
EVUSHELD injektioneste, liuos 150 mg + 150 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi pahvikotelo sisältää kaksi injektiopulloa:
Yksi tiksagevimabi-injektiopullo sisältää 150 mg tiksagevimabia 1,5 ml:ssa (100 mg/ml) (tixagevimab).
Yksi silgavimabi-injektiopullo sisältää 150 mg silgavimabia 1,5 ml:ssa (100 mg/ml) (cilgavimab).
Tiksagevimabi ja silgavimabi on tuotettu kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Altistumista edeltävä ennaltaehkäisy
EVUSHELD on tarkoitettu käytettäväksi ennen altistumista COVID‑19-taudin ennaltaehkäisyyn aikuisille ja vähintään 12-vuotiaille nuorille, jotka painavat vähintään 40 kg (ks. kohdat Annostus ja antotapa, Farmakodynamiikka ja Farmakokinetiikka).
Hoito
EVUSHELD on tarkoitettu aikuisille ja nuorille (ikä vähintään 12 vuotta ja paino vähintään 40 kg) COVID‑19-taudin hoitoon, kun potilas ei tarvitse lisähappea ja vaikean COVID‑19-taudin kehittymisen riski on suurentunut (ks. kohdat Annostus ja antotapa, Farmakodynamiikka ja Farmakokinetiikka).
Annostus ja antotapa
Lääke on annettava olosuhteissa, joissa vaikeiden yliherkkyysreaktioiden, kuten anafylaksian, hoito on mahdollista. Lääkkeenannon jälkeen valmistetta saanutta henkilöä on tarkkailtava paikallisen hoitokäytännön mukaisesti.
Annostus
Altistumista edeltävä ennaltaehkäisy
Suositeltu annos aikuisille ja vähintään 12-vuotiaille, vähintään 40 kg painaville nuorille on 150 mg tiksagevimabia ja 150 mg silgavimabia (taulukko 1) annettuna kahtena erillisenä peräkkäisenä injektiona lihakseen.
Toistuvasta annostelusta ei ole saatavilla turvallisuutta ja tehoa koskevia tietoja.
Hoito
Suositeltu annos aikuisille ja vähintään 12-vuotiaille, vähintään 40 kg painaville nuorille on 300 mg tiksagevimabia ja 300 mg silgavimabia (taulukko 1) annettuna kahtena erillisenä peräkkäisenä injektiona lihakseen.
EVUSHELD on annettava mahdollisimman pian positiivisen SARS‑CoV‑2-virustestituloksen jälkeen ja 7 päivän kuluessa COVID‑19-taudin oireiden alkamisesta (ks. kohta Farmakodynamiikka).
Taulukko 1 Suositeltu annos
Käyttöaihe | EVUSHELD-annos tiksagevimabi + silgavimabi | Vasta-aineannos | Tarvittavien injektiopullojen määräa | Injektiopullosta vedettävä määrä |
Altistumista edeltävä ennaltaehkäisy | 150 mg + 150 mg (1 EVUSHELD-pahvikotelo) | tiksagevimabi, 150 mg | 1 injektiopullo (tummanharmaa korkki) | 1,5 ml |
silgavimabi, 150 mg | 1 injektiopullo (valkoinen korkki) | 1,5 ml | ||
Hoito | 300 mg + 300 mg (2 EVUSHELD-pahvikoteloa) | tiksagevimabi, 300 mg | 2 injektiopulloa (tummanharmaa korkki) | 3,0 ml |
silgavimabi, 300 mg | 2 injektiopulloa (valkoinen korkki) | 3,0 ml |
a Jokaisessa injektiopullossa on hieman ylimääräistä nestettä, jotta injektiopullosta voidaan vetää 150 mg:n (1,5 ml:n) annos.
Iäkkäät
Annoksen muuttaminen ei ole tarpeen (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Annoksen muuttaminen ei ole tarpeen vähintään 12-vuotiailla, vähintään 40 kg painavilla nuorilla (ks. kohta Farmakokinetiikka). EVUSHELD-valmisteen turvallisuutta ja tehoa alle 12 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Injektiona lihakseen.
Tiksagevimabi ja silgavimabi on annettava erillisinä peräkkäisinä injektioina lihakseen. Ne annetaan eri injektiokohtiin kahteen eri lihakseen, mieluiten pakaralihaksiin.
Yksi pahvikotelo sisältää kaksi injektiopulloa:
- tiksagevimabi-injektioneste, liuos (tummanharmaa korkki)
- silgavimabi-injektioneste, liuos (valkoinen korkki).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen käsittelystä ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttaville aineille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyys (myös anafylaksia)
EVUSHELD-valmisteen annon jälkeen on ilmoitettu vakavia yliherkkyysreaktioita, kuten anafylaktisia reaktioita (ks. kohta Haittavaikutukset). Jos kliinisesti merkittävän yliherkkyysreaktion tai anafylaksian merkkejä ja oireita ilmenee, on välittömästi keskeytettävä lääkkeen anto ja aloitettava asianmukaiset lääkehoidot ja/tai tukihoidot.
Kardiovaskulaariset ja/tai tromboemboliset tapahtumat
PROVENT-tutkimuksessa vakavia sydämeen liittyviä tai tromboembolisia haittatapahtumia ilmeni EVUSHELD-hoitohaarassa useammalla tutkittavalla kuin lumehaarassa (1,6 % vs. 0,9 %). Suurimmalla osalla osallistujista oli kardiovaskulaarisia riskitekijöitä ja/tai anamneesissa sydän- ja verisuonitauti, mikä saattoi selittää kyseisten tapahtumien esiintyvyyden.
Syy-yhteyttä EVUSHELD-valmisteen käytön ja näiden tapahtumien välillä ei ole varmistettu.
EVUSHELD-hoidon riskit ja hyödyt on arvioitava ennen hoidon aloittamista henkilöillä, joilla on suuri kardiovaskulaaristen tai tromboembolisten tapahtumien riski. Potilaille on kerrottava kardiovaskulaarisiin tapahtumiin viittaavista merkeistä ja oireista (erityisesti rintakipu, hengenahdistus, huonovointisuus, heikotus tai huimaus), ja heitä on neuvottava hakeutumaan välittömästi lääkärin hoitoon, jos tällaisia oireita ilmenee.
Kliinisesti merkittävät verenvuotohäiriöt
Kuten muitakin lihakseen annettavia injektioita annettaessa, EVUSHELD-valmisteen antamisessa on noudatettava varovaisuutta, jos potilaalla on trombosytopenia tai jokin hyytymishäiriö.
Viruslääkeresistenssi
Kliiniset tutkimukset EVUSHELD-valmisteella tehtiin alfa-, beeta-, gamma- ja deltavarianttien ollessa vallitsevia. Tiksagevimabin ja silgavimabin teho on epävarma joitakin sellaisia kiertäviä SARS-CoV-2-variantteja vastaan, joiden herkkyys on heikentynyt in vitro (ks. kohta Farmakodynamiikka).
PROVENT-tutkimuksen kliinisten tietojen perusteella suojan kesto EVUSHELD-kerta-annoksen (150 mg tiksagevimabia ja 150 mg silgavimabia) antamisen jälkeen on arviolta vähintään 6 kuukautta. Koska in vitro ‑neutralointivaikutuksen on havaittu heikentyneen omikron-alavariantteja BA.1, BA.1.1 (BA.1+R346K), BA.4 ja BA.5 vastaan, EVUSHELD-valmisteen antaman suojan kesto näitä alavariantteja vastaan on tällä hetkellä tuntematon.
COVID-19-rokotteet
Altistumista edeltävä ennaltaehkäisy EVUSHELD-valmisteella ei korvaa rokotusta henkilöillä, joille suositellaan COVID-19-rokotusta.
Yhteisvaikutukset
Farmakokineettiset yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty ihmisillä.
