkkään aromataasinestäjään satunnaistetussa, avoimessa, vaiheen III kliinisessä monikeskustutkimuksessa miehillä ja pre- ja postmenopausaalisilla naisilla, joilla oli suuren uusiutumisriskin hormonireseptoripositiivinen, HER2-negatiivinen varhaisvaiheen rintasyöpä. Syövän anatomisen levinneisyysasteen oli oltava II tai III riippumatta imusolmukestatuksesta. Syövän vaaditut ominaisuudet olivat:
- anatominen levinneisyysaste IIB–III tai
-
anatominen levinneisyysaste IIA ja joko
- imusolmukestatus positiivinen tai
- imusolmukestatus negatiivinen ja
- histologinen gradus 3 tai
- histologinen gradus 2, jos jompikumpi seuraavista kriteereistä täyttyi:
- Ki67 ≥ 20 %
- suuri riski geeniprofilointitestauksen mukaan.
Miehet ja premenopausaaliset naiset saivat myös gosereliinia. NATALEE-tutkimukseen otettiin potilaita, joilla oli TNM-kriteerien mukaan arvioituna imusolmukkeisiin levinnyt tauti; tai ei leviämistä imusolmukkeisiin ja joko kasvaimen koko > 5 cm tai kasvaimen koko 2–5 cm ja histologisesti joko gradus 2 (ja genomin perusteella suuri riski tai Ki67 ≥ 20 %) tai gradus 3.
Yhteensä 5 101 potilasta (joista 20 miehiä) satunnaistettiin suhteessa 1:1 saamaan joko 400 mg:n Kisqali-annosten ja aromataasinestäjän yhdistelmää (n = 2 549) tai pelkkää aromataasinestäjää (n = 2 552). Hoitoon satunnaistaminen stratifioitiin seuraavien tekijöiden mukaan: anatominen levinneisyysaste (ryhmä II [n = 2 154 (42,2 %)] vs. ryhmä III [n = 2 947 (57,8 %)]), aiempi hoito (neoadjuvantti-/adjuvanttisolunsalpaajahoito: kyllä [n = 4 432 (86,9 %)] vs. ei [n = 669 (13,1 %)]), vaihdevuosistatus (miehet ja premenopausaaliset naiset [n = 2 253 (44,2 %)] vs. postmenopausaaliset naiset [n = 2 848 (55,8 %)]), maantieteellinen alue (Pohjois-Amerikka/Länsi-Eurooppa/Oseania [n = 3 128 (61,3 %)] vs. muu maailma [n = 1 973 (38,7 %)]). Potilaille annettiin suun kautta 400 mg Kisqalia kerran vuorokaudessa 21 perättäisenä päivänä, minkä jälkeen pidettiin 7 päivän tauko, sekä 2,5 mg letrotsolia tai 1 mg anastrotsolia suun kautta kerran vuorokaudessa 28 päivän ajan. Gosereliinia annettiin 3,6 mg:n annos ihon alle injektoitavana implantaattina kunkin 28‑päiväisen hoitojakson päivänä 1. Kisqali-hoitoa jatkettiin 3 vuoden ajan satunnaistamispäivästä (noin 39 hoitojakson ajan).
Tutkimukseen otettujen potilaiden mediaani-ikä oli 52 v (vaihteluväli 24–90). Potilaista 15,2 % oli 65-vuotiaita tai vanhempia, ja 123 potilasta (2,4 %) oli 75-vuotiaita ja vanhempia. Mukana oli valkoihoisia (73,4 %), aasialaisia (13,2 %) ja mustia tai afrikkalaistaustaisia amerikkalaisia (1,7 %) potilaita. Kaikkien potilaiden ECOG-toimintakykyluokka oli 0 tai 1. Kaiken kaikkiaan 88,2 % potilaista oli saanut solunsalpaajahoitoa neoadjuvantti- tai adjuvanttihoitona ja 71,6 % oli saanut endokriinista hoitoa neoadjuvantti- tai adjuvanttihoitona tutkimukseenottoa edeltäneiden 12 kk:n aikana.
Tutkimuksen ensisijainen päätetapahtuma oli invasiivisen syövän suhteen tautivapaa elossaolo (invasive disease-free survival, iDFS). Sen määritelmänä oli aika, joka kului satunnaistamisesta jonkin seuraavista ensimmäiseen esiintymiseen: invasiivisen rintasyövän paikallinen uusiutuminen, invasiivisen rintasyövän alueellinen uusiutuminen, etäpesäkkeinen uusiutuminen, mistä tahansa syystä johtuva kuolema, kontralateraalinen invasiivinen rintasyöpä tai toinen primaarinen invasiivinen syöpä, joka oli muu kuin rintasyöpä (lukuun ottamatta ihon tyvisolusyöpää ja levyepiteelikarsinoomaa).
Tutkimuksen ensisijainen päätetapahtuma saavutettiin primaarianalyysissa (tiedonkeruun katkaisupäivä 11.1.2023). Kisqali-valmisteen ja aromataasinestäjän yhdistelmää saaneilla potilailla todettiin iDFS-ajassa tilastollisesti merkitsevä kohenema (riskitiheyssuhde 0,748; 95 % lv 0,618; 0,906; yksitahoisen stratifioidun log-rank-testin p-arvo 0,0014) verrattuna pelkkää aromataasinestäjää saaneisiin. Anatomisen levinneisyysasteen, vaihdevuosistatuksen, maantieteellisen alueen, imusolmukestatuksen, iän, etnisen taustan ja aiempien adjuvantti-/neoadjuvanttikemoterapia- tai hormonihoitojen alaryhmissä havaittiin kaikissa johdonmukaisia tuloksia.
Myöhemmän analyysin (tiedonkeruun katkaisupäivä 21.7.2023) tiedoista esitetään yhteenveto taulukossa 8; iDFS-ajan Kaplan–Meier-käyrä esitetään kuvassa 1. Tutkimusryhmien yhdistetty hoidon keston mediaani lopullisen iDFS-analyysin ajankohtana oli noin 30 kk ja iDFS-seurannan mediaanikesto oli 33,3 kk. Kokonaiselossaolon (OS) tiedot eivät ole valmiit. Yhteensä 172 potilasta (3,5 %) oli kuollut (ribosiklibia saaneista 83/2 525 ja pelkkää aromataasinestäjää saaneista 89/2 442; riskitiheyssuhde 0,892; 95 % lv 0,661; 1,203).
Taulukko 8 NATALEE – Hoidon tehoa kuvaavat tulokset (iDFS) tutkijan arvion mukaan (koko analyysipopulaatio) (tiedonkeruun katkaisupäivä 21.7.2023)
Kisqali + aromataasinestäjä* N = 2 549 | Aromataasinestäjä N = 2 552 | |
| Invasiivisen syövän suhteentautivapaa elossaolo (iDFSa) | ||
| Tapahtuman kokeneiden potilaiden määrä (n, %) | 226 (8,9 %) | 283 (11,1 %) |
| Riskitiheyssuhde (95 % lv) | 0,749 (0,628; 0,892) | |
| p-arvob | 0,0006 | |
| iDFS 36 kk:n kohdalla (%, 95 % lv) | 90,7 (89,3; 91,8) | 87,6 (86,1; 88,9) |
Lv = luottamusväli; N = potilasmäärä. a iDFS:n määritelmänä oli aika, joka kului satunnaistamisesta jonkin seuraavista ensimmäiseen esiintymiseen: invasiivisen rintasyövän paikallinen uusiutuminen, invasiivisen rintasyövän alueellinen uusiutuminen, etäpesäkkeinen uusiutuminen, mistä tahansa syystä johtuva kuolema, kontralateraalinen invasiivinen rintasyöpä tai toinen primaarinen invasiivinen syöpä, joka oli muu kuin rintasyöpä (lukuun ottamatta ihon tyvisolusyöpää ja levyepiteelikarsinoomaa). b Nimellinen p-arvo perustuu yksitahoiseen stratifioituun log-rank-testiin. * Letrotsoli tai anastrotsoli | ||
Kuva 1 NATALEE – iDFS-ajan Kaplan–Meier-kuvio tutkijan arvion mukaan (tiedonkeruun katkaisupäivä 21.7.2023)
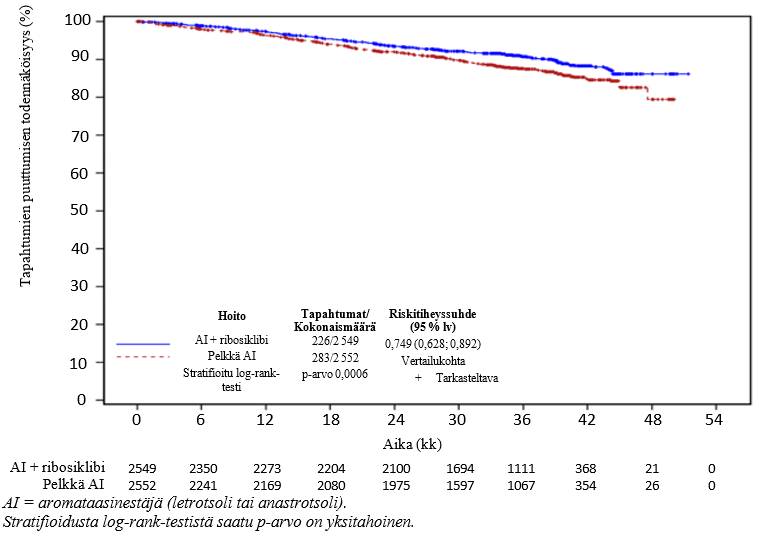
Levinneen syövän suhteen tautivapaan elossaolon (distant disease-free survival, DDFS) tapahtumia oli Kisqali-valmisteen ja aromataasinestäjän yhdistelmää saaneilla 204 (8,0 %) ja pelkkää aromataasinestäjää saaneilla 256 (10 %) (riskitiheyssuhde 0,749, 95 % lv: 0,623; 0,900).
Edennyt rintasyöpä
CLEE011A2301-tutkimus (MONALEESA-2)
Kisqalin ja letrotsolin yhdistelmää verrattiin pelkkään letrotsoliin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen III kliinisessä monikeskustutkimuksessa postmenopausaalisilla naisilla, joilla oli hormonireseptoripositiivinen, HER2-negatiivinen, pitkälle edennyt rintasyöpä ja jotka eivät olleet saaneet aiempaa hoitoa pitkälle edenneeseen tautiin.
