
TALZENNA kapseli, kova 0,25 mg, 1 mg
Vaikuttavat aineet ja niiden määrät
Talzenna 0,1 mg kovat kapselit
Yksi kova kapseli sisältää talatsoparibitosylaattia määrän, joka vastaa 0,1 mg talatsoparibia.
Talzenna 0,25 mg kovat kapselit
Yksi kova kapseli sisältää talatsoparibitosylaattia määrän, joka vastaa 0,25 mg talatsoparibia.
Talzenna 0,35 mg kovat kapselit
Yksi kova kapseli sisältää talatsoparibitosylaattia määrän, joka vastaa 0,35 mg talatsoparibia.
Talzenna 0,5 mg kovat kapselit
Yksi kova kapseli sisältää talatsoparibitosylaattia määrän, joka vastaa 0,5 mg talatsoparibia.
Talzenna 1 mg kovat kapselit
Yksi kova kapseli sisältää talatsoparibitosylaattia määrän, joka vastaa 1 mg talatsoparibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, kova (kapseli).
Kliiniset tiedot
Käyttöaiheet
Rintasyöpä
Talzenna on tarkoitettu monoterapiana aikuispotilaille, joilla on ituradan BRCA1- tai BRCA2-mutaatio ja HER2-negatiivinen paikallisesti edennyt tai metastasoitunut rintasyöpä. Potilaan tulee olla saanut edeltävästi antrasykliiniä ja/tai taksaania joko (neo)adjuvanttiasetelmassa tai paikallisesti edenneen tai metastasoituneen taudin hoitona, paitsi silloin kun nämä hoidot eivät sovi potilaalle (ks. kohta Farmakodynamiikka). Hormonireseptori (HR) ‑positiivista rintasyöpää sairastavan tulee olla saanut edeltävästi hormonaalista hoitoa, paitsi silloin kun tämä hoito ei sovi potilaalle.
Eturauhassyöpä
Talzenna on tarkoitettu käytettäväksi yhdessä entsalutamidin kanssa metastasoitunutta kastraatioresistenttiä eturauhassyöpää (mCRPC) sairastaville aikuispotilaille, kun solunsalpaajahoito ei ole kliinisesti aiheellista.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Talzenna-hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Potilasvalinta
Rintasyöpä
Talzenna-hoitoon soveltuvuuden tulee perustua rintasyöpäpotilaalla todettuun tunnetusti sairautta aiheuttavaan (patogeeniseen) tai mahdollisesti sairautta aiheuttavaan ituradan BRCA-mutaatioon. BRCA-mutaatio tulee osoittaa validoidulla tutkimusmenetelmällä kokeneessa laboratoriossa.
Potilaalle, jolla on BRCA-mutaatio, tulee antaa perinnöllisyysneuvontaa paikallisia ohjeita noudattaen.
Eturauhassyöpä
Talzenna-hoitoon soveltuvien potilaiden valinta ei edellytä kasvainten mutaatiotestausta hoidettaessa metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavia potilaita.
Annostus
Talzenna monoterapiana (rintasyöpä)
Suositeltu annostus on 1 mg talatsoparibia kerran vuorokaudessa. Hoitoa tulee jatkaa, kunnes tauti etenee tai ilmenee haittavaikutuksia, jotka estävät lääkkeen käytön.
Talzenna yhdessä entsalutamidin kanssa (eturauhassyöpä)
Suositeltu annostus on 0,5 mg talatsoparibia yhdistettynä 160 mg:aan entsalutamidia kerran vuorokaudessa. Hoitoa tulee jatkaa, kunnes tauti etenee tai ilmenee haittavaikutuksia, jotka estävät lääkkeen käytön.
Lääkkeellistä kastraatiota luteinisoivaa hormonia vapauttavan hormonin (LHRH) analogilla on jatkettava hoidon aikana potilailla, joita ei ole kastroitu kirurgisesti.
Katso entsalutamidin valmisteyhteenvedosta suositeltu annostus.
Annoksen jääminen väliin
Jos potilas oksentaa tai unohtaa ottaa Talzenna-annoksen, lisäannosta ei tule ottaa. Seuraava lääkemääräyksen mukainen annos tulee ottaa tavanomaiseen aikaan.
Annoksen muuttaminen
Haittavaikutusten hallinnassa tulee harkita hoidon tilapäistä keskeyttämistä tai annoksen pienentämistä haittavaikutuksen vaikeusasteen ja sen kliinisen ilmenemisen mukaisesti (ks. taulukko 1). Suositukset annoksen pienentämiseksi käytettäessä talatsoparibia monoterapiana (rintasyöpä) ja yhdessä entsalutamidin kanssa (eturauhassyöpä) on esitetty taulukoissa 2 ja 3.
Täydellinen verenkuva tulee määrittää ennen talatsoparibihoidon aloittamista ja sen jälkeen kuukausittain sekä kliinisen tarpeen mukaan (ks. taulukko 1 ja kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukko 1.Annosmuutokset haittavaikutusten vuoksi
| Jatka Talzenna-hoitoa vasta, kun arvo on korjaantunut | Aloita Talzenna-hoito uudestaan | |
| Hemoglobiini < 80 g/l | ≥ 90 g/l | Aloita Talzenna-hoito uudestaan yhtä annostasoa alemmalla annoksella |
| Verihiutalemäärä < 50 x 109/l | ≥ 75 x 109/l | |
| Neutrofiilimäärä < 1,0 x 109/l | ≥ 1,5 x 109/l | |
| Muu kuin hematologinen haittavaikutus, vaikeusaste 3 tai 4 | ≤ vaikeusaste 1 | Harkitse Talzenna-hoidon aloittamista uudestaan yhtä annostasoa alemmalla annoksella tai lopeta hoito |
Taulukko 2.Annoksen pienentäminen käytettäessä talatsoparibia monoterapiana (rintasyöpä)
| Talatsoparibin annostaso (rintasyöpä) | |
| Suositeltu aloitusannos | 1 mg kerran vuorokaudessa |
| Ensimmäinen alennettu annostaso | 0,75 mg kerran vuorokaudessa |
| Toinen alennettu annostaso | 0,5 mg kerran vuorokaudessa |
| Kolmas alennettu annostaso | 0,25 mg kerran vuorokaudessa |
Taulukko 3.Annoksen pienentäminen käytettäessä talatsoparibia yhdessä entsalutamidin kanssa (eturauhassyöpä)
| Talatsoparibin annostaso (eturauhassyöpä) | |
| Suositeltu aloitusannos | 0,5 mg kerran vuorokaudessa |
| Ensimmäinen alennettu annostaso | 0,35 mg kerran vuorokaudessa |
| Toinen alennettu annostaso | 0,25 mg kerran vuorokaudessa |
| Kolmas alennettu annostaso | 0,1 mg kerran vuorokaudessa |
Katso ohjeet annoksen muuttamiseen entsalutamidin valmisteyhteenvedosta, jos ilmenee entsalutamidiin liitettävissä olevia haittavaikutuksia.
0,1 mg:n kapselin käyttötarkoitus on tukea annosmuutoksia eikä se ole vaihdettavissa muiden vahvuuksien kanssa.
Samanaikainen hoito P‑glykoproteiinin (P‑gp) estäjillä
Talzenna monoterapiana (rintasyöpä)
Voimakkaat P‑gp:n estäjät saattavat suurentaa talatsoparibialtistusta. Voimakkaiden P‑gp:n estäjien samanaikaista käyttöä talatsoparibihoidon aikana tulee välttää. Samanaikaista antoa tulee harkita vasta mahdollisten hyötyjen ja riskien huolellisen arvioinnin jälkeen. Jos voimakkaan P‑gp:n estäjän samanaikaista antoa ei voida välttää, Talzennan annos tulee pienentää yhtä annostasoa alempaan annokseen. Kun voimakkaan P‑gp:n estäjän käyttö lopetetaan, Talzennan annos tulee suurentaa (P‑gp:n estäjän 3–5 puoliintumisajan jälkeen) annokseen, jota potilas sai ennen voimakkaan P‑gp:n estäjän käytön aloittamista (ks. kohta Yhteisvaikutukset).
Talzenna yhdessä entsalutamidin kanssa (eturauhassyöpä)
P‑gp:n estäjien samanaikaisen annon vaikutusta talatsoparibialtistukseen annettaessa talatsoparibia yhdessä entsalutamidin kanssa ei ole tutkittu. Tästä syystä P‑gp:n estäjien samanaikaista käyttöä talatsoparibihoidon aikana tulee välttää (ks. kohta Yhteisvaikutukset).
Erityisryhmät
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä maksan vajaatoiminta (bilirubiini ≤ 1 × viitevälin yläraja-arvo [upper limit of normal, ULN] ja ASAT [aspartaattiaminotransferaasi] > ULN tai bilirubiini > 1,0–1,5 × ULN ja mikä tahansa ASAT-arvo), keskivaikea maksan vajaatoiminta (bilirubiini > 1,5–3,0 × ULN ja mikä tahansa ASAT-arvo) tai vaikea maksan vajaatoiminta (bilirubiini > 3,0 × ULN ja mikä tahansa ASAT-arvo) (ks. kohta Farmakokinetiikka). Talzenna-valmisteen käyttöä yhdessä entsalutamidin kanssa ei suositella potilaille, joilla on vaikea maksan vajaatoiminta (Child-Pugh-luokitus C), koska näillä potilailla farmakokinetiikkaa ja turvallisuutta ei ole osoitettu (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Rintasyöpä
Annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä munuaisten vajaatoiminta (kreatiniinipuhdistuma [CrCl] 60−89 ml/min). Jos potilaalla on keskivaikea munuaisten vajaatoiminta (CrCl 30−59 ml/min), suositeltu Talzennan aloitusannos on 0,75 mg kerran vuorokaudessa. Jos potilaalla on vaikea munuaisten vajaatoiminta (CrCl 15–29 ml/min), suositeltu Talzennan aloitusannos on 0,5 mg kerran vuorokaudessa. Talzenna-hoitoa ei ole tutkittu potilailla, joiden CrCl on alle 15 ml/min tai jotka tarvitsevat hemodialyysia (ks. kohta Farmakokinetiikka).
Eturauhassyöpä
Annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä munuaisten vajaatoiminta (kreatiniinipuhdistuma [CrCl] 60−89 ml/min). Jos potilaalla on keskivaikea munuaisten vajaatoiminta (CrCl 30−59 ml/min), suositeltu Talzennan annos on 0,35 mg kerran vuorokaudessa yhdessä kerran vuorokaudessa suun kautta otettavan entsalutamidin kanssa. Jos potilaalla on vaikea munuaisten vajaatoiminta (CrCl 15–29 ml/min), suositeltu Talzennan annos on 0,25 mg kerran vuorokaudessa yhdessä kerran vuorokaudessa otettavan entsalutamidin kanssa. Talzenna-hoitoa ei ole tutkittu potilailla, joiden CrCl on alle 15 ml/min tai jotka tarvitsevat hemodialyysia (ks. kohta Farmakokinetiikka).
Iäkkäät
Annoksen muuttaminen ei ole tarpeen iäkkäiden ( ≥ 65-vuotiaiden) potilaiden hoidossa (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Talzennan turvallisuutta ja tehoa lasten ja nuorten (< 18-vuotiaiden) hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Talzenna otetaan suun kautta. Kosketusta kapselin sisällön kanssa on vältettävä, joten kapselit on nieltävä kokonaisina eikä niitä saa avata tai liuottaa. Kapselit voidaan ottaa joko ruoan kanssa tai tyhjään mahaan (ks. kohta Farmakokinetiikka).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Imetys (ks. kohta Raskaus ja imetys).
Varoitukset ja käyttöön liittyvät varotoimet
Myelosuppressio
Talatsoparibilla hoidetuilla potilailla on raportoitu myelosuppressiivisia vaikutuksia, mukaan lukien anemiaa, leukopeniaa/neutropeniaa ja/tai trombosytopeniaa (ks. kohta Haittavaikutukset). Talatsoparibihoitoa ei saa aloittaa ennen kuin potilas on toipunut aiemman hoidon aiheuttamasta hematologisesta toksisuudesta (vaikeusasteelle ≤ 1).
