
IMBRUVICA tabletti, kalvopäällysteinen 140 mg, 280 mg, 420 mg, 560 mg
Vaikuttavat aineet ja niiden määrät
IMBRUVICA 140 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 140 mg ibrutinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi 140 mg:n kalvopäällysteinen tabletti sisältää 28 mg laktoosimonohydraattia.
IMBRUVICA 280 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 280 mg ibrutinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi 280 mg:n kalvopäällysteinen tabletti sisältää 56 mg laktoosimonohydraattia.
IMBRUVICA 420 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 420 mg ibrutinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi 420 mg:n kalvopäällysteinen tabletti sisältää 84 mg laktoosimonohydraattia.
IMBRUVICA 560 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 560 mg ibrutinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi 560 mg:n kalvopäällysteinen tabletti sisältää 112 mg laktoosimonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
IMBRUVICA on tarkoitettu annettavaksi yhdistelmänä rituksimabin, syklofosfamidin, doksorubisiinin, vinkristiinin ja prednisolonin (IMBRUVICA + R‑CHOP) kanssa vuorotellen R‑DHAP-hoidon (tai R‑DHAOx-hoidon) kanssa ilman IMBRUVICA-valmistetta, ja sen jälkeen annettavaksi IMBRUVICA-monoterapiana aiemmin hoitamattoman manttelisolulymfooman hoitoon aikuispotilaille, jotka soveltuisivat autologiseen kantasolusiirtoon.
IMBRUVICA on tarkoitettu monoterapiana aikuispotilaiden uusiutuneen tai hoitoon reagoimattoman manttelisolulymfooman hoitoon.
IMBRUVICA on tarkoitettu monoterapiana tai yhdistelmänä rituksimabin tai obinututsumabin tai venetoklaksin kanssa aikuispotilaiden aiemmin hoitamattoman kroonisen lymfaattisen leukemian hoitoon (ks. kohta Farmakodynamiikka).
IMBRUVICA on tarkoitettu monoterapiana tai yhdistelmänä bendamustiinin ja rituksimabin kanssa kroonisen lymfaattisen leukemian hoitoon aikuispotilaille, jotka ovat saaneet aiemmin vähintään yhtä hoitoa.
IMBRUVICA on tarkoitettu monoterapiana Waldenströmin makroglobulinemian hoitoon aikuispotilaille, jotka ovat saaneet aiemmin vähintään yhtä hoitoa, tai ensisijaiseksi hoidoksi potilaille, joille kemoimmunoterapia ei sovi. IMBRUVICA on tarkoitettu yhdistelmänä rituksimabin kanssa Waldenströmin makroglobulinemian hoitoon aikuispotilaille.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Hoito tällä lääkevalmisteella pitää aloittaa ja toteuttaa syöpälääkkeiden käyttöön perehtyneen lääkärin valvonnassa.
Annostus
Manttelisolulymfooma
Aiemmin hoitamatonta manttelisolulymfoomaa sairastavien aikuispotilaiden hoito
Suositeltu annos aiemmin hoitamattoman manttelisolulymfooman hoitoon on 560 mg ibrutinibia kerran vuorokaudessa (ks. taulukko 1).
| Taulukko 1: IMBRUVICA-hoito-ohjelma aiemmin hoitamattoman manttelisolulymfooman hoitoon | |||
| Hoito | Hoitosyklin numero | Hoito | IMBRUVICA |
| Osa I* | 1, 3, 5 | IMBRUVICA yhdistelmänä R-CHOP-hoidon kanssa§ | Päivinä 1–19 |
| 2, 4, 6 | R-DHAP-hoito#§ | Ilman IMBRUVICA-valmistetta | |
| Osa II± | IMBRUVICA | Päivittäin 24 kuukauden ajan | |
R-CHOP = rituksimabi, syklofosfamidi, doksorubisiini, vinkristiini ja prednisoloni; R-DHAP = rituksimabi, deksametasoni, sytarabiini, sisplatiini
*6 hoitosykliä, kukin hoitosykli on 21 päivää.
§Ks. kunkin lääkevalmisteen annostusta koskevat tiedot niiden valmisteyhteenvedosta.
#Vaihdettavissa R-DHAOx-hoitoon (rituksimabi, deksametasoni, sytarabiini, oksaliplatiini)§.
±Hoito aloitetaan, kun verenkuva on korjautunut. Rituksimabi voidaan lisätä kansallisten hoitosuositusten mukaisesti.
Uusiutunutta tai hoitoon reagoimatonta manttelisolulymfoomaa sairastavien aikuispotilaiden hoito
Suositusannos aiemmin hoidetun manttelisolulymfooman hoitoon on 560 mg ibrutinibia kerran vuorokaudessa monoterapiana. Hoitoa IMBRUVICA-monoterapialla pitää jatkaa, kunnes sairaus etenee tai potilas ei enää siedä hoitoa.
Krooninen lymfaattinen leukemia ja Waldenströmin makroglobulinemia
Suositusannos kroonisen lymfaattisen leukemian ja Waldenströmin makroglobulinemian hoitoon joko monoterapiana tai yhdistelmähoidossa on 420 mg kerran vuorokaudessa (tiedot yhdistelmähoidoista, ks. kohta Farmakodynamiikka).
IMBRUVICA-hoitoa monoterapiana tai yhdistelmänä CD20-vasta-aineiden kanssa pitää jatkaa, kunnes tauti etenee tai potilas ei enää siedä hoitoa. Annettaessa IMBRUVICA-valmistetta yhdistelmänä venetoklaksin kanssa kroonisen lymfaattisen leukemian hoitoon IMBRUVICA-valmistetta annetaan monoterapiana 3 hoitosykliä (yksi hoitosykli on 28 päivää), minkä jälkeen annetaan 12 hoitosykliä IMBRUVICA-valmistetta ja venetoklaksia yhdistelmähoitona. Katso venetoklaksin tarkat annostustiedot venetoklaksin valmisteyhteenvedosta.
Annettaessa IMBRUVICA-valmistetta yhdistelmänä CD20-vasta-aineiden kanssa suositellaan, että IMBRUVICA annetaan ennen CD20-vasta-ainetta, jos ne annetaan samana päivänä.
Annoksen muuttaminen
Kohtalaiset ja voimakkaat CYP3A4:n estäjät voivat suurentaa altistusta ibrutinibille (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Ibrutinibiannos pitää pienentää 280 mg:aan kerran vuorokaudessa, jos samanaikaisesti käytetään kohtalaisia CYP3A4:n estäjiä.
Ibrutinibiannos pitää pienentää 140 mg:aan kerran vuorokaudessa tai hoito keskeyttää enintään 7 päivän ajaksi, jos samanaikaisesti käytetään voimakkaita CYP3A4:n estäjiä.
IMBRUVICA-hoito pitää keskeyttää, jos potilaalle kehittyy jotakin seuraavista tai hänellä esiintyy seuraavien pahenemista: 2. asteen sydämen vajaatoiminta, 3. asteen sydämen rytmihäiriöt, ≥ 3. asteen ei-hematologinen toksisuus, 3. asteen tai vaikeampiasteinen neutropenia, johon liittyy infektio tai kuumetta tai 4. asteen hematologinen toksisuus. Kun toksisuuden oireet ovat lieventyneet 1. asteeseen tai lähtötilanteeseen (potilas toipunut), jatka IMBRUVICA-hoitoa suositeltuina annoksina jäljempänä olevien taulukoiden mukaisesti.
Seuraavassa esitetään annoksen muutossuositukset sydämeen liittymättömien tapahtumien yhteydessä:
| Tapahtumat† | Toksisuuden esiintyminen | Annosmuutos manttelisolulymfooman hoidossa potilaan toipumisen jälkeen | Annosmuutos kroonisen lymfaattisen leukemian/Waldenströmin makroglobulinemian hoidossa potilaan toipumisen jälkeen |
3. tai 4. asteen ei-hematologinen toksisuus
3. tai 4. asteen neutropenia, johon liittyy infektio tai kuumetta
4. asteen hematologinen toksisuus | Ensimmäinen kerta* | jatka hoitoa 560 mg:n vuorokausiannoksella | jatka hoitoa 420 mg:n vuorokausiannoksella |
| Toinen kerta | jatka hoitoa 420 mg:n vuorokausiannoksella | jatka hoitoa 280 mg:n vuorokausiannoksella | |
| Kolmas kerta | jatka hoitoa 280 mg:n vuorokausiannoksella | jatka hoitoa 140 mg:n vuorokausiannoksella | |
| Neljäs kerta | lopeta IMBRUVICA-hoito | lopeta IMBRUVICA-hoito | |
| † Vaikeusasteluokitus perustuu NCI-CTCAE (National Cancer Institute-Common Terminology Criteria for Adverse Events) ‑kriteereihin tai kroonisessa lymfaattisessa leukemiassa / pienilymfosyyttisessa lymfoomassa hematologisen toksisuuden IWCLL (International Workshop on Chronic Lymphocytic Leukemia) ‑kriteereihin. * Aloitettaessa hoitoa uudelleen jatka hyöty-riskiarvion perusteella samalla tai pienemmällä annoksella. Jos toksisuus uusiutuu, pienennä vuorokausiannosta 140 mg:lla. | |||
Seuraavassa esitetään annoksen muutossuositukset sydämen vajaatoiminnan tai sydämen rytmihäiriöiden yhteydessä:
| Tapahtumat | Toksisuuden esiintyminen | Annosmuutos manttelisolulymfooman hoidossa potilaan toipumisen jälkeen | Annosmuutos kroonisen lymfaattisen leukemian/Waldenströmin makroglobulinemian hoidossa potilaan toipumisen jälkeen |
| 2. asteen sydämen vajaatoiminta | Ensimmäinen kerta | jatka hoitoa 420 mg:n vuorokausiannoksella | jatka hoitoa 280 mg:n vuorokausiannoksella |
| Toinen kerta | jatka hoitoa 280 mg:n vuorokausiannoksella | jatka hoitoa 140 mg:n vuorokausiannoksella | |
| Kolmas kerta | lopeta IMBRUVICA-hoito | ||
| 3. asteen sydämen rytmihäiriöt | Ensimmäinen kerta | jatka hoitoa 420 mg:n vuorokausiannoksella† | jatka hoitoa 280 mg:n vuorokausiannoksella† |
| Toinen kerta | lopeta IMBRUVICA-hoito | ||
3. tai 4. asteen sydämen vajaatoiminta 4. asteen sydämen rytmihäiriöt | Ensimmäinen kerta | lopeta IMBRUVICA-hoito | |
| † Tee hyöty-riskiarvio ennen hoidon jatkamista. | |||
Annoksen unohtuminen
Jos annosta ei oteta tavanomaisena ajankohtana, se voidaan ottaa mahdollisimman pian samana päivänä, ja seuraavana päivänä palataan normaaliin hoitoaikatauluun. Potilas ei saa ottaa ylimääräisiä tabletteja unohtuneen annoksen korvaamiseksi.
Erityispotilasryhmät
Iäkkäät
Annosta ei tarvitse muuttaa erikseen iäkkäille potilaille (≥ 65-vuotiaat).
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavilla potilailla ei ole tehty spesifisiä kliinisiä tutkimuksia. IMBRUVICA-valmistetta koskeneissa kliinisissä tutkimuksissa oli mukana lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavia potilaita. Lievää tai keskivaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma yli 30 ml/min) sairastavien potilaiden annosta ei tarvitse muuttaa. Potilaan riittävästä nesteytyksestä pitää huolehtia, ja seerumin kreatiniinipitoisuuksia pitää seurata säännöllisin väliajoin. Anna IMBRUVICA-hoitoa vaikeaa munuaisten vajaatoimintaa (kreatiniinipuhdistuma < 30 ml/min) sairastaville potilaille vain, jos hoidon hyödyt ovat sen riskejä suuremmat, ja seuraa potilasta tarkoin toksisuuden oireiden havaitsemiseksi. Vaikeaa munuaisten vajaatoimintaa sairastavista tai dialyysihoitoa saavista potilaista ei ole tietoja (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Ibrutinibi metaboloituu maksassa. Maksan vajaatoimintaa koskeneen tutkimuksen tiedot osoittivat, että altistus ibrutinibille suureni (ks. kohta Farmakokinetiikka). Jos potilas sairastaa lievää maksan vajaatoimintaa (Child-Pugh-luokka A), suositeltu annos on 280 mg vuorokaudessa. Jos potilas sairastaa keskivaikeaa maksan vajaatoimintaa (Child-Pugh-luokka B), suositeltu annos on 140 mg vuorokaudessa. Seuraa potilasta IMBRUVICA-hoitoon liittyvän toksisuuden oireiden havaitsemiseksi ja noudata tarvittaessa annosmuutoksista annettua ohjetta. IMBRUVICA-hoitoa ei suositella vaikeaa maksan vajaatoimintaa sairastaville potilaille (Child-Pugh-luokka C).
Vaikea-asteinen sydäntauti
IMBRUVICA-valmistetta koskeneisiin kliinisiin tutkimuksiin ei otettu mukaan vaikea-asteista sydän- ja verisuonitautia sairastavia potilaita.
Pediatriset potilaat
IMBRUVICA-valmistetta ei suositella 0–18 vuoden ikäisten lasten ja nuorten hoitoon, sillä sen tehoa ei ole varmistettu. Kypsää B-soluista non-Hodgkin-lymfoomaa sairastavista potilaista saatavissa olevat tiedot on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka.
Antotapa
IMBRUVICA otetaan suun kautta vesilasillisen kanssa kerran päivässä aina suunnilleen samaan aikaan päivästä. Tabletit niellään kokonaisina veden kera eikä niitä saa rikkoa eikä pureskella. IMBRUVICA-tabletteja ei saa ottaa greippimehun eikä pomeranssin kanssa (ks. kohta Yhteisvaikutukset).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Mäkikuismaa sisältävien valmisteiden käyttö on vasta-aiheista IMBRUVICA-hoitoa saaville.
Varoitukset ja käyttöön liittyvät varotoimet
Verenvuotoon liittyvät tapahtumat
IMBRUVICA-hoitoa saaneilla potilailla on raportoitu verenvuototapahtumia, joihin on saattanut liittyä trombosytopeniaa. Nämä ovat olleet lieviä verenvuototapahtumia, kuten ruhjeita, nenäverenvuotoa ja petekioita, sekä vakavia, jotkin kuolemaan johtaneita, verenvuototapahtumia, kuten maha-suolikanavan verenvuotoa, kallonsisäistä verenvuotoa ja verivirtsaisuutta.
Varfariinia tai muita K-vitamiinin antagonisteja ei saa käyttää samaan aikaan IMBRUVICA-hoidon kanssa.
Antikoagulanttien tai verihiutaleiden toimintaa estävien lääkevalmisteiden (verihiutaleiden aggregaation estäjien) samanaikainen käyttö IMBRUVICA-tablettien kanssa lisää vakavien verenvuotojen riskiä. Antikoagulanttien käytössä havaittiin suurempi vakavien verenvuotojen riski kuin verihiutaleiden aggregaation estäjien käytössä. Arvioi antikoagulanttien ja verihiutaleiden aggregaation estäjien riskit ja hyödyt, jos niitä käytetään samanaikaisesti IMBRUVICA-tablettien kanssa. Tarkkaile verenvuodon oireita ja löydöksiä.
Ravintolisiä, kuten kalaöljyä ja E-vitamiinivalmisteita, pitää välttää.
IMBRUVICA-hoito pitää keskeyttää vähintään 3–7 päiväksi ennen leikkausta ja leikkauksen jälkeen riippuen siitä, millainen leikkaus tehdään ja millainen verenvuotoriski siihen liittyy.
Verenvuotoihin liittyvien tapahtumien mekanismia ei tunneta täysin. Potilaita, joilla on synnynnäinen verenvuototaipumus, ei ole tutkittu.
Leukostaasi
IMBRUVICA-hoitoa saaneilla potilailla on raportoitu leukostaasia. Suuri lymfosyyttimäärä verenkierrossa (> 400 000/mikrol) saattaa lisätä tätä riskiä. Harkitse IMBRUVICA-hoidon keskeyttämistä tilapäisesti. Potilaan tilaa on seurattava tarkoin. Anna potilaalle tarvittavaa elintoimintoja tukevaa hoitoa, kuten nesteytystä ja/tai solumäärää vähentävää hoitoa.
Pernan repeämä
IMBRUVICA-hoidon lopettamisen jälkeen on raportoitu pernan repeämiä. IMBRUVICA-hoitoa keskeytettäessä tai lopetettaessa sairauden tilannetta ja pernan kokoa pitää seurata tarkoin (esim. kliininen tutkimus, ultraäänitutkimus). Jos potilaalle kehittyy vasemmalle ylävatsaan tai olkapäähän kipua, pitää potilaan tila arvioida ja pernan repeämän diagnoosia harkita.
Infektiot
IMBRUVICA-hoitoa saaneilla potilailla on havaittu infektioita (sepsis, neutropeeninen sepsis, bakteeri-, virus- tai sieni-infektiot mukaan lukien). Joidenkin tällaisten infektioiden yhteydessä potilas on joutunut sairaalahoitoon ja kuollut. Useimmilla niistä potilaista, joilla oli kuolemaan johtanut infektio, oli myös neutropenia. Potilasta pitää seurata kuumeen, maksan toimintakokeiden epänormaalien tulosten, neutropenian ja infektioiden havaitsemiseksi, ja tarvittava infektion tarkoituksenmukainen hoito on aloitettava. Jos potilaalla on tavanomaista suurempi opportunististen infektioiden riski, harkitse estohoitoa tavanomaisen hoitokäytännön mukaisesti.
Ibrutinibin käytön jälkeen on raportoitu invasiivisia sieni-infektioita, mukaan lukien aspergilloosia, kryptokokkoosia ja Pneumocystis jirovecii ‑infektioita. Raportoituihin invasiivisiin sieni-infektioihin on liittynyt kuolemia.
Ibrutinibia käyttäneillä potilailla, jotka ovat saaneet samanaikaisesti tai aiemmin immunosuppressiivista hoitoa, on havaittu progressiivista multifokaalista leukoenkefalopatiaa (PML), myös potilaan kuolemaan johtaneina tapauksina. Jos potilaalla on uusia neurologisia, kognitiivisia tai käyttäytymiseen liittyviä oireita tai löydöksiä tai näiden pahenemista, lääkärin on otettava PML huomioon erotusdiagnoosia tehdessään. Jos PML:aa epäillään, tarkoituksenmukaiset diagnostiset tutkimukset pitää tehdä ja keskeyttää hoito, kunnes PML on suljettu pois. Jos varmuutta ei saada, potilas on lähetettävä neurologin tutkittavaksi, ja on harkittava tarkoituksenmukaisia PML:n diagnostisia toimenpiteitä, kuten magneettikuvausta mieluiten varjoainetehosteisena, JC-viruksen DNA:n määrittämistä aivo-selkäydinnesteestä sekä toistuvia neurologisia arvioita.
Maksatapahtumat
IMBRUVICA-hoitoa saaneilla potilailla on esiintynyt maksatoksisuutta, hepatiitti B -viruksen reaktivaatiota ja E-hepatiittia, joka voi olla krooninen. IMBRUVICA-hoitoa saaneilla potilailla on esiintynyt maksan vajaatoimintaa, mukaan lukien kuolemaan johtaneita tapahtumia. Maksan toiminta ja virushepatiittistatus pitää selvittää ennen IMBRUVICA-hoidon aloittamista. Potilaita pitää seurata hoidon aikana säännöllisin väliajoin maksan toimintaa kuvastavien parametrien muutosten havaitsemiseksi. Epideemisen hepatiitin viruskuorman määritys ja serologinen testaus pitää tehdä paikallisten hoito-ohjeistojen mukaisesti, kun se on kliinisesti aiheellista. Potilailla todettujen maksatapahtumien hoidossa on harkittava maksasairauksien asiantuntijan konsultoimista.
Sytopeniat
IMBRUVICA-hoitoa saaneilla potilailla on raportoitu hoidosta aiheutuneita 3. ja 4. asteen sytopenioita (neutropenia, trombosytopenia ja anemia). Täydellinen verenkuva on tutkittava kuukausittain.
Interstitiaalinen keuhkosairaus
IMBRUVICA-hoitoa saaneilla potilailla on raportoitu interstitiaalista keuhkosairautta. Seuraa potilaita interstitiaaliseen keuhkosairauteen viittaavien keuhko-oireiden havaitsemiseksi. Jos oireita kehittyy, keskeytä IMBRUVICA-hoito ja hoida interstitiaalinen keuhkosairaus asianmukaisesti. Jos oireet pitkittyvät, arvioi IMBRUVICA-hoidon riskit ja hyödyt ja noudata annosmuutoksia koskevia ohjeita.
Sydämen rytmihäiriöt ja sydämen vajaatoiminta
IMBRUVICA-hoitoa saaneilla potilailla on esiintynyt kuolemaan johtaneita ja vakavia sydämen rytmihäiriöitä ja sydämen vajaatoimintaa. Tapahtumien, mukaan lukien äkillisten kuolemaan johtavien sydäntapahtumien, riski voi olla suurempi potilailla, jotka ovat iäkkäitä, joiden ECOG (Eastern Cooperative Oncology Group) ‑toimintakykyluokka on ≥ 2 tai joilla on muita samanaikaisia sydänsairauksia. Eteisvärinää, eteislepatusta, kammioperäistä takyarytmiaa ja sydämen vajaatoimintaa on raportoitu etenkin potilailla, joilla on akuutteja infektioita tai sydämeen liittyviä riskitekijöitä, mukaan lukien hypertensio, diabetes, ja joilla on aiemmin ollut sydämen rytmihäiriöitä.
Aiemmat sydänsairaudet ja sydämen toiminta on tutkittava asianmukaisesti kliinisesti ennen IMBRUVICA-hoidon aloittamista. Potilaita pitää seurata hoidon aikana tarkoin, jotta sydämen toiminnan kliininen heikkeneminen havaitaan ja hoidetaan kliinisesti. Jos potilaalla on kardiovaskulaarisia huolenaiheita, harkitse lisätutkimuksia (esim. EKG, sydämen kaikukuva) siten kuin on aiheellista.
Jos potilaalla on sydäntapahtumien oleellisia riskitekijöitä, arvioi hyöty-riskisuhde tarkoin ennen IMBRUVICA-hoidon aloittamista; vaihtoehtoista hoitoa voidaan harkita.
Jos potilaalle kehittyy kammioperäisen takyarytmian oireita ja/tai löydöksiä, IMBRUVICA-hoito pitää keskeyttää tilapäisesti, ja on tehtävä perusteellinen kliininen hyöty-riskiarvio ennen kuin hoitoa voidaan mahdollisesti jatkaa.