EVUSHELD-valmisteen ei odoteta metaboloituvan maksaentsyymien välityksellä eikä eliminoituvan munuaisten kautta. Tiksagevimabi ja silgavimabi eivät erity munuaisten kautta tai metaboloidu sytokromi P450 (CYP) -entsyymien välityksellä; siksi yhteisvaikutukset munuaisten kautta erittyvien lääkevalmisteiden tai CYP-entsyymien substraattien, indusorien tai estäjien kanssa ovat epätodennäköisiä.
Farmakokineettisen mallinnuksen perusteella EVUSHELD-valmisteen annon jälkeisellä COVID‑19-rokotuksella ei ollut kliinisesti merkittävää vaikutusta EVUSHELD-valmisteen puhdistumaan.
Farmakokineettisen mallinnuksen perusteella immuunipuutostiloilla ei ollut kliinisesti merkittävää vaikutusta EVUSHELD-valmisteen puhdistumaan.
Farmakodynaamiset yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty ihmisillä.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja tiksagevimabin ja silgavimabin käytöstä raskaana oleville naisille.
Tiksagevimabilla ja silgavimabilla ei ole tehty ei-kliinisiä lisääntymistoksisuustutkimuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Tiksagevimabilla ja silgavimabilla tehdyissä kudosten ristireaktiivisuutta selvittäneissä tutkimuksissa, joissa käytettiin ihmisen sikiökudoksia, ei havaittu kliinistä huolta herättävää sitoutumista. Ihmisen immunoglobuliini G1 (IgG1) -vasta-aineiden tiedetään läpäisevän istukan, joten tiksagevimabi ja silgavimabi saattavat siirtyä äidiltä kehittyvään sikiöön. Ei tiedetä, koituuko tiksagevimabin ja silgavimabin mahdollisesta siirtymisestä istukan läpi mitään hoidollista hyötyä tai riskiä kehittyvälle sikiölle.
EVUSHELD-valmistetta saa käyttää raskauden aikana vain, jos hoidosta äidille mahdollisesti koituvat hyödyt ovat suuremmat kuin mahdolliset sikiölle aiheutuvat riskit.
Imetys
Ei tiedetä, erittyvätkö tiksagevimabi ja silgavimabi ihmisen rintamaitoon, mutta äidin IgG-vasta-aineiden tiedetään erittyvän rintamaitoon muutamien päivien ajan synnytyksen jälkeen.
Tiksagevimabin ja silgavimabin vaikutus kohdentuu suoraan SARS‑CoV‑2:n piikkiproteiiniin, ja nieltyjen vasta-aineiden systeeminen imeytyminen on lisäksi vähäistä, joten EVUSHELD-valmisteen antamista imetyksen aikana voidaan harkita, jos se on kliinisesti aiheellista.
Hedelmällisyys
Tiksagevimabin ja silgavimabin vaikutuksista ihmisen hedelmällisyyteen ei ole tietoa. Vaikutuksia urosten ja naaraiden hedelmällisyyteen ei ole arvioitu eläinkokeissa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
EVUSHELD-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yhteensä 4 210 aikuista tutkittavaa on saanut 150 mg tiksagevimabia ja 150 mg silgavimabia injisoituna lihakseen vaiheen III tutkimuksissa, joissa arvioitiin ennaltaehkäisevää hoitoa. Yleisimmät haittavaikutukset (≥ 1 %) olivat pistoskohdan reaktiot (1,6 %) ja yliherkkyys (1,0 %).
Yhteensä 452 aikuista tutkittavaa, jotka eivät olleet sairaalahoidossa ja joilla oli lievä tai keskivaikea COVID‑19-tauti, on saanut 300 mg tiksagevimabia ja 300 mg silgavimabia injektiona lihakseen vaiheen III tutkimuksessa, jossa arvioitiin valmisteen käyttöä taudin hoidossa. Turvallisuusprofiili oli kokonaisuutena samankaltainen kuin tutkittavilla, jotka saivat 150 mg tiksagevimabia ja 150 mg silgavimabia tutkimuksissa, joissa arvioitiin ennaltaehkäisevää hoitoa. Yleisin haittavaikutus (≥ 1 %) oli pistoskohdan reaktio (2,4 %).
Taulukossa 2 mainitut haittavaikutukset on lueteltu MedDRA-elinjärjestelmäluokituksen ja esiintymistiheyden mukaan. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1 000), hyvin harvinainen (< 1 / 10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutustaulukko
Taulukko 2 Haittavaikutustaulukko
| MedDRA-elinjärjestelmäluokitus | Haittavaikutus | Esiintymistiheysa |
| Immuunijärjestelmä | Yliherkkyysb | Yleinen |
| Anafylaksiac | Harvinainen | |
| Yleisoireet ja antopaikassa todettavat haitat | Injektioon liittyvä reaktiod | Melko harvinainen |
| Vammat, myrkytykset ja hoitokomplikaatiot | Pistoskohdan reaktioe | Yleinen |
a Esiintymistiheydet perustuvat ennaltaehkäisevää hoitoa arvioineiden tutkimusten yhdistettyihin tietoihin altistuksesta 150 mg:lle tiksagevimabia ja 150 mg:lle silgavimabia.
b Sisältää suositellut termit ihottuma ja nokkosihottuma.
c Havaittu markkinoilletulon / myyntiluvan myöntämisen jälkeen saapuneista ilmoituksista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
d Suositellun termin ”Injektioon liittyvä reaktio” alle luokiteltujen tapahtumien kuvauksiin kuuluivat päänsärky, vilunväristykset, punoitus, epämukava tunne tai aristus pistoskohdan lähellä.
e Sisältää suositellut termit pistoskohdan kipu, pistoskohdan punoitus, pistoskohdan kutina, pistoskohdan reaktio ja pistoskohdan kovettuma.
Pediatriset potilaat
Tietoja pediatrisista potilaista (alle 18-vuotiaista) ei ole saatavilla (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tiksagevimabin ja silgavimabin yliannostukseen ei ole spesifistä hoitoa. Yliannostuksen hoitona on yleinen tukihoito, joka sisältää vitaalitoimintojen seurannan ja potilaan kliinisen tilan tarkkailun.
Kliinisissä tutkimuksissa on annettu enintään 300 mg:n annoksia tiksagevimabia ja 300 mg:n annoksia silgavimabia lihakseen sekä enintään 1 500 mg:n annoksia tiksagevimabia ja 1 500 mg:n annoksia silgavimabia laskimoon, eivätkä ne ole aiheuttaneet annosta rajoittavaa toksisuutta.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunoseerumit ja immunoglobuliinit, antiviraaliset monoklonaaliset vasta-aineet, ATC-koodi: J06BD03
Vaikutusmekanismi
Sekä tiksagevimabi että silgavimabi ovat rekombinantteja humaaneja monoklonaalisia IgG1κ-vasta-aineita, joissa on Fc-alueilla aminohapposubstituutioita, jotka pidentävät vasta-aineen puoliintumisaikaa, vähentävät vasta-aineen efektoritoimintaa ja pienentävät vasta-aineesta riippuvaiseen taudin vaikeutumiseen liittyvää mahdollista riskiä (ks. kohta Prekliiniset tiedot turvallisuudesta). Tiksagevimabi ja silgavimabi voivat sitoutua samanaikaisesti SARS‑CoV‑2-viruksen piikkiproteiinin reseptoria sitovan domeenin (RBD) ei-päällekkäisille alueille. Piikkiproteiiniin sitoutumisen KD (tasapainodissosiaatiovakio) on tiksagevimabilla 2,76 pM, silgavimabilla 13,0 pM ja tiksagevimabin ja silgavimabin yhdistelmällä 13,7 pM. Sitoutuminen estää piikkiproteiinin vuorovaikutuksen ihmisen ACE2-reseptorin kanssa, jolloin viruksen pääsy soluun estyy. Tiksagevimabi, silgavimabi ja niiden yhdistelmä estivät reseptoria sitovan domeenin sitoutumista ihmisen ACE2-reseptoriin siten, että IC50-pitoisuudet olivat 0,32 nM (48 ng/ml, tiksagevimabi), 0,53 nM (80 ng/ml, silgavimabi) ja 0,43 nM (65 ng/ml, tiksagevimabin ja silgavimabin yhdistelmä).