Talatsoparibia saavan potilaan veriarvoja tulee seurata rutiininomaisesti ja potilasta tulee tarkkailla anemiaan, leukopeniaan/neutropeniaan ja/tai trombosytopeniaan liittyvien merkkien ja oireiden varalta. Jos näihin viittaavia laboratorioarvojen muutoksia ilmenee, suositellaan annostuksen muuttamista (annoksen pienentämistä tai hoidon tilapäistä keskeyttämistä) (ks. kohta Annostus ja antotapa). Tukihoitoja, kuten veren‑ ja/tai verihiutaleiden siirtoja ja/tai verisolukasvutekijöiden (colony stimulating factor, CSF) antoa, voidaan käyttää tarpeen mukaan.
Myelodysplastinen oireyhtymä / akuutti myelooinen leukemia
Myelodysplastista oireyhtymää / akuuttia myelooista leukemiaa (MDS/AML) on raportoitu potilailla, jotka saivat poly(adenosiinidifosfaatti-riboosi)-polymeraasin (PARP) estäjiä, mukaan lukien talatsoparibia. Kliinisissä tutkimuksissa talatsoparibilla hoidetuista potilaista MDS:ä/AML:a on raportoitu kaiken kaikkiaan < 1 %:lla potilaista, joilla oli kiinteitä kasvaimia (ks. kohta Haittavaikutukset). MDS:n/AML:n kehittymiseen mahdollisesti myötävaikuttavia tekijöitä ovat aiempi platinaa sisältävä solunsalpaajahoito, muut DNA:ta vaurioittavat aineet tai sädehoito. Täydellinen verenkuva on määritettävä lähtötilanteessa ja hoidon aikana kuukausittain hematologisen toksisuuden merkkien varalta. Jos potilaalla todetaan MDS/AML, talatsoparibihoito tulee lopettaa.
Laskimotromboemboliset tapahtumat
Metastasoitunutta kastraatioresistenttiä eturauhassyöpää (mCRPC) sairastavilla potilailla laskimotromboembolisten tapahtumien esiintyvyys oli suurempi käytettäessä Talzennan ja entsalutamidin yhdistelmähoitoa verrattuna pelkän entsalutamidin käyttöön. Potilaita on seurattava syvän laskimotromboosin ja keuhkoembolian kliinisten merkkien ja oireiden varalta ja hoidettava lääketieteellisesti asianmukaisella tavalla (ks. kohta Haittavaikutukset).
Raskaudenehkäisy naisilla, jotka voivat tulla raskaaksi
Talatsoparibi oli klastogeeninen ihmisen ääreisverenkierron lymfosyyteillä in vitro tehdyssä kromosomipoikkeavuustestissä sekä rottien in vivo luuytimen mikrotumatestissä, mutta se ei ollut mutageeninen Amesin testissä (ks. kohta Prekliiniset tiedot turvallisuudesta). Raskaana olevalle naiselle annettu talatsoparibi voi vahingoittaa sikiötä, ja siten raskaana olevalle on kerrottava sikiölle mahdollisesti aiheutuvasta riskistä (ks. kohta Raskaus ja imetys). Naiset, jotka voivat tulla raskaaksi, eivät saa tulla raskaaksi Talzenna-hoidon aikana eivätkä he saa olla raskaana hoitoa aloitettaessa. Kaikille naisille, jotka voivat tulla raskaaksi, tulee tehdä raskaustesti ennen hoidon aloittamista.
Naispotilaiden on käytettävä erittäin tehokasta ehkäisymenetelmää Talzenna-hoidon aikana ja vähintään 7 kuukauden ajan hoidon päättymisen jälkeen. Koska hormonaalista ehkäisyä ei suositella rintasyöpäpotilaille, tulee käyttää kahta ei-hormonaalista ja toisiaan täydentävää ehkäisymenetelmää (ks. kohta Raskaus ja imetys).
Miespotilaita, joiden kumppani voi tulla raskaaksi tai joiden kumppani on raskaana, on kehotettava käyttämään tehokasta raskaudenehkäisyä (myös vasektomian jälkeen) Talzenna-hoidon aikana ja vähintään 4 kuukauden ajan viimeisen annoksen ottamisen jälkeen.
Yhteisvaikutukset
Talatsoparibi on lääkkeiden kuljettajina toimivien P-glykoproteiinin (P‑gp) ja rintasyövän resistenssiproteiinin (breast cancer resistance protein, BCRP) substraatti, joka eliminoituu muuttumattomana pääasiassa munuaisten kautta.
Aineita, jotka voivat vaikuttaa talatsoparibin plasmapitoisuuteen
P‑gp:n estäjät
Entsalutamidin vaikutus
Samanaikainen anto 160 mg:n entsalutamidiannoksen kanssa suurentaa talatsoparibialtistuksen noin kaksinkertaiseksi. Kun talatsoparibia annetaan 0,5 mg vuorokaudessa yhdessä entsalutamidin kanssa, saavutetaan suunnilleen sama vakaan tilan jäännöspitoisuus (Ctrough), joka on raportoitu käytettäessä talatsoparibia 1 mg vuorokaudessa (ks. kohta Farmakokinetiikka). Kun Talzenna-valmistetta annetaan yhdessä entsalutamidin kanssa, Talzennan aloitusannos on 0,5 mg (ks. kohta Annostus ja antotapa). Muiden kuin 160 mg:n entsalutamidiannosten yhteisvaikutusta talatsoparibin kanssa ei ole määritetty.
Muiden P‑gp:n estäjien samanaikaisen annon vaikutusta talatsoparibialtistukseen annettaessa talatsoparibia yhdessä entsalutamidin kanssa ei ole tutkittu. Jos P‑gp:n estäjien samanaikaista antoa ei voida välttää annettaessa Talzenna-valmistetta yhdessä entsalutamidin kanssa, potilasta on seurattava haittavaikutusten mahdollisen lisääntymisen havaitsemiseksi.
Muiden P‑gp:n estäjien vaikutus
Tiedot lääkeaineiden yhteisvaikutustutkimuksesta, johon osallistuneilla potilailla oli edenneitä kiinteitä kasvaimia, osoittivat, että P‑gp:n estäjän itrakonatsolin (100 mg kahdesti vuorokaudessa) anto toistuvina päivittäisinä annoksina yhdessä talatsoparibin 0,5 mg:n kerta-annoksen kanssa suurensi talatsoparibin kokonaisaltistusta (AUCinf) noin 56 % ja huippupitoisuutta (Cmax) noin 40 % verrattuna pelkän talatsoparibin antoon 0,5 mg:n kerta-annoksena. Myös populaatiofarmakokineettinen analyysi on osoittanut, että voimakkaiden P‑gp:n estäjien samanaikainen anto suurensi talatsoparibialtistusta 45 % verrattuna yksinään annettuun talatsoparibiin.
Muun muassa seuraavien voimakkaiden P‑gp:n estäjien samanaikaista käyttöä on vältettävä: amiodaroni, karvediloli, klaritromysiini, kobisistaatti, darunaviiri, dronedaroni, erytromysiini, indinaviiri, itrakonatsoli, ketokonatsoli, lapatinibi, lopinaviiri, propafenoni, kinidiini, ranolatsiini, ritonaviiri, sakinaviiri, telapreviiri, tipranaviiri ja verapamiili. Jos voimakkaan P‑gp:n estäjän samanaikaista antoa ei voida välttää, Talzennan annosta tulee pienentää (ks. kohta Annostus ja antotapa).
P‑gp:tä indusoivat aineet
Tiedot lääkeaineiden yhteisvaikutustutkimuksesta, johon osallistuneilla potilailla oli edenneitä kiinteitä kasvaimia, osoittivat, että talatsoparibin 1 mg:n kerta-annoksen ja P‑gp:tä indusoivien aineiden toistuvien päivittäisten annosten samanaikainen anto (600 mg rifampisiinia annettuna 30 minuuttia ennen talatsoparibia talatsoparibin antopäivänä) suurensi talatsoparibin Cmax-arvoa noin 37 %, mutta AUCinf pysyi muuttumattomana verrattuna pelkän talatsoparibin antoon 1 mg:n kerta-annoksena. Tämä selittyy todennäköisesti rifampisiinin aiheuttaman P-gp:n induktion ja eston nettovaikutuksesta yhteisvaikutustutkimuksen koeolosuhteissa. Talatsoparibin annosta ei tarvitse muuttaa annettaessa sitä samanaikaisesti rifampisiinin kanssa. Muiden P‑gp:tä indusoivien aineiden vaikutusta talatsoparibialtistukseen ei ole kuitenkaan tutkittu. Muut P‑gp:tä indusoivat aineet (mm. karbamatsepiini, fenytoiini ja mäkikuisma) saattavat pienentää talatsoparibialtistusta.
BCRP:n estäjät
BCRP:n estäjien vaikutusta talatsoparibin farmakokinetiikkaan ei ole tutkittu in vivo. Talatsoparibin ja BCRP:n estäjien samanaikainen anto saattaa suurentaa talatsoparibialtistusta. Voimakkaiden BCRP:n estäjien (mm. kurkumiini ja siklosporiini) samanaikaista antoa on vältettävä. Jos voimakkaiden BCRP:n estäjien samanaikaista antoa ei voida välttää, potilasta tulee seurata haittavaikutusten mahdollisen lisääntymisen varalta.
Mahahapon eritystä vähentävien aineiden vaikutus
Populaatiofarmakokineettisen analyysin mukaan mahahapon eritystä vähentävien aineiden, mukaan lukien protonipumpun estäjien ja histamiini (H2) ‑reseptorin salpaajien, tai muiden mahahapon eritystä vähentävien aineiden samanaikainen anto ei vaikuttanut talatsoparibin imeytymiseen merkittävästi.
Systeeminen hormonaalinen ehkäisy
Talatsoparibin ja suun kautta otettavien ehkäisyvalmisteiden yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Naisten ja miesten raskaudenehkäisy
Naiset, jotka voivat tulla raskaaksi, eivät saa tulla raskaaksi Talzenna-hoidon aikana eivätkä he saa olla raskaana hoitoa aloitettaessa. Kaikille naisille, jotka voivat tulla raskaaksi, tulee tehdä raskaustesti ennen hoidon aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisymenetelmää (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) ennen talatsoparibihoidon aloittamista, hoidon aikana ja 7 kuukauden ajan talatsoparibihoidon päättymisen jälkeen. Koska hormonaalista ehkäisyä ei suositella rintasyöpäpotilaille, tulee käyttää kahta ei-hormonaalista ja toisiaan täydentävää ehkäisymenetelmää. Miespotilaita, joiden kumppani voi tulla raskaaksi tai joiden kumppani on raskaana, on kehotettava käyttämään tehokasta raskaudenehkäisyä (myös vasektomian jälkeen) Talzenna-hoidon aikana ja vähintään 4 kuukauden ajan viimeisen annoksen ottamisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Talzenna-valmisteen käytöstä raskaana olevien naisten hoidossa ei ole olemassa tietoja. Eläinkokeissa on osoitettu alkio‑ ja sikiötoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Talzenna voi vahingoittaa sikiötä, jos sitä annetaan raskaana olevalle naiselle. Talzenna-valmisteen käyttöä ei suositella raskauden aikana eikä naisille, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Imetys
Ei tiedetä, erittyykö talatsoparibi ihmisen rintamaitoon. Rintaruokituille lapsille aiheutuvaa riskiä ei voida sulkea pois, ja siksi rintaruokinta on vasta-aiheista (ks. kohta Vasta-aiheet) Talzenna-hoidon aikana ja vähintään 1 kuukauden ajan viimeisen annoksen ottamisen jälkeen.
Hedelmällisyys
Vaikutuksista potilaiden hedelmällisyyteen ei ole tietoa. Prekliinisten tutkimusten perusteella (kiveksiin kohdistuvat vaikutukset osittain palautuvia ja munasarjoihin kohdistuvat vaikutukset palautuvia) Talzenna saattaa heikentää lisääntymiskykyisten miesten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Talzenna-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Talatsoparibin annon jälkeen voi ilmetä väsymystä/voimattomuutta tai heitehuimausta.
Kun Talzenna-valmistetta käytetään yhdessä entsalutamidin kanssa, katso myös entsalutamidin valmisteyhteenvedosta tiedot entsalutamidin vaikutuksista ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Talzenna-valmisteen yleinen turvallisuusprofiili perustuu yhdistettyihin tietoihin 1 088 potilaasta, mukaan lukien 690 potilasta, jotka saivat talatsoparibia monoterapiana 1 mg:n vuorokaudessa kiinteiden kasvainten kliinisissä tutkimuksissa, ja 398 metastasoitunutta kastraatioresistenttiä eturauhassyöpää (mCRPC) sairastavaa potilasta, jotka saivat 0,5 mg talatsoparibia yhdistettynä 160 mg:aan entsalutamidia TALAPRO‑2-tutkimuksessa.
Näissä kliinisissä tutkimuksissa talatsoparibia saaneiden potilaiden yleisimmät haittavaikutukset (≥ 20 %:lla potilaista) olivat anemia (55,6 %), väsymys (52,5 %), pahoinvointi (35,8 %), neutropenia (30,3 %), trombosytopenia (25,2 %) ja heikentynyt ruokahalu (21,1 %). Yleisimmät talatsoparibiin liittyneet vaikeusasteen ≥ 3 haittavaikutukset (≥ 10 %:lla potilaista) olivat anemia (39,2 %), neutropenia (16,5 %) ja trombosytopenia (11,1 %).
Annostusta muutettiin (annosta pienennettiin tai hoito keskeytettiin tilapäisesti) jonkin haittavaikutuksen vuoksi 58,7 %:lla Talzenna-valmistetta 1 mg:n annoksella monoterapiana saaneista potilaista. Yleisimmät annostuksen muutoksiin johtaneet haittavaikutukset olivat anemia (33,5 %), neutropenia (11,7 %) ja trombosytopenia (9,9 %). Hoito lopetettiin pysyvästi jonkin haittavaikutuksen vuoksi 2,9 %:lla Talzenna-hoitoa saaneista potilaista; yleisin syy oli anemia (0,6 %). Altistuksen keston mediaani oli 5,6 kuukautta (vaihteluväli 0,0–70,2 kuukautta).
Talzennan anto keskeytettiin haittavaikutusten vuoksi 62,1 %:lla metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavista potilasta, jotka saivat Talzenna-valmistetta yhdessä entsalutamidin kanssa; yleisin syy oli anemia (44 %). Talzenna-annosta pienennettiin haittavaikutusten vuoksi 52,8 %:lla potilaista; yleisin syy oli anemia (43,2 %). Talzenna-hoito lopetettiin pysyvästi haittavaikutusten vuoksi 18,8 %:lla potilaista; yleisin syy oli anemia (8,3%). Talatsoparibialtistuksen keston mediaani oli 86 viikkoa (vaihteluväli 0,29–186,14).
Haittavaikutustaulukko
Taulukossa 4 on esitetty haittavaikutusten yhteenveto perustuen yhdistettyihin tietoihin kliinisistä tutkimuksista. Haittavaikutukset on lueteltu elinjärjestelmien ja esiintymistiheyksien mukaan. Esiintymistiheysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10) ja melko harvinainen (≥ 1/1 000, < 1/100). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutusten vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 4.Haittavaikutukset perustuen 8 tutkimuksen yhdistettyihin tietoihin (n = 1 088)
| Elinjärjestelmä | Kaikki vaikeusasteet | Vaikeusaste 3 | Vaikeusaste 4 |
| Esiintymistiheys | n (%) | n (%) | n (%) |
| Suositeltu termi | |||
| Hyvän- ja pahanlaatuiset sekä luokittelemattomat kasvaimet (mukaan lukien kystat ja polyypit) | |||
| Melko harvinainen | |||
| Myelodysplastinen oireyhtymä / akuutti myelooinen leukemiaa | 2 (0,2) | 1 (< 0,1) | 1 (< 0,1) |
| Veri ja imukudos | |||
| Hyvin yleinen | |||
| Trombosytopeniab | 274 (25,2) | 88 (8,1) | 33 (3,0) |
| Anemiac | 605 (55,6) | 411 (37,8) | 16 (1,5) |
| Neutropeniad | 330 (30,3) | 163 (15,0) | 17 (1,6) |
| Leukopeniae | 195 (17,9) | 52 (4,8) | 2 (0,2) |
| Yleinen | |||
| Lymfopeniaf | 88 (8,1) | 37 (3,4) | 4 (0,4) |
| Aineenvaihdunta ja ravitsemus | |||
| Hyvin yleinen | |||
| Ruokahalun heikkeneminen | 230 (21,1) | 11 (1,0) | 0 (0,0) |
| Hermosto | |||
| Hyvin yleinen | |||
| Heitehuimaus | 157 (14,4) | 4 (0,4) | 1 (< 0,1) |
| Päänsärky | 207 (19,0) | 8 (0,7) | N/A |
| Yleinen | |||
| Makuhäiriö | 68 (6,3) | 0 (0,0) | 0 (0,0) |
| Verisuonisto | |||
| Yleinen | |||
| Laskimon tromboembolia*g | 36 (3,3 %) | 23 (2,1 %) | 2 (0,2 %) |
| Ruoansulatuselimistö | |||
| Hyvin yleinen | |||
| Oksentelu | 167 (15,3) | 9 (0,8) | 0 (0,0) |
| Ripuli | 205 (18,8) | 4 (0,4) | 0 (0,0) |
| Pahoinvointi | 389 (35,8) | 10 (0,9) | N/A |
| Vatsakipuh | 162 (14,9) | 12 (1,1) | N/A |
| Yleinen | |||
| Suutulehdus | 54 (5,0) | 0 (0,0) | 0 (0,0) |
| Dyspepsia | 69 (6,3) | 0 (0,0) | N/A |
| Iho ja ihonalainen kudos | |||
| Hyvin yleinen | |||
| Alopesia | 189 (17,4) | N/A | N/A |
| Yleisoireet ja antopaikassa todettavat haitat | |||
| Hyvin yleinen | |||
| Väsymysi | 571 (52,5) | 58 (5,3) | N/A |
Lyhenteet: n = potilaiden lukumäärä, N/A = ei sovellu. * Vaikeusasteen 5 haittavaikutuksia raportoitiin. a. Katso myös kohta Varoitukset ja käyttöön liittyvät varotoimet. b. Sisältää seuraavat suositellut termit: trombosytopenia ja verihiutalemäärän väheneminen. c. Sisältää seuraavat suositellut termit: anemia, hematokriitin lasku, hemoglobiinin lasku ja punasolumäärän väheneminen. d. Sisältää seuraavat suositellut termit: neutropenia ja neutrofiilimäärän väheneminen. e. Sisältää seuraavat suositellut termit: leukopenia ja valkosolumäärän väheneminen. f. Sisältää seuraavat suositellut termit: lymfosyyttimäärän väheneminen ja lymfopenia. g. Sisältää seuraavat suositellut termit: keuhkoembolia, syvä laskimotromboosi, laskimoembolia ja laskimotromboosi. Ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet. h. Sisältää seuraavat suositellut termit: vatsakipu, ylävatsakipu, epämiellyttävät tuntemukset vatsassa ja alavatsakipu. i. Sisältää seuraavat suositellut termit: väsymys ja voimattomuus. | |||
Valikoitujen haittavaikutusten kuvaus
Myelosuppressio
Myelosuppressioon liittyviä haittavaikutuksia (anemia, neutropenia ja trombosytopenia) raportoitiin hyvin yleisesti potilailla, joita hoidettiin talatsoparibilla. Myelosuppressioon liittyviä tapahtumia (vaikeusaste 3/vaikeusaste 4) raportoitiin (%:lla potilaista) seuraavasti: anemia 37,8 %/1,5 %, neutropenia 15,0 %/1,6 % ja trombosytopenia 8,1 %/3,0 %. Yhtään kuolemantapausta ei raportoitu myelosuppressioon liittyvien haittavaikutusten yhteydessä.
Monoterapiatutkimuksissa (1 mg/vrk saaneet potilaat) yleisimpiä myelosuppressioon liittyviä haittatapahtumia, jotka johtivat annostuksen muutoksiin, olivat anemia (33,5 %), neutropenia (11,7 %) ja trombosytopenia (9,9 %), joita raportoitiin enintään noin 30 %:lla talatsoparibia 1 mg/vrk saaneista potilaista. Tutkimuslääkkeen käytön pysyvään lopettamiseen johti anemia, jota raportoitiin 0,6 %:lla potilaista.
Metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavilla potilailla, jotka saivat talatsoparibia yhdessä entsalutamidin kanssa, anemia johti talatsoparibin annon keskeyttämiseen 44,0 %:lla potilaista, neutrofiilimäärän väheneminen 13,6 %:lla ja verihiutalemäärän väheneminen 7,8 %:lla. Kaiken kaikkiaan 42,5 % potilaista tarvitsi verensiirtoja. Yleisin verensiirto oli punasolutiiviste (39,2 %). Hoidon keskeytti anemian vuoksi 8,3 %, neutropenian vuoksi 3,3 % ja trombosytopenian vuoksi 0,5 % potilaista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Talatsoparibin yliannostuksesta on vain vähän kokemusta. Haittavaikutuksia ei raportoitu potilaalla, joka otti vahingossa kolmekymmentä 1 mg:n talatsoparibikapselia tutkimuspäivänä 1 ja jolle tehtiin heti mahalaukun tyhjennys. Yliannostuksen oireita ei ole varmuudella osoitettu. Yliannostustapauksessa talatsoparibihoito on lopetettava ja lääkärin on harkittava mahalaukun tyhjennystä. Potilaalle tulee antaa yleistä elintoimintoja tukevaa sekä oireenmukaista hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, muut antineoplastiset lääkeaineet, ATC-koodi: L01XK04
Vaikutusmekanismi
Talatsoparibi on poly-ADP-riboosipolymeraasi (PARP) -entsyymien (PARP1 [IC50 = 0,7 nM] ja PARP2 [IC50 = 0,3 nM]) estäjä. PARP-entsyymit osallistuvat solun DNA:n vauriovasteen signaalireitteihin, kuten DNA:n korjaamiseen, geenitranskriptioon ja solukuolemaan. PARP-estäjien syöpäsoluja tuhoava vaikutus perustuu kahteen mekanismiin: PARP:n katalyyttisen aktiivisuuden estoon ja PARP:n vangitsemiseen, jolloin PARP-estäjään sitoutunut PARP-proteiini ei pääse irtoamaan DNA:n vauriokohdasta. Nämä estävät DNA:n korjauksen, kahdentumisen ja transkription, mikä puolestaan johtaa apoptoosiin ja/tai solukuolemaan. Syöpäsolulinjoissa, joissa DNA-vaurioita korjaavien geenien toiminnassa on poikkeavuutta, talatsoparibi yksinään annettuna lisää γH2AX:n (DNA-kaksoiskierteen katkosten merkkiaine) määrää, ja edelleen vähentää solujen proliferaatiota ja lisää apoptoosia. Talatsoparibilla havaittiin kasvainta estävää aktiivisuutta myös BRCA-mutatoituneessa siirrännäiskasvainmallissa, joka oli peräisin aiemmin platinapohjaista hoitoa saaneelta rintasyöpäpotilaalta, ja androgeenireseptoripositiivisen eturauhassyövän siirrännäiskasvainmallissa. Näissä siirrännäiskasvainmalleissa talatsoparibi vähensi kasvaimen kasvua, suurensi γH2AX:n pitoisuutta ja lisäsi apoptoosia.
Yhdistetyn PARP:n ja androgeenireseptorin (AR) aktiivisuuden eston aikaan saama kasvaimen kasvua ehkäisevä vaikutus perustuu seuraaviin mekanismeihin: AR-signaloinnin esto estää homologisen rekombinaation korjausmekanismin (HRR, homologous recombination repair) geenien, mukaan lukien BRCA1:n, ilmentymistä, mistä seuraa herkkyys PARP:n estolle. AR:n maksimaalisen toiminnan on osoitettu edellyttävän PARP1:n aktiivisuutta, joten PARP:n esto voi vähentää AR-signalointia ja lisätä herkkyyttä AR-signaloinnin estäjille. AR:n eston kliiniseen resistenssiin liittyy joskus RB1- ja BRCA2-geenien samanaikainen deleetio, mikä puolestaan liittyy herkkyyteen PARP-estolle.
Sydämen elektrofysiologia
Talatsoparibin vaikutusta sydämen repolarisaatioon arvioitiin 37 potilaalla, joilla oli edenneitä kiinteitä kasvaimia, käyttäen ajan suhteen kaltaistettuja elektrokardiogrammeja. Arviointi tehtiin määrittämällä sykkeen mukaan korjatun QT-ajan (QTc) muutokset lähtötilanteesta suhteessa vastaaviin talatsoparibin pitoisuuksiin plasmassa. Talatsoparibi ei pidentänyt QTc-aikaa kliinisesti merkittävissä määrin käytettäessä monoterapiana suositeltua enimmäisannosta 1 mg kerran vuorokaudessa.
Kliininen teho ja turvallisuus
Ituradan BRCA-mutaatio (gBRCAm) ja HER2-negatiivinen paikallisesti edennyt tai metastasoitunut rintasyöpä
EMBRACA-tutkimus
EMBRACA oli avoin, satunnaistettu kahden hoitohaaran monikeskustutkimus, jossa Talzenna-hoitoa verrattiin solunsalpaajahoitoon (kapesitabiini, eribuliini, gemsitabiini tai vinorelbiini). Tutkimuspotilailla oli ituradan BRCA‑mutaatio ja HER2‑negatiivinen paikallisesti edennyt tai metastasoitunut rintasyöpä, ja potilaat olivat saaneet edeltävästi enintään kolme solunsalpaajahoitolinjaa metastasoituneen tai paikallisesti edenneen taudin hoitoon. Edeltäviin hoitoihin tuli sisältyä antrasykliini ja/tai taksaani neoadjuvantti‑, adjuvantti‑ ja/tai metastasoituneessa asetelmassa, elleivät nämä hoidot olleet vasta-aiheisia. Jos potilas oli saanut platinapohjaista hoitoa edenneen rintasyövän hoitoon, merkkejä taudin etenemisestä tämän hoidon aikana ei sallittu. Aiempaa PARP-estäjähoitoa ei sallittu.
EMBRACA-tutkimukseen satunnaistetuista 431 potilaasta 408 potilaalla (95 %) vahvistettiin tautia aiheuttava tai mahdollisesti tautia aiheuttava ituradan BRCA-mutaatio keskuslaboratoriossa tehdyssä kliinisen tutkimuksen määrityksessä. Näistä potilaista 354 potilaalla (82 %) vahvistettiin ituradan BRCA-mutaatio BRACAnalysis CDx ‑menetelmällä. BRCA-mutaatiopositiivisten osuus (mutaatio rintasyövän alttiusgeeni 1:n [BRCA1] tai alttiusgeeni 2:n [BRCA2] suhteen) oli samankaltainen hoitohaarojen välillä.
Yhteensä 431 potilasta satunnaistettiin suhteessa 2:1 saamaan joko Talzenna 1 mg:n kapseleita kerran vuorokaudessa tai solunsalpaajahoitoa vakioannoksina, kunnes sairaus eteni tai ilmeni haittavaikutuksia, jotka estivät lääkkeen käytön. EMBRACA-tutkimukseen satunnaistetuista 431 potilaasta 287 potilasta satunnaistettiin Talzenna-haaraan ja 144 potilasta solunsalpaajahaaraan. Satunnaistetut potilaat ositettiin metastasoituneeseen tautiin edeltävästi saatujen solunsalpaajahoitolinjojen lukumäärän (0 vs. 1, 2 tai 3), kasvaimen kolmoisnegatiivisuuden (TNBC [triple-negative breast cancer] vs. muu kuin TNBC) ja raportoitujen keskushermoston metastaasien (kyllä vs. ei) perusteella.
Potilaiden demografiset, lähtötilanteeseen ja tautiin liittyvät tekijät olivat yleisesti samankaltaiset tutkimuksen hoitohaarojen välillä (ks. taulukko 5).
| Taulukko 5. Demografiset, lähtötilanteeseen ja tautiin liittyvät tekijät – EMBRACA‑tutkimus | ||
Talatsoparibi (n = 287) | Solunsalpaaja (n = 144) | |
| Mediaani ikä (vuotta [vaihteluväli]) | 45,0 (27,0–84,0) | 50,0 (24,0–88,0) |
| Ikäluokka (vuotta), n (%) | ||
| < 50 | 182 (63,4 %) | 67 (46,5 %) |
| 50 – < 65 | 78 (27,2 %) | 67 (46,5 %) |
| ≥ 65 | 27 (9,4 %) | 10 (6,9 %) |
| Sukupuoli, n (%) | ||
| Nainen | 283 (98,6 %) | 141 (97,9 %) |
| Mies | 4 (1,4 %) | 3 (2,1 %) |
| Etninen tausta, n (%) | ||
| Aasialainen | 31 (10,8 %) | 16 (11,1 %) |
| Tummaihoinen tai afroamerikkalainen | 12 (4,2 %) | 1 (0,7 %) |
| Valkoihoinen | 192 (66,9 %) | 108 (75,0 %) |
| Muu | 5 (1,7 %) | 1 (0,7 %) |
| Ei raportoitu | 47 (16,4 %) | 18 (12,5 %) |
| ECOG-suorituskykyluokka, n (%) | ||
| 0 | 153 (53,3 %) | 84 (58,3 %) |
| 1 | 127 (44,3 %) | 57 (39,6 %) |
| 2 | 6 (2,1 %) | 2 (1,4 %) |
| Tieto puuttuu | 1 (0,3 %) | 1 (0,7 %) |
| Hormonireseptoristatus, n (%) | ||
| HER2-positiivinen | 0 (0,0 %) | 0 (0,0 %) |
| Kolmoisnegatiivinen | 130 (45,3 %) | 60 (41,7 %) |
| Hormonireseptoripositiivinen (ER‑positiivinen tai PgR‑positiivinen) | 157 (54,7 %) | 84 (58,3 %) |
| BRCA-status keskus‑ tai paikallisessa laboratoriossa tehdyn määrityksen mukaan, n (%) | 287 (100,0 %) | 144 (100,0 %) |
| BRCA1‑mutaatio positiivinen | 133 (46,3 %) | 63 (43,8 %) |
| BRCA2‑mutaatio positiivinen | 154 (53,7 %) | 81 (56,3 %) |
| Aika rintasyövän primaaridiagnoosista edenneen rintasyövän toteamiseen (vuotta) | ||
| n | 286 | 144 |
| Mediaani | 1,9 | 2,7 |
| Minimi; maksimi | 0; 22 | 0; 24 |
| Aikakategoriat rintasyövän primaaridiagnoosista edenneen rintasyövän toteamiseen | ||
| < 12 kuukautta | 108 (37,6 %) | 42 (29,2 %) |
| ≥ 12 kuukautta | 178 (62,0 %) | 102 (70,8 %) |
| Paikallisesti edenneen tai metastasoituneen taudin hoitoon edeltävästi saatujen solunsalpaajahoitolinjojen lukumäärä | ||
| Keskiarvo (keskihajonta) | 0,9 (1,01) | 0,9 (0,89) |
| Mediaani | 1 | 1 |
| Minimi; maksimi | 0; 4 | 0; 3 |
| Potilaat, jotka olivat saaneet edeltävästi solunsalpaajahoitoa paikallisesti edenneen tai metastasoituneen taudin hoitoon, n (%) | ||
| Ei aiempaa solunsalpaajahoitoa | 111 (38,7 %) | 54 (37,5 %) |
| 1 aiempi hoitolinja | 107 (37,3 %) | 54 (37,5 %) |
| 2 aiempaa hoitolinjaa | 57 (19,9 %) | 28 (19,4 %) |
| 3 aiempaa hoitolinjaa | 11 (3,8 %) | 8 (5,6 %) |
| ≥ 4 aiempaa hoitolinjaa | 1 (0,3 %) | 0 (0,0 %) |
| Potilaat, jotka olivat saaneet edeltävästi seuraavia hoitoja, n (%) | ||
| Taksaani | 262 (91,3 %) | 130 (90,3 %) |
| Antrasykliini | 243 (84,7 %) | 115 (79,9 %) |
| Platina | 46 (16,0 %) | 30 (20,8 %) |
Lyhenteet: BRCA = rintasyövän alttiusgeeni, ER = estrogeenireseptori, HER2 = ihmisen epidermaalisen kasvutekijän reseptori 2, n = potilaiden lukumäärä, PgR = progesteronireseptori.
Tehon ensisijainen päätetapahtuma oli taudin etenemisestä vapaa elinaika (progression-free survival, PFS) kiinteiden kasvainten vasteen arviointiin tarkoitettujen RECIST (Response Evaluation Criteria in Solid Tumors) -kriteerien (v. 1.1) mukaisesti. Hoitovasteet arvioi sokkoutettu riippumaton keskitetty arvioijataho (blinded independent central review, BICR). Toissijaiset päätetapahtumat olivat objektiivisen vasteen saaneiden osuus (objective response rate, ORR), kokonaiselinaika (overall survival, OS), turvallisuus ja farmakokinetiikka.
Tutkimus osoitti, että Talzenna-hoito paransi tilastollisesti merkitsevästi taudin etenemisestä vapaata elinaikaa, joka oli tehon ensisijainen päätetapahtuma, verrattuna solunsalpaajahoitoon. Kokonaiselinajassa ei todettu tilastollisesti merkitsevää vaikutusta sen lopullisessa analyysissä. Taulukossa 6 on yhteenveto EMBRACA-tutkimuksen tehoa koskevista tuloksista. Taudin etenemisestä vapaata elinaikaa kuvaavat Kaplan-Meierin käyrät on esitetty kuvassa 1 ja kokonaiselinaikaa kuvaavat käyrät kuvassa 3.
| Taulukko 6. Yhteenveto tehoa koskevista tuloksista – EMBRACA-tutkimus* | ||
| Talatsoparibi | Solunsalpaaja | |
| Taudin etenemisestä vapaa elinaika (PFS) riippumattomasti arvioituna (BICR) | n = 287 | n = 144 |
| Tapahtumat, lukumäärä (%) | 186 (65 %) | 83 (58 %) |
| Mediaani (95 %:n luottamusväli), kk | 8,6 (7,2–9,3) | 5,6 (4,2–6,7) |
| Riskitiheyksien suhdea (95 %:n luottamusväli) | 0,54 (0,41–0,71) | |
| 2‑suuntainen p‑arvob | p < 0,0001 | |
| Kokonaiselinaika (OS) (loppuanalyysi)c | n = 287 | n = 144 |
| Tapahtumat, lukumäärä (%) | 216 (75,3 %) | 108 (75 %) |
| Mediaani (95 %:n luottamusväli), kk | 19,3 (16,6–22,5) | 19,5 (17,4–22,4) |
| Riskitiheyksien suhdea (95 %:n luottamusväli) | 0,85 (0,67–1,07)c | |
| 2‑suuntainen p‑arvob | p = 0,1693 | |
| Objektiiviset vasteet tutkijan arvioimanad,e | n = 219 | n = 114 |
| Vasteen saaneiden osuus (ORR), % (95 %:n luottamusväli) | 62,6 (55,8–69,0) | 27,2 (19,3–36,3) |
| Ristitulosuhde (Odds Ratio) (95 %:n luottamusväli) | 4,99 (2,93–8,83) | |
| 2‑suuntainen p‑arvof | p < 0,0001 | |
| Vasteen kesto tutkijan arvioimanad | n = 137 | n = 31 |
| Mediaani (kvartiiliväli), kk | 5,4 (2,8–11,2) | 3,1 (2,4–6,7) |
Lyhenteet: BICR = sokkoutettu riippumaton keskitetty arvioijataho, n = potilaiden lukumäärä, PARP = poly(adenosiinidifosfaatti-riboosi)-polymeraasi, PFS = taudin etenemisestä vapaa elinaika, RECIST 1.1 = kiinteiden kasvainten vastearviointiin tarkoitetut kriteerit, versio 1.1.
* PFS, ORR ja vasteen kesto perustuvat tiedonkeruun katkaisupäivään 15. syyskuuta 2017 sekä talatsoparibihaaran 13,0 kuukauden (95 %:n luottamusväli: 11,1–18,4) ja solunsalpaajahaaran 7,2 kuukauden (95 %:n luottamusväli: 4,6–11,1) PFS:n seuranta-aikoihin (mediaani). OS perustuu tiedonkeruun katkaisupäivään 30. syyskuuta 2019 sekä talatsoparibihaaran 44,9 kuukauden (95 %:n luottamusväli: 37,9–47,0) ja solunsalpaajahaaran 36,8 kuukauden (95 %:n luottamusväli: 34,3–43,0) seuranta-aikoihin (mediaani).
a. Riskitiheyksien suhde perustui ositettuun Coxin regressiomalliin, jossa hoito oli ainoa kovariaatti (ositustekijät: edeltävien solunsalpaajahoitolinjojen lukumäärä, kasvaimen kolmoisnegatiivisuuden status, aiemmin todettu keskushermoston metastaasi), ja suhteessa solunsalpaajahoitoon riskitiheyksien suhde < 1 suosi talatsoparibia.
b. Ositettu log-rank-testi.
c. Lopullisen OS-analyysin hetkellä 46,3 % talatsoparibihaaraan ja 41,7 % solunsalpaajahaaraan satunnaistetuista potilaista oli saanut tutkimushoidon jälkeistä platinahoitoa ja vastaavasti 4,5 % ja 32,6 % oli saanut tutkimushoidon jälkeistä hoitoa jollakin PARP:n estäjällä.
d. Arvio koski koko tutkimuspopulaatiota (intent-to-treat, ITT) niiden potilaiden osalta, joiden kasvaintauti oli mitattavissa ja jotka saivat objektiivisen vasteen. Täydellisen vasteen saaneiden osuus oli talatsoparibiryhmässä 5,5 % ja solunsalpaajaryhmässä 0 %.
e. RECIST 1.1 ‑kriteerien mukaan, täydellisten tai osittaisten vasteiden vahvistusta ei edellytetty.
f. Ositettu Cochran-Mantel‑Haenszel-testi.
Kuva 1. Taudin etenemisestä vapaata elinaikaa kuvaavat Kaplan-Meierin käyrät – EMBRACA-tutkimus
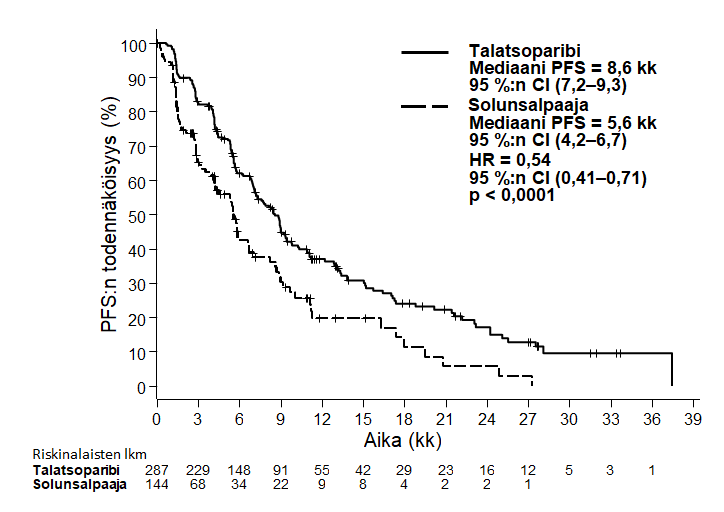
Lyhenteet: CI = luottamusväli, HR = riskitiheyksien suhde, PFS = taudin etenemisestä vapaa elinaika.
Hoitovaikutuksen (PFS) johdonmukaisuutta tutkittiin ennalta määritellyissä alaryhmissä, jotka perustuivat potilaiden ennusteeseen liittyviin tekijöihin ja lähtötilanteen ominaisuuksiin. Taudin etenemisen tai kuoleman riskin pieneneminen todettiin talatsoparibia saaneiden eduksi kaikissa tutkituissa potilasalaryhmissä. Tulokset olivat siten johdonmukaisia koko potilasjoukkoa koskevan tuloksen kanssa (kuva 2).
Kuva 2. Forest plot -kuva keskeisten alaryhmien PFS-analyyseistä – EMBRACA-tutkimus
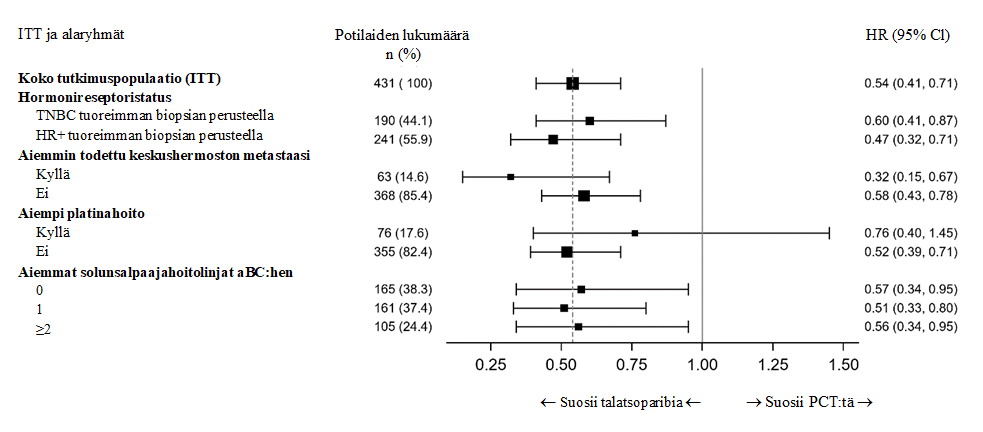
Lyhenteet: ITT = hoitoaikeen mukainen, TNBC = kolmoisnegatiivinen rintasyöpä, HR+ = hormonireseptoripositiivinen, aBC= edennyt rintasyöpä, HR = riskitiheyksien suhde, CI = luottamusväli, PCT = tutkijalääkärin valitsema solunsalpaajahoito.
Kuva 3. Kokonaiselinaikaa kuvaavat Kaplan-Meierin käyrät – EMBRACA-tutkimus
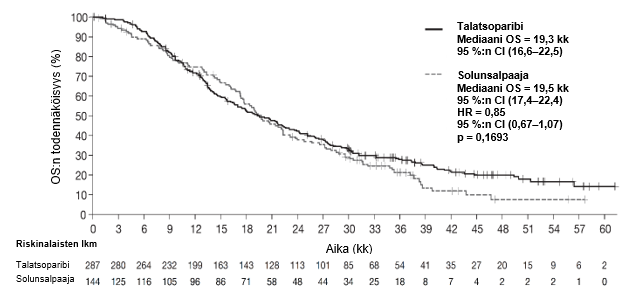
Lyhenteet: CI = luottamusväli, OS = kokonaiselinaika.
Primaarianalyysin p-arvo perustui ositettuun log-rank-testiin.
Metastasoitunut kastraatioresistentti eturauhassyöpä (mCRPC)
TALAPRO‑2-tutkimus
TALAPRO‑2 oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu tutkimus, johon osallistuneet metastasoitunutta kastraatioresistenttiä eturauhassyöpää (mCRPC) sairastavat potilaat (n = 805) satunnaistettiin (1:1) joko saamaan 0,5 mg Talzenna-valmistetta kerran vuorokaudessa yhdistettynä 160 mg:aan entsalutamidia kerran vuorokaudessa tai vertailuhaaraan, jossa potilaat saivat lumelääkettä yhdistettynä 160 mg:aan entsalutamidia kerran vuorokaudessa. Kaikki potilaat saivat gonadotropiinien vapauttajahormonin (GnRH) analogia tai vaihtoehtoisesti heille oli aiemmin tehty bilateraalinen orkiektomia, ja vaatimuksena oli taudin eteneminen aikaisemman androgeenideprivaatiohoidon aikana. Aikaisempi metastasoituneeseen kastraatioherkkään eturauhassyöpään (mCSPC) annettu abirateronihoito tai taksaanipohjainen solunsalpaajahoito oli sallittu.
Satunnaistaminen ositettiin seuraavilla perusteilla: (1) aiempi abirateronihoito tai taksaanipohjainen solunsalpaajahoito vs. ei tällaisia aiempia hoitoja ja (2) HRR-geenien mutaatiostatus, joka testattiin prospektiivisesti uuden sukupolven sekvensoinnilla kasvainkudoksen FoundationOne CDx -menetelmällä tai kiertävän kasvain-DNA:n (ctDNA) FoundationOne Liquid CDx -menetelmällä; potilaat, joilla oli kasvaimen HRR-geenien mutaatioita (ATM, ATR, BRCA1, BRCA2, CDK12, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2 tai RAD51C) vs. potilaat, joilla ei ollut kasvaimen HRR-geenien mutaatioita tai joiden mutaatiostatus ei ollut tiedossa.
Mediaani ikä oli 71 vuotta (vaihteluväli 36–91) kummassakin haarassa; 62 % oli valkoihoisia, 31 % aasialaisia ja 2 % tummaihoisia. Suurimmalla osalla potilaista (66 %) kummassakin haarassa suorituskyvyn ECOG-luokitus oli 0. Talzenna-hoitoa saaneista potilaista niiden potilaiden osuus, joilla oli lähtötilanteessa sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) arvioimana RECIST 1.1 ‑kriteerien mukaan mitattavissa oleva sairaus, oli 30 %. Potilaista 28 % oli aiemmin saanut abirateronihoitoa tai taksaanipohjaista solunsalpaajahoitoa. 20 %:lla oli kasvaimen HRR-geenien mutaatioita, ja 80 %:lla ei ollut kasvaimen HRR-geenien mutaatioita tai geenimutaatiostatus ei ollut tiedossa.
Tehon ensisijainen päätetapahtuma oli radiologisesti todettu taudin etenemisestä vapaa elinaika (rPFS) sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) arvioimana RECIST 1.1 ‑kriteerien ja PCWG3 (Prostate Cancer Clinical Trials Working Group Criteria 3) (luusto) ‑kriteerien mukaan. Kokonaiselinaika (OS) oli alfa-kontrolloitu toissijainen päätetapahtuma.
Talzenna-valmisteen ja entsalutamidin yhdistelmän osoitettiin tilastollisesti merkitsevästi parantavan radiologisesti todettua taudin etenemisestä vapaata elinaikaa (rPFS) ja kokonaiselinaikaa (OS) BICR:n arvioimana verrattuna lumelääkkeen ja entsalutamidin yhdistelmään. Tutkijan arvioiman rPFS:n herkkyysanalyysi oli yhdenmukainen BICR:n arvioiman rPFS:n tulosten kanssa.
Tehoa koskevat tulokset TALAPRO‑2-tutkimuksesta on esitetty taulukoissa 7 ja 8 ja kuvissa 4, 5, 6 ja 7.
| Taulukko 7. Yhteenveto tehoa koskevista tuloksista – TALAPRO‑2 (mCRPC)* | ||
Talatsoparibi + entsalutamidi N = 402 | Lumelääke + entsalutamidi N = 403 | |
| Radiologisesti todettu taudin etenemisestä vapaa elinaika (rPFS) riippumattomasti arvioituna (BICR) | ||
| Tapahtumat, lukumäärä (%) | 151 (37,6) | 191 (47,4) |
| Mediaani (95 %:n luottamusväli), kk | NR (27,5–NR) | 21,9 (16,6–25,1) |
Riskitiheyksien suhde (95 %:n luottamusväli)a p‑arvob | 0,627 (0,506–0,777) p < 0,0001 | |
| Kokonaiselinaika (OS) | ||
| Tapahtumat, lukumäärä (%) | 211 (52,5) | 243 (60,3) |
| Mediaani (95 %:n luottamusväli), kk | 45,8 (39,4–50,8) | 37 (34,1–40,4) |
Riskitiheyksien suhde (95 %:n luottamusväli)a p‑arvob | 0,796 (0,661–0,958) p = 0,0155 | |
Lyhenteet: BICR = sokkoutettu riippumaton keskitetty arvioijataho, HRR (homologous recombination repair) = homologisen rekombinaation korjausmekanismi, mCRPC = metastasoitunut kastraatioresistentti eturauhassyöpä, n = potilaiden lukumäärä, NHT = uusi hormonihoito, NR (not reached) = ei saavutettu.
* rPFS perustuu tiedonkeruun katkaisupäivään 16. elokuuta 2022 sekä talatsoparibi + entsalutamidi ‑haaran 24,9 kuukauden (95 %:n luottamusväli: 24,7–25,3) ja lumelääke + entsalutamidi ‑haaran 24,6 kuukauden (95 %:n luottamusväli: 22,1–24,9) rPFS:n seuranta-aikoihin (mediaani). OS perustuu tiedonkeruun katkaisupäivään 3. syyskuuta 2024 sekä talatsoparibi + entsalutamidi ‑haaran 52,5 kuukauden (95 %:n luottamusväli: 49,9–53,4) ja lumelääke + entsalutamidi ‑haaran 53,0 kuukauden (95 %:n luottamusväli: 50,6–54,0) kokonaiselinajan seuranta-aikoihin (mediaani).
a. Riskitiheyksien suhde perustuu Coxin suhteellisen riskin malliin ositettuna aikaisemman NHT-hoidon (abirateroni) tai kastraatioherkkään metastasoituneeseen eturauhassyöpään annetun taksaanipohjaisen solunsalpaajahoidon (kyllä vs. ei) ja HRR-mutaatiostatuksen (mutaatioita vs. ei mutaatioita / ei tiedossa) mukaan; suhde < 1 puoltaa talatsoparibia.
b. Log-rank-testin p‑arvot (2‑suuntaiset) ositettuina aikaisemman NHT-hoidon (abirateroni) tai kastraatioherkkään eturauhassyöpään annetun taksaanipohjaisen solunsalpaajahoidon ja HRR-mutaatiostatuksen mukaan.
Lopullisen kokonaiselinajan analyysin toteutushetkellä radiologisesti todetun taudin etenemisestä vapaan elinajan (rPFS) mediaani oli 33,1 kuukautta (95 %:n luottamusväli: 27,4–39,0) potilailla, jotka saivat Talzenna-valmistetta yhdistelmänä entsalutamidin kanssa, ja 19,5 kuukautta (95 %:n luottamusväli: 16,6–24,7) potilailla, jotka saivat lumelääkettä yhdistelmänä entsalutamidin kanssa (riskitiheyksien suhde = 0,667, 95 %:n luottamusväli: 0,551–0,807).
| Taulukko 8. Yhteenveto tehoa koskevista tuloksista alaryhmäanalyysien mukaisesti – TALAPRO‑2 (mCRPC)* | ||
| Talatsoparibi + entsalutamidi | Lumelääke + entsalutamidi | |
| HRRm-alaryhmien analyysita | ||
| HRRm | n = 85 | n = 82 |
| Radiologisesti todettu taudin etenemisestä vapaa elinaika (rPFS) riippumattomasti arvioituna (BICR) | ||
| Tapahtumat, lukumäärä (%) | 37 (43,5) | 49 (59,7) |
| Mediaani (95 %:n luottamusväli), kk | 27,9 (16,8–NR) | 13,8 (10,9–19,5) |
| Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,424 (0,275–0,653) | |
| Kokonaiselinaika (OS) | ||
| Tapahtumat, lukumäärä (%) | 41 (48,2) | 55 (67,1) |
| Mediaani (95 %:n luottamusväli), kk | 45,8 (36,4–NR) | 30,1 (25,6–38,2) |
| Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,524 (0,348–0,787) | |
| Ei HRRm | n = 207 | n = 219 |
| Radiologisesti todettu taudin etenemisestä vapaa elinaika (rPFS) riippumattomasti arvioituna (BICR) | ||
| Tapahtumat, lukumäärä (%) | 73 (35,3) | 95 (43,4) |
| Mediaani (95 %:n luottamusväli), kk | NR (25,8–NR) | 22,4 (16,6–NR) |
| Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,695 (0,511–0,944) | |
| Kokonaiselinaika (OS) | ||
| Tapahtumat, lukumäärä (%) | 112 (54,1) | 133 (60,7) |
| Mediaani (95 %:n luottamusväli), kk | 45 (34,7–53,3) | 37,4 (31,8–41,4) |
| Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,817 (0,635–1,053) | |
| BRCAm-alaryhmien analyysita | ||
| BRCAm | n = 27 | n = 32 |
| Radiologisesti todettu taudin etenemisestä vapaa elinaika (rPFS) riippumattomasti arvioituna (BICR) | ||
| Tapahtumat, lukumäärä (%) | 8 (29,6) | 22 (68,7) |
| Mediaani (95 %:n luottamusväli), kk | NR (16,8–NR) | 11 (7,4–24,6) |
| Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,232 (0,101–0,529) | |
| Kokonaiselinaika (OS) | ||
| Tapahtumat, lukumäärä (%) | 14 (51,9) | 23 (71,9) |
| Mediaani (95 %:n luottamusväli), kk | 36,9 (24,9–NR) | 26,1 (15,2–35,4) |
| Riskitiheyksien suhde (95 %:n luottamusväli)b | 0,556 (0,285–1,085) | |
Lyhenteet: BICR = sokkoutettu riippumaton keskitetty arvioijataho, BRCAm = mutaatio rintasyöpäalttiusgeenissä, ctDNA = kiertävä kasvain-DNA, HRRm = mutaatio homologisen rekombinaation korjausmekanismin (HRR) geenissä, mCRPC = metastasoitunut kastraatioresistentti eturauhassyöpä, n = potilaiden lukumäärä, NHT = uusi hormonihoito, NR (not reached) = ei saavutettu.
* rPFS perustuu tiedonkeruun katkaisupäivään 16. elokuuta 2022 sekä talatsoparibi + entsalutamidi ‑haaran 24,9 kuukauden (95 %:n luottamusväli: 24,7–25,3) ja lumelääke + entsalutamidi ‑haaran 24,6 kuukauden (95 %:n luottamusväli: 22,1–24,9) rPFS:n seuranta-aikoihin (mediaani). OS perustuu tiedonkeruun katkaisupäivään 3. syyskuuta 2024 sekä talatsoparibi + entsalutamidi ‑haaran 52,5 kuukauden (95 %:n luottamusväli: 49,9–53,4) ja lumelääke + entsalutamidi ‑haaran 53,0 kuukauden (95 %:n luottamusväli: 50,6–54,0) kokonaiselinajan seuranta-aikoihin (mediaani).
a. Johdettu prospektiivisista kasvainkudoksesta mitatuista tuloksista (tulokset tiedossa ennen satunnaistamista) ja prospektiivisista verestä mitatuista ctDNA-tuloksista (tulokset tiedossa ennen satunnaistamista).
b. Riskitiheyksien suhde perustuu Coxin suhteellisen riskin malliin ositettuna aikaisemman NHT-hoidon (abirateroni) tai kastraatioherkkään eturauhassyöpään annetun solunsalpaajahoidon (kyllä vs. ei) mukaan; suhde < 1 puoltaa talatsoparibia.
Lopullisen kokonaiselinajan analyysin toteutushetkellä HRRm-alaryhmässä radiologisesti todetun taudin etenemisestä vapaan elinajan (rPFS) mediaani oli 27,7 kuukautta (95 %:n luottamusväli: 19,3–38,4) potilailla, jotka saivat Talzenna-valmistetta yhdistelmänä entsalutamidin kanssa, ja 13,8 kuukautta (95 %:n luottamusväli: 10,8–19,3) potilailla, jotka saivat lumelääkettä yhdistelmänä entsalutamidin kanssa (riskitiheyksien suhde = 0,454, 95 %:n luottamusväli: 0,305–0,674); Ei HRRm ‑alaryhmässä rPFS:n mediaani oli 33,2 kuukautta (95 %:n luottamusväli: 25,9–44,2) potilailla, jotka saivat Talzenna-valmistetta yhdistelmänä entsalutamidin kanssa, ja 22,1 kuukautta (95 %:n luottamusväli: 16,6–30,4) potilailla, jotka saivat lumelääkettä yhdistelmänä entsalutamidin kanssa (riskitiheyksien suhde = 0,740, 95 %:n luottamusväli: 0,565–0,969); BRCAm-alaryhmässä rPFS:n mediaania ei saavutettu (95 %:n luottamusväli: 16,8–NR) potilailla, jotka saivat Talzenna-valmistetta yhdistelmänä entsalutamidin kanssa, ja rPFS:n mediaani oli 11 kuukautta (95 %:n luottamusväli: 5,9–13,8) potilailla, jotka saivat lumelääkettä yhdistelmänä entsalutamidin kanssa (riskitiheyksien suhde = 0,259, 95 %:n luottamusväli: 0,120–0,558).
Kuva 4.Radiologisesti todettua taudin etenemisestä vapaata elinaikaa (rPFS) kuvaavat Kaplan-Meierin käyrät riippumattomasti arvioituna (BICR) – TALAPRO‑2 (mCRPC)
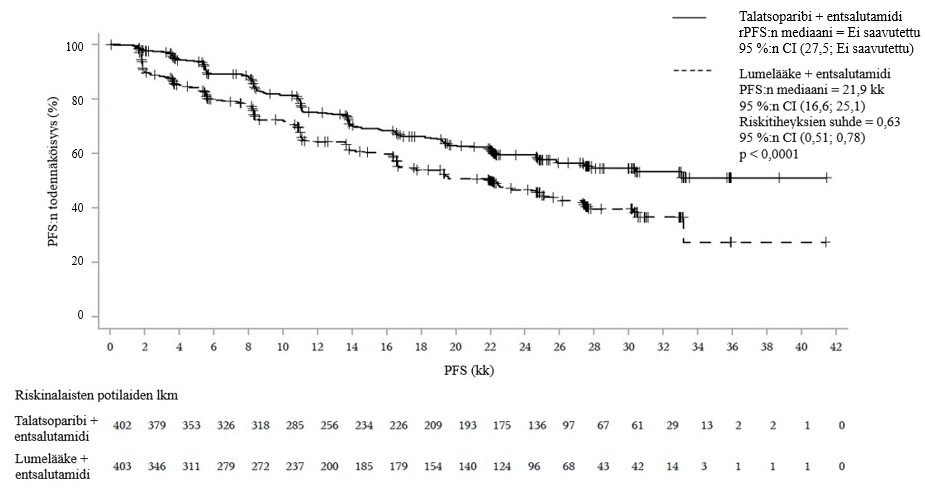
Lyhenteet: BICR = sokkoutettu riippumaton keskitetty arvioijataho, CI = luottamusväli, mCRPC = metastasoitunut kastraatioresistentti eturauhassyöpä, PFS = taudin etenemisestä vapaa elinaika, rPFS = radiologisesti todettu taudin etenemisestä vapaa elinaika.
Kuva 5.Forest plot ‑kuva keskeisten alaryhmien rPFS-analyyseistä – TALAPRO‑2 (mCRPC)
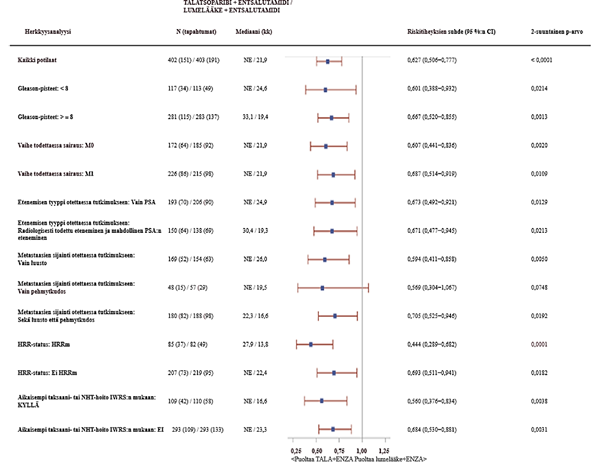
Lyhenteet: CI = luottamusväli, ctDNA = kiertävä kasvain-DNA, ENZA= entsalutamidi, HRR = homologisen rekombinaation korjausmekanismi, HRRm = mutaatio homologisen rekombinaation korjausmekanismin (HRR) geenissä, IWRS (Interactive Web Response System) = interaktiivinen internetpohjainen vastausjärjestelmä, mCRPC = metastasoitunut kastraatioresistentti eturauhassyöpä, N = potilaiden lukumäärä, NE = ei arvioitavissa / ei saavutettu, NHT = uusi hormonihoito, PBO = lumelääke, PSA = prostataspesifinen antigeeni, rPFS = radiologisesti todettu taudin etenemisestä vapaa elinaika, TALA = talatsoparibi.
Kaikkien potilaiden riskitiheyksien suhde perustuu Coxin malliin ositettuna satunnaistamisen ositustekijöillä. Kaikilla alaryhmillä riskitiheyksien suhde perustuu osittamattomaan Coxin maliin, jossa hoito on ainoa kovariaatti. Riskitiheyksien suhde < 1 suosii talatsoparibia.
HRR-status on johdettu prospektiivisista kasvainkudoksesta mitatuista tuloksista ja prospektiivisista verestä mitatuista ctDNA-tuloksista.
Kuva 6.Kokonaiselinaikaa (OS) kuvaavat Kaplan-Meierin käyrät – TALAPRO‑2 (mCRPC)
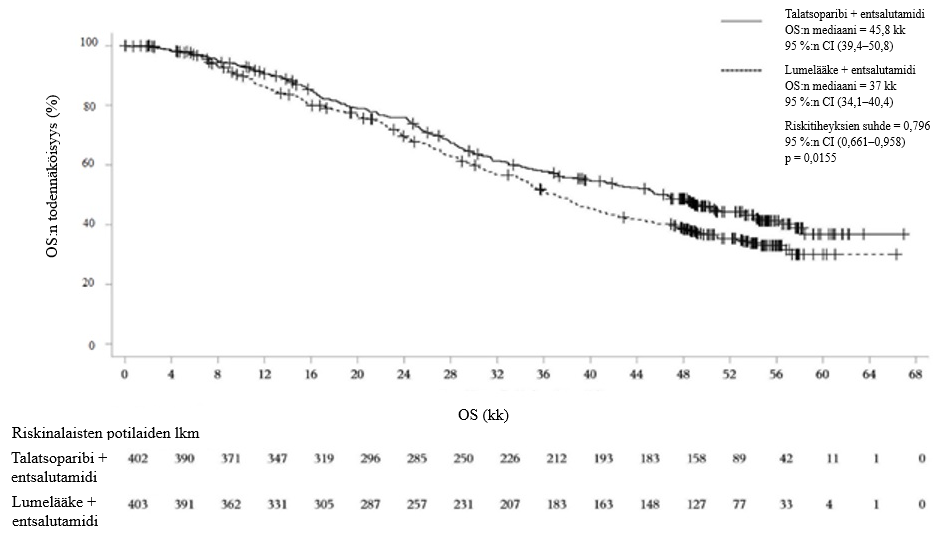
Lyhenteet: CI = luottamusväli; mCRPC = metastasoitunut kastraatioresistentti eturauhassyöpä; OS = kokonaiselinaika.
Kuva 7.Forest plot ‑kuva keskeisten alaryhmien OS-analyyseistä – TALAPRO‑2 (mCRPC)
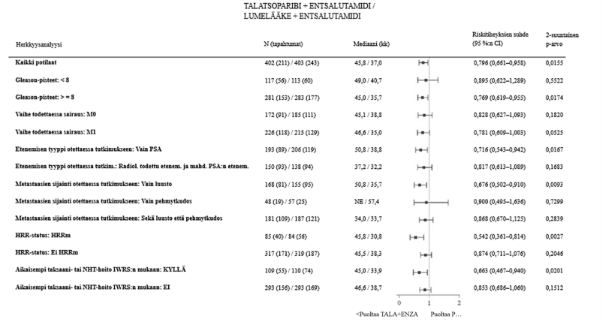
Lyhenteet: CI = luottamusväli; ENZA = entsalutamidi; HRR = homologisen rekombinaation korjausmekanismi, HRRm = mutaatio homologisen rekombinaation korjausmekanismin (HRR) geenissä, IWRS (Interactive Web Response System) = interaktiivinen internetpohjainen vastausjärjestelmä, mCRPC = metastasoitunut kastraatioresistentti eturauhassyöpä, N = potilaiden lukumäärä, NE = ei arvioitavissa / ei saavutettu, NHT = uusi hormonihoito, OS = kokonaiselinaika, PSA = prostataspesifinen antigeeni, TALA = talatsoparibi.
Kaikkien potilaiden riskitiheyksien suhde perustuu Coxin malliin ositettuna satunnaistamisen ositustekijöillä. Kaikilla alaryhmillä riskitiheyksien suhde perustuu osittamattomaan Coxin maliin, jossa hoito on ainoa kovariaatti.
Prosenttiosuudet on laskettu kunkin hoitoryhmän koko analyysijoukon potilaiden lukumäärän (N) perusteella.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset talatsoparibin käytöstä rintasyövän ja eturauhassyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Talatsoparibialtistus suureni yleisesti suhteessa annokseen annettaessa päivittäin toistuvia 0,025–2 mg:n annoksia. Kun rintasyöpäpotilaat saivat monoterapiana toistuvia 1 mg:n talatsoparibiannoksia päivittäin, talatsoparibin vakaassa tilassa mitattu AUC-arvon (plasman pitoisuus-aikakuvaajan alle jäävä pinta-ala) geometrinen keskiarvo (variaatiokerroin [CV] %) oli 126 (107) – 208 (37) ng•h/ml ja huippupitoisuus plasmassa (Cmax) 11 (90) – 19 (27) ng/ml. Kun metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastaville potilaille annettiin talatsoparibia 0,5 mg suun kautta kerran vuorokaudessa yhdessä entsalutamidin kanssa, vakaassa tilassa mitattu Ctrough-arvon geometrinen keskiarvo (CV %) oli eri tutkimuskäynneillä 3,29–3,68 ng/ml (45–48 %). Tämä on yhteneväinen rintasyöpäpotilailla havaitun arvon 3,53 (61 %) ng/ml kanssa, kun rintasyöpäpotilaille annettiin talatsoparibia monoterapiana 1 mg kerran vuorokaudessa. Toistuvalla päivittäisellä annostuksella plasman talatsoparibipitoisuus saavutti vakaan tilan 2–3 viikossa, kun talatsoparibia annettiin monoterapiana, ja noin 9 viikossa, kun talatsoparibia annettiin yhdessä entsalutamidin kanssa. Kun talatsoparibia annettiin toistuvasti monoterapiana 1 mg kerran vuorokaudessa suun kautta, kertymissuhteen mediaani oli 2,3–5,2. Talatsoparibi on P-glykoproteiinin (P-gp) ja rintasyövän resistenssiproteiinin (breast cancer resistance protein, BCRP) kuljettajaproteiinien substraatti.
Imeytyminen
Annettaessa talatsoparibia suun kautta mediaaniaika plasman huippupitoisuuden (Cmax) saavuttamiseen (Tmax) oli yleensä 1–2 tuntia. Absoluuttista biologista hyötyosuutta ihmisellä ei ole tutkittu. Virtsaan erittymistä koskevien tietojen perusteella absoluuttinen biologinen hyötyosuus on kuitenkin vähintään 41 % ja imeytynyt fraktio vähintään 69 % (ks. Eliminaatio). Mahahapon eritystä vähentävien aineiden ei odoteta vaikuttavan talatsoparibialtistukseen merkittävästi, koska talatsoparibin liukoisuus on riittävä pH-arvoilla 1–6,8. Keskeisessä kliinisessä tutkimuksessa 28 % potilaista käytti mahahapon eritystä vähentäviä aineita, pääasiassa protonipumpun estäjiä.
Ruoan vaikutus
Ruoan nauttiminen hidasti talatsoparibin imeytymistä, mutta ei vähentänyt talatsoparibin imeytynyttä määrää. Kun talatsoparibin kerta-annos otettiin suun kautta runsaasti rasvaa ja kaloreja sisältävän ruoan (noin 827 kaloria, 57 % rasvaa) kanssa, talatsoparibin keskimääräinen plasman huippupitoisuus (Cmax) pieneni noin 46 % ja mediaaniaika plasman huippupitoisuuden saavuttamiseen (Tmax) siirtyi 1 tunnista 4 tuntiin, mutta kokonaisaltistus (AUCinf) ei muuttunut. Näiden tulosten perusteella Talzenna-kapselin voi ottaa joko ruoan kanssa tai tyhjään mahaan (ks. kohta Annostus ja antotapa).
Jakautuminen
Talatsoparibin keskimääräinen näennäinen jakautumistilavuus (Vss/F) tutkitussa populaatiossa oli 420 l. Talatsoparibi sitoutuu plasman proteiineihin in vitro noin 74‑prosenttisesti pitoisuudesta riippumatta pitoisuusvälillä 0,01–1 µM. Munuaisten tai maksan vajaatoiminta ei näytä vaikuttavan talatsoparibin sitoutumiseen proteiineihin, sillä sitoutumattoman talatsoparibin osuudessa (fu; keskiarvo) ihmisen plasmassa in vivo ei ilmennyt selkeää muutostrendiä munuaisten tai maksan toiminnan heikentyessä.
Biotransformaatio
Talatsoparibi metaboloituu ihmisillä maksassa hyvin vähäisessä määrin. Kun ihmisille annettiin 1 mg:n kerta-annos [14C]talatsoparibia suun kautta, ei plasmasta todettu yhtään merkittävää kiertävää metaboliittia. Talatsoparibi oli ainoa plasmasta todettu kiertävä lääkeperäinen aine. Virtsasta tai ulosteista ei mitattu yhtään metaboliittia, joka olisi yksinään muodostanut yli 10 % annetusta annoksesta.
Talatsoparibi ei kliinisesti merkityksellisinä pitoisuuksina estänyt sytokromi (CYP) ‑entsyymejä CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ja CYP3A4/5 eikä indusoinut sytokromientsyymejä CYP1A2, CYP2B6 tai CYP3A4 in vitro.
Talatsoparibi ei kliinisesti merkityksellisinä pitoisuuksina estänyt yhtään merkittävää suoliston, maksan tai munuaisten membraanikuljettajaproteiinia (P‑gp, BCRP, orgaanisten anionien kuljettajapolypeptidit [OATP]1B1 ja OATP1B3, orgaanisten kationien kuljettajat [OCT]1 ja OCT2, orgaanisten anionien kuljettajat [OAT]1 ja OAT3, sappisuolapumppu [BSEP], MATE1 ja MATE2-K [multidrug and toxin extrusion]) in vitro.
Talatsoparibi ei kliinisesti merkityksellisinä pitoisuuksina estänyt yhtään merkittävää uridiinidifosfaattiglukuronosyylitransferaasin (UGT) isoformia (1A1, 1A4, 1A6, 1A9, 2B7 ja 2B15) in vitro.
Eliminaatio
Talatsoparibi eliminoituu pääasiassa muuttumattomana munuaisten kautta (passiivinen suodatus ja aktiivinen sekreetio). P‑gp osallistuu todennäköisesti talatsoparibin aktiiviseen sekreetioon munuaisissa. Syöpäpotilailla talatsoparibin terminaalisen puoliintumisajan keskiarvo (± keskihajonta) plasmassa oli 90 (± 58) tuntia ja näennäisen oraalisen puhdistuman (CL/F) populaatiokeskiarvo (yksilöiden välinen vaihtelu) oli 6,5 l/h (31 %). Kun kuudelle naispotilaalle annettiin kerta-annos [14C]talatsoparibia suun kautta, koko annetusta radioaktiivisesta annoksesta mitattiin keskimäärin 69 % (±8,6 %) virtsasta ja 20 % (±5,5 %) ulosteista. Suurin osa talatsoparibista, 55 % annetusta annoksesta, erittyi muuttumattomana virtsaan. Ulosteisiin talatsoparibista erittyi muuttumattomana 14 % annetusta annoksesta.
Erityiset potilasryhmät
Ikä, sukupuoli ja paino
Populaatiofarmakokineettinen analyysi, jonka tarkoituksena oli selvittää iän (vaihteluväli 18–88 vuotta), sukupuolen (53 miestä ja 437 naista) ja painon (vaihteluväli 35,7–162 kg) vaikutusta talatsoparibin farmakokinetiikkaan, toteutettiin 490 syöpäpotilaan tiedoista. Potilaat saivat talatsoparibia 1 mg päivittäin monoterapiana. Tulokset osoittivat, ettei iällä, sukupuolella ja painolla ollut kliinisesti merkittävää vaikutusta talatsoparibin farmakokinetiikkaan.
Etninen tausta
Populaatiofarmakokineettinen analyysi 490 potilaasta, jotka saivat talatsoparibia 1 mg päivittäin monoterapiana, osoitti, että talatsoparibin Cl/F (oraalinen puhdistuma) oli suurempi aasialaisilla kuin muun etnisen taustan potilailla. Aineistossa 41 potilasta oli aasialaisia ja 449 potilasta ei-aasialaisia (361 oli valkoihoisia, 16 tummaihoisia, 9 muita ja 63 potilaan etnistä taustaa ei ollut raportoitu). Aasialaisilla potilailla todettiin ei-aasialaisiin verrattuna 19 % pienempi altistus (AUC).
Pediatriset potilaat
Talatsoparibin farmakokinetiikkaa ei ole tutkittu < 18‑vuotiailla potilailla.
Munuaisten vajaatoiminta
Talatsoparibi monoterapiana
Tiedot farmakokineettisestä tutkimuksesta edennyttä syöpää sairastavilla potilailla, joilla oli todettu eri vaikeusasteen munuaisten vajaatoiminta, osoittivat, että toistuvien kerran päivässä annettujen talatsoparibiannosten jälkeen talatsoparibin kokonaisaltistus (AUC0-24) suureni 92 % keskivaikeaa munuaisten vajaatoimintaa (laskennallinen glomerulussuodosnopeus [eGFR] 30–59 ml/min) sairastavilla ja 169 % vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min) sairastavilla verrattuna potilaisiin, joiden munuaisten toiminta oli normaali (eGFR ≥ 90 ml/min). Talatsoparibin huippupitoisuus (Cmax) suureni 90 % keskivaikeaa ja 107 % vaikeaa munuaisten vajaatoimintaa sairastavilla verrattuna potilaisiin, joiden munuaisten toiminta oli normaali. Talatsoparibialtistus oli samankaltainen potilailla, joilla oli joko lievä munuaisten vajaatoiminta (eGFR 60–89 ml/min) tai joiden munuaisten toiminta oli normaali. Lisäksi populaatiofarmakokineettinen analyysi, jossa oli 490 potilasta, osoitti, että talatsoparibin Cl/F pieneni 14 % lievää ja 37 % keskivaikeaa munuaisten vajaatoimintaa sairastavilla verrattuna potilaisiin, joiden munuaisten toiminta oli normaali (kreatiniinipuhdistuma [CrCl] ≥ 90 ml/min), mikä johti 17 % suurempaan AUC-arvoon lievää munuaisten vajaatoimintaa sairastavilla ja 59 % suurempaan AUC-arvoon keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla. Aineistossa 132 potilaalla oli lievä munuaisten vajaatoiminta (CrCl 60−89 ml/min), 33 potilaalla keskivaikea munuaisten vajaatoiminta (CrCl 30−59 ml/min) ja yhdellä potilaalla vaikea munuaisten vajaatoiminta (CrCl < 30 ml/min). Talatsoparibin farmakokinetiikkaa ei ole tutkittu potilailla, jotka tarvitsevat hemodialyysia (ks. kohta Annostus ja antotapa).
Talatsoparibi yhdessä entsalutamidin kanssa
Populaatiofarmakineettisessa analyysissa oli mukana 412 metastasoitunutta kastraatioresistenttiä eturauhassyöpää sairastavaa potilasta, jotka saivat talatsoparibia yhdessä entsalutamidin kanssa. Aineistossa 152 potilaalla oli lievä munuaisten vajaatoiminta (CrCl 60−89 ml/min), 72 potilaalla keskivaikea munuaisten vajaatoiminta (CrCl 30−59 ml/min) ja 2 potilaalla vaikea munuaisten vajaatoiminta (CrCl < 30 ml/min). Talatsoparibin Cl/F pieneni 8 % lievää ja 27 % keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (vastaa AUC-arvon suurenemista 9 % ja 37 %) verrattuna potilaisiin, joiden munuaisten toiminta oli normaali. Talatsoparibin farmakokinetiikkaa ei ole tutkittu potilailla, jotka tarvitsevat hemodialyysia (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Talatsoparibi monoterapiana
Populaatiofarmakokineettinen analyysi 490 potilaasta, jotka saivat päivittäin 1 mg:n talatsoparibia monoterapiana, osoitti, ettei lievä maksan vajaatoiminta vaikuta talatsoparibin farmakokinetiikkaan. Aineistossa 118 potilaalla oli lievä maksan vajaatoiminta (bilirubiini ≤ 1,0 × viitevälin yläraja-arvo [upper limit of normal, ULN] ja ASAT-arvo > ULN tai bilirubiini > 1,0–1,5 × ULN ja mikä tahansa ASAT-arvo). Talatsoparibin farmakokinetiikkaa selvitettiin myös toisessa farmakokineettisessä tutkimuksessa sekä potilailla, joiden maksan toiminta oli normaali, että potilailla, joilla oli lievä, keskivaikea tai vaikea maksan vajaatoiminta (keskivaikea: bilirubiini > 1,5–3,0 × ULN ja mikä tahansa ASAT-arvo; vaikea: bilirubiini > 3,0 × ULN ja mikä tahansa ASAT-arvo). Populaatiofarmakokineettinen analyysi, jossa käytettiin tästä farmakokineettisestä tutkimuksesta saatuja tietoja, osoitti, ettei lievällä, keskivaikealla tai vaikealla maksan vajaatoiminnalla ollut merkittävää vaikutusta talatsoparibin farmakokinetiikkaan (ks. kohta Annostus ja antotapa).
Talatsoparibi yhdessä entsalutamidin kanssa
Talatsoparibin farmakokinetiikkaa, kun se annetaan yhdessä entsalutamidin kanssa, ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Karsinogeenisuus
Talatsoparibilla ei ole tehty karsinogeenisuustutkimuksia.
Genotoksisuus
Talatsoparibi ei ollut mutageeninen bakteerien käänteismutaatiotestissä (Amesin testi). Talatsoparibi oli klastogeeninen ihmisen ääreisverenkierron lymfosyyteillä in vitro tehdyssä kromosomipoikkeavuustestissä sekä rotilla in vivo tehdyssä mikrotumatestissä altistuksilla, jotka vastasivat kliinisesti merkityksellisiä annoksia. Havainto klastogeenisuudesta on linjassa talatsoparibin farmakologisen vaikutuksen, sen aiheuttaman perimän epästabiliteetin kanssa. Nämä viittaavat mahdolliseen genotoksisuuteen ihmisellä.
Toistuvan annon toksisuus
Rotilla ja koirilla tehdyissä toistuvan annon toksisuustutkimuksissa päälöydökset subterapeuttisilla altistuksilla olivat luuytimen hyposellulaarisuus ja tähän liittyvä annoksesta riippuvainen hematopoieettisten solujen määrän väheneminen, useiden elinten imukudoksen depleetio ja kivesten, lisäkivesten ja siementiehyiden surkastuminen ja/tai rappeumamuutokset. Muita löydöksiä suuremmilla altistuksilla olivat annoksesta riippuvainen apoptoosin/nekroosin lisääntyminen maha-suolikanavassa, maksassa ja munasarjoissa. Useimmat näistä histopatologisista löydöksistä olivat yleensä palautuvia. Kiveslöydökset olivat osittain palautuvia 4 viikon kuluttua annostelun lopettamisesta. Toksisuuslöydökset ovat johdonmukaisia huomioiden talatsoparibin farmakologia ja kudoksiin jakautuminen.
Kehitystoksisuus
Rotilla tehdyssä alkioiden/sikiöiden kehitystä koskeneessa tutkimuksessa talatsoparibi aiheutti alkioiden ja sikiöiden kuolemia, sikiöiden epämuodostumia (silmämunan painumista, pienisilmäisyyttä, rintalastahalkioita, kaulanikamakaaren yhteensulautumia) ja luuston rakenteen variaatioita emon systeemisen AUC24-altistuksen ollessa noin 0,09‑kertainen verrattuna merkitykselliseen altistukseen ihmisillä käytettäessä suositeltua annosta.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Silikonoitu mikrokiteinen selluloosa (mikrokiteinen selluloosa ja piidioksidi)
0,1 mg:n kapselin kuori
Hypromelloosi
Titaanidioksidi (E171)
0,25 mg:n kapselin kuori
Hypromelloosi
Keltainen rautaoksidi (E172)
Titaanidioksidi (E171)
0,35 mg:n kapselin kuori
Hypromelloosi
Keltainen rautaoksidi (E172)
Titaanidioksidi (E171)
0,5 mg:n kapselin kuori
Hypromelloosi
Punainen rautaoksidi (E172)
Titaanidioksidi (E171)
1 mg:n kapselin kuori
Hypromelloosi
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Titaanidioksidi (E171)
Painomuste
Shellakka (E904)
Propyleeniglykoli (E1520)
Ammoniumhydroksidi (E527)
Musta rautaoksidi (E172)
Kaliumhydroksidi (E525)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TALZENNA kapseli, kova
0,25 mg (L:ei) 30 kpl (1774,42 €)
1 mg (L:ei) 30 kpl (4986,21 €)
PF-selosteen tieto
Talzenna 0,1 mg kovat kapselit
Suurtiheyspolyeteenistä (HDPE) valmistettu purkki ja polypropeenista (PP) valmistettu suljin, jossa on kuumainduktiosinetti. Pakkauskoko: 30 kapselin HDPE-purkki kotelossa.
Talzenna 0,25 mg kovat kapselit
Suurtiheyspolyeteenistä (HDPE) valmistettu purkki ja polypropeenista (PP) valmistettu suljin, jossa on kuumainduktiosinetti. Pakkauskoko: 30 kapselin HDPE-purkki kotelossa.
Polyvinyylikloridista/polyvinylideenikloridista (PVC/PVdC) valmistettu perforoitu yksittäispakattu läpipainopakkaus, jossa on alumiinifoliosta tehty repäisykalvo. Pakkauskoot: 30 (30 x 1 kapseli), 60 (60 x 1 kapseli) ja 90 (90 x 1 kapseli) kapselin läpipainopakkaukset, joissa kapselit ovat yksittäispakattuina.
Talzenna 0,35 mg kovat kapselit
Suurtiheyspolyeteenistä (HDPE) valmistettu purkki ja polypropeenista (PP) valmistettu suljin, jossa on kuumainduktiosinetti. Pakkauskoko: 30 kapselin HDPE-purkki kotelossa.
Talzenna 0,5 mg kovat kapselit
Suurtiheyspolyeteenistä (HDPE) valmistettu purkki ja polypropeenista (PP) valmistettu suljin, jossa on kuumainduktiosinetti. Pakkauskoko: 30 kapselin HDPE-purkki kotelossa.
Talzenna 1 mg kovat kapselit
Suurtiheyspolyeteenistä (HDPE) valmistettu purkki ja polypropeenista (PP) valmistettu suljin, jossa on kuumainduktiosinetti. Pakkauskoko: 30 kapselin HDPE-purkki kotelossa.
Polyvinyylikloridista/polyvinylideenikloridista (PVC/PVdC) valmistettu perforoitu yksittäispakattu läpipainopakkaus, jossa on alumiinifoliosta tehty repäisykalvo. Pakkauskoko: 30 (30 × 1 kapseli) kapselin läpipainopakkaukset, joissa kapselit ovat yksittäispakattuina.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Talzenna 0,1 mg kovat kapselit
Läpinäkymätön, noin 14 mm × 5 mm kova kapseli, jossa on valkoinen kansiosa (merkintä ”Pfizer” mustalla) ja valkoinen runko-osa (merkintä ”TLZ 0.1” mustalla).
Talzenna 0,25 mg kovat kapselit
Läpinäkymätön, noin 14 mm × 5 mm kova kapseli, jossa on kermanvalkoinen kansiosa (merkintä ”Pfizer” mustalla) ja valkoinen runko-osa (merkintä ”TLZ 0.25” mustalla).
Talzenna 0,35 mg kovat kapselit
Läpinäkymätön, noin 14 mm × 5 mm kova kapseli, jossa on kermanvalkoinen kansiosa (merkintä ”Pfizer” mustalla) ja kermanvalkoinen runko-osa (merkintä ”TLZ 0.35” mustalla).
Talzenna 0,5 mg kovat kapselit
Läpinäkymätön, noin 14 mm × 5 mm kova kapseli, jossa on vaaleanpinkki kansiosa (merkintä ”Pfizer” mustalla) ja valkoinen runko-osa (merkintä ”TLZ 0.5” mustalla).
Talzenna 1 mg kovat kapselit
Läpinäkymätön, noin 14 mm × 5 mm kova kapseli, jossa on vaaleanpunainen kansiosa (merkintä ”Pfizer” mustalla) ja valkoinen runko-osa (merkintä ”TLZ 1” mustalla).
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TALZENNA kapseli, kova
0,25 mg 30 kpl
1 mg 30 kpl
- Ylempi erityiskorvaus (100 %). Talatsoparibi: Aikuispotilaiden hoito, kun ituradan BRCA-mutaatio ja HER2-negatiivinen paikallisesti edennyt tai etäpesäkkeinen rintasyöpä erityisin edellytyksin (1540).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Talatsoparibi: Aikuispotilaiden hoito, kun ituradan BRCA-mutaatio ja HER2-negatiivinen paikallisesti edennyt tai etäpesäkkeinen rintasyöpä erityisin edellytyksin (3070).
ATC-koodi
L01XK04
Valmisteyhteenvedon muuttamispäivämäärä
19.06.2025
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com