
COSENTYX injektioneste, liuos, esitäytetty ruisku 75 mg
Vaikuttavat aineet ja niiden määrät
Yksi esitäytetty ruisku sisältää 75 mg sekukinumabia 0,5 ml:ssa.
Sekukinumabi on yhdistelmä-DNA-tekniikalla valmistettu ihmisen monoklonaalinen vasta-aine, joka on tuotettu kiinanhamsterin munasarjasoluissa (CHO).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Läiskäpsoriaasi pediatrisilla potilailla
Cosentyx on tarkoitettu keskivaikean tai vaikean läiskäpsoriaasin hoitoon vähintään 6-vuotiaille lapsille ja nuorille, joille harkitaan systeemistä hoitoa.
Juveniili idiopaattinen artriitti (JIA)
Entesiitteihin liittyvä artriitti (ERA)
Cosentyx on tarkoitettu käytettäväksi yksinään tai yhdessä metotreksaatin kanssa aktiivisen entesiitteihin liittyvän artriitin hoitoon vähintään 6-vuotiaille potilaille, joilla tavanomainen hoito on tuottanut riittämättömän vasteen tai ollut huonosti siedetty (ks. kohta Farmakodynamiikka).
Juveniili nivelpsoriaasi (JPsA)
Cosentyx on tarkoitettu käytettäväksi yksinään tai yhdessä metotreksaatin kanssa aktiivisen juveniilin nivelpsoriaasin hoitoon vähintään 6-vuotiaille potilaille, joilla tavanomainen hoito on tuottanut riittämättömän vasteen tai ollut huonosti siedetty (ks. kohta Farmakodynamiikka).
Ehto
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Cosentyx on tarkoitettu käytettäväksi käyttöaiheidensa mukaisten sairauksien diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus
Läiskäpsoriaasi pediatrisilla potilailla (vähintään 6-vuotiaat lapset ja nuoret)
Suositeltu annos perustuu painoon (taulukko 1) ja annetaan injektiona ihon alle aluksi viikoilla 0, 1, 2, 3 ja 4. Tämän jälkeen injektio annetaan kerran kuukaudessa ylläpitohoitona. Kukin 75 mg:n annos annetaan yhtenä 75 mg:n injektiona ihon alle. Kukin 150 mg:n annos annetaan yhtenä 150 mg:n injektiona ihon alle. Kukin 300 mg:n annos annetaan yhtenä 300 mg:n injektiona ihon alle tai kahtena 150 mg:n injektiona ihon alle.
Taulukko 1 Suositeltu annos pediatristen potilaiden läiskäpsoriaasin hoitoon
Paino annosteluajankohtana | Suositeltu annos |
< 25 kg | 75 mg |
25 – < 50 kg | 75 mg |
≥ 50 kg | 150 mg (*voidaan suurentaa 300 mg:aan) |
*Osa potilaista voi saada lisähyötyä suuremmasta annoksesta.
Juveniili idiopaattinen artriitti (JIA)
Entesiitteihin liittyvä artriitti (ERA) ja juveniili nivelpsoriaasi (JPsA)
Suositeltu annos perustuu painoon (taulukko 2) ja annetaan injektiona ihon alle viikoilla 0, 1, 2, 3 ja 4. Tämän jälkeen injektio annetaan kerran kuukaudessa ylläpitohoitona. Kukin 75 mg:n annos annetaan yhtenä 75 mg:n injektiona ihon alle. Kukin 150 mg:n annos annetaan yhtenä 150 mg:n injektiona ihon alle.
Taulukko 2 Suositeltu annos juveniilin idiopaattisen artriitin hoitoon
Paino annosteluajankohtana | Suositeltu annos |
< 50 kg | 75 mg |
≥ 50 kg | 150 mg |
Cosentyx voi olla saatavilla eri vahvuuksina ja/tai lääkemuotoina yksilöllisestä hoitotarkoituksesta riippuen.
Kaikkien edellä mainittujen käyttöaiheiden kohdalla saatavilla olevat tiedot viittaavat siihen, että kliininen vaste saavutetaan tavallisesti 16 viikon kuluessa hoidon aloittamisesta. Hoidon lopettamista on harkittava, jos potilas ei ole saanut vastetta 16 viikon hoidon aikana. Osalla potilaista aluksi saatu osittainen vaste saattaa myöhemmin parantua, kun hoitoa jatketaan yli 16 viikon ajan.
Cosentyx-valmisteen turvallisuutta ja tehoa alle 6 vuoden ikäisten lasten läiskäpsoriaasin ja juveniilin idiopaattisen artriitin (alatyypit ”entesiitteihin liittyvä artriitti” ja ”juveniili nivelpsoriaasi”) hoidossa ei ole varmistettu.
Cosentyx-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten muiden käyttöaiheiden hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Erityisryhmät
Munuaisten/maksan vajaatoiminta
Cosentyx-valmistetta ei ole tutkittu näissä potilasryhmissä. Annossuosituksia ei voida tehdä.
Antotapa
Cosentyx annetaan injektiona ihon alle. Jos mahdollista, pistoskohdaksi ei tule valita ihoaluetta, jossa on psoriaasia. Esitäytettyä ruiskua ei saa ravistaa.
Jos lääkäri katsoo sen asianmukaiseksi, potilas itse tai huoltaja voi pistää Cosentyx-annoksen, kun häntä on neuvottu oikeasta ihon alle pistämisen tekniikasta. Lääkärin tulee kuitenkin varmistaa potilaan asianmukainen seuranta. Potilaita tai huoltajia on neuvottava pistämään koko Cosentyx-injektionestemäärä pakkausselosteen ohjeiden mukaisesti. Tarkat ohjeet valmisteen antoon on kerrottu pakkausselosteessa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Kliinisesti merkittävä aktiivinen infektio, esim. aktiivinen tuberkuloosi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Sekukinumabi saattaa lisätä infektioiden riskiä. Markkinoille tulon jälkeen sekukinumabia saaneilla potilailla on todettu vakavia infektioita. Varovaisuutta on noudatettava, kun harkitaan sekukinumabin käyttöä potilaille, joilla on krooninen infektio tai joilla on ollut toistuvia infektioita.
Potilaita on neuvottava hakeutumaan lääkärin hoitoon, jos infektion oireita tai merkkejä ilmenee. Jos potilaalle kehittyy vakava infektio, häntä on seurattava huolellisesti eikä sekukinumabia saa antaa ennen kuin infektio on parantunut.
Kliinisissä tutkimuksissa sekukinumabia saaneilla potilailla on havaittu infektioita (ks. kohta Haittavaikutukset). Useimmat infektiot olivat lieviä tai keskivaikeita ylähengitystieinfektioita, kuten nenän ja nielun tulehduksia, eivätkä ne vaatineet hoidon keskeyttämistä.
Kliinisissä psoriaasitutkimuksissa sekukinumabin vaikutusmekanismiin liittyviä ei-vakavia mukokutaanisia kandidiaasi-infektioita ilmoitettiin useammin sekukinumabiryhmässä (3,55 infektiota 100 potilasvuotta kohti 300 mg sekukinumabia saaneilla) kuin lumelääkeryhmässä (1,00 infektiota 100 potilasvuotta kohti) (ks. kohta Haittavaikutukset).
Tuberkuloosi
Tuberkuloosia (aktiivista infektiota ja/tai latenttia uudelleen aktivoitumista) on ilmoitettu sekukinumabilla hoidettavilla potilailla. Potilaat on tutkittava tuberkuloosi-infektion varalta ennen sekukinumabihoidon aloittamista. Sekukinumabia ei saa antaa potilaille, joilla on aktiivinen tuberkuloosi (ks. kohta Vasta-aiheet). Potilaille, joilla on latentti tuberkuloosi, on harkittava tuberkuloosilääkitystä hoitosuositusten mukaisesti ennen sekukinumabin aloittamista. Sekukinumabia saavia potilaita on seurattava aktiivisen tuberkuloosin oireiden ja löydösten varalta.
Tulehdukselliset suolistosairaudet (mukaan lukien Crohnin tauti ja haavainen paksusuolitulehdus)
Tulehduksellisten suolistosairauksien puhkeamista tai pahenemista on raportoitu sekukinumabihoidon aikana (ks. kohta Haittavaikutukset). Sekukinumabia ei suositella potilaille, joilla on tulehduksellisia suolistosairauksia. Jos potilaalla ilmenee tulehduksellisen suolistosairauden oireita tai merkkejä tai havaitaan olemassa olevan tulehduksellisen suolistosairauden pahenemista, tulee sekukinumabihoito lopettaa ja aloittaa asianmukainen lääkehoito.
Yliherkkyysreaktiot
Sekukinumabia saaneilla potilailla on todettu harvinaisina tapauksina anafylaktisia reaktioita ja angioedemaa. Jos potilaalla ilmenee anafylaktinen reaktio, angioedeema tai muita vakavia allergisia reaktioita, sekukinumabin antaminen on keskeytettävä välittömästi ja on aloitettava asianmukainen hoito.
Lateksille herkät henkilöt
Esitäytetyn Cosentyx 75 mg ruiskun irrotettava neulansuojus sisältää luonnonkumin (lateksin) johdannaista. Irrotettavassa neulansuojuksessa ei ole toistaiseksi havaittu luonnonkumia (lateksia). Esitäytetyn Cosentyx 75 mg ruiskun käyttöä lateksille herkille henkilöille ei ole tutkittu ja tästä syystä mahdollisten yliherkkyysreaktioiden riskiä ei voida täysin sulkea pois.
Rokotukset
Eläviä rokotteita ei saa antaa sekukinumabi-hoidon aikana.
Sekukinumabia saaville potilaille voidaan antaa inaktivoituja tai muita kuin eläviä rokotteita. Kun terveille vapaehtoisille annettiin tutkimuksessa meningokokki- ja inaktivoitu influenssarokotus, sekukinumabia 150 mg saaneiden ryhmässä ja lumelääkeryhmässä niiden koehenkilöiden osuus, joille kehittyi riittävä immuunivaste, oli samansuuruinen. Riittävä immuunivaste meningokokki- ja influenssarokotteelle määriteltiin vasta-ainetiitterien suurenemiseksi vähintään nelinkertaisiksi. Tiedot viittaavat siihen, että sekukinumabi ei heikennä humoraalista immuunivastetta meningokokki- ja influenssarokotteille.
On suositeltavaa, että pediatriset potilaat saavat kaikki iänmukaiset, ajankohtaisten rokotussuositusten mukaiset rokotukset ennen Cosentyx-hoidon aloittamista.
Samanaikainen immunosuppressiivinen hoito
Psoriaasitutkimuksissa ei ole arvioitu sekukinumabin turvallisuutta ja tehoa, kun sitä käytetään samanaikaisesti immunosuppressiivisten lääkkeiden, kuten biologisten lääkkeiden, tai valohoidon kanssa. Artriittitutkimuksissa (joihin osallistui myös psoriaasiartriittia ja selkärankareumaa sairastavia potilaita) sekukinumabia on annettu samanaikaisesti metotreksaatin (MTX), sulfasalatsiinin ja/tai kortikosteroidien kanssa. Varovaisuutta on noudatettava harkittaessa muiden immunosuppressiivisten lääkkeiden ja sekukinumabin samanaikaista käyttöä (ks. myös kohta Yhteisvaikutukset).
Hepatiitti B:n uudelleenaktivoituminen
Hepatiitti B -virus (HBV) saattaa uudelleenaktivoitua sekukinumabihoitoa saavilla potilailla. Immunosuppressantteja koskevien hoitosuositusten mukaisesti potilaiden testaamista HBV-infektion varalta on harkittava ennen sekukinumabihoidon aloittamista. Potilaita, joilla on positiivinen HBV-serologiatulos, on seurattava HBV:n uudelleenaktivoitumisen kliinisten ja laboratoriolöydösten ilmaantumisen varalta sekukinumabihoidon aikana. Jos HBV aktivoituu uudelleen sekukinumabihoidon aikana, hoidon lopettamista on harkittava ja potilaille on annettava hoitosuositusten mukaista hoitoa.
Yhteisvaikutukset
Eläviä rokotteita ei saa antaa sekukinumabi-hoidon aikana (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yhteisvaikutuksia ei havaittu sekukinumabilla ja midatsolaamilla (CYP3A4-substraatti) tutkimuksessa, jossa aikuispotilailla oli läiskäpsoriaasi.
Niveltulehdusta koskevissa tutkimuksissa (joihin osallistui nivelpsoriaasia ja aksiaalista spondylartriittia sairastavia potilaita) ei todettu yhteisvaikutuksia, kun sekukinumabia annettiin samanaikaisesti metotreksaatin ja/tai kortikosteroidien kanssa.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja vähintään 20 viikkoa hoidon päättymisen jälkeen.
Raskaus
Ei ole riittävästi tietoa sekukinumabin käytöstä raskaana oleville naisille.
Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi sekukinumabin käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö sekukinumabi ihmisen rintamaitoon. Immunoglobuliinit erittyvät ihmisen rintamaitoon eikä ole tiedossa imeytyykö sekukinumabi systeemisesti rintaruokinnassa. Koska sekukinumabi saattaa aiheuttaa haittavaikutuksia imetettävälle lapselle, on päätettävä lopetetaanko rintaruokinta hoidon ajaksi ja vähintään 20 viikon ajaksi hoidon päättymisen jälkeen vai lopetetaanko Cosentyx-hoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Sekukinumabin mahdollista vaikutusta ihmisen hedelmällisyyteen ei ole tutkittu. Eläinkokeissa ei ole havaittu suoria tai epäsuoria haitallisia vaikutuksia hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Cosentyx-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitettuja haittavaikutuksia ovat ylähengitystieinfektiot (17,1 %) (yleisimmin nenän ja nielun tulehdus ja nuha).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa ja markkinoille tulon jälkeen raportoidut haittavaikutukset (taulukko 3) on lueteltu MedDRA-elinjärjestelmäluokituksen mukaisesti. Kunkin elinjärjestelmäluokan haittavaikutukset on järjestetty yleisyysjärjestykseen yleisimmästä alkaen. Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä. Lisäksi haittavaikutukset on luokiteltu esiintymistiheyden mukaan seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000) ja hyvin harvinainen (< 1/10 000); ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Eri käyttöaiheita (läiskäpsoriaasi, nivelpsoriaasi, aksiaalinen spondylartriitti, hidradenitis suppurativa [HS‑tauti] ja muut autoimmuunisairaudet) koskevissa sokkoutetuissa ja avoimissa kliinisissä tutkimuksissa sekukinumabi-hoitoa on saanut yli 20 000 potilasta, mikä vastaa 34 908 potilasvuoden altistusta. Näistä potilaista yli 14 000 sai sekukinumabi-hoitoa vähintään yhden vuoden ajan. Sekukinumabin turvallisuusprofiili on yhdenmukainen kaikissa indikaatioissa.
Taulukko 3 Luettelo kliinisissä tutkimuksissa ja markkinoille tulon jälkeen todetuista haittavaikutuksista1)
| Elinjärjestelmä | Esiintymistiheys | Haittavaikutus |
| Infektiot | Hyvin yleinen | Ylähengitystieinfektiot |
| Yleinen | Huuliherpes | |
| Melko harvinainen | Suun kandidiaasi | |
| Korvakäytävätulehdus | ||
| Alahengitysteiden infektiot | ||
| Jalkasilsa | ||
| Tuntematon | Limakalvojen ja ihon kandidiaasi (mukaan lukien ruokatorven kandidiaasi) | |
| Veri ja imukudos | Melko harvinainen | Neutropenia |
| Immuunijärjestelmä | Harvinainen | Anafylaktiset reaktiot |
| Angioedeema | ||
| Hermosto | Yleinen | Päänsärky |
| Silmät | Melko harvinainen | Sidekalvotulehdus |
| Hengityselimet, rintakehä ja välikarsina | Yleinen | Vetinen nuha |
| Ruoansulatuselimistö | Yleinen | Ripuli |
| Pahoinvointi | ||
| Melko harvinainen | Tulehduksellinen suolistosairaus (IBD) | |
| Iho ja ihonalainen kudos | Yleinen | Ekseema |
| Melko harvinainen | Urtikaria | |
| Dyshidroottinen ekseema | ||
| Harvinainen | Kesivä ihottuma2) | |
| Allerginen verisuonitulehdus | ||
| Tuntematon | Haavautuva ihotulehdus (pyoderma gangraenosum) | |
| Yleisoireet ja antopaikassa todettavat haitat | Yleinen | Väsymys |
1) Lumelääkekontrolloidut kliiniset tutkimukset (vaihe III) läiskäpsoriaasi‑, nivelpsoriaasi-, selkärankareuma-, nr-axSpA- ja hidradenitis suppurativa ‑potilailla, jotka saivat 300 mg:n, 150 mg:n tai 75 mg:n annosta tai lumelääkettä enintään 12 viikkoa jatkuneen (psoriaasin) hoidon ajan tai 16 viikkoa jatkuneen (nivelpsoriaasin, selkärankareuman, non-radiografisen aksiaalisen spondylartriitin tai hidradenitis suppurativan) hoidon ajan 2) Tapauksia on raportoitu psoriaasipotilailla | ||
Valikoitujen haittavaikutusten kuvaus
Infektiot
Läiskäpsoriaasipotilailla tehtyjen kliinisten tutkimusten lumelääkekontrolloidussa vaiheessa (yhteensä 1 382 potilasta, jotka saivat sekukinumabi-hoitoa, ja 694 potilasta, jotka saivat lumelääkettä, enintään 12 viikon ajan) infektioita ilmoitettiin 28,7 %:lla sekukinumabi-hoitoa saaneista ja 18,9 %:lla lumelääkettä saaneista potilaista. Useimmat infektiot olivat ei-vakavia ja vaikeusasteeltaan lieviä tai keskivaikeita ylähengitystieinfektioita, kuten nenän ja nielun tulehduksia, eivätkä ne vaatineet hoidon keskeyttämistä. Limakalvojen tai ihon kandidiaasin esiintyvyys suureni, kuten vaikutusmekanismin perusteella oli odotettavissa, mutta tapaukset olivat vaikeusasteeltaan lieviä tai keskivaikeita, ei-vakavia, reagoivat vakiintuneeseen hoitoon eikä hoitoa tarvinnut keskeyttää niiden vuoksi. Vakavia infektioita ilmeni 0,14 %:lla sekukinumabi-hoitoa ja 0,3 %:lla lumelääkettä saaneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Koko hoitojakson aikana (yhteensä 3 430 potilasta, jotka saivat sekukinumabi-hoitoa ja joista suurin osa sai hoitoa enintään 52 viikon ajan) infektioita ilmoitettiin 47,5 %:lla sekukinumabi-hoitoa saaneista (seurannassa 0,9 potilasvuotta kohti). Vakavia infektioita ilmoitettiin 1,2 %:lla sekukinumabi-hoitoa saaneista potilaista (seurannassa 0,015 potilasvuotta kohti).
Infektioita havaittiin kliinisissä nivelpsoriaasitutkimuksissa ja aksiaalisen spondylartriitin (selkärankareuman ja non-radiografisen aksiaalisen spondylartriitin) kliinisissä tutkimuksissa saman verran kuin psoriaasitutkimuksissa.
Potilaat, joilla on hidradenitis suppurativa, ovat alttiimpia infektioille. Hidradenitis suppurativaa sairastavilla potilailla tehtyjen kliinisten tutkimusten lumelääkekontrolloidussa vaiheessa (yhteensä 721 potilasta, jotka saivat sekukinumabi-hoitoa, ja 363 potilasta, jotka saivat lumelääkettä, enintään 16 viikon ajan) infektioita esiintyi lukumääräisesti enemmän kuin psoriaasitutkimuksissa (30,7 %:lla sekukinumabi-hoitoa saaneista vs. 31,7 %:lla lumelääkettä saaneista potilaista). Useimmat infektiot eivät olleet vakavia vaan vaikeusasteeltaan lieviä tai keskivaikeita eivätkä vaatineet hoidon lopettamista tai keskeyttämistä.
Neutropenia
Kliinisissä vaiheen III psoriaasitutkimuksissa neutropeniaa todettiin useammin sekukinumabi- kuin lumelääkeryhmässä, mutta useimmat tapaukset olivat lieviä, ohimeneviä ja palautuvia. Neutropeniaa, jossa arvo oli < 1,0⎼0,5 x 109/l (CTCAE-luokka 3), ilmoitettiin 18:lla potilaalla 3 430:stä (0,5 %) sekukinumabia saaneesta potilaasta, mutta 15:ssä tapauksessa 18:sta ei havaittu korrelaatiota sekukinumabi-annokseen tai infektioiden esiintymisen ajankohtaan. Vakavampia neutropeniatapauksia ei havaittu. Muut kolme tapausta olivat ei-vakavia infektioita, jotka reagoivat vakiintuneeseen hoitoon, eivätkä edellyttäneet sekukinumabi-hoidon keskeyttämistä.
Neutropeniaa ilmeni nivelpsoriaasin, aksiaalisen spondylartriitin (selkärankareuman ja non-radiografisen aksiaalisen spondylartriitin) ja hidradenitis suppurativan yhteydessä saman verran kuin psoriaasin yhteydessä.
On raportoitu harvinaisia neutropeniatapauksia, joissa arvo on < 0,5 x 109/l (CTCAE-luokka 4).
Immunogeenisuus
Kliinisissä psoriaasi‑ ja nivelpsoriaasitutkimuksissa, aksiaalisen spondylartriitin (selkärankareuman ja non-radiografisen aksiaalisen spondylartriitin) ja hidradenitis suppurativan tutkimuksissa alle 1 % sekukinumabi-hoitoa saaneista potilaista kehitti vasta-aineita sekukinumabille enintään 52 viikkoa kestäneen hoidon aikana. Noin puolet hoidon aiheuttamista lääkeaineelle kehittyneistä vasta-aineista oli neutraloivia, mutta tätä ei voitu yhdistää tehon heikentymiseen tai farmakokineettisiin poikkeavuuksiin.
Pediatriset potilaat
Haittavaikutukset vähintään 6-vuotiailla pediatrisilla läiskäpsoriaasipotilailla
Sekukinumabin turvallisuutta arvioitiin kahdessa vaiheen III tutkimuksessa pediatrisilla läiskäpsoriaasipotilailla. Ensimmäinen tutkimus (pediatrinen tutkimus 1) oli kaksoissokkoutettu, lumekontrolloitu tutkimus 162:lla vaikeaa läiskäpsoriaasia sairastavalla potilaalla, jotka olivat iältään 6 – < 18 vuotta. Toinen tutkimus (pediatrinen tutkimus 2) oli avoin tutkimus 84:llä keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavalla potilaalla, jotka olivat iältään 6 – < 18 vuotta. Näiden kahden tutkimuksen tuottama turvallisuusprofiili oli yhdenmukainen aikuisten läiskäpsoriaasipotilaiden turvallisuusprofiilin kanssa.
Haittavaikutukset juveniilia idiopaattista artriittia sairastavilla pediatrisilla potilailla
Sekukinumabin turvallisuutta arvioitiin myös vaiheen III tutkimuksessa 86:lla potilaalla, jotka sairastivat juveniilia idiopaattista artriittia (entesiitteihin liittyvä artriitti tai juveniili nivelpsoriaasi) ja olivat iältään 2 – < 18 vuotta. Tämän tutkimuksen tuottama turvallisuusprofiili oli yhdenmukainen aikuispotilaiden turvallisuusprofiilin kanssa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa annettiin jopa 30 mg/kg:n annoksia (noin 2 000‑3 000 mg) laskimoon ilman annosta rajoittavaa toksisuutta. Yliannostustapauksissa suositellaan potilaan tarkkailua haittavaikutusten oireiden tai merkkien varalta ja sopivan oireenmukaisen hoidon antamista välittömästi.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC10
Vaikutusmekanismi
Sekukinumabi on ihmisen monoklonaalinen IgG1/κ-vasta-aine, joka sitoutuu selektiivisesti tulehdusta aiheuttavaan sytokiiniin interleukiini 17A:han (IL‑17A) neutraloiden sen. Sekukinumabi vaikuttaa sitoutumalla IL‑17A:han ja estämällä sen kiinnittymisen IL‑17-reseptoriin, joka ilmentyy erilaisissa solutyypeissä, kuten keratinosyyteissä. Tämän seurauksena sekukinumabi estää tulehdusta aiheuttavien sytokiinien, kemokiinien ja kudosvaurion välittäjäaineiden vapautumista ja vähentää IL‑17A-välitteistä vaikutusta autoimmuuni- ja tulehdussairauksiin. Kliinisesti merkitykselliset sekukinumabipitoisuudet saavuttavat ihon ja vähentävät paikallisia tulehdusmerkkiaineita. Sekukinumabihoidon suorana seurauksena läiskäpsoriaasileesioille tyypillinen punoitus, paksuus ja hilseily vähenevät.
IL‑17A on luontainen sytokiini, joka on osallisena normaalin tulehdus- ja immuunivasteen kehittymisessä. IL‑17A:lla on keskeinen tehtävä läiskäpsoriaasin, nivelpsoriaasin ja aksiaalisen spondylartriitin (selkärankareuman ja non-radiografisen aksiaalisen spondylartriitin) synnyssä. Sitä tuotetaan läiskäpsoriaasipotilaiden vaurioituneella iholla enemmän kuin terveellä iholla ja sitä muodostuu enemmän nivelpsoriaasipotilaiden synoviaalikudoksessa. IL-17:ää tuottavia soluja oli myös huomattavasti enemmän selkärankareumapotilaiden fasettinivelten subkondraalisessa luuytimessä. Non-radiografista aksiaalista spondylartriittia sairastavilla potilailla on myös todettu suurentuneita määriä IL-17A-sytokiinia tuottavia lymfosyyttejä. IL-17A:n esto on osoittautunut tehokkaaksi selkärankareuman hoidossa, mikä vahvistaa, että tällä sytokiinilla on keskeinen merkitys aksiaalisessa spondylartriitissa.
Farmakodynaamiset vaikutukset
Alussa kokonais-IL‑17A:n (vapaan ja sekukinumabiin sitoutuneen IL‑17A:n) pitoisuus seerumissa suurenee potilailla, jotka saavat sekukinumabia. Tämän jälkeen pitoisuus pienenee hitaasti sekukinumabiin sitoutuneen IL‑17A:n vähentyneen puhdistuman seurauksena, mikä viittaa siihen, että sekukinumabi sitoo selektiivisesti vapaata IL‑17A:ta, jolla on keskeinen tehtävä läiskäpsoriaasin synnyssä.
Sekukinumabilla tehdyssä tutkimuksessa läiskäpsoriaasipotilaiden vaurioituneella iholla runsaina havaittavien infiltroivien epidermaalisten neutrofiilien ja erilaisten neutrofiileihin liittyvien merkkiaineiden määrä oli selvästi vähentynyt, kun hoitoa oli jatkettu yhden tai kahden viikon ajan.
Sekukinumabin on osoitettu pienentävän tulehduksen merkkiaineena toimivan C-reaktiivisen proteiinin pitoisuutta (1−2 viikon sisällä hoidosta).
Kliininen teho ja turvallisuus
Läiskäpsoriaasi aikuisilla
Sekukinumabin turvallisuutta ja tehoa arvioitiin neljässä satunnaistetussa, kaksoissokkoutetussa, lumelääkekontrolloidussa vaiheen III tutkimuksessa. Tutkimuksiin osallistui keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastavia potilaita, joille harkittiin valohoitoa tai systeemistä hoitoa [ERASURE, FIXTURE, FEATURE, JUNCTURE]. Sekukinumabin 150 mg:n ja 300 mg:n annoksen tehoa verrattiin lumelääkkeeseen tai etanerseptiin. Lisäksi yhdessä tutkimuksessa verrattiin pitkäkestoista hoito-ohjelmaa hoito-ohjelmaan, jossa lääkehoito toistettiin tarpeen vaatiessa [SCULPTURE].
Lumelääkekontrolloituihin tutkimuksiin mukaan otetuista 2 403 potilaasta 79 % ei ollut aiemmin saanut hoitoa biologisilla lääkkeillä, 45 %:lla hoito muilla kuin biologisilla lääkkeillä ei ollut tehonnut ja 8 %:lla hoito biologisilla lääkkeillä ei ollut tehonnut (6 %:lla TNF-salpaajahoito ei ollut tehonnut ja 2 %:lla p40-salpaajahoito ei ollut tehonnut). Suunnilleen 15‑25 %:lla vaiheen III tutkimuksiin osallistuneista potilaista oli lähtötilanteessa nivelpsoriaasi.
Psoriaasitutkimuksessa 1 (ERASURE) arvioitiin 738 potilasta. Sekukinumabi-hoitoon satunnaistetut potilaat saivat joko 150 mg:n tai 300 mg:n annoksen viikolla 0, 1, 2, 3 ja 4 ja sen jälkeen samaa annosta kerran kuukaudessa. Psoriaasitutkimuksessa 2 (FIXTURE) arvioitiin 1 306 potilasta. Sekukinumabi-hoitoon satunnaistetut potilaat saivat joko 150 mg:n tai 300 mg:n annoksen viikolla 0, 1, 2, 3 ja 4 ja sen jälkeen samaa annosta kerran kuukaudessa. Etanerseptiryhmään satunnaistetut potilaat saivat 50 mg:n annoksen kahdesti viikossa 12 viikon ajan ja sen jälkeen 50 mg kerran viikossa. Sekä tutkimuksessa 1 että tutkimuksessa 2 lumelääkeryhmään satunnaistetut potilaat, jotka eivät olleet saaneet vastetta viikkoon 12 mennessä, siirrettiin saamaan sekukinumabia (joko 150 mg tai 300 mg) viikolla 12, 13, 14 ja 15 ja sen jälkeen samaa annosta kerran kuukaudessa viikosta 16 lähtien. Kaikkia potilaita seurattiin enintään 52 viikon ajan ensimmäisen tutkimuslääkeannoksen antamisen jälkeen.
Psoriaasitutkimuksessa 3 (FEATURE) arvioitiin esitäytetyllä ruiskulla itse 12 viikon ajan annetun sekukinumabin turvallisuutta, siedettävyyttä ja käytettävyyttä plaseboon nähden 177 potilaalla. Psoriaasitutkimuksessa 4 (JUNCTURE) arvioitiin esitäytetyllä kynällä itse 12 viikon ajan annetun sekukinumabin turvallisuutta, siedettävyyttä ja käytettävyyttä plaseboon nähden 182 potilaalla. Sekä tutkimuksessa 3 että tutkimuksessa 4 sekukinumabi-hoitoon satunnaistetut potilaat saivat joko 150 mg:n tai 300 mg:n annoksen viikolla 0, 1, 2, 3 ja 4 ja sen jälkeen samaa annosta kerran kuukaudessa. Osa potilaista satunnaistettiin myös saamaan lumelääkettä viikolla 0, 1, 2, 3 ja 4 ja sen jälkeen samaa annosta kerran kuukaudessa.
Psoriaasitutkimuksessa 5 (SCULPTURE) arvioitiin 966 potilasta. Kaikki potilaat saivat sekukinumabia 150 mg tai 300 mg viikolla 0, 1, 2, 3, 4, 8 ja 12 ja sen jälkeen heidät satunnaistettiin saamaan joko samansuuruista ylläpitoannosta kerran kuukaudessa viikosta 12 lähtien tai tarpeen vaatiessa toistettavaa hoitoa samalla annoksella. Tarpeen vaatiessa toteutettua hoitoa saaneiden ryhmään satunnaistettujen potilaiden vaste ei säilynyt riittävänä ja siksi suositellaan lääkkeen antoa säännöllisesti kerran kuukaudessa.
Lumelääke- ja aktiivikontrolloitujen tutkimusten rinnakkaisia ensisijaisia päätemuuttujia olivat niiden potilaiden osuus, jotka saavuttivat PASI 75 -vasteen ja IGA mod 2011 -asteikon mukaisen tuloksen oireeton tai lähes oireeton viikolla 12 verrattuna lumelääkkeeseen (ks. taulukot 4 ja 5). Kaikissa tutkimuksissa 300 mg:n annoksella saavutetut ihon parantumista koskeneet tulokset olivat paremmat erityisesti oireeton- tai lähes oireeton -tuloksen suhteen kaikkien tehoa mittaavien päätemuuttujien osalta, jotka olivat PASI 90, PASI 100 ja 0- tai 1-vaste IGA mod 2011-asteikolla, ja maksimivaikutus todettiin viikolla 16. Siksi suositellaan 300 mg:n annosta.
Taulukko 4 Yhteenveto kliinisistä vasteista PASI 50/75/90/100 ja IGA⃰ mod 2011 oireeton tai lähes oireeton psoriaasitutkimuksissa 1, 3 ja 4 (ERASURE, FEATURE ja JUNCTURE)
| Viikko 12 | Viikko 16 | Viikko 52 | |||||
Lume lääke | 150 mg | 300 mg | 150 mg | 300 mg | 150 mg | 300 mg | |
| Tutkimus 1 | |||||||
| Potilaiden määrä | 246 | 244 | 245 | 244 | 245 | 244 | 245 |
| PASI 50 -vaste, n (%) | 22 (8,9 %) | 203 (83,5 %) | 222 (90,6 %) | 212 (87,2 %) | 224 (91,4 %) | 187 (77 %) | 207 (84,5 %) |
| PASI 75 -vaste, n (%) | 11 (4,5 %) | 174 (71,6 %)** | 200 (81,6 %)** | 188 (77,4 %) | 211 (86,1 %) | 146 (60,1 %) | 182 (74,3 %) |
| PASI 90 -vaste, n (%) | 3 (1,2 %) | 95 (39,1 %)** | 145 (59,2 %)** | 130 (53,5 %) | 171 (69,8 %) | 88 (36,2 %) | 147 (60,0 %) |
| PASI 100 -vaste, n (%) | 2 (0,8 %) | 31 (12,8 %) | 70 (28,6 %) | 51 (21,0 %) | 102 (41,6 %) | 49 (20,2 %) | 96 (39,2 %) |
| IGA mod 2011 oireeton tai lähes oireeton -vaste, n (%) | 6 (2,40 %) | 125 (51,2 %)** | 160 (65,3 %)** | 142 (58,2 %) | 180 (73,5 %) | 101 (41,4 %) | 148 (60,4 %) |
| Tutkimus 3 | |||||||
| Potilaiden määrä | 59 | 59 | 58 | - | - | - | - |
| PASI 50 -vaste, n (%) | 3 (5,1 %) | 51 (86,4 %) | 51 (87,9 %) | - | - | - | - |
| PASI 75 -vaste, n (%) | 0 (0,0 %) | 41 (69,5 %)** | 44 (75,9 %)** | - | - | - | - |
| PASI 90 -vaste, n (%) | 0 (0,0 %) | 27 (45,8 %) | 35 (60,3 %) | - | - | - | - |
| PASI 100 -vaste, n (%) | 0 (0,0 %) | 5 (8,5 %) | 25 (43,1 %) | - | - | - | - |
| IGA mod 2011 oireeton tai lähes oireeton -vaste, n (%) | 0 (0,0 %) | 31 (52,5 %)** | 40 (69,0 %)** | - | - | - | - |
| Tutkimus 4 | |||||||
| Potilaiden määrä | 61 | 60 | 60 | - | - | - | - |
| PASI 50 -vaste, n (%) | 5 (8,2 %) | 48 (80,0 %) | 58 (96,7 %) | - | - | - | - |
| PASI 75 -vaste, n (%) | 2 (3,3 %) | 43 (71,7 %)** | 52 (86,7 %)** | - | - | - | - |
| PASI 90 -vaste, n (%) | 0 (0,0 %) | 24 (40,0 %) | 33 (55,0 %) | - | - | - | - |
| PASI 100 -vaste, n (%) | 0 (0,0 %) | 10 (16,7 %) | 16 (26,7 %) | - | - | - | - |
| IGA mod 2011 oireeton tai lähes oireeton -vaste, n (%) | 0 (0,0 %) | 32 (53,3 %)** | 44 (73,3 %)** | - | - | - | - |
* IGA mod 2011 on asteikko, joka muodostuu 5 kategoriasta, jotka ovat 0 = oireeton, 1 = lähes oireeton 2 = lievä, 3 = keskivaikea tai 4 = vaikea. Asteikko kuvaa lääkärin kokonaisarviota psoriaasin vaikeusasteesta ja arvioi erityisesti paksuutta, punoitusta ja hilseilyä. Hoidon onnistumisen aste oireeton tai lähes oireeton tarkoitti, ettei merkkejä psoriaasista ollut tai psoriaasileesioiden väri vaihteli normaalista vaaleanpunaiseen, läiskä ei ollut paksuuntunut ja pesäkkeiden hilseilyä ei ollut lainkaan tai sitä oli erittäin vähän. ** p-arvot lumelääkkeeseen nähden ja monivertailukorjatut p-arvot: p < 0,0001. | |||||||
Taulukko 5 Yhteenveto kliinisistä vasteista, psoriaasitutkimus 2 (FIXTURE)
| Viikko 12 | Viikko 16 | Viikko 52 | ||||||||
| Lumelääke | 150 mg | 300 mg | Etanersepti | 150 mg | 300 mg | Etanersepti | 150 mg | 300 mg | Etanersepti | |
| Potilaiden määrä | 324 | 327 | 323 | 323 | 327 | 323 | 323 | 327 | 323 | 323 |
| PASI 50 -vaste, n (%) | 49 (15,1 %) | 266 (81,3 %) | 296 (91,6 %) | 226 (70,0 %) | 290 (88,7 %) | 302 (93,5 %) | 257 (79,6 %) | 249 (76,1 %) | 274 (84,8 %) | 234 (72,4 %) |
| PASI 75 -vaste, n (%) | 16 (4,9 %) | 219 (67,0 %)** | 249 (77,1 %)** | 142 (44,0 %) | 247 (75,5 %) | 280 (86,7 %) | 189 (58,5 %) | 215 (65,7 %) | 254 (78,6 %) | 179 (55,4 %) |
| PASI 90 -vaste, n (%) | 5 (1,5 %) | 137 (41,9 %) | 175 (54,2 %) | 67 (20,7 %) | 176 (53,8 %) | 234 (72,4 %) | 101 (31,3 %) | 147 (45,0 %) | 210 (65,0 %) | 108 (33,4 %) |
| PASI 100 -vaste, n (%) | 0 (0 %) | 47 (14,4 %) | 78 (24,1 %) | 14 (4,3 %) | 84 (25,7 %) | 119 (36,8 %) | 24 (7,4 %) | 65 (19,9 %) | 117 (36,2 %) | 32 (9,9 %) |
| IGA mod 2011 oireeton tai lähes oireeton vaste, n (%) | 9 (2,8 %) | 167 (51,1 %)** | 202 (62,5 %)** | 88 (27,2 %) | 200 (61,2 %) | 244 (75,5 %) | 127 (39,3 %) | 168 (51,4 %) | 219 (67,8 %) | 120 (37,2 %) |
** p-arvot vs. etanersepti: p = 0,0250
Psoriaasilisätutkimuksessa (CLEAR) arvioitiin 676 potilasta. Tutkimuksessa saavutettiin sekukinumabi 300 mg annoksella ensisijainen ja toissijaiset päätetapahtumat (superioriteetti): sekukinumabiryhmässä useampi saavutti PASI 90 -vasteen viikolla 16 (ensisijainen päätetapahtuma), sai PASI 75 -vasteen nopeammin (mitattuna viikolla 4), sekä useammalla oli pitkäaikainen PASI 90 -vaste viikolla 52, verrattuna ustekinumabiryhmään. Sekukinumabin parempi teho ustekinumabiin verrattuna havaittiin aikaisessa vaiheessa ja jatkui viikolle 52 asti seuraavien päätemuuttujien osalta: PASI 75/90/100 -vasteen saavuttaneiden osuus, ja 0 tai 1 pistettä IGA mod 2011-asteikolla (”oireeton” tai ”lähes oireeton”) saaneiden osuus (taulukko 6).
Taulukko 6 Yhteenveto kliinisistä vasteista, CLEAR-tutkimus
| Viikko 4 | Viikko 16 | Viikko 52 | ||||
| Sekukinumabi 300 mg | Ustekinumabi* | Sekukinumabi 300 mg | Ustekinumabi* | Sekukinumabi 300 mg | Ustekinumabi* | |
| Potilaiden määrä | 334 | 335 | 334 | 335 | 334 | 335 |
| PASI 75 -vaste, n (%) | 166 (49,7%)** | 69 (20,6%) | 311 (93,1%) | 276 (82,4%) | 306 (91,6%) | 262 (78,2%) |
| PASI 90 -vaste, n (%) | 70 (21,0%) | 18 (5,4%) | 264 (79,0%)** | 192 (57,3%) | 250 (74,9%)*** | 203 (60,6%) |
| PASI 100 -vaste, n (%) | 14 (4,2%) | 3 (0,9%) | 148 (44,3%) | 95 (28,4%) | 150 (44,9%) | 123 (36,7%) |
| IGA mod 2011 oireeton tai lähes oireeton vaste, n (%) | 128 (38,3%) | 41 (12,2%) | 278 (83,2%) | 226 (67,5%) | 261 (78,1%) | 213 (63,6%) |
* Sekukinumabilla hoidetut potilaat saivat 300 mg annoksen viikoilla 0, 1, 2, 3 ja 4, sekä 4 viikon välein viikolle 52 asti. Ustekinumabilla hoidetut potilaat saivat 45 mg tai 90 mg annoksen viikoilla 0 ja 4, sekä 12 viikon välein viikolle 52 asti (annostelu painon mukaan hyväksytyn annostuksen mukaisesti)
** p‑arvot verrattuna ustekinumabiin: p<0,0001 ensisijaiselle päätetapahtumalle (PASI 90-vaste viikolla 16) ja toissijaiselle päätetapahtumalle (PASI 75-vaste viikolla 4)
*** p‑arvot verrattuna ustekinumabiin: p=0,0001 toissijaiselle päätetapahtumalle (PASI 90-vaste viikolla 52)
Sekukinumabi oli tehokas systeemisille ja biologisille hoidoille naiiveilla potilailla, aiemmin biologisilla lääkkeillä / TNF-salpaajilla hoidetuilla potilailla sekä potilailla, joilla hoito biologisilla lääkkeillä / TNF-salpaajilla ei ollut tehonnut. Potilailla, joilla oli samanaikaista psoriartriittia lähtötilanteessa, PASI75 vasteen saavuttaneiden potilaiden osuus oli samansuuruinen kuin läiskäpsoriaasipotilaiden kokonaispopulaatiossa.
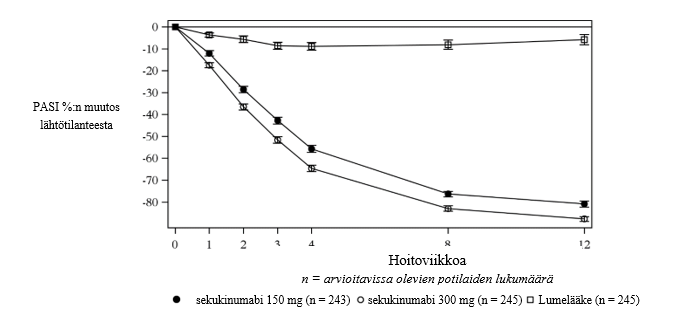
Sekukinumabin vaikutus alkoi nopeasti ja annoksella 300 mg keskimääräinen PASI-vaste pieneni 50 % viikkoon 3 mennessä.
Kuva 1 Keskimääräisen PASI-arvon prosentuaalinen muutos ajan suhteen lähtötilanteesta tutkimuksessa 1 (ERASURE)
Läiskäpsoriaasin erityiset esiintymismuodot
Kahdessa lumelääkekontrolloidussa lisätutkimuksessa havaittiin paranemista sekä kynsipsoriaasissa (TRANSFIGURE, 198 potilasta) että kämmenten ja jalkapohjien psoriaasissa (GESTURE, 205 potilasta). TRANSFIGURE-tutkimuksessa sekukinumabi oli lumelääkettä parempi viikolla 16 (46,1% 300 mg annoksella, 38,4% 150 mg annoksella ja 11,7% lumelääkkeellä) arvioitaessa merkitsevää paranemista lähtötilanteeseen verrattuna Nail Psoriasis Severity Index (NAPSI %) –asteikolla potilailla, jotka sairastivat keskivaikeaa tai vaikeaa läiskäpsoriaasia, johon liittyy kynsioireita. GESTURE-tutkimuksessa sekukinumabi oli lumelääkettä parempi viikolla 16 (33,3% 300 mg annoksella, 22,1% 150 mg annoksella ja 1,5% lumelääkkeellä) arvioitaessa merkitsevää paranemista 0- tai 1-vasteessa ppIGA mod 2011-asteikolla (”oireeton” tai ”lähes oireeton”) potilailla, jotka sairastivat keskivaikeaa tai vaikeaa kämmenten ja jalkapohjien psoriaasia.
Lumelääkekontrolloidussa tutkimuksessa arvioitiin 102 potilasta, joilla oli keskivaikea tai vaikea päänahan psoriaasi (oirepisteet asteikoilla Psoriasis Scalp Severity Index (PSSI) ≥12, IGA mod 2011 päänahan osalta ≥3 ja vähintään 30 % päänahasta on psoriaasin peitossa). Sekukinumabi 300 mg oli lumelääkettä parempi viikolla 12, mikä näkyi merkitsevänä paranemisena lähtötilanteeseen verrattuna niiden potilaiden osuudessa, jotka saavuttivat PSSI 90 -vasteen (52,9 % 300 mg annoksella ja 2,0 % lumelääkkeellä) ja niiden potilaiden osuudessa, jotka saivat 0 tai 1 pistettä päänahan IGA mod 2011 -asteikolla (56,9 % 300 mg annoksella ja 5,9% lumelääkkeellä). Molempien päätetapahtumien tulokset säilyivät sekukinumabihoitoa saavilla potilailla viikolle 24 asti.
Elämänlaatu / potilaiden ilmoittamat vaikutukset
DLQI-mittarilla (Dermatology Life Quality Index) osoitettiin tilastollisesti merkitsevää paranemista lähtötilanteesta viikolla 12 verrattuna lumelääkkeeseen (tutkimukset 1−4). Viikolla 12 todettu keskimääräinen pisteiden alenema (paraneminen) DLQI-pistearvoissa lähtötilanteesta vaihteli arvosta ‑10,4 arvoon -11,6 sekukinumabin 300 mg:n annoksella, arvosta ‑7,7 arvoon ‑10,1 sekukinumabin 150 mg:n annoksella ja arvosta ‑1,1 arvoon ‑1,9 lumelääkkeellä. Nämä tulokset säilyivät 52 viikon ajan (tutkimukset 1 ja 2).
Tutkimuksiin 1 ja 2 osallistuneista 40 % täytti psoriaasin oirepäiväkirjaa (Psoriasis Symptom Diary©). Kussakin näistä tutkimuksista päiväkirjaa täyttäneillä tutkittavilla osoitettiin lumelääkkeeseen nähden tilastollisesti merkitsevää paranemista viikolla 12 potilaiden ilmoittamien oireiden suhteen, joita olivat kutina, kipu ja hilseily.
DLQI-mittarilla osoitettiin tilastollisesti merkitsevää paranemista lähtötilanteesta sekukinumabihoitoa saaneilla potilailla verrattuna ustekinumabihoitoa (CLEAR) saaneisiin potilaisiin viikolla 4. Tulokset säilyivät 52 viikon ajan.
Psoriaasin oirepäiväkirjalla osoitettiin tilastollisesti merkitsevää paranemista potilaan itse raportoimissa oireissa (kutina, kipu ja hilseily) sekukinumabihoitoa saaneilla potilailla verrattuna ustekinumabihoitoa saaneisiin potilaisiin viikolla 16 ja 52 (CLEAR).
Päänahan psoriaasitutkimuksessa potilaan itse raportoimissa oireissa (päänahan kutina, kipu ja hilseily) osoitettiin tilastollisesti merkitsevää paranemista (vähenemistä) lähtötilanteesta verrattuna lumelääkettä saaneisiin potilaisiin viikolla 12.
Pediatriset potilaat
Läiskäpsoriaasi pediatrisilla potilailla
Sekukinumabin on osoitettu lievittävän oireita ja löydöksiä ja parantavan terveyteen liittyvää elämänlaatua vähintään 6-vuotiailla pediatrisilla läiskäpsoriaasipotilailla (ks. taulukot 8 ja 10).
Vaikea läiskäpsoriaasi
Sekukinumabin tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa, lume- ja etanerseptikontrolloidussa vaiheen III tutkimuksessa 6 – < 18-vuotiailla vaikeaa läiskäpsoriaasia sairastavilla pediatrisilla potilailla, joille harkittiin systeemistä hoitoa. Läiskäpsoriaasi määriteltiin vaikeaksi, jos PASI-arvo oli ≥ 20, IGA mod 2011 ‑aste 4 ja oireiden kattavuus ≥ 10 % ihon pinta-alasta. Potilaista noin 43 % oli saanut aiemmin valohoitoa, 53 % tavanomaista systeemistä hoitoa ja 3 % biologisia lääkkeitä ja 9 %:lla oli samanaikainen nivelpsoriaasi.
Pediatrisessa psoriaasitutkimuksessa 1 arvioitiin 162 potilasta, jotka satunnaistettiin saamaan pieniannoksista sekukinumabia (75 mg < 50 kg painaville ja 150 mg ≥ 50 kg painaville), suuriannoksista sekukinumabia (75 mg < 25 kg painaville, 150 mg ≥ 25 kg – < 50 kg painaville ja 300 mg ≥ 50 kg painaville) tai lumelääkettä viikoilla 0, 1, 2, 3 ja 4 ja tämän jälkeen samaa annosta 4 viikon välein tai etanerseptia. Etanerseptiryhmään satunnaistettujen potilaiden annostus oli 0,8 mg/kg viikossa (enintään 50 mg). Potilaiden paino- ja ikäjakauma satunnaistamisvaiheessa on esitetty taulukossa 7.
Taulukko 7 Potilaiden paino- ja ikäjakauma pediatrisessa psoriaasitutkimuksessa 1
| Satunnaistamisen osite | Kuvaus | Sekukinumabi, pieni annos n = 40 | Sekukinumabi, suuri annos n = 40 | Lumelääke n = 41 | Etanersepti n = 41 | Yhteensä N = 162 |
| Ikä | 6 – < 12 v | 8 | 9 | 10 | 10 | 37 |
| ≥ 12 – < 18 v | 32 | 31 | 31 | 31 | 125 | |
| Paino | < 25 kg | 2 | 3 | 3 | 4 | 12 |
| ≥ 25 – < 50 kg | 17 | 15 | 17 | 16 | 65 | |
| ≥ 50 kg | 21 | 22 | 21 | 21 | 85 |
Lumelääkeryhmään satunnaistetut potilaat, jotka eivät olleet saaneet vastetta viikkoon 12 mennessä, siirrettiin joko pieniannoksisen tai suuriannoksisen sekukinumabihoidon ryhmään (annos perustui painoryhmään). Tutkimuslääkettä annettiin viikoilla 12, 13, 14 ja 15, ja sen jälkeen samaa annosta annettiin 4 viikon välein viikosta 16 lähtien. Rinnakkaisia ensisijaisia päätemuuttujia olivat niiden potilaiden osuus, jotka saavuttivat PASI 75 ‑vasteen ja IGA mod 2011 ‑asteikon mukaisen tuloksen oireeton tai lähes oireeton (0 tai 1) viikolla 12.
Pieni- ja suuriannoksisen sekukinumabihoidon teho oli samankaltainen rinnakkaisten ensisijaisten päätemuuttujien osalta 12-viikkoisen lumekontrolloidun vaiheen aikana. Kummankin sekukinumabiannoksen kannalta suotuisat ristitulosuhde-estimaatit olivat tilastollisesti merkitseviä PASI 75 ‑vasteen ja IGA mod 2011 ‑vasteen (tulos 0 tai 1) osalta.
Tehoa ja turvallisuutta seurattiin kaikilla potilailla 52 viikon ajan ensimmäisen annoksen jälkeen. PASI 75- ja IGA mod 2011 ‑asteikon oireeton tai lähes oireeton (0 tai 1) -vasteet saavuttaneiden potilaiden osuudessa havaittiin eroavaisuutta sekukinumabihoitoryhmien ja lumeryhmän välillä ensimmäisellä lähtötilanteen jälkeisellä käynnillä viikon 4 kohdalla. Ero oli suurentunut viikon 12 kohdalla. Vaste säilyi koko 52 viikon jakson ajan (ks. taulukko 8). Myös suurempi osuus PASI 50-, PASI 90- ja PASI 100 ‑vasteita ja CDLQI-pistemäärä (Children’s Dermatology Life Quality Index) 0 tai 1 -tuloksia säilyi koko 52 viikon ajanjakson ajan.
Lisäksi PASI 75-, IGA 0 tai 1- ja PASI 90 ‑vasteiden osuudet viikkojen 12 ja 52 kohdalla olivat sekä pieniannoksisen että suuriannoksisen sekukinumabihoidon ryhmässä suuremmat kuin etanerseptiryhmässä (ks. taulukko 8).
Viikon 12 jälkeen teho pieniannoksisen ja suuriannoksisen sekukinumabihoidon ryhmissä oli samankaltainen, mutta suuren annoksen teho oli suurempi ≥ 50 kg painavilla potilailla. Pienen annoksen ja suuren annoksen turvallisuusprofiilit olivat samankaltaiset ja yhdenmukaiset aikuisten turvallisuusprofiilin kanssa.
Taulukko 8 Yhteenveto kliinisestä vasteesta vaikean pediatrisen psoriaasin hoidossa viikkojen 12 ja 52 kohdalla (pediatrinen psoriaasitutkimus 1)*
| Vaste-kriteeri | Hoitovertailu | Tutkimusvalmiste | Verrokki | Ristitulosuhde- | |
| Tutkimusvalmiste vs. verrokki | n**/m (%) | n**/m (%) | estimaatti (95 % lv) | p-arvo | |
| Viikon 12 kohdalla*** | |||||
| PASI 75 | Sekukinumabi, pieni annos vs. lumelääke | 32/40 (80,0) | 6/41 (14,6) | 25,78 (7,08; 114,66) | < 0,0001 |
| Sekukinumabi, suuri annos vs. lumelääke | 31/40 (77,5) | 6/41 (14,6) | 22,65 (6,31; 98,93) | < 0,0001 | |
| Sekukinumabi, pieni annos vs. etanersepti | 32/40 (80,0) | 26/41 (63,4) | 2,25 (0,73; 7,38) | ||
| Sekukinumabi, suuri annos vs. etanersepti | 31/40 (77,5) | 26/41 (63,4) | 1,92 (0,64; 6,07) | ||
| IGA 0/1 | Sekukinumabi, pieni annos vs. lumelääke | 28/40 (70,0) | 2/41 (4,9) | 51,77 (10,02; 538,64) | < 0,0001 |
| Sekukinumabi, suuri annos vs. lumelääke | 24/40 (60,0) | 2/41 (4,9) | 32,52 (6,48; 329,52) | < 0,0001 | |
| Sekukinumabi, pieni annos vs. etanersepti | 28/40 (70,0) | 14/41 (34,1) | 4,49 (1,60; 13,42) | ||
| Sekukinumabi, suuri annos vs. etanersepti | 24/40 (60,0) | 14/41 (34,1) | 2,86 (1,05; 8,13) | ||
| PASI 90 | Sekukinumabi, pieni annos vs. lumelääke | 29/40 (72,5) | 1/41 (2,4) | 133,67 (16,83; 6 395,22) | < 0,0001 |
| Sekukinumabi, suuri annos vs. lumelääke | 27/40 (67,5) | 1/41 (2,4) | 102,86 (13,22; 4 850,13) | < 0,0001 | |
| Sekukinumabi, pieni annos vs. etanersepti | 29/40 (72,5) | 12/41 (29,3) | 7,03 (2,34; 23,19) | ||
| Sekukinumabi, suuri annos vs. etanersepti | 27/40 (67,5) | 12/41 (29,3) | 5,32 (1,82; 16,75) | ||
| Viikon 52 kohdalla | |||||
| PASI 75 | Sekukinumabi, pieni annos vs. etanersepti | 35/40 (87,5) | 28/41 (68,3) | 3,12 (0,91; 12,52) | |
| Sekukinumabi, suuri annos vs. etanersepti | 35/40 (87,5) | 28/41 (68,3) | 3,09 (0,90; 12,39) | ||
| IGA 0/1 | Sekukinumabi, pieni annos vs. etanersepti | 29/40 (72,5) | 23/41 (56,1) | 2,02 (0,73; 5,77) | |
| Sekukinumabi, suuri annos vs. etanersepti | 30/40 (75,0) | 23/41 (56,1) | 2,26 (0,81; 6,62) | ||
| PASI 90 | Sekukinumabi, pieni annos vs. etanersepti | 30/40 (75,0) | 21/41 (51,2) | 2,85 (1,02; 8,38) | |
| Sekukinumabi, suuri annos vs. etanersepti | 32/40 (80,0) | 21/41 (51,2) | 3,69 (1,27; 11,61) | ||
* Arvojen puuttuminen tulkittiin siten, että vastetta ei saatu. ** n = vasteen saaneiden lukumäärä, m = arvioitavissa olevien potilaiden lukumäärä *** Laajennettu käynnin aikaikkuna viikon 12 kohdalla. Ristitulosuhde, 95 % luottamusväli ja p-arvo perustuvat eksaktiin logistiseen regressiomalliin, jossa tekijöitä olivat hoitoryhmä, lähtötilanteen painoluokka ja ikäluokka. | |||||
Terveyteen liittyvän elämänlaadun paranemista (mittarina CDLQI-pistemäärä 0 tai 1) ilmoitettiin suuremmalla määrällä sekukinumabia saaneista pediatrisista potilaista kuin lumelääkettä saaneista viikon 12 kohdalla (pienen annoksen ryhmässä 44,7 %, suuren annoksen ryhmässä 50 % ja lumelääkeryhmässä 15 %). Molemmat sekukinumabiannosryhmät olivat 52 viikon ajan numeerisesti suurempia kuin etanerseptiryhmä (pienen annoksen ryhmä 60,6 %, suuren annoksen ryhmä 66,7 % ja etanerseptiryhmä 44,4 %).
Keskivaikea tai vaikea läiskäpsoriaasi
Sekukinumabin ennakoitiin olevan tehokas pediatristen potilaiden keskivaikean läiskäpsoriaasin hoidossa aikuispotilaiden keskivaikean ja vaikean läiskäpsoriaasin hoidossa osoitetun tehon ja altistus-vastesuhteen perusteella ja koska taudinkulku, patofysiologia ja lääkkeen vaikutus ovat samojen altistustasojen osalta aikuisilla ja pediatrisilla potilailla samankaltaiset.
Lisäksi sekukinumabin tehoa ja turvallisuutta arvioitiin avoimessa, kaksiryhmäisessä, rinnakkaisryhmillä toteutetussa vaiheen III monikeskustutkimuksessa 6 – < 18-vuotiailla keskivaikeaa tai vaikeaa läiskäpsoriaasia (PASI-arvo ≥ 12, IGA mod 2011‑arvo ≥ 3 ja oireiden kattavuus ≥ 10 % ihon pinta-alasta) sairastavilla pediatrisilla potilailla, joille harkittiin systeemistä hoitoa.
Pediatrisessa psoriaasitutkimuksessa 2 arvioitiin 84 potilasta, jotka satunnaistettiin saamaan pieniannoksista sekukinumabia (75 mg < 50 kg painaville ja 150 mg ≥ 50 kg painaville) tai suuriannoksista sekukinumabia (75 mg < 25 kg painaville, 150 mg ≥ 25 kg – < 50 kg painaville ja 300 mg ≥ 50 kg painaville) viikoilla 0, 1, 2, 3 ja 4 ja tämän jälkeen samaa annosta 4 viikon välein. Potilaiden paino- ja ikäjakauma satunnaistamisvaiheessa on esitetty taulukossa 9.
Taulukko 9 Potilaiden paino- ja ikäjakauma pediatrisessa psoriaasitutkimuksessa 2
| Alaryhmät | Kuvaus | Sekukinumabi, pieni annos n = 42 | Sekukinumabi, suuri annos n = 42 | Yhteensä N = 84 |
| Ikä | 6 – < 12 v | 17 | 16 | 33 |
| ≥ 12 – < 18 v | 25 | 26 | 51 | |
| Paino | < 25 kg | 4 | 4 | 8 |
| ≥ 25 – < 50 kg | 13 | 12 | 25 | |
| ≥ 50 kg | 25 | 26 | 51 |
Rinnakkaisia ensisijaisia päätemuuttujia olivat niiden potilaiden osuus, jotka saavuttivat PASI 75 ‑vasteen ja IGA mod 2011 ‑asteikon mukaisen tuloksen oireeton tai lähes oireeton (0 tai 1) viikon 12 kohdalla.
Sekukinumabin pienen annoksen ja suuren annoksen teho oli samankaltainen ja rinnakkaisten ensisijaisten päätemuuttujien osalta tilastollisesti merkitsevästi parempi kuin vertailuna käytetty historiallinen lumelääkevaste. Positiivisen hoitovaikutuksen arvioitu posteriorinen todennäköisyys oli 100 %.
Tehoa seurattiin potilailla 52 viikon ajan ensimmäisen antokerran jälkeen. Teho (määritelmä PASI 75 ‑vaste ja IGA mod 2011 ‑tulos oireeton tai lähes oireeton [0 tai 1]) havaittiin jo ensimmäisellä lähtötilanteen jälkeisellä käynnillä viikon 2 kohdalla, ja PASI 75 ‑vasteen ja IGA mod 2011 ‑tuloksen oireeton tai lähes oireeton (0 tai 1) saavuttaneiden potilaiden osuus suureni 24 viikon ajanjakson ajan säilyen viikolle 52 asti. Myös PASI 90- ja PASI 100 ‑vasteiden lisääntymistä havaittiin viikon 12 kohdalla, ja lisääntyminen jatkui 24 viikon ajanjakson ajan säilyen viikolle 52 asti (ks. taulukko 10).
Pienen annoksen ja suuren annoksen turvallisuusprofiilit olivat samankaltaiset ja yhdenmukaiset aikuisten turvallisuusprofiilin kanssa.
Taulukko 10 Yhteenveto kliinisestä vasteesta keskivaikean ja vaikean pediatrisen psoriaasin hoidossa viikkojen 12 ja 52 kohdalla (pediatrinen psoriaasitutkimus 2)*
| Viikko 12 | Viikko 52 | |||
Sekukinumabi, pieni annos | Sekukinumabi, suuri annos | Sekukinumabi, pieni annos | Sekukinumabi, suuri annos | |
| Potilasmäärä | 42 | 42 | 42 | 42 |
| PASI 75 ‑vaste n (%) | 39 (92,9 %) | 39 (92,9 %) | 37 (88,1 %) | 38 (90,5 %) |
| IGA mod 2011 ‑tulos oireeton tai lähes oireeton n (%) | 33 (78,6 %) | 35 (83,3 %) | 36 (85,7 %) | 35 (83,3 %) |
| PASI 90 ‑vaste n (%) | 29 (69 %) | 32 (76,2 %) | 32 (76,2 %) | 35 (83,3 %) |
| PASI 100 ‑vaste n (%) | 25 (59,5 %) | 23 (54,8 %) | 22 (52,4 %) | 29 (69,0 %) |
| * Arvojen puuttuminen tulkittiin siten, että vastetta ei saatu. | ||||
Tulokset pediatristen potilaiden keskivaikean ja vaikean läiskäpsoriaasin hoidossa vahvistivat edellä mainittuja ennakko-oletuksia, jotka perustuivat aikuispotilaiden hoidossa osoitettuun tehoon ja altistus-vastesuhteeseen.
Pienen annoksen ryhmässä CDLQI-pistemäärän 0 tai 1 saavutti viikon 12 kohdalla 50 % potilaista ja viikon 52 kohdalla 70,7 % potilaista. Suuren annoksen ryhmässä CDLQI-pistemäärän 0 tai 1 saavutti viikon 12 kohdalla 61,9 % potilaista ja viikon 52 kohdalla 70,3 % potilaista.
Juveniili idiopaattinen artriitti (JIA)
Entesiitteihin liittyvä artriitti (ERA) ja juveniili nivelpsoriaasi (JPsA)
Sekukinumabin tehoa ja turvallisuutta arvioitiin kolmiosaisessa, kaksoisokkoutetussa, lumekontrolloidussa, tapahtumalähtöisessä, satunnaistetussa vaiheen III tutkimuksessa 86 potilaalla (ikä 2 – < 18 vuotta), joilla oli aktiivinen entesiitteihin liittyvä artriitti (ERA) tai juveniili nivelpsoriaasi (JPsA). Diagnoosi perustui juveniilia idiopaattista artriittia (JIA) koskeviin muokattuihin International League of Associations for Rheumatology (ILAR) -liiton luokittelukriteereihin. Tutkimukseen kuului avoin osio (osa 1), jossa kaikki potilaat saivat sekukinumbia viikolle 12 saakka. Potilaat, joilla todettiin JIA ACR 30 -vaste viikolla 12 siirtyivät kaksoissokkoutettuun vaiheeseen (osa 2). Siinä heidät satunnaistettiin suhteessa 1:1 joko jatkamaan sekukinumabihoitoa tai saamaan lumehoitoa (hoidon satunnaistettu lopettaminen) viikolle 104 saakka tai pahenemisvaiheen puhkeamiseen saakka. Potilaat, joilla esiintyi pahenemisvaihe, siirtyivät avoimeen tutkimukseen (osa 3) saamaan sekukinumabihoitoa viikolle 104 saakka.
Potilaiden JIA-alatyypit tutkimukseenottovaiheessa olivat seuraavanlaiset: 60,5 %:lla oli ERA ja 39,5 %:lla JPsA, ja vähintään yksi taudin etenemiseen vaikuttava reumalääke tai vähintään yksi tulehduskipulääke oli joko tuottanut riittämättömän vasteen tai ollut huonosti siedetty. Lähtötilanteessa metotreksaatin käyttöä ilmoitettiin 65,1 %:lla potilaista; (63,5 %:lla [33/52] ERA-potilaista ja 67,6 %:lla [23/34] JPsA-potilaista). Samanaikaista sulfasalatsiinihoitoa sai 12 ERA-potilasta 52:sta (23,1 %). Lähtötilanteessa < 50 kg painaneet potilaat (n = 30) saivat 75 mg:n annoksen ja ≥ 50 kg painaneet potilaat (n = 56) 150 mg:n annoksen. Ikä oli lähtötilanteessa 2–17 vuotta; 3 potilasta oli 2 – < 6-vuotiaita, 22 potilasta 6 – < 12-vuotiaita ja 61 potilasta 12 – < 18-vuotiaita. Lähtötilanteen JADAS-27-pistemäärä (Juvenile Arthritis Disease Activity Score) oli 15,1 (keskihajonta: 7,1).
Ensisijainen päätetapahtuma oli pahenemisvaiheen puhkeamiseen kulunut aika hoidon satunnaistetussa lopettamisvaiheessa (osa 2). Taudin pahenemisvaiheen määritelmänä oli ≥ 30 % pahenema vähintään kolmen JIA ACR -vastekriteerin osalta kuudesta ja ≥ 30 % paranema enintään yhden JIA ACR -vastekriteerin osalta kuudesta ja vähintään kaksi aktiivista niveltä.
Osan 1 lopussa 75 potilaalla 86:sta (87,2 %) esiintyi JIA ACR 30 -vaste, ja he siirtyivät osaan 2.
Tutkimuksessa saavutettiin ensisijainen päätetapahtuma, sillä taudin pahenemisvaiheen puhkeamiseen kulunut aika osassa 2 oli tilastollisesti merkitsevästi pidempi sekukinumabihoitoa saaneilla kuin lumehoitoa saaneilla. Pahenemisvaiheen puhkeamisriski oli tutkimuksen osassa 2 sekukinumabia saaneilla 72 % pienempi kuin lumehoitoa saaneilla (riskitiheyssuhde = 0,28, 95 % lv: 0,13–0,63, p < 0,001) (kuva 2 ja taulukko 11). Osassa 2 pahenemisvaihe esiintyi kaikkiaan 21:llä lumehoitoa saaneista (11 JPsA-potilaalla ja 10 ERA-potilaalla) ja 10:llä sekukinumabia saaneista (4 JPsA-potilaalla ja 6 ERA-potilaalla).
Kuva 2 Kaplan–Meier-estimaatit taudin pahenemisvaiheen puhkeamiseen kuluvasta ajasta tutkimuksen osassa 2
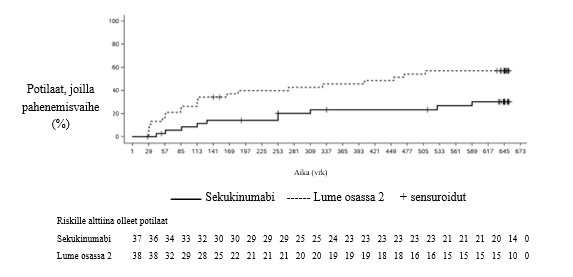
Taulukko 11 Elossaoloanalyysi taudin pahenemisvaiheeseen kuluneen ajan osalta – tutkimuksen osa 2
Sekukinumabi (N = 37) | Lumelääke tutkimuksen osassa 2 (N = 38) | |
| Pahenemisvaiheiden määrä tutkimuksen osan 2 lopussa, n (%) | 10 (27,0) | 21 (55,3) |
| Kaplan–Meier-estimaatit: | ||
| Mediaani, vrk (95 % lv) | NC (NC, NC) | 453,0 (114,0; NC) |
| Osuus potilaista, joilla ei pahenemisvaiheita 6 kk kohdalla (95 % lv) | 85,8 (69,2, 93,8) | 60,1 (42,7, 73,7) |
| Osuus potilaista, joilla ei pahenemisvaiheita 12 kk kohdalla (95 % lv) | 76,7 (58,7, 87,6) | 54,3 (37,1, 68,7) |
| Osuus potilaista, joilla ei pahenemisvaiheita 18 kk kohdalla (95 % lv) | 73,2 (54,6, 85,1) | 42,9 (26,7, 58,1) |
| Riskitiheyssuhde vs. lume: Estimaatti (95 % lv) | 0,28 (0,13, 0,63) | |
| Stratifioidun log-rank-testin p-arvo | < 0,001** | |
Analyysi tehtiin kaikkien sellaisten satunnaistettujen potilaiden osalta, jotka saivat vähintään yhden annoksen tutkimuslääkettä tutkimuksen osassa 2. Sekukinumabi: kaikki potilaat, jotka eivät saaneet lainkaan lumetta. Lume tutkimuksen osassa 2: kaikki potilaat, jotka saivat lumetta tutkimuksen osassa 2 ja sekukinumabia tutkimuksen muussa vaiheessa / muissa vaiheissa. NC = Ei laskettavissa (non calculable). ** = Tilastollinen merkitsevyys yksitahoisella 0,025:n merkitsevyystasolla. | ||
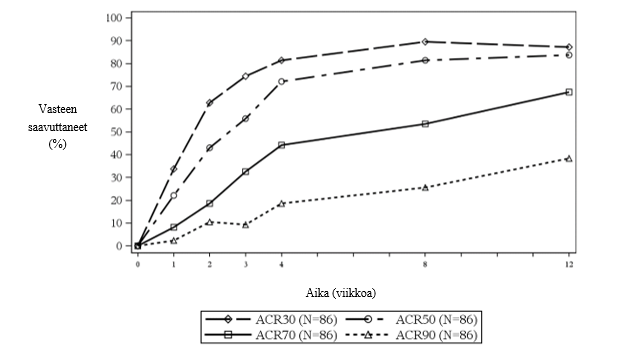
Tutkimuksen avoimessa osassa 1 kaikki potilaat saivat sekukinumbia viikolle 12 saakka. Viikolla 12 JIA ACR 50 -vasteen saavutti 83,7 % lapsista, JIA ACR 70 -vasteen 67,4 % lapsista ja JIA ACR 90 ‑vasteen 38,4 % lapsista (kuva 3). Sekukinumabin vaikutus alkoi jo viikolla 1. Viikolla 12 JADAS-27-pistemäärä oli 4,64 (keskihajonta: 4,73). JADAS-27-pisteiden keskiarvon lasku lähtötilanteesta oli -10,487 (keskihajonta: 7,23).
Kuva 3Tutkittavien JIA ACR 30/50/70/90 -vaste viikkoon 12 mennessä tutkimuksen osassa 1*
* Arvojen puuttuminen tulkittiin siten, että vastetta ei saatu.
Tiedot 2 - < 6-vuotiaiden ikäryhmästä ovat epävarmoja, koska tutkimukseen osallistui vähäinen määrä alle 6-vuotiaita potilaita.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Cosentyx-valmisteen käytöstä vastasyntyneistä alle 6-vuotiaisiin ulottuvien pediatristen potilasryhmien läiskäpsoriaasin hoidossa ja vastasyntyneistä alle 2-vuotiaisiin ulottuvien pediatristen potilasryhmien kroonisen idiopaattisen artriitin hoidossa (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Useimmat havaitut farmakokineettiset ominaisuudet olivat samankaltaiset sekä läiskäpsoriaasissa, nivelpsoriaasissa että selkärankareumassa.
Pediatriset potilaat
Läiskäpsoriaasi
Kahdessa pediatrisessa tutkimuksessa keskivaikeaa tai vaikeaa läiskäpsoriaasia sairastaville potilaille (ikä 6 – < 18 v) annettiin sekukinumabia suositellun pediatrisen hoito-ohjelman mukaisesti. Viikon 24 kohdalla ≥ 25 kg – < 50 kg painavien potilaiden vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli 19,8 ± 6,96 mikrog/ml (n = 24) heidän saatuaan 75 mg sekukinumabia, ja ≥ 50 kg painavien potilaiden minimipitoisuuksien keskiarvo ± keskihajonta oli 27,3 ± 10,1 mikrog/ml (n = 36) heidän saatuaan 150 mg sekukinumabia. Alle 25 kg painavien potilaiden (n = 8) vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli viikon 24 kohdalla 32,6 ± 10,8 mikrog/ml 75 mg:n annoksen jälkeen.
Juveniili idiopaattinen artriitti
Pediatrisessa tutkimuksessa sekukinumabia annettiin entesiitteihin liittyvää artriittia tai juveniilia nivelpsoriaasia sairastaville potilaille (ikä 2 – < 18 v) suositellun pediatrisen hoito-ohjelman mukaisesti. Viikon 24 kohdalla < 50 kg painavien potilaiden vakaan tilan minimipitoisuuksien keskiarvo ± keskihajonta oli 25,2 ± 5,45 mikrog/ml (n = 10) ja ≥ 50 kg painavien potilaiden 27,9 ± 9,57 mikrog/ml (n = 19).
Aikuiset
Imeytyminen
Terveille vapaaehtoisille nestemäisenä lääkemuotona ihon alle annetun 300 mg:n kerta-annoksen jälkeen sekukinumabin huippupitoisuus seerumissa oli 43,2 ± 10,4 mikrog/ml, kun annoksen antamisesta oli kulunut 2⎼ 14 vuorokautta.
Populaatiofarmakokineettisen analyysin perusteella läiskäpsoriaasipotilaille kerta-annoksena ihon alle annetun 150 mg:n annoksen jälkeen sekukinumabin huippupitoisuus seerumissa oli 13,7 ± 4.8 mikrog/ml ja 300 mg:n annoksen jälkeen 27,3 ± 9,5 mikrog/ml, kun annoksen antamisesta oli kulunut 5⎼ 6 vuorokautta.
Populaatiofarmakokineettisen analyysin perusteella ensimmäisen kuukauden aikana annettujen aloitusvaiheen viikoittaisten annosten jälkeen aika maksimipitoisuuden saavuttamiseen oli 31‑34 vuorokautta.
Simuloidun tiedon perusteella vakaan tilan huippupitoisuus (Cmax,ss) ihon alle annetun 150 mg:n annoksen jälkeen oli 27,6 mikrog/ml ja 300 mg:n annoksen jälkeen 55,2 mikrog/ml. Populaatiofarmakokineettinen analyysi viittaa siihen, että vakaa tila saavutetaan 20 viikon kuluttua, kun käytetään kuukausittain annettavia annoksia.
Populaatiofarmakokineettinen analyysi osoitti, että ylläpitohoidon aikana kuukausittain annetun lääkkeen huippupitoisuus seerumissa ja pitoisuuspinta-ala (AUC) suurenivat kaksinkertaisiksi verrattuna kerta-annoksen jälkeen potilailla todettuun altistukseen.
Populaatiofarmakokineettinen analyysi osoitti, että sekukinumabi imeytyi siten, että sen absoluuttinen biologinen hyötyosuus läiskäpsoriaasipotilailla oli keskimäärin 73 %. Kaikissa tutkimuksissa laskettiin absoluuttisen biologisen hyötyosuuden arvoja, jotka olivat 60⎼77 %.
Populaatiofarmakokineettisen mallin perusteella sekukinumabin hyötyosuus nivelpsoriaasipotilailla oli 85 %.
Yksittäisen 300 mg esitäytetyllä ruiskulla annetun ihonalaisen injektion systeeminen sekukinumabialtistus läiskäpsoriaasipotilailla oli samanlainen kuin aiemmin kahdella 150 mg injektiolla havaittu.
Jakautuminen
Läiskäpsoriaasipotilailla kerta-annoksena laskimoon annon jälkeen keskimääräinen loppuvaiheen jakautumistilavuus (Vz) oli 7,10⎼8,60 litraa, mikä viittaa siihen, että sekukinumabi jakautuu vain vähäisessä määrin kehon ääreisosiin.
Biotransformaatio
Suurin osa IgG:stä poistuu solunsisäisen pilkkoutumisen kautta nestefaasin tai reseptorivälitteisen endosytoosin jälkeen.
Eliminaatio
Läiskäpsoriaasipotilailla kerta-annoksena laskimoon annon jälkeen keskimääräinen systeeminen puhdistuma (CL) oli 0,13⎼0,36 l/vrk. Populaatiofarmakokineettisessä analyysissä keskimääräinen systeeminen puhdistuma (CL) läiskäpsoriaasipotilailla oli 0,19 l/vrk. Sukupuoli ei vaikuttanut systeemiseen puhdistumaan. Puhdistuma oli annoksesta ja ajasta riippuvainen.
Populaatiofarmakokineettisen analyysin perusteella arvioitu keskimääräinen eliminaation puoliintumisaika läiskäpsoriaasipotilailla oli 27 vuorokautta ja vaihteli 18:sta 46:een vuorokauteen psoriaasitutkimuksissa, joissa lääke annettiin laskimoon.
Lineaarisuus/ei-lineaarisuus
Sekukinumabin yksittäisen ja toistuvien annosten farmakokinetiikkaa läiskäpsoriaasipotilailla selvitettiin useissa tutkimuksissa laskimoon annetuilla annoksilla, jotka vaihtelivat annoksesta 1 x 0,3 mg/kg annokseen 3 x 10 mg/kg, ja ihon alle annetuilla annoksilla, jotka vaihtelivat annoksesta 1 x 25 mg toistuviin 300 mg:n annoksiin. Altistus muuttui annoksen mukaan kaikilla annostuksilla.
Erityisryhmät
Potilaat, joilla on munuaisten tai maksan vajaatoiminta
Farmakokineettisiä tietoja ei ole saatavilla potilaista, joilla on munuaisten tai maksan vajaatoiminta. Sekukinumabi on monoklonaalinen IgG-vasta-aine ja pilkkoutumattoman sekukinumabin munuaispuhdistuman odotetaan olevan vähäistä ja vähämerkityksistä. IgG eliminoituu pääasiassa pilkkoutumisen kautta eikä maksan vajaatoiminnan odoteta vaikuttavan sekukinumabin puhdistumaan.
Painon vaikutus farmakokinetiikkaan
Sekukinumabin puhdistuma ja jakautumistilavuus kasvavat painon kasvaessa.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta ja lisääntymistoksisuutta koskevien konventionaalisten tutkimusten tulokset tai kudosten ristireagointia koskevien tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille (aikuisille tai pediatrisille potilaille).
Eläimillä ei ole tehty tutkimuksia, joissa olisi arvioitu sekukinumabin karsinogeenisuutta.
Farmaseuttiset tiedot
Apuaineet
Trehaloosidihydraatti
Histidiini
Histidiinihydrokloridimonohydraatti
Metioniini
Polysorbaatti 80
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
2 vuotta
Tarvittaessa Cosentyx-valmistetta voidaan säilyttää kertaluonteisesti jääkaapin ulkopuolella, huoneenlämmössä (alle 30 °C) korkeintaan 4 päivää.
Säilytys
Säilytä jääkaapissa (2 °C - 8 °C). Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
COSENTYX injektioneste, liuos, esitäytetty ruisku
75 mg (L:ei) 1 kpl (turvaneula, 0,5 ml (150 mg/ml)) (325,35 €)
PF-selosteen tieto
Cosentyx 75 mg injektioneste esitäytetty ruisku toimitetaan esitäytetyssä 0,5 ml:n lasiruiskussa, jossa on silikonilla päällystetty, bromobutyylikumista valmistettu männän pysäytin, kiinteä 27G x ½″ neula ja styreenibutadieenikuminen kova neulansuojus sekä polykarbonaatista valmistettu automaattinen neulanpistosuoja.
Cosentyx 75 mg injektioneste esitäytetty ruisku on saatavilla yksikköpakkauksissa, joissa on 1 esitäytetty ruisku, ja monipakkauksissa, joissa on 3 (3 yhden ruiskun pakkausta) esitäytettyä ruiskua.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Liuos on kirkas ja väritön tai hieman kellertävä neste.
Käyttö- ja käsittelyohjeet
Cosentyx 75 mg injektioneste toimitetaan kerta-antoon tarkoitetussa esitäytetyssä ruiskussa, joka on tarkoitettu henkilökohtaiseen käyttöön. Ota ruisku jääkaapista 20 minuuttia ennen pistämistä ja anna sen lämmetä huoneenlämpöiseksi.
Esitäytetty ruisku tulisi tarkistaa silmämääräisesti ennen käyttöä. Nesteen pitää olla kirkasta. Väri voi vaihdella värittömästä hieman kellertävään. Saatat havaita pienen ilmakuplan, mikä on normaalia. Älä käytä, jos neste sisältää selvästi näkyviä hiukkasia, on sameaa tai selvästi ruskeaa.
Yksityiskohtaiset käyttöohjeet on kerrottu pakkausselosteessa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
COSENTYX injektioneste, liuos, esitäytetty ruisku
75 mg 1 kpl
- Alempi erityiskorvaus (65 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli ja tosilitsumabi (tulehdukselliset reumasairaudet): Nivelreuman, juveniilin polyartriitin, psoriaasiin liittyvän niveltulehduksen, selkärankareuman tai edellä mainittuja niveltulehduksia läheisesti muistuttavan niveltulehduksen hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman hoito erityisin edellytyksin (281).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli, tosilitsumabi ja ustekinumabi (tulehdukselliset reumasairaudet): Eräiden reumasairauksien hoito erityisin edellytyksin / Adalimumabi: Uveiitin hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman ja jättisoluarteriitin hoito erityisin edellytyksin (313), Adalimumabi, bimekitsumabi, brodalumabi, etanersepti, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sekukinumabi, sertolitsumabipegoli, tildrakitsumabi ja ustekinumabi (ihopsoriaasi): Vaikean kroonisen ihopsoriaasin hoito erityisin edellytyksin (319).
ATC-koodi
L04AC10
Valmisteyhteenvedon muuttamispäivämäärä
16.10.2025
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com