
RIVASTOR depotlaastari 13,3 mg/24 h
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Vaikuttavat aineet ja niiden määrät
Yksi depotlaastari vapauttaa 24 tunnin kuluessa 13,3 mg rivastigmiinia, on kooltaan 12,8 cm2 ja sisältää 19,2 mg rivastigmiinia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Depotlaastari
Kliiniset tiedot
Käyttöaiheet
Lievän ja kohtalaisen vaikean Alzheimerin taudin oireenmukainen hoito.
Ehto
Hoidon aloittavan ja sitä valvovan lääkärin tulee olla perehtynyt Alzheimerin taudin (dementian) diagnosointiin ja hoitoon.
Annostus ja antotapa
Lääkehoidon aloittavan ja sitä valvovan lääkärin tulee olla perehtynyt Alzheimerin taudin (dementian) diagnosointiin ja hoitoon. Diagnoosi tulee asettaa vallitsevien ohjeiden mukaan. Kuten muutkin dementiapotilaille annettavat hoidot, rivastigmiinihoidon saa aloittaa vain, jos käytettävissä on henkilö, joka huolehtii säännöllisestä lääkkeenannosta ja hoidon seurannasta.
Annostus
Depotlaastarit | Rivastigmiinin vapautumisnopeus 24 tunnin kuluessa in vivo |
Rivastor 4,6 mg/24 tuntia* | 4,6 mg |
Rivastor 9,5 mg/24 tuntia* | 9,5 mg |
Rivastor 13,3 mg/24 tuntia | 13,3 mg |
* Annostuksia 4,6 mg/24 h ja 9,5 mg/24 h ei voida toteuttaa tällä valmisteella. Jos sairaustilan hoito edellyttää kyseisiä vahvuuksia, on käytettävä sellaisia Rivastor-depotlaastareita, joiden vahvuus on 4,6 mg/24 h tai 9,5 mg/24 h.
Alkuannos
Hoito aloitetaan 4,6 mg/24 tuntia ‑depotlaastareilla.
Ylläpitoannos
Jos potilas on hoitavan lääkärin arvion mukaan sietänyt vähintään neljä viikkoa jatkunutta hoitoa hyvin, 4,6 mg/24 tuntia ‑annos suurennetaan tasolle 9,5 mg/24 tuntia, joka on suositeltu päivittäinen tehokas annos. Hoitoa jatketaan tällä annoksella niin kauan kuin siitä on potilaalle terapeuttista hyötyä.
Annoksen suurentaminen
9,5 mg/24 tuntia on suositeltu päivittäinen tehokas annos, ja ylläpitohoitoa on jatkettava niin kauan kuin potilaalle on siitä terapeuttista hyötyä. Jos hoito annoksella 9,5 mg/24 tuntia on hyvin siedetty ja jatkunut vähintään kuusi kuukautta, hoitava lääkäri voi harkita annoksen suurentamista tasolle 13,3 mg/24 tuntia potilailla, joilla on ilmennyt merkittävää kognition (esim. mitattu laskuna MMSE-pisteissä) ja/tai toimintakyvyn heikentymistä (perustuen lääkärin arvioon) käytettäessä suositeltua tehokasta ylläpitovuorokausiannosta, 9,5 mg/24 tuntia (ks. kohta Farmakodynamiikka).
Rivastigmiinin kliinistä hyötyä on arvioitava säännöllisesti. Hoidon lopettamista on myös harkittava, jos näyttöä terapeuttisesta vaikutuksesta optimaaliannoksella ei enää havaita.
Jos potilaalle kehittyy ruoansulatuskanavan haittavaikutuksia, on hoito keskeytettävä tilapäisesti, kunnes haittavaikutukset häviävät. Jos hoito keskeytyy enintään kolmeksi päiväksi, voidaan depotlaastarien käyttöä jatkaa samalla annoksella. Muussa tapauksessa hoito aloitetaan uudelleen 4,6 mg/24 tuntia ‑depotlaastareilla.
Kapselien tai oraaliliuoksen vaihtaminen depotlaastareihin
Suun kautta ja ihon läpi annettavilla rivastigmiinivalmisteilla aikaansaadaan samankaltainen altistus (ks. kohta Farmakokinetiikka). Näin ollen rivastigmiinikapseleita tai -oraaliliuosta käyttävät potilaat voivat siirtyä käyttämään Rivastor-depotlaastareita seuraavaan tapaan:
- Jos potilaan oraalinen rivastigmiiniannos on 3 mg/vrk, hän voi siirtyä käyttämään 4,6 mg/24 tuntia ‑depotlaastareita.
- Jos potilaan oraalinen rivastigmiiniannos on 6 mg/vrk, hän voi siirtyä käyttämään 4,6 mg/24 tuntia ‑depotlaastareita.
- Jos potilaan oraalinen rivastigmiiniannos on 9 mg/vrk ja annos on vakiintunut ja hyvin siedetty, hän voi siirtyä käyttämään 9,5 mg/24 tuntia ‑depotlaastareita. Jos oraalinen annos 9 mg/vrk ei ole vakiintunut tai siedettävyys on huono, on suositeltavaa siirtyä käyttämään 4,6 mg/24 tuntia ‑depotlaastareita.
- Jos potilaan oraalinen rivastigmiiniannos on 12 mg/vrk, hän voi siirtyä käyttämään 9,5 mg/24 tuntia ‑depotlaastareita.
4,6 mg/24 tuntia ‑depotlaastareihin siirtymisen jälkeen annosta voidaan suurentaa tasolle 9,5 mg/24 tuntia, jos potilas on käyttänyt 4,6 mg/24 tuntia ‑laastareita vähintään neljän viikon ajan ja hoito on hyvin siedetty. 9,5 mg/24 tuntia on suositeltava tehokas annos.
Ensimmäinen depotlaastari tulisi kiinnittää viimeisen suun kautta otetun annoksen jälkeisenä päivänä.
Erityisryhmät
- Pediatriset potilaat: Ei ole asianmukaista käyttää rivastigmiinia pediatrisille potilaille Alzheimerin tautiin.
- Potilaat, joiden paino on alle 50 kg: Annoksen titraamisessa suositeltua tehokasta annosta (9,5 mg/24 tuntia) suuremmaksi on noudatettava erityistä varovaisuutta, jos potilaan paino on alle 50 kg (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Näillä potilailla saattaa esiintyä enemmän haittavaikutuksia, ja hoidon keskeyttäminen haittavaikutusten takia saattaa olla todennäköisempää.
- Maksan vajaatoiminta: Koska lievää tai kohtalaista maksan vajaatoimintaa sairastavilla potilailla on todettu altistuksen suurenemista peroraalisia valmisteita käytettäessä, on suosituksia annoksen säätämisestä yksilöllisen sietokyvyn mukaan seurattava tarkasti. Kliinisesti merkittävää maksan vajaatoimintaa sairastavilla potilailla voi esiintyä enemmän annosriippuvaisia haittavaikutuksia. Tutkimuksia ei ole tehty vaikeaa maksan vajaatoimintaa sairastavilla potilailla. Näiden potilaiden kohdalla tulee noudattaa erityistä varovaisuutta annosta säädettäessä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
- Munuaisten vajaatoiminta: Munuaisten vajaatoiminta ei vaadi annoksen muuttamista (ks. kohta Farmakokinetiikka).
Antotapa
Depotlaastari kiinnitetään kerran vuorokaudessa joko ylä- tai alaselkään, olkavarteen tai rintakehään puhtaalle, kuivalle, karvattomalle, vahingoittumattomalle terveelle iholle siten, että tiukat vaatteet eivät hankaa sitä. Depotlaastaria ei tulisi kiinnittää reiteen eikä vatsan alueelle, sillä rivastigmiinin biologisen hyötyosuuden on todettu pienenevän, jos laastari kiinnitetään näille alueille.
Depotlaastaria ei saa kiinnittää punoittavalle, ärtyneelle eikä vahingoittuneelle iholle. Täysin samaa kiinnityskohtaa ei tulisi käyttää uudelleen 14 päivään ihoärsytyksen riskin pitämiseksi mahdollisimman pienenä.
Potilaita ja hoitajia on ohjeistettava tärkeistä annostusohjeista:
- Edellisen päivän depotlaastari on poistettava joka kerta ennen uuden depotlaastarin laittamista paikoilleen (ks. kohta Yliannostus).
- Depotlaastari on vaihdettava uuteen 24 tunnin jälkeen. Käytössä saa olla vain yksi depotlaastari kerrallaan (ks. kohta Yliannostus).
- Depotlaastaria on painettava kämmenellä ihoon vähintään 30 sekunnin ajan, kunnes depotlaastarin reunat kiinnittyvät hyvin.
- Jos depotlaastari irtoaa, uusi laastari kiinnitetään loppupäivän ajaksi. Se on vaihdettava uuteen normaaliin aikaan seuraavana päivänä.
- Depotlaastaria voidaan käyttää kaikissa normaalielämän tilanteissa, mukaan lukien kylvettäessä ja lämpimällä ilmalla.
- Depotlaastaria ei saa altistaa pitkäaikaisesti ulkoisille lämmönlähteille (esim. liialliselle auringolle, saunalle, solariumille).
- Depotlaastaria ei saa leikata osiin.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle (rivastigmiinille), muille karbamaattijohdoksille tai kohdassa Apuaineet mainituille apuaineille.
Aikaisempi allergiseen kosketusihottumaan viittaava reaktio laastarin kiinnityskohdassa käytettäessä rivastigmiinilaastareita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Magneettikuvaus tai sydämen sähköinen rytminsiirto (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Yleensä haittavaikutuksia ilmenee enemmän ja ne ovat vaikeampia suuremmilla annoksilla kuin pienemmillä. Tämä ilmenee etenkin annosta muutettaessa. Jos hoito keskeytetään yli kolmen päivän ajaksi, se tulisi aloittaa uudelleen 4,6 mg/24 tuntia ‑depotlaastareilla.
Lääkevalmisteen väärinkäyttö ja annostusvirheestä johtuva yliannostus
Rivastigmiinidepotlaastareiden väärinkäyttö ja annostusvirheet ovat johtaneet vakaviin haittavaikutuksiin; jotkin tapauksista vaativat sairaalahoitoa, ja harvoin johtivat kuolemaan (ks. kohta Yliannostus). Suurin osa lääkevalmisteen väärinkäyttö- ja annostusvirhetapauksista oli seurausta siitä, että edellistä depotlaastaria ei irrotettu ennen uuden laastarin laittamista ja siitä, että useita depotlaastareita käytettiin yhtä aikaa. Potilaita ja heidän hoitajiaan on ohjeistettava tärkeistä rivastigmiinidepotlaastarin annostusohjeista (ks. kohta Annostus ja antotapa).
Ruoansulatuskanavan häiriöt
Annoksesta riippuvaisia ruoansulatuskanavan häiriöitä, kuten pahoinvointia, oksentelua ja ripulia, saattaa esiintyä hoidon alussa ja/tai annosta suurennettaessa (ks. kohta Haittavaikutukset). Näitä haittavaikutuksia esiintyy useammin naisilla. Potilaita, joille kehittyy pitkittyneestä oksentelusta tai ripulista johtuvan nestehukan oireita ja löydöksiä, voidaan hoitaa antamalla laskimoon nesteitä ja pienentämällä annosta tai keskeyttämällä hoito, jos tila tunnistetaan ja hoidetaan nopeasti. Nestehukan seuraukset voivat olla vakavia.
Painonlasku
Alzheimerin tautia sairastavat potilaat saattavat menettää painoaan koliiniesteraasin estäjien, kuten rivastigmiinin, käytön aikana. Potilaan painoa on tarkkailtava rivastigmiinidepotlaastarien käytön aikana.
Bradykardia
Elektrokardiogrammissa havaittua QT-ajan pitenemistä voi ilmetä potilaille, joita hoidetaan tietyillä koliiniesteraasinestäjillä, kuten rivastigmiinillä. Rivastigmiini saattaa aiheuttaa bradykardiaa, joka on kääntyvien kärkien takykardian riskitekijä etupäässä potilailla, joilla on muitakin riskitekijöitä. Varovaisuutta on noudatettava potilailla, joilla on tai joiden suvuissa on esiintynyt QTc-ajan pitenemistä tai joilla on suurempi riski kääntyvien kärkien takykardian kehittymiselle. Tällaisia ovat esimerkiksi potilaat, joilla on kompensoitumaton sydämen vajaatoiminta, äskettäinen sydäninfarkti, bradyarytmia, alttius hypokalemialle tai hypomagnesemialle tai jotka käyttävät samanaikaisesti QT‑ajan pitenemistä ja/tai kääntyvien kärkien takykardiaa aiheuttavia lääkevalmisteita. Kliinistä seurantaa, mukaan lukien EKG-rekisteröintiä, voidaan tarvita (ks. kohdat Yhteisvaikutukset ja Haittavaikutukset).
Muut haittavaikutukset
Varovaisuutta on noudatettava, jos Rivastor-depotlaastareita määrätään
- potilaille, joilla on sairas sinus -oireyhtymä tai sydämen johtumishäiriöitä (sinus-eteiskatkos, eteis-kammiokatkos) (ks. kohta Haittavaikutukset)
- potilaille, joilla on aktiivinen mahahaava tai pohjukaissuolihaava tai alttius tämäntyyppisiin sairauksiin, sillä rivastigmiini saattaa lisätä mahahapon eritystä (ks. kohta Haittavaikutukset)
- potilaille, joilla on alttius virtsateiden tukokselle ja kouristuskohtauksille, sillä kolinomimeettiset aineet saattavat indusoida tai pahentaa näitä sairauksia
- potilaille, joilla on ollut astma tai obstruktiivinen keuhkosairaus.
Ihoreaktiot laastarin kiinnityskohdassa
Ihoreaktiot laastarin kiinnityskohdassa ovat mahdollisia käytettäessä rivastigmiinilaastareita. Nämä reaktiot ovat yleensä lieviä tai kohtalaisia. Potilaita ja heitä hoitavia henkilöitä on ohjeistettava asianmukaisesti.
Tällaiset ihoreaktiot eivät ole itsessään osoitus herkistymisestä, mutta rivastigmiinilaastareiden käyttö saattaa johtaa allergiseen kosketusihottumaan.
Allergista kosketusihottumaa on epäiltävä, jos laastarin kiinnityskohdassa ilmenevä ihoreaktio ei rajoitu vain laastarin kokoiselle alueelle; jos potilaalla todetaan näyttöä tavallista voimakkaammasta paikallisreaktiosta (esim. paheneva punoitus, turvotus, näppylät, vesirakkulat); tai jos oireet eivät merkittävästi lievity 48 tunnin kuluessa laastarin poistamisen jälkeen. Tällaisissa tapauksissa hoito on keskeytettävä (ks. kohta Vasta-aiheet).
Jos potilaalle kehittyy rivastigmiinilaastarin käytön yhteydessä allergiseen kosketusihottumaan viittaavia reaktioita laastarin kiinnityskohdassa ja hän on edelleen rivastigmiinihoidon tarpeessa, hänen hoitonsa voidaan vaihtaa suun kautta otettavaan rivastigmiiniin ainoastaan negatiivisen allergiatestin jälkeen ja tiiviissä lääkärin valvonnassa. On mahdollista, että jotkut rivastigmiinille rivastigmiinilaastareiden käytön yhteydessä herkistyneet potilaat eivät voi käyttää rivastigmiinia missään muodossa.
Lääkkeen markkinoilletulon jälkeen lääkkeen antotavasta (suun kautta, ihon läpi) riippumatonta rivastigmiinin käyttöön liittyvää allergista dermatiittia (laaja-alaista) on raportoitu harvoin. Tällaisissa tapauksissa hoito on lopetettava (ks. kohta Vasta-aiheet).
Magneettikuvaus ja sydämen sähköinen rytminsiirto
Rivastor-depotlaastarin taustakerros sisältää alumiinia. Ihon palamisen välttämiseksi Rivastor on poistettava, jos potilaalle tehdään magneettikuvaus (MRI) tai sydämen sähköinen rytminsiirto.
Muut varoitukset ja varotoimet
Rivastigmiini saattaa pahentaa tai aiheuttaa ekstrapyramidaalisia oireita.
Rivastor-depotlaastarien käsittelyn jälkeen on vältettävä kosketusta silmiin (ks. kohta Prekliiniset tiedot turvallisuudesta). Kädet on pestävä saippualla ja vedellä depotlaastarin irroittamisen jälkeen. Silmäkontaktitapauksissa tai jos silmät punoittavat depotlaastarin käsittelyn jälkeen, silmät on huuhdeltava välittömästi runsaalla vedellä, ja mikäli tilanne ei korjaannu, on otettava yhteys lääkäriin.
Erityisryhmät
- Alle 50 kg painavilla potilailla voi esiintyä enemmän haittavaikutuksia, ja haittavaikutukset saattavat johtaa heillä useammin hoidon keskeyttämiseen (ks. kohta Annostus ja antotapa). Säädä annosta varovasti, ja seuraa näitä potilaita haittavaikutusten varalta (esim. liiallinen pahoinvointi tai oksentelu). Harkitse ylläpitoannoksen pienentämistä 4,6 mg/24 tuntia ‑depotlaastariin, jos tällaisia haittavaikutuksia esiintyy.
- Maksan vajaatoiminta: Kliinisesti merkitsevää maksan vajaatoimintaa sairastavilla potilailla voi esiintyä enemmän haittavaikutuksia. Suosituksia annoksen säätämisestä yksilöllisen sietokyvyn mukaan on seurattava tarkasti. Tutkimuksia ei ole tehty vaikeaa maksan vajaatoimintaa sairastavilla potilailla. Näiden potilaiden kohdalla tulee noudattaa erityistä varovaisuutta annoksen säätämisessä (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Yhteisvaikutukset
Rivastigmiinidepotlaastareilla ei ole tehty spesifisiä yhteisvaikutustutkimuksia.
Rivastigmiini on koliiniesteraasin estäjä, joten se saattaa voimistaa suksinyylikoliinin kaltaisten lihasrelaksanttien vaikutuksia anestesian aikana. Varovaisuuteen on syytä anestesia-ainetta valittaessa. Annoksen sovittamista tai lääkityksen tilapäistä keskeyttämistä voidaan tarvittaessa harkita.
Farmakodynaamisten vaikutustensa ja mahdollisten additiivisten vaikutustensa vuoksi rivastigmiinia ei saa antaa samanaikaisesti muiden kolinomimeettisten aineiden kanssa. Rivastigmiini voi häiritä antikolinergisten lääkkeiden (esim. oksibutyniini, tolterodiini) vaikutusta.
Additiivisia bradykardiaan (joka voi aiheuttaa pyörtymisen) johtavia vaikutuksia on raportoitu eri beetasalpaajien (mukaan lukien atenololi) ja rivastigmiinin yhteiskäytössä. Kardiovaskulaaristen beetasalpaajien käyttöön odotetaan liittyvän suurimman riskin, mutta myös muita beetasalpaajia käyttäneisiin potilaisiin liittyviä raportteja on saatu. Tämän vuoksi varovaisuutta tulee noudattaa, kun rivastigmiinia käytetään yhdessä beetasalpaajien tai muiden bradykardiaa aiheuttavien lääkkeiden kanssa (esim. ryhmän III rytmihäiriölääkkeet, kalsiuminestäjät, digitalisglykosidit, pilokarpiini).
Koska bradykardia muodostaa riskitekijän kääntyvien kärkien takykardian esiintymiselle, rivastigmiinin yhdistämistä lääkkeisiin, jotka voivat aiheuttaa QT-ajan pitenemistä ja kääntyvien kärkien takykardiaa, on seurattava huolellisesti. Näihin lääkkeisiin kuuluvat psykoosilääkkeet, eli jotkin fentiatsiinit (klooripromatsiini, levomepromatsiini), bentsamidit (sulpiridi, sultopridi, amisulpridi, tiapridi, veralipridi), pimotsidi, haloperidoli ja droperidoli sekä sisapridi, sitalopraami, difemaniili, i.v. erytromysiini, halofantriini, mitsolastiini, metadoni, pentamidiini ja moksifloksasiini. Lisäksi voidaan tarvita kliinistä seurantaa (EKG).
Terveillä tutkittavilla ei esiintynyt farmakokineettisiä yhteisvaikutuksia tutkimuksissa, joissa suun kautta annettavaa rivastigmiinia käytettiin yhdessä digoksiinin, varfariinin, diatsepaamin tai fluoksetiinin kanssa. Suun kautta annettu rivastigmiini ei vaikuttanut varfariinin aikaansaamaan protrombiiniajan pitenemiseen. Digoksiinin ja suun kautta annettavan rivastigmiinin samanaikainen käyttö ei vaikuttanut haitallisesti sydämen johtumisaikaan.
Rivastigmiinin antaminen samanaikaisesti usein käytettyjen lääkevalmisteiden, kuten antasidien, antiemeettien, diabeteslääkkeiden, keskushermostoon vaikuttavien verenpainelääkkeiden, kalsiuminestäjien, inotrooppisten aineiden, angina pectoris -lääkkeiden, tulehduskipulääkkeiden (NSAIDien), estrogeenien, kipulääkkeiden, bentsodiatsepiinien ja antihistamiinien, kanssa ei aiheuttanut muutoksia rivastigmiinin kinetiikassa eikä suurentanut kliinisesti merkityksellisten haittavaikutusten riskiä.
Rivastigmiinin metaboloitumista ajatellen metaboliset yhteisvaikutukset muiden lääkevalmisteiden kanssa näyttävät epätodennäköisiltä, vaikkakin rivastigmiini saattaa estää muiden aineiden butyryylikoliiniesteraasivälitteisen metabolian.
Raskaus ja imetys
Raskaus
Rivastigmiini ja/tai metaboliitit läpäisivät istukan tiineillä eläimillä. Ei ole tiedossa, tapahtuuko vastaavaa ihmisillä. Saatavana ei ole kliinistä tietoa raskaudenaikaisista altistuksista. Rotilla tehdyissä peri- ja postnataalisissa tutkimuksissa havaittiin pidentynyt tiineysaika. Rivastigmiinia ei pidä käyttää raskauden aikana, mikäli käyttö ei ole selvästi välttämätöntä.
Imetys
Eläimillä rivastigmiini kulkeutuu maitoon. Ei tiedetä, erittyykö rivastigmiini ihmisillä äidinmaitoon. Tämän takia rivastigmiinia ei pidä käyttää imetyksen aikana.
Hedelmällisyys
Rotilla ei havaittu hedelmällisyyteen tai lisääntymiskykyyn vaikuttavia rivastigmiinin aiheuttamia haittavaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Rivastigmiinin vaikutuksia ihmisen hedelmällisyyteen ei tiedetä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Alzheimerin tauti saattaa aiheuttaa ajokyvyn asteittaista heikkenemistä tai vaikeuttaa koneidenkäyttökykyä. Lisäksi rivastigmiini voi aiheuttaa pyörtymisiä tai deliriumia. Tämän seurauksena rivastigmiinillä on vähäinen tai kohtalainen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Hoitavan lääkärin tulisi siksi rutiininomaisesti arvioida rivastigmiinia saavien dementiapotilaiden ajokyky ja kyky käyttää tarkkuutta vaativia koneita.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Ihoreaktiot laastarin kiinnityskohdassa (yleensä lievä tai keskivaikea laastarin kiinnityskohdan punoitus) ovat yleisimpiä rivastigmiinidepotlaastareiden käytön yhteydessä esiintyvistä haittavaikutuksista. Seuraavaksi yleisimpiä haittavaikutuksia ovat ruoansulatuskanavaan liittyvät haittavaikutukset, kuten pahoinvointi ja oksentelu.
Taulukossa 1 haittavaikutukset on listattu MedDRA:n elinjärjestelmä- ja yleisyysluokituksen mukaan. Yleisyydet on määritelty seuraavan jaottelun mukaisesti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutustaulukko
Taulukossa 1 on esitetty ne haittavaikutukset, joita raportoitiin 1 670:llä Alzheimerin tautia sairastaneella potilaalla rivastigmiinidepotlaastareilla tehdyissä satunnaistetuissa, kaksoissokkoutetuissa lume‑ ja aktiivikontrolloiduissa 24–48 viikon pituisissa kliinisissä tutkimuksissa sekä valmisteen markkinoilletulon jälkeisissä tiedoissa.
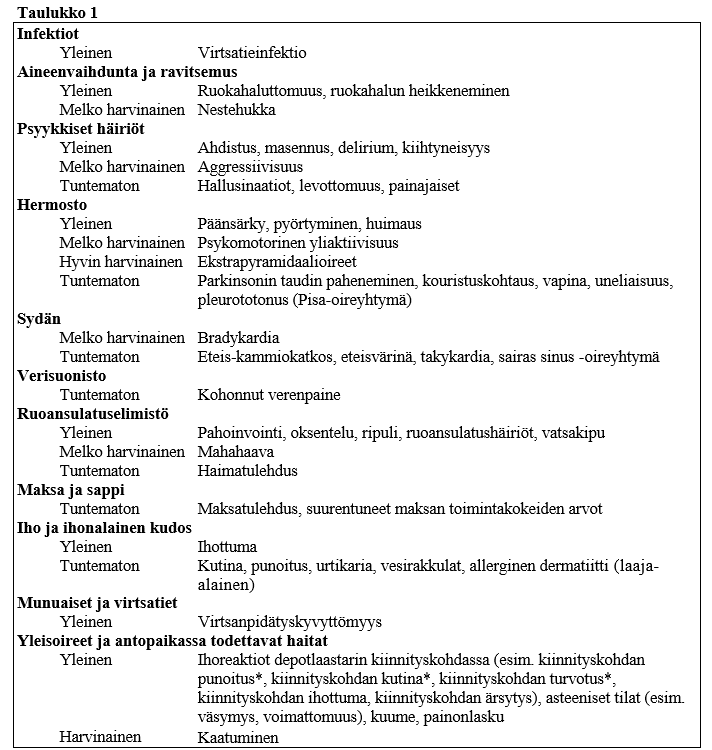
* 24 viikkoa kestäneessä japanilaisilla potilailla tehdyssä kontrolloidussa tutkimuksessa kiinnityskohdan punoituksen, kiinnityskohdan turvotuksen sekä kiinnityskohdan kutinan raportoitiin olevan ”hyvin yleinen”.
Tiettyjen haittavaikutusten kuvaus
Kun yllä mainitussa lumekontrolloidussa tutkimuksessa käytettiin 13,3 mg/24 tuntia ‑annosta suurempia annoksia, unettomuutta ja sydämen vajaatoimintaa todettiin useammin kuin 13,3 mg/24 tuntia ‑ryhmässä tai lumeryhmässä. Tämä viittaa annos-vaikutussuhteen olemassaoloon. Rivastigmiini 13,3 mg/24 tuntia ‑depotlaastariryhmässä näitä tapahtumia ei kuitenkaan esiintynyt sen useammin kuin lumeryhmässäkään.
Seuraavia haittavaikutuksia on todettu vain rivastigmiinikapselien ja -oraaliliuoksen käytön yhteydessä eikä siis rivastigmiinidepotlaastareilla tehdyissä kliinisissä tutkimuksissa: huonovointisuus, sekavuus, lisääntynyt hikoilu (yleinen), pohjukaissuolihaavat, angina pectoris (harvinainen), ruoansulatuskanavan verenvuodot (hyvin harvinainen) ja joissakin tapauksissa voimakas oksentelu, johon liittyi ruokatorven repeämä (yleisyys tuntematon).
Ihoärsytys
Kaksoissokkoutetuissa kontrolloiduissa kliinisissä tutkimuksissa kiinnityskohdan reaktiot olivat vaikeusasteeltaan enimmäkseen lieviä tai kohtalaisia. Kiinnityskohdan ihoreaktiot johtivat hoidon lopettamiseen ≤ 2,3 %:lla rivastigmiinidepotlaastareita käyttävistä potilaista. Hoidon lopettamiseen johtavien kiinnityskohdan reaktioiden ilmaantuvuus oli suurempi aasialaisessa populaatiossa: 4,9 % kiinalaisilla ja 8,4 % japanilaisilla.
Kahdessa 24 viikon pituisessa, kaksoissokkoutetussa, lumekontrolloidussa kliinisessä tutkimuksessa ihoreaktiot mitattiin jokaisen käynnin yhteydessä erityisellä ihoärsytysasteikolla. Rivastigmiinidepotlaastareita käyttäneillä potilailla ihoärsytys oli vaikeusasteeltaan enimmäkseen vähäistä tai lievää. Ihoärsytys luokiteltiin vaikeaksi ≤ 2,2 %:lla potilaista näissä tutkimuksissa ja ≤ 3,7 %:lla potilaista japanilaisessa tutkimuksessa, jossa käytettiin rivastigmiinidepotlaastareita.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet
Suun kautta otettavaan rivastigmiinihoitoon liittyneisiin tahattomiin yliannostustapauksiin ei ole useimmissa tapauksissa liittynyt kliinisiä löydöksiä tai oireita, ja lähes kaikki nämä potilaat jatkoivat rivastigmiinihoitoa 24 tunnin kuluttua yliannostuksesta.
Kolinergista toksisuutta, johon liittyy keskivaikeissa myrkytyksissä havaittuja muskariinisia oireita, on raportoitu. Tällaisia oireita ovat mm. mioosi; punastuminen; ruoansulatushäiriöt, mukaan lukien vatsakipu, pahoinvointi, oksentelu ja ripuli; bradykardia; bronkospasmi ja lisääntynyt bronkiaalinen eritys; liikahikoilu; virtsan‑ ja/tai ulosteenpidätyskyvyttömyys; kyynelvuoto; hypotensio ja liiallinen syljeneritys.
Vaikeammissa tapauksissa voi kehittyä nikotiinisia vaikutuksia, kuten lihasheikkoutta, lihasten nykimistä, kouristuskohtauksia ja hengityksen pysähtyminen, joka voi johtaa kuolemaan.
Lisäksi markkinoilletulon jälkeisissä tapauksissa on esiintynyt huimausta, vapinaa, päänsärkyä, uneliaisuutta, sekavuutta, verenpaineen nousua, hallusinaatioita ja huonovointisuutta. Rivastigmiinidepotlaastareiden väärinkäytöstä/annostusvirheistä (käytössä useampi kuin 1 laastari samaan aikaan) seuranneita yliannostustapauksia on raportoitu markkinoilletulon jälkeen sekä harvoin kliinisissä lääketutkimuksissa.
Hoito
Koska rivastigmiinin puoliintumisaika plasmassa on noin 3,4 tuntia ja asetyylikoliiniesteraasia estävän vaikutuksen kesto on noin 9 tuntia, oireettomassa yliannostustapauksessa suositellaan, että kaikki Rivastor-depotlaastarit irrotetaan välittömästi, eikä seuraavien 24 tunnin aikana saa kiinnittää uutta depotlaastaria. Mikäli yliannostukseen liittyy vaikeaa pahoinvointia ja oksentelua, on harkittava antiemeettien antamista. Muita haittavaikutuksia on hoidettava tarvittaessa oireenmukaisesti.
Suurissa yliannostuksissa voidaan käyttää atropiinia. Suositettu alkuannos on 0,03 mg/kg atropiinisulfaattia laskimoon, minkä jälkeen annoksen suuruus määräytyy kliinisen vasteen mukaan. Skopolamiinin käyttöä antidoottina ei suositella.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: masennuslääkkeet ja keskushermostoa stimuloivat lääkeaineet, antikoliiniesteraasit. ATC-koodi: N06DA03
Rivastigmiini on karbamaatteihin kuuluva asetyyli- ja butyryylikoliiniesteraasin estäjä, jonka uskotaan edistävän kolinergista neurotransmissiota hidastamalla toimintakykyisten kolinergisten neuronien vapauttaman asetyylikoliinin pilkkoutumista. Rivastigmiini saattaa siten lievittää Alzheimerin tautiin liittyvään dementiaan liittyviä kolinergisvälitteisiä kognitiivisia häiriöitä.
Rivastigmiini liittyy kohde-entsyymeihinsä kovalenttisella sidoksella, ja tästä muodostuva kompleksi inaktivoi entsyymit tilapäisesti. Terveillä nuorilla miehillä 3 mg:n peroraalinen annos heikentää asetyylikoliiniesteraasin (AChE) aktiivisuutta aivo-selkäydinnesteessä noin 40 % ensimmäisen 1,5 tunnin kuluessa annoksen antamisesta. Entsyymiaktiivisuus palautuu lähtötasolle noin 9 tunnin kuluttua maksimaalisen estovaikutuksen saavuttamisesta. Alzheimerin tautia sairastavilla potilailla peroraalisen rivastigmiinin AChE-aktiivisuutta estävä vaikutus aivo-selkäydinnesteessä oli annoksesta riippuvainen annostasolle 6 mg kahdesti vuorokaudessa asti, mikä on suurin tutkittu annostus. Peroraalisen rivastigmiinin butyryylikoliiniesteraasiaktiivisuutta estävä vaikutus aivo-selkäydinnesteessä 14 Alzheimer-potilaalla oli samankaltainen kuin AChE:n aktiivisuutta estävä vaikutus.
Alzheimerin tautiin liittyvää dementiaa koskevat kliiniset tutkimukset
Rivastigmiinidepotlaastarien teho Alzheimerin tautia sairastavilla potilailla on osoitettu 24 viikkoa kestäneessä kaksoissokkoutetussa, lumekontrolloidussa ydintutkimuksessa ja sen avoimessa jatkovaiheessa sekä 48 viikon pituisessa, kaksoissokkoutetussa, vertailuvalmisteella tehdyssä tutkimuksessa.
24 viikon pituinen lumekontrolloitu tutkimus
Lumekontrolloituun tutkimukseen osallistuneiden potilaiden MMSE-pistemäärä (Mini-Mental State Examination) oli 10–20. Rivastigmiinin teho osoitettiin riippumattomilla, alaspesifisillä arviointimenetelmillä, joita käytettiin 24 viikon hoitojakson aikana määräajoin. Näihin menetelmiin kuuluvat ADAS-Cog (Alzheimer’s Disease Assessment Scale – Cognitive subscale, kognitiivisen suorituskyvyn mittari), ADCS-CGIC (Alzheimer’s Disease Cooperative Study – Clinician’s Global Impression of Change, lääkärin potilaasta tekemä monipuolinen yleisarviointi, johon sisältyy myös potilaasta huolehtivan henkilön antamia tietoja), sekä ADCS-ADL (Alzheimer’s Disease Cooperative Study – Activities of Daily Living, potilaasta huolehtivan henkilön tekemä arviointi päivittäisistä toimista, joihin kuuluvat henkilökohtainen hygienia, syöminen, pukeutuminen, kotiaskareet kuten ostokset, orientaatiokyvyn säilyminen sekä taloudellisten asioiden hoito). Näistä kolmesta arviointimenetelmästä viikon 24 kohdalla saadut tulokset on esitetty yhteenvetona taulukossa 2.
Taulukko 2
| ITT-LOCF-populaatio | Rivastigmiini- depotlaastarit 9,5 mg/24 tuntia n = 251 | Rivastigmiini- kapselit 12 mg/vrk n =256 | Lumelääke n = 282 |
| ADAS-Cog | |||
| (n = 248) | (n = 253) | (n = 281) | |
Lähtötilanteen keskiarvo ± keskihajonta | 27,0 ± 10,3 | 27,9 ± 9,4 | 28,6 ± 9,9 |
| Keskimääräinen muutos viikon 24 kohdalla ± keskihajonta | −0,6 ± 6,4 | −0,6 ± 6,2 | 1,0 ± 6,8 |
| p-arvo vs. lumelääke | 0,005*1 | 0,003*1 | |
| ADCS-CGIC | |||
| (n = 248) | (n = 253) | (n = 278) | |
| Keskiarvo ± keskihajonta | 3,9 ± 1,20 | 3,9 ± 1,25 | 4,2 ± 1,26 |
| p‑arvo vs. lumelääke | 0,010*2 | 0,009*2 | |
| ADCS-ADL | |||
| (n = 247) | (n = 254) | (n = 281) | |
Lähtötilanteen keskiarvo ± keskihajonta | 50,1 ± 16,3 | 49,3 ± 15,8 | 49,2 ± 16,0 |
| Keskimääräinen muutos viikon 24 kohdalla ± keskihajonta | ‒0,1 ± 9,1 | ‒0,5 ± 9,5 | −2,3 ± 9,4 |
| p‑arvo vs. lumelääke | 0,013*1 | 0,039*1 |
* p ≤ 0,05 vs. lumelääke
ITT: hoitoaikomus; LOCF: viimeisimmän havaintoarvon eteenpäinsiirtämismenettely
1 Perustuu ANCOVA-malliin, jossa tekijöinä hoito ja maa, kovariaattina lähtötilanteessa mitattu arvo. Negatiivinen ADAS-Cog-muutos osoittaa paranemista. Positiivinen ADCS-ADL-muutos osoittaa paranemista.
2 Perustuu CMH-testiin (van Elterenin testi), jaottelu maan perusteella. ADCS-CGIC-pistearvo < 4 osoittaa paranemista.
Tulokset potilaista, joilla todettiin 24 viikon pituisessa lumekontrolloidussa tutkimuksessa kliinisesti merkitsevä hoitovaste, on esitetty taulukossa 3. Kliinisesti merkitsevä paraneminen määriteltiin teoriassa vähintään 4 pisteen paranemiseksi ADAS-Cog-pisteissä, ADCS-CGIG-pistearvon pysymiseksi vähintään lähtöarvossa, ja ADCS-ADL-pistearvon pysymiseksi vähintään lähtöarvossa.
Taulukko 3
| Potilaat, joilla todettiin kliinisesti merkitsevä vaste (%) | |||
| ITT-LOCF-populaatio | Rivastigmiini- depotlaastarit 9,5 mg/24 tuntia n = 251 | Rivastigmiini- kapselit 12 mg/vrk n = 256 | Lumelääke n = 282 |
| Vähintään 4 pisteen paraneminen ADAS-Cog- pisteissä ja ADCS-CGIG- pistearvon ja ADCS-ADL- pistearvon pysyminen vähintään lähtöarvossa | 17,4
| 19,0
| 10,5 |
| p‑arvo vs. lumelääke | 0,037* | 0,004* | |
* p ≤ 0,05 vs. lumelääke
Lokeromallin mukaan 9,5 mg/24 tuntia ‑depotlaastareilla saavutettu altistus oli samankaltainen kuin 12 mg:n/vrk peroraalisella annoksella saavutettu altistus.
48 viikon pituinen aktiivisella vertailuvalmisteella kontrolloitu tutkimus
Aktiivisella vertailuvalmisteella toteutettuun tutkimukseen osallistui potilaita, joiden MMSE-lähtöpisteet olivat 10–24. Tutkimuksen tarkoituksena oli verrata 13,3 mg/24 tuntia ‑depotlaastarin ja 9,5 mg/24 tuntia ‑depotlaastarin tehoa 48 viikon pituisen kaksoissokkoutetun hoitovaiheen aikana Alzheimerin tautia sairastavilla potilailla, joilla osoitettiin toimintakyvyn ja kognition heikentymistä 24–48 viikon pituisen avoimen aloitushoitovaiheen jälkeen. Avoimessa vaiheessa käytettiin ylläpitohoitona 9,5 mg/24 tuntia ‑depotlaastaria. Toimintakyvyn heikkenemistä arvioi tutkija, ja kognition heikkenemisen määritelmänä oli MMSE-pisteiden pieneneminen ≥ 2 pisteellä edellisestä käynnistä tai ≥ 3 pisteellä lähtötilanteesta. Tehoa arvioitiin ADAS-Cog (Alzheimer’s Disease Assessment Scale – Cognitive subscale, kognitiivisen suorituskyvyn mittari) sekä ADCS-IADL (Alzheimer’s Disease Cooperative Study – Instrumental Activities of Daily Living, välineellisten päivittäistoimintojen mittari) -työkaluilla, joiden avulla arvioidaan välineellisten toimintojen suorittamista, kuten talousasioiden hoitoa, ruuanvalmistusta, kaupassa käyntiä, kykyä orientoitua ympäristöön ja kykyä selviytyä ilman valvontaa. Taulukossa 4 esitetään yhteenvetona 48 viikon tulokset näillä kahdella asteikolla arvioituna.
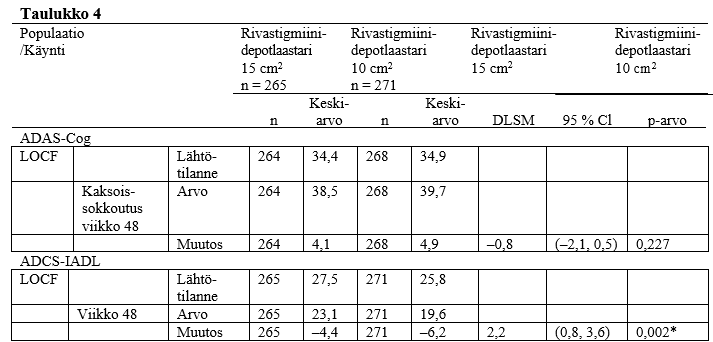
CI = luottamusväli.
DLSM = pienimmän neliösumman keskiarvojen ero.
LOCF = viimeisimmän havaintoarvon eteenpäinsiirtämismenettely.
ADAS-Cog-pisteet: Negatiivinen DLSM-muutos tarkoittaa, että tilanne koheni rivastigmiini 15 cm2 ‑depotlaastareilla enemmän kuin rivastigmiini 10 cm2 -depotlaastareilla.
ADCS-IADL-pisteet: Positiivinen DLSM-muutos tarkoittaa, että tilanne koheni rivastigmiini 15 cm2 ‑depotlaastareilla enemmän kuin rivastigmiini 10 cm2 -depotlaastareilla.
N on niiden potilaiden lukumäärä, joista on saatavana lähtötilanteen arvio (aiemman, avoimen vaiheen viimeinen arvio) ja vähintään 1 arvio lähtötilanteen jälkeen (LOCF-arviota varten).
DLSM, 95 %:n CI ja p-arvot perustuvat maan ja ADAS-Cog-lähtöpisteiden mukaan korjattuun ANCOVA-malliin (kovarianssianalyysiin).
* p < 0,05
Lähde: Tutkimus D2340 – taulukko 11‑6 ja taulukko 11‑7
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset rivastigmiinivalmisteen käytöstä kaikkien pediatristen potilasryhmien hoidossa Alzheimerin taudissa (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Rivastigmiini imeytyy rivastigmiinidepotlaastareista hitaasti. Mitattavia plasman lääkeainepitoisuuksia on havaittavissa vasta 0,5–1 tunnin kuluttua ensimmäisestä annoksesta. Cmax saavutetaan 10–16 tunnissa. Huippupitoisuuden saavuttamisen jälkeen plasman lääkeainepitoisuudet pienenevät hitaasti jäljellä olevan 24 tunnin annostelujakson aikana. Mikäli annostelua jatketaan (kuten vakaassa tilassa) ja depotlaastari vaihdetaan uuteen, plasman lääkeainepitoisuuksien hidas lasku jatkuu aluksi vielä keskimäärin noin 40 minuutin ajan, kunnes uudesta depotlaastarista imeytyy lääkeainetta nopeammin kuin sitä eliminoituu, ja pitoisuudet plasmassa lähtevät taas nousuun ja saavuttavat uuden huippuarvon noin 8 tunnin kuluttua. Vakaassa tilassa pienimmät lääkeainepitoisuudet ovat noin 50 % huippupitoisuuksista, toisin kuin suun kautta tapahtuvassa annostelussa, jossa lääkeainepitoisuudet pienenevät lähes olemattomiksi annosten välillä. Rivastigmiinialtistus (Cmax ja AUC) suureni suhteettomasti 2,6‑kertaiseksi, kun annos suurennettiin 4,6 mg/24 tuntia ‑depotlaastarista 9,5 mg/24 tuntia ‑depotlaastariin, ja 4,9‑kertaiseksi, kun annos suurennettiin 13,3 mg/24 tuntia ‑depotlaastariin, joskin vaikutus on lievempi kuin peroraalisen lääkemuodon kohdalla. Huippupitoisuuksien ja pienimpien pitoisuuksien suhteellista eroa ([Cmax - Cmin]/Cavg) mittaava vaihteluindeksi (FI) oli rivastigmiini 4,6 mg/24 tuntia ‑depotlaastarilla 0,58, rivastigmiini 9,5 mg/24 tuntia ‑depotlaastarilla 0,77 ja rivastigmiini 13,3 mg/24 tuntia ‑depotlaastarilla 0,72. Huippupitoisuuksien ja pienimpien pitoisuuksien välinen ero oli siis huomattavasti pienempi kuin peroraalisen lääkemuodon kohdalla (FI = 3,96 [6 mg/vrk] ja 4,15 [12 mg/vrk]).
Depotlaastarista 24 tunnin aikana vapautuva rivastigmiiniannos (mg/24 tuntia) ei vastaa suoraan kapselin sisältämää rivastigmiiniannosta (mg) plasmassa 24 tunnin aikana saavutettavien rivastigmiinipitoisuuksien suhteen.
Rivastigmiinin kerta-annoksen farmakokineettisten parametrien (normalisoitu painoon perustuvan annoksen suhteen) vaihtelu potilaiden välillä oli ihon läpi annostelussa 43 % (Cmax) ja 49 % (AUC0-24h) ja suun kautta annostelussa 74 % (Cmax) ja 103 % (AUC0-24h). Alzheimerin tautia sairastavilla potilailla tehdyssä vakaan tilan tutkimuksessa potilaiden välinen vaihtelu oli depotlaastarien käytön jälkeen enintään 45 % (Cmax) ja 43 % (AUC0-24h) ja peroraalisen lääkemuodon käytön jälkeen enintään 71 % (Cmax) ja 73 % (AUC0-24h).
Vakaassa tilassa vaikuttavalle aineelle altistumisen (rivastigmiini ja NAP226-90-metaboliitti) ja potilaan painon välillä havaittiin yhteys Alzheimerin tautia sairastavilla potilailla. 65 kg painavaan potilaaseen verrattuna 35 kg painavan potilaan rivastigmiinipitoisuudet vakaassa tilassa ovat noin kaksi kertaa suuremmat ja 100 kg painavan potilaan rivastigmiinipitoisuudet noin puolta pienemmät. Painon vaikutus potilaan altistukseen vaikuttavalle aineelle viittaa siihen, että hyvin pienipainoisten potilaiden annoksen suurentaminen vaatii erityistä varovaisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Suurin altistus (AUC∞) rivastigmiinille (ja NAP266‑90-metaboliitille) saavutettiin, kun depotlaastari kiinnitettiin yläselkään, rintakehään tai olkavarteen. Altistus oli noin 20–30 % pienempi, kun depotlaastari kiinnitettiin vatsan alueelle tai reiteen.
Alzheimerin tautia sairastavilla potilailla ei todettu merkityksellistä rivastigmiinin tai NAP226-90- metaboliitin kertymistä plasmaan, mutta depotlaastarihoidon toisena päivänä plasmasta mitatut lääkepitoisuudet olivat ensimmäisen hoitopäivän pitoisuuksia suuremmat.
Jakautuminen
Rivastigmiini sitoutuu heikosti (noin 40‑prosenttisesti) plasman proteiineihin. Se läpäisee hyvin veri‑aivoesteen, ja sen näennäinen jakautumistilavuus on 1,8–2,7 l/kg.
Biotransformaatio
Rivastigmiini metaboloituu nopeasti ja suuressa määrin, ja sen eliminaation näennäinen puoliintumisaika plasmassa on noin 3,4 tuntia depotlaastarin irrottamisen jälkeen. Imeytymisnopeus rajoitti eliminaatiota (flip-flop-kinetiikka), mikä selittää sen, miksi t½ oli depotlaastarien käytön jälkeen (3,4 h) pidempi kuin peroraalisen tai laskimoon annettavan hoidon jälkeen (1,4–1,7 h). Rivastigmiini metaboloituu pääasiassa koliiniesteraasivälitteisen hydrolyysin kautta NAP226-90-metaboliitiksi. Tällä metaboliitilla on hyvin heikko asetyylikoliiniesteraasia estävä vaikutus (< 10 %) in vitro.
In vitro ‑tutkimuksista saadun näytön perusteella farmakokineettisiä yhteisvaikutuksia ei ole odotettavissa yhteiskäytössä lääkeaineiden kanssa, jotka metaboloituvat seuraavien sytokromi-isoentsyymien kautta: CYP1A2, CYP2D6, CYP3A4/5, CYP2E1, CYP2C9, CYP2C8, CYP2C19 tai CYP2B6. Eläinkokeista saadun näytön perusteella tärkeimmät sytokromi P450 -isoentsyymit osallistuvat hyvin vähäisessä määrin rivastigmiinin metaboliaan. Rivastigmiinin kokonaispuhdistuma plasmasta oli noin 130 l/h laskimoon annetun 0,2 mg:n annoksen jälkeen ja laski tasolle 70 l/h laskimoon annetun 2,7 mg:n annoksen jälkeen. Tämä on yhdenmukainen havaintojen kanssa, jotka koskevat rivastigmiinin eliminaation saturaatiosta johtuvaa epälineaarista, suhteetonta farmakokinetiikan kasvua.
Metaboliitin ja alkuperäisen lääkeaineen AUC∞-suhde oli depotlaastarin käytön jälkeen noin 0,7 ja peroraalisen annostelun jälkeen noin 3,5. Tämä osoittaa, että metabolia jää transdermaalisen hoidon kohdalla huomattavasti vähäisemmäksi kuin peroraalisen hoidon kohdalla. NAP226‑90-metaboliittia muodostuu depotlaastarien käytön yhteydessä vähemmän kuin suun kautta toteutettavan hoidon yhteydessä luultavasti siksi, että presysteemistä metaboliaa (ensikierron metaboliaa maksassa) ei tapahdu.
Eliminaatio
Muuttumattomassa muodossa olevan rivastigmiinin pitoisuudet virtsassa jäävät häviävän pieniksi. Depotlaastarien käytön yhteydessä pääasiallinen eliminaatioreitti on metaboliittien erittyminen munuaisten kautta. Peroraalisen 14C‑rivastigmiinin annon jälkeen lääkeaine eliminoitui munuaisten kautta nopeasti ja lähes täydellisesti (> 90 %) 24 tunnin kuluessa. Alle 1 % annetusta annoksesta erittyy ulosteeseen.
Populaatiofarmakokineettinen analyysi osoitti, että nikotiinin käyttö lisää rivastigmiinin oraalista puhdistumaa 23 % Alzheimerin tautia sairastavilla potilailla (n = 75 tupakoijaa ja 549 tupakoimatonta), kun rivastigmiinia otetaan kapseleina suun kautta enintään 12 mg vuorokaudessa.
Erityisryhmät
Iäkkäät
Ikä ei vaikuttanut rivastigmiinialtistukseen, kun Alzheimerin tautia sairastavia potilaita hoidettiin rivastigmiinidepotlaastareilla.
Maksan vajaatoiminta
Rivastigmiinidepotlaastarien käyttöä ei ole tutkittu henkilöillä, joilla on maksan vajaatoiminta. Peroraalisen annon jälkeen rivastigmiinin Cmax oli noin 60 % suurempi ja rivastigmiinin AUC oli yli kaksinkertainen lievää tai kohtalaista maksan vajaatoimintaa sairastavilla potilailla terveisiin henkilöihin verrattuna.
3 mg:n ja 6 mg:n peroraalisen kerta-annoksen jälkeen keskimääräinen rivastigmiinin puhdistuma oli noin 46–63 % pienempi lievää tai kohtalaista maksan vajaatoimintaa sairastavilla potilailla (n = 10, Child-Pughin luokitus 5–12, biopsialla todennettu) kuin terveillä yksilöillä (n = 10).
Munuaisten vajaatoiminta
Rivastigmiinidepotlaastarien käyttöä ei ole tutkittu henkilöillä, joilla on munuaisten vajaatoiminta. Kreatiniinipuhdistumalla ei ollut selvää vaikutusta rivastigmiinin tai sen metaboliitin vakaan tilan pitoisuuksiin joukkoanalyysissä. Annosta ei ole tarpeen muuttaa munuaisten vajaatoimintapotilaille (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Suun kautta ja paikallisesti annosteltavalla rivastigmiinilla tehdyissä toistuvan altistuksen toksisuustutkimuksissa hiirillä, rotilla, kaniineilla, koirilla ja minisioilla havaittiin vain liialliseen farmakologiseen aktiivisuuteen liittyviä vaikutuksia. Kohde-elintoksisuutta ei havaittu. Suun kautta tapahtuvaa ja paikallista annostelua jouduttiin rajoittamaan tutkimuksissa käytettyjen eläinmallien sensitiivisyyden takia.
Rivastigmiini ei ollut mutageeninen tavanomaisissa in vitro‑ ja in vivo ‑testeissä, paitsi kromosomipoikkeavuustestissä ihmisen perifeerisen veren lymfosyyteillä annoksella, joka oli yli 104 kertaa suurempi kuin odotettavissa oleva kliininen altistus. Mikrotumatesti in vivo oli negatiivinen. Päämetaboliitti NAP226‑90 ei myöskään osoittautunut genotoksiseksi.
Karsinogeenisuuteen viittaavia löydöksiä ei saatu suurimmalla siedetyllä peroraalisella ja paikallisesti annetulla annoksella tehdyissä hiiritutkimuksissa eikä suurimmalla siedetyllä peroraalisella annoksella tehdyssä rottatutkimuksessa. Altistus rivastigmiinille ja sen metaboliiteille oli suurin piirtein samaa luokkaa kuin rivastigmiinikapselien ja -depotlaastarien suurimmilla annoksilla saavutettu altistus ihmisillä.
Eläimillä rivastigmiini läpäisee istukan ja kulkeutuu maitoon. Tiineillä rotilla ja kaniineilla tehdyt peroraaliset tutkimukset eivät antaneet viitteitä rivastigmiinin teratogeenisuudesta. Peroraalisissa tutkimuksissa uros- ja naarasrotilla ei havaittu hedelmällisyyteen tai lisääntymiskykyyn vaikuttavia rivastigmiinin aiheuttamia haittavaikutuksia vanhempien eikä näiden jälkeläisten sukupolvessa. Tiineillä eläimillä ei ole tehty spesifisiä ihotutkimuksia.
Rivastigmiinidepotlaastarit eivät olleet fototoksisia, eikä niiden katsottu olevan herkistäviä. Joissakin muissa ihotoksisuutta selvittäneissä tutkimuksissa koe-eläimillä (myös verrokeilla) todettiin lievää ihoärsytystä. Tämän perusteella rivastigmiinidepotlaastarit saattavat aiheuttaa potilaille lievää ihon punoitusta.
Kaniineilla tehdyssä tutkimuksessa identifioitiin rivastigmiinin mahdollisesti aiheuttama lievä silmän/limakalvon ärsytys. Potilaan tai potilasta hoitavan henkilön on siis vältettävä kosketusta silmiin depotlaastarin käsittelyn jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Farmaseuttiset tiedot
Apuaineet
Aktiivinen kerros:
- poly [(2‑etyyliheksyyli)akrylaatti, vinyyliasetaatti]
Liimamatriksikerros:
- keskisuuren molekyylipainon polyisobuteeni
- suuren molekyylipainon polyisobuteeni
- piidioksidi, kolloidinen, vedetön
- parafiini, kevyt nestemäinen
Taustakalvo:
- polyeteeni‑/kestomuovihartsi‑/alumiinipinnoitettu polyesterikalvo
Repäisykerros:
- polyesterikalvo, fluoropolymeeripinnoitettu
Oranssi painomuste
Yhteensopimattomuudet
Depotlaastarin kiinnityskohdassa ei pidä käyttää emulsiovoiteita, kosteusvoiteita eikä talkkia tai puuteria, sillä ne voivat heikentää laastarin kiinnitystä.
Kestoaika
2 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi lämpötilan suhteen erityisiä säilytysolosuhteita.
Poista depotlaastari annospussista vasta juuri ennen käyttöä.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
RIVASTOR depotlaastari
13,3 mg/24 h (L:ei) 30 kpl (43,90 €), 90 kpl (3x30) (107,62 €)
PF-selosteen tieto
Lapsiturvalliset annospussit on valmistettu paperi‑/polyeteenitereftalaatti‑/alumiini‑/polyakryylinitriili‑ tai paperi/polyeteenitereftalaatti/polyeteeni/alumiini/LasPolD-monikerroslaminaattimateriaalista. Yksi annospussi sisältää yhden depotlaastarin.
Saatavana on 7 tai 30 annospussia sisältäviä pakkauksia sekä 60 (2 x 30) tai 90 (3 x30) annospussia sisältäviä kerrannaispakkauksia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Ohut, pyöreänmallinen matriksityyppinen depotlaastari, jonka halkaisija on noin 4,0 cm ja jossa on kolme kerrosta. Taustakerroksen ulkopinta on kellanruskea, ja siinä on painatus ”RIV‑TDS 13.3 mg/24 h”.
Käyttö- ja käsittelyohjeet
Käytetyt depotlaastarit tulee taittaa kahtia liimapuoli sisäänpäin, laittaa takaisin alkuperäiseen annospussiinsa ja hävittää turvallisesti ja poissa lasten ulottuvilta ja näkyviltä. Käytetyt ja käyttämättä jääneet depotlaastarit on hävitettävä paikallisten vaatimusten mukaisesti tai palautettava apteekkiin.
Korvattavuus
RIVASTOR depotlaastari
13,3 mg/24 h 30 kpl, 90 kpl
- Peruskorvaus (40 %).
ATC-koodi
N06DA03
Valmisteyhteenvedon muuttamispäivämäärä
08.09.2025
Yhteystiedot
 ORION OYJ ORION PHARMA
ORION OYJ ORION PHARMA Orionintie 1, PL 65
02101 Espoo
010 4261
www.orion.fi
etunimi.sukunimi@orionpharma.com