Antiviraalinen vaikutus
Vero E6 -soluilla tehdyssä SARS‑CoV‑2-viruksen neutralointitestissä tiksagevimabi, silgavimabi ja niiden yhdistelmä neutraloivat SARS‑CoV‑2-virusta (USA-WA1/2020-isolaattia) seuraavasti: tiksagevimabin EC50-arvo oli 60,7 pM (9 ng/ml), silgavimabin EC50-arvo oli 211,5 pM (32 ng/ml) ja yhdistelmän EC50-arvo oli 65,9 pM (10 ng/ml). Nämä in vitro -arvot vastaavat in vivo kliinisesti tehokkaita 2,2 mikrog/ml:n EVUSHELD-pitoisuuksia seerumissa.
Viruslääkeresistenssi
SARS‑CoV‑2-viruksille tai rekombinanteille VSV-viruksille, jotka koodaavat SARS‑CoV‑2-viruksen piikkiproteiinia (pseudoviruksille), tehtiin sarjasiirrostus (serial passage) soluviljelmissä, joissa oli pelkkää tiksagevimabia, pelkkää silgavimabia tai tiksagevimabia ja silgavimabia yhdistelmänä. Kun soluviljelmissä oli silgavimabia, siirrostuksen jälkeen havaittiin immuunisuojaa väistäviä variantteja (escape variants). Niitä ei kuitenkaan havaittu, kun viljelmissä oli tiksagevimabia tai sekä tiksagevimabia että silgavimabia.
Neutralointitesteissä, joissa käytetyissä rekombinanteissa SARS‑CoV‑2-pseudoviruksissa oli kiertävässä SARS‑CoV‑2-viruksessa tunnistettuja yksittäisiä piikkiproteiinin substituutioita, heikentynyttä herkkyyttä pelkälle tiksagevimabille havaittiin varianteilla, joissa oli F486S (> 600‑kertainen) ja F486V (121–149‑kertainen), ja heikentynyttä herkkyyttä pelkälle silgavimabille havaittiin varianteilla, joissa oli R346I (> 200‑kertainen), K444E (> 200‑kertainen), K444Q (> 200‑kertainen) ja K444R (> 200‑kertainen).
Tiksagevimabin ja silgavimabin yhdistelmällä säilyi täydellinen tai lähes täydellinen neutralointivaikutus sellaisia pseudoviruksia ja/tai eläviä SARS‑CoV‑2-variantteja vastaan, joissa oli mikä tahansa huolestuttavissa virusvarianteissa alfa (B.1.1.7), beeta (B.1.351), gamma (P.1), delta (B.1.617.2), delta [+K417N] (AY.1/AY.2) tai omikron (BA.2) todettu piikkiproteiinin substituutio. Piikkiproteiinia ilmentävien pseudotyyppisten viruksen kaltaisten partikkelien ja aitojen SARS‑CoV‑2-virusvarianttien omikron BA.1 (B.1.1.529) ja omikron BA.1.1 (B.1.1.529 [+R346K]) herkkyys tiksagevimabin ja silgavimabin yhdistelmälle oli heikentynyt (taulukko 3).
Parhaillaan kerätään tietoja sen selvittämiseksi, miten aidoilla SARS‑CoV‑2-viruksilla ja pseudotyyppisillä viruksen kaltaisilla partikkeleilla tehdyissä määrityksissä havaittu pieni aktiivisuuden väheneminen mahdollisesti korreloi kliinisten tulosten kanssa.
Taulukko 3 Tiedot tiksagevimabin ja silgavimabin yhdistelmän kyvystä neutraloida pseudovirusta ja aitoa SARS‑CoV‑2-virusta SARS‑CoV‑2-variantin substituutioiden mukaan
| Pango-kehityslinja ja piikkiproteiinin substituutiot | Testatut tyypilliset reseptoria sitovan domeenin substituutiot | Herkkyyden heikentymisen kerrannaismuutos a | IC50 (ng/ml) | ||
| Pseudovirus b | Elävä virus c | Pseudovirus b | Elävä virus c | ||
| Huolestuttavat virusvariantit | |||||
| B.1.1.7 (alfa, Iso-Britannia) | N501Y | 1,0–5,2 | 0,5–1,4 | 1,1–9,0 | 4–39,5 |
| B.1.351 (beeta, Etelä-Afrikka) | K417N:E484K:N501Y | 2,5–5,5 | 0,9–3,8 | 5,6–11,4 | 6,5–256 |
| P.1 (gamma, Brasilia) | K417T:E484K:N501Y | 0,8–1,7 | 0,4–2,0 | 1,8–2,7 | 3,2–8 |
| B.1.617.2 (delta, Intia) | L452R:T478K | 1–1,2 | 0,6–1,0 | 1,9–2,2 | 3–7,5 |
| AY.1/AY.2 (delta [+K417N], Intia) | K417N:L452R:T478K | 1,0 | Ei määritetty | 1,9 | Ei määritetty |
| B.1.1.529 omikron, BA.1 (Botswana) | G339D:S371L:S373P: S375F:K417N:N440K: G446S:S477N:T478K: E484A:Q493R:G496S: Q489R:N501Y:Y505H | 132–183 | 12–30 | 51–277 | 147–278 |
| Omikron BA.1.1 (useita maita) | G339D:R346K:S371L: S373P: S375F:K417N: N440K:G446S:S477N: T478K:E484A:Q493R: G496S:Q489R:N501Y: Y505H | 424 | 176 | 466 | 1 147 |
| Omikron BA.2 (useita maita) | G339D:S371F:S373P: S375F:T376A:D405N: R408S:K417N:N440K:S477N: T478K:E484A: Q493R:Q498R:N501Y: Y505H:H655Y:N679K:P681H:N764K | 3,2 | 5,4 | 9,8 | 35 |
| Omikron BA.2.12.1 (Yhdysvallat) | G339D:S371F:S373P: S375F:T376A:D405N: R408S:K417N:N440K: L452Q:S477N+T478K: E484A:Q493R:Q498R: N501Y: Y505H | 5 | Ei määritetty | 10,7 | Ei määritetty |
| Omikron BA.3 (useita maita) | G339D:S371F:S373P: S375F:D405N:K417N: N440K:G446S:S477N: T478K:E484A:Q493R: Q498R:N501Y:Y505H | 16 | Ei määritetty | 34,5 | Ei määritetty |
| Omikron BA.4 (useita maita) | G339D:S371F:S373P: S375F:T376A:D405N: R408S:K417N:N440K: L452R:S477N:T478K: E484A:F486V:Q498R: N501Y:Y505H | 33–65 | Ei määritetty | 65–69,4 | Ei määritetty |
| Omikron BA.5 (useita maita) | G339D:S371F:S373P: S375F:T376A:D405N: R408S:K417N:N440K: L452R:S477N:T478K: E484A:F486V:Q498R: N501Y:Y505H | 33–65 | 4,2–16 | 65–69,4 | 56,6–229 |
| Tehostetun seurannan alaiset variantit | |||||
| B.1.525 (eeta, useita maita) | E484K | 1,8–3,1 | Ei määritetty | 5–9,5 | Ei määritetty |
| B.1.526 (ioota, Yhdysvallat) | Ε484Κ | 0,8–3,4 | 0,3–1,8 | 1,9–5,2 | 1,0–7,0 |
| B.1.617.1 (kappa, Intia) | L452R:E484Q | 0,9–3,4 | 0,5–1,3 | 2,5–5,1 | 2,0–5,0 |
| C.37 (lambda, Peru) | L452Q:F490S | 0,7 | Ei määritetty | 1,1 | Ei määritetty |
| B.1.621 (myy, Kolumbia) | R346K:E484K:N501Y | 7,5 | Ei määritetty | 17,3 | Ei määritetty |
| Seurattavat signaalit | |||||
| B.1.427 / B.1.429 (epsilon, Yhdysvallat) | L452R | 0,8–2,9 | 1,3–3,5 | 1,0–4,5 | 5,0–14,0 |
| R.1 (useita maita) | E484K | 3,5 | Ei määritetty | 4,6 | Ei määritetty |
| B.1.1.519 (useita maita) | T478K | 1,0-1,4 | Ei määritetty | 2,0-2,3 | Ei määritetty |
| C.36.3 (useita maita) | R346S:L452R | 2,3 | Ei määritetty | 3,9 | Ei määritetty |
| B.1.214.2 (useita maita) | Q414K:N450K | 0,8 | Ei määritetty | 1,6 | Ei määritetty |
| B.1.619.1 (useita maita) | N440K:E484K | 3,3 | Ei määritetty | 7,6 | Ei määritetty |
| Signaalit, joiden seuranta on lopetettu | |||||
| P.2 (zeeta, Brasilia) | E484K | 2,9 | Ei määritetty | 10,4 | Ei määritetty |
| B.1.616 (Ranska) | V483A | 0,4–0,5 | Ei määritetty | 1,1–1,2 | Ei määritetty |
| A.23.1 (UK) | V367F | 0,4 | Ei määritetty | 0,5 | Ei määritetty |
| A.27 (useita maita) | L452R:N501Y | 0,8 | Ei määritetty | 1,8 | Ei määritetty |
| AV.1 (useita maita) | N439K:E484K | 5,9 | Ei määritetty | 13,0 | Ei määritetty |
- Heikentyneen in vitro tehon vaihteluväli useissa samanaikaisten substituutioiden sarjoissa ja/tai tutkimuslaboratorioissa, joissa käytetään tutkimuskäyttöön soveltuvan tasoisia määrityksiä; monoklonaalisen vasta-aineen IC50-arvon (pitoisuus, joka vaaditaan infektioiden vähentämiseen 50 %:lla) keskimääräinen kerrannaismuutos villityypin vertailukantaan nähden.
