
TUKYSA tabletti, kalvopäällysteinen 50 mg, 150 mg
Vaikuttavat aineet ja niiden määrät
TUKYSA 50 mg tabletit, kalvopäällysteiset
Yksi kalvopäällysteinen tabletti sisältää 50 mg tukatinibia.
TUKYSA 150 mg tabletit, kalvopäällysteiset
Yksi kalvopäällysteinen tabletti sisältää 150 mg tukatinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi 150 mg:n kalvopäällysteinen tabletti sisältää 27,64 mg natriumia ja 30,29 mg kaliumia.
300 mg:n annos TUKYSA-valmistetta sisältää 55,3 mg natriumia ja 60,6 mg kaliumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
TUKYSA on tarkoitettu paikallisesti levinneen tai metastasoituneen HER2-positiivisen rintasyövän hoitoon yhdessä trastutsumabin ja kapesitabiinin kanssa aikuispotilailla, jotka ovat saaneet vähintään kaksi aikaisempaa anti-HER2-hoitoa sisältävää hoito-ohjelmaa.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
TUKYSA-hoidon saa aloittaa ja hoitoa saa valvoa vain lääkäri, jolla on kokemusta syöpälääkkeiden antamisesta.
Annostus
Suositeltu annos on 300 mg tukatinibia (kaksi 150 mg:n tablettia) kahdesti päivässä jatkuvasti yhdessä trastutsumabin ja kapesitabiinin kanssa. Näiden valmisteiden annokset on esitetty taulukossa 1. Katso lisätiedot samanaikaisesti annettavien trastutsumabin ja kapesitabiinin valmisteyhteenvedoista. Hoitoon kuuluvat lääkkeet voidaan antaa missä tahansa järjestyksessä.
Taulukko 1: Suositellut annokset
Hoito | Annos | Hoitopäivät | Ajoitus suhteessa ruokailuun |
Tukatinibi | 300 mg suun kautta kahdesti päivässä | Jatkuvasti | Ruoan kanssa tai ilman ruokaa |
Kapesitabiini | 1000 mg/m2 suun kautta kahdesti päivässä | Päivinä 1–14 kussakin 21 päivän jaksossa | 30 minuutin sisällä aterian jälkeen |
Trastutsumabi | |||
Laskimoon | Ei oleellinen | ||
Aloitusannos | 8 mg/kg laskimoon | Päivä 1 | |
Seuraavat annokset | 6 mg/kg laskimoon | 21 päivän välein | |
TAI Ihon alle | 600 mg ihon alle | 21 päivän välein |
TUKYSA-hoitoa jatketaan sairauden etenemiseen tai kunnes ilmenee toksisuutta, joka ei ole hyväksyttävissä.
Unohtunut annos
Jos annos jää välistä, potilaan on otettava seuraava annos tavanomaiseen aikaan annostussuunnitelman mukaisesti.
Annosmuutokset
Taulukoissa 2 ja 3 on esitetty tukatinibin suositellut annosmuutokset potilaille, joilla esiintyy haittavaikutuksia (ks. kohta Haittavaikutukset). Katso tiedot annoksen muuttamisesta samanaikaisesti annettavien trastutsumabin ja kapesitabiinin valmisteyhteenvedoista, jos haittavaikutusten epäillään johtuvan kyseisistä valmisteista.
Taulukko 2: Tukatinibin suositellut annosvähennykset haittavaikutusten yhteydessä
Annostaso | Tukatinibiannos |
Suositeltu aloitusannos | 300 mg kahdesti päivässä |
Ensimmäinen annosvähennys | 250 mg kahdesti päivässä |
Toinen annosvähennys | 200 mg kahdesti päivässä |
Kolmas annosvähennys | 150 mg kahdesti päivässä1 |
1 TUKYSA-hoito on lopetettava kokonaan, jos potilas ei siedä 150 mg:n annosta suun kautta kahdesti päivässä.
Taulukko 3: Tukatinibin suositellut annosmuutokset haittavaikutusten yhteydessä
Haittavaikutus | Vaikeusaste1 | Tukatinibin annosmuutos |
Ripuli | Vaikeusaste 1 ja 2 | Annoksen muuttaminen ei ole tarpeen. |
Vaikeusaste 3 ilman ripulilääkettä | Aloita soveltuva lääkitys tai lisää lääkitystä. Keskeytä tukatinibin antaminen, kunnes vaikeusaste ≤ 1, ja jatka sitten tukatinibihoitoa samalla annoksella. | |
Vaikeusaste 3 ripulilääkkeen kanssa | Aloita soveltuva lääkitys tai lisää lääkitystä. Keskeytä tukatinibin antaminen, kunnes vaikeusaste ≤ 1, ja jatka sitten tukatinibihoitoa seuraavaksi pienemmällä annoksella. | |
Vaikeusaste 4 | Lopeta tukatinibihoito pysyvästi. | |
ALAT-, ASAT- tai kokonaisbilirubiinitason nousu2 | Vaikeusasteen 1 bilirubiinitason nousu (> ULN – 1,5 x ULN) | Annoksen muuttaminen ei ole tarpeen. |
Vaikeusasteen 2 bilirubiinitason nousu (> 1,5–3 × ULN) | Keskeytä tukatinibin antaminen, kunnes vaikeusaste ≤ 1, ja jatka sitten tukatinibihoitoa samalla annoksella. | |
Vaikeusasteen 3 ALAT- tai ASAT-tason nousu (> 5–20 × ULN) TAI Vaikeusasteen 3 bilirubiinitason nousu (> 3–10 × ULN) | Keskeytä tukatinibin antaminen, kunnes vaikeusaste ≤ 1, ja jatka sitten tukatinibihoitoa seuraavaksi pienemmällä annoksella. | |
Vaikeusasteen 4 ALAT- tai ASAT-tason nousu (> 20 × ULN) TAI Vaikeusasteen 4 bilirubiinitason nousu (> 10 × ULN) | Lopeta tukatinibihoito pysyvästi. | |
ALAT- tai ASAT-taso > 3 × ULN JA Bilirubiini > 2 × ULN | Lopeta tukatinibihoito pysyvästi. | |
Muut haittavaikutukset | Vaikeusasteet 1 ja 2 | Annoksen muuttaminen ei ole tarpeen. |
Vaikeusaste 3 | Keskeytä tukatinibin antaminen, kunnes vaikeusaste ≤ 1, ja jatka sitten tukatinibihoitoa seuraavaksi pienemmällä annoksella. | |
Vaikeusaste 4 | Lopeta tukatinibihoito pysyvästi. |
- Vaikeusasteet perustuvat NCI-CTCAE-luokituksen (National Cancer Institute Common Terminology Criteria for Adverse Events) versioon 4.03.
- Lyhenteet: ULN = viitealueen yläraja (upper limit of normal); ALAT = alaniiniaminotransferaasi; ASAT = aspartaattiaminotransferaasi
Samanaikainen CYP2C8:n estäjien anto
Samanaikaista voimakkaiden CYP2C8:n estäjien käyttöä on vältettävä. Jos samanaikaista voimakkaan CYP2C8:n estäjän käyttöä ei voida välttää, tukatinibin aloitusannosta on laskettava 100 mg:aan suun kautta kahdesti päivässä. Kun voimakkaan CYP2C8:n estäjän ottamisen keskeyttämisestä on kulunut kolme eliminaation puoliintumisaikaa, tukatinibihoitoa jatketaan sillä annoksella, joka oli käytössä ennen estäjähoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja kohta Yhteisvaikutukset). Seurantaa TUKYSA-valmisteen toksisuuden varalta on lisättävä, jos käytetään samanaikaisesti kohtalaisia CYP2C8:n estäjiä.
