
VYNDAQEL kapseli, pehmeä 61 mg
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi pehmeä kapseli sisältää 61 mg mikronoitua tafamidiisia.
Apuaine, jonka vaikutus tunnetaan
Yksi pehmeä kapseli sisältää enintään 44 mg sorbitolia (E 420).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, pehmeä.
Kliiniset tiedot
Käyttöaiheet
Vyndaqel on tarkoitettu villityypin tai perinnöllisen transtyretiinivälitteisen amyloidoosin hoitoon aikuispotilaille, joilla on kardiomyopatia (ATTR-CM).
Ehto
Hoito on aloitettava amyloidoosin tai kardiomyopatian hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito on aloitettava amyloidoosin tai kardiomyopatian hoitoon perehtyneen lääkärin valvonnassa.
Kun sairausepäily herää tietynlaisen sairaushistorian tai sydämen vajaatoiminnan tai kardiomyopatian merkkejä omaavan potilaan kohdalla, on amyloidoosin tai kardiomyopatian hoitoon perehtyneen lääkärin ennen tafamidiisihoidon aloittamista tehtävä etiologinen diagnoosi ATTR-CM:n vahvistamiseksi ja AL-amyloidoosin poissulkemiseksi asianmukaisilla arviointimenetelmillä, kuten luustokartoituksella (gammakuvaus) ja veri-/virtsakokeella ja/tai näytepalan histologisella tutkimuksella ja transtyretiini (TTR) -genotyypityksellä villityypin tai perinnöllisen muodon selvittämiseksi.
Annostus
Suositeltu annos on yksi Vyndaqel 61 mg kapseli (tafamidiisi) suun kautta kerran päivässä (ks. kohta Farmakodynamiikka).
Vyndaqel 61 mg (tafamidiisi) vastaa 80 mg:aa tafamidiisimeglumiinia. Tafamidiisi ja tafamidiisimeglumiini eivät ole keskenään vaihdettavissa milligrammamäärän perusteella (ks. kohta Farmakokinetiikka).
Vyndaqel-hoito on aloitettava mahdollisimman pian, taudin varhaisvaiheessa, jotta siitä saatava kliininen hyöty taudin etenemiseen olisi mahdollisimman suuri. Jos amyloidiin liittyvä sydänvaurio on pidemmälle edennyt, esim. NYHA-luokkaa III, päätöksen hoidon aloittamisesta tai sen jatkamisesta tulee perustua amyloidoosin tai kardiomyopatian hoitoon perehtyneen lääkärin huolelliseen harkintaan (ks. kohta Farmakodynamiikka). Kliinistä tietoa NYHA-luokan IV potilaiden hoidosta on vain rajallisesti.
Jos potilas oksentaa pian Vyndaqel-kapselin ottamisen jälkeen ja hän oksentaa ulos ehjän kapselin, potilaan on otettava uusi Vyndaqel-annos, jos mahdollista. Jos kapselia ei havaita oksentamisen yhteydessä, niin uuden annoksen ottaminen ei ole tarpeen ja Vyndaqel-hoitoa jatketaan seuraavana päivänä tavanomaiseen tapaan.
Erityisryhmät
Iäkkäät
Annostusta ei tarvitse muuttaa iäkkäitä potilaita (≥ 65‑vuotiaat) hoidettaessa (ks. kohta Farmakokinetiikka).
Maksan ja munuaisten vajaatoiminta
Munuaisten vajaatoimintaa tai lievää tai keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden annostusta ei tarvitse muuttaa. Vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma enintään 30 ml/min) sairastavista potilaista on saatavilla vain vähän tietoa. Tafamidiisia ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla, joten heitä hoidettaessa tulee noudattaa varovaisuutta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Ei ole asianmukaista käyttää tafamidiisia pediatrisille potilaille.
Antotapa
Suun kautta.
Pehmeät kapselit niellään kokonaisina. Niitä ei saa murskata eikä paloitella. Vyndaqel-kapselin voi ottaa ruokailun yhteydessä tai tyhjään mahaan.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Naisten, jotka voivat tulla raskaaksi, on käytettävä asianmukaista raskauden ehkäisyä tafamidiisin käytön aikana ja jatkettava asianmukaisen ehkäisyn käyttöä vielä yhden kuukauden ajan tafamidiisihoidon lopettamisen jälkeen (ks. kohta Raskaus ja imetys).
Tafamidiisi on lisättävä transtyretiinivälitteistä amyloidoosia sairastavien potilaiden tavanomaiseen hoitoon. Lääkärin on seurattava potilaan tilaa ja jatkettava muun hoidon tarpeen arviointia, myös elinsiirron tarvetta, osana tavanomaista hoitoa. Koska elinsiirron jälkeisestä tafamidiisihoidosta ei ole tietoja, tafamidiisihoito on lopetettava, jos potilaalle tehdään elinsiirto.
Maksan toimintakoearvot saattavat kohota ja tyroksiiniarvot saattavat laskea (ks. kohdat Yhteisvaikutukset ja Haittavaikutukset).
Tafamidiisin ja rintasyövän resistenssiproteiinin (breast cancer resistance protein, BCRP) substraattien samanaikainen anto voi lisätä altistusta BCRP:n substraatille. Samanaikaisen annon yhteydessä potilasta on seurattava BCRP:n substraattiin liittyvien haittavaikutusten varalta ja BCRP:n substraatin annoksen pienentämistä voidaan harkita (ks. kohta Yhteisvaikutukset).
Tämä lääkevalmiste sisältää enintään 44 mg sorbitolia per kapseli. Sorbitoli on fruktoosin lähde.
Sorbitolia (tai fruktoosia) sisältävien muiden valmisteiden samanaikaisen annon sekä ravinnosta saatavan sorbitolin (tai fruktoosin) additiivinen vaikutus on huomioitava.
Suun kautta otettavien lääkevalmisteiden sorbitoli saattaa vaikuttaa muiden suun kautta otettavien lääkkeiden biologiseen hyötyosuuteen.
Yhteisvaikutukset
Terveillä vapaaehtoisilla koehenkilöillä tehdyssä kliinisessä tutkimuksessa 20 mg tafamidiisimeglumiinia ei estänyt eikä indusoinut sytokromi P450 ‑entsyymiä CYP3A4.
In vitro -olosuhteissa tafamidiisi estää BCRP-effluksikuljettajaproteiinia IC50-arvolla 1,16 µM ja voi aiheuttaa lääkeaineiden välisiä yhteisvaikutuksia kliinisesti merkityksellisillä pitoisuuksilla tämän kuljettajaproteiinin substraattien (esim. metotreksaatti, rosuvastatiini, atorvastatiini, apiksabaani, rivaroksabaani, imatinibi) kanssa. Terveillä osallistujilla tehdyssä kliinisessä tutkimuksessa altistus rosuvastatiinille (BCRP:n substraatti) kasvoi useiden päivittäisten 61 mg:n tafamidiisiannosten jälkeen noin 2‑kertaiseksi. Tästä syystä potilasta on seurattava BCRP:n substraattiin liittyvien haittavaikutusten varalta, jos BCRP:n substraattia käytetään samanaikaisesti tafamidiisin kanssa. BCRP:n substraatin annoksen pienentämistä voidaan harkita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Samoin tafamidiisi estää orgaanisten anionien kuljettajaproteiinia OAT1 IC50-arvolla 2,9 µM ja orgaanisten anionien kuljettajaproteiinia OAT3 IC50-arvolla 2,36 µM ja voi aiheuttaa lääkeaineiden välisiä yhteisvaikutuksia kliinisesti merkittävillä pitoisuuksilla näiden kuljettajaproteiinien substraattien (esim. tulehduskipulääkkeet, bumetadini, furosemidi, lamivudiini, metotreksaatti, oseltamiviiri, tenofoviiri, gansikloviiri, adefoviiri, sidofoviiri, tsidovudiini, tsalsitabiini) kanssa. In vitro ‑tietojen perusteella OAT1‑ ja OAT3‑substraattien AUC-arvon maksimaalisen ennustetun muutoksen todettiin olevan alle 1,25 tafamidiisiannoksella 61 mg ja siksi tafamidiisin aikaansaaman OAT1‑ ja OAT3-kuljettajaproteiinien eston ei odoteta aiheuttavan kliinisesti merkittäviä yhteisvaikutuksia.
Yhteisvaikutustutkimuksia ei ole tehty muiden lääkevalmisteiden tafamidiisiin kohdistuvien vaikutusten selvittämiseksi.
Laboratoriotutkimusten poikkeama
Tafamidiisi saattaa alentaa kokonaistyroksiini-pitoisuutta seerumissa ilman, että siihen liittyy vapaan tyroksiinin (T4) tai tyreotropiinin (TSH) muutoksia. Kokonaistyroksiini-arvoa koskeva havainto johtuu todennäköisesti tyroksiinin vähentyneestä sitoutumisesta transtyretiiniin (TTR) tai tyroksiinin syrjäytymisestä siitä, koska tafamidiisin sitoutumisaffiniteetti TTR-tyroksiinireseptoriin on suuri. Kilpirauhasen toimintahäiriöön sopivia vastaavia kliinisiä löydöksiä ei ole havaittu.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä raskauden ehkäisyä tafamidiisihoidon aikana sekä kuukauden ajan hoidon päättymisen jälkeen, koska lääkeaineen puoliintumisaika on pitkä.
Raskaus
Ei ole olemassa tietoja tafamidiisin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu kehitystoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Tafamidiisihoitoa ei suositella raskauden aikana eikä naisille, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Olemassa olevat tiedot koe-eläimistä ovat osoittaneet tafamidiisin erittyvän rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. Tafamidiisia ei pidä käyttää rintaruokinnan aikana.
Hedelmällisyys
Ei-kliinisissä tutkimuksissa ei havaittu hedelmällisyyden heikkenemistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Farmakodynaamisen ja farmakokineettisen profiilin perusteella tafamidiisilla ei uskota olevan haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Turvallisuustiedot perustuvat tietoihin, jotka on saatu 176 potilaalta, joilla on ATTR-CM, ja jotka altistettiin päivittäiselle 80 mg:n tafamidiisimeglumiiniannokselle (= 4 x 20 mg) 30 kuukauden lumekontrolloidussa tutkimuksessa (ks. kohta Farmakodynamiikka).
Haittatapahtumien esiintyvyys oli 80 mg:lla tafamidiisimeglumiinia hoidetuilla potilailla tavallisesti samaa luokkaa ja verrattavissa lumelääkettä saaneisiin potilaisiin.
Seuraavia haittatapahtumia raportoitiin useammin tafamidiisimeglumiinia 80 mg saaneilla potilailla kuin lumelääkettä saaneilla potilailla: ilmavaivat [8 potilasta (4,5 %) versus 3 potilasta (1,7 %)] ja maksan toimintakoearvojen kohoaminen [6 potilasta (3,4 %) versus 2 potilasta (1,1 %)]. Syy-yhteyttä ei ole todettu.
Turvallisuustietoja 61 mg:n tafamidiisiannoksesta on saatavissa avoimesta pitkäkestoisesta jatkotutkimuksesta.
Haittavaikutustaulukko
Haittavaikutukset on lueteltu seuraavassa MedDRA:n elinjärjestelmä- ja yleisyysluokituksen mukaan. Yleisyysluokissa on käytetty tavanomaisia termejä: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10) ja melko harvinainen (≥ 1/1 000, < 1/100). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä. Jäljempänä olevassa taulukossa luetellut haittavaikutukset perustuvat transtyretiinivälitteistä amyloidista kardiomyopatiaa (ATTR-CM) sairastavilla tutkittavilla tehtyjen kumulatiivisten kliinisten tutkimusten tietoihin.
| Elinjärjestelmäluokka | Yleinen |
| Ruoansulatuselimistö | Ripuli |
| Iho ja ihonalainen kudos | Ihottuma Kutina |
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet
Yliannostuksesta on hyvin vähän käytännön kokemusta. Kliinisissä tutkimuksissa kaksi potilasta, joilla oli diagnosoitu transtyretiinivälitteinen amyloidinen kardiomyopatia (ATTR‑CM), otti vahingossa suun kautta yhden 160 mg:n tafamidiisimeglumiiniannoksen ilman, että siihen liittyi haittatapahtumia. Suurin tafamidiisimeglumiiniannos, joka kliinisessä tutkimuksessa on annettu terveille vapaaehtoisille koehenkilöille, oli 480 mg:n kerta-annos. Tällä annoksella raportoitiin yksi hoitoon liittynyt haittatapahtuma, joka oli lievä luomirauhastulehdus.
Hoito
Yliannostustapauksessa potilaalle on annettava tavanomaista elintoimintoja tukevaa hoitoa tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut hermostoon vaikuttavat lääkeaineet, ATC-koodi: N07XX08
Vaikutusmekanismi
Tafamidiisi on transtyretiinin (TTR) selektiivinen stabiloija. Tafamidiisi sitoutuu TTR:ään tyroksiinin sitoutumiskohtiin stabiloiden tetrameerin ja hidastaen sen hajoamista monomeereiksi. Tämä on amyloidogeenisen prosessin nopeutta rajoittava vaihe.
Farmakodynaamiset vaikutukset
Transtyretiinivälitteinen amyloidoosi on vaikeasti invalidisoiva sairaus, joka johtuu erilaisten liukenemattomien fibrillaaristen proteiinien, eli amyloidin, kertymisestä kudoksiin sellaisina määrinä, jotka heikentävät kudosten normaalia toimintaa. Transtyretiinitetrameerin pilkkoutuminen monomeereiksi on transtyretiinivälitteisen amyloidoosin patogeneesin etenemisnopeutta rajoittava vaihe. Laskostuneet monomeerit käyvät läpi osittaisen denaturaation, jossa niistä muodostuu väärin laskostuneita monomeerisia amyloidogeenisia välimuotoja. Nämä välimuodot muodostavat rakenteeltaan virheellisiä liukoisia oligomeereja, profilamentteja, filamentteja ja amyloidisäikeitä. Tafamidiisi sitoutuu negatiivisella ko-operatiivisuudella transtyretiinin natiivin tetrameerinmuodon kahteen tyroksiinisitoutumiskohtaan ja estää siten sen hajoamista monomeereiksi. Tafamidiisia käytetään ATTR-CM-potilaille, koska se estää TTR-tetrameerin hajoamista.
Farmakodynaamisena markkerina käytettiin TTR:n stabiloitumismääritystä, jolla arvioitiin TTR-tetrameerin stabiiliutta.
Tafamidiisi stabiloi sekä villityypin että 14 muun TTR-variantin tetrameerirakenteet kliinisissä testeissä, jotka tehtiin kerran päivässä tapahtuneen tafamidiisin annostelun jälkeen. Tafamidiisi stabiloi myös ex vivo testatut 25 TTR-tetrameerivarianttia, mikä osoittaa 40 amyloidogeenisen TTR-genotyypin TTR:n stabiloitumisen.
Kansainvälisessä, kaksoissokkoutetussa, lumekontrolloidussa, satunnaistetussa monikeskustutkimuksessa (ks. kohta Kliininen teho ja turvallisuus) TTR:n havaittiin stabiloituneen ensimmäisen kuukauden jälkeen ja vaikutus säilyi 30. kuukauden loppuun saakka.
Sydämen vajaatoimintaan liittyvät biomarkkerit (NT‑proBNP ja troponiini I) suosivat Vyndaqel-valmistetta lumelääkkeeseen verrattuna.
Kliininen teho ja turvallisuus
Teho osoitettiin kansainvälisessä, kaksoissokkoutetussa, lumekontrolloidussa, satunnaistetussa monikeskustutkimuksessa, jossa oli 3 hoitoryhmää ja 441 potilaista, joilla oli joko villityypin tai perinnöllinen ATTR‑CM.
Potilaat satunnaistettiin saamaan joko tafamidiisimeglumiinia 20 mg (n = 88) tai 80 mg (annettuna neljänä 20 mg:n tafamidiisimeglumiinikapselina) (n = 176) tai samannäköistä lumelääkettä (n = 177) kerran päivässä tavanomaisen hoidon (esim. diureettien) lisäksi 30 kuukauden ajan. Hoidon määräytyminen stratifiointiin sen mukaan, oliko potilaalla TTR-geenivariantti vai ei sekä lähtötilanteessa todetun sairauden vaikeusasteen (NYHA-luokka) mukaan. Taulukossa 1 on kuvailtu potilaiden demografiset tiedot ja ominaisuudet lähtötilanteessa.
Taulukko 1: Potilaiden demografiset tiedot ja ominaisuudet lähtötilanteessa
| Ominaisuus | Yhdistetyt tafamidiisiryhmät N = 264 | Lumelääke N = 177 |
| Ikä — vuosia | ||
| Keskiarvo (keskihajonta) | 74,5 (7,2) | 74,1 (6,7) |
| Mediaani (minimi, maksimi) | 75 (46, 88) | 74 (51, 89) |
| Sukupuoli — lukumäärä (%) | ||
| Miehet | 241 (91,3) | 157 (88,7) |
| Naiset | 23 (8,7) | 20 (11,3) |
| TTR‑genotyyppi — lukumäärä (%) | ||
| ATTRm | 63 (23,9) | 43 (24,3) |
| ATTRwt | 201 (76,1) | 134 (75,7) |
| NYHA-luokka — lukumäärä (%) | ||
| NYHA-luokka I | 24 (9,1) | 13 (7,3) |
| NYHA-luokka II | 162 (61,4) | 101 (57,1) |
| NYHA-luokka III | 78 (29,5) | 63 (35,6) |
Lyhenteet: ATTRm = mutatoitunut transtyretiiniamyloidi, ATTRwt = villityypin transtyretiiniamyloidi, NYHA = New York Heart Association.
Ensisijaisessa analyysissa määritettiin hierarkisesti kokonaiskuolleisuus sen syistä riippumattomasti sekä sydän- ja verisuoniperäisten sairaalahoitojen esiintyvyys käyttäen Finkelstein-Schoenfeld (F‑S) ‑menetelmää. Jälkimmäinen määritellään kertoina, joina potilas joutuu sairaalahoitoon (otetaan hoitoon sairaalan vuodeosastolle) sydän‑ ja verisuonisairauteen liittyvän syyn takia. Menetelmässä verrattiin pareittain kutakin potilaista kaikkia potilaita vastaan jokaisessa tutkimusotannassa. Tässä hierarkisessa mallissa määritettiin ensin kuolleisuus sen syystä riippumattomasti, ja sen jälkeen sydän‑ ja verisuonisairaudesta johtuneiden sairaalahoitojen määrät silloin, kun potilaita ei voitu erotella pelkästään kuoleman perusteella.
Tämä analyysi osoitti, että kokonaiskuolleisuus sekä sydän‑ ja verisuoniperäisten sairaalahoitojaksojen esiintyvyys vähenivät merkitsevästi (p = 0,0006) yhdistetyissä 20 mg:n ja 80 mg:n annosryhmissä verrattuna lumelääkeryhmään (taulukko 2).
Taulukko 2:Ensisijainen analyysi, jossa käytettiinFinkelstein-Schoenfeld (F-S) ‑menetelmää kokonaiskuolleisuuden sekä sydän‑ ja verisuoniperäisten sairaalahoitojaksojen määrittämiseen
| Ensisijainen analyysi | Yhdistetyt tafamidiisiryhmät N = 264 | Lumelääke-ryhmä N = 177 |
| 30 kuukauden kohdalla elossa* olleiden lukumäärä (%) | 186 (70,5) | 101 (57,1) |
| Sydän‑ ja verisuoniperäisiä sairaalahoitojaksoja keskimäärin 30 kuukauden aikana (potilasta ja vuotta kohti) niillä, jotka olivat elossa 30 kuukauden† jälkeen | 0,297 | 0,455 |
| F‑S-menetelmällä saatu p‑arvo | 0,0006 | |
* Sydämensiirtoa ja sydämen mekaanisen apuvälineen implantaatiota on pidetty merkkeinä lähestyvästä loppuvaiheesta. Kyseisiä potilaita on käsitelty analyysissa samoin kuin kuolleita. Näitä potilaita ei siis ole huomioitu ”30 kuukauden jälkeen elossa olevien” -lukumäärässä, vaikka he olisivatkin olleet elossa 30 kuukauden elossaolostatuksen seurannan arviointihetkellä.
† Deskriptiivinen keskiarvo elossa olevista 30 kuukauden kohdalla.
Ensisijaisen analyysin yksittäisten komponenttien (kokonaiskuolleisuus ja sairaalahoitoon joutuminen sydän‑ ja verisuoniperäisten syiden vuoksi) analyysi osoitti myös näiden komponenttien merkittävää alentumista tafamidiisilla lumelääkkeeseen verrattuna.
Kokonaiskuolleisuuden riskisuhde (Hazard ratio) oli Coxin suhteellisessa riskitiheyksien mallissa yhdistetyille tafamidiisiryhmille 0,698 (95 %:n luottamusväli 0,508, 0,958), osoittaen kokonaiskuolleisuuden alentumisen 30,2 %:lla lumelääkkeeseen verrattuna (p = 0,0259). Kuva 1: Kaplan-Meier-kuvaajassa on esitetty kokonaiskuolleisuus suhteessa kuluneeseen aikaan.
Kuva 1: Kokonaiskuolleisuus*
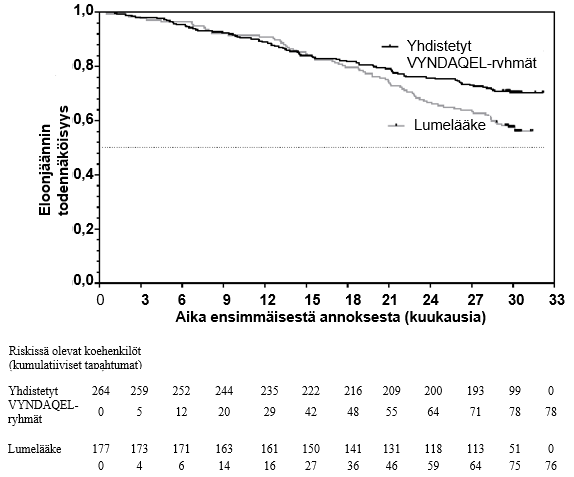
* Sydänsiirteitä ja sydämen mekaanisia apuvälineitä käsiteltiin samoin kuin kuolemaa. Riskisuhde (Hazard ratio) Coxin suhteellisen riskin mallilla, missä on tekijöinä käytetty TTR-genotyyppiä (variantti ja villityyppi) ja New York Heart Association (NYHA) lähtötilanteen luokittelua (NYHA I ja II -luokat yhdessä ja NYHA III).
Tafamidiisilla havaittiin merkittävästi vähemmän sydän‑ ja verisuoniperäisistä syistä johtuneita sairaalahoitojaksoja kuin lumelääkkeellä: riski pieneni 32,4 % (taulukko 3).
Taulukko 3: Sydän‑ ja verisuoniperäisten sairaalahoitojaksojen esiintyvyys
Yhdistetyt tafamidiisiryhmät N = 264 | Lumelääkeryhmä N = 177 | |
| Yhteensä (%) potilaita, joilla sydän‑ ja verisuoniperäisiä sairaalahoitojaksoja | 138 (52,3) | 107 (60,5) |
| Sydän‑ ja verisuoniperäisiä sairaalahoitojaksoja vuotta kohti* | 0,4750 | 0,7025 |
| Hoitoero yhdistettyjen tafamidiisiryhmien ja lumelääkeryhmän välillä (suhteellisen riskin suhdeluku)* | 0,6761 | |
| p‑arvo* | < 0,0001 | |
Lyhenne: NYHA = New York Heart Association.
* Tämä analyysi perustui Poissonin regressiomalliin, jonka tekijöinä olivat termit hoito, TTR-genotyyppi (variantti ja villityyppi), New York Heart Association (NYHA) ‑luokitus (NYHA-luokat I ja II yhdistettyinä ja NYHA‑luokka III) lähtötilanteessa, hoidon ja TTR-genotyypin yhteisvaikutus ja hoidon ja lähtötilanteen NYHA-luokituksen yhteisvaikutus.
Tafamidiisihoidon vaikutusta toimintakykyyn ja terveydentilaan arvioitiin 6 minuutin kävelytestillä (6MWT) ja kardiomyopatiaa koskevasta kyselylomakkeesta (Kansas City Cardiomyopathy Questionnaire-Overall Summary [KCCQ‑OS]) saatavalla pistemäärällä (koostuu kokonaisoireiden, fyysisten rajoitteiden, elämänlaadun ja sosiaalisten rajoitteiden osa-alueista). Merkittävä tafamidiisia suosiva hoitovaikutus havaittiin ensimmäisen kerran kuukauden 6 kohdalla, ja se säilyi yhdenmukaisena 30. kuukauden loppuun asti sekä 6 minuutin kävelytestillä että KCCQ‑OS‑pistemäärällä arvioituna (taulukko 4).
Taulukko 4: 6MWT ja KCCQ‑OS ja komponenttien osapistemäärät
| Pääte-tapahtumat | Lähtötilanteen keskiarvo (keskihajonta) | Muutos lähtötilanteesta 30. kuukauteen, LS-keskiarvo (keskivirhe) | Hoitoero lume-lääkkeeseen nähden, LS-keskiarvo (95 %:n luottamusväli) | p-arvo | ||
Yhdistetyt tafamidiisi-ryhmät N = 264 | Lume-lääke N = 177 | Yhdistetyt tafamidiisi-ryhmät | Lumelääke | |||
| 6MWT* (metriä) | 350,55 (121,30) | 353,26 (125,98) | -54,87 (5,07) | -130,55 (9,80) | 75,68 (57,56, 93,80) | p < 0,0001 |
| KCCQ-OS* | 67,27 (21,36) | 65,90 (21,74) | -7,16 (1,42) | -20,81 (1,97) | 13,65 (9,48, 17,83) | p < 0,0001 |
* Mitä suurempi arvo, sitä parempi terveydentila.
Lyhenteet: 6MWT = 6 minuutin kävelytesti; KCCQ-OS = Kansas City Cardiomyopathy Questionnaire-Overall Summary; LS (least squares) = pienimmän neliösumman menetelmä.
Tulokset F–S‑menetelmästä, jolla kuvattiin yhdistetyn päätetapahtuman ja sen komponenttien (kokonaiskuolleisuus sekä sydän‑ ja verisuoniperäisten sairaalahoitojaksojen esiintyvyys) voittosuhdetta (win ratio), suosivat yhdenmukaisesti tafamidiisia verrattuna lumelääkkeeeseen annoksittain ja kaikissa alaryhmissä (villityyppi, variantti, NYHA-luokat I & II ja III); lukuun ottamatta sydän‑ ja verisuoniperäisten sairaalahoitojaksojen esiintyvyyttä NYHA-luokassa III (kuva 2), jossa esiintyvyys on suurempi tafamidiisiryhmässä kuin lumelääkeryhmässä (ks. kohta Annostus ja antotapa). Myös 6MWT‑ ja KCCQ‑OS-analyysit suosivat tafamidiisia jokaisessa alaryhmässä.
Kuva 2: F–S‑menetelmän tulokset ja komponentit alaryhmittäin ja annoksittain
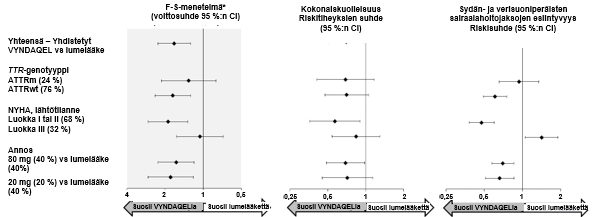
Lyhenteet: ATTRm = mutatoitunut transtyretiiniamyloidi, ATTRwt = villityypin transtyretiiniamyloidi, F-S = Finkelstein‑Schoenfeld, CI = luottamusväli.
* F-S‑tulokset on esitetty voittosuhteena (win ratio) (perustuu kokonaiskuolleisuuteen ja sydän‑ ja verisuoniperäisten sairaalahoitojaksojen esiintyvyys). Voittosuhde on hoitoa saaneiden potilasparien ”voittojen” lukumäärä jaettuna lumelääkettä saaneiden potilasparien ”voittojen” lukumäärällä.
Sydänsiirteitä ja sydämen mekaanisia apuvälineitä käsiteltiin samoin kuin kuolemaa.
Kun F-S-menetelmää käytettiin jokaiseen annosryhmään erikseen, tafamidiisi vähensi yhteenlaskettua kokonaiskuolleisuutta ja sydän- ja verisuoniperäisten sairaalajaksojen esiintyvyyttä sekä 80 mg:n (P=0,0030) että 20 mg:n (p=0,0048) annoksilla lumelääkkeeseen verrattuna. Ensisijaisanalyysin tulokset sekä 6MWT‑ ja KCCQ‑OS-analyysien tulokset 30 kuukauden kohdalla, olivat tilastollisesti merkitseviä sekä 80 mg että 20 mg tafamidiisimeglumiiniannoksella lumelääkkeeseen verrattuna ja molemmilla annoksilla tulos oli samankaltainen.
Tehotietoja 61 mg:n tafamidiisi-annokselle ei ole saatavissa, koska sitä ei ole arvioitu satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa faasi 3 tutkimuksessa. Tafamidiisi 61 mg:n suhteellinen biologinen hyötyosuus on samaa luokkaa kuin 80 mg:lla tafamidiisimeglumiinia vakaassa tilassa (ks. kohta Farmakokinetiikka).
Korjatun QT-ajan (QTc) ei osoitettu pidentyvän, kun terveille vapaaehtoisille koehenkilöille annettiin suun kautta supraterapeuttinen 400 mg:n kerta-annos tafamidiisimeglumiiniliuosta.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset tafamidiisin käytöstä transtyretiinivälitteisen amyloidoosin hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Kun pehmeä kapseli otetaan kerran päivässä suun kautta paaston jälkeen, ajan mediaani (tmax) maksimaalisen huippupitoisuuden saavuttamiseen (Cmax) on 61 mg:n tafamidiisiannoksella 4 tuntia ja 80 mg:n (4 x 20 mg) tafamidiisimeglumiiniannoksella 2 tuntia. Lääkkeen ottaminen runsasrasvaisen, hyvin kaloripitoisen aterian yhteydessä muutti imeytymisnopeutta, mutta ei imeytymisastetta. Nämä tulokset tukevat sitä, että tafamidiisi voidaan ottaa ruokailun yhteydessä tai tyhjään mahaan.
Jakautuminen
Tafamidiisi sitoutuu voimakkaasti (> 99 %) plasman proteiineihin. Näennäinen jakautumistilavuus vakaassa tilassa on 18,5 litraa.
Tafamidiisin sitoutumisastetta plasman proteiineihin on arvioitu eläimen ja ihmisen plasmalla. Tafamidiisin affiniteetti TTR:ään on suurempi kuin albumiiniin. Siksi tafamidiisi sitoutuu plasmassa todennäköisemmin TTR:ään huolimatta siitä, että albumiinin pitoisuus plasmassa on huomattavasti suurempi kuin TTR:n pitoisuus plasmassa (600 μM versus 3,6 μM).
Biotransformaatio ja eliminaatio
Ei ole selvää näyttöä, että tafamidiisi erittyy ihmisellä sappeen. Prekliiniset tiedot viittaavat siihen, että tafamidiisi metaboloituu glukuronidaation kautta ja erittymällä sappeen. Tämä biotransformaatioreitti on ihmisellä todennäköinen, koska noin 59 % annetusta kokonaisannoksesta on havaittavissa ulosteessa ja noin 22 % virtsassa. Populaatiofarmakokineettisten tulosten perusteella tafamidiisin näennäinen oraalinen puhdistuma on 0,263 l/h ja keskimääräinen puoliintumisaika populaatiossa on noin 49 tuntia.
Annoksen ja ajan lineaarisuus
Altistusta kerran päivässä annostellulle tafamidiisimeglumiinille lisättiin nostamalla yksittäistä annosta 480 mg:aan asti ja vastaavasti usealla annoksella annokseen 80 mg/vrk. Yleisesti ottaen altistuksen lisäys oli suhteessa tai lähes suhteessa annokseen ja tafamidiisin puhdistuma säilyi vakaana.
Tafamidiisi 61 mg:n suhteellinen biologinen hyötyosuus on samaa luokkaa kuin 80 mg:lla tafamidiisimeglumiinia vakaassa tilassa. Tafamidiisi ja tafamidiisimeglumiini eivät ole keskenään vaihdettavissa milligrammamäärän perusteella.
Farmakokineettiset parametrit olivat samankaltaiset tafamidiisimeglumiinin 20 mg:n kerta-annoksen ja toistuvan annon yhteydessä, mikä viittaa siihen, ettei tafamidiisin metabolian induktiota tai estymistä tapahdu.
Tulokset 14 vuorokauden ajan kerran päivässä annetusta 15–60 mg:n tafamidiisimeglumiini-oraaliliuoksen annoksesta osoittivat, että vakaa tila saavutettiin 14. hoitopäivään mennessä.
Erityisryhmät
Maksan vajaatoiminta
Farmakokineettiset tiedot viittasivat siihen, että systeeminen altistus tafamidiisimeglumiinille pieneni (noin 40 %) ja kokonaispuhdistuma suureni (0,52 l/h versus 0,31 l/h) potilailta, joilla oli keskivaikea maksan vajaatoiminta (Child-Pugh-pisteet 7–9, nämä arvot mukaan lukien) verrattuna terveisiin koehenkilöihin, koska tafamidiisin sitoutumaton fraktio oli suurempi. Koska keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden TTR-pitoisuus oli pienempi kuin terveillä koehenkilöillä, annoksen muuttaminen ei ole tarpeen, koska tafamidiisin ja sen kohdeproteiinin, TTR:n, stoikiometria on riittävä TTR-tetrameerin stabiloimiseksi. Vaikeaa maksan vajaatoimintaa sairastavien potilaiden altistusta tafamidiisille ei tunneta.
Munuaisten vajaatoiminta
Tafamidiisia ei ole arvioitu erityisesti munuaisten vajaatoimintaa sairastavia potilaita koskevassa tutkimuksessa. Kreatiniinipuhdistuman vaikutusta tafamidiisin farmakokinetiikkaan arvioitiin populaatiofarmakokineettisessä analyysissa potilailla, joiden kreatiniinipuhdistuma oli yli 18 ml/min. Farmakokineettiset arviot eivät osoittaneet mitään eroa tafamidiisin näennäisessä oraalisessa puhdistumassa potilailla, joiden kreatiniinipuhdistuma oli alle 80 ml/min, verrattuna niihin, joiden kreatiniinipuhdistuma oli vähintään 80 ml/min. Annoksen muuttamista munuaisten vajaatoiminnan yhteydessä ei katsota tarpeelliseksi.
Iäkkäät
Populaatiofarmakokineettisten tulosten perusteella ≥ 65‑vuotiaiden tutkimuspotilaiden näennäinen oraalinen puhdistuma oli vakaassa tilassa keskimäärin 15 % pienempi kuin alle 65‑vuotiailla tutkimuspotilailla. Puhdistumassa havaittu ero suurentaa keskimääräistä Cmax-arvoa ja AUC-arvoa kuitenkin alle 20 % nuorempiin tutkimuspotilaisiin verrattuna, mikä ei ole kliinisesti merkittävää.
Farmakokineettiset/farmakodynaamiset suhteet
In vitro ‑tiedot viittaavat siihen, että tafamidiisi ei estä merkittävästi sytokromi P450 ‑entsyymejä CYP1A2, CYP3A4, CYP3A5, CYP2B6, CYP2C8, CYP2C9, CYP2C19 ja CYP2D6. Tafamidiisin ei odoteta aiheuttavan kliinisesti merkityksellisiä lääkeaineiden välisiä yhteisvaikutuksia CYP1A2:n, CYP2B6:n tai CYP3A4:n induktion vuoksi.
In vitro ‑tutkimukset viittaavat siihen, ettei tafamidiisi todennäköisesti aiheuta systeemisesti kliinisesti merkityksellisinä pitoisuuksina lääkeaineiden välisiä yhteisvaikutuksia UDP-glukuronosyylitransferaasin (UGT) substraattien kanssa. Tafamidiisi saattaa estää UGT1A1:n aktiivisuutta suolistossa.
Tafamidiisin kyky estää kliinisesti merkityksellisillä pitoisuuksilla seuraavia on osoitettu vähäiseksi: MDR1-proteiinin (Multi-Drug Resistant Protein; tunnetaan myös nimellä P‑glykoproteiini; P‑gp) esto systeemisesti ja ruoansulatuskanavassa, orgaanisten kationien kuljettajan 2 (OCT2), kuljettajaproteiinien MATE1 ja MATE2K (multidrug and toxin extrusion transporter), orgaanisten anionien kuljettajapolypeptidien 1B1 (OATP1B1) ja OATP1B3 esto.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, hedelmällisyyttä ja alkion varhaiskehitystä, genotoksisuutta ja karsinogeenisuutta koskevien konventionaalisten tutkimusten non-kliiniset tulokset eivät viittaa erityiseen ihmisiin kohdistuvaan vaaraan. Toistuvan altistuksen aiheuttamaa toksisuutta ja karsinogeenisuutta koskevissa tutkimuksissa maksan havaittiin olevan kohde-elin tutkituilla lajeilla. Maksavaikutuksia esiintyi altistuksilla, jotka vastasivat suurin piirtein ihmisen vakaan tilan AUC-arvoa, joka saavutetaan tafamidiisin 61 mg:n hoitoannoksella.
Kaneilla tehdyssä kehitystoksisuutta selvittäneessä tutkimuksessa havaittiin vähäisiä luuston epämuodostumia ja muutoksia, muutamilla naarailla keskenmenoja, alkioiden ja sikiöiden eloonjäännin vähenemistä ja sikiön painon alenemista, kun altistus oli noin ≥ 2,1‑kertainen verrattuna ihmisen vakaan tilan AUC-arvoon, joka saavutetaan tafamidiisin 61 mg:n hoitoannoksella.
Tafamidiisilla tehdyssä rotan pre- ja postnataalista kehitystä selvittäneessä tutkimuksessa havaittiin poikasten eloonjäännin vähenemistä ja poikasten painon alenemista, kun emolle annettu annos oli tiineyden ja imetyksen aikana 15 mg/kg/vrk ja 30 mg/kg/vrk. Urospoikasten painon alenemiseen liittyi viivästynyt sukupuolinen kypsyminen (preputiaalinen separaatio) annoksella 15 mg/kg/vrk. Heikentynyt suorituskyky oppimista ja muistia testaavassa vesilabyrintissa havaittiin annoksella 15 mg/kg/vrk. F1-sukupolven jälkeläisten elinkykyisyyden ja kasvun suhteen haitaton annostaso (NOAEL) emolle oli tiineyden ja imetyksen aikana 5 mg/kg/vrk (vastaa ihmisen annosta 0,8 mg/kg/vrk), joka vastaa suurin piirtein tafamidiisin 61 mg:n hoitoannosta.
Farmaseuttiset tiedot
Apuaineet
Kapselin kuori
Liivate (E 441)
Glyseroli (E 422)
Punainen rautaoksidi (E 172)
Sorbitaani
Sorbitoli (E 420)
Mannitoli (E 421)
Puhdistettu vesi
Kapselin sisältö
Makrogoli 400 (E 1521)
Polysorbaatti 20 (E 432)
Povidoni (K‑arvo 90)
Butyylihydroksitolueeni (E 321)
Painomuste (valkoinen Opacode)
Etanoli
Isopropyylialkoholi
Puhdistettu vesi
Makrogoli 400 (E 1521)
Polyvinyyliasetaattiftalaatti
Propyleeniglykoli (E 1520)
Titaanidioksidi (E 171)
Ammoniumhydroksidi (E 527) 28 %
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
2 vuotta
Säilytys
Ei ohjeita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VYNDAQEL kapseli, pehmeä
61 mg (L:ei) 30 x 1 fol (8125,60 €)
PF-selosteen tieto
PVC/PA/Al/PVC-Al perforoitu kerta-annosläpipainolevy.
Pakkauskoot: 30 x 1 pehmeää kapselia ja kerrannaispakkaus, joka sisältää 90 pehmeää kapselia (kolme 30 x 1 kapselin pakkausta).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Punertavanruskea, läpikuultava, pitkänomainen (noin 21 mm) kapseli, johon on painettu valkoisella ”VYN 61”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VYNDAQEL kapseli, pehmeä
61 mg 30 x 1 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Akoramidiisi ja tafamidiisi: Transtyretiinivälitteisen amyloidoosin hoito erityisin edellytyksin (3011).
ATC-koodi
N07XX08
Valmisteyhteenvedon muuttamispäivämäärä
12.03.2026
Yhteystiedot
 PFIZER OY
PFIZER OY Tietokuja 4
00330 Helsinki
09 430 040
www.pfizer.fi
etunimi.sukunimi@pfizer.com