
PHESGO injektioneste, liuos 600/600 mg, 1200/600 mg
Vaikuttavat aineet ja niiden määrät
Phesgo 600 mg/600 mg injektioneste, liuos
Yksi 10 ml:n injektiopullo liuosta sisältää 600 mg pertutsumabia ja 600 mg trastutsumabia.
Yksi ml liuosta sisältää 60 mg pertutsumabia ja 60 mg trastutsumabia.
Phesgo 1 200 mg/600 mg injektioneste, liuos
Yksi 15 ml:n injektiopullo liuosta sisältää 1 200 mg pertutsumabia ja 600 mg trastutsumabia.
Yksi ml liuosta sisältää 80 mg pertutsumabia ja 40 mg trastutsumabia.
Pertutsumabi ja trastutsumabi ovat nisäkkään (kiinanhamsterin munasarja) soluissa rekombinantti-deoksiribonukleiinihappo (DNA)-teknologialla tuotettuja humanisoituja monoklonaalisia immunoglobuliini (Ig)G1-vasta-aineita.
Apuaine, jonka vaikutus tunnetaan
Yksi 15 ml:n Phesgo-injektiopullo sisältää 6 mg polysorbaattia 20.
Yksi 10 ml:n Phesgo-injektiopullo sisältää 4 mg polysorbaattia 20.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos
Kliiniset tiedot
Käyttöaiheet
Varhaisvaiheen rintasyöpä
Phesgo on tarkoitettu käytettäväksi yhdistelmänä solunsalpaajahoidon kanssa
- HER2‑positiivista, paikallisesti edennyttä, inflammatorista tai varhaisvaiheen rintasyöpää sairastavien aikuispotilaiden neoadjuvanttihoitoon, kun taudin uusiutumisriski on suuri (ks. kohta Farmakodynamiikka)
- varhaisvaiheen HER2‑positiivista rintasyöpää sairastavien aikuispotilaiden adjuvanttihoitoon, kun taudin uusiutumisriski on suuri (ks. kohta Farmakodynamiikka).
Metastasoitunut rintasyöpä
Phesgo on tarkoitettu käytettäväksi yhdistelmänä dosetakselin kanssa HER2‑positiivista metastasoitunutta tai paikallisesti uusiutunutta leikkaushoitoon soveltumatonta rintasyöpää sairastavien aikuispotilaiden hoitoon, kun potilas ei ole aiemmin saanut metastasoituneen taudin hoitoon anti‑HER2‑hoitoa eikä solunsalpaajahoitoa.
Ehto
Tämä valmiste on reseptilääke, jonka määräämiseen liittyy rajoitus, ja hoidon saa aloittaa vain syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa. Valmisteen antajan tulee olla terveydenhuollon ammattilainen, jolla on valmius anafylaksian hoitoon, ja ensiapuvälineiden on oltava saatavilla.
Annostus ja antotapa
Phesgo-hoidon saa aloittaa vain syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa. Phesgo-valmisteen saa antaa anafylaksian hoitoon perehtynyt terveydenhuollon ammattilainen paikassa, jossa elvytysvälineet ovat välittömästi saatavilla.
Kun pertutsumabia sisältävä hoito on saatu turvallisesti käyntiin, lääkäri voi päättää, voidaanko Phesgo-hoito toteuttaa siten, että terveydenhuollon ammattilainen antaa sen muualla kuin hoitopaikassa (esim. potilaan kotona) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Lääkitysvirheiden välttämiseksi on tärkeää varmistaa injektiopullon etiketistä, että käyttökuntoon saatettava ja annettava lääkevalmiste on Phesgo.
Laskimoon annettavaa pertutsumabia ja trastutsumabia parhaillaan saavat potilaat voidaan siirtää Phesgo-hoidolle. Siirtymistä laskimoon annettavista pertutsumabista ja trastutsumabista Phesgo-hoitoon (tai päinvastoin) selvitettiin MO40628-tutkimuksessa (ks. kohdat Haittavaikutukset ja Farmakodynamiikka).
Annostus
Phesgo-hoitoa saavilla potilailla on oltava HER2‑positiivinen kasvain, mikä määritellään validoidulla immunohistokemiallisella menetelmällä (IHC3+) ja/tai in situ ‑hybridisaatiomenetelmällä (ISH) määritettynä suhdelukuna ≥ 2.
Tarkkojen ja toistettavissa olevien tulosten varmistamiseksi, edellä mainitut testit on suoritettava erikoistuneessa laboratoriossa, jossa käytettyjen menetelmien asianmukainen validointi on taattu. Ks. validoidun HER2-testimenetelmän pakkausselosteesta tarkat ohjeet määrityksen toteuttamisesta ja tulkinnasta.
Suositeltu Phesgo-annos varhaisvaiheen ja metastasoituneen rintasyövän hoitoon, ks. taulukko 1.
Taulukko 1. Suositeltu Phesgo-annos ja antotapa
| Annos (painosta riippumatta) | Ihon alle annettavan injektion kesto (noin) | Tarkkailuaikaab | |
| Aloitusannos | 1 200 mg pertutsumabia / 600 mg trastutsumabia | 8 minuuttia | 30 minuuttia |
| Ylläpitoannos (kolmen viikon välein) | 600 mg pertutsumabia / 600 mg trastutsumabia | 5 minuuttia | 15 minuuttia |
aPotilaita pitää tarkkailla injektioon liittyvien reaktioiden ja yliherkkyysreaktioiden havaitsemiseksi.
bTarkkailu pitää aloittaa Phesgo-injektion annon jälkeen ja saattaa loppuun ennen sen jälkeen annettavaa solunsalpaajahoitoa.
Potilaan saadessa jotakin taksaania Phesgo pitää antaa ennen taksaania.
Phesgo-valmisteen kanssa annettavan dosetakselihoidon suositeltu aloitusannos on 75 mg/m2, joka voidaan sen jälkeen suurentaa annokseen 100 mg/m2 valitun hoito-ohjelman mukaan ja sen mukaan, miten potilas sietää aloitusannoksen. Dosetakselia voidaan vaihtoehtoisesti antaa alusta lähtien annoksina 100 mg/m2 kolmen viikon välein, tällöinkin valitun hoito-ohjelman mukaan. Jos noudatetaan karboplatiinia sisältävää hoito-ohjelmaa, suositeltu dosetakseliannos on aina 75 mg/m2 (ei annoksen suurentamista). Phesgo-valmisteen kanssa adjuvanttihoitona annettavan paklitakselihoidon suositeltu annos on 80 mg/m2 kerran viikossa 12 viikon hoitosykleinä.
Potilaan saadessa jotakin antrasykliiniä sisältävää hoitoa Phesgo pitää antaa koko antrasykliinihoito-ohjelman päätyttyä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Metastasoitunut rintasyöpä
Phesgo pitää antaa yhdistelmänä dosetakselin kanssa. Phesgo-hoitoa voidaan jatkaa niin kauan, kunnes tauti etenee tai ilmaantuu haittavaikutuksia, jotka eivät ole hallittavissa, vaikka dosetakselihoito lopetettaisiin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varhaisvaiheen rintasyöpä
Neoadjuvanttihoidossa Phesgo-valmistetta annetaan yhdistelmänä solusalpaajahoidon kanssa 3−6 hoitosykliä osana varhaisvaiheen rintasyövän kokonaishoitoa (ks. kohta Farmakodynamiikka).
Adjuvanttihoidossa Phesgo-valmistetta annetaan yhteensä yhden vuoden ajan (18 hoitosykliin asti tai kunnes tauti uusiutuu tai ilmaantuu haittavaikutuksia, jotka eivät ole hallittavissa, riippumatta siitä, mikä näistä tapahtuu ensin) osana varhaisvaiheen rintasyövän kokonaishoitoa ja riippumatta leikkauksen ajankohdasta. Hoitoon pitää kuulua tavanomainen antrasykliiniä ja/tai taksaania sisältävä solunsalpaajahoito. Phesgo-hoito pitää aloittaa ensimmäisen taksaania sisältävän hoitosyklin 1. päivänä ja sitä pitää jatkaa, vaikka solunsalpaajahoito lopetettaisiin.
Annoksen viivästyminen tai antamatta jääminen
Jos kahden peräkkäisen injektion välinen aika on
- alle 6 viikkoa, Phesgo-ylläpitoannos 600 mg/600 mg pitää antaa mahdollisimman pian. Sen jälkeen jatketaan hoitoa kolmen viikon välein.
- 6 viikkoa tai enemmän, pitää antaa uusi Phesgo-aloitusannos 1 200 mg/600 mg, jonka jälkeen hoitoa jatketaan Phesgo 600 mg/600 mg ‑ylläpitoannoksilla kolmen viikon välein.
Annosmuutokset
Phesgo-annoksen pienentämistä ei suositella. Phesgo-hoito voi olla tarpeen keskeyttää lääkärin harkinnan mukaan.
Potilaan hoitoa voidaan jatkaa silloinkin, jos hänellä esiintyy solunsalpaajahoidosta aiheutunutta korjautuvaa luuydinlamaa, mutta potilasta on seurattava tällöin tarkoin, jotta neutropenian ilmaantuminen voidaan havaita.
Dosetakselin ja muiden solunsalpaajien annosmuutokset, ks. kyseisen valmisteen valmisteyhteenveto.
Sydämen vasemman kammion vajaatoiminta
Phesgo-hoito pitää keskeyttää vähintään 3 viikoksi, jos potilaalle ilmaantuu kongestiiviseen sydämen vajaatoimintaan viittaavia oireita ja löydöksiä. Phesgo-hoito pitää lopettaa, jos oireinen sydämen vajaatoiminta varmistuu (ks. lisätietoja kohdasta Varoitukset ja käyttöön liittyvät varotoimet).
Metastasoitunutta rintasyöpää sairastavat potilaat
Potilaan vasemman kammion ejektiofraktion (LVEF) pitää olla ennen hoitoa ≥ 50 %. Phesgo-hoito pitää keskeyttää vähintään 3 viikon ajaksi, jos
- LVEF pienenee alle 40 %:n
- LVEF on 40–45 % ja se on pienentynyt ≥ 10 prosenttiyksikköä hoitoa edeltäneistä arvoista.
Phesgo-hoitoa voidaan jatkaa, jos LVEF on palautunut > 45 %:iin tai 40–45 %:iin, ja ero hoitoa edeltäneisiin arvoihin on < 10 prosenttiyksikköä.
Varhaisvaiheen rintasyöpää sairastavat potilaat
Potilaan vasemman kammion ejektiofraktion (LVEF) pitää olla ennen hoitoa ≥ 55 % (≥ 50 % antrasykliinisolunsalpaajahoidon annon päättymisen jälkeen, jos sitä annetaan).
Phesgo-hoito pitää keskeyttää vähintään 3 viikon ajaksi, jos
LVEF pienenee alle 50 %:n ja se on pienentynyt ≥ 10 prosenttiyksikköä hoitoa edeltäneistä arvoista.
Phesgo-hoitoa voidaan jatkaa, jos LVEF on palautunut ≥ 50 %:iin tai ero hoitoa edeltäneisiin arvoihin on < 10 prosenttiyksikköä.
Erityiset potilasryhmät
Iäkkäät
Phesgo-hoidon tehossa ei havaittu yleisesti eroja ≥ 65-vuotiaiden ja < 65-vuotiaiden potilaiden välillä. Phesgo-annosta ei tarvitse muuttaa iältään ≥ 65-vuotiailla potilailla. Yli 75-vuotiaista potilaista on vähän tietoja saatavilla.
Ks. iäkkäiden potilaiden hoidon turvallisuuden arviointi kohdasta Haittavaikutukset.
Munuaisten vajaatoiminta
Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden Phesgo-annosta ei tarvitse muuttaa. Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden hoitoon ei voida antaa annossuosituksia, koska saatavissa olevat farmakokineettiset tiedot ovat vähäisiä (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Phesgo-valmisteen turvallisuutta ja tehoa ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla. Maksan vajaatoimintaa sairastavien potilaiden Phesgo-annosta ei todennäköisesti tarvitse muuttaa. Erityisiä annosmuutoksia ei suositella (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Phesgo-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu. Phesgo-valmisteen käyttö ei ole asianmukaista rintasyövän hoidossa tässä ikäryhmässä.
Siirtyminen laskimoon annettavista pertutsumabista ja trastutsumabista Phesgo-hoitoon
- jos pertutsumabia ja trastutsumabia laskimoon saavien potilaiden viimeisimmästä annoksesta on alle 6 viikkoa, Phesgo pitää antaa ylläpitoannoksena 600 mg pertutsumabia / 600 mg trastutsumabia, ja seuraavat annokset annetaan 3 viikon välein
- jos pertutsumabia ja trastutsumabia laskimoon saavien potilaiden viimeisimmästä annoksesta on 6 viikkoa tai enemmän, Phesgo pitää antaa aloitusannoksena 1 200 mg pertutsumabia / 600 mg trastutsumabia, jonka jälkeen seuraavilla antokerroilla annetaan ylläpitoannos 600 mg pertutsumabia / 600 mg trastutsumabia 3 viikon välein.
Antotapa
Phesgo pitää antaa vain injektiona ihon alle. Phesgo ei ole tarkoitettu annettavaksi laskimoon.
Injektio annetaan eri antokerroilla vuorotellen vasempaan ja oikeaan reiteen. Uusi injektio on annettava vähintään 2,5 cm:n etäisyydelle edellisestä injektiokohdasta terveelle iholle, eikä sitä saa koskaan antaa punoittavalle, mustelmaiselle, aristavalle tai kovettuneelle ihoalueelle. Annosta ei saa jakaa kahteen ruiskuun eikä kahteen antokohtaan. Phesgo-valmisteen kanssa samanaikaisesti käytettävät muut ihon alle annettavat lääkkeet tulisi mieluiten injisoida eri antokohtiin.
Aloitusannos annetaan 8 minuutin kestoisena, ja ylläpitoannos annetaan 5 minuutin kestoisena.
Potilasta suositellaan tarkkailemaan 30 minuutin ajan Phesgo-aloitusannoksen annon jälkeen ja 15 minuutin ajan ylläpitoannoksen jälkeen injektioon liittyvien reaktioiden havaitsemiseksi (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Injektioon liittyvät reaktiot
Jos potilaalle ilmaantuu injektioon liittyviä oireita, injektion antonopeutta voidaan hidastaa tai anto keskeyttää (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja kohta Haittavaikutukset). Systeemisiä oireita voidaan lievittää myös antamalla hoitona happea, beeta-agonisteja, antihistamiineja, nopeaa laskimoon annettavaa nesteytystä ja kuumelääkkeitä.
Yliherkkyysreaktiot/anafylaksia
Injektion anto on keskeytettävä heti pysyvästi, jos potilaalle ilmaantuu NCI‑CTCAE-luokituksen gradus 4 reaktio (anafylaksia), bronkospasmi tai akuutti hengitysvaikeusoireyhtymä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja kohta Haittavaikutukset).
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen käsittelystä ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttaville aineille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi annetun lääkevalmisteen nimi ja eränumero tulee dokumentoida selkeästi.
Sydämen vasemman kammion toimintahäiriö (mukaan lukien kongestiivinen sydämen vajaatoiminta)
HER2-aktiivisuutta salpaavien lääkevalmisteiden, mukaan lukien pertutsumabin ja trastutsumabin, käytön yhteydessä on raportoitu vasemman kammion ejektiofraktion (LVEF) pienenemistä. Oireisen vasemman kammion systolisen toimintahäiriön (kongestiivisen sydämen vajaatoiminnan) ilmaantuvuus oli pertutsumabia yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa saaneilla potilailla suurempi verrattuna trastutsumabia ja solunsalpaajahoitoa saaneisiin potilaisiin. Oireista sydämen vajaatoimintaa raportoitiin useimmiten adjuvanttihoidon yhteydessä potilailla, jotka saivat antrasykliiniä sisältävää solunsalpaajahoitoa (ks. kohta Haittavaikutukset). Laskimoon annettavien pertutsumabin sekä trastutsumabin ja solunsalpaajahoidon yhdistelmää koskeneiden tutkimusten perusteella potilailla, jotka ovat aikaisemmin saaneet antrasykliinejä tai sädehoitoa rintakehän alueelle, saattaa olla suurempi vasemman kammion ejektiofraktion pienenemisen riski.
Phesgo-valmisteella tehtyyn varhaisvaiheen rintasyövän (neo)adjuvanttihoitoa koskeneeseen FEDERICA-pivotaalitutkimukseen ei otettu mukaan potilaita, joilla oli aiemmin ollut vakava sydänsairaus tai muita vakavia sairauksia tai joilla oli aiemmin ollut kammioperäisiä rytmihäiriöitä tai joilla oli kammioperäisten rytmihäiriöiden riskitekijöitä.
Phesgo-valmistetta ei ole tutkittu seuraavissa potilasryhmissä: hoitoa edeltävä vasemman kammion ejektiofraktio < 55 % (varhaisvaiheen rintasyöpä) tai < 50 % (metastasoitunut rintasyöpä); potilaalla aikaisempi kongestiivinen sydämen vajaatoiminta; tila, joka saattaa heikentää vasemman kammion toimintaa (esim. huonossa hoitotasapainossa oleva verenpainetauti, äskettäin sairastettu sydäninfarkti, vakavat hoitoa vaativat sydämen rytmihäiriöt) tai aiempi antrasykliinihoito, joka vastaa kumulatiivisesti > 360 mg/m2 doksorubisiinia tai vastaavaa. Pertutsumabin ja trastutsumabin ja solunsalpaajahoidon yhdistelmää ei ole myöskään tutkittu potilailla, joilla vasemman kammion ejektiofraktio on pienentynyt < 50 %:iin aiemman trastutsumabia sisältäneen liitännäishoidon aikana.
Vasemman kammion ejektiofraktio on tutkittava ennen Phesgo-hoidon aloittamista sekä säännöllisesti hoidon aikana (esim. kerran neoadjuvanttihoidon aikana ja 12 viikon välein adjuvanttihoidon aikana ja jos tauti on metastasoitunut), jotta varmistetaan, että vasemman kammion ejektiofraktio on normaaliarvojen puitteissa. Jos vasemman kammion ejektiofraktio on pienentynyt kuten kohdassa Annostus ja antotapa on mainittu eikä kohenemista todeta tai jos se on seuraavalla arviointikerralla pienentynyt edelleen, Phesgo-hoidon lopettamista on harkittava vakavasti, ellei hyötyjen arvioida olevan riskejä suuremmat yksittäiselle potilaalle.
Sydänriski pitää ottaa tarkoin huomioon ja arvioida yksilöllisesti potilaan hoitotarpeen suhteen ennen kuin Phesgo-valmistetta käytetään yhdistelmänä jonkin antrasykliinin kanssa. HER2‑reseptoriin vaikuttavien lääkeaineiden ja antrasykliinien farmakologisten vaikutusten perusteella sydäntoksisuuden riskin voidaan olettaa olevan suurempi käytettäessä Phesgo-valmistetta ja antrasykliinejä samanaikaisesti kuin peräkkäin.
Phesgo-valmisteen (yhdistelmänä jonkin taksaanin kanssa) käyttöä doksorubisiinihoidon jälkeen kahdessa antrasykliinipohjaisessa yhdistelmähoidossa on tutkittu FEDERICA-tutkimuksessa, kun taas laskimoon annettavan pertutsumabin (yhdistelmänä trastutsumabin ja jonkin taksaanin kanssa) käyttöä monien antrasykliinipohjaisten hoitojen epirubisiini- tai doksorubisiiniosan jälkeen on tutkittu APHINITY- ja BERENICE-tutkimuksissa. Tietoja laskimoon annettavien pertutsumabin ja trastutsumabin yhdistelmän ja antrasykliinin samanaikaisen käytön turvallisuudesta on vain rajallisesti saatavilla. Laskimoon annettavaa pertutsumabin ja trastutsumabin yhdistelmää annettiin TRYPHAENA-tutkimuksessa samanaikaisesti epirubisiinin kanssa, jota käytettiin osana FEC-hoitoa (5‑fluorourasiili, epirubisiini, syklofosfamidi) (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). Hoitoa saivat vain potilaat, jotka eivät olleet aiemmin saaneet solunsalpaajahoitoa, ja he saivat pieniä kumulatiivisia epirubisiiniannoksia (enintään 300 mg/m2). Tässä tutkimuksessa sydäntä koskeva turvallisuus oli samankaltainen kuin silloin, kun potilaat saivat samaa hoitoa, mutta pertutsumabi annettiin FEC-solunsalpaajahoidon jälkeen.
Injektioon liittyvät reaktiot / infuusioon liittyvät reaktiot
Phesgo-hoitoon on liittynyt injektioon liittyviä reaktioita (ks. kohta Haittavaikutukset). Injektioon liittyviksi reaktioiksi määriteltiin mikä tahansa systeeminen reaktio, johon liittyy oireina mm. kuumetta, vilunväreitä, päänsärkyä 24 tunnin kuluessa todennäköisesti Phesgo-valmisteen antamisen jälkeen tapahtuvan sytokiinien vapautumisen vuoksi. Potilasta on suositeltavaa tarkkailla aloitusannoksen annon aikana ja 30 minuutin ajan sen jälkeen sekä Phesgo-ylläpitoannosten annon aikana ja 15 minuutin ajan niiden jälkeen. Jos merkittävä injektioreaktio ilmaantuu, injektionopeutta on hidastettava tai injektion anto on keskeytettävä. Potilaalle on tällöin annettava asianmukaista hoitoa. Potilas on tutkittava ja hänen tilaansa on seurattava tarkoin, kunnes oireet ja löydökset häviävät täysin. Jos injektioreaktio on vaikea-asteinen, hoidon lopettamista pysyvästi pitää harkita. Tätä koskevan kliinisen arvion pitää perustua edellisen reaktion vaikeusasteeseen ja haittavaikutukseen annettuun hoitoon saatuun vasteeseen (ks. kohta Annostus ja antotapa). Kuolemaan johtaneita injektioreaktioita ei ole havaittu Phesgo-valmisteen käytössä, mutta hoidossa on oltava varovainen, sillä laskimoon annettavan pertutsumabin ja trastutsumabin ja solunsalpaajahoidon yhdistelmän käyttöön on liittynyt kuolemaan johtaneita infuusioreaktioita.
Yliherkkyysreaktiot/anafylaksia
Potilaita pitää seurata tarkoin yliherkkyysreaktioiden havaitsemiseksi. Vaikea-asteisia yliherkkyysreaktiota, mukaan lukien anafylaksiaa ja kuolemaan johtaneita tapauksia, on havaittu käytettäessä pertutsumabia yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa (ks. kohta Haittavaikutukset). Suurin osa anafylaktisista reaktioista ilmeni ensimmäisten 6-8 hoitosyklin aikana, kun pertutsumabia ja trastutsumabia annettiin yhdistelmänä solunsalpaajahoidon kanssa. Lääkevalmisteet tällaisten reaktioiden hoitamiseen sekä ensiapuvälineet on oltava välittömästi saatavilla.
Annettaessa valmiste muualla kuin hoitopaikassa, paikallisen tavanomaisen hoitokäytännön mukaiset asianmukaiset lääkkeet yliherkkyysreaktioiden hoitoon (reaktion vaikeusasteesta ja tyypistä riippuen esim. adrenaliini, beeta‑agonistit, antihistamiinit ja kortikosteroidit) on oltava välittömästi saatavilla.
Phesgo-hoito on lopetettava pysyvästi, jos potilaalle ilmaantuu NCI‑CTCAE-luokituksen gradus 4 yliherkkyysreaktioita (anafylaksia), bronkospasmi tai akuutti hengitysvaikeusoireyhtymä (ks. kohta Annostus ja antotapa). Phesgo on vasta-aiheista potilaille, joiden tiedetään olevan yliherkkiä pertutsumabille, trastutsumabille tai kohdassa Apuaineet mainituille apuaineille (ks. kohta Vasta-aiheet).
Kuumeinen neutropenia
Phesgo-valmistetta yhdistelmänä jonkin taksaanin kanssa saaneilla potilailla on tavanomaista suurempi kuumeisen neutropenian riski.
Laskimoon annettavilla pertutsumabilla, trastutsumabilla ja dosetakselilla hoidetuilla potilailla on suurentunut kuumeisen neutropenian riski verrattuna potilaisiin, joita hoidetaan lumelääkkeellä, trastutsumabilla ja dosetakselilla. Riski on suurentunut erityisesti kolmen ensimmäisen hoitosyklin aikana (ks. kohta Haittavaikutukset). Metastasoitunutta rintasyöpää koskeneessa CLEOPATRA-tutkimuksessa neutrofiilien määrät olivat alhaisimmillaan samanlaiset sekä pertutsumabia saaneilla potilailla että lumelääkehoitoa saaneilla potilailla. Kuumeisen neutropenian suurempi esiintyvyys pertutsumabihoitoa saaneissa potilaissa liittyi limakalvotulehdusten ja ripulin korkeampaan esiintyvyyteen. Limakalvotulehdusten ja ripulin oireenmukaista hoitoa pitää harkita. Kuumeisen neutropenian haittatapahtumia ei raportoitu dosetakselin annon lopettamisen jälkeen.
Ripuli
Phesgo saattaa aiheuttaa vaikean ripulin. Ripuli on yleisintä samanaikaisen taksaania sisältävän hoidon kanssa. Iäkkäillä potilailla (≥ 65-vuotiailla) ripulin riski on suurempi kuin nuoremmilla potilailla (< 65-vuotiailla). Ripuli hoidetaan tavanomaisten hoitokäytäntöjen ja ohjeistojen mukaisesti. Varhaista hoitoa loperamidilla, nesteytyksellä ja elektrolyyttien korvaushoidolla pitää harkita, etenkin iäkkäille potilaille sekä silloin, jos ripuli on vaikea-asteista tai pitkittyy. Jos potilaan tila ei lievity, on harkittava Phesgo-hoidon keskeyttämistä. Kun ripuli on saatu hallintaan, Phesgo-hoidon voi aloittaa uudestaan.
Keuhkojen toimintaan liittyvät tapahtumat
Vaikeita keuhkojen toimintaan liittyviä tapahtumia on raportoitu trastutsumabin käytön yhteydessä myyntiluvan myöntämisen jälkeen. Nämä tapahtumat ovat satunnaisesti johtaneet kuolemaan. Lisäksi on esiintynyt interstitiaalista keuhkosairautta, mukaan lukien keuhkoinfiltraatteja, akuuttia hengitysvaikeusoireyhtymää, keuhkokuumetta, keuhkotulehdusta, pleuraeffuusiota, hengitysvaikeuksia, akuuttia keuhkopöhöä ja hengityksen vajaatoimintaa. Interstitiaalisen keuhkosairauden riskitekijöitä ovat aiemmat tai samanaikaiset hoidot muilla antineoplastisilla lääkeaineilla, kuten taksaaneilla, gemsitabiinilla, vinorelbiinilla, ja sädehoito. Nämä tapahtumat voivat esiintyä osana infuusioon liittyvää reaktiota tai ilmaantua viiveellä. Keuhkojen toimintaan liittyvien reaktioiden riski saattaa olla keskimääräistä suurempi niillä potilailla, joilla on lepohengenahdistusta edenneen taudin komplikaatioiden tai muun samanaikaisen sairauden takia. Siksi näitä potilaita ei pidä hoitaa Phesgo-valmisteella. Varovaisuutta on noudatettava erityisesti keuhkotulehduspotilailla, jotka saavat samanaikaisesti taksaaneja.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Tämä lääkevalmiste sisältää polysorbaattia 20. Yksi 15 ml:n injektiopullo liuosta sisältää 6 mg polysorbaattia 20. Yksi 10 ml:n injektiopullo liuosta sisältää 4 mg polysorbaattia 20. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Varsinaisia yhteisvaikutustutkimuksia ei ole tehty.
Pertutsumabi
Metastasoitunutta rintasyöpää koskeneen satunnaistetun CLEOPATRA-pivotaalitutkimuksen 37 potilaan osatutkimuksessa ei todettu farmakokineettisiä yhteisvaikutuksia pertutsumabin ja trastutsumabin tai pertutsumabin ja dosetakselin välillä. Pertutsumabin ja trastutsumabin tai pertutsumabin ja dosetakselin välillä ei myöskään todettu lääkkeiden välisiä yhteisvaikutuksia farmakokineettisessä populaatioanalyysissä. NEOSPHERE- ja APHINITY-tutkimusten farmakokineettiset tiedot varmistivat, ettei lääkkeiden välisiä yhteisvaikutuksia esiinny.
Pertutsumabin vaikutusta samanaikaisesti annettujen solunsalpaajien (dosetakselin, paklitakselin, gemsitabiinin, kapesitabiinin, karboplatiinin ja erlotinibin) farmakokinetiikkaan selvitettiin viidessä tutkimuksessa. Pertutsumabin ja näiden lääkeaineiden välillä ei todettu viitteitä farmakokineettisistä yhteisvaikutuksista. Pertutsumabin farmakokinetiikka oli näissä tutkimuksissa vastaava kuin tutkimuksissa, joissa sitä tutkittiin ainoana lääkeaineena.
Trastutsumabi
Varsinaisia yhteisvaikutustutkimuksia ei ole tehty. Kliinisissä tutkimuksissa ei ole havaittu kliinisesti merkityksellisiä yhteisvaikutuksia trastutsumabin ja muiden samanaikaisesti käytettyjen lääkevalmisteiden välillä.
Trastutsumabin vaikutus muiden antineoplastisten aineiden farmakokinetiikkaan
HER2‑positiivista metastasoitunutta rintasyöpää sairastavilla naisilla tehtyjen tutkimusten BO15935 ja M77004 farmakokineettiset tiedot viittasivat siihen, että altistus paklitakselille ja doksorubisiinille (ja niiden päämetaboliiteille 6‑α‑hydroksyylipaklitakselille [POH] ja doksorubisinolille [DOL]) ei muuttunut trastutsumabin läsnä ollessa (8 mg/kg tai 4 mg/kg aloitusannoksena laskimoon, minkä jälkeen 6 mg/kg kerran kolmessa viikossa tai 2 mg/kg kerran viikossa laskimoon). Trastutsumabi saattaa kuitenkin suurentaa kokonaisaltistusta doksorubisiinin yhdelle metaboliitille (7‑deoksi‑13‑dihydrodoksorubisinoni, D7D). D7D:n biologista aktiivisuutta ja tämän metaboliitin pitoisuuden suurenemisen kliinistä vaikutusta ei tunneta.
Tiedot HER2‑positiivista metastasoitunutta rintasyöpää sairastavilla japanilaisilla naisilla tehdystä tutkimuksesta JP16003 (yhden hoitoryhmän tutkimus, jossa annettiin trastutsumabia aloitusannos 4 mg/kg laskimoon ja 2 mg/kg viikossa laskimoon sekä dosetakselia 60 mg/m2 laskimoon) viittasivat siihen, että trastutsumabin samanaikainen antaminen ei vaikuttanut dosetakselikerta-annoksen farmakokinetiikkaan. Tutkimus JP19959 oli tutkimuksen BO18255 (ToGA) osatutkimus, joka tehtiin pitkälle edennyttä mahasyöpää sairastavilla japanilaisilla mies- ja naispotilailla kapesitabiinin ja sisplatiinin farmakokinetiikan tutkimiseksi, kun niitä käytettiin trastutsumabin kanssa tai ilman trastutsumabia. Tämän osatutkimuksen tulokset viittaavat siihen, että sisplatiinin tai sisplatiinin ja trastutsumabin yhdistelmän samanaikainen käyttö ei muuttanut altistusta kapesitabiinin biologisesti aktiivisille metaboliiteille (esim. 5‑FU:lle). Kapesitabiinin pitoisuudet kuitenkin suurenivat ja puoliintumisaika piteni, kun sitä käytettiin yhdistelmänä trastutsumabin kanssa. Tiedot viittaavat myös siihen, että kapesitabiinin tai kapesitabiinin ja trastutsumabin yhdistelmän samanaikainen käyttö ei vaikuttanut sisplatiinin farmakokinetiikkaan.
Farmakokineettiset tiedot tutkimuksesta H4613g/GO01305, joka toteutettiin metastasoitunutta tai paikallisesti edennyttä leikkaushoitoon soveltumatonta HER2‑positiivista syöpää sairastavilla potilailla, viittasivat siihen, ettei trastutsumabi vaikuttanut karboplatiinin farmakokinetiikkaan.
Antineoplastisten aineiden vaikutus trastutsumabin farmakokinetiikkaan
Dosetakselin samanaikaisen käytön ei todettu vaikuttavan HER2‑positiivista metastasoitunutta rintasyöpää sairastavilla japanilaisilla naisilla (tutkimus JP16003) trastutsumabin farmakokinetiikkaan, kun trastutsumabin simuloituja ja havaittuja pitoisuuksia seerumissa vertailtiin trastutsumabimonoterapian jälkeen (aloitusannos 4 mg/kg, minkä jälkeen 2 mg/kg kerran viikossa laskimoon). Kahdesta vaiheen II tutkimuksesta (BO15935 ja M77004) ja yhdestä vaiheen III tutkimuksesta (H0648g), joissa potilaat saivat samanaikaisesti trastutsumabia ja paklitakselia, sekä kahdesta vaiheen II tutkimuksesta, joissa trastutsumabi annettiin monoterapiana (W016229 ja MO16982), HER2‑positiivista metastasoitunutta rintasyöpää sairastavista naisista saatujen farmakokineettisten tulosten vertailu osoittaa, että trastutsumabin yksilölliset ja keskimääräiset pienimmät pitoisuudet seerumissa vaihtelivat tutkimusten sisällä ja tutkimusten välillä mutta samanaikaisesti annetulla paklitakselilla ei todettu olevan selkeää vaikutusta trastutsumabin farmakokinetiikkaan.
Tutkimuksesta M77004, jossa HER2‑positiivista metastasoitunutta rintasyöpää sairastavat naiset saivat samanaikaisesti trastutsumabia, paklitakselia ja doksorubisiinia, sekä tutkimuksista, joissa trastutsumabia annettiin monoterapiana (H0649g) tai yhdistelmänä antrasykliinien sekä syklofosfamidin tai paklitakselin (tutkimus H0648g) kanssa, saatujen trastutsumabin farmakokinetiikkaa koskevien tietojen vertailu viittasi siihen, etteivät doksorubisiini ja paklitakseli vaikuta trastutsumabin farmakokinetiikkaan.
Tutkimuksen H4613g/GO01305 farmakokineettiset tiedot viittasivat siihen, ettei karboplatiini vaikuttanut trastutsumabin farmakokinetiikkaan.
Anastrotsolin samanaikainen anto ei näyttänyt vaikuttavan trastutsumabin farmakokinetiikkaan.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy
Naisten, jotka voivat tulla raskaaksi, pitää käyttää tehokasta raskauden ehkäisyä Phesgo-hoidon aikana ja seitsemän kuukauden ajan viimeisen annoksen jälkeen.
Raskaus
Pertutsumabilla tehdyissä eläinkokeissa on todettu lisääntymistoksisuutta. On vain vähän tietoja pertutsumabin käytöstä raskaana oleville naisille.
Eläinkokeiden perusteella ei tiedetä, voiko trastutsumabi vaikuttaa lisääntymiskykyyn (ks. kohta Prekliiniset tiedot turvallisuudesta). Trastutsumabia myyntiluvan myöntämisen jälkeen saaneilla raskaana olevilla naisilla on kuitenkin raportoitu sikiön munuaisten kasvun ja/tai toiminnan heikkenemistä, johon on liittynyt lapsiveden niukkuutta, mistä on toisinaan aiheutunut sikiön kuolemaan johtanut keuhkojen vajaakehitys.
Edellä mainittujen eläinkokeiden ja myyntiluvan myöntämisen jälkeisten tietojen perusteella Phesgo-valmisteen käyttöä raskauden aikana pitää välttää, ellei mahdollinen hyöty äidille ole sikiölle mahdollisesti aiheutuvaa riskiä suurempi. Raskaaksi tuleville naisille pitää kertoa sikiön vahingoittumisen mahdollisuudesta. Jos raskaana oleva nainen saa Phesgo-hoitoa tai jos potilas tulee raskaaksi Phesgo-hoidon aikana tai 7 kuukauden kuluessa viimeisestä Phesgo-annoksesta, moniammatillisen tiimin on suositeltavaa seurata raskautta tarkasti.
Imetys
Phesgo-hoidon aikana ja vähintään seitsemään kuukauteen viimeisen hoitoannoksen jälkeen ei pidä imettää, koska ihmisen IgG erittyy rintamaitoon eikä mahdollisesta imeytymisestä tai lapselle aiheutuvasta haitasta ole tietoja.
Hedelmällisyys
Pertutsumabi
Eläimillä ei ole tehty erityisiä hedelmällisyystutkimuksia pertutsumabin vaikutusten selvittämiseksi. Cynomolgus-apinoilla tehdyissä enimmillään kuusi kuukautta kestäneissä toistuvan altistuksen aiheuttamaa toksisuutta koskeneissa pertutsumabitutkimuksissa ei havaittu urosten ja naaraiden lisääntymiselimiin kohdistuvia haittavaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Trastutsumabi
Cynomolgus-apinoilla trastutsumabilla tehdyissä lisääntymistutkimuksissa ei havaittu cynomolgus-apinanaaraiden hedelmällisyyden heikkenemistä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Phesgo-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn (ks. kohta Haittavaikutukset). Jos potilaalle ilmaantuu injektioon liittyviä reaktioita tai huimausta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), häntä on neuvottava välttämään autolla ajoa tai koneiden käyttöä, kunnes oireet häviävät.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Phesgo-hoitoa tai laskimoon annettavaa pertutsumabia yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa saaneilla potilailla yleisimmin (≥ 30 %) raportoituja haittavaikutuksia olivat alopesia, ripuli, pahoinvointi, anemia, voimattomuus ja nivelkipu.
Phesgo-hoitoa tai laskimoon annettavaa pertutsumabia yhdistelmänä trastutsumabin kanssa saaneilla potilailla yleisimmin raportoituja vakavia haittavaikutuksia (≥ 1 %) olivat kuumeinen neutropenia, sydämen vajaatoiminta, kuume, neutropenia, neutropeeninen sepsis, pienentynyt neutrofiilimäärä ja keuhkokuume.
Phesgo-valmisteen turvallisuusprofiili oli yleisesti yhdenmukainen laskimoon annettavien pertutsumabin ja trastutsumabin yhdistelmän tunnetun turvallisuusprofiilin kanssa, minkä lisäksi haittavaikutuksena on esiintynyt injektiokohdan reaktioita (15,3 % vs. 0,4 %).
FEDERICA-pivotaalitutkimuksessa vakavat haittavaikutukset jakautuivat tasaisesti Phesgo-hoitohaaran ja laskimoon annettavien pertutsumabin ja trastutsumabin yhdistelmää saaneen hoitohaaran välillä. Seuraavia haittavaikutuksia raportoitiin yleisemmin (≥ 5 %) Phesgo-hoidossa kuin laskimoon annettavien pertutsumabin ja trastutsumabin yhdistelmästä koostuneessa hoidossa: alopesia 79 % vs 73 %, lihassärky 27 % vs 20,6 % ja hengenahdistus 12,1 % vs 6 %.
Haittavaikutustaulukko
Pertutsumabin turvallisuutta yhdistelmänä trastutsumabin kanssa on tutkittu HER2‑positiivista rintasyöpää sairastavilla potilailla pivotaalitutkimuksissa (n = 3834) CLEOPATRA, NEOSPHERE, TRYPHAENA, APHINITY ja FEDERICA. Turvallisuusprofiili oli yleisesti yhdenmukainen, vaikka haittavaikutusten esiintyvyys ja yleisimmät haittavaikutukset vaihtelivat riippuen siitä, annettiinko pertutsumabi ja trastutsumabi yhdistelmänä solunsalpaajahoidon kanssa vai ilman solunsalpaajaa.
Taulukon 2 ensimmäisessä sarakkeessa esitetään haittavaikutukset, joita on raportoitu käytettäessä pertutsumabia yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa jäljempänä mainituissa kliinisissä pivotaalitutkimuksissa (n = 3834) ja valmisteen markkinoille tulon jälkeen. Koska pertutsumabia käytetään yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa, haittavaikutuksen syy-yhteyttä tiettyyn lääkevalmisteeseen on vaikea varmistaa. Kahdessa muussa sarakkeessa on tiedot FEDERICA-tutkimuksen (n = 243) Phesgo-haarassa raportoiduista haittavaikutuksista annettaessa Phesgo-valmistetta solunsalpaajan kanssa ja monoterapiana.
- CLEOPATRA (n = 453), jossa metastasoitunutta rintasyöpää sairastaville potilaille annettiin pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa
- NEOSPHERE (n = 309) ja TRYPHAENA (n = 218), joissa paikallisesti edennyttä, inflammatorista tai varhaisvaiheen rintasyöpää sairastaville potilaille annettiin neoadjuvanttihoitona pertutsumabia yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa
- APHINITY (n = 2364), jossa varhaisvaiheen rintasyöpää sairastaville potilaille annettiin adjuvanttihoitona pertutsumabia yhdistelmänä trastutsumabin ja antrasykliiniä sisältävän tai sisältämättömän, taksaania sisältävän solunsalpaajahoidon kanssa
- FEDERICA (n = 243), jossa varhaisvaiheen rintasyöpää sairastaville potilaille annettiin ensin Phesgo-hoitoa tai laskimoon annettavaa pertutsumabia ja trastutsumabia (n = 247) yhdistelmänä solunsalpaajahoidon kanssa (neoadjuvanttivaihe) ja sen jälkeen monoterapiana (adjuvanttivaihe).
Haittavaikutukset luetellaan seuraavassa MedDRA-elinjärjestelmän ja esiintymistiheysluokan mukaisesti:
- hyvin yleinen (≥ 1/10)
- yleinen (≥ 1/100, < 1/10)
- melko harvinainen (≥ 1/1 000, < 1/100)
- harvinainen (≥ 1/10 000, < 1/1 000)
- hyvin harvinainen (< 1/10 000)
- tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa ja elinjärjestelmässä haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 2 Yhteenveto haittavaikutuksista kliinisissä pivotaalitutkimuksissa^, ^^ sekä valmisteen markkinoille tulon jälkeen† pertutsumabia ja trastutsumabia saaneilla potilailla
| N = 3834^ | N = 243^^ | ||
Pertutsumabi + trastutsumabi | Phesgo yhdistelmänä solunsalpaajahoidon kanssa | Phesgo-monoterapia | |
Haittavaikutus (MedDRA preferred term ‑termi) Elinjärjestelmäluokka | Esiintyvyysluokka | Esiintyvyysluokka | Esiintyvyysluokka |
| Veri ja imukudos | |||
| Neutropenia | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Anemia | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Kuumeinen neutropenia* | Hyvin yleinen | Yleinen | Tuntematon |
| Leukopenia | Hyvin yleinen | Yleinen | Yleinen |
| Sydän | |||
| Sydämen vasemman kammion toimintahäiriö** | Yleinen | Melko harvinainen | Melko harvinainen |
| Sydämen vajaatoiminta** | Yleinen | Melko harvinainen | Yleinen |
| Silmät | |||
| Lisääntynyt kyynelvuoto | Hyvin yleinen | Yleinen | Melko harvinainen |
| Ruoansulatuselimistö | |||
| Ripuli | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| Pahoinvointi | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Oksentelu | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Stomatiitti | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Ummetus | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Dyspepsia | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Vatsakipu | Hyvin yleinen | Yleinen | Yleinen |
| Yleisoireet ja antopaikassa todettavat haitat | |||
| Väsymys | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Limakalvojen tulehdus | Hyvin yleinen | Hyvin yleinen | Melko harvinainen |
| Voimattomuus | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| Kuume | Hyvin yleinen | Yleinen | Yleinen |
| Raajojen turvotus | Hyvin yleinen | Yleinen | Yleinen |
| Injektiokohdan reaktio°°° | Hyvin yleinen | Yleinen | Hyvin yleinen |
| Immuunijärjestelmä | |||
| Yliherkkyys*° | Yleinen | Melko harvinainen | Tuntematon |
| Lääkeaineyliherkkyys*° | Yleinen | Melko harvinainen | Melko harvinainen |
| Anafylaktinen reaktio*° | Melko harvinainen | Tuntematon | Tuntematon |
| Sytokiinioireyhtymä° | Harvinainen | Tuntematon | Tuntematon |
| Infektiot | |||
| Nasofaryngiitti | Hyvin yleinen | Yleinen | Yleinen |
| Ylempien hengitysteiden infektio | Yleinen | Yleinen | Yleinen |
| Kynnenvierustulehdus | Yleinen | Yleinen | Yleinen |
| Aineenvaihdunta ja ravitsemus | |||
| Heikentynyt ruokahalu | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Tuumorilyysioireyhtymä† | Harvinainen | Tuntematon | Tuntematon |
| Luusto, lihakset ja sidekudos | |||
| Nivelkipu | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| Lihaskipu | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Raajakipu | Hyvin yleinen | Yleinen | Yleinen |
| Hermosto | |||
| Makuhäiriö | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Päänsärky | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Perifeerinen sensorinen neuropatia | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Perifeerinen neuropatia | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Heitehuimaus | Hyvin yleinen | Yleinen | Yleinen |
| Parestesiat | Hyvin yleinen | Yleinen | Yleinen |
| Psyykkiset häiriöt | |||
| Unettomuus | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Hengityselimet, rintakehä ja välikarsina | |||
| Nenäverenvuoto | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Yskä | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Hengenahdistus | Hyvin yleinen | Yleinen | Yleinen |
| Interstitiaalinen keuhkosairaus°° | Melko harvinainen | Tuntematon | Tuntematon |
| Iho ja ihonalainen kudos | |||
| Alopesia | Hyvin yleinen | Hyvin yleinen | Melko harvinainen |
| Ihottuma | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Ihon kuivuminen | Hyvin yleinen | Hyvin yleinen | Yleinen |
| Kynsien häiriöt | Hyvin yleinen | Yleinen | Yleinen |
| Kutina | Hyvin yleinen | Yleinen | Yleinen |
| Verisuonisto | |||
| Kuumat aallot | Hyvin yleinen | Yleinen | Hyvin yleinen |
^ Yhdistetyt tiedot CLEOPATRA-tutkimuksen koko hoitojaksosta (tietojen keruun katkaisupäivä 11. helmikuuta 2014; pertutsumabihoitosyklien lukumäärän mediaani oli 24), NEOSPHERE- ja TRYPHAENA-tutkimusten neoadjuvanttihoitojaksoista (NEOSPHERE-tutkimuksessa pertutsumabihoitosyklien lukumäärän mediaani kaikissa tutkimushaaroissa oli 4; TRYPHAENA-tutkimuksessa pertutsumabihoitosyklien lukumäärän mediaani kaikissa tutkimushaaroissa oli 3–6); APHINITY-tutkimuksen hoitojaksosta (pertutsumabihoitosyklien lukumäärän mediaani oli 18) ja FEDERICA-tutkimuksen koko hoitojaksosta (Phesgo-hoitosyklien lukumäärän mediaani oli 18).
^^Phesgo-hoitoa koskevat tiedot FEDERICA-tutkimuksen koko hoitojaksosta (Phesgo-hoitosyklien lukumäärän mediaani oli 18).
* Myös kuolemaan johtaneita haittavaikutuksia on raportoitu.
** Kaikkien 5 tutkimuksen (CLEOPATRA, NEOSPHERE, TRYPHAENA, APHINITY, FEDERICA) koko hoitojakso. Vasemman kammion toimintahäiriöiden ja kongestiivisen sydämen vajaatoiminnan ilmaantuvuus kuvastavat yksittäisissä tutkimuksissa raportoituja MedDRA Preferred Terms ‑termien mukaisia haittavaikutuksia.
° Yleisimmin raportoituja lääketieteellisiä käsitteitä koskevia termejä olivat anafylaktinen reaktio ja injektioon/infuusioon liittyvä reaktio, jotka kuvataan tarkemmin kohdassa Valikoitujen haittavaikutusten kuvaus.
°° FeDeriCa-tutkimuksessa ei raportoitu yhtään interstitiaalista keuhkosairautta koskevaa tapahtumaa, mutta tällaisia tapahtumia on havaittu trastutsumabihoidon yhteydessä.
°°° Havaittu vain Phesgo-valmisteen käytössä (liittyen ihon alle annosteluun). Adjuvanttivaiheessa havaittu suurempi esiintyvyys liittyy pidempään hoitojaksoon, kun Phesgo-valmiste annetaan monoterapiana.
† Pertutsumabin ja trastutsumabin (laskimoon) markkinoille tulon jälkeen raportoidut haittavaikutukset.
Valikoitujen haittavaikutusten kuvaus
Vasemman kammion toimintahäiriö
Phesgo
Oireisen sydämen vajaatoiminnan (NYHA-luokka III tai IV) ja siihen liittyneen vasemman kammion ejektiofraktion pienenemisen vähintään 10 prosenttiyksikköä lähtötilanteesta ja < 50 %:iin ilmaantuvuus oli FEDERICA-pivotaalitutkimuksen neoadjuvanttivaiheen aikana (samanaikaisesti solunsalpaajahoidon kanssa annettuna) 0,4 % Phesgo-hoitoa saaneilla potilailla vs 0 % laskimoon annettavia pertutsumabia ja trastutsumabia saaneilla potilailla. Oireista sydämen vajaatoimintaa kokeneista potilaista yksikään Phesgo-hoitoa saaneista potilaista ei ollut toipunut tietojen keruun katkaisupäivänä ja yhden potilaan Phesgo-hoito päättyi oireisen sydämen vajaatoiminnan vuoksi. Oireisen sydämen vajaatoiminnan ja vasemman kammion ejektiofraktion pienenemisen vähintään 10 prosenttiyksikköä lähtötilanteesta ja < 50 %:iin ilmaantuvuus oli adjuvanttivaiheessa (annettaessa Phesgo-valmistetta yksinään) samankaltainen kuin seurantavaiheessa. Oireetonta tai lievästi oireista (NYHA-luokka II) vasemman kammion ejektiofraktion pienenemistä vähintään 10 prosenttiyksikköä lähtötilanteesta ja < 50 %:iin (varmistettu sekundaarisella vasemman kammion ejektiofraktiolla) ei raportoitu Phesgo-hoitoa saaneilla potilailla, mutta raportoitiin 0,4 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia neoadjuvanttivaiheen aikana saaneista potilaista (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Oireetonta tai lievästi oireista (NYHA-luokka II) vasemman kammion ejektiofraktion pienenemistä vähintään 10 prosenttiyksikköä lähtötilanteesta ja < 50 %:iin (varmistettu sekundaarisella vasemman kammion ejektiofraktiolla) ei raportoitu adjuvanttivaiheessa kummassakaan hoitohaarassa. Seurantavaiheessa tämäntyyppinen sydäntapahtuma todettiin 1,6 %:lla Phesgo-hoitoa saaneista potilaista ja 3,6 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista.
Laskimoon annettava pertutsumabi yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa
Vasemman kammion toimintahäiriöiden ilmaantuvuus oli CLEOPATRA-pivotaalitutkimuksen hoitojakson aikana suurempi lumehoitoa saaneessa ryhmässä (8,6 %) kuin pertutsumabihoitoa saaneessa ryhmässä (6,6 %). Myös oireisen vasemman kammion toimintahäiriön ilmaantuvuus oli pertutsumabihoitoa saaneessa ryhmässä pienempi (lumehoitoa saaneessa ryhmässä 1,8 % vs. pertutsumabihoitoa saaneessa ryhmässä 1,5 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilaat saivat neoadjuvanttihoitoa koskeneessa NEOSPHERE-tutkimuksessa neoadjuvanttihoitona neljä pertutsumabihoitosykliä. Vasemman kammion toimintahäiriön ilmaantuvuus (koko hoitojakson aikana) oli pertutsumabi-, trastutsumabi- ja dosetakselihoitoa saaneessa ryhmässä suurempi (7,5 %) verrattuna trastutsumabi- ja dosetakselihoitoa saaneeseen ryhmään (1,9 %). Yhdellä pertutsumabi- ja trastutsumabihoitoa saaneen ryhmän potilaalla oli oireinen vasemman kammion toimintahäiriö.
Vasemman kammion toimintahäiriön ilmaantuvuus (koko hoitojakson aikana) oli neoadjuvanttihoitoa koskeneessa TRYPHAENA-tutkimuksessa pertutsumabin ja trastutsumabin yhdistelmää sekä FEC-hoitoa (5‑fluorourasiili, epirubisiini, syklofosfamidi) ja sen jälkeen pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa saaneessa ryhmässä 8,3 %, pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa ja sen jälkeen FEC-hoitoa saaneessa ryhmässä 9,3 % ja pertutsumabia yhdistelmänä TCH-hoidon (dosetakseli, karboplatiini ja trastutsumabi) kanssa saaneessa ryhmässä 6,6 %. Oireisen vasemman kammion toimintahäiriön (kongestiivisen sydämen vajaatoiminnan) ilmaantuvuus oli pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa ja sen jälkeen FEC-hoitoa saaneessa ryhmässä 1,3 % (tässä ei ole mukana potilasta jolla oli oireinen vasemman kammion toimintahäiriö FEC-hoidon aikana ennen kuin potilas sai pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa) ja pertutsumabia yhdistelmänä TCH-hoidon kanssa saaneessa ryhmässä myös 1,3 %. Pertutsumabia yhdistelmänä trastutsumabin ja FEC-hoidon kanssa ja sen jälkeen pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa saaneessa ryhmässä yhdelläkään potilaalla ei ollut oireista vasemman kammion toimintahäiriötä.
NYHA-luokan III/IV oireisen vasemman kammion toimintahäiriön (NCI‑CTCAE-luokituksen [v.4] mukainen kongestiivinen sydämen vajaatoiminta) ilmaantuvuus oli BERENICE-tutkimuksen neoadjuvanttijaksossa lyhyen antovälin doksorubisiini- ja syklofosfamidihoitoa (AC-hoitoa) ja sen jälkeen pertutsumabia yhdistelmänä trastutsumabin ja paklitakselin kanssa saaneessa ryhmässä 1,5 %, mutta oireista vasemman kammion toimintahäiriötä ei esiintynyt yhdelläkään potilaalla (0 %) FEC-hoitoa ja sen jälkeen pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa saaneessa ryhmässä. Oireettoman vasemman kammion toimintahäiriön ilmaantuvuus (NCI‑CTCAE-luokituksen [v.4] mukainen ejektiofraktion pieneneminen) oli lyhyen antovälin AC-hoitoa ja sen jälkeen pertutsumabia yhdistelmänä trastutsumabin ja paklitakselin kanssa saaneessa ryhmässä 7 % ja FEC-hoitoa ja sen jälkeen pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa saaneessa ryhmässä 3,5 %.
Oireisen sydämen vajaatoiminnan (NYHA-luokka III tai IV) ja siihen liittyneen vasemman kammion ejektiofraktion pienenemisen vähintään 10 prosenttiyksikköä lähtötilanteesta ja < 50 %:iin ilmaantuvuus oli APHINITY-tutkimuksessa < 1 % (0,6 %:lla pertutsumabihoitoa saaneista potilaista vs 0,3 %:lla lumehoitoa saaneista potilaista). Oireista sydämen vajaatoimintaa kokeneista potilaista tietojen keruun katkaisuajankohtana oli toipunut 46,7 % pertutsumabihoitoa saaneista potilaista ja 57,1 % lumehoitoa saaneista potilaista (toipuminen oli määritelty kahdeksi peräkkäiseksi yli 50 %:n vasemman kammion ejektiofraktion mittaustulokseksi). Valtaosa tapahtumista raportoitiin antrasykliinihoitoa saaneilla potilailla. Oireetonta tai lievästi oireista (NYHA-luokka II) vasemman kammion ejektiofraktion pienenemistä vähintään 10 prosenttiyksikköä lähtötilanteesta ja < 50 %:iin raportoitiin 2,7 %:lla pertutsumabihoitoa saaneista potilaista ja 2,8 %:lla lumehoitoa saaneista potilaista. Näistä potilaista 79,7 % pertutsumabihoitoa saaneista potilaista ja 80,6 % lumehoitoa saaneista potilaista oli tietojen keruun katkaisuajankohtana toipunut.
Injektioon/infuusioon liittyvät reaktiot
Phesgo
FEDERICA-pivotaalitutkimuksessa injektioon/infuusioon liittyviksi reaktioiksi määriteltiin mikä tahansa 24 tunnin kuluessa Phesgo-valmisteen tai laskimoon annettavan pertutsumabin ja trastutsumabin yhdistelmän antamisen jälkeen raportoitu systeeminen reaktio (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Neoadjuvanttivaiheessa injektioon liittyviä reaktioita raportoitiin 0,4 %:lla Phesgo-hoitoa saaneista potilaista ja infuusioon liittyviä reaktioita raportoitiin 10,7 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista. Adjuvanttivaiheessa Phesgo-hoitoa saaneilla potilailla ei raportoitu injektioon liittyviä reaktioita ja 1,6 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista raportoitiin infuusioon liittyviä reaktioita. Phesgo-hoidossa tai laskimoon annettavia pertutsumabia ja trastutsumabia käytettäessä valtaosa systeemisistä injektio-/infuusioreaktioista ilmeni vilunväristyksinä, pahoinvointina tai oksenteluna.
Injektiokohdan reaktioiksi määriteltiin mikä tahansa 24 tunnin kuluessa Phesgo-valmisteen antamisesta raportoitu paikallinen reaktio. Niitä raportoitiin neoadjuvanttivaiheessa 6,9 %:lla ja adjuvanttivaiheessa 12,9 %:lla Phesgo-hoitoa saaneista potilaista. Kaikki olivat graduksen 1 tai 2 tapahtumia. Valtaosa Phesgo-hoidossa havaituista paikallisista injektiokohdan reaktioista ilmeni joko injektiokohdan kipuna tai injektiokohdan punoituksena.
Laskimoon annettava pertutsumabi yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa
Antoon liittyväksi reaktioksi määriteltiin pivotaalitutkimuksissa mikä tahansa tapahtuma, joka raportoitiin yliherkkyydeksi, anafylaktiseksi reaktioksi, akuutiksi infuusioreaktioksi tai sytokiinioireyhtymäksi, joka esiintyi infuusion aikana tai infuusion antopäivänä. CLEOPATRA-pivotaalitutkimuksessa pertutsumabialoitusannos annettiin päivää ennen trastutsumabia ja dosetakselia, jotta pertutsumabiin liittyviä reaktioita voitiin tutkia. Ensimmäisenä päivänä, jolloin annettiin vain pertutsumabia, infuusioon liittyvien reaktioiden kokonaisesiintyvyys oli 9,8 % lumehoitoa saaneessa ryhmässä ja 13,2 % pertutsumabihoitoa saaneessa ryhmässä, ja suurin osa infuusioreaktioista oli lieviä tai keskivaikeita. Yleisimmät infuusioon liittyvät reaktiot (≥ 1 %) pertutsumabihoitoa saaneessa ryhmässä olivat kuume, vilunväristykset, väsymys, päänsärky, voimattomuus, yliherkkyys ja oksentelu.
Toisen hoitosyklin aikana, jolloin kaikkia lääkevalmisteita annettiin samana päivänä, yleisimmät (≥ 1 %) infuusioon liittyvät reaktiot pertutsumabihoitoa saaneessa ryhmässä olivat väsymys, lääkeyliherkkyys, makuaistin häiriöt, yliherkkyys, lihaskipu ja oksentelu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pertutsumabia annettiin neoadjuvantti- ja adjuvanttihoitoa koskeneissa tutkimuksissa samana päivänä kuin muutakin tutkimushoitoa. Infuusioon liittyviä reaktioita esiintyi ensimmäisenä pertutsumabihoidon (yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa) antopäivänä 18,6–25 %:lla potilaista. Haittavaikutuksen tyyppi ja vaikeusaste olivat yhdenmukaiset CLEOPATRA-tutkimuksessa havaittujen kanssa, ja valtaosa reaktioista oli vaikeusasteeltaan lieviä tai keskivaikeita.
Yliherkkyysreaktiot/anafylaksia
Phesgo
FEDERICA-pivotaalitutkimuksessa HER2-kohdennettuun hoitoon liittyneiden yliherkkyytenä/anafylaksiana raportoitujen tapahtumien kokonaisesiintyvyys oli Phesgo-hoitoa saaneilla potilailla 1,2 % ja laskimoon annettavia pertutsumabia ja trastutsumabia saaneilla potilailla 0,8 %; yksikään tapahtumista ei ollut NCI‑CTCAE (versio 4.0) ‑luokituksen gradus 3‑4 reaktio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Yhdellä potilaalla esiintyi ensimmäisessä hoitosyklissä yliherkkyyttä / anafylaktinen tapahtuma Phesgo-valmisteen annon aikana tai heti annon jälkeen, mikä johti hoidon lopettamiseen (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.).
Neoadjuvanttivaiheen aikana lääkeyliherkkyyttä ilmeni 0,4 %:lla Phesgo-hoitoa saaneista potilaista ja 0,4 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista. Adjuvanttivaiheessa lääkeyliherkkyyttä ilmeni 0,4 %:lla Phesgo-hoitoa saaneista potilaista ja yhdelläkään laskimoon annettavia pertutsumabia ja trastutsumabia saaneella potilaalla ei ilmennyt yliherkkyyttä eikä lääkeyliherkkyyttä.
Laskimoon annettava pertutsumabi yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa
Metastasoitunutta rintasyöpää koskeneessa pivotaalitutkimuksessa CLEOPATRA tutkijoiden raportoimien yliherkkyys-/anafylaksiatapahtumien kokonaisesiintyvyys koko hoitojakson aikana oli 9,3 % lumehoitoa saaneessa ryhmässä ja 11,3 % pertutsumabihoitoa saaneessa ryhmässä. Näistä 2,5 % lumehoitoa saaneessa ryhmässä ja 2 % pertutsumabihoitoa saaneessa ryhmässä oli vaikeusasteeltaan NCI‑CTCAE-luokituksen mukaisia gradus 3–4 haittavaikutuksia. Lumehoitoa saaneessa ryhmässä 2 potilaalla ja pertutsumabihoitoa saaneessa ryhmässä 4 potilaalla esiintyi tapahtumia, jotka tutkija kuvasi anafylaksiaksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Suurin osa yliherkkyysreaktioista oli vaikeusasteeltaan lieviä tai keskivaikeita ja ne hävisivät hoidon avulla. Suurimman osan reaktioista arvioitiin tutkimushoitoon tehtyjen muutosten perusteella johtuneen dosetakseli-infuusioista.
Yliherkkyys-/anafylaktiset tapahtumat olivat neoadjuvantti- ja adjuvanttihoitoa koskeneissa tutkimuksissa samankaltaisia kuin CLEOPATRA-tutkimuksessa. NEOSPHERE-tutkimuksessa kahdella pertutsumabia ja dosetakselia saaneen ryhmän potilaalla esiintyi anafylaksiaa. Yliherkkyyden/anafylaksian kokonaisesiintyvyys oli sekä TRYPHAENA- että APHINITY-tutkimuksessa suurin pertutsumabia ja TCH-hoitoa saaneessa ryhmässä (TRYPHAENA-tutkimuksessa 13,2 % ja APHINITY-tutkimuksessa 7,6 %). Näistä 2,6 % TRYPHAENA-tutkimuksessa ja 1,3 % APHINITY-tutkimuksessa oli vaikeusasteeltaan NCI‑CTCAE-luokituksen gradus 3–4.
Kuumeinen neutropenia
Phesgo
Kuumeista neutropeniaa (gradus 3 tai 4) esiintyi FEDERICA-pivotaalitutkimuksen neoadjuvanttivaiheen aikana 6,6 %:lla Phesgo-hoitoa saaneista potilaista ja 5,6 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista. Adjuvantttivaiheen aikana ei ilmennyt kuumeista neutropeniaa koskevia tapahtumia (gradus 3 tai 4).
Laskimoon annettavilla pertutsumabilla ja trastutsumabilla tehdyissä pivotaalitutkimuksissa kuumeisen neutropenian (gradus 3 tai 4) ilmaantuvuuden havaittiin olevan suurempi laskimoon annettavia pertutsumabia ja trastutsumabia saaneilla aasialaisilla potilailla (13 %), ja kuumeisen neutropenian ilmaantuvuus myös Phesgo-valmistetta ihon alle neoadjuvanttivaiheen aikana saaneilla aasialaisilla potilailla oli samalla tavoin suurempi (13,7 %). Adjuvanttivaiheen aikana kummassakaan hoitohaarassa ei ilmennyt kuumeista neutropeniaa koskevia tapahtumia (gradus 3 tai 4).
Laskimoon annettava pertutsumabi yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa
Suurimmalla osalla CLEOPATRA-pivotaalitutkimuksen kummankin hoitoryhmän potilaista esiintyi vähintään yksi leukopeniatapahtuma (63 %:lla pertutsumabihoitoryhmän potilaista ja 58,3 %:lla lumehoitoryhmän potilaista), ja suurin osa näistä oli neutropeenisia tapahtumia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kuumeista neutropeniaa esiintyi 13,7 %:lla pertutsumabihoitoa saaneista potilaista ja 7,6 %:lla lumehoitoa saaneista potilaista. Kummassakin hoitoryhmässä niiden potilaiden osuus, joilla esiintyi kuumeista neutropeniaa, oli suurin ensimmäisen hoitosyklin aikana ja väheni sen jälkeen tasaisesti. Kuumeisen neutropenian esiintymistiheyden todettiin kummassakin hoitoryhmässä olevan suurentunut aasialaisilla potilailla verrattuna muihin etnisiin ryhmiin ja muilta maantieteellisiltä alueilta peräisin oleviin potilaisiin. Aasialaisilla potilailla kuumeisen neutropenian esiintyvyys oli suurempi pertutsumabihoitoa saaneessa ryhmässä (25,8 %) verrattuna lumehoitoa saaneeseen ryhmään (11,3 %).
Kuumeista neutropeniaa esiintyi NEOSPHERE-tutkimuksessa 8,4 %:lla pertutsumabia, trastutsumabia ja dosetakselia neoadjuvanttihoitona saaneista potilaista verrattuna 7,5 %:iin trastutsumabia ja dosetakselia saaneista potilaista. TRYPHAENA-tutkimuksessa kuumeista neutropeniaa esiintyi 17,1 %:lla pertutsumabin ja TCH-hoidon yhdistelmää neoadjuvanttihoitona saaneista potilaista verrattuna 9,3 %:iin neoadjuvanttihoitona pertutsumabia, trastutsumabia ja dosetakselia ja sen jälkeen FEC-hoitoa saaneista potilaista. Kuumeisen neutropenian ilmaantuvuus oli TRYPHAENA-tutkimuksessa suurempi kuusi hoitosykliä pertutsumabia saaneilla potilailla verrattuna kolme hoitosykliä pertutsumabia saaneisiin potilaisiin annetusta solunsalpaajahoidosta riippumatta. Neutropenian ja kuumeisen neutropenian ilmaantuvuus oli kummassakin neoadjuvanttitutkimuksessa, samoin kuin CLEOPATRA-tutkimuksessa, suurempi aasialaisilla potilailla verrattuna muihin potilaisiin. Kuumeista neutropeniaa esiintyi NEOSPHERE-tutkimuksessa 8,3 %:lla pertutsumabia, trastutsumabia ja dosetakselia neoadjuvanttihoitona saaneista aasialaisista potilaista verrattuna 4 %:iin aasialaisista potilaista, jotka saivat neoadjuvanttihoitona trastutsumabia ja dosetakselia.
Kuumeista neutropeniaa esiintyi APHINITY-tutkimuksessa 12,1 %:lla pertutsumabihoitoa saaneista potilaista ja 11,1 %:lla lumehoitoa saaneista potilaista. Kuumeisen neutropenian ilmaantuvuuden havaittiin CLEOPATRA-, TRYPHAENA- ja NEOSPHERE-tutkimusten tavoin olleen APHINITY-tutkimuksessa suurempi pertutsumabihoitoa saaneilla aasialaisilla potilailla verrattuna muihin etnisiin ryhmiin kuuluviin potilaisiin (pertutsumabihoitoa saaneilla potilailla 15,9 % ja lumehoitoa saaneilla potilailla 9,9 %).
Ripuli
Phesgo
Ripulia esiintyi FEDERICA-pivotaalitutkimuksen neoadjuvanttivaiheen aikana 60,5 %:lla Phesgo-hoitoa saaneista potilaista ja 54,8 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista. Graduksen ≥ 3 ripulia raportoitiin Phesgo-haarassa 6,6 %:lla potilaista vs. 4 %:lla potilaista laskimoon annettavien pertutsumabin ja trastutsumabin yhdistelmää saaneessa haarassa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.).
Adjuvanttivaiheen aikana ripulia ilmeni 17,7 %:lla Phesgo-hoitoa saaneista potilaista ja 20,6 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista. Graduksen ≥ 3 ripulia raportoitiin Phesgo-haarassa 0 %:lla potilaista verrattuna 1,2 %:iin potilaista laskimoon annettavia pertutsumabia ja trastutsumabia saaneessa haarassa.
Laskimoon annettava pertutsumabi yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa
Ripulia esiintyi metastasoitunutta rintasyöpää koskeneessa CLEOPATRA-pivotaalitutkimuksessa 68,4 %:lla pertutsumabihoitoa saaneista potilaista ja 48,7 %:lla lumehoitoa saaneista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Useimmat tapahtumat olivat vaikeusasteeltaan lieviä tai kohtalaisia, ja niitä esiintyi muutaman ensimmäisen hoitosyklin aikana. NCI‑CTCAE-luokituksen mukaisen gradus 3–4 ripulin ilmaantuvuus oli 9,3 % pertutsumabihoitoa saaneilla potilailla verrattuna 5,1 %:iin lumehoitoa saaneilla potilailla. Ripuli kesti pisimmillään (mediaani) 18 vuorokautta pertutsumabihoitoa saaneilla potilailla ja 8 vuorokautta lumehoitoa saaneilla potilailla. Potilaat saivat hyvän vasteen ennakoivasti aloitettuun ripulilääkehoitoon.
Ripulia esiintyi NEOSPHERE-tutkimuksessa 45,8 %:lla pertutsumabia, trastutsumabia ja dosetakselia neoadjuvanttihoitona saaneista potilaista verrattuna 33,6 %:iin trastutsumabia ja dosetakselia saaneista potilaista. TRYPHAENA-tutkimuksessa ripulia esiintyi 72,3 %:lla pertutsumabin ja TCH-hoidon yhdistelmää neoadjuvanttihoitona saaneista potilaista verrattuna 61,4 %:iin neoadjuvanttihoitona pertutsumabia, trastutsumabia ja dosetakselia ja sen jälkeen FEC-hoitoa saaneista potilaista. Useimmat tapahtumat olivat kummassakin tutkimuksessa vaikeusasteeltaan lieviä tai keskivaikeita.
Ripulin ilmaantuvuuden raportoitiin olleen APHINITY-tutkimuksessa suurempi pertutsumabihoitoa saaneilla potilailla (71,2 %) verrattuna lumehoitoa saaneisiin potilaisiin (45,2 %). Graduksen ≥ 3 ripulia raportoitiin pertutsumabihaarassa 9,8 %:lla potilaista vs. 3,7 %:iin lumehaaran potilaista. Valtaosa raportoiduista tapahtumista oli vaikeusasteeltaan gradus 1 tai 2. Ripulin (kaikki gradukset) suurin ilmaantuvuus raportoitiin kohdennetun hoidon ja taksaanisolunsalpaajahoidon yhdistelmän käytön aikana (61,4 %:lla pertutsumabihaaran potilaista vs. 33,8 %:lla lumehoitohaaran potilaista). Ripulin ilmaantuvuus oli huomattavasti pienempi solunsalpaajahoidon lopettamisen jälkeen, ja sitä esiintyi kohdennetun solunsalpaajahoitojakson jälkeen 18,1 %:lla pertutsumabihaaran potilaista vs. 9,2 %:lla lumehoitohaaran potilaista.
Ihottuma
Phesgo
Ihottumaa esiintyi FEDERICA-pivotaalitutkimuksen neoadjuvanttivaiheen aikana 10,7 %:lla Phesgo-hoitoa saaneista potilaista ja 15,5 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista. Adjuvanttivaiheen aikana ihottumaa esiintyi 8,2 %:lla Phesgo-hoitoa saaneista potilaista ja 8,7 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista. Valtaosa ihottumista oli vaikeusasteeltaan gradus 1 tai 2.
Laskimoon annettava pertutsumabi yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa
Ihottumaa esiintyi metastasoitunutta rintasyöpää koskeneessa CLEOPATRA-pivotaalitutkimuksessa 51,7 %:lla pertutsumabihoitoa saaneista potilaista verrattuna 38,9 %:iin lumehoitoa saaneista potilaista. Useimpien tapahtumien vaikeusaste oli gradus 1 tai 2, ne esiintyivät kahden ensimmäisen hoitosyklin aikana ja ne vastasivat hyvin hoitosuositusten mukaiseen hoitoon, kuten aknen paikalliseen tai suun kautta otettavaan hoitoon.
Ihottumaa esiintyi NEOSPHERE-tutkimuksessa 40,2 %:lla pertutsumabia, trastutsumabia ja dosetakselia neoadjuvanttihoitona saaneista potilaista verrattuna 29 %:iin trastutsumabia ja dosetakselia saaneista potilaista. TRYPHAENA-tutkimuksessa ihottumaa esiintyi 36,8 %:lla pertutsumabin ja TCH-hoidon yhdistelmää neoadjuvanttihoitona saaneista potilaista verrattuna 20 %:iin neoadjuvanttihoitona pertutsumabia, trastutsumabia ja dosetakselia ja sen jälkeen FEC-hoitoa saaneista potilaista. Ihottuman ilmaantuvuus oli annetusta solunsalpaajahoidosta riippumatta suurempi kuusi hoitosykliä pertutsumabia saaneilla potilailla verrattuna kolme hoitosykliä pertutsumabia saaneisiin potilaisiin.
Ihottumaa esiintyi APHINITY-tutkimuksessa haittavaikutuksena 25,8 %:lla pertutsumabihoitohaaran potilaista vs. 20,3 %:lla lumehoitohaaran potilaista. Valtaosa ihottumista oli vaikeusasteeltaan gradus 1 tai 2.
Laboratorioarvojen poikkeavuudet
Phesgo
NCI CTCAE-luokituksen (v. 4) mukaisen gradus 3–4 neutropenian ilmaantuvuus oli FEDERICA-pivotaalitutkimuksen neoadjuvanttivaiheen aikana vastaavaa kummassakin hoitoryhmässä (13,6 %:lla Phesgo-hoitoa saaneista potilaista ja 13,9 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista). Adjuvanttivaiheessa ilmaantuvuus oli merkittävästi vähäisempää (0,8 %:lla Phesgo-hoitoa saaneista potilaista ja 0 %:lla laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista.
Laskimoon annettava pertutsumabi yhdistelmänä trastutsumabin ja solunsalpaajahoidon kanssa
NCI‑CTCAE-luokituksen (v. 3) mukaisen gradus 3–4 neutropenian ilmaantuvuus oli metastasoitunutta rintasyöpää koskeneessa CLEOPATRA-pivotaalitutkimuksessa vastaavaa kummassakin hoitoryhmässä (86,3 %:lla pertutsumabihoitoa saaneista potilaista ja 86,6 %:lla lumehoitoa saaneista potilaista, mukaan lukien gradus 4 neutropenia 60,7 %:lla pertutsumabihoitoa saaneista potilaista ja 64,8 %:lla lumehoitoa saaneista potilaista).
NCI‑CTCAE-luokituksen (v. 3) gradus 3–4 neutropenian ilmaantuvuus NEOSPHERE-tutkimuksessa pertutsumabia, trastutsumabia ja dosetakselia neoadjuvanttihoitona saaneille potilaille oli 74,5 %, mistä graduksen 4 neutropeniaa oli 50,9 %, verrattuna 84,5 %:n ilmaantuvuuteen trastutsumabia ja dosetakselia saaneilla potilailla, mistä graduksen 4 neutropeniaa oli 60,2 %. TRYPHAENA-tutkimuksessa NCI‑CTCAE-luokituksen (v. 3) graduksen 3–4 neutropenian ilmaantuvuus oli pertutsumabin ja TCH-hoidon yhdistelmää neoadjuvanttihoitona saaneilla potilailla 85,3 %, mistä graduksen 4 neutropeniaa oli 66,7 %, ja neoadjuvanttihoitona pertutsumabia, trastutsumabia ja dosetakselia ja sen jälkeen FEC-hoitoa saaneilla potilailla ilmaantuvuus oli 77 %, mistä graduksen 4 neutropeniaa oli 59,5 %.
NCI‑CTCAE-luokituksen (v. 4) gradus 3–4 neutropenian ilmaantuvuus APHINITY-tutkimuksessa pertutsumabia, trastutsumabia ja solunsalpaajahoitoa saaneille potilaille oli 40,6 % verrattuna 39,1 %:n ilmaantuvuuteen lumelääkettä, trastutsumabia ja solunsalpaajahoitoa saaneilla potilailla. Graduksen 4 neutropenian ilmaantuvuus oli 28,3 % pertutsumabia, trastutsumabia ja solunsalpaajahoitoa saaneille potilaille ja 26,5 % lumelääkettä, trastutsumabia ja solunsalpaajahoitoa saaneille potilaille.
Immunogeenisuus
Muiden hoidossa käytettävien proteiinien tavoin immuunivaste pertutsumabiin ja trastutsumabiin on Phesgo-hoitoa saaneilla potilailla mahdollinen.
FEDERICA-tutkimuksessa laskimoon annettavia pertutsumabia ja trastutsumabia saaneilla potilailla hoidonaikaisten vasta-aineiden ilmaantuvuus pertutsumabia vastaan oli 10,6 % (26/245) ja trastutsumabia vastaan 0,4 % (1/245). Niistä potilaista, joilla todettiin vasta-aineita pertutsumabia vastaan, kolmella potilaalla havaittiin neutraloivia vasta-aineita pertutsumabia vastaan.
Phesgo-hoitoa saaneilla potilailla hoidonaikaisten vasta-aineiden ilmaantuvuus pertutsumabia vastaan oli 12,9 % (31/241), trastutsumabia vastaan 2,1 % (5/241) ja vorhyaluronidaasi alfaa vastaan 6,3 % (15/238). Näistä potilaista kahdella potilaalla havaittiin neutraloivia vasta-aineita pertutsumabia vastaan ja yhdellä potilaalla havaittiin neutraloivia vasta-aineita trastutsumabia vastaan.
Phesgo-hoidon jälkeen pertutsumabia, trastutsumabia tai vorhyaluronidaasi alfaa vastaan kehittyvien vasta-aineiden kliinistä merkitystä ei tiedetä.
Siirtyminen laskimoon annettavista pertutsumabista ja trastutsumabista Phesgo-hoitoon (tai päinvastoin)
MO40628-tutkimuksessa selvitettiin laskimoon annettavista pertutsumabista ja trastutsumabista ihon alle annettavaan Phesgo-hoitoon (hoitohaara A) tai päinvastoin (hoitohaara B) siirtymisen turvallisuutta. Ensisijaisena tavoitteena oli arvioida potilaiden mieltymyksiä Phesgo-hoitoa kohtaan (ks. tutkimusasetelman yksityiskohdat kohdasta Farmakodynamiikka).
Haittavaikutusten ilmaantuvuus hoitohaarassa A syklien 1–3 aikana (laskimoon annettava hoito) oli 77,5 % (62 potilaalla 80 potilaasta) verrattuna sykleihin 4–6 (ihon alle annettava hoito), jolloin ilmaantuvuus oli 72,5 % (58 potilaalla 80 potilaasta).
Haittavaikutusten ilmaantuvuus hoitohaarassa B syklien 1–3 aikana (ihon alle annettava hoito) oli 77,5 % (62 potilaalla 80 potilaasta) verrattuna sykleihin 4–6 (laskimoon annettava hoito), jolloin ilmaantuvuus oli 63,8 % (51 potilaalla 80 potilaasta), lähinnä koska paikallisten injektiokohdan reaktioiden (kaikki asteen 1 tai 2) ilmaantuvuus oli suurempi Phesgo-hoidon aikana. Vakavia haittavaikutuksia, vaikeusasteen 3 haittavaikutuksia ja hoidon keskeytyksiä haittojen vuoksi oli vähän (< 6%) ja yhtä paljon ennen siirtymisiä annosmuodosta toiseen (syklien 1–3 aikana) kuin siirtymisten jälkeen (syklien 4–6 aikana).
Vaikeusasteen 4 tai 5 haittavaikutuksia ei raportoitu.
Iäkkäät potilaat
Phesgo-valmisteen turvallisuudessa ei FEDERICA-tutkimuksessa yleisesti havaittu eroja ≥ 65‑vuotiaiden ja < 65-vuotiaiden potilaiden välillä.
Pertutsumabia koskeneissa kliinisissä pivotaalitutkimuksissa, joissa pertutsumabin ja trastutsumabin yhdistelmää annettiin laskimoon, ruokahalun heikentymistä, anemiaa, painon laskua, voimattomuutta, makuhäiriöitä, perifeeristä neuropatiaa, hypomagnesemiaa ja ripulia esiintyi ≥ 65-vuotiailla potilailla (n = 418) ≥ 5 % yleisemmin kuin < 65-vuotiailla potilailla (n = 2 926).
Iältään > 75-vuotiaista Phesgo-hoitoa tai laskimoon annettavia pertutsumabia ja trastutsumabia saaneista potilaista on vähän kliinisistä tutkimuksista saatuja tietoja. Valmisteen markkinoille tulon jälkeiset tiedot eivät osoita eroja pertutsumabin ja trastutsumabin yhdistelmän turvallisuudessa ≥ 65‑vuotiaille ja < 65-vuotiaille potilaille.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Suurin tutkittu Phesgo-annos on 1 200 mg pertutsumabia ja 600 mg trastutsumabia. Yliannoksen yhteydessä potilasta on tarkkailtava haittavaikutusten oireiden ja löydösten havaitsemiseksi sekä aloitettava sopiva oireenmukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, monoklonaaliset vasta-aineet, ATC-koodi: L01FY01
Vaikutusmekanismi
Phesgo sisältää pertutsumabia ja trastutsumabia, joista saadaan tämän lääkevalmisteen terapeuttinen vaikutus. Valmiste sisältää lisäksi vorhyaluronidaasi alfaa, joka on ihon alle yhdistelmävalmisteena annettavien aineiden dispersiota ja imeytymistä lisäävä entsyymi.
Pertutsumabi ja trastutsumabi ovat humanisoituja, rekombinantteja, monoklonaalisia IgG1:n vasta-aineita, joiden vaikutus kohdentuu ihmisen epidermaalisen kasvutekijän reseptoriin 2 (HER2). Kumpikin aine sitoutuu kilpailematta eri HER2‑aladomeeneihin, ja niillä on toisiaan täydentävä HER2-signaalinvälitystä häiritsevä vaikutusmekanismi:
- Pertutsumabin erityisenä vaikutuskohteena on ihmisen epidermaalisen kasvutekijän reseptori 2:n (HER2) solunulkoinen dimerisaatiodomeeni (aladomeeni II) ja siten se estää HER2:n ligandiriippuvaisen heterodimerisaation muiden HER-sukuisten reseptorien, kuten epidermaalisen kasvutekijän reseptorin (EGFR) HER3 ja HER4 kanssa. Tämän seurauksena pertutsumabi estää ligandin aktivoimaa solunsisäistä signaalinvälitystä kahden pääsiallisen signalointireitin kautta, joita ovat mitogeenien aktivoimat proteiinikinaasit (MAP) ja fosfoinositidi-3-kinaasi (PI3K). Näiden signalointireittien estyminen voi johtaa vastaavasti solun kasvun pysähtymiseen ja apoptoosiin.
- Trastutsumabi sitoutuu HER2‑proteiinin solunulkoisen domeenin aladomeeniin IV ja siten inhiboi ligandista riippumatonta HER2‑välitteistä proliferaatiota sekä HER2‑proteiinia yli-ilmentävien ihmisen syöpäsolujen eloonjääntisignaaleja.
Kumpikin aine toimii lisäksi vasta-aineriippuvaisen soluvälitteisen sytotoksisuuden (antibody‑dependent cell‑mediated cytotoxicity, ADCC) välittäjänä. In vitro-olosuhteissa sekä pertutsumabi että trastutsumabi käynnistää ADCC:n ensisijaisesti niitä syöpäsoluja vastaan, jotka yli-ilmentävät HER2:ta, verrattuna niihin syöpäsoluihin, jotka eivät yli-ilmennä HER2:ta.
Kliininen teho ja turvallisuus
Tässä kohdassa esitetään kliininen kokemus pertutsumabia ja trastutsumabia yhdistelmänä sisältävän Phesgo-valmisteen käytöstä sekä laskimoon annettavan pertutsumabin käytöstä yhdistelmänä trastutsumabin kanssa potilaille, jotka sairastavat HER2:ta yli-ilmentävää varhaisvaiheen tai metastasoitunutta rintasyöpää.
Kliininen kokemus Phesgo-yhdistelmävalmisteen käytöstä varhaisvaiheen HER2‑positiivisen rintasyövän hoidossa
Phesgo-valmisteesta saatu kliininen kokemus perustuu HER2-reseptoria yli-ilmentävää varhaisvaiheen rintasyöpää sairastavilla potilailla tehdyn vaiheen III kliinisen tutkimuksen (FEDERICA WO40324) ja vaiheen II kliinisen tutkimuksen (PHRANCESCA MO40628) tietoihin. HER2-reseptorin yli-ilmentyminen määritettiin keskuslaboratoriossa. HER2-reseptorin yli-ilmentymiseksi määriteltiin jäljempänä mainitussa tutkimuksessa immunohistokemiallinen värjäytymistulos 3+ (ICH 3+) tai ISH-testauksen monistumasuhdeluku ≥ 2.
FEDERICA (WO40324)
FEDERICA oli avoin, satunnaistettu monikeskustutkimus, jossa oli mukana 500 varhaisvaiheen HER2‑positiivista rintasyöpää sairastavaa potilasta, joiden rintasyöpä oli leikkaukseen soveltuva tai paikallisesti edennyt (mukaan lukien inflammatorinen), ja kasvaimen koko oli > 2 cm tai syöpä oli todettu levinneeksi imusolmukkeisiin neoadjuvantti- ja adjuvanttivaiheessa. Potilaat satunnaistettiin saamaan 8 sykliä neoadjuvanttisolunsalpaajahoitoa sekä samanaikaisesti 4 sykliä joko Phesgo-hoitoa tai laskimoon annettavia pertutsumabia ja trastutsumabia sykleissä 5–8. Tutkijat valitsivat potilaille yksilöllisesti toisen kahdesta neoadjuvanttisolunsalpaajahoidosta:
- 4 sykliä doksorubisiinia (60 mg/m2) ja syklofosfamidia (600 mg/m2) joka toinen viikko, ja sen jälkeen paklitakselia (80 mg/m2) viikoittain 12 viikon ajan
- 4 sykliä doksorubisiinia (60 mg/m2) ja syklofosfamidia (600 mg/m2) joka kolmas viikko, ja sen jälkeen neljä sykliä dosetakselia (75 mg/m2 ensimmäisessä syklissä, minkä jälkeen 100 mg/m2 seuraavissa sykleissä tutkijan harkinnan mukaan) kolmen viikon välein.
Leikkauksen jälkeen potilaat jatkoivat vielä 14 syklin ajan ennen leikkausta käyttämäänsä Phesgo-hoitoa tai laskimoon annettavia pertutsumabia ja trastutsumabia siten, että he saivat 18 sykliä HER2-kohdennettua hoitoa. Potilaat saivat paikallisen käytännön mukaisesti adjuvanttihoitona myös sädehoitoa ja hormonaalista hoitoa. Adjuvanttihoitojakson aikana laskimoon annettavasta trastutsumabista oli sallittua siirtyä ihon alle annettavan trastutsumabin käyttöön tutkijan harkinnan mukaan. HER2‑kohdennettua hoitoa annettiin kolmen viikon välein taulukon 3 mukaan seuraavasti:
Taulukko 3. Phesgo-valmisteen, laskimoon annettavan pertutsumabin, laskimoon annettavan trastutsumabin ja ihon alle annettavan trastutsumabin annostus ja annostelumuoto
| Lääkevalmisteet | Annostelumuoto | Annos | |
| Aloitusannos | Ylläpitoannos | ||
| Phesgo | Injektio ihon alle | 1 200 mg/600 mg | 600 mg/600 mg |
| Pertutsumabi | Infuusio laskimoon | 840 mg | 420 mg |
| Trastutsumabi | Infuusio laskimoon | 8 mg/kg | 6 mg/kg |
| Trastutsumabi | Injektio ihon alle | 600 mg | |
FEDERICA-tutkimuksessa oli tavoitteena osoittaa pertutsumabin vertailukelpoisuus (non‑inferiority) syklissä 7 (eli ennen syklin 8 annosta) pienimmän seerumin pertutsumabipitoisuuden (Ctrough) perusteella vertailtaessa Phesgo-valmisteen sisältämää pertutsumabia laskimoon annettavaan pertutsumabiin (ensisijainen päätetapahtuma). Keskeisiä toissijaisia päätetapahtumia ensisijaisen analyysin ajankohtana olivat trastutsumabin vertailukelpoisuus (non‑inferiority) syklissä 7 pienimmän seerumin trastutsumabipitoisuuden (Ctrough) perusteella vertailtaessa Phesgo-valmisteen sisältämää trastutsumabia laskimoon annettavaan trastutsumabiin, tehoa (paikallisesti arvioitu täydellinen patologinen hoitovaste, tpCR) koskevaa sekä turvallisuutta koskevia hoitotuloksia. Muut toissijaiset päätetapahtumat olivat pitkäaikainen turvallisuus ja kliiniset hoitotulokset (elossaoloaika ilman invasiivista tautia ja kokonaiselossaoloaika). Demografiset ominaisuudet olivat hyvin tasapainossa kahden hoitohaaran välillä, ja tutkimuksessa hoitoa saaneiden potilaiden iän mediaani oli 51 vuotta. Valtaosalla potilaista oli hormonireseptoripositiivinen tauti (61,2 %) ja/tai imusolmukkeisiin levinnyt tauti (57,6 %), ja valtaosa oli valkoihoisia (65,8 %).
Pertutsumabi- ja trastutsumabialtistusten vertailukelpoisuus (non‑inferiority) Phesgo-valmisteen käytössä, ks. kohta Farmakokinetiikka. Turvallisuusprofiili, ks. kohta Haittavaikutukset.
Toissijaisen tehon päätetapahtuman (paikallisesti arvioitu patologinen kokonaisvaste [tpCR], joksi määriteltiin, ettei rinnassa ja kainalossa ollut invasiivista tautia [ypT0/is, ypN0]) analyysi esitetään taulukossa 4. Taulukossa 4 esitetään myös tulokset elossaoloajasta ilman invasiivista tautia ja kokonaiselossaoloajasta (kliinisten tietojen keruun katkaisupäivämäärä 2. kesäkuuta 2023) tehdystä loppuanalyysista ja 51 kuukauden (mediaani) seurannasta.
Taulukko 4. Tehoa koskeva yhteenveto
Phesgo (n = 248) | Laskimoon annettava pertutsumabin ja trastutsumabin yhdistelmä (n = 252) | |
| Täydellinen patologinen hoitovaste (tpCR) | ||
| n | 248 | 252 |
| tpCR (ypT0/is, ypN0) | 148 (59,7 %) | 150 (59,5 %) |
| 95 %:n luottamusväli1 | (53,28; 65,84) | (53,18; 65,64) |
| Elossaoloaika ilman invasiivista tautia (iDFS) | ||
| n | 234 | 239 |
| Potilaita, joilla tapahtuma (%) | 26 (11,1 %) | 23 (9,6 %) |
| Osittamaton riskitiheyksien suhde (95 %:n luottamusväli) | 1,13 (0,64; 1,97) | |
| Kokonaiselossaoloaika (OS) | ||
| n | 248 | 252 |
| Potilaita, joilla tapahtuma (%) | 14 (5,6 %) | 12 (4,8 %) |
| Riskitiheyksien suhde2 (95 %:n luottamusväli) | 1,26 (0,58; 2,72) | |
1 Yhden näytteen 95 %:n luottamusväli Pearson–Clopperin binomijakaumalla
2 Analyysi ositettu keskeisten hormonireseptorien statuksen, kliinisen levinneisyysasteen ja solunsalpaajahoidon mukaan
PHRANCESCA (MO40628)
Tutkimuksessa MO40628 selvitettiin laskimoon annettavista pertutsumabista ja trastutsumabista ihon alle annettavaan Phesgo-hoitoon tai päinvastoin siirtymisen turvallisuutta (ks. kohta Haittavaikutukset). Tutkimuksen ensisijainen tavoite oli arvioida, käyttävätkö potilaat mieluummin laskimoon annettavaa vai ihon alle annettavaa hoitoa: 85 % potilaista käyttivät mielummin ihon alle annettavaa ja 13,8 % mielummin laskimoon annettavaa hoitoa, ja 1,2 % potilaista ei ollut ensisijaista vaihtoehtoa.
Tässä kahden hoitohaaran vaihtovuoroisessa tutkimuksessa oli mukana yhteensä 160 potilasta: 80 potilasta satunnaistettiin hoitohaaraan A (3 sykliä pertutsumabia ja trastutsumabia laskimoon, minkä jälkeen 3 sykliä Phesgo-hoitoa) ja 80 potilasta satunnaistettiin hoitohaaraan B (3 sykliä Phesgo-hoidolla, minkä jälkeen 3 sykliä pertutsumabia ja trastutsumabia laskimoon). Primaarianalyysissä mediaanialtistus pertutsumabin ja trastutsumabin liitännäishoidolle (sekä laskimonsisäisesti että subkutaanisesti) ) oli 11 sykliä (vaihteluväli 6 syklistä 15 sykliin).
Kliininen kokemus laskimoon annettavasta pertutsumabin ja trastutsumabin yhdistelmästä HER2‑positiivisen rintasyövän hoidossa
Kliininen kokemus laskimoon annettavan pertutsumabin käytöstä yhdistelmänä trastutsumabin kanssa perustuu tietoihin, jotka on saatu varhaisvaiheen rintasyöpää koskeneista kahdesta satunnaistetusta vaiheen II neoadjuvanttitutkimuksesta (toinen kontrolloitu), satunnaistamattomasta vaiheen II neoadjuvanttitutkimuksesta, satunnaistetusta vaiheen III adjuvanttitutkimuksesta ja satunnaistetusta vaiheen III tutkimuksesta sekä metastasoitunutta rintasyöpää koskeneesta yhden hoitohaaran vaiheen II tutkimuksesta. HER2-reseptorin yli-ilmentyminen määritettiin keskuslaboratoriossa. HER2-reseptorin yli-ilmentymiseksi määriteltiin edellä mainituissa tutkimuksissa immunohistokemiallinen värjäytymistulos 3+ (ICH 3+) tai ISH-testauksen monistumasuhdeluku ≥ 2.
Varhaisvaiheen rintasyöpä
Neoadjuvanttihoito
Paikallisesti edenneeseen ja inflammatoriseen rintasyöpään katsotaan neoadjuvanttihoidossa liittyvän suuri riski hormonireseptoristatuksesta riippumatta. Varhaisvaiheen rintasyövän riskiarviossa pitää ottaa huomioon kasvaimen koko, erilaistumisaste, hormonireseptoristatus ja etäpesäkkeet imusolmukkeissa.
Käyttö rintasyövän neoadjuvanttihoitoon perustuu patologisella kokonaisvasteella osoitettuun kokonaisvasteen paranemiseen sekä taudin etenemisvapaan ajan pitenemissuuntaukseen. Nämä eivät kuitenkaan varmista eivätkä mittaa tarkasti hyötyä pitkäaikaisen hoitotuloksen, kuten kokonaiselossaoloajan tai taudin etenemisvapaan ajan, suhteen.
NEOSPHERE (WO20697)
NEOSPHERE on pertutsumabilla toteutettu vaiheen II, satunnaistettu, kontrolloitu, monikansallinen monikeskustutkimus, jossa oli mukana 417 äskettäin diagnosoitua varhaisvaiheen, inflammatorista tai paikallisesti edennyttä HER2‑positiivista rintasyöpää (T2‑4d, primaarikasvaimen läpimitta > 2 cm) sairastavaa aikuista naispotilasta, jotka eivät olleet saaneet aiemmin trastutsumabi-, solunsalpaaja- tai sädehoitoa. Tutkimukseen ei otettu mukaan potilaita, joilla oli metastasoitunut rintasyöpä, rintasyöpä kummassakin rinnassa, kliinisesti merkittäviä sydämen riskitekijöitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) tai vasemman kammion ejektiofraktio oli < 55 %. Suurin osa potilaista oli alle 65-vuotiaita.
Potilaat satunnaistettiin ennen leikkausta neljän hoitosyklin ajaksi yhteen seuraavista neljästä neoadjuvanttihoitoryhmästä:
- trastutsumabi yhdistelmänä dosetakselin kanssa
- pertutsumabi yhdistelmänä trastutsumabin ja dosetakselin kanssa
- pertutsumabi yhdistelmänä trastutsumabin kanssa
- pertutsumabi yhdistelmänä dosetakselin kanssa.
Satunnaistaminen oli ositettu rintasyövän tyypin (leikattavissa oleva, paikallisesti edennyt tai inflammatorinen) ja estrogeeni (ER) tai progesteroni (PgR) positiivisuuden mukaan.
Pertutsumabia annettiin laskimoon aloitusannos 840 mg, minkä jälkeen annettiin 420 mg kolmen viikon välein. Trastutsumabia annettiin laskimoon aloitusannos 8 mg/kg, minkä jälkeen annettiin 6 mg/kg kolmen viikon välein. Dosetakselia annettiin laskimoon aloitusannos 75 mg/m2, minkä jälkeen annettiin 75 mg/m2 tai 100 mg/m2 (jos potilas sieti hoidon) 3 viikon välein. Kaikki potilaat saivat leikkauksen jälkeen 3 hoitosykliä 5‑fluorourasiilia (600 mg/m2), epirubisiinia (90 mg/m2) ja syklofosfamidia (600 mg/m2) (FEC-hoitoa) laskimoon kolmen viikon välein sekä trastutsumabia laskimoon kolmen viikon välein, kunnes hoitoa oli annettu vuoden ajan. Pelkästään pertutsumabin ja trastutsumabin yhdistelmää ennen leikkausta saaneet potilaat saivat leikkauksen jälkeen sekä FEC-hoitoa että dosetakselia.
Ensisijainen päätetapahtuma oli rinnan täydellinen patologinen hoitovaste (pCR, pathological complete response) (ypT0/is). Toissijaisia tehon päätetapahtumia olivat kliininen vasteluku, rintaa säästävien leikkausten osuus (vain T2–3-kasvaimet), taudista vapaa elossaolo (DFS) ja taudin etenemisvapaa aika (PFS). Muita eksploratiivisia pCR-lukuja oli mm. taudin leviäminen imusolmukkeisiin (ypT0/isN0 ja ypT0N0).
Demografiset ominaisuudet olivat hyvin tasapainossa (iän mediaani oli 49–50 vuotta, suurin osa [71 %] potilaista oli valkoihoisia), ja kaikki potilaat olivat naisia. Kaikkiaan 7 %:lla potilaista oli inflammatorinen rintasyöpä, 32 %:lla potilaista oli paikallisesti edennyt rintasyöpä ja 61 %:lla potilaista oli leikattavissa oleva rintasyöpä. Kussakin hoitoryhmässä noin puolella potilaista oli hormonireseptoripositiivinen tauti (määriteltiin ER-positiiviseksi ja/tai PgR-positiiviseksi).
Tehon tulokset on esitetty taulukossa 5. Pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa saaneilla potilailla havaittiin pCR-luvun (ypT0/is) tilastollisesti merkitsevä paraneminen verrattuna trastutsumabia ja dosetakselia saaneisiin potilaisiin (45,8 % vs 29 %, p‑arvo = 0,0141). Tulosten havaittiin olevan yhdenmukaisia pCR:n määritelmästä riippumatta. pCR-luvussa todetun eron katsottiin todennäköisesti muodostuvan kliinisesti merkittäväksi eroksi pitkän aikavälin hoitotuloksessa, mitä tuki myös myönteinen kehitys taudin etenemisvapaassa ajassa (PFS) (riskitiheyksien suhde 0,69; 95 %:n luottamusväli 0,34; 1,40) ja taudittomassa elossaoloajassa (DFS) (riskitiheyksien suhde 0,60; 95 %:n luottamusväli 0,28; 1,27).
pCR-luvut sekä pertutsumabihoidosta (pertutsumabi yhdistelmänä trastutsumabin ja dosetakselin kanssa verrattuna trastutsumabia ja dosetakselia saaneisiin potilaisiin) saatu hyöty olivat pienemmät siinä potilasjoukossa, joilla oli hormonireseptoripositiivisia kasvaimia (ero rintarauhasen pCR-luvussa 6 %), kuin potilasjoukossa, joilla oli hormonireseptorinegatiivisia kasvaimia (ero rintarauhasen pCR-luvussa 26,4 %).
pCR-luvut olivat samankaltaiset potilasjoukoissa, joilla oli leikattavissa oleva tai paikallisesti edennyt tauti. Inflammatorista rintasyöpää sairastavia potilaita oli varmojen päätelmien tekemiseksi liian vähän, mutta pCR-luku oli suurempi potilailla, jotka saivat pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa.
TRYPHAENA (BO22280)
TRYPHAENA on vaiheen II satunnaistettu, kliininen monikeskustutkimus, jossa oli mukana 225 paikallisesti edennyttä, leikattavissa olevaa tai inflammatorista HER2‑positiivista rintasyöpää (T2-4d; primaarikasvaimen läpimitta > 2 cm) sairastavaa aikuista naispotilasta, jotka eivät olleet aiemmin saaneet trastutsumabi-, solunsalpaaja- tai sädehoitoa. Tutkimukseen ei otettu mukaan potilaita, joilla oli metastasoitunut rintasyöpä, rintasyöpä kummassakin rinnassa, kliinisesti merkittäviä sydämen riskitekijöitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) tai vasemman kammion ejektiofraktio oli < 55 %. Suurin osa potilaista oli alle 65-vuotiaita. Potilaat satunnaistettiin ennen leikkausta yhteen seuraavista kolmesta neoadjuvanttihoitoryhmästä:
- 3 hoitosykliä FEC-hoitoa, minkä jälkeen 3 hoitosykliä dosetakselia, joista kaikki annettiin samanaikaisesti pertutsumabin ja trastutsumabin kanssa
- 3 hoitosykliä pelkästään FEC-hoitoa, minkä jälkeen 3 hoitosykliä dosetakselia, joka annettiin samanaikaisesti trastutsumabin ja pertutsumabin kanssa
- 6 hoitosykliä TCH-hoitoa (dosetakseli, karboplatiini ja trastutsumabi) yhdistelmänä pertutsumabin kanssa.
Satunnaistaminen oli ositettu rintasyövän tyypin (leikattavissa oleva, paikallisesti edennyt tai inflammatorinen) ja ER- ja/tai PgR-positiivisuuden perusteella.
Pertutsumabia annettiin laskimoon aloitusannos 840 mg, minkä jälkeen annettiin 420 mg kolmen viikon välein. Trastutsumabia annettiin laskimoon aloitusannos 8 mg/kg, minkä jälkeen annettiin 6 mg/kg kolmen viikon välein. FEC-hoitoa (5‑fluorourasiili [500 mg/m2], epirubisiini [100 mg/m2], syklofosfamidi [600 mg/m2]) annettiin laskimoon 3 hoitosykliä kolmen viikon välein. Dosetakselia annettiin infuusiona laskimoon aloitusannos 75 mg/m2 kolmen viikon välein, ja annos voitiin suurentaa tutkijalääkärin harkinnan mukaan annokseen 100 mg/m2, jos potilas sieti aloitusannoksen hyvin. Pertutsumabia yhdistelmänä TCH-hoidon kanssa saaneessa ryhmässä dosetakseli annettiin kuitenkin laskimoon annoksena 75 mg/m2 (annoksen suurentaminen ei ollut sallittua), ja karboplatiinia (AUC 6) annettiin laskimoon kolmen viikon välein. Kaikki potilaat saivat leikkauksen jälkeen trastutsumabia, kunnes hoitoa oli annettu vuoden ajan.
Tämän tutkimuksen ensisijainen päätetapahtuma oli sydämeen liittyvä turvallisuus tutkimuksen neoadjuvanttihoitojakson aikana. Toissijaisia tehon päätetapahtumia olivat rinnan pCR-luku (ypT0/is), tauditon elossaoloaika (DFS), taudin etenemisvapaa aika (PFS) ja kokonaiselossaoloaika (OS).
Demografiset ominaisuudet olivat hoitoryhmien välillä hyvin tasapainossa (iän mediaani oli 49–50 vuotta, suurin osa [77 %] potilaista oli valkoihoisia), ja kaikki potilaat olivat naisia. Kaikkiaan 6 %:lla potilaista oli inflammatorinen rintasyöpä, 25 %:lla potilaista oli paikallisesti edennyt rintasyöpä ja 69 %:lla potilaista oli leikattavissa oleva rintasyöpä. Kussakin hoitoryhmässä noin puolella potilaista oli ER-positiivinen ja/tai PgR-positiivinen tauti.
Kaikissa kolmessa hoitoryhmässä havaittiin suuret pCR-luvut verrattuna sellaisista samankaltaisista hoito-ohjelmista julkaistuihin tietoihin, joihin ei kuulunut pertutsumabia (ks. taulukko 5). Tulosten havaittiin olevan yhdenmukaisia käytetystä pCR:n määritelmästä riippumatta. pCR-luvut olivat pienemmät potilasjoukossa, jolla oli hormonireseptoripositiivisia kasvaimia (vaihteluväli 46,2–50 %), kuin potilasjoukossa, jolla oli hormonireseptorinegatiivisia kasvaimia (vaihteluväli 65–83,8 %).
pCR-luvut olivat samankaltaisia potilailla, joilla oli leikattavissa oleva tai paikallisesti edennyt tauti. Inflammatorista rintasyöpää sairastavia potilaita oli varmojen päätelmien tekemiseksi liian vähän.
Taulukko 5. NEOSPHERE (WO20697) ja TRYPHAENA (BO22280): Tehoa koskevat tiedot (Intent-to-treat-potilaat)
| NEOSPHERE (WO20697) | TRYPHAENA (BO22280) | ||||||
| Parametri | Trastu-tsumabi + dosetakseli N = 107 | Pertu-tsumabi + trastu-tsumabi + dosetakseli N = 107 | Pertu-tsumabi + trastu-tsumabi N = 107 | Pertu-tsumabi + dose-takseli N = 96 | Pertu-tsumabi + trastu-tsumabi + FEC-hoito◊ pertu-tsumabi + trastu-tsumabi + dosetakseli N = 73 | FEC-hoito◊ pertu-tsumabi + trastu-tsumabi + dosetakseli N = 75 | Pertu-tsumabi + TCH-hoito N = 77 |
rinnan pCR-luku (ypT0/is) n (%) [95 %:n luottamus-väli]1 | 31 (29 %) [20,6; 38,5] | 49 (45,8 %) [36,1; 55,7] | 18 (16,8 %) [10,3; 25,3] | 23 (24 %) [15,8; 33,7] | 45 (61,6 %) [49,5; 72,8] | 43 (57,3 %) [45,4; 68,7] | 51 (66,2 %) [54,6; 76,6] |
pCR-lukujen ero2 [95 %:n luottamus-väli]3 | +16,8 % [3,5; 30,1] | -12,2 % [-23,8; -0,5] | -21,8 % [-35,1; -8,5] | NA | NA | NA | |
| p‑arvo (CMH-testin Simesin korjaus)4 | 0,0141 (vs. trastu-tsumabi + dosetakseli) | 0,0198 (vs. trastu-tsumabi + dosetakseli) | 0,0030 (vs. pertu-tsumabi+ trastutsumabi + dosetakseli) | NA | NA | NA | |
rinnan ja imusolmuk-keen pCR-luku (ypT0/is N0) n (%) [95 %:n luottamus-väli] | 23 (21,5 %) [14,1; 30,5] | 42 (39,3 %) [30,3; 49,2] | 12 (11,2 %) [5,9; 18,8] | 17 (17,7 %) [10,7; 26,8] | 41 (56,2 %) [44,1; 67,8] | 41 (54,7 %) [42,7; 66,2] | 49 (63,6 %) [51,9; 74,3] |
ypT0 N0 n (%) [95 %:n luottamus-väli] | 13 (12,1 %) [6,6; 19,9] | 35 (32,7 %) [24; 42,5] | 6 (5,6 %) [2,1; 11,8] | 13 (13,2 %) [7,4; 22] | 37 (50,7 %) [38,7; 62,6] | 34 (45,3 %) [33,8; 57,3] | 40 (51,9 %) [40,3; 63,5] |
| Kliininen vaste5 | 79 (79,8 %) | 89 (88,1 %) | 69 (67,6 %) | 65 (71,4 %) | 67 (91,8 %) | 71 (94,7 %) | 69 (89,6 %) |
FEC-hoito: 5‑fluorourasiili, epirubisiini, syklofosfamidi; TCH-hoito: dosetakseli, karboplatiini ja trastutsumabi, CMH: Cochran–Mantel–Haenszel
1. Yhden näytteen 95 %:n luottamusväli Pearson–Clopperin binomijakaumalla.
2. Pertutsumabi+trastutsumabi+dosetakseli- ja pertutsumabi+trastutsumabihoitoa verrataan trastutsumabi+dosetakselihoitoon, kun taas pertutsumabi+dosetakselihoitoa verrataan pertutsumabi+trastutsumabi+dosetakselihoitoon.
3. Kahden vasteluvun eron likimääräinen 95 %:n luottamusväli Hauck–Andersonin menetelmällä.
4. p‑arvo Cochran–Mantel–Haenszelin testillä, johon on tehty Simesin monikerroinkorjaus.
5. Kliininen vaste tarkoittaa potilaita, joilla on neoadjuvanttihoitojakson aikana paras täydellinen tai osittainen kokonaisvaste (rinnan primaarimuutoksessa).
BERENICE (WO29217)
BERENICE on vaiheen II satunnaistamaton, avoin, monikansallinen monikeskustutkimus, joka tehtiin 401:llä HER2‑positiivista paikallisesti edennyttä inflammatorista tai varhaisvaiheen rintasyöpää sairastavalla potilaalla (joiden kasvainten läpimitta oli > 2 cm tai joiden tauti oli levinnyt imusolmukkeisiin).
Potilaista muodostettiin BERENICE-tutkimuksessa kaksi rinnakkaisryhmää. Potilaat, joille trastutsumabin ja antrasykliinin/taksaanipohjaisen solunsalpaajahoidon yhdistelmästä koostuvan neoadjuvanttihoidon katsottiin sopivan, kohdennettiin saamaan ennen leikkausta toista seuraavista kahdesta hoidosta:
- kohortti A: 4 hoitosykliä, joissa annettiin lyhyen antovälin doksorubisiini- ja syklofosfamidihoitoa kahden viikon välein, jonka jälkeen 4 hoitosykliä pertutsumabia yhdistelmänä trastutsumabin ja paklitakselin kanssa
- kohortti B: 4 hoitosykliä FEC-hoitoa, jonka jälkeen 4 hoitosykliä pertutsumabia yhdistelmänä trastutsumabin ja dosetakselin kanssa.
Kaikki potilaat saivat leikkauksen jälkeen pertutsumabia ja trastutsumabia laskimoon kolmen viikon välein, kunnes hoitoa oli annettu 1 vuoden ajan.
BERENICE-tutkimuksen ensisijainen päätetapahtuma on sydämeen liittyvä turvallisuus tutkimuksen neoadjuvanttijakson aikana. Sydämeen liittyvää turvallisuutta koskeva ensisijainen päätetapahtuma (eli NYHA-luokan III/IV vasemman kammion toimintahäiriöiden ilmaantuvuus ja sydämen vasemman kammion ejektiofraktion pieneneminen) oli yhdenmukainen neoadjuvanttihoidosta aiemmin saatujen tietojen kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Adjuvanttihoito
Adjuvanttihoidossa varhaisvaiheen HER2‑positiivista rintasyöpää sairastaviksi potilaiksi, joilla on suuri syövän uusiutumisriski, on APHINITY-tutkimuksen tietojen perusteella määritelty ne potilaat, joiden syöpä on levinnyt imusolmukkeisiin tai joilla on hormonireseptorinegatiivinen tauti.
APHINITY (BO25126)
APHINITY on satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen III monikeskustutkimus, jossa oli mukana 4804 varhaisvaiheen HER2‑positiivista rintasyöpää sairastavaa potilasta, joiden primaarikasvain oli poistettu leikkauksella ennen satunnaistamista. Potilaat satunnaistettiin sen jälkeen saamaan pertutsumabia tai lumelääkettä yhdistelmänä adjuvanttihoitona annetun trastutsumabin ja solunsalpaajahoidon kanssa. Tutkijat valitsivat potilaille yksilöllisesti yhden seuraavista antrasykliiniä sisältävistä tai sisältämättömistä solunsalpaajahoidoista:
- 3 tai 4 hoitosykliä FEC-hoitoa tai 5‑fluorourasiilia, doksorubisiinia ja syklofosfamidia (FAC), jonka jälkeen 3 tai 4 hoitosykliä dosetakselia tai 12 hoitosyklin ajan viikoittain paklitakselia
- 4 hoitosykliä AC-hoitoa tai epirubisiinia ja syklofosfamidia (EC-hoitoa), jonka jälkeen 3 tai 4 hoitosykliä dosetakselia tai 12 hoitosyklin ajan viikoittain paklitakselia
- 6 hoitosykliä dosetakselia yhdistelmänä karboplatiinin kanssa.
Pertutsumabi ja trastutsumabi annettiin laskimoon (ks. kohta Annostus ja antotapa) kolmen viikon välein ensimmäisen taksaania sisältävän hoitosyklin 1. hoitopäivästä lähtien yhteensä 52 viikon ajan (enintään 18 hoitosykliä) tai kunnes tauti uusiutui, potilas peruutti suostumuksensa tutkimukseen osallistumisesta tai potilaalle ilmaantui haittavaikutuksia, jotka eivät olleet hallittavissa. Potilaille annettiin tavanomaisia annoksia 5‑fluorourasiilia, epirubisiinia, doksorubisiinia, syklofosfamidia, dosetakselia, paklitakselia ja karboplatiinia. Solunsalpaajahoidon päätyttyä potilaat saivat sädehoitoa ja/tai hormonihoitoa paikallisen kliinisen hoitokäytännön mukaan.
Tutkimuksen ensisijainen päätetapahtuma oli elossaolo ilman invasiivista tautia (invasive disease-free survival, IDFS), joksi määriteltiin aika satunnaistamisesta invasiivisen rintasyövän paikalliseen tai alueelliseen ensimmäiseen uusiutumiseen samassa rinnassa, uusiutuminen etäpesäkkeenä, invasiivinen rintasyöpä toisessa rinnassa tai kuolema mistä tahansa syystä. Toissijaisia tehon päätetapahtumia olivat elossaolo ilman invasiivista tautia, mukaan lukien uusi muu primaarisyöpä, kokonaiselossaoloaika (OS), tauditon elossaoloaika (DFS), taudin uusiutumisvapaa aika (RFI) ja etäpesäkkeiden uusiutumisvapaa aika (DRFI).
Demografiset tiedot olivat kahden hoitohaaran kesken hyvin tasapainossa. Iän mediaani oli 51 vuotta, ja yli 99 % potilaista oli naisia. Valtaosalla potilaista oli imusolmukkeisiin levinnyt (63 %) ja/tai hormonireseptoripositiivinen tauti (64 %), ja valtaosa oli valkoihoisia (71 %).
45,4 kuukauden (mediaani) seurannan jälkeen APHINITY-tutkimuksessa osoitettiin taudin uusiutumisen tai kuoleman riskin vähentyneen 19 % (riskisuhde [HR] = 0,81; 95 %:n luottamusväli 0,66; 1,00; p‑arvo 0,0446) pertutsumabia saaneilla potilailla verrattuna potilaisiin, jotka satunnaistettiin saamaan lumehoitoa.
Yhteenveto tutkimuksen APHINITY tehoa koskevista tuloksista esitetään taulukossa 6 ja kuvassa 1.
Taulukko 6 Kokonaisteho: hoitoaikeen mukainen (ITT) potilasjoukko
Pertutsumabi + trastutsumabi + solunsalpaajahoito N = 2400 | Lumelääke + trastutsumabi + solunsalpaajahoito N = 2404 | |
| Ensisijainen päätetapahtuma | ||
| Elossaolo ilman invasiivista tautia | ||
| Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 171 (7,1 %) | 210 (8,7 %) |
| Riskisuhde (HR) (95 %:n luottamusväli) | 0,81 (0,66; 1,00) | |
| p‑arvo (Log‑Rank-testi, ositettu1) | 0,0446 | |
| 3 vuoden jaksoja ilman tapahtumia2 [95 %:n luottamusväli] | 94,1 (93,1; 95) | 93,2 (92,2; 94,3) |
| Toissijaiset päätetapahtumat1 | ||
| Elossaolo ilman invasiivista tautia, mukaan lukien uusi muu primaarikasvain | ||
| Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 189 (7,9 %) | 230 (9,6 %) |
| Riskisuhde (HR) (95 %:n luottamusväli) | 0,82 (0,68; 0,99) | |
| p‑arvo (Log‑Rank-testi, ositettu1) | 0,0430 | |
| 3 vuoden jaksoja ilman tapahtumia2 [95 %:n luottamusväli] | 93,5 (92,5; 94,5) | 92,5 (91,4; 93,6) |
| Tauditon elossaolo | ||
| Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 192 (8 %) | 236 (9,8 %) |
| Riskisuhde (HR) (95 %:n luottamusväli) | 0,81 (0,67; 0,98) | |
| p‑arvo (Log‑Rank-testi, ositettu1) | 0,0327 | |
| 3 vuoden jaksoja ilman tapahtumia2 [95 %:n luottamusväli] | 93,4 (92,4; 94,4) | 92,3 (91,2; 93,4) |
| Kokonaiselossaolo3 | ||
| Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 80 (3,3 %) | 89 (3,7 %) |
| Riskisuhde (HR) (95 %:n luottamusväli) | 0,89 (0,66; 1,21) | |
| p‑arvo (Log‑Rank-testi, ositettu1) | 0,4673 | |
| 3 vuoden jaksoja ilman tapahtumia2 [95 %:n luottamusväli] | 97,7 (97; 98,3) | 97,7 (97,1; 98,3) |
Lyhenteet (taulukko 6): HR:riskisuhde; CI: luottamusväli
1. Kaikki analyysit ositettiin imusolmukkeiden statuksen, tutkimussuunnitelman version, keskeisten hormonireseptorien statuksen ja adjuvanttisolunsalpaajahoidon mukaan.
2. 3 vuoden jaksot ilman tapahtumia saatiin Kaplan–Meierin estimaateista.
3. Ensimmäisen välianalyysin tiedot.
Kuva 1 Elossaolo ilman invasiivista tautia: Kaplan-Meier-käyrä
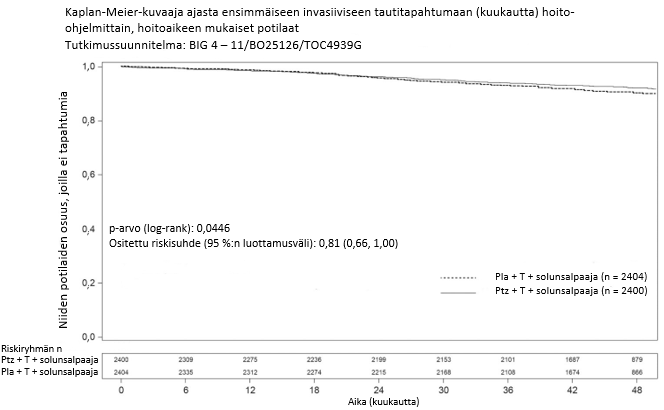
Pla = lumelääke; Ptz = pertutsumabi; T = trastutsumabi.
Elossaolon ilman invasiivista tautia estimaatti 4 vuoden hoidon kohdalla oli pertutsumabihoitoa saaneessa ryhmässä 92,3 % verrattuna 90,6 %:iin lumehoitoa saaneessa ryhmässä. Seurannan mediaani arvion ajankohtana oli 45,4 kuukautta.
Alaryhmäanalyysin tulokset
Pertutsumabin hyödyt olivat primaarianalyysin ajankohtana selkeämmät niiden potilaiden alaryhmissä, joilla uusiutumisriski oli suuri: imusolmukkeisiin levinnyt tai hormonireseptorinegatiivinen tauti (ks. taulukko 7).
Taulukko 7 Alaryhmien tehoa koskevat tulokset imusolmukkeiden statuksen ja hormonireseptorien statuksen mukaan1
| Potilasjoukko | Elossaolon ilman invasiivista tautia koskevien tapahtumien lkm /N yhteensä (%) | Osittamaton riskisuhde (95 %:n luottamusväli) | |
| Pertutsumabi + trastutsumabi + solunsalpaajahoito | Lumelääke + trastutsumabi + solunsalpaajahoito | ||
| Imusolmukkeiden status | |||
| Positiivinen | 139/1503 (9,2 %) | 181/1502 (12,1 %) | 0,77 (0,62; 0,96) |
| Negatiivinen | 32/897 (3,6 %) | 29/902 (3,2 %) | 1,13 (0,68; 1,86) |
| Hormonireseptorien status | |||
| Negatiivinen | 71/864 (8,2 %) | 91/858 (10,6 %) | 0,76 (0,56; 1,04) |
| Positiivinen | 100/1536 (6,5 %) | 119/1546 (7,7 %) | 0,86 (0,66; 1,13) |
1 Ennalta määritellyt alaryhmäanalyysit ilman korjausta useiden vertailujen suhteen, jonka vuoksi tulokset katsotaan deskriptiivisiksi.
Elossaolon ilman invasiivista tautia estimaatit alaryhmässä, jossa potilaiden tauti oli levinnyt imusolmukkeisiin, olivat 3 vuoden hoidon kohdalla pertutsumabihoitoa saaneilla potilailla 92 % verrattuna 90,2 %:iin lumehoitoa saaneilla potilailla ja 4 vuoden hoidon kohdalla pertutsumabihoitoa saaneilla potilailla 89,9 % verrattuna 86,7 %:iin lumehoitoa saaneilla potilailla. Elossaolon ilman invasiivista tautia estimaatit alaryhmässä, jossa potilaiden tauti ei ollut levinnyt imusolmukkeisiin, olivat 3 vuoden hoidon kohdalla pertutsumabihoitoa saaneilla potilailla 97,5 % verrattuna 98,4 %:iin lumehoitoa saaneilla potilailla ja 4 vuoden hoidon kohdalla pertutsumabihoitoa saaneilla potilailla 96,2 % verrattuna 96,7 %:iin lumehoitoa saaneilla potilailla. Elossaolon ilman invasiivista tautia estimaatit hormonireseptorinegatiivisten potilaiden alaryhmässä olivat 3 vuoden hoidon kohdalla pertutsumabihoitoa saaneilla potilailla 92,8 % verrattuna 91,2 %:iin lumehoitoa saaneilla potilailla ja 4 vuoden hoidon kohdalla pertutsumabihoitoa saaneilla potilailla 91 % verrattuna 88,7 %:iin lumehoitoa saaneilla potilailla. Elossaolon ilman invasiivista tautia estimaatit hormonireseptoripositiivisten potilaiden alaryhmässä olivat 3 vuoden hoidon kohdalla pertutsumabihoitoa saaneilla potilailla 94,8 % verrattuna 94,4 %:iin lumehoitoa saaneilla potilailla ja 4 vuoden hoidon kohdalla pertutsumabihoitoa saaneilla potilailla 93 % verrattuna 91,6 %:iin lumehoitoa saaneilla potilailla.
Potilaiden raportoimat hoitotulokset
Toissijaisiin päätetapahtumiin kuului potilaiden raportoiman yleisen terveydentilan, rooli- ja fyysisen toimintakyvyn sekä hoidon oireiden arviointi EORTC QLQ-C30- ja EORTC QLQ-BR23 kyselyiden avulla. Potilaiden raportoimien hoitotulosten analyysissä 10 pisteen ero katsottiin kliinisesti merkittäväksi.
Potilaiden fyysistä toimintakykyä, yleistä terveydentilaa ja ripulia koskevissa pisteissä todettiin kummassakin hoitohaarassa kliinisesti merkittävä muutos solunsalpaajahoidon aikana. Fyysistä toimintakykyä kuvaava pisteiden lasku kyseisenä ajankohtana lähtötilanteeseen verrattuna oli pertutsumabihoitohaarassa keskimäärin ‑10,7 (95 %:n luottamusväli ‑11,4; ‑10) ja lumehoitohaarassa ‑10,6 (95 %:n luottamusväli ‑11,4; ‑9,9); yleistä terveydentilaa kuvaavien pisteiden lasku oli pertutsumabihoitohaarassa ‑11,2 (95 %:n luottamusväli ‑12,2; ‑10,2) ja lumehoitohaarassa ‑10,2 (95 %:n luottamusväli ‑11,1; ‑9,2). Ripulin oireita kuvaavat pisteet suurenivat pertutsumabihoitohaarassa tasolle +22,3 (95 %:n luottamusväli 21; 23,6) ja lumehoitohaarassa tasolle +9,2 (95 %:n luottamusväli 8,2; 10,2).
Kummankin hoitohaaran fyysistä toimintakykyä ja yleistä terveydentilaa kuvaavat pisteet palautuivat tämän jälkeen kohdennetun hoidon aikana lähtötilanteen tasolle. Ripulin oireet palautuivat pertutsumabihoitohaarassa lähtötilanteen tasolle HER2-hoidon jälkeen. Pertutsumabin lisääminen trastutsumabin ja solunsalpaajahoidon yhdistelmään ei vaikuttanut tutkimuksen aikana potilaiden yleiseen roolitoimintakykyyn.
Metastasoitunut rintasyöpä
Pertutsumabi yhdistelmänä trastutsumabin ja dosetakselin kanssa
CLEOPATRA (WO20698) on satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen III kliininen monikeskustutkimus, joka toteutettiin 808 metastasoitunutta tai paikallisesti uusiutunutta leikkaushoitoon soveltumatonta HER2‑positiivista rintasyöpää sairastavalla potilaalla. Tutkimukseen ei otettu mukaan potilaita, joilla oli kliinisesti merkityksellisiä sydämeen liittyviä riskitekijöitä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Koska tutkimukseen ei otettu mukaan potilaita, joilla oli etäpesäkkeitä aivoissa, pertutsumabin vaikutuksesta etäpesäkkeisiin aivoissa ei ole tietoja saatavissa. Vain hyvin rajoitettu määrä tietoa on saatavilla potilaista, joilla on leikkaushoitoon soveltumaton, paikallisesti uusiutunut tauti. Potilaat satunnaistettiin suhteessa 1:1 saamaan hoitona lumelääkkeen, trastutsumabin ja dosetakselin yhdistelmää tai pertutsumabin, trastutsumabin ja dosetakselin yhdistelmää.
Pertutsumabi ja trastutsumabi annettiin vakioannoksina kolmen viikon välein. Potilas sai pertutsumabi- ja trastutsumabihoitoa, kunnes tauti eteni, potilas perui suostumuksensa tutkimukseen osallistumiseen tai hänelle ilmaantui haittavaikutuksia, jotka eivät olleet hoidettavissa. Dosetakselihoito aloitettiin annoksella 75 mg/m2 infuusiona laskimoon kolmen viikon välein vähintään 6 hoitosyklin ajan. Dosetakseliannos voitiin suurentaa tutkijan harkinnan mukaan annokseen 100 mg/m2, jos potilas sieti alkuannoksen hyvin.
Tutkimuksen ensisijainen päätetapahtuma oli taudin etenemisvapaa aika (PFS), jonka arvioi riippumaton arviointilautakunta (independent review facility, IRF), ja joka määriteltiin ajaksi satunnaistamispäivästä taudin etenemiseen tai (mistä tahansa syystä tapahtuneeseen) kuolemaan, jos potilas kuoli 18 viikon kuluessa kasvaimen viimeisimmästä tutkimuskerrasta. Toissijaisia tehon päätetapahtumia olivat kokonaiselossaoloaika (OS), (tutkijan arvioima) taudin etenemisvapaa aika (PFS), objektiivinen vasteluku (ORR), vasteen kesto ja aika oireiden etenemiseen FACT B Quality of Life ‑elämänlaatukyselyn perusteella.
Kummassakin hoitoryhmässä noin puolella potilaista oli hormonireseptoripositiivinen tauti (määriteltiin estrogeenireseptoripositiiviseksi [ER‑positiiviseksi] ja/tai progesteronireseptoripositiiviseksi [PgR-positiiviseksi]) ja kummassakin hoitoryhmässä noin puolet potilaista oli saanut aiemmin adjuvantti- tai neoadjuvanttihoitoa. Suurin osa näistä potilaista oli saanut aiemmin antrasykliiniä ja 11 % kaikista potilaista oli saanut aiemmin trastutsumabia. Yhteensä 43 % kummankin hoitoryhmän potilaista oli saanut aiemmin sädehoitoa. Potilaiden vasemman kammion ejektiofraktion mediaani ennen hoitoa oli 65 % kummassakin ryhmässä (vaihteluväli 50–88 %).
Hoidon tehon tulokset CLEOPATRA-tutkimuksessa on esitetty yhteenvetona taulukossa 8. Riippumattoman arviointilautakunnan arvioima taudin etenemisvapaa aika oli pertutsumabihoitoa saaneessa ryhmässä tilastollisesti merkitsevästi pidempi kuin lumelääkehoitoa saaneessa ryhmässä. Tulokset tutkijan arvioimasta taudin etenemisvapaasta ajasta olivat samankaltaiset kuin riippumattoman arviointilautakunnan arvioimat taudin etenemisvapaan ajan tulokset.
Taulukko 8. Yhteenveto hoidon tehosta CLEOPATRA-tutkimuksessa
| Parametri | Lumelääke + trastutsumabi + dosetakseli n = 406 | Pertutsumabi + trastutsumabi + dosetakseli n = 402 | Riskisuhde (HR) (95 %:n luottamusväli) | p‑arvo |
Taudin etenemisvapaa aika (riippumaton arvio) ensisijainen päätetapahtuma* Niiden potilaiden lukumäärä, joilla tapahtuma esiintyi Kuukautta (mediaani) | 242 (59 %) 12,4 | 191 (47,5 %) 18,5 | 0,62 (0,51; 0,75) | < 0,0001 |
Kokonaiselossaoloaika - toissijainen päätetapahtuma** Niiden potilaiden lukumäärä, joilla tapahtuma esiintyi Kuukautta (mediaani) | 221 (54,4 %) 40,8 | 168 (41,8 %) 56,5 | 0,68 (0,56; 0,84) | 0,0002 |
Objektiivinen vasteluku (ORR)^ - toissijainen päätetapahtuma Niiden potilaiden lukumäärä, joilla tauti oli mitattavissa Vasteen saaneiden lukumäärä*** Objektiivisen vasteluvun 95 %:n luottamusväli Täydellinen vaste Osittainen vaste Stabiili tauti Etenevä tauti | 336 233 (69,3 %) [64,1; 74,2] 14 (4,2 %) 219 (65,2 %) 70 (20,8 %) 28 (8,3 %) | 343 275 (80,2 %) [75,6; 84,3] 19 (5,5 %) 256 (74,6 %) 50 (14,6 %) 13 (3,8 %) | Objektiivisen vasteluvun ero: 10,8 % (4,2; 17,5) | 0,0011 |
Vasteen kesto †^ n = Viikkoa (mediaani) Mediaanin 95 %:n luottamusväli | 233 54,1 [46; 64] | 275 87,6 [71; 106] |
*Taudin etenemisvapaan ajan ensisijainen analyysi, tiedonkeruun katkaisupäivä 13. toukokuuta 2011.
** Tapahtumaperusteinen lopullinen kokonaiselossaoloaika, tiedonkeruun katkaisupäivä 11. helmikuuta 2014.
*** Potilaan paras kokonaisvaste RECIST-luokituksen mukainen vahvistettu täydellinen tai osittainen vaste.
† Arvioitu potilailla, joiden paras kokonaisvaste on täydellinen vaste tai osittainen vaste.
^ Objektiivinen vasteluku ja vasteen kestoaika perustuvat riippumattoman arviointilautakunnan tekemään arvioon kasvaimesta.
Tulokset olivat yhdenmukaisia ennalta määritellyissä potilaiden alaryhmissä, mukaan lukien alaryhmissä, jotka perustuivat ositustekijöinä käytettyihin maantieteelliseen alueeseen ja aiempaan adjuvantti-/neoadjuvanttihoitoon tai de novo metastasoituneeseen rintasyöpään (ks. kuva 2). Eksploratiivisessa post hoc ‑analyysissa todettiin, että trastutsumabia aiemmin saaneiden potilaiden (n = 88) riippumattoman arviointilautakunnan arvioima taudin etenemisvapaan ajan riskisuhde oli 0,62 (95 %:n luottamusväli 0,35, 1,07) verrattuna riskisuhteeseen 0,60 (95 %:n luottamusväli 0,43, 0,83) potilailla, jotka olivat saaneet aiempaa hoitoa, joka ei sisältänyt trastutsumabia (n = 288).
Kuva 2. Riippumattoman arviointilautakunnan arvioima taudin etenemisvapaa aika potilasryhmittäin
Kokonaiselossaolon tapahtumaperusteinen loppuanalyysi tehtiin, kun 389 potilasta oli kuollut (221 lumehoitoa saaneessa ryhmässä ja 168 pertutsumabihoitoa saaneessa ryhmässä). Pertutsumabihoitoa saaneen ryhmän kokonaiselossaolon tilastollisesti merkitsevä hyöty, joka oli aikaisemmin havaittu kokonaiselossaolon välianalyysissa (vuoden kuluttua ensisijaisen tehon analyysista), oli säilynyt (riskisuhde 0,68, p = 0,0002 log‑rank-testi). Ajan mediaani kuolemaan oli lumehoitoa saaneessa ryhmässä 40,8 kuukautta ja pertutsumabihoitoa saaneessa ryhmässä 56,5 kuukautta (ks. taulukko 8, kuva 3).
Tutkimuksen lopussa, kun 515 potilasta oli kuollut (280 lumehoitoa saaneessa ryhmässä ja 235 pertutsumabihoitoa saaneessa ryhmässä), tehtiin kokonaiselinajan deskriptiivinen analyysi. Analyysi osoitti, että pertutsumabihoitoa saaneen ryhmän kokonaiselinajan tilastollisesti merkitsevä hyöty oli ajan mittaan 99 kuukauden (mediaani) seuranta-ajan jälkeen säilynyt (riskisuhde 0,69, log‑rank-testin p < 0,0001; aika [mediaani] kuolemaan 40,8 kuukautta [lumehoitoa saanut ryhmä] verrattuna 57,1 kuukauteen [pertutsumabihoitoa saanut ryhmä]). 8 vuoden kohdalla elinajan estimaatit (landmark) olivat pertutsumabihoitoa saaneessa ryhmässä 37 % ja lumehoitoa saaneessa ryhmässä 23 %.
Kuva 3. Tapahtumaperusteisen kokonaiselinajan Kaplan-Meier-käyrä.
T = trastutsumabi; D = dosetakseli.
Näiden kahden hoitoryhmän välillä ei havaittu tilastollisesti merkitseviä eroja terveyteen liittyvässä elämänlaadussa, jota arvioitiin FACT-B TOI-PFB -pisteytyksellä.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Phesgo-valmisteen käytöstä rintasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Pertutsumabin ensisijaisen päätetapahtuman eli pertutsumabin Ctrough-arvon hoitosyklissä 7 (eli ennen hoitosykliä 8) farmakokineettiset tulokset olivat vertailukelpoiset (non‑inferiority) verrattaessa Phesgo-valmisteen sisältämää pertutsumabia (geometrinen keskiarvo 88,7 mikrog/ml) laskimoon annettavaan pertutsumabiin (geometrinen keskiarvo 72,4 mikrog/ml), jolloin geometristen keskiarvojen suhde oli 1,22 (90 %:n luottamusväli: 1,14–1,31). Phesgo-valmisteen sisältämän pertutsumabin ja laskimoon annettavan pertutsumabin suhteen geometrisen keskiarvon kaksitahoisen 90 %:n luottamusvälin alaraja oli 1,14 eli suurempi kuin ennalta määritelty marginaali 0,8.
Toissijaisen päätetapahtuman eli trastutsumabin Ctrough-arvon hoitosyklissä 7 (eli ennen hoitosykliä 8) farmakokineettiset tulokset olivat vertailukelpoiset (non‑inferiority) verrattaessa Phesgo-valmisteen sisältämää trastutsumabia (geometrinen keskiarvo 57,5 mikrog/ml) laskimoon annettavaan trastutsumabiin (geometrinen keskiarvo 43,2 mikrog/ml), jolloin geometristen keskiarvojen suhde oli 1,33 (90 %:n luottamusväli: 1,24–1,43).
Imeytyminen
Phesgo-valmisteen sisältämän pertutsumabin huippupitoisuuden mediaani (Cmax) seerumissa oli 157 mikrog/ml ja aika huippupitoisuuden saavuttamiseen (Tmax) oli 3,82 vuorokautta. Populaatiofarmakokineettiseen analyysiin perustuva absoluuttinen biologinen hyötyosuus oli 0,712 ja ensimmäisen asteen imeytymisnopeus (Ka) on 0,348 (1/vrk).
Phesgo-valmisteen sisältämän trastutsumabin huippupitoisuuden mediaani (Cmax) seerumissa oli 114 mikrog/ml ja aika huippupitoisuuden saavuttamiseen (Tmax) oli ja 3,84 vuorokautta. Populaatiofarmakokineettiseen analyysiin perustuva absoluuttinen biologinen hyötyosuus oli 0,771 ja ensimmäisen asteen imeytymisnopeus (Ka) on 0,404 (1/vrk).
Jakautuminen
Populaatiofarmakokineettisen analyysin perusteella Phesgo-valmisteen sisältämän pertutsumabin jakautumistilavuus keskustilaan (Vc) tyypillisellä potilaalla oli 2,77 litraa.
Populaatiofarmakokineettisen analyysin perusteella ihon alle annetun trastutsumabin jakautumistilavuus keskustilaan (Vc) tyypillisellä potilaalla oli 2,91 litraa.
Biotransformaatio
Phesgo-valmisteen metaboliaa ei ole tutkittu suoranaisesti. Vasta-aineet poistuvat elimistöstä pääasiassa kataboloitumalla.
Eliminaatio
Populaatiofarmakokineettisen analyysin perusteella Phesgo-valmisteen sisältämän pertutsumabin puhdistuma oli 0,163 l/vrk ja eliminaation puoliintumisaika (t1/2) oli noin 24,3 vuorokautta.
Populaatiofarmakokineettisen analyysin perusteella Phesgo-valmisteen sisältämän trastutsumabin puhdistuma oli 0,111 l/vrk. Arvion mukaan trastutsumabipitoisuuden < 1 mikrog/ml (noin 3 % potilasjoukon ennustetusta Cmin,ss-arvosta tai noin 97 % poistumasta) on saavuttanut 7 kuukautta viimeisen annoksen jälkeen vähintään 95 % potilaista.
Iäkkäät
Phesgo-valmisteen farmakokinetiikkaa ei ole tutkittu iäkkäillä potilailla.
Phesgo-valmisteen sisältämästä pertutsumabista ja laskimoon annetusta pertutsumabista tehdyissä populaatiofarmakokineettisissä analyyseissä iän ei todettu vaikuttavan merkittävästi pertutsumabin farmakokinetiikkaan.
Ihon alle tai laskimoon annetusta trastutsumabista tehdyissä populaatiofarmakokineettisissä analyyseissä iän ei todettu vaikuttavan trastutsumabin ominaisuuksiin.
Munuaisten vajaatoiminta
Phesgo-valmisteen farmakokinetiikkaa ei ole tutkittu munuaisten vajaatoimintaa sairastavilla potilailla.
Phesgo-valmisteen sisältämästä pertutsumabista ja laskimoon annetusta pertutsumabista tehtyjen populaatiofarmakokineettisten analyysien perusteella osoitettiin, että munuaisten vajaatoiminta ei vaikuta pertutsumabialtistukseen. Populaatiofarmakokineettisissä analyyseissa oli kuitenkin vain vähän tietoja vaikeaa munuaisten vajaatoimintaa sairastavista potilaista.
Ihon alle tai laskimoon annetusta trastutsumabista tehdyissä populaatiofarmakokineettisissä analyyseissa munuaisten vajaatoiminnan ei havaittu vaikuttavan trastutsumabin poistumiseen elimistöstä.
Maksan vajaatoiminta
Maksan vajaatoimintaa sairastavilla potilailla ei ole tehty varsinaisia farmakokineettisiä tutkimuksia. Phesgo-valmisteen sisältämän pertutsumabin populaatiofarmakokineettisten analyysien perusteella lievän maksan vajaatoiminnan ei osoitettu vaikuttavan pertutsumabialtistukseen. Populaatiofarmakokineettisissä analyyseissä oli kuitenkin vain vähän tietoja lievää maksan vajaatoimintaa sairastavista potilaista. IgG1-molekyylit, kuten pertutsumabi ja trastutsumabi, kataboloituvat laajasti jakautuneiden proteolyyttisten entsyymien välityksellä, mikä ei rajoitu vain maksakudokseen. Maksan toiminnan muutokset eivät siten todennäköisesti vaikuta pertutsumabin ja trastutsumabin eliminaatioon.
Prekliiniset tiedot turvallisuudesta
Ihon alle annettavan pertutsumabin, trastutsumabin ja vorhyaluronidaasi alfan yhdistelmällä ei ole tehty varsinaisia tutkimuksia.
Pertutsumabi
Eläimillä ei ole tehty erityisiä hedelmällisyystutkimuksia pertutsumabin vaikutusten selvittämiseksi. Cynomolgus-apinoilla tehtyjen toistuvan altistuksen aiheuttamaa toksisuutta selvittäneiden tutkimusten perusteella ei voida tehdä täsmällisiä johtopäätöksiä urosten lisääntymisjärjestelmään kohdistuvista haittavaikutuksista.
Lisääntymistoksisuustutkimuksia on tehty tiineillä cynomolgus-apinoilla (gestaatiopäivinä 19–50), jolloin aloitusannos oli 30–150 mg/kg, minkä jälkeen annettiin kerran kahdessa viikossa annos 10−100 mg/kg. Näistä annoksista aiheutunut kliinisesti oleellinen altistus oli huippupitoisuuden (Cmax) perusteella 2,5–20 kertaa suurempi kuin ihmiselle ihon alle annettavaksi suositellulla annoksella. Pertutsumabin anto laskimoon gestaatiopäivinä 19–50 (organogeneesin aikana) aiheutti alkiotoksisuutta, johon liittyi annosriippuvaista alkio- ja sikiökuolemien lisääntymistä gestaatiopäivien 25–70 välillä. Alkioista tai sikiöistä kuoli 33 %, kun pertutsumabia annettiin kerran kahdessa viikossa tiineille naarasapinoille annoksilla 10 mg/kg, ja vastaavasti 50 % kuoli annoksilla 30 mg/kg kerran kahdessa viikossa ja 85 % annoksilla 100 mg/kg kerran kahdessa viikossa (annokset olivat huippupitoisuuden [Cmax] perusteella 4–35 kertaa suurempia kuin ihmiselle suositeltu annos). Kun gestaatiopäivänä 100 tehtiin keisarileikkaus, kaikissa pertutsumabiannosryhmissä todettiin sikiöveden niukkuutta, vähentynyt keuhkojen ja munuaisten suhteellinen paino sekä mikroskooppista näyttöä munuaisten hypoplasiasta, joka oli yhdenmukaista munuaisten kehityksen viivästymisen kanssa. Sikiöveden niukkuudesta aiheutuneeksi katsotun sikiön kasvun heikkenemisen lisäksi havaittiin keuhkojen hypoplasiaa (yhdellä kuudesta 30 mg/kg ryhmässä ja yhdellä kahdesta 100 mg/kg ryhmässä), kammioväliseinän vikoja (yhdellä kuudesta 30 mg/kg ryhmässä), ohut kammioväliseinä (yhdellä kahdesta 100 mg/kg ryhmässä) ja vähäisiä luustovikoja (ulkoisia, kolmella kuudesta 30 mg/kg ryhmässä). Kaikkien hoitoryhmien jälkeläisillä raportoitiin pertutsumabialtistukseksi 29–40 % emolla seerumissa gestaatiopäivänä 100 todetusta pitoisuudesta.
Cynomolgus-apinat (binding species) sietivät hyvin ihon alle annettavan pertutsumabin (250 mg/kg/viikko neljän viikon ajan) ja laskimoon annettavan pertutsumabin (enintään 150 mg/kg viikossa enintään 26 viikon ajan), lukuun ottamatta ripulin ilmaantumista. Laskimoon annettavan pertutsumabin annoksilla 15 mg/kg ja sitä suuremmilla annoksilla havaittiin ajoittaista lievää hoitoon liittynyttä ripulia. Osalla apinoista valmisteen pitkäaikainen (26 viikoittaista annosta) anto johti vaikea-asteiseen sekretoriseen ripuliin. Ripuli hoidettiin (lukuun ottamatta yhtä, annoksia 50 mg/kg/annos saanutta eläintä, joka oli lopetettava) tukihoidolla, kuten laskimoon annettavalla nestekorvaushoidolla.
Trastutsumabi
Cynomolgus-apinoilla tehdyissä lisääntymistutkimuksissa, joissa käytettiin jopa 16-kertaisia laskimoon annettavia annoksia verrattuna ihmisille Phesgo-valmisteessa annettavaan trastutsumabin ylläpitoannokseen (600 mg), ei havaittu lääkkeen vaikuttavan haitallisesti apinoiden fertiliteettiin eikä aiheuttavan haittaa niiden sikiöille. Trastutsumabin todettiin siirtyvän istukan läpi tiineyden alku- (päivinä 20–50) ja loppuvaiheessa (päivinä 120–150).
Jopa kuusi kuukautta kestäneissä toksisuustutkimuksissa ei havaittu akuuttia eikä toistuvista annoksista johtuvaa toksisuutta. Lisääntymiseen liittyvää toksisuutta ei myöskään todettu tutkimuksissa, joissa selvitettiin teratogeenisuutta, vaikutuksia naaraan fertiliteettiin ja tiineyden loppuvaiheen toksisuutta/aineen siirtymistä istukan läpi. Trastutsumabi ei ole genotoksinen. Toksisuutta ei myöskään todettu trehaloosia koskeneessa tutkimuksessa (tärkeä apuaine valmisteessa).
Trastutsumabin karsinogeenista potentiaalia tai vaikutuksia urosten hedelmällisyyteen ei ole selvitetty pitkäaikaistutkimuksissa eläimillä.
Cynomolgus-apinoilla tehdyssä tutkimuksessa, jossa laskimoon annettu trastutsumabiannos oli 16‑kertainen verrattuna ihmisille Phesgo-valmisteessa annettavaan trastsutsumabin 600 mg:n ylläpitoannokseen, havaittiin trastutsumabin erittyvän synnytyksen jälkeen imettävien apinoiden maitoon. Altistumisen trastutsumabille kohdussa ja apinanpoikasten seerumissa havaittu trastutsumabi eivät vaikuttaneet haitallisesti eläinten kasvuun tai kehitykseen niiden syntymästä yhden kuukauden ikään saakka.
Hyaluronidaasi
Hyaluronidaasia esiintyy useimmissa ihmiselimistön kudoksissa. Prekliinisten konventionaalisten toistuvan altistuksen toksisuutta selvittäneiden tutkimusten, myös turvallisuusfarmakologisten päätetapahtumien, tulokset viittaavat siihen, ettei rekombinantista ihmisen hyaluronidaasista ole erityistä haittaa ihmiselle. Lisääntymistoksisuutta koskevat toksikologiset tutkimukset vorhyaluronidaasi alfalla osoittivat, että suuri systeeminen altistus on hiirelle alkio- ja sikiötoksinen, mutta teratogeenisuutta ei todettu.
Ihon alle annettavalla trastutsumabilla tehtiin kaniineilla kerta-annostutkimus ja Cynomolgus-apinoilla tehtiin 13 viikon ajan toistetuilla annoksilla toteutettu toksisuustutkimus. Kaniineilla tehty tutkimus toteutettiin erityisesti paikallisen siedettävyyden selvittämiseksi. 13 viikon pituinen tutkimus tehtiin sen varmistamiseksi, että antoreitin muutos ihonalaiseen antoon ja apuaineen, vorhyaluronidaasi alfan, käyttö eivät vaikuta trastutsumabin turvallisuuteen liittyviin ominaisuuksiin. Ihon alle annettava trastutsumabin lääkemuoto oli paikallisesti ja systeemisesti hyvin siedetty.
Farmaseuttiset tiedot
Apuaineet
Vorhyaluronidaasi alfa
L‑histidiini
L‑histidiinihydrokloridimonohydraatti
α,α‑trehaloosidihydraatti
Sakkaroosi
L‑metioniini
Polysorbaatti 20 (E432)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Phesgo on käyttövalmis liuos eikä sitä saa sekoittaa muiden valmisteiden kanssa eikä laimentaa muihin valmisteisiin.
Kestoaika
18 kuukautta.
Injektiopullosta ruiskuun siirretty lääkevalmiste on fysikaalisesti ja kemiallisesti stabiili 28 päivää 2 °C – 8 °C:n lämpötilassa valolta suojattuna ja 24 tuntia (kumulatiivinen aika injektiopullossa ja ruiskussa) vallitsevassa lämpötilassa (enintään 30 °C) hajapäivänvalossa.
Phesgo ei sisällä antimikrobista säilytysainetta, joten lääkevalmiste on mikrobiologiselta kannalta käytettävä heti. Jos lääkevalmistetta ei käytetä heti, ovat käytönaikaiset säilytysajat ja ‑olosuhteet käyttäjän vastuulla eivätkä saisi tavallisesti ylittää 24 tuntia 2 °C – 8 °C:ssa, ellei ruiskua ole valmisteltu kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Avatun lääkevalmisteen säilytys, ks. kohdat Kestoaika ja Käyttö- ja käsittelyohjeet.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
PHESGO injektioneste, liuos
600/600 mg (L:ei) 1 kpl (5684,22 €)
1200/600 mg (L:ei) 1 kpl (9253,79 €)
PF-selosteen tieto
Phesgo 600 mg/600 mg injektioneste, liuos
Yksi 15 ml:n, borosilikaattilasinen, tyypin I injektiopullo, jossa on fluororesiinikalvolla laminoitu kumitulppa. Injektiopullo sisältää 10 ml liuosta (600 mg pertutsumabia ja 600 mg trastutsumabia). Tulppa on sinetöity alumiinikorkilla, ja sitä suojaa oranssi, muovinen, irti napsautettava (flip‑off) korkki.
Phesgo 1 200 mg/600 mg injektioneste, liuos
Yksi 20 ml:n, borosilikaattilasinen, tyypin I injektiopullo, jossa on fluororesiinikalvolla laminoitu kumitulppa. Injektiopullo sisältää 15 ml liuosta (1 200 mg pertutsumabia ja 600 mg trastutsumabia). Tulppa on sinetöity alumiinikorkilla, ja sitä suojaa vihreä, muovinen, irti napsautettava (flip‑off) korkki.
Valmisteen kuvaus:
Kirkas tai hieman maitomainen, väritön tai hieman ruskehtava liuos, jonka pH on 5,2–5,8, osmolaliteetti on 270–370 mosm/kg (vahvuus 1200 mg/600 mg) ja 275–375 mosm/kg (vahvuus (600 mg/600 mg).
Käyttö- ja käsittelyohjeet
Phesgo on tarkistettava silmämääräisesti ennen antoa, ettei siinä ole hiukkasia eikä värimuutoksia havaittavissa. Jos injektiopullossa havaitaan hiukkasia tai värimuutoksia, se on hävitettävä paikallisten ohjeistojen mukaisesti.
Injektiopulloa ei saa ravistaa.
Phesgo-valmisteen injektiopullosta vetämiseen ja injektion antoon ihon alle tarvitaan ruisku, siirtoneula ja injektioneula. Phesgo voidaan antaa injektiona käyttämällä hypodermista injektioneulaa, jonka koko on 25G–27G ja pituus on 3/8"(10 mm) – 5/8"(16 mm). Phesgo on yhteensopiva ruostumattoman teräksen, polypropeenin, polykarbonaatin, polyeteenin, polyuretaanin, polyvinyylikloridin ja fluoratun eteenipolypropeenin kanssa.
Phesgo ei sisällä antimikrobista säilytysainetta, joten lääkevalmiste on mikrobiologiselta kannalta käytettävä heti. Jos valmistetta ei käytetä heti, käyttökuntoon saattamisen tulee tapahtua kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa. Kun liuos on siirretty ruiskuun, siirtoneulan tilalle suositellaan vaihtamaan ruiskun suojatulppa, jotta vältetään liuoksen kuivuminen ruiskun sisälle ja lääkevalmisteen laadun vaarantuminen. Merkitse ruisku kiinnittämällä siihen irti vedettävä tarra. Ruiskuun on kiinnitettävä juuri ennen lääkkeen antoa hypoderminen injektioneula, minkä jälkeen tilavuudeksi säädetään 15 ml käytettäessä Phesgo 1 200 mg/600 mg ‑valmistetta tai 10 ml käytettäessä Phesgo 600 mg/600 mg ‑valmistetta.
Phesgo on tarkoitettu yhtä käyttökertaa varten. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
PHESGO injektioneste, liuos
600/600 mg 1 kpl
1200/600 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FY01
Valmisteyhteenvedon muuttamispäivämäärä
22.09.2025
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com