
ENSPRYNG injektioneste, liuos, esitäytetty ruisku 120 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Yleinen
Vaikuttavat aineet ja niiden määrät
Yksi esitäytetty ruisku sisältää 120 mg satralitsumabia 1 ml:ssa.
Satralitsumabi tuotetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Enspryng on tarkoitettu monoterapiana tai yhdistelmänä immunosuppressiivisen lääkityksen kanssa neuromyelitis optica -kirjon häiriön (NMOSD) hoitoon aikuisille sekä 12-vuotialle ja sitä vanhemmille lapsille ja nuorille, jotka ovat akvaporiini-4 IgG (AQP4-IgG) -vasta-aineseropositiivisia (ks. kohta Farmakodynamiikka).
Ehto
Hoito on aloitettava valmisteen käyttöaiheissa mainittujen sairauksien hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito pitää aloittaa neuromyelitis optican (NMO) tai neuromyelitis optica -kirjon häiriön (NMOSD) hoitoon perehtyneen lääkärin valvonnassa.
Annostus
Enspryng-valmistetta voidaan käyttää monoterapiana tai yhdistelmänä suun kautta otettavien kortikosteroidien, atsatiopriinin tai mykofenolaattimofetiilin kanssa (ks. kohta Farmakodynamiikka). Annostus ≥ 40 kg painaville ≥ 12-vuotiaille lapsille ja nuorille on sama kuin aikuisille.
Aloitusannokset
Suositeltu aloitusannos on 120 mg pistoksena ihon alle kahden viikon välein kolmen ensimmäisen antokerran ajan (ensimmäinen annos viikolla 0, toinen annos viikolla 2 ja kolmas annos viikolla 4).
Ylläpitoannokset
Suositeltu ylläpitoannos on 120 mg pistoksena ihon alle neljän viikon välein.
Hoidon kesto
Enspryng on tarkoitettu pitkäaikaishoitoon.
Annoksen viivästyminen tai antamatta jääminen
Jos pistos jää antamatta minkä tahansa muun syyn kuin suurentuneen maksaentsyymipitoisuuden vuoksi, on pistos annettava taulukon 1 mukaisesti.
Taulukko 1. Annossuositukset annoksen viivästyttyä tai jäätyä antamatta
Viimeisimmän annoksen antamisesta kulunut aika | Suositus annoksen viivästyttyä tai jäätyä antamatta |
Antamatta jäänyt aloitusannos tai alle 8 viikkoa viivästynyt annos ylläpitojaksossa | Suositeltu annos pitää antaa mahdollisimman pian odottamatta seuraavaan suunniteltuun antoajankohtaan. Aloitusjakso Jos toinen aloitusannos viivästyy tai jää antamatta, annos pitää antaa mahdollisimman pian ja sen jälkeen kolmas eli viimeinen aloitusannos 2 viikkoa myöhemmin. Jos kolmas aloitusannos viivästyy tai jää antamatta, annos pitää antaa mahdollisimman pian ja ensimmäinen ylläpitoannos 4 viikkoa myöhemmin. Ylläpitojakso Kun viivästynyt tai antamatta jäänyt annos on annettu, annostusta jatketaan 4 viikon välein. |
8 viikosta alle 12 viikkoon | Suositeltu annos pitää antaa viikoilla 0* ja 2 ja sen jälkeen 4 viikon välein. |
12 viikkoa tai pidempään | Suositeltu annos pitää antaa viikoilla 0*, 2 ja 4 ja sen jälkeen 4 viikon välein. |
* Viikko 0 tarkoittaa ensimmäistä antokertaa sen jälkeen, kun annos on jäänyt antamatta.
Ohjeet annosmuutoksiin maksaentsyymien poikkeavuuksien yhteydessä
Jos alaniiniaminotransferaasin (ALAT) tai aspartaattiaminotransferaasin (ASAT) pitoisuus kohoaa tasolle > 5 x viitearvojen ylärajan (upper limit of normal, ULN) ja siihen liittyy minkä tahansa suuruista bilirubiinipitoisuuden kohoamista, hoito on lopetettava, eikä hoidon aloittamista uudelleen suositella.
Jos ALAT- tai ASAT-arvo kohoaa tasolle > 5 x ULN eikä siihen liity bilirubiinipitoisuuden kohoamista, hoito pitää lopettaa. Kun ALAT- ja ASAT-arvot ovat normalisoituneet, hoitoa voidaan jatkaa annoksella 120 mg pistoksena ihon alle neljän viikon välein potilaskohtaisen hyöty-riskiarvion perusteella. Jos hoitoa päätetään jatkaa, maksa-arvoja on seurattava tarkoin, ja jos ALAT- tai ASAT-arvo ja/tai bilirubiinipitoisuus suurenee tämän jälkeen, hoito pitää lopettaa, eikä hoidon aloittamista uudelleen suositella (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Taulukko 2. Suositeltu annos jatkettaessa hoitoa maksan transaminaasipitoisuuden kohoamisen jälkeen
Viimeisimmän annoksen antamisesta kulunut aika | Hoidon jatkamiseen suositeltu annos |
Alle 12 viikkoa | Hoitoa pitää jatkaa suositellulla annoksella 4 viikon välein. |
12 viikkoa tai pidempään | Hoitoa pitää jatkaa suositellulla annoksella viikoilla 0*, 2 ja 4 ja sen jälkeen 4 viikon välein. |
* Viikko 0 tarkoittaa ensimmäistä antokertaa hoidon jatkamisen jälkeen.
Ohjeet annosmuutoksiin neutropenian yhteydessä
Jos neutrofiilimäärä on alle 1,0 x 109/l, ja se varmistuu uusintamäärityksessä, hoito pitää keskeyttää, kunnes neutrofiilimäärä on > 1,0 x 109/l.
Ohjeet annosmuutoksiin trombosyyttien vähyyden yhteydessä
Jos trombosyyttimäärä on alle 75 x 109/l ja varmistettu uusintamäärityksessä, hoito pitää keskeyttää, kunnes trombosyyttimäärä on ≥ 75 x 109/l.
Erityiset potilasryhmät
Pediatriset potilaat
Annostus ≥ 40 kg painaville ≥ 12-vuotiaille lapsille ja nuorille on sama kuin aikuisille (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Satralitsumabin turvallisuutta ja tehoa < 40 kg:n painoisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Iäkkäät
Iältään ≥ 65-vuotiaiden potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Satralitsumabin turvallisuutta ja tehoa ei ole virallisesti tutkittu munuaisten vajaatoimintaa sairastavilla potilailla. Lievää munuaisten vajaatoimintaa sairastavien potilaiden annoksen muuttamista ei suositella (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Satralitsumabin turvallisuutta ja tehoa ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla. Tietoja ei ole saatavilla (ks. kohta Farmakokinetiikka).
Satralitsumabihoidon aikana on havaittu maksaentsyymipitoisuuksien kohoamista (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Annosmuutokset, ks. edellä kohta Ohjeet annosmuutoksiin maksaentsyymien poikkeavuuksien yhteydessä.
Antotapa
Satralitsumabiannos 120 mg annetaan pistoksena ihon alle käyttämällä kerta-annoksen sisältävää esitäytettyä ruiskua. Esitäytetyn ruiskun koko sisältö (1 ml) pitää antaa.
Suositeltuja pistoskohtia ovat vatsa ja reisi. Pistoskohtia pitää vaihdella, eikä pistosta saa koskaan antaa luomiin, arpiin eikä ihoalueille, jolla on aristusta, mustelma, punoitusta, kovettuma tai joka ei ole ehjä.
Tarkat ohjeet satralitsumabin antamiseen ovat pakkausselosteen lopussa.
Pistoksen antaminen kotona (potilas ja/tai potilasta hoitava henkilö)
Ensimmäinen pistos pitää antaa pätevän terveydenhuollon ammattilaisen valvonnassa.
Saatuaan riittävän opastuksen, miten pistos valmistellaan ja pistetään, voi aikuinen potilas / potilasta hoitava henkilö antaa muut annokset kotona, jos se on hoitavan lääkärin mielestä mahdollista ja jos aikuinen potilas / potilasta hoitava henkilö suoriutuu pistostekniikasta.
Jos potilaalle kehittyy vakavan allergisen reaktion oireita, potilaan / potilasta hoitavan henkilön pitää kääntyä heti lääkärin puoleen ja varmistaa lääkäriltä, jatketaanko hoitoa vai ei.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Jos potilaalla on aktiivinen infektio, satralitsumabin antamista on siirrettävä myöhemmäksi, kunnes infektio on hallinnassa (ks. kohta Annostus ja antotapa).
Tarkkaavaisuutta suositellaan, jotta infektiot havaitaan ja diagnosoidaan viiveettä satralitsumabi-hoitoa saavilla potilailla. Jos potilaalle kehittyy vakava tai opportunistinen infektio, hoitoa pitää siirtää myöhemmäksi ja asianmukainen hoito pitää aloittaa seurantaa jatkaen. Potilaita pitää kehottaa hakeutumaan pikaisesti lääkärin hoitoon, jos heille ilmaantuu infektion oireita ja löydöksiä, jotta infektiot havaitaan ajoissa. Potilaille pitää antaa potilaskortti.
Rokotukset
Eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältäviä rokotteita ei pidä antaa samanaikaisesti satralitsumabin kanssa, sillä tämän kliinistä turvallisuutta ei ole varmistettu. Eläviä taudinaiheuttajia sisältävien rokotteiden ja satralitsumabihoidon aloittamisen välisen ajan tulee olla immuniteettia muuntavia tai immunosuppressiivisia lääkkeitä koskevien voimassa olevien rokotusohjeistojen mukainen.
Rokotuksen vaikutuksista satralitsumabihoitoa saaviin potilaisiin ei ole tietoja saatavilla. Kaikkia potilaita suositellaan saattamaan kaikki rokotuksensa ajan tasalle voimassa olevien rokotusohjeistojen mukaisesti ennen satralitsumabihoidon aloittamista.
Maksaentsyymit
Satralitsumabihoidossa on havaittu lievää ja keskivaikeaa maksan transaminaasipitoisuuden kohoamista, useimmiten alle 5 x ULN (ks. kohta Haittavaikutukset).
ALAT- ja ASAT-arvoja pitää seurata ensimmäisten kolmen hoitokuukauden ajan neljän viikon välein, sen jälkeen vuoden ajan kolmen kuukauden välein, ja sen jälkeen kliinisen tarpeen mukaan.
Jos potilaan ALAT- tai ASAT-arvo on > 5 x ULN, satralitsumabihoito pitää lopettaa (ks. kohta Annostus ja antotapa).
Neutrofiilimäärä
Satralitsumabihoidon jälkeen on todettu neutrofiilimäärän vähenemistä (ks. kohta Haittavaikutukset). Neutrofiilimäärää pitää seurata 4–8 viikkoa hoidon aloittamisen jälkeen ja sen jälkeen kliinisen tarpeen mukaan. Suositukset hoidon keskeyttämisestä, ks. kohta Annostus ja antotapa.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty.
Populaatiofarmakokineettisissä analyyseissä atsatiopriinin, suun kautta otettavien kortikosteroidien tai mykofenolaattimofetiilin ei havaittu vaikuttaneen satralitsumabin puhdistumaan.
Sekä in vitro- että in vivo -tutkimukset ovat osoittaneet, että sytokiinit, kuten IL-6, estävät spesifisten maksan CYP450-entsyymien (CYP1A2, CYP2C9, CYP2C19 ja CYP3A4) ilmentymistä.
Jos potilas käyttää myös CYP450 3A4:n, 1A2:n, 2C9:n tai 2C19:n substraatteja ja etenkin sellaisia, joiden terapeuttinen indeksi on kapea (kuten varfariini, karbamatsepiini, fenytoiini ja teofylliini), on satralitsumabihoitoa aloitettaessa tai lopetettaessa noudatettava varovaisuutta, ja niiden annosta on tarvittaessa muutettava.
Satralitsumabin pitkän terminaalisen puoliintumisajan vuoksi sen vaikutukset voivat jatkua useita viikkoja hoidon lopettamisen jälkeen.
Raskaus ja imetys
Raskaus
Satralitsumabin käytöstä raskauden aikana ei ole tietoja saatavissa. Apinoilla tehdyissä kokeissa ei ole havaittu lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Varotoimena on suositeltavaa välttää Enspryng-valmisteen käyttöä raskauden aikana.
Imetys
Ei tiedetä, erittyykö satralitsumabi ihmisen rintamaitoon. Ihmisen IgG:n tiedetään erittyvän rintamaitoon ensimmäisinä synnytyksen jälkeisinä päivinä, minkä jälkeen pitoisuudet pienenevät vähäisiksi. Näin ollen riskiä rintaruokituille vauvoille tänä lyhyenä ajanjaksona ei voida sulkea pois. Enspryng-valmisteen käyttöä voidaan harkita myöhemmin imetyksen aikana vain, jos se on kliinisesti tarpeen.
Hedelmällisyys
Satralitsumabin vaikutuksesta ihmisen hedelmällisyyteen ei ole kliinisiä tietoja saatavissa. Eläinkokeissa ei todettu urosten tai naaraiden hedelmällisyyden heikentymistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Enspryng-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin raportoituja haittavaikutuksia olivat päänsärky (19,2 %), nivelkipu (13,5 %), pienentynyt veren valkosolumäärä (13,5 %), hyperlipidemia (13,5 %) ja injektioon liittyvät reaktiot (12,5 %).
Haittavaikutustaulukko
Taulukossa 3 on yhteenveto haittavaikutuksista, joita on raportoitu kliinisissä tutkimuksissa käytettäessä satralitsumabia monoterapiana tai yhdistelmänä immunosuppressiivisen lääkityksen kanssa.
Kliinisissä tutkimuksissa todetut haittavaikutukset (taulukko 3) luetellaan MedDRA-elinjärjestelmäluokituksen mukaisesti. Haittavaikutukset esitetään haittatapahtumien lukumääränä 100 potilasvuotta kohden ja esiintyvyyden mukaan. Kunkin haittavaikutuksen esiintymistiheysluokka perustuu esiintyvyyteen ja seuraavaan esitystapaan: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000).
Taulukko 3. Haittavaikutukset
| Elinjärjestelmäluokka | Esiintyvyys | |
| Hyvin yleinen | Yleinen | |
| Veri ja imukudos | Hypofibrinogenemia | |
| Aineenvaihdunta ja ravitsemus | Hyperlipidemia | |
| Psyykkiset häiriöt | Unettomuus | |
| Hermosto | Päänsärky | Migreeni |
| Sydän | Bradykardia | |
| Verisuonisto | Hypertensio | |
| Hengityselimet, rintakehä ja välikarsina | Allerginen nuha | |
| Ruoansulatuselimistö | Gastriitti | |
| Iho ja ihonalainen kudos | Ihottuma, kutina | |
| Luusto, lihakset ja sidekudos | Nivelkipu | Luuston ja lihasten jäykkyys |
| Yleisoireet ja antopaikassa todettavat haitat | Injektioon liittyvät reaktiot | Raajojen turvotus |
| Tutkimukset | Pienentynyt veren valkosolumäärä | Pienentynyt neutrofiilimäärä, pienentynyt trombosyyttimäärä, suurentunut transaminaasipitoisuus, suurentunut veren bilirubiinipitoisuus, painon nousu |
Valikoitujen haittavaikutusten kuvaus
Pistoskohdan reaktiot
Satralitsumabihoitoa saaneilla potilailla raportoidut injektioon liittyneet reaktiot olivat pääasiassa lieviä tai keskivaikeita ja ilmenivät useimmiten 24 tunnin kuluessa pistoksen jälkeen. Yleisimmin raportoituja systeemisiä oireita olivat ripuli ja päänsärky. Yleisimmin raportoituja paikallisia pistoskohdan reaktioita olivat kasvojen punoitus, ihon punoitus, kutina, ihottuma ja kipu.
Paino
Painon havaittiin nousseen kaksoissokkoutetun jakson aikana ≥ 15 % lähtöpainosta 3,8 %:lla satralitsumabihoitoa (monoterapiana tai yhdistelmänä immunosuppressiivisen lääkityksen kanssa) saaneista potilaista verrattuna 2,7 %:iin lumelääkettä (tai lumelääkettä ja immunosuppressiivista lääkitystä) saaneista potilaista.
Laboratorioarvojen poikkeavuudet
Neutrofiilit
Neutrofiilimäärän havaittiin pienentyneen kaksoissokkoutetun jakson aikana 31,7 %:lla satralitsumabihoitoa (monoterapiana tai yhdistelmänä immunosuppressiivisen lääkityksen kanssa) saaneista potilaista verrattuna 21,6 %:iin lumelääkettä (tai lumelääkettä ja immunosuppressiivista lääkitystä) saaneista potilaista. Neutrofiilien väheneminen oli useimmiten ohimenevää tai jaksottaista.
Neutrofiilimäärä alle 1 x 109/l todettiin 9,6 %:lla satralitsumabihoitoa saaneista potilaista verrattuna 5,4 %:iin lumelääkettä (tai lumelääkettä ja immunosuppressiivista lääkitystä) saaneista.
Trombosyytit
Trombosyyttimäärän havaittiin pienentyneen (alle 150 × 109/l) kaksoissokkoutetun jakson aikana 24,0 %:lla satralitsumabihoitoa (monoterapiana tai yhdistelmänä immunosuppressiivisen lääkityksen kanssa) saaneista potilaista verrattuna 9,5 %:iin lumelääkettä tai lumelääkettä ja immunosuppressiivista lääkitystä saaneista potilaista. Pienentyneisiin trombosyyttimääriin ei liittynyt verenvuototapahtumia.
Trombosyyttimäärät pienenivät yleensä tilapäisesti, eivätkä ne pienentyneet alle 75 × 109/l.
Maksaentsyymit
Kohonneita ALAT- ja ASAT-arvoja havaittiin kaksoissokkoutetun jakson aikana 27,9 %:lla (ALAT) ja 18,3 %:lla (ASAT) satralitsumabihoitoa (monoterapiana tai yhdistelmänä immunosuppressiivisen lääkityksen kanssa) saaneista potilaista verrattuna 12,2 %:iin (ALAT) ja 13,5 %:iin (ASAT) lumelääkettä tai lumelääkettä ja immunosuppressiivista lääkitystä saaneista potilaista. Arvot kohosivat useimmiten tasolle alle 3 x ULN, kohoaminen oli ohimenevää ja hävisi satralitsumabihoitoa keskeyttämättä.
ALAT- tai ASAT-arvojen kohoamista tasolle > 3 x ULN todettiin 2,9 %:lla (ALAT) ja 1,9 %:lla (ASAT) satralitsumabihoitoa (monoterapiana tai yhdistelmänä immunosuppressiivisen lääkityksen kanssa) saaneista potilaista. Näihin arvojen kohoamisiin ei liittynyt kokonaisbilirubiinipitoisuuden suurenemista.
Yhdellä potilaalla (1 %) ALAT-arvon havaittiin kohonneen 4 viikkoa hoidon aloittamisen jälkeen tasolle yli 5 x ULN, kun potilas sai satralitsumabivalmistetta yhdistelmänä immunosuppressiivisen lääkityksen kanssa; arvo normalisoitui hoidon lopettamisen jälkeen eikä tämän potilaan satralitsumabihoitoa enää jatkettu (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Lipidiparametrit
Kokonaiskolesterolipitoisuus kohosi kaksoissokkoutetun hoitojakson aikana tasolle yli 7,75 mmol/l 10,6 %:lla satralitsumabihoitoa (monoterapiana tai yhdistelmänä immunosuppressiivisen lääkityksen kanssa) saavista potilaista verrattuna 1,4 %:iin lumelääkettä (tai lumelääkettä ja immunosuppressiivista lääkitystä) saaneista potilaista; triglyseridipitoisuus kohosi tasolle yli 3,42 mmol/l 20,2 %:lla satralitsumabihoitoa saaneista potilaista verrattuna 10,8 %:iin lumelääkettä saaneista potilaista.
Pediatriset potilaat
Satralitsumabin turvallisuutta ja tehoa on tutkittu yhdeksällä ≥ 12-vuotiaalla lapsella. Haittavaikutusten esiintyvyys, tyyppi ja vaikeusaste ovat lapsilla 12 vuoden iästä lähtien oletettavasti samat kuin aikuisilla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannoksen yhteydessä potilasta on seurattava tarkoin, hoidettava oireenmukaisesti, ja tarpeen mukaan on aloitettava tukitoimenpiteet.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: immunosuppressantit, interleukiinin estäjät, ATC-koodi: L04AC19.
Vaikutusmekanismi
Satralitsumabi on rekombinantti humanisoitu immunoglobuliini G2:n (IgG2) monoklonaalinen vasta-aine (mAb), joka sitoutuu liukoiseen ja kalvoon sitoutuneeseen ihmisen IL-6-reseptoriin (IL-6R). Satralitsumabi estää näiden reseptorien kautta tapahtuvaa IL-6:n (downstream) signaalinvälitystä.
IL-6:n pitoisuudet NMO:ta tai NMOSD:tä sairastavien potilaiden aivo-selkäydinnesteessä ja seerumissa lisääntyvät, kun tauti on aktiivinen. IL-6 on osallisena NMO:n ja NMOSD:n patogeneesissä ja vaikuttaa mm. seuraaviin: B-solujen aktivaatio, B-solujen erilaistuminen plasmablasteiksi ja patologisten autovasta-aineiden tuotanto, esim. pääasiassa keskushermoston astrosyyttien ilmentämää akvaporiinikanavan proteiinia AQP4:ää vastaan, Th17-solujen aktivaatio ja erilaistuminen, säätelijä-T-solujen inhibitio ja muutokset veri-aivoesteen läpäisevyydessä.
Farmakodynaamiset vaikutukset
NMO:ta tai NMOSD:tä koskeneissa kliinisissä satralitsumabitutkimuksissa havaittiin C-reaktiivisen proteiinin (CRP), fibrinogeenin ja komplementin (C3, C4 ja CH50) laskua.
Kliininen teho ja turvallisuus
Satralitsumabin tehoa ja turvallisuutta arvioitiin kahdessa kliinisessä vaiheen III pivotaalitutkimuksessa potilailla, joilla oli NMOSD (diagnosoitu AQP4-IgG-seropositiivisena tai -seronegatiivisena NMO:na [Wingerchuck 2006 -kriteerit] tai AQP4-IgG-seropositiivisena NMOSD:na [Wingerchuk 2007 -kriteerit]) .
Tutkimuksessa BN40898 oli mukana iältään 12–74-vuotiaita NMOSD:ta sairastavia aikuisia ja nuoria, jotka saivat immunosuppressiivista lääkitystä vakioannoksina ja joilla oli ollut vähintään kaksi pahenemisvaihetta seulontaa edeltäneiden kahden vuoden aikana (joista vähintään yksi pahenemisvaihe seulontaa edeltäneiden 12 kuukauden aikana) ja joiden EDSS-pisteet (expanded disability status scale) olivat 0–6,5. Tutkimuksessa BN40900 oli puolestaan mukana iältään 18–74-vuotiaita aikuisia, jotka eivät saaneet perushoitona immunosuppressiivista lääkitystä ja joilla oli ollut vähintään yksi pahenemisvaihe tai ensimmäinen episodi seulontaa edeltäneiden 12 kuukauden aikana ja joiden EDSS-pisteet olivat 0–6,5.
Kummassakin tutkimuksessa oli noin 30 % AQP4-IgG-seronegatiivista NMO-potilaista.
Tehon arviointiperusteena oli kummassakin tutkimuksessa aika ensimmäiseen pahenemisvaiheeseen, jonka vahvisti riippumaton arviointitoimikunta (Clinical Endpoint Committee, CEC). Pahenemisvaiheeksi määriteltiin EDSS-pisteiden (Expanded Disability Status Scale) ja FSS-kriteerien (functional system score) ennalta määritelty huononeminen 7 päivän kuluessa potilaan raportoimien oireiden perusteella arvioituna (vahvistettu pahenemisvaihe).
Tutkimus BN40898 (tunnetaan myös nimillä SA-307JG tai SAkuraSky)
Tutkimus BN40898 oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu kliininen monikeskustutkimus, jossa arvioitiin satralitsumabin tehoa yhdistettynä vakioannoksina käytettyyn immunosuppressiiviseen lääkitykseen (suun kautta otettavat kortikosteroidit enintään annoksina 15 mg/vrk [prednisoloniekvivalentteina], atsatiopriini enintään annoksina 3 mg/kg/vrk tai mykofenolaattimofetiili enintään 3000 mg/vrk; nuoret saivat atsatiopriinin ja suun kautta otettavien kortikosteroidien tai mykofenolaattimofetiilin ja suun kautta otettavien kortikosteroidien yhdistelmää). Tutkimuksen kaksoissokkoutetussa vaiheessa oli mukana 83 AQP4-IgG-seropositiivista ja -seronegatiivista potilasta (76 aikuista ja 7 nuorta). Potilaat saivat kolme ensimmäistä 120 mg:n satralitsumabikerta-annosta tai kaltaistettua lumelääkettä pistoksena vatsan tai reiden alueen ihon alle kahden viikon välein ensimmäisten neljän viikon ajan ja sen jälkeen neljän viikon välein.
Tutkimusasetelma ja tutkimuspotilasjoukon lähtötilanteen ominaisuudet esitetään taulukossa 4.
Taulukko 4. Tutkimuksen BN40898 tutkimusasetelma ja AQP4-IgG-seropositiivisten potilaiden lähtötilanteen ominaisuudet
Tutkimuksen nimi | Tutkimus BN40898 (AQP4-IgG-seropositiiviset: N = 55; ITT*-potilaat N = 83) | |
Tutkimusasetelma | ||
Tutkimuspotilasjoukko | Nuoret ja aikuiset NMO- tai NMOSD-potilaat, jotka saavat vakioannoksina immunosuppressiivista lääkitystä. Ikä 12–74 vuotta, seulontaa edeltäneiden 2 vuoden aikana ≥ 2 relapsia (vähintään yksi pahenemisvaihe seulontaa edeltäneiden 12 kuukauden aikana), EDSS-pisteet 0–6,5 | |
Tehoa arvioivan tutkimuksen kesto | Tapahtumaperusteinen** (26 vahvistettua pahenemisvaihetta) Seuranta-ajan mediaani: satralitsumabi 139,4 viikkoa, lumelääke 40,2 viikkoa (ITT-ryhmässä: satralitsumabi 115,1 viikkoa ja lumelääke 42,5 viikkoa) | |
Hoitoryhmät, satunnaistaminen 1:1 | Ryhmä A: 120 mg satralitsumabia ihon alle Ryhmä B: lumelääke | |
Lähtötilanteen ominaisuudet AQP4-IgG-seropositiivisilla potilailla | Satralitsumabi + immunosuppressiivinen lääkitys (n = 27) | Lumelääke + immunosuppressiivinen lääkitys (n = 28) |
Diagnoosi, n (%): NMO NMOSD | 19 (70,4) 8 (29,6) | 14 (50,0) 14 (50,0) |
Ikä keskimäärin vuosina (keskihajonta) (min-–maks.) | 44,4 (15,7) (13–73) | 43,4 (12,9) (14–65) |
Iäkkäät (≥ 65 vuotta), n (%) | 3 (11,1) | 1 (3,6) |
Nuoret (≥ 12 – < 18 vuotta), n (%) | 1 (3,7) | 2 (7,1) |
Sukupuolijakauma n (%) miehiä / n (%) naisia | 0 / 27 (100) | 0 / 28 (100) |
Immunosuppressiivinen lääkitys, n (%): Suun kautta otettavat kortikosteroidit Atsatiopriini Mykofenolaattimofetiili Atsatiopriini + suun kautta otettavat kortikosteroidit*** Mykofenolaattimofetiili + suun kautta otettavat kortikosteroidit*** | 14 (51,9) 11 (40,7) 1 (3,7) 0 1 (3,7) | 13 (46,4) 11 (39,3) 3 (10,7) 0 1 (3,6) |
* Intention-to-treat (ITT)
** Varahoitoa (rescue therapy) saaneet potilaat, joilla ei ollut vahvistettuja pahenemisvaiheita, saivat tulla mukaan tutkimuksen avoimeen jatkovaiheeseen, ja heidät sensuroitiin tehoa koskevasta ensisijaisesta analyysista
*** Nuorille potilaille sallittu yhdistelmä
Tutkimus BN40900 (tunnetaan myös nimellä SA-309JG tai SAkuraStar)
Tutkimus BN40900 oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu kliininen monikeskustutkimus, jossa satralitsumabimonoterapian tehoa verrattiin lumelääkkeeseen. Tutkimuksessa oli mukana 95 AQP4-IgG-seropositiivista ja -seronegatiivista aikuista potilasta. Potilaat saivat kolme ensimmäistä 120 mg:n satralitsumabikerta-annosta tai kaltaistettua lumelääkettä pistoksena vatsan tai reiden alueen ihon alle kahden viikon välein ensimmäisten neljän viikon ajan ja sen jälkeen neljän viikon välein.
Tutkimusasetelma ja tutkimuspotilasjoukon lähtötilanteen ominaisuudet esitetään taulukossa 5.
Taulukko 5. Tutkimuksen BN40900 tutkimusasetelma ja AQP4-IgG-seropositiivisten potilaiden lähtötilanteen ominaisuudet
Tutkimuksen nimi | Tutkimus BN40900 (AQP4-IgG-seropositiiviset: N = 64; ITT*-potilaat: N = 95) | |
Tutkimusasetelma | ||
Tutkimuspotilasjoukko | Aikuiset NMO- tai NMOSD-potilaat Ikä 18–74 vuotta, seulontaa edeltäneiden 12 kuukauden aikana ≥ 1 pahenemisvaihe tai ensimmäinen episodi, EDSS-pisteet 0–6,5. Potilaat olivat joko saaneet estohoitoa NMOSD:n pahenemisvaiheeseen tai olivat hoito-naiiveja | |
Tehoa arvioivan tutkimuksen kesto | Tapahtumaperusteinen (44 vahvistettua pahenemisvaihetta tai 1,5 vuotta viimeisen mukaan otetun potilaan satunnaistamisesta, kumpi tapahtuu ensin) Seuranta-ajan mediaani: satralitsumabi 96,7 viikkoa, lumelääke 60,1 viikkoa (ITT-ryhmässä: satralitsumabi 95,4 viikkoa ja lumelääke 60,5 viikkoa) | |
Hoitoryhmät, satunnaistaminen 2:1 | Monoterapia: Ryhmä A: 120 mg satralitsumabia ihon alle Ryhmä B: lumelääke | |
Lähtötilanteen ominaisuudet AQP4-IgG-seropositiivisilla potilailla | Satralitsumabi (n = 41) | Lumelääke (n = 23) |
Diagnoosi, n (%): NMO NMOSD | 26 (63,4) 15 (36,6) | 15 (65,2) 8 (34,8) |
Ikä keskimäärin vuosina (keskihajonta) (min.–maks.) | 46,0 (12,0) (22–70) | 40,1 (11,5) (20–56) |
Iäkkäät (≥ 65 vuotta), n (%) | 1 (2,4) | 0 |
Sukupuolijakauma n (%) miehiä / n (%) naisia | 10 (24,4) / 31 (75,6) | 1 (4,3) / 22 (95,7) |
* Intention-to-treat (ITT)
Ensisijainen teho
AQP4-IgG-seropositiivisilla potilailla vahvistetun pahenemisvaiheen suhteellinen riski pieneni BN40898-tutkimuksessa 79 % (riskitiheyssuhde (hazard ratio; HR) [95 %:n luottamusväli]: 0,21 [0,06–0,75]) ja BN40900-tutkimuksessa 74 % (HR [95 %:n luottamusväli]: 0,26-[0,11–0,63]) (ks. kuvat 1 ja 2). Kun tutkimusten BN40898 ja BN40900 tiedot yhdistettiin, satralitsumabihoito yhdistettynä immunosuppressiiviseen lääkitykseen tai satralitsumabimonoterapia pienensivät AQP4-IgG-seropositiivisilla potilailla vahvistetun pahenemisvaiheen kokonaisriskiä 75 % (HR [95 %:n luottamusväli]; 0,25 (0,12–0,50]). Viikolla 48 AQP4-IgG-seropositiivisista potilaista 85,7 %:lla satralitsumabihoidon ja immunosuppressiivisen lääkityksen yhdistelmää tai satralitsumabimonoterapiaa saaneista potilaista ei esiintynyt vahvistettua pahenemisvaihetta verrattuna 58,7 %:iin lumeryhmän potilaista. Viikolla 96 AQP4-IgG-seropositiivisista potilaista 81,4 %:lla satralitsumabihoidon ja immunosuppressiivisen lääkityksen yhdistelmää tai satralitsumabimonoterapiaa saaneista potilaista ei esiintynyt vahvistettua pahenemisvaihetta verrattuna 47,2 %:iin lumeryhmän potilaista. AQP4-IgG-seronegatiivisilla potilailla teho ei ollut merkittävä.
Kuva 1. Tutkimus BN40898: ensimmäiseen vahvistettuun pahenemisvaiheeseen kulunut aika AQP4-IgG-seropositiivisilla potilailla kaksoissokkoutetun jakson aikana
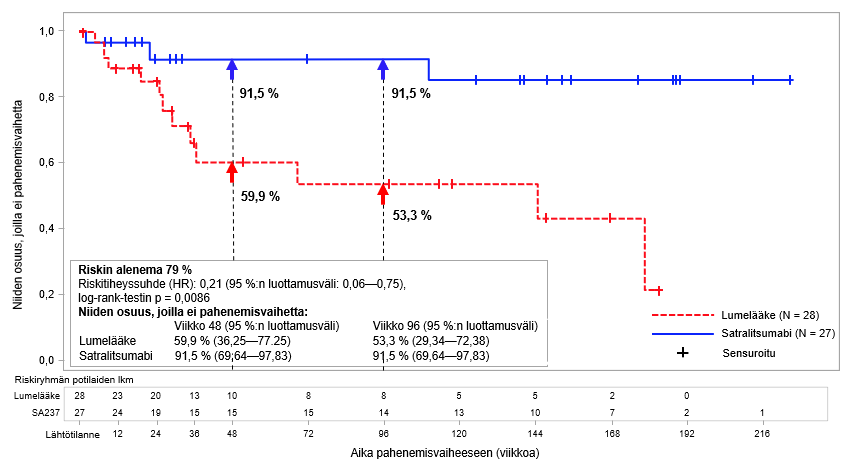
Kuva 2. Tutkimus BN40900: ensimmäiseen vahvistettuun pahenemisvaiheeseen kulunut aika AQP4-IgG-seropositiivisilla potilailla kaksoissokkoutetun jakson aikana

Satralitsumabihoito vähensi AQP4-IgG-seropositiivisilla potilailla vuotuistettua vahvistettujen pahenemisvaiheiden määrää BN40898-tutkimuksessa 88 % (esiintyvyyssuhde [rate ratio; RR]) = 0,122; 95 %:n luottamusväli: 0,027–0,546; p = 0,0039) ja BN40900-tutkimuksessa 90 % (RR = 0,096; 95 %:n luottamusväli: 0,020–0,473; p = 0,0086) lumelääkkeeseen verrattuna.
Satralitsumabihoitoa saaneiden AQP4-IgG-seropositiivisten potilaiden varahoitojen (rescue therapy; esim. kortikosteroidit, laskimoon annettava immunoglobuliini ja/tai afereesi [mukaan lukien plasmafereesi tai plasmanvaihto]) tarve väheni BN40898-tutkimuksessa 61 % (vetokertoimien suhde [odds ratio; OR]) = 0,3930; 95 %:n luottamusväli: 0,1343–1,1502; p = 0,0883) ja BN40900-tutkimuksessa 74 % (OR = 0,2617; 95 %:n luottamusväli: 0,0862–0,7943; p = 0,0180) lumehoitoa saaneisiin potilaisiin verrattuna.
Satralitsumabihoito vähensi AQP4-IgG-seropositiivisilla potilailla vaikea-asteisten pahenemisvaiheiden riskiä BN40898-tutkimuksessa 85 % (kaksoissokkoutetun jakson aikana varmistettuun vaikea-asteiseen relapsiin kulunut aika; HR = 0,15; 95 %:n luottamusväli: 0,02–1,25; p = 0,0441) ja BN40900-tutkimuksessa 79 % (HR = 0,21; 95 %:n luottamusväli: 0,05–0,91; p = 0,0231) lumelääkehoitoon verrattuna. Vaikea-asteiseksi pahenemisvaiheeksi määriteltiin EDSS-pisteiden suureneminen ≥ 2 pistettä edellisestä EDSS-arviosta.
Keskeiset toissijaiset päätetapahtumat
Tutkimuksissa BN40898 ja BN40900 muutosta lähtötilanteesta viikkoon 24 koskien kipua tai uupumusta ei saavutettu.
Avoin jatkotutkimus
Pitkäaikaisempien tietojen, mukaan lukien avoin jatkotutkimus (perustuen varahoidolla hoidettuihin pahenemisvaiheisiin), analyysi osoitti, että 120 viikon satralitsumabihoidon jälkeen AQP4-IgG-seropositiivisista potilaista pahenemisvaihetta ei edelleen ollut 58 %:lla satralitsumabiyhdistelmähoitoa saaneista ja 73 %:lla monoterapiaa saaneista.
Immunogeenisuus
Lääkevasta-aineita havaittiin vaiheen III tutkimuksessa BN40898 (yhdistelmähoito immunosuppressiivisen lääkityksen kanssa) ja vaiheen III tutkimuksessa BN40900 (monoterapia) kaksoissokkoutetun jakson aikana 41 %:lla (BN4089) ja 71 %:lla (BN40900) satralitsumabihoitoa saaneista potilaista. Lääkevasta-aineiden kykyä neutraloida satralitsumabin sitoutumista ei tunneta.
Lääkevasta-ainepositiivisilla potilailla altistus oli pienempi, mutta lääkevasta-aineet eivät vaikuttaneet turvallisuuteen eikä niillä ollut selkeää vaikutusta tehoon tai farmakodynaamisiin merkkiaineisiin.
Satralitsumabihoito vähensi vaiheen III tutkimuksissa vahvistetun pahenemisvaiheen riskiä yhtä paljon, vaikka lääkevasta-aineiden esiintyvyydessä oli tutkimusten välillä eroja.
Pediatriset potilaat
Tutkimuksen BN40898 kaksoissokkoutetussa jaksossa oli mukana 7 nuorta potilasta. Heidän ikänsä oli keskimäärin 15,4 vuotta, ja painon mediaani oli 79,6 kg. Valtaosa oli tyttöjä (n = 6). Neljä potilasta oli valkoihoisia, kaksi oli mustaihoisia/afroamerikkalaisia ja yksi oli aasialainen. Kolme (42,9 %) nuorta potilasta todettiin seulonnassa AQP4-IgG-seropositiivisiksi (2 lumeryhmässä ja 1 satralitsumabiryhmässä). Kaksoissokkoutetun jakson aikana kolmesta lumeryhmän nuoresta yhdellä ja neljästä satralitsumabiryhmän nuoresta yhdelle ilmaantui vahvistettu pahenemisvaihe. Ensisijaisen päätetapahtuman eli ensimmäisen vahvistettuun pahenemisvaiheeseen kuluneen ajan riskitiheyssuhdetta ei laskettu tässä alaryhmässä pienen otoskoon vuoksi. Tutkimuksen avoimeen jaksoon otettiin lisäksi mukaan kaksi nuorta potilasta.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Enspryng-valmisteen käytöstä NMOSD:n hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Satralitsumabin farmakokinetiikkaa on tutkittu sekä japanilaisilla että valkoihoisilla terveillä vapaaehtoisilla ja NMO- ja NMOSD-potilailla. Farmakokinetiikka määritettiin suositeltuja annoksia saaneiden NMO:ta ja NMOSD:ta potilaiden perusteella 154 potilaan tietokannasta populaatiofarmakokineettisellä analyysilla.
Satralitsumabin pitoisuus-aikasuhdetta NMO- ja NMOSD-potilailla kuvaa tarkasti populaatiofarmakokineettinen kaksitilamalli, jossa on rinnan lineaarinen ja kohdevälitteinen (Michaelis-Mentenin) eliminaatio ja ihon alle antoa koskeva ensimmäisen asteen imeytyminen. Satralitsumabin puhdistuma- ja tilavuusparametrit skaalautuvat allometrisesti painon mukaan (voimafunktiona, jossa puhdistuman kiinteä voimakerroin on 0,75 ja tilavuusparametrien kiinteä voimakerroin on 1). Painon osoitettiin olevan merkittävä kovariaatti, sillä 123 kg:n painoisten potilaiden (painojakautuman 97,5. persentiili) puhdistuma lisääntyi 71,3 %:lla ja Vc 105 %:lla verrattuna 60 kg painoiseen potilaaseen.
Vakaan tilan farmakokinetiikka saavutettiin aloitusjakson (8 viikkoa) jälkeen siten, että Cmin-arvo (keskiarvo [± keskihajonta]) oli 19,7 (12,2) mikrog/ml, Cmax-arvo (keskiarvo [± keskihajonta]) oli 31,5 (14,9) mikrog/ml ja AUC-arvo (keskiarvo [± keskihajonta]) oli 737 (386) mikrog.ml/vrk.
Imeytyminen
Satralitsumabin absorptionopeusvakio oli 0,0104 h, mikä vastaa noin 3 vuorokauden (66 tuntia) imeytymisen puoliintumisaikaa käytettäessä suositeltua annosta (ks. kohta Annostus ja antotapa). Biologinen hyötyosuus oli suuri (85,4 %).
Jakautuminen
Satralitsumabin jakautuminen on kaksivaiheinen. Sentraalinen jakautumistilavuus oli 3,46 l ja perifeerinen jakautumistilavuus oli 2,07 l. Tilojen välinen puhdistuma oli 14 ml/h.
Biotransformaatio
Satralitsumabin metaboliaa ei ole tutkittu suoraan, sillä monoklonaaliset vasta-aineet poistuvat elimistöstä pääasiassa kataboloitumalla.
Eliminaatio
Satralitsumabin kokonaispuhdistuma on pitoisuusriippuvainen. Lineaariseksi puhdistumaksi arvioidaan 2,50 ml/h (NMO- ja NMOSD-potilailla suositeltua annosta käytettäessä vastaa vakaassa tilassa noin puolta kokonaispuhdistumasta). Vaiheen 3 tutkimusten yhdistettyjen tietojen perusteella tähän liittyvä terminaalinen t1/2 on noin 30 vuorokautta (vaihteluväli 22–37 vuorokautta).
Erityiset potilasryhmät
NMO:ta ja NMOSD:ta sairastavien aikuisten potilaiden populaatiofarmakokineettiset analyysit osoittivat, että ikä, sukupuoli ja etninen tausta eivät vaikuta merkittävästi satralitsumabin farmakokinetiikkaan. Paino vaikuttaa satralitsumabin farmakokinetiikkaan, mutta minkään näiden demografisten tietojen perusteella ei suositella annosmuutoksia.
Pediatriset potilaat
Aikuisten annostusta saaneista kahdeksasta nuoresta (iältään 13–17-vuotiaasta) potilaasta saadut tiedot osoittavat, että satralitsumabin populaatiofarmakokineettiset parametrit eivät poikkea merkittävästi aikuisten potilaiden vastaavista parametreista. Annosmuutos ei näin ollen ole tarpeen.
Iäkkäät
Satralitsumabin farmakokinetiikan tutkimiseksi iältään ≥ 65-vuotiailla potilailla ei ole tehty varsinaisia tutkimuksia. Kliinisissä tutkimuksissa BN40898 ja BN40900 oli mukana iältään 65–74-vuotiaita NMO:ta ja NMOSD:ta sairastavia potilaita.
Munuaisten vajaatoiminta
Varsinaista tutkimusta munuaisten vajaatoiminnan vaikutuksesta satralitsumabin farmakokinetiikkaan ei ole tehty. Vaiheen III tutkimuksissa oli kuitenkin mukana lievää munuaisten vajaatoimintaa (kreatiniinipuhdistuma ≥ 50 ml/min – < 80 ml/min) sairastavia potilaita. Populaatiofarmakokineettisen analyysin perusteella munuaisten vajaatoiminta ei vaikuta satralitsumabin farmakokinetiikkaan, mikä on yhdenmukainen havainto satralitsumabin tunnetun puhdistumamekanismin kanssa. Annosmuutos ei näin ollen ole tarpeen.
Maksan vajaatoiminta
Maksan vajaatoiminnan vaikutusta satralitsumabin farmakokinetiikkaan ei ole selvitetty varsinaisissa tutkimuksissa (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Karsinogeenisuus
Satralitsumabin karsinogeenisuuden varmistamiseksi ei ole tehty karsinogeenisuustutkimuksia jyrsijöillä. Cynomolgus-apinoilla tehdyssä 6 kuukauden pituisessa pitkäaikaistoksisuutta selvittäneessä tutkimuksessa ei havaittu proliferatiivisia leesioita.
Genotoksisuus
Satralitsumabin mutageenisuuden varmistamiseksi ei ole tehty tutkimuksia. Vasta-aineilla ei odoteta olevan vaikutuksia DNA:han.
Lisääntymistoksisuus
Tiineiden apinoiden ja niiden jälkeläisten prenataalinen satralitsumabihoito ja postnataalinen altistus satralitsumabille eivät aiheuttaneet haittavaikutuksia emoille, sikiön kehitykseen, tiineyden lopputulokseen eikä poikasen eloonjääntiin ja kehitykseen (mukaan lukien oppimiskykyyn).
Satralitsumabipitoisuus nisämaidossa oli hyvin pieni (< 0,9 % vastaavasta pitoisuudesta emon plasmassa).
Hedelmällisyys
Apinoille annetusta pitkäaikaisesta satralitsumabihoidosta ei todettu vaikutuksia urosten eikä naaraiden lisääntymiselimiin.
Sytokiinien vapautumisoireyhtymä
Ihmisen verellä tehtyjen in vitro -tutkimusten perusteella tulehdusta edistävien sytokiinien vapautumisriskin sytokiinien ilmaantuvuuden ja lisääntymisen suhteen katsotaan olevan satralitsumabin käytössä vähäinen.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Asparagiinihappo
Arginiini
Poloksameeri 188
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä. Älä käytä ruiskua, jos se on jäätynyt.
Pidä ruisku aina kuivana.
Pidä esitäytetty ruisku ulkopakkauksessa. Herkkä valolle. Herkkä kosteudelle.
Avaamaton ulkopakkauksessa säilytetty ruisku voi olla poissa jääkaappisäilytyksestä alle 30 °C:ssa yhden enintään 8 päivän pituisen jakson. Huoneenlämmössä säilyttämisen jälkeen valmistetta ei saa laittaa takaisin jääkaappiin vaan se pitää joko käyttää tai hävittää.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ENSPRYNG injektioneste, liuos, esitäytetty ruisku
120 mg (L:ei) 1 kpl (1 ml (120 mg/ml)) (9050,63 €)
PF-selosteen tieto
1 ml liuosta esitäytetyssä ruiskussa (polymeeria), jossa on ruostumattomasta teräksestä valmistettu kiinteä neula sekä klooratusta butyylikumista ja polypropeenista valmistettu jäykkä neulansuojus ja joka on suljettu klooratusta butyylikumista valmistetulla männän tulpalla. Esitäytetyssä ruiskussa on etiketti, automaattinen turvamekanismi, männän varsi ja sormituet.
Pakkauskoot: 1 esitäytetty ruisku ja 3 esitäytetyn ruiskun monipakkaus (3 x 1). Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Väritön tai hieman keltainen neste. Liuoksen pH on noin 6,0 ja osmolaalisuus noin 310 mosm/kg.
Käyttö- ja käsittelyohjeet
Jääkaapista otettu sinetöity ulkopakkaus avataan, ja esitäytetty ruisku nostetaan varovasti pois ulkopakkauksesta siten, että ruiskua pidellään sen säiliöstä. On tärkeää antaa esitäytetyn ruiskun lämmetä huoneenlämpöiseksi 30 minuutin ajan ennen pistoksen antamista.
Jos liuos on sameaa, värjäytynyttä tai siinä on näkyviä hiukkasia tai jos esitäytetyn ruiskun jokin osa vaikuttaa vioittuneelta, lääkevalmistetta ei pidä käyttää.
Pistos on annettava heti korkin irrottamisen jälkeen ja viimeistään 5 minuutin kuluttua korkin irrottamisesta, jotta vältetään lääkevalmisteen kuivuminen ja neulan tukkeutuminen. Jos esitäytettyä ruiskua ei käytetä 5 minuutin kuluessa korkin irrottamisen jälkeen, ruisku on hävitettävä laittamalla se pistävälle ja viiltävälle jätteelle tarkoitettuun astiaan, ja käyttöön on otettava uusi esitäytetty ruisku.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ENSPRYNG injektioneste, liuos, esitäytetty ruisku
120 mg 1 kpl
- Ei korvausta.
ATC-koodi
L04AC19
Valmisteyhteenvedon muuttamispäivämäärä
10.02.2026
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com