
VYEPTI infuusiokonsentraatti, liuosta varten 100 mg, 300 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Vyepti 100 mg infuusiokonsentraatti, liuosta varten
Jokainen injektiopullo konsentraattia sisältää 100 mg eptinetsumabia millilitraa kohti.
Vyepti 300 mg infuusiokonsentraatti, liuosta varten
Jokainen injektiopullo konsentraattia sisältää 300 mg eptinetsumabia 3 millilitraa kohti.
Eptinetsumabi on humanisoitu monoklonaalinen vasta-aine, joka tuotetaan Pichia pastoris -hiivasoluista.
Apuaine(et), joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 40,5 mg sorbitolia per millilitra ja 0,15 mg polysorbaatti 80:tä per millilitra.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Vyepti on tarkoitettu migreenin estohoitoon aikuisilla, joilla on vähintään 4 migreenipäivää kuukaudessa.
Ehto
Hoidon saa aloittaa migreenin toteamiseen ja hoitoon perehtynyt lääkäri.
Annostus ja antotapa
Hoidon aloittaa migreenin toteamiseen ja hoitoon perehtynyt terveydenhuollon ammattilainen. Vyepti-infuusion aloittaa ja sitä valvoo terveydenhuollon ammattilainen.
Annostus
Suositeltu annos on 100 mg, joka annetaan infuusiona laskimoon 12 viikon välein. Jotkut potilaat saattavat hyötyä 300 mg:n annoksesta, joka annetaan infuusiona laskimoon 12 viikon välein (ks. kohta Farmakodynamiikka).
Annoksen suurentamisen tarve tulee arvioida 12 viikon kuluessa hoidon aloittamisesta. Annostusta vaihdettaessa uuden hoito-ohjelman mukainen ensimmäinen annos annetaan hoidon seuraavana annostuspäivänä.
Hoidon kokonaishyöty ja jatkaminen arvioidaan 6 kuukauden kuluttua hoidon aloittamisesta. Päätös hoidon jatkamisesta tehdään kunkin potilaan kohdalla yksilöllisesti.
Erityiset potilasryhmät
Iäkkäät (vähintään 65-vuotiaat)
Vyepti-valmisteen käytöstä ≥ 65-vuotiailla potilailla on vähän tietoja. Annosta ei tarvitse muuttaa iäkkäillä potilailla, koska ikä ei vaikuta eptinetsumabin farmakokinetiikkaan.
Munuaisten vajaatoiminta / maksan vajaatoiminta
Annosta ei tarvitse muuttaa potilailla, joilla on munuaisten vajaatoiminta tai maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Vyepti-valmisteen turvallisuutta ja tehoa 6–18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei tällä hetkellä ole saatavilla.
Ei ole asianmukaista käyttää Vyepti-valmistetta alle 6 vuoden ikäisillä lapsilla migreenin estohoitoon.
Antotapa
Vyepti annetaan laskimoon vasta laimentamisen jälkeen.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vyepti annetaan laimentamisen jälkeen noin 30 minuutin kestoisena infuusiona.
Hoitavan terveydenhuollon ammattilaisen täytyy tarkkailla tai valvoa potilaita infuusion aikana ja sen jälkeen tavallisen kliinisen käytännön mukaisesti.
Vyepti-valmistetta ei saa antaa bolusinjektiona.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun lääkevalmisteen nimi ja eränumero dokumentoitava selkeästi.
Potilaat, joilla kardiovaskulaarisia, neurologisia tai psykiatrisia sairauksia
Potilaita, joilla oli aiempia kliinisesti merkittäviä kardiovaskulaarisia sairauksia (esim. iskeeminen sydänsairaus, kardiovaskulaarinen sairaus, vaskulaarinen iskemia tai tromboembolisia tapahtumia), ei otettu mukaan kliinisiin tutkimuksiin (ks. kohta Farmakodynamiikka). Potilaista, joilla on kardiovaskulaarinen sairaus ja/tai diabeteksen, verenkiertosairauksien ja hyperlipidemian tyyppisiä kardiovaskulaaristen sairauksien riskitekijöitä, on saatavilla vähän turvallisuustietoja.
Potilaita, joilla on aiempia neurologisia sairauksia, tai potilaita, joilla on kontrolloimattomia ja/tai hoitamattomia psyykkisiä sairauksia, ei otettu mukaan kliinisiin tutkimuksiin. Näistä potilaista on saatavilla vähän turvallisuustietoja.
Vakava yliherkkyys
Vakavia yliherkkyysreaktioita, muun muassa anafylaktisia reaktioita, on ilmoitettu ja voi kehittyä minuuttien kuluessa infuusiosta. Useimmat yliherkkyysreaktiot tapahtuivat infuusion aikana eivätkä olleet vakavia (ks. kohta Haittavaikutukset). Jos vakava yliherkkyysreaktio tapahtuu, täytyy Vyepti-valmisteen antaminen lopettaa välittömästi ja aloittaa asianmukainen hoito. Jos yliherkkyysreaktio ei ole vakava, hoidon jatkaminen Vyepti-valmisteella on hoitavan lääkärin harkinnassa huomioiden hyödyt ja riskit yksittäiselle potilaalle.
Apuaineet
Vyepti sisältää sorbitolia (E420). Tätä lääkevalmistetta ei saa antaa potilaille, joilla on perinnöllinen fruktoosi-intoleranssi (HFI), ellei se ole aivan välttämätöntä.
Jokaiselta potilaalta täytyy ottaa tarkat tiedot perinnöllisen fruktoosi-intoleranssin oireista ennen tämän lääkevalmisteen antamista.
Yhteisvaikutukset
Sytokromi P450 -entsyymit eivät metaboloi eptinetsumabia. Siksi eptinetsumabin yhteisvaikutuksia samanaikaisesti käytettyjen lääkkeiden kanssa, jotka ovat sytokromi P450 -entsyymien substraatteja, induktoreja tai estäjiä, pidetään epätodennäköisinä.
Raskaus ja imetys
Raskaus
On vain vähän tietoja eptinetsumabin käytöstä raskaana oleville naisille. Eptinetsumabilla tehdyissä eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Ihmisen IgG:n tiedetään läpäisevän istukkaesteen, joten eptinetsumabi saattaa siirtyä äidistä kehittyvään sikiöön.