Yhteensä 668 potilasta satunnaistettiin suhteessa 1:1 saamaan joko 600 mg:n Kisqali-annosten ja letrotsolin yhdistelmää (n = 334) tai lumelääkkeen ja letrotsolin yhdistelmää (n = 334). Heidät stratifioitiin maksa- ja/tai keuhkometastaasien suhteen (Kyllä [n = 292 (44 %)]) vs. Ei [n = 376 (56 %)]) . Ryhmien demografiset tiedot ja taudin lähtötilannetiedot olivat tasapainossa ja verrattavissa. Potilaille annettiin suun kautta 600 mg/vrk Kisqalia 21 perättäisenä päivänä, minkä jälkeen pidettiin 7 päivän tauko, sekä 2,5 mg letrotsolia kerran vuorokaudessa 28 päivän ajan. Potilaat eivät saaneet siirtyä lumelääkeryhmästä Kisqali-ryhmään tutkimuksen aikana eivätkä taudin etenemisen jälkeen.
Tutkimukseen otettujen potilaiden mediaani-ikä oli 62 v (vaihteluväli 23–91). 44,2 % potilaista oli 65-vuotiaita tai vanhempia, ja 69 potilasta oli yli 75-vuotiaita. Mukana oli valkoihoisia (82,2 %), aasialaisia (7,6 %) ja mustia (2,5 %) potilaita. Kaikkien potilaiden ECOG-toimintakykyluokka oli 0 tai 1. Kisqali-ryhmässä 46,6 % potilaista oli saanut solunsalpaajahoitoa neoadjuvantti- tai adjuvanttihoitona ja 51,3 % oli saanut hormonitoimintaa estävää hoitoa neoadjuvantti- tai adjuvanttihoitona ennen tutkimukseenottoa. 34,1 % potilaista oli de novo ‑potilaita. 22,0 %:lla potilaista oli tautimuutoksia vain luustossa, ja 58,8 %:lla oli viskeraalinen tauti. Jos potilas oli saanut aiemmin anastrotsolia tai letrotsolia (neo)adjuvanttihoitona, tämän hoidon päättymisestä oli tullut kulua vähintään 12 kk ennen tutkimukseen satunnaistamista.
Ensisijainen analyysi
Tutkimuksen ensisijainen päätetapahtuma saavutettiin suunnitellussa välianalyysissä, joka tehtiin, kun 80 % tavoitelluista RECIST v 1.1 -kriteerien (Response Evaluation Criteria in Solid Tumors) mukaisista etenemättömyysajan tapahtumista (PFS) oli havaittu. Tapahtumat perustuivat tutkijan arvioon koko populaatiossa (kaikkien satunnaistettujen potilaiden joukossa), ja ne vahvistettiin tekemällä sokkoutettu, riippumaton keskitetty radiologinen arviointi.
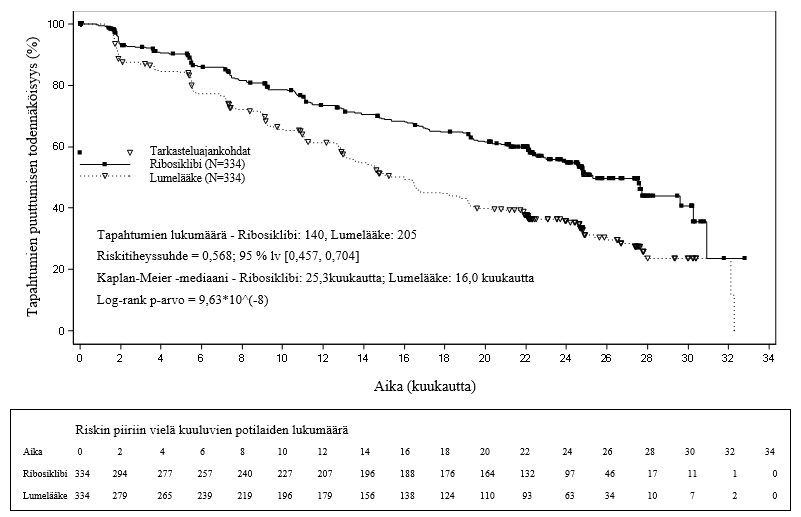
Hoidon tehoa kuvaavat tulokset osoittivat Kisqali- ja letrotsolihoitoa saaneilla potilailla tilastollisesti merkitsevän etenemättömyysajan (PFS) parantumisen verrattuna lumelääke- ja letrotsolihoitoa saaneisiin koko analyysipopulaatiossa (riskitiheyssuhde 0,556; 95 % lv 0,429; 0,720, yksitahoisen stratifioidun log-rank-testin p-arvo 0,00000329). Hoitovaikutus oli kliinisesti merkittävä.
Yleisen terveydentilan / elämänlaadun tiedoissa ei todettu merkittävää eroa Kisqali- ja letrotsoliryhmän ja lumelääke- ja letrotsoliryhmän välillä.
Taulukoissa 9 ja 10 esitetään tuoreemmat, päivitetyt tehotiedot (tiedonkeruun katkaisupäivä 2.1.2017).
PFS-ajan mediaani oli ribosiklibi- ja letrotsoliryhmässä 25,3 kk (95 % lv 23,0; 30,3) ja lumelääke- ja letrotsoliryhmässä 16,0 kk (95 % lv 13,4; 18,2). Ribosiklibi- ja letrotsoliryhmässä 54,7 %:lla potilaista arvioitiin, että tauti ei ollut edennyt 24 kk kohdalla; lumelääke- ja letrotsoliryhmässä vastaava osuus oli 35,9 %.
Taulukko 9 MONALEESA-2 – Hoidon tehoa kuvaavat tulokset (etenemättömyysaika) tutkijan radiologisen arvion mukaan (tiedonkeruun katkaisupäivä 2.1.2017)
| Päivitetty analyysi | ||
Kisqali + letrotsoli N = 334 | Lumelääke + letrotsoli N = 334 | |
| Etenemättömyysaika | ||
| Etenemättömyysajan mediaani [kk] (95 % lv) | 25,3 (23,0; 30,3) | 16,0 (13,4; 18,2) |
| Riskitiheyssuhde (95 % lv) | 0,568 (0,457; 0,704) | |
| p-arvoa | 9,63×10-8 | |
Lv = luottamusväli; N = potilasmäärä a p-arvo perustuu yksitahoiseen stratifioituun log-rank-testiin. | ||
Kuva 2 MONALEESA-2 – Etenemättömyysajan Kaplan–Meier-kaavio tutkijan arvion mukaan (tiedonkeruun katkaisupäivä 2.1.2017)
Hoitovaikutuksen sisäisen johdonmukaisuuden selvittämiseksi aineistosta tehtiin useita etukäteen määriteltyjä etenemättömyysajan alaryhmäanalyysejä ennusteeseen vaikuttavien tekijöiden ja lähtötasotietojen pohjalta. Alaryhmien määrittelyperusteena olivat ikä, etninen tausta, aiempi adjuvantti- tai neoadjuvanttisolunsalpaajahoito tai hormonaalinen hoito, maksa- ja/tai keuhkoaffisio ja pelkkiä luustomuutoksia aiheuttanut etäpesäkkeinen tauti. Kaikissa eri alaryhmissä todettiin, että taudin etenemisen tai kuoleman riski oli Kisqali-letrotsoliryhmässä pienempi. Tämä oli ilmeistä potilailla, joilla oli maksa- ja/tai keuhkoetäpesäkkeitä (HR 0,561 [95 % lv 0,424; 0,743], etenemättömyysajan mediaani [mPFS] 24,8 kk ribosiklibi- ja letrotsoliryhmässä vs. 13,4 kk pelkkää letrotsolia saaneilla), ja potilailla, joilla ei ollut maksa- ja/tai keuhkoetäpesäkkeitä (HR 0,597 [95 % lv 0,426; 0,837], mPFS 27,6 kk Kisqali- ja letrotsoliryhmässä vs. 18,2 kk pelkkää letrotsolia saaneilla).
Kokonaisvasteprosentteja ja kliinisiä hyötyprosentteja koskevat päivitetyt tulokset esitetään taulukossa 10.
Taulukko 10 MONALEESA-2 – Tutkimuksen hoidon tehoa kuvaavat tulokset (kokonaisvasteprosentti, kliininen hyötyprosentti) tutkijan arvion mukaan (tiedonkeruun katkaisupäivä 2.1.2017)
| Analyysi | Kisqali plus letrotsoli (%, 95 % lv) | Lumelääke plus letrotsoli (%, 95 % lv) | p-arvoc |
| Koko analyysipopulaatio | N = 334 | N = 334 | |
| Kokonaisvasteprosenttia | 42,5 (37,2; 47,8) | 28,7 (23,9; 33,6) | 9,18 × 10-5 |
| Kliininen hyötyprosenttib | 79,9 (75,6; 84,2) | 73,1 (68,3; 77,8) | 0,018 |
| Potilaat, joiden tauti oli mitattavissa | n = 257 | n = 245 | |
| Kokonaisvasteprosenttia | 54,5 (48,4; 60,6) | 38,8 (32,7, 44,9) | 2,54 × 10-4 |
| Kliininen hyötyprosenttib | 80,2 (75,3; 85,0) | 71,8 (66,2; 77,5) | 0,018 |
a Kokonaisvasteprosentti (ORR) = täydellisen vasteen + osittaisen vasteen saavuttaneiden potilaiden osuus b Kliininen hyötyprosentti (CBR) = täydellisen vasteen + osittaisen vasteen (+ taudin etenemisen pysähtymisen tai epätäydellisen vasteen / ≥ 24 viikon pituisen taudin etenemättömyyden) saavuttaneiden potilaiden osuus c p-arvot perustuvat yksitahoiseen Cochran–Mantel–Haenszelin khiin neliötestiin | |||
Lopullinen kokonaiselossaoloaika-analyysi
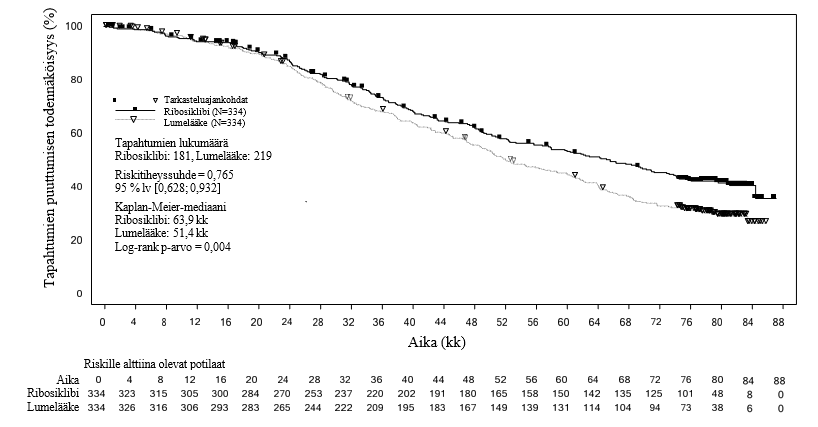
Lopullisen kokonaiselossaoloaika-analyysin tulokset koko tutkimuspopulaatiosta esitetään taulukossa 11 ja kuvassa 3.
Taulukko 11 MONALEESA‑2 – Tehotulokset (OS) (tiedonkeruun katkaisupäivä 10.6.2021)
| Kokonaiselossaoloaika, koko tutkimuspopulaatio | Kisqali plus letrotsoli N = 334 | Lumelääke plus letrotsoli N = 334 |
| Tapahtumien määrä – n [%] | 181 (54,2) | 219 (65,6) |
| Mediaani OS [kuukautta] (95 % lv) | 63,9 (52,4; 71,0) | 51,4 (47,2; 59,7) |
| Riskitiheyssuhdea (95 % lv) | 0,765 (0,628; 0,932) | |
| p-arvob | 0,004 | |
| OS tapahtumien puuttumisen osuus, (%) (95 % lv) | ||
| 24 kuukautta | 86,6 (82,3; 89,9) | 85,0 (80,5; 88,4) |
| 60 kuukautta | 52,3 (46,5; 57,7) | 43,9 (38,3; 49,4) |
| 72 kuukautta | 44,2 (38,5; 49,8) | 32,0 (26,8; 37,3) |
lv=luottamusväli a Riskitiheyssuhde perustuu stratifioituun Coxin suhteellisten riskitiheyksien malliin b p‑arvo perustuu 1-tahoiseen log‑rank-testiin (p< 0,0219 tehon paremmuuden raja-arvo). Stratifiointitekijöitä olivat keuhko- ja/tai maksaetäpesäkkeiden tila IRT-järjestelmän mukaan | ||
Kuva 3 MONALEESA‑2 – Kokonaiselossaoloajan Kaplan-Meier-kuvio koko populaatiossa (tiedonkeruun katkaisupäivä 10.6.2021)
Log-rank testin ja Coxin suhteellisten riskitiheyksien mallin stratifiointitekijöitä olivat keuhko- ja/tai maksaetäpesäkkeet IRT-järjestelmän mukaan.
Yksitahoinen p-arvo perustuu stratifioituun log-rank-testiin.
CLEE011E2301-tutkimus (MONALEESA‑7)
Kisqali‑valmistetta arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen III kliinisessä monikeskustutkimuksessa pre‑ ja perimenopausaalisilla naisilla, joilla oli hormonireseptoripositiivinen, HER2‑negatiivinen pitkälle edennyt rintasyöpä. Tutkimuksessa Kisqali‑valmistetta annettiin yhdessä ei-steroidisen aromataasinestäjän tai tamoksifeenin sekä gosereliinin kanssa ja tätä yhdistelmää verrattiin lumelääkkeen ja ei-steroidisen aromataasinestäjän tai tamoksifeenin sekä gosereliinin yhdistelmään. MONALEESA-7-tutkimuksen potilaat eivät olleet saaneet aiemmin endokriinista hoitoa pitkälle edenneen rintasyövän hoitoon.
Yhteensä 672 potilasta satunnaistettiin suhteessa 1:1 saamaan joko Kisqali-valmistetta (600 mg) ja ei-steroidista aromataasinestäjää / tamoksifeenia sekä gosereliinia (n = 335) tai lumelääkettä ja ei-steroidista aromataasinestäjää / tamoksifeenia sekä gosereliinia (n = 337). Stratifiointitekijöinä olivat maksa- ja/tai keuhkoetäpesäkkeet (Kyllä [n = 344 (51,2 %)] vs. Ei [n = 328 (48,8 %)]), aiempi solunsalpaajahoito pitkälle edenneen taudin hoitoon (Kyllä [n = 120 (17,9 %)] vs. Ei [n = 552 (82,1 %)]) ja yhdistelmässä käytetty endokriininen hoito (ei-steroidinen aromataasinestäjä ja gosereliini [n = 493 (73,4 %)] vs. tamoksifeeni ja gosereliini [n = 179 (26,6 %)]). Tutkimusryhmien demografiset tiedot ja taudin lähtötilannetiedot olivat tasapainossa ja vertailukelpoiset. Kisqali-valmistetta annettiin suun kautta 600 mg:n vuorokausiannoksina 21 perättäisen päivän ajan, minkä jälkeen pidettiin 7 päivän pituinen hoitotauko. Samanaikaisesti annettiin ei-steroidista aromataasinestäjää (letrotsoli, 2,5 mg, tai anastrotsoli, 1 mg) tai tamoksifeenia (20 mg) suun kautta kerran vuorokaudessa 28 päivän ajan ja gosereliinia (3,6 mg) ihon alle 28 päivän välein, kunnes tauti eteni tai ilmeni sietämätöntä toksisuutta. Potilaiden siirtyminen lumelääkeryhmästä Kisqali-ryhmään ei ollut sallittua tutkimuksen aikana eikä taudin etenemisen jälkeen. Yhdistelmähoidon endokriinisten valmisteiden vaihtaminen ei myöskään ollut sallittua.
Tutkimukseen otettujen potilaiden mediaani-ikä oli 44 v (vaihteluväli 25–58), ja 27,7 % potilaista oli alle 40-vuotiaita. Valtaosa mukaan otetuista potilaista oli valkoihoisia (57,7 %), aasialaisia (29,5 %) tai mustia (2,8 %), ja lähes kaikkien potilaiden (99,0 %) ECOG-toimintakykyluokka oli lähtötilanteessa 0 tai 1. Ennen tutkimukseenottoa näistä 672 potilaasta 14 % oli saanut aiempaa solunsalpaajahoitoa etäpesäkkeisen taudin hoitoon, 32,6 % oli saanut solunsalpaajahoitoa adjuvanttihoitona ja 18,0 % neoadjuvanttihoitona; 39,6 % oli saanut endokriinista hoitoa adjuvanttihoitona ja 0,7 % neoadjuvanttihoitona. E2301‑tutkimuksessa 40,2 %:lla potilaista oli de novo etäpesäkkeinen tauti, 23,7 %:lla oli vain luustoa affisioiva tauti ja 56,7 %:lla oli viskeraalinen tauti.
Tutkimuksen ensisijainen päätetapahtuma saavutettiin ensisijaisessa analyysissä, joka tehtiin, kun tutkimuksen koko analyysipopulaatiossa (kaikki satunnaistetut potilaat) oli tapahtunut 318 etenemättömyysaika‑ eli PFS-tapahtumaa tutkijan arvion mukaan RECIST v1.1 -kriteereillä arvioituna. Sokkoutettuun, riippumattomaan keskitettyyn radiologiseen arviointiin perustuvat PFS-tulokset tukivat ensisijaisia tehotuloksia. Ensisijaisen PFS‑analyysin ajankohtana seurannan mediaanikesto oli 19,2 kk.
Koko tutkimuspopulaation tehotulokset osoittivat, että etenemättömyysaika oli tilastollisesti merkitsevästi parempi Kisqali-valmistetta, ei-steroidista aromataasinestäjää / tamoksifeenia ja gosereliinia saaneilla potilailla verrattuna lumelääkettä, ei-steroidista aromataasinestäjää / tamoksifeenia ja gosereliinia saaneisiin (riskitiheyssuhde 0,553, 95 % lv 0,441; 0,694, 1‑tahoisen stratifioidun log‑rank-testin p‑arvo 9,83 x 10‑8) ja yhdistelmällä oli kliinisesti merkittävä terapeuttinen vaikutus. Etenemättömyysajan mediaani oli 23,8 kk (95 % lv 19,2; ei arvioitavissa) Kisqali-valmistetta ja ei-steroidista aromataasinestäjää / tamoksifeenia sekä gosereliinia saaneilla potilailla ja 13,0 kk (95 % lv 11,0; 16,4) lumelääkettä, ei-steroidista aromataasinestäjää / tamoksifeenia ja gosereliinia saaneilla.
PFS-tulosten jakaumasta esitetään yhteenveto PFS-ajan Kaplan‑Meier-käyrässä kuvassa 4.
Kuva 4 MONALEESA‑7 ‑ Etenemättömyysajan Kaplan-Meier-kuvio koko populaatiossa tutkijan arvion mukaan
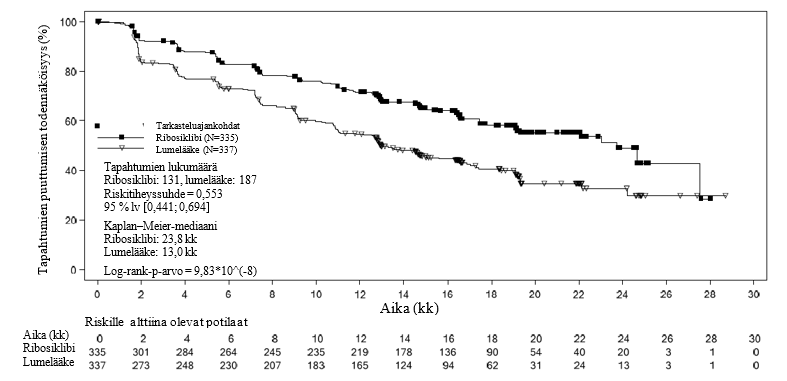
Sokkoutettuun, riippumattomaan keskitettyyn radiologiseen arviointiin perustuvat etenemättömyysaikatulokset sattumanvaraisesti valitusta osapopulaatiosta, johon kuului noin 40 % satunnaistetuista potilaista, tukivat tutkijan arvioon perustuvia ensisijaisia tehotuloksia (riskitiheyssuhde 0,427; 95 % lv 0,288; 0,633).
Ensisijaisen PFS-analyysin ajankohtana kokonaiselossaolon tiedot eivät olleet valmiit, ja kun tutkimuksessa oli tapahtunut 89 kuolemaa (13 %) (HR 0,916 [95 % lv 0,601; 1,396]).
Tutkijan arvioon perustuva RECIST v1.1 -kriteereillä arvioitu kokonaisvasteprosentti (ORR) oli Kisqali-ryhmässä suurempi (40,9 %; 95 % lv 35,6; 46,2) kuin lumelääkeryhmässä (29,7 %; 95 % lv 24,8; 34,6, p = 0,00098). Havaittu kliininen hyötyprosentti oli Kisqali-ryhmässä suurempi (79,1 %; 95 % lv 74,8; 83,5) kuin lumelääkeryhmässä (69,7 %; 95 % lv 64,8; 74,6, p = 0,002).
Ennalta määritellyssä 495 potilaan joukkoon perustuvassa alaryhmäanalyysissä potilailla, joille oli annettu Kisqali‑valmistetta tai lumelääkettä yhdessä ei-steroidisen aromataasinestäjän ja gosereliinin kanssa, PFS-ajan mediaani oli 27,5 kk (95 % lv 19,1; ei arvioitavissa) Kisqali-valmistetta ja ei-steroidista aromataasinestäjää saaneessa alaryhmässä ja 13,8 kk (95 % lv 12,6; 17,4) lumelääkettä ja ei-steroidista aromataasinestäjää saaneessa alaryhmässä [HR: 0,569; 95 % lv 0,436; 0,743]. Tehotulosten yhteenveto esitetään taulukossa 12 ja PFS-ajan Kaplan‑Meier-käyrä esitetään kuvassa 5.
Taulukko 12 MONALEESA‑7 ‑ Tehotulokset (etenemättömyysaika) ei-steroidista aromataasinestäjää saaneilla potilailla
Kisqali + ei-steroidinen aromataasinestäjä + gosereliini N = 248 | Lumelääke + ei-steroidinen aromataasinestäjä + gosereliini N = 247 | |
| Etenemättömyysaikaa | ||
| Etenemättömyysajan mediaani [kk] (95% lv) | 27,5 (19,1; ei arvioitavissa) | 13,8 (12,6; 17,4) |
| Riskitiheyssuhde (95 % lv) | 0,569 (0,436; 0,743) | |
Lv = luottamusväli; N = potilasmäärä. a Etenemättömyysaika perustuu tutkijan radiologiseen arvioon | ||
Kuva 5 MONALEESA‑7 – Tutkijan arvioon perustuva etenemättömyysajan Kaplan‑Meier-kuvio ei-steroidista aromataasinestäjää saaneilla potilailla
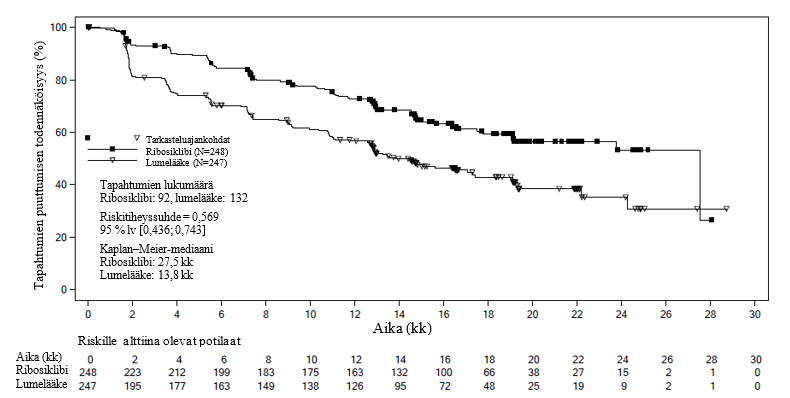
Tehotulokset eli tutkijan arvioon ja RECIST v1.1 -kriteereihin perustuvat kokonaisvasteprosentit ja kliiniset hyötyprosentit esitetään taulukossa 13.
Taulukko 13 MONALEESA‑7 ‑ Tutkijan arvioon perustuvat tehotulokset (kokonaisvasteprosentti, kliininen hyötyprosentti) ei-steroidista aromataasinestäjää saaneilla potilailla
| Analyysi | Kisqali + ei-steroidinen aromataasinestäjä + gosereliini (%, 95 % lv) | Lumelääke + ei-steroidinen aromataasinestäjä + gosereliini (%, 95 % lv) |
| Koko analyysipopulaatio | N = 248 | N = 247 |
| Kokonaisvasteprosenttia | 39,1 (33,0; 45,2) | 29,1 (23,5; 34,8) |
| Kliininen hyötyprosenttib | 80,2 (75,3; 85,2) | 67,2 (61,4; 73,1) |
| Potilaat, joiden tauti oli mitattavissa | n = 192 | n = 199 |
| Kokonaisvasteprosenttia | 50,5 (43,4; 57,6) | 36,2 (29,5; 42,9) |
| Kliininen hyötyprosenttib | 81,8 (76,3; 87,2) | 63,8 (57,1; 70,5) |
a Kokonaisvasteprosentti: täydellisen vasteen + osittaisen vasteen saavuttaneiden potilaiden osuus b Kliininen hyötyprosentti: täydellisen vasteen + osittaisen vasteen (+ taudin etenemisen pysähtymisen tai ei-täydellisen vasteen / taudin etenemättömyyden ≥ 24 viikon ajalta) saavuttaneiden potilaiden osuus | ||
Kisqali-valmistetta ja ei-steroidista aromataasinestäjää saaneen alaryhmän tulokset olivat johdonmukaiset eri alaryhmissä, joiden määrittelyperusteena olivat ikä, etninen tausta, aiemmat adjuvantti/neoadjuvanttisolunsalpaajahoidot tai hormonihoidot, maksa‑ ja/tai keuhkoaffisio ja luustoon rajoittunut etäpesäkkeinen tauti.
Tarkempi päivitys kokonaiselossaoloaikaa koskeviin tuloksiin (tiedonkeruun katkaisupäivä 30.11.2018) esitetään taulukossa 14 ja kuvissa 6 ja 7.
Toisessa kokonaiselossaoloaikaa koskevassa analyysissä saavutettiin tutkimuksen keskeinen toissijainen päätetapahtuma, joka osoitti kokonaiselossaoloajan paranevan tilastollisesti merkittävästi.
Taulukko 14 MONALEESA‑7 – Tehotulokset (OS) (tiedonkeruun katkaisupäivä 30.11.2018)
| Päivitetty analyysi | ||
| Kokonaiselossaoloaika, koko tutkimuspopulaatio | Kisqali 600 mg N=335 | Lumelääke N=337 |
| Tapahtumien määrä – n [%] | 83 (24,8) | 109 (32,3) |
| Mediaani OS [kuukautta] (95% lv) | Ei arvioitavissa (ei arvioitavissa; ei arvioitavissa) | 40,9 (37,8; ei arvioitavissa) |
| Riskitiheyssuhde (95% Iv) | 0,712 (0,535; 0,948) | |
| p‑arvoa | 0,00973 | |
| Kokonaiselossaoloaika, NSAI alaryhmä | Kisqali 600 mg n = 248 | Lumelääke n = 247 |
| Tapahtumien määrä – n [%] | 61 (24,6) | 80 (32,4) |
| Mediaani OS [kuukautta] (95% CI) | Ei arvioitavissa (ei arvioitavissa; ei arvioitavissa) | 40,7 (37,4; ei arvioitavissa) |
| Riskitiheyssuhde (95% lv) | 0,699 (0,501; 0,976) | |
lv=luottamusväli, N=potilasmäärä; ap‑arvo perustuu 1-tahoiseen log‑rank-testiin, jonka stratifiointitekijöitä olivat keuhko- ja/tai maksaetäpesäkkeet, aiempi solunsalpaajahoito pitkälle edenneen taudin hoitoon sekä yhdistelmässä käytetty IRT-järjestelmään (automaattinen satunnaistamis- ja logistiikkajärjestelmä) perustuva endokriininen hoito. | ||
Kuva 6 MONALEESA-7 – Kokonaiselossaoloajan lopullisen analyysin Kaplan Meier-kaavio (tiedonkeruun katkaisupäivä 30.11.2018)
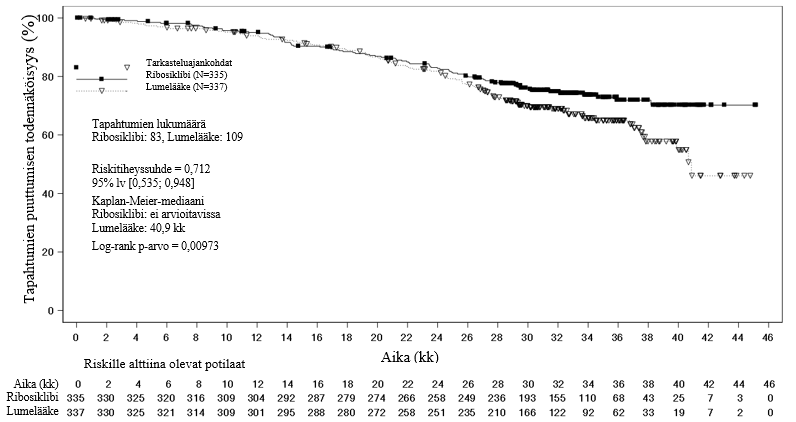
Log-rank-testin ja Coxin mallin stratifiointitekijöitä olivat keuhko- ja/tai maksaetäpesäkkeet, aiempi solunsalpaajahoito pitkälle edenneen taudin hoitoon sekä yhdistelmässä käytetty IRT-järjestelmään perustuva endokriininen hoito.
Kuva 7 MONALEESA-7 – Lopullisen kokonaiselossaoloajan analyysin Kaplan-Meier-kuvio potilailla, jotka saivat ei-steroidista aromataasin estäjää (tiedonkeruun katkaisupäivä 30.11.2018)
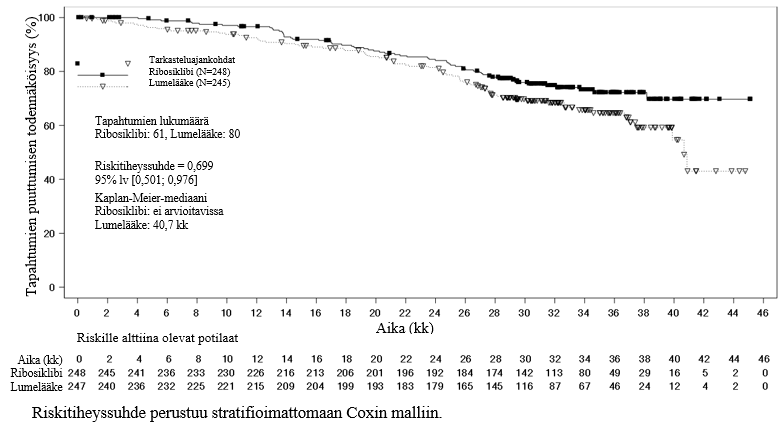
Lisäksi taudinetenemisen todennäköisyys seuraavan linjan hoidossa tai kuolemalle (PFS2) oli alhaisempi tutkimuksessa ribosiklibia saaneiden potilaiden kohdalla verrattuna lumelääkettä saaneisiin potilaisiin koko tutkimuspopulaatiossa (riskitiheyssuhde 0,692, 95 % lv 0,548; 0,875). PFS2-mediaani oli 32,3 kuukautta (95 % lv 27,6; 38,3) lumelääkeryhmässä ja sitä ei saavutettu ribosiklibi ryhmässä (95 % lv 39,4; ei arvioitavissa). Samankaltaisia tuloksia havaittiin ei-steroidista aromataasin estäjää saaneiden potilaiden alaryhmässä (riskitiheyssuhde 0,660, 95 % lv 0,503; 0,868), jossa PFS2-mediaani oli 32,3 kuukautta (95 % lv 26,9; 38,3) lumelääkettä saaneilla potilailla verrattuna ribosiklibia saaneisiin potilaisiin, jossa tätä ei saavutettu (95 % lv 39,4; ei arvioitavissa).
CLEE011F2301-tutkimus (MONALEESA‑3)
Kisqalin ja fulvestrantin yhdistelmää verrattiin pelkkään fulvestranttiin suhteessa 2:1 satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen III kliinisessä monikeskustutkimuksessa 726 postmenopausaalisella naisella, joilla oli hormonireseptoripositiivinen, HER2‑negatiivinen, pitkälle edennyt rintasyöpä ja jotka eivät olleet saaneet aiempaa endokriinista hoitoa tai olivat saaneet aiemmin vain yhden endokriinisen hoitolinjan.
Tutkimukseen otettujen potilaiden mediaani-ikä oli 63 v (vaihteluväli 31–89). Potilaista 46,7 % oli täyttänyt 65 vuotta ja 13,8 % oli täyttänyt 75 vuotta. Mukaan otetut potilaat olivat valkoihoisia (85,3 %), aasialaisia (8,7 %) tai mustia (0,7 %), ja melkein kaikkien potilaiden (99,7 %) ECOG-toimintakykyluokka oli lähtötilanteessa 0 tai 1. Tutkimukseen otettiin ensilinjan tai toisen linjan potilaita (19,1 %:lla potilaista oli de novo etäpesäkkeinen tauti). Ennen tutkimukseenottoa 42,7 % potilaista oli saanut solunsalpaajahoitoa adjuvanttihoitona ja 13,1 % neoadjuvanttihoitona; 58,5 % oli saanut endokriinista hoitoa adjuvanttihoitona ja 1,4 % neoadjuvanttihoitona; ja 21 % oli saanut aiemmin endokriinista hoitoa pitkälle edenneen rintasyövän hoitoon. F2301‑tutkimuksessa 21,2 %:lla potilaista oli luustoon rajoittunut tauti ja 60,5 %:lla oli viskeraalinen tauti.
Ensisijainen analyysi
Tutkimuksen ensisijainen päätetapahtuma saavutettiin ensisijaisessa analyysissä, joka tehtiin, kun tutkimuksen koko analyysipopulaatiossa (kaikki satunnaistetut potilaat, tiedonkeruun katkaisupäivä 3.11.2017) oli tapahtunut 361 etenemättömyysaika‑ eli PFS-tapahtumaa tutkijan arvion mukaan RECIST v1.1 -kriteereillä arvioituna. Seurannan mediaanikesto ensisijaisen PFS‑analyysin ajankohtana oli 20,4 kk.
Ensisijaiset tehotulokset osoittivat, että PFS-aika oli Kisqali-valmistetta ja fulvestranttia saaneilla tilastollisesti merkitsevästi parempi kuin lumelääkettä ja fulvestranttia saaneilla koko analyysipopulaatiossa (riskitiheyssuhde 0,593, 95 % lv 0,480; 0,732, 1‑tahoisen stratifioidun log‑rank-testin p‑arvo 4,1 x 10‑7) ja taudin etenemisen tai kuoleman suhteellinen riski oli Kisqali- ja fulvestranttiryhmässä arviolta 41 % pienempi.
40-prosenttisen kuvantamisosapopulaation sattumanvarainen keskitetty auditointi (sokkoutettu riippumaton keskitetty radiologinen arviointi) tuki ensisijaisia tehotuloksia (riskitiheyssuhde 0,492, 95 % lv 0,345; 0,703).
PFS-tulosten deskriptiivinen päivitys tehtiin toisen kokonaiselossaoloaikaa koskevan välianalyysin ajankohtana. Koko populaation ja aiempaan endokriiniseen hoitoon perustuvien alaryhmien päivitetyistä PFS-tuloksista esitetään yhteenveto taulukossa 15 ja Kaplan‑Meier-käyrä esitetään kuvassa 8.
Taulukko 15 MONALEESA‑3 (F2301) – Päivitetyt etenemättömyysaikatulokset tutkijan arvion mukaan (tiedonkeruun katkaisupäivä 3.6.2019)
Kisqali + fulvestrantti N = 484 | Lume + fulvestrantti N = 242 | ||
| Etenemättömyysaika koko tutkimuspopulaatiossa | |||
| Tapahtumien määrä – n [%] | 283 (58,5) | 193 (79,8) | |
| Etenemättömyysajan mediaani [kk] (95 % lv) | 20,6 (18,6; 24,0) | 12,8 (10,9; 16,3) | |
| Riskitiheyssuhde (95 % lv) | 0,587 (0,488, 0,705) | ||
| Ensilinjan potilaiden alaryhmäa | Kisqali + fulvestrantti n = 237 | Lume + fulvestrantti n = 128 | |
| Tapahtumien määrä – n [%] | 112 (47,3) | 95 (74,2) | |
| Etenemättömyysajan mediaani [kk] (95% lv) | 33,6 (27,1; 41,3) | 19,2 (14,9; 23,6) | |
| Riskitiheyssuhde (95 % lv) | 0,546 (0,415; 0,718) | ||
| Toisen linjan potilaiden tai varhaisen relapsin alaryhmäb | Kisqali + fulvestrantti n = 237 | Lume + fulvestrantti n = 109 | |
| Tapahtumien määrä – n [%] | 167 (70,5) | 95 (87,2) | |
| Etenemättömyysajan mediaani [kk] (95% lv) | 14,6 (12,5; 18,6) | 9,1 (5,8; 11,0) | |
| Riskitiheyssuhde (95 % lv) | 0,571 (0,443; 0,737) | ||
Lv = luottamusväli a Potilaat, joilla oli pitkälle edennyt de novo ‑rintasyöpä ja jotka eivät olleet saaneet aiempaa endokriinista hoitoa, ja potilaat, joilla tauti uusiutui 12 kuukauden kuluttua endokriinisen (neo)adjuvanttihoidon päättymisestä. b Potilaat, joilla tauti uusiutui adjuvanttihoidon aikana tai 12 kuukauden kuluttua endokriinisen (neo)adjuvanttihoidon päättymisestä, ja potilaat, joilla tauti eteni yhden pitkälle edenneeseen tautiin annetun endokriinisen hoitolinjan jälkeen. | |||
Kuva 8 MONALEESA‑3 (F2301) ‑ Etenemättömyysajan Kaplan-Meier-kuvio tutkijan arvion mukaan (koko analyysipopulaatio) (tiedonkeruun katkaisupäivä 3.6.2019)

Tehotulokset eli tutkijan arvioon ja RECIST v1.1 -kriteereihin perustuvat kokonaisvasteprosentit ja kliiniset hyötyprosentit esitetään taulukossa 16.
Taulukko 16 MONALEESA‑3 ‑ Tehotulokset (kokonaisvasteprosentti, kliininen hyötyprosentti) tutkijan arvion mukaan (tiedonkeruun katkaisupäivä 3.11.2017)
| Analyysi | Kisqali + fulvestrantti (%, 95 % lv) | Lume + fulvestrantti (%, 95 % lv) |
| Koko analyysipopulaatio | N = 484 | N = 242 |
| Kokonaisvasteprosenttia | 32,4 (28,3; 36,6) | 21,5 (16,3; 26,7) |
| Kliininen hyötyprosenttib | 70,2 (66,2; 74,3) | 62,8 (56,7; 68,9) |
| Potilaat, joiden tauti oli mitattavissa | n = 379 | n = 181 |
| Kokonaisvasteprosenttia | 40,9 (35,9; 45,8) | 28,7 (22,1; 35,3) |
| Kliininen hyötyprosenttib | 69,4 (64,8; 74,0) | 59,7 (52,5; 66,8) |
a Kokonaisvasteprosentti: täydellisen vasteen + osittaisen vasteen saavuttaneiden potilaiden osuus b Kliininen hyötyprosentti: täydellisen vasteen + osittaisen vasteen (+ taudin etenemisen pysähtymisen tai epätäydellisen vasteen / taudin etenemättömyyden ≥ 24 viikon ajalta) saavuttaneiden potilaiden osuus. | ||
Riskisuhteet, jotka perustuivat Kisqali‑valmisteen ja fulvestrantin yhdistelmää saaneiden potilaiden joukossa tehtyihin ennalta määriteltyihin alaryhmäanalyyseihin, osoittivat hoidon tuottaneen hyötyä johdonmukaisesti eri alaryhmissä. Alaryhmien ryhmittelyperusteina olivat ikä, aiempi hoito (varhainen / pitkälle edennyt tilanne), aiempi adjuvantti/neoadjuvanttisolunsalpaajahoito tai hormonaalinen hoito, maksa‑ ja/tai keuhkoaffisio ja luustoon rajoittunut etäpesäkkeinen tauti.
Kokonaiselossaoloaikaa koskeva analyysi
Toisessa kokonaiselossaoloaikaa koskevassa analyysissä saavutettiin tutkimuksen toissijainen päätetapahtuma, joka osoitti kokonaiselossaoloajan paranevan tilastollisesti merkitsevästi.
Koko tutkimuspopulaatiota koskevan lopullisen kokonaiselossaoloaika-analyysin ja alaryhmäanalyysien tulokset esitetään taulukossa 17 ja kuvassa 9.
Taulukko 17 MONALEESA-3 (F2301) – Tehotulokset (OS) (tiedonkeruun katkaisupäivä 3.6.2019)
| Kisqali + fulvestrantti | Lume + fulvestrantti | |
| Koko tutkimuspopulaatio | N = 484 | N = 242 |
| Tapahtumien määrä – n [%] | 167 (34,5) | 108 (44,6) |
| Mediaani OS [kuukautta] (95 % lv) | Ei arvioitavissa (ei arvioitavissa, ei arvioitavissa) | 40 (37, ei arvioitavissa) |
| Riskitiheyssuhde (95 % lv)a | 0,724 (0,568; 0,924) | |
| p-arvob | 0,00455 | |
| Ensilinjan potilaiden alaryhmä | n = 237 | n = 128 |
| Tapahtumien määrä – n [%] | 63 (26,6) | 47 (36,7) |
| Riskitiheyssuhde (95 % lv)c | 0,700 (0,479; 1,021) | |
| Toisen linjan potilaiden tai varhaisen relapsin alaryhmä | n = 237 | n = 109 |
| Tapahtumien määrä – n [%] | 102 (43,0) | 60 (55,0) |
| Riskitiheyssuhde (95 % lv)c | 0,730 (0,530, 1,004) | |
a Riskitiheyssuhde perustuu Coxin suhteellisten riskitiheyksien malliin, jonka stratifiointitekijöitä olivat keuhko- ja/tai maksaetäpesäkkeet ja aiempi endokriininen hoito. b 1-tahoinen p‑arvo perustuu log-rank-testiin, jonka stratifiointitekijöitä olivat keuhko- ja/tai maksaetäpesäkkeet ja aiempi IRT-järjestelmään perustuva endokriininen hoito. P-arvo on 1-tahoinen, ja vertailukohtana on kynnysarvo 0,01129, joka määritettiin Lan–DeMetsin (O’Brien–Flemingin) alfavirheen korjausfunktiolla (kokonaismerkitsevyystaso 0,025). c Riskitiheyssuhde perustuu stratifioimattomaan Coxin suhteellisten riskitiheyksien malliin. | ||
Kuva 9 MONALEESA-3 (F2301) – Kokonaiselossaoloajan Kaplan–Meier-kaavio (koko analyysipopulaatio) (tiedonkeruun katkaisupäivä 3.6.2019)
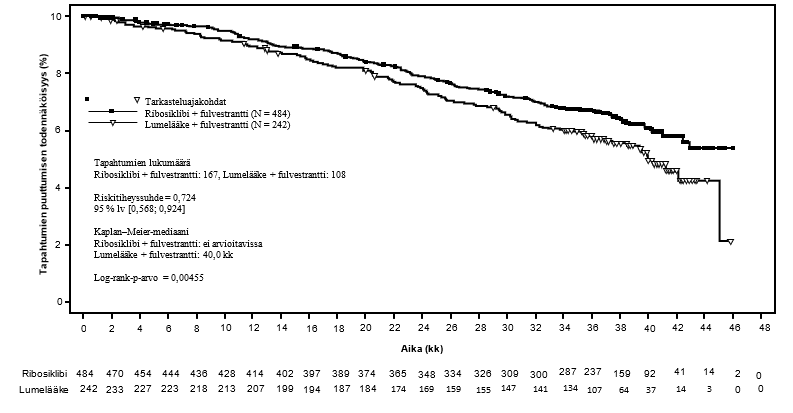
Log-rank-testin ja Coxin mallin stratifiointitekijöitä olivat keuhko- ja/tai maksaetäpesäkkeet, aiempi solunsalpaajahoito pitkälle edenneen taudin hoitoon sekä yhdistelmässä käytetty IRT-järjestelmään perustuva endokriininen hoito.
Taudin etenemiseen kulunut aika seuraavan linjan hoidossa tai kuolemaan kulunut aika (PFS2) oli Kisqali-ryhmässä pidempi verrattuna lumelääkeryhmään koko tutkimuspopulaatiossa (riskitiheyssuhde 0,670, 95 % lv 0,542; 0,830). PFS2-mediaani oli 39,8 kuukautta (95 % lv 32,5; ei arvioitavissa) Kisqali-ryhmässä ja 29,4 kk (95 % lv 24,1; 33,1) lumelääkeryhmässä.
Iäkkäät potilaat
Merkittävä osuus potilaista, jotka saivat Kisqalia tutkimuksissa MONALEESA-2 ja MONALEESA-3 oli täyttänyt 65 vuotta ja 75 vuotta (ks. kohta Farmakodynamiikka). Kisqali-valmisteen turvallisuudessa ja vaikuttavuudessa ei havaittu yleisiä eroja näiden iäkkäiden potilaiden ja nuorempien potilaiden välillä (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoimintapotilaat
Kolmessa avaintutkimuksessa (MONALEESA-2, MONALEESA-3 ja MONALEESA-7) ribosiklibihoitoa sai 510 (53,8 %) potilasta, joilla oli normaali munuaisten toiminta, 341 (36 %) potilasta, joilla oli lievä munuaisten vajaatoiminta, ja 97 (10,2 %) potilasta, joilla oli keskivaikea munuaisten vajaatoiminta. Tutkimuksiin ei otettu yhtään vaikeaa munuaisten vajaatoimintaa sairastavaa potilasta. PFS-tulokset olivat johdonmukaiset lievää ja keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla, jotka saivat ribosiklibia 600 mg:n aloitusannoksella, verrattuna potilaisiin, joiden munuaisten toiminta oli normaali. Turvallisuusprofiili oli yleisesti ottaen johdonmukainen kaikissa munuaistoimintakohorteissa (ks. kohta Haittavaikutukset).
Pediatriset potilaat
Tutkimus CLEE011Q12101
Vaiheen I/II monikeskustutkimuksessa arvioitiin ribosiklibin ja topotekaani + temotsolomidi -yhdistelmän tehoa ja turvallisuutta pediatrisilla potilailla (4–<18 vuotta, n=10) ja nuorilla aikuisilla (18–20 vuotta, n=2), joilla oli uusiutunut tai refraktaarinen (r/r) neuroblastooma tai muita kiinteitä kasvaimia (kuten alveolaarinen rabdomyosarkooma / rabdomyosarkooma, pahanlaatuinen gliooma, pahanlaatuinen astrosytooma, glioblastooma ja medulloblastooma). Vaiheen I annoshakuvaiheessa suoritettiin kolme kohorttia, joissa tutkittiin seuraavat annostusohjelmat (28 päivän hoitosykli): ribosiklibi 200 mg/m²/päivä (päivät 1–21) yhdessä topotekaanin 0,75 mg/m²/päivä ja temotsolomidin 150 mg/m²/päivä (päivät 1–5) kanssa (samanaikainen annostelu); ribosiklibi 100 mg/m²/päivä (päivät 6–21) topotekaanin 0,75 mg/m²/päivä ja temotsolomidin 150 mg/m²/päivä jälkeen (päivät 1–5) (peräkkäinen annostelu); ribosiklibi 100 mg/m²/päivä (päivät 1–14) yhdessä topotekaanin 0,75 mg/m²/päivä ja temotsolomidin 100 mg/m²/päivä (päivät 1–5) kanssa (samanaikainen annostelu). Potilaiden mediaani-ikä oli 14 vuotta (n=12) ja heistä 7 oli tyttöjä. Objektiivista hoitovastetta ei havaittu. Suurinta siedettyä annosta ja suositeltua vaiheen II annosta ei voitu määrittää tässä tutkimuksessa annosta rajoittavan toksisuuden ilmenemisen vuoksi. Uusia turvallisuussignaaleja ei havaittu.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Kisqali-valmisteen käytöstä rintasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Ribosiklibin farmakokinetiikkaa arvioitiin pitkälle edennyttä syöpää sairastavilla potilailla, jotka saivat 50–1 200 mg:n vuorokausiannoksia suun kautta. Terveet henkilöt saivat suun kautta 400–600 mg:n kerta-annoksia tai toistuvia 400 mg:n vuorokausiannoksia 8 päivän ajan.
Imeytyminen
Ribosiklibin absoluuttisen biologisen hyötyosuuden geometrinen keskiarvo oli terveillä henkilöillä 65,8 % suun kautta saadun 600 mg:n kerta-annoksen jälkeen.
Kun ribosiklibi otettiin suun kautta, Cmax-pitoisuuden saavuttamiseen kulunut aika (tmax) oli 1–4 h. Ribosiklibialtistus (Cmax ja AUC) suureni hieman enemmän kuin suhteessa annokseen testatulla annosalueella (50–1 200 mg). Toistuvan kerran vuorokaudessa tapahtuneen annostelun jälkeen vakaa tila saavutettiin yleensä 8 päivän jälkeen, ja ribosiklibin kumuloitumisen kumulaatiokertoimen geometrinen keskiarvo oli 2,51 (vaihteluväli 0,97–6,40).
Ruoan vaikutus
600 mg:n ribosiklibikerta-annoksen otto kalvopäällysteisinä tabletteina suun kautta runsasrasvaisen, runsaskalorisen aterian kanssa ei vaikuttanut ribosiklibin imeytymisnopeuteen eikä sen imeytymisprosenttiin verrattuna tilanteeseen, jossa lääke otettiin paaston jälkeen.
Jakautuminen
Ribosiklibi sitoutuu ihmisellä plasman proteiineihin in vitro noin 70-prosenttisesti pitoisuuksista riippumatta (pitoisuusalueella 10–10 000 ng/ml). Ribosiklibi jakautuu yhtäläisesti veren punasoluihin ja plasmaan, ja veren ja plasman lääkepitoisuuksien suhde on keskimäärin 1,04 in vivo. Vakaan tilan näennäinen jakautumistilavuus (Vss/F) oli populaatiofarmakokinetiikan analyysin perusteella 1 090 l.
Biotransformaatio
In vitro- ja in vivo ‑tutkimukset viittaavat siihen, että ribosiklibi poistuu ihmisillä pääosin CYP3A4-välitteisen maksametabolian kautta. Kun ihmiselle annettiin suun kautta 600 mg:n kerta-annos [14C]-ribosiklibia, ribosiklibin ensisijaiset metaboliareitit olivat oksidaatio (dealkylaatio, C- ja/tai N-oksigenaatio, oksidaatio (-2H)) ja niiden yhdistelmät. Ribosiklibin faasin I metaboliittien faasin II konjugaattien kohdalla kyseessä olivat N-asetylaatio, sulfaatio, kysteiinikonjugaatio, glykosylaatio ja glukuronidaatio. Ribosiklibi oli tärkein plasmassa kiertävä lääkeperäinen fraktio. Tärkeimpiä kiertäviä metaboliitteja olivat M13 (CCI284, N-hydroksylaatio), M4 (LEQ803, N-demetylaatio) ja M1 (toissijainen glukuronidi). Ribosiklibin kliininen vaikutus (farmakologinen vaikutus ja turvallisuus) johtui lähinnä kanta-aineesta, ja kiertävien metaboliittien osuus oli erittäin pieni.
Ribosiklibi metaboloitui voimakkaasti, ja 17,3 % annoksesta erittyi muuttumattomana ulosteeseen ja 12,1 % virtsaan. LEQ803-metaboliittia esiintyi merkittävinä määrinä ulosteessa ja virtsassa, ja noin 13,9 % annetusta annoksesta erittyi tämän metaboliitin muodossa ulosteeseen ja 3,74 % virtsaan. Sekä ulosteessa että virtsassa todettiin pieniä määriä useita muita metaboliitteja (≤ 2,78 % annetusta annoksesta).
Eliminaatio
Plasman efektiivisen puoliintumisajan geometrinen keskiarvo (kumulaatiokertoimen perusteella) oli 32,0 h (63 % CV) ja näennäisen peroraalisen puhdistuman (CL/F) geometrinen keskiarvo taas 25,5 l/h (66 % CV) vakaassa tilassa 600 mg:n annoksilla pitkälle edennyttä syöpää sairastavilla potilailla. Populaatiofarmakokinetiikan analyysin perusteella ribosiklibialtistus on annoksen ollessa sama todennäköisesti hieman pienempi varhaisvaiheen rintasyöpää sairastavilla kuin edennyttä rintasyöpää sairastavilla. Plasman näennäisen terminaalisen puoliintumisajan (t1/2) geometrinen keskiarvo vaihteli välillä 29,7–54,7 h ja ribosiklibin CL/F-arvon geometrinen keskiarvo vaihteli välillä 39,9–77,5 l/h terveillä henkilöillä tehdyissä tutkimuksissa 600 mg:n annoksilla.
Ribosiklibi ja sen metaboliitit eliminoituvat pääasiassa ulosteeseen, ja pieni osa erittyy munuaisten kautta. 6 terveellä miestutkittavalla 91,7 % koko annetusta radioaktiivisuusannoksesta erittyi 22 päivän kuluessa, kun miehet saivat suun kautta kerta-annoksen [14C]-ribosiklibia. Tärkein eliminaatioreitti oli erittyminen ulosteeseen (69,1 %). Virtsaan erittyi 22,6 % annoksesta.
Lineaarisuus/ei-lineaarisuus
Ribosiklibialtistus (Cmax ja AUC) suureni hieman enemmän kuin suhteessa annokseen 50–1 200 mg:n annosalueella kerta-annosten ja toistuvien annosten jälkeen. Analyysia rajoittaa se, että useimpien annoskohorttien otoskoot olivat pienet ja valtaosa tiedoista perustuu 600 mg:n annoskohorttiin.
Erityisryhmät
Munuaisten vajaatoiminta
Munuaisten toiminnan vaikutusta ribosiklibin farmakokinetiikkaan arvioitiin munuaisten vajaatoimintatutkimuksessa, johon osallistui 14 tervettä henkilöä, joilla oli normaali munuaisten toiminta (absoluuttinen glomerulusten suodatusnopeus [aGFR] ≥ 90 ml/min), 8 henkilöä, joilla oli lievä munuaisten vajaatoiminta (aGFR 60 – < 90 ml/min), 6 henkilöä, joilla oli keskivaikea munuaisten vajaatoiminta (aGFR 30 – < 60 ml/min), 7 henkilöä, joilla oli vaikea munuaisten vajaatoiminta (aGFR 15 – < 30 ml/min), ja 3 henkilöä, joilla oli loppuvaiheen munuaissairaus (ESRD) (aGFR < 15 ml/min), annostuksella yksi 400 mg:n kerta-annos ribosiklibia.
Verrattuna altistukseen normaalin munuaistoiminnan potilailla AUCinf suureni lievää munuaisten vajaatoimintaa sairastavilla 1,6‑kertaisesti, keskivaikeaa munuaisten vajaatoimintaa sairastavilla 1,9‑kertaisesti ja vaikeaa munuaisten vajaatoimintaa sairastavilla 2,7‑kertaisesti; Cmax puolestaan suureni lievää munuaisten vajaatoimintaa sairastavilla 1,8-kertaisesti, keskivaikeaa munuaisten vajaatoimintaa sairastavilla 1,8‑kertaisesti ja vaikeaa munuaisten vajaatoimintaa sairastavilla 2,3‑kertaisesti. Suurella osuudella ribosiklibia koskevien teho- ja turvallisuustutkimusten osallistujista oli lievä munuaisten vajaatoiminta (ks. kohta Farmakodynamiikka). Siksi munuaisten vajaatoimintatutkimukseen osallistuneiden, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavien tutkittavien tietoja verrattiin myös yhdistettyihin tietoihin tutkittavista, joilla munuaisten toiminta oli normaali tai munuaisten vajaatoiminta oli lievä. Verrattuna yhdistettyihin tietoihin tutkittavista, joilla munuaisten toiminta oli normaali tai munuaisten vajaatoiminta lievä, AUCinf suureni keskivaikeaa munuaisten vajaatoimintaa sairastavilla 1,6‑kertaisesti ja vaikeaa munuaisten vajaatoimintaa sairastavilla 2,2‑kertaisesti ja Cmax keskivaikeaa munuaisten vajaatoimintaa sairastavilla 1,5‑kertaisesti ja vaikeaa munuaisten vajaatoimintaa sairastavilla 1,9‑kertaisesti. Loppuvaiheen munuaissairautta sairastavien osalta kerrannaisuuseroa ei laskettu tutkittavien pienen määrän vuoksi, mutta tulokset viittaavat samankaltaiseen tai hieman suurempaan ribosiklibialtistuksen suurenemiseen kuin vaikeaa munuaisten vajaatoimintaa sairastavilla.
Munuaisten toiminnan vaikutusta ribosiklibin farmakokinetiikkaan arvioitiin myös edennyttä tai etäpesäkkeistä rintasyöpää sairastaneilla potilailla, jotka osallistuivat teho- ja turvallisuustutkimuksiin aloitusannoksella 600 mg (ks. kohta Farmakodynamiikka). Edennyttä tai etäpesäkkeistä rintasyöpää sairastaneita potilaita koskevien tutkimusten farmakokineettisten tietojen alaryhmäanalyysissa, kun ribosiklibia annettiin 600 mg:n kerta-annoksena tai toistuvina annoksina suun kautta, ribosiklibin AUCinf ja Cmax lievää (n = 57) tai keskivaikeaa (n = 14) munuaisten vajaatoimintaa sairastavilla potilailla olivat verrattavissa potilaisiin, joilla munuaisten toiminta oli normaali (n = 86). Tämä viittaa siihen, että lievällä tai keskivaikealla munuaisten vajaatoiminnalla ei ole kliinisesti merkittävää vaikutusta ribosiklibialtistukseen.
Maksan vajaatoiminta
Syöpää sairastamattomilla henkilöillä, joilla oli maksan vajaatoiminta, tehdyn farmakokinetiikan tutkimuksen perusteella lievä maksan vajaatoiminta ei vaikuttanut ribosiklibialtistukseen (ks. kohta Annostus ja antotapa). Ribosiklibin keskialtistus suureni alle 2-kertaiseksi potilailla, joilla oli keskivaikea maksan vajaatoiminta (geometristen keskiarvojen suhde [GMR] Cmax-arvolle 1,44 ja AUCinf-arvolle 1,28) tai vaikea maksan vajaatoiminta (GMR Cmax-arvolle 1,32 ja AUCinf-arvolle 1,29) (ks. kohta Annostus ja antotapa).
Populaatiofarmakokinetiikan analyysissä, johon osallistui 160 edennyttä tai etäpesäkkeistä rintasyöpää sairastavaa, maksatoiminnaltaan normaalia potilasta ja 47 potilasta, joilla oli lievä maksan vajaatoiminta, lievä maksan vajaatoiminta ei vaikuttanut ribosiklibialtistukseen, mikä tukee maksan vajaatoimintaa koskeneen spesifisen tutkimuksen löydöksiä. Ribosiklibia ei ole tutkittu rintasyöpäpotilailla, joilla on keskivaikea tai vaikea maksan vajaatoiminta.
Iän, sukupuolen ja etnisen taustan vaikutus
Populaatiofarmakokinetiikan analyysissä todettiin, että ikä, paino ja sukupuoli eivät vaikuttaneet systeemiseen ribosiklibialtistukseen kliinisesti oleellisella tavalla, joka olisi edellyttänyt annosmuutoksia. Tieto etnisen taustan aiheuttamista farmakokinetiikan eroista ei riitä johtopäätösten tekemiseen.
Pediatriset potilaat
Ribosiklibin farmakokinetiikkaa pediatrisilla potilailla (n=10) ja nuorilla aikuisilla (n=2), joilla oli uusiutunut tai refraktaarinen neuroblastooma tai muita kiinteitä kasvaimia, arvioitiin Kisqalin ja topotekaani + temotsolomidi -yhdistelmän (joko samanaikainen tai peräkkäinen annostelu) vaiheen I/II-tutkimuksen annoshakuvaiheessa (ks. kohta Farmakodynamiikka). Ribosiklibialtistus suureni, kun annos nousi 100:sta 200 mg/m2/päivä, ja potilaiden Tmax-arvo oli 1–4 h. Ribosiklibiannoksella 100 mg/m2/päivä yhdessä topotekaanin ja temotsolomidin samanaikaisen annostelun kanssa ribosiklibin vakaan tilan näennäisen puhdistuman (CL/F) geometrinen keskiarvo oli 35,4 l/h (69 % CV) ja kumulaatiokertoimen geometrinen keskiarvo oli 1,06 (53,9 % CV) (n=5).
In vitro‑yhteisvaikutustiedot
Ribosiklibin vaikutus sytokromi P450 ‑entsyymitoimintaan
Kliinisesti merkitykselliset ribosiklibipitoisuudet estivät palautuvasti CYP1A2-, CYP2E1- ja CYP3A4/5-toimintaa ja estivät aikariippuvaisesti CYP3A4/5-toimintaa in vitro. In vitro ‑arvioinnit viittaavat siihen, että ribosiklibi ei estä CYP2A6-, CYP2B6-, CYP2C8-, CYP2C9-, CYP2C19- eikä CYP2D6-toimintaa kliinisesti merkityksellisinä pitoisuuksina. Ribosiklibilla ei ole potentiaalia estää aikariippuvaisesti CYP1A2-, CYP2C9- eikä CYP2D6-toimintaa.
In vitro ‑tiedot viittaavat siihen, että ribosiklibilla ei ole potentiaalia indusoida UGT-entsyymitoimintaa eikä CYP-isoentsyymien CYP2C9, CYP2C19 ja CYP3A4 toimintaa PXR-välitteisesti. Kisqali ei siis todennäköisesti vaikuta näiden entsyymien substraatteihin. In vitro -tiedot eivät riitä sulkemaan pois ribosiklibin potentiaalia indusoida CYP2B6-toimintaa CAR-välitteisesti.
Kuljettajaproteiinien vaikutus ribosiklibiin
Ribosiklibi on P-gp:n substraatti in vitro, mutta massatase-menetelmällä saatujen tietojen perusteella P-gp:n tai BCRP:n esto ei todennäköisesti vaikuta ribosiklibialtistukseen hoitoannoksia käytettäessä. Ribosiklibi ei ole maksan soluunoton kuljettajaproteiinien OATP1B1, OATP1B3 eikä OCT-1 substraatti in vitro.
Ribosiklibin vaikutus kuljettajaproteiineihin
In vitro ‑arvioinnit viittaavat siihen, että ribosiklibi saattaa estää P-gp-, BRCP-, OATP1B1/1B3-, OCT1-, OCT2-, MATE1- ja BSEP-kuljettajaproteiinien toimintaa. Ribosiklibi ei estänyt OAT1-, OAT3- eikä MRP2-toimintaa kliinisesti merkityksellisinä pitoisuuksina in vitro.
Prekliiniset tiedot turvallisuudesta
Farmakologinen turvallisuus
In vivo ‑sydänturvallisuustutkimuksissa koirilla todettiin annos- ja pitoisuusriippuvaista QTc-ajan pitenemistä altistuksilla, jotka todennäköisesti saavutettaisiin potilaiden elimistöissä 600 mg:n suositusannoksilla. Suuret altistukset (noin 5 kertaa oletettu kliininen Cmax) voivat myös indusoida ennenaikaisia kammiosupistuksia.
Toistuvien annosten toksisuus
Toistuvilla annoksilla tehdyissä toksisuustutkimuksissa (hoitoaikataulu: 3 hoitoviikkoa / 1 tauko‑viikko), jotka kestivät rotalla enintään 27 viikkoa ja koiralla enintään 39 viikkoa, todettiin maksan ja sappiteiden olevan ribosiklibitoksisuuden ensisijainen kohde-elin (proliferaatiomuutokset, kolestaasi, hiekkamaiset saostumat sappirakossa ja sappisaostumien aiheuttama sappitieobstruktio). Toistuvien annosten tutkimuksissa ribosiklibin farmakologiseen vaikutukseen liittyviä kohde-elimiä ovat luuydin (hyposellulaarisuus), lymfaattiset elimet (imukudoskato), suolen limakalvo (atrofia), iho (atrofia), luu (vähentynyt luunmuodostus), munuainen (tubulusten epiteelisolujen samanaikainen degeneraatio ja regeneraatio) ja kivekset (atrofia). Kaikki muutokset lukuun ottamatta kiveksissä todettuja atrofisia muutoksia korjautuivat täysin 4 viikon hoidottoman jakson jälkeen. Kivesten atrofisten muutosten korjautuminen oli trendinomaista. Toksisuustutkimuksissa eläinten ribosiklibialtistus oli (AUC-arvon perusteella) yleensä pienempi tai vastaava kuin toistuvia 600 mg/vrk annoksia saavilla potilailla.
Lisääntymistoksisuus/Hedelmällisyys
Ribosiklibi osoittautui rotalla ja kanilla sikiötoksiseksi ja teratogeeniseksi annoksilla, jotka eivät olleet toksisia emolle. Prenataalisen altistuksen jälkeen rotalla todettiin kiinnittyneiden alkioiden kuolemien lisääntynyttä ilmaantuvuutta ja sikiöiden painon pienentymistä ja kaneilla todettiin teratogeenisuutta, kun AUC-arvon perusteella laskettu altistus oli pienempi kuin ihmisen altistus (rotta) tai 1,5-kertainen verrattuna ihmisen altistukseen (kani) edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla potilailla suurimmilla suositusannoksilla (600 mg/vrk).
Rotalla todettiin sikiöiden painon laskua ja samanaikaisia luustomuutoksia, joiden katsottiin olevan ohimeneviä ja/tai liittyvän sikiöiden pienempään painoon. Kanilla todettiin alkion- ja sikiönkehitykseen kohdistuvia haittavaikutuksia, jotka ilmenivät sikiöanomalioiden ilmaantuvuuden suurenemisena (epämuodostumat ja ulkoiset, viskeraaliset ja luustomuutokset) ja vaikuttivat sikiöiden kasvuun (sikiöiden pienempi paino). Näitä löydöksiä olivat mm. keuhkolohkojen pieneneminen/pieni koko ja lisäverisuoni aortankaaressa ja palleatyrä, puuttuva keuhkojen lobus accessorius tai (osittain) fuusioituneet keuhkolohkot, keuhkojen lobus accessoriuksen pieneneminen/pieni koko (30 ja 60 mg/kg), ylimääräiset/rudimentaariset 13. kylkiluut, epämuotoinen kieliluu ja vähemmän jäseniä peukalossa. Alkio- tai sikiökuolleisuudesta ei todettu näyttöä.
Naarasrotilla tehdyssä hedelmällisyystutkimuksessa ribosiklibi ei vaikuttanut lisääntymistoimintoihin, hedelmällisyyteen eikä alkioiden varhaiskehitykseen, kun annos oli enintään 300 mg/kg/vrk (jolloin altistus on AUC-arvon perusteella laskettuna todennäköisesti pienempi tai sama kuin potilaiden kliininen altistus suurimmilla suositusannoksilla eli 600 mg:n vuorokausiannoksilla).
Ribosiklibia ei ole arvioitu urosten hedelmällisyyttä koskeneissa tutkimuksissa. Rotalla ja koiralla tehdyissä toksisuustutkimuksissa ilmoitettiin kuitenkin atrofisia kivesmuutoksia, kun altistus oli AUC-arvon perusteella laskettuna pienempi tai sama kuin ihmisen kliininen altistus suurimmilla suositelluilla vuorokausiannoksilla eli 600 mg:n vuorokausiannoksilla. Vaikutukset saattavat liittyä kivesten itusolujen proliferaatiota estävään välittömään vaikutukseen, joka johtaa siementiehyiden atrofiaan.
Ribosiklibi ja sen metaboliitit erittyvät herkästi rotan maitoon. Maidon ribosiklibialtistus oli plasman altistusta suurempi.
Genotoksisuus
Genotoksisuustutkimuksissa ei todettu näyttöä siitä, että ribosiklibilla olisi genotoksisuuspotentiaalia, kun asiaa arvioitiin in vitro ‑koeasetelmissa bakteereilla ja in vitro- ja in vivo ‑koeasetelmissa nisäkkäillä joko metabolian aktivaatiota käyttäen tai ilman metabolian aktivaatiota.
Karsinogeenisuus
Ribosiklibin karsinogeenisuutta arvioitiin kaksi vuotta kestäneessä tutkimuksessa rotilla.
Ribosiklibin annostelu suun kautta kahden vuoden ajan aiheutti lisääntyneitä endometriumin epiteelin kasvaimia ja glandulaarista ja levyepiteelissä esiintyvää hyperplasiaa kohdussa/kohdunkaulassa naarasrotilla annoksella ≥300 mg/kg/vrk sekä kilpirauhasten follikulaarisia kasvaimia urosrotilla annoksella 50 mg/kg/vrk. Keskimääräinen altistuminen vakaassa tilassa (AUC0-24h) naaras- ja urosrotilla, joilla todettiin neoplastisia muutoksia oli naarasrotilla 1,2- ja urosrotilla 1,4-kertainen verrattuna siihen, mitä saavutettiin potilailla suositellulla 600 mg/vrk annoksella. Keskimääräinen altistuminen vakaassa tilassa (AUC0-24h) naaras- ja urosrotilla, joilla todettiin neoplastisia muutoksia oli naarasrotilla 2,2- ja urosrotilla 2,5-kertainen verrattuna siihen, mitä saavutettiin potilailla suositellulla 400 mg/vrk annoksella.
Lisäksi havaittiin ei-neoplastisia proliferatiivisia muutoksia, joita olivat lisääntyneet maksan kasvupesäkkeet (basofiilisiä ja kirkassoluisia) annoksella ≥5 mg/kg/vrk sekä kivesten interstitiaalisolujen (Leydig) hyperplasia urosrotilla annoksella 50 mg/kg/vrk.
Urosrottien kilpirauhaslöydösten mekanismiin liittyy todennäköisesti jyrsijäspesifinen mikrosomaalinen entsyymi-induktio maksassa, jolla ei katsota olevan merkitystä ihmisille. Vaikutukset kohtuun/kohdunkaulaan ja kivesten interstitiaalisiin (Leydig) soluihin liittyvät pitkittyneeseen hypoprolaktinemiaan. Tämä on seurausta aivolisäkkeen laktotrofisen solutoiminnan CDK4:n välittämästä estymisestä, mikä muuttaa hypotalamus-aivolisäke-sukupuolirauhasakselia.
Estrogeenin/ progesteronin pitoisuuden mahdollinen suureneminen ihmisillä tällä mekanismilla kompensoituisi yhdistelmänä käytettävän anti-estrogeenihoidon estrogeenisynteesiä estävällä vaikutuksella, sillä ihmisillä Kisqali-valmistetta käytetään yhdistelmänä estrogeenitasoa alentavan lääkityksen kanssa.
Tällä vaikutusmekanismilla ei odoteta olevan vaikutusta ihmisissä, kun otetaan huomioon jyrsijöiden ja ihmisten väliset oleelliset erot liittyen prolaktiinin synteesiin ja rooliin.
Tutkimukset nuorilla eläimillä
Nuorilla rotilla, joille annettiin ribosiklibia suun kautta synnytyksen jälkeisestä päivästä 14 (vastaa ihmisellä 1 vuoden ikää) päivään 63, havaittiin ainoastaan naarailla pieniä, palautuvia reisiluun pituuden lyhenemiä (-3 % – -4 % verrokeihin nähden). Suurin annos, joka ei aiheuta haittavaikutuksia (NOAEL) oli koirailla 150 mg/kg/päivä ja naarailla 150/300 mg/kg/päivä (annoksen nostaminen synnytyksen jälkeisenä päivänä 32). NOAEL‑tasolla todettu altistus oli koirailla 5–22‑kertainen ja naarailla 4–6‑kertainen verrattuna pediatristen potilaiden altistukseen (AUC₀–₂₄h) annoksella 100 mg/m²/vrk.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mikrokiteinen selluloosa
Krospovidoni, tyyppi A
Matalasubstituutioasteinen hydroksipropyyliselluloosa
Magnesiumstearaatti
Vedetön kolloidinen piidioksidi
Kalvopäällyste
Musta rautaoksidi (E172)
Punainen rautaoksidi (E172)
Soijalesitiini (E322)
Polyvinyylialkoholi (osittain hydrolysoitu)
Talkki
Titaanidioksidi (E171)
Ksantaanikumi
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Vnr 10 77 85 ja 53 97 93:1 vuosi.
Vnr 40 87 19 ja 44 98 83: 2 vuotta.
Säilytys
Vnr 10 77 85 ja 53 97 93:
Apteekki: Säilytä jääkaapissa (2 °C – 8 °C) enintään 10 kuukautta.
Potilas: Säilytä alle 25 °C enintään 2 kuukautta. Säilytä alkuperäispakkauksessa.
Vnr 40 87 19 ja 44 98 83:
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
KISQALI tabletti, kalvopäällysteinen
200 mg (L:ei) 42 fol (1683,10 €), 42 fol (1683,10 €), 63 fol (2456,57 €)
PF-selosteen tieto
PVC/PCTFE-materiaalista (polyvinyylikloridi/polyklooritrifluoroetyleeni) tai PA/Al/PVC‑materiaalista (polyamidi/alumiini/polyvinyylikloridi) valmistetut läpipainopakkaukset, joissa 14 tai 21 kalvopäällysteistä tablettia.
Yksikköpakkaukset, joissa 21, 42 tai 63 kalvopäällysteistä tablettia, ja monipakkaukset, joissa 63 (3 pakkausta à 21 kpl), 126 (3 pakkausta à 42 kpl) tai 189 (3 pakkausta à 63 kpl) kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Vaalean harmahtavan violetti, jakouurteeton, pyöreä, pyöristetty, viistoreunainen (läpimitta noin 11,1 mm), toisella puolella kaiverrus ”RIC” ja toisella puolella kaiverrus ”NVR”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
KISQALI tabletti, kalvopäällysteinen
200 mg 42 fol, 42 fol, 63 fol
- Ylempi erityiskorvaus (100 %). Ribosiklibi: Hormonireseptoripositiivisen ja HER2-negatiivisen rintasyövän hoito erityisin edellytyksin (1514).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Ribosiklibi: Hormonireseptoripositiivisen ja HER2- negatiivisen paikallisesti edenneen tai etäpesäkkeisen rintasyövän hoito erityisin edellytyksin (3002).
ATC-koodi
L01EF02
Valmisteyhteenvedon muuttamispäivämäärä
19.03.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com