Jos potilaalla on ennestään antikoagulanttihoitoa vaativa eteisvärinä, on harkittava muita hoitovaihtoehtoja IMBRUVICA-valmisteen sijasta. Jos potilaalle kehittyy eteisvärinä IMBRUVICA-hoidon aikana, tromboembolisen sairauden riski on arvioitava perusteellisesti. Jos riski on suuri ja vaihtoehtoiset hoidot eivät sovi IMBRUVICA-valmisteen sijasta, on harkittava tarkkaan kontrolloitua antikoagulanttihoitoa.
Potilaita pitää seurata IMBRUVICA-hoidon aikana sydämen vajaatoiminnan oireiden ja löydösten havaitsemiseksi. Sydämen vajaatoiminta on joissakin näistä tapauksista hävinnyt tai lieventynyt IMBRUVICA-hoidon lopettamisen tai annoksen pienentämisen jälkeen.
Aivoverisuonitapahtumat
IMBRUVICA-hoitoa saaneilla potilailla on raportoitu aivoverisuonitapahtumista, ohimenevistä aivoverenkiertohäiriöistä ja iskeemisistä aivohalvauksista, myös kuolemaan johtaneista tapauksista, joihin on tai ei ole liittynyt samanaikainen eteisvärinä ja/tai hypertensio. Niissä tapauksissa, joissa raportoitiin latenssi, iskeemisten keskushermoston verisuonisairauksien ilmeneminen IMBRUVICA-hoidon aloittamisen jälkeen kesti useimmiten useita kuukausia (yli kuukauden 78 prosentissa tapauksista ja yli kuusi kuukautta 44 prosentissa tapauksista). Tämän takia potilaita on seurattava säännöllisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet Sydämen rytmihäiriöt ja hypertensio sekä kohta Haittavaikutukset).
Tuumorilyysioireyhtymä
IMBRUVICA-hoidon yhteydessä on raportoitu tuumorilyysioireyhtymää. Tuumorilyysin suhteen riskipotilaita ovat ne, joiden kasvaintaakka on suuri ennen hoitoa. Potilaita pitää seurata tarkoin, ja tarkoituksenmukaisiin varotoimiin pitää ryhtyä.
Ei‑melanoottinen ihosyöpä
IMBRUVICA-hoitoa saaneilla potilailla raportoitiin ei-melanoottisia ihosyöpiä yleisemmin kuin vertailuvalmisteilla hoitoa saaneilla potilailla vaiheen 3 yhdistetyissä, satunnaistetuissa vertailututkimuksissa. Potilaita pitää seurata ei-melanoottisen ihosyövän ilmaantumisen havaitsemiseksi.
Hypertensio
IMBRUVICA-hoitoa saaneilla potilailla on esiintynyt hypertensiota (ks. kohta Haittavaikutukset). Seuraa IMBRUVICA-hoitoa saavan potilaan verenpainetta säännöllisesti, ja aloita verenpainelääkitys tai säädä sitä IMBRUVICA-hoidon aikana tarpeen mukaan.
Hemofagosyyttinen lymfohistiosytoosi (HLH)
IMBRUVICA-hoitoa saaneilla potilailla on raportoitu hemofagosyyttistä lymfohistiosytoosia (mukaan lukien kuolemaan johtaneita tapauksia). Hemofagosyyttinen lymfohistiosytoosi on henkeä uhkaava patologinen immuuniaktivaatio-oireyhtymä, jolle tyypillisiä kliinisiä oireita ja löydöksiä ovat erittäin voimakkaat systeemiset tulehdukset. Hemofagosyyttiselle lymfohistiosytoosille tyypillistä ovat kuume, pernan ja maksan suurentuneisuus, hypertriglyseridemia, seerumin suuri ferritiinipitoisuus ja sytopeniat. Potilaille pitää kertoa hemofagosyyttisen lymfohistiosytoosin oireet. Jos potilaalle kehittyy patologisen immuuniaktivaation varhaisia ilmenemismuotoja, potilas on tutkittava heti, ja hemofagosyyttisen lymfohistiosytoosin diagnoosi on huomioitava.
Lääkkeiden yhteisvaikutukset
IMBRUVICA-valmisteen ja voimakkaiden tai kohtalaisten CYP3A4:n estäjien samanaikainen käyttö saattaa suurentaa ibrutinibialtistusta ja lisätä siten toksisuusriskiä. CYP3A4:n induktorien samanaikainen käyttö saattaa sitä vastoin pienentää IMBRUVICA-altistusta, jolloin riskinä saattaa olla tehon puuttuminen. IMBRUVICA-valmisteen ja voimakkaiden CYP3A4:n estäjien ja voimakkaiden tai kohtalaisten CYP3A4:n induktorien samanaikaista käyttöä on siksi vältettävä aina kun se on mahdollista, ja samanaikaista käyttöä pitää harkita vain, jos mahdolliset hyödyt ovat selvästi mahdollisia riskejä suuremmat. Jos CYP3A4:n estäjien käyttö on välttämätöntä, potilasta pitää seurata tarkoin IMBRUVICA-valmisteesta aiheutuvan toksisuuden oireiden havaitsemiseksi (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset). Jos CYP3A4:n induktorien käyttö on välttämätöntä, potilasta pitää seurata tarkoin IMBRUVICA-valmisteen tehon puuttumisesta aiheutuvien oireiden havaitsemiseksi.
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisymenetelmää IMBRUVICA-hoidon aikana (ks. kohta Raskaus ja imetys).
Apuaineet, joiden vaikutus tunnetaan
Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Yksi kalvopäällysteinen tabletti sisältää alle 1 mmol natriumia (23 mg) eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Ibrutinibi metaboloituu ensisijaisesti sytokromi P450 ‑entsyymin 3A4 (CYP3A4) välityksellä.
Lääkeaineet, jotka saattavat suurentaa ibrutinibin pitoisuutta plasmassa
IMBRUVICA-valmisteen ja CYP3A4:ää voimakkaasti ja kohtalaisesti estävien lääkevalmisteiden samanaikainen käyttö voi suurentaa ibrutinibialtistusta, joten voimakkaita CYP3A4:n estäjiä pitää välttää.
Voimakkaat CYP3A4:n estäjät
Hyvin voimakkaan CYP3A4:n estäjän, ketokonatsolin, samanaikainen antaminen 18 paastonneelle, terveelle tutkittavalle suurensi altistusta ibrutinibille (Cmax-arvon 29-kertaiseksi ja AUC-arvon 24-kertaiseksi). Paastotilassa tehdyt simulaatiot viittasivat siihen, että voimakas CYP3A4:n estäjä klaritromysiini saattaa suurentaa ibrutinibin AUC-arvon 14-kertaiseksi. Voimakkaan CYP3A4:n estäjän, vorikonatsolin, samanaikainen anto IMBRUVICA-valmisteen ruokailun yhteydessä ottaville B-solusyöpää sairastaville potilaille suurensi Cmax-arvon 6,7-kertaiseksi ja AUC-arvon 5,7-kertaiseksi. Voimakkaita CYP3A4:n estäjiä (esim. ketokonatsolia, indinaviiria, nelfinaviiria, ritonaviiria, sakinaviiria, klaritromysiiniä, telitromysiiniä, itrakonatsolia, nefatsodonia, kobisistaattia, vorikonatsolia ja posakonatsolia) on vältettävä. Jos hyöty on riskejä suurempi ja jonkin voimakkaan CYP3A4:n estäjän käyttö on välttämätöntä, pienennä IMBRUVICA-annos 140 mg:aan estäjän käytön ajaksi tai keskeytä IMBRUVICA-hoito tilapäisesti (7 päiväksi tai lyhyemmäksi aikaa). Seuraa potilasta tarkoin toksisuuden havaitsemiseksi ja noudata tarvittaessa annosmuutoksista annettua ohjetta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kohtalaiset CYP3A4:n estäjät
CYP3A4:n estäjän, erytromysiinin, samanaikainen anto IMBRUVICA-valmisteen ruokailun yhteydessä ottaville B-solusyöpää sairastaville potilaille suurensi Cmax-arvon 3,4-kertaiseksi ja AUC-arvon 3,0-kertaiseksi. Jos kohtalaisen CYP3A4:n estäjän (esim. flukonatsoli, erytromysiini, amprenaviiri, aprepitantti, atatsanaviiri, siprofloksasiini, kritsotinibi, diltiatseemi, fosamprenaviiri, imatinibi, verapamiili, amiodaroni ja dronedaroni) käyttö on aiheellista, pienennä IMBRUVICA-annos 280 mg:aan estäjän käytön ajaksi. Seuraa potilasta tarkoin toksisuuden havaitsemiseksi ja noudata tarvittaessa annosmuutoksista annettua ohjetta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Heikot CYP3A4:n estäjät
Paastotilassa tehdyt simulaatiot viittasivat siihen, että heikot CYP3A4:n estäjät atsitromysiini ja fluvoksamiini saattavat suurentaa ibrutinibin AUC-arvon alle 2-kertaiseksi. Annosta ei tarvitse muuttaa, jos hoitoon yhdistetään heikkoja estäjiä. Seuraa potilasta tarkoin toksisuuden havaitsemiseksi ja noudata tarvittaessa annosmuutoksista annettua ohjetta.
Greippimehun (sisältää CYP3A4:n estäjiä) samanaikainen antaminen kahdeksalle terveelle tutkittavalle suurensi altistusta ibrutinibille (Cmax-arvon noin nelinkertaiseksi ja AUC-arvon noin kaksinkertaiseksi). Greippihedelmien ja pomeranssin syömistä IMBRUVICA-hoidon aikana pitää välttää, sillä ne sisältävät kohtalaisia CYP3A4:n estäjiä (ks. kohta Annostus ja antotapa).
Lääkeaineet, jotka saattavat pienentää ibrutinibin pitoisuutta plasmassa
IMBRUVICA-valmisteen ja CYP3A4:n induktorien samanaikainen käyttö voi pienentää ibrutinibipitoisuutta plasmassa.
Voimakkaan CYP3A4:n induktorin, rifampisiinin, samanaikainen antaminen 18 paastonneelle, terveelle tutkittavalle pienensi altistusta ibrutinibille (Cmax-arvoa 92 % ja AUC-arvoa 90 %). Vältä voimakkaiden tai kohtalaisten CYP3A4:n induktorien (esim. karbamatsepiinin, rifampisiinin, fenytoiinin) samanaikaista käyttöä. Mäkikuismaa sisältävät valmisteet ovat vasta-aiheisia IMBRUVICA-hoidon aikana, koska hoidon teho saattaa heikentyä. Harkitse vaihtoehtoisia lääkeaineita, jotka indusoivat CYP3A4:ää heikommin. Jos hyöty on riskejä suurempi ja jonkin voimakkaan tai kohtalaisen CYP3A4:n induktorin käyttö on välttämätöntä, seuraa potilasta tarkoin hoidon tehon puuttumisen havaitsemiseksi (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet). Heikkoja induktoreita voi käyttää samanaikaisesti IMBRUVICA-valmisteen kanssa, mutta potilaita on seurattava mahdollisen hoidon tehon puuttumisen havaitsemiseksi.
Ibrutinibin liukoisuus on pH:sta riippuvaista siten, että suuremman pH-arvon yhteydessä liukoisuus on vähäisempää. Kun terveille tutkittaville annettiin paastotilassa 560 mg:n kerta-annos ibrutinibia sen jälkeen, kun he olivat käyttäneet omepratsolia annoksina 40 mg kerran päivässä 5 päivän ajan, havaittiin pienempi huippupitoisuus (Cmax) (ks. kohta Farmakokinetiikka). Siitä ei ole näyttöä, että pienemmällä huippupitoisuudella olisi kliinistä merkitystä, ja keskeisissä kliinisissä tutkimuksissa mahalaukun pH:ta suurentavia lääkevalmisteita (esim. protonipumpun estäjiä) on käytetty ilman rajoituksia.
Lääkeaineet, joiden pitoisuutta plasmassa ibrutinibi saattaa muuttaa
Ibrutinibi on P-gp:n ja rintasyövän resistenssiproteiinin (breast cancer resistance protein, BCRP) estäjä in vitro. Koska tästä yhteisvaikutuksesta ei ole kliinisiä tietoja saatavissa, ei voida sulkea pois sitä, että ibrutinibi voi terapeuttisen annoksen jälkeen estää P-gp:tä ja BCRP:tä suolistossa. Jotta mahdollinen yhteisvaikutus maha-suolikanavassa voidaan minimoida, sellaiset suun kautta otettavat P-gp:n tai BCRP:n substraatit, joiden terapeuttinen leveys on pieni, kuten digoksiini tai metotreksaatti, on otettava vähintään 6 tuntia ennen IMBRUVICA-valmistetta tai 6 tuntia sen jälkeen. Ibrutinibi saattaa estää BCRP:tä myös maksassa ja lisätä altistusta sellaisille lääkevalmisteille, joihin kohdistuu maksassa BCRP-välitteinen ulospumppausvaikutus (effluksi), kuten rosuvastatiinille.
Tutkimuksissa, joissa kroonista lymfaattista leukemiaa sairastaville potilaille annettiin ibrutinibin (420 mg) ja venetoklaksin (400 mg) yhdistelmää, venetoklaksialtistuksen havaittiin suurentuneen (AUC:n perusteella noin 1,8-kertaiseksi) venetoklaksimonoterapiaa koskeviin tietoihin verrattuna.
B-solusyöpiä sairastavilla potilailla tehdyssä yhteisvaikutustutkimuksessa 560 mg:n ibrutinibikerta-annoksella ei ollut kliinisesti merkittävää vaikutusta CYP3A4:n substraatin midatsolaamin altistukseen. Samassa tutkimuksessa 2 viikon ibrutinibihoidolla 560 mg:n vuorokausiannoksina ei ollut kliinisesti oleellista vaikutusta ehkäisytablettien (etinyyliestradioli ja levonorgestreeli), CYP3A4:n substraatti midatsolaamin eikä CYP2B6:n substraatti bupropionin farmakokinetiikkaan.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi/Ehkäisy naisille
Eläimillä on todettu, että IMBRUVICA saattaa raskauden aikana käytettynä vahingoittaa sikiötä. Naisten pitää välttää raskaaksi tulemista IMBRUVICA-hoidon aikana ja 3 kuukautta hoidon päättymisen jälkeen. Naisten, jotka voivat tulla raskaaksi, on siksi käytettävä erittäin tehokasta ehkäisyä IMBRUVICA-hoidon aikana ja kolmen kuukauden ajan hoidon päättymisen jälkeen.
Raskaus
IMBRUVICA on vasta-aiheista raskauden aikana. IMBRUVICA-valmisteen käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imetys
Ei tiedetä, erittyvätkö ibrutinibi tai sen metaboliitit ihmisillä äidinmaitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. Imetys on lopetettava IMBRUVICA-hoidon ajaksi.
Hedelmällisyys
Uros- tai naarasrotilla ei havaittu vaikutuksia hedelmällisyyteen tai lisääntymiskykyyn suurimmallakaan testatulla annoksella 100 mg/kg/vrk (ihmisen vastaava annos [Human Equivalent Dose, HED] 16 mg/kg/vrk) (ks. kohta Prekliiniset tiedot turvallisuudesta). Ibrutinibin vaikutuksista ihmisen hedelmällisyyteen ei ole tietoa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
IMBRUVICA-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Joillakin IMBRUVICA-hoitoa saaneilla potilailla on raportoitu uupumusta, huimausta ja voimattomuutta, mikä pitää ottaa huomioon arvioitaessa potilaan ajokykyä tai koneidenkäyttökykyä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin esiintyneitä haittavaikutuksia (≥ 20 %,) olivat ripuli, neutropenia, tuki- ja liikuntaelinperäinen kipu, verenvuodot (esim. mustelmat), ihottuma, pahoinvointi, trombosytopenia, nivelsärky ja ylähengitystieinfektio. Yleisimpiä asteen 3/4 haittavaikutuksia (≥ 5 %) olivat neutropenia, lymfosytoosi, trombosytopenia, hypertensio ja keuhkokuume.
Haittavaikutustaulukko
Ibrutinibihoitoa B-solusyöpien hoitoon saaneilla potilailla esiintyneet ja valmisteen markkinoille tulon jälkeen ilmenneet haittavaikutukset luetellaan seuraavassa elinjärjestelmän ja esiintymistiheyden mukaan ryhmiteltyinä. Esiintyvyydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
B-solusyöpiä koskeva yhteenveto
Turvallisuusprofiili perustuu neljässä vaiheen 2 kliinisessä tutkimuksessa ja kahdeksassa satunnaistetussa vaiheen 3 tutkimuksessa IMBRUVICA-hoitoa saaneen 1 981 potilaan yhdistettyihin tietoihin sekä valmisteen markkinoilletulon jälkeisiin kokemuksiin. Yhdistetyissä tiedoissa ei ole mukana TRIANGLE-tutkimuksen tietoja ja ne esitetään erikseen taulukossa 3. Kliinisissä tutkimuksissa manttelisolulymfoomaan hoitoa saaneet potilaat saivat IMBRUVICA-valmistetta 560 mg kerran vuorokaudessa, ja kliinisissä tutkimuksissa krooniseen lymfaattiseen leukemiaan tai Waldenströmin makroglobulinemiaan hoitoa saaneet potilaat saivat IMBRUVICA-valmistetta 420 mg kerran vuorokaudessa. Kaikki kliinisiin tutkimuksiin osallistuneet potilaat saivat IMBRUVICA-valmistetta, kunnes tauti eteni tai potilas ei enää sietänyt hoitoa; poikkeuksena tästä olivat IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää koskeneet tutkimukset, joissa potilaiden hoitojakso oli määräaikainen (tutkimukset CLL3011 ja PCYC-1142-CA). IMBRUVICA-hoidon keston mediaani yhdistetyssä tietoaineistossa oli 14,7 kuukautta. Krooniseen lymfaattiseen leukemiaan / pienilymfosyyttiseen lymfoomaan annetun hoidon keston mediaani oli 14,7 kuukautta (enintään 52 kuukautta), manttelisolulymfooman hoidossa se oli 11,7 kuukautta (enintään 28 kuukautta) ja Waldenströmin makroglobulinemian hoidossa se oli 21,6 kuukautta (enintään 37 kuukautta).
| Taulukko 2: B-solusyöpiä sairastavilla potilailla kliinisissä tutkimuksissa tai valmisteen markkinoille tulon jälkeisessä seurannassa raportoidut haittavaikutukset† | ||||
| Elinjärjestelmä | Esiintyvyys (kaikki asteet) | Haittavaikutus | Kaikki asteet (%) | Aste ≥ 3 (%) |
| Infektiot | Hyvin yleinen | Keuhkokuume*# Ylempien hengitysteiden infektio Ihoinfektio* | 12 21 15 | 7 1 2 |
| Yleinen | Sepsis*# Virtsatieinfektio Sinuiitti* | 3 9 9 | 3 1 1 | |
| Melko harvinainen | Kryptokokki-infektiot* | < 1 | 0 | |
| Pneumocystis-infektiot* # | < 1 | < 1 | ||
| Aspergillus-infektiot* | < 1 | < 1 | ||
| Hepatiitti B ‑viruksen reaktivaatio@ # | < 1 | < 1 | ||
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Yleinen | Ei-melanoottinen ihosyöpä* Tyvisolusyöpä Okasolusyöpä | 5 3 1 | 1 < 1 < 1 |
| Veri ja imukudos | Hyvin yleinen | Neutropenia* Trombosytopenia* Lymfosytoosi* | 39 29 15 | 31 8 11 |
| Yleinen | Kuumeinen neutropenia Leukosytoosi | 4 4 | 4 4 | |
| Harvinainen | Leukostaasioireyhtymä | < 1 | < 1 | |
| Immuunijärjestelmä | Yleinen | Interstitiaalinen keuhkosairaus*,# | 2 | < 1 |
| Aineenvaihdunta ja ravitsemus | Yleinen | Hyperurikemia | 9 | 1 |
| Melko harvinainen | Tuumorilyysioireyhtymä | 1 | 1 | |
| Hermosto | Hyvin yleinen | Huimaus Päänsärky | 12 19 | < 1 1 |
| Yleinen | Perifeerinen neuropatia* | 7 | < 1 | |
| Melko harvinainen | Aivoverisuonitapahtuma# Ohimenevä aivoverenkiertohäiriö Iskeeminen aivohalvaus# | < 1 < 1 < 1 | < 1 < 1 < 1 | |
| Silmät | Yleinen | Näön sumeneminen | 6 | 0 |
| Melko harvinainen | Silmäverenvuoto‡ Uveiitti* | < 1 < 1 | 0 0 | |
| Sydän | Yleinen | Sydämen vajaatoiminta*, # Eteisvärinä | 2 8 | 1 4 |
| Melko harvinainen | Kammioperäinen takyarytmia*,# Sydämenpysähdys# | 1 < 1 | < 1 < 1 | |
| Verisuonisto | Hyvin yleinen | Verenvuoto*# Mustelmat* Hypertensio* | 35 27 18 | 1 < 1 8 |
| Yleinen | Nenäverenvuoto Petekiat | 9 7 | < 1 0 | |
| Melko harvinainen | Subduraalihematooma# | 1 | < 1 | |
| Ruoansulatuselimistö | Hyvin yleinen | Ripuli Oksentelu Stomatiitti* Pahoinvointi Ummetus Dyspepsia | 47 15 17 31 16 11 | 4 1 1 1 < 1 < 1 |
| Maksa ja sappi | Melko harvinainen | Maksan vajaatoiminta*,# | < 1 | < 1 |
| Iho ja ihonalainen kudos | Hyvin yleinen | Ihottuma* | 34 | 3 |
| Yleinen | Nokkosihottuma Punoitus Kynsien haurastuminen | 1 3 4 | < 1 < 1 0 | |
| Melko harvinainen | Angioedeema Pannikuliitti* Neutrofiiliset dermatoosit* Pyogeeninen granulooma Ihovaskuliitti | < 1 < 1 < 1 < 1 < 1 | < 1 < 1 < 1 0 0 | |
| Harvinainen | Stevens–Johnsonin oireyhtymä | < 1 | < 1 | |
| Luusto, lihakset ja sidekudos | Hyvin yleinen | Nivelkipu Lihasspasmit Tuki- ja liikuntaelinperäinen kipu* | 24 15 36 | 2 < 1 3 |
| Munuaiset ja virtsatiet | Yleinen | Akuutti munuaisvaurio# | < 2 | < 1 |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Kuume Perifeerinen turvotus | 19 16 | 1 1 |
| Tutkimukset | Hyvin yleinen | Suurentunut veren kreatiniinipitoisuus | 10 | < 1 |
† Esiintyvyydet on pyöristetty lähimpään kokonaislukuun. * Sisältää useita haittavaikutustermejä. ‡ Joihinkin tapauksiin liittyy näönmenetys. # Sisältää kuolemaan johtaneita tapahtumia. @ Valikoiduista haittavaikutuksista käytetty alemman tason termi. | ||||
Yhteenveto aiemmin hoitamatonta manttelisolulymfoomaa sairastavista potilaista, jotka soveltuivat autologiseen kantasolusiirtoon
Turvallisuusprofiili perustuu vaiheen 3 TRIANGLE-tutkimuksessa IMBRUVICA-hoitoa saaneista 265 potilaasta (IMBRUVICA-ryhmä) saatuihin tietoihin. Potilaat saivat 560 mg IMBRUVICA-valmistetta kerran vuorokaudessa TRIANGLE-hoito-ohjelman mukaisesti (ks. kohta Farmakodynamiikka). IMBRUVICA-ryhmässä hoidon keston mediaani oli 28,5 kuukautta.
Taulukko 3: TRIANGLE-tutkimuksen IMBRUVICA-ryhmässä raportoidut haittavaikutukset† | ||||
|---|---|---|---|---|
| N = 265 | |||
Elinjärjestelmä | Esiintyvyys | Haittavaikutus | Kaikki asteet (%) | Aste ≥ 3 (%) |
| Infektiot | Hyvin yleinen | Keuhkokuume* # | 16 | 9 |
| Ihoinfektio* | 12 | 3 | ||
Yleinen
| Ylempien hengitysteiden infektio | 6 | < 1 | |
| Sepsis* | 2 | 2 | ||
| Virtsatieinfektio | 6 | < 1 | ||
| Sinuiitti* | 6 | 1 | ||
| Melko harvinainen | Aspergillus-infektiot* | 1 | < 1 | |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Yleinen | Ei-melanoottinen ihosyöpä* | 1 | < 1 |
| Tyvisolusyöpä | 1 | < 1 | ||
| Veri ja imukudos | Hyvin yleinen | Trombosytopenia* | 69 | 61 |
| Neutropenia* | 63 | 60 | ||
| Kuumeinen neutropenia | 14 | 14 | ||
| Yleinen | Leukosytoosi | 3 | 1 | |
| Immuunijärjestelmä | Yleinen | Interstitiaalinen keuhkosairaus* | 5 | 1 |
| Aineenvaihdunta ja ravitsemus | Yleinen | Hyperurikemia | 8 | 3 |
| Tuumorilyysioireyhtymä* | 3 | 3 | ||
| Hermosto | Hyvin yleinen | Perifeerinen neuropatia* | 35 | 3 |
| Päänsärky | 11 | 1 | ||
| Yleinen | Huimaus | 6 | < 1 | |
| Melko harvinainen | Ohimenevä aivoverenkiertohäiriö | 1 | 0 | |
| Silmät | Melko harvinainen | Näön sumeneminen | 1 | 0 |
| Silmäverenvuoto | < 1 | 0 | ||
| Sydän | Yleinen | Eteisvärinä | 10 | 4 |
| Sydämen vajaatoiminta* | 2 | 0 | ||
| Verisuonisto | Hyvin yleinen | Verenvuoto* | 14 | 2 |
| Hypertensio* | 14 | 5 | ||
| Yleinen | Mustelmat* | 8 | 1 | |
| Nenäverenvuoto | 6 | 1 | ||
| Petekiat | 3 | 0 | ||
| Ruoansulatuselimistö | Hyvin yleinen | Pahoinvointi | 32 | 4 |
| Ripuli | 28 | 5 | ||
| Oksentelu | 18 | 4 | ||
| Stomatiitti* | 11 | 2 | ||
| Ummetus | 17 | < 1 | ||
| Yleinen | Dyspepsia | 8 | 0 | |
| Iho ja ihonalainen kudos | Hyvin yleinen | Ihottuma* | 23 | 2 |
| Yleinen | Punoitus | 5 | 0 | |
| Kynsien haurastuminen | 2 | 0 | ||
| Melko harvinainen | Nokkosihottuma | < 1 | 0 | |
| Angioedeema | 1 | 0 | ||
| Ihovaskuliitti | < 1 | 0 | ||
| Pannikuliitti* | 1 | 0 | ||
| Luusto, lihakset ja sidekudos | Hyvin yleinen | Tuki- ja liikuntaelinperäinen kipu* | 19 | 2 |
| Yleinen | Lihasspasmit | 9 | 1 | |
| Nivelkipu | 8 | 1 | ||
| Munuaiset ja virtsatiet | Hyvin yleinen | Akuutti munuaisvaurio | 11 | 5 |
| Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Kuume | 22 | 2 |
| Yleinen | Perifeerinen turvotus | 5 | 0 | |
| Tutkimukset | Hyvin yleinen | Suurentunut veren kreatiniinipitoisuus | 16 | 1 |
† Esiintyvyydet on pyöristetty lähimpään kokonaislukuun. * Termit edellyttivät ryhmittelyä. # Sisältää kuolemaan johtaneita tapahtumia. | ||||
Valikoitujen haittavaikutusten kuvaus
Hoidon lopettaminen ja annoksen pienentäminen haittavaikutusten vuoksi
B-solusyöpien hoitoon IMBRUVICA-valmistetta saaneista 1 981 potilaasta 6 % lopetti hoidon pääasiassa haittavaikutusten vuoksi. Tällaisia haittavaikutuksia olivat keuhkokuume, eteisvärinä, neutropenia, ihottuma, trombosytopenia ja verenvuodot. Annoksen pienentämiseen johtaneita haittavaikutuksia esiintyi noin 8 %:lla potilaista. Vaiheen 3 TRIANGLE-tutkimuksessa oli mukana 265 aiemmin hoitamatonta manttelisolulymfoomaa sairastavaa potilasta, jotka soveltuivat autologiseen kantasolusiirtoon, ja siinä havaittiin, että 13 % IMBRUVICA-ryhmän potilaista lopetti hoidon haittavaikutusten vuoksi. Tällaisia haittavaikutuksia olivat neutropenia, keuhkokuume, eteisvärinä, akuutti munuaisvaurio, ripuli, ihottuma ja interstitiaalinen keuhkosairaus. Annoksen pienentämiseen johtaneita haittavaikutuksia ilmeni IMBRUVICA-ryhmässä noin 12 %:lla potilaista.
Iäkkäät
IMBRUVICA-hoitoa saaneista 1 981 potilaasta 50 % oli 65-vuotiaita tai vanhempia.
Asteen 3 tai vaikeampiasteista keuhkokuumetta (11 %:lla ≥ 65-vuotiaista potilaista verrattuna 4 %:iin < 65-vuotiaista potilaista) ja trombosytopeniaa (11 %:lla ≥ 65-vuotiaista potilaista verrattuna 5 %:iin < 65-vuotiaista potilaista) esiintyi yleisemmin iäkkäillä IMBRUVICA-hoitoa saaneilla potilailla.
Pitkäaikaisturvallisuus
Viiden vuoden ajalta 1 284 potilaasta saadut pitkäkestoisen IMBRUVICA-hoidon turvallisuutta koskevat tiedot (aiemmin hoitamaton krooninen lymfaattinen leukemia / pienilymfosyyttinen lymfooma n = 162, uusiutunut/hoitoon reagoimaton krooninen lymfaattinen leukemia / pienilymfosyyttinen lymfooma n = 646, uusiutunut / hoitoon reagoimaton manttelisolulymfooma n = 370 ja Waldenströmin makroglobulinemia n = 106) analysoitiin. Krooniseen lymfaattiseen leukemiaan / pienilymfosyyttiseen lymfoomaan annetun hoidon keston mediaani oli 51 kuukautta (vaihteluväli 0,2–98 kuukautta). 70 % potilaista sai hoitoa yli 2 vuoden ajan, ja 52 % potilaista sai hoitoa yli 4 vuoden ajan. Manttelisolulymfoomaan annetun hoidon keston mediaani oli 11 kuukautta (vaihteluväli 0–87 kuukautta). 31 % potilaista sai hoitoa yli 2 vuoden ajan, ja 17 % potilaista sai hoitoa yli 4 vuoden ajan. Waldenströmin makroglobulinemiaan annetun hoidon keston mediaani oli 47 kuukautta (vaihteluväli 0,3–61 kuukautta). 78 % potilaista sai hoitoa yli 2 vuoden ajan, ja 46 % potilaista sai hoitoa yli 4 vuoden ajan. IMBRUVICA-valmisteelle altistuneiden potilaiden tunnettu kokonaisturvallisuusprofiili pysyi yhdenmukaisena, paitsi että hypertension vallitsevuus lisääntyi. Uusia turvallisuutta koskevia huolenaiheita ei tunnistettu. 3. asteen tai vaikeampiasteisen hypertension vallitsevuus oli 4 % (vuosina 0–1), 7 % (vuosina 1–2), 9 % (vuosina 2–3), 9 % (vuosina 3–4) ja 9 % (vuosina 4–5). Viiden vuoden aikana sen kokonaisilmaantuvuus oli 11 %.
Pediatriset potilaat
Turvallisuusarvio perustuu tietoihin, jotka saatiin vaiheen 3 tutkimuksesta. Siinä uusiutunutta tai hoitoon reagoimatonta kypsää B-soluista non-Hodgkin-lymfoomaa sairastaville pediatrisille tai nuorille aikuisille potilaille (iältään 3–19 vuotta) annettiin IMBRUVICA-valmisteen ja runkohoidon eli joko rituksimabista, ifosfamidista, karboplatiinista, etoposidista ja deksametasonista koostuvan hoito-ohjelman (RICE) tai rituksimabista, vinkristiinistä, ifosfamidista, karboplatiinista, idarubisiinista ja deksametasonista koostuvan hoito-ohjelman (RVICI) yhdistelmää tai pelkästään runkohoitoa (ks. kohta Farmakodynamiikka). Tässä tutkimuksessa ei havaittu uusia haittavaikutuksia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
IMBRUVICA-yliannoksen vaikutuksista on vähän tietoa. Vaiheen 1 tutkimuksessa, jossa potilaat saivat enimmillään annoksen 12,5 mg/kg/vrk (1 400 mg/vrk), ei saavutettu suurinta siedettyä annosta. Eräässä toisessa tutkimuksessa yhdelle terveelle tutkittavalle, joka sai annoksen 1 680 mg, ilmaantui korjautuvaa asteen 4 maksaentsyymipitoisuuksien suurenemista (aspartaattiaminotransferaasi [ASAT] ja alaniiniaminotransferaasi [ALAT]). IMBRUVICA-valmisteelle ei ole spesifistä vasta-ainetta. Jos potilas nielee suositeltua annosta suuremman määrän lääkettä, häntä on tarkkailtava ja hänelle on annettava elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Solunsalpaajat, proteiinikinaasin estäjät, ATC-koodi: L01EL01.
Vaikutusmekanismi
Ibrutinibi on voimakas Brutonin tyrosiinikinaasin (BTK) pienimolekyylinen estäjä. Ibrutinibi muodostaa kovalenttisen sidoksen BTK:n aktiivisen kohdan kysteiinitähteeseen (Cys481), mikä johtaa BTK:n entsymaattisen aktiivisuuden pitkäkestoiseen estymiseen. BTK kuuluu Tec-kinaasiryhmään ja on B-soluantigeenireseptori- (BCR) ja sytokiinireseptorireittien tärkeä signalointimolekyyli. BCR-reitti liittyy useiden B-solusyöpien, kuten manttelisolulymfooman, diffuusin suurisoluisen B-solulymfooman, follikulaarisen lymfooman ja kroonisen lymfaattisen leukemian, patogeneesiin. BTK:n keskeinen rooli signaalinvälityksessä B-solujen pinnalla sijaitsevien reseptorien kautta johtaa B-soluliikenteelle, kemotaksikselle ja adheesiolle välttämättömien reittien aktivoitumiseen. Prekliinisissä tutkimuksissa on osoitettu, että ibrutinibi estää tehokkaasti pahanlaatuisten B-solujen proliferaation ja eloonjäännin in vivo sekä solujen migraation ja substraattien adheesion in vitro.
Ibrutinibin ja venetoklaksin yhdistelmä lisäsi prekliinisissä kasvainmalleissa syöpäsolujen apoptoosia ja esti kasvaimen kasvua enemmän kuin kumpikaan vaikuttava aine yksinään. Ibrutinibin aiheuttama BTK:n estyminen lisää kroonisen lymfaattisen leukemian tautisolujen riippuvuutta BCL‑2:sta, joka on solujen eloonjäämiseen liittyvä reaktiotie, kun taas venetoklaksi estää BCL‑2:ta ja siten aiheuttaa syöpäsolujen apoptoosin.
Lymfosytoosi
Noin kolmella potilaalla neljästä kroonista lymfaattista leukemiaa sairastavasta IMBRUVICA-hoitoa saaneesta potilaasta on havaittu hoidon aloittamisen jälkeen palautuva lymfosyyttimäärän lisääntyminen (eli ≥ 50 %:n lisääntyminen hoitoa edeltävästä tilanteesta ja absoluuttinen määrä > 5 000/mikrol), mihin liittyi usein lymfadenopatian väheneminen. Tällainen vaikutus on havaittu myös noin kolmanneksella uusiutunutta tai hoitoon reagoimatonta manttelisolulymfoomaa sairastavista IMBRUVICA-hoitoa saaneista potilaista. Havaittu lymfosytoosi on farmakodynaaminen vaikutus eikä sitä pidä katsoa taudin etenemiseksi, jos potilaalla ei ole muita kliinisiä löydöksiä. Lymfosytoosi ilmaantuu kummassakin tautityypissä tyypillisesti ensimmäisen IMBRUVICA-hoitokuukauden aikana ja häviää manttelisolulymfoomaa sairastavilla potilailla tyypillisesti 8,0 viikon (mediaani) kuluessa ja kroonista lymfaattista leukemiaa sairastavilla potilailla 14 viikon kuluessa. Joidenkin potilaiden verenkierrossa on havaittu lymfosyyttimäärän huomattavaa suurenemista (esim. > 400 000/mikrol).
Lymfosytoosia ei havaittu IMBRUVICA-hoitoa saaneilla Waldenströmin makroglobulinemiaa sairastavilla potilailla.
Trombosyyttiaggregaatio in vitro
Ibrutinibilla todettiin in vitro ‑tutkimuksessa kollageenin indusoiman trombosyyttiaggregaation estymistä. Ibrutinibilla ei todettu merkittävää trombosyyttiaggregaation estymistä käytettäessä muita trombosyyttiaggregaation aktivaattoreita.
Vaikutus QT/QTc‑aikaan ja sydämen sähköfysiologiaan
Ibrutinibin vaikutusta QTc‑aikaan selvitettiin 20 terveellä miehellä ja naisella tehdyssä satunnaistetussa, kaksoissokkoutetussa, kattavassa QT‑tutkimuksessa, jossa tutkittavat saivat lumevalmistetta tai vaikuttavaa hoitoa. Terapeuttisia annoksia suuremmat 1 680 mg:n ibrutinibiannokset eivät pidentäneet QTc‑aikaa kliinisesti oleellisesti. Ibrutinibin ja lumelääkkeen lähtötilanteen suhteen korjatun keskimääräisen eron kaksitahoisen 90 %:n luottamusvälin suurin yläraja oli alle 10 ms. Samassa tutkimuksessa havaittiin pitoisuudesta riippuvaista QTc‑ajan lyhenemistä (‑5,3 ms [90 %:n luottamusväli: ‑9,4; ‑1,1], kun huippupitoisuus [Cmax] oli terapeuttista annosta suuremman 1 680 mg:n annoksen yhteydessä 719 ng/ml).
Kliininen teho ja turvallisuus
Manttelisolulymfooma
Yhdistelmähoito aiemmin hoitamatonta manttelisolulymfoomaa sairastavilla potilailla, jotka soveltuivat autologiseen kantasolusiirtoon
IMBRUVICA-valmisteen turvallisuutta ja tehoa potilaille, joilla oli aiemmin hoitamaton manttelisolulymfooma ja jotka soveltuivat autologiseen kantasolusiirtoon, arvioitiin satunnaistetussa, vaiheen 3 avoimessa, kolmen ryhmän monikeskustutkimuksessa (TRIANGLE). TRIANGLE-tutkimuksessa satunnaistettiin 870 potilasta suhteessa 1:1:1 saamaan jotakin seuraavista hoidoista:
- IMBRUVICA-ryhmä: induktiohoitona 560 mg IMBRUVICA-valmistetta päivittäin (päivät 1–19) yhdistelmänä R‑CHOP-hoidon kanssa kolmen 21 päivän pituisen hoitosyklin ajan (hoitosyklit 1, 3, 5), jotka vuorottelivat kolmen 21 päivän pituisen R‑DHAP-hoitosyklin kanssa (hoitosyklit 2, 4, 6), minkä jälkeen 2 vuoden ajan 560 mg IMBRUVICA-valmistetta päivittäin
- IMBRUVICA-hoidon + autologisen kantasolusiirron ryhmä: induktiohoitona 560 mg IMBRUVICA-valmistetta päivittäin (päivät 1–19) yhdistelmänä R‑CHOP-hoidon kanssa kolmen 21 päivän pituisen hoitosyklin ajan (hoitosyklit 1, 3, 5), jotka vuorottelivat kolmen 21 päivän pituisen R‑DHAP-hoitosyklin kanssa (hoitosyklit 2, 4, 6), minkä jälkeen suuriannoksinen solunsalpaajahoito ja autologinen kantasolusiirto ja sen jälkeen 2 vuoden ajan 560 mg IMBRUVICA-valmistetta päivittäin
- autologisen kantasolusiirron ryhmä: induktiohoitona R‑CHOP-hoito kolmen 21 päivän pituisen hoitosyklin ajan (hoitosyklit 1, 3, 5), jotka vuorottelivat kolmen 21 päivän pituisen R‑DHAP-hoitosyklin kanssa (hoitosyklit 2, 4, 6), minkä jälkeen suuriannoksinen solunsalpaajahoito ja autologinen kantasolusiirto (vertailuryhmä).
Tehon analyysit tehtiin koko analyysitietueen 809 potilaan potilasjoukosta 3 hoitoryhmän pareittaisina vertailuina: IMBRUVICA + autologinen kantasolusiirto vs. autologinen kantasolusiirto, IMBRUVICA vs. autologinen kantasolusiirto ja IMBRUVICA + autologinen kantasolusiirto vs. IMBRUVICA. Koko analyysitietueen potilasjoukossa oli potilaita, jotka olivat joko antaneet nimenomaisen suostumuksen tietojensa sisällyttämiseen EU:n yleisen tietosuoja-asetuksen (EU General Data Protection Regulation) mukaisesti tai olivat kuolleet. Esitettävät tiedot on saatu vain IMBRUVICA (N = 265) ‑ryhmästä ja autologisen kantasolusiirron (N = 268) ryhmästä.
R-CHOP‑induktiohoito (375 mg/m2 rituksimabia päivänä 0 tai 1, 750 mg/m2 syklofosfamidia päivänä 1, 50 mg/m2 doksorubisiinia päivänä 1, 1,4 mg/m2 ja enintään 2 mg vinkristiiniä päivänä 1 ja 100 mg prednisonia päivinä 1–5), joka vuorotteli R-DHAP-hoidon (375 mg/m2 rituksimabia päivänä 0 tai 1, 40 mg deksametasonia päivinä 1–4, 2 x 2 g/m2 sytarabiinia 12 tunnin välein päivänä 2, 100 mg/m2 sisplatiinia [vaihtoehtoisesti 130 mg/m² oksaliplatiinia] päivänä 1 ja 5 mikrog/kg granulosyyttiryhmiä stimuloivaa kasvutekijää päivästä 6 alkaen veren valkosolumäärän korjautumiseen saakka) kanssa oli sama kaikissa kolmessa hoitoryhmässä. Rituksimabiylläpitohoito oli sallittu kaikissa hoitoryhmissä (59,7 % IMBRUVICA-ryhmässä, 62,5 % autologisen kantasolusiirron ryhmässä) kansallisten hoitosuositusten mukaisesti.
Iän mediaani oli 57 vuotta (vaihteluväli: 27–65 vuotta), 78 % oli miehiä ja 99 % oli valkoihoisia. Yhdeksälläkymmenelläkahdeksalla prosentilla potilaista ECOG-toimintakykyluokka oli lähtötilanteessa 0 tai 1. Lähtötilanteessa 86 %:lla oli Ann Arbor ‑levinneisyysluokan IV sairaus; 57 %:lla oli pienen riskin, 28 %:lla oli keskisuuren riskin ja 15 %:lla oli suuren riskin MIPI-riskipisteytys (MCL International Prognostic Index). Potilaista 11,6 %:lla oli blastoidinen tai pleomorfinen histologia. P53:n ilmentymä arvioitiin 64,6 %:lta potilaista; ilmentymä > 50 % todettiin 14,1 %:lla näistä potilaista. Ki-67:n proliferaatioindeksi arvioitiin 88,3 %:lta potilaista, ja 32,9 %:lla näistä potilaista proliferaatioindeksi oli > 30 %.
Vastetta kasvaimessa arvioitiin päivitettyjen IWG:n (International Working Group) non‑Hodgkin-lymfoomaa (NHL) koskevien kriteerien (2007) perusteella. Ensisijainen päätetapahtuma oli elossaoloaika ilman hoidon epäonnistumista (failure-free survival, FFS), jonka määritelmä oli satunnaistamisesta induktiohoitona annetun kemoimmunoterapian päättyessä todettuun vakaaseen tautiin, sairauden etenemiseen tai mistä syystä tahansa tapahtuneeseen kuolemaan kulunut aika, sen mukaan, mikä näistä toteutui ensimmäisenä.
TRIANGLE-tutkimuksen tehon tulokset 54,9 kuukauden (mediaani) seurannan ajankohtana esitetään taulukossa 4. Elossaoloaikaa ilman hoidon epäonnistumista koskeva Kaplan–Meier-käyrä esitetään kuvassa 1 ja kokonaiselossaoloajan Kaplan–Meier-käyrä esitetään kuvassa 2.
| Taulukko 4: Aiemmin hoitamatonta manttelisolulymfoomaa sairastavien potilaiden tehon tulokset (TRIANGLE) (koko analyysitietueen potilasjoukko) | ||
| Päätetapahtuma | IMBRUVICA-ryhmä N = 268 | Autologisen kantasolusiirron ryhmä N = 269 |
| Elossaoloaika ilman hoidon epäonnistumista± | ||
| Tapahtumien lukumäärä (%) | 61 (22,8 %) | 87 (32,3 %) |
| Vakaa sairaus induktiohoidon päättyessä | 1 (0,4 %) | 5 (1,9 %) |
| Sairauden eteneminen | 49 (18,3 %) | 60 (22,3 %) |
| Kuolemaan liittyviä tapahtumia | 11 (4,1 %) | 22 (8,2 %) |
| Mediaani (95 %:n luottamusväli), kuukautta | NE (NE; NE) | NE (NE; NE) |
IMBRUVICA- vs. autologisen kantasolusiirron ryhmä (p-arvo)* | 0,639 (0,428; 0,953) (0,0068) | |
| Kokonaiselossaolo§ | ||
| Kuolemien lukumäärä (%) | 33 (12,3 %) | 60 (22,3 %) |
| IMBRUVICA- vs. autologisen kantasolusiirron ryhmä Riskisuhde (HR) (95 %:n luottamusväli) (p-arvo)* | 0,522 (0,341; 0,799) (0,0023) | |
Kokonaisvasteluku (%)§ (95 %:n luottamusväli) | 258 (96,3 %) (93,3 %; 98,2 %) | 248 (92,2 %) (88,3 %; 95,1 %) |
Täydellinen vaste (%)§ (95 %:n luottamusväli) | 180 (67,2 %) (61,2 %; 72,8 %) | 174 (64,7 %) (58,7 %; 70,4 %) |
NE = ei arvioitavissa (not estimable); HR = riskisuhde (hazard ratio) (perustuu osittamattomaan Coxin regressiomalliin); RR = suhteellinen riski (relative risk) ±Elossaoloaikaa ilman hoidon epäonnistumista koskevia tuloksia ei ole vakioitu tyypin 1 virheen suhteen, sillä nämä analyysit on saatu myyntilupatarkoituksiin tehdyistä täydentävistä analyyseista. *Kaksitahoiset p-arvot on saatu osittamattomalla log-rank-testillä; p-arvojen testauksen peruste oli p < 0,0167. §Esitetyt tulokset on saatu deskriptiivisestä analyysista. | ||
Kuva 1: TRIANGLE-tutkimuksessa European MCL Networkin arvioimaa elossaoloaikaa ilman hoidon epäonnistumista koskeva Kaplan–Meier-käyrä (koko analyysitietueen potilasjoukko)*
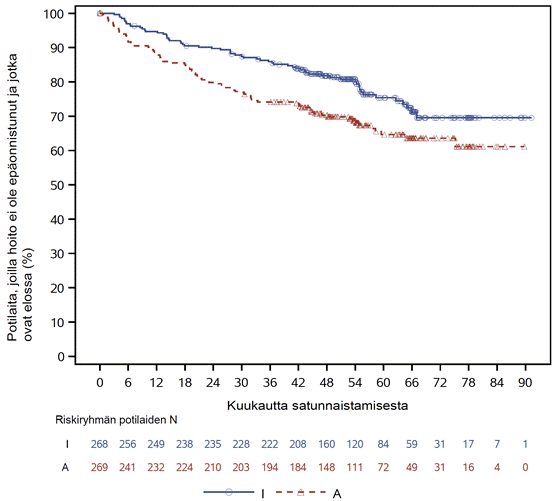
* I = IMBRUVICA; A = autologinen kantasolusiirto
Kuva 2: TRIANGLE-tutkimuksen kokonaiselossaoloa§ koskeva Kaplan–Meier-käyrä (koko analyysitietueen potilasjoukko)*
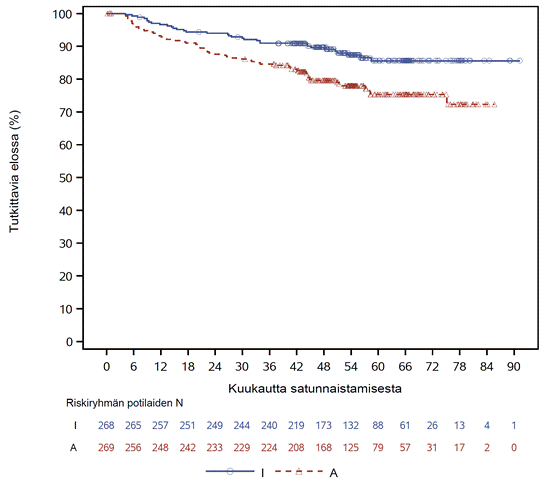
*I = IMBRUVICA; A = autologinen kantasolusiirto
§Esitetyt tulokset on saatu deskriptiivisestä analyysista
Manttelisolulymfoomaa sairastavat potilaat, jotka olivat saaneet vähintään yhtä aiempaa hoitoa
Monoterapia
IMBRUVICA-valmisteen turvallisuutta ja tehoa uusiutunutta tai hoitoon reagoimatonta manttelisolulymfoomaa sairastavilla potilailla arvioitiin 111 potilaalla yhdessä avoimessa vaiheen 2 monikeskustutkimuksessa (PCYC‑1104‑CA). Potilaiden iän mediaani oli 68 vuotta (vaihteluväli: 40–84 vuotta), ja miehiä oli 77 % potilaista ja valkoihoisia 92 %. Potilasta ei otettu mukaan tutkimukseen, jos ECOG‑suorituskykyluokka oli 3 tai suurempi. Ajan mediaani diagnoosista oli 42 kuukautta, ja aiempien hoitokertojen lukumäärän mediaani oli 3 (vaihteluväli: 1–5 hoitoa). Aiempia hoitoja olivat suuriannoksinen solunsalpaajahoito 35 %:lla, bortetsomibihoito 43 %:lla, lenalidomidihoito 24 %:lla ja autologinen tai allogeeninen kantasolusiirto 11 %:lla potilaista. Lähtötilanteen seulonnassa 39 %:lla potilaista oli suurikokoinen kasvain (≥ 5 cm), 49 %:lla oli suuren riskin tautia osoittavat MIPI-pisteet (Simplified MCL International Prognostic Index, MIPI) ja 72 %:lla oli pitkälle edennyt tauti (tauti levinnyt imusolmukkeiden ulkopuolelle ja/tai luuytimeen).
IMBRUVICA-hoitoa annettiin suun kautta 560 mg:n annoksina kerran vuorokaudessa, kunnes tauti eteni tai potilaalle ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Kasvaimessa todettua vastetta arvioitiin muokattujen non-Hodgkin-lymfoomaa koskevien International Working Group (IWG) ‑kriteerien perusteella. Tämän tutkimuksen ensisijainen päätetapahtuma oli tutkijan arvioima kokonaisvasteluku (overall response rate, ORR). Vasteet IMBRUVICA-hoitoon esitetään taulukossa 5.
| Taulukko 5: Uusiutunutta tai hoitoon reagoimatonta manttelisolulymfoomaa sairastavien potilaiden kokonaisvasteluku (ORR) ja vasteen kestoaika (duration of response, DOR) (tutkimus PCYC‑1104‑CA) | |
Yhteensä N = 111 | |
| ORR (%) | 67,6 |
| 95 %:n luottamusväli (%) | (58,0; 76,1) |
| Täydellinen vaste (CR) (%) | 20,7 |
| Osittainen vaste (PR) (%) | 46,8 |
| Vasteen kestoajan mediaani (CR+PR) (kuukautta) | 17,5 (15,8; NR) |
| Ajan mediaani vasteen alkamiseen, kuukautta (vaihteluväli) | 1,9 (1,4–13,7) |
| Ajan mediaani täydellisen vasteen (CR) saavuttamiseen, kuukautta (vaihteluväli) | 5,5 (1,7–11,5) |
| CR = täydellinen vaste (complete response); PR = osittainen vaste (partial response); NR = ei saavutettu (not reached) | |
Riippumattoman arviointikomitean (Independent Review Committee, IRC) arvioidessa tehon tietoja edelleen kokonaisvasteluvuksi todettiin 69 %, jolloin täydellisen vasteen osuus oli 21 % ja osittaisen vasteen osuus 48 %. IRC:n arvioima vasteen kestoajan mediaani oli 19,6 kuukautta.
IMBRUVICA-valmisteen kokonaisvaste oli riippumaton aiemmasta hoidosta, bortetsomibi ja lenalidomidi mukaan lukien, tai taustalla olevista riski-/ennustetekijöistä, kasvaimen suurikokoisuudesta, sukupuolesta tai iästä.
IMBRUVICA-valmisteen turvallisuus ja teho osoitettiin satunnaistetussa vaiheen 3 avoimessa monikeskustutkimuksessa, jossa oli mukana 280 manttelisolulymfoomaa sairastavaa potilasta, jotka olivat saaneet vähintään yhtä aiempaa hoitoa (tutkimus MCL3001). Potilaat satunnaistettiin suhteessa 1:1 saamaan joko IMBRUVICA-hoitoa suun kautta 560 mg:n annoksina kerran vuorokaudessa 21 päivän ajan tai temsirolimuusia laskimoon 175 mg:n annoksina ensimmäisen hoitosyklin päivinä 1, 8 ja 15, ja sen jälkeen 75 mg:n annoksina jokaisen seuraavan 21 päivän pituisen hoitosyklin päivinä 1, 8 ja 15. Hoitoa jatkettiin kummassakin hoitoryhmässä, kunnes tauti eteni tai potilaalle ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Potilaiden iän mediaani oli 68 vuotta (vaihteluväli: 34; 88 vuotta), ja miehiä oli 74 % potilaista ja valkoihoisia 87 %. Ajan mediaani diagnoosista oli 43 kuukautta, ja aiempien hoitokertojen lukumäärän mediaani oli 2 (vaihteluväli: 1–9 hoitoa). Aiempia hoitoja olivat suuriannoksinen solunsalpaajahoito 51 %:lla, bortetsomibihoito 18 %:lla, lenalidomidihoito 5 %:lla ja kantasolusiirto 24 %:lla. Lähtötilanteen seulonnassa 53 %:lla potilaista oli suurikokoinen kasvain (≥ 5 cm), 21 %:lla oli suuren riskin tautia osoittavat MIPI-pisteet (Simplified MCL International Prognostic Index, MIPI), 60 %:lla tauti oli levinnyt imusolmukkeiden ulkopuolelle ja 54 %:lla luuytimeen.
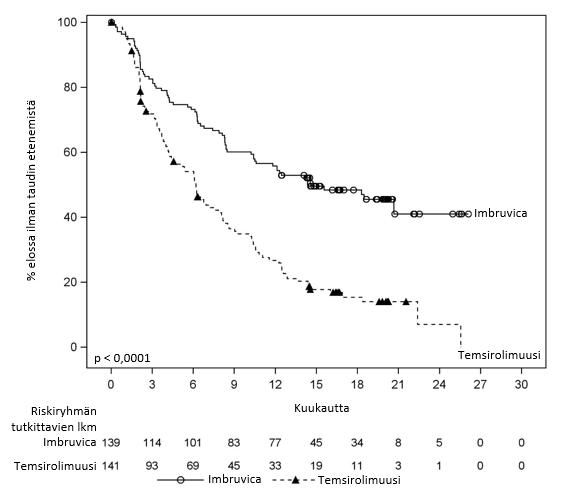
Riippumaton arviointikomitea arvioi taudin etenemättömyysaikaa (progession‑free survival, PFS) muokattujen non-Hodgkin-lymfoomaa koskevien International Working Group (IWG) ‑kriteerien perusteella. Tutkimuksen MCL3001 tehoa koskevat tulokset esitetään taulukossa 6 ja taudin etenemättömyysajan Kaplan–Meier-käyrä on kuvassa 3.
| Taulukko 6: Uusiutunutta tai hoitoon reagoimatonta manttelisolulymfoomaa sairastavien potilaiden hoidon tehoa koskevat tulokset (tutkimus MCL3001) | |||
| Päätetapahtuma | IMBRUVICA N = 139 | Temsirolimuusi N = 141 | |
| Taudin etenemättömyysaikaa | |||
| Taudin etenemättömyysajan mediaani (95 %:n luottamusväli) (kuukautta) | 14,6 (10,4; ei arvioitavissa) | 6,2 (4,2; 7,9) | |
| Riskisuhde (HR) = 0,43 [95 %:n luottamusväli: 0,32; 0,58] | |||
| Kokonaisvasteluku (%) | 71,9 | 40,4 | |
| p-arvo | p < 0,0001 | ||
| a Riippumattoman arviointikomitean (IRC) arvio. | |||
Lymfooman oireet pahenivat kliinisesti merkityksellisesti pienemmällä osalla ibrutinibihoitoa saaneista potilaista verrattuna temsirolimuusia saaneisiin potilaisiin (27 % versus 52 %), ja aika oireiden pahenemiseen oli ibrutinibiryhmässä pidempi kuin temsirolimuusiryhmässä (riskisuhde [HR] 0,27, p < 0,0001).
Kuva 3: Tutkimuksen MCL3001 taudin etenemättömyysajan (hoitoaikeen mukainen potilasjoukko) Kaplan–Meier-käyrä
Krooninen lymfaattinen leukemia
Aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavat potilaat
Monoterapia
Vaiheen 3 avoimessa, satunnaistetussa monikeskustutkimuksessa (PCYC‑1115‑CA) IMBRUVICA-valmistetta verrattiin klorambusiiliin aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavien 65‑vuotiaiden tai vanhempien potilaiden hoidossa. Iältään 65–70-vuotiailla potilailla piti olla vähintään yksi samanaikainen sairaus, joka sulki pois ensilinjan kemoimmunoterapian fludarabiinilla, syklofosfamidilla ja rituksimabilla. Potilaat (n = 269) satunnaistettiin suhteessa 1:1 saamaan joko IMBRUVICA-valmistetta 420 mg päivittäin, kunnes sairaus eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä, tai klorambusiilia enintään 12 hoitosykliä aloitusannoksella 0,5 mg/kg kunkin 28 päivän pituisen hoitosyklin päivinä 1 ja 15. Annosta voitiin suurentaa siedettävyyden perusteella annokseen 0,8 mg/kg saakka. Kun sairauden eteneminen oli varmistunut, klorambusiilia saaneiden potilaiden oli mahdollista siirtyä ibrutinibihoitoon.
Potilaiden iän mediaani oli 73 vuotta (vaihteluväli: 65–90 vuotta), ja miehiä oli 63 % ja valkoihoisia 91 % potilaista. Yhdeksälläkymmenelläyhdellä prosentilla potilaista lähtötilanteen ECOG-suorituskykyluokka oli 0 tai 1, ja 9 %:lla ECOG-suorituskykyluokka oli 2. Tutkimukseen otettiin mukaan 269 kroonista lymfaattista leukemiaa sairastavaa potilasta. Potilaista 45 %:lla oli lähtötilanteessa pitkälle edennyt tauti (Rai-luokituksen aste III tai IV), 35 %:lla potilaista oli vähintään yksi kasvain (≥ 5 cm), 39 %:lla oli lähtötilanteessa anemia, 23 %:lla oli lähtötilanteessa trombosytopenia, 65 %:n β2-mikroglobuliinipitoisuus oli yli 3,5 mg/l, 47 %:n CrCL oli < 60 ml/min, 20 %:lla potilaista oli 11q-deleetio, 6 %:lla potilaista oli 17p-deleetio/kasvainproteiinin 53 (TP53) mutaatio, ja 44 %:lla potilaista oli mutatoitumaton immunoglobuliinigeenin raskaan ketjun vaihtuva alue (IGHV-geeni).
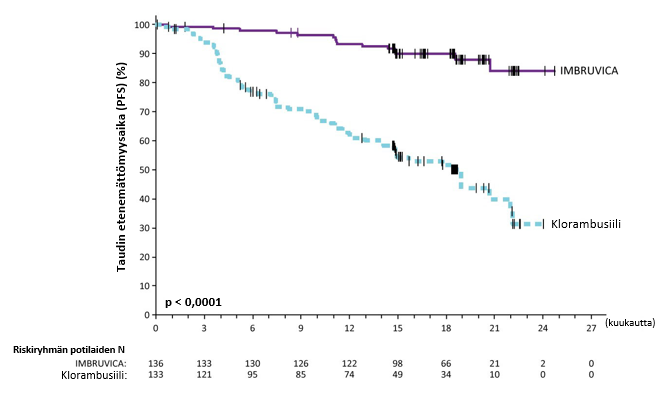
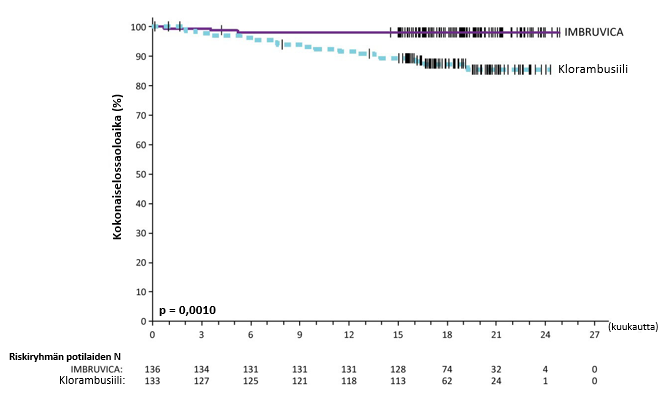
Riippumaton arviointikomitea (IRC) arvioi taudin etenemättömyyttä IWCLL (International Workshop on CLL) ‑kriteerien mukaisesti. Arviointi osoitti, että kuoleman ja taudin etenemisen riski väheni IMBRUVICA-ryhmässä tilastollisesti merkitsevästi 84 %. Tutkimuksen PCYC‑1115‑CA hoidon tehoa koskevat tulokset esitetään taulukossa 7, taudin etenemättömyysajan Kaplan–Meier-käyrä esitetään kuvassa 4 ja kokonaiselossaoloajan Kaplan–Meier-käyrä esitetään kuvassa 5.
Hoitoaikeen mukaisen (ITT) potilasjoukon trombosyytti- tai hemoglobiinipitoisuudet paranivat ibrutinibihoidossa pitkäkestoisesti ja tilastollisesti merkitsevästi verrattuna klorambusiilihoitoon. Jos potilaalla oli sytopenioita lähtötilanteessa, seuraavat hematologiset arvot paranivat pitkäkestoisesti: trombosyytit 77,1 % (ibrutinibi) versus 42,9 % (klorambusiili); hemoglobiini 84,3 % (ibrutinibi) versus 45,5 % (klorambusiili).
| Taulukko 7: Tutkimuksen PCYC‑1115‑CA tehoa koskevat tulokset | ||
| Päätetapahtuma | IMBRUVICA N = 136 | Klorambusiili N = 133 |
| Taudin etenemättömyysaikaa | ||
| Tapahtumien lukumäärä (%) | 15 (11,0) | 64 (48,1) |
| Mediaani (95 %:n luottamusväli), kk | Ei saavutettu | 18,9 (14,1; 22,0) |
| Riskisuhde (HR) (95 %:n luottamusväli) | 0,161 (0,091; 0,283) | |
| Kokonaisvastelukua (CR + PR) | 82,4 % | 35,3 % |
| P‑arvo | < 0,0001 | |
| Kokonaiselossaoloaikab | ||
| Kuolemien lukumäärä (%) | 3 (2,2) | 17 (12,8) |
| Riskisuhde (HR) (95 %:n luottamusväli) | 0,163 (0,048; 0,558) | |
CR = täydellinen vaste (complete response); PR = osittainen vaste (partial response) a Riippumattoman arviointikomitean (IRC) arvio, seuranta-ajan mediaani 18,4 kuukautta. b Kokonaiselossaoloajan mediaania ei saavutettu kummassakaan ryhmässä, kokonaiselossaoloajan p < 0,005. | ||
Kuva 4: Tutkimuksen PCYC‑1115‑CA taudin etenemättömyysajan (hoitoaikeen mukainen potilasjoukko) Kaplan‑Meier-käyrä
Kuva 5: Tutkimuksen PCYC‑1115‑CA kokonaiselossaoloajan (hoitoaikeen mukainen potilasjoukko) Kaplan‑Meier-käyrä
Tutkimuksessa PCYC-1115-CA ibrutinibin hoitoteho oli yhdenmukainen kaikilla suuren riskin potilailla, joilla oli 17p-deleetio/TP53-mutaatio, 11q-deleetio ja/tai mutatoitumaton IGHV-geeni.
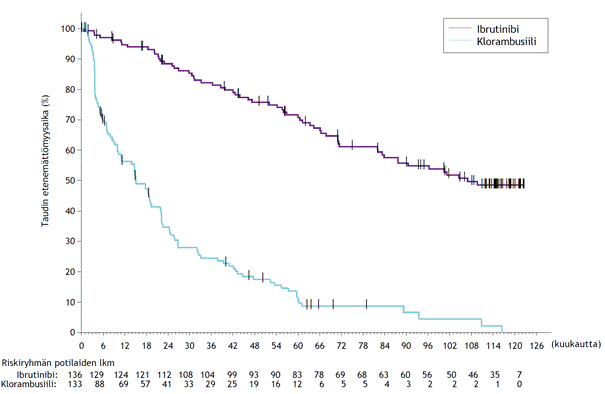
Loppuanalyysi > 9 vuoden (115 kuukauden) mediaaniseurannan aikapisteessä
Tutkimuksen PCYC-1115-CA ja sen jatkotutkimuksen tutkimuksenaikaisen seuranta-ajan mediaanin ollessa 115 kuukautta IMBRUVICA-ryhmän potilailla havaittiin tutkijan arvion perusteella kuoleman tai taudin etenemisen riskin 85 %:n vähenemä. Tutkijan arvioiman taudin etenemättömyysajan mediaani oli IMBRUVICA-ryhmässä 107 kuukautta ja klorambusiiliryhmässä 15 kuukautta (riskisuhde [HR] = 0,155 [95 %:n luottamusväli (0,110; 0,220)]). Taudin etenemättömyysajan päivitetty Kaplan–Meier-käyrä on kuvassa 6. Kokonaisvasteluvun paraneminen ibrutinibiryhmässä (91,2 %) verrattuna klorambusiiliryhmään (36,8 %) säilyi. Täydellisen vasteen (CR ja CRi) luku suureni IMBRUVICA-ryhmässä ensisijaisen analyysin ja tutkimuksen päättymisen välillä 11 %:sta 36 %:iin. 108 kuukauden kokonaiselossaolon Kaplan–Meierin kiintopiste-estimaatti IMBRUVICA-ryhmässä oli 68,0 %.
Kuva 6: Tutkimuksen PCYC‑1115‑CA taudin etenemättömyysajan 115 kuukauden seurannan (hoitoaikeen mukainen potilasjoukko) Kaplan–Meier-käyrä
Yhdistelmähoito
IMBRUVICA-valmisteen turvallisuutta ja tehoa aiemmin hoitamattomien kroonista lymfaattista leukemiaa / pienilymfosyyttistä lymfoomaa sairastavien potilaiden hoitoon arvioitiin lisäksi satunnaistetussa, avoimessa vaiheen 3 monikeskustutkimuksessa (PCYC-1130-CA), jossa IMBRUVICA-valmisteen ja obinututsumabin yhdistelmää verrattiin klorambusiilin ja obinututsumabin yhdistelmään. Tutkimukseen mukaan otetut potilaat olivat 65-vuotiaita tai vanhempia tai < 65-vuotiaita, joilla oli muita samanaikaisia sairauksia, heikentynyt munuaisten toiminta (osoitettu kreatiniinipuhdistumana < 70 ml/min) tai 17p-deleetio/TP53-mutaatio. Potilaat (n = 229) satunnaistettiin suhteessa 1:1 saamaan joko 420 mg IMBRUVICA-valmistetta päivittäin, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä, tai klorambusiilia annoksina 0,5 mg/kg kunkin 28 päivän pituisen hoitosyklin päivinä 1 ja 15 kuuden hoitosyklin ajan. Kummankin ryhmän potilaat saivat 1 000 mg obinututsumabia ensimmäisen hoitosyklin päivinä 1, 8 ja 15, ja sen jälkeen viiden seuraavan hoitosyklin ensimmäisenä päivänä (yhteensä kuuden 28 päivän pituisen hoitosyklin ajan). Ensimmäinen obinututsumabiannos jaettiin päivälle 1 (100 mg) ja päivälle 2 (900 mg).
Potilaiden iän mediaani oli 71 vuotta (vaihteluväli: 40–87 vuotta), ja miehiä oli 64 % potilaista ja valkoihoisia 96 %. Kaikkien potilaiden ECOG-toimintakykyluokka oli lähtötilanteessa 0 (48 %) tai 1–2 (52 %). Potilaista 52 %:lla oli lähtötilanteessa pitkälle edennyt tauti (Rai-luokituksen aste III tai IV), 32 %:lla potilaista oli suurikokoinen kasvain (≥ 5 cm), 44 %:lla oli lähtötilanteessa anemia, 22 %:lla oli lähtötilanteessa trombosytopenia, 28 %:n kreatiniinipuhdistuma (CrCL) oli < 60 ml/min ja vanhusten sairauksien kasautumisasteikon (Cumulative Illness Rating Score for Geriatrics, CIRS-G) mediaani oli 4 (vaihteluväli: 0–12). Lähtötilanteessa 65 %:lla potilaista oli krooninen lymfaattinen leukemia / pienilymfosyyttinen lymfooma, johon liittyi suuren riskin tekijöitä (17p-deleetio/TP53-mutaatio [18 %], 11q-deleetio [15 %] tai mutatoitumaton IGHV-geeni [54 %]).
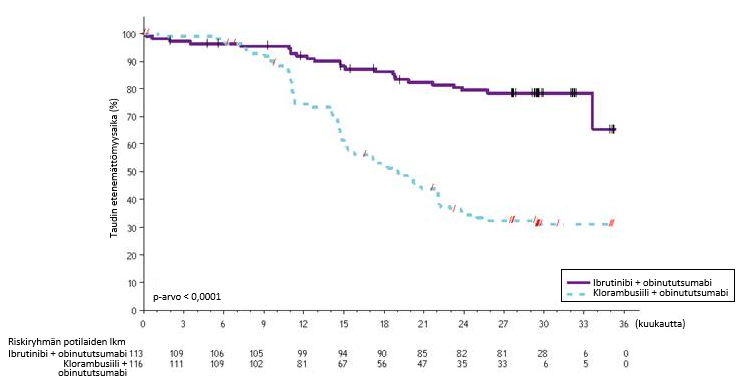
Riippumattoman arviointikomitean IWCLL-kriteerien mukaisesti tekemä arvio taudin etenemättömyysajasta osoitti, että kuoleman tai taudin etenemisen riski väheni IMBRUVICA-hoitoryhmässä tilastollisesti merkitsevästi 77 %. Tutkimuksenaikaisen seuranta-ajan ollessa 31 kuukautta (mediaani) taudin etenemättömyysajan mediaania ei ollut saavutettu IMBRUVICA-valmisteen ja obinututsumabin yhdistelmää saaneessa ryhmässä, ja se oli 19 kuukautta klorambusiilin ja obinututsumabin yhdistelmää saaneessa ryhmässä. Tutkimuksen PCYC‑1130‑CA tehoa koskevat tulokset esitetään taulukossa 8, ja taudin etenemättömyysajan Kaplan–Meier-käyrä on kuvassa 7.
| Taulukko 8: Tutkimuksen PCYC-1130-CA tehoa koskevat tulokset | ||
| Päätetapahtuma | IMBRUVICA + obinututsumabi N = 113 | Klorambusiili + obinututsumabi N = 116 |
| Taudin etenemättömyysaikaa | ||
| Tapahtumien lukumäärä (%) | 24 (21,2) | 74 (63,8) |
| Mediaani (95 %:n luottamusväli), kk | Ei saavutettu | 19,0 (15,1; 22,1) |
| Riskisuhde (HR) (95 %:n luottamusväli) | 0,23 (0,15; 0,37) | |
| Kokonaisvastelukua (%) | 88,5 | 73,3 |
| CRb | 19,5 | 7,8 |
| PRc | 69,0 | 65,5 |
CR = täydellinen vaste (complete response); PR = osittainen vaste (partial response). a Riippumattoman arviointikomitean (IRC) arvio. b Mukana IMBRUVICA-valmisteen ja obinututsumabin yhdistelmää saaneesta ryhmästä 1 potilas, jolla täydellinen vaste, mutta luuydin toipui epätäydellisesti (incomplete marrow recovery, CRi). c PR = PR + nPR. | ||
Kuva 7: Tutkimuksen PCYC-1130-CA taudin etenemättömyysajan (hoitoaikeen mukainen potilasjoukko) Kaplan–Meier-käyrä
Ibrutinibihoidon teho oli yhdenmukainen koko suuren riskin kroonista lymfaattista leukemiaa / pienilymfosyyttistä lymfoomaa sairastavassa potilasjoukossa (17p-deleetio/TP53-mutaatio, 11q-deleetio tai mutatoitumaton IGHV-geeni), jossa taudin etenemättömyysajan riskisuhde (HR) oli 0,15 (95 %:n luottamusväli [0,09; 0,27]), kuten taulukossa 9 esitetään. Taudin etenemättömyysajan 2 vuoden estimaatit suuren riskin kroonista lymfaattista leukemiaa / pienilymfosyyttistä lymfoomaa sairastavassa potilasjoukossa olivat IMBRUVICA-valmisteen ja obinututsumabin yhdistelmää saaneilla 78,8 % (95 %:n luottamusväli [67,3; 86,7]) ja klorambusiilin ja obinututsumabin yhdistelmää saaneilla 15,5 % (95 %:n luottamusväli [8,1; 25,2]).
| Taulukko 9: Taudin etenemättömyysajan alaryhmäanalyysi (tutkimus PCYC-1130-CA) | |||
| N | Riskisuhde (HR) | 95 %:n luottamusväli | |
| Kaikki tutkittavat | 229 | 0,231 | 0,145; 0,367 |
| Suuri riski (17p-deleetio/TP53-mutaatio/11q-deleetio/mutatoitumaton IGHV-geeni) | |||
| Kyllä | 148 | 0,154 | 0,087; 0,270 |
| Ei | 81 | 0,521 | 0,221; 1,231 |
| 17p-deleetio/TP53-mutaatio | |||
| Kyllä | 41 | 0,109 | 0,031; 0,380 |
| Ei | 188 | 0,275 | 0,166; 0,455 |
| FISH | |||
| 17p-deleetio | 32 | 0,141 | 0,039; 0,506 |
| 11q-deleetio | 35 | 0,131 | 0,030; 0,573 |
| Muut | 162 | 0,302 | 0,176; 0,520 |
| Mutatoitumaton IGHV-geeni | |||
| Kyllä | 123 | 0,150 | 0,084; 0,269 |
| Ei | 91 | 0,300 | 0,120; 0,749 |
| Ikä | |||
| < 65 | 46 | 0,293 | 0,122; 0,705 |
| ≥ 65 | 183 | 0,215 | 0,125; 0,372 |
| Suurikokoinen kasvain | |||
| < 5 cm | 154 | 0,289 | 0,161; 0,521 |
| ≥ 5 cm | 74 | 0,184 | 0,085; 0,398 |
| Rai-luokituksen aste | |||
| 0/I/II | 110 | 0,221 | 0,115; 0,424 |
| III/IV | 119 | 0,246 | 0,127; 0,477 |
| Krooniseen munuaisten vajaatoimintaan liittyvä ECOG-toimintakykyluokka | |||
| 0 | 110 | 0,226 | 0,110; 0,464 |
| 1–2 | 119 | 0,239 | 0,130; 0,438 |
| Riskisuhde (HR) perustuu osittamattomaan analyysiin | |||
Kaikkien vaikeusasteiden infuusioon liittyviä reaktioita havaittiin 25 %:lla IMBRUVICA-valmisteen ja obinututsumabin yhdistelmää saaneista potilaista ja 58 %:lla klorambusiilin ja obinututsumabin yhdistelmää saaneista potilaista. 3. asteen tai vaikeampiasteisia tai vakavia infuusioon liittyneitä reaktioita havaittiin 3 %:lla IMBRUVICA-valmisteen ja obinututsumabin yhdistelmää saaneista potilaista ja 9 %:lla klorambusiilin ja obinututsumabin yhdistelmää saaneista potilaista.
IMBRUVICA-valmisteen turvallisuutta ja tehoa aiemmin hoitamattomilla kroonista lymfaattista leukemiaa tai pienilymfosyyttistä lymfoomaa sairastavilla potilailla arvioitiin edelleen satunnaistetussa, avoimessa vaiheen 3 monikeskustutkimuksessa (E1912), jossa IMBRUVICA-valmisteen ja rituksimabin yhdistelmää (IR) verrattiin kemoimmunoterapiaan fludarabiinilla, syklofosfamidilla ja rituksimabilla (FCR). Tutkimuksen otettiin mukaan aiemmin hoitamattomia kroonista lymfaattista leukemiaa tai pienilymfosyyttistä lymfoomaa sairastavia potilaita, jotka olivat 70-vuotiaita tai nuorempia. Potilaita, joilla oli 17p-deleetio, ei otettu tutkimukseen mukaan. Potilaat (n = 529) satunnaistettiin suhteessa 2:1 saamaan joko IR- tai FCR-hoitoa. IMBRUVICA-valmistetta annettiin päivittäin 420 mg:n annoksina, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Fludarabiinia annettiin annoksina 25 mg/m2, ja syklofosfamidia annettiin annoksina 250 mg/m2 hoitosykleissä 1–6 päivinä 1, 2 ja 3. Rituksimabihoito aloitettiin IR-ryhmässä hoitosyklissä 2 ja FCR-ryhmässä hoitosyklissä 1, ja sitä annettiin annoksena 50 mg/m2 ensimmäisen hoitosyklin päivänä 1, annoksena 325 mg/m2 ensimmäisen hoitosyklin päivänä 2 ja annoksena 500 mg/m2 päivänä 1 viiden seuraavan hoitosyklin aikana. Yhteensä annettiin 6 hoitosykliä. Yksi hoitosykli oli 28 päivää.
Potilaiden iän mediaani oli 58 vuotta (vaihteluväli: 28–70 vuotta), ja miehiä oli 67 % potilaista ja valkoihoisia 90 %. Kaikkien potilaiden ECOG-toimintakykyluokka oli lähtötilanteessa 0 tai 1 (98 %) tai 2 (2 %). Potilaista 43 %:lla oli lähtötilanteessa pitkälle edennyt tauti (Rai-luokituksen aste III tai IV), ja 59 %:lla potilaista oli krooninen lymfaattinen leukemia / pienilymfosyyttinen lymfooma, johon liittyi suuren riskin tekijöitä (TP53-mutaatio [6 %], 11q-deleetio [22 %] tai mutatoitumaton IGHV-geeni [53 %]).
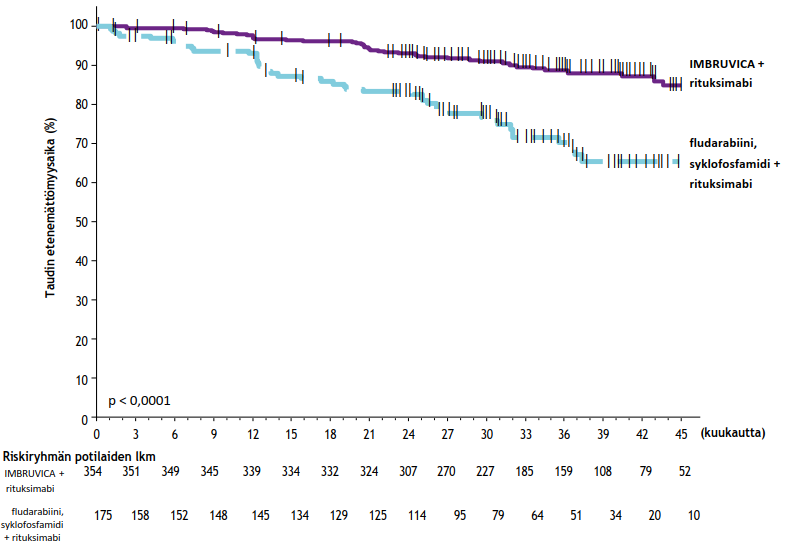
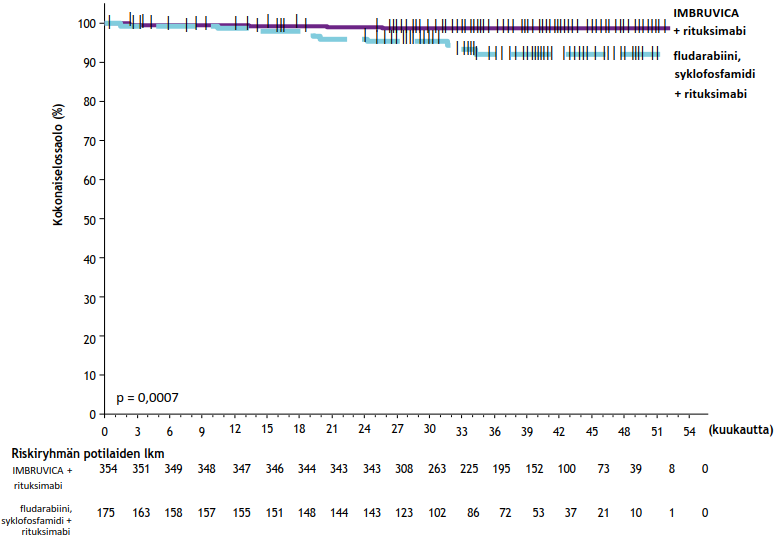
E1912-tutkimuksen tehon tulokset tutkimuksenaikaisen seuranta-ajan ollessa 37 kuukautta (mediaani) esitetään taulukossa 10. IWCLL-kriteerien mukaisesti arvioidun taudin etenemättömyysajan Kaplan–Meier-käyrä on kuvassa 8 ja kokonaiselossaoloajan Kaplan–Meier-käyrä kuvassa 9.
| Taulukko 10: Tutkimuksen E1912 tehoa koskevat tulokset | ||
| Päätetapahtuma | Ibrutinibi + rituksimabi (IR) N = 354 | Fludarabiini, syklofosfamidi ja rituksimabi (FCR) N = 175 |
| Taudin etenemättömyysaika | ||
| Tapahtumien lukumäärä (%) | 41 (12) | 44 (25) |
| Taudin eteneminen | 39 | 38 |
| Kuolemia | 2 | 6 |
| Mediaani (95 %:n luottamusväli), kk | Ei arvioitavissa (49,4; ei arvioitavissa) | Ei arvioitavissa (47,1; ei arvioitavissa) |
| Riskisuhde (95 %:n luottamusväli) | 0,34 (0,22; 0,52) | |
| p-arvoa | < 0,0001 | |
| Kokonaiselossaolo | ||
| Kuolemien lukumäärä (%) | 4 (1) | 10 (6) |
| Riskisuhde (95 %:n luottamusväli) | 0,17 (0,05; 0,54) | |
| p-arvoa | 0,0007 | |
| Kokonaisvastelukub (%) | 96,9 | 85,7 |
a p-arvo perustuu osittamattomaan log-rank-testiin. b Tutkijan arvio. | ||
Kuva 8: Tutkimuksen E1912 taudin etenemättömyysajan (hoitoaikeen mukainen potilasjoukko) Kaplan–Meier-käyrä
Ibrutinibihoidon teho oli yhdenmukainen koko suuren riskin kroonista lymfaattista leukemiaa / pienilymfosyyttistä lymfoomaa sairastavassa potilasjoukossa (TP53-mutaatio, 11q-deleetio tai mutatoitumaton IGHV-geeni), jossa taudin etenemättömyysajan riskisuhde (HR) oli 0,23 (95 %:n luottamusväli [0,13; 0,40]), p < 0,0001, kuten taulukossa 11 esitetään. Taudin etenemättömyysajan 3 vuoden estimaatit suuren riskin kroonista lymfaattista leukemiaa / pienilymfosyyttistä lymfoomaa sairastavassa potilasjoukossa olivat IR-hoitoryhmässä 90,4 % (95 %:n luottamusväli [85,4; 93,7]) ja FCR-hoitoryhmässä 60,3 % (95 %:n luottamusväli [46,2; 71,8]).
| Taulukko 11: Taudin etenemättömyysajan alaryhmäanalyysi (tutkimus E1912) | |||
| N | Riskisuhde (HR) | 95 %:n luottamusväli | |
| Kaikki tutkittavat | 529 | 0,340 | 0,222; 0,522 |
| Suuri riski (TP53-mutaatio/11q-deleetio/mutatoitumaton IGHV-geeni) | |||
| Kyllä | 313 | 0,231 | 0,132; 0,404 |
| Ei | 216 | 0,568 | 0,292; 1,105 |
| 11q-deleetio | |||
| Kyllä | 117 | 0,199 | 0,088; 0,453 |
| Ei | 410 | 0,433 | 0,260; 0,722 |
| Mutatoitumaton IGHV-geeni | |||
| Kyllä | 281 | 0,233 | 0,129; 0,421 |
| Ei | 112 | 0,741 | 0,276; 1,993 |
| Suurikokoinen kasvain | |||
| < 5 cm | 316 | 0,393 | 0,217; 0,711 |
| ≥ 5 cm | 194 | 0,257 | 0,134; 0,494 |
| Rai-luokituksen aste | |||
| 0/I/II | 301 | 0,398 | 0,224; 0,708 |
| III/IV | 228 | 0,281 | 0,148; 0,534 |
| ECOG-toimintakykyluokka | |||
| 0 | 335 | 0,242 | 0,138; 0,422 |
| 1–2 | 194 | 0,551 | 0,271; 1,118 |
| Riskisuhde (HR) perustuu osittamattomaan analyysiin | |||
Kuva 9: Tutkimuksen E1912 kokonaiselossaolon (hoitoaikeen mukainen potilasjoukko) Kaplan–Meier-käyrä
Määräaikainen yhdistelmähoito
Satunnaistetussa, avoimessa vaiheen 3 tutkimuksessa (CLL3011) arvioitiin IMBRUVICA-valmisteen ja venetoklaksin määräaikaisen yhdistelmähoidon turvallisuutta ja tehoa klorambusiilin ja obinututsumabin yhdistelmään verrattuna aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavilla potilailla. Tutkimukseen otettiin mukaan aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavia potilaita, jotka olivat iältään vähintään 65-vuotiaita, sekä iältään alle 65‑vuotiaita aikuisia potilaita, joiden CIRS-pisteet olivat > 6 tai joiden kreatiniinipuhdistuma (CrCL) oli ≥ 30 – < 70 ml/min. Potilaita, joilla oli 17p‑deleetio tai tiedossa olevia TP53-mutaatioita, ei otettu tutkimukseen mukaan. Potilaat (n = 211) satunnaistettiin suhteessa 1:1 joko IMBRUVICA-valmisteen ja venetoklaksin yhdistelmähoitoon tai klorambusiilin ja obinututsumabin yhdistelmähoitoon. IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää saaneet potilaat saivat 3 hoitosykliä IMBRUVICA-monoterapiaa ja sen jälkeen 12 hoitosykliä IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää (viiden viikon annostitrausohjelma mukaan lukien). Yksi hoitosykli oli 28 päivää. IMBRUVICA-valmistetta annettiin päivittäin 420 mg:n annoksena. Venetoklaksia annettiin päivittäin; hoito aloitettiin 20 mg:lla yhden viikon ajan, jonka jälkeen annettiin annoksia 50 mg, 100 mg ja 200 mg kutakin viikon ajan ja sen jälkeen suositeltua 400 mg:n vuorokausiannosta. Potilaat, jotka satunnaistettiin klorambusiilin ja obinututsumabin yhdistelmää saavaan ryhmään, saivat 6 hoitosykliä. Ensimmäisessä hoitosyklissä obinututsumabia annettiin 1 000 mg:n annoksena päivinä 1, 8 ja 15. 2.-6. hoitosyklissä annettiin 1 000 mg obinututsumabia päivänä 1. Klorambusiilia annettiin annoksena 0,5 mg/kg 1.–6. hoitosyklin päivinä 1 ja 15. Potilaita, joilla varmistui IWCLL-kriteerien perusteella sairauden eteneminen kumman tahansa määräaikaisen yhdistelmähoidon päätyttyä, voitiin hoitaa IMBRUVICA-monoterapialla.
Potilaiden iän mediaani oli 71 vuotta (vaihteluväli 47–93 vuotta), ja miehiä oli 58 % ja valkoihoisia 96 % potilaista. Kaikkien potilaiden lähtötilanteen ECOG-toimintakykyluokka oli 0 (35 %), 1 (53 %) tai 2 (12 %). 18 %:lla potilaista krooniseen lymfaattiseen leukemiaan liittyi lähtötilanteessa 11q‑deleetio ja 52 %:lla oli mutatoitumaton immunoglobuliinigeenin raskaan ketjun vaihtuva alue (IGHV-geeni).
Arvioitaessa lähtötilanteessa tuumorilyysioireyhtymän riskiä, 25 %:lla potilaista oli suuri kasvaintaakka. Hoidon alussa annettujen kolmen IMBRUVICA-monoterapiahoitosyklin jälkeen 2 %:lla potilaista oli suuri kasvaintaakka. Suureksi kasvaintaakaksi määriteltiin jonkin imusolmukkeen koko ≥ 10 cm tai jonkin imusolmukkeen koko ≥ 5 cm ja absoluuttinen lymfosyyttimäärä ≥ 25 × 109/l.
CLL3011-tutkimuksessa riippumattoman arviointikomitean IWCLL-kriteerien mukaan arvioimat tutkimuksen tehon tulokset seuranta-ajan ollessa 28 kuukautta (mediaani) esitetään taulukossa 12, taudin etenemättömyysajan Kaplan-Meier-käyrä esitetään kuvassa 10 ja minimaalisen jäännöstaudin (minimal residual disease, MRD) suhteen negatiivisten osuudet esitetään taulukossa 13.
| Taulukko 12: Tutkimuksen CLL3011 tehoa koskevat tulokset | ||
| Päätetapahtumaa | IMBRUVICA + venetoklaksi N = 106 | Klorambusiili + obinututsumabi N = 105 |
| Taudin etenemättömyysaika | ||
| Tapahtumien lukumäärä (%) | 22 (20,8) | 67 (63,8) |
| Mediaani (95 %:n luottamusväli), kk | Ei arvioitavissa (31,2; ei arvioitavissa) | 21,0 (16,6; 24,7) |
| Riskisuhde (95 %:n luottamusväli) | 0,22 (0,13; 0,36) | |
| p-arvob | < 0,0001 | |
| Täydellisen vasteen osuus(%)c | 38,7 | 11,4 |
| 95 %:n luottamusväli | (29,4; 48,0) | (5,3; 17,5) |
| p-arvod | < 0,0001 | |
| Kokonaisvasteluku (%)e | 86,8 | 84,8 |
| 95 %:n luottamusväli | (80,3; 93,2) | (77,9; 91,6) |
a Riippumattoman arviointikomitean arvio b p-arvo saatu ositetulla log-rank-testillä c Sisältää IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää saaneen ryhmän 3 potilasta, joilla täydellinen vaste, jossa luuydin palautunut epätäydellisesti (CRi) d p-arvo saatu Cochran-Mantel-Haenszelin khi-neliötestillä e Kokonaisvaste = CR + CRi + nPR + PR CR = täydellinen vaste (complete response); CRi = täydellinen vaste, jossa luuydin palautunut epätäydellisesti (complete response with incomplete marrow recovery); nPR = nodulaarinen osittainen vaste (nodular partial response); PR = osittainen vaste (partial response) | ||
Kuva 10: Tutkimuksen CLL3011 kroonista lymfaattista leukemiaa sairastavien potilaiden (hoitoaikeen mukainen potilasjoukko) taudin etenemättömyysajan Kaplan-Meierin käyrä
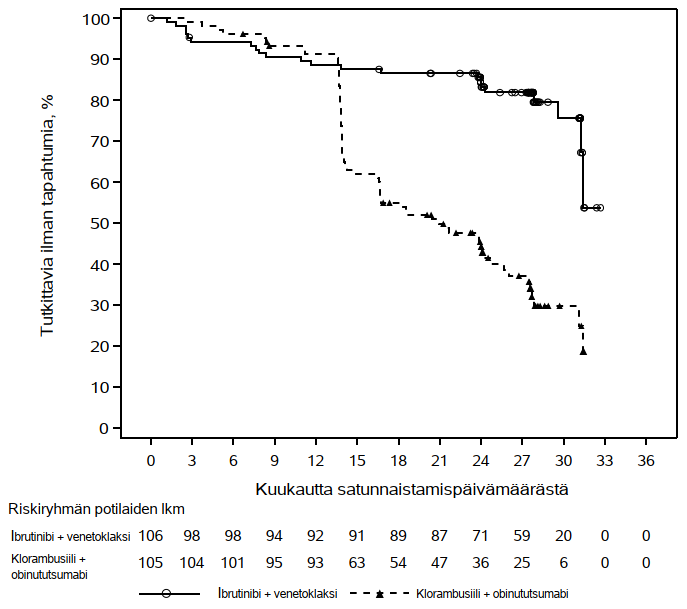
IMBRUVICA-valmisteen ja venetoklaksin yhdistelmän hoitoteho oli yhdenmukainen koko suuren riskin kroonista lymfaattista leukemiaa sairastavassa potilasjoukossa (TP53-mutaatio, 11q-deleetio tai mutatoitumaton IGHV-geeni), ja taudin etenemättömyysajan riskisuhde (HR) oli 0,23 (95 %:n luottamusväli [0,13; 0,41]).
Kokonaiselossaoloaikaa (OS) koskevat tiedot eivät olleet kypsät. Tutkimuksen seuranta-ajan ollessa 28 kuukautta (mediaani) hoitoryhmien välillä ei ollut merkittävää eroa, ja yhteensä 23 tutkittavaa oli kuollut: 11 (10,4 %) IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää saaneessa ryhmässä ja 12 (11,4 %) klorambusiilin ja obinututsumabin yhdistelmää saaneessa ryhmässä, joten kokonaiselossaolon riskisuhde (HR) oli 1,048 (95 %:n luottamusväli [0,454; 2,419]). 6 kuukauden lisäseurannan jälkeen IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää saaneessa ryhmässä oli raportoitu 11 (10,4 %) kuolemaa ja klorambusiilin ja obinututsumabin yhdistelmää saaneessa ryhmässä 16 (15,2 %) kuolemaa, joten kokonaiselossaolon riskisuhteen estimaatti oli 0,760 (95 %:n luottamusväli [0,352; 1,642]).
| Taulukko 13: Tutkimuksen CLL3011 minimaalisen jäännöstaudin (MRD) suhteen negatiivisten osuudet | ||||
| NGS-menetelmäa | Virtaussytometriab | |||
| IMBRUVICA + venetoklaksi N = 106 | Klorambusiili + obinututsumabi N = 105 | IMBRUVICA + venetoklaksi N = 106 | Klorambusiili + obinututsumabi N = 105 | |
| Minimaalisen jäännöstaudin suhteen negatiivisten osuus | ||||
| Luuydin, n (%) | 59 (55,7) | 22 (21,0) | 72 (67,9) | 24 (22,9) |
| 95 %:n luottamusväli | (46,2; 65,1) | (13,2; 28,7) | (59,0; 76,8) | (14,8; 30,9) |
| p-arvo | < 0,0001 | |||
| Ääreisveri, n (%) | 63 (59,4) | 42 (40,0) | 85 (80,2) | 49 (46,7) |
| 95 %:n luottamusväli | (50,1; 68,8) | (30,6; 49,4) | (72,6; 87,8) | (37,1; 56,2) |
| Minimaalisen jäännöstaudin suhteen negatiivisten osuus kolme kuukautta hoidon päättymisen jälkeen | ||||
| Luuydin, n (%) | 55 (51,9) | 18 (17,1) | 60 (56,6) | 17 (16,2) |
| 95 %:n luottamusväli | (42,4; 61,4) | (9,9; 24,4) | (47,2; 66,0) | (9,1; 23,2) |
| Ääreisveri, n (%) | 58 (54,7) | 41 (39,0) | 65 (61,3) | 43 (41,0) |
| 95 %:n luottamusväli | (45,2; 64,2) | (29,7; 48,4) | (52,0; 70,6) | (31,5; 50,4) |
p-arvot saatu Cochran-Mantel-Haenszelin khi-neliötestillä. NGS-menetelmään perustuva luuytimen minimaalisen jäännöstaudin suhteen negatiivisten osuuden p-arvo oli minimaalisen jäännöstaudin primaarianalyysi. a Perustuu NGS-menetelmän (clonoSEQ) raja-arvoon 10-4 b Minimaalinen jäännöstauti arvioitiin keskuslaboratoriossa tehdyllä ääreisveren tai luuytimen virtaussytometrialla. Negatiivisen statuksen määritelmä oli < 1 KLL-tautisolua per 10 000 leukosyyttiä (< 1 × 104). KLL = krooninen lymfaattinen leukemia NGS-menetelmä = next-generation sequencing | ||||
Kaksitoista kuukautta hoidon päättymisen jälkeen ääreisveren minimaalisen jäännöstaudin suhteen negatiivisten osuus IMBRUVICA-valmisteen ja venetoklaksin yhdistelmähoitoa saaneista potilaista oli NGS-menetelmän perusteella 49,1 % (52/106) ja virtaussytometrian perusteella 54,7 % (58/106) ja klorambusiilin ja obinututsumabin yhdistelmähoitoa saaneessa ryhmässä kyseinen osuus oli vastaavana ajankohtana NGS-menetelmän perusteella 12,4 % (13/105) ja virtaussytometrian perusteella 16,2 % (17/105).
Tuumorilyysioireyhtymää raportoitiin 6 potilaalla klorambusiilin ja obinututsumabin yhdistelmää saaneessa ryhmässä, mutta sitä ei raportoitu IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää saaneessa ryhmässä.
64 kuukauden mediaaniseuranta-aika
CLL3011-tutkimuksen aikaisen mediaaniseuranta-ajan ollessa 64,0 kuukautta tutkijan arvioon perustuvan kuoleman tai sairauden etenemisen riskin vähenemä oli 73 % IMBRUVICA-ryhmän potilailla. Taudin etenemättömyysajan riskisuhde oli 0,267 (95 %:n luottamusväli [0,182; 0,393], nimellinen p-arvo < 0,0001, tyypin 1 virhettä ei kontrolloitu). IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää saaneessa ryhmässä oli 20 kuolemaa (18,9 %), ja klorambusiilin ja obinututsumabin yhdistelmää saaneessa ryhmässä oli 40 kuolemaa (38,1 %), jolloin vastaava riskisuhde oli 0,462 (95 %:n luottamusväli 0,269; 0,791, nimellinen p-arvo 0,0039, tyypin 1 virhettä ei kontrolloitu). IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää saaneessa ryhmässä ei ollut saavutettu seuraavaan hoitoon kuluneen ajan mediaania ja klorambusiilin ja obinututsumabin yhdistelmää saaneessa ryhmässä se oli 65 kuukautta (riskisuhde = 0,233; 95 %:n luottamusväli: 0,130; 0,416), ja 15,1 % IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää saaneen ryhmän tutkittavista ja 43,8 % klorambusiilin ja obinututsumabin yhdistelmää saaneen ryhmän tutkittavista oli aloittanut seuraavan linjan syöpähoidon.
Kuvassa 11 esitetään kokonaiselossaolon Kaplan-Meierin käyrä.
Kuva 11: Tutkimuksen CLL3011 kroonista lymfaattista leukemiaa / pienilymfosyyttistä lymfoomaa sairastavien potilaiden (hoitoaikeen mukainen potilasjoukko) kokonaiselossaoloajan 64 kuukauden seurannan Kaplan Meier-käyrä
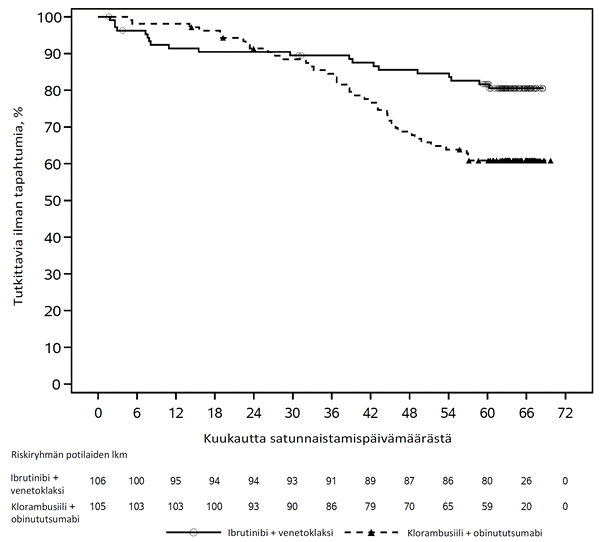
Määräaikaisen IMBRUVICA-valmisteen ja venetoklaksin yhdistelmähoidon turvallisuutta ja tehoa aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavilla potilailla arvioitiin lisäksi vaiheen 2 kahden kohortin monikeskustutkimuksessa (PCYC-1142-CA). Tutkimukseen otettiin mukaan aiemmin hoitamatonta kroonista lymfaattista leukemiaa sairastavia potilaita, jotka olivat 70-vuotiaita tai nuorempia. Tutkimuksessa oli mukana 323 potilasta, joista 159 potilasta otettiin mukaan hoito-ohjelmaan, jonka kesto oli vakio ja joka käsitti 3 hoitosykliä IMBRUVICA-monoterapiaa ja sen jälkeen 12 hoitosykliä IMBRUVICA-valmisteen ja venetoklaksin yhdistelmähoitoa (viiden viikon annostitrausohjelma mukaan lukien). Yksi hoitosykli oli 28 päivää. IMBRUVICA-valmistetta annettiin päivittäin 420 mg:n annoksena. Venetoklaksia annettiin päivittäin; hoito aloitettiin 20 mg:lla yhden viikon ajan, minkä jälkeen annettiin annoksia 50 mg, 100 mg ja 200 mg kutakin viikon ajan ja sen jälkeen suositeltua 400 mg:n vuorokausiannosta. Potilaita, joilla varmistui IWCLL-kriteerien perusteella sairauden eteneminen määräaikaisen yhdistelmähoidon päätyttyä, voitiin hoitaa uudelleen IMBRUVICA-monoterapialla.
Potilaiden iän mediaani oli 60 vuotta (vaihteluväli 33–71 vuotta), ja miehiä oli 67 % ja valkoihoisia 92 % potilaista. Kaikkien potilaiden lähtötilanteen ECOG-toimintakykyluokka oli 0 (69 %) tai 1 (31 %). Lähtötilanteessa 13 %:lla potilaista oli 17p-deleetio, 18 %:lla oli 11q-deleetio, 17 %:lla 17p‑deleetio/TP53-mutaatio, 56 %:lla mutatoitumaton IGHV-geeni ja 19 %:lla kompleksinen karyotyyppi. Arvioitaessa lähtötilanteessa tuumorilyysioireyhtymän riskiä 21 %:lla potilaista oli suuri kasvaintaakka.
Hoidon alussa annettujen kolmen IMBRUVICA-monoterapiahoitosyklin jälkeen 1 %:lla potilaista oli suuri kasvaintaakka. Suureksi kasvaintaakaksi määriteltiin jonkin imusolmukkeen koko ≥ 10 cm tai jonkin imusolmukkeen koko ≥ 5 cm ja absoluuttinen lymfosyyttimäärä ≥ 25 × 109/l.
PCYC‑1142-CA-tutkimuksessa riippumattoman arviointikomitean IWCLL-kriteerien mukaan arvioimat tehon tulokset tutkimuksen seuranta-ajan ollessa 28 kuukautta (mediaani) esitetään taulukossa 14 ja minimaalisen jäännöstaudin (MRD) suhteen negatiivisten osuudet esitetään taulukossa 15.
| Taulukko 14: Tutkimuksen PCYC 1142-CA tehon tulokset (määräaikaisen hoidon kohortti) | ||
| Päätetapahtumaa | IMBRUVICA + venetoklaksi | |
| Ei 17p-deleetiota (N = 136) | Kaikki (N = 159) | |
| Kokonaisvasteluku, n (%)b | 130 (95,6) | 153 (96,2) |
| 95 %:n luottamusväli (%) | (92,1; 99,0) | (93,3; 99,2) |
| Täydellisen vasteen osuus, n (%)c | 83 (61,0) | 95 (59,7) |
| 95 %:n luottamusväli (%) | (52,8; 69,2) | (52,1; 67,4) |
| Täydellisen vasteen kesto (mediaani), kk (vaihteluväli)d | Ei arvioitavissa (0,03+; 24,9+) | Ei arvioitavissa (0,03+; 24,9+) |
a Riippumattoman arviointikomitean arvio b Kokonaisvaste = CR + CRi + nPR + PR c Sisältää 3 potilasta, joilla täydellinen vaste, jossa luuydin palautunut epätäydellisesti (CRi) d Plusmerkki (+) tarkoittaa sensuroitua havaintoa CR = täydellinen vaste (complete response); CRi = täydellinen vaste, jossa luuydin palautunut epätäydellisesti (complete response with incomplete marrow recovery); nPR = nodulaarinen osittainen vaste (nodular partial response); PR = osittainen vaste (partial response) | ||
| Taulukko 15: Tutkimuksen PCYC 1142-CA minimaalisen jäännöstaudin suhteen negatiivisten osuudet (määräaikaisen hoidon kohortti) | ||
| Päätetapahtuma | IMBRUVICA + venetoklaksi | |
| Ei 17p-deleetiota (N = 136) | Kaikki (N = 159) | |
| Minimaalisen jäännöstaudin suhteen negatiivisten osuus | ||
| Luuydin, n (%) | 84 (61,8) | 95 (59.7) |
| 95 %:n luottamusväli | (53,6; 69,9) | (52.1, 67.4) |
| Ääreisveri, n (%) | 104 (76,5) | 122 (76.7) |
| 95 %:n luottamusväli | (69,3; 83,6) | (70.2, 83.3) |
| Minimaalisen jäännöstaudin suhteen negatiivisten osuus kolme kuukautta hoidon päättymisen jälkeen | ||
| Luuydin, n (%) | 74 (54,4) | 83 (52,2) |
| 95 %:n luottamusväli | (46,0; 62,8) | (44,4; 60,0) |
| Ääreisveri, n (%) | 78 (57,4) | 90 (56,6) |
| 95 %:n luottamusväli | (49,0; 65,7) | (48,9; 64.3) |
Minimaalinen jäännöstauti arvioitiin keskuslaboratoriossa tehdyllä ääreisveren tai luuytimen virtaussytometrialla. Negatiivisen statuksen määritelmä oli < 1 KLL-tautisolua per 10 000 leukosyyttiä (< 1 × 104). KLL = krooninen lymfaattinen leukemia | ||
Tutkimuksessa PCYC-1142-CA riippumattoman arviointikomitean arvioon perustuva kokonaisvasteluku potilailla, joilla oli 17p-deleetio/TP53-mutaatio (n = 27), oli 96,3 %; täydellisen vasteen saaneiden osuus oli 55,6 % ja täydellisen vasteen keston mediaania ei saavutettu (vaihteluväli 4,3–22,6 kuukautta). Minimaalisen jäännöstaudin suhteen negatiivisten osuus potilaista, joilla oli 17p-deleetio/TP53-mutaatio kolme kuukautta hoidon päättymisen jälkeen, oli luuytimen osalta 40,7 % ja ääreisveren osalta 59,3 %.
IMBRUVICA-valmisteen ja venetoklaksin yhdistelmää saaneilla potilailla ei raportoitu tuumorilyysioireyhtymää.
Vähintään yhtä aiempaa hoitoa saaneet kroonista lymfaattista leukemiaa sairastavat potilaat
Monoterapia
IMBRUVICA-valmisteen turvallisuus ja teho kroonista lymfaattista leukemiaa sairastavilla potilailla osoitettiin yhdessä kontrolloimattomassa tutkimuksessa ja yhdessä satunnaistetussa, kontrolloidussa tutkimuksessa. Avoimessa monikeskustutkimuksessa (PCYC‑1102‑CA) oli mukana 51 uusiutunutta tai hoitoon reagoimatonta kroonista lymfaattista leukemiaa sairastavaa potilasta, jotka saivat lääkettä 420 mg kerran vuorokaudessa. IMBRUVICA-valmistetta annettiin, kunnes tauti eteni tai potilaalle ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Iän mediaani oli 68 vuotta (vaihteluväli: 37–82 vuotta), ajan mediaani diagnoosista oli 80 kuukautta ja aiempien hoitokertojen lukumäärän mediaani oli 4 (vaihteluväli: 1−12 hoitoa). Aiempia hoitoja olivat nukleosidianalogihoito 92,2 %:lla, rituksimabihoito 98,0 %:lla, hoito alkyloivilla aineilla 86,3 %:lla, bendamustiinihoito 39,2 %:lla ja ofatumumabihoito 19,6 %:lla potilaista. Lähtötilanteessa 39,2 %:lla potilaista oli Rai-luokituksen mukaan asteen 4 tauti, 45,1 %:lla oli suurikokoinen kasvain (≥ 5 cm), 35,3 %:lla oli 17p-deleetio ja 31,4 %:lla oli 11q-deleetio.
Tutkijat ja riippumaton arviointikomitea arvioivat kokonaisvasteluvun IWCLL 2008 ‑kriteereiden mukaisesti. Riippumaton arviointikomitea arvioi, että 51:n uusiutunutta tai hoitoon reagoimatonta tautia sairastavan potilaan kokonaisvasteluku 16,4 kuukauden (mediaani) seurannan jälkeen oli 64,7 % (95 %:n luottamusväli: 50,1 %; 77,6 %), joista kaikki olivat osittaisia vasteita. Kokonaisvasteluku, mihin sisältyy osittainen vaste, johon liittyy lymfosytoosi, oli 70,6 %. Ajan mediaani vasteen alkamiseen oli 1,9 kuukautta. Vasteen kestoaika oli 3,9 – yli 24,2 kuukautta. Vasteen kestoajan mediaania ei saavutettu.
Satunnaistetussa, avoimessa, vaiheen 3 monikeskustutkimuksessa, jossa IMBRUVICA-valmistetta verrattiin ofatumumabiin (PCYC‑1112‑CA), oli mukana uusiutunutta tai hoitoon reagoimatonta kroonista lymfaattista leukemiaa sairastavia potilaita. Potilaat (n = 391) satunnaistettiin suhteessa 1:1 saamaan joko IMBRUVICA-valmistetta 420 mg päivittäin, kunnes tauti eteni tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä, tai enintään 12 annosta ofatumumabia (300/2 000 mg). Taudin edettyä 57 ofatumumabiryhmään satunnaistettua potilasta siirtyi IMBRUVICA-hoitoon taudin edettyä. Potilaiden iän mediaani oli 67 vuotta (vaihteluväli: 30–88 vuotta), ja miehiä oli 68 % potilaista ja valkoihoisia 90 %. Kaikkien potilaiden lähtötilanteen ECOG-suorituskykyluokka oli 0 tai 1. Ajan mediaani diagnoosista oli 91 kuukautta, ja aiempien hoitokertojen lukumäärän mediaani oli 2 (vaihteluväli: 1–13 hoitoa). Lähtötilanteessa 58 %:lla potilaista oli vähintään yksi ≥ 5 cm:n kasvain. Kolmellakymmenelläkahdella prosentilla potilaista oli 17p-deleetio (näistä potilaista 50 %:lla oli 17p-deleetio/TP53-mutaatio), 24 %:lla oli 11q-deleetio, ja 47 %:lla oli mutatoitumaton IGHV-geeni.
Riippumattoman arviointikomitean IWCLL-kriteerien mukaisesti arvioima taudin etenemättömyysaika (progression free survival, PFS) osoitti kuoleman riskin tai taudin etenemisriskin pienentyneen IMBRUVICA-hoitoryhmässä tilastollisesti merkitsevästi 78 %. Kokonaiselossaoloajan (overall survival, OS) analyysi osoitti kuoleman riskin pienentyneen IMBRUVICA-hoitoryhmässä tilastollisesti merkitsevästi 57 %. Tutkimuksen PCYC‑1112‑CA tehon tulokset esitetään taulukossa 16.
| Taulukko 16: Kroonista lymfaattista leukemiaa sairastavien potilaiden tehon tulokset (tutkimus PCYC‑1112‑CA) | ||
| Päätetapahtuma | IMBRUVICA N = 195 | Ofatumumabi N = 196 |
| Taudin etenemättömyysaika (progression-free survival, PFS) mediaani | Ei saavutettu | 8,1 kuukautta |
| Riskisuhde (hazard ratio, HR) = 0,215 [95 %:n luottamusväli: 0,146; 0,317] | ||
| Kokonaiselossaoloaikaa | Riskisuhde (hazard ratio, HR) = 0,434 [95 %:n luottamusväli: 0,238; 0,789]b Riskisuhde (hazard ratio, HR) = 0,387 [95 %:n luottamusväli: 0,216; 0,695]c | |
| Kokonaisvastelukud, e (%) | 42,6 | 4,1 |
| Kokonaisvasteluku, mukaan lukien osittainen vaste, johon liittyy lymfosytoosi (%) | 62,6 | 4,1 |
PR = osittainen vaste (partial response) a Kummassakaan hoitoryhmässä ei saavutettu kokonaiselossaoloajan mediaania, kokonaiselossaoloajan p < 0,005. b Potilaat, jotka oli satunnaistettu saamaan ofatumumabia, arvioitiin tällöin IMBRUVICA-hoitoa aloitettaessa. c Herkkyysanalyysi, jossa ofatumumabihoidosta pois siirtyneitä potilaita ei arvioitu ensimmäisen IMBRUVICA-annoksen ottamispäivänä. d IRC:n mukaan. Vasteen varmistaminen edellytti TT-kuvauksen uusimista. e Kaikki saavutetut osittaiset vasteet; p < 0,0001 kokonaisvasteluvun suhteen. Seuranta-ajan mediaani tutkimuksen aikana = 9 kuukautta | ||
Teho oli samankaltainen potilaiden kaikissa tutkituissa osajoukoissa, mukaan lukien potilaat, joilla oli tai ei ollut 17p-deleetio, joka oli ennalta määritelty ositustekijä (taulukko 17).
| Taulukko 17: Etenemättömyysajan analyysi osajoukoittain (tutkimus PCYC‑1112‑CA) | |||
| N | Riskisuhde | 95 % CI | |
| Kaikki tutkittavat | 391 | 0,210 | (0,143; 0,308) |
| 17P-deleetio | |||
| Kyllä | 127 | 0,247 | (0,136; 0,450) |
| Ei | 264 | 0,194 | (0,117; 0,323) |
| Puriinianalogihoitoon reagoimaton sairaus | |||
| Kyllä | 175 | 0,178 | (0,100; 0,320) |
| Ei | 216 | 0,242 | (0,145; 0,404) |
| Ikä | |||
| < 65 | 152 | 0,166 | (0,088; 0,315) |
| ≥ 65 | 239 | 0,243 | (0,149; 0,395) |
| Aikaisempien hoitojen lukumäärä | |||
| < 3 | 198 | 0,189 | (0,100; 0,358) |
| ≥ 3 | 193 | 0,212 | (0,130; 0,344) |
| Suurikokoinen kasvain | |||
| < 5 cm | 163 | 0,237 | (0,127; 0,442) |
| ≥ 5 cm | 225 | 0,191 | (0,117; 0,311) |
| Riskisuhde perustuu osittamattomaan analyysiin | |||
Taudin etenemättömyysajan (PFS) Kaplan-Meier-käyrä esitetään kuvassa 12.
Kuva 12: Tutkimuksen PCYC‑1112-CA taudin etenemättömyysajan (hoitoaikeen mukainen potilasryhmä, ITT) Kaplan-Meier-käyrä
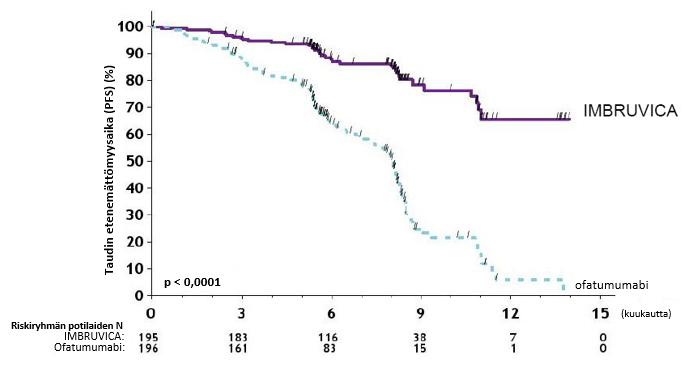
Loppuanalyysi 65 kuukauden seurannan jälkeen
Tutkimuksen PCYC-1112-CA tutkimuksenaikaisen seuranta-ajan mediaani oli 65 kuukautta, ja IMBRUVICA-ryhmän potilailla havaittiin tutkijan arvion perusteella kuoleman tai taudin etenemisen riskin 85 %:n vähenemä. Tutkijan IWCLL-kriteerien mukaan arvioiman taudin etenemättömyysajan mediaani oli IMBRUVICA-ryhmässä 44,1 kuukautta (95 %:n luottamusväli [38,47; 56,18]) ja ofatumumabiryhmässä 8,1 kuukautta (95 %:n luottamusväli [7,79; 8,25]); riskisuhde (HR) = 0,15 (95 %:n luottamusväli [0,11; 0,20]). Taudin etenemättömyysajan päivitetty Kaplan–Meier-käyrä on kuvassa 13. Tutkijan arvioima kokonaisvasteluku oli IMBRUVICA-ryhmässä 87,7 % verrattuna 22,4 %:iin ofatumumabiryhmässä. Loppuanalyysissa yhteensä 196 ofatumumabihoitoryhmään alun perin satunnaistetuista tutkittavista 133 tutkittavaa (67,9 %) siirtyi ibrutinibiristikkäishoitoon. Tutkijan IWCLL-kriteerien mukaan arvioima etenemättömyysaika (mediaani) ennen toista taudin etenemistä (PFS2, aika satunnaistamisesta ensimmäisen antineoplastisen hoidon jälkeiseen etenemättömyysajan tapahtumaan) oli IMBRUVICA-ryhmässä 65,4 kuukautta (95 %:n luottamusväli [51,61; ei arvioitavissa]) ja ofatumumabiryhmässä 38,5 kuukautta (95 %:n luottamusväli [19,98; 47,24]); riskisuhde (HR) = 0,54 (95 %:n luottamusväli [0,41; 0,71]). IMBRUVICA-ryhmässä kokonaiselossaoloajan mediaani oli 67,7 kuukautta (95 %:n luottamusväli [61,0; ei arvioitavissa]).
Tutkimuksessa PCYC-1112-CA ibrutinibin hoitoteho oli yhdenmukainen kaikilla suuren riskin potilailla, joilla oli 17p-deleetio/TP53-mutaatio, 11q-deleetio ja/tai mutatoitumaton IGHV-geeni.
Kuva 13: Tutkimuksen PCYC‑1112‑CA taudin etenemättömyysajan 65 kuukauden seurannan (hoitoaikeen mukainen potilasjoukko) loppuanalyysin Kaplan–Meier-käyrä
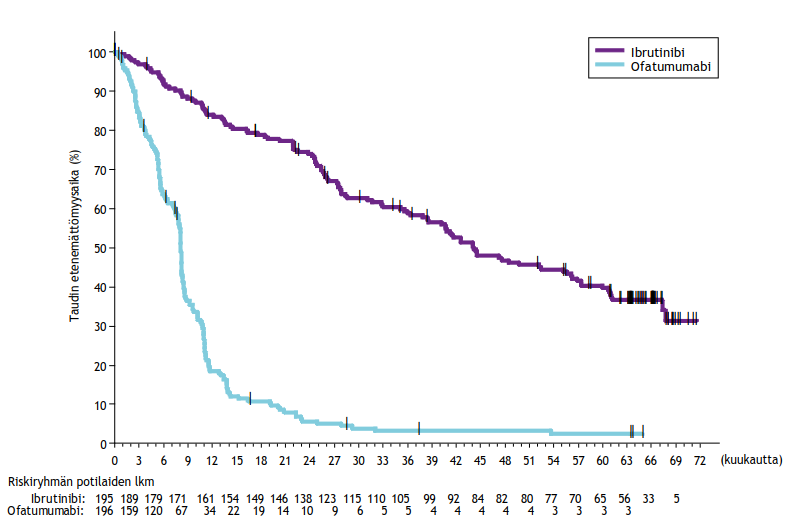
Yhdistelmähoito
IMBRUVICA-valmisteen turvallisuutta ja tehoa krooniseen lymfaattiseen leukemiaan aiemmin hoitoa saaneille potilaille arvioitiin edelleen satunnaistetussa, kaksoissokkoutetussa, vaiheen 3 monikeskustutkimuksessa, jossa IMBRUVICA-valmistetta yhdistelmänä bendamustiinin ja rituksimabin (BR) kanssa verrattiin lumevalmisteen, bendamustiinin ja rituksimabin yhdistelmään (tutkimus CLL3001). Potilaat (n = 578) satunnaistettiin suhteessa 1:1 saamaan joko IMBRUVICA-valmistetta 420 mg:n annoksina päivittäin tai lumevalmistetta yhdistelmänä bendamustiinin ja rituksimabin kanssa, kunnes tauti eteni tai potilaalle ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Kaikki potilaat saivat bendamustiinin ja rituksimabin yhdistelmää enintään kuusi 28 päivän pituista hoitosykliä. Bendamustiinia annettiin annoksina 70 mg/m2 laskimoon 30 minuutin kestoisina infuusioina hoitosyklin 1 päivinä 2 ja 3 sekä hoitosyklien 2–6 päivinä 1 ja 2. Hoitosyklejä annettiin enintään 6. Rituksimabia annettiin annoksena 375 mg/m2 ensimmäisen hoitosyklin päivänä 1 ja annoksena 500 mg/m2 hoitosyklien 2–6 päivänä 1. Yhdeksänkymmentä potilasta, jotka satunnaistettiin saamaan lumevalmisteen, bendamustiinin ja rituksimabin yhdistelmää, siirtyi IMBRUVICA-hoitoon sen jälkeen, kun riippumaton arviointikomitea oli vahvistanut taudin etenevän. Potilaiden iän mediaani oli 64 vuotta (vaihteluväli: 31–86 vuotta), ja miehiä oli 66 % potilaista ja valkoihoisia 91 %. Kaikkien potilaiden ECOG-toimintakykyluokka oli lähtötilanteessa 0 tai 1. Ajan mediaani diagnoosista oli 6 vuotta, ja aiempien hoitokertojen lukumäärän mediaani oli 2 (vaihteluväli: 1–11 hoitoa). Lähtötilanteessa 56 %:lla potilaista oli vähintään yksi ≥ 5 cm:n kasvain, ja 26 %:lla potilaista oli 11q-deleetio.
Riippumaton arviointikomitea arvioi taudin etenemättömyysaikaa (progression free survival, PFS) IWCLL-kriteerien mukaisesti. Tutkimuksen CLL3001 tehoa koskevat tulokset esitetään taulukossa 18.
| Taulukko 18: Kroonista lymfaattista leukemiaa sairastavien potilaiden hoidon tehoa koskevat tulokset (tutkimus CLL3001) | ||
| Päätetapahtuma | IMBRUVICA + BR N = 289 | Lumevalmiste + BR N = 289 |
| Taudin etenemättömyysaikaa | ||
| Mediaani (95 %:n luottamusväli), kuukautta | Ei saavutettu | 13,3 (11,3; 13,9) |
| Riskisuhde (HR) = 0,203 [95 %:n luottamusväli: 0,150; 0,276] | ||
| Kokonaisvastelukub % | 82,7 | 67,8 |
| Kokonaiselossaoloaika (Overall Survival, OS)c | Riskisuhde (HR) = 0,628 [95 %:n luottamusväli: 0,385; 1,024] | |
a Riippumattoman arviointikomitean (IRC) arvio. b Riippumattoman arviointikomitean (IRC) arvio, kokonaisvasteluku (ORR) (täydellinen vaste, täydellinen vaste, jossa luuydin palautunut epätäydellisesti, nodulaarinen osittainen vaste, osittainen vaste). c Kummassakaan hoitoryhmässä ei saavutettu kokonaiselossaoloajan mediaania. | ||
Waldenströmin makroglobulinemia
Monoterapia
IMBRUVICA-valmisteen turvallisuutta ja tehoa Waldenströmin makroglobulinemian (IgM‑immunoglobuliinia tuottava lymfoplasmasyyttinen lymfooma) hoidossa arvioitiin 63 aiempaa hoitoa saaneella potilaalla avoimessa, yhden ryhmän monikeskustutkimuksessa. Potilaiden iän mediaani oli 63 vuotta (vaihteluväli: 44–86 vuotta), ja miehiä oli 76 % potilaista ja valkoihoisia 95 %. Kaikkein potilaiden ECOG-suorituskykyluokka oli lähtötilanteessa 0 tai 1. Ajan mediaani diagnoosista oli 74 kuukautta, ja aiempien hoitokertojen lukumäärän mediaani oli 2 (vaihteluväli: 1–11 hoitoa). Seerumin IgM-arvon mediaani oli lähtötilanteessa 35 g/l, ja 60 %:lla potilaista oli anemia (hemoglobiini ≤ 110 g/l eli 6,8 mmol/l).
IMBRUVICA-hoitoa annettiin suun kautta 420 mg:n annoksina kerran vuorokaudessa, kunnes tauti eteni tai potilaalle ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Tämän tutkimuksen ensisijainen päätetapahtuma oli tutkijan arvioima kokonaisvasteluku (overall response rate, ORR). Kokonaisvastelukua ja vasteen kestoaikaa (duration of response, DOR) arvioitiin kolmannessa Waldenströmin makroglobulinemiaa käsitelleessä kansainvälisessä työryhmässä (the Third International Workshop of Waldenström’s Macroglobulinaemia) käyttöön otettujen kriteerien mukaisesti. Vasteet IMBRUVICA-hoitoon esitetään taulukossa 19.
| Taulukko 19: Waldenströmin makroglobulinemiaa sairastavien potilaiden kokonaisvasteluku (ORR) ja vasteen kestoaika (duration of response, DOR) | |
| Yhteensä (N = 63) | |
| ORR (%) | 87,3 |
| 95 %:n luottamusväli (%) | (76,5; 94,4) |
| Erittäin hyvä osittainen vaste (VGPR) (%) | 14,3 |
| Osittainen vaste (PR) (%) | 55,6 |
| Vähäinen vaste (MR) (%) | 17,5 |
| Vasteen kestoajan mediaani, kuukautta (vaihteluväli) | NR (0,03+, 18,8+) |
NR = ei saavutettu (not reached); MR = vähäinen vaste (minor response); PR = osittainen vaste (partial response); VGPR = erittäin hyvä osittainen vaste (very good partial response); ORR = MR+PR+VGPR Seuranta-ajan mediaani tutkimuksen aikana = 14,8 kuukautta | |
Ajan mediaani vasteen alkamiseen oli 1,0 kuukausi (vaihteluväli: 0,7–13,4 kuukautta).
Myös riippumaton arviointikomitea (Independent Review Committee, IRC) arvioi tehoa koskevat tulokset, ja kokonaisvasteluvuksi todettiin 83 %, jolloin erittäin hyvän osittaisen vasteen osuus oli 11 % ja osittaisen vasteen osuus 51 %.
Yhdistelmähoito
IMBRUVICA-valmisteen turvallisuutta ja tehoa Waldenströmin makroglobulinemian hoidossa arvioitiin lisäksi aiemmin hoitamattomilla tai aiempaa hoitoa saaneilla Waldenströmin makroglobulinemiaa sairastavilla potilailla satunnaistetussa, kaksoissokkoutetussa vaiheen 3 monikeskustutkimuksessa, jossa IMBRUVICA-valmisteen ja rituksimabin yhdistelmää verrattiin lumelääkkeen ja rituksimabin yhdistelmään (PCYC‑1127‑CA). Potilaat (n = 150) satunnaistettiin suhteessa 1:1 saamaan joko 420 mg IMBRUVICA-valmistetta päivittäin tai lumelääkettä yhdistelmänä rituksimabin kanssa, kunnes tauti eteni tai potilaalle ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Rituksimabia annettiin viikoittain annos 375 mg/m2 neljänä peräkkäisenä viikkona (viikot 1–4). Tämän jälkeen annettiin toinen hoitojakso, jossa rituksimabia annettiin viikoittain neljänä peräkkäisenä viikkona (viikot 17–20).
Potilaiden iän mediaani oli 69 vuotta (vaihteluväli: 36–89 vuotta), ja miehiä oli 66 % potilaista ja valkoihoisia 79 %. Yhdeksälläkymmenelläkolmella prosentilla potilaista ECOG-toimintakykyluokka oli lähtötilanteessa 0 tai 1, ja 7 %:lla potilaista ECOG-toimintakyky oli lähtötilanteessa 2. Neljäkymmentäviisi prosenttia potilaista ei ollut saanut aiempaa hoitoa, ja 55 % potilaista oli saanut aiempaa hoitoa. Ajan mediaani diagnoosista oli 52,6 kuukautta (aiemmin hoitamattomat potilaat = 6,5 kuukautta, ja aiempaa hoitoa saaneet potilaat = 94,3 kuukautta). Aiempaa hoitoa saaneiden potilaiden aiempien hoitojen lukumäärän mediaani oli 2 (vaihteluväli: 1–6 hoitoa). Seerumin IgM-arvon mediaani oli lähtötilanteessa 32 g/l (vaihteluväli: 6–83 g/l), ja 63 %:lla potilaista oli anemia (hemoglobiiniarvo ≤ 110 g/l eli 6,8 mmol/l). MYD88 L265P ‑mutaatioita oli 77 %:lla potilaista, niitä ei ollut 13 %:lla potilaista, ja 9 %:lla mutaatiostatus ei ollut arvioitavissa.
Ensisijaisessa analyysissa seuranta-ajan mediaani oli 26,5 kuukautta ja riippumattoman arviointikomitean arvioima etenemättömyysajan (PFS) riskisuhde oli 0,20 (95 %:n luottamusväli [0,11; 0,38]). Taudin etenemättömyysajan riskisuhteet aiemmin hoitamattomilla potilailla, aiempaa hoitoa saaneilla potilailla sekä potilailla, joilla oli tai ei ollut MYD88 L265P ‑mutaatioita, olivat yhdenmukaiset hoitoaikeen mukaiselle potilasjoukolle raportoidun taudin etenemättömyysajan riskisuhteen kanssa.
3. tai 4. asteen infuusioon liittyviä reaktioita havaittiin 1 %:lla IMBRUVICA-valmisteen ja rituksimabin yhdistelmää saaneista potilaista ja 16 %:lla lumelääkkeen ja rituksimabin yhdistelmää saaneista potilaista.
Kasvainsolujen äkillistä lisääntymistä, joka oli havaittavissa IgM-pitoisuuden suurenemisena, esiintyi 8,0 %:lla tutkittavista IMBRUVICA-valmisteen ja rituksimabin yhdistelmää saaneessa ryhmässä ja 46,7 %:lla tutkittavista lumelääkkeen ja rituksimabin yhdistelmää saaneessa ryhmässä.
Loppuanalyysi 63 kuukauden seurannan jälkeen
63 kuukauden kokonaisseuranta-ajan tehoa koskevat, riippumattoman arviointikomitean PCYC‑1127‑CA-tutkimuksen loppuanalyysin ajankohtana arvioimat tulokset esitetään taulukossa 20, ja etenemättömyysajan (PFS) Kaplan‑Meierin käyrät esitetään kuvassa 14. Etenemättömyysajan riskisuhteet aiemmin hoitamattomien potilaiden (0,31 [95 %:n luottamusväli: 0,14; 0,69]) ja aiemmin hoidettujen potilaiden (0,22 [95 %:n luottamusväli: 0,11; 0,43]) osalta olivat yhdenmukaiset hoitoaikeen mukaisen (ITT) potilasjoukon etenemättömyysajan riskisuhteen kanssa.
| Taulukko 20: Tutkimuksen PCYC‑1127‑CA tehoa koskevat tulokset (loppuanalyysi*) | ||
| Päätetapahtuma | IMBRUVICA + rituksimabi N = 75 | Lumelääke + rituksimabi N = 75 |
| Taudin etenemättömyysaikaa, b | ||
| Tapahtumien lukumäärä (%) | 22 (29) | 50 (67) |
| Mediaani (95 %:n luottamusväli), kk | Ei saavutettu | 20,3 (13,0; 27,6) |
| Riskisuhde (HR) (95 %:n luottamusväli) | 0,25 (0,15; 0,42) | |
| p‑arvo | < 0,0001 | |
| Aika seuraavaan hoitoon | ||
| Mediaani (95 %:n luottamusväli), kk | Ei saavutettu | 18,1 (11,1; 33,1) |
| Riskisuhde (HR) (95 %:n luottamusväli) | 0,1 (0,05; 0,21) | |
| Paras kokonaisvasteluku (%) | ||
| CR | 1,3 | 1,3 |
| VGPR | 29,3 | 4,0 |
| PR | 45,3 | 25,3 |
| MR | 16,0 | 13,3 |
| Kokonaisvastelukuc (CR, VGPR, PR, MR) (%) | 69 (92,0) | 33 (44,0) |
| Kokonaisvasteen keston mediaani, kk (vaihteluväli) | Ei saavutettu (2,7; 58,9+) | 27,6 (1,9; 55,9+) |
| Vasteluku (CR, VGPR, PR)c, d (%) | 57 (76,0) | 23 (30,7) |
| Vasteen keston mediaani, kk (vaihteluväli) | Ei saavutettu (1,9+; 58,9+) | Ei saavutettu (4,6; 49,7+) |
| Hemoglobiiniarvon pitkäkestoinen paraneminenc, e (%) | 77,3 | 42,7 |
CR = täydellinen vaste (complete response); MR = vähäinen vaste (minor response); PR = osittainen vaste (partial response); VGPR = erittäin hyvä osittainen vaste (very good partial response) * Seuranta-ajan mediaani tutkimuksen aikana = 49,7 kuukautta. a Riippumattoman arviointikomitean (IRC) arvio. b 4 vuoden etenemättömyysajan estimaatit olivat IMBRUVICA-valmistetta ja rituksimabia saaneessa ryhmässä 70,6 % (95 %:n luottamusväli: 58,1; 80,0) vs lumelääkettä ja rituksimabia saaneessa ryhmässä 25,3 % (95 %:n luottamusväli: 15,3; 36,6). c Vastelukuun liittyvä p‑arvo oli < 0,0001. d Aiemmin hoitamattomien potilaiden vasteluku oli IMBRUVICA-valmistetta ja rituksimabia saaneessa ryhmässä 76 % vs lumelääkettä ja rituksimabia saaneessa ryhmässä 41 %, ja aiemmin hoitoa saaneiden potilaiden vasteluku oli IMBRUVICA-valmistetta ja rituksimabia saaneessa ryhmässä 76 % vs lumelääkettä ja rituksimabia saaneessa ryhmässä 22 %. e Määritelty ≥ 20 g/l lisäykseksi lähtötilanteesta lähtötilanteen arvosta riippumatta tai suurenemiseksi arvoon > 110 g/l sekä ≥ 5 g/l lisäykseksi, jos lähtötilanteen arvo oli ≤ 110 g/l. | ||
Kuva 14: Tutkimuksen PCYC‑1127‑CA taudin etenemättömyysajan (hoitoaikeen mukainen potilasjoukko) Kaplan‑Meier-käyrät (loppuanalyysi)
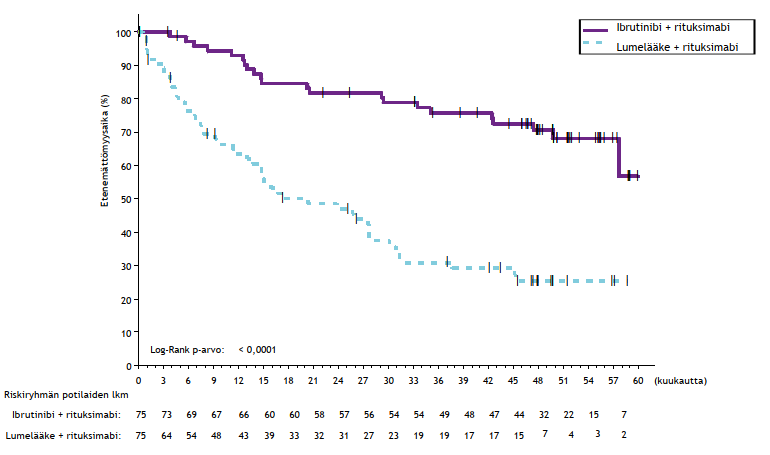
Tutkimuksessa PCYC-1127-CA oli erillinen monoterapiaryhmä, jossa oli 31 Waldenströmin makroglobulinemiaa sairastavaa potilasta. Nämä potilaat olivat saaneet aiempaa hoitoa, heille aiemmin annettu rituksimabia sisältänyt hoito oli epäonnistunut, ja he saivat IMBRUVICA-monoterapiaa. Potilaiden iän mediaani oli 67 vuotta (vaihteluväli: 47–90 vuotta). Kahdeksallakymmenelläyhdellä prosentilla potilaista ECOG-toimintakykyluokka oli lähtötilanteessa 0 tai 1, ja 19 %:lla potilaista ECOG-toimintakyky oli lähtötilanteessa 2. Aiempien hoitojen lukumäärän mediaani oli 4 (vaihteluväli: 1–7 hoitoa). Tutkimuksen PCYC-1127-CA monoterapiaryhmässä 61 kuukauden kokonaisseurannan aikana havaittu vasteluku oli riippumattoman arviointikomitean arvion perusteella 77 % (täydellinen vaste 0 %, erittäin hyvä osittainen vaste 29 %, osittainen vaste 48 %). Vasteen keston mediaani oli 33 kuukautta (vaihteluväli 2,4–60,2+ kuukautta). Monoterapiaryhmässä kokonaisvasteluku oli riippumattoman arviointikomitean arvion perusteella 87 % (täydellinen vaste 0 %, erittäin hyvä osittainen vaste 29 %, osittainen vaste 48 %, vähäinen vaste 10 %). Kokonaisvasteen keston mediaani oli 39 kuukautta (vaihteluväli 2,07–60,2+ kuukautta).
Pediatriset potilaat
IMBRUVICA-valmisteen turvallisuutta, tehoa ja farmakokinetiikkaa arvioitiin uusiutunutta tai hoitoon reagoimatonta kypsää B-soluista non-Hodgkin-lymfoomaa sairastavilla pediatrisilla ja nuorilla aikuisilla potilailla kaksiosaisessa avoimessa vaiheen 3 monikeskustutkimuksessa (LYM3003), jossa IMBRUVICA-valmistetta annettiin yhdistelmänä runkohoitona annetun rituksimabista, ifosfamidista, karboplatiinista, etoposidista ja deksametasonista koostuneen hoito-ohjelman (RICE) tai rituksimabista, vinkristiinistä, ifosfamidista, karboplatiinista, idarubisiinista ja deksametasonista koostuneen hoito-ohjelman (RVICI) kanssa.
Tutkimuksen 1. osassa (21 iältään 3–17-vuotiasta potilasta) arvioitiin 2. osassa käytettävää annosta (51 iältään 3–19-vuotiasta potilasta) (ks. kohta Farmakokinetiikka).
Tutkimuksen 2. osassa potilaat satunnaistettiin suhteessa 2:1 saamaan joko IMBRUVICA-valmistetta annoksena 440 mg/m2 päivittäin (iältään alle 12-vuotiaita) tai 329 mg/m2 (iältään 12-vuotiaita ja vanhempia) yhdessä runkohoidon kanssa tai pelkästään runkohoitoa, kunnes jokin seuraavista toteutui: 3 hoitosykliä annettu kokonaan, kantasolusiirto, taudin eteneminen tai toksisuus, joka ei ollut hyväksyttävissä. Ensisijaista päätetapahtumaa eli elossaoloa ilman tapahtumia (event-free survival, EFS) koskevaa paremmuutta ei saavutettu, mikä viittaa siihen, ettei ibrutinibin lisäämisestä RICE- tai RVICI-hoitoon ole lisähyötyä (ks. kohta Annostus ja antotapa).
Farmakokinetiikka
Imeytyminen
Suun kautta otettu ibrutinibi imeytyy nopeasti, ja Tmax:n mediaani on 1–2 tuntia. Absoluuttinen biologinen hyötyosuus paastotilassa (n = 8) oli 2,9 % (90 %:n luottamusväli = 2,1–3,9), ja se kaksinkertaistui, kun lääke otettiin aterian yhteydessä. Ibrutinibin farmakokinetiikka ei ole merkittävästi erilainen erilaisia B-solusyöpiä sairastavilla potilailla. Ibrutinibialtistus suurenee annoksilla 840 mg:aan saakka. 560 mg:n annoksia käyttäneillä potilailla havaittu vakaan tilan AUC on (keskiarvo ± keskihajonta) 953 ± 705 ng∙h/ml. Paastotilassa otetun ibrutinibin altistus oli noin 60 % (AUClast) verrattuna altistukseen, kun lääke otettiin 30 minuuttia ennen runsasrasvaista aamiaista tai 30 minuuttia (ei paastotilassa) tai 2 tuntia tällaisen aamiaisen jälkeen.
Ibrutinibin liukoisuus on pH:sta riippuvaista siten, että suuremman pH-arvon yhteydessä liukoisuus on vähäisempää. Kun terveille tutkittaville annettiin paastotilassa 560 mg:n kerta-annos ibrutinibia sen jälkeen, kun he olivat käyttäneet omepratsolia annoksina 40 mg kerran päivässä 5 päivän ajan, geometristen keskiarvojen suhteet (90 %:n luottamusväli) pelkän ibrutinibin käyttöön verrattuna olivat AUC0-24-arvon osalta 83 % (68–102 %), AUClast-arvon osalta 92 % (78–110 %) ja Cmax-arvon osalta 38 % (26–53 %).
Jakautuminen
Ibrutinibista 97,3 % sitoutui palautuvasti ihmisen plasman proteiineihin in vitro, mikä ei ollut pitoisuuksilla 50–1 000 ng/ml konsentraatiosta riippuvainen. Vakaan tilan näennäinen jakautumistilavuus (Vd,ss/F) oli noin 10 000 l.
Metabolia
Ibrutinibi metaboloituu pääasiassa CYP3A4:n välityksellä, jolloin muodostuu dihydrodiolimetaboliitti, jolla on noin 15 kertaa heikompi BTK:ta estävä vaikutus ibrutinibiin verrattuna. CYP2D6:n osallistuminen ibrutinibin metaboliaan vaikuttaa olevan hyvin vähäistä.
Näin ollen varotoimet eri CYP2D6-genotyyppien suhteen eivät ole tarpeen.
Eliminaatio
Näennäinen puhdistuma (CL/F) on noin 1 000 l/h. Ibrutinibin puoliintumisaika on 4–13 tuntia.
Terveille tutkittaville annetusta radioaktiivisesti merkitystä [14C]‑ibrutinibikerta-annoksesta noin 90 % radioaktiivisuudesta erittyi 168 tunnin kuluessa, jolloin suurin osa (80 %) erittyi ulosteisiin ja < 10 % virtsaan. Radioaktiivisesti merkitystä lääkeaineesta noin 1 % erittyi muuttumattomana ibrutinibina ulosteisiin, mutta ei yhtään virtsaan.
Erityispotilasryhmät
Iäkkäät potilaat
Populaatiofarmakokinetiikka osoitti, että ikä ei vaikuta merkittävästi ibrutinibin puhdistumaan verenkierrosta.
Pediatriset potilaat
Farmakokineettiset tiedot osoittavat, että uusiutunutta tai hoitoon reagoimatonta kypsää B-soluista non-Hodgkin-lymfoomaa sairastavilla iältään 12-vuotiailla ja vanhemmilla lapsilla, jotka saivat päivittäin annoksen 329 mg/m2, sekä iältään 3-vuotiaista alle 12-vuotiaisiin päivittäin annoksen 440 mg/m2 saaneilla ibrutinibialtistus oli yleensä samansuuruinen kuin päivittäin 560 mg:n annoksia saaneilla aikuisilla potilailla havaittu altistus.
Sukupuoli
Populaatiofarmakokineettiset tiedot osoittivat, että sukupuoli ei vaikuta merkittävästi ibrutinibin puhdistumaan verenkierrosta.
Rotu
Tiedot ovat riittämättömiä, jotta voitaisiin arvioida rodun mahdollista vaikutusta ibrutinibin farmakokinetiikkaan.
Paino
Populaatiofarmakokineettiset tiedot osoittivat, että painolla (vaihteluväli: 41–146 kg; keskiarvo [keskihajonta]: 83 [19] kg) oli vähäinen vaikutus ibrutinibin puhdistumaan.
Munuaisten vajaatoiminta
Ibrutinibin munuaispuhdistuma on hyvin vähäistä, sillä virtsaan erittyy metaboliitteina < 10 % annoksesta. Munuaisten vajaatoimintaa sairastavilla henkilöillä ei ole tähän mennessä tehty spesifisiä tutkimuksia. Vaikeaa munuaisten vajaatoimintaa sairastavista tai dialyysihoitoa saavista potilaista ei ole tietoja (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Ibrutinibi metaboloituu maksassa. Maksan vajaatoimintaa koskenut tutkimus tehtiin syöpää sairastamattomilla tutkittavilla, joille annettiin paastotilassa 140 mg:n kerta-annos lääkevalmistetta. Maksan vajaatoiminnan vaikutuksessa oli huomattavia yksilöllisiä eroja, mutta ibrutinibialtistuksen (AUClast-arvon) havaittiin suurentuneen lievää maksan vajaatoimintaa sairastavilla tutkittavilla (n = 6, Child–Pugh-luokka A) keskimäärin 2,7-kertaiseksi, keskivaikeaa maksan vajaatoimintaa sairastavilla (n = 10, Child–Pugh-luokka B) keskimäärin 8,2-kertaiseksi ja vaikeaa maksan vajaatoimintaa sairastavilla (n = 8, Child–Pugh-luokka C) keskimäärin 9,8-kertaiseksi. Myös vapaan ibrutinibin osuus plasmassa suureni vajaatoiminnan vaikeusasteen myötä, jolloin lisäys oli lievää maksan vajaatoimintaa sairastavilla 3,0 %, keskivaikeaa maksan vajaatoimintaa sairastavilla 3,8 % ja vaikeaa maksan vajaatoimintaa sairastavilla 4,8 % verrattuna 3,3 %:iin tässä tutkimuksessa kaltaistetuilla terveillä verrokeilla. Altistuksen (AUCunbound, last-arvon) sitoutumattomalle ibrutinibille arvioidaan suurenevan vastaavasti lievää maksan vajaatoimintaa sairastavilla tutkittavilla 4,1-kertaiseksi, keskivaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla 9,8-kertaiseksi ja vaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla 13-kertaiseksi (ks. kohta Annostus ja antotapa).
Samanaikainen käyttö kuljetusproteiinien substraattien/estäjien kanssa
In vitro ‑tutkimukset osoittivat, että ibrutinibi ei ole P-gp:n eikä OCT2:ta lukuun ottamatta muiden merkittävien kuljetusproteiinien substraatti. Dihydrodiolimetaboliitti ja muut metaboliitit ovat P-gp:n substraatteja. Ibrutinibi on P-gp:n ja BCRP:n estäjä in vitro (ks. kohta Yhteisvaikutukset).
Prekliiniset tiedot turvallisuudesta
Rotalla ja koiralla tehdyissä 13 viikkoa kestäneissä tutkimuksissa havaittiin seuraavia haittavaikutuksia. Ibrutinibin todettiin aiheuttavan sekä rotille että koirille kummallekin lajille haittavaikutuksettomina (No Observed Adverse Effect Level, NOAEL) annoksina 30 mg/kg/vrk maha-suolikanavan vaikutuksia (pehmeät ulosteet/ripuli ja/tai tulehdus) ja lymfoidista vajetta. Kliinisen annoksen 560 mg/vrk keskialtistuksen (AUC) perusteella AUC-suhteet NOAEL-annoksilla olivat urosrotilla 2,6 ja naarasrotilla 21, ja uroskoirilla 0,4 ja naaraskoirilla 1,8. Pienimmän havaittavan vaikutuksen (Lowest Observed Effect Level, LOEL) aiheuttavan annoksen (60 mg/kg/vrk) marginaali on koiralla 3,6-kertainen (urokset) ja 2,3-kertainen (naaraat). Rottakokeissa urosrotalla havaittiin keskivaikeaa haiman rauhasrakkulasoluatrofiaa (katsotaan haitalliseksi) annoksilla ≥ 100 mg/kg (AUC-altistusmarginaali 2,6-kertainen), mutta sitä ei havaittu naarailla annoksiin 300 mg/kg/vrk saakka tutkittuna (AUC-altistusmarginaali 21,3-kertainen). Kun naarasrotat saivat annoksia ≥ 100 mg/kg/vrk (AUC-altistusmarginaali 20,3-kertainen), niillä havaittiin hohkaluun ja tiivisluun lievää vähenemistä. Kaikki maha-suolikanavan, lymfoidiset ja luuston löydökset korjautuivat 6–13 viikon palautumisjakson aikana. Haiman löydökset korjautuivat osittain vastaavan palautumisjakson aikana.
Nuorilla eläimillä ei ole tehty toksisuustutkimuksia.
Karsinogeenisuus/geenitoksisuus
Ibrutinibi ei ollut karsinogeeninen 6 kuukautta kestäneessä siirtogeenisillä (Tg.rasH2) hiirillä tehdyssä tutkimuksessa, jossa suun kautta annetut annokset olivat enintään 2 000 mg/kg/vrk. Altistusmarginaali oli tällöin noin 23-kertainen (urokset) – 37-kertainen (naaraat) ibrutinibiannoksia 560 mg/vrk käyttäneiden ihmisten AUC:hen verrattuna.
Ibrutinibilla ei ole bakteeri-, nisäkässolu- tai hiirikokeiden perusteella geenitoksisia ominaisuuksia.
Lisääntymistoksisuus
Tiineille rotille annettuna ibrutinibiin liittyi annoksella 80 mg/kg/vrk lisääntynyttä implantaation jälkeistä alkiokuolleisuutta ja viskeraalisten (sydämen ja suurten verisuonten) epämuodostumien ja luustomuutosten lisääntymistä altistusmarginaalilla, joka oli 14-kertainen 560 mg:n vuorokausiannoksia käyttäneiden potilaiden AUC:hen verrattuna. Ibrutinibiin liittyi annoksilla ≥ 40 mg/kg/vrk sikiön painon vähenemistä (AUC-suhde ≥ 5,6 verrattuna potilaiden käyttämään 560 mg:n vuorokausiannokseen). Sikiön NOAEL oli näin ollen 10 mg/kg/vrk (noin 1,3 kertaa ibrutinibin AUC annoksella 560 mg vuorokaudessa) (ks. kohta Raskaus ja imetys).
Tiineille kaniineille annettuna ibrutinibiin liittyi annoksella 15 mg/kg/vrk tai enemmän luuston epämuodostumia (yhteensulautuneet kehittyvän rintalastan osat), ja annoksella 45 mg/kg/vrk lisääntynyttä implantaation jälkeistä alkiokuolleisuutta. Ibrutinibi aiheutti kaniineille epämuodostumia annoksella 15 mg/kg/vrk (noin 2,0-kertainen altistus [AUC] manttelisolulymfoomaa sairastaville potilaille annettaviin ibrutinibiannoksiin 560 mg/vrk nähden ja 2,8-kertainen altistus kroonista lymfaattista leukemiaa tai Waldenströmin makroglobulinemiaa sairastavien potilaiden ibrutinibiannoksiin 420 mg/vrk nähden). Sikiön NOAEL oli näin ollen 5 mg/kg/vrk (noin 0,7 kertaa ibrutinibin AUC annoksella 560 mg vuorokaudessa) (ks. kohta Raskaus ja imetys).
Hedelmällisyys
Uros- tai naarasrotilla ei havaittu vaikutuksia hedelmällisyyteen tai lisääntymiskykyyn suurimmallakaan testatulla annoksella 100 mg/kg/vrk (ihmisen vastaava annos [Human Equivalent Dose, HED] 16 mg/kg/vrk).
Farmaseuttiset tiedot
Apuaineet
Tablettiydin
Vedetön kolloidinen piidioksidi
Kroskarmelloosinatrium
Laktoosimonohydraatti
Magnesiumstearaatti
Mikrokiteinen selluloosa
Povidoni
Natriumlauryylisulfaatti (E487)
Kalvopäällyste
IMBRUVICA 140 mg kalvopäällysteiset tabletit ja IMBRUVICA 420 mg kalvopäällysteiset tabletit
Makrogoli
Polyvinyylialkoholi
Talkki
Titaanidioksidi (E171)
Musta rautaoksidi (E172)
Keltainen rautaoksidi (E172)
IMBRUVICA 280 mg kalvopäällysteiset tabletit
Makrogoli
Polyvinyylialkoholi
Talkki
Titaanidioksidi (E171)
Musta rautaoksidi (E172)
Punainen rautaoksidi (E172)
IMBRUVICA 560 mg kalvopäällysteiset tabletit
Makrogoli
Polyvinyylialkoholi
Talkki
Titaanidioksidi (E171)
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
IMBRUVICA tabletti, kalvopäällysteinen
140 mg (L:kyllä) 28 fol (1974,54 €)
280 mg (L:kyllä) 28 fol (3768,48 €)
420 mg (L:kyllä) 28 fol (5562,42 €)
560 mg (L:kyllä) 28 fol (7356,35 €)
PF-selosteen tieto
Kaksi polyvinyylikloridilaminaatista (PVC) ja polyklooritrifluorieteenistä (PCTFE) / alumiinista valmistettua läpipainopakkausta, joissa kummassakin on 7 kalvopäällysteistä tablettia ja jotka on pakattu yhteen kartonkiseen taskupakkaukseen. Yksi kartonkikotelo sisältää (28 kalvopäällysteistä tablettia) 2 taskupakkausta.
Kaksi polyvinyylikloridilaminaatista (PVC) ja polyklooritrifluorieteenistä (PCTFE) / alumiinista valmistettua läpipainopakkausta, joissa kummassakin on 5 kalvopäällysteistä tablettia ja jotka on pakattu yhteen kartonkiseen taskupakkaukseen. Yksi kartonkikotelo sisältää (30 kalvopäällysteistä tablettia) 3 taskupakkausta.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
IMBRUVICA 140 mg kalvopäällysteiset tabletit
Keltaisenvihreä tai vihreä pyöreä tabletti (9 mm), jonka toiselle puolelle on kaiverrettu ”ibr” ja vastakkaiselle puolelle ”140”.
IMBRUVICA 280 mg kalvopäällysteiset tabletit
Purppuranvärinen pitkänomainen tabletti (pituus 15 mm ja leveys 7 mm), jonka toiselle puolelle on kaiverrettu ”ibr” ja vastakkaiselle puolelle ”280”.
IMBRUVICA 420 mg kalvopäällysteiset tabletit
Keltaisenvihreä tai vihreä pitkänomainen tabletti (pituus 17,5 mm ja leveys 7,4 mm), jonka toiselle puolelle on kaiverrettu ”ibr” ja vastakkaiselle puolelle ”420”.
IMBRUVICA 560 mg kalvopäällysteiset tabletit
Keltainen tai oranssi pitkänomainen tabletti (pituus 19 mm ja leveys 8,1 mm), jonka toiselle puolelle on kaiverrettu ”ibr” ja vastakkaiselle puolelle ”560”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
IMBRUVICA tabletti, kalvopäällysteinen
140 mg 28 fol
280 mg 28 fol
420 mg 28 fol
560 mg 28 fol
- Ylempi erityiskorvaus (100 %). Ibrutinibi: Kroonisen lymfaattisen leukemian, manttelisolulymfooman ja Waldenströmin makroglobulinemian hoito erityisin edellytyksin (1512).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Ibrutinibi: Aikuispotilaiden kroonisen lymfaattisen leukemian, uusiutuneen tai hoitoon reagoimattoman manttelisolulymfooman tai Waldenströmin makroglobulinemian hoito erityisin edellytyksin (390).
ATC-koodi
L01EL01
Valmisteyhteenvedon muuttamispäivämäärä
13.02.2026
Yhteystiedot
 JANSSEN-CILAG OY
JANSSEN-CILAG OY PL 15
02621 Espoo
020 753 1300
innovativemedicine.jnj.com/finland
jacfi@its.jnj.com