Varmuuden vuoksi Vyepti-valmisteen käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei ole tietoja eptinetsumabin erittymisestä ihmisen rintamaitoon, vaikutuksista imeväiseen tai vaikutuksista maidontuotantoon. Ihmisen IgG:n tiedetään erittyvän rintamaitoon muutaman ensimmäisen päivän aikana synnytyksen jälkeen ja vähenevän sitten nopeasti pieneen pitoisuuteen; näin ollen riskiä imeväiselle ei voida sulkea pois tällä lyhyellä ajanjaksolla. Sen jälkeen eptinetsumabin käyttöä voidaan harkita imetyksen aikana vain, jos se on kliinisesti tarpeen.
Hedelmällisyys
Eptinetsumabin vaikutusta ihmisen hedelmällisyyteen ei ole arvioitu. Eptinetsumabilla tehdyissä eläinkokeissa ei ole havaittu naaraiden ja urosten hedelmällisyyteen kohdistuvaa vaikutusta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Vyepti-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yli 5 900 potilasta on hoidettu VYEPTI-valmisteella kliinisissä tutkimuksissa. Näistä noin 3 000 potilasta on altistunut valmisteelle vähintään 6 kuukauden ajan ja 1 600 potilasta on altistunut valmisteelle vähintään 1 vuoden ajan.
Yleiset haittavaikutukset migreenin estohoidon lumekontrolloiduissa kliinisissä tutkimuksissa olivat yliherkkyys ja infuusioon liittyvät reaktiot (ks. jäljempänä). Useimmat yliherkkyysreaktiot tapahtuivat infuusion aikana eivätkä olleet vakavia. Infuusiokohtaan liittyviä haittatapahtumia esiintyi harvoin ja samassa suhteessa VYEPTI-valmistetta ja lumelääkettä saaneilla potilailla (< 1 %) ilman ilmeistä yhteyttä VYEPTI-annokseen. Useimmin esiintynyt infuusiokohtaan liittyvä haittatapahtuma oli infuusiokohdan ekstravasaatio, jota esiintyi ≤ 0,5 %:lla VYEPTI-valmistetta ja lumelääkettä saaneista potilaista.
Taulukoitu luettelo haittavaikutuksista
Haittavaikutukset kliinisistä tutkimuksista ja markkinoille saattamisen jälkeisestä kokemuksesta (taulukko 1) on luokiteltu MedDRA:n elinjärjestelmäluokituksen ja esiintyvyyksien mukaisesti. Esiintyvyydet on arvioitu seuraavan käytännön mukaisesti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000).
Taulukko 1: Luettelo haittavaikutuksista
| Elinjärjestelmäluokka | Haittavaikutuksen suositettava termi | Esiintyvyys |
| Immuunijärjestelmä | Yliherkkyysreaktiot | Yleinen |
| Anafylaktinen reaktio | Melko harvinainen | |
| Yleisoireet ja antopaikassa todettavat haitat | Infuusioon liittyvä reaktio Uupumus | Yleinen |
Valikoitujen haittavaikutusten kuvaus
Yliherkkyys ja infuusioon liittyvät reaktiot
Vakavia yliherkkyysreaktioita, muun muassa anafylaktisia reaktioita, on ilmoitettu ja voi kehittyä minuuttien kuluessa infuusiosta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Raportoituihin anafylaktisiin reaktioihin on kuulunut hypotension oireita ja hengitysvaikeuksia, ja ne ovat johtaneet VYEPTI-hoidon keskeyttämiseen. Kliinisissä tutkimuksissa muita yliherkkyysreaktioita, muun muassa angioedeemaa, nokkosihottumaa, punoitusta, ihottumaa ja kutinaa, raportoitiin noin 4 %:lla potilaista, jotka saivat 300 mg:n annosta, 3 %:lla potilaista, jotka saivat 100 mg:n annosta, ja 2 %:lla potilaista, jotka saivat lumelääkettä (suolaliuosta).
Muita eptinetsumabi-infuusion yhteydessä ilmoitettuja oireita ovat mm. hengitystieoireet (nenän tukkoisuus, nuha, nielun ärsytys, yskä, aivastelu, hengenahdistus) ja uupumus (ks. alla). Useimmat näistä tapahtumista eivät olleet vakavia, ja ne olivat luonteeltaan ohimeneviä.
Uupumus
Noin 3 % eptinetsumabia saaneista potilaista ja 2 % lumelääkettä saaneista potilaista koki uupumusta lumekontrolloiduissa kliinisissä tutkimuksissa. Uupumus oli yleisintä ensimmäisenä infuusiopäivänä. Ensimmäisen viikon ja seuraavien infuusioiden jälkeen uupumuksen esiintymistä raportoitiin vähemmän ja lumelääkkeeseen verrattavasti.
Immunogeenisuus
Keskeisissä lumekontrolloiduissa tutkimuksissa, joissa hoidon keston vaihteluväli oli 3–12 kuukautta, lääkevasta-aineiden ilmaantuvuus oli 15,4 % ja neutraloivien vasta-aineiden ilmaantuvuus oli 6,3 %. Lääkevasta-aineiden ilmaantuvuus oli suurimmillaan viikolla 24 ja väheni sitten vakaasti jopa jokaisen seuraavan 12 viikon välein annetun annoksen jälkeen.
Pitkäkestoisessa, toistuvan altistuksen tutkimuksissa, jossa hoito kesti vähintään 1 vuoden ajan, lääkevasta-aineiden ilmaantuvuus vaihteli välillä 18–21 % ja neutraloivien vasta-aineiden ilmaantuvuus vaihteli välillä 4–7 %. PREVAIL-tutkimuksessa, jossa potilaat saivat enintään 8 infuusiota, lääkevasta-aineita kehittyi 18 %:lle potilaista ja neutraloivia vasta-aineita 7 %:lle potilaista. 5,3 % potilaista oli lääkevasta-ainepositiivisia viikolla 48, 4 % oli lääkevasta-ainepositiivisia viikolla 72 ja kaikki potilaat yhtä seurannasta poisjäänyttä lukuun ottamatta olivat lääkevasta-ainenegatiivisia viikolla 104 (tutkimuksen viimeinen arviointi).