Erityisryhmät
Iäkkäät
Annoksen muuttaminen ei ole tarpeen ≥ 65‑vuotiaille potilaille (ks. kohta Farmakokinetiikka). Tukatinibia ei ole tutkittu yli 80‑vuotiailla potilailla.
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastaville potilaille (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen lievää tai keskivaikeaa maksan vajaatoimintaa sairastaville potilaille (ks. kohta Farmakokinetiikka). Vaikeaa maksan vajaatoimintaa (Child‑Pugh C) sairastaville potilaille suositellaan pienennettyä aloitusannosta, 200 mg suun kautta kahdesti päivässä.
Pediatriset potilaat
TUKYSA-valmisteen turvallisuutta ja tehoa pediatristen potilaiden hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
TUKYSA otetaan suun kautta. Tabletit nielaistaan kokonaisina eikä niitä saa pureskella, murskata tai jakaa ennen nielaisemista (ks. kohta Farmakokinetiikka).
TUKYSA otetaan noin 12 tunnin välein, samaan aikaan joka päivä, joko ruoan kanssa tai ilman. TUKYSA voidaan ottaa samaan aikaan kapesitabiinin kanssa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Laboratoriokokeet
ALAT-, ASAT- ja bilirubiinitason nousu
Tukatinibihoidon aikana on raportoitu ALAT-, ASAT- ja bilirubiinitason nousua (ks. kohta Haittavaikutukset). ALAT-, ASAT- ja kokonaisbilirubiinitasoa on seurattava kolmen viikon välein tai kliinisen tarpeen mukaan. Haittavaikutuksen vaikeusasteesta riippuen ensin on keskeytettävä tukatinibihoito, sitten pienennettävä annosta tai lopetettava hoito kokonaan (ks. kohta Annostus ja antotapa).
Kreatiniinin nousu potilailla, joilla ei ole munuaisten vajaatoimintaa
Seerumin kreatiniinitason nousua (keskimääräinen nousu 30 %) on havaittu. Nousu johtuu kreatiniinin tubuluskuljetuksen estämisestä, joka ei vaikuta glomerulusten toimintaan (ks. kohta Haittavaikutukset). Munuaisten toiminnan seuraamiseen voidaan harkita vaihtoehtoisia markkereita (esim. BUN, kystatiini C tai laskettu glomerulusten suodatusnopeus [GFR]), jotka eivät perustu kreatiniiniin.
Ripuli
Tukatinibihoidon aikana on raportoitu ripulia, mukaan lukien vakavia tapahtumia, kuten kuivumista, hypotensiota, akuuttia munuaisvauriota ja kuolemantapauksia (ks. kohta Haittavaikutukset). Jos ilmenee ripulia, ripulilääkkeitä annetaan kliinisen indikaation mukaan. Vaikeusasteen ≥ 3 ripulissa ensin on keskeytettävä tukatinibihoito, sitten pienennettävä annosta tai lopetettava tukatinibihoito kokonaan (ks. kohta Annostus ja antotapa). Lääkitys on aloitettava ripeästi myös, jos potilaalla esiintyy samanaikaisesti sitkeää vaikeusasteen 2 ripulia ja vaikeusasteen ≥ 2 pahoinvointia ja/tai oksentelua. Infektioperäiset syyt on suljettava pois diagnostisilla testeillä kliinisen indikaation perusteella, jos potilaalla on vaikeusasteen 3 tai 4 ripuli tai minkä tahansa vaikeusasteen ripuli, johon liittyy komplisoivia tekijöitä (kuivuminen, kuume, neutropenia).
Alkio- ja sikiötoksisuus
Eläinkokeissa tehtyjen löydösten ja tukatinibin vaikutusmekanismin perusteella on mahdollista, että valmiste aiheuttaa haittaa sikiöille, kun sitä annetaan raskaana oleville naisille. Eläinten lisääntymistutkimuksissa tukatinibin antaminen tiineille kaniineille organogeneesin aikana aiheutti sikiöille epämuodostumia, kun emojen saamat annokset vastasivat suositusannoksen aiheuttamaa kliinistä altistumista.
Raskaana oleville naisille on kerrottava potentiaalisista sikiöön kohdistuvista riskeistä. Naisia, jotka voivat tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä hoidon aikana ja vähintään yhden viikon ajan viimeisen hoitoannoksen saamisen jälkeen (ks. kohta Raskaus ja imetys). Miespotilaita, joiden naispuoliset kumppanit voivat tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisymenetelmää hoidon aikana ja vähintään yhden viikon ajan viimeisen hoitoannoksen saamisen jälkeen.
Herkät CYP3A:n substraatit
Tukatinibi on voimakas CYP3A:n estäjä. Tukatinibilla voi olla yhteisvaikutuksia CYP3A:n metaboloimien lääkevalmisteiden kanssa, mikä voi johtaa näiden lääkevalmisteiden kohonneisiin pitoisuuksiin plasmassa (ks. kohta Yhteisvaikutukset). Kun tukatinibia annetaan samanaikaisesti muiden lääkevalmisteiden kanssa, niiden valmisteyhteenvedoista on katsottava lisätietoja samanaikaista CYP3A:n estäjien antamista koskevista suosituksista. Tukatinibihoidon aikana on vältettävä samanaikaista CYP3A:n substraattien käyttöä silloin, kun hyvin pienet pitoisuusmuutokset voivat johtaa vakaviin tai henkeä uhkaaviin haittavaikutuksiin. Jos samanaikaista käyttöä ei ole mahdollista välttää, samanaikaisesti käytettävän CYP3A:n substraatin annostusta on pienennettävä valmisteyhteenvedon mukaisesti.
P‑gp:n substraatit
P‑gp:n substraatin ja tukatinibin samanaikainen käyttö aiheutti P‑gp:n substraatin plasmapitoisuuden nousua, mikä voi lisätä P‑gp:n substraatin aiheuttamaa toksisuutta. P‑gp:n substraatin (mukaan lukien herkät suoliston substraatit, kuten dabigatraani) annoksen pienentämistä on harkittava samanaikaisesti otettavan lääkevalmisteen valmisteyhteenvedon mukaisesti, ja P-gp:n substraatteja annettaessa on noudatettava varovaisuutta silloin, kun hyvin pienet pitoisuusmuutokset voivat johtaa vakavaan tai henkeä uhkaavaan toksisuuteen.
Voimakkaat CYP3A:n indusoijat / kohtalaiset CYP2C8:n indusoijat
Voimakkaiden CYP3A:n tai kohtalaisten CYP2C8:n indusoijien ja tukatinibin samanaikainen käyttö pienensi tukatinibin pitoisuuksia, mikä voi heikentää tukatinibin vaikutusta. Samanaikaista voimakkaiden CYP3A:n indusoijien tai kohtalaisten CYP2C8:n indusoijien käyttöä on vältettävä.
Voimakkaat / kohtalaiset CYP2C8:n estäjät
Tukatinibin ja voimakkaan CYP2C8:n estäjän samanaikainen käyttö nosti tukatinibin pitoisuuksia, mikä voi lisätä tukatinibin aiheuttaman toksisuuden riskiä. Samanaikaista käyttöä voimakkaiden CYP2C8:n estäjien kanssa on vältettävä (ks. kohta Annostus ja antotapa).
Kohtalaisten CYP2C8:n estäjien samanaikaisen käytön vaikutuksesta tukatinibipitoisuuksiin ei ole kliinisiä tietoja. Seurantaa tukatinibin toksisuuden varalta on lisättävä, jos käytetään samanaikaisesti kohtalaisia CYP2C8:n estäjiä.
Apuaineita koskevat tiedot
Tämä lääkevalmiste sisältää 55,3 mg natriumia per 300 mg:n annos. Tämä vastaa 2,75 %:a suositellusta natriumin enimmäisvuorokausiannoksesta aikuisille.
Tämä lääkevalmiste sisältää 60,6 mg kaliumia per 300 mg:n annos. Potilaiden, joilla on munuaisten vajaatoimintaa tai ruokavalion kaliumrajoitus (ruokavalio, jonka kaliumpitoisuus on pieni), on otettava tämä huomioon.
Yhteisvaikutukset
Tukatinibi metaboloituu pääasiassa CYP2C8:n välityksellä. Tukatinibi on metaboliavälitteinen CYP3A:n estäjä, joka estää metformiinin ja kreatiniinin kuljettajaproteiinien toimintaa munuaisissa. Tukatinibi on P‑gp:n substraatti.
Muiden lääkevalmisteiden vaikutukset tukatinibiin
CYP3A:n/CYP2C8:n indusoijat
Kliinisessä yhteisvaikutustutkimuksessa havaittiin, että yksittäinen 300 mg:n tukatinibiannos yhdessä rifampisiinin (voimakas CYP3A:n ja kohtalainen CYP2C8:n indusoija) kanssa johti tukatinibipitoisuuksien pienenemiseen (0,6‑kertainen Cmax [90 %:n CI: 0,5; 0,8] ja 0,5‑kertainen kokonaisaltistus [AUC] [90 %:n CI: 0,4; 0,6]). Samanaikaista voimakkaan CYP3A:n tai kohtalaisen CYP2C8:n indusoijan (esim. rifampisiini, fenytoiini, mäkikuisma tai karbamatsepiini) antamista on vältettävä, sillä se voi johtaa tukatinibin vaikutuksen heikkenemiseen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
CYP2C8:n estäjät
Kliinisessä yhteisvaikutustutkimuksessa havaittiin, että yksittäinen 300 mg:n tukatinibiannos yhdessä gemfibrotsiilin (voimakas CYP2C8:n estäjä) kanssa annettuna johti tukatinibipitoisuuksien kohoamiseen (1,6‑kertainen Cmax [90 %:n CI: 1,5; 1,8] ja 3,0‑kertainen kokonaisaltistus [AUC] [90 %:n CI: 2,7; 3,5]). Samanaikaista voimakkaiden CYP2C8:n estäjien (esim. gemfibrotsiili) antamista on vältettävä, sillä se voi lisätä tukatinibiin liittyvän toksisuuden riskiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
CYP3A:n estäjät
Kliinisessä yhteisvaikutustutkimuksessa havaittiin, että yksittäinen 300 mg:n tukatinibiannos yhdessä itrakonatsolin (voimakas CYP3A:n estäjä) kanssa annettuna johti tukatinibipitoisuuksien kohoamiseen (1,3‑kertainen Cmax [90 %:n CI: 1,2; 1,4] ja 1,3‑kertainen kokonaisaltistus [AUC] [90 %:n CI: 1,3; 1,4]). Annoksen muuttaminen ei ole tarpeen.
Protonipumpun estäjät
Tukatinibia koskevissa kliinisissä yhteisvaikutustutkimuksissa ei havaittu yhteisvaikutuksia, kun tukatinibia annettiin yhdessä omepratsolin (protonipumpun estäjä) kanssa. Annoksen muuttaminen ei ole tarpeen.
Tukatinibin vaikutukset muihin lääkevalmisteisiin
CYP3A:n substraatit
Tukatinibi on voimakas CYP3A:n estäjä. Kliinisessä yhteisvaikutustutkimuksessa havaittiin, että tukatinibin ja midatsolaamin (herkkä CYP3A:n substraatti) samanaikainen anto johti midatsolaamipitoisuuksien kohoamiseen (3,0‑kertainen Cmax [90 %:n CI: 2,6; 3,4] ja 5,7‑kertainen kokonaisaltistus [AUC] [90 %:n CI: 5,0; 6,5]). Tukatinibin ja herkän CYP3A:n substraatin (esim. alfentaniili, avanafiili, buspironi, darifenasiini, darunaviiri, ebastiini, everolimuusi, ibrutinibi, lomitapidi, lovastatiini, midatsolaami, naloksegoli, sakinaviiri, simvastatiini, sirolimuusi, takrolimuusi, tipranaviiri, triatsolaami ja vardenafiili) samanaikainen anto voi lisätä systeemistä altistusta, mikä voi lisätä CYP3A:n substraattiin liittyvää toksisuutta. CYP3A:n substraattien samanaikaista käyttöä on vältettävä tukatinibihoidon aikana silloin, kun hyvin pienet pitoisuusmuutokset voivat johtaa henkeä uhkaavaan toksisuuteen. Jos samanaikaista käyttöä ei ole mahdollista välttää, samanaikaisesti käytettävän CYP3A:n substraatin annostusta on pienennettävä valmisteyhteenvedon mukaisesti.
P‑gp:n substraatit
Kliinisessä yhteisvaikutustutkimuksessa havaittiin, että tukatinibin ja digoksiinin (herkkä P‑gp:n substraatti) samanaikainen anto johti digoksiinipitoisuuksien kohoamiseen (2,4‑kertainen Cmax [90 %:n CI: 1,9; 2,9] ja 1,5‑kertainen kokonaisaltistus [AUC] [90 %:n CI: 1,3; 1,7]). P‑gp:n substraatin ja tukatinibin samanaikainen käyttö voi suurentaa P‑gp:n substraatin plasmapitoisuuksia, mikä voi johtaa P‑gp:n substraattiin liittyvän toksisuuden lisääntymiseen. P‑gp:n substraatin (mukaan lukien herkät suoliston substraatit, kuten dabigatraani) annoksen pienentämistä on harkittava samanaikaisesti otettavan lääkevalmisteen valmisteyhteenvedon mukaisesti, ja P-gp:n substraatteja annettaessa on noudatettava varovaisuutta silloin, kun hyvin pienet pitoisuusmuutokset voivat johtaa vakavaan tai henkeä uhkaavaan toksisuuteen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
CYP2C8:n substraatit
Kliinisessä yhteisvaikutustutkimuksessa havaittiin, että tukatinibin ja repaglinidin (CYP2C8:n substraatti) samanaikainen anto johti repaglidinipitoisuuksien kohoamiseen (1,7‑kertainen Cmax [90 %:n CI: 1,4; 2,1] ja 1,7‑kertainen kokonaisaltistus [AUC] [90 %:n CI: 1,5; 1,9]). Annoksen muuttaminen ei ole tarpeen.
MATE1:n/2K:n substraatit
Kliinisessä yhteisvaikutustutkimuksessa havaittiin, että tukatinibin ja metformiinin (MATE1:n/2K:n substraatti) samanaikainen anto johti metformiinipitoisuuksien kohoamiseen (1,1‑kertainen Cmax [90 %:n CI: 1,0; 1,2] ja 1,4‑kertainen kokonaisaltistus [AUC] [90 %:n CI: 1,2; 1,5]). Tukatinibi vähensi metformiinin munuaispuhdistumaa ilman GFR:ään kohdistuvaa vaikutusta joheksolin puhdistuman ja seerumin kystatiini C:n avulla mitattuna. Annoksen muuttaminen ei ole tarpeen.
CYP2C9:n substraatit
Tukatinibia koskevissa kliinisissä yhteisvaikutustutkimuksissa ei havaittu yhteisvaikutuksia, kun tukatinibia annettiin yhdessä tolbutamiinin (herkkä CYP2C9:n substraatti) kanssa. Annoksen muuttaminen ei ole tarpeen.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehillä ja naisilla
Eläinkokeissa tehtyjen löydösten perusteella tukatinibilla voi olla haitallisia farmakologisia vaikutuksia, kun sitä annetaan raskaana oleville naisille ja/tai sikiöille/imeväisille. Naisia, jotka voivat tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä hoidon aikana ja vähintään yhden viikon ajan hoidon päättymisen jälkeen. Miespotilaita, joiden naispuoliset kumppanit voivat tulla raskaaksi, on neuvottava käyttämään tehokasta ehkäisyä hoidon aikana ja vähintään yhden viikon ajan hoidon päättymisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Lue lisätietoja trastutsumabin ja kapesitabiinin valmisteyhteenvetojen kohdista Raskaus ja imetys.
Raskaus
Ei ole olemassa tietoja tukatinibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). TUKYSA-valmistetta ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa tukatinibilla. Naiset, jotka voivat tulla raskaaksi, on tutkittava raskauden varalta ennen tukatinibihoidon aloittamista. Jos potilas tulee raskaaksi hoidon aikana, hänelle on kerrottava sikiöön/imeväiseen kohdistuvista potentiaalisista haitoista.
Imetys
Ei tiedetä, erittyvätkö tukatinibi/metaboliitit ihmisen rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Rintaruokinta on lopetettava TUKYSA-hoidon ajaksi. Rintaruokintaa voidaan jatkaa yhden viikon kuluttua hoidon loppumisen jälkeen.
Hedelmällisyys
Miesten tai naisten hedelmällisyyttä koskevia tutkimuksia ei ole tehty. Eläinkokeissa tehtyjen löydösten perusteella tukatinibi voi heikentää lisääntymiskykyisten naisten hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
TUKYSA-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Potilaan kliininen tila on huomioitava arvioitaessa potilaan kykyä suorittaa tehtäviä, joissa vaaditaan harkintakykyä sekä motorisia ja kognitiivisia kykyjä.
Haittavaikutukset
Turvallisuusprofiilin yhteisveto
Yleisimmin raportoidut vaikeusasteen 3 ja 4 haittavaikutukset (≥ 5 %) hoidon aikana olivat ripuli (13 %) sekä ALAT- (6 %) ja ASAT-tason nousu (5 %).
Vakavia haittavaikutuksia, joita olivat ripuli (4 %), oksentelu (3 %) ja pahoinvointi (2 %), esiintyi 29 %:lla tukatinibihoitoa saaneista potilaista.
TUKYSA-hoidon keskeyttämiseen johtaneita haittavaikutuksia esiintyi 6 %:lla potilaista, ja yleisimmät hoidon keskeyttämiseen johtaneet haittavaikutukset olivat ripuli (1 %) ja ALAT-tason nousu (1 %). TUKYSA-annoksen pienentämiseen johtaneita haittavaikutuksia esiintyi 23 %:lla potilaista, ja yleisimmät annoksen pienentämiseen johtaneet haittavaikutukset olivat ripuli (6 %) sekä ALAT- (5 %) ja ASAT-tason nousu (4 %).
Haittavaikutustaulukko
Seuraavat tiedot perustuvat kahdessa kliinisessä tutkimuksessa (HER2CLIMB ja ONT-380-005) saatuihin tietoihin 431 potilaasta, jotka olivat altistuneet TUKYSA-valmisteelle ja joilla oli paikallisesti levinnyt ja leikkaushoitoon soveltumaton tai metastasoitunut HER2-positiivinen rintasyöpä ja joita hoidettiin TUKYSA-valmisteella yhdessä trastutsumabin ja kapesitabiinin kanssa (ks. kohta Farmakodynamiikka). Keskimääräinen altistumisaika TUKYSA-valmisteelle oli näissä tutkimuksissa 7,4 kuukautta (vaihteluväli: < 0,1–43,6).
Hoidon aikana havaitut haittavaikutukset on lueteltu yleisyysluokittain. Yleisyysluokat ovat seuraavat: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 4: Haittavaikutukset
Elinjärjestelmä | Yleisyys | Haittavaikutus |
Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Nenäverenvuoto |
Ruoansulatuselimistö | Hyvin yleinen | Ripuli, pahoinvointi, oksentelu, stomatiitti1 |
Iho ja ihonalainen kudos | Hyvin yleinen | Ihottuma2 |
Luusto, lihakset ja sidekudos | Hyvin yleinen | Nivelkipu |
Tutkimukset | Hyvin yleinen | ASAT-tason nousu, ALAT-tason nousu, veren bilirubiinitason nousu3, laihtuminen |
- Stomatiitti mukaan lukien suutulehdus, suunielun kipu, haavaumat suussa, suukipu, haavaumat huulissa, kielikipu, rakkulat kielessä tai huulissa, suun alueen tuntohäiriö, haavaumat kielessä, aftat
- Ihottuma mukaan lukien makulopapulaarinen ihottuma, ihottuma, aknetyyppinen ihottuma, eryteema, täpläinen ihottuma, näppyläinen ihottuma, märkärakkulainen ihottuma, kutiava ihottuma, punoittava ihottuma, ihon hilseily, nokkosihottuma, allerginen ihottuma, kämmenien punoitus, jalkapohjien punoitus, ihon toksisuus
- Veren bilirubiinitason nousu mukaan lukien hyperbilirubinemia
Valikoitujen haittavaikutusten kuvaus
ALAT-, ASAT- tai bilirubiinitason nousu
HER2CLIMB-tutkimuksessa 41 %:lla potilaista, joita hoidettiin tukatinibin, trastutsumabin ja kapesitabiinin yhdistelmällä, esiintyi ALAT-, ASAT tai bilirubiinitason nousua. 9 %:lla potilaista esiintyi vähintään vaikeusasteen 3 tapahtumia. ALAT-, ASAT- ja bilirubiinitason nousu johti annoksen pienentämiseen 9 %:lla potilaista ja hoidon keskeyttämiseen 1,5 %:lla potilaista. Mediaaniaika minkä tahansa vaikeusasteen ALAT-, ASAT- tai bilirubiinitason nousun ilmaantumiseen oli 37 vuorokautta; 84 % tapahtumista oli ohimeneviä, ja oireiden lievittymisen mediaaniaika oli 22 vuorokautta. Seurantaa ja annoksen muuttamista (ml. keskeyttäminen) on harkittava (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ripuli
HER2CLIMB-tutkimuksessa 82 %:lla potilaista, joita hoidettiin tukatinibin, trastutsumabin ja kapesitabiinin yhdistelmällä, esiintyi ripulia. 13 %:lla potilaista esiintyi vähintään vaikeusasteen 3 ripulia. Tutkimuksen aikana kuoli kaksi potilasta, joilla oli vaikeusasteen 4 ripuli, ja ripuli oli kuolemaan myötävaikuttava tekijä. Ripuli johti annoksen pienentämiseen 6 %:lla potilaista ja hoidon keskeyttämiseen 1 %:lla potilaista. Mediaaniaika minkä tahansa vaikeusasteen ripulin ilmaantumiseen oli 12 vuorokautta; 81 % ripulitapahtumista oli ohimeneviä, ja oireiden lievittymisen mediaaniaika oli kahdeksan vuorokautta. Ripulilääkevalmisteiden profylaktinen käyttö ei ollut tarpeen. Ripulilääkkeitä käytettiin alle puolessa hoitosykleistä, joissa raportoitiin ripulitapahtumia. Ripulilääkityksen mediaanikesto oli kolme vuorokautta per hoitosykli (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kreatiniinin nousu potilailla, joilla ei ole munuaisten vajaatoimintaa
Tukatinibihoitoa saaneilla potilailla on havaittu seerumin kreatiniinitason nousua, joka johtuu kreatiniinin tubuluskuljetuksen estämisestä, joka ei vaikuta glomerulusten toimintaan. Kliinisissä tutkimuksissa havaittiin seerumin kreatiniinitason nousua (keskimääräinen nousu 30 %) ensimmäisen tukatinibihoitosyklin aikana. Kreatiniinitasot pysyivät kohonneina mutta vakaina koko hoidon ajan ja palautuivat ennalleen hoidon päätyttyä.
Erityisryhmät
Iäkkäät
HER2CLIMB-tutkimuksessa tukatinibia saaneista potilaista 82 potilasta oli ≥ 65-vuotiaita, ja heistä kahdeksan potilasta ≥ 75-vuotiaita. Haittavaikutusten esiintyvyys ≥ 65‑vuotiailla potilailla oli 34 % ja < 65-vuotiailla potilailla 28 %. ≥ 75-vuotiaita potilaita oli liian vähän turvallisuuserojen arvioimista varten.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Spesifistä vastalääkettä ei ole olemassa, ja hemodialyysin hyötyä tukatinibin yliannostuksen hoidossa ei tunneta. Yliannostustapauksessa tukatinibihoito on keskeytettävä ja annettava tarvittavaa tukihoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset aineet, proteiinikinaasin estäjät, ATC-koodi: L01EH03.
Vaikutusmekanismi
Tukatinibi on reversiibeli, potentti ja selektiivinen HER2‑tyrosiinikinaasin estäjä. Solusignalointia koskevissa tutkimuksissa tukatinibi oli > 1 000 kertaa selektiivisempi HER2:ta kohtaan kuin epidermaalista kasvutekijäreseptoria kohtaan. Tukatinibi estää HER2- ja HER3-fosforylaatiota in vitro, mistä seuraa fosforylaatiota seuraavan solusignaloinnin ja solujen lisääntymisen estyminen, ja indusoi HER2-positiivisten syöpäsolujen kuolemaa. Tukatinibi estää HER2-positiivisten kasvainten kasvua in vivo, ja tukatinibin ja trastutsumabin yhdistelmän on osoitettu ehkäisevän kasvainten kasvua in vitro ja in vivo tehokkaammin kuin kyseisten lääkevalmisteiden yksinään.
Farmakodynaamiset vaikutukset
Sydämen elektrofysiologia
Toistuva 300 mg:n tukatinibiannosten antaminen kahdesti päivässä ei vaikuttanut terveiden tutkittavien QTc-aikaan TQT-tutkimuksessa.
Kliininen teho ja turvallisuus
Tukatinibin tehoa yhdessä trastutsumabin ja kapesitabiinin kanssa arvioitiin satunnaistetussa, lumelääkekontrolloidussa, maailmanlaajuisessa kaksoissokkotutkimuksessa, jossa oli aktiivinen vertailuvalmiste (HER2CLIMB). Tutkimukseen otetuilla potilailla oli paikallisesti levinnyt ja leikkaushoitoon soveltumaton tai metastasoitunut HER2-positiivinen rintasyöpä, ja heillä oli tai ei ollut aivometastaaseja, ja heitä oli hoidettu aikaisemmin trastutsumabilla, pertutsumabilla ja trastutsumabiemtansiinilla (T-DM1) yksinään tai yhdistelmänä neoadjuvantti-, adjuvantti- tai metastaasihoitona. HER2:n yli-ilmentyminen tai amplifikaatio varmistettiin keskitetysti laboratorioanalyysien avulla.
Potilaat, joilla oli metastaaseja aivoissa (mukaan lukien potilaat, joilla oli hoitamattomia tai eteneviä leesioita), soveltuivat osallistumaan tutkimukseen, mikäli he olivat neurologisesti stabiileja eivätkä tarvinneet välitöntä aivojen sädehoitoa tai leikkausta. Välitöntä paikallista interventiota tarvitseville potilaille voitiin antaa paikallishoitoa, minkä jälkeen heidän oli mahdollista osallistua tutkimukseen. Tutkimukseen osallistui potilaita, joilla oli hoitamattomia metastaaseja aivoissa, ja potilaita, joiden aivometastaaseja oli hoidettu. Nämä metastaasit olivat joko pysyneet ennallaan tai alkaneet edetä edellisen säde- tai leikkaushoidon jälkeen. Tutkimuksesta suljettiin ulos potilaat, jotka saivat systeemisiä kortikosteroideja (kokonaisvuorokausiannos ≥ 2 mg deksametasonia tai vastaavaa valmistetta) keskushermostometastaasien aiheuttamien oireiden hallintaan < 28 vuorokautta ennen ensimmäistä tutkimuslääkeannosta. Tutkimuksesta suljettiin pois myös potilaat, joiden sairaus oli leptomeningeaalinen. HER2-tyrosiinikinaasin estäjillä aiemmin hoidetut potilaat suljettiin pois tutkimuksesta lukuun ottamatta niitä potilaita, jotka olivat saaneet lapatinibia ≤ 21 vuorokauden ajan ja joiden hoito oli keskeytetty muista kuin taudin etenemiseen tai vakavaan toksisuuteen liittyvistä syistä. Potilaille, joilla oli hormonireseptoripositiivisia kasvaimia, ei sallittu estrogeenideprivaatiota samanaikaisena hoitona, poikkeuksena gonadotropiinia vapauttavien hormonien agonistit, joita käytetään munasarjasuppressioon premenopausaalisilla naisilla.
Yhteensä 612 potilasta satunnaistettiin suhteessa 2:1 saamaan tukatinibia yhdessä trastutsumabin ja kapesitabiinin kanssa (N = 410) tai lumelääkettä trastutsumabin ja kapesitabiinin kanssa (N = 202). Satunnaistaminen ositettiin aivoissa olevien metastaasien (kyllä vs. ei), ECOG (Eastern Cooperative Oncology Group) -toimintakykyluokan (0 vs. 1) ja alueen (Yhdysvallat, Kanada, muu maailma) suhteen.
Eri tutkimushaarojen potilasdemografiat olivat tasapainossa. Mediaani-ikä oli 54 vuotta (vaihteluväli 25–82); 116 potilasta (19 %) oli vähintään 65 vuoden ikäisiä. Potilaista 444 oli valkoihoisia (73 %) ja 607 naisia (99 %). 314 potilaan (51 %) ECOG-toimintakykyluokka oli 1 ja 298 potilaan (49 %) ECOG-toimintakykyluokka oli 0. Potilaista 60 %:lla oli estrogeeni- ja/tai progesteronireseptoripositiivinen sairaus. Potilaista 48 %:lla oli tai oli ollut metastaaseja aivoissa. Näistä potilaista 23 %:lla metastaasit olivat hoitamattomia, 40 %:lla metastaaseja oli hoidettu ja ne olivat stabiileja ja 37 %:lla metastaasien oli todettu röntgentutkimuksen perustella etenevän hoidosta huolimatta. Lisäksi 49 %:lla potilaista oli keuhkometastaaseja, 35 %:lla metastaaseja maksassa ja 14 %:lla ihometastaaseja. Potilaiden aiemmin saamien systeemisten hoitojen mediaanilukumäärä oli 4 (vaihteluväli 2–17) ja systeemisten metastaasihoitojen mediaanilukumäärä oli 3 (vaihteluväli 1–14). Kaikki potilaat olivat saaneet aiemmin trastutsumabiin perustuvia hoitoja ja trastutsumabiemtansiinia, ja kaikki potilaat kahta potilasta lukuun ottamatta olivat saaneet aiemmin pertutsumabiin perustuvaa hoitoa.
Tukatinibia tai lumelääkettä annettiin 300 mg suun kautta kahdesti päivässä taudin etenemiseen tai ei-hyväksyttävissä olevan toksisuuden ilmaantumiseen asti. Trastutsumabia annettiin laskimoon 8 mg/kg:n latausannoksena hoitosyklin 1 päivänä 1, ja tämän jälkeen 6 mg/kg:n ylläpitoannoksena jokaisen 21 päivää kestävän hoitosyklin päivänä 1. Vaihtoehtoinen trastutsumabiannos oli 600 mg:n kiinteä annos ihon alle jokaisen 21 päivää kestäneen hoitosyklin päivänä 1. Kapesitabiinia annettiin 1 000 mg/m2 suun kautta kahdesti päivässä jokaisen 21 päivää kestäneen hoitosyklin päivinä 1–14.
Ensisijainen päätetapahtuma oli etenemisvapaa elinaika (PFS) BICR (Blinded Independent Central Review) -arvion mukaan ensimmäisillä 480 satunnaistetulla potilaalla. Tässä populaatiossa tukatinibialtistuksen mediaanikesto oli 7,3 kuukautta (vaihteluväli < 0,1–35,1 kuukautta) tukatinibia + trastutsumabia + kapesitabiinia saaneiden potilaiden haarassa ja lumelääkealtistuksen 4,4 kuukautta (vaihteluväli < 0,1–24,0) lumelääkettä + trastutsumabia + kapesitabiinia saaneiden potilaiden haarassa. Trastutsumabin ja kapesitabiinin altistuksen suhteen havaittiin samankaltaisia eroja.
Toissijaiset päätetapahtumat arvioitiin kaikilta satunnaistetuilta potilailta (N = 612), niihin sisältyi kokonaiselinaika (OS), etenemisvapaa elinaika, kun potilaalla oli aiemmin ollut tai oli metastaaseja aivoissa (PFSBrainMets), ja vahvistettu objektiivinen kokonaisvaste (ORR).
Ensisijaisen ja tärkeimmän toissijaisen päätemuuttujan tulokset olivat johdonmukaisia ennalta määriteltyjen alaryhmien välillä: hormonireseptoristatus, aiemmat tai nykyiset metastaasit aivoissa, ECOG-toimintakykyluokka ja alue. Tutkijan määrittelemä PFS vastasi BICR-arviota.
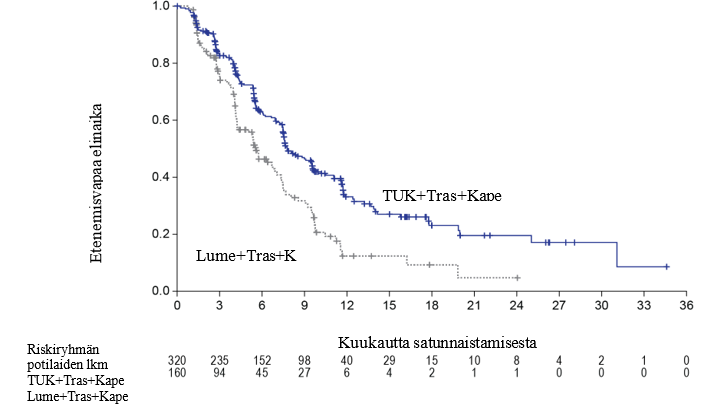
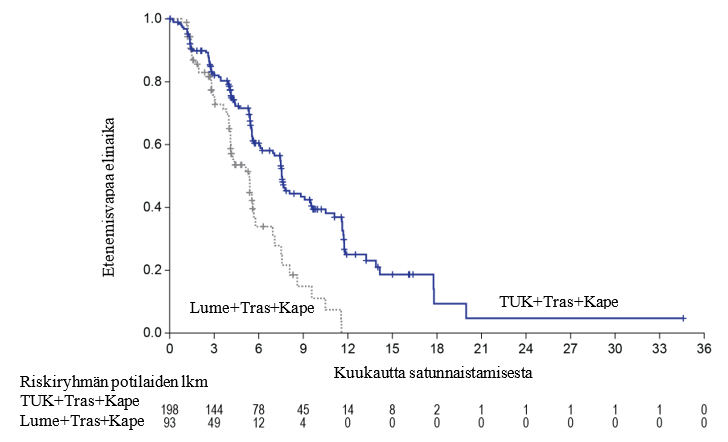
Ensisijaisen analyysin tehoa koskevat tulokset on esitetty taulukossa 5 ja kuvissa 1 ja 2.
Taulukko 5: Tehoa koskevat tulokset HER2CLIMB-tutkimuksessa (ensisijainen analyysi)
| Tukatinibi + trastutsumabi + kapesitabiini | Lumelääke + trastutsumabi + kapesitabiini | |
| PFS1 | N = 320 | N = 160 |
| Tapahtumien lukumäärä (%) | 178 (56) | 97 (61) |
| Riskisuhde (95 %:n CI)2 | 0,54 (0,42; 0,71) | |
| P‑arvo3 | < 0,00001 | |
| Mediaani (kuukautta) (95 %:n CI4) | 7,8 (7,5; 9,6) | 5,6 (4,2; 7,1) |
| Kokonaiselinaika (OS) | N = 410 | N = 202 |
| Kuolemien lukumäärä, n (%) | 130 (32) | 85 (42) |
| Riskisuhde (95 %:n CI)2 | 0,66 (0,50; 0,87) | |
| P-arvo3 | 0,00480 | |
| Kokonaiselinajan mediaani, kuukautta (95 %:n CI) | 21,9 (18,3; 31,0) | 17,4 (13,6; 19,9) |
| PFSBrainMets4 | N = 198 | N = 93 |
| Tapahtumien lukumäärä (%) | 106 (53,5) | 51 (54,8) |
| Riskisuhde (95 %:n CI)2 | 0,48 (0,34; 0,69) | |
| P-arvo3 | < 0,00001 | |
| Mediaani (kuukautta) (95 %:n CI) | 7,6 (6,2; 9,5) | 5,4 (4,1; 5,7) |
| Objektiivinen kokonaisvaste potilailla, joilla mitattavissa oleva sairaus | N = 340 | N = 171 |
| ORR (95 %:n CI)5 | 40,6 (35,3; 46,0) | 22,8 (16,7; 29,8) |
| P-arvo6 | 0,00008 | |
| CR (%) | 3 (0,9) | 2 (1,2) |
| PR (%) | 135 (39,7) | 37 (21,6) |
| Vasteen kesto | ||
| Vasteen mediaanikesto kuukausina (95 %:n CI) 7 | 8,3 (6,2; 9,7) | 6,3 (5,8; 8.9) |
BICR = sokkoutettu, riippumaton ja keskitetty arvioijataho (blinded independent central review); CI = luottamusväli (confidence interval); PFS = etenemisvapaa elinaika (progression-free survival), OS = kokonaiselinaika (overall survival), ORR = objektiivinen kokonaisvaste (objective response rate), CR = täydellinen vaste (complete response), PR = osittainen vaste (partial response).
- Ensisijainen PFS-analyysi ensimmäisillä 480 satunnaistetulla potilaalla. PFS perustuu Kaplan-Meier-estimaatteihin.
- Riskisuhde ja 95 %:n luottamusvälit perustuvat ositettuun Coxin regressiomalliin, joka huomioi ositustekijät (aiemmat tai nykyiset metastaasit aivoissa, ECOG-toimintakykyluokka ja maantieteellinen alue).
- Kaksipuoleinen p‑arvo perustuu uudelleensatunnaistamismenetelmään ositettujen tekijöiden suhteen.
- Analyysi sisältää potilaat, joilla oli aiemmin ollut tai oli lähtötilanteessa parenkymaalisia metastaaseja aivoissa, mukaan lukien kohdeleesiot ja muut kuin kohdeleesiot. Ei sisällä potilaita, joilla oli vain duraalisia leesioita.
- Kaksipuoleinen 95 %:n tarkka luottamusväli on määritelty Clopper-Pearsonin laskutavalla.
- Cochran–Mantel–Haenszelin testi ositettujen tekijöiden kontrollointia varten (aiemmat tai nykyiset metastaasit aivoissa, ECOG-toimintakykyluokka ja maantieteellinen alue).
- Laskettu komplementaarisella log-log-muunnosmenetelmällä.
Kuva 1. Kaplan‑Meier-kuvaajat etenemisvapaasta elinajasta (BICR-arvion mukaan)
Kuva 2. Kaplan‑Meier-kuvaajat etenemisvapaasta kokonaiselinajasta (BICR-arvion mukaan) potilailla, joilla oli metastaaseja aivoissa
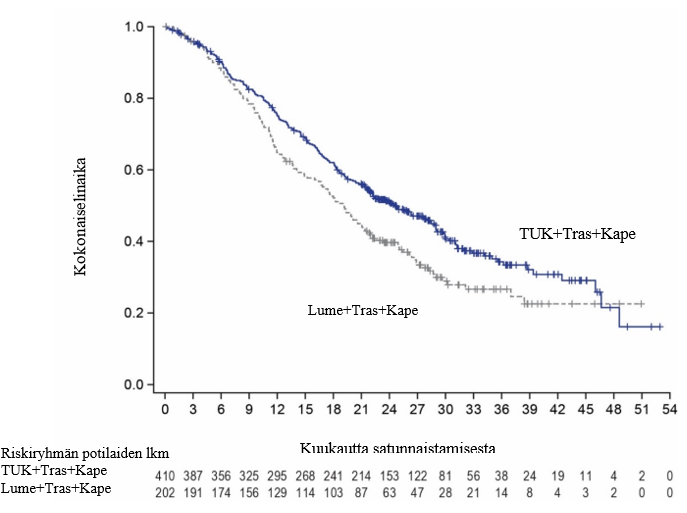
Tutkimussuunnitelman mukaisesti lopullinen kokonaiselinajan analyysi tehtiin noin kahden vuoden kuluttua viimeisen potilaan satunnaistamisesta 370 tapahtuman perusteella. Tämä vastasi 29,6 kuukauden mediaanikestoista seurantaa. Kokonaiselinajan mediaani oli 24,7 kuukautta (95 %:n luottamusväli: 21,6; 28,9) tukatinibia + trastutsumabia + kapesitabiinia saaneiden potilaiden haarassa ja 19,2 kuukautta (95 %:n luottamusväli: 16,4; 21,4) lumelääkettä + trastutsumabia + kapesitabiinia saaneiden potilaiden haarassa (riskisuhde [HR] = 0,725; 95 %:n luottamusväli: 0,585; 0,898). Lopullinen kokonaiselinajan analyysi on esitetty kuvassa 3.
Kuva 3. Kaplan‑Meier-kuvaajat kokonaiselinajasta (lopullinen analyysi)
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset TUKYSA-valmisteen käytöstä rintasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Plasman tukatinibialtistus (AUCinf ja Cmax) nousi annosta vastaavasti, kun suun kautta otettiin 50–300 mg:n annoksia (0,17–1‑kertaiset annokset suositukseen nähden). Tukatinibin AUC-arvo nousi 1,7‑kertaiseksi ja Cmax 1,5‑kertaiseksi, kun tukatinibia annettiin 300 mg kahdesti päivässä 14 päivän ajan. Vakaa tila saavutettiin noin neljässä päivässä.
Imeytyminen
Yhden suun kautta annettavan 300 mg:n tukatinibiannoksen jälkeinen mediaaniaika plasman huippupitoisuuteen oli noin 2,0 tuntia (vaihteluväli 1,0–4,0 tuntia).
Ruoan vaikutus
Kun 11 tutkittavalle, jotka olivat syöneet runsaasti rasvaa sisältävän aterian (noin 58 % rasvaa, 26 % hiilihydraattia ja 16 % proteiinia), annettiin yksittäinen tukatinibiannos, keskimääräinen AUCinf oli 1,5‑kertaa korkeampi, Tmax muuttui 1,5 tunnista 4,0 tuntiin ja Cmax pysyi ennallaan. Ruoan vaikutus tukatinibin farmakokinetiikkaan ei ollut kliinisesti merkityksellinen, joten tukatinibia voidaan ottaa ruoan kanssa tai ilman ruokaa.
Jakautuminen
Tukatinibin näennäinen jakautumistilavuus on noin 1 670 l terveillä tutkittavilla yhden 300 mg:n annoksen jälkeen. Sitoutuminen plasman proteiineihin oli 97,1 % kliinisesti relevanteilla pitoisuuksilla.
Biotransformaatio
Tukatinibi metaboloituu pääasiassa CYP2C8:n välityksellä ja pienemmässä määrin CYP3A:n ja aldehydioksidaasin välityksellä.
Lääkkeiden in vitro -yhteisvaikutustutkimukset
Tukatinibi on CYP2C8:n ja CYP3A:n substraatti.
Tukatinibi on CYP2C8:n ja CYP3A:n reversiibeli estäjä ja CYP3A:n ajasta riippuvainen estäjä kliinisesti merkityksellisinä pitoisuuksina.
Tukatinibilla on alhainen potentiaali toimia CYP1A2:n, CYP2B6:n, CYP2C9:n, CYP2C19:n, CYP2D6:n ja UGT1A1:n estäjänä kliinisesti merkityksellisinä pitoisuuksina.
Tukatinibi on P‑gp:n BCRP:n substraatti. Tukatinibi ei ole OAT1:n, OAT3:n, OCT1:n, OCT2:n, OATP1B1:n, OATP1B3:n, MATE1:n, MATE2-K:n tai BSEP:n substraatti.
Tukatinibi estää MATE1/MATE2‑K-välitteistä metformiinin kuljetusta ja OCT2/MATE1-välitteistä kreatiniinin kuljetusta. Kliinisissä tutkimuksissa havaittu seerumin kreatiniinitason nousu tukatinibihoidon aikana johtuu kreatiinin tubuluserityksen estymisestä OCT2:n ja MATE1:n kautta.
Eliminaatio
Yhden suun kautta annettavan 300 mg:n tukatinibiannoksen puoliintumisajan mediaani plasmasta oli noin 8,5 tuntia ja näennäinen puhdistuma 148 l/h terveillä tutkittavilla.
Eritys
Tukatinibi eliminoituu pääasiassa maksan ja sappiteiden kautta, eikä se eliminoidu merkittävissä määrin munuaisten kautta. Yhden suun kautta annetun 300 mg:n 14C-tukatinibiannoksen jälkeen noin 85,8 % radiomerkitystä annoksesta löytyi ulosteista (15,9 % annoksesta muuttumattomana tukatinibina) ja 4,1 % virtsasta, joten yhteensä 89,9 % annoksesta saatiin talteen 312 tunnin sisällä antamisen jälkeen. Noin 75,6 % radioaktiivisuudesta säilyi muuttumattomana plasmassa, 19 % liittyi tunnistettuihin metaboliitteihin ja 5 % oli vapaata.
Erityisryhmät
Populaatiofarmakokineettisten analyysien perusteella ikä (< 65 vuotta [N = 211]; ≥ 65 vuotta [N = 27]), albumiini (25,0–52,0 g/l), kreatiniinipuhdistuma (CLcr 60–89 ml/min [N = 89]; CLcr 30–59 ml/min [N = 5]), kehonpaino (40,7–138,0 kg) ja rotu (valkoinen [N = 168], musta [N = 53] tai aasialainen [N = 10]) eivät vaikuta tukatinibialtistukseen kliinisesti merkityksellisesti. Vaikeaa munuaisten vajaatoimintaa sairastavista tutkittavista ei ole olemassa tietoja.
Munuaisten vajaatoiminta
Tukatinibin farmakokinetiikkaa ei ole arvioitu erillisessä munuaisten vajaatoimintaa käsittelevässä tutkimuksessa.
Maksan vajaatoiminta
Lievällä (Child–Pugh A) ja kohtalaisella (Child‑Pugh B) maksan vajaatoiminnalla ei ollut kliinisesti merkittävää vaikutusta tukatinibialtistukseen. Tukatinibin AUCinf nousi 1,6‑kertaiseksi vaikeaa maksan vajaatoimintaa (Child‑Pugh C) sairastavilla tutkittavilla verrattuna tutkittaviin, joiden maksan toiminta oli normaalia. Ei ole olemassa tietoja vaikeaa maksan vajaatoimintaa sairastavista rintasyöpäpotilaista.
Prekliiniset tiedot turvallisuudesta
Tukatinibilla ei ole tehty karsinogeenisuutta koskevia tutkimuksia.
Tukatinibi ei ollut klastogeeninen tai mutageeninen tavanomaisissa genotoksisuustutkimuksissa.
Toistuvan annoksen toksisuutta koskevissa tutkimuksissa rotilla havaittiin keltarauhasen pienenemistä, munasarjan välisolujen lisääntymistä, kohdun surkastumista ja emätinerityksen lisääntymistä annoksilla ≥ 6 mg/kg/vrk kahdesti päivässä, joka vastaa 0,09‑kertaisesti ihmisen altistusta AUC0‑12:n perusteella suositellulla annoksella. Uros- tai naaraspuolisten jaavanmakakien tai urospuolisten rottien lisääntymiselimissä ei havaittu histologisia vaikutuksia annoksilla, jotka johtivat enintään 8‑kertaiseen (apinoilla) tai 13‑kertaiseen (rotilla) altistukseen suhteessa ihmisen altistukseen suositusannoksella AUC0‑12:n perusteella.
Kaniineilla ja rotilla on tehty embryofetaaliseen kehitykseen kohdistuvia tutkimuksia. Tiineillä kaniineilla havaittiin lisääntynyttä resorptiota, elävänä syntyneiden poikasten osuuden pienenemistä sekä sikiöiden luuston ja sisäelinten sekä ulkoisia epämuodostumia annoksella ≥ 90 mg/kg/vrk. Emon altistus tällä annoksella vastaa noin ihmisen saamaa altistusta AUC:hen perustuvalla suositusannoksella. Tiineillä rotilla havaittiin emon painon alenemista ja painon nousua annoksella ≥ 90 mg/kg/vrk. Sikiöillä havaittiin painon alenemista ja viivästynyttä ossifikaatiota annoksella ≥ 120 mg/kg/vrk. Emon altistus tällä annoksella on noin kuusi kertaa suurempi kuin ihmisen saama altistus suositusannoksella AUC:n perusteella.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Kopovidoni (E1208)
Krospovidoni (E1202)
Natriumkloridi
Kaliumkloridi (E508)
Natriumvetykarbonaatti (E500)
Piioksidi, kolloidinen, vedetön (E551)
Magnesiumstearaatti
Mikrokiteinen selluloosa
Kalvopäällyste
Poly(vinyylialkoholi) (E1203)
Titaanidioksidi (E171)
Makrogoli 4000 (E1521)
Talkki (E553b)
Keltainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TUKYSA tabletti, kalvopäällysteinen
50 mg (L:ei) 88 fol (2338,00 €)
150 mg (L:ei) 84 fol (6358,60 €)
PF-selosteen tieto
OPA/ALU/PVC-läpipainopakkaus, joka on suljettu alumiinifoliolla.
TUKYSA 50 mg tabletit, kalvopäällysteiset
Yksi pahvipakkaus sisältää 88 kalvopäällysteistä tablettia (11 läpipainolevyä, joista kukin sisältää 8 tablettia).
TUKYSA 150 mg tabletit, kalvopäällysteiset
Yksi pahvipakkaus sisältää 84 kalvopäällysteistä tablettia (21 läpipainolevyä, joista kukin sisältää 4 tablettia).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
TUKYSA 50 mg tabletit, kalvopäällysteiset
Pyöreä, keltainen kalvopäällysteinen tabletti, jonka toisella puolella on merkintä "TUC" ja toisella puolella "50". 50 mg:n tabletin halkaisija on noin 8 mm.
TUKYSA 150 mg tabletit, kalvopäällysteiset
Soikea, keltainen kalvopäällysteinen tabletti, jonka toisella puolella on merkintä "TUC" ja toisella puolella "150". 150 mg:n tabletti on noin 17 mm pitkä ja noin 7 mm leveä.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TUKYSA tabletti, kalvopäällysteinen
50 mg 88 fol
150 mg 84 fol
- Ylempi erityiskorvaus (100 %). Tukatinibi: HER2-positiivisen rintasyövän hoito aikuisille yhdistelmänä trastutsumabin ja kapesitabiinin kanssa erityisin edellytyksin (1544).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Tukatinibi: HER2-positiivisen rintasyövän hoito aikuisille yhdistelmänä trastutsumabin ja kapesitabiinin kanssa erityisin edellytyksin (3065).
ATC-koodi
L01EH03
Valmisteyhteenvedon muuttamispäivämäärä
24.11.2025
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com