
MINJUVI kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 200 mg
Huomioitavaa

Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi injektiopullo kuiva‑ainetta sisältää 200 mg tafasitamabia.
Käyttövalmiiksi saattamisen jälkeen yksi millilitra liuosta sisältää 40 mg tafasitamabia.
Tafasitamabi on humanisoitu CD19‑spesifinen monoklonaalinen immunoglobuliini G:n (IgG:n) alaluokan vasta‑aine. Tafasitamabia valmistetaan nisäkässoluissa (kiinanhamsterin munasarjasoluissa) yhdistelmä‑DNA‑tekniikalla.
Apuaine(et), joiden vaikutus tunnetaan
Yksi MINJUVI‑injektiopullo sisältää 7,4 mg natriumia ja 1,0 mg polysorbaatti 20:tä. Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kuiva‑aine välikonsentraatiksi infuusionestettä varten, liuos (kuiva‑aine välikonsentraattia varten).
Valkoinen tai hieman kellertävä kylmäkuivattu jauhe.
Kliiniset tiedot
Käyttöaiheet
MINJUVI on tarkoitettu käytettäväksi lenalidomidin kanssa ja sen jälkeen MINJUVI‑monoterapiana aikuispotilailla, joilla on uusiutunut tai hoitoon reagoimaton diffuusi suurisoluinen B‑solulymfooma (DLBCL) ja jotka eivät sovellu autologiseen kantasolusiirtoon (ASCT).
MINJUVI on tarkoitettu käytettäväksi yhdessä lenalidomidin ja rituksimabin kanssa vähintään yhden systeemisen hoitolinjan jälkeen aikuispotilailla, joilla on uusiutunut tai hoitoon reagoimaton follikulaarinen lymfooma (FL) (aste 1–3a).
Ehto
Valmistetta saa määrätä vain syöpäpotilaiden hoitoon perehtynyt lääkäri ja sitä saa antaa vain syöpäpotilaiden hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Annostus ja antotapa
MINJUVI‑annoksen antaa syöpäpotilaiden hoitoon erikoistunut terveydenhuollon ammattilainen.
Suositeltu esilääkitys
Infuusioon liittyvien reaktioiden vähentämiseen tarkoitettu esilääkitys on annettava vähintään 30 minuuttia ja korkeintaan 2 tuntia ennen tafasitamabi‑infuusiota. Potilaille, jotka eivät saa infuusioon liittyviä reaktioita ensimmäisen 3 infuusion aikana, esilääkitys on valinnaista myöhempien infuusioiden yhteydessä.
Esilääkitys voi sisältää kuumetta alentavia lääkkeitä (esim. parasetamoli), histamiini H1 ‑reseptorin salpaajia (esim. difenhydramiini), histamiini H2 ‑reseptorin salpaajia (esim. simetidiini) ja/tai glukokortikosteroideja (esim. metyyliprednisoloni).
Infuusioon liittyvien reaktioiden hoito
Jos infuusioon liittyviä reaktioita esiintyy (vakavuusaste vähintään 2), infuusio on keskeytettävä. Lisäksi on aloitettava oireiden mukainen lääkinnällinen hoito. Kun oireet on hoidettu tai niiden vakavuusaste on laskenut tasolle 1, MINJUVI‑infuusiota voidaan jatkaa alennetulla infuusionopeudella (katso taulukko 1).
Jos potilaalla on ollut infuusioon liittyvä reaktio, jonka vakavuusaste on 1–3, ennen seuraavaa tafasitamabi‑infuusioita on annettava esilääkitys.
Yhdistelmähoito lenalidomidin kanssa
Koska MINJUVI on tarkoitettu käytettäväksi yhdessä lenalidomidin kanssa, katso profylaktisia antitromboottisia valmisteita koskevat suositukset lenalidomidin valmisteyhteenvedosta.
Annostus
Suositeltu annos aikuispotilaille, joilla on uusiutunut tai hoitoon reagoimaton DLBCL
Suositeltu MINJUVI‑annos on 12 mg painokiloa kohden ja se annetaan laskimonsisäisenä infuusiona seuraavan aikataulun mukaisesti:
- Sykli 1: infuusio hoitosyklin päivinä 1, 4, 8, 15 ja 22.
- Syklit 2 ja 3: infuusio hoitosyklin päivinä 1, 8, 15 ja 22.
- Syklistä 4 eteenpäin kunnes sairaus etenee: infuusio syklien päivinä 1 ja 15.
Hoitosyklin pituus on 28 päivää.
Lisäksi potilaiden pitää ottaa lenalidomidikapseleita suositellulla päivittäisellä 25 mg:n aloitusannoksella kunkin syklin päivinä 1–21. Aloitusannosta ja sen jälkeisiä annoksia voidaan muuttaa lenalidomidin valmisteyhteenvedon mukaisesti.
Minjuvi‑valmisteen ja lenalidomidin yhdistelmää annetaan korkeintaan 12 syklin ajan.
Lenalidomidihoito on lopetettava viimeistään 12 yhdistelmähoitosyklin jälkeen. Potilaan Minjuvi‑infuusioita jatketaan monoterapiana jokaisen 28‑päiväisen hoitosyklin päivinä 1 ja 15, kunnes sairaus etenee tai toksisuus muuttuu kestämättömäksi.
Suositeltu annos aikuispotilaille, joilla on uusiutunut tai hoitoon reagoimaton FL, vähintään yhden aiemman systeemisen hoitolinjan jälkeen
Suositeltu MINJUVI-annos on 12 mg painokiloa kohden ja se annetaan laskimonsisäisenä infuusiona seuraavan aikataulun mukaisesti:
- Syklit 1–3: infuusio hoitosyklin päivinä 1, 8, 15 ja 22.
- Syklit 4–12: infuusio hoitosyklin päivinä 1 ja 15.
Hoitosyklin pituus on 28 päivää.
Suositeltu rituksimabin aloitusannos on 375 mg/m2 ja se annetaan laskimonsisäisenä infuusiona seuraavan aikataulun mukaisesti:
- Sykli 1: hoitosyklin päivinä 1, 8, 15 ja 22.
- Syklit 2–5: infuusio hoitosyklin päivänä 1.
Hoitosyklin pituus on 28 päivää. Katso laskimoon annettavien rituksimabilääkemuotojen valmisteyhteenvedosta tiedot valmisteen antotavasta ja esilääkityksestä sekä profylaktisista lääkkeistä.
Lisäksi potilaiden pitää ottaa lenalidomidikapseleita suositellulla päivittäisellä 20 mg:n aloitusannoksella kunkin 28 päivän syklin päivinä 1–21. Aloitusannosta ja sen jälkeisiä annoksia voidaan muuttaa lenalidomidin valmisteyhteenvedon mukaisesti.
MINJUVI-valmisteen ja lenalidomidin sekä rituksimabin yhdistelmää annetaan korkeintaan 12 syklin ajan MINJUVI-valmisteen ja lenalidomidin osalta ja korkeintaan 5 syklin ajan rituksimabin osalta. Rituksimabihoito on lopetettava5 yhdistelmähoitosyklin jälkeen. Potilaan MINJUVI-infuusioita jatketaan suun kautta otettavan lenalidomidin kanssa korkeintaan kahdenteentoista sykliin asti. Hoito tafasitamabilla ja lenalidomidillaon lopetettava viimeistään 12 syklin jälkeen.
Annoksen muutokset
Taulukossa 1 esitetään tehtävät MINJUVI-annosmuutokset haittavaikutuksia havaittaessa. Lenalidomidin annosmuutosten osalta pyydetään tutustumaan myös lenalidomidin valmisteyhteenvetoon.
Taulukko 1. Annosmuutokset haittavaikutuksia havaittaessa
| Haittavaikutus | Vakavuus | Annosmuutos |
| Infuusioon liittyvät reaktiot | Vakavuusaste 2 (kohtalainen) |
|
| Vakavuusaste 3 (vaikea) |
| |
| Vakavuusaste 4 (hengenvaarallinen) |
| |
| Myelosuppressio | Verihiutalemäärä alle 50 000/µl |
|
Neutrofiilimäärä alle 1 000/µl vähintään 7 päivän ajan tai Neutrofiilimäärä alle 1 000/µl ja kehon lämpötila nousee vähintään 38 °C:seen tai Neutrofiilimäärä alle 500/µl |
Jatka MINJUVI-hoitoa samalla annoksella ja lenalidomidihoitoa alennetulla annoksella, jos neutrofiilimäärä palaa tasolle ≥ 1 000/µl. Katso tarkempia tietoja lenalidomidiannoksen muuttamisesta lenalidomidin valmisteyhteenvedosta. |
Erityispotilasryhmät
Pediatriset potilaat
MINJUVI‑valmisteen turvallisuutta ja tehoa alle 18‑vuotiaiden lasten hoidossa ei ole varmistettu.
Tietoja ei ole saatavilla.
Iäkkäät
Annoksen muuttaminen ei ole tarpeen iäkkäillä potilailla (≥ 65‑vuotiaat).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä tai kohtalainen munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka). Vaikeaa munuaisten vajaatoimintaa sairastaville potilaille ei ole annostelusuositustietoja.
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen potilailla, joilla on lievä maksan vajaatoiminta (ks. kohta Farmakokinetiikka). Kohtalaista tai vaikeaa maksan vajaatoimintaa sairastaville potilaille ei ole annostelusuositustietoja.
Antotapa
MINJUVI‑valmiste on tarkoitettu laskimonsisäiseen käyttöön käyttövalmiiksi saattamisen ja laimennuksen jälkeen.
- Syklin 1 ensimmäisessä infuusiossa laskimonsisäisen infuusion nopeuden on oltava 70 ml tunnissa ensimmäisten 30 minuutin ajan. Sen jälkeen nopeutta lisätään niin, että koko ensimmäinen infuusio voidaan suorittaa 2,5 tunnissa.
- Kaikki sen jälkeiset infuusiot on annettava 1,5–2 tunnissa.
- Haittavaikutustapauksessa on harkittava taulukossa 1 mainittuja annosmuutoksia.
- Minjuvi‑valmistetta ei saa antaa yhdessä muiden lääkevalmisteiden kanssa saman infuusioletkun kautta.
- Minjuvi‑valmistetta ei saa antaa laskimonsisäisenä pistoksena eikä bolusinjektiona.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infuusioon liittyvät reaktiot
Infuusioon liittyvät reaktiot ovat mahdollisia ja niistä on raportoitu useammin ensimmäisen infuusion yhteydessä (ks. kohta Haittavaikutukset). Potilaita on seurattava tarkoin koko infuusion ajan. Potilaita on ohjeistettava ottamaan yhteyttä terveydenhuollon ammattilaiseen, jos heillä on merkkejä tai oireita infuusioon liittyvistä reaktioista, kuten kuumetta, vilunväristyksiä, ihottumaa tai hengitysvaikeuksia 24 tunnin kuluessa infuusiosta. Potilaille on annettava esilääkitystä ennen tafasitamabi‑infuusion aloittamista. Riippuen infuusioon liittyvän reaktion vakavuudesta tafasitamabi‑infuusio on keskeytettävä tai lopetettava ja soveltuva lääkinnällinen hoito aloitettava (ks. kohta Annostus ja antotapa).
Myelosuppressio
Tafasitamabihoito voi aiheuttaa vakavaa ja/tai vaikeaa myelosuppressiota, mukaan lukien neutropeniaa, trombosytopeniaa ja anemiaa (ks. kohta Haittavaikutukset). Täydellinen verenkuva on määritettävä ennen jokaista hoitosykliä koko hoidon ajan. Jos haittavaikutus on vakava, tafasitamabi‑infuusio on keskeytettävä (katso taulukko 1). Katso tarkempia tietoja lenalidomidiannoksen muuttamisesta lenalidomidin valmisteyhteenvedosta.
Neutropenia
Neutropeniasta, mukaan lukien febriilistä neutropeniasta, on raportoitu tafasitamabihoidon yhteydessä. Hoitona on harkittava granulosyyttipesäkkeitä stimuloivien tekijöiden (G‑CSF:iden) antamista erityisesti, jos potilaiden neutropenia on vakavuusasteeltaan 3 tai 4. Kehittyvän infektion oireisiin on varauduttava, ja mahdolliset infektiot on arvioitava ja hoidettava.
Trombosytopenia
Trombosytopeniasta on raportoitu tafasitamabihoidon yhteydessä. Samanaikaisten lääkevalmisteiden käytön keskeyttämistä on harkittava, jos ne saattavat lisätä verenvuotoriskiä (esim. verihiutaleiden estäjät, antikoagulantit). Potilaita pitää ohjeistaa ilmoittamaan mustelmien tai verenvuodon merkeistä ja oireista välittömästi.
Infektiot
Tafasitamabilla hoidetuilla potilailla on esiintynyt vakavia ja kuolemaan johtavia infektioita, mukaan lukien opportunistisia infektioita. Tafasitamabia voi antaa potilaille, joilla on aktiivinen infektio ainoastaan, jos infektiota hoidetaan asianmukaisesti ja se on hyvin hallinnassa. Potilailla, joilla on aiemmin ollut toistuvia tai kroonisia infektioita, voi olla kohonnut infektioriski ja heitä on valvottava sen mukaisesti.
Potilaita pitää ohjeistaa ottamaan yhteyttä terveydenhuollon ammattilaiseen, jos heillä on kuumetta tai muita mahdollisen infektion merkkejä, kuten vilunväristyksiä, yskää tai kipua virtsaamisen yhteydessä.
Progressiivinen multifokaalinen leukoenkefalopatia
Progressiivista multifokaalista leukoenkelofalopatiaa (PML) on raportoitu tafasitamabia sisältävän yhdistelmähoidon yhteydessä. Potilaita on seurattava PML:ään mahdollisesti viittaavien uusien tai pahenevien neurologisten oireiden ja löydösten varalta. PML:n oireet ovat epäspesifisiä ja voivat vaihdella infektoituneesta aivoalueesta riippuen. Oireita voivat olla mm. psyykkisen tilan muutokset, muistihäiriöt, puhevaikeudet, motoriset häiriöt (hemipareesi tai monopareesi), raajan ataksia, kävelyataksia ja näköhäiriöt, kuten hemianopsia ja diplopia. Jos potilaalla epäillään PML:ää, tafasitamabin anto on keskeytettävä välittömästi ja lähetettä neurologille on harkittava. Asianmukaisia diagnostisia toimia saattavat olla esim. magneettikuvaus, JC‑viruksen DNA:n määritys aivo‑selkäydinnesteestä ja toistuvat neurologiset arvioinnit. Jos PML todetaan, tafasitamabihoito on lopetettava pysyvästi.
Tuumorilyysioireyhtymä
Tuumorilyysioireyhtymän riski voi olla kohonnut potilailla, joilla on merkittävä tuumorikuorma ja nopeasti proliferoituva kasvain. Tuumorilyysioireyhtymää on raportoitu tafasitamabihoidon aikana. Soveltuvat toimenpiteet / ennaltaehkäisevä hoito on toteutettava paikallisten ohjeiden mukaan ennen tafasitamabihoidon toteuttamista. Potilaita on seurattava tarkasti tuumorilyysioireyhtymän varalta tafasitamabihoidon aikana.
CD19-negatiivinen tai CD20-negatiivinen tauti
CD19-negatiivisten tai CD20-negatiivisten FL-potilaiden hoidosta tafasitamabilla yhdessä lenalidomidin ja rituksimabin kanssa ei ole. On mahdollista, että CD19-negatiivista tai CD20-negatiivista FL:aa sairastavat potilaat hyötyvät hoidosta vähemmän kuin CD19-positiivista ja CD20-positiivista FL:aa sairastavat potilaat. Riskit ja hyödyt, jotka liittyvät CD19-negatiivista tai CD20-negatiivista FL:aa sairastavien potilaiden hoitamiseen tafasitamabilla yhdessä lenalidomidin ja rituksimabin kanssa, on arvioitava.
Rokotukset
Rokotusta elävillä rokotteilla tafasitamabihoidon jälkeen ei ole tutkittu, eikä eläviä rokotteita suositella annettavaksi samanaikaisesti tafasitamabihoidon kanssa.
Apuaineet
Tämä lääkevalmiste sisältää 37,0 mg natriumia per 5 injektiopulloa (83 kg painavan potilaan annos), mikä vastaa 1,85 %:ia WHO:n suositellusta 2 g:n natriumin päivittäisestä enimmäissaannista aikuisille.
Tämä lääkevalmiste sisältää 5,0 mg polysorbaatti 20:tä per 5 injektiopulloa. Polysorbaatti 20 saattaa aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Raskaus ja imetys
Tafasitamabihoitoa yhdessä lenalidomidin kanssa ei saa aloittaa naispotilailla, ennen kuin raskauden mahdollisuus on suljettu pois. Tutustu myös lenalidomidin valmisteyhteenvetoon.
Naiset, jotka voivat tulla raskaaksi / ehkäisy
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä tafasitamabihoidon aikana ja vähintään 3 kuukauden ajan hoidon päättymisen jälkeen.
Raskaus
Tafasitamabilla ei ole toteutettu lisääntymis‑ ja kehitystoksisuuteen liittyviä tutkimuksia.
Ei ole olemassa tietoja tafasitamabin käytöstä raskaana oleville naisille. IgG:n tiedetään kuitenkin kulkeutuvan istukan läpi, ja tafasitamabi voi aiheuttaa sikiön B‑solujen vähenemistä valmisteen farmakologisten ominaisuuksien perusteella (ks. kohta Farmakodynamiikka). Vastasyntyneitä, jotka ovat altistuneet valmisteelle raskauden aikana, tulisi seurata B‑solujen vähenemisen varalta, ja elävillä rokotteilla tehtäviä rokotuksia tulisi siirtää, kunnes vastasyntyneen B‑solutasot ovat palanneet normaalitasolle (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tafasitamabin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Lenalidomidi voi aiheuttaa haittaa alkiolle tai sikiölle ja sen käyttö on vasta‑aiheista raskauden aikana sekä naisilla, jotka voivat tulla raskaaksi, paitsi jos kaikki lenalidomidin raskaudenestämisohjelman ehdot täyttyvät.
Imetys
Ei tiedetä, erittyykö tafasitamabi ihmisen rintamaitoon. Äidin IgG:n tiedetään kuitenkin erittyvän rintamaitoon. Tietoja tafasitamabin käytöstä imettävillä naisilla ei ole, eikä riskiä imetettäville lapsille voida sulkea pois. Naisia pitää ohjeistaa välttämään imetystä tafasitamabihoidon aikana ja vähintään 3 kuukauden ajan viimeisen annoksen antamisesta.
Hedelmällisyys
Spesifisiä eläinkokeita tafasitamabin vaikutuksista hedelmällisyyteen ei ole tehty. Toistuvan annoksen toksisuustutkimuksissa eläimillä ei havaittu haitallisia vaikutuksia urosten tai naarasten lisääntymiselimiin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
MINJUVI‑valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneiden käyttökykyyn tai vaikutus on vähäinen. Tafasitamabia käyttävillä potilailla on kuitenkin raportoitu väsymystä, mikä on otettava huomioon ajettaessa ja koneita käytettäessä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Potilaat, joilla on uusiutunut tai hoitoon reagoimaton DLBCL
Tafasitamabin turvallisuutta DLBCL-potilailla tutkittiin avoimessa yksihaaraisessa vaiheen 2 monikeskustutkimuksessa, L-MIND, 81 tutkittavalla, joilla oli uusiutunut tai hoitoon reagoimaton DLBCL. Potilaat saivat 12 mg/kg tafasitamabia laskimonsisäisesti yhdistettynä lenalidomidiin korkeintaan 12 syklin ajan, minkä jälkeen tafasitamabimonoterapiaa jatkettiin, kunnes sairaus eteni tai haittavaikutukset muuttuivat liian voimakkaiksi. Tafasitamabialtistuksen mediaanikesto oli 7,7 kuukautta.
Yleisimmät haittavaikutukset olivat infektiot (73 %), neutropenia (51 %), astenia (40 %), anemia (36 %), ripuli (36 %), trombosytopenia (31 %), yskä (26 %), perifeerinen ödeema (24 %), kuume (24 %) ja ruokahaluttomuus (22 %).
Yleisimmät vakavat haittavaikutukset olivat infektiot (26 %), mukaan lukien keuhkokuume (7 %), ja febriili neutropenia (6 %).
Tafasitamabihoito lopetettiin haittavaikutuksen takia pysyvästi 15 %:lla potilaista. Yleisimmät haittavaikutukset, jotka johtivat tafasitamabihoidon pysyvään lopettamiseen, olivat infektiot ja infestaatiot (5 %), hermojärjestelmän häiriöt (2,5 %) sekä hengityselinten, rintaontelon ja välikarsinan sairaudet (2,5 %).
Haittavaikutuksesta johtuvan annoksen muutoksen tai keskeyttämisen esiintyvyys oli 65 %. Yleisimmät haittavaikutukset, jotka johtivat tafasitamabihoidon keskeyttämiseen, olivat verenkierto‑ ja imukudosjärjestelmän häiriöt (41 %).
Potilaat, joilla on uusiutunut tai hoitoon reagoimaton FL, vähintään yhden aiemman systeemisen hoitolinjan jälkeen
Tafasitamabin turvallisuutta FL-potilailla tutkittiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen 3 monikeskustutkimuksessa, inMIND, 652 tutkittavalla, joista 546:lla oli uusiutunut tai hoitoon reagoimaton (R/R) follikulaarinen lymfooma ja 106:lla uusiutunut tai hoitoon reagoimaton marginaalivyöhykkeen lymfooma. Potilaat saivat tafasitamabia 12 mg/kg (n = 327) tai lumelääkettä (n = 325) laskimonsisäisesti sekä rituksimabia 375 mg/m2 laskimonsisäisesti (korkeintaan 5 syklin ajan) ja lenalidomidia 20 mg suun kautta (korkeintaan 12 syklin ajan). Tafasitamabihoito lopetettiin 12 syklin jälkeen. Tafasitamabia saaneista potilaista 83 % sai valmistetta vähintään 6 kuukauden ajan. Tafasitamabialtistuksen mediaanikesto oli 322 päivää.
Yleisimmät inMIND-tutkimuksessa havaitut haittavaikutukset olivat infektiot (68 %), mukaan lukien virusinfektiot (41 %) ja bakteeri-infektiot (27 %), neutropenia (57 %), ihottuma (36,4 %), heikkous (34,9 %), kuume (19 %), trombosytopenia (17 %), anemia (17 %), infuusioon liittyvä reaktio (15,9 %), kutina (15,6 %) ja päänsärky (10,4 %).
Yleisimmät vakavat haittavaikutukset olivat infektiot (26 %), mukaan lukien virusinfektiot (13 %) ja bakteeri-infektiot (6 %), kuumeinen neutropenia (2,8 %), akuutti munuaisvaurio (2,8 %) ja kuume (1,8 %).
Tafasitamabihoito lopetettiin haittavaikutuksen takia pysyvästi 11,6 %:lla potilaista. Yleisimmät haittavaikutukset, jotka johtivat tafasitamabihoidon pysyvään lopettamiseen, olivat virusinfektiot (2,4 %), mukaan lukien COVID-19 (1,5 %), ja COVID-19 pneumonia (1,2 %), infuusioon liittyvä reaktio (0,9 %) ja kuume (0,9 %).
Haittavaikutuksesta johtuvan tafasitamabiannoksen muutoksen tai keskeyttämisen esiintyvyys oli 74,9 %. Yleisimmät haittavaikutukset, jotka johtivat tafasitamabihoidon muutokseen tai keskeyttämiseen, olivat neutropenia (38,8 %) ja virusinfektiot (23,9 %), mukaan lukien COVID-19 (21,1 %), ja COVID-19 pneumonia (3,7 %).
Haittavaikutustaulukko
Tafasitamabin kliinisessä tutkimuksessa havaitut haittavaikutukset on lueteltu MedDRA:n elinjärjestelmäluokkien ja esiintymistiheyden perusteella jaoteltuina.
Kliinisestä tutkimuksesta saadut haittavaikutusten esiintymistiheydet perustuvat kaikista syistä johtuvien haittavaikutusten esiintymistiheyksiin, jossa osa haittatapahtumista saattaa johtua muista syistä kuin lääkevalmisteesta, kuten sairaudesta, muista lääkkeistä tai muista tutkimukseen liittymättömistä syistä. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan pienenevässä järjestyksessä.
Taulukko 2. Haittavaikutukset potilailla, joilla oli uusiutunut tai hoitoon reagoimaton DLBCL ja jotka saivat tafasitamabia yhdessä lenalidomidin kanssa kliinisessä tutkimuksessa MOR208C203 (L‑MIND)
| Elinjärjestelmä | Esiintymistiheys | Haittavaikutukset |
| Infektiot ja infestaatiot | Hyvin yleiset | Bakteeri-, virus ja sieni-infektiot+, mukaan lukien opportunistiset kuolemaan johtavat infektiot, (esim. bronkopulmonaalinen aspergilloosi, keuhkoputkentulehdus, keuhkokuume ja virtsatieinfektio) |
| Yleiset | Sepsis (mukaan lukien neutropeninen sepsis) | |
| Hyvän- ja pahanlaatuiset kasvaimet ja tarkemmin määrittelemättömät (mukaan lukien kystat ja polyypit) | Yleiset | Tyvisolukarsinooma |
| Veri ja imukudos | Hyvin yleiset | Febriili neutropenia+, neutropenia+, trombosytopenia+, anemia, leukopenia+ |
| Yleiset | Lymfopenia | |
| Immuunijärjestelmä | Yleiset | Hypogammaglobulinemia |
| Aineenvaihdunta ja ravitsemus | Hyvin yleiset | Hypokalemia, ruokahaluttomuus |
| Yleiset | Hypokalsemia, hypomagnesemia | |
| Hermosto | Yleiset | Päänsärky, parestesia, dysgeusia |
| Hengityselimet, rintakehä ja välikarsina | Hyvin yleiset | Hengenahdistus, yskä |
| Yleiset | Kroonisen obstruktiivisen keuhkosairauden paheneminen, nenän tukkoisuus | |
| Ruoansulatuselimistö | Hyvin yleiset | Ripuli, ummetus, oksentelu, pahoinvointi, vatsakipu |
| Maksa ja sappi | Yleiset | Hyperbilirubinemia, kohonnut transaminaasitaso (sisältää kohonneet ALT- ja/tai AST-tasot), kohonnut gammaglutamyylitransferaasitaso |
| Iho ja ihonalainen kudos | Hyvin yleiset | Ihottuma (sisältää erityyppiset ihottumat, kuten tavallinen ihottuma, makulopapulaarinen ihottuma, pruriittinen ihottuma, punoittava ihottuma) |
| Yleiset | Kutina, alopesia, eryteema, hyperhidroosi | |
| Luusto, lihakset ja sidekudos | Hyvin yleiset | Selkäkipu, lihaskrampit |
| Yleiset | Nivelkipu, raajakipu, luusto- ja lihaskivut | |
| Munuaiset ja virtsatiet | Yleiset | Veren kreatiniinipitoisuuden nousu |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleiset | Astenia++, perifeerinen ödeema, kuume |
| Yleiset | Limakalvotulehdus | |
| Tutkimukset | Yleiset | Painon lasku, kohonnut c-reaktiivisen proteiinin taso |
| Vammat, myrkytykset ja hoitokomplikaatiot | Yleiset | Infuusioon liittyvät reaktiot |
+ Lisätietoa tästä haittavaikutuksesta on alla.
++ Astenia sisältää astenian, väsymyksen ja huonovointisuuden.
Verrattuna esiintyvyyksiin yhdessä samanaikaisen lenalidomidihoidon kanssa tafasitamabimonoterapian ei‑hematologiset haittavaikutukset vähenivät vähintään 10 % ruokahaluttomuuden, astenian, hypokalemian, ummetuksen, pahoinvoinnin, lihaskramppien, hengenahdistuksen ja c‑reaktiivisen proteiinin tasojen kohoamisen osalta.
Taulukko 3: Haittavaikutukset potilailla, joilla oli uusiutunut tai hoitoon reagoimaton FL ja jotka saivat tafasitamabia yhdessä rituksimabin ja lenalidomidin kanssa kliinisessä tutkimuksessa INCMOR 0208-301 (inMIND)
| Elinjärjestelmä/haittavaikutus | Kaikkien vakavuusasteiden esiintyvyys | Vakavuusasteiden 3–4a esiintyvyys |
| Infektiot | ||
| Virusinfektiotb | Hyvin yleinen | Hyvin yleinen |
| Bakteeri-infektiotc | Hyvin yleinen | Yleinen |
| Pneumonia | Hyvin yleinen | Yleinen |
| Keuhkoputkentulehdus | Yleinen | – |
| Sepsis | Yleinen | Melko harvinainen |
| Veri ja imukudos | ||
| Neutropeniad | Hyvin yleinen | Hyvin yleinen |
| Trombosytopeniae | Hyvin yleinen | Yleinen |
| Anemiaf | Hyvin yleinen | Yleinen |
| Kuumeinen neutropenia | Yleinen | Yleinen |
| Leukopenia | Yleinen | Melko harvinainen |
| Aineenvaihdunta ja ravitsemus | ||
| Tuumorilyysioireyhtymä | Melko harvinainen | Melko harvinainen |
| Hermosto | ||
| Päänsärky | Hyvin yleinen | Melko harvinainen |
| Ruoansulatuselimistö | ||
| Ripuli | Hyvin yleinen | Melko harvinainen |
| Ummetus | Hyvin yleinen | Melko harvinainen |
| Vatsakipug | Hyvin yleinen | – |
| Iho ja ihonalainen kudos | ||
| Ihottumah | Hyvin yleinen | Yleinen |
| Kutina | Hyvin yleinen | Melko harvinainen |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Heikkousi | Hyvin yleinen | Yleinen |
| Kuume | Hyvin yleinen | Yleinen |
| Vilunväristykset | Yleinen | – |
| Tutkimukset | ||
| Kohonnut ALAT-arvo | Yleinen | Melko harvinainen |
| Kohonnut ASAT-arvo | Yleinen | Melko harvinainen |
| Vammat, myrkytykset ja hoitokomplikaatiot | ||
| Infuusioon liittyvä reaktio | Hyvin yleinen | Melko harvinainen |
| ||
Valikoitujen haittavaikutusten kuvaukset
Myelosuppressio
Tafasitamabihoito voi aiheuttaa vakavaa tai vaikeaa myelosuppressiota, mukaan lukien neutropeniaa, trombosytopeniaa ja anemiaa (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
L‑MIND‑tutkimuksessa myelosuppressiota (eli neutropeniaa, febriiliä neutropeniaa, trombosytopeniaa, leukopeniaa, lymfopeniaa tai anemiaa) esiintyi 65,4 %:lla tafasitamabilla hoidetuista potilaista. Myelosuppressio johti tafasitamabihoidon keskeyttämiseen 41 %:ssa tapauksista ja tafasitamabihoidon lopettamiseen 1,2 %:ssa tapauksista.
InMIND-tutkimuksessa myelosuppressiota (eli neutropeniaa, kuumeista neutropeniaa, trombosytopeniaa, leukopeniaa, lymfopeniaa tai anemiaa) esiintyi 63,3 %:lla tafasitamabi-, lenalidomidi- ja rituksimabihoitoa saaneista potilaista (tafasitamabiryhmä) ja 63,1 %:lla lenalidomidi- ja rituksimabihoitoa saaneista potilaista (lumelääkeryhmä). Vakavuusasteen 4 hematologisia haittavaikutuksia olivat neutropenia, trombosytopenia ja kuumeinen neutropenia. Myelosuppressio johti tafasitamabihoidon keskeyttämiseen 42,8 %:ssa tapauksista ja tafasitamabihoidon lopettamiseen 1,5 %:ssa tapauksista.
Myelosuppressiota hallittiin pienentämällä lenalidomidiannosta tai keskeyttämällä lenalidomidihoito, keskeyttämällä tafasitamabihoito ja/tai rituksimabihoito. Vaikeaa neutropeniaa hallittiin lisäksi antamalla potilaalle G‑CSF:ää (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Neutropenia / febriili neutropenia
L-MIND-tutkimuksessa neutropenian esiintymistiheys oli 51 %. Vakavuusasteen 3 tai 4 neutropenian esiintymistiheys oli 49 % ja vakavuusasteen 3 tai 4 febriilin neutropenian 12 %. Neutropeniaan liittyvän haittavaikutuksen mediaanikesto oli 8 päivää (vaihteluväli 1–222 päivää). Mediaaniaika neutropenian ensimmäiseen ilmenemiseen hoidon aloituksen jälkeen oli 49 päivää (vaihteluväli 1–994 päivää).
Neutropenian esiintyvyys oli inMIND-tutkimuksessa 56,9 % tafasitamabiryhmässä (tafasitamabi, lenalidomidi ja rituksimabi) ja 54,2 % lumelääkeryhmässä (lenalidomidi ja rituksimabi). Vakavuusasteen 3 tai 4 neutropenian esiintyvyys oli 46,8 % tafasitamabiryhmässä ja 45,5 % lumelääkeryhmässä. Vakavuusasteen 3 tai 4 kuumeisen neutropenian esiintyvyys oli 4,3 % tafasitamabiryhmässä ja 3,4 % lumelääkeryhmässä. Neutropeniaan liittyvän haittavaikutuksen mediaanikesto oli 11 päivää (vaihteluväli 1–433 päivää). Kuumeisen neutropenian mediaanikesto oli 5 päivää (vaihteluväli 1–57 päivää); mediaaniaika neutropenian ensimmäiseen ilmenemiseen hoidon aloituksen jälkeen oli 57 päivää (vaihteluväli 1–338 päivää) ja mediaaniaika kuumeisen neutropenian ensimmäiseen ilmenemiseen hoidon aloituksen jälkeen oli 77 päivää (vaihteluväli 3–304 päivää).
Trombosytopenia
L-MIND-tutkimuksessa trombosytopenian esiintymistiheys oli 31 %. Vakavuusasteen 3 tai 4 trombosytopenian esiintymistiheys oli 17 %. Trombosytopeniaan liittyvän haittavaikutuksen mediaanikesto oli 11 päivää (vaihteluväli 1–470 päivää). Mediaaniaika trombosytopenian ensimmäiseen ilmenemiseen hoidon aloituksen jälkeen oli 71 päivää (vaihteluväli 1–358 päivää).
Trombosytopenian esiintyvyys oli inMIND-tutkimuksessa 17,1 % tafasitamabiryhmässä (tafasitamabi, lenalidomidi ja rituksimabi) ja 20,6 % lumelääkeryhmässä (lenalidomidi ja rituksimabi). Vakavuusasteen 3 tai 4 trombosytopenian esiintyvyys oli 6,4 % tafasitamabiryhmässä ja 9,8 % lumelääkeryhmässä. Trombosytopenian mediaanikesto oli 16 päivää (vaihteluväli 2–434 päivää); mediaaniaika trombosytopenian ensimmäiseen ilmenemiseen hoidon aloituksen jälkeen oli 33 päivää (vaihteluväli 1–324 päivää).
Anemia
L-MIND-tutkimuksessa anemian esiintymistiheys oli 36 %. Vakavuustason 3 tai 4 anemian esiintymistiheys oli 7 %. Anemiaan liittyvän haittavaikutuksen mediaanikesto oli 15 päivää (vaihteluväli 1–535 päivää). Mediaaniaika anemian ensimmäiseen esiintymiseen aloituksen jälkeen oli 49 päivää (vaihteluväli 1 – 1 129 päivää).
Kun L‑MIND‑tutkimuksen potilaat siirtyivät tafasitamabi‑ ja lenalidomidin yhdistelmähoitovaiheesta laajennettuun tafasitamabimonoterapiavaiheeseen, hematologisten tapahtumien esiintyvyys väheni vähintään 20 % neutropenian, trombosytopenian ja anemian osalta. Tafasitamabimonoterapian yhteydessä ei ilmoitettu febriilin neutropenian tapauksista (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Anemian esiintymistiheys oli inMIND-tutkimuksessa 17,1 % tafasitamabiryhmässä (tafasitamabi, lenalidomidi ja rituksimabi) ja 14,5 % lumelääkeryhmässä (lenalidomidi ja rituksimabi). Vakavuusasteen 3 tai 4 anemian esiintymistiheys oli 6,4 % tafasitamabiryhmässä ja 6,5 % lumelääkeryhmässä. Anemiaan liittyvän haittavaikutuksen mediaanikesto oli 23 päivää (vaihteluväli 1–432 päivää); mediaaniaika anemian ensimmäiseen ilmenemiseen hoidon aloituksen jälkeen oli 49 päivää (vaihteluväli 1–274 päivää).
Infektiot
L‑MIND‑tutkimuksessa infektioita havaittiin 73 %:lla tutkituista. Vakavuusasteen 3 tai 4 infektioiden esiintymistiheys oli 28 %. Yleisimmät vähintään vakavuusasteen 3 infektiot olivat keuhkokuume (7 %), hengitysteiden infektiot (4,9 %), virtsatieinfektiot (4,9 %) ja sepsis (4,9 %). Infektiot johtivat kuolemaan < 1 %:lla potilaista (keuhkokuume) viimeistään 30 päivän kuluttua viimeisestä hoitokerrasta.
Vakavuusasteen 3 tai 4 infektioiden ensimmäisen ilmenemisen mediaaniaika oli 62,5 päivää (4 – 1 014 päivää). Infektioiden mediaanikesto oli 11 päivää (1–392 päivää).
Infektiot johtivat tafasitamabihoidon keskeyttämiseen 27 %:ssa tapauksista ja tafasitamabihoidon lopettamiseen 4,9 %:ssa tapauksista.
Infektioiden esiintymistiheys inMIND-tutkimuksessa oli 52,3 % tafasitamabiryhmässä (tafasitamabi, lenalidomidi ja rituksimabi) ja 45,2 % lumelääkeryhmässä (lenalidomidi ja rituksimabi). Virusinfektioiden esiintymistiheys oli 41,3 % tafasitamabiryhmässä ja 32 % lumelääkeryhmässä. Bakteeri-infektioiden esiintymistiheys oli 27,2 % tafasitamabiryhmässä ja 25,2 % lumelääkeryhmässä. Vakavuusasteen 3 tai 4 virusinfektioiden esiintymistiheys oli 11,6 % tafasitamabiryhmässä ja 4,6 % lumelääkeryhmässä. Vakavuusasteen 3 tai 4 bakteeri-infektioiden esiintymistiheys oli 7,6 % tafasitamabiryhmässä ja 7,7 % lumelääkeryhmässä. Infektiot johtivat kuolemaan 3 potilaalla tafasitamabiryhmässä (kaksi COVID-19-tapausta ja yksi sepsis).
Vähintään vakavuusasteen 3 infektioiden ilmenemisen mediaaniaika oli 10 päivää (2–311 päivää).
Infektioiden hallintaa koskevat suositukset ovat kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Infuusioon liittyvät reaktiot
L‑MIND‑tutkimuksessa infuusioon liittyviä reaktioita havaittiin 6 %:lla tutkituista. Kaikki infuusioon liittyvät reaktiot olivat vakavuudeltaan astetta 1 ja ne hälvenivät tapahtumapäivänä. 80 % näistä reaktioista tapahtui syklin 1 tai 2 aikana.
Infuusioon liittyvien reaktioiden esiintymistiheys inMIND-tutkimuksessa oli 15,9 % tafasitamabiryhmässä (tafasitamabi, lenalidomidi ja rituksimabi) ja 15,1 % lumelääkeryhmässä (lenalidomidi ja rituksimabi). Vakavuusasteen 3 infuusioon liittyvien reaktioiden esiintymistiheys oli 6,1 % tafasitamabiryhmässä. Infuusioon liittyvien reaktioiden esiintymistiheys oli tafasitamabiryhmässä 15,3 % hoitosyklin 1 aikana, 1,3 % hoitosyklin 2 aikana ja 0,3 % hoitosyklin 3 aikana.
Oireisiin kuuluivat mm. vilunväristykset, punoitus, hengenahdistus, korkea verenpaine ja ihottuma (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Immunogeenisuus
Tafasitamabilla ensimmäisissä kliinisissä tutkimuksissa hoidetuilla 245 tutkittavalla ei havaittu hoidon aikana ilmestyviä tai lisääntyneitä tafasitamabin vasta‑aineita. Aiemmin syntyneitä tafasitamabin vasta‑aineita havaittiin 17:llä tutkittavalla 245:stä (6,9 %), millä ei ollut vaikutusta tafasitamabin farmakokinetiikkaan, tehoon tai turvallisuuteen.
Lääkevasta-aineet (ADA:t) testattiin 327 potilaalta, joilla oli uusiutunut tai hoitoon reagoimaton follikulaarinen lymfooma tai uusiutunut tai hoitoon reagoimaton marginaalivyöhykkeen lymfooma ja jotka saivat tafasitamabia inMIND-tutkimuksessa. Tafasitamabihoitoon liittyvien lääkevasta-aineiden esiintyvyys oli 0,9 % (3/327) siltatyyppisessä ELISA-määrityksessä.
Neutraloivia vasta-aineita ei havaittu. Lääkevasta-aineilla ei ollut ilmeistä kliinisesti merkitsevää vaikutusta tafasitamabin farmakokinetiikkaan, farmakodynamiikkaan, turvallisuuteen tai tehoon hoidon 322,5 päivän mediaanikeston aikana.
Erityispotilasryhmät
Iäkkäät
L‑MIND‑tutkimuksessa hoidetuista 81 tutkittavasta 56 oli yli 65‑vuotiaita (69 %). Yli 65‑vuotiailla tutkittavilla oli numeerisesti korkeampi hoitoon liittyvien vakavien haittavaikutusten esiintyvyys (55 %) verrattuna ≤ 65‑vuotiaisiin (44 %).
Tafasitamabihoitoa inMIND-tutkimuksessa saaneista 274:stä FL-potilaasta 50 % oli ≥ 65-vuotiaita ja 20 % oli ≥ 75-vuotiaita. Näiden potilaiden ja nuorempien potilaiden välillä ei havaittu kliinisesti merkitseviä eroja turvallisuudessa tai tehossa, mutta joidenkin ikääntyneiden potilaiden suurempi herkkyys on silti mahdollista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‑haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksissa potilaita on valvottava tarkasti haittavaikutusten oireiden varalta, ja potilaalle on tarpeen mukaan annettava yleistä elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Solunsalpaajat, monoklonaaliset vasta‑aineet, ATC‑koodi: L01FX12.
Vaikutusmekanismi
Tafasitamabi on Fc‑tehostettu monoklonaalinen vasta‑aine, joka kohdistuu pre‑B‑lymfosyyttien ja kypsien B‑lymfosyyttien pinnalla ilmentyvään CD19‑antigeeniin.
Tafasitamabi sitoutuu CD19‑antigeeniin ja edistää B‑solujen hajoamista seuraavilla tavoilla:
- immuuniefektorisolujen, kuten luonnollisten tappajasolujen, γδ‑T‑solujen ja fagosyyttien, osallistaminen
- solukuoleman (apoptoosin) suora induktio
Fc‑modifikaatio saa aikaan tehostetun vasta‑aineriippuvaisen solusytotoksisuuden ja vasta‑aineriippuvaisen solujen fagosytoosin.
Farmakodynaamiset vaikutukset
Tafasitamabi indusoi ääreisverenkierron B-solumäärien laskua. Potilailla, joilla on uusiutunut tai refraktorinen DLBCL. L‑MIND‑tutkimuksessa B‑solumäärä laski kahdeksan päivän hoidon jälkeen 97 %:a verrattuna lähtötilanteeseen. B‑solujen enimmäislasku, joka oli noin 100 % (mediaani), saavutettiin 16 viikon hoidon kuluessa.
Potilailla, joilla on uusiutunut tai hoitoon reagoimaton follikulaarinen lymfooma, ja joilla oli hoidon alussa havaittavissa olevia B-soluja, B-solumäärä laski suositellun tafasitamabiannoksen antamisen jälkeen kahdeksan päivän hoidon jälkeen tasolle, jota ei voitu havaita. Vaje säilyi potilailla, joiden hoitoa jatkettiin.
Vaikka B‑solujen katoaminen perifeerisestä verestä on huomattava farmakodynaaminen vaikutus, se ei korreloi suoraan kiinteiden elinten tai pahanlaatuisten kertymien B‑solujen katoamisen kanssa.
Kliininen teho
Uusiutunut tai hoitoon reagoimaton DLBCL
Avoimessa yksihaaraisessa monikeskustutkimuksessa, L‑MIND, tutkittiin tafasitamabin ja lenalidomidin yhdistelmähoitoa, jota seurasi tafasitamabimonoterapia. Tutkimus toteutettiin aikuisilla tutkittavilla, joilla oli 1–3 systeemisen DLBCL‑hoidon jälkeen uusiutunut tai hoitoon reagoimaton DLBCL ja jotka eivät olleet tutkimuksen aloittamisen aikana soveltuvia korkea‑annoksiseen kemoterapiaan ja autologiseen kantasolusiirtohoitoon tai jotka olivat kieltäytyneet autologisesta kantasolusiirtohoidosta. CD20‑vasta‑aineeseen kohdistuvan hoidon oli oltava osa jotakin potilaan aiemmin saamaa systeemistä hoitoa. Tutkimukseen ei otettu potilaita, joilla oli vakava maksan vajaatoiminta (seerumin kokonaisbilirubiini yli 3 mg/dl), potilaita, joilla oli munuaisten vajaatoiminta (CrCL alle 60 ml/min), eikä potilaita, joilla oli aiemmin todettuja tai seulonnan aikana havaittuja merkkejä kliinisesti merkittävistä sydän‑ ja verisuonisairauksista, keskushermojärjestelmän sairauksista ja/tai muista systeemisistä sairauksista. Myös sellaiset DLBCL‑potilaat, joilla oli tunnettu geenien uudelleenjärjestäytyminen ”double hit” tai ”triple hit”, suljettiin pois tutkimuksesta seulonnan aikana.
Ensimmäisten kolmen syklin aikana potilaat saivat 12 mg/kg tafasitamabia infuusiona 28‑päiväisen syklin päivinä 1, 8, 15 ja 22. Lisäksi 1. syklin päivänä 4 annettiin kyllästysannos. Tämän jälkeen tafasitamabia annettiin jokaisen syklin päivinä 1 ja 15, kunnes tauti eteni. Esilääkitystä, johon kuuluivat kuumetta alentavat lääkkeet, histamiini H1‑ ja H2‑reseptorin salpaajia ja glukokortikosteroideja, annettiin 30–120 minuuttia ennen kolmea ensimmäistä tafasitamabi‑infuusiota.
Potilaat ottivat itse 25 mg lenalidomidia päivittäin jokaisen 28‑päiväisen syklin päivinä 1–21 korkeintaan 12 syklin ajan.
L‑MIND‑tutkimukseen ja sen turvallisuutta koskevaan analyysiin otettiin mukaan yhteensä 81 potilasta. Mediaani‑ikä oli 72 vuotta (vaihteluväli 41–86 vuotta). 89 % potilaista oli valkoisia ja 54 % oli miehiä. 81 potilaasta 74:llä (91,4 %), ECOG‑toimintakykypisteet olivat 0 tai 1, ja 7 potilaalla (8,6 %) ECOG‑pisteet olivat 2. Aiempien hoitojen lukumäärän mediaani oli kaksi (vaihteluväli 1–4). 40 potilasta (49,4 %) oli saanut aiemmin yhtä hoitoa ja 35 potilasta (43,2 %) oli saanut aiemmin kahta hoitoa. Viisi potilasta (2 %) oli saanut aiemmin kolmea eri hoitoa, ja 1 (1,24 %) potilas oli aiemmin saanut neljää eri hoitoa. Kaikki potilaat olivat saaneet aiemmin CD20‑vasta‑ainetta sisältävää hoitoa. Kahdeksalla potilaalla oli matala‑asteisesta lymfoomasta DLBCL:ksi muuntuneen syövän diagnoosi. 15 potilaalla (18,5 %) oli primaarihoidolle refraktorinen sairaus, 36 potilasta (44,4 %) oli ollut refraktorisia viimeisimmälle edeltävälle hoidolle ja 34 potilasta (42,0 %) oli ollut refraktorisia rituksimabille. Yhdeksän potilasta (11,1 %) oli aiemmin saanut autologisen kantasolusiirron (ASCT). Pääasialliset syyt sille, etteivät potilaat soveltuneet ASCT‑hoitoon, olivat ikä (45,7 %), refraktorisuus toisen linjan kemoterapialle (23,5 %), komorbiditeetit (13,6 %) ja kieltäytyminen korkean annoksesta kemoterapiasta tai ASCT‑hoidosta (16,0 %).
Yksi potilas sai tafasitamabia, mutta ei lenalidomidia. Muut 80 potilasta saivat vähintään yhden annoksen tafasitamabia ja lenalidomidia. Kaikilla L‑MIND‑tutkimukseen mukaan otetuilla potilailla on paikalliseen patologiaan perustuva DLBCL‑diagnoosi. Kuitenkin keskuspatologian tarkastuksen mukaan 10 potilaalla ei voitu luokitella DLBCL‑diagnoosia.
Hoidon mediaanikesto oli 9,2 kuukautta (vaihtelualue: 0,23–54,67 kuukautta). 32 potilasta (39,5 %) osallistui kaikkiin 12 tafasitamabisykliin ja 30 potilasta (37,0 %) osallistui kaikkiin 12 lenalidomidisykliin.
Ensisijainen tehon päätetapahtuma oli paras mahdollinen kokonaishoitovaste (ORR), joka määriteltiin täydellisen vasteen tai osittaisen vasteen saaneina tutkittavina riippumattoman arviointikomitean (IRC) näkemyksen mukaisesti. Muut tehon päätepisteet sisälsivät vasteen keston (DOR), etenemisvapaan elossa olon (PFS) ja kokonaiselinajan (OS). Tehoa koskevat tulokset on esitetty yhteenvetona taulukossa 4.
Taulukko 4. Tehoa koskevat tulokset potilailla, joilla oli uusiutunut tai refraktorinen diffuusi suurisoluinen B‑solulymfooma, MOR208C203 (L‑MIND) ‑tutkimuksessa
| Tehon muuttuja | Tafasitamabi + lenalidomidi (N = 81 [ITT]*) | |
Tiedonkeruun päättymispäivä 30. marraskuuta 2019 (24 kuukauden analyysi) | Tiedonkeruun päättymispäivä 30. lokakuuta 2020 (35 kuukauden analyysi) | |
| Ensisijainen päätetapahtuma | ||
| Paras mahdollinen kokonaishoitovaste (riippumattoman arviointikomitean mukaan) | ||
Kokonaishoitovaste, n (%) (95 %:n luottamusväli) | 46 (56,8) [45,3; 67,8] | 46 (56,8) [45,3; 67,8] |
Täydellinen vaste, n (%) (95 %:n luottamusväli) | 32 (39,5) [28,8; 51,0] | 32 (39,5) [28,8; 51,0] |
Osittainen vaste, n (%) (95 %:n luottamusväli) | 14 (17,3) [9,8; 27,3] | 14 (17,3) [9,8; 27,3] |
| Keskeiset toissijaiset päätetapahtumat | ||
| Vasteen kokonaiskesto (täydellinen ja osittainen vaste)a | ||
Mediaani, kuukautta (95 %:n luottamusväli) | 34,6 [26,1; NR] | 43,9 [26,1; NR] |
ITT = hoitoaie; NR = ei saavutettu
*Yksi potilas sai vain tafasitamabia
CI: Binomiaalinen tarkka luottamusväli, joka on laskettu Clopper‑Pearson‑menetelmällä
a Kaplan‑Meierin estimaatit
Kokonaiselinaika (OS) oli tutkimuksen toissijainen päätepiste. Seuranta‑ajan mediaani oli 42,7 kuukautta (95 %:n luottamusväli 38,0; 47,2) ja kokonaiselinajan mediaani oli 31,62 kuukautta (95 %:n luottamusväli: 18,3; ei saavutettu).
Niistä kahdeksasta tutkittavasta, joilla aiempi hidaskasvuinen lymfooma oli muuttunut DLBCL:ksi, seitsemällä oli objektiivinen vaste (kolmella täydellinen vaste, neljällä osittainen vaste); yhden tutkittavan paras vaste tafasitamabi‑ ja lenalidomidihoitoon oli vakaa sairaus.
Uusiutunut tai hoitoon reagoimaton FL vähintään yhden aiemman systeemisen hoitolinjan jälkeen
Tafasitamabin turvallisuutta yhdessä lenalidomidin ja rituksimabin kanssa käytettynä tutkittiin uusiutunutta tai hoitoon reagoimatonta follikulaarista lymfoomaa sairastavilla potilailla satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen 3 tutkimuksessa (inMIND; INCMOR 0028-301).
Tutkimukseen otettiin aikuispotilaita, joiden ikä oli vähintään 18 vuotta, joilla oli histologisesti vahvistettu asteen 1–3a follikulaarinen lymfooma (WHO 2016) ja joiden sairaus oli uusiutunut tai lakannut reagoimasta hoitoon vähintään yhden aiemman systeemisen hoitolinjan jälkeen, mukaan lukien CD20-vasta-aineeseen kohdistuva hoito. Lisäksi GELF-kriteerien käyttöä suositeltiin tutkijoille sellaisten FL-potilaiden tunnistamiseksi, jotka tarvitsivat hoitoa. Sisäänottokriteerien mukaisesti kaikkien tutkimukseen otettujen potilaiden oli oltava paikallisesti tai keskitetysti tehdyn patologisen arvioinnin perusteella dokumentoidusti CD20- ja CD19-positiivisia. Tutkimukseen ei otettu potilaita, joilla oli keskushermostoinvaasio tai joille oli aiemmin tehty allogeeninen kantasolu siirto.
Kaikkiaan 548 potilasta, joilla oli uusiutunut tai hoitoon reagoimaton follikulaarinen lymfooma, satunnaistettiin suhteessa 1:1 saamaan joko tafasitamabia yhdessä lenalidomidin ja rituksimabin kanssa tai lumelääkettä ja R2:ta korkeintaan kahdentoista 28 päivän hoitosyklin ajan. Satunnaistaminen ositettiin ryhmiin sen mukaan, oliko sairaus edennyt 24 kuukauden kuluessa sen diagnosoinnista (POD24) (kyllä tai ei), oliko potilaalla saatu vaste aiempaan CD20-vasta-aineeseen kohdistuvan hoidon aikana (kyllä tai ei) ja mikä oli aiempien hoitolinjojen lukumäärä (< 2 tai ≥ 2).
Hoitoryhmissä käytetyt annostukset olivat seuraavat:
- Tafasitamabi 12 mg/kg laskimonsisäisesti (hoitosyklien 1–3 päivinä 1, 8, 15 ja 22 ja hoitosyklien 4–12 päivinä 1 ja 15) ja lenalidomidi 20 mg suun kautta kerran päivässä (hoitosyklien 1–12 päivinä 1–21) sekä rituksimabi 375 mg/m² laskimonsisäisesti (hoitosyklin 1 päivinä 1, 8, 15 ja 22 ja hoitosyklien 2–5 päivänä 1).
- Lumelääke laskimonsisäisesti (hoitosyklien 1–3 päivinä 1, 8, 15 ja 22 ja hoitosyklien 4–12 päivinä 1 ja 15) ja lenalidomidi 20 mg suun kautta kerran päivässä (hoitosyklien 1–12 päivinä 1–21) sekä rituksimabi 375 mg/m² laskimonsisäisesti (hoitosyklin 1 päivinä 1, 8, 15 ja 22 ja hoitosyklien 2–5 päivänä 1).
Lähtötilanteen demografiset tiedot ja sairauden ominaisuudet olivat yleisesti ottaen samankaltaiset molemmissa hoitoryhmissä. InMIND-tutkimukseen otettujen 548:n potilaan, joilla oli uusiutunut tai hoitoon reagoimaton FL, mediaani-ikä oli 64 vuotta (vaihteluväli 31–88 vuotta), 54,6 % oli miehiä ja 79,9 % oli valkoisia. Mediaaniaika diagnoosin tekemisestä oli 5,3 vuotta (vaihteluväli 0–34 vuotta). Useimpien tutkittavien (56,8 %) sairauden Ann Arbor levinneisyysluokitus oli IV tutkimukseen ottohetkellä. Noin puolella potilaista oli FLIPI-pistemäärän perusteella arvioituna suuririskinen sairaus, ja useimmat potilaat täyttivät vähintään yhden suuren kasvainkuorman GELF-kriteereistä. Useimpien tutkittavien ECOG-toimintakykypistemäärä oli 0 (66,4 %), ja 37,8 %:lla tutkittavista oli laaja tautimassa (> 7 cm).
Valtaosa (54,7 %) tutkittavista oli saanut yhden edeltävän systeemisen syöpähoitolinjan hoidon; aiempien hoitojen mediaanilukumäärä oli 1 (vaihteluväli 1–10), 209 tutkittavalla (38,1 %) ei ollut saatu vastetta viimeiseen edeltävään hoitoon. Kaikki FL-populaation tutkittavat olivat saaneet aiemmin CD20-vasta-ainetta sisältävää hoitoa; useimmat tutkittavat olivat saaneet yhden (61,3 %) tai kaksi (24,8 %) aiempaa hoitoa, joihin sisältyi CD20-vasta-aine. Kaksi tutkittavaa, jotka molemmat kuuluivat lumelääkeryhmään, oli saanut aiempaa CD19-epitooppiin kohdistuvaa hoitoa. Aiempia hoitoja olivat R-CHOP (23,9 % tutkittavista), R-CHOP +R-ylläpito (27,9 % tutkittavista), R-bendamustiini (21,7 % tutkittavista), rituksimabimonoterapia (17,2 % tutkittavista), R-bendamustiini +R-ylläpito (12,2 % tutkittavista), R-CVP (6,8 % tutkittavista) ja R-CVP +R-ylläpito (5,8 % tutkittavista). Aiemman autologisen kantasolusiirron oli saanut 28 tutkittavaa (5,1 %).
Yksi kolmasosa tutkittavista (34,3 %) ei ollut reagoinut CD20-kohdistuvaan hoitoon ja 31,6 %:lla sairaus oli edennyt 24 kuukauden kuluessa sen diagnosoinnista (POD24).
Hoitoa sai kaikkiaan 546 tutkittavaa (99,6 %), joilla oli uusiutunut tai hoitoon reagoimaton FL: 273 tutkittavaa (100,0 %) tafasitamabi+R2 ryhmässä ja 273 tutkittavaa (99,3 %) lumelääke+R2 ryhmässä.
Ensisijainen tehon päätetapahtuma oli etenemisvapaa elossaolo (PFS) FL-populaatiossa tutkijan arvioimana. Tällä tarkoitettiin aikaa satunnaistamisesta sairauden dokumentoituun etenemiseen tai mistä tahansa syystä johtuvaan kuolemaan. Tärkeimpiä toissijaisia päätetapahtumia olivat kokonaiselinaika (OS) FL-populaatiossa sekä PET-CR-osuus tutkijan arvioimana FDG:lle herkässä FL-populaatiossa, jolla tarkoitettiin täydellistä metabolista vastetta koska tahansa hoidon aloittamisen jälkeen. PFS-seurannan mediaanikesto oli 14,3 kuukautta (95 %:n luottamusväli: 11,8; 15) tafasitamabiryhmässä ja 14,1 kuukautta (95 %:n luottamusväli: 11,5; 15) lumelääkeryhmässä.
Tehoa koskevat tulokset on esitetty taulukossa 5 ja kuvassa 1.
Taulukko 5: Tehoa koskevat tulokset INCMOR 0208-301 (inMIND) tutkimuksessa
| Päätetapahtuma | Tafasitamabi ja lenalidomidi + rituksimabi (N = 273) | Lumelääke ja lenalidomidi + rituksimabi (N = 275) |
| Etenemisvapaa elossaoloa, b | ||
| Potilaat, joilla tapahtuma toteutui, n (%) | 75 (27,5) | 131 (47,6) |
PFS:n mediaani (kuukautta) (95 %:n luottamusväli)c | 22,4 (19,2; NE) | 13,9 (11,5; 16,4) |
| Riskitiheyssuhde (HR)d (95 %:n luottamusväli) | 0,43 (0,32; 0,58) | |
| p-arvo | < 0,0001 | |
| Potilaat, joilla lähtötilanteessa FDG-herkkä PET-kuvausa | (N = 251) | (N = 254) |
| PET-CR-osuus (95 %:n luottamusväli)e, f | 49,4 (43,1; 55,8) | 39,8 (33,7; 46,1) |
| Kerroinsuhde (95 %:n luottamusväli) | 1,5 (1,04; 2,13) | |
| p-arvo | 0,0286 | |
NE = ei voitu arvioida.
- Tutkijan arvioima
- Cheson 2014 ‑vastekriteerien mukaisesti
- Kaksisuuntaiset 95 %:n luottamusvälit perustuvat Brookmeyer–Crowley-menetelmään.
- Riskitiheyssuhde (HR) perustuu ositettuun Coxin suhteelliseen riskimalliin.
- PET-CR-osuudella tarkoitettiin niiden FDG:lle herkkien FL-populaation potilaiden osuutta, joilla saavutettiin Luganon luokituksella arvioituna täydellinen metabolinen vaste koska tahansa hoidon aloittamisen jälkeen, niistä potilaista, joille oli suoritettu taudin osoittava PET-kuvaus lähtötilanteessa. Potilaat, joille ei tehty PET-arviointia lähtötilanteen jälkeen tai joilla ei saavutettu täydellistä metabolista vastetta, luokiteltiin ei-täydellisen hoitovasteen potilaiksi.
- 95 %:n luottamusvälit perustuvat Clopper–Pearson-menetelmään.
Kuva 1: Kaplan–Meier-käyrä: etenemisvapaa elossaolo tutkijan arvioimana inMIND-tutkimuksessa
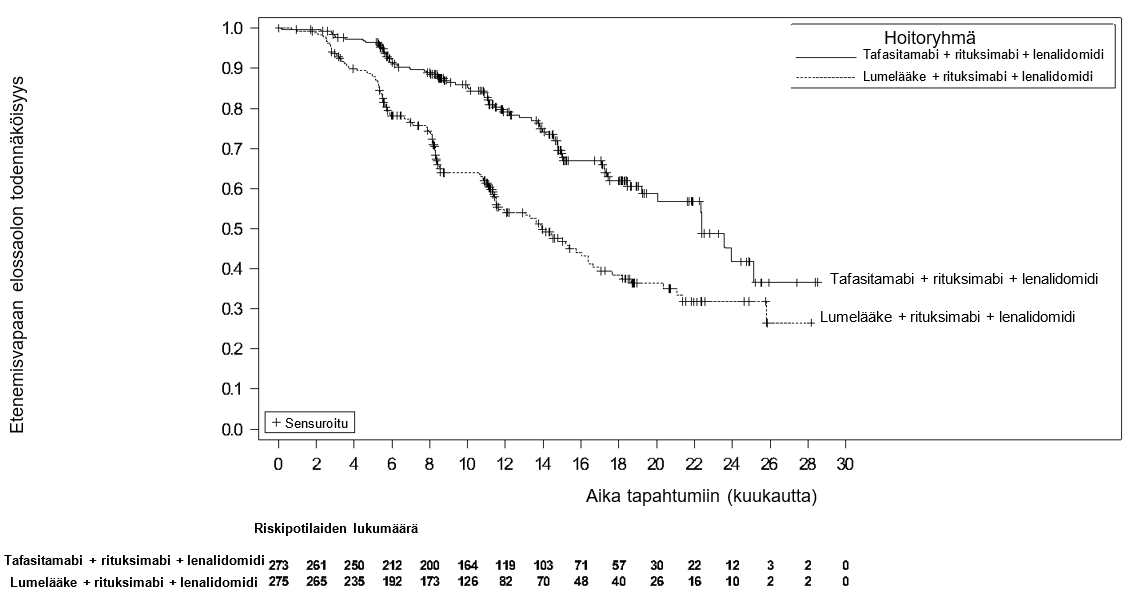
Välianalyysissa keskeistä toissijaista päätetapahtumaa, OS:aa, ei pystytty vielä määrittämään, eikä OS:n mediaania ollut saavutettu kummassakaan hoitoryhmässä (ositettu riskisuhde 0,587 (95 %:n luottamusväli: 0,306, 1,128); p-arvo 0,1061).
Iäkkäät
L-MIND-tutkimuksen ITT‑joukossa 36 tutkittavaa 81:sta oli ≤ 70‑vuotiaita ja 45 tutkittavaa 81:sta oli yli 70‑vuotiaita.
Uusiutunutta tai hoitoon reagoimatonta follikulaarilymfooma sairastavien, tafasitamabihoitoa inMIND-tutkimuksessa saaneiden 273 potilaan ryhmässä 178 oli ≤ 70-vuotiaita ja 95 oli > 70 vuotiaita.
Yleisiä eroja ei havaittu 70‑vuotiailla ja sitä vanhemmilla tutkittavilla verrattuna alle 70‑vuotiaisiin tutkittaviin.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset MINJUVI‑ valmisteen käytöstä kaikkien pediatristen potilasryhmien hoidossa diffuusin suurisoluisen kypsien B-soluneoplasmojen osalta (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Tämä lääkevalmiste on saanut ns. ehdollisen myyntiluvan. Se tarkoittaa, että lääkevalmisteesta odotetaan uutta tietoa.
Euroopan lääkevirasto arvioi vähintään kerran vuodessa tätä lääkevalmistetta koskevat uudet tiedot, ja tarvittaessa tämä valmisteyhteenveto päivitetään.
Farmakokinetiikka
Absorptio, jakautuminen, biotransformaatio ja eliminaatio dokumentoitiin populaatiofarmakokineettisen analyysin pohjalta.
Imeytyminen
Tafasitamabin matalimman seerumipitoisuuden keskiarvo (± standardipoikkeama) oli 178,4 (± 66) μg/ml viikoittaisten laskimonsisäisten 12 mg/kg annosten jälkeen sykleillä 1–3. Kun annos annettiin 14 päivän välein sykleillä 4-6, matalimman seerumipitoisuuden keskiarvo oli 163,2 (± 74,3) μg/ml. Tafasitamabin seerumipitoisuuden keskimääräinen enimmäisarvo oli 488,4 (± 126,6) μg/ml.
Jakautuminen
Tafasitamabin jakautumisen kokonaistilavuus vakaassa tilassa oli 7,11 l.
Biotransformaatio
Tafasitamabin tarkkaa metaboliatietä ei ole määritetty. Tafasitamabi on ihmisen monoklonaalinen IgG‑vasta‑aine ja sen odotetaan endogeenisten immunoglobuliinien tavoin hajoavan lyhyiksi peptideiksi ja aminohapoiksi katabolisten reittien kautta.
Eliminaatio
Tafasitamabin puhdistuma oli 0,44 l/vrk ja terminaalisen eliminaation puoliintumisaika oli 13,4 vuorokautta. Pitkäaikaisessa seurannassa tafasitamabin puhdistuman todettiin hidastuvan kahden vuoden kuluessa arvoon 0,29 l/vrk.
Erityispotilasryhmät
Ikä, paino, sukupuoli, kasvaimen koko, sairauden tyyppi, B‑solumäärät tai absoluuttiset lymfosyyttimäärät, lääkkeen vasta‑ainetasot, laktaattidehydrogenaasitasot ja seerumin albumiinitasot eivät vaikuttaneet olennaisesti tafasitamabin farmakokinetiikkaan. Etnisen taustan vaikutus tafasitamabin farmakokinetiikkaan on tuntematon.
Munuaisten vajaatoiminta
Munuaisten vajaatoiminnan vaikutusta ei testattu muodollisesti erillisissä kliinisissä tutkimuksissa. Tafasitamabin farmakokinetiikassa ei kuitenkaan havaittu kliinisesti merkittäviä eroja tutkittavilla, joilla oli lievä, keskivaikea tai vaikea munuaisten vajaatoiminta (kreatiniinin poistuma CrCL ≥ 15 ja < 90 ml/min Cockcroft‑Gaultin kaavalla arvioituna). Vakavan munuaisten vajaatoiminnan vaikutus (CrCL alle 15 ml/min) on tuntematon.
Maksan vajaatoiminta
Maksan vajaatoiminnan vaikutusta ei testattu muodollisesti erillisissä kliinisissä tutkimuksissa. Tafasitamabin farmakokinetiikassa ei kuitenkin havaittu kliinisesti merkittäviä eroja tutkittavilla, joilla oli lievä tai keskivaikea maksan vajaatoiminta (kokonaisbilirubiini ≤ normaalin yläraja (ULN) ja aspartaattiaminotransferaasi (AST) > ULN tai kokonaisbilirubiini 1-3 kertaa ULN ja mikä tahansa AST). Kohtalaisen tai vakavan maksan vajaatoiminnan (kokonaisbilirubiini yli 3 kertaa ULN ja mikä tahansa AST) vaikutus on tuntematon.
Prekliiniset tiedot turvallisuudesta
Prekliiniset tiedot eivät osoittaneet erityisiä vaaroja ihmisille.
Toistuvan annoksen toksisuustutkimukset
Tafasitamabin on osoitettu olevan erittäin spesifinen B‑solujen CD19‑antigeenille. Toksisuustutkimuksissa jaavanmakakeille annettiin tafasitamabia laskimonsisäisesti. Tutkimuksissa ei havaittu muita vaikutuksia kuin odotettua farmakologista B‑solujen katoamista perifeerisestä verestä ja imukudoksista. Nämä muutokset korjaantuivat hoidon lopettamisen jälkeen.
Mutageenisuus/karsinogeenisuus
Koska tafasitamabi on monoklonaalinen vasta‑aine, sille ei ole tehty genotoksisuus‑ eikä karsinogeenisuustutkimuksia. Nämä testit eivät ole oleellisia tämän molekyylin aiotuille käyttöaiheille.
Lisääntymistoksisuus
Tafasitamabilla ei ole toteutettu lisääntymis‑ eikä kehitystoksisuuteen liittyviä tutkimuksia, ei myöskään spesifisiä tutkimuksia sen vaikutuksista hedelmällisyyteen. Jaavanmakakiurosten tai ‑naaraiden lisääntymiselimissä ei kuitenkaan havaittu haitallisia vaikutuksia 13 viikon toistuvan annoksen toksisuustutkimuksessa, kuten ei myöskään naaraiden kuukausikierron pituudessa.
Farmaseuttiset tiedot
Apuaineet
Natriumsitraattidihydraatti
Sitruunahappomonohydraatti
Trehaloosidihydraatti
Polysorbaatti 20
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Ei havaittuja yhteensopimattomuuksia tavallisten infuusiomateriaalien kanssa.
Kestoaika
Avaamaton injektiopullo
6 vuotta
Käyttökuntoon saatettu valmiste (ennen laimentamista)
Kemiallisen ja fysikaalisen käytönaikaisen stabiiliuden on osoitettu olevan 30 päivää 2–8 °C:ssa tai 24 tuntia 25 °C:ssa.
Mikrobiologisista syistä käyttökuntoon saatettu valmiste on käytettävä heti. Ellei valmistetta käytetä heti, käytönaikaiset säilytysajat ja käyttöä edeltävät olosuhteet ovat käyttäjän vastuulla. Näiden odotetaan olevan normaalisti korkeintaan 24 tuntia 2–8 °C:n lämpötilassa, jollei käyttövalmiiksi saattaminen ole tapahtunut kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa. Ei saa jäätyä. Ei saa ravistaa.
Laimennettu liuos (infuusiota varten)
Kemiallisen ja fysikaalisen käytönaikaisen stabiiliuden on osoitettu olevan 14 päivää 2–8 °C:ssa ja sen jälkeen korkeintaan 24 tuntia korkeintaan 25 °C:ssa.
Mikrobiologisista syistä laimennettu liuos on käytettävä heti. Mikäli valmistetta ei käytetä heti, säilytysajat ja ‑olosuhteet ennen käyttöä ovat käyttäjän vastuulla. Näiden odotetaan olevan normaalisti korkeintaan 24 tuntia 2–8 °C:n lämpötilassa, ellei laimennusta ole toteutettu kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa. Ei saa jäätyä. Ei saa ravistaa.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
MINJUVI kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
200 mg (L:ei) 1 kpl (1002,98 €)
PF-selosteen tieto
200 mg tafasitamabia sisältävä lasinen injektiopullo (tyypin I kirkasta lasia), jossa on butyylikumitulppa, alumiinisuljin ja muovinen repäisykorkki. Pakkauskoko: 1 injektiopullo.
Valmisteen kuvaus:
Valkoinen tai hieman kellertävä kylmäkuivattu jauhe.
Käyttö- ja käsittelyohjeet
MINJUVI toimitetaan steriilissä, säilöntäaineettomassa ja kertakäyttöisessä injektiopullossa.
MINJUVI on saatettava käyttökuntoon ja laimennettava ennen suonensisäistä infuusiota.
Käytä soveltuvaa aseptista tekniikkaa käyttökuntoon saattamisen ja laimentamisen aikana.
Ohjeet käyttökuntoon saattamiseen
- Määritä tafasitamabiannos potilaan painon mukaan kertomalla 12 mg potilaan painolla (kg). Laske sitten tarvittava tafasitamabi‑injektiopullojen määrä (jokainen injektiopullo sisältää 200 mg tafasitamabia) (ks. kohta Annostus ja antotapa).
- Käytä steriiliä ruiskua ja lisää 5,0 ml steriiliä injektionesteisiin tarkoitettua vettä jokaiseen Minjuvi‑injektiopulloon. Ohjaa nestevirta jokaisen injektiopullon seinämiä pitkin, älä suoraan kylmäkuivattuun jauheeseen.
- Liuota kylmäkuivattu jauhe nesteeseen pyörittämällä injektiopulloa varovaisesti sivusuunnassa. Älä ravista injektiopulloa voimakkaasti. Älä poista injektiopullon sisältöä, ennen kuin kaikki kiinteä aine on liuennut täysin. Kylmäkuivatun jauheen pitäisi liueta 5 minuutin kuluessa.
- Käyttökuntoon saatetun liuoksen pitäisi olla väritöntä tai kellahtavaa. Varmista silmämääräisesti ennen jatkamista, että liuoksessa ei ole kiinteää ainetta eikä värimuutoksia. Jos liuos on sameaa, siinä on värimuutoksia tai se sisältää näkyviä partikkeleita, hävitä injektiopullo.
Laimennusohjeet
- Ota käyttöön infuusiopussi, joka sisältää 250 ml injektioihin tarkoitettua natriumkloridiliuosta 9 mg/ml (0,9 %).
- Laske tarvittava käyttökuntoon saatettavan, 40 mg/ml tafasitamabiliuoksen määrä. Ota tätä vastaava määrä nestettä infuusiopussista ja hävitä pussista poistettu neste.
- Ota lasketun tilavuuden (ml) verran käyttökuntoon saatettua tafasitamabiliuosta injektiopulloista ja lisää se varovaisesti natriumkloridia 9 mg/ml (0,9 %) sisältävään infuusiopussiin. Hävitä injektiopulloon mahdollisesti jäänyt tafasitamabi.
- Lopullisen laimennetun liuoksen kokonaispitoisuuden tulisi olla 2–8 mg/ml tafasitamabia.
- Sekoita infuusiopussia kääntämällä sitä varovaisesti ylösalaisin. Älä ravista.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
MINJUVI kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
200 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FX12
Valmisteyhteenvedon muuttamispäivämäärä
18.12.2025
Yhteystiedot
Paasheuvelweg 25
1105 BP Amsterdam
Netherlands
09 7479 0132
eumedinfo@incyte.com