- Tutkitut pseudovirukset ilmensivät SARS‑CoV‑2-viruksen koko muuntunutta piikkiproteiinia ja yksittäisiä tyypillisiä piikkiproteiinin substituutioita, paitsi L452Q-substituutiota. Niihin kuuluivat alfa (+L455F, E484K, F490S, Q493R ja/tai S494P) ja delta (+K417N), joissa oli myös tässä mainittuja muita reseptoria sitovan domeenin substituutioita, joita näissä kehityslinjoissa ei enää havaita tai havaitaan vain hyvin vähän.
- Tutkitut aidot SARS‑CoV‑2-virukset ilmensivät koko muuntunutta piikkiproteiinia. Niihin kuului alfa (+E484K tai S494P), jossa oli myös tässä mainittuja muita reseptoria sitovan domeenin substituutioita, joita näissä kehityslinjoissa ei enää havaita tai havaitaan vain hyvin vähän.
Ei tiedetä, miten pseudovirusten tai aidon SARS‑CoV‑2:n neutralointiherkkyyttä koskevat tiedot korreloivat kliinisen tuloksen kanssa.
PROVENT-tutkimuksessa sairauskäynneillä kerätyt sekvensointitiedot olivat saatavilla 21 tutkittavalta, joilla oli oireinen COVID‑19-infektio (7 tutkittavista sai tiksagevimabia ja silgavimabia ja 14 tutkittavista sai lumelääkettä). Kun tarkasteltiin ≥ 25 %:n alleeliosuuksia, yleisimmin havaitut huolestuttavat virusvariantit tai tehostetun seurannan alaiset variantit olivat alfavariantti (yhteensä 5 tapahtumaa; kaikki lumeryhmässä) ja deltavariantti (yhteensä 7 tapahtumaa; 6 lumeryhmässä ja 1 EVUSHELD-ryhmässä). Lisäksi alkuperäisen kannan sekvenssejä havaittiin 7 (3 lumeryhmässä ja 4 EVUSHELD-ryhmässä).
On mahdollista, että sellaisilla varianteilla, joihin liittyy resistenssi tiksagevimabin ja silgavimabin yhdistelmälle, saattaa olla ristiresistenssiä muille monoklonaalisille vasta-aineille, joiden vaikutus kohdentuu SARS‑CoV‑2-viruksen reseptoria sitovaan domeeniin. Tiksagevimabin ja silgavimabin yhdistelmän vaikutus säilyi sellaisia pseudoviruksia vastaan, joissa oli yksittäisiä SARS‑CoV‑2-viruksen piikkiproteiinin substituutioita (E484D/K/Q, F490S, Q493R, S494P, K417E/N, D420N, K444Q, V445A, Y453F, L455F, N460K/S/T, F486V ja Q493K). Näitä substituutioita on tunnistettu muiden SARS‑CoV‑2-viruksen piikkiproteiinin reseptoria sitovaan domeeniin kohdentuvien monoklonaalisten vasta-aineiden aiheuttamaa neutraloitumista väistävissä varianteissa.
TACKLE-tutkimuksessa lähtötilanteen käynnillä kerättyä sekvensointitietoa oli saatavilla 748 tutkittavalta (382 näistä tutkittavista sai tiksagevimabia ja silgavimabia ja 367 sai lumelääkettä). Kun tarkasteltiin ≥ 25 %:n alleeliosuuksia, niiden tutkittavien osuus, joilla todettiin huolestuttava virusvariantti tai tehostetun seurannan alainen variantti, oli eri hoitoryhmissä samaa luokkaa. Tutkittavilla todettuja tällaisia variantteja olivat alfavariantti, beetavariantti, gammavariantti, deltavariantti, lambdavariantti ja myyvariantti.
Farmakodynaamiset vaikutukset
PROVENT-tutkimuksessa 150 mg:n tiksagevimabiannoksen ja 150 mg:n silgavimabiannoksen lihakseen antamisen jälkeen neutraloivien vasta-aineiden GMT-arvot (titterien geometriset keskiarvot) olivat 8 päivän kohdalla 19‑kertaiset, 29 päivän kohdalla 23‑kertaiset, 58 päivän kohdalla 18‑kertaiset, 92 päivän kohdalla 14‑kertaiset, 183 päivän kohdalla 6‑kertaiset ja 366 päivän kohdalla 3‑kertaiset verrattuna COVID-19-potilaiden toipilasplasmasta mitattuihin GMT-arvoihin (GMT = 30,8).
Kun TACKLE-tutkimuksessa annettiin tutkittaville 300 mg tiksagevimabia ja 300 mg silgavimabia kerta-annoksena lihakseen, EVUSHELD-ryhmässä havaittiin lumeryhmään verrattuna yli viisinkertainen neutraloivien vasta-aineiden GMT-arvojen suureneminen päivään 169 mennessä: 16‑kertainen päivänä 6, 14‑kertainen päivänä 15, 22‑kertainen päivänä 29, 18‑kertainen päivänä 85 ja 5,3‑kertainen päivänä 169.
Immunogeenisyys
Kun tutkittaville annettiin PROVENT-tutkimuksessa kerta-annos EVUSHELD-valmistetta (150 mg tiksagevimabia ja 150 mg silgavimabia), hoidon aikana ilmeneviä vasta-aineita tiksagevimabille havaittiin 7,6 %:lla (234/3 085) EVUSHELD-valmistetta saaneista potilaista, joilta lääkevasta-aineita voitiin arvioida. Vasta-aineita silgavimabille havaittiin 11,3 %:lla (341/3 024) näistä potilaista ja vasta-aineita EVUSHELD-valmisteelle 13,1 %:lla (403/3 086) näistä potilaista.
Kun tutkittaville annettiin TACKLE-tutkimuksessa kerta-annos EVUSHELD-valmistetta (300 mg tiksagevimabia ja 300 mg silgavimabia), hoidon aikana ilmeneviä vasta-aineita tiksagevimabille havaittiin 7,3 %:lla (27/372) EVUSHELD-valmistetta saaneista potilaista, joilta lääkevasta-aineita voitiin arvioida. Vasta-aineita silgavimabille havaittiin 12,7 %:lla (46/363) näistä potilaista ja vasta-aineita EVUSHELD-valmisteelle 14,5 %:lla (54/373) näistä potilaista.
