
KESIMPTA injektioneste, liuos, esitäytetty kynä 20 mg
Vaikuttavat aineet ja niiden määrät
Kesimpta 20 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 20 mg ofatumumabia 0,4 millilitrassa liuosta (50 mg/ml).
Kesimpta 20 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kynä sisältää 20 mg ofatumumabia 0,4 millilitrassa liuosta (50 mg/ml).
Ofatumumabi on täysin humaani monoklonaalinen vasta‑aine, joka valmistetaan hiiren solulinjassa (NS0) yhdistelmä‑DNA‑tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Injektioneste, liuos (injektioneste), esitäytetty kynä (Sensoready‑kynä)
Kliiniset tiedot
Käyttöaiheet
Kesimpta on tarkoitettu relapsoivan MS‑taudin (RMS) hoitoon aikuispotilailla, joilla tauti on aktiivinen kliinisten tai kuvantamisella havaittujen piirteiden perusteella (ks. kohta Farmakodynamiikka).
Ehto
Neurologisten sairauksien hoitoon perehtyneen lääkärin on aloitettava hoito.
Annostus ja antotapa
Neurologisten sairauksien hoitoon perehtyneen lääkärin on aloitettava hoito.
Annostus
Suositeltu ofatumumabiannos on 20 mg injektiona ihon alle:
- aloitusannokset viikoilla 0, 1 ja 2, minkä jälkeen
- annostelu kuukauden välein alkaen viikolta 4.
Väliin jääneet annokset
Jos injektio jää väliin, injektio on otettava mahdollisimman pian eikä odottaa seuraavaan aikataulun mukaiseen annokseen asti. Myöhempien annosten osalta noudatetaan suositeltuja injektiovälejä.
Erityisryhmät
Yli 55‑vuotiaat aikuiset
Yli 55‑vuotiailla MS‑potilailla ei ole tehty tutkimuksia. Saatavilla olevien vähäisten kliinisten tietojen perusteella yli 55‑vuotiaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annosta ei todennäköisesti tarvitse muuttaa munuaisten vajaatoimintapotilaille (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei todennäköisesti tarvitse muuttaa maksan vajaatoimintapotilaille (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Kesimpta‑valmisteen turvallisuutta ja tehoa 0–18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Tämä lääkevalmiste on tarkoitettu potilaan itse pistettäväksi ihon alle.
Ihon alle annettavien injektioiden tavanomaiset antokohdat ovat vatsa, reisi ja olkavarren ulkosyrjä.
Ensimmäinen injektio annostellaan terveydenhuollon ammattilaisen valvonnassa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pakkausselosteessa on valmisteen antoa koskevat kattavat ohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Vaikea immuunipuutos (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vakava aktiivinen infektio, kunnes se on parantunut (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tiedossa oleva aktiivinen maligniteetti.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Systeemiset injektioon liittyvät reaktiot
Potilaille on kerrottava, että systeemisiä injektioon liittyviä reaktioita saattaa ilmaantua, yleensä 24 tunnin kuluessa ja valtaosin ensimmäisen injektion jälkeen (ks. kohta Haittavaikutukset). Relapsoivan MS‑taudin (RMS) kliinisissä tutkimuksissa yleisimmin havaittuja oireita olivat kuume, päänsärky, lihassärky, vilunväristykset, uupumus, pahoinvointi ja oksentelu, ja ne olivat pääasiassa (99,8 %) lieviä tai keskivaikeita. Kliinisissä RMS-tutkimuksissa ei raportoitu hengenvaarallisia injektioon liittyviä systeemisiä oireita (ks. kohta Haittavaikutukset).
Markkinoille tulon jälkeen raportoituja muita injektioon liittyviä systeemisiä oireita ovat ihottuma, nokkosihottuma, hengenahdistus ja angioedeema (esim. kielen, nielun tai kurkunpään turvotus) sekä harvinaiset tapaukset, jotka raportoitiin anafylaksiana. Vaikka jotkut tapaukset olivat vakavia ja johtivat ofatumumabihoidon keskeyttämiseen, oli myös vakavia tapauksia, joissa potilaat pystyivät jatkamaan ofatumumabihoitoa ilman uusia tapauksia.
Jotkin injektioon liittyvät systeemiset oireet voivat olla kliinisesti erottamattomia tyypin 1 akuuteista yliherkkyysreaktioista (IgE-välitteiset). Yliherkkyysreaktio voi ilmaantua minkä tahansa injektion yhteydessä, vaikka tyypillisesti sitä ei ilmaannu ensimmäisen injektion yhteydessä. Seuraavien injektioiden yhteydessä aiemmin koettuja oireita vakavampien oireiden tai uusien vakavien oireiden vuoksi on harkittava mahdollista yliherkkyysreaktiota. Potilaita, joilla tiedetään olevan IgE-välitteinen yliherkkyys ofatumumabille, ei saa hoitaa ofatumumabilla (ks. kohta Vasta-aiheet).
Relapsoivaa MS‑tautia koskeneissa kliinisissä tutkimuksissa esilääkityksestä steroidilla todettiin olevan vain vähäistä hyötyä. Jos injektioon liittyviä reaktioita ilmenee, niitä voidaan hoitaa oireenmukaisesti. Näin ollen esilääkityksen käyttö ei ole tarpeen.
Ensimmäinen injektio annostellaan asianmukaisen koulutuksen saaneen terveydenhuollon ammattilaisen valvonnassa (ks. kohta Annostus ja antotapa).
Paikalliset injektiokohtaan liittyvät reaktiot
Kliinisissä tutkimuksissa havaittuja paikallisia injektiokohtaan liittyviä reaktioita olivat punoitus, turvotus, kutina ja kipu (ks. kohta Haittavaikutukset).
Infektiot
Potilaan immuunijärjestelmän tila on suositeltavaa arvioida ennen hoidon aloittamista.
Vaikutustavan ja saatavilla olevan kliinisen kokemuksen perusteella ofatumumabiin voi liittyä suurentunut infektioriski (ks. kohta Haittavaikutukset).
Jos potilaalla on aktiivinen infektio, annostelua on lykättävä, kunnes infektio on parantunut.
Ofatumumabia ei saa antaa, jos potilaalla on vaikea immuunipuutos (esim. merkittävä neutropenia tai lymfosytopenia).
Progressiivinen multifokaalinen leukoenkefalopatia
JC-virusinfektioita ja niiden seurauksena progressiivista multifokaalista leukoenkefalopatiaa (PML) on havaittu potilailla, jotka ovat saaneet CD20 vasta-aineita, muita MS hoitoja tai huomattavasti suurempia ofatumumabiannoksia syöpätautien hoidossa. Tästä syystä lääkärien on oltava tarkkaavaisia aiemman PML-historian ja mahdollisesti PML:ään viittaavien kliinisten oireiden ja magneettikuvauslöydösten suhteen. Jos PML:ää epäillään, ofatumumabihoito on keskeytettävä, kunnes PML on suljettu pois.
Hepatiitti B ‑viruksen uudelleenaktivoituminen.
Anti‑CD20‑vasta‑aineita saaneilla potilailla on esiintynyt hepatiitti B:n uudelleenaktivoitumista, joka on joissain tapauksissa johtanut fulminanttiin hepatiittiin, maksan vajaatoimintaan ja kuolemaan.
Potilaita, joilla on aktiivinen hepatiitti B-tauti, ei tule hoitaa ofatumumabilla. Kaikki potilaat on seulottava HBV‑infektion varalta ennen hoidon aloittamista. Seulontaan on sisällyttävä vähintään hepatiitti B:n pinta‑antigeenin (HBsAg) ja hepatiitti B:n ydinvasta‑aineen (HBcAb) testaus. Testausta voidaan täydentää muilla asianmukaisilla markkereilla paikallisten hoitosuositusten mukaisesti. Jos serologisen hepatiitti B ‑testin tulos on positiivinen (joko HBsAg tai HBcAb), maksasairauksien asiantuntijaa on konsultoitava ennen hoidon aloittamista ja potilasta on seurattava ja hoidettava paikallisten hoitokäytäntöjen mukaisesti hepatiitti B:n uudelleen aktivoitumisen ehkäisemiseksi.
Vaikeasti immuunipuutteisten potilaiden hoito
Hoitoa ei saa antaa vaikeasti immuunipuutteisille potilaille ennen kuin immuunipuutostila on korjautunut (ks. kohta Vasta-aiheet).
Ofatumumabin ja muiden immunosuppressiivisten lääkeaineiden samanaikainen käyttö ei ole suositeltavaa lukuun ottamatta pahenemisvaiheiden oireenmukaiseen hoitoon käytettäviä kortikosteroideja.
Rokotukset
Kaikki elävillä tai elävillä heikennetyillä rokotteilla toteutettavat rokotukset on annettava rokotussuositusten mukaisesti vähintään 4 viikkoa ennen ofatumumabihoidon aloittamista ja, jos mahdollista, inaktivoiduilla rokotteilla toteutettavat rokotukset vähintään 2 viikkoa ennen ofatumumabihoidon aloittamista.
Ofatumumabi voi vaikuttaa inaktivoitujen rokotteiden tehoon.
Elävillä tai elävillä heikennetyillä rokotteilla toteutettavien rokotusten turvallisuutta ofatumumabihoidon jälkeen ei ole tutkittu. Elävien tai elävien heikennettyjen rokotteiden antoa ei suositella hoidon aikana eikä hoidon päättymisen jälkeen ennen kuin B‑soluarvo on korjautunut (ks. kohta Yhteisvaikutukset). Vaiheen III tutkimuksista saatujen tietojen perusteella B-soluarvon korjautumisen mediaaniaika viitealueen alarajalle (määritelmän mukaan 40 solua/µl) tai lähtötilanteen tasolle on 24,6 viikkoa hoidon lopettamisen jälkeen (ks. kohta Farmakodynamiikka).
Rokotus vauvoilla, joiden äidit ovat saaneet ofatumumabihoitoa raskauden aikana
Jos äiti on saanut ofatumumabihoitoa raskauden aikana, vauvalle ei saa antaa eläviä eikä eläviä heikennettyjä rokotteita ennen kuin on vahvistettu, että B‑soluarvo on normaalialueella. B‑solukato näillä vauvoilla voi lisätä elävien tai elävien heikennettyjen rokotteiden riskejä.
Inaktivoituja rokotteita voidaan antaa tarpeen mukaan ennen kuin on vahvistettu, että B‑soluarvo on normaalialueella, mutta rokotteen tuottaman immuunivasteen arviointia (ml. pätevän erikoislääkärin konsultointia) on kuitenkin harkittava suojaavan immuunivasteen muodostumisen määrittämiseksi (ks. kohta Raskaus ja imetys).
Apuaineet, joiden vaikutus tunnetaan
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Polysorbaatit
Tämä lääkevalmiste sisältää 0,08 mg polysorbaatti 80:ä per annos. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty, sillä CYP450‑entsyymeihin, muihin metaboloiviin entsyymeihin tai kuljettajaproteiineihin liittyvät yhteisvaikutukset eivät ole todennäköisiä.
Rokotukset
Elävien, elävien heikennettyjen tai inaktivoitujen rokotteiden turvallisuutta tai kykyä tuottaa primaarinen tai sekundaarinen vaste ofatumumabihoidon aikana ei ole tutkittu. Vaste rokotukselle voi heikentyä B‑solukadon yhteydessä. On suositeltava, että potilaat saavat rokotussarjat kokonaisuudessaan ennen ofatumumabihoidon aloittamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Muut immunosuppressiiviset tai immunomoduloivat hoidot
Additiivisten immuunijärjestelmään kohdistuvien vaikutusten riski on otettava huomioon, jos immunosuppressiivista hoitoa ja ofatumumabia käytetään samanaikaisesti.
Jos ofatumumabihoito aloitetaan muiden pitkäkestoisia immuunivaikutuksia aiheuttavien immunosuppressiivisten hoitojen jälkeen tai tällainen immunosuppressiivinen hoito aloitetaan ofatumumabihoidon jälkeen, kyseisten lääkevalmisteiden käytön kesto ja vaikutustapa on otettava huomioon mahdollisten additiivisten immunosuppressiivisten vaikutusten takia (ks. kohta Farmakodynamiikka).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä (menetelmät, joihin liittyvä raskausprosentti on alle 1 %) Kesimpta‑hoidon aikana ja 2 kuukautta viimeisen Kesimpta‑annoksen jälkeen.
Raskaus
On vain vähän tietoja ofatumumabin käytöstä raskaana oleville naisille. Eläinkokeiden löydösten perusteella ofatumumabi voi läpäistä istukan ja aiheuttaa sikiölle B‑solukadon (ks. kohta Prekliiniset tiedot turvallisuudesta). Teratogeenisuutta ei havaittu, kun tiineille apinoille annettiin ofatumumabia laskimoon organogeneesin aikana.
Ohimenevää perifeeristä B‑solukatoa ja lymfosytopeniaa on raportoitu imeväisillä, joiden äidit ovat altistuneet muille anti‑CD20‑vasta‑aineille raskauden aikana. B‑solukadon mahdollista kestoa ofatumumabille in utero altistuneilla imeväisillä ja B‑solukadon vaikutusta rokotteiden turvallisuuteen ja tehoon ei tunneta (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Ofatumumabihoitoa on vältettävä raskauden aikana, ellei mahdollinen hyöty äidille ole suurempi kuin mahdollinen riski sikiölle.
Ofatumumabin vaikutusten selvittämiseksi raskaana olevilla naisilla terveydenhuoltohenkilöstöä kehotetaan ilmoittamaan kaikki hoidon aikana tai 2 kuukauden kuluessa viimeisestä ofatumumabiannoksesta ilmenevät raskaustapaukset ja ‑komplikaatiot myyntiluvan haltijan paikalliselle edustajalle, jotta kyseisiä potilaita voidaan seurata PRIM‑ohjelman (PRegnancy outcomes Intensive Monitoring) puitteissa. Lisäksi kaikki raskauteen liittyvät haittatapahtumat on ilmoitettava seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Imetys
Ofatumumabin käytöstä imettäville naisille on vain vähän tietoja. Ei ole olemassa tietoja ofatumumabin vaikutuksesta maidon tuotantoon. Ihmisillä IgG‑vasta‑aineita erittyy äidinmaitoon muutaman päivän ajan synnytyksen jälkeen, minkä jälkeen pitoisuudet pienenevät pian matalalle tasolle. Julkaistujen tietojen perusteella äidinmaidossa olevia vasta-aineita ei siirry vastasyntyneen tai imetettävän vauvan verenkiertoon merkittäviä määriä.
Havainnoivassa tutkimuksessa todettiin, että ofatumumabin pitoisuus äidinmaidossa oli yleensä matala. Ofatumumabihoitoa saaneiden imettävien naisten äidinmaidossa ofatumumabin keskimääräinen pitoisuus (Cavg) ja huippupitoisuus (Cmax) olivat alle 0,02 µg/ml.
Samassa havainnoivassa tutkimuksessa viidellä imetettävällä vauvalla, joiden B-soluarvot olivat saatavilla, B-solujen tasot olivat normaalit. Kahdeksan imetettävää vauvaa sai eläviä rokotteita imetyksen aikaisen altistuksen aikana tai sen jälkeen ilman komplikaatioita. Imetettävillä vauvoilla (enintään 24 kuukauden ikään asti) ei todettu poikkeavuuksia infektioiden esiintymisessä, antibioottien käytössä tai sairaalahoidon tarpeessa, eikä kehitysviiveitä esiintynyt.
Näin ollen ensimmäisten syntymän jälkeisten päivien aikana imetettävälle lapselle koituvaa riskiä ei voida sulkea pois. Tämän jälkeen ofatumumabia voidaan käyttää imetyksen aikana, jos se on kliinisesti tarpeen. Jos potilas on saanut ofatumumabihoitoa raskauden viimeisiin kuukausiin asti, imetys voidaan kuitenkin aloittaa heti synnytyksen jälkeen.
Hedelmällisyys
Ofatumumabin vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa.
Ei‑kliiniset tiedot eivät viittaa vaaraan ihmisille uros‑ ja naarasapinoilla arvioitujen hedelmällisyyden parametrien perusteella.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kesimpta‑valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Tärkeimmät ja yleisimmin ilmoitetut haittavaikutukset ovat ylähengitystieinfektiot (39,4 %), systeemiset injektioon liittyvät reaktiot (20,6 %), injektiokohdan paikalliset reaktiot (10,9 %) ja virtsatieinfektiot (11,9 %) (lisätiedot, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja alakohta ”Valikoitujen haittavaikutusten kuvaus” jäljempänä).
Haittavaikutustaulukko
Relapsoivaa MS‑tautia koskevissa kliinisissä avaintutkimuksissa sekä markkinoille tulon jälkeen ofatumumabin käytön yhteydessä ilmoitetut haittavaikutukset on lueteltu MedDRA‑elinjärjestelmäluokituksen mukaisesti taulukossa 1. Kunkin elinjärjestelmäluokan haittavaikutukset esitetään yleisyysjärjestyksessä yleisimmistä alkaen. Kunkin yleisyysluokan haittavaikutukset esitetään vakavuusjärjestyksessä vakavimmasta alkaen. Kunkin haittavaikutuksen kohdalla mainittava yleisyysluokka perustuu seuraavaan käytäntöön: hyvin yleinen (≥1/10), yleinen (≥1/100, <1/10), melko harvinainen (≥1/1 000, <1/100), harvinainen (≥1/10 000, <1/1 000), hyvin harvinainen (<1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1 Haittavaikutustaulukko
Infektiot | ||
Hyvin yleinen | Ylähengitystieinfektiot1 Virtsatieinfektiot2 | |
Yleinen | Huuliherpes | |
Immuunijärjestelmä | ||
Tuntematon | Yliherkkyysreaktiot3 | |
Ruoansulatuselimistö | ||
Yleinen | Pahoinvointi, oksentelu4 | |
Maksa ja sappi | ||
Yleinen | Maksaentsyymien nousu5 | |
Yleisoireet ja antopaikassa todettavat haitat | ||
Hyvin yleinen | Injektiokohdan reaktiot (paikalliset) | |
Tutkimukset | ||
Yleinen | Veren immunoglobuliini M -pitoisuuden pieneneminen | |
Vammat, myrkytykset ja hoitokomplikaatiot | ||
Hyvin yleinen | Injektioon liittyvät reaktiot (systeemiset) | |
1 Haittavaikutusten yleisyyden määrityksessä käytettiin termien ryhmittämistä, ja ryhmä sisältää seuraavat: nasofaryngiitti, ylähengitystieinfektio, influenssa, sinuiitti, faryngiitti, nuha, virusperäinen ylähengitystieinfektio, tonsilliitti, akuutti sinuiitti, faryngotonsilliitti, laryngiitti, streptokokkifaryngiitti, virusperäinen nuha, bakteerisinuiitti, bakteeritonsilliitti, virusfaryngiitti, virustonsilliitti, krooninen sinuiitti, nenän herpes, trakeiitti. 2 Haittavaikutusten yleisyyden määrityksessä käytettiin termien ryhmittämistä, ja ryhmä sisältää seuraavat: virtsatieinfektio, kystiitti, virtsateiden Escherichia-infektio, oireeton bakteriuria, bakteriuria. 3 Raportoitu markkinoille tulon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) 4 Pahoinvointia ja oksentelua on ilmoitettu systeemisten injektioon liittyvien reaktioiden yhteydessä (ks. alla ja kohta Varoitukset ja käyttöön liittyvät varotoimet) 5 Haittavaikutusten yleisyyden määrityksessä käytettiin termien ryhmittämistä, ja ryhmä sisältää seuraavat: alaniiniaminotransferaasin nousu, aspartaattiaminotransferaasin nousu, maksaentsyymiarvojen nousu, transaminaasien nousu, maksan toimintakokeiden tulosten kohoaminen, poikkeava maksan toiminta, poikkeavat tulokset maksan toimintakokeissa. | ||
Valikoitujen haittavaikutusten kuvaus
Infektiot
Vaiheen III kliinisissä RMS‑tutkimuksissa infektioiden ja vakavien infektioiden kokonaisesiintymistiheydet olivat ofatumumabia saaneilla potilailla samaa luokkaa kuin teriflunomidia saaneilla (infektioiden esiintymistiheydet 51,6 % vs. 52,7 % ja vakavien infektioiden esiintymistiheydet 2,5 % vs. 1,8 %). Kaksi potilasta (0,2 %) lopetti tutkimushoidon ja 11 potilasta (1,2 %) keskeytti sen väliaikaisesti vakavan infektion takia.
Ylähengitystieinfektiot
Kyseisissä tutkimuksissa 39,4 %:lle ofatumumabia saaneista potilaista kehittyi ylähengitystieinfektio. Teriflunomidia saaneilla potilailla määrä oli 37,8 %. Infektiot olivat valtaosin lieviä tai keskivaikeita, ja useimmiten kyseessä oli nasofaryngiitti, ylähengitystieinfektio tai influenssa.
Systeemiset injektioon liittyvät reaktiot
Vaiheen III kliinisissä RMS‑tutkimuksissa injektioon liittyviä systeemisiä reaktioita ilmoitettiin 20,6 %:lla ofatumumabia saaneista potilaista.
Systeemiset injektioon liittyvien reaktioiden ilmaantuvuus oli suurin ensimmäisen injektion yhteydessä (14,4 %) ja merkitsevästi pienempi myöhempien injektioiden yhteydessä (4,4 % toisen injektion yhteydessä ja < 3 % kolmannesta injektiosta alkaen). Systeemiset injektioon liittyvät reaktiot olivat vaikeusasteeltaan yleensä (99,8 %) lieviä tai keskivaikeita. Kahdella (0,2 %) ofatumumabia saaneista MS‑potilaista ilmoitettiin vakava injektioon liittyvä reaktio. Reaktiot eivät kuitenkaan olleet henkeä uhkaavia. Yleisimmin ilmoitettuja oireita (≥ 2 %) olivat kuume, päänsärky, lihaskipu, vilunväristykset ja väsymys. Muita ilmoitettuja oireita olivat pahoinvointi (1,7 %) ja oksentelu (0,6 %).
Paikalliset injektiokohdan reaktiot
Vaiheen III kliinisissä RMS‑tutkimuksissa injektiokohdan paikallisia reaktioita ilmoitettiin 10,9 %:lla ofatumumabia saaneista potilaista.
Paikalliset injektiokohdan reaktiot olivat hyvin yleisiä. Ne olivat kaikki vaikeusasteeltaan lieviä tai keskivaikeita ja ei‑vakavia. Yleisimmin ilmoitettuja oireita (≥ 2 %) olivat punoitus, kipu, kutina ja turvotus.
Laboratorioarvojen poikkeavuudet
Immunoglobuliinit
Vaiheen III kliinisten RMS‑tutkimusten aikana havaittiin immunoglobuliini M (IgM) ‑keskiarvon pienenemistä (30,9 % pienenemä 48 viikon kuluttua ja 38,8 % pienenemä 96 viikon kuluttua), johon ei todettu liittyvän infektioiden (ei myöskään vakavien) riskiä.
Potilaista 14,3 %:lla ofatumumabihoito johti IgM‑arvon pienenemiseen alle arvon 0,34 g/l.
Ofatumumabiin liittyi immunoglobuliini G (IgG) –pitoisuuden keskiarvon ohimenevä 4,3 % pienenemä 48 hoitoviikon jälkeen mutta 2,2 % suurenema 96 viikon jälkeen.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‑haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa MS‑potilaille on annettu enimmillään 700 mg annoksia eikä annosta rajoittavaa toksisuutta ole havaittu. Yliannostustapauksissa on suositeltavaa seurata potilasta haittavaikutusoireiden ja ‑löydösten varalta ja aloittaa tarvittaessa asianmukainen oireenmukainen hoito.
Ofatumumabia on käytetty aiemmin kroonista lymfaattista leukemiaa (KLL) koskevissa käyttöaiheissa enintään 2 000 mg annoksina infuusiona laskimoon. Injektiona ihon alle annettavaa ofatumumabia ei ole arvioitu eikä hyväksytty kyseisissä käyttöaiheissa, eikä sitä saa käyttää onkologisiin käyttöaiheisiin.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: immunosuppressantit, monoklonaaliset vasta-aineet, ATC‑koodi: L04AG12
Vaikutusmekanismi
Ofatumumabi on täysin humaani monoklonaalinen immunoglobuliini G1 –luokan (IgG1) anti‑CD20‑vasta‑aine. CD20‑molekyyli on solukalvon läpäisevä fosfoproteiini, joka ilmentyy B‑lymfosyyteissä pre‑B‑lymfosyyttivaiheesta alkaen aina kypsiin B‑lymfosyytteihin asti. Myös pieni osuus aktivoituneista T‑soluista ilmentää CD20‑molekyyliä. Ofatumumabin ihonalainen antoreitti ja myöhempi vapautuminen/absorptio kudoksesta mahdollistavat asteittaisen vuorovaikutuksen B-solujen kanssa.
Ofatumumabin sitoutuminen CD20:een indusoi CD20+‑B‑solujen hajoamisen valtaosin komplementtiriippuvaisen sytotoksisuuden (CDC) kautta ja vähäisemmässä määrin vasta‑aineriippuvaisen soluvälitteisen sytotoksisuuden (ADCC) kautta. Ofatumumabin on myös osoitettu indusoivan solujen hajoamista sekä runsaasti että niukasti CD20‑molekyyliä ilmentävissä soluissa. Ofatumumabi vähentää myös CD20‑molekyyliä ilmentäviä T‑soluja.
Farmakodynaamiset vaikutukset
B‑solujen depleetio
Kun kliinisissä RMS‑tutkimuksissa käytettiin ofatumumabia 20 mg 4 viikon välein aloitusannosten (20 mg päivinä 1, 7 ja 14) jälkeen, anto johti nopeaan ja pitkäkestoiseen B‑solumäärän pienenemiseen alle viitealueen alarajan (määritelmän mukaan 40 solua/µl) jo kahden viikon kuluttua hoidon aloittamisesta. Ennen viikolla 4 alkaneen ylläpitovaiheen aloittamista B‑solujen kokonaisarvo < 10 solua/mikrol saavutettiin 94 %:lla potilaista. Määrä suureni 98 %:iin viikon 12 kohdalla ja säilyi jopa 120 viikkoa (tutkimushoidon ajan).
B‑soluarvon korjautuminen
Tiedot vaiheen III kliinisistä RMS‑tutkimuksista osoittavat, että B‑soluarvon korjautuminen viitealueen alarajalle tai lähtöarvoon kesti 24,6 viikkoa (mediaani) hoidon lopettamisen jälkeen. Farmakokineettinen B‑solumallinnus ja B‑soluarvon korjautumisen simulaatio vahvistavat tiedot, ja niiden perusteella ennustettu aika (mediaani), jonka kuluessa B‑soluarvo on korjautunut viitealueen alarajalle, on 23 viikkoa hoidon lopettamisen jälkeen.
Immunogeenisuus
Vaiheen III RMS‑tutkimuksissa hoidon indusoimien vasta‑aineiden ilmaantuvuus lääkkeelle oli ofatumumabia saaneilla potilailla 0,2 % (2/914) eikä yhdelläkään potilaalla tunnistettu hoitoa tehostavia tai neutraloivia lääkevasta‑aineita. Positiivisten lääkevasta‑ainelöydösten vaikutusta ofatumumabin farmakokinetiikkaan, turvallisuusprofiiliin tai B‑solujen kinetiikkaan ei voitu arvioida lääkevasta‑aineiden matalan esiintyvyyden takia.
Kliininen teho ja turvallisuus
Ofatumumabin tehoa ja turvallisuutta arvioitiin kahdessa satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa vaiheen III avaintutkimuksessa, joiden tutkimusasetelmat olivat identtiset (tutkimus 1 [ASCLEPIOS I] ja tutkimus 2 [ASCLEPIOS II]). Potilaat olivat iältään 18–55‑vuotiaita ja sairastivat relapsoivaa MS‑tautia (RMS). Potilaiden toiminnanvajausta mittaava EDSS‑arvo (Expanded Disability Status Scale) oli seulontahetkellä 0–5,5, ja potilailla oli ollut vähintään yksi dokumentoitu pahenemisvaihe edeltävän vuoden aikana tai kaksi pahenemisvaihetta edeltävien kahden vuoden aikana tai gadoliniumilla tehostuva (Gd+) muutos magneettikuvauksessa edeltävän vuoden aikana. Tutkimukseen osallistui äskettäin diagnosoituja potilaita ja potilaita, jotka vaihtoivat nykyisestä hoidosta tutkimusvalmisteeseen.
Näissä kahdessa tutkimuksessa 927 RMS‑potilasta (ASCLEPIOS I) ja 955 RMS‑potilasta (ASCLEPIOS II) satunnaistettiin saamaan suhteessa 1:1 joko 20 mg ofatumumabia injektiona ihon alle 4 viikon välein alkaen viikosta 4 aloitusannosten jälkeen (kolme 20 mg annosta viikossa ensimmäisten 14 päivän aikana päivinä 1, 7 ja 14) tai teriflunomidia 14 mg kapseleina suun kautta kerran vuorokaudessa. Potilaat saivat myös toisen hoitoryhmän tutkimuslääkettä vastaavaa lumelääkettä sokkouttamisen varmistamiseksi (kaksoislumeasetelma).
Hoidon kesto vaihteli yksilöllisesti tutkimuksen päättymiskriteerien täyttymisajankohdan perusteella. Kummassakin tutkimuksessa hoidon keston mediaani oli 85 viikkoa; 33,0 % ofatumumabiryhmän potilaista vs. 23,2 % teriflunomidiryhmän potilaista sai hoitoa yli 96 viikon ajan.
Nämä kaksi tutkimusta ja hoitoryhmät olivat demografisilta ja lähtötilanteen ominaisuuksiltaan samankaltaisia (ks. taulukko 2). Ikäkeskiarvo oli 38 vuotta, taudin keskikesto 8,2 vuotta ensimmäisen oireen ilmaantumisesta ja EDSS‑keskiarvo 2,9; 40 % potilaista ei ollut saanut aiemmin taudinkulkuun vaikuttavaa hoitoa ja 40 %:lla oli todettu Gd‑tehostuvia T1‑muutoksia lähtötilanteen magneettikuvauksessa.
Ensisijainen tehon päätetapahtuma oli kummassakin tutkimuksessa EDSS‑arvoon perustuva vahvistettujen pahenemisvaiheiden vuotuinen määrä (ARR). Tärkeimpiä toissijaisia tehon päätetapahtumia olivat EDSS‑asteikolla mitattuun toiminnanvajauksen pahenemiseen kuluva aika (vahvistettiin 3 kuukauden ja 6 kuukauden kohdalla). Pahenemiseksi määriteltiin EDSS‑lähtöarvon 0 suurenema ≥ 1,5:llä, lähtöarvon 1–5 suurenema ≥ 1:llä tai lähtöarvon ≥ 5,5 suurenema ≥ 0,5:llä. Muita tärkeimpiä toissijaisia päätetapahtumia olivat Gd‑tehostuvien T1‑muutosten määrä per magneettikuvaus, uusien tai suurenevien T2‑muutosten vuotuinen määrä ja neurofilamentin kevytketjun (NfL) pitoisuus seerumissa. Toiminnanvajaukseen liittyviä tärkeimpiä toissijaisia päätetapahtumia arvioitiin ASCLEPIOS‑tutkimusten 1 ja 2 yhdistettyjen tietojen meta‑analyysissä, kuten tutkimussuunnitelmissa oli määritelty.
Taulukko 2 Demografiset ja lähtötilanteen ominaisuudet
| Ominaisuudet | Tutkimus 1 (ASCLEPIOS I) | Tutkimus 2 (ASCLEPIOS II) | ||
| Ofatumumabi (N = 465) | Teriflunomidi (N = 462) | Ofatumumabi (N = 481) | Teriflunomidi (N = 474) | |
| Ikä (keskiarvo ± keskihajonta; vuotta) | 39 ± 9 | 38 ± 9 | 38 ± 9 | 38 ± 9 |
| Sukupuoli (naisia; %) | 68,4 | 68,6 | 66,3 | 67,3 |
| MS‑taudin toteamisesta kulunut aika (keskiarvo/mediaani; vuotta) | 5,77 / 3,94 | 5,64 / 3,49 | 5,59 / 3,15 | 5,48 / 3,10 |
| Aiemmin taudinkulkuun vaikuttavaa hoitoa saaneita (%) | 58,9 | 60,6 | 59,5 | 61,8 |
| Pahenemisvaiheiden määrä edeltävien 12 kk aikana | 1,2 | 1,3 | 1,3 | 1,3 |
| EDSS‑arvo (keskiarvo/mediaani) | 2,97 / 3,00 | 2,94 / 3,00 | 2,90 / 3,00 | 2,86 / 2,50 |
| T2‑muutosten kokonaistilavuuden keskiarvo (cm3) | 13,2 | 13,1 | 14,3 | 12,0 |
| Potilaita, joilla Gd+ T1‑muutoksia (%) | 37,4 | 36,6 | 43,9 | 38,6 |
| Gd+ T1‑muutosten määrä (keskiarvo) | 1,7 | 1,2 | 1,6 | 1,5 |
Molempien tutkimusten tehotulosten yhteenveto esitetään taulukossa 3, kuvassa 1 ja kuvassa 2.
Ofatumumabi vähensi pahenemisvaiheiden vuotuista määrää merkitsevästi teriflunomidiin verrattuna kummassakin vaiheen III tutkimuksessa, 50,5 % ASCLEPIOS I ‑tutkimuksessa ja 58,4 % ASCLEPIOS II ‑tutkimuksessa.
Yhdistettyjen tietojen ennalta määritelty meta‑analyysi osoitti, että teriflunomidiin verrattuna ofatumumabi pienensi merkitsevästi sekä 3 kuukauden vahvistetun toiminnanvajauksen etenemisen riskiä (34,3 %) ja 6 kuukauden vahvistetun toiminnanvajauksen etenemisen riskiä (32,4 %) (ks. kuva 1).
Ofatumumabi vähensi merkitsevästi Gd‑tehostuvien T1‑muutosten määrää, 95,9 %, ja uusien tai suurenevien T2‑muutosten määrää, 83,5 %, verrattuna teriflunomidiin (arvot edustavat vähenemisen keskiarvoja yhdistettyjä tutkimustietoja tarkasteltaessa).
Ofatumumabi pienensi NfL-pitoisuutta merkitsevästi teriflunomidiin verrattuna ensimmäisestä arvioinnista lähtien 3 kuukauden kohdalla (ks. taulukko 3 ja kuva 2).
Eksploratiivisessa alaryhmäanalyysissa kahdesta vaiheen III tutkimuksesta ofatumumabilla todettiin samankaltainen vaikutus keskeisiin tehotuloksiin verrattuna teriflunomidiin sukupuolen, iän, painon, aiemman (ei-steroidipohjaisen) MS-lääkityksen, lähtötilanteen toiminnanvajauksen ja tautiaktiivisuuden mukaan jaetuissa alaryhmissä.
Taulukko 3 Yleiskatsaus vaiheen III RMS‑tutkimusten tärkeimpiin tuloksiin
| Päätetapahtumat | Tutkimus 1 (ASCLEPIOS I) | Tutkimus 2 (ASCLEPIOS II) | ||
Ofatumumabi 20 mg (n = 465) | Teriflunomidi 14 mg (n = 462) | Ofatumumabi 20 mg (n = 481) | Teriflunomidi 14 mg (n = 474) | |
| Päätetapahtumat erillisissä tutkimuksissa | ||||
| Pahenemisvaiheiden vuotuinen määrä (ARR) (ensisijainen päätetapahtuma)1 | 0,11 | 0,22 | 0,10 | 0,25 |
| Määrän pienenemä | 50,5 % (p < 0,001) | 58,4 % (p < 0,001) | ||
| Gd‑tehostuvien T1‑muutosten keskimäärä per magneettikuvaus | 0,0115 | 0,4555 | 0,0317 | 0,5172 |
| Suhteellinen vähenemä | 97,5 % (p < 0,001) | 93,9 % (p < 0,001) | ||
| Uusien tai suurenevien T2‑muutosten vuotuinen määrä | 0,72 | 4,00 | 0,64 | 4,16 |
| Suhteellinen vähenemä | 81,9 % (p < 0,001) | 84,6 % (p < 0,001) | ||
| NfL 3 kuukauden kohdalla (pg/ml) | 8,80 | 9,41 | 8,92 | 10,02 |
| Suhteellinen vähenemä | 7 % (p = 0,011) | 11 % (p < 0,001) | ||
| Ennalta määriteltyihin meta‑analyyseihin perustuvat päätetapahtumat | ||||
3 kuukauden vahvistettu toiminnanvajauksen eteneminen, osuus potilaista2 Riskin pienenemä | 10,9 % ofatumumabi vs. 15,0 % teriflunomidi 34,3 % (p = 0,003) | |||
6 kuukauden vahvistettu toiminnanvajauksen eteneminen, osuus potilaista2 Riskin pienenemä | 8,1 % ofatumumabi vs. 12,0 % teriflunomidi 32,4 % (p = 0,012) | |||
1 Vahvistetut pahenemisvaiheet (joihin liittyy kliinisesti merkittävä EDSS‑arvon muutos). 2 Kaplan–Meier‑estimaatti 24 kuukauden kohdalla. 3 ja 6 kuukauden vahvistettu toiminnanvajauksen eteneminen arvioitiin kyseisten kahden vaiheen III tutkimuksen yhdistettyjen tietojen prospektiivisesti suunnitellun analyysin perusteella. Etenemiseksi määriteltiin kliinisesti merkittävä EDSS‑arvon suurenema, joka kesti vähintään 3 tai 6 kuukautta. Kliinisesti merkittäväksi EDSS‑arvon suurenemaksi määriteltiin vähintään 1,5 pisteen suurenema EDSS‑lähtöarvon ollessa 0 pistettä, vähintään 1,0 pisteen suurenema EDSS‑lähtöarvon ollessa 1,0–5,0 pistettä ja vähintään 0,5 pisteen suurenema EDSS‑lähtöarvon ollessa 5,5 pistettä tai enemmän. | ||||
Kuva 1 Aika ensimmäiseen 3 kuukauden vahvistettuun toiminnanvajauksen etenemiseen eri hoitoryhmissä (ASCLEPIOS‑tutkimus 1 ja ‑tutkimus 2 yhdistettynä, koko analyysipopulaatio)
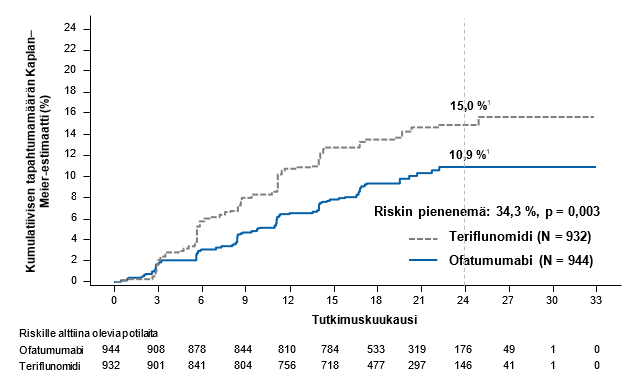
1 Käyrien yllä olevat luvut ovat Kaplan–Meier‑estimaatteja tapahtuman riskistä 24 kuukauden kohdalla (merkitty pystysuoralla katkoviivalla).
Kuva 2 Seerumin NfL-pitoisuudet eri hoitoryhmissä (ASCLEPIOS-tutkimus 1 ja tutkimus 2 yhdistettynä, koko analyysipopulaatio)
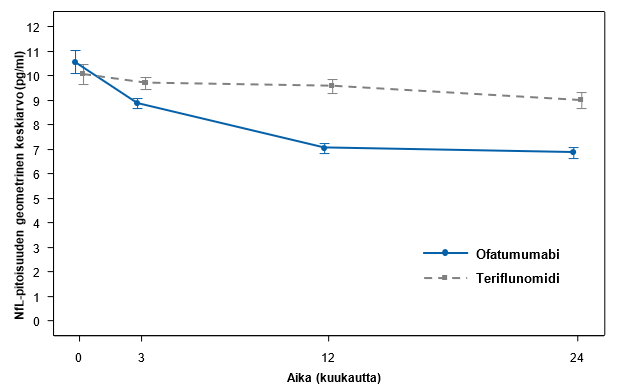
Viivakuviot esittävät toistettujen mittausten malliin perustuvaa mukautettua geometrista keskiarvoa (luottamusväli 95 %) kussakin aikapisteessä. Lähtötilanteen geometriset keskiarvot on johdettu seerumin NfL-pitoisuuksien raaka-arvojen luonnollisen logaritmin eksponentoidusta aritmeettisesta keskiarvosta.
Vaiheen III tutkimuksissa haittatapahtuman kokeneiden potilaiden määrä (83,6 % vs. 84,2 %) ja hoidon lopettamiseen johtaneiden haittatapahtumien määrä (5,7 % vs. 5,2 %) olivat ofatumumabi‑ ja teriflunomidiryhmissä samaa luokkaa.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Kesimpta‑valmisteen käytöstä multippeliskleroosin hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Ihon alle annetulla ofatumumabilla on pitkittynyt vapautumis-/absorptioprofiili (Tmax 4,3 päivää) ja se imeytyy valtaosin imukudoksen kautta.
Kuukausittainen 20 mg annos ihon alle tuottaa AUCtau‑keskiarvon 483 mikrog*h/ml ja Cmax‑keskiarvon 1,43 mikrog/ml vakaassa tilassa.
Jakautuminen
Vakaan tilan jakautumistilavuus oli arviolta 5,42 litraa, kun ofatumumabia annettiin toistuvasti ihon alle 20 mg annoksina.
Biotransformaatio
Ofatumumabi on proteiini, jonka oletettu metaboliareitti on pilkkoutuminen pieniksi peptideiksi ja aminohapoiksi kaikkialla elimistössä esiintyvien proteolyyttisten entsyymien toimesta.
Eliminaatio
Kuten muutkin IgG‑molekyylit, ofatumumabi eliminoituu kahdella tavalla: kohdemolekyylin kautta välittyvän reitin kautta (sitoutuminen B‑soluihin) ja kohdemolekyylistä riippumattoman reitin kautta (ei‑spesifisen endosytoosin ja tätä seuraavan solunsisäisen katabolian kautta). B‑solujen läsnäolo lähtötilanteessa johtaa kohdemolekyylin kautta välittyvän ofatumumabin puhdistuman suurempaan osuuteen hoidon alussa. Ofatumumabin anto johtaa voimakkaaseen B‑solukatoon, mikä johtaa kokonaispuhdistuman pienenemiseen.
Vakaan tilan puoliintumisaika oli arviolta noin 11 vrk, kun ofatumumabia annettiin toistuvasti ihon alle 20 mg annoksina.
Lineaarisuus/ei‑lineaarisuus
Ofatumumabin farmakokinetiikka oli ei‑lineaarinen, mikä johtui sen puhdistuman vähenemisestä ajan mittaan.
Erityisryhmät
Yli 55‑vuotiaat aikuiset
Yli 55‑vuotiailla potilailla ei ole tehty spesifisiä ofatumumabia koskevia farmakokineettisiä tutkimuksia vähäisen kliinisen kokemuksen takia (ks. kohta Annostus ja antotapa).
Pediatriset potilaat
Tutkimuksissa ei ole arvioitu ofatumumabin farmakokinetiikkaa alle 18‑vuotiailla pediatrisilla potilailla.
Sukupuoli
Sukupuolella oli vähäinen (12 %) vaikutus ofatumumabin sentraaliseen jakautumistilavuuteen tutkimusten välisessä populaatioanalyysissä; naispotilailla havaittiin suurempia Cmax‑ ja AUC‑arvoja (48 % analysoiduista potilaista oli miehiä ja 52 % naisia). Vaikutusten ei katsota olevan kliinisesti merkittäviä eikä annoksen muuttamista suositella.
Paino
Tutkimusten välisen populaatioanalyysin tulosten perusteella todettiin, että paino oli ofatumumabialtistuksen (Cmax ja AUC) kovariaatti RMS-potilailla. Paino ei kuitenkaan vaikuttanut kliinisissä tutkimuksissa arvioituihin turvallisuuden eikä tehon mittareihin, joten annosta ei tarvitse muuttaa.
Munuaisten vajaatoiminta
Ofatumumabia ei ole tutkittu spesifisesti munuaisten vajaatoimintapotilailla.
Kliinisiin tutkimuksiin otettiin potilaita, joilla oli lievä munuaisten vajaatoiminta. Keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastavista ei ole kokemusta. Ofatumumabi ei kuitenkaan erity virtsan kautta, joten annosta ei todennäköisesti tarvitse muuttaa munuaisten vajaatoimintapotilailla.
Maksan vajaatoiminta
Ofatumumabia ei ole tutkittu maksan vajaatoimintapotilailla.
Monoklonaalisten vasta‑aineiden kuten ofatumumabin maksametabolia on merkityksetöntä, joten maksan vajaatoiminta ei vaikuttane ofatumumabin farmakokinetiikkaan. Näin ollen annosta ei todennäköisesti tarvitse muuttaa maksan vajaatoimintapotilaille.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten (mukaan lukien turvallisuusfarmakologisten) tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Ofatumumabin karsinogeenisuutta tai mutageenisuutta ei ole tutkittu. Vasta‑aineena ofatumumabilla ei todennäköisesti ole suoraa vuorovaikutusta DNA:n kanssa.
Alkion‑ ja sikiönkehitystä koskevat tutkimukset ja laajennetut pre‑ ja postnataalista kehitystä koskevat tutkimukset (ePPND) apinoilla osoittivat, että tiineyden aikainen altistus laskimoon annetulle ofatumumabille ei aiheuttanut toksisuutta emolle, teratogeenisuutta eikä haittavaikutuksia alkion‑ tai sikiönkehitykseen eikä pre‑ tai postnataaliseen kehitykseen.
Kyseisissä tutkimuksissa ofatumumabia havaittiin sikiöiden ja poikasten veressä. Tämä vahvistaa, että ofatumumabi läpäisee istukan ja sikiön altistus ofatumumabille on havaittavissa vielä postnataalisesti (syynä monoklonaalisen vasta‑aineen pitkä puoliintumisaika). Altistus suurille ofatumumabiannoksille tiineyden aikana johti odotetusti CD20+‑B‑solukatoon emoilla ja niiden sikiöillä ja poikasilla sekä pernan painon pienenemiseen (ei histologista korrelaattia) sikiöillä ja keyhole limpet ‑kotilon hemosyaniinin (KLH) tuottaman humoraalisen immuunivasteen heikentymiseen poikasilla. Kaikki muutokset korjautuivat 6 kuukauden postnataalikauden aikana. Poikasilla havaittiin kuolleisuutta postnataalikauden varhaisvaiheessa, kun annos oli 160‑kertainen terapeuttiseen annokseen verrattuna (AUC:n perusteella). Kuolleisuus johtui todennäköisesti mahdollisista immunomodulaatioon liittyvistä infektioista. Poikasten ePPND‑tutkimuksessa ofatumumabin farmakologiseen vaikutukseen liittyvä NOAEL‑annos (pitoisuus, jonka yhteydessä ei todettu mitään haitallista vaikutusta) tuotti AUC‑arvon perusteella vähintään 22‑kertaisen turvallisuusmarginaalin, kun emon NOAEL‑altistusta verrataan ihmisen altistukseen käytettäessä terapeuttista annostusta 20 mg kuukaudessa.
Spesifisessä apinoiden hedelmällisyyttä koskeneessa tutkimuksessa urosten ja naaraiden hedelmällisyyden päätetapahtumiin ei kohdistunut vaikutuksia.
Farmaseuttiset tiedot
Apuaineet
L‑arginiini
Natriumasetaattitrihydraatti
Natriumkloridi
Polysorbaatti 80 (E433)
Dinatriumedetaattidihydraatti
Kloorivetyhappo (pH:n säätelyyn)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Säilytys
Kesimpta 20 mg injektioneste, liuos, esitäytetty ruisku
Säilytä jääkaapissa (2 °C – 8 °C). Ei saa jäätyä.
Tarvittaessa Kesimpta-valmistetta voidaan säilyttää kertaluonteisesti jääkaapin ulkopuolella, huoneenlämmössä (alle 30 °C) korkeintaan 7 päivää. Jos sitä ei käytetä tänä aikana, Kesimpta voidaan palauttaa jääkaappiin korkeintaan 7 päivän ajaksi.
Pidä esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Kesimpta 20 mg injektioneste, liuos, esitäytetty kynä
Säilytä jääkaapissa (2 °C – 8 °C). Ei saa jäätyä.
Tarvittaessa Kesimpta-valmistetta voidaan säilyttää kertaluonteisesti jääkaapin ulkopuolella, huoneenlämmössä (alle 30 °C) korkeintaan 7 päivää. Jos sitä ei käytetä tänä aikana, Kesimpta voidaan palauttaa jääkaappiin korkeintaan 7 päivän ajaksi.
Pidä esitäytetty kynä ulkopakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
KESIMPTA injektioneste, liuos, esitäytetty kynä
20 mg (L:ei) 1 kpl (0,4 ml, 50 mg/ml) (1256,14 €)
PF-selosteen tieto
Kesimpta 20 mg injektioneste, liuos, esitäytetty ruisku
Kesimpta toimitetaan kertakäyttöisessä lasiruiskussa, jossa on ruostumattomasta teräksestä valmistettu neula, männän pysäytin ja jäykkä neulansuojus. Ruiskun osia ovat lisäksi mäntä ja turvasuojus.
Kesimpta on saatavana yksikköpakkauksissa, joissa on 1 esitäytetty ruisku, ja monipakkauksissa, joissa on 3 esitäytettyä ruiskua (3 yhden ruiskun pakkausta).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Kesimpta 20 mg injektioneste, liuos, esitäytetty kynä
Kesimpta toimitetaan kertakäyttöisessä lasiruiskussa, jossa on ruostumattomasta teräksestä valmistettu neula, männän pysäytin ja jäykkä neulansuojus. Ruisku on asennettu autoinjektoriin.
Kesimpta on saatavana yksikköpakkauksissa, joissa on 1 esitäytetty kynä, ja monipakkauksissa, joissa on 3 esitäytettyä kynää (3 yhden kynän pakkausta).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Liuos on kirkasta tai hieman opaalinhohtoista ja väritöntä tai hieman rusehtavankeltaista.
Käyttö- ja käsittelyohjeet
Esitäytetyn ruiskun käsittelyohjeet
Ennen injektiota esitäytetty ruisku on otettava jääkaapista lämpenemään huoneenlämpöiseksi noin 15–30 minuutiksi. Esitäytetty ruisku on pidettävä alkuperäisessä kotelossa käyttöön asti, ja neulan suojuksen saa poistaa vasta juuri ennen lääkkeen pistämistä. Ennen käyttöä liuos on tarkastettava silmämääräisesti tarkistusikkunasta. Esitäytettyä ruiskua ei saa käyttää, jos neste sisältää näkyviä hiukkasia tai on täysin sameaa.
Pakkausselosteessa on valmisteen antoa koskevat kattavat ohjeet.
Esitäytetyn kynän käsittelyohjeet
Ennen injektiota esitäytetty kynä on otettava jääkaapista lämpenemään huoneenlämpöiseksi noin 15–30 minuutiksi. Esitäytetty kynä on pidettävä alkuperäisessä kotelossa käyttöön asti, ja korkin saa poistaa vasta juuri ennen lääkkeen pistämistä. Ennen käyttöä liuos on tarkastettava silmämääräisesti tarkistusikkunasta. Esitäytettyä kynää ei saa käyttää, jos neste sisältää näkyviä hiukkasia tai on täysin sameaa.
Pakkausselosteessa on valmisteen antoa koskevat kattavat ohjeet.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
KESIMPTA injektioneste, liuos, esitäytetty kynä
20 mg 1 kpl
- Ylempi erityiskorvaus (100 %). Dimetyylifumaraatti, diroksimeelifumaraatti, glatirameeriasetaatti, interferoni beeta, ofatumumabi ja ponesimodi: Aaltoilevan tai aaltoilevaan läheisesti rinnastettavan MS-taudin hoito erityisin edellytyksin (157).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Dimetyylifumaraatti, diroksimeelifumaraatti, glatirameeriasetaatti, interferoni beeta, ofatumumabi ja ponesimodi: Aaltoilevan ja aaltoilevaan läheisesti rinnastettavan MS-taudin hoito erityisin edellytyksin (303).
ATC-koodi
L04AG12
Valmisteyhteenvedon muuttamispäivämäärä
19.06.2026
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com