Kliinisissä tutkimuksissa eptinetsumabin jäännöspitoisuus plasmassa oli matalampi potilailla, joille kehittyi eptinetsumabin vasta-aineita. Eptinetsumabin vasta-aineiden kehittymisen vaikutuksesta tehoon tai turvallisuuteen ei ollut näyttöä kliinisissä tutkimuksissa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Ihmisille on annettu laskimonsisäisesti annoksia 1 000 mg:aan asti ilman siedettävyysongelmia tai kliinisesti merkittäviä haittavaikutuksia.
Yliannostuksessa potilasta hoidetaan oireenmukaisesti ja tukitoimenpiteitä tehdään tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: kipulääkkeet, kalsitoniinigeeniin liittyvän peptidin (CGRP) antagonistit, ATC-koodi: N02CD05.
Vaikutusmekanismi
Eptinetsumabi on rekombinantti humanisoitu immunoglobuliini G1 (IgG1) -vasta-aine, joka sitoutuu ihmisen kalsitoniinigeeniin liittyvän peptidin (CGRP) α- ja β-muotojen ligandiin pienellä pikomolaarisella affiniteetilla (4 ja 3 pM Kd tässä järjestyksessä). Eptinetsumabi estää CGRP-reseptorien aktivoitumista ja siten migreenikohtausten alkamiseen liittyvää fysiologisten tapahtumien alavirran kaskadia.
Eptinetsumabi estää α- ja β-CGRP-välitteistä neurogeenistä tulehdusta ja verisuonten laajenemista.
Eptinetsumabi on erittäin selektiivinen (> 100 000-kertainen verrattuna lähisukuisiin neuropeptideihin amyliiniin, kalsitoniiniin, adrenomedulliiniin ja intermediiniin).
Kliininen teho ja turvallisuus
Vyepti-valmistetta (eptinetsumabi) arvioitiin migreenin estohoitona kahdessa lumekontrolloidussa avaintutkimuksessa: PROMISE 1 tehtiin episodista migreeniä sairastavilla potilailla (n = 888) ja PROMISE 2 kroonista migreeniä sairastavilla potilailla (n = 1 072). Osallistuneilla potilailla oli ollut migreeni (auraoirein tai ilman) vähintään 12 kuukauden ajan kansainvälisen päänsärkysairauksien luokituksen diagnostisten kriteerien mukaan (ICHD-II tai III).
PROMISE 1: episodinen migreeni
PROMISE 1 oli rinnakkaisryhmillä toteutettu, kaksoissokkoutettu, lumekontrolloitu tutkimus, jossa arvioitiin Vyepti-valmisteen tehoa ja turvallisuutta episodisen migreenin estohoitona aikuisilla. 665 potilasta satunnaistettiin saamaan lumelääkettä (N = 222), 100 mg eptinetsumabia (N = 221) tai 300 mg eptinetsumabia (N = 222) 12 viikon välein 48 viikon ajan (4 infuusiota). Episodisen migreenin määritelmänä oli ≥ 4 ja ≤ 14 päänsärkypäivää, joista vähintään 4 oli migreenipäiviä kullakin 28 päivän jaksolla 3 kuukauden aikana ennen seulontaa ja vahvistettuna lähtötasojakson aikana. Potilaille sallittiin tutkimuksen aikana samanaikainen migreenin tai päänsäryn kohtauslääkitys, myös migreenin täsmälääkitys (esim. triptaanit ja ergotamiinijohdannaiset). Muiden migreenin estohoitojen säännöllistä käyttöä (yli 7 päivää kuukaudessa) ei sallittu.
Ensisijainen tehon päätetapahtuma oli muutos lähtötasosta keskimääräisissä kuukausittaisissa migreenipäivissä (MMD) viikoilla 1–12. Tärkeimpiä toissijaisia päätetapahtumia olivat ≥ 50 %:n ja ≥ 75 %:n vasteen migreenihoitoon saaneiden osuudet, jotka määriteltiin niiden potilaiden osuutena, joiden migreenipäivät vähenivät vähintään määrätyllä prosenttimäärällä viikoilla 1–12, ≥ 75 %:n vasteen migreenihoitoon saaneiden osuus viikoilla 1–4 ja niiden potilaiden prosenttimäärä, joilla oli migreeni ensimmäisen annostuksen jälkeisenä päivänä (päivä 1).
Potilaiden keskimääräinen ikä oli 40 vuotta (vaihteluväli: 18–71 vuotta), 84 % oli naisia ja 84 % oli valkoihoisia. Lähtötasolla migreenipäivien keskimääräinen määrä kuukaudessa oli 8,6 ja niiden potilaiden osuus, joilla oli migreeni tiettynä päivänä, oli 31 %; luvut olivat samanlaiset eri hoitoryhmissä.
Keskimääräisten kuukausittaisten migreenipäivien väheneminen lumelääkkeeseen verrattuna havaittiin kummallakin annoksella annostelun jälkeen ensimmäisestä päivästä alkaen.
