
SHINGRIX injektiokuiva-aine ja suspensio, suspensiota varten
Vaikuttavat aineet ja niiden määrät
Käyttökuntoon saattamisen jälkeen yksi annos (0,5 ml) sisältää:
Varicella zoster -viruksen1 glykoproteiini E -antigeenia2,3 50 mikrogrammaa
1 Varicella zoster ‑virus = VZV
2 Mukana oleva AS01B-adjuvantti sisältää seuraavia aineita:
Quillaja saponaria Molina -kasviuute, fraktio 21 (QS-21) 50 mikrogrammaa
3-O-desasyyli-4’-monofosforyylilipidi A (MPL) Salmonella minnesota -mikrobista 50 mikrogrammaa
3 Glykoproteiini E (gE) on valmistettu kiinanhamsterin munasarjasoluissa (CHO) yhdistelmä-DNA-tekniikalla.
Apuaine(et), joiden vaikutus tunnetaan
Yksi annos sisältää 0,08 mg polysorbaatti 80:tä (E433) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektiokuiva-aine ja suspensio suspensiota varten.
Kliiniset tiedot
Käyttöaiheet
Shingrix on tarkoitettu vyöruusun (herpes zoster, HZ) ja postherpeettisen neuralgian (PHN) ehkäisyyn
- 50 vuotta täyttäneillä aikuisilla;
- 18 vuotta täyttäneillä aikuisilla, joilla on suurentunut vyöruusun riski.
Shingrix-rokotteen käytön tulee perustua virallisiin suosituksiin.
Annostus ja antotapa
Annostus
Perusrokotussarjaan kuuluu kaksi 0,5 ml:n annosta: aloitusannos ja 2 kuukauden kuluttua annettava toinen annos.
Jos rokotussarjaan on välttämätöntä saada joustavuutta, toinen annos voidaan antaa 2–6 kuukauden kuluttua ensimmäisestä annoksesta (ks. kohta Farmakodynamiikka).
Jos jokin sairaus tai hoito aiheuttaa tai saattaa aiheuttaa potilaalle immuunivajavuuden tai immunosuppression ja potilas hyötyisi lyhyemmästä rokotussarjasta, toinen annos voidaan antaa 1–2 kuukauden kuluttua ensimmäisestä annoksesta (ks. kohta Farmakodynamiikka).
Perusrokotussarjan jälkeisten tehosteannosten tarvetta ei ole selvitetty (ks. kohta Farmakodynamiikka).
Shingrix voidaan antaa saman rokotussarjan mukaan henkilöille, jotka ovat aiemmin saaneet rokotuksen elävällä heikennetyllä HZ -rokotteella (ks. kohta Farmakodynamiikka).
Shingrix ei ole tarkoitettu primaari-infektion (vesirokon eli varicella zosterin) ehkäisyyn.
Pediatriset potilaat
Shingrix-rokotteen turvallisuutta ja tehoa lasten ja nuorten hoidossa ei ole varmistettu.
Tietoja ei ole saatavilla.
Antotapa
Vain injektiona lihakseen, mieluiten olkavarren hartialihakseen.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttaville aineille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Ennen rokottamista
Kuten kaikkia pistettäviä rokotteita annettaessa, saatavilla on aina oltava asianmukainen hoito- ja seurantavalmius siltä varalta, että rokotteen annon jälkeen kehittyy anafylaktinen reaktio.
Kuten muidenkin rokotteiden kohdalla, Shingrix-rokotusta on lykättävä, mikäli potilaalla on vaikea akuutti kuumetauti. Vähäinen infektio kuten nuhakuume ei kuitenkaan edellytä rokotuksen lykkäämistä.
Kuten muidenkaan rokotteiden yhteydessä, ei kaikille rokotetuille välttämättä kehity suojaavaa immuunivastetta.
Rokote on tarkoitettu vain estohoitoon eikä jo kehittyneen kliinisen taudin hoitoon.
Shingrix-rokotetta ei saa antaa suoneen eikä ihon sisään.
Ihon alle anto ei ole suositeltavaa.
Valmisteen anto virheellisesti ihon alle voi johtaa ohimenevien paikallisreaktioiden lisääntymiseen.
Shingrix on annettava varoen, jos potilaalla on trombosytopenia tai mikä tahansa hyytymishäiriö. Näillä potilailla voi esiintyä verenvuotoa, kun valmiste on annettu lihakseen.
Pyörtymistä voi esiintyä minkä tahansa rokotuksen jälkeen tai jopa ennen rokotusta. Kyseessä on psykogeeninen reaktio neulanpistoon. Tähän voi liittyä monia neurologisia löydöksiä kuten ohimeneviä näköhäiriöitä, parestesiaa ja toonis-kloonisia raajaliikkeitä potilaan toipuessa. On tärkeää ryhtyä varotoimiin pyörtymisestä johtuvien vammojen ehkäisemiseksi.
Saatavilla ei ole turvallisuus-, immunogeenisuus- eikä tehotietoa, joka tukisi Shingrix-rokotteen annoksen korvaamista toisen vyöruusurokotteen annoksella.
Apuaineet, joiden vaikutus tunnetaan
Polysorbaatti 80
Tämä lääkevalmiste sisältää 0,08 mg polysorbaatti 80:tä per annos. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Kalium
Tämä lääkevalmiste sisältää kaliumia alle 1 mmol (39 mg) per annos, eli sen voidaan sanoa olevan ”kaliumiton”.
Yhteisvaikutukset
Shingrix voidaan antaa samanaikaisesti kausi-influenssarokotteen (inaktivoitu, adjuvantiton), 23-valenttisen pneumokokkipolysakkaridirokotteen (PPV23), 13-valenttisen pneumokokkikonjugaattirokotteen (PCV13), kurkkumätä-jäykkäkouristus-soluton hinkuyskä-rokotteen (matala antigeenipitoisuus) (dTap), koronavirus (COVID-19) mRNA-rokotteen tai RSV-rokotteen (respiratory syncytial virus; rekombinantti, adjuvanttia sisältävä) kanssa. Rokotteet on annettava eri pistoskohtiin.
Haittavaikutuksista kuumetta ja vilunväristyksiä esiintyi yleisemmin, kun PPV23-rokote annosteltiin samanaikaisesti Shingrix-rokotteen kanssa (16 % ja 21 %) verrattuna siihen, kun Shingrix-rokotetta annettiin yksinään (7 % molemmille haittavaikutuksille).
Yksinään annetun Shingrix-valmisteen yhteydessä hyvin yleisinä ilmoitettujen systeemisten haittavaikutusten (ks. taulukko 1; esim. lihaskipu 32,9 %, uupumus 32,2 % ja päänsärky 26,3 %) ja melko harvinaisena haittavaikutuksena ilmoitetun nivelkivun esiintymistiheydet suurenivat ≥ 50-vuotiailla aikuisilla, kun Shingrix annettiin samanaikaisesti COVID-19 mRNA -rokotteen kanssa (lihaskipu 64 %, uupumus 51,7 %, päänsärky 39 %, nivelkipu 30,3 %).
Shingrix-rokotteen samanaikaista käyttöä muiden kuin edellä lueteltujen rokotteiden kanssa ei ole tutkittu.
Raskaus ja imetys
Raskaus
Shingrix-valmisteen käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria haitallisia vaikutuksia raskauteen, alkion/sikiön kehitykseen, synnytykseen tai postnataaliseen kehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Varmuuden vuoksi Shingrix-valmisteen käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Äidille annettavan Shingrix-valmisteen vaikutusta imetettäviin vauvoihin ei ole tutkittu.
Ei tiedetä, erittyykö Shingrix ihmisillä äidinmaitoon.
Hedelmällisyys
Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria vaikutuksia urosten eikä naaraiden hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Shingrix-valmisteella voi olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn 2–3 päivää rokotuksen jälkeen. Uupumusta ja yleistä sairaudentunnetta voi ilmetä annon jälkeen (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin ilmoitettuja haittavaikutuksia 50 vuotta täyttäneillä aikuisilla olivat pistoskohdan kipu (yhteensä 68,1 %/annos; vaikeita tapauksia 3,8 %/annos), lihaskipu (yhteensä 32,9 %/annos; vaikeita tapauksia 2,9 %/annos), uupumus (yhteensä 32,2 %/annos; vaikeita tapauksia 3,0 %/annos) ja päänsärky (yhteensä 26,3 %/annos; vaikeita tapauksia 1,9 %/annos). Nämä reaktiot eivät useimmiten olleet pitkäkestoisia (mediaanikesto 2–3 vrk). Vaikeiksi ilmoitetut reaktiot kestivät 1–2 vrk.
Turvallisuusprofiili 18 vuotta täyttäneillä immuunipuutteisilla (sairauden tai hoidon aiheuttama immuunivajavuus tai immunosuppressio) aikuisilla vastasi 50 vuotta täyttäneiden aikuisten profiilia. Tietoja on rajallisesti 18–49-vuotiaista ei-immuunipuutteisista aikuisista, joilla vyöruusun riski on suurentunut.
Joidenkin haittavaikutusten ilmaantuvuus oli kaiken kaikkiaan suurempi nuoremmissa ikäryhmissä:
- Tutkimukset 18 vuotta täyttäneillä immuunipuutteisilla aikuisilla (yhdistetty analyysi): injektiokohdan kivun, uupumuksen, lihaskivun, päänsäryn, vilunväreiden ja kuumeen ilmaantuvuus oli 18–49-vuotiailla aikuisilla suurempi kuin 50 vuotta täyttäneillä.
- Tutkimukset 50 vuotta täyttäneillä aikuisilla (yhdistetty analyysi): lihaskivun, uupumuksen, päänsäryn, vilunväreiden, kuumeen ja ruoansulatuskanavan oireiden ilmaantuvuus oli 50–69-vuotiailla aikuisilla suurempi kuin 70 vuotta täyttäneillä.
Haittavaikutustaulukko
Jäljempänä esitettävä turvallisuusprofiili perustuu yhdistettyyn analyysiin 5 887:stä iältään 50–69-vuotiaasta aikuispotilaasta ja 8 758:sta 70 vuotta täyttäneestä aikuispotilaasta, jotka osallistuivat lumekontrolloituihin kliinisiin tutkimuksiin. Näistä 14 645 aikuisesta 7 408 otettiin pitkäaikaisseurantaa koskeneeseen jatkotutkimukseen. Seuranta kesti noin 11 vuotta rokotuksen jälkeen.
Kliinisissä tutkimuksissa määritetty 18 vuotta täyttäneiden immuunipuutteisten aikuisten (1 587 tutkittavaa) turvallisuusprofiili vastaa jäljempänä olevan taulukon 1 tietoja.
Markkinoille saattamisen jälkeisen valvonnan aikana raportoidut haittavaikutukset on myös taulukoitu alle.
Ilmoitetut haittavaikutukset on lueteltu seuraavien yleisyysluokkien mukaisesti:
Hyvin yleinen (≥ 1/10)
Yleinen (≥ 1/100, < 1/10)
Melko harvinainen (≥ 1/1 000, < 1/100)
Harvinainen (≥ 1/10 000, < 1/1 000)
Hyvin harvinainen (< 1/10 000)
Haittavaikutusten esiintyvyyden vakavuusaste on esitetty alenevassa järjestyksessä.
Taulukko 1: Haittavaikutukset
| Elinjärjestelmä1 | Yleisyys | Haittavaikutukset |
| Veri ja imukudos | Melko harvinainen | lymfadenopatia |
| Immuunijärjestelmä | Harvinainen | yliherkkyysreaktiot, mukaan lukien ihottuma, urtikaria ja angioedeema2 |
| Hermosto | Hyvin yleinen | päänsärky |
| Hyvin harvinainen | Guillain–Barrén oireyhtymä3 | |
| Ruoansulatuselimistö | Hyvin yleinen | ruoansulatuskanavan oireet (mm. pahoinvointi, oksentelu, ripuli ja/tai vatsakipu) |
| Luusto, lihakset ja sidekudos | Hyvin yleinen | lihaskipu |
| Melko harvinainen | nivelkipu | |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | pistoskohdan reaktiot (kuten kipu, punoitus, turvotus), uupumus, vilunväristykset, kuume |
| Yleinen | pistoskohdan kutina, yleinen sairaudentunne | |
| Melko harvinainen | pistoskohdan kovettuminen |
1 MedDRA (Medical Dictionary for Regulatory Activities) ‑termien mukaisesti
2Spontaanisti raportoidut haittavaikutukset
3 Katso ‘valikoitujen haittavaikutusten kuvaus’
Valikoitujen haittavaikutusten kuvaus
Guillain–Barrén oireyhtymän riskiä koskeneet markkinoilletulon jälkeiset havainnointitutkimukset
Yhdysvalloissa toteutettiin ≥ 65-vuotiailla kaksi keskenään samankaltaista markkinoilletulon jälkeistä havainnointitutkimusta. Näissä tutkimuksissa Guillain–Barrén oireyhtymän riski oli suurentunut 42 vuorokauden seurannassa minkä tahansa Shingrix-annoksen jälkeen (arvio 3–7 ylimääräistä tapausta miljoonaa annettua rokoteannosta kohden). Jatkoanalyyseissa todettiin, että Guillain–Barrén oireyhtymän riski oli suurentunut ensimmäisen Shingrix-annoksen jälkeen (arvio 6–12 ylimääräistä tapausta miljoonaa annettua rokoteannosta kohden) mutta ei toisen annoksen jälkeen.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksia ei ole raportoitu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: rokotteet, varicella zoster -rokotteet, ATC-koodi: J07BK03
Vaikutusmekanismi
Shingrix-valmisteessa VZV:n spesifinen antigeeni (gE) on yhdistetty adjuvanttiin (AS01B). Näin pyritään indusoimaan antigeenispesifinen soluvälitteinen ja humoraalinen immuunivaste henkilöillä, jotka ovat entuudestaan immuuneja VZV:lle.
Ei-kliiniset tiedot osoittavat, että AS01B aktivoi paikallisesti ja ohimenevästi synnynnäistä immuniteettia spesifisten molekyylitason reittien kautta. Tämä edistää gE-peräisiä antigeeneja esittelevien solujen rekrytointia ja aktivoitumista imusolmukkeessa, jonne imuneste laskee, mikä taas johtaa gE-spesifisten CD4+-T-solujen ja vasta-aineiden muodostukseen. AS01B-adjuvantin tehostevaikutus perustuu liposomimuodossa olevan MPL:n ja QS-21:n väliseen vuorovaikutukseen.
Shingrix-valmisteen kliininen teho
Teho vyöruusua (herpes zoster) ja postherpeettistä neuralgiaa (PHN) vastaan
Kahdessa vaiheen III lumekontrolloidussa, havainnoijasokkoutetussa Shingrix-valmisteen tehotutkimuksessa 50 vuotta täyttäneille aikuisille annettiin 2 annosta, joiden antoväli oli 2 kuukautta:
- ZOE-50 (Zoster-006): koko rokotettu kohortti (TVC) käsitti 15 405 aikuista, joiden ikä oli ≥ 50 vuotta ja jotka saivat vähintään yhden annoksen joko Shingrix-valmistetta (N = 7 695) tai lumerokotetta (N = 7 710).
- ZOE-70 (Zoster-022): koko rokotettu kohortti (TVC) käsitti 13 900 aikuista, joiden ikä oli ≥ 70 vuotta ja jotka saivat vähintään yhden annoksen joko Shingrix-valmistetta (N = 6 950) tai lumerokotetta (N = 6 950).
Näitä tutkimuksia ei suunniteltu osoittamaan valmisteen tehoa hauraiden henkilöiden alaryhmissä kuten potilailla, joilla on useita oheissairauksia. Tällaisia tutkittavia ei kuitenkaan suljettu pois kyseisistä tutkimuksista.
Kahdessa vaiheen III lumekontrolloidussa, havainnoijasokkoutetussa Shingrix-valmisteen tehotutkimuksessa 18 vuotta täyttäneille immuunipuutteisille aikuisille annettiin 2 annosta, joiden antoväli oli 1–2 kuukautta:
- Zoster-002: koko rokotettu kohortti (TVC) käsitti 1 846 autologisen veren kantasolusiirron saajaa, jotka saivat vähintään yhden annoksen joko Shingrix-valmistetta (N = 922) tai lumerokotetta (N = 924) 50–70 vrk kantasolusiirron jälkeen. Tutkittavista 21,3% (Shingrix) ja 20,5% (lumelääke) saivat vähintään yhtä immunosuppressiivista (IS) lääkehoitoa (vähintään yhden päivän ajan) veren kantasolusiirron jälkeen, jopa 30 päivän ajan toisesta annoksesta (TVC). Tutkittavista 53,1%:lla (Shingrix) ja 53,4%:lla (lumelääke) oli perussairautena multippeli myelooma (MM) ja 46,9%:lla (Shingrix) ja 46,6%:lla (lumelääke) jokin muu diagnoosi.
- Zoster-039: koko rokotettu kohortti (TVC) käsitti 562 tutkittavaa, joilla oli hematologinen maligniteetti ja jotka saivat vähintään yhden annoksen joko Shingrix-valmistetta (N = 283) tai lumerokotetta (N = 279) syöpähoitojakson aikana (37 %) tai koko syöpähoitojakson päättymisen jälkeen (63 %). Tutkittavista 70,7%:lla (Shingrix) ja 71,3%:lla (lumelääke) oli perussairautena multippeli myelooma (MM) tai jokin muu sairaus, 14,5%:lla (Shingrix) ja 14,0%:lla (placebo) non-Hodgin-B-solulymfooma (NHBLC) ja 14,8%:lla (Shingrix) ja 14,7%:lla (placebo) krooninen lymfaattinen leukemia (CLL).
Näitä tutkimuksia ei suunniteltu arvioimaan samanaikaisen IS lääkehoidon tai tiettyjen IS lääkehoitojen vaikutusta rokotteen tehoon. Useimmat rokotettavat eivät saaneet IS lääkehoitoa rokotushetkellä (ks. tiedot yllä). Tutkituissa populaatioissa ei käytetty kaikkia IS hoitotyyppejä.
Vyöruusun ja postherpeettisen neuralgian ilmaantuvuutta sekä rokotteen tehoa arvioitiin muokatussa koko rokotetussa kohortissa (modified Total Vaccinated Cohort, mTVC) eli kohortissa, josta suljettiin pois aikuiset, jotka eivät saaneet toista rokoteannosta tai joilla todettiin vahvistetusti vyöruusu yhden kuukauden kuluessa toisesta annoksesta.
Shingrix pienensi merkitsevästi vyöruusun ilmaantuvuutta verrattuna lumerokotteeseen:
- 50 vuotta täyttäneillä aikuisilla (ZOE-50): 6 tapausta vs. 210 tapausta;
- 70 vuotta täyttäneillä aikuisilla (ZOE-50- ja ZOE-70-tutkimusten yhdistetyt tiedot): 25 tapausta vs. 284 tapausta;
- 18 vuotta täyttäneillä aikuisilla, jotka olivat saaneet autologisen veren kantasolusiirron (Zoster-002): 49 tapausta vs. 135 tapausta;
- 18 vuotta täyttäneillä aikuisilla, joilla oli hematologinen maligniteetti (Zoster-039): 2 tapausta vs. 14 tapausta. Rokotteen teho laskettiin jälkianalyysissä.
Tulokset rokotteen tehosta vyöruusua vastaan esitetään taulukossa 2.
Taulukko 2: Shingrix-valmisteen teho vyöruusua vastaan (muokattu koko rokotettu kohortti)
Ikä (vuotta) | Shingrix | Lume | Rokotteen teho (%) [95 % lv] | ||||
| Arviointikelpoisia tutkittavia | Vyöruusutapauksia | Ilmaantuvuus / 1 000 henkilövuotta | Arviointikelpoisia tutkittavia | Vyöruusutapauksia | Ilmaantuvuus / 1 000 henkilövuotta | ||
| ZOE-50* | |||||||
| ≥ 50 | 7 344 | 6 | 0,3 | 7 415 | 210 | 9,1 | 97,2 [93,7; 99,0] |
| 50–59 | 3 492 | 3 | 0,3 | 3 525 | 87 | 7,8 | 96,6 [89,6; 99,4] |
| ≥ 60 | 3 852 | 3 | 0,2 | 3 890 | 123 | 10,2 | 97,6 [92,7; 99,6] |
| 60–69 | 2 141 | 2 | 0,3 | 2 166 | 75 | 10,8 | 97,4 [90,1; 99,7] |
| ZOE-50- ja ZOE-70-tutkimusten yhdistetyt tiedot** | |||||||
| ≥ 70 | 8 250 | 25 | 0,8 | 8 346 | 284 | 9,3 | 91,3 [86,8; 94,5] |
| 70–79 | 6 468 | 19 | 0,8 | 6 554 | 216 | 8,9 | 91,3 [86,0; 94,9] |
| ≥ 80 | 1 782 | 6 | 1,0 | 1 792 | 68 | 11,1 | 91,4 [80,2; 97,0] |
| Zoster-002*** (autologisen veren kantasolusiirron saaneet potilaat#) | |||||||
| ≥ 18 | 870 | 49 | 30,0 | 851 | 135 | 94,3 | 68,2 [55,5; 77,6] |
| 18–49 | 213 | 9 | 21,5 | 212 | 29 | 76,0 | 71,8 [38,7; 88,3] |
| ≥ 50 | 657 | 40 | 33,0 | 639 | 106 | 100,9 | 67,3 [52,6; 77,9] |
| Zoster-039 (potilaat, joilla oli hematologisia maligniteetteja#) | |||||||
| ≥ 18 | 259 | 2 | 8,5 | 256 | 14 | 66,2 | 87,2**** [44,2; 98,6] |
lv luottamusväli
* Seuranta-ajan mediaanikesto 3,1 vuotta
** Seuranta-ajan mediaanikesto 4,0 vuotta
70 vuotta täyttäneitä potilaita koskevat tiedot perustuvat ZOE-50- ja ZOE-70-tutkimusten ennalta määriteltyihin yhdistettyihin analyyseihin (mTV-kohortti), sillä nämä analyysit tarjoavat vakaimmat arviot rokotteen tehosta tässä ikäryhmässä.
*** Seuranta-ajan mediaanikesto 21 kuukautta
**** Rokotteen teho laskettiin jälkianalyysissä; seuranta-ajan mediaanikesto 11,1 kuukautta
# Paikallisen hoitokäytännön mukainen profylaktinen viruslääkitys sallittiin.
ZOE-50- ja ZOE-70-tutkimuksiin otettiin noin 13 000 tutkittavaa, joilla oli perussairauksia, mm. sairauksia, joiden yhteydessä vyöruusun riski on suurentunut. Jälkianalyysi rokotteen tehosta vahvistettua vyöruusua vastaan yleisiä perussairauksia (kroonista munuaistautia, keuhkoahtaumatautia, sepelvaltimotautia, masennusta tai diabetesta) sairastaneilla tutkittavilla viittaa siihen, että rokotteen teho näissä potilasryhmissä vastaa sen yleistä tehoa vyöruusua vastaan.
Shingrix pienensi merkitsevästi postherpeettisen neuralgian (PHN) ilmaantuvuutta verrattuna lumerokotteeseen:
- 50 vuotta täyttäneillä aikuisilla (ZOE-50): 0 tapausta vs. 18 tapausta;
- 70 vuotta täyttäneillä aikuisilla (ZOE-50- ja ZOE-70-tutkimusten yhdistetyt tiedot): 4 tapausta vs. 36 tapausta;
18 vuotta täyttäneillä aikuisilla, jotka olivat saaneet autologisen veren kantasolusiirron (Zoster-002): 1 tapaus vs. 9 tapausta.
Tulokset rokotteen tehosta postherpeettistä neuralgiaa vastaan esitetään taulukossa 3.
Taulukko 3: Shingrix-valmisteen teho postherpeettistä neuralgiaa vastaan (mTVC)
Ikä (vuotta) | Shingrix | Lume | Rokotteen teho (%) [95 % lv] | ||||
| Arviointikelpoisia tutkittavia | PHN*-tapauksia | Ilmaantuvuus / 1 000 henkilövuotta | Arviointikelpoisia tutkittavia | PHN*-tapauksia | Ilmaantuvuus / 1 000 henkilövuotta | ||
| ZOE-50** | |||||||
| ≥ 50 | 7 340 | 0 | 0,0 | 7 413 | 18 | 0,6 | 100 [77,1; 100] |
| 50–59 | 3 491 | 0 | 0,0 | 3 523 | 8 | 0,6 | 100 [40,8; 100] |
| ≥ 60 | 3 849 | 0 | 0,0 | 3 890 | 10 | 0,7 | 100 [55,2; 100] |
| 60–69 | 2 140 | 0 | 0,0 | 2 166 | 2 | 0,2 | 100§ [< 0; 100] |
| ZOE-50- ja ZOE-70-tutkimusten yhdistetyt tiedot*** | |||||||
| ≥ 70 | 8 250 | 4 | 0,1 | 8 346 | 36 | 1,2 | 88,8 [68,7; 97,1] |
| 70–79 | 6 468 | 2 | 0,1 | 6 554 | 29 | 1,2 | 93,0 [72,4; 99,2] |
| ≥ 80 | 1 782 | 2 | 0,3 | 1 792 | 7 | 1,1 | 71,2§ [< 0; 97,1] |
| Zoster-002**** (autologisen veren kantasolusiirron saaneet potilaat #) | |||||||
| ≥ 18 | 870 | 1 | 0,5 | 851 | 9 | 4,9 | 89,3 [22,5; 99,8] |
| 18–49 | 213 | 0 | 0,0 | 212 | 1 | 2,2 | 100,0§ [< 0; 100,0] |
| ≥ 50 | 657 | 1 | 0,7 | 639 | 8 | 5,8 | 88,0 [10,4; 99,8] |
* PHN määriteltiin vyöruusuun liittyväksi kivuksi, jonka vaikeusaste oli ≥ 3 (asteikolla 0–10) ja joka jatkui tai kehittyi yli 90 päivän kuluttua vyöruusuihottuman alusta; arvioinnissa käytettiin Zoster Brief Pain Inventory ‑mittaria (ZBPI)
lv luottamusväli
** Seuranta-ajan mediaanikesto 4,1 vuotta
*** Seuranta-ajan mediaanikesto 4,0 vuotta
70 vuotta täyttäneitä potilaita koskevat tiedot perustuvat ZOE-50- ja ZOE-70-tutkimusten ennalta määriteltyihin yhdistettyihin analyyseihin (mTV-kohortti), sillä nämä analyysit tarjoavat vakaimmat arviot rokotteen tehosta tässä ikäryhmässä.
**** Seuranta-ajan mediaanikesto 21 kuukautta
§ Ei tilastollisesti merkitsevä
# Paikallisen hoitokäytännön mukainen profylaktinen viruslääkitys sallittiin.
Shingrix-valmisteen hyödyt PHN:n ehkäisyssä johtuvat oletettavasti rokotteen vyöruusua ehkäisevästä vaikutuksesta. PHN:n ilmaantuvuuden pienenemistä vahvistettua vyöruusua sairastaneilla ei pystytty osoittamaan, sillä rokoteryhmässä oli vähän vyöruusutapauksia.
Neljäntenä vuonna rokotuksen jälkeen teho vyöruusua vastaan oli 50 vuotta täyttäneillä aikuisilla 93,1 % (95 % lv: 81,2; 98,2) (ZOE-50) ja 70 vuotta täyttäneillä aikuisilla 87,9 % (95 % lv: 73,3; 95,4) (ZOE-50- ja ZOE-70-tutkimusten yhdistetyt tiedot).
Zoster-002-tutkimuksen seuranta-aikana, joka alkoi 1 kuukauden kuluttua annoksesta 2 (eli noin 6 kuukauden kuluttua autologisesta veren kantasolusiirrosta) ja jatkui, kunnes autologisesta kantasolusiirrosta oli kulunut 1 vuosi (suurimman vyöruusuriskin ajanjakso), valmisteen teho vyöruusua vastaan oli 76,2 % (95 % lv: 61,1; 86,0).
Teho muita vyöruusuun liittyviä komplikaatioita kuin postherpeettistä neuralgiaa vastaan
Arvioituja vyöruusuun liittyviä komplikaatioita (muita kuin postherpeettinen neuralgia) olivat vyöruusuvaskuliitti, disseminoitunut tauti, silmänseudun vyöruusu, neurologinen tauti (mukaan lukien aivohalvaus) ja viskeraalinen tauti. ZOE-50- ja ZOE-70-tutkimusten yhdistetyssä analyysissa Shingrix vähensi merkitsevästi näitä vyöruusuun liittyviä komplikaatioita. 50 vuotta täyttäneillä aikuisilla komplikaatiot vähenivät 93,7 % (95 % lv: 59,5; 99,9) (1 tapaus vs. 16 tapausta) ja 70 vuotta täyttäneillä aikuisilla taas 91,6 % (95 % lv: 43,3; 99,8) (1 tapaus vs. 12 tapausta). Tutkimusten aikana ei ilmoitettu yhtään viskeraalista tautitapausta eikä yhtään aivohalvaustapausta.
Zoster-002-tutkimuksessa Shingrix vähensi vyöruusuun liittyviä komplikaatioita merkitsevästi, 77,8 % (95 % lv: 19,0; 96,0), 18 vuotta täyttäneillä tutkittavilla, jotka olivat saaneet autologisen veren kantasolusiirron (3 tapausta vs. 13 tapausta).
Zoster-002-tutkimuksessa Shingrix vähensi merkitsevästi myös vyöruusuun liittyviä sairaalahoitojaksoja, 84,7 % (95 % lv: 32,1; 96,6) (2 tapausta vs. 13 tapausta).
Shingrix-valmisteen teho vyöruusuun liittyvään kipuun
Yleisesti ottaen ZOE-50- ja ZOE-70-tutkimuksissa Shingrix-valmistetta saaneilla todettiin yleisenä trendinä vähemmän vaikeaa vyöruusuun liittyvää kipua kuin lumerokotetta saaneilla. Rokotteen teho vyöruusun ehkäisyssä on hyvä, joten läpilyöntitapauksia kertyi vähän eikä tutkimuksen näiden tavoitteiden suhteen pystytty tekemään varmoja johtopäätöksiä.
ZOE-50-tutkimuksen ja ZOE-70-tutkimuksen yhdistetyistä tiedoista todettiin, että Shingrix vähensi merkitsevästi vyöruusuun liittyvää kipulääkityksen käyttöä 39,0 % (95 % lv: 11,9; 63,3) ja lyhensi käytön kestoa merkitsevästi 50,6 % (95 % lv: 8,8; 73,2) 70 vuotta täyttäneillä aikuisilla, joilla oli vähintään yksi vahvistettu vyöruusuepisodi. Kipulääkityksen mediaanikesto oli Shingrix-ryhmässä 32,0 vrk ja lumeryhmässä 44,0 vrk.
Tutkittavilla, joilla oli vähintään yksi vahvistettu vyöruusuepisodi, Shingrix pienensi merkitsevästi keskimääräisen kivun maksimipistearvoa verrattuna lumerokotteeseen koko vyöruusuepisodin aikana (50 vuotta täyttäneillä [ZOE-50] keskiarvo 3,9 vs. 5,5 ja p-arvo 0,049; 70 vuotta täyttäneillä [ZOE-50- ja ZOE-70-tutkimusten yhdistetyt tiedot] keskiarvo 4,5 vs. 5,6 ja p-arvo 0,043). Lisäksi Shingrix pienensi merkitsevästi pahimman kivun maksimipisteitä verrattuna lumerokotteeseen (keskiarvo 5,7 vs. 7,0, p-arvo 0,032) koko vyöruusuepisodin aikana 70 vuotta täyttäneillä tutkittavilla (ZOE-50- ja ZOE-70-tutkimusten yhdistetyt tiedot).
Tautitaakkapisteissä otetaan huomioon vyöruusun ilmaantuvuus ja akuutin ja kroonisen vyöruusukivun vaikeusaste ja kesto 6 kuukauden kuluessa ihottuman alkamisesta.
Teho tautitaakan vähentämisessä oli 50 vuotta täyttäneillä 98,4 % (95 % lv: 92,2; 100) (ZOE-50) ja 70 vuotta täyttäneillä 92,1 % (95 % lv: 90,4; 93,8) (ZOE-50- ja ZOE-70-tutkimusten yhdistetyt tiedot).
Zoster-002-tutkimuksessa Shingrix lyhensi vaikeimman ”pahimman” vyöruusuun liittyvän kivun kestoa merkitsevästi, 38,5 % (95 % lv: 11,0; 57,6), 18 vuotta täyttäneillä tutkittavilla, jotka olivat saaneet autologisen veren kantasolusiirron ja joilla oli vähintään yksi vahvistettu vyöruusuepisodi. Shingrix pienensi merkitsevästi keskimääräisen kivun maksimipistearvoa verrattuna lumerokotteeseen koko vyöruusuepisodin aikana (keskiarvo = 4,7 vs. 5,7, p-arvo = 0,018) pahimman kivun maksimipisteitä verrattuna lumerokotteeseen koko vyöruusuepisodin aikana (keskiarvo = 5,8 vs. 7,1, p-arvo = 0,011).
Niiden tutkittavien prosenttiosuus, joilla oli Zoster-002-tutkimuksessa vähintään yksi vahvistettu vyöruusuepisodi ja jotka käyttivät vähintään yhtä kipulääkettä, oli Shingrix-ryhmässä 65,3 % ja lumeryhmässä 69,6 %. Kipulääkkeen käytön keston mediaani oli Shingrix-ryhmässä 21,5 vrk ja lumeryhmässä 47,5 vrk.
Lisäksi Zoster-002-tutkimuksessa teho tautitaakan vähentämisessä oli 82,5 % (95% lv: 73,6 %, 91,4 %).
Pitkäaikaisteho vyöruusua (herpes zoster), postherpeettistä neuralgiaa (PHN) ja muita vyöruusuun liittyviä komplikaatioita kuin postherpeettistä neuralgiaa vastaan
ZOE‑50- ja ZOE‑70-tutkimuksissa mukana olleilla 50 vuotta täyttäneillä aikuisilla toteutettiin Shingrix-valmistetta koskenut vaiheen IIIb avoin pitkäaikaisseurantatutkimus (Zoster‑049). Osallistujat otettiin tutkimukseen noin 5 vuoden kuluttua siitä, kun he olivat saaneet Shingrix-valmistetta ZOE‑50‑ tai ZOE‑70-tutkimuksessa. Tutkimuksesta suljettiin tutkimukseenottovaiheessa pois aikuiset, joille oli kehittynyt immuunivajavuus tai immunosuppressio. Tehoanalyysin koko rokotettu kohortti (TVC) käsitti 7 408 tutkittavaa (50,6 % ZOE‑50‑ ja ZOE‑70-tutkimusten tehon TV‑kohortin 14 645 tutkittavasta). Tehon pitkäkestoisuutta immuunipuutos-/immunosuppressiopopulaatiossa ei tunneta.
Rokotteen teho vyöruusua, postherpeettistä neuralgiaa ja muita vyöruusuun liittyviä komplikaatioita kuin postherpeettistä neuralgiaa vastaan analysoitiin kuvailevasti mTV-kohortissa (eli kohortissa, josta suljettiin pois tutkittavat, jotka eivät saaneet toista rokoteannosta alkuperäistutkimuksissa tai joilla todettiin vahvistetusti vyöruusu yhden kuukauden kuluessa toisesta annoksesta). Tehoarvioinnin perusteena käytettiin ensimmäistä tai ainoaa tapahtumaa. Näin ollen henkilöt, joilla oli ZOE‑50‑ tai ZOE‑70-tutkimuksen aikana esiintynyt vyöruusu, postherpeettinen neuralgia tai muu vyöruusuun liittyvä komplikaatio kuin postherpeettinen neuralgia, suljettiin pois kyseistä tapahtumaa koskeneista Zoster‑049-tutkimuksen aikaisista tehoanalyyseista. Rokotteen Zoster‑049-tutkimuksen aikaisen tehon arviointiin käytetyt vertailuryhmän ilmaantuvuusprosenttien arviot olivat historiallisia ja perustuivat ZOE‑50- ja ZOE‑70-tutkimusten lumeryhmien tietoihin.
Taulukossa 4 esitetään tulokset Shingrix-valmisteen pitkäaikaistehosta vyöruusua vastaan noin 5 vuodesta noin 11 vuoteen rokotuksen jälkeen.
Taulukko 4: Shingrix-valmisteen pitkäaikaisteho vyöruusua vastaan (mTVC) noin 5 vuodesta noin 11 vuoteen rokotuksen jälkeen
Ikä rokotushetkellä (vuotta) | Shingrix | Lume / Historiallinen verrokki* | Rokotteen teho** (%) [95 % lv] | ||||
| Arviointikelpoisia tutkittavia | Vyöruusutapauksia | Ilmaantuvuus / 1 000 henkilövuotta | Arviointikelpoisia tutkittavia | Vyöruusutapauksia | Ilmaantuvuus / 1 000 henkilö-vuotta | ||
| Zoster‑049-tutkimuksen aika | |||||||
| ≥ 50 | 7 258 | 69 | 1,8 | 7 258 | 341 | 8,7 | 79,8 [73,7; 84,6] |
| 50–59 | 2 043 | 12 | 1,0 | 2 043 | 90 | 7,7 | 86,7 [75,6; 93,4] |
| 60–69 | 1 242 | 9 | 1,3 | 1 242 | 70 | 10,1 | 87,1 [74,2; 94,4] |
| ≥ 70 | 3 973 | 48 | 2,4 | 3 973 | 179 | 8,8 | 73,2 [62,9; 80,9] |
lv luottamusväli
* ZOE‑50- tai ZOE‑70-tutkimuksen lumeryhmän tietoja käytettiin vuosien 1–4 analyysiin sekä historiallisten vertailutietojen muodostamiseen vuoden 6 ja sen jälkeisten vuosien analyysiin Zoster‑049-tutkimuksessa
** Kuvaileva tehoanalyysi
mTV-kohortin seuranta Zoster‑049-tutkimuksessa alkoi 5,6 vuoden (mediaani) kuluttua ZOE‑50- tai ZOE‑70-tutkimuksessa annetusta rokotuksesta ja päättyi 11,4 vuoden (mediaani) kuluttua rokotuksesta.
11. vuonna rokotuksen jälkeen teho vyöruusua vastaan oli 50 vuotta täyttäneillä tutkittavilla (Shingrix-ryhmä: N = 5 849) 82,0 % (95 % lv: 63,0; 92,2), 50–59-vuotiailla tutkittavilla (Shingrix-ryhmä: N = 1 883) 86,7 % (95 % lv: 42,7; 98,5), 60–69-vuotiailla tutkittavilla (Shingrix-ryhmä: N = 1 075) 100,0 % (95 % lv: 65,1; 100,0) ja 70 vuotta täyttäneillä tutkittavilla (Shingrix-ryhmä: N = 2 891) 72,0 % (95 % lv: 33,4; 89,8).
Taulukossa 5 esitetään tulokset Shingrix-valmisteen pitkäaikaistehosta postherpeettistä neuralgiaa vastaan noin 5 vuodesta noin 11 vuoteen rokotuksen jälkeen.
Taulukko 5: Shingrix-valmisteen pitkäaikaisteho postherpeettistä neuralgiaa vastaan (mTVC) noin 5 vuodesta noin 11 vuoteen rokotuksen jälkeen
Ikä rokotushetkellä (vuotta) | Shingrix | Lume / Historiallinen verrokki* | Rokotteen teho*** (%) [95 % lv] | ||||
| Arviointikelpoisia tutkittavia | PHN**-tapauksia | Ilmaantuvuus / 1 000 henkilövuotta | Arviointikelpoisia tutkittavia | PHN**-tapauksia | Ilmaantuvuus / 1 000 henkilövuotta | ||
| Zoster‑049-tutkimuksen aika | |||||||
| ≥ 50 | 7 271 | 4 | 0,1 | 7 271 | 32 | 0,8 | 87,5 [64,8; 96,8] |
| 50–59 | 2 046 | 0 | 0,0 | 2 046 | 7 | 0,6 | 100 [46,6; 100] |
| 60–69 | 1 243 | 1 | 0,1 | 1 243 | 2 | 0,3 | 50,0 [< 0; 99,2] |
| ≥ 70 | 3 982 | 3 | 0,1 | 3 982 | 23 | 1,1 | 87,0 [56,8; 97,5] |
lv luottamusväli
* ZOE‑50- tai ZOE‑70-tutkimuksen lumeryhmän tietoja käytettiin vuosien 1–4 analyysiin sekä historiallisten vertailutietojen muodostamiseen vuoden 6 ja sen jälkeisten vuosien analyysiin Zoster‑049-tutkimuksessa
** PHN määriteltiin vyöruusuun liittyväksi kivuksi, jonka vaikeusaste oli ≥ 3 (asteikolla 0–10) ja joka jatkui tai kehittyi yli 90 päivän kuluttua vyöruusuihottuman alusta; arvioinnissa käytettiin Zoster Brief Pain Inventory ‑mittaria (ZBPI)
*** Kuvaileva tehoanalyysi
mTV-kohortin seuranta Zoster‑049-tutkimuksessa alkoi 5,6 vuoden (mediaani) kuluttua ZOE‑50- tai ZOE‑70-tutkimuksessa annetusta rokotuksesta ja päättyi 11,4 vuoden (mediaani) kuluttua rokotuksesta.
Shingrix-valmisteen teho muita vyöruusuun liittyviä komplikaatioita kuin postherpeettistä neuralgiaa vastaan Zoster‑049-tutkimuksen aikana oli 50 vuotta täyttäneillä aikuisilla 91,7 % (95 % lv: 43,7; 99,8; 1 tapaus vs. 12 tapausta) ja 70 vuotta täyttäneillä aikuisilla 88,9 % (95 % lv: 19,8; 99,8; 1 tapaus vs. 9 tapausta).
Tutkittavat, joilla oli anamneesissa vyöruusu ennen rokotusta
Vaiheen III satunnaistetussa, lumekontrolloidussa, havainnoijasokkoutetussa kliinisessä monikeskustutkimuksessa (Zoster‑062) vähintään 50 vuotta täyttäneet aikuiset, joilla oli anamneesissa vyöruusu (parantunut >6 kk ennen tutkimukseenottoa), satunnaistettiin saamaan 2 annosta Shingrix-valmistetta tai lumerokotetta 2–6 kuukauden välein. Tutkimuksessa 1 426 tutkittavaa sai vähintään yhden annoksen Shingrix-valmistetta (N = 714) tai lumerokotetta (N = 712). 1 286 tutkittavaa suoritti tutkimuksen loppuun (vähimmäisseuranta-aika 26 kk).
Uusiutuneen vyöruusun ilmaantuvuus (Shingrix verrattuna lumerokotteeseen) arvioitiin muokatussa altistuneessa populaatiossa (mES; N = 1 350), johon kuuluivat kaksi annosta Shingrix-valmistetta (N = 668) tai lumerokotetta (N = 682) saaneet, joilla ei ollut todettu vahvistetusti vyöruusua 30 vuorokauden kuluessa toisen annoksen saamisesta. Tutkimuksesta saatujen tietojen mukaan vyöruusun uusiutumisriski ei ole Shingrix-rokotuksen jälkeen suurentunut henkilöillä, joilla on anamneesissa vyöruusu (0 vyöruusutapausta Shingrix-ryhmässä ja 8 vyöruusutapausta lumeryhmässä; uusiutuneen vyöruusun ilmaantuvuuksien suhde [Shingrix vs. lume] 0,00 [95 % lv 0,00; 0,46]).
Shingrix-valmisteen immunogeenisuus
Suojaan korreloivaa immunologista korrelaattia ei ole määritelty, joten vyöruusulta suojaavaa immuunivasteen tasoa ei tunneta.
Shingrix-valmisteen (2 annosta, joiden antoväli 2 kuukautta) tuottamia immuunivasteita arvioitiin 50 vuotta täyttäneillä aikuisilla vaiheen III tehotutkimusten ZOE-50 (humoraalinen immuniteetti ja soluvälitteinen immuniteetti) ja ZOE-70 (humoraalinen immuniteetti) potilasjoukon osajoukossa. Taulukoissa 6 ja 7 esitetään tiedot Shingrix-rokotuksen tuottamista gE-spesifisistä immuunivasteista (humoraalinen ja soluvälitteinen immuniteetti).
Taulukko 6: Shingrix-valmisteen humoraalinen immunogeenisuus 50 vuotta täyttäneillä aikuisilla (immunogeenisuuden tutkimussuunnitelman mukainen kohortti)
| Anti-gE immuunivaste^ | ||||||||
| Ikäryhmä (vuotta) | Kuukausi 3* | Kuukausi 38** | ||||||
| N | Geom. ka (mIU/ml) (95 % lv) | Pitoisuuksien moninkertaistuminen rokotusta edeltävästä tilanteesta, kertaluvun mediaani (K1; K3) | N | Geom. ka (mIU/ml) (95 % lv) | Pitoisuuksien moninkertaistuminen rokotusta edeltävästä tilanteesta, kertaluvun mediaani (K1; K3) | |||
| ZOE-50 | ||||||||
| ≥ 50 | 1070 | 52 376,6 (50 264,1; 54 577,9) | 41,9 (20,8; 86,9) | 967 | 11 919,6 (11 345,6; 12 522,7) | 9,3 (4,9; 19,5) | ||
| ZOE-50- ja ZOE-70-tutkimusten yhdistetyt tiedot | ||||||||
| ≥ 70 | 742 | 49 691,5 (47 250,8; 52 258,2) | 34,3 (16,7; 68,5) | 648 | 10 507,7 (9 899,2; 11 153,6) | 7,2 (3,5; 14,5) | ||
^ Anti-gE immuunivaste= anti-gE vasta-aineiden pitoisuudet, mitattuna anti-gE ELISA-tutkimuksella (gE-vasta-aineiden entsyymivälitteinen immunosorbenttimääritys)
* Kuukausi 3 = 1 kuukausi annoksesta 2
** Kuukausi 38 = 3 vuotta annoksesta 2
N Arviointikelpoisten tutkittavien määrä tiettynä ajankohtana (pitoisuuksien geometrista keskiarvoa varten)
lv luottamusväli
geom. ka = pitoisuuksien geometrinen keskiarvo
K1; K3 Ensimmäinen ja kolmas kvartiili
Taulukko 7: Shingrix-valmisteen soluvälitteinen immunogeenisuus 50 vuotta täyttäneillä aikuisilla (immunogeenisuuden tutkimussuunnitelman mukainen kohortti)
| gE-spesifinen CD4[2+]-T-soluvaste^ | ||||||
| Ikäryhmä (vuotta) | Kuukausi 3* | Kuukausi 38** | ||||
| N | Frekvenssin mediaani (K1; K3) | Frekvenssin moninkertaistuminen rokotusta edeltävästä tilanteesta, kertaluvun mediaani (K1; K3) | N | Frekvenssin mediaani (K1; K3) | Frekvenssin moninkertaistuminen rokotusta edeltävästä tilanteesta, kertaluvun mediaani (K1; K3) | |
| ZOE-50 | ||||||
| ≥ 50 | 164 | 1 844,1 (1 253,6; 2 932,3) | 24,6 (9,9; 744,2) | 152 | 738,9 (355,7; 1 206,5) | 7,9 (2,7; 31,6) |
| ≥ 70*** | 52 | 1 494,6 (922,9; 2 067,1) | 33,2 (10,0; 1 052,0) | 46 | 480,2 (196,1; 972,4) | 7,3 (1,7; 31,6) |
^ gE-spesifinen CD4[2+]-T-soluvaste = gE-spesifinen CD4+-T-solutoiminta, mitattuna ICS-testillä (solunsisäinen sytokiinivärjäys) (CD4[2+]-T-solut = CD4+-T-soluja, jotka ilmentävät vähintään kahta neljästä valikoidusta immuunimarkkerista)
* Kuukausi 3 = 1 kuukausi annoksesta 2
** Kuukausi 38 = 3 vuotta annoksesta 2
N Arviointikelpoisten tutkittavien määrä tiettynä ajankohtana (frekvenssin mediaania varten)
K1; K3 Ensimmäinen ja kolmas kvartiili
*** gE-spesifisten CD4[2+]-solujen tiedot 70 vuotta täyttäneiden ikäryhmässä perustuvat vain ZOE-50-tutkimukseen, sillä ZOE-70-tutkimuksessa ei arvioitu CD4+-T-solutoimintaa
Shingrix-valmisteen (2 annosta, joiden antoväli 1–2 kuukautta) tuottamaa humoraalista immuniteettivastetta ja soluvälitteistä immuniteettivastetta arvioitiin 18 vuotta täyttäneillä immuunipuutteisilla aikuisilla:
- yhdessä vaiheen I/II tutkimuksessa: Zoster-015 (tutkittavat HIV-positiivisia; valtaosalla (76,42 %) tila vakaa retroviruslääkehoidolla (kestänyt vähintään vuoden) ja CD4-T-soluarvo ≥ 200/mm3);
- yhdessä vaiheen II/III tutkimuksessa: Zoster-028 (kiinteään kasvaimeen solunsalpaajahoitoa saavia potilaita);
- kolmessa vaiheen III tutkimuksessa: Zoster-002 (autologisen veren kantasolusiirron saaneita, jotka oli rokotettu siirteen saannin jälkeen), Zoster-039 (potilailla oli hematologinen maligniteetti, ja rokote annettiin syöpähoitojakson aikana tai koko syöpähoitojakson päättymisen jälkeen) ja Zoster-041 (munuaissiirteen saaneita, jotka käyttivät rokotusajankohtana pitkäaikaista immunosuppressiivista hoitoa).
Taulukoissa 8 ja 9 esitetään tiedot Shingrix-rokotuksen tuottamista gE-spesifisistä immuunivasteista (humoraalinen ja soluvälitteinen immuniteetti) kaikissa tutkituissa immuunipuutteisissa populaatioissa.
Taulukko 8: Shingrix-valmisteen humoraalinen immunogeenisuus 18 vuotta täyttäneillä immuunipuutteisilla aikuisilla (immunogeenisuuden tutkimussuunnitelman mukainen kohortti)
| Anti-gE-immuunivaste^ | |||||
| Kuukausi 3 | Kuukausi 13/18/25 | ||||
| N | Geom. ka (mIU/ml) (95 % lv) | Pitoisuuksien moninkertaistuminen rokotusta edeltävästä tilanteesta, kertaluvun mediaani (K1; K3) | N | Geom. ka (mIU/ml) (95 % lv) | pitoisuuksien moninkertaistuminen rokotusta edeltävästä tilanteesta, kertaluvun mediaani (K1; K3) |
| Zoster-002 (autologisen veren kantasolusiirron saaneet) | |||||
| 82 | 12 753,2 (7 973,0; 20 399,4) | 14,1 (1,7; 137,0) | 54 | Kuukausi 13: 3 183,8 (1 869,8; 5 421,2) | Kuukausi 13: 2,7 (1,0; 24,0) |
| 39 | Kuukausi 25: 2 819,0 (1 387,1; 5 729,1) | Kuukausi 25: 1,3 (0,6; 44,7) | |||
| Zoster-028 (potilaat, joilla oli kiinteä kasvain) | |||||
| 87 | 18 291,7 (14 432,1; 23 183,5) | 21,5 (7,0; 45,2) | 68 | Kuukausi 13: 4 477,3 (3 482,4; 5 756,3) | Kuukausi 13: 4,1 (2,1; 7,9) |
| Zoster-039 (potilaat, joilla oli hematologinen maligniteetti) | |||||
| 217 | 13 445,6 (10 158,9; 17 795,6) | 17,2 (1,4; 87,4) | 167 | Kuukausi 13: 5 202,7 (4 074,8; 6 642,8) | Kuukausi 13: 5,1 (1,1; 17,0) |
| Zoster-041 (munuaissiirteen saaneet) | |||||
| 121 | 19 163,8 (15 041,5; 24 416,0) | 15,1 (6,1; 35,0) | 111 | Kuukausi 13: 8 545,1 (6 753,7; 10 811,5) | Kuukausi 13: 6,5 (3,1; 13,3) |
| Zoster-015 (HIV-positiiviset tutkittavat) | |||||
| 53 | 42 723,6 (31 233,0; 58 441,6) | 40,9 (18,8; 93,0) | 49 | Kuukausi 18: 25 242,2 (19 618,9; 32 477,3) | Kuukausi 18: 24,0 (9,8; 39,7) |
^ Anti-gE immuunivaste = anti-gE-vasta-aineiden pitoisuudet, mitattuna anti-gE ELISA -tutkimuksella (gE-vasta-aineiden entsyymivälitteinen immunosorbenttimääritys)
N Arviointikelpoisten tutkittavien määrä tiettynä ajankohtana (pitoisuuksien geometrista keskiarvoa varten)
lv luottamusväli
geom. ka pitoisuuksien geometrinen keskiarvo
K1; K3 Ensimmäinen ja kolmas kvartiili
Zoster-028 -tutkimuksessa geometrinen keskiarvo 1 kk kuluttua toisesta annoksesta oli 22 974,3 (19 080,0; 27 663,5) tutkittavilla, jotka saivat ensimmäisen Shingrix annoksen vähintään 10 päivää ennen solunsalpaajahoitojaksoa (PreChemo -ryhmä) ja 9 328,0 (4 492,5; 19 368,2) tutkittavilla, jotka saivat ensimmäisen Shingrix annoksen solunsalpaajahoitojakson aikana (OnChemo – ryhmä). Zoster-039 tutkimuksessa geometrinen keskiarvo 1 kk kuluttua toisesta annoksesta oli 19 934,7 (14 674,1; 27 081,2) tutkittavilla, jotka saivat ensimmäisen Shingrix annoksen koko syöpähoitojakson päättymisen jälkeen ja 5 777,4 (3 342,5; 9 985,9) tutkittavilla, jotka saivat ensimmäisen Shingrix annoksen syöpähoitojakson aikana. Tehoon vaikuttamisen (lyhyt- tai pitkäaikaisvaikutukset) kliinistä merkitystä ei tunneta.
Taulukko 9: Shingrix-valmisteen soluvälitteinen immunogeenisuus 18 vuotta täyttäneillä immuunipuutteisilla aikuisilla (immunogeenisuuden tutkimussuunnitelman mukainen kohortti)
| gE-spesifinen CD4[2+]-T-soluvaste^ | |||||
| Kuukausi 3 | Kuukausi 13/18/25 | ||||
| N | Frekvenssin mediaani (K1; K3) | Frekvenssin moninkertaistuminen rokotusta edeltävästä tilanteesta, kertaluvun mediaani (K1; K3) | N | Frekvenssin mediaani (K1; K3) | Frekvenssin moninkertaistuminen rokotusta edeltävästä tilanteesta, kertaluvun mediaani (K1; K3) |
| Zoster-002 (autologisen veren kantasolusiirron saaneet) | |||||
| 51 | 6 644,9 (1 438,3; 13 298,6) | 109,0 (34,4; 2 716,4) | 32 | Kuukausi 13: 1 706,4 (591,4; 5 207,0) | Kuukausi 13: 43,6 (13,1; 977,8) |
| 30 | Kuukausi 25: 2 294,4 (455,2; 3 633,2) | Kuukausi 25: 50,9 (15,3; 515,2) | |||
| Zoster-028 (potilaat, joilla oli kiinteä kasvain) | |||||
| 22 | 778,8 (393,1; 1 098,2) | 4,9 (1,7; 33,0) | 18 | Kuukausi 13: 332,9 (114,9; 604,6) | Kuukausi 13: 2,0 (1,3; 5,2) |
| Zoster-039 (potilaat, joilla oli hematologinen maligniteetti) | |||||
| 53 | 3 081,9 (1 766,2; 7 413,6) | 45,9 (16,4; 2 221,9) | 44 | Kuukausi 13: 1 006,7 (416,0; 3 284,5) | Kuukausi 13: 21,4 (7,5; 351,4) |
| Zoster-041 (munuaissiirteen saaneet) | |||||
| 32 | 2 149,0 (569,4; 3 695,1) | 47,7 (14,7; 439,6) | 33 | Kuukausi 13: 1 066.,3 (424,8; 1 481,5) | Kuukausi 13: 16,9 (5,9; 211,4) |
| Zoster-015 (HIV-positiiviset tutkittavat) | |||||
| 41 | 2 809,7 (1 554,5; 4 663,7) | 23,4 (8,5; 604,1) | 49 | Kuukausi 18: 1 533,0 (770,0; 2 643,1) | Kuukausi 18: 12,0 (5,7; 507,0) |
^ gE-spesifinen CD4[2+]-T-soluvaste = gE-spesifinen CD4+-T-solutoiminta, mitattuna ICS-testillä (solunsisäinen sytokiinivärjäys) (CD4[2+]-T-solut = CD4+-T-soluja, jotka ilmentävät vähintään kahta neljästä valikoidusta immuunimarkkerista)
N Arviointikelpoisten tutkittavien määrä tiettynä ajankohtana (frekvenssin mediaania varten)
K1; K3 Ensimmäinen ja kolmas kvartiili
* Soluvälitteisen immuniteetin tutkimista varten verinäytteitä otettiin vain tutkittavilta, jotka saivat ensimmäisen Shingrix-annoksen 8–30 vrk ennen solunsalpaajahoitojakson alkua (tutkimuksen suurin ryhmä)
Immunogeenisuus tutkittavilla, jotka saivat 2 annosta Shingrix-valmistetta 6 kuukauden välein
Tehoa ei ole arvioitu tilanteessa, jossa rokote annetaan kuukausina 0 ja 6.
Vaiheen III avoimessa kliinisessä tutkimuksessa (Zoster-026), jossa 238 iältään ≥ 50-vuotiasta aikuista satunnaistettiin suhteessa 1:1 saamaan 2 annosta Shingrix-valmistetta joko 2 kuukauden tai 6 kuukauden välein, humoraalinen immuunivaste oli rokotteen kuukausina 0 ja 6 saaneilla vähintään samanveroinen kuin rokotteen kuukausina 0 ja 2 saaneilla todettu vaste. Anti-gE-vasta-aineiden geometrinen keskiarvo 1 kk kuluttua viimeisestä rokoteannoksesta antoaikataululla 0, 6 kk oli 38 153,7 mIU/ml (95 % lv 34 205,8; 42 557,3) ja antoaikataululla 0, 2 kk 44 376,3 mIU/ml (95 % lv 39 697,0; 49 607,2).
Immunogeenisuus henkilöillä, jotka ovat aiemmin saaneet rokotuksen elävällä heikennetyllä herpes zoster (HZ) -rokotteella
Vaiheen III avoimessa kliinisessä monikeskustutkimuksessa (Zoster-048) arvioitiin Shingrix-rokotteen käyttöä kahden rokotuksen (kahden kuukauden välein) sarjana 215 iältään ≥ 65-vuotiaalla aikuisella, joilla oli ≥ 5 vuotta aiempi rokotushistoria elävällä heikennetyllä HZ -rokotteella, verrattuna 215 vastaavaan henkilöön, jotka eivät olleet koskaan saaneet elävää heikennettyä HZ-rokotetta. Aikaisempi rokotus elävällä heikennetyllä HZ-rokotteella ei vaikuttanut Shingrixin tuottamaan immuunivasteeseen.
Immunogeenisuuden pitkäkestoisuus
Immunogeenisuuden pitkäkestoisuutta arvioitiin tutkittavien osajoukossa vaiheen IIIb avoimessa pitkäaikaisseurantatutkimuksessa (Zoster‑049), jossa oli mukana ZOE‑50- tai ZOE‑70-tutkimuksessa mukana olleita 50 vuotta täyttäneitä aikuisia. Vuonna 12 rokotuksen jälkeen anti‑gE-vasta-aineiden pitoisuus oli 435 arviointikelpoisella tutkittavalla 5,8 kertaa (95 % lv: 5,2; 6,4) suurempi (suurenemisen geometrinen keskiarvo) rokotusta edeltävään tasoon verrattuna. 73 arviointikelpoisen tutkittavan gE‑spesifisten CD4[2+]-T‑solujen mediaanifrekvenssi vuonna 12 rokotuksen jälkeen oli säilynyt rokotusta edeltävää tasoa suurempana.
Immunogeenisuuden pitkäkestoisuutta arvioitiin vaiheen IIIb avoimessa tutkimuksessa (Zoster‑073). Tutkimuksessa oli mukana 68 Zoster‑041-tutkimuksessa mukana ollutta, 18 vuotta täyttänyttä, munuaissiirteen saanutta tutkittavaa, jotka käyttivät pitkäaikaista immunosuppressiivista hoitoa. Zoster‑073-tutkimus aloitettiin 4–6 vuoden kuluttua Zoster‑041-tutkimuksessa saadusta rokotuksesta. Kuukautena 24 (noin 6–8 vuoden kuluttua annoksesta 2) anti‑gE-vasta-aineiden pitoisuus oli 49 arviointikelpoisella tutkittavalla 2,4 kertaa (95 % lv: 1,6; 3,7) suurempi (suurenemisen geometrinen keskiarvo) rokotusta edeltävään tasoon verrattuna. Soluvälitteisen immuniteetin alajoukon 19 arviointikelpoisen tutkittavan gE‑spesifisten CD4[2+]‑T‑solujen mediaanifrekvenssi kuukautena 24 oli säilynyt rokotusta edeltävää tasoa suurempana.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Shingrix-valmisteen käytöstä varicella zoster ‑viruksen reaktivaation ehkäisyssä yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Ei oleellinen.
Prekliiniset tiedot turvallisuudesta
Akuuttia ja toistuvan altistuksen aiheuttamaa toksisuutta, paikallista siedettävyyttä, farmakologista turvallisuutta sydämen, verisuoniston ja hengityselimistön kannalta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
Kuiva-aine (gE-antigeeni):
Sakkaroosi
Polysorbaatti 80 (E 433)
Natriumdivetyfosfaattidihydraatti (E 339)
Dikaliumfosfaatti (E 340)
Suspensio (AS01B-adjuvantti):
Dioleoyylifosfatidyylikoliini (E 322)
Kolesteroli
Natriumkloridi
Vedetön dinatriumfosfaatti (E 339)
Kaliumdivetyfosfaatti (E 340)
Injektionesteisiin käytettävä vesi
Adjuvanttia koskien ks. kohta Vaikuttavat aineet ja niiden määrät.
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
42 kuukautta
Käyttökuntoon saattamisen jälkeen
Valmisteen on osoitettu säilyvän käytön aikana kemiallisesti ja fysikaalisesti stabiilina 24 tunnin ajan 30 °C:n lämpötilassa.
Mikrobiologiselta kannalta rokote tulee käyttää välittömästi. Jos valmistetta ei käytetä välittömästi, käytönaikainen säilytysaika ja käyttöä edeltävät säilytysolosuhteet ovat käyttäjän vastuulla ja ovat normaalisti enintään 6 tuntia 2–8 °C:n lämpötilassa.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SHINGRIX injektiokuiva-aine ja suspensio, suspensiota varten
1 annos (212,26 €), 10 x 1 annosta (1917,15 €)
PF-selosteen tieto
- Kuiva-aine 1 annosta varten injektiopullossa (tyypin I lasia), jossa on tulppa (butyylikumia)
- Suspensio 1 annosta varten injektiopullossa (tyypin I lasia), jossa on tulppa (butyylikumia).
Shingrix on saatavilla pakkauksessa, jossa on 1 injektiopullo kuiva-ainetta ja 1 injektiopullo suspensiota, sekä pakkauksessa, jossa on 10 injektiopulloa kuiva-ainetta ja 10 injektiopulloa suspensiota.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kuiva-aine on valkoista.
Suspensio on opalisoivaa, väritöntä tai vaalean rusehtavaa nestettä.
Käyttö- ja käsittelyohjeet
Shingrix on pakattu kuiva-ainetta (antigeenia) sisältävään injektiopulloon, jossa on ruskea irti napsautettava (flip-off) korkki, ja suspensiota (adjuvanttia) sisältävään injektiopulloon, jossa on sinivihreä irti napsautettava (flip-off) korkki.
Kuiva-aine ja suspensio on saatettava käyttökuntoon ennen antoa.
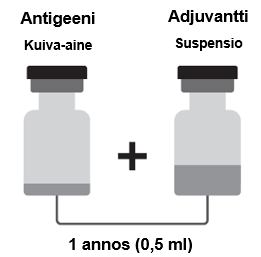
Kuiva-aine ja suspensio on tarkastettava silmämääräisesti vieraiden hiukkasten ja/tai ulkonäkömuutosten varalta. Jos jompaakumpaa näistä todetaan, rokotetta ei saa saattaa käyttökuntoon.
Shingrix-rokotteen valmistelu
Shingrix saatetaan käyttökuntoon ennen antoa.
1. Suspensiota sisältävän injektiopullon koko sisältö vedetään ruiskuun sopivan kokoisella neulalla (21G–25G).
2. Ruiskun koko sisältö lisätään injektiopulloon, jossa kuiva-aine on.
3. Ravistellaan varovasti, kunnes kuiva-aine on liuennut täysin.
Käyttökuntoon saatettu rokote on opalisoivaa, väritöntä tai vaalean rusehtavaa nestettä.
Käyttökuntoon saatettu rokote on tarkastettava silmämääräisesti vieraiden hiukkasten ja/tai ulkonäkömuutosten varalta. Jos jompaakumpaa näistä todetaan, rokotetta ei saa antaa.
Käyttökuntoon saattamisen jälkeen rokote tulee antaa heti; mikäli tämä ei ole mahdollista, rokote säilytetään jääkaapissa (2 °C – 8 °C). Jos sitä ei käytetä 6 tunnissa, se on hävitettävä.
Ennen antoa
1. Käyttökuntoon saatettua rokotetta sisältävän injektiopullon koko sisältö vedetään ruiskuun.
2. Neula vaihdetaan eli rokotteen antoon käytetään uutta neulaa.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SHINGRIX injektiokuiva-aine ja suspensio, suspensiota varten
1 annos, 10 x 1 annosta
- Ei korvausta.
ATC-koodi
J07BK03
Valmisteyhteenvedon muuttamispäivämäärä
12.02.2026
Yhteystiedot
 GLAXOSMITHKLINE OY
GLAXOSMITHKLINE OY Porkkalankatu 20 A
00180 Helsinki
010 303 030
www.glaxosmithkline.fi