
SPINRAZA injektioneste, liuos 12 mg, 28 mg, 50 mg
Vaikuttavat aineet ja niiden määrät
Spinraza 12 mg
Yksi 5 ml:n kertakäyttöinen injektiopullo sisältää nusinerseeninatriumia määrän, joka vastaa 12 mg:aa nusinerseeniä.
Yksi ml sisältää 2,4 mg nusinerseeniä.
Spinraza 28 mg
Yksi 5 ml:n kertakäyttöinen injektiopullo sisältää nusinerseeninatriumia määrän, joka vastaa 28 mg:aa nusinerseeniä.
Yksi ml sisältää 5,6 mg nusinerseeniä.
Spinraza 50 mg
Yksi 5 ml:n kertakäyttöinen injektiopullo sisältää nusinerseeninatriumia määrän, joka vastaa 50 mg:aa nusinerseeniä.
Yksi ml sisältää 10 mg nusinerseeniä.
Apunaineet, joiden vaikutus tunnetaan
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos.
Kliiniset tiedot
Käyttöaiheet
Spinraza on tarkoitettu kromosomiin 5q liittyvän spinaalisen lihasatrofian hoitoon.
Ehto
Spinraza-hoidon saa aloittaa vain lääkäri, joka on perehtynyt spinaalisen lihasatrofian hoitoon.
Annostus ja antotapa
Spinraza-hoidon saa aloittaa vain lääkäri, joka on perehtynyt spinaalisen lihasatrofian (spinal muscular atrophy, SMA) hoitoon.
Hoitopäätöksen on perustuttava yksilölliseen asiantuntija-arvioon hoidon oletettavista hyödyistä kyseiselle henkilölle, ja niitä on arvioitava nusinerseenihoidon mahdollisiin riskeihin nähden.
Spinrazan käyttöä ei ole tutkittu potilailla, joilla on syntyessään vaikea hypotonia ja hengityksen vajaatoiminta, ja vaikean survival motor neuron (SMN) -proteiinin puutoksen vuoksi nämä potilaat eivät ehkä saa hoidosta kliinisesti merkittävää hyötyä.
Annostus
Valmisteesta on käytettävissä kaksi annostusohjelmaa: matala-annoksinen 12 mg ohjelma ja korkea-annoksinen 50/28 mg ohjelma. Spinraza-hoito pitää aloittaa mahdollisimman pian diagnoosin jälkeen.
Matala-annoksinen ohjelma
Tämä annostus aloitetaan 12 mg:n latausannoksella päivinä 0, 14, 28 ja 63, minkä jälkeen annetaan 12 mg:n ylläpitoannos 4 kuukauden välein.
Korkea-annoksinen ohjelma
50 mg:n latausannos annetaan päivänä 0 ja päivänä 14. Tämän jälkeen annetaan 28 mg:n ylläpitoannos 4 kuukauden välein.
Vaihtaminen matala-annoksisesta ohjelmasta korkea-annoksiseen ohjelmaan
Potilaille, joita hoidetaan parhaillaan 12 mg:n Spinraza-annoksilla, voidaan vaihtaa 50/28 mg:n annostus yhdellä 50 mg:n latausannoksella, joka annetaan aikaisintaan 4 kuukautta (+/– 14 päivää) viimeisen 12 mg:n annoksen jälkeen. Tämän jälkeen annetaan 28 mg:n ylläpitoannos 4 kuukauden välein.
Hoidon kesto
Hoidon jatkamisen tarve on tarkistettava säännöllisesti ja arvioitava yksilöllisesti potilaan kliinisen tilan ja hoitovasteen mukaan.
Annoksen unohtuminen tai myöhästyminen
Jos jokin lataus- tai ylläpitoannoksista unohtuu tai myöhästyy, Spinrazaa pitää antaa alla olevien taulukon 1 (12 mg:n annostusohjelma) ja taulukon 2 (50/28 mg:n annostusohjelma) mukaisesti.
Taulukko 1: Suositukset koskien myöhästynyttä tai unohtunutta annosta 12 mg:n annostuksen yhteydessä
| Myöhästynyt tai unohtunut annos | Annoksen ajoittaminen | |
| Latausannos | ||
Esim. jos kolmas latausannos on annettu 30 päivää myöhässä päivänä 58 (eikä alkuperäisen suunnitelman mukaisesti päivänä 28), neljäs latausannos pitää antaa 35 päivää myöhemmin päivänä 93 (eikä alkuperäisen suunnitelman mukaisesti päivänä 63) ja ylläpitoannos 4 kuukautta sen jälkeen. | ||
| Ylläpitoannos | Annoksen ajoittaminen | |
| > 4 – < 8 kuukautta edellisestä annoksesta |
| |
| ≥ 8 – < 16 kuukautta edellisestä annoksesta |
| |
| ≥ 16 – < 40 kuukautta edellisestä annoksesta |
| |
| ≥ 40 kuukautta edellisestä annoksesta |
| |
| *Edellä mainittujen suositusten jälkeen annetaan ylläpitoannos 4 kuukauden kuluttua edellisestä annoksesta ja sen jälkeen toistetaan 4 kuukauden välein. | ||
Taulukko 2: Suositukset koskien myöhästynyttä tai unohtunutta annosta 50/28 mg:n annostuksen yhteydessä
| Myöhästynyt tai unohtunut annos | Annoksen antamisajankohta |
| Toinen latausannos | |
| |
| Ylläpitoannos | |
| < 8 kuukautta edellisestä annoksesta |
|
| 8 – < 12 kuukautta edellisestä annoksesta |
|
| ≥ 12 kuukautta edellisestä annoksesta |
|
Erityisryhmät
Munuaisten vajaatoiminta
Nusinerseeniä ei ole tutkittu munuaisten vajaatoimintaa sairastavilla potilailla. Turvallisuutta ja tehoa munuaisten vajaatoimintaa sairastavien potilaiden hoidossa ei ole varmistettu. Näitä potilaita on seurattava huolellisesti.
Maksan vajaatoiminta
Nusinerseeniä ei ole tutkittu maksan vajaatoimintaa sairastavilla potilailla. Nusinerseeni ei metaboloidu maksan sytokromi P450 (CYP450) -entsyymijärjestelmän kautta, joten maksan vajaatoimintaa sairastavien potilaiden annoksen muuttaminen ei todennäköisesti ole tarpeen (ks. kohdat Yhteisvaikutukset ja Farmakokinetiikka).
Antotapa
Spinraza annetaan lannepistolla selkäydinnesteeseen.
Hoitoa saavat antaa vain lannepiston tekemiseen perehtyneet terveydenhuollon ammattilaiset.
Spinraza annetaan spinaalineulaa käyttämällä 1–3 minuutin kestoisena bolusinjektiona selkäydinnesteeseen. Injektiota ei saa antaa ihoalueelle, jossa on merkkejä infektiosta tai inflammaatiosta. Ennen Spinrazan antamista potilaalta on suositeltavaa poistaa injisoitavaa Spinraza-tilavuutta vastaava tilavuus aivo-selkäydinnestettä.
Sedaatio saattaa olla tarpeen ennen Spinrazan antoa riippuen potilaan kliinisestä tilasta.
Annettaessa Spinrazaa selkäydinnesteeseen, etenkin nuorille potilaille ja skolioosipotilaille, apuna voidaan käyttää ultraääntä (tai muuta kuvantamismenetelmää), ks. käyttöohjeet kohdasta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Lannepistotoimenpide
Lannepistotoimenpiteeseen liittyy haittavaikutusriski (esim. araknoidiitti, päänsärky, selkäkipu, oksentelu, ks. kohta Haittavaikutukset). Tähän antoreittiin saattaa liittyä vaikeuksia hyvin nuorilla potilailla ja skolioosipotilailla. Spinrazan intratekaalisen annon apuna voidaan käyttää lääkärin harkinnan mukaan ultraääntä tai muita kuvantamismenetelmiä. Jos araknoidiittia epäillään, araknoidiitti ja tulehduksen laajuus on varmistettava magneettikuvauksella. Jos araknoidiitti todetaan, kyseistä injektiokohtaa ei saa käyttää, ennen kuin paikallisen tulehduksen mahdollisuus on suljettu pois.
Trombosytopenia ja veren hyytymiseen liittyvät poikkeavuudet
Muiden antisense-oligonukleotidien ihon alle tai laskimoon tapahtuneen annon jälkeen on havaittu veren hyytymiseen liittyviä poikkeavuuksia ja trombosytopeniaa, mukaan lukien akuuttia vaikea‑asteista trombosytopeniaa. Ennen Spinrazan antoa suositellaan kliinisen tarpeen mukaan laboratoriokokeita veren trombosyyttipitoisuuden ja hyytymisen selvittämiseksi.
Munuaistoksisuus
Muiden antisense-oligonukleotidien ihon alle ja laskimoon tapahtuneen annon jälkeen on havaittu munuaistoksisuutta. Virtsan proteiinipitoisuus suositellaan tarkistamaan laboratoriokokeen avulla (suositeltavin näyte on aamun ensimmäinen virtsa) kliinisen tarpeen mukaan. Jos virtsan proteiinipitoisuus on jatkuvasti koholla, on harkittava lisätutkimuksia.
Hydrokefalus
Ahtaumattoman hydrokefaluksen tapauksia, jotka eivät liittyneet meningiittiin tai verenvuotoon, on markkinoille tulon jälkeisessä käytössä raportoitu nusinerseenihoitoa 12 mg:n annoksina saaneilla potilailla. Joillekin potilaille oli asennettu ventrikuloperitoneaalinen suntti. Potilaille, joiden tajunnan taso on alentunut, on harkittava tutkimuksia hydrokefaluksen poissulkemiseksi. Nusinerseenihoidon hyötyjä ja riskejä potilaille, joilla on ventrikuloperitoneaalinen suntti, ei tällä hetkellä tunneta ja hoidon ylläpidon tarve on huolellisesti arvioitava.
Apuaineet
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 5 ml:n injektiopullo, eli sen voidaan sanoa olevan ”natriumiton”.
Kalium
Tämä lääkevalmiste sisältää kaliumia alle 1 mmol (39 mg) per 5 ml:n injektiopullo, eli sen voidaan sanoa olevan ”kaliumiton”.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. In vitro -tutkimukset osoittivat, että nusinerseeni ei indusoi eikä estä CYP450-välitteistä metaboliaa. In vitro -tutkimukset osoittivat olevan epätodennäköistä, että kilpailu plasman proteiineihin sitoutumisesta, kilpailu kuljettajaproteiinien kanssa tai kuljettajaproteiinien esto johtaisi yhteisvaikutuksiin nusinerseenin kanssa.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja nusinerseenin käytöstä raskaana oleville naisille. Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi nusinerseenin käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyvätkö nusinerseeni/metaboliitit ihmisen rintamaitoon.
Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko nusinerseenihoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Eläimillä tehdyissä toksisuustutkimuksissa ei havaittu vaikutuksia urosten tai naaraiden hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta). Tietoja mahdollisista vaikutuksista ihmisen hedelmällisyyteen ei ole saatavilla.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Nusinerseenillä ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmät nusinerseenin lannepistolla antoon liittyneet haittavaikutukset olivat päänsärky, oksentelu ja selkäkipu.
Kokemus 12 mg:n annostusohjelman käytöstä
12 mg:n Spinraza-annoksen turvallisuutta arvioitiin kliinisissä tutkimuksissa perustuen kahteen faasin 3 tutkimukseen, joissa oli mukana SMA:ta sairastavia vauvoja (CS3B) ja lapsia (CS4), yhteen faasin 2 tutkimukseen, jossa oli mukana SMA:ta sairastavia vauvoja ja lapsia (CS7), sekä avoimiin tutkimuksiin, joissa oli mukana vielä oireettomia geneettisen SMA‑diagnoosin saaneita vauvoja (CS5) sekä SMA:ta sairastavia vauvoja ja lapsia. Tutkimukseen CS11 otettiin mukaan imeväisiässä ja myöhemmin sairastuneita potilaita, mukaan lukien potilaita, jotka olivat suorittaneet tutkimukset CS3B, CS4 ja CS12.
Hoitoa enintään 12 mg:n Spinraza-annoksilla sai yhteensä 385 SMA:ta sairastavaa potilasta, ja tutkimuksessa mukana olon kokonaiskesto oli 1–3 940 vuorokautta (> 10 vuotta) (mediaani 2 388 vuorokautta).
Kokemus 50/28 mg:n annostusohjelman käytöstä
50/28 mg:n Spinraza-annoksen turvallisuutta SMA:ta sairastaville vauvoille, lapsille ja aikuisille arvioitiin tutkimuksessa SM203 ja tutkimuksessa SM302 oireisilla SMA:ta sairastavilla potilailla, joiden ikä ensimmäisen tutkimuksessa annetun annoksen antoajankohtana oli 14 vuorokautta – 65 vuotta.
Hoitoa 50 mg:n tai 28 mg:n Spinraza-annoksilla sai yhteensä 128 SMA:ta sairastavaa potilasta, ja tutkimuksessa mukana olon kokonaiskesto oli 13–1 521 vuorokautta (> 4 vuotta) (mediaani 740,5 vuorokautta).
Haittavaikutustaulukko
Nusinerseenin turvallisuuden arviointi perustuu kliinisissä tutkimuksissa olleiden potilaiden tietoihin sekä myyntiluvan myöntämisen jälkeiseen lääketurvatoimintaan. Nusinerseenin antoon liittyneet haittavaikutukset on esitetty taulukossa 3.
Haittavaikutusten arviointi perustuu seuraaviin esiintymistiheyttä koskeviin tietoihin:
Hyvin yleinen (≥1/10)
Tuntematon (koska saatavissa oleva tieto ei riitä arviointiin)
Taulukko 3: Spinrazan antoon liittyvät haittavaikutukset
| MedDRA-elinjärjestelmäluokka | Haittavaikutus | Esiintymistiheysluokka |
| Infektiot | Meningiitti | Tuntematon |
| Immuunijärjestelmä | Yliherkkyys* | Tuntematon |
| Hermosto | Päänsärky** Aseptinen meningiitti Araknoidiitti | Hyvin yleinen Tuntematon Tuntematon |
| Ruoansulatuselimistö | Oksentelu** | Hyvin yleinen |
| Luusto, lihakset ja sidekudos | Selkäkipu** | Hyvin yleinen |
| Yleisoireet ja antopaikassa todettavat haitat | Kuume | Hyvin yleinen |
*esim. angioedeema, urtikaria ja ihottuma.
**Lannepistotoimenpiteeseen liittyviksi katsotut haittavaikutukset. Näiden haittavaikutusten voidaan katsoa olevan lannepiston jälkeisen oireyhtymän ilmentymiä. Näitä haittavaikutuksia on raportoitu tutkimuksessa CS4 (myöhemmin alkaneet SMA-oireet) 12 mg:n Spinraza-annoksia saaneilla tutkittavilla (n = 84) vähintään 5 % yleisemmin kuin lumetoimenpiteitä saaneilla verrokeilla.
Ahtaumattoman hydrokefaluksen tapauksia on havaittu valmisteen markkinoille tulon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Valittujen haittavaikutusten kuvaus
Nusinerseenin antoon lannepistolla liittyviä haittavaikutuksia on havaittu. Niitä on raportoitu useimmiten 72 tunnin kuluessa toimenpiteestä. Näiden tapahtumien ilmaantuvuus ja vaikeusaste olivat yhdenmukaisia lannepiston yhteydessä tavallisesti esiintyvien tapahtumien kanssa. Nusinerseenin kliinisissä tutkimuksissa ei ole havaittu lannepiston vakavia komplikaatioita, kuten vakavia infektioita.
Joitain lannepistoon yleisesti liittyviä haittavaikutuksia (esim. päänsärky ja selkäkipu) ei voitu arvioida nusinerseenille altistetuilla pikkulapsilla kommunikoinnin rajoitusten vuoksi tässä ikäryhmässä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksia, joihin liittyi haittavaikutuksia, ei raportoitu kliinisissä tutkimuksissa.
Yliannostustapauksessa hoitoon pitää kuulua elintoimintoja tukeva hoito ja terveydenhuollon ammattilaisen konsultointi, ja potilaan kliinistä tilaa on seurattava huolellisesti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut tuki- ja liikuntaelinten sairauksien lääkkeet, ATC-koodi: M09AX07
Vaikutusmekanismi
Nusinerseeni on antisense-oligonukleotidi, joka lisää eksoni 7:n inkluusion osuutta survival motor neuron 2 –geenin (SMN2-geenin) lähetti-RNA:n (mRNA) transkripteissa sitoutumalla SMN2‑geenin esiaste-mRNA:n introni 7:ssä sijaitsevaan silmukointia vaimentavaan intronin kohtaan (intronic splice silencing site, ISS‑N1). Sitoutumalla antisense-oligonukleotidi syrjäyttää silmukointitekijöitä, jotka normaalisti estävät silmukointia. Näiden tekijöiden syrjäyttäminen johtaa eksoni 7:n säilymiseen SMN2-geenin mRNA:ssa, jolloin tuotettu SMN2-geenin mRNA voidaan kääntää toimivaksi, normaalipituiseksi SMN-proteiiniksi.
SMA on etenevä neuromuskulaarinen sairaus, joka johtuu mutaatioista SMN1-geenin kromosomissa 5q. Toinen geeni, SMN2, joka sijaitsee lähellä SMN1:tä, vastaa pienestä määrästä SMN-proteiinin tuotantoa. SMA on kliininen tautikirjo, jossa pienempi SMN2-geenin kopioiden määrä ja oireiden alkaminen nuoremmalla iällä on yhdistetty vaikeampaan taudinkuvaan.
Plasman neurofilamentin kevytketju (Nf-L)
Aksonaalisen vaurion osoittavan veren merkkiaineen neurofilamentin kevytketjun (Nf-L) pitoisuus plasmassa mitattiin tutkimuksessa SM203 lähtötilanteessa ja useissa aikapisteissä. Kyseisessä tutkimuksessa arvioitiin 50/28 mg:n annostusta imeväisiässä ja myöhemmin alkanutta SMA:ta sairastavilla potilailla.
Plasman Nf-L-pitoisuudet pienenivät 50/28 mg:n annosryhmässä nopeammin kuin 12 mg:n annosryhmässä, sillä pitoisuus pieneni lähtötilanteesta päivään 64 mennessä 50/28 mg:n annosryhmässä 88 % ja 12 mg:n annosryhmässä 77 % (50/28 mg:n ryhmän vs. 12 mg:n ryhmän geometristen keskiarvojen suhteiden ero: 49 % [p = 0,0020]) (kuva 1).
Myös aiemmin hoitamattomilla myöhemmin alkanutta SMA:ta sairastavilla potilailla plasman Nf-L-pitoisuudet pienenivät lähtötilanteesta päivään 64 mennessä 50/28 mg:n annosryhmässä 66 % ja 12 mg:n annosryhmässä 42 % (50/28 mg:n annosryhmän vs. 12 mg:n annosryhmän geometristen keskiarvojen suhteiden ero: 42 % [p = 0,0495]).
Kuva 1: Tutkimuksen SM203 osa B ANCOVA-analyysillä moni-imputointia käyttäen saatu imeväisiässä alkanutta SMA:ta koskeva plasman Nf-L-pitoisuuden pienimmän neliösumman keskiarvon suhde lähtötilanteeseen (95 %:n luottamusväli) käynneittäin: hoitoaikeen mukainen potilasjoukko (ITT), kaltaistettu lumetoimenpidejoukko

Päivä 64 on toissijainen päätetapahtuma 12 mg:n ja 50/28 mg:n annostusten vertailussa
Päivä 183 on toissijainen päätetapahtuma kaltaistetun lumetoimenpidejoukon ja 50/28 mg:n ryhmän vertailussa
Immunogeenisuus
Lääkevasta-aineet vaikuttivat vähentävän Spinrazan puhdistumaa plasmasta. Lääkevasta-aineilla ei ole havaittu Spinraza-valmisteen 12 mg:n ja 50/28 mg:n annostuksia käytettäessä selkeitä vaikutuksia plasman Nf-L-pitoisuuteen eikä kliinisen toiminnan mittareihin. Lääkevasta-aineilla ei ollut vaikutusta turvallisuuteen, kun mittarina käytettiin haittatapahtumien (kuten yliherkkyyden, anafylaktisen reaktion ja angioedeeman) ilmaantuvuutta.
Kliininen teho ja turvallisuus
Oireiset potilaat, jotka saivat hoitoa Spinraza-valmisteen 50/28 mg:n annostusohjelmalla
Tutkimuksen SM203 osa B oli 50/28 mg:n nusinerseeniannoksen turvallisuuden ja tehon satunnaistettu, kaksoissokkoutettu arviointi aiemmin hoitamattomilla imeväisiässä ja myöhemmin alkanutta SMA:ta sairastavilla potilailla. Osalla B oli osoitusvoimaa tehon arvioimiseksi imeväisiässä alkavaa tautimuotoa sairastavilla potilailla vertailtaessa 50/28 mg:n annosryhmää ennalta määritettyyn tutkimuksen CS3B kaltaistettuun lumetoimenpideryhmään. Tutkimuksen SM203 osan B 12 mg:n annostuksesta saatiin tueksi näyttöä, mutta tutkimuksella ei ollut riittävästi osoitusvoimaa tilastollisesti merkitsevien hoitoerojen havaitsemiseksi niiden välillä, jotka satunnaistettiin saamaan 50/28 mg tai 12 mg nusinerseeniä. Osa C oli avoin turvallisuuden ja tehon arviointi imeväisiässä tai myöhemmin alkanutta SMA:ta sairastavilla lapsilla ja aikuisilla, jotka olivat siirtyneet 12 mg:n annostusohjelmasta 50/28 mg:n annostusohjelmaan.
Sairastuminen imeväisiässä
Tutkimuksen SM203 osassa B imeväisiässä SMA:han sairastuneet potilaat (kaksi SMN2-kopiota; oireet ilmaantuvat ennen 6 kuukauden ikää) satunnaistettiin suhteessa 2:1 saamaan joko 50/28 mg:n tai 12 mg:n annostusta. Ennalta määritellyt analyysit kaltaistivat kaksikymmentä CS3B-tutkimuksessa lumetoimenpiteitä saaneista 37 potilaasta sekä lähtötilanteen sairauden keston että CHOP INTEND ‑pisteiden (Children’s Hospital of Philadelphia Infant Test for Neuromuscular Disease) samankaltaisuuksien perusteella. Ensisijainen päätetapahtuma oli imeväisiässä sairastuneiden potilaiden CHOP INTEND ‑pisteiden muutos päivän 183 aikapisteessä 50/28 mg:n ryhmässä verrattuna tähän tutkimuksesta CS3B kaltaistettuun lumetoimenpideryhmään.
Tutkimuksen SM203 imeväisiässä sairastuneiden kohortin tutkittavat ositettiin iän mukaan, kun heidät satunnaistettiin tasapainoisten 50/28 mg:n ja 12 mg:n ryhmien muodostamiseksi. Keskeiset demografiset ominaisuudet lähtötilanteessa (ikä ensimmäisen annoksen ajankohtana, ikä seulonta-ajankohtana, ikä oireiden alkaessa, SMN2-kopioiden lukumäärä ja lähtötilanteen motorinen toiminta) olivat tasapainossa 50/28 mg:n ryhmän, 12 mg:n ryhmän ja kaltaistetun lumetoimenpideryhmän välillä. Suhteessa tutkimuksen CS3B imeväisiässä sairastuneisiin nähden, tutkimukseen SM203 otettujen potilaiden sairaus eteni nopeammin ja potilaiden sairaus oli edennyt pidemmälle, näiden potilaiden sairauden kesto oli lyhyempi (aika oireiden alkamisesta seulontaan) ja CHOP INTEND ‑pisteet olivat lähtötilanteessa pienemmät. Ennalta määritelty kaltaistaminen tutkimuksen CS3B lumetoimenpiteitä saaneen vertailuryhmän alaryhmään auttoi minimoimaan tätä epäsuhtaa jonkin verran. Sairauden keskimääräinen (keskihajonta) kesto lähtötilanteessa oli kuitenkin edelleen lyhyempi ja lähtötilanteen CHOP INTEND ‑pisteet olivat 50/28 mg:n ja 12 mg:n ryhmissä edelleen pienemmät suhteessa tutkimuksen CS3B kaltaistettuun lumetoimenpideryhmään (taulukko 4). Muut keskeiset lähtötilanteen demografiset ominaisuudet (ikä ensimmäisen annoksen ajankohtana, ikä seulonta-ajankohtana, ikä oireiden alkaessa, SMN2-kopioiden lukumäärä ja lähtötilanteen motorinen toiminta) olivat tasapainossa 50/28 mg:n, 12 mg:n ja kaltaistetun lumetoimenpideryhmän välillä.
Taulukko 4: Potilaiden ominaisuudet tutkimuksen SM203 osan B lähtötilanteessa
| Potilaiden ominaisuudet | Spinraza 50/28 mg (n = 50) | Spinraza 12 mg (n = 25) | CS3B:n kaltaistettu lumetoimenpide (n = 20) |
| Iän lähtötilanteen mediaani (vaihteluväli) ensimmäisen annoksen saamisen ajankohtana (viikkoa) | 18,4 (2–33) | 15,9 (3–31) | 22,2 (4–34) |
| Iän lähtötilanteen keskiarvo (keskihajonta) oireiden alkaessa (viikkoa) | 7,5 (5,26) | 5,8 (4,44) | 8,8 (5,11) |
| Sairauden keston (oireiden alkamisesta seulontaan kuluneen ajan) lähtötilanteen keskiarvo (keskihajonta) (viikkoa) | 9,6 (5,29) | 9,2 (6,11) | 11,1 (4,92) |
| CHOP INTEND ‑pisteiden lähtötilanteen keskiarvo (pistettä) | 20,9 (10,23) | 19,9 (9,63) | 23,6 (5,84) |
Ensisijainen päätetapahtuma eli CHOP INTEND ‑pisteiden keskimääräinen muutos lähtötilanteesta päivään 183 mennessä oli 50/28 mg:n ryhmässä tilastollisesti merkitsevästi suurempi (15,1 pisteen paraneminen) verrattuna kaltaistettuun lumetoimenpideryhmään (11,1 pisteen huononeminen) (pienimmän neliösumman keskiarvon ero: 26,19 pistettä [95 %:n luottamusväli: 20,7–31,7], p < 0,0001).
CHOP INTEND ‑pisteiden muutos lähtötilanteesta päivään 302 oli 50/28 mg:n ryhmässä numeerisesti suurempi kuin 12 mg:n ryhmässä sijavertailun (ranks) eron perusteella, mutta tämä ero ei joint rank test ‑testin perusteella ollut tilastollisesti merkitsevä (sijavertailun pienimmän neliösumman keskiarvon ero [1,00 {95 %:n luottamusväli: ‑9,290–11,299}]; joint rank test ‑testin p = 0,8484). Pienimmän neliösumman keskimääräinen muutos lähtötilanteesta päivään 302 oli ANCOVA-analyysin ja moni-imputaation perusteella numeerisesti suurempi 12 mg:n ryhmässä; 50/28 mg:n ryhmässä todettiin 19,6 pisteen paraneminen ja 12 mg:n ryhmässä 21,6 pisteen paraneminen (95 %:n luottamusväli: 16,5–22,8) (pienimmän neliösumman keskiarvojen ero –1,94 [7,77–3,88]).
Täydentävässä analyysissä, joka oli samankaltainen kuin tutkimuksen CS3B ensisijainen päätetapahtuma, 60 % potilaista 50/28 mg:n ryhmässä ja 44 % potilaista 12 mg:n ryhmässä täytti päivänä 302 HINE-testin osan 2 (HINE‑2) vasteen saaneen määritelmän. HINE-2-testissä motorisen kehitystason muutos lähtötilanteesta päivään 302 oli 50/28 mg:n ryhmässä numeerisesti suurempi (5,9 pisteen paraneminen) kuin 12 mg:n ryhmässä (5,3 pisteen paraneminen) (pienimmän neliösumman keskiarvon ero 0,58 [1,89; 3,04]), mutta nämä erot eivät olleet tilastollisesti merkitseviä.
Lumetoimenpiteeseen verrattuna tilastollisesti merkitsevästi suurempi osuus potilaista 50/28 mg:n ryhmässä täytti päivänä 183 HINE-testin osan 2 (HINE-2) vasteen saanutta koskevan määritelmän (58 % vs. 0 %; p < 0,0001) (taulukko 5).
Taulukko 5: Motoriset hoitotulokset 50/28 mg:n ryhmässä vs. 12 mg:n ryhmässä ja 50/28 mg:n ryhmässä vs. kaltaistetussa lumetoimenpideryhmässä – tutkimuksen SM203 osan B imeväisiässä sairastuneet
| Tehon muuttuja | Spinraza50/28 mg:n ryhmä (n = 50) | Kaltaistettu lumetoimenpideryhmä tutkimuksesta CS3B (n = 20) | Ryhmien välinen ero (95 %:n luottamusväli) |
CHOP-INTEND Muutosta lähtötilanteesta päivään 183 koskevien sijavertailtujen pisteiden pienimmän neliösumman keskiarvo (95 %:n luottamusväli) Pienimmän neliösumman keskiarvon muutos (95 %:n luottamusväli) lähtötilanteesta päivään 1831,2 | 42,9 (38,7–47,2) 15,1 (12,4–17,8) | 16,9 (10,1–23,7) ‑11,1 (‑15,9 – ‑6,2) | 26,06 (17,94–34,17) p < 0,00013 26,1 (20,7–31,7)2 |
HINE-2-vasteen saanut5 Motorisen kehitystason suhteen vasteen saaneiden kriteerit päivänä 183 täyttäneiden osuus | 29 (58 %) | 0 (0 %) | 58 % (39,5–71,8)4 p < 0,0001 |
HINE-2-kokonaispisteet Muutosta lähtötilanteesta päivään 183 koskevien sijavertailtujen pisteiden pienimmän neliösumman keskiarvo (95 %:n luottamusväli) HINE-2-kokonaispisteiden pienimmän neliösumman keskiarvon muutos (95 %:n luottamusväli) lähtötilanteesta päivään 1831,2 | 43,1 (39,0–47,2) 3,7 (3,0–4,4) | 16,5 (9,9–23,0) ‑0,2 (‑1,5–1,0) | 26,67 (18,81–35,53) p < 0,00013 3,94 (2,46–5,42)2 |
1ANCOVA-analyysiä ja moni-imputaatiota käytetty
2 Pienimmän neliösumman keskiarvon ero
3 Joint rank test ‑testi
4 Fisherin tarkka testi
5 Vasteen saaneen määritelmä: tässä ensisijaisessa analyysissä vasteen saamiseksi määriteltiin ≥ 2 pisteen lisäys (tai maksimipisteet) kyvyssä potkia tai ≥ 1 pisteen lisäys pään hallintaa, kääntymistä, istumista, ryömimistä, seisomista tai kävelemistä koskevassa motorisessa kehitystasossa sekä useammassa motorisen kehitystason luokassa paranemista kuin huononemista.
Kuoleman tai pysyvän ventilaation riskin todettiin vähentyneen 50/28 mg:n ryhmässä 29,9 % verrattuna 12 mg:n ryhmään (p = 0,2775) ja nimellisesti tilastollisesti merkitsevästi 68 % verrattuna kaltaistettuun lumetoimenpideryhmään (p = 0,0006). 50/28 mg:n ryhmässä ei saavutettu kuolemaan tai pysyvään ventilaatioon kuluneen ajan mediaania, mutta 12 mg:n ryhmässä se oli 24,7 viikkoa ja kaltaistetussa lumetoimenpideryhmässä se oli 19,1 viikkoa. Kokonaiselossaoloa koskevat havainnot olivat samankaltaisia (kuva 2).
Kuva 2 osa B: Imeväisiässä SMA:han sairastuminen: kuolemaan tai pysyvään ventilaatioon kulunutta aikaa kuvaavat Kaplan–Meier-käyrät (EAC:n arvioimat tapahtumat): hoitoaikeen mukainen potilasjoukko (ITT), kaltaistettu lumetoimenpidejoukko
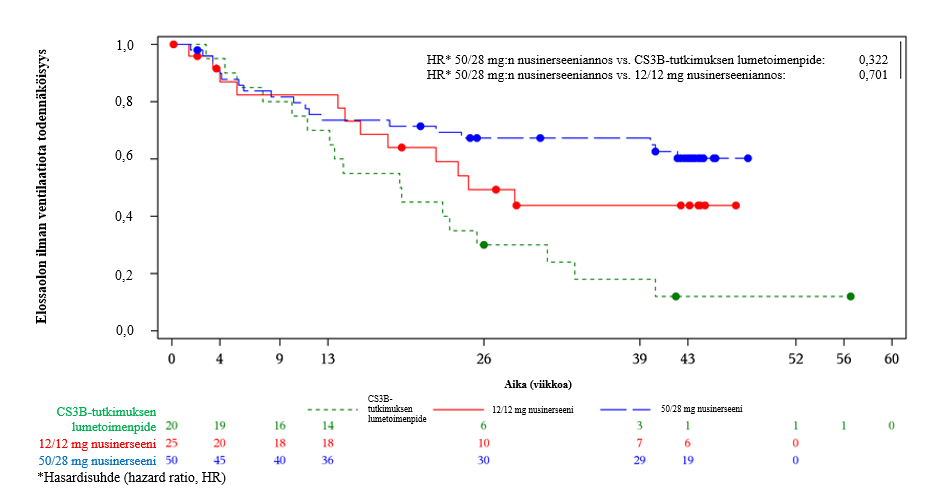
Sairastuminen myöhemmin
Tutkimuksen SM203 osassa B oli mukana 24 potilasta, joiden SMA-oireet olivat alkaneet myöhemmin (suurimmalla osalla oli kolme SMN2-kopiota; oireet olivat ilmaantuneet yli 6 kuukauden iässä) ja jotka satunnaistettiin suhteessa 2:1 saamaan 50/28 mg:n annostusta (n = 16) tai 12 mg:n annostusta (n = 8). Analyysit oli ennalta määritelty vertaamaan 50/28 mg:n annostusta tutkimuksen CS4 kaltaistettuun lumetoimenpideryhmään. Analyyseillä ei ollut osoitusvoimaa merkitsevien erojen havaitsemiseksi hoitoryhmien välillä.
50/28 mg:n ryhmän, kaltaistettua hoitoa saaneen ryhmän ja kaltaistetun lumetoimenpideryhmän demografiset ominaisuudet lähtötilanteessa olivat yleisesti ottaen tasapainossa, lukuun ottamatta ikää ensimmäisen annoksen antoajankohtana. Iän keskiarvo (keskihajonta) ensimmäisen annoksen antoajankohtana oli 50/28 mg:n ryhmässä 6,1 (3,0) vuotta, 12 mg:n ryhmässä 5,7 (3,0) vuotta ja kaltaistetussa lumetoimenpideryhmässä 5,13 (1,8) vuotta.
HFMSE-pisteiden muutos lähtötilanteesta päivään 302 (pienimmän neliösumman keskiarvo [95 %:n luottamusväli]) oli 50/28 mg:n ryhmässä (3,3 [1,5–5,0]) numeerisesti suurempi kuin 12 mg:n ryhmässä (2,6 [0,2–5,1]) (pienimmän neliösumman keskiarvon ero: 0,63 (–2,5–3,8; p = 0,70). HFMSE-pisteiden muutos lähtötilanteesta päivään 279 oli 50/28 mg:n ryhmässä myös numeerisesti suurempi kuin tutkimuksesta CS4 kaltaistetussa lumetoimenpideryhmässä (pienimmän neliösumman keskiarvon ero: 3,2 (0,2–6,2); p = 0,037).
Muutetusta yläraajan moduulitestistä (Revised Upper Limb Module, RULM) saatujen pisteiden muutos lähtötilanteesta päivään 302 (pienimmän neliösumman keskiarvo [95 %:n luottamusväli]) oli 50/28 mg:n ryhmässä (2,5 [0,7–4,2]) numeerisesti suurempi kuin 12 mg:n ryhmässä (1,8 [–0,8–4,4]), mutta ero ei ollut tilastollisesti merkitsevä (p = 0,66). RULM-pisteiden muutos lähtötilanteesta päivään 279 oli sekin 50/28 mg:n ryhmässä numeerisesti suurempi kuin tutkimuksesta CS4 kaltaistetussa lumetoimenpideryhmässä (pienimmän neliösumman keskiarvon ero: 1,7 (–0,2–3,5); p = 0,076).
Tutkimuksen SM203 osa C oli avoin kohortti. Siihen otettiin 40 potilasta, joiden ikä oli 4–65 vuotta, joilla oli 1–4 SMN2-kopiota ja jotka olivat siirtyneet 12 mg:n annostuksesta 50/28 mg:n annostukseen. Potilaat saivat yhden 50 mg:n annoksen ja sen jälkeen kaksi 28 mg:n ylläpitoannosta (4 kuukauden välein).
Kaksi potilasta (5 %) oli sairastunut imeväisiässä ja 38 potilasta (95 %) oli sairastunut myöhemmin. Kuusitoista potilasta oli alle 18-vuotiaita ja 24 potilasta oli yli 18-vuotiaita saadessaan 50 mg:n latausannoksen. Iän mediaani (vaihteluväli) SMA:n oireiden ilmaantuessa oli 24 (4–192) kuukautta. 12 mg:n Spinraza-annostusohjelman keston mediaani (vaihteluväli) oli 3,9 vuotta (1–5). Kaksikymmentäyksi potilasta (53 %) pystyi lähtötilanteessa liikkumaan 15 askelta itsenäisesti.
Tutkittavien HFMSE-pisteet paranivat lähtötilanteesta päivään 302 mennessä keskimäärin 1,8 pistettä (keskihajonta 3,99), ja aikuisten alaryhmässä (n = 24) pisteet paranivat keskimäärin 2,3 pistettä (keskihajonta 3,95). Kaikkiaan 53 %:lla potilaista (n = 38) HFMSE-pisteiden todettiin parantuneen lähtötilanteesta päivään 302 mennessä.
Tutkittavien RULM-pisteet paranivat lähtötilanteesta päivään 302 mennessä keskimäärin 1,2 pistettä (keskihajonta 2,14), ja aikuisten alaryhmässä pisteet paranivat 0,9 pistettä (keskihajonta 1,89). Niistä, joilla pisteiden paraneminen oli mahdollista (lähtötilanteen pisteet maksimipisteitä pienemmät; n = 26), 62 %:lla RULM-pisteet suurenivat lähtötilanteesta päivään 302 mennessä.
Oireiset potilaat, jotka saivat hoitoa Spinraza-valmisteen 12 mg:n annostusohjelmalla
Sairastuminen imeväisiässä
Tutkimus CS3B (ENDEAR) oli satunnaistettu, kaksoissokkoutettu, lumetoimenpiteillä kontrolloitu faasin 3 tutkimus, jossa oli mukana 121 SMA-diagnoosin saanutta ≤ 7 kuukauden ikäistä vauvaa, joilla oli oireita (oireet alkaneet ennen 6 kuukauden ikää). Tutkimuksessa CS3B oli tarkoitus arvioida Spinrazan vaikutusta motorisiin toimintoihin ja elossaoloaikaan. Potilaat satunnaistettiin suhteessa 2:1 saamaan joko 12 mg Spinrazaa (hyväksytyn annostusohjelman mukaisesti) tai lumetoimenpiteitä. Hoidon kesto oli 6–442 päivää.
SMA:n kliinisten oireiden ja löydösten ilmaantuessa 12 mg:n Spinraza-annoksia saaneiden potilaiden iän mediaani oli 6,5 viikkoa ja lumetoimenpiteitä saaneiden 8 viikkoa; 99 %:lla potilaista oli kaksi SMN2-geenin kopiota, minkä vuoksi todennäköisimpänä pidettiin tyypin I SMA:n kehittymistä. Spinraza‑hoitoa saaneiden potilaiden iän mediaani ensimmäisen annoksen yhteydessä oli 164,5 vuorokautta ja kontrolliryhmän potilaiden iän mediaani ensimmäisen lumetoimenpiteen kohdalla 205 vuorokautta. Sairauden piirteet olivat lähtötilanteessa yleisesti ottaen samankaltaisia 12 mg:n Spinraza-annoksia saaneilla ja lumetoimenpiteitä saaneilla potilailla. Seuraavia esiintyi lähtötilanteessa kuitenkin prosentuaalisesti useammalla 12 mg:n Spinraza-annoksia saaneista kuin lumetoimenpiteitä saaneista potilaista: paradoksaalinen hengitys (89 % vs. 66 %), keuhkokuume tai hengitystieoireet (35 % vs. 22 %), nielemis- tai syömisvaikeudet (51 % vs. 29 %) ja avustetun hengityksen tarve (26 % vs. 15 %).
Loppuanalyysissa tilastollisesti merkitsevästi suurempi prosenttiosuus 12 mg:n Spinraza-annostusta saaneen ryhmän potilaista täytti motorisen kehitystason saavuttamisen määritelmän verrattuna lumetoimenpideryhmän potilaisiin (51 % vs. 0 %) (p < 0,0001). Ensisijaisena päätetapahtumana arvioitiin aika kuolemaan tai jatkuvaan ventilaatioon (≥ 16 tuntia ventilaatiota/vrk yhtäjaksoisesti > 21 vuorokauden ajan ilman akuuttia, korjautuvaa tapahtumaa tai trakeostomiaa). 12 mg:n Spinraza-annostusta saaneessa ryhmässä havaittiin lumetoimenpideryhmään verrattuna tilastollisesti merkitseviä vaikutuksia elossaoloaikaan ilman tapahtumia, kokonaiselossaoloaikaan, motorisen kehitystason saavuttamisen määritelmän täyttäneiden potilaiden osuuteen sekä niiden potilaiden prosenttiosuuteen, joiden CHOP INTEND ‑testin pisteet paranivat lähtötilanteesta vähintään 4 pistettä (taulukko 6).
Tehoa koskeneessa potilasjoukossa 18 potilasta (25 %) 12 mg:n Spinraza-annostusta saaneessa ryhmässä ja 12 potilasta (32 %) lumetoimenpideryhmässä tarvitsi jatkuvaa ventilaatiota. Näistä potilaista 6 (33 %) 12 mg:n Spinraza-annostusta saaneessa ryhmässä ja 0 (0 %) lumetoimenpideryhmässä täytti tutkimussuunnitelmassa määritellyt motorisen kehitystason saavuttamisen kriteerit.
Taulukko 6:Ensisijaiset ja toissijaiset päätetapahtumat loppuanalyysissa - tutkimus CS3B
| Tehon muuttuja | 12 mg:n Spinraza-annoksia saaneet potilaat | Lumetoimenpiteitä saaneet potilaat |
| Elossaoloaika | ||
| Elossaoloaika ilman tapahtumia2 | ||
| Kuolleiden tai jatkuvaa ventilaatiota saaneiden potilaiden lukumäärä | 31 (39 %) | 28 (68 %) |
| Riskitiheyksien suhde (95 %:n luottamusväli) | 0,53 (0,32–0,89) | |
| p‑arvo6 | p = 0,0046 | |
| Kokonaiselossaoloaika2 | ||
| Kuolleiden potilaiden lukumäärä | 13 (16 %) | 16 (39 %) |
| Riskitiheyksien suhde (95 %:n luottamusväli) | 0,37 (0,18–0,77) | |
| p‑arvo6 | p = 0,0041 | |
| Motoriset toiminnot | ||
| Motoriset kehitystasot3 | ||
| Niiden potilaiden osuus, jotka täyttivät motorisen kehitystason saavuttamisen etukäteen määritellyt kriteerit (HINE-testin osa 2)4,5 | 37 (51 %)1 p < 0,0001 | 0 (0 %) |
| Osuus päivänä 183 | 41 % | 5 % |
| Osuus päivänä 302 | 45 % | 0 % |
| Osuus päivänä 394 | 54 % | 0 % |
| Niiden potilaiden osuus, joiden motorisen kehitystason saavuttamisen kokonaispisteet paranivat | 49 (67 %) | 5 (14 %) |
| Niiden potilaiden osuus, joiden motorisen kehitystason saavuttamisen kokonaispisteet huononivat | 1 (1 %) | 8 (22 %) |
| CHOP INTEND3 | ||
| Niiden potilaiden osuus, joiden pisteet paranivat 4 pistettä | 52 (71 %) p < 0,0001 | 1 (3 %) |
| Niiden potilaiden osuus, joiden pisteet huononivat 4 pistettä | 2 (3 %) | 17 (46 %) |
| Niiden potilaiden osuus, joiden pisteet paranivat | 53 (73 %) | 1 (3 %) |
| Niiden potilaiden osuus, joiden pisteet huononivat | 5 (7 %) | 18 (49 %) |
1Tutkimus CS3B lopetettiin, kun ensisijaisen päätetapahtuman tilastollisessa välianalyysissa saatiin positiivinen tulos (tilastollisesti merkitsevästi suurempi prosenttiosuus 12 mg:n Spinraza-annostusta saaneen ryhmän potilaista täytti motorisen kehitystason saavuttamisen määritelmän verrattuna lumetoimenpideryhmän potilaisiin (41 % vs. 0 %, p < 0,0001).
2Elossaoloaika ilman tapahtumia ja kokonaiselossaoloaika arvioitiin loppuanalyysissä käyttämällä hoitoaikeen mukaista potilasjoukkoa (ITT-populaatiota) (12 mg Spinraza-valmistetta n = 80, lumetoimenpiteet n = 41).
3CHOP INTEND -pisteitä ja motorisia kehitystasoja koskevat analyysit tehtiin loppuanalyysissä käyttämällä tehoa koskenutta potilasjoukkoa (12 mg Spinraza-valmistetta n = 73, lumetoimenpiteet n = 37).
4Arvioitiin viimeisellä päivien 183, 302 ja 394 tutkimuskäynneistä.
5HINE-testin (Hammersmith Infant Neurological Examination) 2. osan mukaan eli ≥ 2 pisteen paraneminen (tai enimmäispisteet) potkimiskyvyssä TAI ≥ 1 pisteen paraneminen jossakin seuraavista motorisista kehitystasoista: pään hallinta, kääntyminen, istuminen, ryömiminen, seisominen tai käveleminen JA useammassa motorisen kehitystason kategoriassa paranemista kuin huonontumista, määriteltiin vasteen saaneeksi tässä ensisijaisessa analyysissa.
6Perustuu sairauden keston mukaan stratifioituun log-rank-testiin
CHOP INTEND ‑pisteiden paraneminen on esitetty kuvassa 3 (kunkin tutkittavan muutos lähtötilanteesta).
Kuva 3: CHOP INTEND ‑pisteiden muutos lähtötilanteesta viimeiseen päivien 183, 302 ja 394 tutkimuskäynneistä – ENDEAR /CS3B-tutkimus (tehoa koskeva potilasjoukko)
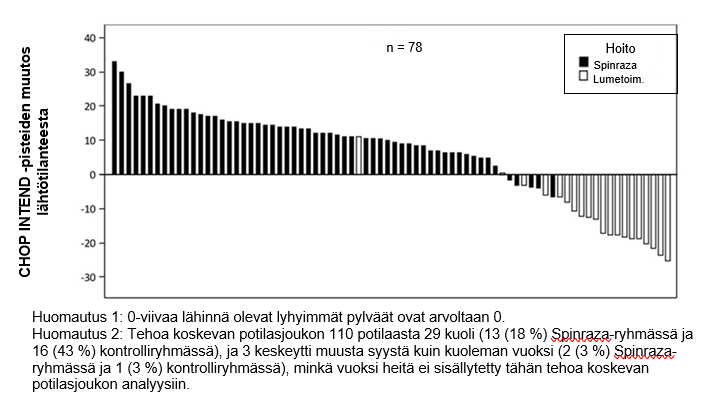
Näiden potilaiden pitkäkestoisen seurannan mahdollistamiseksi tutkimuksen CS3B lopussa 89 potilasta (Spinraza: n = 65, lumetoimenpiteet: n = 24) otettiin mukaan tutkimukseen CS11 (SHINE). Tutkimus CS11 on avoin jatkotutkimus SMA-potilaille, jotka ovat aiemmin osallistuneet muihin Spinraza-tutkimuksiin. Potilaat, jotka satunnaistettiin saamaan Spinraza-hoitoa tutkimuksessa CS3B, mukaan lukien Spinraza-hoidon jatkovaiheessa tutkimuksessa CS11, saivat lääkitystä 6–3 043 vuorokauden ajan (mediaani 2 443 vuorokautta). Potilaat, jotka satunnaistettiin tutkimuksessa CS3B saamaan lumelääkettä ja aloittivat Spinraza-hoidon tutkimuksessa CS11, saivat lääkitystä 65–2 520 vuorokauden ajan (mediaani 2 090 vuorokautta).
Motoristen toimintojen paranemista havaittiin potilailla, jotka jatkoivat Spinraza-hoidon saamista tutkimuksesta CS3B, sekä potilailla, joille aloitettiin Spinraza-hoito tutkimuksessa CS11 (kuva 5). Suurin hyöty havaittiin potilailla, joiden hoito alkoi aiemmin. Suurin osa potilaista, joille oli aloitettu Spinraza-hoito joko tutkimuksessa CS3B tai tutkimuksessa CS11, oli elossa viimeisen käynnin ajankohtana.
Tutkimuksessa CS3B Spinraza-hoidon aloittaneiden potilaiden iän mediaani oli 5,5 kuukautta (vaihteluväli 1,7–14,9 kuukautta). Spinraza-hoidon aloittamisen jälkeen, mukaan lukien hoidon jatkovaihe tutkimuksessa CS11, kuolemaan tai jatkuvan ventilaation aloittamiseen kuluneen ajan mediaani oli 1,4 vuotta. Tutkimuksen CS11 lopussa 81 potilaasta 60 (74 %) oli elossa ja 81 potilaasta 41 (51 %) oli elossa eikä täyttänyt tutkimuksen CS11 jatkuvaa ventilaatiota koskevaa määritelmää. HINE-2-testin motorisen kehitystason keskimääräinen kokonaispistearvo suureni 5,3 pistettä (keskihajonta 4,6; n = 52) Spinraza-hoidon aloittamisen ja päivän 394 seurantakäynnin välisenä aikana ja CHOP INTEND -pistearvo suureni 18,4 pistettä (keskihajonta 14,7; n = 38) Spinraza-hoidon aloittamisen ja päivän 2 198 seurantakäynnin välisenä aikana.
Tutkimuksessa CS3B lumelääkkeeseen satunnaistettujen ja tutkimuksessa CS11 Spinraza-hoidon aloittaneiden potilaiden iän mediaani oli 17,8 kuukautta (vaihteluväli 10,1–23,0 kuukautta). Ennen Spinraza-hoidon aloittamista 24 potilaasta 12 (50 %) täytti tutkimuksen CS11 jatkuvaa ventilaatiota koskevan määritelmän. Tutkimuksessa CS11 tapahtuneen Spinraza-hoidon aloittamisen jälkeen kuolemaan tai jatkuvan ventilaation aloittamiseen kuluneen ajan mediaani oli 2,76 vuotta. Tutkimuksen CS11 lopussa 24 potilaasta 19 (79 %) oli elossa ja 12 potilaasta 6 (50 %) oli elossa ilman jatkuvaa ventilaatiota. Motorisen kehitystason keskimääräinen kokonaispistearvon havaittiin parantuneen 1,4 pistettä (keskihajonta 1,8; n = 12) tutkimuksen CS11 lähtötilanteen ja päivän 394 seurantakäynnin välisenä aikana ja CHOP INTEND ‑pistearvon havaittiin parantuneen 11,5 pistettä (keskihajonta 12,2; n = 10) tutkimuksen CS11 lähtötilanteen ja päivän 2 198 seurantakäynnin välisenä aikana.
Näitä tuloksia tukee avoin faasin 2 tutkimus, jossa oli mukana SMA-diagnoosin saaneita potilaita, joilla oli oireita (CS3A). Kliinisten oireiden ja löydösten ilmaantuessa iän mediaani oli 56 vuorokautta, ja potilailla oli joko kaksi SMN2-geenin kopiota (n = 17) tai kolme SMN2-geenin kopiota (n = 2) (yhdellä potilaalla SMN2-geenin kopioiden määrä oli tuntematon). Tämän tutkimuksen potilaille todennäköisimpänä pidettiin tyypin I SMA:n kehittymistä. Iän mediaani ensimmäisen annoksen yhteydessä oli 162 vuorokautta.
Ensisijainen päätetapahtuma oli niiden potilaiden osuus, joiden pisteet paranivat yhdessä tai useammassa motorisen kehitystason kategoriassa [HINE-testin 2. osan mukaan eli ≥ 2 pisteen paraneminen (tai enimmäispisteet) potkimiskyvyssä tai tahdonalaisessa tarttumisessa tai ≥ 1 pisteen paraneminen jossakin seuraavista motorisista kehitystasoista: pään hallinta, kääntyminen, istuminen, ryömiminen, seisominen tai käveleminen]. Tutkimuksen 20 potilaasta 12 (60 %) saavutti ensisijaisen päätetapahtuman, joka oli ajan kuluessa tapahtunut paraneminen keskimääräisen motorisen kehitystason saavuttamisessa. Keskimääräiset CHOP INTEND -pisteet paranivat ajan kuluessa lähtötilanteesta päivään 1072 (keskimääräinen muutos oli 21,30). Viimeisellä tutkimuskäynnillä 20 potilaasta 11 (55 %) saavutti CHOP INTEND -pisteitä koskevan päätetapahtuman eli kokonaispisteiden paranemisen ≥ 4 pistettä. Tutkimukseen otetusta 20 tutkittavasta 11 (55 %) oli elossa eikä tarvinnut jatkuvaa ventilaatiota viimeisellä käynnillä. Neljä potilasta täytti jatkuvan ventilaation kriteerit ja viisi potilasta kuoli tutkimuksen aikana.
Sairastuminen myöhemmin
Tutkimus CS4 (CHERISH) oli satunnaistettu, kaksoissokkoutettu, lumetoimenpiteillä kontrolloitu faasin 3 tutkimus 126 oireisella potilaalla, joiden SMA-oireet olivat alkaneet myöhemmin (6 ikäkuukauden jälkeen). Potilaat satunnaistettiin suhteessa 2:1 saamaan joko 12 mg Spinrazaa (3 latausannosta ja niiden jälkeen ylläpitoannoksia 6 kuukauden välein) tai lumetoimenpiteitä 324−482 päivän hoidon ajan. Seulonnassa iän mediaani oli 3 vuotta, ja SMA:n kliinisten oireiden ja löydösten ilmaantuessa iän mediaani oli 11 kuukautta. Suurimmalla osalla potilaista (88 %) on kolme SMN2-geenin kopiota (8 %:lla on kaksi kopiota, 2 %:lla neljä kopiota ja 2 %:lla tuntematon määrä kopioita). Lähtötilanteessa potilaiden keskimääräinen Hammersmith Functional Motor Scale Expanded (HFMSE) -pisteluku oli 21,6 ja muutetun yläraajan moduulitestin (RULM) keskimääräinen pisteluku 19,1. Kaikki potilaat kykenivät istumaan ilman tukea, mutta yksikään potilaista ei kyennyt kävelemään ilman tukea. Tämän tutkimuksen potilaille pidettiin todennäköisimpänä tyypin II tai III SMA:n kehittymistä. Sairauden piirteet olivat lähtötilanteessa yleisesti ottaen samankaltaisia. Poikkeuksen muodosti epätasapaino niiden potilaiden osuudessa, jotka olivat jossakin vaiheessa kyenneet seisomaan ilman tukea (13 % potilaista 12 mg:n Spinraza-annostusta saaneessa ryhmässä ja 29 % lumetoimenpideryhmässä) tai kävelemään tuettuna (24 % potilaista 12 mg:n Spinraza-annostusta saaneessa ryhmässä ja 33 % lumetoimenpideryhmässä).
Loppuanalyysissa havaittiin tilastollisesti merkitsevää paranemista HFMSE-pisteissä lähtötilanteesta kuukauteen 15 hoitoa 12 mg:n Spinraza-annostuksella saaneessa ryhmässä verrattuna lumetoimenpideryhmään (taulukko 7, kuva 4). Analyysi tehtiin ITT-potilasjoukosta (12 mg:n Spinraza-annostus: n = 84; lumetoimenpiteet: n = 42), ja niiden potilaiden lähtötilanteen jälkeiset HFMSE-tiedot, jotka eivät olleet käyneet kuukauden 15 tutkimuskäynnillä, korvattiin käyttämällä useiden puuttuvien tietojen korvausmenetelmää. Niiden ITT-potilasjoukon potilaiden analyysissa, joista kuukauden 15 arvot olivat olemassa, tulokset olivat yhdenmukaisia ja tilastollisesti merkitseviä. Niistä potilaista, joista kuukauden 15 arvot olivat olemassa, suuremmalla osuudella 12 mg:n Spinraza-annostusta saaneista potilaista todettiin paranemista (73 % vs. 41 %), ja pienemmällä osuudella 12 mg:n Spinraza-annostusta saaneista potilaista todettiin huononemista (23 % vs. 44 %) HFMSE-kokonaispistemäärässä verrattuna lumetoimenpiteitä saaneisiin potilaisiin. Toissijaiset päätetapahtumat, mukaan lukien toimintakyvyn mittaukset ja WHO:n määrittelemien motoristen kehitystasojen saavuttamista koskeva arviointi, testattiin muodollisesti tilastollisesti ja tulokset on esitetty taulukossa 7.
Hoidon aloittaminen nopeammin oireiden alkamisen jälkeen johti nopeampaan ja suurempaan paranemiseen motorisissa toiminnoissa kuin hoidon aloittaminen myöhemmin. Hoitoa saaneet potilaat hyötyivät kuitenkin kummassakin tapauksessa lumetoimenpiteitä saaneisiin potilaisiin verrattuna.
Taulukko 7:Ensisijaiset ja toissijaiset päätetapahtumat loppuanalyysissa - tutkimus CS41
| 12 mg:n Spinraza-annostusta saaneet potilaat | Lumetoimenpiteitä saaneet potilaat | |
| HFMSE-pisteet | ||
| HFMSE-kokonaispisteiden muutos lähtötilanteesta kuukauden 15 kohdalla1,2,3 | 3,9 (95 %:n luottamusväli: 3,0–4,9) p = 0,0000001 | -1,0 (95 %:n luottamusväli: -2,5; 0,5) |
| Niiden potilaiden osuus, joiden pisteet paranivat vähintään 3 pistettä lähtötilanteesta kuukauteen 152 | 56,8 % (95 %:n luottamusväli: 45,6; 68,1) p = 0,00065 | 26,3 % (95 %:n luottamusväli: 12,4; 40,2) |
| RULM | ||
| RULM-kokonaispisteiden keskimääräinen muutos lähtötilanteesta kuukauteen 152,3 | 4,2 (95 %:n luottamusväli: 3,4; 5,0) p = 0,00000016 | 0,5 (95 %:n luottamusväli: -0,6; 1,6) |
| WHO:n määrittelemät motoriset kehitystasot | ||
| Niiden potilaiden osuus, jotka olivat kuukauden 15 kohdalla saavuttaneet uusia motorisia kehitystasoja4 | 19,7 % (95 %:n luottamusväli: 10,9; 31,3) p = 0,0811 | 5,9 % (95 %:n luottamusväli: 0,7; 19,7) |
1 Tutkimus CS4 lopetettiin, kun ensisijaisen päätetapahtuman tilastollisessa välianalyysissa saatiin positiivinen tulos (tilastollisesti merkitsevää paranemista lähtötilanteen HFMSE-pistemäärästä havaittiin 12 mg:n Spinraza-annostusta saaneissa potilaissa verrattuna lumetoimenpiteitä saaneisiin potilaisiin (12 mg:n Spinraza-annostus vs. lumetoimenpiteet: 4,0 vs. ‑1,9; p = 0,0000002)
2 Arvioitu käyttämällä ITT-potilasjoukkoa (12 mg:n Spinraza-annostus n = 84; lumetoimenpiteet n = 42); tiedot potilaista, jotka eivät olleet käyneet kuukauden 15 tutkimuskäynnillä, korvattiin käyttämällä useiden puuttuvien tietojen korvausmenetelmää
3 Pienimmän neliösumman keskiarvo
4 Arvioitu käyttämällä kuukauden 15 tehoa koskevaa potilasjoukkoa (12 mg:n Spinraza-annostus n = 66; lumetoimenpiteet n = 34); tietojen puuttuessa analyysit perustuvat korvattuihin tietoihin.
5 Perustuu logistiseen regressioon: hoidon teho ja säätö kunkin potilaan iän mukaan seulontavaiheessa ja HFMSE-pisteiden mukaan lähtötilanteessa
6 Nimellinen p-arvo
Kuva 4: Keskimääräinen muutos HFMSE-pisteissä ajan kuluessa lähtötilanteesta loppuanalyysiin (ITT) – CS4-tutkimus 1,2
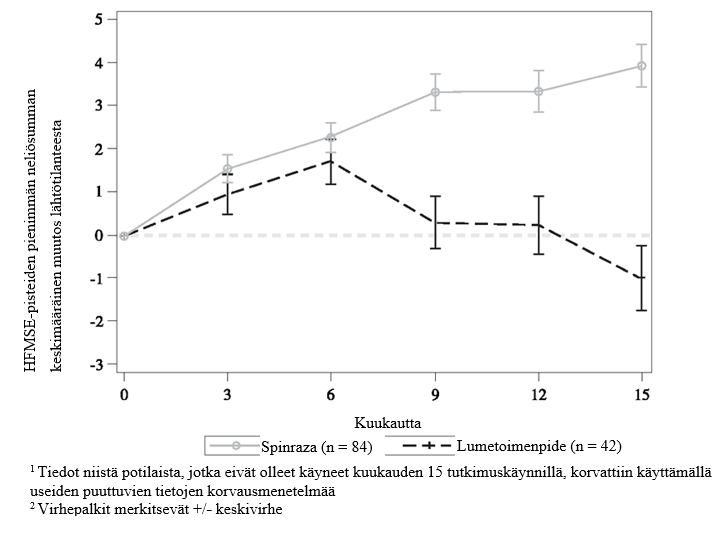
Tutkimuksen CS4 (CHERISH) päätyttyä 125 potilasta (83 Spinraza-ryhmässä ja 42 lumeryhmässä) otettiin mukaan tutkimukseen CS11 (SHINE), jossa kaikki potilaat saivat 12 mg:n Spinraza-annoksia. Suurimmalla osalla 12 mg:n Spinraza-annoksia saaneista potilaista motoriset toiminnot vakaantuivat tai paranivat. Suurin hyöty havaittiin potilailla, joiden hoito alkoi aiemmin.
Tutkimuksessa CS4 Spinraza-hoidon aloittaneiden potilaiden iän mediaani oli 4,1 vuotta (vaihteluväli 2,1–9,2 vuotta). Spinraza-hoidon aloittamisen jälkeen, mukaan lukien hoidon jatkovaihe tutkimuksessa CS11, potilaiden saaman lääkityksen keston mediaani oli 7,2 vuotta (vaihteluväli 1,3–8,4 vuotta). Päivän 2 070 seurantakäynnillä keskimääräinen HFMSE-pistearvo oli suurentunut 1,3 pistettä (keskihajonta 9,4; n = 54) ja keskimääräinen RULM-pistearvo oli suurentunut 6,4 pistettä (keskihajonta 6,5; n = 54).
Tutkimuksessa CS4 lumehoitoon satunnaistetut potilaat aloittivat tutkimuksessa CS11 Spinraza-hoidon 4,9 vuoden (mediaani) ikäisinä (vaihteluväli 3,3–9,0 vuotta). Tutkimuksessa CS11 tapahtuneen Spinraza-hoidon aloittamisen jälkeen potilaiden saaman lääkityksen keston mediaani oli 5,8 vuotta (vaihteluväli 2,7–6,7 vuotta). Päivän 2 070 seurantakäynnillä keskimääräinen HFMSE-pistearvo oli pienentynyt 1,3 pistettä (keskihajonta 9,3; n = 22) ja keskimääräinen RULM-pistearvo oli suurentunut 4,2 pistettä (keskihajonta 4,4; n = 23).
Samanikäisten ja kliinisiltä ominaisuuksiltaan samankaltaisten hoitamattomien potilaiden luonnolliseen taudinkulkuun sitä vastoin liittyy etenevää motoristen toimintojen heikkenemistä ajan mittaan, ja HFMSE-pistearvon on arvioitu pienenevän keskimäärin 6,6 pistettä samankaltaisen 5 vuoden ajanjakson aikana.
Näitä tuloksia tukee kaksi avointa tutkimusta (tutkimukset CS2 ja CS12). Analyysi käsitti 28 potilasta, jotka saivat ensimmäisen annoksensa tutkimuksessa CS2, minkä jälkeen heidät siirrettiin jatkotutkimukseen CS12. Tutkimuksiin otettiin mukaan potilaita, joiden ikä ensimmäisen annoksen ajankohtana oli 2–15 vuotta. Näistä 28 potilaasta kolme oli vähintään 18-vuotiaita viimeisen tutkimuskäyntinsä ajankohtana. 28 potilaasta yhdellä oli kaksi SMN2-geenin kopiota, 21:llä kolme kopiota ja 6:lla neljä kopiota.
Potilaita arvioitiin 3 vuoden hoitojakson ajan. Jatkuvaa parantumista todettiin niillä tyypin II SMA:ta sairastavilla, joiden HFMSE-pisteet olivat parantuneet lähtötilanteesta keskimäärin 5,1 pistettä (keskihajonta 4,05; n = 11) päivänä 253 ja 9,1 pistettä (keskihajonta 6,61; n = 9) päivänä 1 050. Kokonaispisteiden keskiarvo oli 26,4 (keskihajonta 11,91) päivänä 253 ja 31,3 (keskihajonta 13,02) päivänä 1 050 ja tasannevaiheita ei havaittu. Tyypin III SMA:ta sairastavien potilaiden HFMSE-pisteet olivat 1 050 päivän hoidon jälkeen parantuneet lähtötilanteesta keskimäärin 1,3:lla (keskihajonta 1,87; n = 16) päivänä 253 ja 1,2:lla (keskihajonta 4,64; n = 11) päivänä 1 050. Kokonaispisteiden keskiarvo oli 49,8 (keskihajonta 12,46) päivänä 253 ja 52,6 (keskihajonta 12,78) päivänä 1 050.
Tyypin II SMA:ta sairastavilla potilailla tehdyssä yläraajan moduulitestissä pisteiden todettiin parantuneen keskimäärin 1,9:llä (keskihajonta 2,68, n = 11) tutkimuspäivänä 253 ja 3,5:llä (keskihajonta 3,32, n = 9) tutkimuspäivänä 1 050. Keskimääräiset kokonaispisteet olivat 13,8 (keskihajonta 3,09) tutkimuspäivänä 253 ja 15,7 (keskihajonta 1,92) tutkimuspäivänä 1 050.
Vain kävelykykyisille potilaille tehtiin 6 minuutin kävelytesti (6MWT). Kävelytestin tulos oli parantunut näillä potilailla keskimäärin 28,6 metriä (keskihajonta 47,22; n = 12) päivänä 253 ja 86,5 metriä (keskihajonta 40,58; n = 8) päivänä 1 050. 6MWT-kävelytestin keskimääräinen tulos oli 278,5 metriä (keskihajonta 206,46) päivänä 253 ja 333,6 metriä (keskihajonta 176,47) päivänä 1 050. Kaksi aiemmin itsenäiseen kävelyyn kykenemätöntä potilasta (tyyppi III) saavutti itsenäisen kävelykyvyn, ja yksi aiemmin kävelykyvytön potilas (tyyppi II) saavutti itsenäisen kävelykyvyn.
Kliininen lisätutkimus CS7 (EMBRACE) avattiin potilaille, jotka eivät soveltuneet osallistumaan tutkimukseen CS3B eivätkä tutkimukseen CS4 seulontaiän tai SMN2-geenin kopioluvun perusteella. CS7 on satunnaistettu, kaksoissokkoutettu, lumetoimenpiteillä kontrolloitu faasin 2 tutkimus oireisilla potilailla, joilla on diagnosoitu imeväisiässä alkanut SMA (≤ 6 kuukautta) tai myöhemmin alkanut SMA (> 6 kuukautta) ja 2 tai 3 SMN2-geenin kopiota (osa 1), jota seuraa pitkäkestoinen avoin jatkovaihe (osa 2). Tutkimuksen 1. osassa potilaiden seuranta-ajan mediaani oli 302 päivää.
Kaikki 12 mg:n Spinraza-annostusta saaneet potilaat olivat elossa tutkimuksen 1. osan ennenaikaisen lopettamisen ajankohtana, yksi kontrolliryhmän potilas kuitenkin kuoli tutkimuspäivänä 289. Lisäksi yksikään potilas 12 mg:n Spinraza- tai lumetoimenpideryhmässä ei tarvinnut jatkuvaa ventilaatiota. Niistä 13 potilaasta, joilla oli imeväisiässä alkanut SMA, 9 potilaasta 7 (78 %, 95 %:n luottamusväli: 45–94) 12 mg:n Spinraza-ryhmässä ja 4 potilaasta 0 (0 %; 95 %:n luottamusväli: 0–60) lumetoimenpideryhmässä täyttivät motorisen kehitystason saavuttamisen vasteen kriteerit (HINE-testin 2. osan mukaan: ≥ 2 pisteen paraneminen (tai enimmäispisteet) potkimiskyvyssä TAI ≥ 1 pisteen paraneminen jossakin seuraavista motorisista kehitystasoista: pään hallinta, kääntyminen, istuminen, ryömiminen, seisominen tai käveleminen ja useammassa motorisen kehitystason kategoriassa paranemista kuin huonontumista). Niistä 8 potilaasta, joilla oli myöhemmin alkanut SMA, 5 potilaasta 4 (80 %, 95 %:n luottamusväli: 38–96) 12 mg:n Spinraza-ryhmässä ja 3 potilaasta 2 (67 %; 95 %:n luottamusväli: 21–94) lumetoimenpideryhmässä täytti tämän vasteen määritelmän.
Aikuiset
Tosielämän (real world, RW) kliiniset havainnot tukevat nusinerseenin tehoa motorisen toiminnan vakauttamisessa ja parantamisessa joillakin tyypin II ja III SMA-aikuispotilailla.
12 mg:n nusinerseenihoidon 14. kuukauteen mennessä potilaiden määrä, joilla oli kliinisesti merkittävä parannus lähtötilanteesta HFMSE-pisteissä (≥ 3 pistettä) oli 53 potilasta 129:stä sekä potilaiden määrä, joilla oli kliinisesti merkittävä parannus RULM-pisteissä (≥ 2 pistettä) oli 28 potilasta 70:stä ja kävelevien potilaiden osalta 25 potilasta 49:stä 6MWT-testissä (≥ 30 metriä).
Tutkimuksen SM203 osassa C tehoa ja turvallisuutta arvioitiin potilailla, jotka olivat siirtyneet 50/28 mg:n Spinraza-annostukseen; siihen osallistui 24 aikuista (≥ 18-vuotiaita), joita oli hoidettu 12 mg:n annoksilla 3,9 vuotta (mediaani). Siirtymisen jälkeen tutkittavien HFMSE-pisteet paranivat aikuisten alaryhmässä (n = 24) keskimäärin 2,3 pistettä (keskihajonta 3,95). Aikuisten alaryhmässä tutkittavien RULM-pisteet paranivat 0,9 pistettä (keskihajonta 1,89). Lisäksi 14 aikuisella 23 aikuisesta (61 %) HFMSE-pisteet paranivat lähtötilanteesta päivään 302 mennessä ja kahdeksalla aikuisella 12 aikuisesta (67 %), joilla ei ollut RULM-maksimipisteitä lähtötilanteessa, pisteet paranivat päivään 302 mennessä.
Aikuisväestön turvallisuustiedot ovat yhdenmukaiset nusinerseenin tunnetun turvallisuusprofiilin ja SMA:n perussairauteen liittyvien liitännäissairauksien kanssa.
Oireettomat imeväisikäiset
Tutkimus CS5 (NURTURE) on avoin tutkimus, johon otettiin mukaan enintään 6 viikon ikäisiä vielä oireettomia geneettisen SMA-diagnoosin saaneita vauvoja. Tämän tutkimuksen potilaille todennäköisimpänä pidettiin tyypin I tai II SMA:n kehittymistä. Iän mediaani ensimmäisen annoksen ajankohtana oli 22 vuorokautta.
Välianalyysin ajankohtana potilaiden tutkimukseen osallistumisen mediaani oli 48,3 kuukautta (36,6–57,1 kuukautta) ja potilaiden iän mediaani viimeisellä käynnillä oli 46,0 kuukautta (34,0–57,1 kuukautta). Välianalyysin kohdalla kaikki 25 potilasta (kaksi SMN2-geenin kopiota n = 15, kolme SMN2-geenin kopiota n = 10) olivat elossa eivätkä tarvinneet jatkuvaa ventilaatiota. Ensisijaista päätetapahtumaa, joka oli aika kuolemaan tai hengitysinterventioon (joksi määriteltiin invasiivinen tai ei-invasiivinen ventilaatio ≥ 6 tuntia/vrk ≥ 7 peräkkäisenä päivänä tai trakeostomia), ei voitu määrittää, koska tapauksia oli liian vähän. Neljä potilasta (kaksi SMN2-geenin kopiota) tarvitsi hengitysintervention (> 6 tuntia/vrk ≥ 7 peräkkäisenä päivänä), heistä jokaisella ventilaatiotuki aloitettiin akuutin paranevan sairauden aikana.
Potilaat saavuttivat kehitystasoja, joiden saavuttaminen on epätavallista tyypin I tai II SMA:ssa ja jotka ovat yhdenmukaisempia normaalin kehityksen kanssa. Välianalyysin kohdalla kaikki 25 potilasta (100 %) olivat saavuttaneet WHO:n määritelmän mukaisen motorisen kehitystason istuminen ilman tukea, 23 (92 %) potilasta käveli tuettuna ja 22 (88 %) potilasta käveli itse. 21 (84 %) potilasta saavutti suurimman mahdollisen CHOP INTEND -pistearvon 64. Viimeisellä käynnillä (päivänä 788) kaikilla potilailla oli kyky imeä ja niellä ja 22 (88 %) vauvoista sai HINE-testin 1. osassa maksimipisteet.
Potilaat, joille kehittyi kliinisesti ilmeinen SMA, arvioitiin päivän 700 tutkimuskäynnillä. Tutkimussuunnitelmassa määritellyt kliinisesti ilmeisen SMA:n kriteerit olivat viidennen WHO:n persentiilin alittava iän mukaan korjattu paino, painokehityskäyrän kahden tai useamman persentiilin lasku, perkutaanisen mahaletkun asettaminen ja/tai kyvyttömyys saavuttaa iän mukaan odotettuja WHO:n määrittelemiä motorisia kehitystasoja (istuminen ilman tukea, seisominen tuettuna, käsien ja polvien avulla ryömiminen, käveleminen tuettuna, seisominen itsenäisesti ja käveleminen itsenäisesti). Päivänä 700 kliinisesti ilmeisen SMA:n kriteerit täyttivät 7 potilasta niistä 15 potilaasta (47 %), joilla oli kaksi SMN2-geenin kopiota, ja 0 potilasta niistä 5 potilaasta (0 %), joilla oli kolme SMN2-geenin kopiota. Nämä potilaat saivat kuitenkin lisää painoa ja saavuttivat WHO:n määrittelemiä motorisia kehitystasoja, mikä ei ole yhdenmukaista tyypin I SMA:n kanssa. Vertailu SMA:han imeväisiässä sairastuneiden oireisten potilaiden ja vielä oireettomien SMA‑diagnoosin saaneiden potilaiden motoristen kehitystasojen saavuttamisesta esitetään kuvassa 5.
Kuva 5: HINE-testin mukaisten motoristen kehitystasojen saavuttaminen suhteessa tutkimuspäiviin tutkimuksissa CS3B (hoito ja lumetoimenpiteet), CS3A, CS5 ja CS11
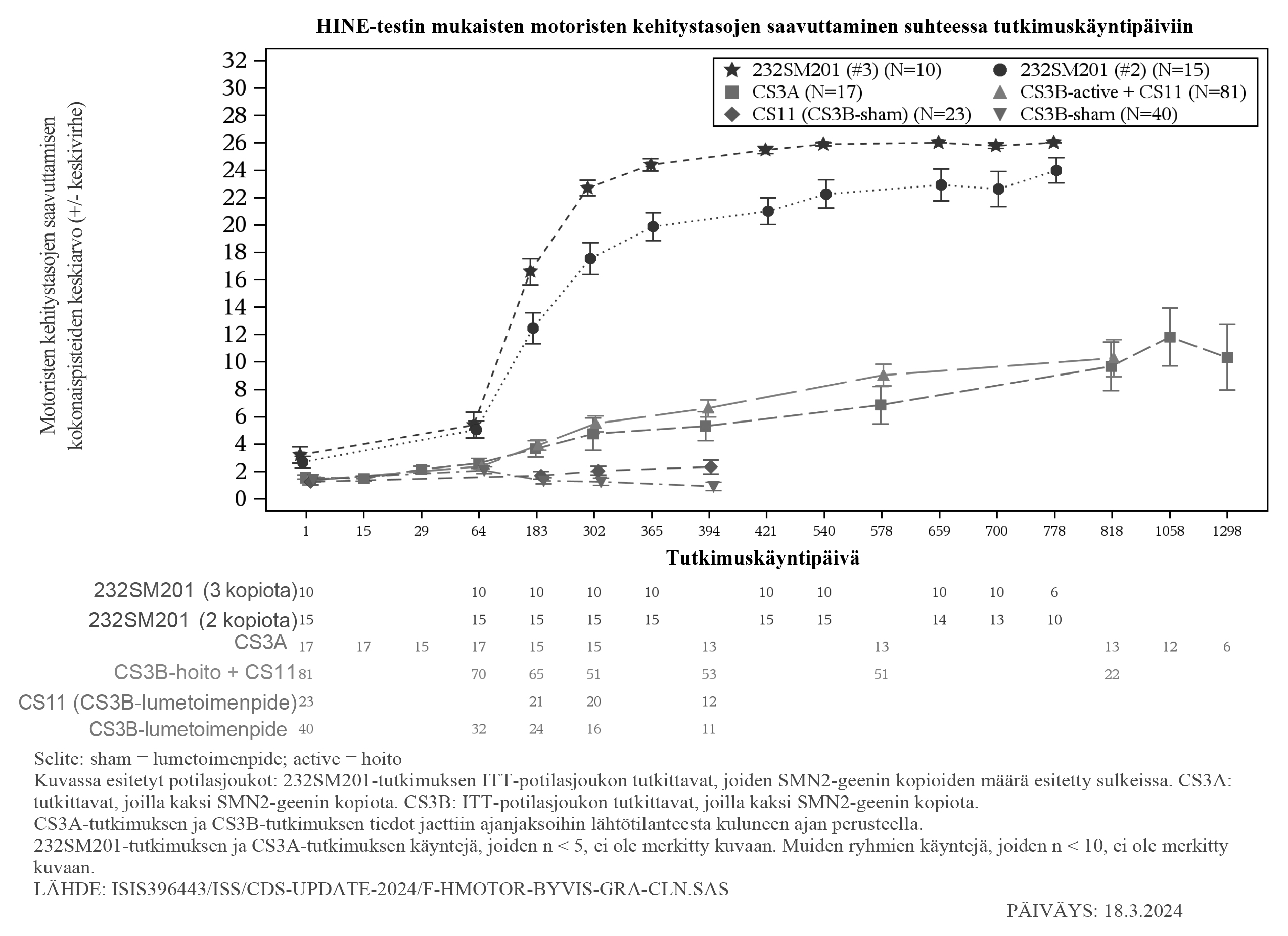
Farmakokinetiikka
Selkäydinesteeseen injektiona annettujen yksittäisten ja useiden nusinerseeniannosten farmakokinetiikkaa arvioitiin SMA-diagnoosin saaneilla pediatrisilla ja aikuisilla potilailla.
Imeytyminen
Intratekaalinen nusinerseeni-injektio aivo-selkäydinnesteeseen mahdollistaa nusinerseenin täydellisen jakautumisen aivo-selkäydinnesteestä keskushermoston kohdekudoksiin.
Aivo-selkäydinnestettä koskevia kumulatiivisia farmakokineettisiä tietoja arvioitiin 12 mg:n annostuksen osalta pitkäkestoisessa altistustutkimuksessa CS11, ja ne osoittivat, että imeväisiässä tai myöhemmin alkanutta SMA:ta sairastavilla potilailla aivo-selkäydinnesteen lääkeainepitoisuuksien kumuloituminen jatkui noin 9 vuoden ajan.
Aivo-selkäydinnesteen lääkeainepitoisuudessa havaittiin samankaltaista kertymistä tutkimuksessa SM203 myös 50/28 mg:n annostuksen osalta ja tutkimuksessa SM302 farmakokinetiikasta aivo-selkäydinnesteessä kerättyjen tietojen kattaman koko ajanjakson ajan.
Intratekaalisesti annetun nusinerseenin pienimmät pitoisuudet plasmassa olivat suhteellisen pieniä verrattuna pienimpiin pitoisuuksiin aivo-selkäydinnesteessä. Plasman Tmax-arvojen mediaani vaihteli välillä 1,7–6,0 tuntia. Plasman keskimääräiset Cmax- ja AUC-arvot suurenivat arvioidulla annosalueella suunnilleen suhteessa annokseen. Altistusta plasmassa mittaavat arvot (Cmax ja AUC) eivät kumuloituneet useiden annosten jälkeen.
Jakautuminen
Potilaiden ruumiinavaustiedot (n = 3) osoittavat intratekaalisesti annetun nusinerseenin jakautuvan laajasti keskushermostoon, missä terapeuttiset pitoisuudet saavutetaan selkäytimen kohdekudoksissa. Nusinerseeniä havaittiin myös selkäytimen ja aivojen hermosoluissa ja muissa solutyypeissä sekä perifeerisissä kudoksissa, kuten luustolihaksissa, maksassa ja munuaisissa.
Biotransformaatio
Nusinerseeni metaboloituu hitaasti, pääasiassa eksonukleaasi (3’ ja 5’) ‑välitteisen hydrolyysin kautta, eikä se ole CYP450-entsyymien substraatti, estäjä eikä indusoija.
Eliminaatio
Keskimääräisen terminaalisen eliminaation puoliintumisajan aivo-selkäydinnesteessä arvioidaan olevan noin 20 kuukautta. Ensisijaisen eliminaatioreitin oletetaan olevan nusinerseenin ja sen metaboliittien erittyminen virtsaan.
Yhteisvaikutukset
In vitro -tutkimukset osoittivat, että nusinerseeni ei indusoi eikä estä CYP450-välitteistä oksidatiivista metaboliaa eikä sillä sen vuoksi odoteta olevan yhteisvaikutuksia näitä metaboliareittejä käyttävien lääkevalmisteiden kanssa. Nusinerseeni ei ole seuraavien substraatti eikä estäjä ihmisellä: BCRP, P‑gp, OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3 ja BSEP-kuljettajaproteiinit.
Ominaisuudet erityisryhmillä
Munuaisten ja maksan vajaatoiminta
Nusinerseenin farmakokinetiikkaa ei ole tutkittu munuaisten tai maksan vajaatoimintaa sairastavilla potilailla. Maksan tai munuaisten vajaatoiminnan vaikutusta kovariaatteina ei pystytty arvioimaan tarkasti populaatiofarmakokineettisessä mallissa, koska kliinisesti merkityksellistä maksan tai munuaisten vajaatoimintaa sairastavia potilaita oli vähän. Populaatiofarmakokineettiset analyysit eivät osoittaneet selvää vastaavuussuhdetta maksan ja munuaisten kliinis-kemiallisten merkkiaineiden ja tutkittavien välisen vaihtelun välillä.
Rotu
Suurin osa tutkituista potilaista oli valkoihoisia. Populaatiofarmakokineettinen analyysi viittaa siihen, että etninen alkuperä ei todennäköisesti vaikuta nusinerseenin farmakokinetiikkaan.
Prekliiniset tiedot turvallisuudesta
Genotoksisuus/karsinogeenisuus
Nusinerseenin genotoksisuudesta ei ole näyttöä.
Nusinerseeniä annettiin karsinogeenisuustutkimuksessa uros- ja naarashiirille 2 vuoden ajan injektioina ihon alle annoksina 0, 5, 15 tai 50 mg/kg kahden viikon välein. Verisuonikasvainten (yhdistetyt tiedot hemangioomasta ja hemangiosarkoomasta) ilmaantuvuuden havaittiin lisääntyneen käytettäessä annosta 50 mg/kg.
Nusinerseenista aiheutuneista onkogeenisistä vaikutuksista ei havaittu näyttöä, kun annos oli enintään 15 mg/kg. Vuosiannoksen perusteella tähän annokseen liittyi seerumin AUC0-24-arvo, joka on 30-kertainen kliiniseen seerumin altistukseen nähden käytettäessä 28 mg:n ylläpitoannosta.
Lisääntymistoksisuus
Nusinerseeniä annettiin lisääntymistoksisuustutkimuksissa hiirille ja kaniineille ihon alle. Vaikutuksia urosten tai naaraiden hedelmällisyyteen, alkion tai sikiön kehitykseen tai tiineydenaikaiseen tai syntymänjälkeiseen kehitykseen ei havaittu.
Toksisuus
Toistuvan altistuksen (yhdistetty 6 viikkoa ja 13 viikkoa, 14 viikkoa sekä 53 viikkoa) aiheuttamaa toksisuutta koskevissa nuorilla cynomolgus-apinoilla tehdyissä tutkimuksissa nusinerseenin antamisesta selkäydinnesteeseen ei arvioiduilla annoksilla aiheutunut haitallisia toksisia vaikutuksia.
Akuuttia, ohimenevää alempien selkäydinrefleksien puutosta, rajoittunutta raajojen käyttöä ja/tai koordinoimattomia liikkeitä ilmeni joillakin yksittäisillä apinoilla. Näitä vaikutuksia havaittiin useiden tuntien kuluessa annon jälkeen, ne hävisivät 48 tunnin kuluessa eikä niitä katsottu haitallisiksi.
Nuorilla apinoilla tehdyssä 53 viikon pituisessa toksikologiatutkimuksessa nusinerseeniä tutkittiin annostasoilla enintään 4 mg/annos, joka vastaa potilailla 40 mg:aa per selkäydinnesteeseen annettava annos ja josta yhden vuoden aikana saatava kumulatiivinen annos on 6,2 kertaa suurempi kuin nusinerseenin kliininen 28 mg:n ylläpitoannos (kolme ylläpitoannosta vuodessa).
Farmaseuttiset tiedot
Apuaineet
Natriumdivetyfosfaattidihydraatti
Dinatriumfosfaatti
Natriumkloridi
Kaliumkloridi
Kalsiumklorididihydraatti
Magnesiumkloridiheksahydraatti
Natriumhydroksidi (pH:n säätöön)
Kloorivetyhappo (pH:n säätöön)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
5 vuotta
Säilytys
Säilytä jääkaapissa (2 °C - 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Jos jääkaappisäilytys ei ole mahdollinen, Spinraza voidaan säilyttää alkuperäispakkauksessa valolta suojattuna enintään 30 °C:n lämpötilassa enintään 14 vuorokauden ajan.
Avaamaton Spinraza-injektiopullo voidaan ennen antoa poistaa jääkaapista ja tarvittaessa palauttaa sinne. Jos injektiopullo poistetaan alkuperäispakkauksesta, säilytysaika poissa jääkaapista ei saa ylittää yhteensä 30 tuntia silloin, kun lämpötila on enintään 25 °C.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SPINRAZA injektioneste, liuos
12 mg (L:ei) 5 ml (2,4 mg/ml) (94757,88 €)
28 mg (L:ei) 5 ml (5,6 mg/ml) (94757,88 €)
50 mg (L:ei) 5 ml (10 mg/ml) (169068,60 €)
PF-selosteen tieto
Spinraza 12 mg
Kertakäyttöisessä tyypin I lasisessa injektiopullossa, jossa on harmaa bromobutyylikumitulppa, alumiinisinetti ja muovikorkki, on 12 mg nusinerseeniä 5 ml:ssa keinotekoista aivo-selkäydinnestettä.
Pakkauskoko: yhden injektiopullon pakkaus.
Spinraza 28 mg
Kertakäyttöisessä tyypin I lasisessa injektiopullossa, jossa on harmaa bromobutyylikumitulppa, punainen alumiinisinetti ja muovikorkki, on 28 mg nusinerseeniä 5 ml:ssa keinotekoista aivo-selkäydinnestettä.
Pakkauskoko: yhden injektiopullon pakkaus.
Spinraza 50 mg
Kertakäyttöisessä tyypin I lasisessa injektiopullossa, jossa on harmaa bromobutyylikumitulppa, sininen alumiinisinetti ja muovikorkki, on 50 mg nusinerseeniä 5 ml:ssa keinotekoista aivo-selkäydinnestettä.
Pakkauskoko: yhden injektiopullon pakkaus.
Valmisteen kuvaus:
Kirkas, väritön liuos, jonka pH-arvo on noin 7,2.
Käyttö- ja käsittelyohjeet
Vain kertakäyttöön yhdelle potilaalle. Ei saa laimentaa.
Ohjeet lääkevalmisteen käyttökuntoon saattamiseen ennen antoa
1. Spinraza-injektiopullo pitää tarkistaa ennen antoa, ettei liuoksessa ole hiukkasia. Jos hiukkasia havaitaan ja/tai injektiopullossa oleva neste ei ole kirkasta ja väritöntä, injektiopulloa ei saa käyttää.
2. Intratekaalisesti annettavan Spinrazan valmistamisessa on käytettävä aseptista tekniikkaa.
3. Ennen antoa injektiopullo otetaan jääkaapista, ja sen annetaan lämmetä huoneenlämpöiseksi (25 °C) ulkoisia lämmönlähteitä käyttämättä.
4. Jos liuosta ei käytetä eikä injektiopulloa avata, se pitää palauttaa jääkaappiin (ks. kohta Säilytys).
5. Poista muovikorkki juuri ennen antoa, työnnä ruiskun neula injektiopulloon sinetin keskikohdan läpi, ja vedä tarvittava määrä liuosta ruiskuun. Spinrazaa ei saa laimentaa. Ulkoisten suodattimien käyttö ei ole tarpeen.
6. Jos ruiskuun vedettyä liuosta ei käytetä 6 tunnin kuluessa, se on hävitettävä.
7. Injektiopullossa oleva käyttämätön lääkevalmiste on hävitettävä, ja jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SPINRAZA injektioneste, liuos
12 mg 5 ml
28 mg 5 ml
50 mg 5 ml
- Ei korvausta.
ATC-koodi
M09AX07
Valmisteyhteenvedon muuttamispäivämäärä
09.01.2026
Yhteystiedot
Bertel Jungin aukio 5 C
02600 Espoo
0207 401 200
www.biogen.fi
www.MS-nyt.fi