Ei ole havaittu näyttöä lääkevasta-aineista, jotka vaikuttaisivat millään tavoin farmakokinetiikkaan tai turvallisuuteen.
Kliininen teho
COVID-19-taudin ennaltaehkäisy
PROVENT oli vaiheen III satunnaistettu (2:1), kaksoissokkoutettu, lumekontrolloitu kliininen tutkimus, jossa tutkittiin EVUSHELD-valmisteen käyttöä altistumista edeltävään COVID‑19-taudin ennaltaehkäisyyn vähintään 18-vuotiailla aikuisilla. Tutkittavilla katsottiin olevan suurentunut riski saada riittämätön vaste aktiiviseen immunisaatioon (syitä olivat vähintään 60 vuoden ikä, samanaikainen sairaus, aiemmin todettu krooninen sairaus, immuunipuutos tai rokotteen puutteellinen siedettävyys) tai suurentunut SARS‑CoV‑2-infektion riski (syynä oli olinpaikka tai olosuhteet mukaanottohetkellä: esimerkiksi terveydenhuollon työntekijät, kuten pitkäaikaishoitolaitosten työntekijät, sekä suuren riskin oloissa toimivat teollisuustyöntekijät tai henkilöt, jotka asuvat tiiviisti lähekkäin, esimerkiksi opiskelija-asuntolassa tai armeijan kasarmissa). Tutkittavat saivat joko 150 mg tiksagevimabia ja 150 mg silgavimabia tai lumelääkettä annettuna kahtena erillisenä pistoksena lihakseen. Tutkimuksesta suljettiin pois tutkittavat, joilla oli anamneesissa laboratoriossa vahvistettu SARS‑CoV‑2-infektio tai joilla saatiin seulontavaiheessa positiivinen SARS‑CoV‑2-vasta-ainetulos.
Lähtötilanteen demografiset tiedot olivat EVUSHELD-valmistetta saaneiden ryhmässä ja lumeryhmässä hyvin samankaltaiset. Tutkittavien mediaani-ikä oli 57 vuotta (24 % tutkittavista oli vähintään 65-vuotiaita ja 4 % tutkittavista vähintään 75-vuotiaita), 46 % tutkittavista oli naisia, 73 % oli valkoihoisia, 3 % oli aasialaisia, 17 % oli mustaihoisia/afrikkalaisamerikkalaisia ja 15 % oli taustaltaan espanjankielisistä maista tai latinalaisamerikkalaisia. 5 197 tutkittavasta 78 %:lla oli lähtötilanteessa samanaikaisia sairauksia tai ominaisuuksia, joihin liittyi vaikean COVID‑19-taudin suurentunut riski, kuten lihavuus (42 %), diabetes (14 %), sydän- tai verisuonitauti (8 %), syöpä, mukaan lukien aiemmin sairastettu syöpä (7 %), keuhkoahtaumatauti (5 %), krooninen munuaistauti (5 %), krooninen maksasairaus (5 %), immunosuppressiivinen lääkitys (3 %) ja immuunivastetta heikentävä tauti (< 1 %).
Primaarianalyysi käsitti 5 172 tutkittavaa, jotka olivat lähtötilanteessa SARS‑CoV‑2 RT‑PCR‑negatiivisia. Näistä 3 441 sai EVUSHELD-valmistetta ja 1 731 lumelääkettä. EVUSHELD pienensi SARS‑CoV‑2-viruksen suhteen RT‑PCR-positiivisen oireisen sairauden (COVID‑19:n) riskiä merkitsevästi (p-arvo < 0,001) lumelääkkeeseen verrattuna (taulukko 4). Lääkkeenannon jälkeisen seuranta‑ajan mediaani oli 83 päivää.
Taulukko 4COVID‑19-taudin ilmaantuvuus
| N | Tapausten määräa, n (%) | Suhteellisen riskin vähenemä, % (95 %:n luottamusväli) | |
| EVUSHELDb | 3 441 | 8 (0,2 %) | 77 % (46 ; 90) |
| Lumelääke | 1 731 | 17 (1,0 %) |
N = analyysiin otettujen tutkittavien määrä.
a Ensisijainen päätemuuttuja; tutkittava määriteltiin COVID‑19‑tapaukseksi, jos tutkittavan ensimmäinen SARS‑CoV‑2-viruksen suhteen RT‑PCR-positiivinen oireinen sairastuminen ilmeni lääkkeen annon jälkeen ja ennen päivää 183.
b 150 mg tiksagevimabia ja 150 mg silgavimabia.
Teho oli johdonmukainen kaikissa ennalta määritellyissä alaryhmissä; alaryhmien määrittelykriteereitä olivat mm. ikä, sukupuoli, etninen tausta ja lähtötilanteessa todetut samanaikaiset sairaudet tai ominaisuudet, joihin liittyi suurentunut vaikean COVID‑19‑taudin riski.
EVUSHELD-valmistetta saaneiden tutkittavien joukossa ei ollut yhtään vaikeaa/kriittistä COVID‑19-tapahtumaa (määritelmänä oli SARS‑CoV‑2-viruksen suhteen RT-PCR-positiivinen oireinen sairaus, jossa ilmeni vähintään joko keuhkokuume [kuume, yskä, takypnea tai hengenahdistus sekä keuhkoinfiltraatteja] tai hypoksemia [SpO2 < 90 % huoneilmassa ja/tai vaikea hengitysvaikeus] ja pistemäärä WHO:n asteikolla [WHO Clinical Progression Scale] vähintään 5), kun taas lumelääkettä saaneilla ilmeni yksi tapaus (0,1 %).
Päivitettyjen turvallisuutta ja tehoa koskevien jälkianalyysien laatimiseksi tehtiin ylimääräinen tiedonkeruun katkaisu; seuranta-ajan mediaani oli 6,5 kuukautta sekä EVUSHELD-ryhmässä että lumeryhmässä. SARS‑CoV‑2-viruksen suhteen RT‑PCR-positiivisen oireisen sairauden suhteellisen riskin vähenemä oli 83 % (95 %:n luottamusväli 66 ; 91); EVUSHELD-ryhmässä tapahtumia ilmeni 11 tutkittavalla 3 441:sta (0,3 %) ja lumelääkeryhmässä 31 tutkittavalla 1 731:sta (1,8 %), ks. kuva 1. EVUSHELD-valmistetta saaneiden tutkittavien joukossa ei ilmennyt yhtään vaikeaa/kriittistä COVID‑19-tapahtumaa, kun taas lumelääkettä saaneiden tutkittavien joukossa ilmeni viisi tapahtumaa.
Eksploratiivisissa analyyseissä, joissa tarkasteltiin kaikkia tutkittavia, jotka saivat EVUSHELD-valmistetta tai lumelääkettä (mukaan lukien 25:tä tutkittavaa, joiden havaittiin myöhemmin olleen lähtötilanteessa SARS-CoV-2-viruksen suhteen RT-PCR‑positiivisia), SARS‑CoV‑2-viruksen suhteen RT‑PCR‑positiivisen oireisen taudin suhteellisen riskin vähenemä oli 78 % (95 %:n luottamusväli 59 ; 88), kun seuranta-ajan mediaani oli 6,5 kuukautta; tällöin EVUSHELD-ryhmässä oli ilmennyt 14 tapahtumaa / 3 460 tutkittavaa (0,4 %) ja lumeryhmässä 31 tapahtumaa / 1 737 tutkittavaa (1,8 %).
