
TRODELVY kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 200 mg
Huomioitavaa
▼Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi kuiva-aineinjektiopullo sisältää 200 mg sasitutsumabigovitekaania.
Käyttökuntoon saattamisen jälkeen yksi ml liuosta sisältää 10 mg sasitutsumabigovitekaania.
Sasitutsumabigovitekaani on Trop‑2:een kohdennettu vasta-ainekonjugoitu solunsalpaaja (antibody-drug conjugate, ADC). Sasitutsumabi on Trop‑2:n tunnistava humanisoitu monoklonaalinen vasta-aine (hRS7 IgG1κ), ja pieni molekyyli, SN‑38, on topoisomeraasi I:n estäjä, joka on kovalenttisesti sitoutunut vasta-aineeseen hydrolysoituvalla liitäntäosalla. Kuhunkin vasta-ainemolekyyliin on sitoutunut noin 7–8 SN‑38-molekyyliä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Kliiniset tiedot
Käyttöaiheet
Kolmoisnegatiivinen rintasyöpä
Trodelvy on tarkoitettu:
-
monoterapiana leikkaukseen soveltumattoman paikallisesti edenneen tai metastasoituneen kolmoisnegatiivisen rintasyövän (TNBC) hoitoon aikuispotilaille, jotka eivät ole aiemmin saaneet systeemistä hoitoa metastasoituneeseen tautiin ja joille PD-1- tai PD-L1-estäjähoito ei sovellu (ks. kohta Farmakodynamiikka).
-
monoterapiana leikkaukseen soveltumattoman tai metastasoituneen kolmoisnegatiivisen rintasyövän hoitoon aikuispotilaille, jotka ovat saaneet kahta tai useampaa aikaisempaa systeemistä hoitoa, joista vähintään yksi on annettu pitkälle edenneen taudin hoitoon (ks. kohta Farmakodynamiikka).
HR-positiivinen, HER2-negatiivinen rintasyöpä
Trodelvy monoterapiana on tarkoitettu leikkaukseen soveltumattoman tai metastasoituneen hormonireseptoripositiivisen (HR-positiivisen), HER2-negatiivisen rintasyövän hoitoon aikuispotilaille, jotka ovat saaneet hormonaalista hoitoa ja vähintään kahta muuta systeemistä hoitoa pitkälle edenneeseen tautiin (ks. kohta Farmakodynamiikka).
Ehto
Valmiste on annettava kokeneen terveydenhuollon ammattilaisen valvonnassa sellaisessa hoitopaikassa, jossa kaikki potilaan elvyttämiseen tarvittavat välineet ovat välittömästi saatavilla.
Annostus ja antotapa
Trodelvy-valmistetta saavat määrätä ja antaa potilaalle vain syöpähoitoihin perehtyneet terveydenhuollon ammattilaiset. Valmiste on annettava tiloissa, joissa on käytössä täydellinen elvytysvälineistö.
Annostus
Sasitutsumabigovitekaanin suositeltu annos on 10 mg painokiloa kohti, ja se annetaan infuusiona laskimoon kerran viikossa 21 päivän mittaisten hoitosyklien päivinä 1 ja 8. Hoitoa jatketaan, kunnes tauti etenee tai ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä.
Ehkäisevä hoito
Infuusioon liittyviä reaktioita sekä solunsalpaajahoidon aiheuttamaa pahoinvointia ja oksentelua (CINV) ehkäisevää hoitoa suositellaan aina ennen sasitutsumabigovitekaaniannoksen antamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Neutropenian estohoito
Potilailla, joilla kuumeisen neutropenian riski on suurentunut, on harkittava granulosyyttiryhmiä stimuloivan kasvutekijän (G‑CSF) antamista primaariprofylaksina ensimmäisestä hoitosyklistä alkaen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutusten vuoksi tehtävät annosmuutokset
Haittavaikutusten hoito saattaa vaatia sasitutsumabigovitekaanihoidon lopettamisen tilapäisesti tai pysyvästi, tai annosta saattaa olla tarpeen pienentää. Suositeltu annoksen pienentämissuunnitelma on esitetty taulukossa 1, ja haittavaikutusten ilmetessä suositellut annosmuutokset on esitetty taulukossa 2. Sasitutsumabigovitekaaniannosta ei pidä suurentaa uudelleen, jos annosta on pienennetty haittavaikutusten vuoksi.
Taulukko 1: Annoksen pienentämissuunnitelma
Annoksen pienentämissuunnitelma | Annostaso |
|---|---|
Suositeltu aloitusannos | 10 mg/kg |
Annoksen ensimmäinen pienentämiskerta | Pienennä 7,5 mg:aan/kg |
Annoksen toinen pienentämiskerta | Pienennä 5 mg:aan/kg |
Tarve pienentää annosta edelleen | Lopeta hoito |
Taulukko 2: Haittavaikutusten vuoksi suositellut annosmuutokset
Haittavaikutukset | Vaikeusaste | Annosmuutos |
|---|---|---|
Neutropenia |
|
|
Pahoinvointi/oksentelu/ripuli |
|
|
Infuusioon liittyvä reaktio |
|
|
|
| |
Muut toksiset vaikutukset |
|
|
Erityisryhmät
Iäkkäät
Annosta ei tarvitse muuttaa ≥ 65‑vuotiaille potilaille. Tietoa sasitutsumabigovitekaanin käytöstä ≥ 75‑vuotiaille potilaille on niukasti.
Maksan vajaatoiminta
Aloitusannosta ei tarvitse muuttaa, kun sasitutsumabigovitekaania annetaan potilaille, joilla on lievä maksan vajaatoiminta (bilirubiini ≤ 1,5‑kertainen ja aspartaattiaminotransferaasi [ASAT] / alaniiniaminotransferaasi [ALAT] < 3‑kertainen viitealueen ylärajaan nähden).
Sasitutsumabigovitekaanin turvallisuutta kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavien potilaiden hoidossa ei ole varmistettu. Sasitutsumabigovitekaania ei ole tutkittu potilailla, joilla on jokin seuraavista: seerumin bilirubiini on > 1,5‑kertainen viitealueen ylärajaan nähden tai ASAT tai ALAT on > 3‑kertainen viitealueen ylärajaan nähden potilailla, joiden maksassa ei ole etäpesäkkeitä, tai ASAT tai ALAT on > 5‑kertainen viitealueen ylärajaan nähden potilailla, joiden maksassa on etäpesäkkeitä. Sasitutsumabigovitekaanin käyttöä tällaisille potilaille on vältettävä.
Munuaisten vajaatoiminta
Aloitusannosta ei tarvitse muuttaa, kun sasitutsumabigovitekaania annetaan potilaille, joilla on lievä tai kohtalainen munuaisten vajaatoiminta.
Sasitutsumabigovitekaania ei ole tutkittu potilailla, joilla on vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaissairaus (kreatiniinipuhdistuma < 15 ml/min).
Pediatriset potilaat
Sasitutsumabigovitekaanin turvallisuutta ja tehoa 0–18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Sasitutsumabigovitekaanin saa antaa vain laskimoon. Syöpälääkkeiden käsittelyyn perehtyneen terveydenhuollon ammattilaisen on saatettava se käyttökuntoon ja laimennettava se. Se on annettava laskimoon infuusiona, ei saa antaa laskimoon push- eikä bolusinjektiona.
Ensimmäinen infuusio: annetaan 3 tuntia kestävänä infuusiona.
Seuraavat infuusiot: annetaan 1–2 tuntia kestävinä infuusioina, jos potilas on sietänyt aiemmat infuusiot.
Potilaita on seurattava kunkin infuusion aikana ja vähintään 30 minuuttia kunkin infuusion jälkeen infuusioon liittyviin reaktioihin viittaavien merkkien ja oireiden varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Neutropenia
Sasitutsumabigovitekaani voi aiheuttaa vaikean tai hengenvaarallisen neutropenian (ks. kohta Haittavaikutukset). Sasitutsumabigovitekaanilla tehdyissä kliinisissä tutkimuksissa on havaittu kuolemaan johtaneita infektioita neutropenian yhteydessä, pääasiassa kahden ensimmäisen hoitosyklin aikana.
Potilailla, joilla kuumeisen neutropenian riski on suurentunut, on harkittava granulosyyttiryhmiä stimuloivan kasvutekijän (G‑CSF) antamista primaariprofylaksina ensimmäisestä hoitosyklistä alkaen. Tällaisia potilaita ovat esimerkiksi iäkkäät (erityisesti vähintään 65‑vuotiaat) sekä ne, joilla on aiemmin ollut neutropenia, joilla suorituskykypistemäärä on huono, joilla on jonkin elimen toimintahäiriö (mukaan lukien munuaisten, maksan tai verenkiertoelimistön toimintahäiriö) tai joilla on useita samanaikaisia sairauksia. Neutrofiilien absoluuttista määrää on seurattava hoidon aikana.
Sasitutsumabigovitekaania ei pidä antaa, jos neutrofiilien absoluuttinen määrä on alle 1 500/mm3 minkä tahansa syklin päivänä 1 tai jos neutrofiilien määrä on alle 1 000/mm3 minkä tahansa syklin päivänä 8. Sasitutsumabigovitekaania ei pidä antaa, jos potilaalla on neutropeenista kuumetta. Annosmuutokset saattavat olla tarpeen, jos potilaalla on neutropenia tai kuumeinen neutropenia. Neutropenian hoitoon annetaan G‑CSF:ää, ja seuraavissa hoitosykleissä on harkittava profylaksia kliinisen tarpeen mukaan (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Ripuli
Sasitutsumabigovitekaani voi aiheuttaa vaikean ripulin (ks. kohta Haittavaikutukset). Joissakin tapauksissa ripulin havaittiin johtaneen elimistön kuivumiseen ja tästä johtuvaan akuuttiin munuaisvaurioon. Sasitutsumabigovitekaania ei pidä antaa, jos ripulin vaikeusaste on 3 tai 4 hoidon suunniteltuna ajankohtana, ja hoitoa jatketaan vasta, kun tila on lievittynyt asteeseen ≤ 1 (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).Jos ripulin ilmaantuessa ei ole tiedossa infektiota, joka voisi olla ripulin aiheuttaja, on aloitettava hoito loperamidilla. Myös muita tukitoimia (esim. menetetyn nesteen ja elektrolyyttien korvaaminen) voidaan ottaa käyttöön kliinisen tarpeen mukaan.
Potilaat, joilla sasitutsumabigovitekaanihoito aiheuttaa epänormaalin voimakkaan kolinergisen vasteen (esim. vatsanväänteitä, ripulia tai syljeneritystä), voivat saada asianmukaista hoitoa (esim. atropiinia) seuraavien sasitutsumabigovitekaanihoitojen yhteydessä.
Yliherkkyys
Sasitutsumabigovitekaani voi aiheuttaa vaikeaa ja hengenvaarallista yliherkkyyttä (ks. kohta Haittavaikutukset). Sasitutsumabigovitekaanilla tehdyissä kliinisissä tutkimuksissa on havaittu anafylaktisia reaktioita, ja sasitutsumabigovitekaanin käyttö on vasta-aiheista potilaille, joiden tiedetään olevan yliherkkiä sasitutsumabigovitekaanille (ks. kohta Vasta-aiheet).
Sasitutsumabigovitekaania saaville potilaille suositellaan ennen infuusiota annettavaa esihoitoa, kuten kuumelääkettä, H1- ja H2‑salpaajia tai kortikosteroideja (esim. 50 mg hydrokortisonia tai vastaavaa hoitoa joko suun kautta tai laskimoon). Potilaita on seurattava tarkasti jokaisen sasitutsumabigovitekaani-infuusion ajan ja vähintään 30 minuuttia jokaisen infuusion jälkeen infuusioon liittyvien reaktioiden varalta. Sasitutsumabigovitekaanin infuusionopeutta on hidastettava tai infuusio on keskeytettävä, jos potilaalle kehittyy infuusioon liittyvä reaktio. Sasitutsumabigovitekaanin käyttö on lopetettava pysyvästi, jos potilaalla ilmenee hengenvaarallinen infuusioon liittyvä reaktio (ks. kohta Annostus ja antotapa).
Pahoinvointi ja oksentelu
Sasitutsumabigovitekaani aiheuttaa pahoinvointia (ks. kohta Haittavaikutukset). Solunsalpaajahoidon aiheuttaman pahoinvoinnin ja oksentelun (CINV) ehkäisyyn suositellaan antiemeettistä ennaltaehkäisevää hoitoa kahdella tai kolmella lääkevalmisteella (esim. deksametasoni yhdessä joko 5‑hydroksitryptamiini 3 [5‑HT3] ‑reseptorinsalpaajan tai neurokiniini 1 [NK1] ‑reseptorinsalpaajan sekä tarvittaessa muiden lääkevalmisteiden kanssa).
Sasitutsumabigovitekaania ei pidä antaa, jos hoidon suunniteltuna antoajankohtana ilmenee asteen 3 pahoinvointia tai asteen 3–4 oksentelua, ja hoitoa voidaan jatkaa vain käyttäen ylimääräisiä tukitoimenpiteitä ja vasta sitten, kun tila on lievittynyt asteeseen ≤ 1 (ks. kohta Annostus ja antotapa). Myös muita pahoinvointilääkkeitä ja tukitoimenpiteitä voidaan ottaa käyttöön kliinisen tarpeen mukaan. Kaikille potilaille tulee antaa pahoinvoinnin ja oksentelun ennaltaehkäisyyn ja hoitoon kotona otettavia lääkevalmisteita ja selkeät ohjeet.
Käyttö potilaille, joilla UGT1A1-aktiivisuus on heikentynyt
SN‑38 (sasitutsumabigovitekaanin pienimolekyylinen osa) metaboloituu uridiinidifosfaattiglukuronosyylitransferaasin (UGT1A1) välityksellä. UGT1A1-geenin muunnokset, kuten UGT1A1*28-alleeli, aiheuttavat UGT1A1-entsyymin heikentynyttä aktiivisuutta. UGT1A1*28-alleelin suhteen homotsygoottisilla henkilöillä on suurentunut neutropenian, kuumeisen neutropenian ja anemian riski. Heillä on myös suurentunut sasitutsumabigovitekaanihoidon aloituksen jälkeen ilmenevien haittavaikutusten riski (ks. kohta Haittavaikutukset). Asteen 3–4 neutropeniaa, kuumeista neutropeniaa ja anemiaa havaittiin enemmän UGT1A1*28-alleelin suhteen homotsygoottisilla potilailla ja UGT1A1*6‑alleelin suhteen heterotsygoottisilla potilailla kuin villityypin alleelin suhteen homotsygoottisilla potilailla. Noin 20 % mustaihoisista, 10 % valkoihoisista ja 2 % itäaasialaisista ovat homotsygoottisia UGT1A1*28-alleelin suhteen. Noin 0,2 % mustaihoisista, 0,3 % valkoihoisista ja 27 % itäaasialaisista on heterotsygoottisia UGT1A1*6-alleelin suhteen. UGT1A1*28- ja *6-alleelin lisäksi tietyissä populaatioissa saattaa esiintyä muita heikentyneeseen toimintaan liittyviä alleeleja. Jos potilaan UGT1A1-aktiivisuuden tiedetään olevan heikentynyt, häntä on seurattava tarkasti haittavaikutusten varalta. Jos UGT1A1-status ei ole tiedossa, sen määrittäminen ei ole tarpeen, sillä haittavaikutusten hoito, mukaan lukien suositellut annosmuutokset, on sama kaikilla potilailla.
Alkio- ja sikiötoksisuus
Vaikutusmekanisminsa perusteella sasitutsumabigovitekaani voi olla teratogeeninen ja/tai aiheuttaa alkio- ja sikiökuolleisuutta, kun sitä annetaan raskaana olevalle naiselle. Sasitutsumabigovitekaanin sisältämä komponentti, SN‑38, on genotoksinen, ja genotoksisuus kohdistuu nopeasti jakautuviin soluihin. Raskaana oleville naisille ja naisille, jotka voivat tulla raskaaksi, on kerrottava sikiöön kohdistuvasta mahdollisesta riskistä. Naisilta, jotka voivat tulla raskaaksi, on tarkistettava ennen sasitutsumabigovitekaanihoidon aloitusta, ovatko he raskaana (ks. kohta Raskaus ja imetys).
Natrium
Tämän lääkevalmisteen valmistelussa antoa varten käytetään natriumia sisältävää liuosta (ks. kohta Käyttö- ja käsittelyohjeet). Tämä on otettava huomioon potilaan kaikista natriumin lähteistä saaman natriumin päivittäisen kokonaismäärän suhteen.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
UGT1A1:n estäjät
Sasitutsumabigovitekaanin samanaikainen anto UGT1A1:n estäjien kanssa saattaa suurentaa haittavaikutusten ilmaantuvuutta systeemisen SN‑38-altistuksen mahdollisen suurenemisen takia. Sasitutsumabigovitekaania on käytettävä varoen potilaille, jotka saavat UGT1A1:n estäjiä (esim. propofolia, ketokonatsolia tai EGFR-tyrosiinikinaasin estäjiä).
UGT1A1:n indusorit
SN‑38-altistus saattaa pienentyä, jos potilas saa samanaikaisesti UGT1A1-entsyymien indusoreja. Sasitutsumabigovitekaania on käytettävä varoen potilaille, jotka saavat UGT1A1:n indusoreja (esim. karbamatsepiinia, fenytoiinia, rifampisiinia, ritonaviiria tai tipranaviiria).
Potilaista, jotka saivat UGT1A1:n estäjiä (n = 15) tai indusoreja (n = 7) sasitutsumabigovitekaanihoidon aikana, on saatavilla niukasti tietoja. Näiden tietojen mukaan altistukset vapaalle SN‑38:lle olivat vastaavanlaisia kuin potilailla, jotka eivät saaneet UGT1A1:n estäjiä tai indusoreja.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy miehillä ja naisilla
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja 6 kuukautta viimeisen annoksen saamisen jälkeen.
Miespotilaiden, joiden naiskumppani voi tulla raskaaksi, on käytettävä tehokasta ehkäisyä sasitutsumabigovitekaanihoidon aikana ja 3 kuukautta viimeisen annoksen saamisen jälkeen.
Raskaus
Sasitutsumabigovitekaanin käytöstä raskaana oleville naisille ei ole saatavilla tietoja. Vaikutusmekanisminsa perusteella sasitutsumabigovitekaani voi kuitenkin olla teratogeeninen ja/tai aiheuttaa alkio- ja sikiökuolleisuutta, kun sitä annetaan raskauden aikana. Sasitutsumabigovitekaanin sisältämä komponentti, SN‑38, on genotoksinen, ja genotoksisuus kohdistuu nopeasti jakautuviin soluihin.
Sasitutsumabigovitekaania ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa sasitutsumabigovitekaanilla.
Ennen sasitutsumabigovitekaanihoidon aloittamista naiselle, joka voi tulla raskaaksi, on varmistettava, onko hän raskaana.
Naisten, jotka tulevat raskaaksi, on ilmoitettava asiasta välittömästi lääkärille.
Imetys
Ei tiedetä, erittyvätkö sasitutsumabigovitekaani tai sen metaboliitit ihmisillä äidinmaitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. Imetys on lopetettava sasitutsumabigovitekaanihoidon ajaksi sekä 1 kuukauden ajaksi viimeisen annoksen saamisen jälkeen.
Hedelmällisyys
Eläimillä tehtyjen havaintojen perusteella sasitutsumabigovitekaani saattaa heikentää hedelmällisyyttä naisilla, jotka voivat tulla raskaaksi (ks. kohta Prekliiniset tiedot turvallisuudesta). Sasitutsumabigovitekaanin vaikutuksista ihmisen hedelmällisyyteen ei ole saatavilla tietoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Sasitutsumabigovitekaanilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Se voi aiheuttaa esimerkiksi huimausta tai väsymystä (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Sasitutsumabigovitekaania saaneilla potilailla yleisimmin ilmoitetut haittavaikutukset olivat neutropenia (67,3 %), pahoinvointi (62,1 %), ripuli (59,9 %), väsymys (57,4 %), alopesia (48,3 %), anemia (40,1 %), ummetus (36,7 %), oksentelu (31,3 %) ja vähentynyt ruokahalu (23,3 %).
Yleisimmät vähintään vaikeusasteen 3 haittavaikutukset olivat neutropenia (48,6 %), ripuli (10,0 %), leukopenia (9,6 %), anemia (7,7 %), väsymys (5,8 %), kuumeinen neutropenia (5,6 %) ja lymfopenia (3,1 %).
Sasitutsumabigovitekaania saaneilla potilailla useimmin ilmoitetut vakavat haittavaikutukset olivat kuumeinen neutropenia (4,5 %), ripuli (3,7 %), neutropenia (2,9 %) ja keuhkokuume (2,1 %).%).
Haittavaikutustaulukko
Haittavaikutusten esiintymistiheydet perustuvat neljän kliinisen tutkimuksen yhdistettyihin tietoihin. Tutkimuksiin osallistui 963 potilasta, jotka saivat sasitutsumabigovitekaania 10 mg/painokilo metastasoituneen kolmoisnegatiivisen rintasyövän (triple-negative breast cancer, TNBC) tai HR‑positiivisen, HER2-negatiivisen (HR+/HER2-) rintasyövän hoitoon. Mediaanialtistus sasitutsumabigovitekaanille tässä tietoaineistossa oli 5,29 kuukautta.
Haittavaikutusten esiintymistiheydet perustuvat mistä syystä tahansa ilmaantuneiden haittatapahtumien esiintymistiheyksiin, jolloin osalla tietyn haittavaikutuksen tapahtumista saattaa olla muita aiheuttajia kuin sasitutsumabigovitekaani, kuten sairaus, muut lääkevalmisteet tai tähän hoitoon liittymättömät syyt. Lääkkeen haittavaikutusten vaikeusasteet arvioitiin CTCAE-luokituksen (Common Terminology Criteria for Adverse Events) mukaan seuraavin määritelmin: aste 1 = lievä, aste 2 = keskivaikea, aste 3 = vaikea, aste 4 = hengenvaarallinen ja 5 = kuolema.
Haittavaikutukset on lueteltu elinjärjestelmä- ja esiintymistiheysluokittain. Esiintymistiheysluokat on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1 000), hyvin harvinainen (< 1 / 10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin esiintymistiheysluokassa vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3: Haittavaikutusluettelo
Elinjärjestelmä | Esiintyvyys | Haittavaikutukset |
|---|---|---|
Infektiot | ||
| Hyvin yleinen | Virtsatieinfektio |
Yleinen | Sepsis | |
Veri ja imukudos | ||
| Hyvin yleinen | Neutropenia1 |
Yleinen | Kuumeinen neutropenia | |
Immuunijärjestelmä | ||
| Hyvin yleinen | Yliherkkyys6 |
Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleinen | Vähentynyt ruokahalu |
Yleinen | Elimistön kuivuminen | |
Psyykkiset häiriöt | ||
| Yleinen | Unettomuus |
Hermosto | ||
| Hyvin yleinen | Päänsärky |
Yleinen | Makuhäiriö | |
Verisuonisto | ||
| Yleinen | Hypotensio |
Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleinen | Hengenahdistus7 |
Yleinen | Nenäverenvuoto | |
Ruoansulatuselimistö | ||
| Hyvin yleinen | Ripuli |
Yleinen | Neutropeeninen koliitti8 | |
Melko harvinainen | Enteriitti | |
Iho ja ihonalainen kudos | ||
| Hyvin yleinen | Alopesia |
Yleinen | Makulopapulaarinen ihottuma | |
Luusto, lihakset ja sidekudos | ||
| Hyvin yleinen | Selkäkipu |
Yleinen | Muskuloskeletaalinen rintakipu | |
Munuaiset ja virtsatiet | ||
| Yleinen | Hematuria |
Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleinen | Väsymys9 |
Yleinen | Kipu | |
Tutkimukset | ||
| Hyvin yleinen | Suurentunut veren alkalisen fosfataasin pitoisuus |
| Yleinen | Painon lasku |
Vammat, myrkytykset ja hoitokomplikaatiot | ||
| Yleinen | Infuusioon liittyvä reaktio |
1: Sisältää seuraavat suositellut termit: neutropenia; neutrofiilien määrä alentunut.
2: Sisältää seuraavat suositellut termit: anemia; hemoglobiiniarvo alentunut; punasolujen määrä alentunut.
3: Sisältää seuraavat suositellut termit: leukopenia; valkosolujen määrä alentunut.
4: Sisältää seuraavat suositellut termit: lymfopenia; lymfosyyttien määrä alentunut.
5: Sisältää seuraavat suositellut termit: trombosytopenia; trombosyyttien määrä alentunut.
6: Hoidon annon jälkeisen päivän loppuun mennessä raportoidut yliherkkyystapahtumat. Sisältää seuraavilla suositelluilla termeillä koodatut tapahtumat: ihottuma, infuusioon liittyvä reaktio, yliherkkyys, periorbitaalinen edeema, katetrointikohdan dermatiitti, varjoaineen aiheuttama reaktio, allerginen dermatiitti, kontaktidermatiitti, ekseema, erytematoottinen ihottuma, nokkosihottuma, anafylaktinen reaktio, bronkospasmi, allerginen sidekalvotulehdus, dermatiitti, aknea muistuttava dermatiitti, yleistynyt kesivä punaiho, lääkeyliherkkyys, infuusioon liittyvä yliherkkyysreaktio, makulaarinen ihottuma, makulopapulaarinen ihottuma, papulaarinen ihottuma, märkärakkulainen ihottuma, allerginen nuha, kasvojen turvotus, silmäluomen turpoaminen, turvonnut kieli, tyypin IV yliherkkyysreaktio, yskä ja kutina, yskä ja ihottuma, hengenahdistus ja kutina, hengenahdistus ja ihottuma, hengenahdistus ja hengityksen vinkuminen ja ihottuma sekä hypotensio ja hengenahdistus.
7: Sisältää seuraavat suositellut termit: hengenahdistus; rasitushengenahdistus
8: Sisältää suositellun termin neutropeeninen koliitti sekä umpisuolitulehduksena raportoituja tapahtumia
9: Sisältää seuraavat suositellut termit: väsymys, astenia
Valikoitujen haittavaikutusten kuvaus
Neutropenia
Aika neutropenian (kuumeinen neutropenia mukaan lukien) ilmaantumiseen ensimmäisen hoitosyklin aloitusajankohdasta laskien oli 20 päivää (mediaani). Neutropenian keston mediaani oli 10 päivää.
Neutropeniaa ilmeni 67,3 %:lla sasitutsumabigovitekaania saaneista potilaista, mukaan lukien asteen 3–4 neutropeniaa 48,6 %:lla potilaista. Neutropenia oli syynä annoksen pienentämiseen 14,2 %:lla potilaista. Neutropeeninen koliitti havaittiin 1,3 %:lla potilaista. Neutropeniaan liittyviä kuolemaan johtaneita infektioita (mukaan lukien neutropeeninen koliitti) ilmeni 0,6 %:lla potilaista.
Kuumeista neutropeniaa ilmeni 5,6 %:lla sasitutsumabigovitekaania saaneista potilaista. Kuumeinen neutropenia oli syynä annoksen pienentämiseen 2,7 %:lla potilaista.
Käyttö potilaille, joilla UGT1A1-aktiivisuus on heikentynyt
Asteen 3–4 neutropenian ilmaantuvuus oli 59,6 % potilailla, jotka olivat UGT1A1*28-alleelin suhteen homotsygoottisia, 50,7 % potilailla, jotka olivat UGT1A1*28-alleelin suhteen heterotsygoottisia, ja 44,9 % potilailla, jotka olivat villityypin alleelin suhteen homotsygoottisia. Asteen 3–4 kuumeisen neutropenian ilmaantuvuus oli 10,6 % potilailla, jotka olivat UGT1A1*28-alleelin suhteen homotsygoottisia, 5,9 % potilailla, jotka olivat UGT1A1*28-alleelin suhteen heterotsygoottisia, ja 4,3 % potilailla, jotka olivat villityypin alleelin suhteen homotsygoottisia. Asteen 3–4 anemian ilmaantuvuus oli 10,6 % potilailla, jotka olivat UGT1A1*28-alleelin suhteen homotsygoottisia, 6,4 % potilailla, jotka olivat UGT1A1*28-alleelin suhteen heterotsygoottisia, ja 6,3 % potilailla, jotka olivat villityypin alleelin suhteen homotsygoottisia. UGT1A1*6-alleelin suhteen heterotsygoottisilla potilailla asteen 3–4 neutropenian esiintyvyys oli 53,8 %, kuumeisen neutropenian 7,7 % ja anemian 7,7 %.
Verrattuna potilaisiin, jotka olivat homotsygoottisia villityypin alleelin suhteen, neutropenian ja anemian alkamisen (mediaani) todettiin tapahtuvan aiemmin potilailla, jotka olivat homotsygoottisia tai heterotsygoottisia UGT1A1*28-alleelin suhteen.
Ripuli
Aika ripulin alkamiseen ensimmäisen hoitosyklin aloitusajankohdasta laskien oli 13 päivää (mediaani). Ripulin keston mediaani oli 7 päivää.
Ripulia ilmeni 59,9 %:lla sasitutsumabigovitekaania saaneista potilaista. Asteen 3 tapahtumia ilmeni 10,0 %:lla potilaista. Neljä 963 potilaasta (< 1 %) lopetti hoidon ripulin takia.
Yliherkkyys
Yliherkkyysreaktioita raportoitiin annostelupäivää seuraavan päivän loppuun mennessä 12,8 %:lla sasitutsumabigovitekaania saaneista potilaista. Vähintään asteen 3 yliherkkyyttä ilmeni 0,5 %:lla sasitutsumabigovitekaania saaneista potilaista. Pysyvään sasitutsumabigovitekaanihoidon lopettamiseen johtaneiden yliherkkyysreaktioiden ilmaantuvuus oli 0,2 %.
Immunogeenisuus
Sasitutsumabigovitekaanihoitoa saaneilla potilailla tehdyissä kliinisissä tutkimuksissa 13 potilaalla 1 058:sta (1,2 %) kehittyi vasta-aineita sasitutsumabigovitekaanille valmisteen annon jälkeen. Heistä 10 potilaalla (0,9 %:lla kaikista sasitutsumabigovitekaania saaneista potilaista) oli neutraloivia vasta-aineita sasitutsumabigovitekaania vastaan.
Erityisryhmät
Haittavaikutusten vuoksi hoidon keskeyttäneiden osuus oli suurempi vähintään 65-vuotiailla potilailla (8 %) kuin nuoremmilla potilailla (4 %), joilla oli metastasoitunut rintasyöpä. Vähintään 65-vuotiailla potilailla havaittiin enemmän kuumeista neutropeniaa ja vaikeaa ripulia. Vähintään 65-vuotiailla potilailla havaittiin enemmän kuolemaan johtaneita infektioita. Vakavien haittatapahtumien esiintyvyys oli suurempi vähintään 75-vuotiailla potilailla (52 %) verrattuna vähintään 65-vuotiaisiin potilaisiin (35 %) ja alle 65-vuotiaisiin potilaisiin (25 %), joilla oli metastasoitunut rintasyöpä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa annetut annokset, jotka olivat enintään 18 mg/kg (noin 1,8-kertaisia suurimpaan suositeltuun annokseen 10 mg/painokilo nähden), johtivat vaikean neutropenian suurempaan ilmaantuvuuteen.
Yliannostustapauksessa potilasta on seurattava tarkasti haittavaikutuksiin, erityisesti vaikeaan neutropeniaan, viittaavien merkkien tai oireiden varalta, ja on aloitettava asianmukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, monoklonaaliset vasta-aineet ja vasta-aine–lääke-konjugaatit, muut monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, ATC-koodi: L01FX17.
Vaikutusmekanismi
Sasitutsumabigovitekaani sitoutuu Trop‑2:ta ilmentäviin kasvainsoluihin ja siirtyy solun sisään, minkä jälkeen hydrolysoituvasta liitäntäosasta vapautuu SN‑38:aa. Vuorovaikutuksessa topoisomeraasi I:n kanssa SN‑38 estää topoisomeraasi I:n indusoimien yhden juosteen katkosten liittymisen uudelleen yhteen. Tästä aiheutuva DNA-vaurio johtaa apoptoosiin ja solukuolemaan.
Kliininen teho ja turvallisuus
Kolmoisnegatiivinen rintasyöpä (TNBC)
Aiemmin hoitamaton, leikkaukseen soveltumaton paikallisesti edennyt tai metastasoitunut kolmoisnegatiivinen rintasyöpä potilailla, joille PD-1/PD-L1-estäjähoito ei sovellu (ASCENT-03)
Sasitutsumabigovitekaanin tehoa arvioitiin avoimessa, satunnaistetussa monikeskustutkimuksessa (ASCENT-03) 558 potilaalla, joilla oli leikkaukseen soveltumaton paikallisesti edennyt tai metastasoitunut kolmoisnegatiivinen rintasyöpä (mTNBC), jotka eivät olleet aiemmin saaneet systeemistä hoitoa pitkälle edenneeseen tautiin ja joille PD‑1- tai PD‑L1-estäjähoito ei soveltunut. Tutkimukseen otettiin potilaita, joilla oli PD‑L1-negatiivinen kasvain (määritelmänä kasvaimen CPS < 10 IHC 22C3 ‑määrityksellä), sekä potilaita, joilla oli PD‑L1-positiivinen kasvain (määritelmänä CPS ≥ 10 IHC 22C3 ‑määrityksellä), jos he olivat saaneet PD‑1- tai PD‑L1-estäjää (neo)adjuvanttihoitona tai jos heillä oli samanaikainen PD‑1- tai PD‑L1-estäjähoidon estävä sairaus.
Potilaat olivat saattaneet saada solunsalpaajahoitoa sekä mahdollisesti yhdistelmänä sen kanssa PD‑1- tai PD‑L1-estäjähoitoa ja/tai sädehoitoa varhaisvaiheen TNBC:n hoidossa, mutta systeemisen (neo)adjuvanttihoidon tai leikkauksen – sen mukaan, kummasta oli vähemmän aikaa – ja ensimmäisen paikallisen tai etäpesäkkeisen uusiutumisen välillä täytyi olla kulunut vähintään 6 kuukautta. Aikaisempaa hoitoa topoisomeraasi I:n estäjillä tai topoisomeraasin estäjää sisältävillä vasta-aine–lääke-konjugaateilla ei sallittu.
Potilaat satunnaistettiin (suhteessa 1:1) kahteen ryhmään, josta toinen sai 10 mg/kg sasitutsumabigovitekaania laskimonsisäisenä infuusiona 21 päivän mittaisen hoitosyklin päivinä 1 ja 8 (n = 279) ja toinen lääkärin valitsemaa hoitoa (n = 279). Ennen satunnaistamista tutkija määritteli lääkärin valitsemaksi hoidoksi jonkin seuraavista hoito-ohjelmista: gemsitabiini ja karboplatiini, paklitakseli tai nab-paklitakseli. Satunnaistaminen ositettiin sen mukaan, oliko kyseessä de novo -tapaus, taudin uusiutuminen 6–12 kuukauden kuluessa kuratiivisen hoidon päättymisestä vai taudin uusiutuminen > 12 kuukauden kuluttua kuratiivisen hoidon päättymisestä, sekä maantieteellisen alueen mukaan (Yhdysvallat, Kanada, Länsi-Eurooppa vs. muu maailma).
Hoitoa jatkettiin BICR:n vahvistamaan taudin etenemiseen asti tai kunnes potilaalla ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä, potilas kuoli tai potilas peruutti suostumuksensa. Kasvain kuvannettiin ensimmäisen vuoden aikana 6 viikon välein ja sen jälkeen 12 viikon välein. BICR:n vahvistaman objektiivisen taudin etenemisen ja tutkimushoidon lopettamisen jälkeen potilaat, jotka oli satunnaistettu saamaan lääkärin valitsemaa solunsalpaajahoitoa, pystyivät tämän tutkimuksen vaihtovuoroisessa vaiheessa saamaan sasitutsumabigovitekaania osana tutkimusta, jos he täyttivät kelpoisuusvaatimukset.
Ensisijainen tehoa mittaava päätetapahtuma oli etenemisvapaa elossaoloaika (PFS), jonka arvioi BICR käyttämällä RECIST v1.1 ‑kriteerejä. Muita tehoa mittaavia päätetapahtumia olivat kokonaiselossaoloaika (OS) ja BICR:n arvioima objektiivinen hoitovaste (ORR).
Tutkimuspopulaatiossa iän mediaani oli 55 vuotta (vaihteluväli: 23–86 vuotta), ja 26 % potilaista oli vähintään 65‑vuotiaita; 99,5 % potilaista oli naispuolisia. Suurin osa potilaista oli valkoihoisia (64 %); 23 % oli aasialaisia, 3 % mustia ja 4 % muuta rotua. Potilaista 31 %:lla oli de novo ‑tauti, 21 %:lla oli uusiutunut tauti, jossa tauditon jakso oli 6–12 kuukautta, ja 48 %:lla oli uusiutunut tauti, jossa tauditon jakso oli yli 12 kuukautta. Tutkimukseen osallistuneista potilaista 99,5 %:lla oli PD‑L1-negatiivisia kasvaimia ja 0,5 %:lla PD-L1-positiivisia kasvaimia. Kaikkien potilaiden ECOG-suorituskykypistemäärä oli 0 (66 %) tai 1 (34 %) heidän aloittaessaan tutkimuksessa. Suurimmalla osalla potilaista (97,8 %) oli seulonnan yhteydessä metastasoitunut tauti, ja aivoissa etäpesäkkeitä havaittiin 5 %:lla potilaista. BRCA1/2-mutaatiostatus oli tiedossa 76 %:lla osallistujista, ja näistä tutkituista osallistujista 18 %:lla todettiin BRCA-mutaatioita.
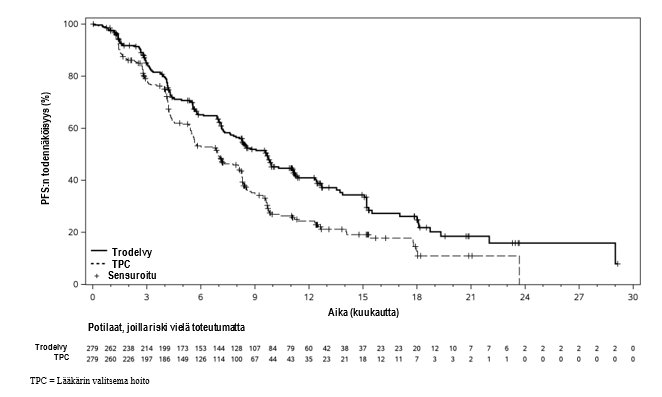
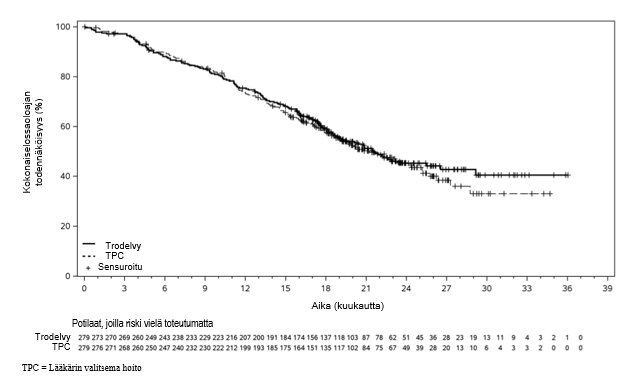
BICR:n arvion mukaan sasitutsumabigovitekaanihoito pidensi PFS-aikaa tilastollisesti merkitsevästi verrattuna lääkärin valitsemaan hoitoon. Kokonaiselossaoloajan välianalyysin laatimishetkellä potilaiden seuranta-ajan mediaani oli 17,7 kuukautta. Tehoa koskevat tulokset on koottu taulukkoon 4, kuvaan 1 ja kuvaan 2.
Taulukko 4. Tehoa mittaavat päätetapahtumat (ASCENT-03)
| TRODELVY N = 279 | Lääkärin valitsema hoito (TPC) N = 279 |
BICR:n arvioima etenemisvapaa elossaoloaika1 | ||
Niiden potilaiden lukumäärä (%), joilla ilmeni tapahtumia | 161 (58 %) | 188 (67 %) |
PFS:n mediaani kuukausina | 9,7 | 6,9 |
Riskitiheyksien suhde (95 %:n CI)2 | 0,62 (0,50, 0,77) | |
p-arvo3 | < 0,0001 | |
BICR:n arvioima objektiivinen hoitovaste1 | ||
ORR (95 %:n CI)4 | 48 % | 46 % |
Kokonaiselossaoloaika5 | ||
Niiden potilaiden lukumäärä (%), joilla ilmeni tapahtumia | 139 (50 %) | 144 (52 %) |
OS:n mediaani kuukausina (95 %:n CI) | 21,5 (18,9; 26,5) | 21,3 (18,7; 25,2) |
Riskitiheyksien suhde (95 %:n CI)2 | 0,95 (0,75, 1,20) | |
-
Perustuu PFS-primaarianalyysiin (tiedonkeruun päättymispäivä 2. huhtikuuta 2025), seuranta-ajan mediaani 13,2 kuukautta.
-
Riskitiheyksien suhde 95 %:n luottamusväleillä perustuu Coxin ositettuun suhteellisten riskitiheyksien malliin, joka on vakioitu satunnaistamisen ositustekijöiden mukaan: taudin ositus (de novo vs. uusiutuminen 6–12 kuukauden kuluessa vs. uusiutuminen > 12 kuukautta kuratiivisen hoidon päättymisestä) ja maantieteellinen alue (Yhdysvallat/Kanada/Länsi-Eurooppa vs. muu maailma).
-
2-puolinen p-arvo perustuu log-rank-testiin, joka on vakioitu satunnaistamisen ositustekijöiden mukaan: taudin ositus (de novo vs. uusiutuminen 6–12 kuukauden kuluessa vs. uusiutuminen > 12 kuukautta kuratiivisen hoidon päättymisestä) ja maantieteellinen alue (Yhdysvallat/Kanada/Länsi-Eurooppa vs. muu maailma).
-
95 %:n luottamusväli perustuu Clopper–Pearsonin tarkkaan menetelmään.
-
Perustuu ensimmäiseen kokonaiselossaoloajan välianalyysiin (tiedonkeruun päättymispäivä 27. lokakuuta 2025), seuranta-ajan mediaani 17,7 kuukautta.
BICR = sokkoutettu, riippumaton, keskitetty asiantuntijaryhmä; CI = luottamusväli
TPC = gemsitabiini ja karboplatiini, paklitakseli tai nab-paklitakseli
Kuva 1: BICR:n arvioima etenemisvapaa elossaoloaika ASCENT-03-tutkimuksessa (tiedonkeruun päättymispäivä 2. huhtikuuta 2025)
Kuva 2: Kokonaiselossaoloaika ASCENT-03-tutkimuksessa (tiedonkeruun päättymispäivä 27. lokakuuta 2025)
Leikkaukseen soveltumaton tai metastasoitunut kolmoisnegatiivinen rintasyöpä (ASCENT)
Sasitutsumabigovitekaanin tehoa ja turvallisuutta arvioitiin ASCENT (IMMU-132-05) ‑tutkimuksessa, joka oli kansainvälinen, avoin, satunnaistettu vaiheen 3 monikeskustutkimus, johon osallistui 529 potilasta, joilla oli leikkaushoitoon soveltumaton paikallisesti edennyt tai metastasoitunut kolmoisnegatiivinen rintasyöpä (mTNBC), joka oli uusiutunut vähintään kahden aiemman (ei ylärajaa) rintasyövän vuoksi annetun solunsalpaajahoidon jälkeen. Rajoittuneemman taudin aiempi adjuvantti- tai neoadjuvanttihoito hyväksyttiin osaksi vaadittuja aiempia hoito-ohjelmia, jos leikkaukseen soveltumaton, paikallisesti levinnyt tai metastasoitunut tauti kehittyi 12 kuukauden kuluessa solunsalpaajahoidon päättymisen jälkeen. Kaikki potilaat olivat saaneet taksaanihoitoa joko adjuvantti- tai neoadjuvanttihoitona tai pitkälle edenneen taudin hoitona, paitsi jos se oli vasta-aiheista tai he eivät sietäneet taksaanihoitoa. Poly-ADP-riboosipolymeraasin (PARP) estäjät olivat sallittuja toisena kahdesta aiemmasta solunsalpaajahoidosta potilailla, joilla oli dokumentoitu ituradan BRCA1/BRCA2-mutaatio.
Potilaat satunnaistettiin (1:1) saamaan joko 10 mg/kg sasitutsumabigovitekaania infuusiona laskimoon 21 päivän mittaisen hoitosyklin päivänä 1 ja päivänä 8 tai lääkärin valitsemaa hoitoa, jota annettiin kehon pinta-alan perusteella ja hyväksyttyjen tuotetietojen mukaisesti. Tutkija valitsi hoidon ennen satunnaistamista seuraavista yhdellä lääkevalmisteella annettavista hoito-ohjelmista: eribuliini (n = 139), kapesitabiini (n = 33), gemsitabiini (n = 38) tai vinorelbiini (paitsi jos potilaalla oli asteen ≥ 2 neuropatia, n = 52). Tutkimukseen soveltuivat potilaat, joilla oli vakaita etäpesäkkeitä aivoissa (potilaat, jotka olivat saaneet esihoitoa, joiden tauti ei ollut etenevä, jotka eivät saaneet kouristuslääkkeitä ja jotka olivat saaneet vakaalla annoksella kortikosteroidia ainakin 2 viikkoa). Magneettikuvausta aivoetäpesäkkeiden määrittämiseksi edellytettiin vain potilailta, joilla tiedettiin tai epäiltiin olevan etäpesäkkeitä aivoissa. Tutkimuksesta suljettiin pois potilaat, joilla tiedettiin olevan Gilbertin oireyhtymä, luustoon rajoittunut tauti, aiemmin todettu epästabiili angina pectoris, sydäninfarkti tai sydämen kongestiivinen vajaatoiminta, aktiivinen krooninen tulehduksellinen suolistotauti tai maha-suolikanavan perforaatio, ihmisen immuunikatovirus (HIV) tai aktiivinen hepatiitti B- tai hepatiitti C ‑infektio, sekä potilaat, jotka olivat saaneet eläviä taudinaiheuttajia sisältävän rokotteen 30 päivän sisällä tai jotka olivat saaneet aiemmin irinotekaania.
Potilaiden hoitoa jatkettiin, kunnes tauti eteni tai ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä. Ensisijainen tehoa mittaava päätetapahtuma oli etenemisvapaa elossaoloaika (PFS) potilailla, joilla ei ollut aivoetäpesäkkeitä lähtötilanteessa (BM-negatiiviset), ja sen mittasi sokkoutettu, riippumaton, keskitetty (BICR) radiologian asiantuntijoista koostuva asiantuntijaryhmä käyttämällä RECIST v1.1 ‑kriteerejä. Toissijaisia tehoa mittaavia päätetapahtumia olivat etenemisvapaa elossaoloaika (PFS) sokkoutetun riippumattoman keskitetyn arvioijatahon arvioimana koko populaatiossa, mukaan lukien kaikki potilaat riippumatta siitä, oliko heillä aivoetäpesäkkeitä, kokonaiselossaoloaika (overall survival, OS), objektiivinen hoitovaste (objective response rate, ORR) ja vasteen kesto (duration of response, DOR).
Primaarianalyysi käsitti 235 BM-negatiivista potilasta sasitutsumabigovitekaaniryhmässä ja 233 BM-negatiivista potilasta lääkärin valitsemaa hoitoa saaneiden ryhmässä. Koko populaatio analyysi käsitti 267 potilasta sasitutsumabigovitekaaniryhmässä ja 262 potilasta lääkärin valitsemaa hoitoa saaneiden ryhmässä.
Koko populaatiossa (n = 529) potilaiden demografiset tiedot ja ominaisuudet lähtötilanteessa olivat: iän mediaani 54 vuotta (vaihteluväli: 27–82 vuotta) ja 81 % < 65‑vuotiaita; 99,6 % naisia; 79 % valkoihoisia; 12 % mustaihoisia; aikaisempien systeemisten hoitojen määrän mediaani oli 4; 69 % oli saanut kahta tai kolmea aiempaa solunsalpaajahoitoa; 31 % oli saanut yli kolmea aiempaa solunsalpaajahoitoa; 42 %:lla oli etäpesäkkeitä maksassa; ja 12 %:lla oli parhaillaan tai oli aiemmin ollut aivoetäpesäkkeitä. 8 %:lla oli positiivinen BRCA1/BRCA2-mutaatiostatus; BRCA-status oli saatavilla 339 potilaalta. Kaikkien potilaiden ECOG-suorituskykypistemäärä oli 0 (43 %) tai 1 (57 %) heidän aloittaessaan tutkimuksessa. Mediaaniaika vaiheen 4 diagnoosista tutkimuksessa aloittamiseen oli 16,2 kuukautta (vaihteluväli: -0,4 – 202,9 kuukautta). Yleisimpiä aiempia solunsalpaajahoitoja olivat syklofosfamidi (83 %), antrasykliini (83 %), mukaan lukien doksorubisiini (53 %), paklitakseli (78 %), karboplatiini (65 %), kapesitabiini (67 %), gemsitabiini (36 %), dosetakseli (35 %) ja eribuliini (33 %). Yhteensä 29 % potilaista oli saanut aiemmin PD-1- tai PD-L1-hoitoa. 13 % sasitutsumabigovitekaaniryhmän potilaista kokonaispopulaatiossa oli saanut vain yhtä aiemman linjan systeemistä hoitoa metastasoituneeseen tautiin.
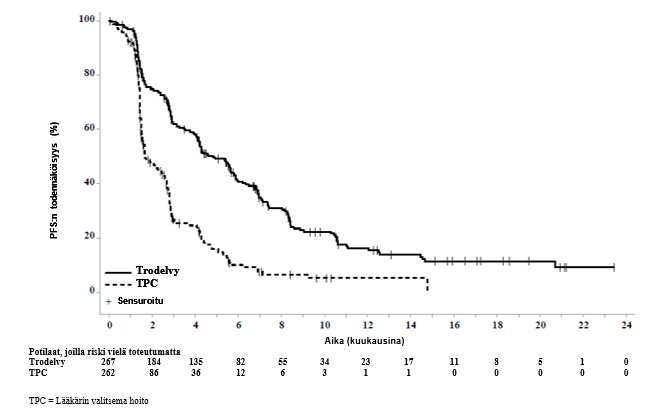
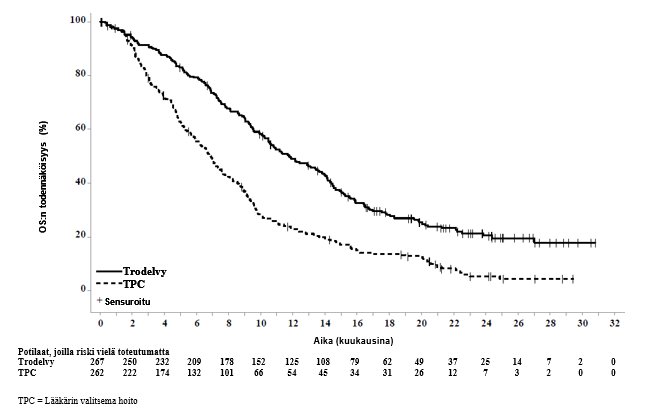
Tehoa koskevat tulokset BM-negatiivisessa populaatiossa osoittivat tilastollisesti merkitsevän sasitutsumabigovitekaanihoidolla saavutetun pitenemisen etenemisvapaassa elossaoloajassa (riskitiheyksien suhde [hazard ratio, HR] 0,41 [n = 468; 95 %:n luottamusväli: 0,32, 0,52; p-arvo: < 0,0001]) ja kokonaiselossaoloajassa (riskitiheyksien suhde 0,48 [n = 468; 95 %:n luottamusväli: 0,38, 0,59; p‑arvo: < 0,0001]) verrattuna lääkärin valitsemaan hoitoon. Etenemisvapaan elossaoloajan mediaani oli 5,6 kuukautta sasitutsumabigovitekaania saaneilla potilailla ja 1,7 kuukautta lääkärin valitsemaa hoitoa saaneilla potilailla, ja kokonaiselossaoloajan mediaani oli 12,1 kuukautta sasitutsumabigovitekaania saaneilla potilailla ja 6,7 kuukautta lääkärin valitsemaa hoitoa saaneilla potilailla.
Etukäteen määritellyssä lopullisessa analyysissä (tiedonkeruun päättymispäivä 11. maaliskuuta 2020) tehoa koskevat tulokset koko populaatiossa olivat yhdenmukaiset BM-negatiivisen populaation kanssa, ja ne on esitetty yhteenvetona taulukossa 5.
Taulukko 5: Tehoa mittaavat päätetapahtumat (koko populaatio) – etukäteen määritelty lopullinen analyysi (ASCENT)
| Etukäteen määritelty lopullinen analyysi | |
| Sasitutsumabigovitekaani | Lääkärin valitsema hoito (TPC) |
Etenemisvapaa elossaoloaika1 | ||
Tapahtumien määrä (%) | 190 (71,2) | 171 (65,3) |
Etenemisvapaan elossaoloajan mediaani kuukausina (95 %:n CI) | 4,8 (4,1, 5,8) | 1,7 (1,5, 2,5) |
Riskitiheyksien suhde (95 %:n CI) | 0,43 (0,35, 0,54) | |
p‑arvo2 | < 0,0001 | |
Kokonaiselossaoloaika (OS) | ||
Kuolemantapausten määrä (%) | 179 (67,0) | 206 (78,6) |
Kokonaiselossaoloajan mediaani kuukausina (95 %:n CI) | 11,8 (10,5, 13,8) | 6,9 (5,9, 7,7) |
Riskitiheyksien suhde (95 %:n CI) | 0,51 (0,41, 0,62) | |
p‑arvo2 | < 0,0001 | |
Kokonaishoitovaste (ORR) | ||
Vasteen saaneiden määrä (%) | 83 (31) | 11 (4) |
Kerroinsuhde (95 %:n CI) | 10,99 (5,66, 21,36) | |
p‑arvo3 | < 0,0001 | |
Täydellinen vaste, n (%) | 10 (4) | 2 (1) |
Osittainen vaste, n (%) | 73 (27) | 9 (3) |
Vasteen kesto (DOR) | ||
Vasteen keston mediaani kuukausina | 6,3 | 3,6 |
1 Etenemisvapaa elossaoloaika on määritelty ajaksi satunnaistamispäivästä ensimmäiseen radiologisesti todettuun taudin etenemispäivään tai mistä tahansa syystä johtuneeseen kuolemaan, sen mukaan, kumpi tapahtuu ensin.
2 Ositettu log-rank-testi vakioituna stratifikaatiotekijöiden mukaan: aiempien solunsalpaajahoitojen määrä, tiedossa olevat aivoetäpesäkkeet potilaiden aloittaessa tutkimuksessa ja alue.
3 Perustuu Cochran–Mantel–Haenszelin testiin.
CI = luottamusväli
Päivitetyssä tehoa koskevassa analyysissä (tietokannan lopullinen lukitus 25. helmikuuta 2021) tulokset olivat yhdenmukaiset etukäteen määritellyn lopullisen analyysin kanssa. BICR:n arvioima etenemisvapaan elossaoloajan (PFS) mediaani oli 4,8 kuukautta sasitutsumabigovitekaania saaneilla potilailla ja 1,7 kuukautta lääkärin valitsemaa hoitoa saaneilla potilailla (riskitiheyksien suhde 0,41, 95 %:n luottamusväli: 0,33, 0,52). Kokonaiselossaoloajan (OS) mediaani oli 11,8 kuukautta sasitutsumabigovitekaania saaneilla potilailla ja 6,9 kuukautta lääkärin valitsemaa hoitoa saaneilla potilailla (riskitiheyksien suhde 0,51, 95 %:n luottamusväli: 0,42, 0,63). Päivitetyt BICR:n arvioiman etenemisvapaan elossaoloajan sekä kokonaiselossaoloajan Kaplan–Meier-käyrät on esitetty kuvissa 3 ja 4.
Kuva 3: Etenemisvapaa elossaoloaika (koko populaatio; tietokannan lopullinen lukitseminen 25. helmikuuta 2021) BICR:n arvioimana ASCENT-tutkimuksessa
Kuva 4: Kokonaiselossaoloaika (koko populaatio; tietokannan lopullinen lukitseminen 25. helmikuuta 2021)ASCENT-tutkimuksessa
Alaryhmäanalyysi
Alaryhmäanalyyseissä etenemisvapaa elossaoloaika ja kokonaiselossaoloaika paranivat sasitutsumabigovitekaania saaneilla potilailla lääkärin valitsemaa hoitoa saaneisiin potilaisiin verrattuna, ja paraneminen oli yhdenmukaista kaikissa potilasryhmissä riippumatta iästä, rodusta, BRCA-statuksesta, aiempien systeemisten hoitojen kokonaismäärästä (2 ja > 2, 2–3 ja > 3) ja aiempien metastasoituneen taudin vuoksi annettujen systeemisten hoitojen määrästä (1 ja > 1), aiemmasta antrasykliini- tai PDL1-hoidosta ja etäpesäkkeistä maksassa.
Aivoetäpesäkkeet
Etenemisvapaan elossaoloajan ja kokonaiselossaoloajan eksploratiivinen analyysi tehtiin potilaista, joilla oli aiemmin hoidettuja, vakaita aivoetäpesäkkeitä. Analyysin mukaan ositettu riskitiheyksien suhde (hazard ratio, HR) oli etenemisvapaalle elossaoloajalle 0,65 (n = 61; 95 %:n luottamusväli: 0,35, 1,22) ja kokonaiselossaoloajalle 0,87 (n = 61; 95 %:n luottamusväli: 0,47, 1,63). Etenemisvapaan elossaoloajan mediaani oli 2,8 kuukautta sasitutsumabigovitekaania saaneilla potilailla ja 1,6 kuukautta lääkärin valitsemaa hoitoa saaneilla potilailla, ja kokonaiselossaoloajan mediaani oli 6,8 kuukautta sasitutsumabigovitekaania saaneilla potilailla ja 7,5 kuukautta lääkärin valitsemaa hoitoa saaneilla potilailla.
Trop-2:n ilmentyminen
Muita alaryhmäanalyysejä tehtiin tehon arvioimiseksi kasvaimen Trop-2:n ilmentymisen tasojen perusteella. Tulokset olivat yhdenmukaiset käytetystä pisteytysmenetelmästä riippumatta. Potilaat, joiden Trop-2-tasot todettiin mataliksi käyttämällä solukalvon H-pisteytystä (membrane H-score) kvartiileittain, hyötyivät sasitutsumabigovitekaanihoidosta enemmän kuin lääkärin valitsemasta hoidosta, minkä osoittivat sekä etenemisvapaa elossaoloaika (riskitiheyksien suhde 0,64; 95 %:n luottamusväli: 0,37, 1,11) että kokonaiselossaoloaika (riskitiheyksien suhde 0,71; 95 %:n luottamusväli: 0,42, 1,21).
HR‑positiivinen, HER2-negatiivinen rintasyöpä
Leikkaukseen soveltumaton tai metastasoitunut HR-positiivinen, HER2-negatiivinen rintasyöpä (TROPiCS-02)
Sasitutsumabigovitekaanin tehoa arvioitiin avoimessa, satunnaistetussa TROPiCS-02-monikeskustutkimuksessa (IMMU-132-09), johon osallistui 543 potilasta. Potilailla oli leikkaukseen soveltumaton, paikallisesti edennyt tai metastasoitunut HR-positiivinen, HER2-negatiivinen (IHC 0, IHC 1+ tai IHC 2+/ISH-) rintasyöpä, joka oli edennyt missä tahansa tautitilanteessa, kun potilas oli saanut CDK 4/6:n estäjää, hormonaalista hoitoa tai taksaania. Potilaat olivat saaneet vähintään kaksi aikaisempaa solunsalpaajahoitoa metastaattiseen tautiin (joista yksi oli voitu antaa neoadjuvantti- tai adjuvanttihoitona, jos tauti eteni tai uusiutui 12 kuukauden kuluessa solunsalpaajahoidon päättymisen jälkeen). Tutkimuksesta suljettiin pois potilaat, joilla oli luustoon rajoittunut tauti, aktiivinen krooninen tulehduksellinen suolistotauti ja aiemmin todettu suolitukos, aiemmin todettu epästabiili angina pectoris tai sydäninfarkti tai sydämen kongestiivinen vajaatoiminta tai aktiivinen hepatiitti B- tai hepatiitti C ‑infektio.
Potilaat satunnaistettiin (1:1) saamaan joko 10 mg/kg sasitutsumabigovitekaania infuusiona laskimoon 21 päivän mittaisen hoitosyklin päivänä 1 ja päivänä 8 (n = 272) tai lääkärin valitsemaa hoitoa (n = 271). Tutkija valitsi hoidon ennen satunnaistamista seuraavista yhdellä lääkevalmisteella annettavista hoito-ohjelmista: eribuliini (n = 130), vinorelbiini (n = 63), gemsitabiini (n = 56) tai kapesitabiini (n = 22). Satunnaistamisen yhteydessä ositustekijöinä olivat metastasoituneeseen tautiin annetut aikaisemmat solunsalpaajahoidot (2 tai 3–4), sisäelinmetastaasi (kyllä tai ei) ja hormonaalisen hoidon käyttö metastasoituneeseen tautiin vähintään 6 kuukauden ajan (kyllä tai ei).
Potilaiden hoitoa jatkettiin, kunnes tauti eteni tai ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä. Ensisijainen tehoa mittaava päätetapahtuma oli etenemisvapaa elossaoloaika (PFS), jonka arvioi BICR käyttämällä RECIST v1.1 ‑kriteerejä. Muita tehoa mittaavia päätetapahtumia olivat kokonaiselossaoloaika (OS), BICR:n arvioima objektiivinen hoitovaste (ORR) ja BICR:n arvioima vasteen kesto (DOR).
Tutkimuspopulaatiossa iän mediaani oli 56 vuotta (vaihteluväli: 27–86 vuotta) ja 26 % potilaista oli vähintään 65-vuotiaita. Lähes kaikki potilaat olivat naispuolisia (99 %). Suurin osa potilaista oli valkoihoisia (67 %); 4 % oli mustaihoisia, 3 % oli aasialaisia ja 26 %:lla rotu ei ollut tiedossa. Potilaiden aiemmin saamien hoitojen mediaanimäärä oli 7 (vaihteluväli: 3–17) aikaisempaa systeemistä hoitoa missä tahansa tautitilanteessa ja 3 (vaihteluväli: 0–8) aikaisempaa systeemistä solunsalpaajahoitoa metastasoituneeseen tautiin. Noin 42 % potilaista oli saanut kahta aikaisempaa solunsalpaajahoitoa metastasoituneeseen tautiin, ja 58 % potilaista oli saanut kolmea tai neljää aikaisempaa solunsalpaajahoitoa. Useimmat potilaat olivat saaneet hormonaalista hoitoa metastasoituneeseen tautiin ≥ 6 kuukauden ajan (86 %). Potilaiden ECOG-suorituskykypistemäärä oli 0 (44 %) tai 1 (56 %). 95 %:lla potilaista oli sisäelinmetastaaseja; 4,6 %:lla potilaista oli stabiileja, aikaisemmin hoidettuja aivometastaaseja.
Tutkimuksessa todettiin, että BICR:n arvioima PFS sekä OS olivat tilastollisesti merkitsevästi paremmat sasitutsumabigovitekaanihoitoa kuin lääkärin valitsemaa hoitoa saaneilla. BICR:n arvioiman PFS:n parantuminen sekä OS:n parantuminen olivat yleisesti yhdenmukaisia etukäteen määritellyissä alaryhmissä. Tehoa koskevat tulokset on esitetty yhteenvetona taulukossa 6.
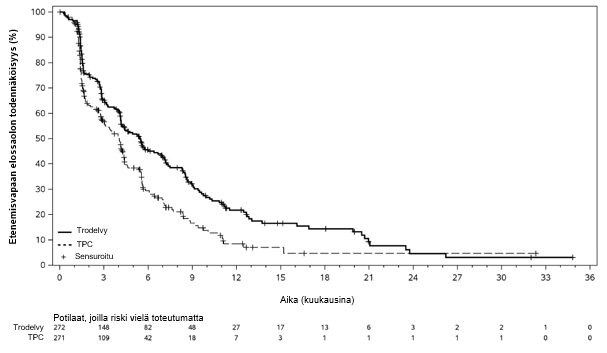
Taulukko 6. Tehoa mittaavat päätetapahtumat – etukäteen määritelty lopullinen analyysi (TROPiCS-02)
| Sasitutsumabigovitekaani | Lääkärin valitsema hoito |
BICR:n arvioima etenemisvapaa elossaoloaika1 | ||
Tapahtumien määrä (%) | 170 (62,5 %) | 159 (58,7 %) |
Etenemisvapaan elossaoloajan mediaani kuukausina (95 %:n CI) | 5,5 (4,2, 7,0) | 4,0 (3,1, 4,4) |
Riskitiheyksien suhde (95 %:n CI) | 0,661 (0,529, 0,826) | |
p-arvo2 | 0,0003 | |
Etenemisvapaan elossaolon osuus 12 kuukauden kohdalla, % (95 %:n CI) | 21,3 (15,2, 28,1) | 7,1 (2,8, 13,9) |
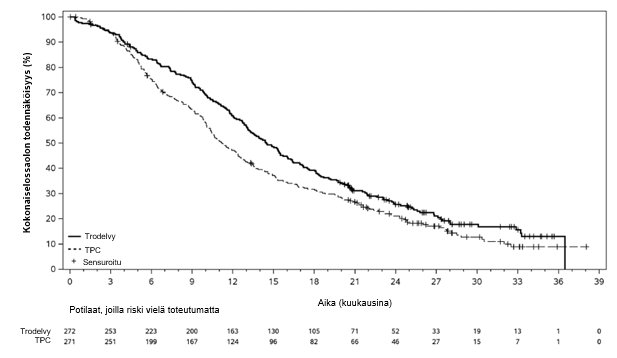
Kokonaiselossaoloaika3 | ||
Tapahtumien määrä (%) | 191 (70,2 %) | 199 (73,4 %) |
Kokonaiselossaoloajan mediaani kuukausina (95 %:n CI) | 14,4 (13,0, 15,7) | 11,2 (10,1, 12,7) |
Riskitiheyksien suhde (95 %:n CI) | 0,789 (0,646, 0,964) | |
p-arvo2 | 0,0200 | |
BICR:n arvioima objektiivinen hoitovaste3 | ||
Vasteen saaneiden määrä (%) | 57 (21,0 %) | 38 (14,0 %) |
Kerroinsuhde (95 %:n CI) | 1,625 (1,034, 2,555) | |
p-arvo | 0,0348 | |
1 Etenemisvapaa elossaoloaika on määritelty ajaksi satunnaistamispäivästä ensimmäiseen radiologisesti todettuun taudin etenemispäivään tai mistä tahansa syystä johtuneeseen kuolemaan, sen mukaan, kumpi tapahtuu ensin (tiedonkeruun katkaisupäivä 3. tammikuuta 2022).
2 Ositettu log-rank-testi vakioituna stratifikaatiotekijöiden mukaan: metastasoituneen taudin aiempien solunsalpaajahoitojen määrä (2 tai 3–4), sisäelinmetastaasi (kyllä tai ei) ja vähintään 6 kuukautta jatkunut hormonaalinen hoito metastasoituneen taudin vuoksi (kyllä tai ei).
3 Perustuu toiseen kokonaiselossaoloajan välianalyysiin (tiedonkeruun katkaisupäivä 1. heinäkuuta 2022).
BICR = sokkoutettu, riippumaton, keskitetty asiantuntijaryhmä; CI = luottamusväli
Päivitetyssä tehoanalyysissä, jossa seurannan keston mediaani oli 12,8 kuukautta (tiedonkeruun katkaisupäivä 1. joulukuuta 2022), tulokset olivat yhdenmukaisia etukäteen määritetyn lopullisen analyysin kanssa. BICR:n arvioiman PFS:n mediaani oli 5,5 kuukautta sasitutsumabigovitekaanilla hoidetuilla potilailla ja 4,0 kuukautta potilailla, jotka saivat lääkärin valitsemaa hoitoa (riskitiheyksien suhde 0,65; 95 %:n luottamusväli: 0,53, 0,81). OS:n mediaani oli vastaavasti 14,5 kuukautta ja 11,2 kuukautta (riskitiheyksien suhde 0,79; 95 %:n luottamusväli: 0,65, 0,95). Päivitetyt BICR:n arvioiman etenemisvapaan elossaoloajan sekä kokonaiselossaoloajan Kaplan–Meier-käyrät on esitetty kuvissa 5 ja 6.
Kuva 5: Etenemisvapaa elossaoloaika BICR:n arvioimana (tiedonkeruun katkaisupäivä 1. joulukuuta 2022) TROPiCS-02-tutkimuksessa
Kuva 6: Kokonaiselossaoloaika (tiedonkeruun katkaisupäivä 1. joulukuuta 2022) TROPiCS-02-tutkimuksessa
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset sasitutsumabigovitekaanin käytöstä rintasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Sasitutsumabigovitekaanin ja SN‑38:n farmakokinetiikkaa seerumissa arvioitiin metastasoitunutta rintasyöpää (mBC) sairastavilla potilailla, jotka saivat sasitutsumabigovitekaania ainoana lääkkeenä annoksella 10 mg/painokilo. Sasitutsumabigovitekaanin ja vapaan SN‑38:n farmakokineettiset parametrit on esitetty taulukossa 7.
Taulukko 7: Yhteenveto sasitutsumabigovitekaanin ja vapaan SN‑38:n farmakokineettisten parametrien keskiarvoista (variaatiokerroin, %)
| Sasitutsumabigovitekaani | Vapaa SN-38 |
Cmax [ng/ml] | 257 107 (17,9 %) | 108,4 (39,4 %) |
AUC0–168 [ng*h/ml] | 12 049 500 (18,5 %) | 3 510,2 (62,6 %) |
*Parametrit arvioitu populaatiofarmakokineettisten analyysien perusteella
Cmax: huippupitoisuus seerumissa 0–168 tunnin kuluttua ensimmäisen annoksen antamisesta
AUC0–168: pitoisuutta seerumissa kuvaavan käyrän alle jäävä pinta-ala 168 tunnin ajalta ensimmäisen annoksen antamisen jälkeen
Jakautuminen
Populaatiofarmakokineettisten analyysien mukaan sasitutsumabigovitekaanin vakaan tilan jakautumistilavuus oli 4,62 l.
Eliminaatio
Populaatiofarmakokineettisten analyysien mukaan sasitutsumabigovitekaanin eliminaation puoliintumisajan (t1/2) mediaani oli 155 tuntia ja vapaan SN‑38:n eliminaation puoliintumisajan mediaani 21,5 tuntia. Sasitutsumabigovitekaanin arvioitu keskimääräinen (%CV) puhdistuma oli 0,132 l/h (19,8 %).
Metabolia
Sasitutsumabigovitekaanilla ei ole tehty metaboliatutkimuksia.
SN‑38 (sasitutsumabigovitekaanin pienimolekyylinen osa) metaboloituu UGT1A1:n välityksellä.
Erityisryhmät
Sasitutsumabigovitekaania saaneilla potilailla tehdyissä farmakokineettisissä analyyseissä ei todettu iän, rodun ja lievän tai kohtalaisen munuaisten vajaatoiminnan vaikuttavan sasitutsumabigovitekaanin farmakokinetiikkaan.
Munuaisten vajaatoiminta
Munuaisten kautta tapahtuvalla eliminaatiolla tiedetään olevan hyvin vähäinen merkitys SN‑38:n, sasitutsumabigovitekaanin pienimolekyylisen osan, poistumiseen. Sasitutsumabigovitekaania koskevia farmakokineettisia tietoja ei ole saatavilla potilailta, joilla on vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaissairaus (kreatiniinipuhdistuma < 15 ml/min).
Maksan vajaatoiminta
Sasitutsumabigovitekaanialtistus on vastaavanlainen potilailla, joilla on lievä maksan vajaatoiminta (bilirubiini ≤ viitealueen yläraja ja ASAT > viitealueen yläraja tai bilirubiini > 1,0‑kertainen ja ≤ 1,5‑kertainen viitealueen ylärajaan nähden ja ASAT-arvo mikä tahansa), kuin potilailla, joiden maksan toiminta on normaalia (bilirubiini ja ASAT ≤ viitealueen yläraja).
Sasitutsumabigovitekaanialtistuksesta ja altistuksesta vapaalle SN-38:lle potilailla, joilla on kohtalainen tai vaikea maksan vajaatoiminta, on rajallisesti tietoja.
Prekliiniset tiedot turvallisuudesta
In vitro ‑mikrotumatestissä nisäkkään soluilla (kiinanhamsterin munasarjasoluilla) SN‑38 oli klastogeeninen, mutta se ei ollut mutageeninen bakteereilla tehdyssä in vitro ‑käänteismutaatiotestissä (Amesin testi).
Cynomolgus-apinoilla tehdyssä toistuvan altistuksen aiheuttamaa toksisuutta koskeneessa tutkimuksessa laskimoon annettu sasitutsumabigovitekaani aiheutti kohdun limakalvon atrofiaa, kohtuverenvuotoa, lisääntynyttä munarakkuloiden atresiaa munasarjoissa ja emättimen epiteelisolujen atrofiaa annoksilla ≥ 60 mg/kg (1,9‑kertaisia ihmisille suositeltuun annokseen 10 mg/kg nähden ruumiinpainon allometrisen skaalauksen perusteella).
Uudella apuaineella, MES:illä, tehtyjen toistuvan altistuksen aiheuttamaa toksisuutta ja genotoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
2-(N-morfolino)etaanisulfonihappo (MES), polysorbaatti 80 (E433), trehaloosidihydraatti
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta.
Käyttökuntoon saattamisen jälkeen
Käyttökuntoon saatettu liuos on käytettävä välittömästi laimennetun liuoksen valmistukseen infuusiota varten. Jos laimennettua liuosta sisältävää infuusiopussia ei käytetä välittömästi, sitä voidaan säilyttää jääkaapissa (2–8° C) enintään 24 tuntia valolta suojattuna.
Säilytys
Säilytä jääkaapissa (2–8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TRODELVY kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
200 mg (L:ei) 1 kpl (1194,20 €)
PF-selosteen tieto
Tyypin I värittömästä, kirkkaasta lasista valmistettu 50 ml:n injektiopullo, jossa on elastomeeributyylitulppa ja joka on suljettu alumiinisella repäisykorkilla ja joka sisältää 200 mg sasitutsumabigovitekaania.
Kukin pakkaus sisältää yhden injektiopullon.
Valmisteen kuvaus:
Luonnonvalkoinen tai kellertävä kuiva-aine.
Käyttö- ja käsittelyohjeet
Trodelvy on sytotoksinen lääkevalmiste. Asianmukaisia käsittely- ja hävittämistoimenpiteitä täytyy noudattaa.
Käyttökuntoon saattaminen
- Laske tarvittava Trodelvy-annos (mg) potilaan painon mukaan.
- Lisää steriilillä ruiskulla hitaasti 20 ml natriumkloridi-injektionestettä (9 mg/ml, 0,9 %) kuhunkin injektiopulloon, jolloin pitoisuudeksi saadaan 10 mg/ml.
- Pyörittele injektiopulloja varovasti ja anna liueta 15 minuutin ajan. Ei saa ravistaa. Valmiste on tarkastettava silmämääräisesti hiukkasten ja värimuutosten varalta ennen antoa. Liuoksen on oltava kirkasta ja keltaista, eikä siinä saa näkyä hiukkasia. Älä käytä käyttökuntoon saatettua liuosta, jos se on sameaa tai siinä näkyy värimuutoksia.
- Käytä välittömästi laimennetun infuusionesteen valmistukseen.
Laimennus
- Laske, kuinka paljon käyttökuntoon saatettua liuosta tarvitaan potilaan painon mukaiseen oikeaan annokseen.
- Määritä oikean annoksen antamiseksi tarvittava lopullinen infuusionesteen määrä. Liuoksen sasitutsumabigovitekaanipitoisuuden on oltava 1,1–3,4 mg/ml.
- Vedä ja hävitä lopullisesta infuusiopussista 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionestettä määrä, joka vastaa tarvittavaa käyttökuntoon saatetun liuoksen määrää.
- Vedä ruiskulla injektiopullo(i)sta laskettu määrä käyttökuntoon saatettua liuosta. Hävitä käyttämätön injektiopulloihin jäänyt liuos.
- Välttääksesi vaahdon muodostumisen ruiskuta tarvittava määrä käyttökuntoon saatettua liuosta hitaasti polyvinyylikloridista, polyolefiinista (polypropeenista ja/tai polyeteenistä) tai eteenivinyyliasetaatista valmistettuun infuusiopussiin. Sisältöä ei saa ravistaa.
- Säädä tarvittaessa infuusiopussissa olevaa nestemäärää 0,9-prosenttisella (9 mg/ml) natriumkloridi-injektionesteellä, jotta pitoisuudeksi saadaan 1,1–3,4 mg/ml. Vain 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionestettä saa käyttää, sillä käyttökuntoon saatetun valmisteen säilyvyyttä ei ole määritetty muilla infuusioliuoksilla.
- Jos laimennettua liuosta sisältävää infuusiopussia ei käytetä välittömästi, sitä voidaan säilyttää jääkaapissa (2–8° C) valolta suojattuna enintään 24 tuntia. Ei saa jäätyä. Jääkaapista ottamisen jälkeen laimennettu liuos on annettava huoneenlämmössä (enintään 25° C:ssä) säilytettynä 8 tunnin sisällä (infuusioaika mukaan lukien).
Anto
- Anna Trodelvy infuusiona laskimoon. Suojaa infuusiopussi valolta. Infuusiopussi tulee peittää annon ajaksi, ja se on pidettävä peitettynä infuusion loppuun saakka. Infuusioletkuja ei tarvitse peittää, eikä ole tarpeen käyttää valolta suojaavia letkuja infuusion aikana.
- Infuusiopumppua voidaan käyttää.
- Älä sekoita Trodelvy-valmistetta muiden lääkevalmisteiden kanssa äläkä anna sitä infuusiona muiden lääkevalmisteiden kanssa.
- Kun infuusio on annettu, huuhtele laskimolinja 20 ml:lla 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionestettä.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TRODELVY kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
200 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FX17
Valmisteyhteenvedon muuttamispäivämäärä
01.06.2026
Yhteystiedot
Karhumäentie 3
01530 Vantaa
Suomi
09 42726918
www.gilead.se/utility/contact
nordics.medinfo@gilead.com