Kuva 1: Kuukausittaisten migreenipäivien keskimääräiset muutokset lähtötasosta PROMISE 1 -tutkimuksessa
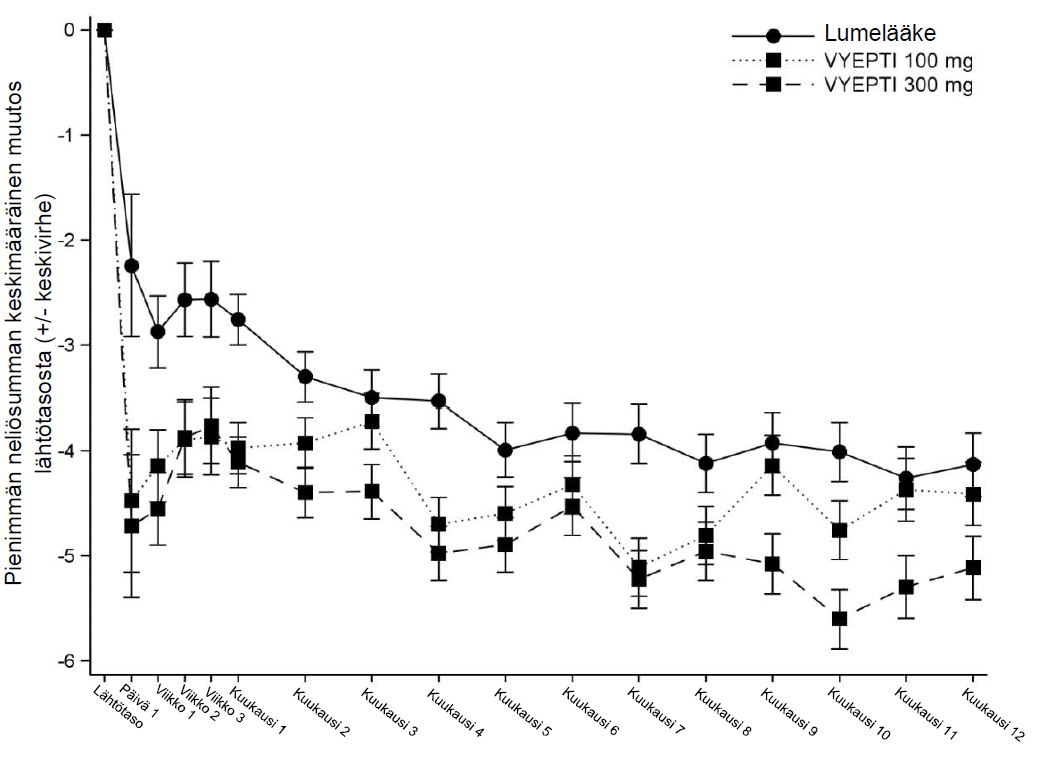
Vyepti = eptinetsumabi
Jokaisessa aikapisteessä käytettiin kovarianssianalyysiä (ANCOVA), joka sisälsi hoidon ja estolääkityksen käytön tekijöinä ja lähtötason migreenipäivät jatkuvana kovariaattina arvioitaessa keskimääräistä muutosta lähtötasosta.
Taulukko 2: Ensisijaisten ja tärkeimpien toissijaisten tehon päätetapahtumien tulokset PROMISE 1 -tutkimuksessa (episodinen migreeni)
Vyepti 100 mg N = 221 | Vyepti N = 222 | Lumelääke N = 222 | |
| Kuukausittaiset migreenipäivät (MMD) – viikot 1–12 | |||
| Lähtötaso | 8,7 | 8,6 | 8,4 |
| Keskimääräinen muutos | -3,9 | -4,3 | -3,2 |
| Ero lumelääkkeeseen | -0,7 | -1,1 | |
| 95 % lv | (-1,3, -0,1) | (-1,7, -0,5) | |
| p-arvo verrattuna lumelääkkeeseen | 0,0182 | 0,0001 | |
| ≥ 75 %:n MMD-vasteen saaneet – viikot 1–4 | |||
| Vasteen saaneet | 30,8 % | 31,5 % | 20,3 % |
| Ero lumelääkkeeseen | 10,5 % | 11,3 % | |
| p-arvo verrattuna lumelääkkeeseen | 0,0112 | 0,0066 | |
| ≥ 75 %:n MMD-vasteen saaneet – viikot 1–12 | |||
| Vasteen saaneet | 22,2 % | 29,7 % | 16,2 % |
| Ero lumelääkkeeseen | 6,0 % | 13,5 % | |
| p-arvo verrattuna lumelääkkeeseen | 0,1126 | 0,0007 | |
| ≥ 50 %:n MMD-vasteen saaneet – viikot 1–12 | |||
| Vasteen saaneet | 49,8 % | 56,3 % | 37,4 % |
| Ero lumelääkkeeseen | 12,4 % | 18,9 % | |
| p-arvo verrattuna lumelääkkeeseen | 0,0085 | 0,0001 | |
PROMISE 2: krooninen migreeni
PROMISE 2 oli rinnakkaisryhmillä toteutettu, kaksoissokkoutettu, lumekontrolloitu globaali tutkimus, jossa arvioitiin Vyepti-valmisteen tehoa ja turvallisuutta kroonisen migreenin estohoitona aikuisilla. Yhteensä 1 072 potilasta satunnaistettiin saamaan lumevalmistetta (N = 366), 100 mg eptinetsumabia (N = 356) tai 300 mg eptinetsumabia (N = 350) 12 viikon välein 24 viikon ajan (2 infuusiota). Kroonisen migreenin määritelmänä oli ≥ 15 – ≤ 26 päänsärkypäivää, joista ≥ 8 arvioitiin migreenipäiviksi 3 kuukauden aikana ennen seulontaa ja vahvistettiin 28 päivän seulontajakson aikana. Tutkimuksen aikana potilaat saivat käyttää migreenin tai päänsäryn kohtaus- tai estolääkitystä vakiintuneella vakaalla hoito-ohjelmalla (pois lukien onabotuliinitoksiini A).
Tutkimusryhmään kuului yhteensä 431 potilasta (40 %), joilla oli kroonisen migreenin ja särkylääkepäänsäryn kaksoisdiagnoosi (liittyen triptaanien, ergotamiinin tai kipulääkkeiden yhdistelmän liikakäyttöön > 10 päivänä kuukaudessa tai asetaminofeenin, asetyylisalisyylihapon tai ei-steroidaalisten tulehduskipulääkkeiden liikakäyttöön ≥ 15 päivänä kuukaudessa) vahvistettuna seulontajakson aikana.
Ensisijainen tehon päätetapahtuma oli muutos lähtötasosta keskimääräisissä kuukausittaisissa migreenipäivissä (MMD) viikoilla 1–12. Tärkeimpiä toissijaisia päätetapahtumia olivat ≥ 50 %:n ja ≥ 75 %:n vasteen migreenihoitoon saaneiden osuudet, jotka määriteltiin niiden potilaiden osuutena, joiden migreenipäivät vähenivät vähintään määrätyllä prosenttimäärällä viikoilla 1–12, ≥ 75 %:n vasteen migreenihoitoon saaneiden osuus viikoilla 1–4, niiden potilaiden prosenttimäärä, joilla oli migreeni annostuksen jälkeisenä päivänä, migreenin esiintyvyyden väheneminen lähtötasosta viikolla 4, päänsärkytestin (HIT-6) kokonaispisteiden muutos lähtötasosta viikolla 12 (vain 300 mg:n annoksella) ja migreenin kohtauslääkkeen keskimääräisten käyttöpäivien muutos lähtötasosta viikoilla 1–12 (vain 300 mg:n annoksella).
Potilaiden keskimääräinen ikä oli 41 vuotta (vaihteluväli: 18–65 vuotta), 88 % oli naisia ja 91 % oli valkoihoisia. 41 % potilaista käytti samanaikaista migreenin estolääkitystä. Lähtötasolla migreenipäivien keskimääräinen määrä kuukaudessa oli 16,1, ja niiden potilaiden osuus, joilla oli migreeni tiettynä päivänä, oli 57,6 %; luvut olivat samanlaiset eri hoitoryhmissä.
Keskimääräisten kuukausittaisten migreenipäivien väheneminen lumelääkkeeseen verrattuna havaittiin kummallakin annoksella annostelun jälkeen ensimmäisestä päivästä alkaen.
Kuva 2: Kuukausittaisten migreenipäivien keskimääräiset muutokset lähtötasosta PROMISE 2 -tutkimuksessa
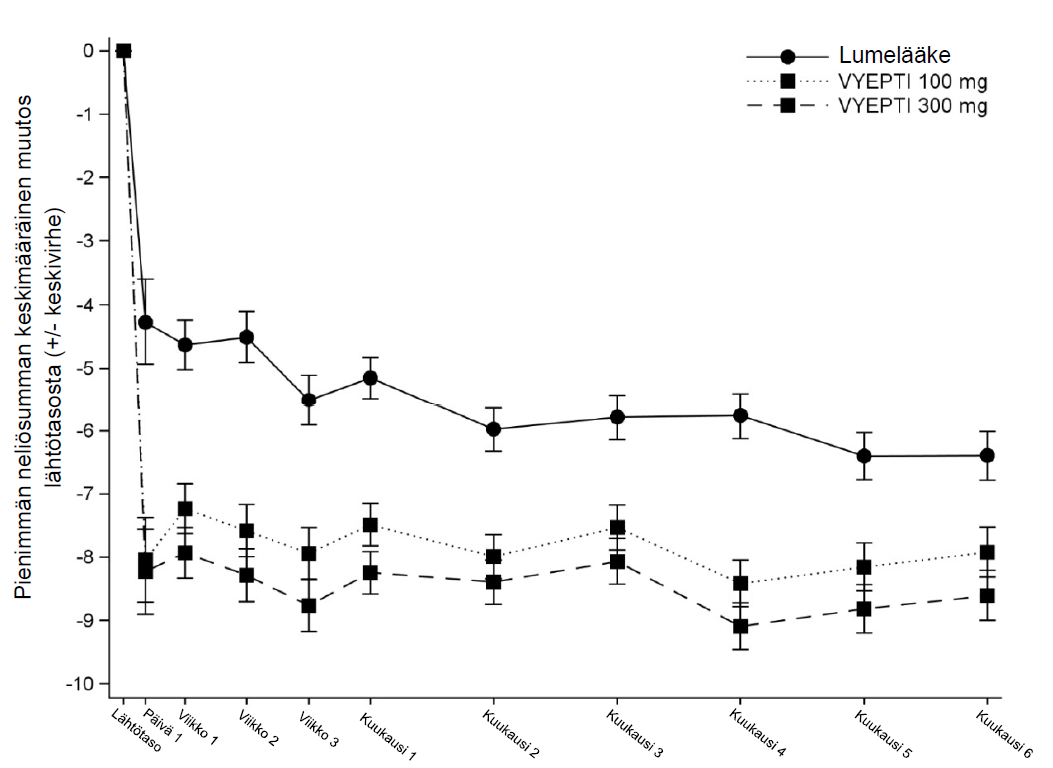
Vyepti = eptinetsumabi
Jokaisessa aikapisteessä käytettiin kovarianssianalyysiä (ANCOVA), joka sisälsi hoidon tekijänä ja lähtötason migreenipäivät jatkuvana kovariaattina arvioitaessa keskimääräistä muutosta lähtötasosta.
Taulukko 3: Ensisijaisten ja tärkeimpien toissijaisten tehon päätetapahtumien tulokset PROMISE 2 -tutkimuksessa (krooninen migreeni)
Vyepti 100 mg N = 356 | Vyepti 300 mg N = 350 | Lumelääke N = 366 | |
| Kuukausittaiset migreenipäivät (MMD) – viikot 1–12 | |||
| Lähtötaso | 16,1 | 16,1 | 16,2 |
| Keskimääräinen muutos | -7,7 | -8,2 | -5,6 |
| Ero lumelääkkeeseen | -2,0 | -2,6 | |
| 95 % lv | (-2,9, -1,2) | (-3,5, -1,7) | |
| p-arvo verrattuna lumelääkkeeseen | < 0,0001 | < 0,0001 | |
| ≥ 75 %:n MMD-vasteen saaneet – viikot 1–4 | |||
| Vasteen saaneet | 30,9 % | 36,9 % | 15,6 % |
| Ero lumelääkkeeseen | 15,3 % | 21,3 % | |
| p-arvo verrattuna lumelääkkeeseen | < 0,0001 | < 0,0001 | |
| ≥ 75 %:n MMD-vasteen saaneet – viikot 1–12 | |||
| Vasteen saaneet | 26,7 % | 33,1 % | 15,0 % |
| Ero lumelääkkeeseen | 11,7 % | 18,1 % | |
| p-arvo verrattuna lumelääkkeeseen | 0,0001 | < 0,0001 | |
| ≥ 50 %:n MMD-vasteen saaneet – viikot 1–12 | |||
| Vasteen saaneet | 57,6 % | 61,4 % | 39,3 % |
| Ero lumelääkkeeseen | 18,2 % | 22,1 % | |
| p-arvo verrattuna lumelääkkeeseen | < 0,0001 | < 0,0001 | |
| HIT-6-pisteet – viikko 12a | |||
| Lähtötaso | 65,0 | 65,1 | 64,8 |
| Keskimääräinen muutos | -6,2 | -7,3 | -4,5 |
| Ero lumelääkkeeseen | -1,7 | -2,9 | |
| 95 % lv | (-2,8, -0,7) | (-3,9, -1,8) | |
| p-arvo verrattuna lumelääkkeeseen | 0,0010 | < 0,0001 | |
| Kohtauslääkkeen käyttöpäivät kuukaudessa – viikot 1–12a,b | |||
| Lähtötaso | 6,6 | 6,7 | 6,2 |
| Keskimääräinen muutos | -3,3 | -3,5 | -1,9 |
| Ero lumelääkkeeseen | -1,2 | -1,4 | |
| 95 % lv | (-1,7, -0,7) | (-1,9, -0,9) | |
| p-arvo verrattuna lumelääkkeeseen | < 0,0001 | < 0,0001 | |
a 100 mg:n annoksen päätetapahtuma ei ollut ennalta määritelty tärkeä toissijainen päätetapahtuma.
b Perustaso oli 28 päivän seulontajakson keskiarvo ennen hoidon saamista.
Potilaat, joilla oli diagnosoitu särkylääkepäänsärky
431 (40 %) potilaalla, joilla oli diagnosoitu särkylääkepäänsärky (MOH) PROMISE-2-tutkimuksessa, kuukausittaisten migreenipäivien (MMD) keskimääräinen muutos lähtötasosta (viikoilla 1–12) oli Vyepti-valmisteen 100 mg:n annoksella -8,4 päivää, Vyepti-valmisteen 300 mg:n annoksella -8,6 päivää ja lumelääkkeellä -5,4 päivää (keskimääräinen ero lumelääkkeeseen -3,0 päivää 100 mg:n annoksella ja -3,2 päivää 300 mg:n annoksella).
DELIVER: Aiempien migreenin estohoitojen epäonnistuminen
VYEPTI-valmistetta on arvioitu tehon ja turvallisuuden tutkimuksessa (DELIVER) episodista (n = 484) ja kroonista (n = 405) migreeniä sairastavilla potilailla, joilla 2–4 aiemman migreenin estolääkeryhmän kokeilu oli kirjattu epäonnistuneeksi. Tutkimuksessa oli 24 viikon pituinen kaksoissokkoutettu, lumekontrolloitu hoitojakso ja 48 viikon pituinen pitkäkestoinen jatkojakso.
Tutkimuksessa havaittiin, että VYEPTI-hoito vähensi viikkoina 1–12 kuukausittaisia migreenipäiviä (MMD) keskimäärin 4,8 päivällä VYEPTI 100 mg -ryhmässä, 5,3 päivällä VYEPTI 300 mg -ryhmässä ja 2,1 päivällä lumelääkeryhmässä. Ero lumelääkkeeseen oli −2,7 päivää (95 %:n luottamusväli: −3,4…−2,0) VYEPTI 100 mg -ryhmässä ja −3,2 päivää (95 %:n luottamusväli: −3,9…−2,5) VYEPTI 300 mg -ryhmässä.
Tutkimuksessa todettiin myös, että 42 %:lla VYEPTI 100 mg -ryhmässä olleista ja 50 %:lla VYEPTI 300 mg -ryhmässä olleista kuukausittaiset migreenipäivät vähenivät vähintään 50 % viikkoina 1–12. Lumelääkeryhmässä olleista 13 % saavutti tämän tasoisen kuukausittaisten migreenipäivien vähenemisen. 16 %:lla VYEPTI 100 mg -ryhmässä olleista ja 19 %:lla VYEPTI 300 mg -ryhmässä olleista kuukausittaiset migreenipäivät vähenivät vähintään 75 % viikkoina 1–12. Lumelääkeryhmässä olleista 2 % saavutti tämän tasoisen kuukausittaisten migreenipäivien vähenemisen.
Lumekontrolloidun hoitojakson aikana todettu teho säilyi jopa 72 viikon VYEPTI-hoidon ajan jatkotutkimusjaksoaikana.
Turvallisuutta koskevat tiedot olivat yhdenmukaisia kohdassa Haittavaikutukset kuvatun VYEPTI-valmisteen turvallisuusprofiilin kanssa.
RELIEF: Estohoidon aloittaminen migreenikohtauksen aikana
VYEPTI-valmistetta arvioitiin tehoa ja turvallisuutta selvittäneessä tutkimuksessa (RELIEF) potilailla, joilla oli 4–15 migreenipäivää kuukaudessa (n = 480). Potilaat saivat VYEPTI-valmistetta tai lumelääkettä 1–6 tunnin sisällä keskivaikean tai vaikean migreenikohtauksen alkamisesta.
Tutkimuksen tulosten perusteella keskivaikean tai vaikean migreenikohtauksen aikana aloitettu VYEPTI-hoito lyhentää tilastollisesti merkitsevästi aikaa päänsärkykivun loppumiseen (p < 0,001; mediaaniaika 4 tuntia vs. 9 tuntia) ja oireiden lievittymiseen häiritsevimpien oireiden osalta (p < 0,001; mediaaniaika 2 tuntia vs. 3 tuntia) lumelääkkeeseen verrattuna potilailla, jotka soveltuvat saamaan migreenin estohoitoa. Tämän lisäksi lumelääkettä saaneisiin verrattuna useammilla VYEPTI-hoitoa saavilla potilailla myös päänsärkykipu (24 % vs. 12 %) ja häiritsevimmät oireet (56 % vs. 36 %) olivat poissa 2 tunnin kuluttua (p < 0,001). Lisäksi ensimmäisten 24 tunnin sisällä infuusion jälkeen harvemmat potilaat tarvitsivat akuuttia kohtauslääkitystä VYEPTI-hoidon jälkeen lumelääkkeeseen verrattuna (p < 0,001).
Turvallisuutta koskevat tiedot olivat yhdenmukaisia kohdassa Haittavaikutukset kuvatun VYEPTI-valmisteen turvallisuusprofiilin kanssa.
PREVAIL: pitkäaikainen tutkimus
Vyepti-valmistetta annettiin 300 mg 12 viikon välein infuusiona laskimoon enintään 96 viikon ajan 128:lle kroonista migreeniä sairastavalle potilaalle. Ensisijainen tavoite oli arvioida pitkäaikaista turvallisuutta toistuvien Vyepti-annosten jälkeen. Toissijaisia tavoitteita olivat Vyepti-valmisteen farmakokinetiikan ja immunogeenisuusprofiilin kuvaaminen (kohta Haittavaikutukset) ja Vyepti-valmisteen hoitotehon arviointi useilla potilaan raportoimilla migreeniin ja elämänlaatuun liittyvillä mittareilla, joihin kuului päänsärkytesti (HIT-6). Potilaiden keskimääräinen ikä oli 41,5 vuotta (vaihteluväli: 18–65 vuotta), 85 % oli naisia, 95 % oli valkoihoisia ja 36 % käytti samanaikaista migreenin estolääkitystä. Migreenipäiviä oli keskimäärin 14,1 päivää 28 päivän jaksolla 3 kuukauden aikana ennen seulontaa. Yhteensä 100 potilasta (78,1 %) suoritti tutkimuksen loppuun (viikolle 104). Lähtötasolla sairaus vaikutti potilaisiin vakavasti, ja HIT-6-kokonaispisteet olivat keskimäärin 65. Keskimääräinen muutos lähtötasosta viikolle 104 oli -9,7 (p < 0,0001). Turvallisuusprofiili vastasi satunnaistetuissa, lumekontrolloiduissa tutkimuksissa havaittuja turvallisuusprofiileja, ja pitkäaikainen vaikutus potilaille olennaisiin tuloksiin havaittiin 96 viikkoon asti.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Vyepti-valmisteen käytöstä hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä migreenin estohoidossa (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Vyepti annetaan laskimoon, joten sen hyötyosuus on 100 %. Eptinetsumabin farmakokinetiikka on lineaarinen ja altistus kasvaa suhteellisesti annoksilla 10–1 000 mg. Vakaa tila saavutetaan ensimmäisen annoksen jälkeen kerran 12 viikon välein annettavalla annostuksella. Mediaaniaika enimmäispitoisuuteen (Cmax) on 30 minuuttia (infuusion päättyminen), ja keskimääräinen terminaalisen eliminaation puoliintumisaika on 27 päivää. Keskimääräinen kertymäsuhde on Cmax-arvon perusteella 1,08 ja AUC0-tau-arvon perusteella 1,15.
Imeytyminen
Vyepti annetaan infuusiona laskimoon, jolloin ohitetaan suonenulkoinen imeytyminen ja hyötyosuus on 100 %. Mediaaniaika huippupitoisuuteen saavutettiin infuusion lopussa (30 minuuttia).
Jakautuminen
Eptinetsumabin sentraalinen jakautumistilavuus (Vc) oli noin 3,7 litraa.
Biotransformaatio
Eptinetsumabin odotetaan hajoavan proteolyyttisten entsyymien vaikutuksesta pieniksi peptideiksi ja aminohapoiksi.
Eliminaatio
Eptinetsumabin ilmeinen puhdistuma oli 0,15 l/vrk, ja terminaalisen eliminaation puoliintumisaika oli noin 27 päivää.
Erityiset potilasryhmät
Populaatiofarmakokineettisessä analyysissa, jossa oli mukana 2 123 koehenkilöä, tutkittiin iän, sukupuolen, etnisen taustan ja painon vaikutusta eptinetsumabin farmakokinetiikkaan. Suhteessa 70 kg painavaan koehenkilöön eptinetsumabin vakaan tilan altistus 190 kg painavalla koehenkilöllä oli enintään 52 % matalampi, kun taas 39 kg painavalla koehenkilöllä se olisi 50 % korkeampi. Vakaan tilan altistuksen arvioinnissa paino ei kuitenkaan vaikuttanut kliiniseen tehoon. Annosta ei tarvitse muuttaa painon perusteella. Ikä (18–71), sukupuoli tai rotu eivät populaatiofarmakokinetiikan perusteella vaikuttaneet eptinetsumabin farmakokinetiikkaan. Siksi annosta ei tarvitse muuttaa.
Munuaisten tai maksan vajaatoiminta
Munuaisten tai maksan vajaatoiminnasta ei ole tehty nimenomaisia tutkimuksia sen arvioimiseksi, vaikuttaako munuaisten tai maksan vajaatoiminta eptinetsumabin farmakokinetiikkaan. Populaatiofarmakokineettinen analyysi Vyepti-valmisteen kliinisten tutkimusten integroiduista tiedoista ei osoittanut munuaisten tai maksan vajaatoimintaa sairastavilla potilailla eroja, jotka edellyttäisivät annoksen muuttamista. Tietoja ei ole potilaista, joilla on vaikea munuaisten vajaatoiminta.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, juveniilia toksisuutta tai lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Genotoksisuus ja karsinogeenisuus
Koska eptinetsumabilla ei todennäköisesti ole välittömiä yhteisvaikutuksia DNA:n tai muun kromosomimateriaalin kanssa, mahdollisen genotoksisuuden arviointi katsottiin tarpeettomaksi eikä sitä suoritettu.
Koska karsinogeenisuusriskiä ei ole tunnistettu laajassa CGRP:n estämiseen liittyvän kirjallisuuden arvioinnissa eikä eptinetsumabiin liittyviä proliferatiivisia löydöksiä havaittu pitkäaikaisissa tutkimuksissa apinoilla, karsinogeenisuuden testaus katsottiin tarpeettomaksi eikä sitä tehty.
Farmaseuttiset tiedot
Apuaineet
Sorbitoli (E420)
L-histidiini
L-histidiinihydrokloridimonohydraatti
Polysorbaatti 80
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
3 vuotta.
Laimentamisen jälkeen Vyepti-valmisteen infuusioliuos (Vyepti ja 0,9-prosenttinen natriumkloridiliuos injektiota varten) täytyy infusoida 8 tunnin kuluessa (ks. kohta Käyttö- ja käsittelyohjeet).
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä eikä ravistaa.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Kun Vyepti on otettu pois jääkaapista, se on käytettävä 7 vuorokauden kuluessa, jos sitä on säilytetty huoneenlämmössä (enintään 25 °C), tai hävitettävä. Jos sitä on säilytetty korkeammassa lämpötilassa tai pidemmän aikaa, se on hävitettävä.
Laimentamisen jälkeen Vyepti-valmisteen infuusioliuosta (Vyepti ja 0,9-prosenttinen natriumkloridiliuos injektiota varten) voidaan säilyttää huoneenlämmössä (alle 25 °C) tai jääkaapissa 2–8 °C:ssa. Laimentamisen jälkeen VYEPTI-valmisteen infuusioliuos täytyy infusoida 8 tunnin kuluessa.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VYEPTI infuusiokonsentraatti, liuosta varten
100 mg (L:ei) 1 kpl (100 mg/ml) (888,51 €)
300 mg (L:ei) 1 kpl (3 ml (100 mg/ml)) (888,51 €)
PF-selosteen tieto
4 ml:n tyypin I lasinen injektiopullo klorobutyylikumitulpalla. Injektiopullon tulppa on valmistettu ilman luonnonkumilateksia.
Vyepti 100 mg infuusiokonsentraatti, liuosta varten
Vyepti on saatavana 1 ja 3 kertakäyttöisen injektiopullon pakkauksissa.
Vyepti 300 mg infuusiokonsentraatti, liuosta varten
Vyepti on saatavana 1 kertakäyttöisen injektiopullon pakkauksessa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Infuusiokonsentraatti, liuosta varten on kirkasta tai himmeän kuultavaa ja väritöntä tai ruskeankeltaista. Sen pH on 5,5–6,1 ja osmolaliteetti 290–350 mOsm/kg.
Käyttö- ja käsittelyohjeet
Tämä lääkevalmiste täytyy laimentaa ennen antamista. Terveydenhuollon ammattilaisen täytyy valmistaa laimennos aseptisella tekniikalla infuusioliuoksen steriiliyden varmistamiseksi.
Tämä lääkevalmiste ei sisällä säilöntäainetta ja on tarkoitettu vain kertakäyttöön. Käyttämättä jäänyt lääkevalmiste täytyy hävittää.
Ennen laimentamista tämä lääkevalmiste (konsentraatti injektiopulloissa) täytyy tarkastaa silmämääräisesti; sitä ei saa käyttää, jos konsentraatissa on näkyviä hiukkasia tai se on sameaa tai siinä on värimuutoksia (muuta kuin kirkasta tai himmeän kuultavaa ja väritöntä tai ruskeankeltaista).
Sekä 100 mg:n että 300 mg:n annokselle käytetään 100 ml:n pussi 9 mg/ml (0,9-prosenttista) natriumkloridiliuosta Vyepti-valmisteen infuusioliuoksen valmistamiseen alla kuvatulla tavalla. Vyepti-valmisteen infuusioliuoksen valmistamiseen ei saa käyttää muita suonensisäisiä laimentimia tai muuta tilavuutta.
Sekoita Vyepti-valmisteen infuusioliuos kokonaan kääntämällä se varovasti ylösalaisin. Ei saa ravistaa.
Laimentamisen jälkeen Vyepti-valmisteen infuusioliuos täytyy infusoida 8 tunnin kuluessa. Tänä aikana Vyepti-valmisteen infuusioliuosta voidaan säilyttää huoneenlämmössä (alle 25 °C) tai jääkaapissa 2–8 °C:ssa. Jos Vyepti-valmisteen infuusioliuosta säilytetään 2–8 °C:ssa, se täytyy lämmittää huoneenlämpöiseksi ennen infusoimista. EI SAA JÄÄTYÄ.
Vyepti-valmisteen 100 mg:n annos
Vyepti-valmisteen infuusioliuos valmistetaan vetämällä 1,0 ml Vyepti-valmistetta yhdestä kertakäyttöisestä 100 mg:n injektiopullosta steriilillä neulalla ja ruiskulla. Injektoi 1,0 ml:n (100 mg) sisältö 100 ml:n pussiin 0,9-prosenttista natriumkloridiliuosta.
Vyepti-valmisteen 300 mg:n annos
Vyepti-valmisteen infuusioliuos valmistetaan vetämällä 1,0 ml Vyepti-valmistetta kolmesta kertakäyttöisestä 100 mg:n injektiopullosta tai 3,0 ml Vyepti-valmistetta yhdestä kertakäyttöisestä 300 mg:n injektiopullosta steriilillä neulalla ja ruiskulla. Injektoi saatu 3,0 ml:n (300 mg) sisältö 100 ml:n pussiin 0,9-prosenttista natriumkloridiliuosta.
Ohjeet infuusion antamiseen
Parenteraaliset lääkevalmisteet täytyy ennen antamista tarkastaa silmämääräisesti hiukkasten ja värimuutosten varalta. Ei saa käyttää, jos nesteessä on näkyviä hiukkasia tai se on sameaa tai siinä on värimuutoksia.
Anna lääkärin määräämä Vyepti-valmisteen 100 mg:n annos tai Vyepti-valmisteen 300 mg:n annos noin 30 minuutin kestoisena infuusiona sen jälkeen, kun injektiopullon sisältö on laimennettu 100 ml:n pussiin 0,9-prosenttista natriumkloridiliuosta. Käytä suonensisäistä infuusiosarjaa, jossa on 0,2 tai 0,22 μm:n sisäänrakennettu tai lisätty suodatin. Kun infuusio on valmis, huuhtele letku 20 ml:lla 0,9-prosenttista natriumkloridiliuosta.
Vyepti-valmistetta ei saa antaa bolusinjektiona.
Infuusiosarjan kautta tai Vyepti-valmisteeseen sekoitettuna ei saa antaa muita lääkkeitä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VYEPTI infuusiokonsentraatti, liuosta varten
100 mg 1 kpl
300 mg 1 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Atogepantti, eptinetsumabi, erenumabi, fremanetsumabi, galkanetsumabi ja rimegepantti (migreenin estohoito): Aikuisten migreenin estohoito erityisin edellytyksin (3007).
ATC-koodi
N02CD05
Valmisteyhteenvedon muuttamispäivämäärä
10.04.2026
Yhteystiedot
Junakatu 9, Logomo Byrå
20100 Turku
02 276 5000
www.lundbeck.fi
suomi@lundbeck.com
www.skitsofreniainfo.fi, www.masennusinfo.fi, www.migreeniinfo.fi