

CALQUENCE kapseli, kova 100 mg
Vaikuttavat aineet ja niiden määrät
Yksi kova kapseli sisältää 100 mg akalabrutinibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kova kapseli (kapseli).
Kliiniset tiedot
Käyttöaiheet
Calquence on tarkoitettu monoterapiana tai yhdistelmänä obinututsumabin kanssa aikuispotilaille aiemmin hoitamattoman kroonisen lymfaattisen leukemian (KLL) hoitoon.
Calquence on tarkoitettu yhdistelmänä venetoklaksin kanssa joko yhdistettynä obinututsumabiin tai ilman obinututsumabia aikuispotilaille aiemmin hoitamattoman kroonisen lymfaattisen leukemian (KLL) hoitoon.
Calquence on tarkoitettu monoterapiana kroonisen lymfaattisen leukemian (KLL) hoitoon aikuispotilaille, jotka ovat aiemmin saaneet vähintään yhtä hoitoa.
Calquence on tarkoitettu yhdistelmänä bendamustiinin ja rituksimabin (BR) kanssa aikuispotilaille aiemmin hoitamattoman manttelisolulymfooman (MCL) hoitoon, kun autologinen kantasolusiirto ei sovellu potilaalle.
Calquence on tarkoitettu monoterapiana uusiutuneen tai refraktaarisen manttelisolulymfooman (MCL) hoitoon aikuispotilaille, joita ei ole aiemmin hoidettu BTK:n estäjällä.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Hoito tällä lääkevalmisteella pitää aloittaa ja toteuttaa syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa.
Annostus
Suositeltu Calquence‑annos monoterapiassa tai yhdistelmähoidossa muiden lääkevalmisteiden kanssa on 100 mg akalabrutinibia kaksi kertaa vuorokaudessa (kokonaisvuorokausiannos 200 mg).
Calquence-valmisteen annosväli on noin 12 tuntia.
Katso yhdistelmähoidoissa käytettävien lääkevalmisteiden annostelutiedot kunkin lääkevalmisteen valmisteyhteenvedosta (tarkemmat tiedot yhdistelmähoidoista, ks. kohta Farmakodynamiikka).
Calquence monoterapiana tai yhdistelmänä obinututsumabin kanssa
Hoitoa Calquence-valmisteella monoterapiana tai yhdistelmänä obinututsumabin kanssa jatketaan, kunnes tauti etenee tai ilmenee toksisuutta, jota ei voida hyväksyä.
Calquence yhdistelmänä venetoklaksin kanssa joko yhdistettynä obinututsumabiin tai ilman obinututsumabia
Hoitoa Calquence-valmisteella yhdistelmänä venetoklaksin kanssa joko yhdistettynä obinututsumabiin tai ilman obinututsumabia jatketaan, kunnes tauti etenee, ilmenee toksisuutta, jota ei voida hyväksyä, tai hoitoa on annettu 14 hoitojakson ajan (yhden hoitojakson pituus on 28 päivää).
Calquence-valmistetta annetaan ensimmäisen hoitojakson päivästä 1 alkaen yhteensä 14 hoitojakson ajan. Venetoklaksia annetaan hoitojakson 3 päivästä 1 alkaen yhteensä 12 hoitojakson ajan aloittaen 20 mg:n annoksella ja suurentaen annosta viikoittain 50 mg:aan, 100 mg:aan, 200 mg:aan ja lopuksi 400 mg:aan.
Jos Calquence-valmistetta annetaan yhdistelmänä venetoklaksin ja obinututsumabin kanssa, obinututsumabia annetaan 100 mg:n annos hoitojakson 2 päivänä 1 ja sen jälkeen 900 mg:n annos, joka voidaan antaa päivänä 1 tai 2. Obinututsumabia annetaan 1 000 mg:n annos hoitojakson 2 päivinä 8 ja 15 ja sen jälkeen 1 000 mg:n annos hoitojaksojen 3–7 päivänä 1. Obinututsumabia annetaan yhteensä 6 hoitojakson ajan.
Calquence yhdistelmänä bendamustiinin ja rituksimabin kanssa
Calquence‑valmistetta annetaan ensimmäisen hoitojakson päivästä 1 alkaen (yhden hoitojakson pituus on 28 päivää), kunnes tauti etenee tai ilmenee toksisuutta, joita ei voida hyväksyä. Bendamustiinia annetaan annoksella 90 mg/m2 kunkin hoitojakson päivinä 1 ja 2 yhteensä 6 hoitojakson ajan. Rituksimabia annetaan annoksella 375 mg/m2 kunkin hoitojakson päivänä 1 yhteensä 6 hoitojakson ajan. Potilaat, joilla todetaan vaste (osittainen vaste [PR] tai täydellinen vaste [CR]) ensimmäisten 6 hoitojakson jälkeen, voivat saada ylläpitohoitona enintään 12 lisäannosta rituksimabia annoksella 375 mg/m2 joka toisen hoitojakson päivänä 1 alkaen hoitojaksosta 8 hoitojaksoon 30 asti.
Annoksen muuttaminen
Haittavaikutukset
Vaikeusasteen ≥ 3 haittavaikutusten vuoksi tehtävät suositellut Calquence-valmisteen annosmuutokset Calquence‑monoterapiaa tai Calquence‑valmistetta yhdistelmänä obinututsumabin kanssa saavilla potilailla on esitetty taulukossa 1.
Vaikeusasteen ≥ 3 haittavaikutusten vuoksi tehtävät suositellut annosmuutokset Calquence‑valmistetta yhdistelmänä bendamustiinin ja rituksimabin kanssa saavilla potilailla on esitetty taulukossa 2.
Taulukko 1. Haittavaikutusten vuoksi tehtävät suositellut annosmuutokset*
| Haittavaikutus | Haittavaikutuksen esiintyminen | Annosmuutos (Aloitusannos = 100 mg noin 12 tunnin välein) |
Asteen 3 trombosytopenia, johon liittyy verenvuotoa, asteen 4 trombosytopenia tai asteen 4 neutropenia, joka jatkuu yli 7 päivää Vähintään asteen 3 ei-hematologinen toksisuus | Ensimmäinen ja toinen | Keskeytä Calquence-hoito. Kun toksisuus on lieventynyt asteeseen 1 tai lähtötasolle, Calquence-hoitoa voidaan jatkaa annostuksella 100 mg noin 12 tunnin välein. |
| Kolmas | Keskeytä Calquence-hoito. Kun toksisuus on lieventynyt asteeseen 1 tai lähtötasolle, Calquence-hoitoa voidaan jatkaa harvemmalla annostuksella eli 100 mg kerran vuorokaudessa. | |
| Neljäs | Lopeta Calquence-hoito. |
*Haittavaikutusten vaikeusasteet NCI:n CTCAE-luokituksen (National Cancer Institute Common Terminology Criteria for Adverse Events) version 4.03 mukaan.
Taulukko 2. Vaikeusasteen ≥ 3 haittavaikutusten* vuoksi tehtävät suositellut annosmuutokset Calquence‑valmistetta yhdistelmänä bendamustiinin ja rituksimabin kanssa saavilla potilailla
| Haittavaikutus | Bendamustiiniannoksen muutos† | Calquence‑annoksen muutos |
|---|---|---|
Neutropenia
| Asteen 3 tai 4 neutropenia‡: Keskeytä bendamustiinihoito. Kun toksisuus on lieventynyt asteeseen ≤ 2 tai lähtötasolle, bendamustiinihoitoa voidaan jatkaa annoksella 70 mg/m2. Lopeta bendamustiinihoito, jos annosta on tarpeen pienentää uudelleen. | Jos asteen 4 neutropenia jatkuu yli 7 päivää, keskeytä Calquence‑hoito. Kun toksisuus on lieventynyt asteeseen ≤ 2 tai lähtötasolle, Calquence‑hoitoa voidaan jatkaa aloitusannoksella (jos haittavaikutus on ilmaantunut 1. kerran) tai harvennetulla antotiheydellä 100 mg kerran vuorokaudessa (jos haittavaikutus on ilmaantunut 2. tai 3. kerran). Lopeta Calquence‑hoito, jos haittavaikutus ilmaantuu 4. kerran. |
Trombosytopenia
| Asteen 3 tai 4 trombosytopenia: Keskeytä bendamustiinihoito. Kun toksisuus on lieventynyt asteeseen ≤ 2 tai lähtötasolle, bendamustiinihoitoa voidaan jatkaa annoksella 70 mg/m2. Lopeta bendamustiinihoito, jos annosta on tarpeen pienentää uudelleen. | Jos kyseessä on asteen 3 trombosytopenia, johon liittyy merkittävää verenvuotoa, tai asteen 4 trombosytopenia, keskeytä Calquence‑hoito. Kun toksisuus on lieventynyt asteeseen ≤ 2 tai lähtötasolle, Calquence‑hoitoa voidaan jatkaa aloitusannoksella (jos haittavaikutus on ilmaantunut 1. kerran) tai harvennetulla antotiheydellä 100 mg kerran vuorokaudessa (jos haittavaikutus on ilmaantunut 2. tai 3. kerran).¶ Lopeta Calquence‑hoito, jos trombosytopenia ja siihen liittyvä merkittävä verenvuoto ilmaantuvat 3. kerran. Lopeta Calquence‑hoito, jos haittavaikutus ilmaantuu 4. kerran. |
| Muu hematologinen asteen 4§ toksisuus tai asteen 3 toksisuus, jota ei saada hallintaan | Keskeytä bendamustiinihoito. Kun toksisuus on lieventynyt asteeseen ≤ 2 tai lähtötasolle, bendamustiinihoitoa voidaan jatkaa annoksella 70 mg/m2. Lopeta bendamustiinihoito, jos annosta on tarpeen pienentää uudelleen. | Keskeytä Calquence‑hoito. Kun toksisuus on lieventynyt asteeseen ≤ 2 tai lähtötasolle, Calquence‑hoitoa voidaan jatkaa aloitusannoksella (jos haittavaikutus on ilmaantunut 1. kerran) tai harvennetulla antotiheydellä 100 mg kerran vuorokaudessa (jos haittavaikutus on ilmaantunut 2. tai 3. kerran). Lopeta Calquence‑hoito, jos haittavaikutus ilmaantuu 4. kerran. |
| Vähintään asteen 3 ei‑hematologiset toksisuudet | Keskeytä bendamustiinihoito. Kun toksisuus on lieventynyt asteeseen 1 tai lähtötasolle, bendamustiinihoitoa voidaan jatkaa annoksella 70 mg/m2. Lopeta bendamustiinihoito, jos annosta on tarpeen pienentää uudelleen. | Keskeytä Calquence‑hoito. Kun toksisuus on lieventynyt asteeseen 2 tai lähtötasolle, Calquence‑hoitoa voidaan jatkaa aloitusannoksella (jos haittavaikutus on ilmaantunut 1. kerran) tai harvennetulla antotiheydellä 100 mg kerran vuorokaudessa (jos haittavaikutus on ilmaantunut 2. kerran).¶ Lopeta Calquence‑hoito, jos haittavaikutus ilmaantuu 3. kerran. |
*Haittavaikutusten vaikeusasteet NCI:n CTCAE‑luokituksen (National Cancer Institute Common Terminology Criteria for Adverse Events) version 4.03 mukaan.
†Jos jotakin toksisuutta ei ole mainittu tässä taulukossa, katso bendamustiinia koskeva paikallinen valmisteyhteenveto.
‡Harkitse myelooisten kasvutekijöiden käyttöä ennen bendamustiiniannoksen muuttamista.
§Asteen 4 lymfopenia on bendamustiini‑ ja rituksimabihoidon odotettavissa oleva seuraus. Lymfopeniasta johtuvia annosmuutoksia on odotettavissa vain, jos lääkäri pitää sitä kliinisesti tärkeänä, esim. lymfopeniaan liittyvien toistuvien infektioiden takia.
¶Annosta voidaan suurentaa uudelleen lääkärin harkinnan mukaan, jos potilas sietää pienennettyä annosta ≥ 4 viikon ajan.
Katso haittavaikutusten hallintaa koskevat lisätiedot Calquence‑valmisteen kanssa yhdistelmänä käytettävien lääkevalmisteiden valmisteyhteenvedoista.
Yhteisvaikutukset
Suositukset, jotka koskevat Calquence-valmisteen käyttöä CYP3A:n estäjien tai indusorien sekä mahahapon eritystä vähentävien lääkeaineiden kanssa, on esitetty taulukossa 3 (ks. kohta Yhteisvaikutukset).
Taulukko 3. Käyttö CYP3A:n estäjien tai indusorien sekä mahahapon eritystä vähentävien lääkeaineiden kanssa
| Samanaikaisesti annettu lääkevalmiste | Suositeltu Calquence-valmisteen käyttö | |
| CYP3A:n estäjät | Voimakas CYP3A:n estäjä | Vältä samanaikaista käyttöä. Jos tällaisia estäjiä käytetään lyhytaikaisesti (kuten infektiolääkkeitä enintään seitsemän päivän ajan), keskeytä Calquence-hoito. |
| Keskivahva CYP3A:n estäjä | Annosta ei muuteta. Potilaan tilaa seurataan tarkasti haittavaikutusten havaitsemiseksi, jos hän käyttää keskivahvaa CYP3A:n estäjää. | |
| Heikko CYP3A:n estäjä | Annosta ei muuteta. | |
| CYP3A:n indusorit | Voimakas CYP3A:n indusori | Vältä samanaikaista käyttöä. |
| Maha-hapon eritystä vähentävät lääkeaineet | Protonipumpun estäjät | Vältä samanaikaista käyttöä. |
| H2-reseptorin salpaajat | Calquence otetaan 2 tuntia ennen H2-reseptorin salpaajan ottamista (tai 10 tuntia sen ottamisen jälkeen). | |
| Antasidit | Näiden lääkevalmisteiden ottamisen välillä pitää olla vähintään 2 tuntia. |
Ottamatta jäänyt annos
Jos Calquence-annoksen ottaminen viivästyy yli 3 tunnilla, potilasta neuvotaan ottamaan seuraava annos sen tavanomaiseen ottoaikaan. Potilaan ei pidä ottaa kaksinkertaista Calquence-annosta korvatakseen unohtuneen annoksen.
Erityisryhmät
Iäkkäät potilaat
Annoksen muuttaminen ei ole tarpeen iäkkäillä (≥ 65-vuotiailla) potilailla (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Spesifisiä kliinisiä tutkimuksia ei ole tehty munuaisten vajaatoimintaa sairastavilla potilailla. Lievää tai kohtalaista munuaisten vajaatoimintaa sairastavia potilaita hoidettiin kliinisissä Calquence-tutkimuksissa. Annoksen muuttaminen ei ole tarpeen lievää tai kohtalaista munuaisten vajaatoimintaa (kreatiniinipuhdistuma yli 30 ml/min) sairastavilla potilailla. Nesteytyksestä on huolehdittava ja seerumin kreatiniinipitoisuus on tutkittava säännöllisesti. Calquence-valmistetta käytetään vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min) sairastavien hoidossa vain siinä tapauksessa, että hoidon hyödyt ovat suuremmat kuin sen riskit, ja tällaisen potilaan tilaa on seurattava tarkasti toksisuuden merkkien havaitsemiseksi. Valmisteen käytöstä vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden tai dialyysipotilaiden hoidossa ei ole tietoja (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Annoksen muuttamista ei suositella, jos potilaalla on lievä tai kohtalainen maksan vajaatoiminta (Child–Pugh-luokka A, Child–Pugh-luokka B tai 1,5–3 kertaa viitealueen ylärajaa [ULN] suurempi kokonaisbilirubiinipitoisuus ja mikä tahansa ASAT-arvo). Kohtalaista maksan vajaatoimintaa sairastavien potilaiden tilaa on kuitenkin seurattava tarkasti toksisuuden merkkien havaitsemiseksi. Calquence-valmisteen käyttöä ei suositella, jos potilaalla on vaikea maksan vajaatoiminta (Child-Pugh-luokka C tai > 3 kertaa viitealueen ylärajaa suurempi kokonaisbilirubiinipitoisuus ja mikä tahansa ASAT-arvo) (ks. kohta Farmakokinetiikka).
Vaikea sydänsairaus
Vaikeaa sydän- ja verisuonitautia sairastavat potilaat suljettiin pois Calquence-valmisteen kliinisistä tutkimuksista.
Pediatriset potilaat
Calquence-valmisteen turvallisuutta ja tehoa 0–18 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Calquence otetaan suun kautta. Kapselit niellään kokonaisina veden kanssa suunnilleen samaan aikaan joka päivä, joko aterian yhteydessä tai tyhjään mahaan (ks. kohta Yhteisvaikutukset). Kapseleita ei saa pureskella, liuottaa eikä avata, sillä tällöin lääkkeen imeytyminen elimistöön saattaa muuttua.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Verenvuoto
Merkittäviä verenvuototapahtumia, mukaan lukien keskushermoston ja maha-suolikanavan verenvuotoa, joista osa on johtanut kuolemaan, on ilmennyt hematologisia maligniteetteja sairastavilla potilailla, jotka ovat saaneet Calquence-valmistetta monoterapiana tai yhdistelmänä muiden lääkevalmisteiden kanssa. Tällaisia tapahtumia on ilmennyt potilailla sekä trombosytopenian yhteydessä että ilman trombosytopeniaa. Yleisesti ottaen verenvuototapahtumat eivät olleet kovin vaikeita; ne olivat esimerkiksi mustelmia ja pieniä verenpurkaumia (ks. kohta Haittavaikutukset).
Verenvuototapahtumien mekanismia ei ymmärretä kunnolla.
Antitromboottisia lääkkeitä käyttävillä potilailla saattaa olla suurentunut verenvuotoriski. Antitromboottisten lääkkeiden käytön yhteydessä on noudatettava varovaisuutta ja harkittava lisäseurantaa verenvuodon merkkien havaitsemiseksi, kun samanaikainen käyttö on lääketieteellisesti välttämätöntä. Varfariinia tai muita K-vitamiiniantagonisteja ei pidä käyttää samanaikaisesti Calquence-valmisteen kanssa.
Harkitse Calquence-valmisteen käytön keskeyttämisen hyötyjä ja riskejä; käyttö olisi hyvä keskeyttää ainakin 3 päiväksi sekä ennen leikkausta että sen jälkeen.
Infektiot
Vakavia infektioita (bakteeri-, virus- ja sieni-infektioita), mukaan lukien kuolemaan johtaneita tapahtumia, on ilmennyt hematologisia maligniteetteja sairastavilla potilailla, jotka ovat saaneet Calquence-valmistetta monoterapiana tai yhdistelmänä muiden lääkevalmisteiden kanssa. Tällaisia infektioita ilmeni pääasiassa ilman neutropeniaa; neutropeenisiä infektioita raportoitiin 10,1 %:lla monoterapiaa saaneista potilaista ja 26,8 %:lla yhdistelmähoitoa saaneista potilaista. Hepatiitti B ‑viruksen (HBV) ja herpes zoster ‑viruksen (HZV) reaktivaation aiheuttamia infektioita, aspergilloosia ja etenevää multifokaalista leukoenkefalopatiaa (PML) on ilmennyt (ks. kohta Haittavaikutukset).
Virusten reaktivaatio
Hepatiitti B ‑viruksen reaktivaatiotapauksia on raportoitu Calquence-valmistetta käyttävillä potilailla. Hepatiitti B ‑viruksen tilanne on selvitettävä ennen Calquence-hoidon aloittamista. Jos potilaan hepatiitti B ‑serologia on positiivinen, ennen hoidon aloittamista on konsultoitava maksasairauksiin erikoistunutta lääkäriä, ja potilasta on tarkkailtava ja hoidettava paikallisten hoito-ohjeiden mukaisesti hepatiitti B ‑viruksen reaktivaation ehkäisemiseksi.
Etenevän multifokaalisen leukoenkefalopatian (PML) tapauksia, mukaan lukien kuolemaan johtaneita tapauksia, on raportoitu Calquence-valmisteen käytön jälkeen. Näissä tapauksissa potilas oli saanut aiemmin tai samanaikaisesti immuunivastetta heikentävää hoitoa. Lääkärien on otettava huomioon PML:n mahdollisuus erotusdiagnostiikassa, jos potilaalle ilmaantuu uusia tai pahenevia neurologisia, kognitiivisia tai käyttäytymiseen liittyviä merkkejä ja oireita. Jos epäillään PML:aa, on tehtävä asianmukaiset diagnostiset arvioinnit ja Calquence-hoito on keskeytettävä, kunnes PML on suljettu pois. Jos asiasta on minkäänlaista epäilystä, potilas on ohjattava neurologin vastaanotolle ja on harkittava asianmukaisia PML:n diagnosoinnin toimenpiteitä. Näitä ovat mm. magneettikuvaus, mieluiten varjoaineella, JC-viruksen DNA:n testaaminen aivo-selkäydinnesteestä sekä toistetut neurologiset arvioinnit.
Tavanomaista estohoitoa on harkittava, jos potilaalla on suurentunut opportunististen infektioiden riski. Potilaan tilaa seurataan infektion merkkien ja oireiden havaitsemiseksi, ja potilaalle annetaan lääketieteellisesti tarkoituksenmukaista hoitoa.
Sytopeniat
Hoidosta aiheutuvia asteen 3 tai 4 sytopenioita, mukaan lukien neutropeniaa, anemiaa ja trombosytopeniaa, ilmeni hematologisia maligniteetteja sairastavilla potilailla, jotka olivat saaneet Calquence-valmistetta monoterapiana tai yhdistelmänä muiden lääkevalmisteiden kanssa. Täydellistä verenkuvaa pitää seurata lääketieteellisen tarpeen mukaan (ks. kohta Haittavaikutukset).
Sekundaarimaligniteetit
Sekundaarimaligniteetteja, mukaan lukien iho- sekä muita syöpäsairauksia, ilmeni hematologisia maligniteetteja sairastavilla potilailla, jotka olivat saaneet Calquence-valmistetta monoterapiana tai yhdistelmänä muiden lääkevalmisteiden kanssa. Ihosyöpiä raportoitiin yleisesti. Potilaiden tilaa on seurattava ihosyöpien ilmaantumisen havaitsemiseksi, ja potilaita on kehotettava suojautumaan auringolta (ks. kohta Haittavaikutukset).
Eteisvärinä
Eteisvärinää/eteislepatusta ilmeni hematologisia maligniteetteja sairastavilla potilailla, jotka olivat saaneet Calquence-valmistetta monoterapiana tai yhdistelmänä muiden lääkevalmisteiden kanssa. Potilaita on seurattava eteisvärinän ja eteislepatuksen oireiden (kuten sydämentykytyksen, huimauksen, pyörtymisen, rintakivun tai hengenahdistuksen) varalta, ja EKG on otettava lääketieteellisen tarpeen mukaan (ks. kohdat Yhteisvaikutukset ja Annostus ja antotapa). Jos potilaalle kehittyy eteisvärinää Calquence-hoidon aikana, tromboembolisen sairauden riski on arvioitava perusteellisesti. Jos potilaalla on suuri tromboembolisen sairauden riski, on harkittava tarkkaan kontrolloitua antikoagulanttihoitoa ja muita hoitovaihtoehtoja Calquence-valmisteen sijaan.
Tuumorilyysioireyhtymä
Calquence‑hoidon yhteydessä on ilmoitettu tuumorilyysioireyhtymää. Jos potilaalla katsotaan olevan tuumorilyysioireyhtymän riski (esim. kookas kasvainmassa [bulky] lähtötilanteessa), potilas on arvioitava mahdollisen tuumorilyysioireyhtymän riskin suhteen ja häntä on seurattava tarkasti kliinisen tarpeen mukaan.
Interstitiaalinen keuhkosairaus / pneumoniitti
Interstitiaalista keuhkosairautta / pneumoniittia on ilmoitettu potilailla, jotka saivat Calquence‑valmistetta yhdistelmänä bendamustiinin ja rituksimabin kanssa manttelisolulymfooman hoitoon. Potilaita on seurattava interstitiaaliseen keuhkosairauteen / pneumoniittiin viittaavien keuhko‑oireiden (esim. yskän, hengenahdistuksen tai hypoksian) varalta, ja interstitiaalinen keuhkosairaus / pneumoniitti on hoidettava kliinisen tarpeen mukaan.
Muut lääkevalmisteet
Voimakkaiden CYP3A:n estäjien samanaikainen käyttö Calquence-valmisteen kanssa saattaa suurentaa akalabrutinibialtistusta ja siten suurentaa toksisuusriskiä. Sen sijaan samanaikainen CYP3A:n indusorien käyttö saattaa pienentää akalabrutinibialtistusta, jolloin vaarana on tehon puute. Samanaikaista käyttöä voimakkaiden CYP3A:n estäjien kanssa on vältettävä. Jos tällaisia estäjiä käytetään lyhytaikaisesti (esimerkiksi infektiolääkkeitä enintään seitsemän päivän ajan), Calquence-hoito on keskeytettävä. Potilaiden tilaa on seurattava tarkasti toksisuuden merkkien havaitsemiseksi, jos käytetään keskivahvaa CYP3A:n estäjää (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset). Samanaikaista käyttöä voimakkaiden CYP3A4:n indusorien kanssa on vältettävä tehon mahdollisen heikkenemisen vuoksi.
Calquence sisältää natriumia
Tämä lääkevalmiste sisältää alle 1 mmol (23 mg) natriumia per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Akalabrutinibi ja sen aktiivinen metaboliitti metaboloituvat pääasiassa sytokromi P450 ‑entsyymin 3A4 (CYP3A4) välityksellä, ja molemmat aineet ovat P‑gp:n ja rintasyövän resistenssiproteiinin (BCRP) substraatteja.
Vaikuttavat aineet, jotka saattavat suurentaa akalabrutinibin pitoisuutta plasmassa
CYP3A:n/P‑gp:n estäjät
Samanaikainen voimakkaan CYP3A:n/P‑gp:n estäjän käyttö (200 mg itrakonatsolia kerran vuorokaudessa 5 päivän ajan) suurensi akalabrutinibin Cmax-arvoa 3,9‑kertaisesti ja AUC-arvoa 5,0‑kertaisesti terveillä tutkittavilla (N = 17).
Samanaikaista käyttöä voimakkaiden CYP3A:n/P‑gp:n estäjien kanssa on vältettävä. Jos käytetään lyhytaikaisesti voimakkaita CYP3A:n/P‑gp:n estäjiä (kuten ketokonatsolia, konivaptaania, klaritromysiinia, indinaviiria, itrakonatsolia, ritonaviiria, telapreviiria, posakonatsolia tai vorikonatsolia), Calquence-hoito on keskeytettävä (ks. kohta Annostus ja antotapa).
Keskivahvojen CYP3A:n estäjien samanaikainen anto (400 mg flukonatsolia kerta-annoksena tai 200 mg isavukonatsolia toistuvina annoksina 5 päivän ajan) terveille tutkittaville suurensi akalabrutinibin Cmax-arvoa ja AUC-arvoa 1,4–2-kertaiseksi samalla kun aktiivisen ACP-5862-metaboliitin Cmax ja AUC pienenivät 0,65–0,88-kertaiseksi verrattuna tilanteeseen, jossa akalabrutinibia annettiin ainoana lääkkeenä. Annosta ei tarvitse muuttaa keskivahvojen CYP3A:n estäjien käytön yhteydessä. Potilaan tilaa seurataan tarkasti haittavaikutusten havaitsemiseksi (ks. kohta Annostus ja antotapa).
Vaikuttavat aineet, jotka saattavat pienentää akalabrutinibin pitoisuutta plasmassa
CYP3A:n indusorit
Samanaikainen voimakkaan CYP3A:n indusorin käyttö (600 mg rifampisiinia kerran vuorokaudessa 9 päivän ajan) pienensi akalabrutinibin Cmax-arvoa 68 % ja AUC-arvoa 77 % terveillä tutkittavilla (N = 24).
Samanaikaista käyttöä voimakkaiden CYP3A:n toiminnan indusorien (kuten fenytoiinin, rifampisiinin tai karbamatsepiinin) kanssa on vältettävä. Samanaikaista hoitoa mäkikuismalla on vältettävä, sillä se saattaa ennalta-arvaamattomasti pienentää akalabrutinibin pitoisuutta plasmassa.
Mahahapon eritystä vähentävät lääkevalmisteet
Akalabrutinibin liukoisuus vähenee pH:n suurenemisen myötä. Akalabrutinibin samanaikainen käyttö antasidin (1 g kalsiumkarbonaattia) kanssa pienensi akalabrutinibin AUC-arvoa 53 % terveillä tutkittavilla. Samanaikainen käyttö protonipumpun estäjän (40 mg omepratsolia 5 päivän ajan) pienensi akalabrutinibin AUC-arvoa 43 %.
Jos hoito mahahapon eritystä vähentävillä lääkkeillä on tarpeen, kannattaa harkita antasidin (esim. kalsiumkarbonaatin) tai H2-reseptorin salpaajan (esim. ranitidiinin tai famotidiinin) käyttöä. Antasideja käytettäessä lääkevalmisteiden ottamisen välillä on oltava vähintään 2 tuntia (ks. kohta Annostus ja antotapa). H2-reseptorin salpaajia käytettäessä Calquence otetaan 2 tuntia ennen H2-reseptorin salpaajan ottamista (tai 10 tuntia sen ottamisen jälkeen).
Koska protonipumpun estäjien vaikutus kestää pitkään, niiden käytön yhteydessä annosten ottaminen eri aikaan ei välttämättä estä yhteisvaikutuksia Calquence-valmisteen kanssa, joten samanaikaista käyttöä on vältettävä (ks. kohta Annostus ja antotapa).
Vaikuttavat aineet, joiden pitoisuuksia plasmassa Calquence saattaa muuttaa
CYP3A:n substraatit
In vitro ‑tietojen perusteella ei voida sulkea pois mahdollisuutta, että akalabrutinibi on CYP3A4:n estäjä suolistossa ja saattaa suurentaa altistusta suoliston CYP3A-metabolialle herkille CYP3A4:n substraateille. Varovaisuutta on noudatettava, jos käytetään samanaikaisesti akalabrutinibia ja suun kautta otettavia CYP3A4:n substraatteja, joilla on kapea terapeuttinen alue (esim. siklosporiinia, ergotamiinia tai pimotsidia).
Akalabrutinibin vaikutus CYP1A2:n substraatteihin
In vitro ‑tutkimusten mukaan akalabrutinibi indusoi CYP1A2:ta. Akalabrutinibin samanaikainen käyttö CYP1A2:n substraattien (kuten teofylliinin tai kofeiinin) kanssa saattaa pienentää altistusta niille.
Akalabrutinibin ja sen aktiivisen metaboliitin ACP‑5862:n vaikutukset lääkeaineiden kuljetusjärjestelmiin
Akalabrutinibi saattaa suurentaa altistusta samanaikaisesti käytetyille BCRP:n substraateille (kuten metotreksaatille) estämällä BCRP:ia suolistossa (ks. kohta Farmakokinetiikka). Maha-suolikanavassa ilmenevien yhteisvaikutusten mahdollisuuden minimoimiseksi suun kautta otettavat BCRP:n substraatit, joilla on kapea terapeuttinen alue, kuten metotreksaatti, on otettava vähintään 6 tuntia ennen akalabrutinibin ottamista tai vähintään 6 tuntia sen ottamisen jälkeen.
ACP‑5862 saattaa suurentaa altistusta samanaikaisesti käytetyille MATE1:n substraateille (kuten metformiinille) estämällä MATE1:tä (ks. kohta Farmakokinetiikka). Jos potilaat käyttävät samanaikaisesti Calquence-valmisteen kanssa lääkevalmisteita, joiden dispositio riippuu MATE1:stä (kuten metformiinia), heidän tilaansa on seurattava muuttuneeseen siedettävyyteen viittaavien merkkien havaitsemiseksi. Siedettävyys voi muuttua sen seurauksena, että altistus Calquence-hoidon aikana samanaikaisesti käytettävälle lääkitykselle suurenee.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisia, jotka voivat tulla raskaaksi, on kehotettava välttämään raskaaksi tulemista Calquence-hoidon aikana.
Raskaus
Akalabrutinibin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Eläinkokeissa tehtyjen havaintojen perusteella akalabrutinibialtistus raskauden aikana saattaa aiheuttaa sikiöön kohdistuvan riskin. Rotilla havaittiin synnytyshäiriöitä (vaikea tai pitkittynyt synnytys), ja valmisteen antamiseen tiineille kaneille liittyi sikiön kasvun hidastumista (ks. kohta Prekliiniset tiedot turvallisuudesta).
Calquence-valmistetta ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa akalabrutinibilla.
Imetys
Ei tiedetä, erittyykö akalabrutinibi ihmisillä äidinmaitoon. Ei ole olemassa tietoa akalabrutinibin vaikutuksista imetettävään lapseen tai maidoneritykseen. Imettävien rottien maidossa havaittiin akalabrutinibia ja sen aktiivista metaboliittia. Imetettävään lapseen kohdistuvia riskejä ei voida sulkea pois. Imettäviä äitejä kehotetaan olemaan imettämättä Calquence-hoidon aikana ja kahden vuorokauden ajan viimeisen annoksen ottamisen jälkeen.
Hedelmällisyys
Calquence-valmisteen vaikutuksista ihmisen hedelmällisyyteen ei ole tietoja. Ei-kliinisessä akalabrutinibitutkimuksessa koiras- ja naarasrotilla ei havaittu hedelmällisyysparametreihin kohdistuvia haitallisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Calquence-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn. Akalabrutinibihoidon aikana on kuitenkin raportoitu uupumusta ja huimausta, ja jos potilaalla on tällaisia oireita, häntä on kehotettava olemaan ajamatta tai käyttämättä koneita, kunnes oireet ovat lievittyneet.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Calquence-monoterapia
Niillä 1 478 potilaalla, jotka saivat Calquence-monoterapiaa, yleisimpiä (≥ 20 %) minkä tahansa vaikeusasteen haittavaikutuksia olivat infektio, ripuli, päänsärky, tuki- ja liikuntaelinperäinen kipu, mustelmat, yskä, nivelkipu, uupumus, pahoinvointi ja ihottuma. Yleisimmin (≥ 5 %) raportoituja vaikeusasteen ≥ 3 haittavaikutuksia olivat infektio, leukopenia, neutropenia, anemia, sekundaarimaligniteetti ja trombosytopenia.
Calquence yhdistelmänä obinututsumabin kanssa
Niillä 223 potilaalla, jotka saivat Calquence-valmistetta yhdistelmänä obinututsumabin kanssa, yleisimpiä (≥ 20 %) minkä tahansa vaikeusasteen haittavaikutuksia olivat infektio, tuki- ja liikuntaelinperäinen kipu, ripuli, päänsärky, leukopenia, neutropenia, yskä, uupumus, nivelkipu, pahoinvointi, huimaus ja ummetus. Yleisimmin (≥ 5 %) raportoituja vaikeusasteen ≥ 3 haittavaikutuksia olivat leukopenia, neutropenia, infektio, trombosytopenia ja anemia.
Calquence yhdistelmänä venetoklaksin kanssa
Niillä 291 potilaalla, jotka saivat Calquence-valmistetta yhdistelmänä venetoklaksin kanssa, yleisimpiä (≥ 20 %) minkä tahansa vaikeusasteen haittavaikutuksia olivat infektiot, neutropenia, päänsärky, mustelmat, ripuli ja tuki- ja liikuntaelinperäinen kipu. Yleisimmin (≥ 5 %) raportoitu vaikeusasteen ≥ 3 haittavaikutus oli neutropenia.
Calquence yhdistelmänä venetoklaksin ja obinututsumabin kanssa
Niillä 284 potilaalla, jotka saivat Calquence-valmistetta yhdistelmänä venetoklaksin ja obinututsumabin kanssa, yleisimpiä (≥ 20 %) minkä tahansa vaikeusasteen haittavaikutuksia olivat infektiot, neutropenia, päänsärky, mustelmat, ripuli, pahoinvointi ja tuki- ja liikuntaelinperäinen kipu. Yleisimmin (≥ 5 %) raportoituja vaikeusasteen ≥ 3 haittavaikutuksia olivat neutropenia ja trombosytopenia.
Calquence yhdistelmänä bendamustiinin ja rituksimabin kanssa
Niillä 297 potilaalla, jotka saivat Calquence‑valmistetta yhdistelmänä bendamustiinin ja rituksimabin kanssa, yleisimpiä (≥ 20 %) minkä tahansa vaikeusasteen haittavaikutuksia olivat neutropenia, pahoinvointi, ihottuma, ripuli, tuki‑ ja liikuntaelinperäinen kipu, päänsärky, uupumus, oksentelu, ummetus, anemia ja trombosytopenia. Yleisimmin (≥ 5 %) raportoituja vaikeusasteen ≥ 3 haittavaikutuksia olivat neutropenia, ihottuma, trombosytopenia, anemia, keuhkokuume, sekundaarimaligniteetit, hypertensio ja muut sekundaarimaligniteetit kuin ei‑melanoottinen ihosyöpä.
Haittavaikutustaulukko
Seuraavissa taulukoissa on esitetty kliinisissä tutkimuksissa todetut haittavaikutukset potilailla, jotka saivat Calquence-valmistetta monoterapiana tai yhdistelmähoitona hematologisten maligniteettien hoitoon. Calquence-monoterapian keston mediaani yhdistetyssä tutkimusaineistossa oli 38,2 kuukautta. Calquence‑hoidon keston mediaani Calquence‑valmistetta yhdistelmänä bendamustiinin ja rituksimabin kanssa saaneilla potilailla oli 28,6 kuukautta. Calquence-hoidon keston mediaani potilailla, jotka saivat Calquence-valmistetta yhdistelmänä venetoklaksin kanssa joko yhdistettynä obinututsumabiin tai ilman obinututsumabia, oli 12,9 kuukautta.
Haittavaikutukset on lueteltu MedDRAn elinjärjestelmäluokituksen mukaisesti. Kussakin elinjärjestelmäluokassa haittavaikutukset on jaoteltu esiintymistiheyden mukaan niin, että yleisimmät haittavaikutukset mainitaan ensin. Lisäksi haittavaikutusten esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 4. Haittavaikutukset* hematologisia maligniteetteja sairastavilla potilailla, jotka ovat saaneet akalabrutinibi-monoterapiaa (N = 1 478)
| MedDRAn elinjärjestelmä-luokka | MedDRA-termi | Kaikki vaikeusasteet (%)
| Vaikeusaste ≥ 3* (%) |
| Infektiot | Ylähengitystieinfektio | Hyvin yleinen (25,8) | 1,2 |
| Keuhkokuume | Hyvin yleinen (15,8) | 8,7 | |
| Sinuiitti | Hyvin yleinen (11,4) | 0,4 | |
| Virtsatieinfektio | Yleinen (9,9) | 1,8 | |
| Keuhkoputkitulehdus | Yleinen (9,7) | 0,6 | |
| Herpesvirusinfektiot† | Yleinen (9,1) | 0,9 | |
| Nenänielutulehdus | Yleinen (8,3) | 0 | |
| Aspergillus-infektiot† | Melko harvinainen (0,7) | 0,6 | |
| Hepatiitti B ‑viruksen reaktivaatio | Melko harvinainen (0,4) | 0,3 | |
| Hyvän- ja pahanlaatuiset kasvaimet |
Sekundaarimaligniteetti† Ei-melanoottinen ihosyöpä† Muu sekundaarimaligniteetti kuin ei-melanoottinen ihosyöpä† | Hyvin yleinen (17,6) Yleinen (9,9) Yleinen (9,7) | 6,7 1,4 5,5 |
| Veri ja imukudos | Neutropenia† | Hyvin yleinen (19,4) | 17,5 |
| Anemia† | Hyvin yleinen (17,1) | 9,5 | |
| Trombosytopenia† | Hyvin yleinen (11,5) | 6,2 | |
| Lymfosytoosi | Melko harvinainen (0,5) | 0,3 | |
| Aineenvaihdunta ja ravitsemus | Tuumorilyysioireyhtymä | Melko harvinainen (0,5) | 0,4 |
| Hermosto | Päänsärky | Hyvin yleinen (36,5) | 1,2 |
| Huimaus | Hyvin yleinen (13,9) | 0,1 | |
| Sydän | Eteisvärinä/eteislepatus† | Yleinen (7,4) | 2,3 |
Verisuonisto | Mustelmien muodostuminen† Ruhjevamma Pienet verenpurkaumat Mustelmat
| Hyvin yleinen (30,9) Hyvin yleinen (20,7) Yleinen (8,9) Yleinen (5,7)
| 0 0 0 0 |
Verenvuoto/verenpurkauma† Maha-suolikanavan verenvuoto Kallonsisäinen verenvuoto | Hyvin yleinen (16,3) Melko harvinainen (0,9)
Melko harvinainen (0,1) | 3,2 0,7
0,1 | |
| Hypertensio† | Hyvin yleinen (11,9) | 4,9 | |
| Nenäverenvuoto | Yleinen (8,0) | 0,3 | |
| Ruoansulatus-elimistö | Ripuli | Hyvin yleinen (36,7) | 2,6 |
| Pahoinvointi | Hyvin yleinen (21,8) | 0,8 | |
| Ummetus | Hyvin yleinen (15,2) | 0,1 | |
| Vatsakipu† | Hyvin yleinen (14,5) | 1,2 | |
| Oksentelu | Hyvin yleinen (14,0) | 0,7 | |
| Iho ja ihonalainen kudos | Ihottuma† | Hyvin yleinen (20,3) | 0,9 |
| Luusto, lihakset ja sidekudos | Tuki- ja liikuntaelinperäinen kipu† | Hyvin yleinen (31,9) | 1,8 |
| Nivelkipu | Hyvin yleinen (24,0) | 0,9 | |
| Yleisoireet ja antopaikassa todettavat haitat | Uupumus | Hyvin yleinen (23,6) | 2,0 |
| Voimattomuus | Yleinen (7,0) | 0,9 | |
Tutkimukset§ (Löydökset perustuvat koetuloksiin)
| Hemoglobiiniarvon pieneneminen± | Hyvin yleinen (47,4) | 10,8 |
| Absoluuttisen neutrofiilimäärän pieneneminen± | Hyvin yleinen (43,9) | 24,0 | |
| Verihiutalemäärän pieneneminen± | Hyvin yleinen (36,9) | 9,5 |
*NCI:n CTCAE-luokituksen (National Cancer Institute Common Terminology Criteria for Adverse Events) version 4.03 mukaan.
†Sisältää useita haittavaikutustermejä.
±Tarkoittaa laboratoriolöydösten ilmaantuvuutta, ei raportoitujen haittatapahtumien ilmaantuvuutta.
§Ilmoitetaan CTCAE-asteina.
Taulukko 5. Haittavaikutukset* hematologisia maligniteetteja sairastavilla potilailla, jotka ovat saaneet akalabrutinibi-yhdistelmähoitoa (N = 1 095)
| Calquence + obinututsumabi N = 223 | Calquence + BR N = 297 | Calquence + venetoklaksi N = 291 | Calquence + venetoklaksi + obinututsumabi N = 284 | |||||
|---|---|---|---|---|---|---|---|---|
| MedDRA‑elinjärjestelmäluokka ja MedDRA‑termi |
Kaikki vaikeusasteet
| Vaikeusaste ≥ 3* (%) |
Kaikki vaikeusasteet
| Vaikeusaste ≥ 3* (%) |
Kaikki vaikeusasteet
| Vaikeusaste ≥ 3* (%) |
Kaikki vaikeusasteet
| Vaikeusaste ≥ 3* (%) |
| Infektiot | ||||||||
| Ylähengitystieinfektio | Hyvin yleinen (31,4) | 1,8 | Hyvin yleinen (18,2) | 0,3 | Yleinen (8,2) | 0,3 | Yleinen (6,3) | 0 |
| Sinuiitti | Hyvin yleinen (15,2) | 0,4 | Yleinen (6,4) | 0 | Yleinen (2,7) | 0 | Yleinen (2,5) | 0 |
| Nenänielutulehdus | Hyvin yleinen (13,5) | 0,4 | Yleinen (5,4) | 0 | Yleinen (1,4) | 0 | Yleinen (1,1) | 0 |
| Virtsatieinfektio | Hyvin yleinen (13) | 0,9 | Hyvin yleinen (11,1) | 1,7 | Yleinen (3,1) | 0 | Yleinen (6,0) | 0,4 |
| Keuhkokuume | Hyvin yleinen (10,8) | 5,4 | Hyvin yleinen (16,2) | 8,8 | Yleinen (3,8) | 1,4 | Yleinen (5,3) | 3,9 |
| Keuhkoputkitulehdus | Yleinen (9,9) | 0 | Yleinen (6,4) | 0,3 | Yleinen (2,1) | 0 | Yleinen (2,5) | 0 |
| Herpesvirusinfektiot† | Yleinen (6,7) | 1,3 | Hyvin yleinen (12,8) | 1,0 | Yleinen (4,8) | 0 | Yleinen (3,5) | 0,4 |
| Etenevä multifokaalinen leukoenkefalopatia | Melko harvinainen (0,4) | 0,4 | Tuntematon | 0 | Tuntematon | 0 | Tuntematon | 0 |
| Hepatiitti B ‑viruksen reaktivaatio | Melko harvinainen (0,9) | 0,1 | Yleinen (1,3) | 0,3 | Tuntematon | 0 | Tuntematon | 0 |
| Aspergillus‑infektiot† | Tuntematon | 0 | Melko harvinainen (0,3) | 0,3 | Tuntematon | 0 | Melko harvinainen (0,4) | 0,4 |
| Hyvän‑ ja pahanlaatuiset kasvaimet | ||||||||
Sekundaarimaligniteetti†
Ei‑melanoottinen ihosyöpä†
Muu sekundaarimaligniteetti kuin ei‑melanoottinen ihosyöpä† | Hyvin yleinen (13)
Yleinen (7,6)
Yleinen (6,3) | 4,0
0,4
3,6 | Hyvin yleinen (17,8)
Hyvin yleinen (11,1)
Yleinen (9,8) | 7,4
2,0
5,4 | Yleinen (5,2)
Yleinen (3,1)
Yleinen (2,7) | 1,7
0
1,7 | Yleinen (4,2)
Yleinen (1,8)
Yleinen (2,5) | 1,8
0,4
1,4 |
| Veri ja imukudos | ||||||||
| Neutropenia† | Hyvin yleinen (31,8) | 30 | Hyvin yleinen (54,9) | 50,2 | Hyvin yleinen (37,1) | 32,3 | Hyvin yleinen (50,4) | 46,1 |
| Trombosytopenia† | Hyvin yleinen (13,9) | 9 | Hyvin yleinen (22,9) | 9,8 | Yleinen (5,8) | 2,1 | Hyvin yleinen (12,3) | 9,2 |
| Anemia† | Hyvin yleinen (11,7) | 5,8 | Hyvin yleinen (24,2) | 9,4 | Yleinen (6,9) | 3,8 | Yleinen (4,6) | 2,1 |
| Lymfosytoosi | Melko harvinainen (0,4) | 0,4 | Melko harvinainen (0,7) | 0 | Tuntematon | 0 | Melko harvinainen (0,7) | 0,4 |
| Aineenvaihdunta ja ravitsemus | ||||||||
| Tuumorilyysioireyhtymä | Yleinen (1,8) | 1,3 | Yleinen (1,3) | 1,3 | Melko harvinainen (0,3) | 0,3 | Melko harvinainen (0,4) | 0,4 |
| Hermosto | ||||||||
| Päänsärky | Hyvin yleinen (43) | 0,9 | Hyvin yleinen (30,3) | 1,3 | Hyvin yleinen (35,1) | 1,4 | Hyvin yleinen (28,2) | 0,4 |
| Huimaus | Hyvin yleinen (23,8) | 0 | Hyvin yleinen (14,5) | 0,7 | Yleinen (5,5) | 0 | Yleinen (6,7) | 0 |
| Sydän | ||||||||
| Eteisvärinä/eteislepatus† | Yleinen (3,1) | 0,9 | Yleinen (6,7) | 4,0 | Melko harvinainen (0,7) | 0,3 | Yleinen (2,1) | 0,7 |
| Verisuonisto | ||||||||
Mustelmien muodostuminen†
Ruhjevamma
Pienet verenpurkaumat
Mustelmat | Hyvin yleinen (38,6)
Hyvin yleinen (27,4)
Hyvin yleinen (11,2)
Yleinen (3,1) | 0
0
0
0 | Hyvin yleinen (14,1)
Hyvin yleinen (11,1)
Yleinen (2,0)
Yleinen (3,0) | 0,3
0
0
0,3 | Hyvin yleinen (20,6)
Hyvin yleinen (14,1)
Yleinen (4,8)
Yleinen (2,7) | 0
0
0
0 | Hyvin yleinen (21,8)
Hyvin yleinen (16,2)
Yleinen (5,3)
Yleinen (3,9) | 0
0
0
0 |
Verenvuoto/verenpurkauma†
Maha‑suolikanavan verenvuoto
Kallonsisäinen verenvuoto | Hyvin yleinen (17,5)
Yleinen (3,6)
Melko harvinainen (0,9) | 1,3
0,9
0 | Hyvin yleinen (15,5)
Melko harvinainen (0,3)
Tuntematon | 1,0
0
0 | Yleinen (8,9)
Melko harvinainen (0,7)
Tuntematon | 0,7
0,3
0 | Yleinen (8,5)
Tuntematon
Tuntematon
| 1,1
0
0 |
| Hypertensio† | Hyvin yleinen (13,5) | 3,6 | Hyvin yleinen (12,5) | 5,7 | Yleinen (4,1) | 2,7 | Yleinen (3,9) | 2,1 |
| Nenäverenvuoto | Yleinen (8,5) | 0 | Yleinen (2,7) | 0 | Yleinen (1,7) | 0 | Yleinen (4,2) | 0 |
| Hengityselimet, rintakehä ja välikarsina | ||||||||
| Pneumoniitti± | ‑ | ‑ | Yleinen (2,4) | 0,3 | - | - | - | - |
| Ruoansulatuselimistö | ||||||||
| Ripuli | Hyvin yleinen (43,9) | 4,5 | Hyvin yleinen (37,4) | 3,0 | Hyvin yleinen (32,6) | 1,7 | Hyvin yleinen (36,3) | 1,4 |
| Pahoinvointi | Hyvin yleinen (26,9) | 0 | Hyvin yleinen (42,8) | 1,3 | Hyvin yleinen (14,8) | 0 | Hyvin yleinen (21,8) | 0,7 |
| Ummetus | Hyvin yleinen (20,2) | 0 | Hyvin yleinen (24,6) | 1,0 | Yleinen (6,5) | 0,3 | Yleinen (8,1) | 0 |
| Oksentelu | Hyvin yleinen (19,3) | 0,9 | Hyvin yleinen (25,6) | 0,7 | Yleinen (5,5) | 0 | Yleinen (6,7) | 0 |
| Vatsakipu† | Hyvin yleinen (14,8) | 1,3 | Hyvin yleinen (12,1) | 2,0 | Yleinen (7,9) | 1,0 | Yleinen (8,1) | 0,7 |
| Iho ja ihonalainen kudos | ||||||||
| Ihottuma† | Hyvin yleinen (30,9) | 1,8 | Hyvin yleinen (39,1) | 9,8 | Hyvin yleinen (12,0) | 0,3 | Hyvin yleinen (16,2) | 1,1 |
| Luusto, lihakset ja sidekudos | ||||||||
| Tuki‑ ja liikuntaelinperäinen kipu† | Hyvin yleinen (44,8) | 2,2 | Hyvin yleinen (34,3) | 3,7 | Hyvin yleinen (24,1) | 0,7 | Hyvin yleinen (21,8) | 1,1 |
| Nivelkipu | Hyvin yleinen (26,9) | 1,3 | Hyvin yleinen (17,5) | 0,7 | Hyvin yleinen (12,7) | 1,0 | Hyvin yleinen (10,9) | 0,4 |
| Yleisoireet ja antopaikassa todettavat haitat | ||||||||
| Uupumus | Hyvin yleinen (30,5) | 1,8 | Hyvin yleinen (29,3) | 2,7 | Hyvin yleinen (14,8) | 0,3 | Hyvin yleinen (14,4) | 0 |
| Voimattomuus | Yleinen (7,6) | 0,4 | Hyvin yleinen (10,4) | 1,0 | Yleinen (4,1) | 0 | Yleinen (3,2) | 0 |
| Tutkimukset¶ | ||||||||
| Absoluuttisen neutrofiilimäärän pieneneminen§ | Hyvin yleinen (57,4) | 35 | Hyvin yleinen (76,8) | 56,6 | Hyvin yleinen (78,0) | 38,1 | Hyvin yleinen (81,7) | 53,5 |
| Verihiutalemäärän pieneneminen§ | Hyvin yleinen (46,2) | 10,8 | Hyvin yleinen (69,4) | 17,8 | Hyvin yleinen (42,6) | 5,2 | Hyvin yleinen (54,9) | 13,7 |
| Hemoglobiiniarvon pieneneminen§ | Hyvin yleinen (43,9) | 9 | Hyvin yleinen (79,5) | 10,8 | Hyvin yleinen (34,7) | 6,5 | Hyvin yleinen (45,8) | 3,5 |
| ALAT‑arvon suureneminen‡ | ‑ | ‑ | Yleinen (9,1) | 4,4 | - | - | - | - |
| ASAT‑arvon suureneminen‡ | ‑ | ‑ | Yleinen (8,1) | 3,0 | - | - | - | - |
*NCI:n CTCAE‑luokituksen (National Cancer Institute Common Terminology Criteria for Adverse Events) version 4.03 mukaan.
†Sisältää useita haittavaikutustermejä.
±Yksi kuolemaan johtanut tapahtuma ilmoitettiin.
§Tarkoittaa laboratoriolöydösten ilmaantuvuutta, ei raportoitujen haittatapahtumien ilmaantuvuutta.
¶Ilmoitetaan CTCAE‑asteina.
‡Haittavaikutus vain ECHO‑tutkimuksen Calquence + BR ‑haarassa.
Valikoitujen haittavaikutusten kuvaus
Vakavat infektiot, kun potilaat saivat Calquence-valmistetta yhdistelmänä venetoklaksin kanssa joko yhdistettynä obinututsumabiin tai ilman obinututsumabia
Calquence-valmistetta yhdistelmänä venetoklaksin kanssa saaneista 291 potilaasta 12,4 %:lla raportoitiin vaikeita (vaikeusasteen ≥ 3) infektioita (useimmin raportoituja olivat COVID‑19 ja COVID‑19-keuhkokuume). Kuolemaan johtaneita infektioita ilmeni 3,1 %:lla potilaista (useimmin raportoituja olivat COVID‑19 ja COVID‑19-keuhkokuume).
Calquence-valmistetta yhdistelmänä venetoklaksin ja obinututsumabin kanssa saaneista 284 potilaasta 23,6 %:lla raportoitiin vaikeita (vaikeusasteen ≥ 3) infektioita (useimmin raportoituja olivat COVID‑19 ja COVID‑19-keuhkokuume). Kuolemaan johtaneita infektioita ilmeni 5,6 %:lla potilaista (useimmin raportoituja olivat COVID‑19 ja COVID‑19-keuhkokuume).
Hoidon lopettaminen ja annoksen pienentäminen haittavaikutusten vuoksi
Calquence-monoterapiaa saaneista 1 478 potilaasta 14,6 %:n raportoitiin lopettaneen hoidon haittavaikutusten vuoksi. Pääasiallisia hoidon lopettamiseen johtaneita haittavaikutuksia olivat mm. keuhkokuume, trombosytopenia ja ripuli. Haittavaikutuksista johtunutta annoksen pienentämistä raportoitiin 5,9 %:lla potilaista. Pääasiallisia annoksen pienentämiseen johtaneita haittavaikutuksia olivat mm. hepatiitti B ‑viruksen reaktivaatio, sepsis ja ripuli.
Calquence-valmistetta yhdistelmänä obinututsumabin kanssa saaneista 223 potilaasta 10,8 %:n raportoitiin lopettaneen Calquence-hoidon haittavaikutusten takia. Pääasiallisia hoidon lopettamiseen johtaneita haittavaikutuksia olivat mm. keuhkokuume, trombosytopenia ja ripuli. Haittavaikutuksista johtunutta annoksen pienentämistä raportoitiin 6,7 %:lla potilaista. Pääasiallisia annoksen pienentämiseen johtaneita haittavaikutuksia olivat mm. neutropenia, ripuli ja oksentelu.
Calquence-valmistetta yhdistelmänä venetoklaksin kanssa saaneista 291 potilaasta 7,6 %:n raportoitiin lopettaneen Calquence-hoidon haittavaikutusten vuoksi ja 5,8 %:lla raportoitiin, että Calquence-annosta oli pienennetty haittavaikutusten vuoksi. Pääasiallisia hoidon lopettamiseen johtaneita haittavaikutuksia olivat mm. COVID‑19-keuhkokuume ja COVID‑19, ja annoksen pienentämiseen johtanut haittavaikutus oli neutropenia.
Calquence-valmistetta yhdistelmänä venetoklaksin ja obinututsumabin kanssa saaneista 284 potilaasta 13,7 %:n raportoitiin lopettaneen Calquence-hoidon haittavaikutusten vuoksi ja 6,3 %:lla raportoitiin, että Calquence-annosta oli pienennetty haittavaikutusten vuoksi. Pääasiallisia hoidon lopettamiseen johtaneita haittavaikutuksia olivat mm. COVID‑19-keuhkokuume ja COVID‑19, ja annoksen pienentämiseen johtanut haittavaikutus oli neutropenia.
Calquence‑valmistetta yhdistelmänä bendamustiinin ja rituksimabin kanssa saaneista 297 potilaasta 42,8 %:n raportoitiin lopettaneen Calquence-hoidon haittavaikutusten vuoksi. Pääasiallisia hoidon lopettamiseen johtaneita haittavaikutuksia olivat mm. COVID‑19, COVID‑19‑keuhkokuume, neutropenia ja keuhkokuume. Haittavaikutuksista johtunutta annoksen pienentämistä raportoitiin 10,1 %:lla potilaista. Pääasiallisia annoksen pienentämiseen johtaneita haittavaikutuksia olivat mm. neutropenia ja pahoinvointi.
Iäkkäät potilaat
Kliinisiin Calquence-monoterapiatutkimuksiin osallistuneista 1 478 potilaasta 42 % oli yli 65‑vuotiaita ja alle 75‑vuotiaita ja 20,6 % oli vähintään 75‑vuotiaita. Kliinisesti merkityksellisiä eroja turvallisuudessa tai tehossa ei havaittu ≥ 65‑vuotiaiden ja sitä nuorempien välillä.
Niistä 223 potilaasta, jotka osallistuivat Calquence-valmisteen ja obinututsumabin yhdistelmähoidolla tehtyihin kliinisiin tutkimuksiin, 47 % oli yli 65‑vuotiaita ja alle 75‑vuotiaita ja 26 % oli vähintään 75‑vuotiaita. Kliinisesti merkityksellisiä eroja turvallisuudessa tai tehossa ei havaittu ≥ 65‑vuotiaiden ja sitä nuorempien välillä.
Niistä 291 potilaasta, jotka saivat Calquence-valmistetta yhdistelmänä venetoklaksin kanssa, 28,9 % oli yli 65‑vuotiaita ja alle 75‑vuotiaita ja 4,5 % oli vähintään 75‑vuotiaita. Kliinisesti merkityksellisiä eroja turvallisuudessa tai tehossa ei havaittu ≥ 65‑vuotiaiden ja sitä nuorempien välillä.
Niistä 284 potilaasta, jotka saivat Calquence-valmistetta yhdistelmänä venetoklaksin ja obinututsumabin kanssa, 24 % oli yli 65‑vuotiaita ja alle 75‑vuotiaita ja 6,3 % oli vähintään 75‑vuotiaita. Kliinisesti merkityksellisiä eroja turvallisuudessa tai tehossa ei havaittu ≥ 65‑vuotiaiden ja sitä nuorempien välillä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Akalabrutinibin yliannostukseen ei ole spesifistä hoitoa, eikä yliannostuksen oireita ole varmistettu. Yliannostustapauksessa potilaan tilaa täytyy seurata tarkasti haittavaikutusten merkkien ja oireiden havaitsemiseksi, ja asianmukainen oireenmukainen hoito on aloitettava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, proteiinikinaasin estäjät, ATC-koodi: L01EL02.
Vaikutusmekanismi
Akalabrutinibi on selektiivinen Brutonin tyrosiinikinaasin (BTK) estäjä. BTK on B-soluantigeenireseptori- (BCR‑) ja sytokiinireseptorireittien signaalimolekyyli. B-soluissa BTK-signaalivälitys saa aikaan B-solujen säilymisen elossa ja proliferaation, ja sitä tarvitaan soluadheesioon, soluliikenteeseen ja solujen kemotaksikseen.
Akalabrutinibi ja sen aktiivinen metaboliitti ACP‑5862 muodostavat kovalenttisen sidoksen BTK:n aktiivisen kohdan kysteiinitähteeseen, mikä johtaa BTK:n palautumattomaan inaktivaatioon, jolloin off-target-yhteisvaikutuksia on erittäin vähän.
Farmakodynaamiset vaikutukset
B-solusyöpiä sairastavilla potilailla, jotka saivat 100 mg akalabrutinibia kaksi kertaa vuorokaudessa, BTK:iin sitoutumisen mediaani vakaassa tilassa ääreisveressä pysyi tasolla ≥ 95 % yli 12 tunnin ajan, minkä ansiosta BTK:n inaktivaatio säilyi koko suositellun annosvälin ajan.
Sydämen elektrofysiologia
Akalabrutinibin vaikutusta QTc-aikaan arvioitiin 46 terveellä mies- ja naistutkittavalla satunnaistetussa, kaksoissokkoutetussa kattavassa QT-tutkimuksessa, jossa käytettiin lumelääkettä ja positiivisia vertailuvalmisteita. Käytettäessä annosta, joka oli suurempi kuin terapeuttinen annos (4 kertaa suositeltu enimmäisannos), Calquence ei pidentänyt QT/QTc-aikaa kliinisesti merkittävästi (esim. enintään 10 ms) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset ja Prekliiniset tiedot turvallisuudesta).
Kliininen teho ja turvallisuus
Aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavat potilaat
Calquence monoterapiana tai yhdistelmänä obinututsumabin kanssa
Calquence-valmisteen turvallisuutta ja tehoa monoterapiana tai yhdistelmänä obinututsumabin kanssa aiemmin hoitamattoman kroonisen lymfaattisen leukemian hoidossa arvioitiin satunnaistetussa, avoimessa vaiheen 3 monikeskustutkimuksessa (ELEVATE-TN), johon osallistui 535 potilasta. Potilaat saivat joko Calquence-valmistetta ja obinututsumabia, Calquence-valmistetta monoterapiana tai obinututsumabia ja klorambusiilia. ELEVATE-TN-tutkimuksessa oli mukana vähintään 65‑vuotiaita potilaita sekä 18–65-vuotiaita potilaita, joilla oli muita samanaikaisia sairauksia. 27,9 %:lla potilaista kreatiniinipuhdistuma oli < 60 ml/min. Alle 65-vuotiaista potilaista 16,1 %:lla CIRS-G-pistemäärän (Cumulative Illness Rating Scale for Geriatrics) mediaani oli 8. Tutkimuksessa potilaille sallittiin antitromboottisten lääkkeiden käyttö. Tutkimuksesta suljettiin pois potilaat, jotka tarvitsivat varfariinia tai vastaavaa K-vitamiiniantagonistia antikoagulaatiohoitona.
Potilaat satunnaistettiin suhteessa 1:1:1 kolmeen hoitohaaraan, joissa he saivat:
- Calquence-valmistetta ja obinututsumabia (Calquence+G): Calquence-valmistetta annettiin 100 mg kaksi kertaa vuorokaudessa hoitojakson 1 päivästä 1 alkaen siihen asti, että tauti eteni tai ilmeni toksisuutta, jota ei voitu hyväksyä. Obinututsumabia annettiin hoitojakson 2 päivästä 1 alkaen enintään 6 hoitojakson ajan. Obinututsumabia annettiin 1 000 mg hoitojakson 2 päivinä 1 ja 2 (100 mg päivänä 1 ja 900 mg päivänä 2), 8 ja 15 ja sen jälkeen 1 000 mg hoitojaksojen 3–7 päivänä 1. Kaikkien hoitojaksojen pituus oli 28 päivää.
- Calquence-valmistetta monoterapiana: Calquence-valmistetta annettiin 100 mg kaksi kertaa vuorokaudessa siihen asti, että tauti eteni tai ilmeni toksisuutta, jota ei voitu hyväksyä.
- obinututsumabia ja klorambusiilia (GClb): Obinututsumabia ja klorambusiilia annettiin enintään 6 hoitojakson ajan. Obinututsumabia annettiin 1 000 mg hoitojakson 1 päivinä 1 ja 2 (100 mg päivänä 1 ja 900 mg päivänä 2), 8 ja 15 ja sen jälkeen 1 000 mg hoitojaksojen 2–6 päivänä 1. Klorambusiilia annettiin 0,5 mg/kg hoitojaksojen 1–6 päivinä 1 ja 15. Kaikkien hoitojaksojen pituus oli 28 päivää.
Potilaat stratifioitiin 17p-deleetion (mutaatio vs. ei mutaatiota), ECOG-toimintakykyluokan (0 tai 1 vs. 2) ja maantieteellisen alueen (Pohjois-Amerikka ja Länsi-Eurooppa vs. muut) mukaan. Kun taudin eteneminen oli vahvistettu, 45 GClb-haaraan satunnaistettua potilasta siirrettiin saamaan Calquence-valmistetta monoterapiana. Taulukossa 6 on esitetty yhteenveto tutkimuspopulaation demografisista tiedoista ja sairauden ominaispiirteistä lähtötilanteessa.
Taulukko 6. Potilaiden ominaisuudet lähtötilanteessa aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavilla potilailla (ELEVATE-TN)
| Ominaisuus | Calquence ja obinututsumabi N = 179 | Calquence-monoterapia N = 179 | Obinututsumabi ja klorambusiili N = 177 |
| Ikä vuosina; mediaani (vaihteluväli) | 70 (41–88) | 70 (44–87) | 71 (46–91) |
| Miehiä; % | 62 | 62 | 59,9 |
| Valkoihoisia; % | 91,6 | 95 | 93,2 |
| ECOG-toimintakykyluokka 0–1; % | 94,4 | 92,2 | 94,4 |
| Diagnoosista kuluneen ajan mediaani (kk) | 30,5 | 24,4 | 30,7 |
| Syöpäsolujen kertyminen imusolmukkeisiin ja muihin elimiin, imusolmukkeet ≥ 5 cm; % | 25,7 | 38 | 31,1 |
| Sytogenetiikka/FISH-kategoria; % | |||
| 17p-deleetio | 9,5 | 8,9 | 9 |
| 11q-deleetio | 17,3 | 17,3 | 18,6 |
| TP53-mutaatio | 11,7 | 10,6 | 11,9 |
| Mutatoitumaton IGHV | 57,5 | 66,5 | 65,5 |
| Kompleksinen karyotyyppi (≥ 3 poikkeavuutta) | 16,2 | 17,3 | 18,1 |
| Rai-luokka; % | |||
| 0 | 1,7 | 0 | 0,6 |
| I | 30,2 | 26,8 | 28,2 |
| II | 20,1 | 24,6 | 27,1 |
| III | 26,8 | 27,9 | 22,6 |
| IV | 21,2 | 20,7 | 21,5 |
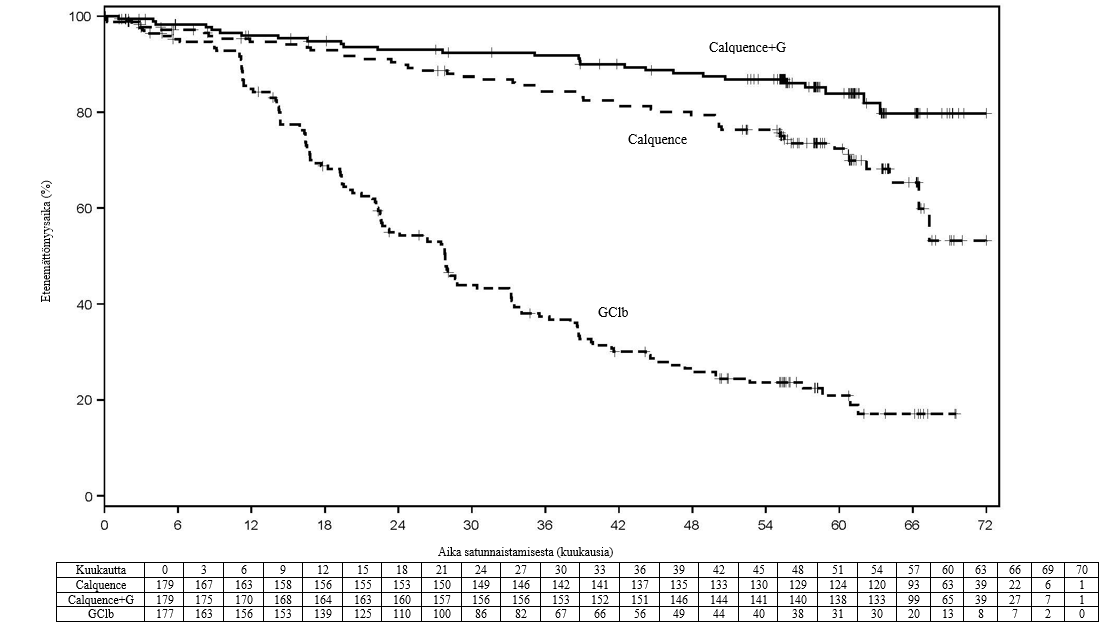
Ensisijainen päätetapahtuma oli etenemättömyysaika (PFS) Calquence+G-haarassa verrattuna GClb-haaraan riippumattoman arviointikomitean (Independent Review Committee, IRC) arvioimana vuoden 2008 IWCLL-kriteerien (International Workshop on Chronic Lymphocytic Leukaemia) mukaisesti niin, että kriteereihin oli sisällytetty hoitoon liittyvää lymfosytoosia koskeva selvitys (Cheson 2012). Seuranta-ajan mediaani oli 28,3 kuukautta, ja IRC:n arvioima etenemättömyysaika osoitti, että taudin etenemisen tai kuoleman riski oli aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavilla potilailla 90 % pienempi Calquence+G-haarassa kuin GClb-haarassa. Tulos oli tilastollisesti merkitsevä. Tehoa koskevat tulokset on esitetty taulukossa 7.
Taulukko 7. IRC:n arvioiden mukaiset tehoa koskevat tulokset kroonista lymfaattista leukemiaa sairastavilla potilailla (ELEVATE-TN)
Calquence ja obinututsumabi N = 179 | Calquence-monoterapia N = 179 | Obinututsumabi ja klorambusiili N = 177 | |
| Etenemättömyysaika* | |||
| Tapahtumien määrä (%) | 14 (7,8) | 26 (14,5) | 93 (52,5) |
| Etenevä tauti, n (%) | 9 (5) | 20 (11,2) | 82 (46,3) |
| Kuolemat (%) | 5 (2,8) | 6 (3,4) | 11 (6,2) |
| Mediaani (95 %:n luottamusväli), kk | Ei saavutettu | Ei saavutettu (34,2, ei saavutettu) | 22,6 (20,2, 27,6) |
| HR† (95 %:n luottamusväli) | 0,10 (0,06, 0,17) | 0,20 (0,13, 0,30) | - |
| p-arvo | < 0,0001 | < 0,0001 | - |
| 24 kk:n arvio, % (95 %:n luottamusväli) | 92,7 (87,4, 95,8) | 87,3 (80,9, 91,7) | 46,7 (38,5, 54,6) |
| Kokonaiselossaoloaikaa | |||
| Kuolemat (%) | 9 (5) | 11 (6,1) | 17 (9,6) |
| HR (95 %:n luottamusväli) † | 0,47 (0,21, 1,06) | 0,60 (0,28, 1,27) | - |
| Paras kokonaisvasteluku (ORR)* (CR + CRi + nPR + PR) | |||
ORR, n (%) (95 %:n luottamusväli) | 168 (93,9) (89,3, 96,5) | 153 (85,5) (79,6, 89,9) | 139 (78,5) (71,9, 83,9) |
| p-arvo | < 0,0001 | 0,0763 | - |
| CR, n (%) | 23 (12,8) | 1 (0,6) | 8 (4,5) |
| CRi, n (%) | 1 (0,6) | 0 | 0 |
| nPR, n (%) | 1 (0,6) | 2 (1,1) | 3 (1,7) |
| PR, n (%) | 143 (79,9) | 150 (83,8) | 128 (72,3) |
HR = riskitiheyksien suhde; CR = täydellinen vaste; CRi = täydellinen vaste ilman veriarvojen täydellistä palautumista; nPR = nodulaarinen osittainen vaste; PR = osittainen vaste.
* IRC:n arvion mukaan.
† Ositetun Coxin verrannollisten riskitiheyksien mallin mukaan.
a Kokonaiselossaoloajan mediaania ei saavutettu molemmissa hoitohaaroissa.
Calquence-hoidon (obinututsumabin kanssa tai ilman obinututsumabia) yhteydessä todettua etenemättömyysaikaa koskevat tulokset olivat yhdenmukaiset kaikissa alaryhmissä, myös suuren riskin ryhmissä. Korkean riskin (17p-deleetio, 11q-deleetio, TP53-mutaatio tai mutatoitumaton IGHV) kroonista lymfaattista leukemiaa sairastavilla tutkittavilla etenemättömyysaikaa koskeva riskitiheyksien suhde oli 0,08 [95 %:n luottamusväli (0,04, 0,15)], kun verrattiin Calquence-valmistetta ja obinututsumabia saaneiden ryhmää obinututsumabia ja klorambusiilia saaneisiin, ja 0,13 [95 %:n luottamusväli (0,08, 0,21)] verrattaessa pelkästään Calquence-valmistetta saaneita obinututsumabia ja klorambusiilia saaneisiin.
Taulukko 8. Etenemättömyysajan alaryhmäanalyysi (ELEVATE-TN-tutkimus)
| Calquence-monoterapia | Calquence+G | |||||
| N | HR | 95 %:n luottamusväli | N | HR | 95 %:n luottamusväli | |
| Kaikki tutkittavat | 179 | 0,20 | (0,13, 0,30) | 179 | 0,10 | (0,06, 0,17) |
| Del 17P | ||||||
| Kyllä | 19 | 0,20 | (0,06, 0,64) | 21 | 0,13 | (0,04, 0,46) |
| Ei | 160 | 0,20 | (0,12, 0,31) | 158 | 0,09 | (0,05, 0,17) |
| TP53-mutaatio | ||||||
| Kyllä | 19 | 0,15 | (0,05, 0,46) | 21 | 0,04 | (0,01, 0,22) |
| Ei | 160 | 0,20 | (0,12, 0,32) | 158 | 0,11 | (0,06, 0,20) |
| Del 17P tai/ja TP53-mutaatio | ||||||
| Kyllä | 23 | 0,23 | (0,09, 0,61) | 25 | 0,10 | (0,03, 0,34) |
| Ei | 156 | 0,19 | (0,11, 0,31) | 154 | 0,10 | (0,05, 0,18) |
| IGHV-mutaatio | ||||||
| Mutatoitunut | 58 | 0,69 | (0,31, 1,56) | 74 | 0,15 | (0,04, 0,52) |
| Mutatoitumaton | 119 | 0,11 | (0,07, 0,19) | 103 | 0,08 | (0,04, 0,16) |
| Del 11q | ||||||
| Kyllä | 31 | 0,07 | (0,02, 0,22) | 31 | 0,09 | (0,03, 0,26) |
| Ei | 148 | 0,26 | (0,16, 0,41) | 148 | 0,10 | (0,05, 0,20) |
| Kompleksinen karyotyyppi | ||||||
| Kyllä | 31 | 0,10 | (0,03, 0,33) | 29 | 0,09 | (0,03, 0,29) |
| Ei | 117 | 0,27 | (0,16, 0,46) | 126 | 0,11 | (0,05, 0,21) |
HR = riskitiheyksien suhde
Pitkän aikavälin tietojen osalta seuranta-ajan mediaani oli 58,2 kuukautta Calquence+G-haarassa, 58,1 kuukautta Calquence-haarassa ja 58,2 kuukautta GClb-haarassa. Tutkijan arvioiman etenemättömyysajan mediaania ei ollut saavutettu Calquence+G-haarassa eikä Calquence-monoterapian haarassa, ja GClb-haarassa se oli 27,8 kuukautta. Tuoreimpaan tiedonkeruun katkaisupäivään mennessä yhteensä 72 potilasta (40,7 %), jotka oli alun perin satunnaistettu GClb-haaraan, oli siirretty saamaan Calquence-monoterapiaa. Kokonaiselossaoloajan mediaania ei ollut saavutettu missään haarassa, ja tutkimuksessa oli tapahtunut yhteensä 76 kuolemaa: 18 (10,1 %) Calquence+G-haarassa, 30 (16,8 %) Calquence-monoterapian haarassa ja 28 (15,8 %) GClb-haarassa.
Taulukko 9. Tutkijan arvioiden mukaiset tehoa koskevat tulokset kroonista lymfaattista leukemiaa sairastavilla potilailla (ELEVATE-TN)
Calquence ja obinututsumabi N = 179 | Calquence-monoterapia N = 179 | Obinututsumabi ja klorambusiili N = 177 | |
|---|---|---|---|
| Etenemättömyysaika | |||
| Tapahtumien määrä (%) | 27 (15,1) | 50 (27,9) | 124 (70,1) |
| Etenevä tauti, n (%) | 14 (7,8) | 30 (16,8) | 112 (63,3) |
| Kuolemat (%) | 13 (7,3) | 20 (11,2) | 12 (6,8) |
| Mediaani (95 %:n luottamusväli), kk* | Ei saavutettu | Ei saavutettu (66,5, ei saavutettu) | 27,8 (22,6, 33,2) |
| HR† (95 %:n luottamusväli) | 0,11 (0,07, 0,16) | 0,21 (0,15, 0,30) | - |
| Kokonaiselossaoloaika | |||
| Kuolemat (%) | 18 (10,1) | 30 (16,8) | 28 (15,8) |
| HR (95 %:n luottamusväli)† | 0,55 (0,30, 0,99) | 0,98 (0,58, 1,64) | - |
HR = riskitiheyksien suhde
*95 %:n luottamusväli perustuu Kaplan–Meier-arvioon.
†Arvio perustuu ositettuun Coxin verrannollisten riskitiheyksien malliin riskitiheyksien suhteen (95 %:n luottamusvälin) osalta; stratifiointitekijänä oli 17p‑deleetio (kyllä vs. ei).
Kuva 1. Tutkijan arvioiman etenemättömyysajan Kaplan–Meier-käyrä kroonista lymfaattista leukemiaa sairastavilla potilailla (ELEVATE-TN, ITT-populaatio)
Aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavat potilaat – määräaikainen hoito
Calquence yhdistelmänä venetoklaksin kanssa joko yhdistettynä obinututsumabiin tai ilman obinututsumabia
Calquence-valmisteen turvallisuutta ja tehoa yhdistelmänä venetoklaksin kanssa (joko yhdistettynä obinututsumabiin tai ilman obinututsumabia) aiemmin hoitamattoman kroonisen lymfaattisen leukemian hoidossa arvioitiin satunnaistetussa, avoimessa vaiheen 3 monikeskustutkimuksessa (AMPLIFY), johon osallistui 867 potilasta. Potilaat saivat joko Calquence-valmisteen ja venetoklaksin yhdistelmää, Calquence-valmisteen, venetoklaksin ja obinututsumabin yhdistelmää tai kemoimmunoterapiaa eli tutkijan valinnan mukaan joko FCR-hoitoa (fludarabiini, syklofosfamidi ja rituksimabi) tai BR-hoitoa (bendamustiini ja rituksimabi). AMPLIFY-tutkimukseen osallistui vähintään 18‑vuotiaita aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavia potilaita, joilla ei ollut 17p-deleetiota tai TP53-mutaatiota. Tutkimuksessa potilaille sallittiin antitromboottisten lääkkeiden käyttö varfariinia ja muita K‑vitamiiniantagonisteja lukuun ottamatta.
Potilaat satunnaistettiin suhteessa 1:1:1 kolmeen hoitohaaraan, joissa he saivat
- Calquence-valmistetta ja venetoklaksia (AV): Calquence-valmistetta annettiin 100 mg kaksi kertaa vuorokaudessa hoitojakson 1 päivästä 1 alkaen yhteensä 14 hoitojakson ajan tai kunnes tauti eteni tai ilmeni toksisuutta, jota ei voitu hyväksyä. Hoitojakson 3 päivänä 1 potilaille aloitettiin 5 viikon mittainen venetoklaksin annostitrausohjelma, jossa valmistetta otettiin kerran vuorokaudessa aloittaen 20 mg:n annoksella ja suurentaen annosta viikoittain 50 mg:aan, 100 mg:aan, 200 mg:aan ja lopuksi 400 mg:aan. Venetoklaksia annettiin yhteensä 12 hoitojakson ajan. Yhden hoitojakson pituus oli 28 päivää.
- Calquence-valmistetta, venetoklaksia ja obinututsumabia (AVO): Calquence-valmistetta annettiin 100 mg kaksi kertaa vuorokaudessa hoitojakson 1 päivästä 1 alkaen yhteensä 14 hoitojakson ajan tai kunnes tauti eteni tai ilmeni toksisuutta, jota ei voitu hyväksyä. Hoitojakson 3 päivänä 1 potilaille aloitettiin 5 viikon mittainen venetoklaksin annostitrausohjelma, jossa valmistetta otettiin kerran vuorokaudessa aloittaen 20 mg:n annoksella ja suurentaen annosta viikoittain 50 mg:aan, 100 mg:aan, 200 mg:aan ja lopuksi 400 mg:aan. Venetoklaksia annettiin yhteensä 12 hoitojakson ajan. Obinututsumabia annettiin 1 000 mg hoitojakson 2 päivänä 1 tai päivinä 1 ja 2 (100 mg päivänä 1 ja 900 mg päivänä 1 tai 2), 8 ja 15 ja sen jälkeen 1 000 mg hoitojaksojen 3–7 päivänä 1. Yhden hoitojakson pituus oli 28 päivää.
- tutkijan valinnan mukaan kemoimmunoterapiaa (FCR/BR):
- fludarabiinia, syklofosfamidia ja rituksimabia (FCR): Fludarabiinia (25 mg/m2) ja syklofosfamidia (250 mg/m2) annettiin päivinä 1–3 enintään 6 hoitojakson ajan. Rituksimabia annettiin annoksella 375 mg/m2 hoitojakson 1 päivänä 1 ja annoksella 500 mg/m2 hoitojaksojen 2–6 päivänä 1. Yhden hoitojakson pituus oli 28 päivää.
- bendamustiinia ja rituksimabia (BR): bendamustiinia annettiin 90 mg/m2 päivinä 1 ja 2 enintään 6 hoitojakson ajan. Rituksimabia annettiin annoksella 375 mg/m2 hoitojakson 1 päivänä 1 ja annoksella 500 mg/m2 hoitojaksojen 2–6 päivänä 1. Yhden hoitojakson pituus oli 28 päivää.
Potilaat stratifioitiin iän (> 65 vuotta tai ≤ 65 vuotta), IGHV-mutaatiostatuksen (mutatoitunut vs. mutatoitumaton), Rai-luokan (suuri riski [≥ 3] vs. ei suurta riskiä) ja maantieteellisen alueen (Pohjois-Amerikka ja Länsi-Eurooppa vs. muut) mukaan. Taulukossa 10 on esitetty yhteenveto tutkimuspopulaation demografisista tiedoista ja sairauden ominaispiirteistä lähtötilanteessa.
Taulukko 10. Potilaiden ominaisuudet lähtötilanteessa aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavilla potilailla (AMPLIFY)
| Ominaisuus | AV N = 291 | AVO N = 286 | FCR/BR N = 290 |
| Ikä vuosina; mediaani (vaihteluväli) | 61 (31–84) | 61 (29–81) | 61 (26–86) |
| Miehiä; % | 61,2 | 69,2 | 63,1 |
| Valkoihoisia; % | 91,1 | 86,7 | 86,9 |
| ECOG-toimintakykyluokka 0–1; % | 90,0 | 95,1 | 90,3 |
| Diagnoosista satunnaistamiseen kuluneen ajan mediaani (kk) | 28,5 | 26,1 | 29,6 |
| Syöpäsolujen kertyminen imusolmukkeisiin ja muihin elimiin, imusolmukkeet ≥ 5 cm; % | 38,8 | 35,0 | 42,8 |
| Sytogenetiikka/FISH-kategoria; % | |||
| 11q-deleetio | 17,5 | 19,6 | 15,9 |
| Kompleksinen karyotyyppi (≥ 3 poikkeavuutta) | 15,5 | 16,1 | 14,5 |
| Mutatoitumaton IGHV; % | 57,4 | 59,1 | 59,3 |
| Rai-luokka; % | |||
| 0 | 1,0 | 0,3 | 1,4 |
| I | 16,2 | 21,3 | 21,4 |
| II | 35,7 | 37,8 | 33,4 |
| III | 23,7 | 17,8 | 20,3 |
| IV | 23,4 | 22,7 | 23,4 |
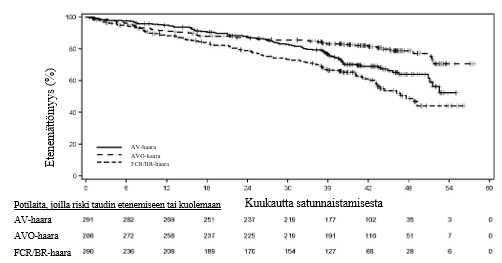
Ensisijainen päätetapahtuma oli IRC:n arvioima etenemättömyysaika AV‑haarassa verrattuna tutkijan valitsemaa kemoimmunoterapiaa (FCR/BR) saaneiden haaraan vuoden 2018 IWCLL-kriteerien mukaisesti arvioituna. Muita tehoa koskevia päätetapahtumia olivat IRC:n arvioima etenemättömyysaika AVO-haarassa verrattuna tutkijan valitsemaa kemoimmunoterapiaa (FCR/BR) saaneiden haaraan ja kokonaiselossaoloaika sekä AV-haarassa verrattuna tutkijan valitsemaa kemoimmunoterapiaa (FCR/BR) saaneiden haaraan että AVO-haarassa verrattuna tutkijan valitsemaa kemoimmunoterapiaa (FCR/BR) saaneiden haaraan.
Tehoa koskevat tulokset on esitetty taulukossa 11. IRC:n arvioiman etenemättömyysajan Kaplan–Meier-käyrä on esitetty kuvassa 2.
Taulukko 11. Tehoa koskevat tulokset aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavilla potilailla (AMPLIFY)
AV N = 291 | AVO N = 286 | FCR/BRa N = 290 | |
|---|---|---|---|
| Etenemättömyysaika* | |||
| Tapahtumien määrä (%) | 89 (30,6) | 56 (19,6) | 95 (32,8) |
| Etenevä tauti, n (%) | 77 (26,5) | 23 (8,0) | 66 (22,8) |
| Kuolemat (%) | 12 (4,1) | 33 (11,5) | 29 (10,0) |
| Mediaani (95 %:n luottamusväli), kk | Ei laskettavissa (51,1, ei laskettavissa) | Ei laskettavissa (ei laskettavissa, ei laskettavissa) | 47,6 (43,3, ei laskettavissa) |
| HR† (95 %:n luottamusväli) | 0,65 (0,49, 0,87) | 0,42 (0,30, 0,59) | - |
| p‑arvo | 0,0038 | < 0,0001 | - |
| Kokonaiselossaoloaikab | |||
| Kuolemat (%) | 23 (7,9) | 37 (12,9) | 44 (15,2) |
| HR† (95 %:n luottamusväli) | 0,42 (0,25, 0,70)c | 0,75 (0,48, 1,16) | - |
| *IRC:n arvion mukaan. †Ositetun Coxin verrannollisten riskitiheyksien mallin mukaan. aTutkijan valinnan mukaan suunniteltiin, että 143 potilaalle annettaisiin FCR-hoitoa ja 147 potilaalle BR-hoitoa. bKokonaiselossaoloaikaa koskevat tiedot 6 kuukauden lisäseuranta-ajan kuluttua etenemättömyysaikaa koskevan välianalyysin jälkeen. cp‑arvo ei ollut merkitsevä monivertailukorjauksen jälkeen. | |||
Kuva 2.IRC:n arvioiman etenemättömyysajan Kaplan–Meier-käyrä kroonista lymfaattista leukemiaa sairastavilla potilailla (AMPLIFY, ITT-populaatio)
Kroonista lymfaattista leukemiaa sairastavat potilaat, jotka olivat saaneet aiemmin vähintään yhtä hoitoa
Calquence-valmisteen tehoa ja turvallisuutta uusiutuneen tai refraktaarisen kroonisen lymfaattisen leukemian hoidossa arvioitiin satunnaistetussa, avoimessa vaiheen 3 monikeskustutkimuksessa (ASCEND). Siihen osallistui 310 potilasta, jotka olivat saaneet aiemmin vähintään yhtä hoitoa (ei BCL-2-estäjiä tai B-solureseptorin estäjiä). Potilaat saivat Calquence-valmistetta monoterapiana tai tutkijan valinnan mukaan joko idelalisibia ja rituksimabia tai bendamustiinia ja rituksimabia. Tutkimuksessa potilaille sallittiin antitromboottisten lääkkeiden käyttö. Tutkimuksesta suljettiin pois potilaat, jotka tarvitsivat varfariinia tai vastaavaa K-vitamiiniantagonistia antikoagulaatiohoitona.
Potilaat satunnaistettiin suhteessa 1:1 saamaan joko:
- Calquence-valmistetta 100 mg kaksi kertaa vuorokaudessa siihen asti, että tauti eteni tai ilmeni toksisuutta, jota ei voitu hyväksyä, tai
- tutkijan valinnan mukaan
- idelalisibia 150 mg kaksi kertaa vuorokaudessa yhdistelmänä rituksimabin kanssa. Rituksimabia annettiin 375 mg/m2:n annos laskimoon ensimmäisen hoitojakson päivänä 1, sitten neljä 500 mg/m2:n annosta laskimoon 2 viikon välein ja sen jälkeen kolme annosta 4 viikon välein, niin että infuusioita oli yhteensä kahdeksan.
- bendamustiinia 70 mg/m2 (kunkin 28 päivän mittaisen hoitojakson päivinä 1 ja 2) yhdistelmänä rituksimabin kanssa (375 mg/m2/500 mg/m2) kunkin 28 päivän mittaisen hoitojakson päivänä 1 (enintään 6 hoitojakson ajan).
Potilaat stratifioitiin 17p-deleetion (mutaatio vs. ei mutaatiota), ECOG-toimintakykyluokan (0 tai 1 vs. 2) ja aiempien hoitojen lukumäärän (1–3 vs. ≥ 4) mukaan. Tutkijan valinnan mukaan joko idelalisibia ja rituksimabia tai bendamustiinia ja rituksimabia saamaan satunnaistetuista potilaista 35 potilasta siirrettiin saamaan Calquence-valmistetta, kun taudin eteneminen oli vahvistettu. Taulukossa 12 on esitetty yhteenveto tutkimuspopulaation demografisista tiedoista ja sairauden ominaispiirteistä lähtötilanteessa.
Taulukko 12. Potilaiden ominaisuudet lähtötilanteessa kroonista lymfaattista leukemiaa (KLL) sairastavilla potilailla (ASCEND)
| Ominaisuus | Calquence-monoterapia N = 155 | Tutkijan valitsema hoito: idelalisibi ja rituksimabi (IR) tai bendamustiini ja rituksimabi (BR) N = 155 |
| Ikä vuosina; mediaani (vaihteluväli) | 68 (32–89) | 67 (34–90) |
| Miehiä; % | 69,7 | 64,5 |
| Valkoihoisia; % | 93,5 | 91,0 |
| ECOG-toimintakykyluokka; % | ||
| 0 | 37,4 | 35,5 |
| 1 | 50,3 | 51,0 |
| 2 | 12,3 | 13,5 |
| Diagnoosista kuluneen ajan mediaani (kk) | 85,3 | 79,0 |
| Syöpäsolujen kertyminen imusolmukkeisiin ja muihin elimiin, imusolmukkeet ≥ 5 cm; % | 49,0 | 48,4 |
| Aiempien KLL-hoitojen määrän mediaani (vaihteluväli) | 1 (1–8) | 2 (1–10) |
| Aiempien KLL-hoitojen määrä; % | ||
| 1 | 52,9 | 43,2 |
| 2 | 25,8 | 29,7 |
| 3 | 11,0 | 15,5 |
| ≥ 4 | 10,3 | 11,6 |
| Sytogenetiikka/FISH-kategoria; % | ||
| 17p-deleetio | 18,1 | 13,5 |
| 11q-deleetio | 25,2 | 28,4 |
| TP53-mutaatio | 25,2 | 21,9 |
| Mutatoitumaton IGHV | 76,1 | 80,6 |
| Kompleksinen karyotyyppi (≥ 3 poikkeavuutta) | 32,3 | 29,7 |
| Rai-luokka; % | ||
| 0 | 1,3 | 2,6 |
| I | 25,2 | 20,6 |
| II | 31,6 | 34,8 |
| III | 13,5 | 11,6 |
| IV | 28,4 | 29,7 |
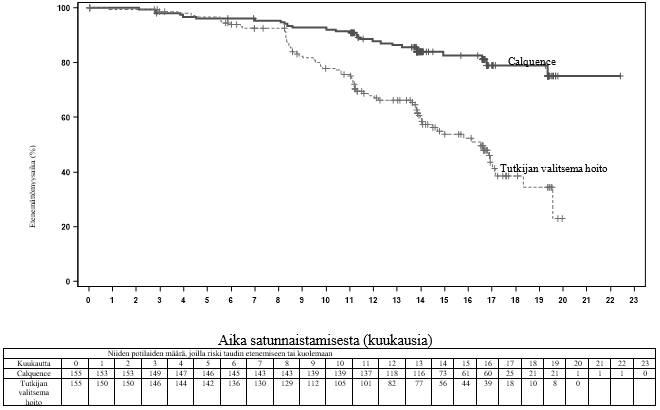
Ensisijainen päätetapahtuma oli IRC:n IWCLL 2008 ‑kriteerien mukaan arvioitu etenemättömyysaika niin, että kriteereihin oli sisällytetty hoitoon liittyvää lymfosytoosia koskeva selvitys (Cheson 2012). Kun seuranta-ajan mediaani oli 16,1 kuukautta, etenemättömyysaika osoitti, että kuoleman tai taudin etenemisen riski oli Calquence-ryhmän potilailla pienentynyt tilastollisesti merkitsevästi 69 %. Tehoa koskevat tulokset on esitetty taulukossa 13. Etenemättömyysajan Kaplan–Meier-käyrä on esitetty kuvassa 3.
Taulukko 13. IRC:n arvioiden mukaiset tehoa koskevat tulokset kroonista lymfaattista leukemiaa sairastavilla potilailla (ASCEND)
| Calquence-monoterapia N = 155 | Tutkijan valitsema hoito: idelalisibi ja rituksimabi (IR) tai bendamustiini ja rituksimabi (BR) N = 155 | |
| Etenemättömyysaika* | ||
| Tapahtumien määrä (%) | 27 (17,4) | 68 (43,9) |
| Etenevä tauti, n (%) | 19 (12,3) | 59 (38,1) |
| Kuolemat (%) | 8 (5,2) | 9 (5,8) |
| Mediaani (95 %:n luottamusväli), kk | Ei saavutettu | 16,5 (14,0, 17,1) |
| HR† (95 %:n luottamusväli) | 0,31 (0,20, 0,49) | |
| p-arvo | < 0,0001 | |
| 15 kk:n arvio, % (95 %:n luottamusväli) | 82,6 (75,0, 88,1) | 54,9 (45,4, 63,5) |
| Kokonaiselossaoloaikaa | ||
| Kuolemat (%) | 15 (9,7) | 18 (11,6) |
| HR (95 %:n luottamusväli) † | 0,84 (0,42, 1,66) | - |
| Paras kokonaisvasteluku (ORR)* (CR + CRi + nPR + PR)** | ||
ORR, n (%) (95 %:n luottamusväli) | 126 (81,3) (74,4, 86,6) | 117 (75,5) (68,1, 81,6) |
| p-arvo | 0,2248 | - |
| CR, n (%) | 0 | 2 (1,3) |
| PR, n (%) | 126 (81,3) | 115 (74,2) |
| Vasteen kesto (DoR) | ||
| Mediaani (95 %:n luottamusväli), kk | Ei saavutettu | 13,6 (11,9, ei saavutettu) |
HR = riskitiheyksien suhde; CR = täydellinen vaste; CRi = täydellinen vaste ilman veriarvojen täydellistä palautumista; nPR = nodulaarinen osittainen vaste; PR = osittainen vaste
* IRC:n arvion mukaan
a Kokonaiselossaoloajan mediaania ei saavutettu molemmissa hoitohaaroissa. Kokonaiselossaoloajan p < 0,6089.
**CRi- ja nPR-arvot ovat 0.
† Ositetun Coxin verrannollisten riskitiheyksien mallin mukaan
Kuva 3. IRC:n arvioiman etenemättömyysajan Kaplan–Meier-käyrä kroonista lymfaattista leukemiaa sairastavilla potilailla (ASCEND, ITT-populaatio)
Calquence-hoidon yhteydessä todettua etenemättömyysaikaa koskevat tulokset olivat yhdenmukaiset kaikissa alaryhmissä, myös suuren riskin ryhmissä. Kroonista lymfaattista leukemiaa sairastavilla tutkittavilla, joilla riski oli suuri (17p-deleetio, 11q-deleetio, TP53-mutaatio ja mutatoitumaton IGHV), etenemättömyysaikaa koskeva riskitiheyksien suhde oli 0,27 [95 %:n luottamusväli (0,17, 0,44)].
Taulukko 14. IRC:n arvioiman etenemättömyysajan alaryhmäanalyysi (ASCEND-tutkimus)
| Calquence-monoterapia | |||
|---|---|---|---|
| N | HR | 95 %:n luottamusväli | |
| Kaikki tutkittavat | 155 | 0,30 | (0,19, 0,48) |
Del 17P Kyllä Ei |
28 127 |
0,21 0,33 |
(0,07, 0,68) (0,21, 0,54) |
TP53-mutaatio Kyllä Ei |
39 113 |
0,24 0,33 |
(0,11, 0,56) (0,20, 0,57) |
Del 17P tai TP53-mutaatio Kyllä Ei |
45 108 |
0,21 0,36 |
(0,09, 0,48) (0,21, 0,61) |
IGHV-mutaatio Mutatoitunut Mutatoitumaton |
33 118 |
0,32 0,32 |
(0,11, 0,94) (0,19, 0,52) |
Del 11q Kyllä Ei |
39 116 |
0,28 0,31 |
(0,11, 0,70) (0,19, 0,53) |
Kompleksinen karyotyyppi Kyllä Ei |
50 97 |
0,32 0,23 |
(0,16, 0,63) (0,12, 0,44) |
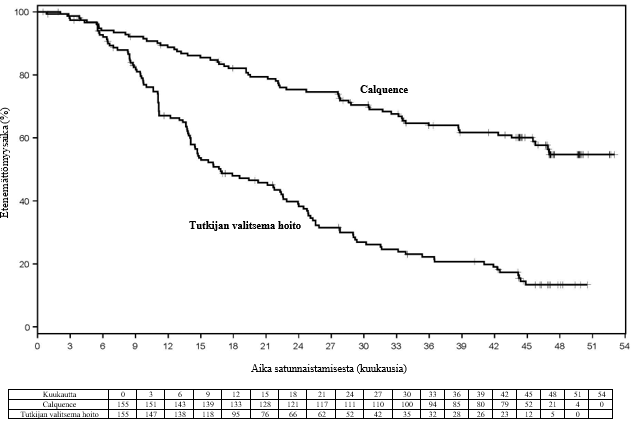
Lopullisen analyysin ajankohtana seurannan keston mediaani oli Calquence-ryhmässä 46,5 kuukautta ja IR/BR-ryhmässä 45,3 kuukautta, ja tutkijan arvioiman taudin etenemisen tai kuoleman riskin todettiin pienentyneen Calquence-ryhmän potilailla 72 %. Tutkijan arvioiman etenemättömyysajan mediaania ei saavutettu Calquence-ryhmässä, ja IR/BR-ryhmässä se oli 16,8 kuukautta. Tutkijan arvioiden mukaiset tehoa koskevat tulokset on esitetty taulukossa 15. Tutkijan arvioiman etenemättömyysajan Kaplan–Meier-käyrä on esitetty kuvassa 4.
Taulukko 15. Lopullisen analyysin tehoa koskevat tulokset tutkijan arvioiden mukaan kroonista lymfaattista leukemiaa sairastavilla potilailla (ASCEND)
Calquence-monoterapia N = 155
| Tutkijan valitsema hoito: idelalisibi ja rituksimabi (IR) tai bendamustiini ja rituksimabi (BR) N = 155 | |
|---|---|---|
| Etenemättömyysaika* | ||
| Tapahtumien määrä (%) | 62 (40,0) | 119 (76,8) |
| Etenevä tauti, n (%) | 43 (27,7) | 102 (65,8) |
| Kuolemat (%) | 19 (12,3) | 17 (11,0) |
| Mediaani (95 %:n luottamusväli), kk | Ei saavutettu | 16,8 (14,1, 22,5) |
| HR† (95 %:n luottamusväli) | 0,28 (0,20, 0,38) | |
| Kokonaiselossaoloaikaa | ||
| Kuolemat (%) | 41 (26,5) | 54 (34,8) |
| HR (95 %:n luottamusväli) † | 0,69 (0,46, 1,04) | - |
HR = riskitiheyksien suhde
* Tutkijan arvion mukaan.
a Kokonaiselossaoloajan mediaania ei saavutettu molemmissa hoitohaaroissa. Kokonaiselossaoloajan p = 0,0783.
† Ositetun Coxin verrannollisten riskitiheyksien mallin mukaan.
Kuva 4. Tutkijan arvioiman etenemättömyysajan Kaplan–Meier-käyrä lopullisen analyysin ajankohtana kroonista lymfaattista leukemiaa sairastavilla potilailla (ASCEND)
Calquence-hoidon yhteydessä todettua tutkijan arvioimaa etenemättömyysaikaa koskevat tulokset lopullisen analyysin ajankohtana olivat yhdenmukaiset kaikissa alaryhmissä, myös suuren riskin ryhmissä, ja yhdenmukaiset ensisijaisen analyysin tulosten kanssa.
Aiemmin hoitamatonta manttelisolulymfoomaa sairastavat potilaat
Calquence‑valmisteen turvallisuutta ja tehoa aiemmin hoitamattoman manttelisolulymfooman hoidossa arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen 3 monikeskustutkimuksessa (ECHO). ECHO‑tutkimukseen osallistui 598 vähintään 65‑vuotiasta potilasta, joilla oli vahvistettu, aiemmin hoitamaton manttelisolulymfooma.
Potilaat satunnaistettiin suhteessa 1:1 kahteen hoitohaaraan, joissa he saivat hoitoa seuraavasti:
- Calquence + bendamustiini ja rituksimabi (Calquence + BR) ‑haara: Calquence‑valmistetta (100 mg) annettiin kahdesti vuorokaudessa hoitojakson 1 päivästä 1 alkaen ilman taukoja. Bendamustiinia (90 mg/m2) annettiin laskimoon 30 minuutin kuluessa päivinä 1 ja 2 kuuden 28 päivän pituisen hoitojakson ajan, ja rituksimabia (375 mg/m2) annettiin laskimoon kuuden 28 päivän pituisen hoitojakson päivänä 1. Calquence + BR ‑hoitoa annettiin enintään 6 hoitojakson ajan (induktiohoito).
- Lumelääke + bendamustiini ja rituksimabi (lumelääke + BR) ‑haara: Lumelääkettä annettiin kahdesti vuorokaudessa hoitojakson 1 päivästä 1 alkaen ilman taukoja. Bendamustiinia (90 mg/m2) annettiin laskimoon 30 minuutin kuluessa päivinä 1 ja 2 kuuden 28 päivän pituisen hoitojakson ajan, ja rituksimabia (375 mg/m2) annettiin laskimoon kuuden 28 päivän pituisen hoitojakson päivänä 1. Lumelääke + BR ‑hoitoa annettiin enintään 6 hoitojakson ajan (induktiohoito).
Calquence‑valmistetta tai lumelääkettä annettiin ilman taukoja, kunnes tauti eteni tai ilmeni toksisuutta, joita ei voitu hyväksyä. Vasteen (PR tai CR) saavuttaneet potilaat saivat induktiohoidon jälkeen ylläpitohoitona enintään 12 lisäannosta rituksimabia annoksella 375 mg/m2 joka toisen hoitojakson päivänä 1 hoitojaksoon 30 asti. Lumelääke + BR ‑haaraan satunnaistetut potilaat, joilla oli vahvistettu etenevä tauti, saivat mahdollisuuden siirtyä saamaan Calquence‑monoterapiaa annostuksella 100 mg kahdesti vuorokaudessa, kunnes tauti eteni toisen kerran tai ilmeni toksisia vaikutuksia, joita ei voitu hyväksyä.
Potilaiden satunnaistamisessa ositustekijöinä olivat maantieteellinen alue (Pohjois‑Amerikka vs. Länsi‑Eurooppa vs. muu) ja yksinkertaistetun MIPI‑riskipisteytyksen (Mantle Cell Lymphoma International Prognostic Index) pistemäärä (0–3 vs. 4–5 vs. 6–11).
Iän mediaani oli 71 vuotta (65–86), 70,7 % oli miehiä, 78,3 % oli valkoihoisia ja 93,1 %:lla ECOG‑toimintakykyluokka oli 0–1. Yksinkertaistetun MIPI‑riskipisteytyksen pistemäärä oli pieni (0–3) 33,1 %:lla potilaista, keskisuuri (4–5) 42,8 %:lla potilaista ja suuri (6–11) 24,1 %:lla potilaista. Yhteensä 37,7 %:lla potilaista kasvaimen koko oli ≥ 5 cm ja 86 %:lla Ann Arbor ‑levinneisyysluokka oli IV. Manttelisolulymfooman aggressiivisista tautimuodoista blastoidia muotoa todettiin 7,7 %:lla potilaista ja pleomorfista muotoa 5,5 %:lla potilaista. Yhteensä 47,8 %:lla potilaista Ki‑67‑indeksi oli ≥ 30 %. Ominaisuudet lähtötilanteessa olivat molemmissa hoitohaaroissa samankaltaiset.
Ensisijainen päätetapahtuma oli etenemättömyysaika (PFS) tutkittavilla, joilla oli aiemmin hoitamaton manttelisolulymfooma. Etenemättömyysajan arvioi riippumaton arviointikomitea (IRC) non‑Hodgkin‑lymfooman vuoden 2014 Lugano‑luokituksen mukaisesti. IRC arvioi myös kokonaisvasteluvun (ORR).
IRC arvioi etenemättömyysajan, kun seuranta‑ajan mediaani oli 49,8 kuukautta.
Kun ensisijaisen etenemättömyysaika‑analyysin jälkeen oli ollut 6 kuukauden lisäseuranta‑aika ja seuranta‑ajan mediaani oli 63,0 kuukautta, kokonaiselossaoloajan mediaania ei ollut saavutettu kummassakaan hoitohaarassa. Kuolemantapauksia oli yhteensä 218: 105 (35,1 %) Calquence + BR ‑haarassa ja 113 (37,8 %) lumelääke + BR ‑haarassa. Tehoa koskevat tulokset on esitetty taulukossa 16. Etenemättömyysajan Kaplan–Meier‑käyrä on esitetty kuvassa 5.
Taulukko 16. Tehoa koskevat tulokset aiemmin hoitamatonta manttelisolulymfoomaa sairastavilla potilailla ECHO‑tutkimuksessa
Calquence + BR N = 299 | Lumelääke + BR N = 299 | |
|---|---|---|
| IRC:n arvioima etenemättömyysaika (PFS) | ||
| Mediaani (95 %:n luottamusväli) | 66,4 (55,1, ei arvioitavissa) | 49,6 (36,0, 64,1) |
| HR (95 %:n luottamusväli) (ositettu)* | 0,73 (0,57, 0,94) | |
| p‑arvo‡ | 0,0160 | |
| IRC:n arvioima kokonaisvasteluku (ORR) | ||
| CR + PR, n (%) | 272 (91,0) | 263 (88,0) |
| 95 %:n luottamusväli | 87,3, 93,8 | 83,9, 91,3 |
| CR, n (%) | 199 (66,6) | 160 (53,5) |
| PR, n (%) | 73 (24,4) | 103 (34,4) |
| p‑arvo | 0,2196 | ‑ |
HR = riskitiheyksien suhde, CR = täydellinen vaste, PR = osittainen vaste.
* Satunnaistamisen ositustekijät: maantieteellinen alue (Pohjois‑Amerikka, Länsi‑Eurooppa, muu) ja yksinkertaistetun MIPI‑riskipisteytyksen pistemäärä (pieni riski [0–3], keskisuuri riski [4–5], suuri riski [6–11]) IXRS‑järjestelmän kautta kerättynä. Arvio perustuu ositettuun Coxin verrannollisten riskitiheyksien malliin riskitiheyksien suhteen (95 %:n luottamusvälin) osalta.
‡ Arvio perustuu ositettuun log rank ‑testiin p‑arvon osalta.
Kuva 5. IRC:n arvioiman etenemättömyysajan Kaplan–Meier‑käyrä aiemmin hoitamatonta manttelisolulymfoomaa sairastavilla potilailla (ECHO)
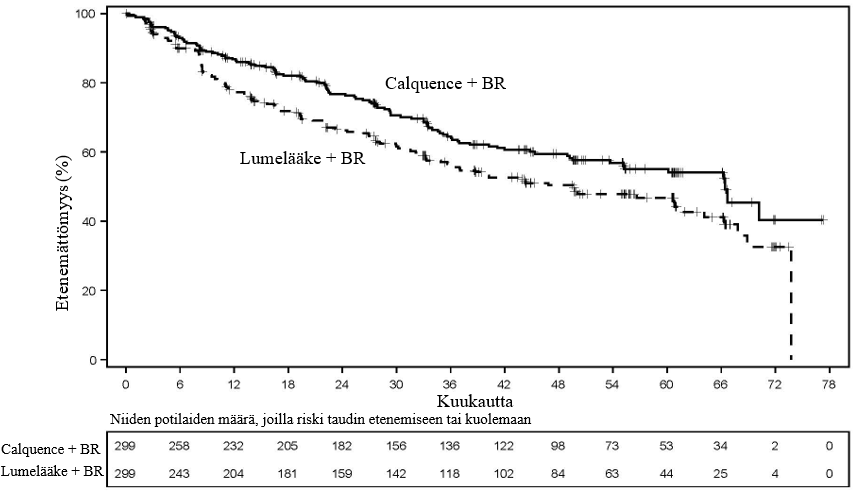
Manttelisolulymfoomaa sairastavat potilaat, jotka olivat saaneet aiemmin vähintään yhtä hoitoa
CALQUENCE-valmisteen turvallisuutta ja tehoa manttelisolulymfooman hoidossa arvioitiin avoimessa, yksihaaraisessa vaiheen 2 monikeskustutkimuksessa (ACE‑LY‑004). Siihen osallistui 124 potilasta, jotka olivat saaneet aiemmin hoitoa. Kaikki potilaat saivat CALQUENCE-valmistetta 100 mg suun kautta kaksi kertaa vuorokaudessa siihen asti, että tauti eteni tai ilmeni toksisuutta, jota ei voitu hyväksyä. Tutkimuksessa ei ollut mukana potilaita, jotka olivat saaneet aiemmin joko BTK:n tai BCL‑2:n estäjiä. Ensisijainen päätetapahtuma oli kokonaisvasteluku (ORR), jonka tutkija arvioi non-Hodgkin-lymfooman Lugano-luokituksen mukaisesti. Lisäksi lopputulosmuuttujana oli vasteen kesto (DoR). Lopulliseen analyysiin (54 kuukauden kohdalla) perustuvat tehoa koskevat tulokset on esitetty taulukossa 17.
Lopullisessa analyysissä potilaiden iän mediaani oli 68 vuotta (vaihteluväli 42–90 vuotta), 79,8 % oli miehiä ja 74,2 % oli valkoihoisia. Lähtötilanteessa 92,8 %:lla potilaista ECOG-toimintakykyluokka oli 0 tai 1. Diagnoosista kuluneen ajan mediaani oli 46,3 kuukautta, ja aiempien hoitojen määrän mediaani oli 2 (vaihteluväli 1–5). Aiempiin hoitoihin laskettiin myös kantasolusiirto, joka oli tehty 17,7 %:lle potilaista. Yleisimpiä aiempia hoitoja olivat CHOP-pohjaiset hoidot (51,6 %) ja Ara‑C (33,9 %). Lähtötilanteessa 37,1 %:lla potilaista oli ainakin yksi kasvain, jonka suurin halkaisija oli ≥ 5 cm, ja 72,6 %:lla oli etäpesäkkeitä imusolmukkeiden ulkopuolella, mukaan lukien luuytimessä (50,8 %:lla). Yksinkertaistetulla MIPI-riskipisteytyksellä (jossa ovat mukana ikä, ECOG-pistemäärä sekä lähtötilanteen laktaattidehydrogenaasipitoisuus ja valkosolujen määrä) riski oli keskisuuri 43,5 %:lla potilaista ja suuri 16,9 %:lla potilaista.
Taulukko 17. Kokonaisvasteluku (ORR) ja vasteen kesto (DoR) (ACE‑LY‑004-tutkimuksessa) manttelisolulymfoomaa sairastavilla potilailla 54 kuukauden kohdalla tehdyssä lopullisessa analyysissä
Tutkijan arvio 54 kuukauden kohdalla N = 124 n (%) (95 %:n luottamusväli*) | |
|---|---|
Kokonaisvasteluku (ORR) | |
Kokonaisvasteluku | 101 (81,5 %) (73,5, 87,9) |
Täydellinen vaste | 59 (47,6 %) (38,5, 56,7) |
Osittainen vaste | 42 (33,9 %) (25,6, 42,9) |
Ei arvioitavissa† | 3 (2,4 %) (0,5, 6,9) |
Vasteen kesto (DoR) | |
Mediaani (kk) | 28.6 (17,5, 39,1) |
*95 %:n eksakti binomiaalinen luottamusväli. †Käsittää tutkittavat, joista ei ollut saatavilla yhtään riittävää taudin arviointia lähtötilanteen jälkeen. | |
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Calquence-valmisteen käytöstä kypsien B‑solukasvainten hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Akalabrutinibin ja sen aktiivisen metaboliitin ACP‑5862:n farmakokinetiikkaa tutkittiin terveillä tutkittavilla ja B-solusyöpiä sairastavilla potilailla. Akalabrutinibin farmakokinetiikka on suhteessa annokseen, ja sekä akalabrutinibin että ACP‑5862:n farmakokinetiikka on lähes lineaarista annosalueella 75–250 mg. Populaatiofarmakokineettinen mallinnus viittaa siihen, että akalabrutinibin ja ACP‑5862:n farmakokinetiikka on samanlaista erilaisia B-solusyöpiä sairastavilla potilailla. Kun käytettiin suositeltua 100 mg:n annosta kaksi kertaa vuorokaudessa B-solusyöpiä (mukaan lukien kroonista lymfaattista leukemiaa) sairastavien potilaiden hoidossa, akalabrutinibin pitoisuutta plasmassa ajan funktiona kuvaavan käyrän alla olevan pinta-alan (AUC24h) geometrinen keskiarvo vakaassa tilassa vuorokauden aikana oli 1 679 ng•h/ml ja enimmäispitoisuus plasmassa (Cmax) 438 ng/ml. ACP‑5862:n vastaavat arvot olivat 4 166 ng•h/ml ja 446 ng/ml.
Imeytyminen
Aika, joka kului huippupitoisuuden saavuttamiseen plasmassa (tmax), oli akalabrutinibilla 0,5–1,5 tuntia ja ACP‑5862:lla 1,0 tuntia. Calquence-valmisteen absoluuttinen hyötyosuus oli 25 %.
Ruuan vaikutus akalabrutinibiin
Akalabrutinibin 75 mg:n kerta-annoksen antaminen terveille tutkittaville runsaasti rasvaa ja energiaa sisältävän aterian yhteydessä (noin 918 kaloria, 59 grammaa hiilihydraatteja, 59 grammaa rasvaa ja 39 grammaa proteiinia) ei vaikuttanut keskimääräiseen AUC-arvoon, kun sitä verrattiin valmisteen antamiseen tyhjään mahaan. Saatu Cmax-arvo pieneni 69 % ja tmax-aika piteni 1–2 tuntia.
Jakautuminen
Akalabrutinibista 99,4 % ja ACP‑5862:sta 98,8 % sitoutui reversiibelisti ihmisen plasman proteiineihin. Akalabrutinibin keskimääräinen veri/plasma-suhde in vitro oli 0,8 ja ACP‑5862:n 0,7. Akalabrutinibin keskimääräinen vakaan tilan jakautumistilavuus (Vss) oli noin 34 l.
Biotransformaatio/metabolia
Akalabrutinibi metaboloituu in vitro pääasiassa CYP3A-entsyymien välityksellä ja vähäisessä määrin glutationikonjugaation ja amidihydrolyysin välityksellä. ACP‑5862 oli pääasiallinen plasmassa todettu metaboliitti, ja se metaboloitui edelleen pääasiassa CYP3A-välitteisen oksidaation kautta. ACP‑5862-altistuksen (AUC) geometrinen keskiarvo oli noin 2–3 kertaa suurempi kuin akalabrutinibialtistus. ACP‑5862:n teho on BTK:n eston suhteen noin 50 % pienempi kuin akalabrutinibin.
In vitro ‑tutkimusten mukaan akalabrutinibi ei estä CYP1A2‑, CYP2B6‑, CYP2C8‑, CYP2C9‑, CYP2C19‑, CYP2D6‑, UGT1A1- tai UGT2B7-entsyymejä kliinisesti merkityksellisinä pitoisuuksina eikä todennäköisesti vaikuta näiden CYP-entsyymien substraattien puhdistumaan.
In vitro ‑tutkimusten mukaan ACP‑5862 ei estä CYP1A2‑, CYP2B6‑, CYP2C8‑, CYP2C9‑, CYP2C19‑, CYP2D6‑, CYP3A4/5‑, UGT1A1- tai UGT2B7-entsyymejä kliinisesti merkityksellisinä pitoisuuksina eikä todennäköisesti vaikuta näiden CYP-entsyymien substraattien puhdistumaan.
Yhteisvaikutukset kuljettajaproteiinien kanssa
In vitro ‑tutkimusten mukaan akalabrutinibi ja ACP‑5862 ovat P‑gp:n ja BCRP:n substraatteja. Samanaikainen käyttö BCRP:n estäjien kanssa ei kuitenkaan todennäköisesti johda kliinisesti merkityksellisiin yhteisvaikutuksiin. Samanaikainen käyttö OATP1B1/1B3:n estäjän (600 mg rifampisiinia, kerta-annos) kanssa suurensi akalabrutinibin Cmax-arvon 1,2‑kertaiseksi ja AUC-arvon 1,4‑kertaiseksi (N = 24, terveet tutkittavat), mikä ei ole kliinisesti merkityksellistä.
Akalabrutinibi ja ACP‑5862 eivät estä P‑gp:tä, OAT1:tä, OAT3:a, OCT2:ta, OATP1B1:tä, OATP1B3:a tai MATE2-K:ta kliinisesti merkityksellisinä pitoisuuksina. Akalabrutinibi saattaa estää BCRP:ia suolistossa, ja ACP‑5862 saattaa estää MATE1:tä kliinisesti merkityksellisinä pitoisuuksina (ks. kohta Yhteisvaikutukset). Akalabrutinibi ei estä MATE1:tä eikä ACP‑5862 estä BCRP:ia kliinisesti merkityksellisinä pitoisuuksina.
Eliminaatio
Suun kautta otetun 100 mg:n kerta-annoksen jälkeen akalabrutinibin terminaalinen eliminaation puoliintumisaika (t1/2) oli 1–2 tuntia. Aktiivisen metaboliitin ACP‑5862:n t1/2 oli noin 7 tuntia.
Suun kautta otetun akalabrutinibin keskimääräinen näennäinen puhdistuma (CL/F) B-solusyöpiä sairastavilla potilailla oli 134 l/h ja ACP‑5862:n 22 l/h.
Kun terveille tutkittaville annettiin 100 mg:n kerta-annos radioaktiivisesti merkittyä [14C]-akalabrutinibia, 84 % annoksesta erittyi ulosteeseen ja 12 % virtsaan; alle 2 % annoksesta poistui elimistöstä muuttumattomana akalabrutinibina.
Erityisryhmät
Populaatiofarmakokineettisen analyysin perusteella potilaan ikä (> 18 vuotta), sukupuoli, rotu (valkoihoinen, afroamerikkalainen) ja paino eivät vaikuttaneet kliinisesti merkityksellisesti akalabrutinibin ja sen aktiivisen metaboliitin ACP‑5862:n farmakokinetiikkaan.
Pediatriset potilaat
Alle 18‑vuotiailla potilailla ei ole tehty Calquence-valmistetta koskevia farmakokineettisiä tutkimuksia.
Munuaisten vajaatoiminta
Akalabrutinibin eliminoituminen munuaisten kautta on erittäin vähäistä. Munuaisten vajaatoimintaa sairastavilla potilailla ei ole tehty farmakokineettistä tutkimusta.
Populaatiofarmakokineettisen analyysin perusteella ei havaittu kliinisesti merkityksellistä eroa farmakokinetiikassa, kun tarkasteltiin 408:aa tutkittavaa, joilla oli lievä munuaisten vajaatoiminta (eGFR 60–89 ml/min/1,73 m2 MDRD-kaavalla arvioituna), 109:ää tutkittavaa, joilla oli kohtalainen munuaisten vajaatoiminta (eGFR 30–59 ml/min/1,73 m2), ja 192:ta tutkittavaa, joilla munuaisten toiminta oli normaalia (eGFR ≥ 90 ml/min/1,73 m2). Akalabrutinibin farmakokinetiikkaa ei ole määritetty potilailla, jotka sairastavat vaikeaa munuaisten vajaatoimintaa (eGFR alle 29 ml/min/1,73 m2) tai dialyysihoitoa vaativaa munuaisten vajaatoimintaa. Kliinisiin tutkimuksiin ei otettu potilaita, joilla kreatiniinipitoisuus oli yli 2,5 kertaa suurempi kuin hoitopaikan käytännön mukainen viitealueen yläraja (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Akalabrutinibi metaboloituu maksassa. Maksan vajaatoimintaa tarkastelevissa tutkimuksissa akalabrutinibialtistus (AUC) suureni 1,9-kertaiseksi lievää maksan vajaatoimintaa (Child–Pugh-luokka A) sairastavilla (N = 6), 1,5‑kertaiseksi kohtalaista maksan vajaatoimintaa (Child–Pugh-luokka B) sairastavilla (N = 6) ja 5,3‑kertaiseksi vaikeaa maksan vajaatoimintaa (Child–Pugh-luokka C) sairastavilla tutkittavilla (N = 8) verrattuna tutkittaviin, joilla maksan toiminta oli normaalia (N = 6). Kohtalaista maksan vajaatoimintaa sairastavilla tutkittavilla lääkkeiden eliminaatiokykyä osoittavat markkerit eivät kuitenkaan muuttuneet merkitsevästi, joten kohtalaisen maksan vajaatoiminnan vaikutus todennäköisesti aliarvioitiin tässä tutkimuksessa. Populaatiofarmakokineettisen analyysin perusteella ei havaittu kliinisesti merkityksellistä eroa, kun verrattiin lievää (N = 79) tai kohtalaista (N = 6) maksan vajaatoimintaa (kokonaisbilirubiini 1,5–3 x viitealueen yläraja ja mikä tahansa ASAT-arvo) sairastavia tutkittavia tutkittaviin, joilla maksan toiminta oli normaalia (N = 613) (kokonaisbilirubiini ja ASAT-arvo viitealueella) (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Karsinogeenisuus
Akalabrutinibilla ei ole tehty karsinogeenisuustutkimuksia.
Genotoksisuus/mutageenisuus/valotoksisuus
Akalabrutinibi ei ollut mutageeninen bakteereilla tehdyssä takaisinmutaatiokokeessa, in vitro ‑kromosomipoikkeamakokeessa tai hiirillä in vivo tehdyssä luuytimen mikrotumatestissä.
3T3-solulinjassa in vitro tehtyjen valotoksisuuskokeiden perusteella akalabrutinibiin katsotaan liittyvän pieni valotoksisuuden riski ihmisellä.
Toistuvan altistuksen aiheuttama toksisuus
Rotilla havaittiin mikroskooppisia, vaikeusasteeltaan minimaalisista lieviin vaihtelevia löydöksiä haimassa (verenvuoto/pigmentti/tulehdus/fibroosi haimasaarekkeessa) kaikilla annostasoilla. Vaikeusasteeltaan minimaalisesta lievään vaihtelevia ei-haitallisia löydöksiä munuaisissa (tubulaarinen basofilia, tubulaarinen regeneraatio ja tulehdus) havaittiin rotilla enintään 6 kuukautta kestäneissä tutkimuksissa, joissa suurin annos, joka ei aiheuttanut havaittavaa haittavaikutusta (NOAEL-taso, No Observed Adverse Effect Level), oli 30 mg/kg/vrk. Keskimääräiset altistukset (AUC) NOAEL-tasolla urosrotilla vastaavat 0,6-kertaista kliinistä altistusta ja naarasrotilla 1-kertaista kliinistä altistusta suositellulla annostuksella 100 mg kaksi kertaa vuorokaudessa. Pienin havaittavan haittavaikutuksen aiheuttava altistustaso (LOAEL, Lowest Observed Adverse Effect Level), jolla havaittiin palautuvia munuaisiin liittyviä (keskivaikea tubulaarinen degeneraatio) ja maksaan liittyviä (yksittäisten hepatosyyttien nekroosi) löydöksiä pitkäkestoisessa rotilla tehdyssä tutkimuksessa, oli 100 mg/kg/vrk, ja sillä altistusmarginaali oli 4,2 kertaa suurempi kuin kliininen altistus käytettäessä suositeltua annosta 100 mg kaksi kertaa vuorokaudessa. Yhdeksän kuukautta kestäneissä koirilla tehdyissä tutkimuksissa NOAEL-taso oli 10 mg/kg/vrk, mikä vastaa altistusta, joka on kolminkertainen kliiniseen AUC:iin nähden käytettäessä suositeltua kliinistä annosta. Koirilla havaittiin annostuksella 30 mg/kg/vrk (9 kertaa kliininen AUC) minimaalista tubulaarista degeneraatiota munuaisissa, vähäistä pernan painon laskua ja ohimenevää minimaalista tai lievää punasolumassan pienenemistä ja ALAT- ja AFOS-arvojen suurenemista. Sydäntoksisuutta havaittiin rotilla (sydänlihaksen verenvuoto, tulehdus ja nekroosi) ja koirilla (perivaskulaarinen/vaskulaarinen inflammaatio) vain niillä eläimillä, jotka kuolivat tutkimusten aikana, kun käytettiin suurinta siedettyä annosta suurempia annoksia. Sydänlöydösten yhteydessä altistus rotilla oli vähintään 6,8-kertainen ja koirilla 25-kertainen verrattuna kliiniseen AUC-arvoon. Sydänlöydösten palautuvuutta ei voitu arvioida, sillä löydöksiä havaittiin vain käytettäessä suurinta siedettyä annosta suurempia annoksia.
Lisääntymistoksikologia
Vaikutuksia hedelmällisyyteen ei havaittu urosrotilla, kun altistus oli 10-kertainen, tai naarasrotilla, kun altistus oli 9-kertainen verrattuna kliiniseen AUC-arvoon käytettäessä suositeltua annosta.
Vaikutuksia alkion ja sikiön kehitykseen ja eloonjäämiseen ei havaittu tiineillä rotilla, kun altistukset olivat noin 9-kertaisia verrattuna potilaiden AUC-arvoon käytettäessä suositeltua annosta 100 mg kaksi kertaa vuorokaudessa. Kahdessa rotilla tehdyssä lisääntymistutkimuksessa havaittiin synnytyshäiriöitä (pitkittynyt/vaikea synnytys), kun altistukset olivat > 2,3-kertaisia verrattuna kliiniseen altistukseen annostuksella 100 mg kaksi kertaa vuorokaudessa. Rotilla sikiön plasmassa vahvistettiin olevan akalabrutinibia ja sen aktiivista metaboliittia. Akalabrutinibia ja sen aktiivista metaboliittia oli imettävien rottien maidossa.
Tiineillä kaneilla tehdyssä alkio-/sikiötutkimuksessa sikiön painon alenemista ja luutumisen viivästymistä havaittiin altistustasoilla, joilla maternaalinen toksisuus oli 2,4-kertainen verrattuna ihmisen AUC-arvoon käytettäessä suositeltua annosta.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Mikrokiteinen selluloosa
Kolloidinen vedetön piidioksidi
Osittain esigelatinoitu maissitärkkelys
Magnesiumstearaatti (E470b)
Natriumtärkkelysglykolaatti
Kapselin kuori
Liivate
Titaanidioksidi (E171)
Keltainen rautaoksidi (E172)
Indigokarmiini (E132)
Painoväri
Shellakka
Musta rautaoksidi (E172)
Propyleeniglykoli (E1520)
Ammoniumhydroksidi
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
CALQUENCE kapseli, kova
100 mg (L:ei) 60 fol (6001,29 €)
PF-selosteen tieto
Alumiini/alumiiniläpipainopakkaukset, joissa on aurinko-/kuusymbolit ja jotka sisältävät 6 tai 8 kovaa kapselia. Pahvikotelossa on 56 tai 60 kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kova kapseli, jossa on keltainen runko ja sininen kansi, koko 1 (20 mm); mustalla merkintä ”ACA 100 mg”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
CALQUENCE kapseli, kova
100 mg 60 fol
- Ylempi erityiskorvaus (100 %). Akalabrutinibi: Kroonisen lymfaattisen leukemian hoito erityisin edellytyksin (1532).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Akalabrutinibi: Aikuisten kroonisen lymfaattisen leukemian hoito erityisin edellytyksin (3059).
ATC-koodi
L01EL02
Valmisteyhteenvedon muuttamispäivämäärä
28.07.2025
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi