
TRIAXIS injektioneste, suspensio, esitäytetty ruisku
Vaikuttavat aineet ja niiden määrät
1 annos (0,5 ml) sisältää:
Difteriatoksoidi Vähintään 2 IU* (2 Lf)
Tetanustoksoidi Vähintään 20 IU* (5 Lf)
Pertussisantigeeneja
Pertussistoksoidi 2,5 mikrogrammaa
Filamenttinen hemagglutiniini 5 mikrogrammaa
Pertaktiini 3 mikrogrammaa
Tyypin 2 ja 3 fimbriat 5 mikrogrammaa
Adsorboitu alumiinifosfaattiin 1,5 mg (0,33 mg Al3+)
* Alempi aktiivisuuden luottamusrajana (p = 0,95) Euroopan farmakopeiassa kuvatulla menetelmällä mitattuna.
Tämä rokote voi sisältää jäämiä formaldehydistä ja glutaraldehydistä joita käytetään valmistusprosessin aikana (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, suspensio, esitäytetty ruisku.
Kliiniset tiedot
Käyttöaiheet
Triaxis (Tdap)on tarkoitettu:
Aktiiviseen immunisaatioon kurkkumätää (difteria), jäykkäkouristusta (tetanus), ja hinkuyskää (pertussis) vastaan 4 vuotta täyttäneille ja sitä vanhemmille tehosteeksi perusrokotesarjan jälkeen.
Antamaan passiivisen suojan hinkuyskää (pertussis) vastaan varhaisessa imeväisiässä, kun äiti on rokotettu raskauden aikana (ks. kohdat Annostus ja antotapa, Raskaus ja imetys ja Farmakodynamiikka).
Triaxis-rokotetta on käytettävä virallisten suositusten mukaisesti.
Annostus ja antotapa
Annostus
Yksi 0,5 ml:n kerta-annos suositellaan annettavaksi kaikille ikäryhmille.
Nuorille ja aikuisille, joiden rokotusstatus jäykkäkouristuksen tai kurkkumädän suhteen on tuntematon tai joilta nämä rokotukset osittain puuttuvat, voidaan antaa yksi annos Triaxis-rokotetta osana rokotusohjelmaa, joka antaa suojan hinkuyskää ja useimmiten myös jäykkäkouristusta ja kurkkumätää vastaan. Yksi lisäannos kurkkumätä- ja jäykkäkouristuskomponentteja (dT) sisältävää rokotetta voidaan antaa yhden kuukauden kuluttua ja sen jälkeen kolmas annos kurkkumätä- tai dT-komponentteja sisältävää rokotetta 6 kuukauden kuluttua ensimmäisestä annoksesta, jotta suoja tautia vastaan on mahdollisimman hyvä (ks. kohta Farmakodynamiikka). Annettavien annosten määrä ja aikataulu on määritettävä paikallisten suositusten mukaisesti.
Triaxis-rokotetta voidaan käyttää uusintarokotukseen 5–10 vuoden välein tehostamaan immuniteettia kurkkumätää, jäykkäkouristusta ja hinkuyskää vastaan (ks. kohta Farmakodynamiikka).
Triaxis-rokotetta voi käyttää jäykkäkouristusvammojen hoitoon samanaikaisesti jäykkäkouristusimmunoglobuliinin kanssa, tai ilman sitä, virallisten suositusten mukaisesti.
Triaxis-rokotetta voidaan antaa raskaana oleville naisille toisen tai kolmannen raskauskolmanneksen aikana antamaan imeväiselle passiivisen suojan hinkuyskää vastaan (ks. kohdat Käyttöaiheet, Raskaus ja imetys ja Farmakodynamiikka).
Antotapa
Yksi annos (0,5 ml) Triaxis-rokotetta annetaan lihakseen. Suositeltu paikka on olkavarsi (deltoidilihas).
Triaxis-rokotetta ei saa antaa pakaroiden alueelle, ihon sisään tai alle (poikkeustilanteissa ihonalaista antoreittiä voidaan kuitenkin tarvittaessa harkita; ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ennen lääkkeen käsittelyä tai antoa huomioon otettavat varotoimet.
Lääkkeen käsittelyohjeet ennen antoa, ks. kohta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Triaxis-rokotetta ei pidä antaa henkilöille, joilla on tunnettu yliherkkyys
- difteria-, tetanus tai pertussis-rokotteille
- jollekin rokotteen ainesosista (ks. kohta Apuaineet)
- jollekin rokotuksen valmistuksen aikana käytetyn aineen jäämälle (formaldehydi ja glutaraldehydi), joita voi olla rokotteessa häviävän pieniä määriä.
Triaxis-rokotetta ei saa antaa henkilöille, jotka ovat saaneet tuntemattomasta syystä johtuvan enkefalopatian 7 päivän kuluessa aiemmasta pertussiskomponetteja sisältävästä rokotteesta.
Muiden rokotteiden tavoin Triaxis-rokotuksen antamistaon lykättävä henkilöillä, joilla on akuutti kuumesairaus. Lievä infektiosairaus ei ole rokotuksen vasta-aihe.
Varoitukset ja käyttöön liittyvät varotoimet
Triaxis-rokotetta ei saa käyttää lasten perusrokotukseen.
Triaxis-rokotteen tehosteannoksen ja edeltävien difteriaa ja/tai tetanusta sisältävien rokotteiden tehosteannosten välisen ajan pituus pitää yleensä olla virallisten suositusten mukainen. Kliiniset tutkimustiedot ovat osoittaneet, ettei tetanusta, difteriaa ja pertussista sisältävien tehosterokotusten antoon vain neljän viikon jälkeen liittynyt kliinisesti merkittäviä eroja haittavaikutusten esiintyvyydessä verrattuna tehosterokotteen antoon viiden vuoden kuluttua edellisen tetanusta ja difteriaa sisältävän rokoteannoksen jälkeen.
Ennen rokotusta
Ennen rokotteen antamista rokotettavan henkilön lääketieteellinen historia on selvitettävä (erityisesti aiemmat rokotukset ja mahdolliset haittatapahtumat). Rokotusohjelmaa on harkittava huolella niiden henkilöiden osalta, jotka ovat saaneet vakavia tai voimakkaita reaktioita 48 tunnin kuluessa samoja komponentteja sisältävistä rokotteista.
Kaikkien injisoitavien rokotteiden tavoin rokotuksen jälkeisen, harvinaisen anafylaktisen reaktion varalta on järjestettävä paikalle tarvittava lääketieteellinen hoito ja valvonta.
Jos Guillain-Barrén oireyhtymää on esiintynyt 6 viikon sisällä aiemman tetanustoksoidia sisältävän rokotteen saamisesta, seuraavan tetanustoksoidia sisältävän rokotteen, kuten Triaxis-rokotteen, antoa on harkittava huolella ja mahdollisia hyötyjä ja riskejä arvioiden.
Triaxis-rokotetta ei pidä antaa henkilöille, joilla on etenevä neurologinen sairaus, hallitsematon epilepsia tai etenevä enkefalopatioa, ennen kuin hoito-ohjelma on vakiintunut ja tila vakaa.
Immunosupressiolääkitys tai immuunipuutostila saattaa heikentää rokotteen immunogeenisuutta. Rokottamista suositellaan siirrettäväksi, kunnes tällainen tila tai lääkitys on päättynyt, mikäli se on käytännössä mahdollista. Kuitenkin HIV-tartunnan saaneet tai kroonista immuunipuutostilaa kuten AIDS:ia sairastavat henkilöt suositellaan rokotettaviksi, vaikka vasta-ainevaste olisikin vajaa.
Rokotuksen antoon liittyvät varotoimet
Älä annostele suonensisäisenä tai ihonalaisena injektiona.
Verenvuotoriskin vuoksi henkilöille, jotka saavat verenohennuslääkkeitä tai joilla on veren hyytymishäiriö, injektiot lihakseen on annettava varoen. Näissä tilanteissa voidaan harkita Triaxis-rokotteen antamista syvälle ihon alle, joskin tällöin riskinä on paikallisten reaktioiden lisääntyminen.
Pyörtymistä (synkopee) voi esiintyä injektioina annettavien rokotteiden, kuten Triaxis-rokotteen, antamisen jälkeen tai jopa ennen rokottamista. Pyörtymisestä johtuvat vammat on estettävä ja tajunnan menetykseen liittyviin reaktioihin on varauduttava varotoimenpiteillä.
Esitäytettyjen ruiskujen (1,5 ml) pehmeät kärkisuojukset sisältävät luonnonkumin (lateksin) johdannaista, joka saattaa aiheuttaa allergisia reaktioita lateksille herkille henkilöille.
Muut varoitukset
Kaikkein muiden rokotteiden tavoin Triaxis-rokote ei ehkä suojaa täysin kaikkia yksilöitä.
Pitkään kestävä kyhmy pistoskohdassa saattaa jäädä kaikkien adsorboitujen rokotteiden jälkeen, etenkin jos rokote annetaan ihonalaiskudoksen pintakerrokseen.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yhteisvaikutukset
Samanaikaista antoa koskevien kliinisten tutkimusten perusteella, Triaxis voidaan antaa samanaikaisesti inaktivoidun influenssarokotteen, hepatiitti B-rokotteen ja inaktivoidun tai oraalisen poliorokotteen kanssa. Lisäksi Triaxis voidaan antaa papilloomavirusrokotteen (HPV) ja/tai meningokokkipolysakkaridikonjugaattirokotteiden (seroryhmät A, C, Y ja W) (MenACYW) kanssa (joko kaikki kolme rokotetta samanaikaisesti tai pareittain) (ks. kohta Haittavaikutukset) paikallisten suositusten mukaisesti.
Samanaikaisen rokotteiden annon yhteydessä injektiot on annettava eri raajoihin.
Yhteisvaikutustutkimuksia muiden rokotteiden, biologisten valmisteiden tai terapeuttisten lääkkeiden kanssa ei ole tehty. Yleisesti hyväksyttyjen rokotusohjeiden mukaisesti Triaxis voidaan kuitenkin antaa samanaikaisesti toisten rokotteiden tai immuuniglobuliinien kanssa eri pistoskohtiin, koska se on inaktivoitu valmiste.
Jos potilas saa immunosuppressiohoitoa, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Raskaus ja imetys
Raskaus
Triaxis-rokotetta voidaan käyttää toisen tai kolmannen raskauskolmanneksen aikana virallisten suositusten mukaisesti (ks. kohta Annostus ja antotapa).
Neljästä satunnaistetusta kontrolloidusta tutkimuksesta (tiedot 310 raskaudesta), yhdestäprospektiivisesta havainnoivasta tutkimuksesta (tiedot 546 raskaudesta), viidestäretrospektiivisesta havainnoivasta tutkimuksesta (tiedot 124 810 raskaudesta) ja toisen tai kolmannen raskauskolmanneksen aikana Triaxis- tai Repevax-rokotteen (Tdap-IPV, sisältää samat määrät jäykkäkouristus-, kurkkumätä- ja hinkuyskäantigeenejä kuin Triaxis) saaneiden naisten passiivisesta seurannasta saaduissa turvallisuustiedoissa ei ole todettu rokotteeseen liittyviä raskauteen tai sikiön/vastasyntyneen terveyteen kohdistuvia haittavaikutuksia. Kuten muidenkaan inaktivoitujen rokotteiden kohdalla, ei ole odotettavissa, että minkään raskauskolmanneksen aikana annettu Triaxis-rokote aiheuttaisi haittaa sikiölle.
Eläinkokeissa ei ole havaittu suoria tai epäsuoria haitallisia vaikutuksia raskauteen, alkion/sikiön kehitykseen, synnytykseen tai postnataaliseen kehitykseen.
Katso kohdasta Farmakodynamiikka tiedot raskauden aikana annetun rokotuksen immuunivasteesta ja tehokkuudesta hinkuyskän estämisessä imeväisillä.
Imetys
Ei tiedetä, erittyvätkö Triaxis-rokotteen vaikuttavat aineet äidinmaitoon, mutta rokotuksen vasta-aineiden on havaittu siirtyvän kaniinien imeväisikäisiin jälkeläisiin. Kahdessa kaniineilla tehdyissä eläinkehityskokeessa ei osoitettu emon rokottamisesta johtuvien vasta-aineiden vaikuttavan haitallisesti jälkeläisten postnataaliseen kehitykseen.
Ihmisillä ei ole kuitenkaan tutkittu, vaikuttaako äideille annettu Triaxis imeväisikäisiin lapsiin. Koska Triaxis on inaktivoitu rokote, riski imeväiselle on epätodennäköinen. Rokotuksen haittoja ja hyötyjä on arvioitava ennen rokotteen antamista imettävälle naiselle.
Hedelmällisyys
Triaxis-rokotetta ei ole arvioitu hedelmällisyystutkimuksissa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia valmisteen vaikutuksesta ajokykyyn tai koneiden käyttökykyyn ei ole tehty. Triaxis-rokotteella ei ole haitallista vaikutusta ajokykyyn ja koneiden käyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kliinisissä tutkimuksissa Triaxis-rokotetta annettiin yhteensä 4 546 tutkittavalle, joista 298 4–6 vuoden ikäistä lasta, 1 313 11–17 vuoden ikäistä nuorta ja 2 935 18–64 vuoden ikäistä aikuista. Yleisimmin ilmoitettuja rokotuksen jälkeisiä reaktioita olivat pistokohdan paikalliset reaktiot (kipu, punotus ja turvotus), joita ilmeni 21–78 prosentilla rokotuksen saaneista, sekä päänsärky ja väsymys, joita ilmeni 16–44 prosentilla rokotuksen saaneista. Löydökset ja oireet olivat yleensä voimakkuudeltaan lieviä ja ilmenivät 48 tunnin kuluessa rokotuksesta. Kaikki reaktiot hävisivät ilman jälkiseurauksia.
Turvallisuusanalyysi, joka koski Triaxis- tai Repevax-rokotteen samanaikaista antoa MenACYW (Menactra tai MenQuadfi)- ja/tai HPV (Gardasil tai Gardasil 9) -rokotteiden kanssa, sisälsi 5 122 tervettä 10–17-vuotiasta miespuolista ja naispuolista henkilöä kuudesta kliinisestä tutkimuksesta.
Repevax- ja Triaxis-rokotteen yleinen turvallisuusprofiili oli samankaltainen, kun kaikki kolme rokotetta annettiin samanaikaisesti verrattuna samanaikaisesti annettuihin pareihin (Triaxis ja Menactra tai Repevax/Triaxis ja Gardasil/Gardasil 9). Samanaikaisesti annettujen Repevax- ja Gardasil/Gardasil 9 -rokotteiden turvallisuusprofiili oli samankaltainen kuin yksinään annetun Gardasil/Gardasil 9 -rokotteen.
Kliinisissä tutkimuksissa havaittiin enemmän injektiokohdan reaktioita (kipu, punoitus, turvotus ja mustelmat) sekä päänsärkyä, huonovointisuutta ja lihaskipua, kun kaikki kolme rokotetta annettiin yhdessä tai pareittain verrattuna siihen, kun ne annettiin yksinään. Kaiken kaikkiaan havaitut erot injektiokohdan reaktioissa olivat < 10 %, kun taas erot päänsäryssä, huonovointisuudessa ja lihaskivussa vaihtelivat < 10 %:sta < 28 %:iin. Kuumeen esiintyvyyden ero kliinisissä tutkimuksissa oli < 2 %.
Suurin osa raportoiduista haittavaikutuksista oli ilmoitusten mukaan voimakkuudeltaan lieviä tai keskivaikeita.
Haittavaikutustaulukko
Haittavaikutukset on lueteltu esiintymistiheytensä mukaan seuraavasti:
Hyvin yleinen (≥1/10)
Yleinen (≥1/100, <1/10)
Melko harvinainen (≥1/1 000, <1/100)
Harvinainen (≥1/10 000, <1/1 000)
Hyvin harvinainen (<1/10 000)
Tuntematon (koska saatavissa oleva tieto ei riitä arviointiin).
Taulukko 1 esittää kliinisissä tutkimuksissa havaittuja haittavaikutuksia ja siihen sisältyvät myös lisähaittatapahtumat, jotka on spontaanisti raportoitu Triaxis-rokotteen myyntiin tulon jälkeen maailmanlaajuisesti.
Koska myyntiin tulon jälkeisistä haittatapahtumista on ilmoitettu vapaaehtoisesti, eikä populaation määrää tiedetä tarkasti, ei ole aina mahdollista arvioida luotettavasti niiden esiintymistiheyttä tai selvittää syy‑yhteyttä rokotteelle altistumiselle. Tästä syystä nämä haittatapahtumat on sisällytetty ”tuntematon”‑kategoriaan.
Taulukko 1: Tutkimuksissa esiintyvät ja maailmanlaajuisen myyntiin tulon jälkeisen kokemuksen osoittamat haittatapahtumat
| Elinjärjestelmäluokitus | Esiintymistiheys | Lapset (4–6 vuoden ikäiset) | Nuoret (11–17 vuoden ikäiset) | Aikuiset (18–64 vuoden ikäiset) |
| Immuunijärjestelmä | tuntematon | yliherkkyys- (anafylaktinen) reaktio (angioödeema, ödeema, ihottuma, hypotensio)* | ||
| Aineenvaihdunta ja ravitsemus | hyvin yleinen | anoreksia (ruokahaluttomuus) | ||
| Hermosto | hyvin yleinen | päänsärky | ||
| tuntematon | parestesia, hypoestesia*, Guillain-Barrén syndrooma, brakiaalinen neuriitti*, kasvohalvaus*, kouristukset*, synkopee*, myeliitti* | |||
| Sydän | tuntematon | myokardiitti* | ||
| Ruoansulatuselimistö | hyvin yleinen | ripuli | ripuli, pahoinvointi | ripuli |
| yleinen | pahoinvointi, oksentelu | oksentelu | pahoinvointi, oksentelu | |
| Iho ja ihonalainen kudos | yleinen | ihottuma | ||
| tuntematon | kutina*, urtikaria* | |||
| Luusto, lihakset ja sidekudos | hyvin yleinen | yleinen kipu tai lihasheikkous, artralgia tai nivelturvotus | yleinen kipu tai lihasheikkous | |
| yleinen | yleinen kipu tai lihasheikkous, artralgia tai nivelten turvotus | artralgia tai nivelten turvotus | ||
| tuntematon | myosiitti* | |||
| Yleisoireet ja antopaikassa todettavat haitat | hyvin yleinen | väsymys/astenia | väsymys/astenia, huonovointisuus, vilunväristykset | väsymys/astenia, huonovointisuus |
| pistoskohdan kipu, pistoskohdan punoitus, pistoskohdan turvotus | ||||
| yleinen | pyreksia, vilunväristykset, aksillaarinen adenopatia | pyreksia, aksillaarinen adenopatia | pyreksia, vilunväristykset, aksillaarinen adenopatia | |
| tuntematon | pistoskohdan mustelma*, pistokohdan steriili abskessi*, pistoskohdan kyhmy* | |||
* haittatapahtumia myyntiin tulon jälkeen
Valittujen haittavaikutusten kuvaus
Yleisoireet ja antopaikassa todettavat haitat:
Laaja-alaisia (> 50 mm) pistoskohtareaktioita kuten raajan laajamittainen turpoaminen pistoskohdasta ensimmäiseen tai toiseen niveleen saakka Triaxis-rokotteen antamisen jälkeen nuorilla ja aikuisilla. Nämä reaktiot ilmenevät yleensä 24–72 tunnin kuluttua rokotuksen annosta ja niihin saattaa liittyä punoitusta, kuumotusta, pistoskohdan arkuutta tai kipua ja ne häviävät itsestään 3–5 päivän kuluessa.
Pediatriset potilaat
Triaxis-rokotteen turvallisuusprofiiliin, kuten se on esitetty Taulukossa 1, sisältyy tietoja 298 4–6 vuoden ikäisen lapsen kliinisestä tutkimuksesta, jotka olivat aikaisemmin saaneet yhteensä 4 annosta, primaarinen immunisaatio mukaan lukien, DTaP-IPV:tä yhdistettynä Hib:hen, noin 2, 4, 6 ja 18 kuukauden ikäisinä. Tässä kliinisessä tutkimuksessa yleisimmät raportoidut haittatapahtumat 14 päivä kuluessa rokotuksesta olivat pistoskohdan kipu (39,6 prosentissa tutkittavista) ja väsymys (31,5 prosentissa tutkittavista).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Ei sovellettava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Hinkuyskärokote yhdistelmävalmisteena toksoidien kanssa, puhdistettu antigeeni
ATC-koodi: J07AJ52
Kliiniset tutkimukset
Immuunivasteet kuukauden kuluttua Triaxis-rokotuksen jälkeen 265 lapsella, 527 nuorella ja 743 aikuisella näkyvät alla olevasta taulukosta.
Taulukko 2 Aikuisten, nuorten ja lasten immuunivaste kuukausi Triaxis-rokotuksen jälkeen
| Vasta-aine | Kriteerit | Lapset (4–6 vuotta)1 (N = 265) % | Nuoret (11–17 vuotta)2 (N = 527) % | Aikuiset (18–64 vuotta)2 (N = 743) % |
| Difteria (SN, IU/ml) | ≥0,1 | 100 | 99,8 | 94,1 |
| Tetanus (ELISA, IU/ml tai EU/ml) | ≥0,1 | 100 | 100 | 100 |
| Pertussis (ELISA, EU/ml) | Tehostevaste3 | |||
| PT FHA PRN FIM | 91,9 88,1 94,6 94,3 | 92,0 85,6 94,5 94,9 | 84,4 82,7 93,8 85,9 |
DTaP: difteriatoksoidi [pediatrinen annos], jäykkäkouristus ja soluton hinkuyskä; ELISA: Enzyme Linked Immnoassay; EU: ELISA-yksikkö; IU: kansainvälinen yksikkö; N: niiden osallistujien määrä, joiden tiedot ovat saatavilla; SN: seroneutralisaatio.
1 Tutkimus Td508 tehtiin Kanadassa ja siihen osallistui 4–6-vuotiaita lapsia.
2 Tutkimus Td506 tehtiin Yhdysvalloissa ja siihen osallistui 11–17-vuotiaita nuoria ja 18–64-vuotiaita aikuisia.
3 Niillä tutkimukseen Td508 osallistuneilla lapsilla, jotka olivat saaneet aikaisemmin DTaP-rokotuksen 2, 4, 6 ja 18 kk iässä, tehostevaste määritellään nelinkertaiseksi nousuksi pertussisvasta-aineiden pitoisuudessa. Tutkimukseen Td506 osallistuneilla nuorilla ja aikuisilla tehostevaste määritellään kaksinkertaiseksi nousuksi tutkittavilla, joilla rokotusta edeltävä pitoisuus oli suuri ja nelinkertaiseksi tutkittavilla, joilla rokotusta edeltävä pertussisvasta-ainepitoisuus oli pieni.
Triaxis-rokotteen turvallisuuden ja immunogeenisuuden aikuisilla ja nuorilla osoitettiin olevan verrattavissa niihin, joita havaittiin yksittäisannoksella aikuisten adsorboidulla difteria-tetanus (Td) rokotteella, joka sisälsi yhtä paljon tetanus- ja difteriatoksoideja.
Serologista korrelaattia hinkuyskältä suojautumiseen ei ole selvitetty. Verrattaessa tietoja Ruotsissa vuosina 1992–1996 tehdyistä hinkuyskän tehokkuustutkimuksista (Sweden I pertussis efficacy trial), jossa perusrokotuksena käytetyllä Sanofi Pasteurin soluton pertussis DTaP -pikkulasten valmisteella suojaustehoksi hinkuyskää vastaan vahvistui 85 %, tämän perusteella Triaxis-rokotteen oletetaan antaneen suojaavan immuunivasteen. Pertussis- vasta-ainetasot kaikille antigeeneille Triaxis-tehosterokoteannoksen jälkeen nuorilla ja aikuisilla ylittivät ne vasta-ainetasot, joita havaittiin tehokkuustutkimukseen liitetyssä, kotitalouksille suunnatussa kyselytutkimuksessa.
Taulukko 3 Nuorilla ja aikuisilla kuukausi Triaxis-annoksen jälkeen mitattu hinkuyskän vasta-aine-GMC-suhde verrattuna vasta-aine-GMC-suhteeseen kuukausi rokotuksen jälkeen Sweden I DTaP-tehokkuustutkimuksen aikana 2, 4 ja 6 kk ikäisillä pikkulapsilla (PPI-populaatio1).
| Nuoret (11-17 vuotta)2 | Aikuiset (18-64 vuotta)2 | ||||
| Triaxis/DTaP3 GMC-kvot 95 % KI4 | Triaxis/DTaP3 GMC-kvot 95 % KI4 | ||||
| Osallistujat | N=524–526 | N=741 | |||
| Anti-PT | 3,6 (2,8, 4,5) | 2,1 (1,6, 2,7) | |||
| Anti-FHA | 5,4 (4,5, 6,5) | 4,8 (3,9, 5,9) | |||
| Anti-PRN | 3,2 (2,5, 4,1) | 3,2 (2,3, 4,4) | |||
| Anti-FIM | 5,3 (3,9, 7,1) | 2,5 (1,8, 3,5) |
DTaP: difteriatoksoidi [pediatrinen annos], jäykkäkouristus ja soluton hinkuyskä; GMC: Geometric Mean Concentration, vasta-ainekonsentraatioiden keskiarvo; N: niiden tutkittavien määrä, joiden tiedot ovat saatavilla; PPI: per protocol immunigenicity, tutkimussuunnitelman mukainen immunogeenisuus
1 Tutkimukseen soveltuvat tutkittavat, joiden immunogeenisuutta koskevat tiedot ovat saatavilla.
2 Tutkimus Td506 tehtiin Yhdysvalloissa ja siihen osallistui 11–17-vuotiaita nuoria ja 18–64-vuotiaita aikuisia. ELISA-yksikköinä mitatut vasta-ainekonsentraatioiden keskiarvot laskettiin erikseen imeväisille, nuorille ja aikuisille.
3 N = 80, niiden imeväisten lukumäärä, jotka saivat DTaP-rokotuksen 2, 4 ja 6 kuukauden ikäisinä ja joista on saatavilla tietoa kolmannen annoksen antamisen jälkeen (Sweden I Efficacy Trial -tutkimuksen seeruminäytteet tutkittiin samanaikaisesti Td506-tutkimuksen kanssa).
4 Vasta-ainekonsentraatioiden keskiarvot eivät olleet Triaxis-rokotteen jälkeen huonompia kuin DTaP-rokotuksen jälkeen (Triaxis-rokotteen GMC-suhteen 95 %:n luottamusvälin alaraja jaettuna DTaP:llä > 0,67).
Vasta-aineiden säilyminen
Serologiset seurantatutkimukset tehtiin 3, 5 ja 10 vuoden kuluttua henkilöillä, jotka olivat aiemmin saaneet yhden Triaxis-rokotteen tehosteannoksen. Serosuojan säilyminen kurkkumädän ja jäykkäkouristuksen suhteen ja seropositiivisuus hinkuyskän suhteen on esitetty yhteenvetona taulukossa 4.
Taulukko 4: Serosuojan/seropositiivisuuden säilyminen (%) lapsilla, nuorilla ja aikuisilla 3, 5 ja 10 vuotta Triaxis-annoksen antamisen jälkeen (PPI-populaatio: Protokollan mukainen immunogeeninen väestö1)
Lapset (4–6 vuotta)2 | Nuoret (11–17 vuotta)3 | Aikuiset (18–64 vuotta)3 | ||||||
| Aika Triaxis-annoksesta | 5 vuotta | 3 vuotta | 5 vuotta | 10 vuotta | 3 vuotta | 5 vuotta | 10 vuotta | |
| Osallistujat | N = 128–150 | N = 300 | N = 204–206 | N = 28–39 | N = 292 | N = 237–238 | N = 120–136 | |
| Vasta-aine Serosuojan/seropositiivisuuden % | ||||||||
Kurkkumätä (SN, IU/ml) | ≥ 0,1 | 86,0 | 97,0 | 95,1 | 94,9 | 81,2 | 81,1 | 84,6 |
| ≥ 0,01 | 100 | 100 | 100 | 100 | 95,2 | 93,7 | 99,3 | |
Jäykkäkouristus (ELISA, IU/ml) | ≥ 0,1 | 97,3 | 100 | 100 | 100 | 99,0 | 97,1 | 100 |
Hinkuyskä (ELISA, /EUml) | seroposi-tiivisuus4 | 63,3 | 97,3 | 85,4 | 82,1 | 94,2 | 89,1 | 85,8 |
| PT | ||||||||
| FHA | 97,3 | 100 | 99,5 | 100 | 99,3 | 100 | 100 | |
| PRN | 95,3 | 99,7 | 98,5 | 100 | 98,6 | 97,1 | 99,3 | |
| FIM | 98,7 | 98,3 | 99,5 | 100 | 93,5 | 99,6 | 98,5 | |
ELISA: Enzyme Linked Immnoassay; EU: ELISA-yksikkö; IU: kansainvälinen yksikkö; N: niiden tutkittavien määrä, joiden tiedot ovat saatavilla; PPI: per protocol immunogenicity, tutkimussuunnitelman mukainen immunogeenisuus; SN: seroneutralisaatio
1 Tutkimukseen soveltuvat tutkittavat, joista oli saatavana ainakin yhtä antigeeniä koskevat immunogeenisuustiedot tiettynä ajankohtana.
2 Tutkimus Td508 tehtiin Kanadassa ja siihen osallistui 4–6-vuotiaita lapsia.
3 Tutkimus Td506 tehtiin Yhdysvalloissa ja siihen osallistui 11–17-vuotiaita nuoria ja 18–64-vuotiaita aikuisia.
4 Niiden tutkittavien prosentuaalinen osuus, joilla oli vasta-aineita ≥ 5 EU/ml PT:lle, ≥ 3 EU/ml FHA:lle ja PRN:lle ja ≥ 17 EU/ml FIM:lle 3 vuoden seurannassa; ≥ 4 EU/ml PT:lle, PRN:lle ja FIM:lle ja ≥ 3 EU/ml FHA:lle 5 ja 10 vuoden seurannassa.
Immunogeenisuus aiemmin rokottamattomilla henkilöillä ja henkilöillä, joiden rokotusstatus on tuntematon
Kun 330:lle vähintään 40‑vuotiaalle aikuiselle, jotka eivät olleet saaneet mitään kurkkumätä- tai jäykkäkouristuskomponentteja sisältävää rokotetta viimeksi kuluneiden 20 vuoden aikana, annettiin yksi annos REPEVAX-rokotetta (Tdap‑IPV; sisältää samat määrät jäykkäkouristus‑, kurkkumätä- ja hinkuyskäantigeenejä kuin Triaxis)
- ≥ 95,8 % aikuisista oli seropositiivisia (≥ 5 EU/ml) kaikille rokotteen sisältämille hinkuyskäantigeeneille muodostuneiden vasta-aineiden suhteen
- serosuoja kurkkumätää vastaan todettiin 82,4 %:lla tutkittavista ≥ 0,1 IU/ml:n kynnysarvolla ja 92,7 %:lla tutkittavista ≥ 0,01 IU/ml:n kynnysarvolla
- serosuoja jäykkäkouristusta vastaan todettiin 98,5 %:lla tutkittavista ≥ 0,1 IU/ml:n kynnysarvolla ja 99,7 %:lla tutkittavista ≥ 0,01 IU/ml:n kynnysarvolla
- serosuoja poliota (tyyppejä 1, 2 ja 3) vastaan todettiin ≥ 98,8 %:lla tutkittavista, kun kynnysarvona oli laimennos ≥ 1:8.
Kun 316 tutkittavalle annettiin kaksi lisäannosta kurkkumätä‑, jäykkäkouristus- ja poliokomponentteja sisältävää rokotetta yhden ja kuuden kuukauden kuluttua ensimmäisen annoksen antamisesta, serosuojan kurkkumätää vastaan saavutti 94,6 % (≥ 0,1 IU/ml) ja 100 % (≥ 0,01 IU/ml), jäykkäkouristusta vastaan 100 % (≥ 0,1 IU/ml) ja poliota (tyyppejä 1, 2 ja 3) vastaan 100 % (laimennos ≥ 1:8).
Immunogeenisuus uusintarokotuksen jälkeen
Triaxis-rokotteen immunogeenisuutta on arvioitu uusintarokotuksen jälkeen, joka annettiin 10 vuoden kuluttua edellisen Triaxis- tai REPEVAX-annoksen antamisesta. Yhden kuukauden kuluttua rokotuksesta vähintään 98,5 % tutkimukseen osallistuneista saavutti serosuojaavat vasta‑ainepitoisuudet (≥ 0,1 IU/ml) kurkkumätää ja jäykkäkouristusta vastaan ja vähintään 84 % saavutti tehostevasteen hinkuyskäantigeeneille. (Hinkuyskän tehostevaste määriteltiin rokotuksen jälkeiseksi vasta-ainepitoisuudeksi, joka oli vähintään 4 kertaa LLOQ (määrityksen alaraja), jos pitoisuus oli ennen rokotusta alle LLOQ:n; vähintään 4 kertaa rokotusta edeltävä taso, jos se oli vähintään LLOQ:n tasolla, mutta alle 4 kertaa LLOQ-tason, tai vähintään 2 kertaa rokotusta edeltävä taso, jos se oli vähintään 4 kertaa LLOQ).
Serologiaseurantaa ja uusintarokotusta koskevien tietojen perusteella Triaxis-valmistetta voidaan käyttää jäykkäkouristus- ja kurkkumätärokotteen (dT) sijasta tehostamaan immuniteettia hinkuyskää vastaan kurkkumädän ja jäykkäkouristuksen lisäksi.
Immunogeenisuus raskaana olevilla naisilla
Hinkuyskän vasta-ainevasteet ovat raskaana olevilla naisilla yleensä samankaltaisia kuin naisilla, jotka eivät ole raskaana. Rokottaminen toisen tai kolmannen raskauskolmanneksen aikana on optimaalista vasta-aineiden siirtymisen kannalta kehittyvään sikiöön.
Immunogeenisuus hinkuyskää vastaan imeväisillä (< 3 kuukauden ikäisillä), joiden äidit on rokotettu raskauden aikana
Kahdesta julkaistusta, satunnaistetusta, kontrolloidusta tutkimuksesta saadut tiedot osoittavat, että hinkuyskän vasta-ainepitoisuudet olivat korkeammat syntymähetkellä ja 2 kuukauden iässä (toisin sanoen ennen ensimmäisten rokotusten antamista) sellaisilla imeväisillä, joiden äidit olivat saaneet Triaxis-rokotteen raskauden aikana verrattuna imeväisiin, joiden äitejä ei ollut rokotettu hinkuyskää vastaan raskauden aikana.
Ensimmäisessä tutkimuksessa 33 raskaana olevaa naista sai Triaxis-rokotteen ja 15 naista sai keittosuolaliuosta sisältävän lumerokotteen raskausviikolla 30–32. Hinkuyskän PT-, FHA-, PRN- ja FIM-antigeenien vasta-ainepitoisuuksien keskiarvot (GMC) (EU/ml) rokotettujen naisten imeväisillä olivat syntymähetkellä: PT 68,8, FHA 234,2, PRN 226,8 ja FIM 1 867,0 ja 2 kuukauden iässä: PT 20,6, FHA 99,1, PRN 75,7 ja FIM 510,4. Verrokkiryhmän imeväisillä vastaavat GMC-lukemat olivat syntymähetkellä: PT 14,0, FHA 25,1, PRN 14,4 ja FIM 48,5 ja 2 kuukauden iässä: PT 5,3, FHA 6,6, PRN 5,2 ja FIM 12,0. GMC-suhteet (Triaxis/verrokkiryhmä) olivat syntymähetkellä: PT 4,9, FHA 9,3, PRN 15,8 ja FIM 38,5 ja 2 kuukauden iässä: PT 3,9, FHA 15,0, PRN 14,6 ja FIM 42,5.
Toisessa tutkimuksessa 134 raskaana olevaa naista sai Triaxis-rokotteen ja 138 sai jäykkäkouristus- ja kurkkumätä-vertailurokotteen keskimäärin raskausviikolla 34,5. Hinkuyskän PT-, FHA-, PRN- ja FIM-antigeenien vasta-aineiden GMC-lukemat (EU/ml) rokotettujen naisten imeväisillä olivat syntymähetkellä: PT 54,2, FHA 184,2, PRN 294,1 ja FIM 939,6 ja 2 kuukauden iässä: PT 14,1, FHA 51,0, PRN 76,8 ja FIM 220,0. Verrokkiryhmän imeväisillä vastaavat GMC-lukemat olivat syntymähetkellä: PT 9,5, FHA 21,4, PRN 11,2 ja FIM 31,5 ja 2 kuukauden iässä: PT 3,6, FHA 6,1, PRN 4,4 ja FIM 9,0. GMC-suhteet (Triaxis/verrokkiryhmä) olivat syntymähetkellä: PT 5,7, FHA 8,6, PRN 26,3 ja FIM 29,8 ja 2 kuukauden iässä: PT 3,9, FHA 8,4, PRN 17,5 ja FIM 24,4.
Näiden korkeampien vasta-ainepitoisuuksien pitäisi tarjota imeväisille passiivinen immuniteetti hinkuyskää vastaan ensimmäisten 2–3 elinkuukauden aikana, kuten havainnoivissa tehokkuutta arvioineissa tutkimuksissa on osoitettu.
Immunogeenisuus imeväisillä ja taaperoilla, joiden äidit on rokotettu raskauden aikana
Useissa julkaistuissa tutkimuksissa arvioitiin imeväisten rutiininomaisen rokottamisen aikaansaamaa immunogeenisuutta raskausaikana Repevax- tai Triaxis-rokotteen saaneiden naisten imeväisillä. Tietoja imeväisten vasteesta hinkuyskän ja muiden kuin hinkuyskän antigeeneille arvioitiin ensimmäisen elinvuoden aikana.
Raskauden aikana annettujen Repevax- tai Triaxis-rokotteiden kautta äidiltä saadut vasta-aineet saattavat heikentää imeväisen immuunivastetta aktiiviselle immunisaatiolle hinkuyskää vastaan. Saatavilla olevien epidemiologisten tutkimusten perusteella tällä immuunivasteen heikentymisellä ei välttämättä ole kliinistä merkitystä.
Useista tutkimuksista saaduissa tiedoissa ei havaittu kliinisesti merkittävää heikentymistä raskauden aikana Repevax- tai Triaxis-rokotteen saaneiden äitien imeväisten tai taaperoikäisten vasteissa kurkkumätä- tai jäykkäkouristusrokotteiden, Haemophilus influenzae tyyppi B -rokotteen, IPV-rokotteen tai pneumokokkirokotteen antigeeneille.
Tehokkuus hinkuyskää vastaan imeväisillä, joiden äidit on rokotettu raskauden aikana
Rokotteen tehokkuutta hinkuyskää vastaan kolmannella raskauskolmanneksella rokotettujen äitien imeväisillä ensimmäisten 2–3 elinkuukauden aikana on arvioitu kolmessa havainnoivassa tutkimuksessa. Rokotteen tehokkuus oli kaiken kaikkiaan > 90 %.
Taulukko 5: Rokotteen tehokkuus (VE) hinkuyskää vastaan pienillä imeväisillä, joiden äidit ovat saaneet Triaxis- tai Repevax-rokotteen raskauden aikana kolmessa retrospektiivisessä tutkimuksessa
| Maa | Rokote | VE (95 %:n luottamusväli) | VE:n arviointimenetelmä | Seuranta-aika imeväisillä |
| Iso-Britannia | Repevax | 93 % (81, 97) | kaltaistamaton tapaus-verrokki | 2 kuukautta |
| Yhdysvallat | Triaxis* | 91,4 % (19,5, 99,1) | kohorttiregressiomalli | 2 kuukautta |
| Iso-Britannia | Repevax | 93 % (89, 95) | seulonta (case-coverage) | 3 kuukautta |
* Noin 99 % naisista sai Triaxis-rokotteen.
Farmakokinetiikka
Rokotteilta ei vaadita farmakokineettisiä tutkimuksia.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, geenitoksisuutta, karsinogeenisuutta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
Fenoksietanoli
Injektionesteisiin käytettävä vesi.
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, Triaxis-rokotetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
4 vuotta.
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Ei saa jäätyä. Hävitä jäätynyt rokote.
Pidä ruisku ulkopakkauksessaan. Herkkä valolle.
Valmisteen säilyvyyttä koskevien tietojen perusteella rokotteen komponentit pysyvät stabiileina ≤ 25 °C:n lämpötilassa 72 tunnin ajan, minkä jälkeen Triaxis pitää joko käyttää tai hävittää. Nämä tiedot on tarkoitettu terveydenhuollon ammattilaisille vain sellaisia tilanteita varten, joissa suositellusta säilytyslämpötilasta joudutaan tilapäisesti poikkeamaan.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TRIAXIS injektioneste, suspensio, esitäytetty ruisku
1 kpl (Adacel poikkeuslupa, 2 erillistä neulaa) (32,80 €), 1 kpl (erillinen neula) (32,80 €), 10 x 1 kpl (ilman neulaa) (-)
PF-selosteen tieto
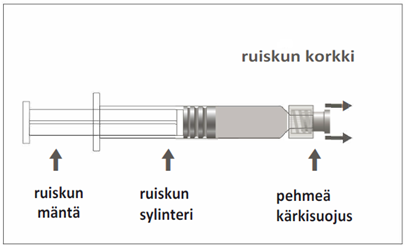
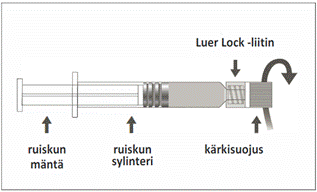
Triaxis 1,5 ml:n esitäytetyissä ruiskuissa, joissa on pehmeä kärkisuojus
0,5 ml suspensiota esitäytetyssä ruiskussa (tyypin I lasia), jossa tulppa (bromibutyylielastomeeri) ja Luer Lock ‑liitin sekä pehmeä kärkisuojus (kumiyhdiste).
Pakkauskoot: 1 tai 10 esitäytettyä ruiskua, joissa ei ole neuloja.
Pakkauskoot: 1 tai 10 esitäytettyä ruiskua, joissa on 1 tai 2 erillistä neulaa (ruostumatonta terästä).
Esitäytettyjen ruiskujen pehmeät kärkisuojukset sisältävät luonnonkumin (lateksin) johdannaista.
Triaxis 1 ml:n esitäytetyissä ruiskuissa, joissa on jäykkä kärkisuojus
0,5 ml suspensiota esitäytetyssä ruiskussa (tyypin I lasia), jossa tulppa (klooributyylielastomeeri) ja Luer Lock ‑liitin sekä jäykkä kärkisuojus (synteettinen isopreenibromibutyyli + polypropeeni).
Pakkauskoot: 1 tai 10 esitäytettyä ruiskua, joissa ei ole neuloja.
Pakkauskoot: 1 esitäytetty ruisku, jossa on 1 tai 2 erillistä neulaa (ruostumatonta terästä).
Pakkauskoot: 10 esitäytettyä ruiskua, joissa on 1 erillinen neula (ruostumatonta terästä).
Pakkauskoot: 1 tai 10 esitäytettyä ruiskua, joissa on turvasuojukset (polykarbonaattia).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Triaxis on samean valkoinen suspensio.
Käyttö- ja käsittelyohjeet
Rokote on ulkonäöltään normaalisti yhtenäinen, samea valkoinen suspensio, joka saattaa sedimentoitua ja johon saattaa muodostua paakkumaisia tai hiutalemaisia kertymiä säilytyksen aikana. Ravista esitäytettyä ruiskua hyvin, jotta suspensio jakautuisi tasaisesti ennen rokotteen antamista. Jos suspensiossa on kertymiä, rokotetta voidaan ravistaa uudelleen, kunnes suspensio on tasaista.
Valmistelut ennen antamista
Esitäytetyssä ruiskussa voi olla Luer Lock ‑liitin, jossa on joko pehmeä kärkisuojus (kuva A) tai jäykkä kärkisuojus (kuva B). Ruisku, jossa on injektioneste, suspensio, on tarkastettava silmämääräisesti ennen antamista. Jos injektionesteessä on vierashiukkasia, ruisku vuotaa, mäntä on aktivoitunut ennenaikaisesti tai kärkisuojus on viallinen, esitäytetty ruisku on hävitettävä. Ruisku on tarkoitettu vain yhtä käyttökertaa varten, eikä sitä saa käyttää uudelleen.
Esitäytetyn Luer Lock ‑ruiskun käyttöohjeet:
Kuva A: Luer Lock ‑ruisku, jossa on pehmeä kärkisuojus
| Vaihe 1: Pidä toisella kädellä kiinni ruiskun korkista (älä pidä kiinni ruiskun männästä tai sylinteristä) ja poista kärkisuojus vetämällä. | 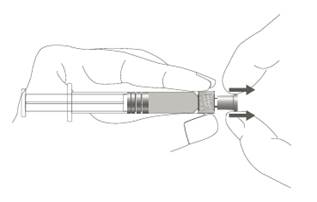 |
| Vaihe 2: Kiinnitä neula ruiskuun kiertämällä neulaa varovasti myötäpäivään, kunnes tunnet vähäisen vastuksen. | 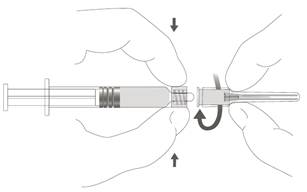 |
Kuva B: Luer Lock ‑ruisku, jossa on jäykkä kärkisuojus
| Vaihe 1: Pidä toisella kädellä kiinni Luer Lock ‑liittimestä (älä pidä kiinni ruiskun männästä tai sylinteristä) ja poista kärkisuojus kiertämällä sitä. | 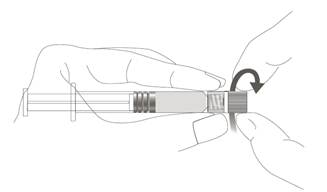 |
| Vaihe 2: Kiinnitä neula ruiskuun kiertämällä neulaa varovasti ruiskun Luer Lock ‑liittimeen, kunnes tunnet vähäisen vastuksen. | 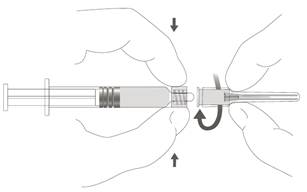 |
Ohjeet turvaneulan käytölle esitäytetyn Luer Lock ‑ruiskun kanssa:
Valmistele Luer Lock ‑ruisku ja neula kiinnittämistä varten noudattaen edellä esitettyjä vaiheita 1 ja 2.
| Kuva C: Turvaneula (kotelossa) | Kuva D: Turvaneulan osat (valmisteltuna käyttöä varten) |
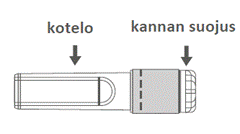 |  |
| Vaihe 3: Poista turvaneulan kotelo vetämällä kohtisuoraan. Neula on turvasuojuksen ja suojaosan sisällä. | |
Vaihe 4: A: Käännä turvasuojusta poispäin neulasta, kohti ruiskun sylinteriä kuvassa esitettyyn asentoon. | 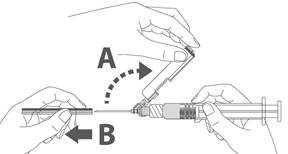 |
Vaihe 5: Kun pistos on annettu, lukitse (aktivoi) turvasuojus käyttämällä jotakin kolmesta (3) yhden käden tekniikasta, jotka on esitetty kuvassa, eli aktivoinnilla pintaa vasten, peukalolla tai sormella. Huomaa: Aktivointi on onnistunut, kun kuulet ja/tai tunnet naksahduksen. | 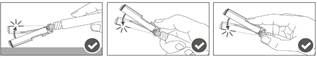 |
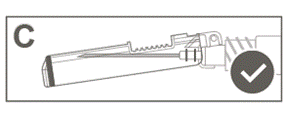
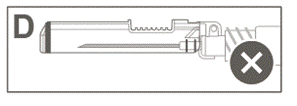
Vaihe 6: Tarkasta turvasuojuksen aktivoituminen silmämääräisesti. Turvasuojuksen on oltava täysin lukkiutunut (aktivoitunut), kuten kuvassa C on esitetty.
Kuvassa D on turvasuojus, joka EI ole täysin lukkiutunut (ei ole aktivoitunut). |
|
| Varoitus: Älä yritä poistaa turvalaitteen lukitusta (peruuttaa turvalaitteen aktivointia) ottamalla neula väkisin pois turvasuojuksesta. | |
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Neuloja ei saa peittää uudestaan korkilla.
Korvattavuus
TRIAXIS injektioneste, suspensio, esitäytetty ruisku
1 kpl, 1 kpl, 10 x 1 kpl
- Ei korvausta.
ATC-koodi
J07AJ52
Valmisteyhteenvedon muuttamispäivämäärä
14.01.2026
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi