
JEMPERLI infuusiokonsentraatti, liuosta varten 500 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Yleinen
Tärkeää turvallisuustietoa immuunivälitteisten haittavaikutusten riskin pienentämiseksi.
Huomioitavaa
▼ Tähän lääkevalmisteeseen kohdistuu lisäseuranta. Tällä tavalla voidaan havaita nopeasti turvallisuutta koskevaa uutta tietoa. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan epäillyistä lääkkeen haittavaikutuksista. Ks. kohdasta Haittavaikutukset, miten haittavaikutuksista ilmoitetaan.
Vaikuttavat aineet ja niiden määrät
Yksi 10 ml:n injektiopullo infuusiokonsentraattia, liuosta varten sisältää 500 mg dostarlimabia.
Yksi millilitra infuusiokonsentraattia, liuosta varten sisältää 50 mg dostarlimabia.
Dostarlimabi on immunoglobuliini G4 (IgG4) ‑isotyypin humanisoitu monoklonaalinen PD‑1:n (programmed cell death protein‑1) vasta‑aine, joka tuotetaan yhdistelmä‑DNA‑tekniikalla nisäkässoluissa (kiinanhamsterin munasarjasoluissa).
Apuaine(et), joiden vaikutus tunnetaan
2 mg polysorbaatti 80:tä per annosyksikkö.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
JEMPERLI on tarkoitettu käytettäväksi yhdistelmähoitona karboplatiinin ja paklitakselin kanssa primaarisen pitkälle edenneen tai uusiutuneen kohdun limakalvon syövän ensilinjan hoitoon aikuispotilaille, joille harkitaan systeemistä hoitoa.
JEMPERLI on tarkoitettu käytettäväksi monoterapiana mismatch repair deficient (dMMR) / microsatellite instability‑high (MSI‑H) ‑tyyppisen, uusiutuneen tai pitkälle edenneen kohdun limakalvon syövän hoitoon aikuispotilaille, kun syöpä on edennyt aiemman platinaa sisältävän hoito‑ohjelman aikana tai sen jälkeen.
Ehto
Valmiste annetaan syövän lääkehoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Hoito tulee aloittaa ja sitä tulee valvoa syövän hoitoon perehtyneen erikoislääkärin toimesta.
Kasvaimen dMMR‑/MSI‑H‑status on määritettävä validoidulla testausmenetelmällä, kuten IHK‑, PCR‑ tai NGS‑menetelmällä* (tiedot tutkimuksissa käytetyistä määrityksistä, ks. kohta Farmakodynamiikka).
*IHK = immunohistokemiallinen; PCR = polymeraasiketjureaktio; NGS = uuden sukupolven sekvensointi.
Annostus
JEMPERLI-yhdistelmähoito karboplatiinin ja paklitakselin kanssa
Kun JEMPERLI-valmistetta annetaan yhdistelmähoitona karboplatiinin ja paklitakselin kanssa, on perehdyttävä yhdistelmähoidossa käytettävien valmisteiden valmisteyhteenvetoihin (ks. myös kohta Farmakodynamiikka).
Suositeltu annos on 500 mg dostarlimabia 3 viikon välein yhdistelmänä 3 viikon välein annettavan karboplatiinin ja paklitakselin kanssa 6 hoitojakson ajan ja tämän jälkeen 1 000 mg dostarlimabia monoterapiana 6 viikon välein kaikkien seuraavien hoitojaksojen ajan.
Hoito-ohjelma yhdistelmähoitona karboplatiinin ja paklitakselin kanssa esitetään taulukossa 1.
Taulukko 1. Hoito-ohjelma: JEMPERLI-yhdistelmähoito karboplatiinin ja paklitakselin kanssa

a Dostarlimabi annetaan ennen karboplatiinia ja paklitakselia samana päivänä.
Dostarlimabin antoa jatketaan suositeltua aikataulua noudattaen, kunnes tauti etenee tai potilaalla ilmenee toksisuutta, jota ei voida hyväksyä, tai enintään kolmen vuoden ajan (ks. kohta Farmakodynamiikka).
JEMPERLI-monoterapia
Suositeltu annos monoterapiassa on 500 mg dostarlimabia 3 viikon välein 4 hoitojakson ajan ja tämän jälkeen 1 000 mg 6 viikon välein kaikkien seuraavien hoitojaksojen ajan.
Hoito‑ohjelma monoterapiana esitetään taulukossa 2.
Taulukko 2. Hoito‑ohjelma: JEMPERLI‑monoterapia

Dostarlimabin antoa jatketaan suositeltua aikataulua noudattaen, kunnes tauti etenee tai potilaalla ilmenee toksisuutta, jota ei voida hyväksyä (ks. kohta Farmakodynamiikka.).
Annosmuutokset
Annoksen pienentämistä ei suositella. Annoksen siirtäminen tai hoidon keskeytys voi olla tarpeen yksilöllisen turvallisuuden ja siedettävyyden perusteella. Haittavaikutusten takia tehtäväksi suositellut muutokset esitetään taulukossa 3.
Tarkemmat ohjeet immuunivälitteisten haittavaikutusten ja infuusioreaktioiden hoitoon on kuvattu kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Taulukko 3. Suositeltavat JEMPERLI‑annosmuutokset | ||
Immuunivälitteiset haittavaikutukset | Vaikeusastea | Annosmuutokset |
Koliitti | 2 tai 3 | Hoito tauotetaan. Hoito aloitetaan uudelleen, kun toksisuus on korjautunut asteen 0 tai 1 tasolle. |
4 | Hoito lopetetaan pysyvästi. | |
Hepatiitti | Aste 2; ASATb tai ALATc > 3–5 × ULNd tai kokonaisbilirubiini > 1,5 – 3 × ULN | Hoito tauotetaan. Hoito aloitetaan uudelleen, kun toksisuus on korjautunut asteen 0 tai 1 tasolle. |
Aste ≥ 3; ASAT tai ALAT > 5 × ULN tai kokonaisbilirubiini > 3 × ULN | Hoito lopetetaan pysyvästi (ks. poikkeus jäljempänä)e. | |
Tyypin 1 diabetes | 3 tai 4 (hyperglykemia) | Hoito tauotetaan. Hoito aloitetaan uudelleen, jos haittaa hoidetaan asianmukaisesti ja potilas on kliinisesti ja metabolisesti stabiili. |
Hypofysiitti tai lisämunuaisten vajaatoiminta | 2, 3 tai 4 | Hoito tauotetaan. Hoito aloitetaan uudelleen, kun toksisuus on korjautunut asteen 0 tai 1 tasolle. Hoito lopetetaan pysyvästi, jos toksisuus uusiutuu tai pahenee riittävän hormonihoidon aikana. |
Kilpirauhasen vajaatoiminta tai liikatoiminta | 3 tai 4 | Hoito tauotetaan. Hoito aloitetaan uudelleen, kun toksisuus on korjautunut asteen 0 tai 1 tasolle. |
Pneumoniitti | 2 | Hoito tauotetaan. Hoito aloitetaan uudelleen, kun toksisuus on korjautunut asteen 0 tai 1 tasolle. Jos asteen 2 toksisuus uusiutuu, hoito lopetetaan pysyvästi. |
3 tai 4 | Hoito lopetetaan pysyvästi. | |
Nefriitti | 2 | Hoito tauotetaan. Hoito aloitetaan uudelleen, kun toksisuus on korjautunut asteen 0 tai 1 tasolle. |
3 tai 4 | Hoito lopetetaan pysyvästi. | |
Eksfoliatiiviset ihoreaktiot (esim. Stevens–Johnsonin oireyhtymä (SJSf), toksinen epidermaalinen nekrolyysi (TENg) ja DRESSh-oireyhtymä) | Epäilty | Hoito tauotetaan epäiltäessä minkä tahansa asteen tilaa. Hoito aloitetaan uudelleen, kun toksisuus on korjautunut asteen 0 tai 1 tasolle, jos tilaa ei ole vahvistettu. |
Vahvistettu | Hoito lopetetaan pysyvästi. | |
Myokardiitti | 2,3 tai 4 | Hoito lopetetaan pysyvästi. |
Vaikea neurologinen toksisuus (myasteeninen oireyhtymä / myasthenia gravis, Guillain–Barrén oireyhtymä, enkefaliitti, transversaalimyeliitti) | 2,3 tai 4 | Hoito lopetetaan pysyvästi. |
Muut immuunivälitteiset haittavaikutukset (mm. myosiitti, sarkoidoosi, autoimmuunihemolyyttinen anemia, pankreatiitti, iridosykliitti, uveiitti, diabeettinen ketoasidoosi, nivelkipu, kiinteän elimen siirteen hyljintä, käänteishyljintä) | 3 | Hoito tauotetaan. Hoito aloitetaan uudelleen, kun toksisuus on korjautunut asteen 0 tai 1 tasolle. |
4 | Hoito lopetetaan pysyvästi. | |
Immuunivälitteisten haittavaikutusten uusiutuminen sen jälkeen, kun haitta on korjautunut asteen ≤ 1 tasolle (pois lukien pneumoniitti, ks. edellä) | 3 tai 4 | Hoito lopetetaan pysyvästi. |
Muut haittavaikutukset | Vaikeusastea | Annosmuutokset |
Infuusioreaktiot | 2 | Hoito tauotetaan. Jos haitta korjautuu 1 tunnin kuluessa tauottamisesta, antoa voidaan jatkaa alkuperäistä 50 % pienemmällä infuusionopeudella, tai antoa voidaan jatkaa esilääkityksen kera, kun oireet ovat hävinneet. Jos asteen 2 haitta uusiutuu riittävän esilääkityksen yhteydessä, hoito lopetetaan pysyvästi. |
3 tai 4 | Hoito lopetetaan pysyvästi. | |
a Toksisuusasteet perustuvat National Cancer Institute ‑organisaation Common Terminology Criteria for Adverse Events ‑kriteereihin (CTCAE, versio 5.0).
b ASAT = aspartaattiaminotransferaasi
c ALAT = alaniiniaminotransferaasi
d ULN = viitealueen yläraja
e Jos potilaalla, jolla on maksametastasointi, on hoidon alkaessa asteen 2 ASAT‑ tai ALAT‑arvon suurenema ja ASAT‑ tai ALAT‑arvo suurenee ≥ 50 % suhteessa lähtötilanteeseen ja suurenema säilyy vähintään 1 viikon ajan, hoito on lopetettava pysyvästi
f SJS = Stevens–Johnsonin oireyhtymä
g TEN = toksinen epidermaalinen nekrolyysi
h DRESS = lääkereaktio, johon liittyy eosinofiliaa ja systeemisiä oireita.
Potilaskortti
Kaikkien JEMPERLI‑valmisteen määrääjien on kerrottava potilaalle potilaskortista ja annettava toimintaohjeet immuunivälitteisten haittavaikutusten minkä tahansa oireen ilmaantumisen varalle. Lääkäri antaa potilaskortin kaikille potilaille.
Erityisryhmät
Iäkkäät
65‑vuotiaille ja sitä iäkkäämmille potilaille ei suositella annoksen muuttamista.
Dostarlimabin käytöstä 75 vuotta täyttäneiden potilaiden hoidossa on niukasti tietoa (ks. kohta Farmakodynamiikka).
Munuaisten vajaatoiminta
Annoksen muuttamista ei suositella, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta. Tietoja on niukasti potilaista, joilla on vaikea munuaisten vajaatoiminta tai loppuvaiheen munuaisten vajaatoiminta ja jotka saavat dialyysihoitoa (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttamista ei suositella potilaille, joilla maksan vajaatoiminta on lievä. Tietoja on niukasti käytöstä potilailla, joilla on keskivaikea maksan vajaatoiminta, eikä lainkaan käytöstä potilailla, joilla on vaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
JEMPERLI‑valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu. Muuten kuin hyväksytyissä käyttöaiheissa JEMPERLI-valmistetta on tutkittu yhdistelmänä niraparibin kanssa 5 – < 18 vuoden ikäisillä lapsilla ja nuorilla, joilla oli uusiutunut tai hoitoresistentti kiinteä kasvain, kuten osteosarkooma tai neuroblastooma, mutta tutkimustiedot olivat rajalliset eikä tutkimustulosten perusteella ollut mahdollista tehdä johtopäätöstä, että tällaisen käytön hyödyt olisivat suuremmat kuin sen riskit. Saatavissa oleva tieto on kuvattu kohdissa Farmakodynamiikka ja Farmakokinetiikka.
Antotapa
JEMPERLI on tarkoitettu annettavaksi vain infuusiona laskimoon. JEMPERLI annetaan infuusiopumpulla laskimoon 30 minuuttia kestävänä infuusiona.
JEMPERLI‑valmistetta ei saa antaa nopeana injektiona / bolusinjektiona laskimoon.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Immuunivälitteiset haittavaikutukset
Immuunivälitteisiä, mahdollisesti vaikeita tai kuolemaan johtavia haittavaikutuksia, voi esiintyä potilailla, joita hoidetaan PD‑1/PD‑L1‑reittiä estävillä vasta‑aineilla, kuten dostarlimabilla. Immuunivälitteiset haittavaikutukset ilmaantuvat yleensä PD‑1/PD‑L1‑reittiä estävillä vasta‑aineilla toteutettavan hoidon aikana, mutta oireita voi ilmaantua myös hoidon päätyttyä. Immuunivälitteisiä haittavaikutuksia voi esiintyä missä tahansa elimessä tai kudoksessa ja useammassa elinjärjestelmässä samanaikaisesti. Tässä kohdassa luetellut tärkeät immuunivälitteiset haittavaikutukset eivät sisällä kaikkia mahdollisia vaikeita ja kuolemaan johtavia immuunivälitteisiä vaikutuksia.
Immuunivälitteisten haittavaikutusten varhainen tunnistaminen ja hoito on keskeistä PD‑1/PD‑L1‑reittiä estävien vasta‑aineiden turvallisen käytön varmistamiseksi. Potilaita on tarkkailtava immuunivälitteisten haittavaikutusten oireiden ja löydösten varalta. Hematologiset ja kliinisen kemian tutkimukset, mukaan lukien maksan, munuaisen ja kilpirauhasen toimintakokeet, on arvioitava lähtötilanteessa ja määräajoin hoidon aikana. Jos immuunivälitteistä haittavaikutusta epäillään, riittävä arviointi, mukaan lukien erikoislääkärin konsultaatio, on varmistettava.
Haittavaikutuksen vaikeusasteesta riippuen dostarlimabihoito on tauotettava tai lopetettava pysyvästi ja annettava kortikosteroideja (1–2 mg/kg/vrk prednisonia tai vastaavaa) tai muuta asianmukaista hoitoa (ks. jäljempänä sekä kohta Annostus ja antotapa). Jos haitta korjautuu asteen ≤ 1 tasolle, aloitetaan kortikosteroidiannoksen purku, ja sitä jatketaan 1 kuukauden ajan tai pidempään. Jos potilaiden immuunivälitteisiä haittavaikutuksia ei saada hallintaan kortikosteroideilla, niukkojen kliinisistä tutkimuksista saatujen tietojen perusteella voidaan harkita muiden systeemisten immunosuppressiivisten lääkeaineiden antoa. Endokrinopatioihin on aloitettava tarvittaessa hormonikorvaushoito.
Dostarlimabihoito on lopetettava pysyvästi, jos mikä tahansa asteen 3 immuunivälitteinen haittavaikutus uusiutuu tai jos potilaalle ilmaantuu mikä tahansa asteen 4 immuunivälitteinen haittavaikutus, ellei kyseessä ole hormonikorvaushoidolla hallintaan saatava endokrinopatia tai ellei taulukossa 3 ole mainittu muuta.
Immuunivälitteinen pneumoniitti
Dostarlimabia saaneilla potilailla on ilmoitettu pneumoniittia (ks. kohta Haittavaikutukset). Potilaita on seurattava pneumoniitin oireiden ja löydösten varalta. Pneumoniittiepäily on vahvistettava röntgenkuvauksella, ja muut syyt on suljettava pois. Pneumoniittitapauksissa dostarlimabihoitoa on muutettava ja potilaalle on annettava kortikosteroidihoitoa (ks. kohta Annostus ja antotapa).
Immuunivälitteinen koliitti
Dostarlimabi voi aiheuttaa immuunivälitteisen koliitin (ks. kohta Haittavaikutukset). Potilaita on seurattava koliitin oireiden ja löydösten varalta, ja jos koliitti ilmaantuu, dostarlimabihoitoa on muutettava ja potilaalle on annettava ripulilääkkeitä ja kortikosteroidihoitoa (ks. kohta Annostus ja antotapa).
Immuunivälitteinen hepatiitti
Dostarlimabi voi aiheuttaa immuunivälitteisen hepatiitin (ks. kohta Haittavaikutukset). Potilaita on seurattava maksatoiminnan muutosten varalta määräajoin kuten aiheellista kliinisen arvioinnin perusteella, ja hepatiitin hoidossa dostarlimabihoitoa on muutettava ja potilaalle on annettava kortikosteroidihoitoa (ks. kohta Annostus ja antotapa).
Immuunivälitteiset endokrinopatiat
Immuunivälitteisiä endokrinopatioita (mukaan lukien kilpirauhasen vajaatoiminta, kilpirauhasen liikatoiminta, tyreoidiitti, hypofysiitti, tyypin 1 diabetes, diabeettinen ketoasidoosi ja lisämunuaisten vajaatoiminta) on ilmoitettu dostarlimabia saaneilla potilailla (ks. kohta Haittavaikutukset).
Kilpirauhasen vajaatoiminta ja liikatoiminta
Immuunivälitteistä kilpirauhasen vajaatoimintaa ja liikatoimintaa (mukaan lukien tyreoidiitti) on esiintynyt dostarlimabia saaneilla potilailla, ja kilpirauhasen liikatoiminnan jälkeen voi esiintyä vajaatoimintaa. Potilaita on seurattava kilpirauhastoimintakokeiden tulosten poikkeavuuksien varalta ennen hoitoa ja määräajoin hoidon aikana sekä kuten aiheellista kliinisen arvioinnin perusteella. Immuunivälitteistä kilpirauhasen vajaatoimintaa ja liikatoimintaa (mukaan lukien tyreoidiitti) tulee hoitaa kohdassa Annostus ja antotapa suositellulla tavalla.
Lisämunuaisten vajaatoiminta
Dostarlimabia saaneilla potilailla on esiintynyt immuunivälitteistä lisämunuaisten vajaatoimintaa. Potilaita on seurattava lisämunuaisten vajaatoiminnan kliinisten oireiden ja löydösten varalta. Oireista lisämunuaisten vajaatoimintaa tulee hoitaa kohdassa Annostus ja antotapa suositellulla tavalla.
Immuunivälitteinen nefriitti
Dostarlimabi voi aiheuttaa immuunivälitteisen nefriitin (ks. kohta Haittavaikutukset). Potilaita on seurattava munuaistoiminnan muutosten varalta, ja nefriitin hoidossa dostarlimabihoitoa on muutettava ja potilaalle on annettava kortikosteroidihoitoa (ks. kohta Annostus ja antotapa).
Ihon immuunivälitteiset haittavaikutukset
Dostarlimabia saaneilla potilailla on ilmoitettu immuunivälitteistä ihottumaa, mukaan lukien pemfigoidia (ks. kohta Haittavaikutukset). Potilaita on seurattava ihottuman oireiden ja löydösten varalta. Stevens-Johnsonin oireyhtymä (SJS), joka voi olla henkeä uhkaava tai kuolemaan johtava, on raportoitu dostarlimabihoidon yhteydessä (ks. kohta Haittavaikutukset). PD-1-estäjähoitoa saaneilla potilailla on raportoitu toksista epidermaalista nekrolyysiä. Potilaita tulee neuvoa vakavien ihon haittavaikutusten (SCAR) oireista ja löydöksistä sekä seurata tarkasti ihoreaktioiden varalta. Potilaita tulee neuvoa hakeutumaan välittömästi lääkäriin, jos he havaitsevat viitteellisiä oireita tai löydöksiä. Epäiltäessä SCAR-reaktiota potilas tulee ohjata erikoislääkärille jatkoarviointia ja hoitoa varten, ja hoito tulee toteuttaa kohdassa Annostus ja antotapa annettujen suositusten mukaisesti.
Varovaisuutta on noudatettava, jos dostarlimabin käyttöä harkitaan potilaalle, jolle on aiemmin ilmaantunut vaikea tai henkeä uhkaava ihon haittavaikutus muun immuunivastetta stimuloivan syöpälääkkeen käytön aikana.
Immuunivälitteinen nivelkipu
Dostarlimabia saaneilla potilailla on ilmoitettu immuunivälitteistä nivelkipua (ks. kohta Haittavaikutukset). Potilaita on seurattava nivelkivun oireiden ja löydösten varalta. Epäilty immuunivälitteinen nivelkipu on vahvistettava ja muut syyt on suljettava pois. Nivelkiputapauksissa dostarlimabihoitoa on muutettava ja potilaalle on annettava kortikosteroidihoitoa (ks. kohta Annostus ja antotapa).
Muut immuunivälitteiset haittavaikutukset
Dostarlimabin vaikutusmekanismista johtuen muita mahdollisia immuunivälitteisiä haittavaikutuksia voi esiintyä, myös mahdollisesti vakavia tapahtumia (esim. myosiitti, myokardiitti, enkefaliitti, demyelinoiva neuropatia [mukaan lukien Guillain–Barrén oireyhtymä], sarkoidoosi). Alle 1 %:lla dostarlimabimonoterapiaa saaneista potilaista ilmoitettuja kliinisesti merkittäviä immuunivälitteisiä haittavaikutuksia ovat enkefaliitti, autoimmuunihemolyyttinen anemia, pankreatiitti, iridosykliitti ja uveiitti. Potilaita on seurattava immuunivälitteisten haittavaikutusten oireiden ja löydösten varalta ja hoidettava kohdassa Annostus ja antotapa kuvatulla tavalla. Kiinteän elimen siirteen hyljintää on ilmoitettu PD‑1:n estäjillä hoidetuilla potilailla markkinoilletulon jälkeen. Dostarlimabihoito voi suurentaa hyljintäriskiä kiinteä elimen siirteen saajilla. Dostarlimabihoidon hyöty verrattuna mahdollisen elinsiirteen hyljinnän riskiin on otettava huomioon kyseisillä potilailla.
Kuolemaan johtavia ja muita vakavia komplikaatioita voi esiintyä, jos potilas saa allogeenisen hematopoieettisen kantasolusiirron ennen PD‑1/PD‑L1‑reittiä estävää vasta‑ainehoitoa tai tällaisen hoidon jälkeen. Siirteisiin liittyviä komplikaatioita ovat hyperakuutti käänteishyljintä, akuutti käänteishyljintä, krooninen käänteishyljintä, maksan veno‑okklusiivinen tauti kevytesihoidon jälkeen ja steroideja vaativa kuumeoireyhtymä (ilman tunnistettua infektion aiheuttajaa). Näitä komplikaatioita voi esiintyä huolimatta PD‑1/PD‑L1:n eston ja allogeenisen hematopoieettisen kantasolusiirron välillä annetusta hoidosta. Potilaita on seurattava tarkasti siirteisiin liittyvien komplikaatioiden merkkien varalta, ja komplikaatioiden hoito on aloitettava ripeästi. Ennen allogeenista hematopoieettista kantasolusiirtoa tai siirron jälkeen annettavan, PD‑1/PD‑L1‑reittiä estävän vasta‑ainehoidon hyödyt ja riskit on punnittava.
Infuusioreaktiot
Dostarlimabi voi aiheuttaa infuusioreaktioita, jotka voivat olla vaikeita (ks. kohta Haittavaikutukset). Jos potilaalle kehittyy vaikea (aste 3) tai henkeä uhkaava (aste 4) infuusioreaktio, infuusio on keskeytettävä ja hoito lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Potilaat, jotka suljettiin pois kliinisistä tutkimuksista
GARNET‑tutkimuksesta suljettiin pois potilaat, joilla oli jokin seuraavista: ECOG (Eastern Cooperative Oncology Group) ‑toimintakykyluokka lähtötilanteessa ≥ 2; hallitsemattomat keskushermoston metastaasit tai meningeaalinen karsinoosi; muu maligniteetti edeltävien 2 vuoden aikana; immuunipuutos tai immunosuppressiivinen hoito 7 vuorokauden aikana ennen 1. annosta; aktiivinen HIV‑, hepatiitti B tai hepatiitti C ‑infektio; aktiivinen autoimmuunisairaus, joka on vaatinut systeemistä hoitoa edeltävien 2 vuoden aikana (pois lukien korvaushoito); interstitiaalinen keuhkosairaus anamneesissa tai rokotus elävällä rokotteella 14 vuorokauden aikana ennen 1. annosta.
RUBY-tutkimuksesta suljettiin pois potilaat, joilla oli jokin seuraavista: samanaikainen maligniteetti, tai aiempi invasiivinen maligniteetti muualla kuin kohdun limakalvossa ja joiden tauditon aika oli ollut < 3 vuotta tai tähän maligniteettiin oli annettu mitä tahansa aktiivihoitoa edeltävien 3 vuoden aikana; hallitsemattomat keskushermoston metastaasit tai meningeaalinen karsinoosi, tai molemmat; anamneesissa HIV‑infektio tai aktiivinen hepatiitti B tai hepatiitti C; immuunipuutos tai immunosuppressiivinen hoito 7 vuorokauden aikana ennen 1. annosta; suureksi katsottu lääketieteellinen riski vakavan, huonossa hoitotasapainossa olevan lääketieteellisen tilan, ei-malignin systeemisen taudin tai systeemistä hoitoa vaativan infektion vuoksi tai rokotus elävällä rokotteella 30 vuorokauden aikana ennen 1. tutkimuslääkeannosta, tutkimushoidon aikana tai viimeistä tutkimuslääkeannosta seuraavien 180 vuorokauden aikana.
Mahdollisen suurentuneen riskin huolellisen arvion jälkeen näille potilaille voidaan dostarlimabia käyttää yhdessä muun asianmukaisen hoidon kanssa.
Polysorbaatti 80 ‑pitoisuus
Tämä lääkevalmiste sisältää polysorbaatti 80:tä (ks. kohta Vaikuttavat aineet ja niiden määrät), joka saattaa aiheuttaa allergisia reaktioita.
Natriumpitoisuus
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 500 mg:n annos eli sen voidaan sanoa olevan ”natriumiton”. Tämä lääkevalmiste voidaan laimentaa 9 mg/ml (0,9 %) NaCl‑infuusionesteeseen. Tämä on otettava huomioon, jos potilaalla on ruokavalion natriumrajoitus (ks. kohta Käyttö- ja käsittelyohjeet).
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Monoklonaaliset vasta‑aineet kuten dostarlimabi eivät ole CYP450-entsyymien tai kuljettajaproteiinien substraatteja. Dostarlimabi ei ole sytokiini eikä todennäköisesti sytokiinimodulaattori. Dostarlimabin ja pienimolekyylisten vaikuttavien aineiden farmakokineettiset yhteisvaikutukset eivät myöskään ole todennäköisiä. Vasta‑aineiden kohdalla ei ole näyttöä yhteisvaikutuksesta, joka tapahtuu lysosomien hajottamistoiminnasta aiheutuvan ei‑spesifisen puhdistuman välityksellä.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy
Dostarlimabin antoon liittyy riski naisilla, jotka voivat tulla raskaaksi. Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä dostarlimabihoidon aikana ja 4 kuukautta viimeisen dostarlimabiannoksen jälkeen.
Raskaus
Dostarlimabin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Vaikutusmekanisminsa perusteella dostarlimabi voi aiheuttaa haitallisia farmakologisia vaikutuksia sikiölle, jos sitä annetaan raskauden aikana.
Dostarlimabia ei ole tutkittu eläinten lisääntymis‑ ja kehitystutkimuksissa. PD‑1/PD‑L1‑reitin esto voi kuitenkin suurentaa kehittyvän sikiön immuunivälitteisen hyljinnän riskiä johtaen sikiökuolemaan (ks. kohta Prekliiniset tiedot turvallisuudesta). Ihmisen immunoglobuliinien (IgG4) tiedetään läpäisevän istukkaesteen, joten dostarlimabi (IgG4) saattaa siirtyä äidistä kehittyvään sikiöön.
JEMPERLI‑valmisteen käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi mutta eivät käytä tehokasta ehkäisyä.
Imetys
Ei tiedetä, erittyvätkö dostarlimabi/metaboliitit ihmisillä äidinmaitoon.
Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois.
JEMPERLI‑valmistetta ei pidä käyttää imetyksen aikana, ja imetystä on vältettävä vähintään 4 kuukautta viimeisen dostarlimabiannoksen jälkeen.
Hedelmällisyys
Dostarlimabin vaikutusta hedelmällisyyteen ei ole tutkittu (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
JEMPERLI‑valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Dostarlimabiin liittyi yleisimmin immuunivälitteisiä haittavaikutuksia. Useimmat niistä, mukaan lukien vaikeat haittavaikutukset, hävisivät asianmukaisen lääketieteellisen hoidon aloittamisen jälkeen tai dostarlimabihoidon tauottamisen jälkeen (ks. ”Valikoitujen haittavaikutusten kuvaus” jäljempänä).
Dostarlimabi monoterapiana
Dostarlimabin turvallisuutta on arvioitu GARNET‑tutkimuksessa 605 potilaalla, jotka saivat dostarlimabia monoterapiana kohdun limakalvon syövän tai muun pitkälle edenneen kiinteän kasvaimen hoitoon. Joukkoon sisältyi 153 potilasta, joilla oli pitkälle edennyt tai uusiutunut dMMR‑/MSI‑H‑tyyppinen kohdun limakalvon syöpä. Potilaat saivat 500 mg annoksia 3 viikon välein 4 hoitojakson ajan ja tämän jälkeen 1 000 mg 6 viikon välein kaikkien seuraavien hoitojaksojen ajan.
Potilailla, joilla oli pitkälle edenneitä tai uusiutuneita kiinteitä kasvaimia (N = 605), yleisimmät haittavaikutukset (> 10 %) olivat anemia (28,6 %), ripuli (26,0 %), pahoinvointi (25,8 %), oksentelu (19,0 %), nivelkipu (17,0 %), kutina (14,2 %), ihottuma (13,2 %), kuume (12,4 %), ASAT-arvon suureneminen (11,2 %) ja kilpirauhasen vajaatoiminta (11,2 %). JEMPERLI‑hoito lopetettiin pysyvästi haittavaikutuksen takia 38 potilaalla (6,3 %); valtaosa tapahtumista oli immuunivälitteisiä. Haittavaikutukset olivat vakavia 11,2 %:lla potilaista; useimmat vakavat haittavaikutukset olivat immuunivälitteisiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
GARNET‑tutkimuksessa dMMR‑/MSI‑H‑tyyppistä kohdun limakalvon syöpää sairastaneiden potilaiden (N = 153) turvallisuusprofiili ei ollut erilainen verrattuna koko monoterapiapopulaatioon, jonka tiedot on esitetty taulukossa 4.
Dostarlimabi yhdistelmähoitona karboplatiinin ja paklitakselin kanssa
Dostarlimabin turvallisuutta on arvioitu RUBY-tutkimuksessa 241 potilaalla, jotka saivat dostarlimabia yhdistelmähoitona karboplatiinin ja paklitakselin kanssa primaarisen pitkälle edenneen tai uusiutuneen kohdun limakalvon syövän hoitoon. Potilaat saivat dostarlimabia 500 mg 3 viikon välein 6 hoitojakson ajan ja tämän jälkeen 1 000 mg 6 viikon välein kaikkien seuraavien hoitojaksojen ajan.
Potilailla, joilla oli primaarinen pitkälle edennyt tai uusiutunut kohdun limakalvon syöpä (N = 241), yleisimmät haittavaikutukset (≥ 10 %) olivat ihottuma (23,2 %), makulopapulaarinen ihottuma (14,5 %), kilpirauhasen vajaatoiminta (14,5 %), kuume (12,9 %), ALAT-arvon suureneminen (12,9 %), ASAT-arvon suureneminen (12,0 %) ja ihon kuivuus (10,0 %). JEMPERLI-hoito lopetettiin pysyvästi lääkkeestä aiheutuneiden haittavaikutusten takia 12 potilaalla (5,0 %); valtaosa tapahtumista oli immuunivälitteisiä. Haittavaikutukset olivat vakavia 5,8 %:lla potilaista. Yleisin (> 1 %) vakava haittavaikutus oli kuume (2,9 %). Yleisin (> 10 %) immuunivälitteinen haittavaikutus oli kilpirauhasen vajaatoiminta (12,0 %), ja yleisin (> 1 %) hoidon lopettamiseen johtanut immuunivälitteinen haittavaikutus oli makulopapulaarinen ihottuma (1,2 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Kliinisissä tutkimuksissa dostarlimabille monoterapiana tai yhdistelmäsolunsalpaajahoidon osana ilmoitetut haittavaikutukset luetellaan taulukossa 4 elinjärjestelmäluokituksen ja esiintymistiheyden mukaan. Ellei toisin mainita dostarlimabi monoterapiana -sarakkeessa esitetyt haittavaikutusten esiintymistiheydet perustuvat kaikista syistä johtuneiden haittatapahtumien esiintymistiheyksiin GARNET‑tutkimuksessa 605:llä dostarlimabimonoterapiaa saaneella potilaalla, joilla oli pitkälle edenneitä tai uusiutuneita kiinteitä kasvaimia. Hoidon keston mediaani tutkimuksessa oli 24 viikkoa (vaihteluväli: 1–229 viikkoa). Ellei toisin ole mainittu, dostarlimabi yhdistelmäsolunsalpaajahoidon osana -sarakkeessa esitetyt haittavaikutusten esiintymistiheydet perustuvat kaikista syistä johtuneiden haittatapahtumien esiintymistiheyksiin RUBY-tutkimuksessa 241:llä dostarlimabia yhdistelmähoitona karboplatiinin ja paklitakselin kanssa saaneella potilaalla, joilla oli primaarinen pitkälle edennyt tai uusiutunut kohdun limakalvon syöpä. Hoidon keston mediaani tutkimuksessa oli 43 viikkoa (vaihteluväli 3–193 viikkoa). Lisätietoja dostarlimabin turvallisuudesta osana yhdistelmähoitoa karboplatiinin ja paklitakselin kanssa löytyy yhdistelmähoidossa käytettävien valmisteiden valmisteyhteenvedoista.
Haittavaikutuksia, joita tiedetään esiintyvän monoterapiana annetun dostarlimabin tai yksinään annetun karboplatiini- tai paklitakselihoidon yhteydessä, voi esiintyä myös näiden lääkevalmisteiden yhdistelmäkäytön yhteydessä, vaikka kyseisiä vaikutuksia ei olisi ilmoitettu dostarlimabin yhdistelmäkäyttöä karboplatiinin ja paklitakselin kanssa käsitelleissä kliinisissä tutkimuksissa. Haittavaikutukset luokitellaan elinjärjestelmäluokituksen ja esiintymistiheyden mukaan. Esiintymistiheydet määritellään seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä yleisyyden arviointiin).
Taulukko 4: Haittavaikutukset dostarlimabia saaneilla potilailla
Dostarlimabi monoterapiana | Dostarlimabi yhdistelmäsolunsalpaajahoidon osana | |
Veri ja imukudos | ||
Hyvin yleinen | Anemiaa | |
Umpieritys | ||
Hyvin yleinen | Kilpirauhasen vajaatoiminta*b | Kilpirauhasen vajaatoimintae |
Yleinen | Kilpirauhasen liikatoiminta*, lisämunuaisten vajaatoiminta* | Kilpirauhasen liikatoiminta |
Melko harvinainen | Tyreoidiitti*c, hypofysiittid | Tyreoidiitti, lisämunuaisten vajaatoiminta |
Aineenvaihdunta ja ravitsemus | ||
Melko harvinainen | Tyypin 1 diabetes, diabeettinen ketoasidoosi | Tyypin 1 diabetes |
Hermosto | ||
Melko harvinainen | Enkefaliitti, myasthenia gravis | Myasteeninen oireyhtymä†, Guillain–Barrén oireyhtymä†f |
Silmät | ||
Melko harvinainen | Uveiittig | Uveiitti |
Sydän | ||
Melko harvinainen | Myokardiitti†h | |
Hengityselimet, rintakehä ja välikarsina | ||
Yleinen | Pneumoniitti*i | Pneumoniitti |
Ruoansulatuselimistö | ||
Hyvin yleinen | Ripuli, pahoinvointi, oksentelu | |
Yleinen | Koliitti*j, pankreatiittik, gastriitti | Koliitti†l, pankreatiitti |
Melko harvinainen | Esofagiitti | Immuunivälitteinen gastriitti†, maha-suolikanavan vaskuliitti† |
Maksa ja sappi | ||
Yleinen | Hepatiitti*m | |
Iho ja ihonalainen kudos | ||
Hyvin yleinen | Ihottuma*n, kutina | Ihottuma o, ihon kuivuus |
Melko harvinainen | Stevens-Johnsonin oireyhtymä† | |
Luusto, lihakset ja sidekudos | ||
Hyvin yleinen | Nivelkipu* | |
Yleinen | Lihaskipu | |
Melko harvinainen | Immuunivälitteinen niveltulehdus, polymyalgia rheumatica, immuunivälitteinen myosiitti | Immuunivälitteinen niveltulehdus, myosiitti† |
Munuaiset ja virtsatiet | ||
Melko harvinainen | Nefriitti*p | |
Yleisoireet ja antopaikassa todettavat haitat | ||
Hyvin yleinen | Kuume | Kuume |
Yleinen | Vilunväristykset | |
Melko harvinainen | Systeeminen tulehdusvaste (SIRS-oireyhtymä)† | |
Tutkimukset | ||
Hyvin yleinen | Transaminaasiarvojen suureneminenq | ALAT-arvon suureneminen, ASAT-arvon suureneminen |
Vammat, myrkytykset ja hoitokomplikaatiot | ||
Yleinen | Infuusioreaktio*r | |
† Sisältää tapahtumat, joita on todettu muissa kliinisissä tutkimuksissa dostarlimabia monoterapiana tai dostarlimabia erilaisten muiden syöpähoitojen kanssa yhdistelmähoitona saaneilla potilailla, joilla oli kiinteä kasvain.
* Ks. kohta Valikoitujen haittavaikutusten kuvaus
a Sisältää anemian ja autoimmuunihemolyyttisen anemian
b Sisältää hypotyreoosin ja autoimmuunihypotyreoosin
c Sisältää tyreoidiitin ja autoimmuunityreoidiitin
d Sisältää hypofysiitin ja lymfosyyttisen hypofysiitin
e Sisältää hypotyreoosin ja immuunivälitteisen hypotyreoosin
f Sisältää Guillain–Barrén oireyhtymän ja demyelinoivan polyneuropatian
g Sisältää uveiitin ja iridosykliitin
h Sisältää myokardiitin ja immuunivälitteisen myokardiitin
i Sisältää pneumoniitin, interstitiaalisen keuhkosairauden ja immuunivälitteisen keuhkosairauden
j Sisältää koliitin, enterokoliitin ja immuunivälitteisen enterokoliitin
k Sisältää pankreatiitin ja akuutin pankreatiitin
l Sisältää koliitin ja enteriitin
m Sisältää hepatiitin, autoimmuunihepatiitin ja sytolyyttisen hepatiitin
n Sisältää ihottuman, makulopapulaarisen ihottuman, eryteeman, makulaarisen ihottuman, kutiavan ihottuman, punoittavan ihottuman, papulaarisen ihottuman, erythema multiformen (monimuotoinen punavihoittuma), ihotoksisuuden, lääkeaineihottuman, toksisen ihomuutoksen, eksfoliatiivisen ihottuman ja pemfigoidin
o Sisältää ihottuman ja makulopapulaarisen ihottuman
p Sisältää nefriitin ja tubulointerstitiaalisen nefriitin
q Sisältää transaminaasiarvojen suurenemisen, ALAT‑arvon suurenemisen, ASAT‑arvon suurenemisen ja suurentuneet transaminaasiarvot
r Sisältää infuusioon liittyvän reaktion ja yliherkkyysreaktion.
Valikoitujen haittavaikutusten kuvaus
Jäljempänä kuvatut valikoidut haittavaikutukset perustuvat monoterapian turvallisuutta koskevan yhdistetyn tietokannan tietoihin dostarlimabin turvallisuudesta 605 potilaalla GARNET‑tutkimuksessa. Potilailla oli kohdun limakalvon syöpä tai muu pitkälle edennyt kiinteä kasvain. Immuunivälitteisiksi haittavaikutuksiksi määriteltiin vähintään asteen 2 tapahtumat; jäljempänä mainituista esiintymistiheyksistä on suljettu pois asteen 1 tapahtumat. Haittavaikutusten hoito‑ohjeet on kuvattu kohdassa Annostus ja antotapa.
Immuunivälitteiset haittavaikutukset (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Immuunivälitteinen pneumoniitti
Immuunivälitteistä pneumoniittia esiintyi 14 potilaalla (2,3 %), mukaan lukien asteen 2 (1,3 %), asteen 3 (0,8 %) ja asteen 4 (0,2 %) pneumoniitti. Pneumoniitin takia dostarlimabihoito lopetettiin 8 potilaalla (1,3 %).
Systeemistä kortikosteroidihoitoa (≥ 40 mg/vrk prednisonia tai vastaavaa) tarvittiin 11 pneumoniittipotilaalla (78,6 %). Pneumoniitti korjautui 11 potilaalla (78,6 %).
Immuunivälitteinen koliitti
Koliittia esiintyi 8 potilaalla (1,3 %), mukaan lukien asteen 2 (0,7 %) ja asteen 3 (0,7 %) koliitti. Koliitti ei johtanut hoidon lopetukseen yhdelläkään potilaalla.
Systeemistä kortikosteroidihoitoa (≥ 40 mg/vrk prednisonia tai vastaavaa) tarvittiin 5 potilaalla (62,5 %). Koliitti korjautui 5 potilaalla (62,5 %).
Immuunivälitteinen hepatiitti
Hepatiittia (aste 3) esiintyi 3 potilaalla (0,5 %). Systeeminen kortikosteroidi (≥ 40 mg/vrk prednisonia tai vastaavaa) oli tarpeen 2 potilaalla (66,7 %). Hepatiitti johti dostarlimabihoidon lopettamiseen 1 potilaalla (0,2 %) ja korjautui 2 potilaalla 3:sta.
Immuunivälitteiset endokrinopatiat
Kilpirauhasen vajaatoimintaa esiintyi 46 potilaalla (7,6 %) (kaikki astetta 2). Kilpirauhasen vajaatoiminta ei johtanut dostarlimabihoidon lopettamiseen, ja korjautui 17 potilaalla (37,0 %).
Kilpirauhasen liikatoimintaa esiintyi 14 potilaalla (2,3 %), mukaan lukien asteen 2 (2,1 %) ja asteen 3 (0,2 %) liikatoiminta. Kilpirauhasen liikatoiminta ei johtanut dostarlimabihoidon lopettamiseen, ja korjautui 10 potilaalla (71,4 %).
Tyreoidiittia (aste 2) esiintyi 3 potilaalla (0,54 %). Tyreoidiitti ei korjautunut yhdessäkään tapauksessa; tyreoidiitti ei johtanut dostarlimabihoidon lopettamiseen.
Lisämunuaisten vajaatoimintaa esiintyi 7 potilaalla (1,2 %), mukaan lukien asteen 2 (0,5 %) ja asteen 3 (0,7 %) vajaatoiminta. Lisämunuaisten vajaatoiminta johti dostarlimabihoidon lopettamiseen 1 potilaalla (0,2 %) ja korjautui 4 potilaalla (57,1 %).
Immuunivälitteinen nefriitti
Nefriittiä (mukaan lukien tubulointerstitiaalista nefriittiä) esiintyi 3 potilaalla (0,5 %); kaikki tapahtumat olivat astetta 2. Systeemistä kortikosteroidihoitoa (≥ 40 mg/vrk prednisonia tai vastaavaa) tarvittiin 2 nefriittipotilaalla (66,7 %). Nefriitti johti dostarlimabihoidon lopettamiseen 1 potilaalla (0,2 %) ja korjautui kaikilla 3 potilaalla.
Immuunivälitteinen ihottuma
Immuunivälitteistä ihottumaa (ihottuma, makulopapulaarinen ihottuma, makulaarinen ihottuma, kutiava ihottuma, pemfigoidi, lääkeaineihottuma, ihotoksisuus, toksinen ihomuutos) esiintyi dostarlimabihoidon yhteydessä 31 potilaalla (5,1 %), joista 9 potilaalla (1,5 %) haitta oli astetta 3. Ihottuman ilmaantumiseen kuluneen ajan mediaani oli 57 vrk (vaihteluväli: 2–1 485 vrk). Systeemistä kortikosteroidihoitoa (≥ 40 mg/vrk prednisonia tai vastaavaa) tarvittiin 9 ihottumapotilaalla (29,0 %). Ihottuma johti dostarlimabihoidon lopettamiseen 1 potilaalla (0,2 %) ja korjautui 24 potilaalla (77,4 %).
Immuunivälitteinen nivelkipu
Immuunivälitteistä nivelkipua esiintyi 34 potilaalla (5,6 %). Asteen 3 immuunivälitteistä nivelkipua ilmoitettiin 5 potilaalla (0,8 %), jotka saivat dostarlimabia. Nivelkivun ilmaantumiseen kuluneen ajan mediaani oli 94,5 vrk (vaihteluväli: 1–840 vrk). Systeemistä kortikosteroidihoitoa (≥ 40 mg/vrk prednisonia tai vastaavaa) tarvittiin 3 nivelkipupotilaalla (8,8 %). Nivelkipu johti dostarlimabihoidon lopettamiseen 1 potilaalla (0,2 %) ja korjautui 19 potilaalla (55,9 %).
Infuusioreaktiot
Infuusioreaktioita (yliherkkyys mukaan lukien) esiintyi 6 potilaalla (1,0 %), mukaan lukien asteen 2 (0,3 %) ja asteen 3 (0,2 %) infuusioreaktiot. Kaikki potilaat toipuivat infuusioreaktiosta.
Immuunivasteen vapauttajien vaikutukset
Seuraavia haittavaikutuksia on raportoitu muiden immuunivasteen vapauttajien käytön yhteydessä: keliakia ja haiman eksokriininen vajaatoiminta. Vastaavia vaikutuksia voi myös esiintyä dostarlimabi-hoidon aikana.
Immunogeenisuus
GARNET-tutkimuksessa dostarlimabille muodostuvia lääkevasta‑aineita tutkittiin 315 potilaalta, jotka olivat saaneet dostarlimabia. Dostarlimabihoidon aikana ilmenneiden lääkevasta‑aineiden ilmaantuvuus oli 2,5 %. Neutraloivia vasta‑aineita havaittiin 1,3 %:lla potilaista. Samanaikainen karboplatiini- ja paklitakselihoito ei vaikuttanut dostarlimabin immunogeenisuuteen. RUBY-tutkimuksessa 225 potilaalla, jotka saivat dostarlimabia yhdistelmähoitona karboplatiinin ja paklitakselin kanssa ja joilla lääkevasta-aineiden esiintymistä voitiin tutkia, ei havaittu dostarlimabihoidon aikana ilmenneitä lääkevasta-aineita tai neutraloivia vasta-aineita.
Potilailla, joille kehittyi vasta‑aineita dostarlimabille, ei havaittu näyttöä dostarlimabin tehon tai turvallisuuden muuttumisesta.
Iäkkäät
605:stä dostarlimabia monoterapiana saaneesta potilaasta 51,6 % oli alle 65-vuotiaita, 36,9 % 65–< 75-vuotiaita ja 11,5 % vähintään 75-vuotiaita. 241:stä dostarlimabia yhdistelmähoitona karboplatiinin ja paklitakselin kanssa saaneesta potilaasta 52,3 % oli alle 65‑vuotiaita, 36,5 % 65–< 75‑vuotiaita ja 11,2 % vähintään 75‑vuotiaita. Turvallisuudessa ei raportoitu eroja iäkkäiden (≥ 65-vuotiaiden) ja nuorempien (< 65-vuotiaiden) potilaiden välillä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‑haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Jos yliannostusta epäillään, potilasta tulee seurata haittavaikutusten oireiden ja löydösten varalta ja asianmukainen oireenmukainen hoito aloittaa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, monoklonaaliset vasta‑aineet ja vasta-ainekonjugoidut lääkkeet, ATC‑koodi: L01FF07
Vaikutusmekanismi
Dostarlimabi on IgG4‑isotyypin humanisoitu monoklonaalinen vasta‑aine, joka sitoutuu PD‑1‑reseptoreihin ja estää niiden sitoutumisvuorovaikutusta PD‑L1‑ ja PD‑L2‑ligandien kanssa. PD‑1‑reittivälitteisen immuunivasteen estyminen johtaa T‑solujen toiminnan kuten proliferaation, sytokiinituotannon ja sytotoksisen aktiviteetin uudelleen aktivoitumiseen. Dostarlimabi voimistaa T‑soluvastetta, mukaan lukien antituumorista immuunivastetta, estämällä PD‑1:n sitoutumisen PD‑L1‑ ja PD‑L2‑ligandeihin. Syngeenisissä hiiren kasvainmalleissa PD‑1‑toiminnan esto vähensi kasvaimen kasvua.
Kliininen teho ja turvallisuus
RUBY: Satunnaistettu kontrolloitu tutkimus dostarlimabin käytöstä yhdistelmähoidossa karboplatiinin ja paklitakselin kanssa aikuispotilailla, joilla on primaarinen pitkälle edennyt tai uusiutunut kohdun limakalvon syöpä
Dostarlimabin tehoa ja turvallisuutta yhdistelmähoidossa karboplatiinin ja paklitakselin kanssa tutkittiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen 3 monikeskustutkimuksessa, johon osallistui primaarista pitkälle edennyttä tai uusiutunutta kohdun limakalvon syöpää sairastavia potilaita.
Potilaat satunnaistettiin (1:1) saamaan joko 500 mg dostarlimabia sekä karboplatiinia AUC 5 mg/ml/min ja paklitakselia 175 mg/m2 3 viikon välein 6 hoitojaksoa ja sen jälkeen 1 000 mg dostarlimabia 6 viikon välein (n = 245) tai lumelääkettä ja karboplatiinia AUC 5 mg/ml/min ja paklitakselia 175 mg/m2 3 viikon välein 6 hoitojaksoa ja sen jälkeen lumelääkettä 6 viikon välein (n = 249). Satunnaistamisessa ryhmät stratifioitiin MMR/MSI-statuksen, aiemman lantion alueen ulkoisen sädehoidon ja taudin tilanteen (uusiutunut, primaarinen levinneisyysluokka III tai primaarinen levinneisyysluokka IV) mukaan. Hoitoa jatkettiin enintään kolme vuotta tai kunnes ilmeni toksisuutta, jota ei voitu hyväksyä, tauti eteni tai tutkija päätti lopettaa hoidon. Kasvaimen tila arvioitiin 6 viikon välein viikkoon 25 asti, 9 viikon välein viikkoon 52 asti ja sen jälkeen 12 viikon välein. Kun seurannan keston mediaani oli 37 kuukautta, 27 potilasta 245:stä dostarlimabia sekä karboplatiinia ja paklitakselia saamaan satunnaistetusta potilaasta oli saanut hoitoa yli 3 vuotta (tiedonkeruun katkaisupäivä 22.9.2023).
Tärkeimmät tutkimukseenottokriteerit olivat FIGO (International Federation of Gynaecology and Obstetrics) -luokituksen mukainen primaarinen levinneisyysluokan III tai IV tauti, mukaan lukien RECIST 1.1 -kriteereillä arvioitavissa tai mitattavissa olevat levinneisyysluokan IIIA–IIIC1 taudit, levinneisyysluokan IIIC1 karsinosarkooma, kirkassoluinen, seroosi tai sekatyyppinen histologia (josta ≥ 10 % karsinosarkoomaa, kirkassoluista tai seroosia) riippumatta siitä, oliko tauti kuvantamalla arvioitavissa tai mitattavissa, levinneisyysluokan IIIC2 tai IV tauti riippumatta siitä, oliko tauti kuvantamalla arvioitavissa tai mitattavissa. Tutkimukseen otettiin myös potilaita, joilla oli ensimmäisen kerran uusiutunut kohdun limakalvon syöpä, jonka hoitomahdollisuudet sädehoidolla tai/ja leikkaushoidolla olivat huonot, mukaan lukien potilaat, joilla oli ensimmäisen kerran uusiutunut tauti ja jotka eivät olleet aiemmin saaneet systeemistä syöpälääkehoitoa, tai jotka olivat saaneet aiemmin systeemistä neoadjuvantti-/adjuvanttihoitoa ja tauti oli uusiutunut tai edennyt ≥ 6 kuukauden kuluttua hoidon päättymisen jälkeen (ensimmäinen uusiutuminen). Potilaat eivät olleet saaneet saada sädehoitoa 21 vuorokauteen ennen tutkimushoidon alkua, lukuun ottamatta palliatiivista sädehoitoa, jonka aikaraja oli yksi viikko ennen tutkimushoidon alkua.
Ensisijaiset tehon tulosmuuttujat olivat tutkijan RECIST 1.1 -kriteerien mukaan arvioima etenemisvapaa elossaolo potilailla, joilla oli dMMR/MSI-H-tyyppinen primaarinen pitkälle edennyt tai uusiutunut kohdun limakalvon syöpä, ja kaikilla potilailla (koko populaatio), joilla oli primaarinen pitkälle edennyt tai uusiutunut kohdun limakalvon syöpä, sekä kokonaiselossaolo kaikilla potilailla (koko populaatio), joilla oli primaarinen pitkälle edennyt tai uusiutunut kohdun limakalvon syöpä.
RUBY-tutkimuksessa tehoa arvioitiin yhteensä 494 potilaalla, joilla oli kohdun limakalvon syöpä. Demografiset tiedot ja potilaiden lähtötilanteen ominaisuudet olivat seuraavanlaiset: iän mediaani oli 65 vuotta (38 % 65–74‑vuotiaita ja 13 % ≥ 75-vuotiaita); 77 % oli valkoihoisia, 12 % mustaihoisia ja 3 % aasialaisia; ECOG‑toimintakykyluokka oli 63 %:lla 0 ja 37 %:lla 1; primaarinen levinneisyysluokan III tauti oli 19 %:lla, primaarinen levinneisyysluokan IV tauti 34 %:lla, ja uusiutunut kohdun limakalvon syöpä 48 %:lla potilaista; endometrioidi karsinooma oli 55 %:lla, sekatyyppinen karsinooma 4 %:lla, karsinosarkooma 9 %:lla, kirkassoluinen karsinooma 3 %:lla, seroosi karsinooma 21 %:lla ja muu 8 %:lla potilaista; aiemmin leikkaushoitoa oli saanut 91 %, sädehoitoa 28 % ja syövän lääkehoitoa 20 % potilaista.
Kasvaimen dMMR‑/MSI‑H‑status määritettiin prospektiivisesti paikallisilla määritysmenetelmillä (IHK, PCR tai NGS) tai keskitetyllä testauksella (IHK), jos tuloksia ei voitu saada paikallisesti.
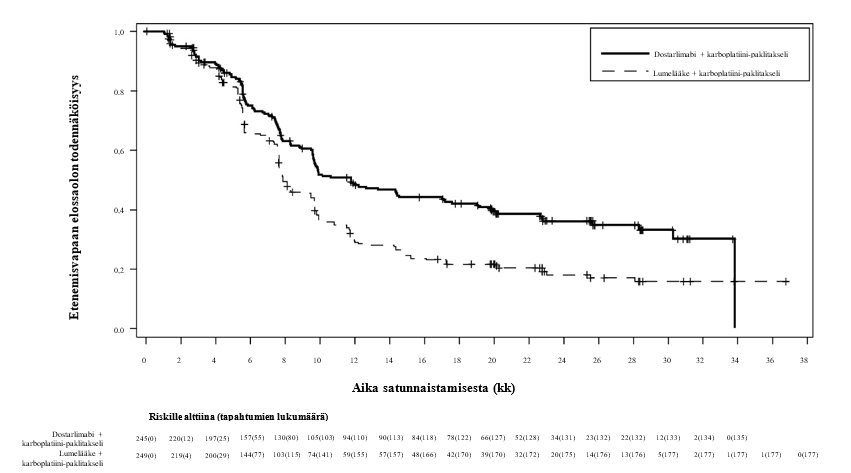
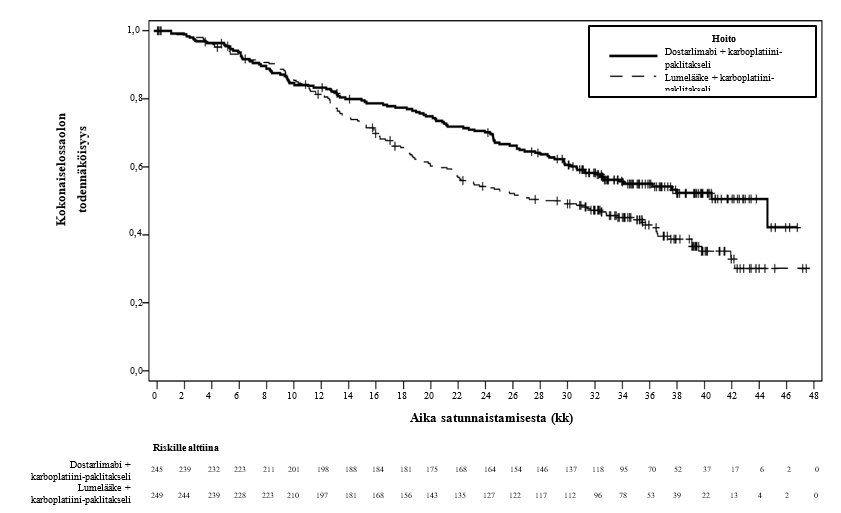
Tehotulokset esitetään taulukossa 5 ja kuvissa 1 ja 2. Etenemisvapaan elossaolon osalta on esitetty tiedot primaarianalyysista, jossa seurannan keston mediaani oli 25 kuukautta. Kokonaiselossaolon tulokset perustuvat toiseen kokonaiselossaolon välianalyysiin, jossa seurannan keston mediaani oli 37 kuukautta. Potilailla, jotka saivat dostarlimabia sekä karboplatiinia ja paklitakselia, todettiin tilastollisesti merkitsevät kohenemat tutkijan arvioimassa etenemisvapaassa elossaolossa (dMMR‑/MSI‑H-populaatio ja koko populaatio) ja kokonaiselossaolossa (koko populaatio) verrattuna potilaisiin, jotka saivat lumelääkettä sekä karboplatiinia ja paklitakselia.
Taulukko 5: Tehotulokset RUBY‑tutkimuksessa koko populaatiossa ja dMMR‑/MSI‑H‑tyyppistä kohdun limakalvon syöpää sairastavilla potilailla
| Koko populaatio | dMMR‑/MSI‑H-populaatio | ||
Päätetapahtuma | Dostarlimabi + karboplatiini-paklitakseli (N = 245) | Lumelääke + karboplatiini-paklitakseli (N = 249) | Dostarlimabi + karboplatiini-paklitakseli (N = 53) | Lumelääke + karboplatiini-paklitakseli (N = 65) |
Etenemisvapaa elossaoloa |
|
|
|
|
Mediaani, kuukausia (95 % lv)b | 11,8 (9,6, 17.1) | 7,9 (7,6, 9,5) | Ei saavutettu (11,8, ES) | 7,7 (5,6, 9,7) |
Tapahtuman kokeneiden lukumäärä (%) | 135 (55,1) | 177 (71,1) | 19 (35,8) | 47 (72,3) |
Riskitiheyssuhde (95 % lv)c | 0,64 (0,51, 0,80) | 0,28 (0,16, 0,50) | ||
p-arvod | < 0,0001 | < 0,0001 | ||
Kokonaiselossaoloe,f |
|
|
|
|
Mediaani, kuukausia (95 % lv)b | 44,6 (32,6, ES) | 28,2 (22,1, 35,6) | Ei saavutettu (ES, ES) | 31,4 (20,3, ES) |
Tapahtuman kokeneiden lukumäärä (%) | 109 (44,5) | 144 (57,8) | 12 (22,6) | 35 (53,8) |
Riskitiheyssuhde (95 % lv)c | 0,69 (0,54, 0,89) | 0,32 (0,17, 0,63) | ||
p-arvod | 0,0020 | Ei saatavillag | ||
Objektiivisten vasteiden osuus (ORR)h |
|
|
|
|
ORR, n (%) (95 % lv) | 149 (70,3) (63,6, 76,3) | 142 (64,8) (58,1, 71,2) | 38 (77,6) (63,4, 88,2) | 40 (69,0) (55,5, 80,5) |
Vasteen kesto (DOR)h,i |
|
|
|
|
Mediaani, kuukausia (95 % lv) | 10,6 (8,2, 17,6) | 6,2 (4,4, 6,7) | Ei saavutettu (10,1, ES) | 5.4 (3,9, 8,1) |
ES: ei saavutettu; lv: luottamusväli.
a Seurannan keston mediaani 25 kk (tiedonkeruun katkaisupäivä 28.9.2022).b Brookmeyer–Crowleyn menetelmän mukaan.
c Perustuu ositettuun Coxin regressiomalliin.
d Yksipuolinen p-arvo perustuu ositettuun log-rank-testiin.
e Kokonaiselossaolo on ensisijainen päätetapahtuma vain koko populaatiossa.
f Seurannan keston mediaani 37 kk (tiedonkeruun katkaisupäivä 22.9.2023).
g Ei tilastollisesti merkitsevä, koska hypoteesin testausta ei suoritettu kokonaiselossaololle dMMR/MSI-H-populaatiossa.
h Tutkijan RECIST 1.1 ‑kriteerien mukaan arvioimana.
i Potilailla, jotka saavuttivat osittaisen tai täydellisen vasteen.
Potilaista, joilla oli mismatch repair proficient (MMRp) / microsatellite stable (MSS) -tyyppinen kohdun limakalvon syöpä (n = 376), tehtiin ennalta määritellyt eksploratiiviset etenemisvapaan elossaolon ja kokonaiselossaolon analyysit. Etenemisvapaan elossaolon riskitiheyssuhde oli 0,76 (95 % lv 0,59; 0,98). Dostarlimabia sekä karboplatiinia ja paklitakselia saaneiden (n = 192) etenemisvapaan elossaolon mediaani oli 9,9 kuukautta ja lumelääkettä sekä karboplatiinia ja paklitakselia saaneiden (n = 184) 7,9 kuukautta (tiedonkeruun katkaisupäivä 28.9.2022). Kokonaiselossaolon riskitiheyssuhde oli 0,79 (95 % lv 0,60; 1,04). Dostarlimabia sekä karboplatiinia ja paklitakselia saaneiden kokonaiselossaolon mediaani oli 34 kuukautta ja lumelääkettä sekä karboplatiinia ja paklitakselia saaneiden 27 kuukautta (tiedonkeruun katkaisupäivä 22.9.2023).
Kuva 1. Kaplan–Meier-kuvaaja tutkijan arvioimasta etenemisvapaasta elossaolosta kaikilla kohdun limakalvon syöpää sairastavilla potilailla (koko populaatio, RUBY-tutkimus)
Kuva 2. Kaplan–Meier-kuvaaja kokonaiselossaolosta kaikilla kohdun limakalvon syöpää sairastavilla potilailla (koko populaatio, RUBY-tutkimus)
GARNET: aikuispotilaat, joilla oli uusiutunut tai pitkälle edennytdMMR/MSI-H-tyyppinen kohdun limakalvon syöpä, joka oli edennyt platinaa sisältävän hoito-ohjelman aikana tai sen jälkeen
Dostarlimabimonoterapian tehoa ja turvallisuutta tutkittiin GARNET‑tutkimuksessa, joka oli kontrolloimaton, avoin, usean rinnakkaiskohortin monikeskustutkimus. GARNET‑tutkimuksessa käytettiin laajennuskohortteja, joiden potilailla oli uusiutunut tai pitkälle edennyt kiinteä kasvain ja joille saatavilla olevat hoitovaihtoehdot olivat rajalliset. Kohorttiin A1 otettiin potilaita, joilla oli dMMR/MSI‑H‑tyyppinen kohdun limakalvon syöpä, joka oli edennyt platinaa sisältävän hoito‑ohjelman aikana tai sen jälkeen.
Potilaat saivat 500 mg dostarlimabia 3 viikon välein 4 hoitojakson ajan ja tämän jälkeen 1 000 mg dostarlimabia 6 viikon välein. Hoitoa jatkettiin, kunnes todettiin toksisuus, jota ei voitu hyväksyä tai taudin eteneminen, kahteen vuoteen asti.
Tärkeimmät tehon tulosmuuttujat olivat objektiivisten vasteiden osuus (ORR) ja vasteen kesto (DOR), jotka sokkoutettu riippumaton radiologiarviointitoimikunta arvioi keskitetysti RECIST v1.1 ‑kriteerien (Response Evaluation Criteria in Solid Tumours) perusteella. Tehopopulaatioon laskettiin potilaat, joilla oli lähtötilanteessa sokkoutetun, riippumattoman, keskitetyn arvioinnin perusteella mitattavissa oleva tauti ja joita seurattiin vähintään 24 viikkoa tai joilla oli vähemmän kuin 24 viikkoa seuranta-aikaa ja joilla hoito lopetettiin haittatapahtuman tai taudin etenemisen takia.
GARNET‑tutkimuksessa tehoa arvioitiin yhteensä 143 potilaalla, joilla oli dMMR‑/MSI‑H‑tyyppinen kohdun limakalvon syöpä. Näillä 143 potilaalla lähtötilanteen ominaisuudet olivat seuraavanlaiset: iän mediaani oli 65 vuotta (52 % vähintään 65‑vuotiaita), 77 % oli valkoihoisia, 3,5 % aasialaisia ja 2,8 % mustaihoisia ja ECOG‑toimintakykyluokka oli 39 %:lla 0 ja 61 %:lla 1. Toteamishetkellä 21 %:lla dMMR‑/MSI‑H‑tyyppistä kohdun limakalvon syöpää sairastavista FIGO‑levinneisyysluokka oli IV. Tutkimukseenottohetkellä (tuorein FIGO‑levinneisyysluokka) 67 %:lla potilaista FIGO‑levinneisyysluokka oli IV. Aiempien hoitojen määrän mediaani oli yksi: 63 % potilaista oli saanut yhtä aiempaa hoitoa, 37 % vähintään kahta aiempaa hoitoa. Potilaista 49 (34 %) oli saanut ennen tutkimukseen osallistumista hoitoa vain neoadjuvantti- tai adjuvanttihoitona.
Kasvaimen dMMR‑/MSI‑H‑status määritettiin prospektiivisesti paikallisella testauksella. Kasvainmateriaalin dMMR‑/MSI‑H‑ekspression toteamiseen käytettiin tutkimuspaikoissa saatavilla olevia, paikallisia diagnostisia määritysmenetelmiä (IHK, PCR tai NGS). Useimmissa paikoissa käytettiin IHK‑menetelmää, sillä se oli yleisimmin saatavilla oleva määritysmenetelmä.
Taulukko 6 sisältää tehotiedot 143 potilaasta. Hoidon kokonaiskeston mediaani oli 34 viikkoa (vaihteluväli 2–220 viikkoa). Dostarlimabia saaneista potilaista 24 % sai hoitoa yli 102 viikon ajan (yli 2 vuotta).
Taulukko 6: Tehotulokset GARNET‑tutkimuksessa dMMR‑/MSI‑H‑tyyppistä kohdun limakalvon syöpää sairastavilla potilailla
Päätetapahtuma | Tulokset (N = 143)a | |
Objektiivisten vasteiden osuus (ORR) |
| |
| ORR, n (%) | 65 (45,5) |
| (95 % lv) | (37,1, 54,0) |
| Täydellisen vasteen osuus, n (%) | 23 (16,1) |
| Osittaisen vasteen osuus, n (%) | 42 (29,4) |
Vasteen kesto (DOR)b |
| |
| Mediaani, kk | Ei saavutettu |
| Vasteen kesto ≥ 12 kk, n (%) | 52 (80,0) |
| Vasteen kesto ≥ 24 kk, n (%) | 29 (44,6) |
Taudin hallintaosuus (DCR)c |
| |
| DCR, n (%) | 86 (60,1) |
| (95 % lv) | (51,6, 68,2) |
Lv: luottamusväli
a Tehotiedot seurannan keston mediaanin ollessa 27,6 kk (tiedonkeruun katkaisupäivä 1.11.2021)b Potilailla, jotka saavuttivat osittaisen tai täydellisen vasteen.
c Sisältää potilaat, jotka saavuttivat täydellisen vasteen, osittaisen vasteen tai vakaan taudin vähintään 12 viikon ajaksi.
Teho ja PD‑L1‑status
Kliinistä aktiivisuutta havaittiin immunohistokemiallisesti määritetystä kasvaimen PD‑L1‑ekspression CPS‑arvosta (Combined Positive Score) riippumatta. PD‑L1‑statuksen ja tehon yhteydestä tehtiin jälkianalyysi GARNET-tutkimuksen kohortin A1 tehopopulaation potilailla, joilta oli saatavilla kudosnäytteitä (N = 81). Tiedonkeräyksen katkaisupäivä oli 1.3.2020. 23 potilaalla, joilla PD‑L1‑CPS‑arvo oli < 1 %, ORR oli 30,4 % (7/23, 95 % lv 13,2; 52,9) ja 58 potilaalla, joilla PD‑L1‑CPS‑arvo oli ≥ 1 %, ORR oli 55,2 % (32/58, 95 % lv 41,5; 68,3).
Iäkkäät potilaat
GARNET-tutkimuksessa dostarlimabia saaneista 108 tehopopulaation potilaasta 50,0 % oli yli 65-vuotiaita.
Tutkimustulosten havaittiin olevan yhdenmukaisia iäkkäillä. Sokkoutetun, riippumattoman, keskitetyn arvioinnin perusteella (95 % lv) ORR oli 42,6 % (29,2 %; 56,8 %) 65‑vuotta täyttäneillä tai sitä vanhemmilla.
Pediatriset potilaat
JEMPERLI-valmisteen ja niraparibin yhdistelmän turvallisuutta, farmakokinetiikkaa ja antituumorivaikutusta arvioitiin vaiheen 1 tutkimuksessa (SCOOP) 47 lapsella ja nuorella, joilla oli uusiutunut tai hoitoresistentti kiinteä kasvain, kuten osteosarkooma tai neuroblastooma. Potilaat olivat 5 – < 18-vuotiaita ja saivat 3 mg/kg tai 7,5 mg/kg (kuitenkin enintään 500 mg) JEMPERLI-valmistetta 3 viikon välein.
Tutkimus keskeytettiin ennenaikaisesti havaittujen toksisten vaikutusten sekä riittämättömän tehon vuoksi. Tutkimuksen annoslaajennusosaan osallistuneilla 17:llä osteosarkooma- tai neuroblastoomapotilaalla ei havaittu tehoa. Lääkeyhdistelmän turvallisuusprofiilia pediatrisilla potilailla ei voida pitää vahvistettuna, koska potilasmäärä oli pieni ja altistus oli vähäinen. Ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Farmakokinetiikka
Dostarlimabin farmakokinetiikkaa arvioitiin sekä monoterapiana että yhdistelmähoitona karboplatiinin ja paklitakselin kanssa.
Dostarlimabin ominaisuuksia monoterapiana ja yhdistelmähoitona karboplatiinin ja paklitakselin kanssa tutkittiin populaatiofarmakokinetiikan analyysissä 869 potilaalla, joilla oli erilaisia kiinteitä kasvaimia. Joukkoon sisältyi 546 potilasta, joilla oli kohdun limakalvon syöpä. Kun dostarlimabia annettiin monoterapiana suositeltuna hoitoannoksena (500 mg laskimoon 3 viikon välein 4 annoksen ajan ja tämän jälkeen 1 000 mg 6 viikon välein) tai yhdistelmähoitona karboplatiinin ja paklitakselin kanssa suositeltuna hoitoannoksena (500 mg laskimoon 3 viikon välein 6 annoksen ajan ja tämän jälkeen 1 000 mg 6 viikon välein), kumulaatio (Cmin) oli noin kaksinkertainen, ja tulos oli yhdenmukainen terminaalisen puoliintumisajan (t1/2) kanssa. Dostarlimabialtistus monoterapiana ja/tai yhdistelmähoitona karboplatiinin ja paklitakselin kanssa oli samankaltainen.
Imeytyminen
Dostarlimabi annetaan laskimoon, minkä takia imeytymistä koskevat arviot eivät ole sovellettavissa.
Jakautuminen
Dostarlimabin keskimääräinen vakaan tilan jakautumistilavuus on noin 5,8 l (variaatiokerroin [CV %] 14,9 %).
Biotransformaatio
Dostarlimabi on terapeuttinen IgG4‑isotyypin monoklonaalinen vasta‑aine, jonka odotetaan kataboloituvan pieniksi peptideiksi, aminohapoiksi ja pieniksi hiilihydraateiksi lysosomien vaikutuksesta pinosytoosin tai reseptorivälitteisen endosytoosin seurauksena. Hajoamistuotteet eliminoituvat erittymällä munuaisten kautta tai palautuvat ravintoainevarastoon aiheuttamatta biologisia vaikutuksia.
Eliminaatio
Keskimääräinen vakaan tilan puhdistuma on 0,007 l/h (CV % 30,2 %). Vakaan tilan t1/2 on 23,2 vrk (CV % 20,8 %).
Dostarlimabin puhdistuman arvioitiin olevan 7,8 % pienempi, kun dostarlimabia annetaan yhdessä karboplatiinin ja paklitakselin kanssa. Vaikutus dostarlimabialtistukseen ei ollut merkittävä.
Lineaarisuus/ei‑lineaarisuus
Altistus (enimmäispitoisuus [Cmax] ja pitoisuus‑aikakäyrän alle jäävä pinta‑ala, [AUC0‑tau] sekä [AUC0‑inf]) oli suunnilleen annosriippuvainen.
Farmakokineettiset/farmakodynaamiset suhteet
Altistuksen tehon ja turvallisuuden suhteiden perusteella tehossa ja turvallisuudessa ei ole kliinisesti merkittäviä eroja, kun altistus dostarlimabille on kaksinkertainen. Täysi reseptorien miehitys suoran PD‑1‑sitoutumisen ja interleukiini‑2 (IL‑2) ‑tuotannon toiminnallisen määrityksen perusteella mitattuna säilyi koko suositellun hoito‑ohjelman annosteluvälin ajan.
Erityisryhmät
Potilastietojen populaatiofarmakokinetiikan analyysi osoittaa, että iällä (vaihteluväli: 24–86 v), sukupuolella, etnisellä taustalla tai kasvaintyypillä ei ole kliinisesti merkittävää vaikutusta dostarlimabin puhdistumaan.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa arvioitiin laskennallisen kreatiniinipuhdistuman (CLCR ml/min) perusteella (normaali munuaistoiminta: CLCR ≥ 90 ml/min, n = 305; lievä vajaatoiminta: CLCR = 60–89 ml/min, n = 397; keskivaikea vajaatoiminta: CLCR = 30–59 ml/min, n = 164; vaikea vajaatoiminta: CLCR = 15–29 ml/min, n = 3 ja loppuvaiheen munuaisten vajaatoiminta: CLCR < 15 ml/min, n = 1). Munuaisten vajaatoiminnan vaikutusta dostarlimabin puhdistumaan arvioitiin populaatiofarmakokinetiikan analyyseissä lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla verrattuna potilaisiin, joiden munuaistoiminta oli normaalia. Dostarlimabin puhdistumassa ei havaittu kliinisesti merkittäviä eroja lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden ja munuaistoiminnaltaan normaalien potilaiden välillä. Tietoja on niukasti potilaista, joilla on vaikea munuaisten vajaatoiminta.
Maksan vajaatoiminta
Maksan vajaatoimintaa arvioitiin Yhdysvaltain National Cancer Institute ‑organisaation maksan toimintahäiriöitä koskevien kriteerien määritelmien mukaan. Perusteina olivat kokonaisbilirubiini‑ ja ASAT‑arvot (normaali maksatoiminta: kokonaisbilirubiini ja ASAT < = viitealueen yläraja (ULN), n = 772; lievä vajaatoiminta: kokonaisbilirubiini > ULN – 1,5 ULN tai ASAT > ULN, n = 92; ja keskivaikea vajaatoiminta: kokonaisbilirubiini > 1,5–3 ULN, mikä tahansa ASAT, n = 5). Maksan vajaatoiminnan vaikutusta dostarlimabin puhdistumaan arvioitiin populaatiofarmakokinetiikan analyyseissä lievää maksan vajaatoimintaa sairastavilla potilailla verrattuna potilaisiin, joiden maksatoiminta oli normaalia. Dostarlimabin puhdistumassa ei havaittu kliinisesti merkittäviä eroja lievää maksan vajaatoimintaa sairastavien potilaiden ja maksatoiminnaltaan normaalien potilaiden välillä. Tietoja on niukasti käytöstä keskivaikean maksan vajaatoiminnan yhteydessä eikä lainkaan käytöstä vaikean maksan vajaatoiminnan yhteydessä.
Pediatriset potilaat
Dostarlimabin farmakokinetiikkaa arvioitiin vaiheen 1 tutkimuksessa (SCOOP) 44 lapsella ja nuorella, joilla oli uusiutunut tai hoitoresistentti kiinteä kasvain. Tutkimus keskeytettiin ennenaikaisesti, joten farmakokinetiikkaa koskevat tiedot ovat rajalliset. 7,5 mg/kg:n annoksia saaneiden osallistujien (N = 28) näytteiden analyysissa dostarlimabialtistus oli yleisesti ottaen samaa luokkaa kuin aikuisilla.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien ei‑kliinisten, enimmillään 3 kuukauden pituisten tutkimusten tulokset jaavanmakakeilla eivät viittaa erityiseen vaaraan ihmisille. Dostarlimabin karsinogeenisuutta tai genotoksisuutta ei ole tutkittu. Dostarlimabilla ei ole tehty lisääntymis‑ ja kehitystoksisuustutkimuksia eläimillä. Tiineyden hiirimalleissa PD‑L1‑signaloinnin eston on osoitettu häiritsevän sikiön sietoa ja johtavan sikiökuolemien lisääntymiseen. Tulosten perusteella on olemassa mahdollinen riski, että dostarlimabin anto raskauden aikana vahingoittaa sikiötä, mukaan lukien keskenmenojen ja kohtukuolemien lisääntyneet määrät.
Apinoilla ei havaittu huomattavia vaikutuksia urosten ja naaraiden sukuelimiin 1 kuukauden ja 3 kuukauden pituisissa, toistuvan altistuksen aiheuttamaa toksisuutta koskeneissa tutkimuksissa. Tulokset eivät välttämättä ole kuitenkaan lainkaan edustavia mahdollisen kliinisen riskin suhteen tutkimuksissa käytettyjen eläinten sukuelinten kehittymättömyyden takia. Tästä syystä hedelmällisyyteen kohdistuvaa toksisuutta ei toistaiseksi tunneta.
Farmaseuttiset tiedot
Apuaineet
Trinatriumsitraattidihydraatti (E331)
Sitruunahappomonohydraatti (E330)
L‑arginiinihydrokloridi
Natriumkloridi
Polysorbaatti 80 (E433)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Avaamaton injektiopullo
5 vuotta
Laimennuksen jälkeen
Jos valmistetta ei käytetä välittömästi, sen on osoitettu säilyvän käytön aikana kemiallisesti ja fysikaalisesti stabiilina 24 tunnin ajan 2–8 °C:ssa ja 6 tunnin ajan huoneenlämmössä (enintään 25 °C) (aika valmistelusta/laimentamisesta annon päättymiseen).
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
JEMPERLI infuusiokonsentraatti, liuosta varten
500 mg (L:ei) 10 ml (8275,42 €)
PF-selosteen tieto
Tyypin I kirkkaasta borosilikaattilasista valmistettu 10 ml:n injektiopullo, jossa on harmaa, fluoropolymeerillä päällystetty klorobutyylielastomeeritulppa ja joka on sinetöity alumiinisella irti napsautettavalla flip‑off‑korkilla. Yksi injektiopullo sisältää 500 mg dostarlimabia.
Yksi kotelo sisältää yhden injektiopullon.
Valmisteen kuvaus:
Kirkas tai hieman opalisoiva, väritön tai keltainen liuos, jossa ei ole käytännössä lainkaan näkyviä hiukkasia.
Infuusiokonsentraatin, liuosta varten pH on noin 6,0 ja osmolaliteetti noin 300 mOsm/kg.
Käyttö- ja käsittelyohjeet
Valmistelu/laimennus
Parenteraaliset lääkevalmisteet on tarkastettava silmämääräisesti hiukkasten ja värimuutosten varalta ennen antoa. JEMPERLI on hieman opalisoivaa, väritöntä tai keltaista liuosta. Hävitä injektiopullo, jos havaitset näkyviä hiukkasia.
JEMPERLI on yhteensopiva infuusiopussin kanssa, joka on valmistettu polyvinyylikloridista (PVC) di(2-etyyliheksyyli)ftalaatin (DEHP) kanssa tai ilman, eteenivinyyliasetaatista, polyeteenista (PE), polypropeenista (PP) tai polyolefiinisekoituksesta (PP+PE), ja PP:stä valmistetun ruiskun kanssa.
500 mg annos: Vedä injektiopullosta 10 ml JEMPERLI-valmistetta ja siirrä infuusiopussiin, joka sisältää 9 mg/ml (0,9 %) NaCl‑injektionestettä tai 50 mg/ml (5 %) glukoosi‑injektionestettä. Laimennetun liuoksen lopullisen pitoisuuden on oltava 2–10 mg/ml. Infuusion kokonaistilavuus ei saa olla yli 250 ml. Tämä saattaa vaatia laimennusaineen poistamista infuusiopussista ennen JEMPERLI-määrän lisäämistä infuusiopussiin.
- Jos esimerkiksi valmistetaan 500 mg:n annos 250 ml:n laimennusainetta sisältävään infuusiopussiin, 2 mg/ml:n pitoisuuden saavuttamiseksi on 250 ml:n infuusiopussista otettava pois 10 ml laimennusainetta. Sitten 10 ml JEMPERLI-valmistetta vedetään injektiopullosta ja siirretään infuusiopussiin.
1000 mg annos: Vedä kahdesta injektiopullosta 10 ml JEMPERLI-valmistetta (yhteensä 20 ml) ja siirrä infuusiopussiin, joka sisältää 9 mg/ml (0,9 %) NaCl‑injektionestettä tai 50 mg/ml (5 %) glukoosi‑injektionestettä. Laimennetun liuoksen lopullisen pitoisuuden on oltava 4–10 mg/ml. Infuusion kokonaistilavuus ei saa olla yli 250 ml. Tämä saattaa vaatia laimennusaineen poistamista infuusiopussista ennen JEMPERLI-määrän lisäämistä infuusiopussiin.
- Jos esimerkiksi valmistetaan 1000 mg:n annos 250 ml:n laimennusainetta sisältävään infuusiopussiin, 4 mg/ml:n pitoisuuden saavuttamiseksi on 250 ml:n infuusiopussista otettava pois 20 ml laimennusainetta. Sitten 10 ml JEMPERLI-valmistetta vedetään kustakin kahdesta injektiopullosta, yhteensä 20 ml ja siirretään infuusiopussiin.
Sekoita laimennettu liuos kääntelemällä pussia varovasti. Älä ravista lopullista infuusiopussia. Hävitä injektiopulloon mahdollisesti käyttämättä jäänyt liuos.
Säilytys
Säilytä valmisteluun asti alkuperäiskotelossa. Herkkä valolle. Valmisteltu annos voidaan säilyttää:
- huoneenlämmössä enintään 25 ºC:ssa enintään 6 tuntia (aika laimennuksesta infuusion päättymiseen)
- jääkaapissa 2–8 ºC:ssa enintään 24 tuntia (aika laimennuksesta infuusion päättymiseen). Jos laimennettua liuosta on säilytetty jääkaapissa, liuoksen on annettava lämmetä huoneenlämpöiseksi ennen antoa.
Anto
Terveydenhuollon ammattilainen antaa JEMPERLI‑valmisteen infuusiopumpulla laskimoon 30 minuuttia kestävänä infuusiona. Putkien tulee olla PVC:tä, platinakovetettua silikonia tai PP:tä; liittimet valmistettu PVC:stä tai polykarbonaatista ja neulat valmistettu ruostumattomasta teräksestä. JEMPERLIN annon aikana on käytettävä A 0,2 tai 0,22 mikronin in-line-tyyppistä polyeetterisulfoni (PES) -suodatinta.
JEMPERLI‑valmistetta ei saa antaa nopeana injektiona / bolusinjektiona laskimoon.
Muita lääkevalmisteita ei saa antaa saman infuusioletkun kautta.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
JEMPERLI infuusiokonsentraatti, liuosta varten
500 mg 10 ml
- Ei korvausta.
ATC-koodi
L01FF07
Valmisteyhteenvedon muuttamispäivämäärä
04.06.2026
Yhteystiedot
 GLAXOSMITHKLINE OY
GLAXOSMITHKLINE OY Porkkalankatu 20 A
00180 Helsinki
010 303 030
www.glaxosmithkline.fi