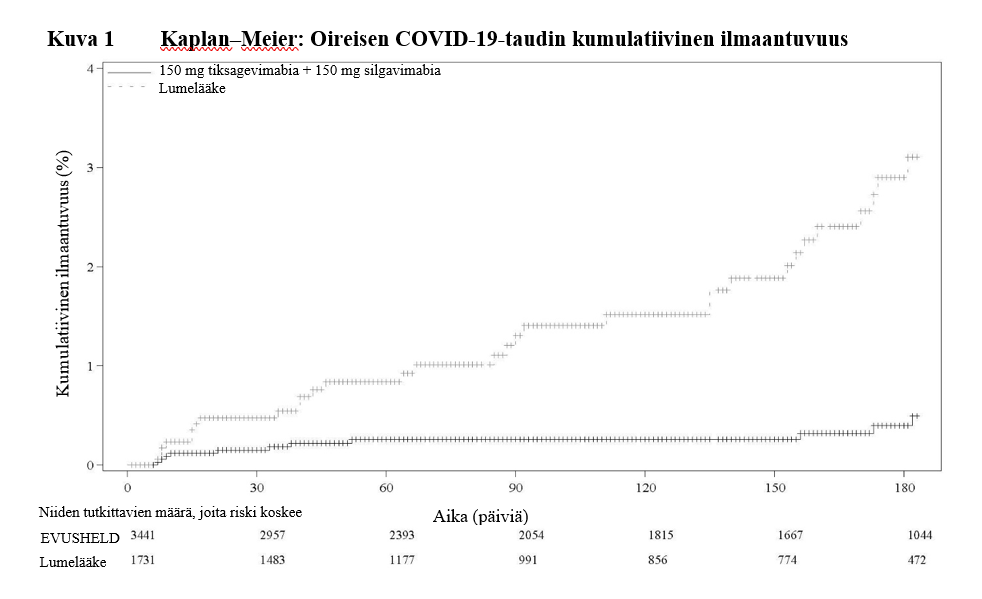
Kuvassa 1 esitettävänä ajanjaksona kiertäneitä vallitsevia SARS‑CoV‑2-variantteja olivat alfa-, beeta-, gamma-, epsilon- ja deltavariantit. Ensisijaisen päätetapahtuman tapahtumien ilmaantuvuuden perusteella tehon kesto oli 6 kuukautta.
Lievän tai keskivaikean COVID-19-taudin hoito
TACKLE oli vaiheen III satunnaistettu (1:1), kaksoissokkoutettu, lumekontrolloitu kliininen tutkimus, jossa tutkittiin EVUSHELD-valmisteen käyttöä aikuisilla potilailla, joilla on lievä tai keskivaikea COVID‑19-tauti. Tutkimukseen otettiin mukaan henkilöitä, jotka eivät olleet saaneet COVID‑19-rokotetta ja eivät olleet sairaalahoidossa COVID‑19-taudin vuoksi sekä joilla oli vähintään yksi COVID‑19-taudin oire, joka oli vaikeusasteeltaan vähintään lievä. Hoito aloitettiin 3 päivän kuluessa SARS‑CoV‑2-virusinfektion suhteen positiivisen näytteen ottamisesta ja enintään 7 päivän kuluessa COVID‑19-taudin oireiden alkamisesta. Tutkittavat saivat tavanomaista hoitoa ja joko 300 mg tiksagevimabia ja 300 mg silgavimabia (N = 413) tai lumelääkettä (N = 421) annettuna kahtena erillisenä pistoksena lihakseen. Ositusperusteina olivat aika oireiden alkamisesta (≤ 5 päivää vs. > 5 päivää) ja vaikean COVID-19-taudin kehittymisen riski (suuri riski vs. pieni riski).
Demografiset tiedot ja taudin ominaisuudet olivat hyvin samankaltaiset sekä hoitoryhmässä että lumeryhmässä. Lähtötilanteessa tutkittavien mediaani-ikä oli 46 vuotta (13 % tutkittavista oli vähintään 65-vuotiaita), 50 % tutkittavista oli naisia, 62 % oli valkoihoisia, 5,6 % oli aasialaisia, 4,0 % oli mustaihoisia ja 52 % oli taustaltaan espanjankielisistä maista tai latinalaisamerikkalaisia. Suurin osa osallistujista (84 %) oli lähtötilanteessa seronegatiivisia, ja 90 %:lla katsottiin olevan suurentunut vaikean COVID‑19-taudin kehittymisen riski. Määritelmän mukaan tähän ryhmään luettiin henkilöt, jotka olivat vähintään 65-vuotiaita satunnaistamishetkellä, tai alle 65-vuotiaat henkilöt, joilla oli vähintään yksi sairaus tai muu tekijä, johon liittyi suurentunut vaikean COVID‑19-taudin kehittymisen riski. Suuren riskin aiheuttaviin samanaikaisiin sairauksiin kuuluivat seuraavat: lihavuus (painoindeksi ≥ 30) (43 %), tupakointi (nykyinen tai aiempi) (40 %), hypertensio (28 %), krooninen keuhkosairaus tai keskivaikea tai vaikea astma (12 %), diabetes (12 %), sydän- tai verisuonitauti (mukaan lukien aiempi aivohalvaus) (9 %), immuunipuutostila (jonka syynä oli kiinteän elimen siirto, veritransplantaatio tai luuytimensiirto, immuunipuutos, HIV-infektio tai kortikosteroidien tai muiden immunosuppressiivisten lääkkeiden käyttö) (5 %), syöpä (4 %), krooninen munuaistauti (2 %) tai krooninen maksasairaus (2 %).
Lähtötilanteessa pistemäärä WHO:n asteikolla [WHO Clinical Progression Scale] oli 88 %:lla potilaista 2 ja 12 %:lla 3, ja COVID‑19-taudin oireiden keston mediaani ennen hoitoa oli 5 päivää.
Ensisijainen tehon arviointiin käytetty päätetapahtuma oli yhdistelmäpäätetapahtuma eli joko vaikea COVID‑19-tauti tai mistä tahansa syystä johtunut kuolema päivään 29 mennessä tutkittavilla, jotka saivat hoitoa 7 päivän kuluessa oireiden alkamisesta eivätkä olleet lähtötilanteessa sairaalahoidossa. Vaikea COVID‑19-tauti määriteltiin taudiksi, jossa ilmeni joko keuhkokuume (kuume, yskä, takypnea tai hengenahdistus sekä keuhkoinfiltraatteja rintakehän röntgenkuvassa tai keuhkojen tietokonetomografiassa) tai hypoksemia (SpO2 < 90 % huoneilmassa ja/tai vaikea hengitysvaikeus) ja pistemäärä WHO:n asteikolla [WHO Clinical Progression Scale] oli vähintään 5. EVUSHELD-valmisteen todettiin vähentävän tilastollisesti merkitsevästi vaikeita COVID‑19-tautitapauksia ja mistä tahansa syystä johtuneita kuolemia lumelääkkeeseen verrattuna (taulukko 5). Pienen otoskoon vuoksi valmisteen tehosta seropositiivisilla potilailla ei pystytä tekemään johtopäätöksiä.
Taulukko 5 Vaikean COVID-19-taudin tai mistä tahansa syystä johtuneen kuoleman ilmaantuvuus päivään 29 mennessä
| Potilasjoukko | Hoito | N | Tapahtumien määrä, n (%) | Suhteellisen riskin vähenemä, % (95 %:n luottamusväli) | p-arvo a |
| Potilaat, jotka eivät olleet sairaalahoidossa ja jotka saivat lääkettä ≤ 7 päivän kuluessa oireiden alkamisesta (muokattu täydellinen analyysijoukko) | EVUSHELDb | 407 | 18 (4,4 %) | 50 % (15; 71) | p = 0,010 |
| Lumelääke | 415 | 37 (8,9 %) | |||
| Kaikki satunnaistetut tutkittavat, sekä sairaalahoitoon otetut että ne, jotka eivät olleet sairaalahoidossa (täydellinen analyysijoukko) | EVUSHELDb | 446 | 24 (5,4 %) | 42 % (5; 64) | p = 0,028 |
| Lumelääke | 444 | 44 (9,2 %) |
N = analyysiin otettujen osallistujien määrä
a. Tulokset CMH-testistä, jossa käytettiin ositustekijänä aikaa oireiden alkamisesta (≤ 5 vs. > 5 päivää) ja riskiä, että tauti etenisi vaikeaksi COVID-19-taudiksi (suuri vs. pieni).
b. 300 mg tiksagevimabia ja 300 mg silgavimabia.
Puuttuvia vastetietoja ei imputoitu.
Potilailla, jotka eivät olleet sairaalahoidossa ja jotka saivat lääkettä 5 päivän kuluessa oireiden alkamisesta, suhteellisen riskin vähenemä oli 67 % (95 %:n luottamusväli 31; 84) (p = 0,002).
Ensisijaisen yhdistelmäpäätetapahtuman tuloksiin vaikutti merkittävästi vaikean COVID‑19-taudin ilmaantuvuus. Päivään 29 mennessä oli ilmoitettu 7 kuolemaa, joista 3 tapahtui EVUSHELD-ryhmässä ja 4 lumeryhmässä. Näistä 7 kuolemasta 2 tapausta ei liittynyt COVID‑19-tautiin. Molemmat näistä tapahtuivat EVUSHELD-ryhmässä, ja ne laskettiin mukaan ensisijaiseen yhdistelmäpäätetapahtumaan.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset EVUSHELD-valmisteen käytöstä COVID‑19-taudin ennaltaehkäisyssä ja hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Tiksagevimabin ja silgavimabin farmakokinetiikat ovat keskenään vastaavanlaiset, lineaariset ja annoksesta riippuvaiset, kun laskimoon annetut kerta-annokset ovat tiksagevimabilla 150 – 1 500 mg ja silgavimabilla 150 – 1 500 mg. Käsitystä tiksagevimabin, silgavimabin ja EVUSHELD-valmisteen annoksesta riippuvaisesta farmakokinetiikasta tukee populaatiofarmakokineettinen analyysi, jossa tarkasteltiin terveitä vapaaehtoisia ja tutkittavia, jotka osallistuivat kolmeen tiksagevimabia ja silgavimabia arvioineeseen vaiheen III tutkimukseen (PROVENT, jossa arvioitiin altistumista edeltävää ennaltaehkäisyä, STORMCHASER, jossa arvioitiin altistumisen jälkeistä ennaltaehkäisyä, ja TACKLE, jossa arvioitiin lievän ja keskivaikean COVID-19-taudin hoitoa), ja jossa arvioitiin lisäksi tietoja viidestä vaiheiden I ja II tutkimuksesta. Tutkimuksissa käytetyt lihakseen annetut annokset vaihtelivat 300 mg:sta (150 mg tiksagevimabia ja 150 mg silgavimabia) 600 mg:aan (300 mg tiksagevimabia ja 300 mg silgavimabia) ja laskimoon annetut annokset 300 mg:sta (150 mg tiksagevimabia ja 150 mg silgavimabia) 3 000 mg:aan (1 500 mg tiksagevimabia ja 1 500 mg silgavimabia).
Imeytyminen
Populaatiofarmakokineettisen mallinnuksen perusteella EVUSHELD-valmisteen huippupitoisuuden (Cmax) ennustettu mediaani seerumissa (90 %:n ennusteväli) 150 mg tiksagevimabia ja 150 mg silgavimabia sisältävän lihakseen annetun kerta-annoksen antamisen jälkeen oli 26,9 mikrog/ml (90 %:n ennusteväli 12,6; 53,7) ja mediaaniaika Cmax-arvon saavuttamiseen (Tmax) oli 19 vuorokautta (90 %:n ennusteväli 5; 45).
EVUSHELD-valmisteen ennustettu Cmax 300 mg tiksagevimabia ja 300 mg silgavimabia sisältävän lihakseen annetun kerta-annoksen antamisen jälkeen oli 53,9 mikrog/ml (90 %:n ennusteväli 25,2; 107,3), ja sen saavuttamiseen kuluneen ajan (Tmax) mediaani oli 19 vuorokautta (90 %:n ennusteväli 5; 46).
Arvioitu absoluuttinen hyötyosuus oli EVUSHELD-valmisteella 67,1 %, tiksagevimabilla 61,5 % ja silgavimabilla 65,8 %.
Jakautuminen
Farmakokineettisen mallinnuksen perusteella sentraalinen jakautumistilavuus oli tiksagevimabilla 3,17 l ja silgavimabilla 3,52 l. Perifeerinen jakautumistilavuus oli tiksagevimabilla 1,77 l ja silgavimabilla 1,82 l.
Biotransformaatio
Tiksagevimabin ja silgavimabin odotetaan hajoavan endogeenisten IgG-vasta-aineiden tavoin pieniksi peptideiksi ja niiden aminohappokomponenteiksi katabolisten reittien kautta.
Eliminaatio
EVUSHELD-valmisteen puhdistuman mediaani (95 %:n luottamusväli) oli 0,050 (0,049; 0,052) l/vrk, tiksagevimabin puhdistuman mediaani 0,046 (0,044; 0,047) l/vrk ja silgavimabin puhdistuman mediaani 0,052 (0,049; 0,054) l/vrk. Yksilöiden välinen vaihtelu oli EVUSHELD-valmisteella 43 %, tiksagevimabilla 41 % ja silgavimabilla 44 %. Arvioitu terminaalisen eliminaation puoliintumisajan mediaani (5. ja 95. persentiili) populaatiossa oli EVUSHELD-valmisteella 79 (46; 101) vuorokautta, tiksagevimabilla 81 (49; 106) vuorokautta ja silgavimabilla 78 (49; 97) vuorokautta.
EVUSHELD-valmisteen pitoisuuden ennustettu mediaani seerumissa 150 mg tiksagevimabia ja 150 mg silgavimabia sisältävän lihakseen annetun kerta-annoksen antamisen jälkeen oli 24,5 mikrog/ml (90 %:n ennusteväli 11,8; 44, 8) päivänä 29 ja 6,2 mikrog/ml (90 %:n ennusteväli 1,8; 14,7) päivänä 183.
EVUSHELD-valmisteen pitoisuuden ennustettu mediaani seerumissa 300 mg tiksagevimabia ja 300 mg silgavimabia sisältävän lihakseen annetun kerta-annoksen antamisen jälkeen oli 49,1 mikrog/ml (90 %:n ennusteväli 23,6; 89,5) päivänä 29 ja 12,5 mikrog/ml (90 %:n ennusteväli 3,6; 29,3) päivänä 183.
Tiksagevimabin ja silgavimabin puhdistumissa ei havaittu kliinisesti merkittäviä eroja TACKLE-tutkimukseen osallistuneiden COVID‑19-tautia sairastaneiden tutkittavien ja ennaltaehkäisevää hoitoa arvioineisiin tutkimuksiin osallistuneiden tutkittavien välillä.
Erityiset potilasryhmät
Munuaisten vajaatoiminta
Ei ole tehty erityisiä tutkimuksia, joilla olisi arvioitu munuaisten vajaatoiminnan vaikutuksia tiksagevimabin ja silgavimabin farmakokinetiikkaan.
Tiksagevimabi ja silgavimabi eivät eliminoidu muuttumattomina virtsaan, joten munuaisten vajaatoiminnan ei odoteta vaikuttavan merkittävästi tiksagevimabi- tai silgavimabialtistukseen. Myöskään dialyysin ei odoteta vaikuttavan tiksagevimabin ja silgavimabin farmakokinetiikkaan.
Populaatiofarmakokineettisen analyysin perusteella tiksagevimabin ja silgavimabin puhdistumissa ei ole eroa munuaisten vajaatoimintaa sairastavilla potilailla (arvioitu lähtötilanteen eGFR-arvon ja kreatiniinipuhdistuman perusteella) verrattuna potilaisiin, joiden munuaiset toimivat normaalisti. Populaatiofarmakokineettisessä mallissa vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden määrä oli liian pieni johtopäätösten tekemiseen.
Maksan vajaatoiminta
Ei ole tehty erityisiä tutkimuksia, joilla olisi arvioitu maksan vajaatoiminnan vaikutuksia tiksagevimabin ja silgavimabin farmakokinetiikkaan. Maksan vajaatoiminnalla odotetaan olevan vähäinen vaikutus tiksagevimabin ja silgavimabin farmakokinetiikkaan.
Tiksagevimabin ja silgavimabin odotetaan kataboloituvan monissa kudoksissa hajoamalla proteolyyttisesti aminohapoiksi ja muuntumalla kierrätysmekanismilla muiksi proteiineiksi. Maksan vajaatoiminnan ei siis odoteta vaikuttavan tiksagevimabi- tai silgavimabialtistukseen.
Iäkkäät
Yhdistetyssä farmakokineettisessä analyysissä mukana olleista tutkittavista 17,6 % (N = 871) oli vähintään 65-vuotiaita ja 3,2 % (N = 156) vähintään 75-vuotiaita. Tiksagevimabin ja silgavimabin farmakokinetiikassa ei ole kliinisesti merkittäviä eroja iäkkäiden (vähintään 65-vuotiaiden) ja nuorempien tutkittavien välillä.
Pediatriset potilaat
Tiksagevimabin ja silgavimabin farmakokinetiikkaa ei ole arvioitu alle 18-vuotiailla.
Suositellun annostusohjelman odotetaan populaatiofarmakokineettisen mallinnuksen ja simulaation perusteella aikaansaavan vähintään 12-vuotiailla ja vähintään 40 kg painavilla nuorilla vastaavanlaisia tiksagevimabi- ja silgavimabialtistuksia seerumissa kuin on havaittu aikuisilla, koska ennaltaehkäisyä ja hoitoa arvioineisiin kliinisiin tutkimuksiin on osallistunut samanpainoisia aikuisia.
Suuri kehonpaino
Populaatiofarmakokineettisen analyysin perusteella havaittiin, että EVUSHELD valmisteen huippupitoisuus seerumissa ja pitoisuus 6 kuukauden kohdalla pienenevät suuremman kehonpainon myötä. Huippupitoisuuden seerumissa ja pitoisuuden 6 kuukauden kohdalla ennustettiin olevan 108 kg (87,5. persentiili) painavalla aikuisella noin 24 % pienempi kuin 81 kg (mediaani) painavalla aikuisella.
Muut erityiset potilasryhmät
Populaatiofarmakokineettisen analyysin perusteella sukupuolella, iällä, rodulla, etnisellä taustalla, sydän- ja verisuonisairauksilla, diabeteksella tai immuunipuutoksella ei ollut kliinisesti merkittäviä vaikutuksia tiksagevimabin tai silgavimabin farmakokinetiikkaan.
Prekliiniset tiedot turvallisuudesta
Tiksagevimabilla ja silgavimabilla ei ole tehty karsinogeenisuutta, mutageenisuutta ja lisääntymistoksisuutta koskevia tutkimuksia.
Ei-kliinisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille perustuen kudoksiin sitoutumista arvioineisiin tutkimuksiin ja jaavanmakakeilla toteutettuun kerta-annoksen toksisuutta arvioineeseen tutkimukseen, jossa selvitettiin myös farmakologista turvallisuutta ja paikallista siedettävyyttä.
Vasta‑ainevälitteinen infektion vaikeutuminen (ADE)
Tiksagevimabin ja silgavimabin kykyä välittää vasta-aineista riippuvaista viruksen pääsyä soluihin arvioitiin Raji-soluissa, jotka ilmensivät Fc-gamma-reseptoria II ja joita inkuboitiin yhdessä rekombinantin viruksen kanssa, johon oli pseudotyypitetty SARS‑CoV‑2-viruksen piikkiproteiini. Arvioinnissa käytetty vasta-ainepitoisuus oli 6,6 nM (1 mikrog/ml) – 824 pM (125 ng/ml). Tiksagevimabi, silgavimabi ja niiden yhdistelmä eivät välittäneet pseudoviruksen pääsyä näihin soluihin.
Vasta-ainevälitteisen infektion vaikeutumisen mahdollisuutta arvioitiin EVUSHELD-valmisteella myös SARS‑CoV‑2-infektion kädellismallissa (muilla kädellisillä kuin ihmisillä). Valmisteen anto suonensisäisesti ennen viruksen inokulaatiota paransi annosriippuvaisesti kaikkia mitattuja hoitotuloksia (viruksen kokonais-RNA-määrää keuhkoissa tai nenän limakalvoilla, TCID50-mittauksiin perustuvia infektiokykyisten virusten määriä keuhkoissa sekä histologisiin mittauksiin perustuvia tietoja keuhkovaurioista ja patologisista muutoksista). Näyttöä taudin vaikeutumisesta ei havaittu millään arvioidulla annoksella, ei myöskään neutraloivaa annosta pienemmillä annoksilla (pienimmillään 0,04 mg/kg).
Farmaseuttiset tiedot
Apuaineet
Histidiini
Histidiinihydrokloridimonohydraatti
Sakkaroosi
Polysorbaatti 80
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo
2 vuotta.
Valmistellut ruiskut
Valmisteltujen ruiskujen sisältö on annettava välittömästi. Jos lääkkeen anto välittömästi ei ole mahdollista, käytönaikaiset säilytysajat ja käyttöä edeltävät säilytysolosuhteet ovat käyttäjän vastuulla, eivätkä ne yleensä saa ylittää neljää tuntia 2–25 °C:n lämpötilassa.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Ei saa jäätyä.
Älä ravista.
Säilytysolosuhteet sen jälkeen, kun neula on viety injektiopulloon ensimmäisen kerran ja ruiskut on valmisteltu, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
EVUSHELD injektioneste, liuos
150 mg + 150 mg 1,5 ml + 1,5 ml (100 mg/ml+100 mg/ml) (-)
PF-selosteen tieto
Tiksagevimabi-injektiopullo
1,5 ml injektionestettä kirkkaassa lasisessa injektiopullossa, jossa on klooributyylielastomeeritulppa ja joka on suljettu tummanharmaalla alumiinisella nostokorkilla.
Silgavimabi-injektiopullo
1,5 ml injektionestettä kirkkaassa lasisessa injektiopullossa, jossa on klooributyylielastomeeritulppa ja joka on suljettu valkoisella alumiinisella nostokorkilla.
Pakkauskoko: Yksi pahvikotelo sisältää 2 injektiopulloa: yhden tiksagevimabi-injektiopullon ja yhden silgavimabi-injektiopullon.
Valmisteen kuvaus:
Kirkas tai opaalinhohtoinen, väritön tai hiukan kellertävä liuos, jonka pH on 6,0.
Käyttö- ja käsittelyohjeet
Käsittelyohjeet
Tätä lääkevalmistetta saavat käsitellä vain terveydenhuollon ammattilaiset, ja heidän on noudatettava aseptista tekniikkaa jokaisen annoksen steriiliyden varmistamiseksi.
Tarkasta injektiopullot silmämääräisesti hiukkasten ja värimuutosten varalta. Sekä tiksagevimabi että silgavimabi ovat kirkkaita tai opaalinhohtoisia, värittömiä tai hiukan kellertäviä liuoksia. Hävitä injektiopullot, jos liuos on sameaa tai siinä näkyy värimuutoksia tai hiukkasia. Älä ravista injektiopulloja.
Jokainen annos tiksagevimabia ja silgavimabia vedetään kahteen erilliseen ruiskuun annettavaksi lihaksensisäisesti kahteen eri lihakseen, mieluiten pakaralihaksiin.
Valmisteltujen ruiskujen säilytysolosuhteet, ks. kohta Kestoaika.
Käyttämätön liuos on hävitettävä.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
EVUSHELD injektioneste, liuos
150 mg + 150 mg 1,5 ml + 1,5 ml
- Ei korvausta.
ATC-koodi
J06BD03
Valmisteyhteenvedon muuttamispäivämäärä
12.10.2023
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi