
DASATINIB KRKA tabletti, kalvopäällysteinen 20 mg, 50 mg, 70 mg
Vaikuttavat aineet ja niiden määrät
Dasatinib Krka 20 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 20 mg dasatinibia.
Apuaine(et), joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 26 mg laktoosia.
Dasatinib Krka 50 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 50 mg dasatinibia.
Apuaine(et), joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 66 mg laktoosia.
Dasatinib Krka 70 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 70 mg dasatinibia.
Apuaine(et), joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 92 mg laktoosia.
Dasatinib Krka 80 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 80 mg dasatinibia.
Apuaine(et), joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 105 mg laktoosia.
Dasatinib Krka 100 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 100 mg dasatinibia.
Apuaine(et), joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 131 mg laktoosia.
Dasatinib Krka 140 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 140 mg dasatinibia.
Apuaine(et), joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 184 mg laktoosia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Dasatinib Krka on tarkoitettu aikuisille potilaille:
- vastadiagnosoidun Philadelphia-kromosomipositiivisen (Ph+) kroonisessa vaiheessa olevan kroonisen myelooisen leukemian hoitoon.
- kroonisessa, akseleraatio- tai blastikriisivaiheessa olevan kroonisen myelooisen leukemian (KML) hoitoon silloin, kun aikaisempi hoito, imatinibi mukaan lukien, ei ole tuottanut tulosta tai potilas ei ole sietänyt sitä.
- Ph+ akuutin lymfaattisen leukemian (ALL) ja lymfaattisen blastikriisivaiheen KML:n hoitoon, kun aikaisempi hoito ei ole tuottanut tulosta tai potilas ei ole sietänyt sitä.
Dasatinib Krka on tarkoitettu pediatrisille potilaille:
- vastadiagnosoidun kroonisessa vaiheessa olevan Ph+ KML:n (Ph+ CP-KML) hoitoon tai kroonisessa vaiheessa olevan Ph+ KML:n hoitoon silloin, kun aikaisempi hoito, imatinibi mukaan lukien, ei ole tuottanut tulosta tai potilas ei ole sietänyt sitä.
- vastadiagnosoidun Ph+ ALL:n hoitoon yhdessä kemoterapian kanssa.
Ehto
Hoidon saa aloittaa leukemian diagnosointiin ja hoitoon perehtynyt lääkäri.
Annostus ja antotapa
Hoidon saa aloittaa leukemian diagnosointiin ja hoitoon perehtynyt lääkäri.
Annostus
Aikuiset
Suositeltu aloitusannos kroonisessa vaiheessa olevan KML:n hoidossa on 100 mg dasatinibia kerran vuorokaudessa.
Suositeltu aloitusannos akseleraatiovaiheessa, myelooisessa tai lymfaattisessa blastikriisivaiheessa (edenneessä vaiheessa) olevan KML:n tai Ph+ ALL:n hoidossa on 140 mg kerran vuorokaudessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat (Ph+ CP-KML ja Ph+ ALL)
Lasten ja nuorten annos määräytyy kehonpainon perusteella (ks. taulukko 1). Dasatinibia otetaan kerran vuorokaudessa suun kautta joko kalvopäällysteisinä dasatinibitabletteina tai dasatinibijauheena oraalisuspensiota varten. Painossa tapahtuvien muutosten vuoksi annos on laskettava uudelleen kolmen kuukauden välein tai tarvittaessa useammin. Tabletteja ei suositella potilaille, jotka painavat alle 10 kg. Näille potilaille on käytettävä jauhetta oraalisuspensiota varten. Annoksen suurentamista tai pienentämistä suositellaan yksilöllisen hoitovasteen ja potilaan sietokyvyn mukaan. Alle 1-vuotiaiden lasten hoidosta Dasatinib Krkalla ei ole kokemusta.
Kalvopäällysteiset dasatinibitabletit ja dasatinibijauhe oraalisuspensiota varten eivät ole bioekvivalentteja. Potilaat, jotka pystyvät nielemään tabletteja ja jotka haluavat vaihtaa dasatinibioraalisuspensiosta dasatinibitabletteihin ja potilaat, jotka eivät pysty nielemään tabletteja ja haluavat vaihtaa tableteista oraalisuspensioon, voivat vaihtaa lääkemuotoa edellyttäen, että lääkemuodon annostussuosituksia noudatetaan.
Pediatrisille potilaille suositeltava vuorokausittainen Dasatinib Krka ‑tablettien aloitusannos on esitetty taulukossa 1.
Taulukko 1: Dasatinib Krka ‑tablettien annostus pediatrisille potilaille, joilla on Ph+ CP-KML tai Ph+ ALL
| Kehonpaino (kg)a | Vuorokausiannos (mg) |
| 10 – alle 20 kg | 40 mg |
| 20 – alle 30 kg | 60 mg |
| 30 – alle 45 kg | 70 mg |
| vähintään 45 kg | 100 mg |
a Tablettia ei suositella potilaille, jotka painavat alle 10 kg. Näille potilaille on käytettävä jauhetta oraalisuspensiosta varten.
Hoidon kesto
Kliinisissä tutkimuksissa aikuisten, joilla oli Ph+ CP-KML, akseleraatiovaiheessa, myelooisessa tai lymfaattisessa blastikriisivaiheessa (edenneessä vaiheessa) oleva KML tai Ph+ ALL, ja pediatristen potilaiden, joilla oli Ph+ CP-KML, dasatinibihoitoa jatkettiin taudin etenemiseen saakka tai siihen saakka, kunnes potilas ei enää sietänyt sitä. Ei ole tutkittu, miten hoidon lopettaminen vaikuttaa pitkän aikavälin hoitotulokseen sen jälkeen, kun on ensin saavutettu sytogeneettinen tai molekulaarinen vaste (mukaan lukien täydellinen sytogeneettinen vaste [CCyR], merkittävä molekulaarinen vaste [MMR] ja molekulaarisen vasteen 4,5 login alenema [MR4,5]).
Kliinisissä tutkimuksissa pediatrisille potilaille, joilla oli Ph+ ALL, annettiin dasatinibihoitoa jatkuvana hoitona lisättynä peräkkäisiin kemoterapiahoito-ohjelman jaksoihin enintään kahden vuoden ajan. Niille potilaille, jotka saavat myöhemmin kantasolujen siirron, dasatinibihoitoa voidaan antaa vielä vuoden ajan kantasolujen siirron jälkeen.
Suositellun annostuksen mahdollistamiseksi Dasatinib Krka ‑valmistetta on saatavilla 20 mg:n, 50 mg:n, 70 mg:n, 80 mg:n, 100 mg:n ja 140 mg:n kalvopäällysteisinä tabletteina. Annoksen suurentamista tai pienentämistä suositellaan hoitovasteen ja potilaan sietokyvyn mukaan.
Annoksen suurentaminen
Kliinisissä, aikuisilla KML ja Ph+ ALL ‑potilailla tehdyissä tutkimuksissa annoksen suurentaminen 140 mg:aan kerran vuorokaudessa (kroonisen vaiheen KML) tai 180 mg:aan kerran vuorokaudessa (edenneen vaiheen KML tai Ph+ ALL) sallittiin potilailla, jotka eivät saavuttaneet hematologista tai sytogeneettistä vastetta suositeltua aloitusannostusta käytettäessä.
Annoksen suurentamista suositellaan taulukon 2 mukaisesti pediatrisille potilaille, joilla on Ph+ CP-KML, jotka eivät saavuta hematologista, sytogeneettistä ja molekulaarista vastetta nykyisissä hoitosuosituksissa mainittuina ajankohtina ja jotka sietävät hoidon.
Taulukko 2: Annoksen suurentaminen pediatrisille potilaille, joilla on Ph+ CP-KML
| Annos (suurin vuorokausiannos) | ||
| Aloitusannos | Suurennettu annos | |
| Tabletit | 40 mg | 50 mg |
| 60 mg | 70 mg | |
| 70 mg | 90 mg | |
| 100 mg | 120 mg | |
Annoksen suurentamista ei suositella pediatrisille Ph+ ALL ‑potilaille, sillä dasatinibihoitoa annetaan näille potilaille yhdessä kemoterapian kanssa.
Annoksen muuttaminen haittavaikutusten takia
Myelosuppressio
Kliinisissä tutkimuksissa hoito keskeytettiin, annosta pienennettiin tai tutkimushoito lopetettiin myelosuppression hoitamiseksi. Trombosyyttien ja punasolujen siirto suoritettiin tarvittaessa. Hematopoieettista kasvutekijää on käytetty potilailla, joilla oli resistentti myelosuppressio.
Ohjeet annoksen muuttamiseen aikuisille on esitetty taulukossa 3 ja pediatrisille potilaille, joilla on Ph+ CP-KML, taulukossa 4. Ohjeet pediatrisille Ph+ ALL ‑potilaille, joita hoidetaan yhdistelmähoidolla kemoterapian kanssa, ovat erillisessä kappaleessa taulukoiden jälkeen.
Taulukko 3: Aikuisten annoksen muuttaminen neutropeniassa ja trombosytopeniassa
Aikuiset, joilla on kroonisen vaiheen KML (aloitusannos 100 mg kerran vuorokaudessa) | B-Neut < 0,5 x 109/l ja/tai verihiutaleita < 50 x 109/l |
| |
Aikuiset, joilla on akseleraatio- ja blastikriisivaiheen KML ja Ph+ ALL (aloitusannos 140 mg kerran vuorokaudessa) | B-Neut < 0,5 x 109/l ja/tai verihiutaleita < 10 x 109/l |
| |
B-Neut: absoluuttinen neutrofiilien määrä
Taulukko 4: Annoksen muuttaminen neutropeniassa ja trombosytopeniassa pediatrisille
potilaille, joilla on Ph+ CP-KML
1. Jos sytopenia jatkuu yli 3 viikkoa, varmista, liittyykö sytopenia leukemiaan (luuydinaspiraatio tai -biopsia). 2. Jos sytopenia ei liity leukemiaan, keskeytä hoito, kunnes B-Neut ≥ 1,0 x 109/l ja verihiutaleita ≥ 75 x 109/l ja jatka alkuperäisellä aloitusannoksella tai pienennetyllä annoksella. 3. Jos sytopenia ilmenee uudelleen, toista luuydinaspiraatio tai -biopsia ja jatka hoitoa pienennetyllä annoksella. | Annos (suurin vuorokausiannos) | |||
Alkuperäinen aloitusannos | Yhden annostason pienennys | Kahden annostason pienennys | ||
| Tabletit | 40 mg | 20 mg | * | |
| 60 mg | 40 mg | 20 mg | ||
| 70 mg | 60 mg | 50 mg | ||
| 100 mg | 80 mg | 70 mg | ||
B-Neut: absoluuttinen neutrofiilien määrä
*pienempää annosta ei ole saatavilla tablettina
Jos asteen ≥ 3 neutropenia tai trombosytopenia ilmenee uudelleen täydellisen hematologisen vasteen (CHR) aikana pediatrisilla potilailla, joilla on Ph+ CP-KML, dasatinibihoito on keskeytettävä, ja sitä voidaan jatkaa myöhemmin pienennetyllä annoksella. Annosta on tarpeen mukaan pienennettävä tilapäisesti, jos potilaalla on kohtalainen sytopenia ja jos potilas on saanut vasteen.
Annoksen muuttamista ei suositella pediatrisille potilaille, joilla on Ph+ ALL, jos hoitoon liittyy hematologista 1.–4. asteen toksisuutta. Jos neutropenian ja/tai trombosytopenian takia seuraava hoitojakso viivästyy yli 14 päivää, Dasatinib Krka ‑hoito on keskeytettävä, ja sitä jatketaan samalla annoksella, kun seuraava hoitojakso aloitetaan. Jos neutropenia ja/tai trombosytopenia jatkuvat ja seuraava hoitojakso viivästyy vielä ylimääräiset 7 päivää, luuydin on arvioitava, jotta voidaan arvioida solukkuutta ja blastien prosenttiosuutta. Jos luuytimen solukkuus on < 10 %, Dasatinib Krka ‑hoito on keskeytettävä, kunnes B-Neut > 500/mikrol (0,5 x 109/l), jolloin hoitoa voidaan jatkaa täydellä annoksella. Jos luuytimen solukkuus on > 10 %, Dasatinib Krka ‑hoidon jatkamista voidaan harkita.
Ei-hematologiset haittavaikutukset
Jos keskivaikeita 2. asteen ei-hematologisia haittavaikutuksia ilmenee dasatinibihoidon aikana, hoito tulee keskeyttää, kunnes haittavaikutus on poistunut tai on palattu lähtötilanteeseen. Hoito tulee aloittaa uudelleen samalla annoksella, jos haittavaikutus esiintyi ensimmäistä kertaa, ja pienennetyllä annoksella haittavaikutuksen uusiutuessa. Jos dasatinibihoidon yhteydessä kehittyy vaikeita 3. tai 4. asteen ei-hematologisia haittavaikutuksia, hoito täytyy keskeyttää, kunnes haittavaikutus on poistunut. Sen jälkeen hoitoa voidaan jatkaa pienennetyllä annoksella ottaen huomioon, kuinka vaikea haittavaikutus alun perin oli. Kroonisessa vaiheessa olevan KML:n hoidossa potilaille, joiden annos oli 100 mg kerran vuorokaudessa, suositellaan annoksen pienentämistä 80 mg:aan kerran vuorokaudessa ja tarvittaessa annoksen pienentämistä edelleen 80 mg:sta kerran vuorokaudessa 50 mg:aan kerran vuorokaudessa. Edenneessä vaiheessa olevan KML:n tai Ph+ ALL:n hoidossa potilaille, joiden annos oli 140 mg kerran vuorokaudessa, suositellaan annoksen pienentämistä 100 mg:aan kerran vuorokaudessa ja tarvittaessa annoksen pienentämistä edelleen 100 mg:sta kerran vuorokaudessa 50 mg:aan kerran vuorokaudessa. Jos pediatrisilla potilailla, joilla on CP-KML, ilmenee ei-hematologisia haittavaikutuksia, on noudatettava yllä kuvattuja hematologisia haittavaikutuksia koskevia annoksen pienentämiseen liittyviä suosituksia. Pediatristen potilaiden, joilla on Ph+ ALL ja joilla on ilmennyt ei-hematologisia haittavaikutuksia, annosta pienennetään tarvittaessa yhdellä annostasolla noudattaen yllä kuvattuja hematologisia haittavaikutuksia koskevia annoksen pienentämiseen liittyviä suosituksia.
Pleuraeffuusio
Jos pleuraeffuusio diagnosoidaan, dasatinibihoito on keskeytettävä, kunnes potilas on tutkittu, oireeton tai on palattu lähtötilanteeseen. Jos pleuraeffuusio ei häviä noin viikon kuluessa, on harkittava diureetti- tai kortikosteroidikuuria tai molempia samanaikaisesti (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Ensimmäisellä kerralla oireiden häviämisen jälkeen dasatinibihoidon uudelleen aloittamista samalla annoksella tulee harkita. Sitä seuraavilla kerroilla (oireiden häviämisen jälkeen) dasatinibihoito tulee aloittaa uudelleen pienennetyllä annoksella (seuraava annostaso). Vakavien (3. tai 4. asteen) haittavaikutusten korjaannuttua hoito voidaan aloittaa uudelleen pienennetyllä annoksella ottaen huomioon, kuinka vaikea haittavaikutus alun perin oli.
Annoksen pienentäminen voimakkaiden CYP3A4:n estäjien samanaikaisen käytön takia
Voimakkaan CYP3A4:n estäjän ja greippimehun samanaikaista käyttöä Dasatinib Krka ‑valmisteen kanssa on vältettävä (ks. kohta Yhteisvaikutukset). Mahdollisuuksien mukaan on valittava jokin muu vaihtoehtoinen samanaikainen lääkitys, joka ei estä entsyymin toimintaa tai estää sitä mahdollisimman vähän. Jos Dasatinib Krka ‑valmistetta on annettava voimakkaan CYP3A4:n estäjän kanssa, annoksen pienentämistä on harkittava seuraavasti:
- 40 mg:aan päivittäin potilaille, jotka ottavat Dasatinib Krka 140 mg -tabletin päivittäin
- 20 mg:aan päivittäin potilaille, jotka ottavat Dasatinib Krka 100 mg -tabletin päivittäin
- 20 mg:aan päivittäin potilaille, jotka ottavat Dasatinib Krka 70 mg -tabletin päivittäin.
Jos potilas käyttää Dasatinib Krka ‑annosta 60 mg tai 40 mg päivittäin, on harkittava Dasatinib Krka ‑annoksen keskeyttämistä, kunnes hoito CYP3A4:n estäjällä lopetetaan, tai vaihtamista pienempään annokseen käyttämällä lääkemuotoa jauhe oraalisuspensiota varten. CYP3A4:n estäjän lopettamisen jälkeen on pidettävä noin 1 viikon lääkityskatko, kunnes Dasatinib Krka ‑hoitoa jatketaan.
Näiden pienennettyjen dasatinibiannosten odotetaan muuttavan käyrän alla olevaa pinta-alaa (AUC) tasolle, joka on havaittavissa ilman CYP3A4:n estäjiä; kliinisiä tietoja ei ole kuitenkaan saatavilla näistä annosmuutoksista niiden potilaiden osalta, jotka saavat voimakkaita CYP3A4:n estäjiä. Jos potilas ei siedä dasatinibihoitoa annoksen pienentämisen jälkeen, on joko hoito voimakkaalla CYP3A4:n estäjällä lopetettava tai dasatinibihoito keskeytettävä, kunnes hoito CYP3A4:n estäjällä lopetetaan. CYP3A4:n estäjän lopettamisen jälkeen on pidettävä noin 1 viikon lääkityskatko, kunnes dasatinibiannosta nostetaan.
Erityisryhmät
Iäkkäät potilaat
Kliinisesti merkittäviä ikään liittyviä farmakokineettisiä eroavaisuuksia ei ole havaittu tässä potilasryhmässä. Annoksen muuttaminen ei ole tarpeen iäkkäillä potilailla.
Maksan vajaatoiminta
Lievää, keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastaville potilaille voidaan antaa suositeltu aloitusannos. Dasatinib Krka ‑valmistetta tulee kuitenkin käyttää varoen maksan vajaatoimintaa sairastaville potilaille (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Kliinisiä tutkimuksia dasatinibin käytöstä ei ole tehty potilailla, joiden munuaisten toiminta on heikentynyt (tutkimuksesta vastadiagnosoidun kroonisen vaiheen KML-potilailla, oli poissuljettu potilaat, joiden seerumin kreatiniinipitoisuus > 3 kertaa normaalialueen yläraja ja tutkimuksista kroonisen vaiheen KML-potilailla, joilla aikaisempi hoito imatinibi mukaan lukien ei tuottanut tulosta tai potilaat eivät sietäneet sitä, oli poissuljettu potilaat, joilla seerumin kreatiniinipitoisuus > 1,5 kertaa normaalialueen yläraja). Koska dasatinibin ja sen metaboliittien munuaispuhdistuma on < 4 %, ei kokonaispuhdistuman odoteta pienenevän munuaisten vajaatoimintaa sairastavilla potilailla.
Antotapa
Dasatinib Krka annostellaan suun kautta.
Kalvopäällysteisiä tabletteja ei saa murskata, jakaa tai pureskella, vaan ne tulee annostuksen tasalaatuisuuden ylläpitämiseksi ja ihokosketuksen välttämiseksi niellä kokonaisina. Kalvopäällysteisiä tabletteja ei saa hajottaa, sillä hajotettuja tabletteja ottavilla potilailla altistus on pienempi kuin niillä, jotka nielevät tabletin kokonaisena. Dasatinibia on saatavilla myös oraalisuspensiona pediatrisille potilaille, joilla on Ph+ CP-KML tai Ph+ ALL, ja aikuispotilaille, joilla on CP-KML ja jotka eivät pysty nielemään tabletteja.
Dasatinib Krka voidaan ottaa joko aterian yhteydessä tai tyhjään mahaan, ja tabletit tulee ottaa johdonmukaisesti joko aamulla tai illalla (ks. kohta Farmakokinetiikka). Dasatinib Krka ‑valmistetta ei saa ottaa greipin tai greippimehun kanssa (ks. kohta Yhteisvaikutukset).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Kliinisesti merkittävät interaktiot
Dasatinibi on sytokromi P450 (CYP) 3A4:n substraatti ja inhibiittori. Siksi yhteisvaikutukset toisten samanaikaisesti annettavien, pääasiassa CYP3A4:n avulla metaboloituvien tai CYP3A4:n aktiivisuuteen vaikuttavien lääkevalmisteiden kanssa ovat mahdollista (ks. kohta Yhteisvaikutukset).
Dasatinibin samanaikainen käyttö voimakkaiden CYP3A4-entsyymiä inhiboivien lääkevalmisteiden tai aineiden kanssa (esim. ketokonatsoli, itrakonatsoli, erytromysiini, klaritromysiini, ritonaviiri, telitromysiini, greippimehu) saattaa lisätä dasatinibialtistusta. Sen vuoksi voimakkaan CYP3A4:n inhibiittorin antoa dasatinibia saaville potilaille ei suositella (ks. kohta Yhteisvaikutukset).
Dasatinibin samanaikainen käyttö CYP3A4-entsyymiä indusoivien lääkevalmisteiden kanssa (esim. deksametasoni, fenytoiini, karbamatsepiini, rifampisiini, fenobarbitaali tai (perinteiset) kasvirohdosvalmisteet, jotka sisältävät Hypericum perforatumia eli mäkikuismaa) saattaa vähentää dasatinibialtistusta huomattavasti ja mahdollisesti lisätä hoidon epäonnistumisen riskiä. Sen vuoksi dasatinibia saavia potilaita tulee hoitaa vaihtoehtoisilla, vähemmän CYP3A4-entsyymiä indusoivilla lääkevalmisteilla (ks. kohta Yhteisvaikutukset).
Samanaikainen dasatinibin ja CYP3A4:n substraatin käyttö saattaa lisätä altistusta CYP3A4:n substraatille. Sen vuoksi varovaisuutta tulee noudattaa annettaessa dasatinibia samanaikaisesti sellaisten CYP3A4:n substraattien kanssa, joilla on kapea terapeuttinen alue, kuten astemitsoli, terfenadiini, sisapridi, pimotsidi, kinidiini, bepridiili tai ergotalkaloidit (ergotamiini, dihydroergotamiini) (ks. kohta Yhteisvaikutukset).
Dasatinibin ja histamiini-2 (H2) ‑antagonistien (esim. famotidiini), protonipumpun estäjien (esim. omepratsoli) tai alumiinihydroksidin/magnesiumhydroksidin samanaikainen käyttö saattaa vähentää dasatinibialtistusta. Siksi H2-antagonisteja ja protonipumpun estäjiä ei suositella ja alumiinihydroksidi/magnesiumhydroksidi-valmisteet tulisi antaa vähintään 2 tuntia ennen dasatinibin antamista tai 2 tuntia dasatinibin antamisen jälkeen (ks. kohta Yhteisvaikutukset).
Erityisryhmät
Kerta-annoksella tehdyn farmakokineettisen tutkimuksen tulosten perusteella lievää, keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastaville potilaille voidaan antaa suositeltu aloitusannos (ks. kohta Farmakokinetiikka). Tässä kliinisessä tutkimuksessa olevien puutteiden vuoksi suositellaan varovaisuutta, kun dasatinibia annetaan maksan vajaatoimintaa sairastaville potilaille.
Tärkeät haittavaikutukset
Myelosuppressio
Dasatinibihoitoon voi liittyä anemiaa, neutropeniaa ja trombosytopeniaa. Näitä ilmenee aiemmin ja useammin potilailla, joilla on edenneen vaiheen KML tai Ph+ ALL, kuin potilailla, joilla on kroonisen vaiheen KML. Jos aikuispotilaalla, jota hoidetaan dasatinibimonoterapialla, on edenneen vaiheen KML tai Ph+ ALL, täydellinen verenkuva (TVK) tulee ottaa viikoittain kahden ensimmäisen kuukauden ajan ja sen jälkeen kuukausittain tai kliinisen tarpeen mukaan. Jos aikuisilla ja pediatrisilla potilailla on kroonisen vaiheen KML, täydellinen verenkuva tulee ottaa kahden viikon välein ensimmäisten 12 viikon ajan, sitten kolmen kuukauden välein tai kliinisen tarpeen mukaan. Pediatrisilta potilailta, joiden Ph+ ALL:aa hoidetaan dasatanibilla yhdessä kemoterapian kanssa, on otettava TVK ennen jokaisen kemoterapiajakson aloittamista ja kliinisen tarpeen mukaan. Kemoterapian konsolidaatiojaksojen aikana TVK on otettava joka toinen päivä veriarvojen palautumiseen asti (ks. kohdat Annostus ja antotapa ja Haittavaikutukset). Myelosuppressio on yleensä palautuva, ja tavallisesti se hoidetaan keskeyttämällä dasatinibihoito väliaikaisesti tai vähentämällä annosta.
Verenvuoto
Kroonisen vaiheen KML-potilaista (n = 548) viidellä dasatinibihoitoa saaneella (1 %) ilmeni 3. tai 4. asteen verenvuoto. Kliinisissä tutkimuksissa potilailla, joilla oli edenneen vaiheen KML ja jotka saivat suositeltua dasatinibiannosta (n = 304), ilmeni vaikeaa keskushermoston verenvuotoa 1 %:lla. Yksi potilas kuoli, ja tapaukseen liittyi yleisten toksisuuskriteereiden (Common Toxicity Criteria, CTC) mukainen 4. asteen trombosytopenia. Asteen 3 ja 4 ruoansulatuskanavan verenvuotoa ilmeni 6 %:lla potilaista, joilla oli edenneen vaiheen KML, ja yleensä se vaati lääkehoidon keskeyttämisen ja verensiirron. Muuta 3. ja 4. asteen verenvuotoa esiintyi 2 %:lla potilaista, joilla oli edenneen vaiheen KML. Useimpiin verenvuotoon liittyvistä haittavaikutuksista näillä potilailla liittyi tyypillisesti 3. ja 4. asteen trombosytopenia (ks. Kohta Haittavaikutukset). Lisäksi in vitro- ja in vivo ‑tutkimuksissa tehty verihiutaleiden määrittäminen viittaa siihen, että dasatinibihoidon vaikutus verihiutaleiden aktivaatioon on palautuva.
Varovaisuutta tulee noudattaa, jos potilaat käyttävät verihiutaleiden toimintaa estäviä lääkevalmisteita
tai antikoagulantteja.
Nesteretentio
Dasatinibin käyttöön liittyy nesteen kertymistä. Faasin III kliinisissä tutkimuksissa vastadiagnosoidun kroonisen vaiheen KML-potilailla raportoitiin 3. tai 4. asteen nesteretentiota 13 potilaalla (5 %) dasatinibiryhmässä ja 2 potilaalla (1 %) imatinibiryhmässä, kun seuranta oli kestänyt vähintään 60 kuukautta (ks. Kohta Haittavaikutukset). Kaikista dasatinibihoitoa saaneista kroonisen vaiheen KML-potilaista vaikeaa nesteretentiota ilmeni 32 potilaalla (6 %), jotka saivat dasatinibia suositusannoksena (n = 548). Kliinisissä tutkimuksissa dasatinibia suositusannoksena saaneilla edenneen vaiheen KML-potilailla tai Ph+ ALL -potilailla (n = 304) raportoitiin 3. ja 4. asteen nesteretentiota 8 %:lla, ja 7 %:lla nesteretentioon liittyi myös 3. ja 4. asteen pleuraeffuusio ja 1 %:lla 3. ja 4. asteen perikardiaalinen effuusio. Näillä potilailla raportoitiin sekä 3. tai 4. asteen keuhkoedeemaa ja keuhkoverenpainetautia kumpaakin 1 %:lla.
Potilaille, jotka saavat pleuraeffuusioon viittaavia oireita, kuten dyspneaa tai kuivaa yskää, tulee suorittaa keuhkojen röntgenkuvaus. Asteen 3 tai 4 pleuraeffuusio saattaa vaatia pleurapunktiota ja happihoitoa. Nesteretentioon liittyviä haittavaikutuksia hoidettiin tyypillisesti tukihoitotoimenpiteillä, diureetti- ja lyhytkestoinen steroidihoito mukaan lukien (ks. Kohdat Annostus ja antotapa ja Haittavaikutukset). Vähintään 65-vuotiailla potilailla esiintyy nuorempia potilaita todennäköisemmin pleuraeffuusiota, dyspneaa, yskää, perikardiaalista effuusiota ja kongestiivista sydämen vajaatoimintaa, ja heitä tulee seurata tarkasti. Kylothorax-tapauksia on myös raportoitu potilailla, joilla on pleuraeffuusio (ks. kohta Haittavaikutukset).
Keuhkovaltimoiden verenpainetauti (pulmonaaliarteriahypertensio, PAH)
Dasatinibihoidon yhteydessä on ilmoitettu haittavaikutuksena keuhkovaltimoiden verenpainetautia (prekapillaarista pulmonaaliarteriahypertensiota, joka on vahvistettu sydämen oikean puolen katetrisaatiolla, ks. Kohta Haittavaikutukset). Ilmoitusten mukaan PAH on ilmennyt dasatinibihoidon aloittamisen jälkeen, yli vuodenkin hoidon jälkeen.
Dasatinibihoito tulisi aloittaa vasta kun on selvitetty, ettei potilaalla ole sydämeen ja keuhkoihin liittyvän sairauden merkkejä ja oireita. Sydämen kaikututkimus on tehtävä hoitoa aloitettaessa jokaiselle potilaalle, jolla on sydänsairauden oireita, ja sen tekemistä on harkittava sellaiselle potilaalle, jolla on sydän- tai keuhkosairauden riskitekijöitä. Jos potilaalla ilmenee dasatinibihoidon aloittamisen jälkeen hengenahdistusta ja väsymystä, näiden oireiden tavalliset aiheuttajat (mm. pleuraeffuusio, keuhkoedeema, anemia, keuhkoinfiltraatti) on poissuljettava. Tutkimusten ajaksi dasatinibihoito on joko keskeytettävä tai dasatinibiannosta on pienennettävä ei-hematologisten haittavaikutusten hoidosta annettujen suositusten mukaisesti (ks. Kohta Annostus ja antotapa). Jos oireille ei löydy selitystä tai hoidon keskeyttäminen tai annoksen pienentäminen ei kohenna potilaan tilaa, PAH:n mahdollisuus on tutkittava. PAH tulee diagnosoida tavanomaisen käytännön mukaisesti. Jos potilaalla vahvistetaan PAH, dasatinibihoito on lopetettava pysyvästi.
Potilasta on seurattava tavanomaisen käytännön mukaisesti. Kun dasatinibihoito on lopetettu, hemodynaamisten ja kliinisten tutkimusten tulokset ovat joillakin dasatinibihoitoa saaneilla PAH-potilailla parantuneet.
QT-ajan piteneminen
In vitro ‑kokeista saadut tiedot viittaavat siihen, että dasatinibi voi pidentää sydämen kammioiden repolarisaatiota (QT-aika) (ks. Kohta Prekliiniset tiedot turvallisuudesta). Faasin III kliinisessä tutkimuksessa, jossa vastadiagnosoidun kroonisen vaiheen KML-potilaista 258 sai dasatinibihoitoa ja 258 imatinibihoitoa, raportoitiin kummassakin ryhmässä 1 potilaalla (< 1 %) haittavaikutuksena QTc-ajan pidentymistä, kun seuranta oli kestänyt vähintään 60 kuukautta. QTcF-ajan muutosten mediaani lähtötasosta oli 3,0 millisekuntia dasatinibihoitoa saaneilla potilailla ja 8,2 millisekuntia imatinibihoitoa saaneilla. Yhdellä potilaalla (< 1 %) kummassakin ryhmässä havaittiin QTcF > 500 millisekuntia. Faasin II kliinisissä tutkimuksissa leukemiaa sairastavilla, dasatinibilla hoidetuilla 865 potilailla keskimääräiset QTc-ajan muutokset lähtötasosta Friderician menetelmällä (QTcF) olivat 4–6 millisekuntia; ylempi 95 %:n luottamusväli kaikille lähtötason keskimääräisille muutoksille oli < 7 millisekuntia (ks. Kohta Haittavaikutukset).
Kliinisissä tutkimuksissa 2 182 potilaalla, joilla aikaisempi hoito imatinibi mukaan lukien ei tuottanut tulosta tai potilaat eivät sietäneet sitä ja jotka saivat dasatinibia, 15 (1 %) QTc-ajan pidentymistapausta raportoitiin haittavaikutukseksi. Kahdellakymmenelläyhdellä näistä potilaista (1 %) esiintyi > 500 millisekunnin QTcF.
Dasatinibia tulee antaa varoen potilaille, joilla QTc on pidentynyt tai se saattaa pidentyä. Näihin kuuluvat potilaat, joilla on hypokalemiaa tai hypomagnesemiaa, potilaat, joilla on synnynnäinen pitkä QT-aika sekä rytmihäiriölääkkeitä tai muita QT-ajan pidentymistä aiheuttavia lääkevalmisteita ja kumulatiivista suuriannoksista antrasykliinihoitoa saavat potilaat. Hypokalemia tai hypomagnesemia tulee korjata ennen dasatinibin antoa.
Sydämeen liittyvät haittavaikutukset
Dasatinibia tutkittiin satunnaistetussa kliinisessä tutkimuksessa 519 potilaalla, joilla oli vastadiagnosoitu kroonisen vaiheen KML, mukaan lukien potilaat, joilla oli aikaisempi sydänsairaus. Dasatinibihoitoa saaneilla potilailla raportoitiin seuraavia sydämeen liittyviä haittavaikutuksia: sydämen vajaatoiminta/sydämen toimintahäiriö, perikardiaalinen effuusio, sydämen rytmihäiriöt, sydämentykytys, QT-ajan pidentyminen ja sydäninfarkti (myös kuolemaan johtaneet tapaukset). Sydämeen liittyviä haittavaikutuksia esiintyi useammin potilailla, joilla oli riskitekijöitä tai aikaisemmin esiintynyt sydänsairaus. Potilaita, joilla on riskitekijöitä (esim. hypertensio, hyperlipidemia, diabetes) tai aikaisempi sydänsairaushistoria (esim. sepelvaltimon perkutaaninen toimenpide, osoitettu sepelvaltimotauti), on huolellisesti seurattava sydämen toimintahäiriöön liittyvien kliinisten merkkien tai oireiden, kuten rintakipu, hengenahdistus tai hikoilu, varalta.
Jos näitä kliinisiä merkkejä tai oireita kehittyy, lääkärin on keskeytettävä dasatinibin antaminen ja harkittava vaihtoehtoisen KML-hoidon tarvetta. Oireiden häviämisen jälkeen on kliinisesti arvioitava dasatinibihoidon uudelleen aloitus. Dasatinibihoitoa voidaan jatkaa alkuperäisellä annoksella lievien/keskivaikeiden haittavaikutusten (≤ 2. asteen) jälkeen ja annostasolla pienennetyllä annoksella vakavien (≥ 3. asteen) haittavaikutusten jälkeen (ks. Kohta Annostus ja antotapa). Hoitoa jatkavia potilaita on seurattava säännöllisin väliajoin.
Kliinisissä tutkimuksissa ei ollut mukana potilaita, joilla oli hoitamaton tai merkittävä kardiovaskulaarisairaus.
Tromboottinen mikroangiopatia (TMA)
BCR-ABL-tyrosiinikinaasin estäjien käyttöön on liittynyt tromboottista mikroangiopatiaa (TMA), ja yksittäisiä tapauksia on ilmoitettu myös dasatinibin käytön yhteydessä (ks. Kohta Haittavaikutukset). Jos dasatinibia saavalla potilaalla havaitaan TMA:han liittyviä laboratoriolöydöksiä tai kliinisiä löydöksiä, dasatinibihoito on keskeytettävä ja TMA:n mahdollisuus on arvioitava huolellisesti, mukaan lukien ADAMTS13-aktiivisuus ja anti-ADAMTS13-vasta-ainemääritys. Jos anti-ADAMTS13-vasta-aineet ovat koholla ja ADAMTS13-aktiivisuus on vähäistä, dasatinibihoitoa ei pidä aloittaa uudelleen.
Hepatiitti B:n uudelleen aktivoituminen
Hepatiitti B:n uudelleen aktivoitumista on tapahtunut kyseisen viruksen pysyvillä kantajilla sen jälkeen, kun potilas on saanut BCR‑ABL-tyrosiinikinaasin estäjiä. Tämä aiheutti joissakin tapauksissa maksan vajaatoimintaa tai fulminanttia hepatiittia, joka johti maksansiirtoon tai kuolemaan.
Potilaat on testattava hepatiitti B -viruksen varalta ennen Dasatinib Krka ‑hoidon aloittamista. Maksasairauksien ja hepatiitti B:n hoitoon perehtyneitä asiantuntijoita on kuultava ennen hoidon aloittamista, jos potilaan hepatiitti B ‑serologia on positiivinen (mukaan lukien potilaat, joilla sairaus on aktiivinen) ja jos potilas saa positiivisen hepatiitti B -testituloksen hoidon aikana. Hepatiitti B ‑viruksen kantajia, jotka tarvitsevat Dasatinib Krka ‑hoitoa, on seurattava tarkasti aktiivisen hepatiitti B ‑virusinfektion oireiden varalta koko hoidon ajan ja useita kuukausia hoidon jälkeen (ks. Kohta Haittavaikutukset).
Vaikutukset pediatristen potilaiden kasvuun ja kehitykseen
Pediatrisilla potilailla tehdyissä dasatinibitutkimuksissa, joihin osallistui imatinibille resistenttejä/intolerantteja pediatrisia Ph+ CP-KML -potilaita ja hoitamattomia pediatrisia Ph+ CP-KML -potilaita, vähintään kahden vuoden hoidon jälkeen 6:lla (4,6 %) potilaalla ilmoitettiin hoidosta johtuvia luiden kasvuun ja kehitykseen liittyviä haittavaikutuksia, joista yksi tapaus oli vaikeusasteeltaan vaikea (asteen 3 kasvuhäiriö). Näissä kuudessa tapauksessa haittavaikutukset olivat epifyysin luutumisen hidastuminen, osteopenia, kasvuhäiriö ja gynekomastia (ks. Kohta Farmakodynamiikka). Tuloksia on vaikeaa tulkita kroonisten sairauksien, kuten KML:n, yhteydessä, ja ne vaativat pitkäaikaista seurantaa.
Tutkimuksissa, joissa tutkittiin dasatinibia yhdessä kemoterapian kanssa ja joihin osallistui vastadiagnosoituja pediatrisia Ph+ ALL ‑potilaita, 1:llä (0,6 %) potilaalla ilmoitettiin enintään kahden vuoden hoidon jälkeen hoidosta johtuvia luiden kasvuun ja kehitykseen liittyviä haittavaikutuksia. Kyseessä oli 1. asteen osteopenia.
Dasatinibilla hoidetuilla pediatrisilla potilailla on kliinisissä tutkimuksissa havaittu kasvuhäiriöitä (ks. kohta Haittavaikutukset). Enintään 2 vuoden hoidon jälkeen on havaittu laskua ennustetussa pituudessa yhtä paljon kuin mitä pelkkää kemoterapiaa käytettäessä on havaittu. Tämä ei ole vaikuttanut ennustettuun painoon tai painoindeksiin, eikä sillä ole ollut yhteyttä hormoniepätasapainoihin tai muihin laboratoriotutkimusten löydöksiin. Pediatristen potilaiden luiden kasvun ja kehityksen seurantaa suositellaan.
Apuaineet
Tämä lääkevalmiste sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Yksi kalvopäällysteinen tabletti sisältää alle 1 mmol natriumia (23 mg) eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Lääkeaineet, jotka saattavat lisätä dasatinibin pitoisuuksia plasmassa
In vitro ‑tutkimukset osoittavat, että dasatinibi on CYP3A4:n substraatti. Dasatinibin ja voimakkaiden CYP3A4-entsyymiä inhiboivien lääkevalmisteiden tai aineiden (esim. ketokonatsoli, itrakonatsoli, erytromysiini, klaritromysiini, ritonaviiri, telitromysiini, greippimehu) käyttö samanaikaisesti saattaa lisätä dasatinibialtistusta. Siksi voimakkaan CYP3A4:n estäjän systeemistä antoa dasatinibia saaville potilaille ei suositella (ks. Kohta Annostus ja antotapa).
In vitro ‑tutkimusten perusteella dasatinibi sitoutuu noin 96-prosenttisesti plasman proteiineihin kliinisesti merkityksellisinä pitoisuuksina. Dasatinibin yhteisvaikutuksia muiden proteiineihin sitoutuvien lääkevalmisteiden kanssa ei ole tutkittu. Syrjäytymisen mahdollisuutta ja sen kliinistä merkitystä ei tunneta.
Lääkeaineet, jotka saattavat pienentää dasatinibin pitoisuuksia plasmassa
Kun dasatinibia annettiin sen jälkeen, kun rifampisiinia, voimakasta CYP3A4-induktoria, oli annettu 600 mg iltaisin 8 päivän ajan, dasatinibin AUC pieneni 82 %. Muut CYP3A4-aktiivisuutta lisäävät lääkevalmisteet (esim. deksametasoni, fenytoiini, karbamatsepiini, fenobarbitaali tai (perinteiset) kasvirohdosvalmisteet, jotka sisältävät Hypericum perforatumia eli mäkikuismaa) saattavat lisätä dasatinibin metaboliaa ja pienentää sen pitoisuutta plasmassa. Sen vuoksi voimakkaiden CYP3A4-induktorien ja dasatinibin samanaikaista käyttöä ei suositella. Potilaiden, joille rifampisiini tai muu CYP3A4-induktori on indisoitu, tulee käyttää vaihtoehtoisia, heikommin entsyymejä indusoivia lääkevalmisteita. Deksametasonin, heikon CYP3A4:n induktorin, samanaikainen käyttö dasatinibin kanssa sallitaan; dasatinibin AUC:n odotetaan pienenevän noin 25 % deksametasonin samanaikaisen käytön takia, mikä ei todennäköisesti ole kliinisesti merkitsevää.
H2-reseptorinsalpaajat ja protonipumpun estäjät
Pitkäaikainen vatsahappojen erityksen estäminen H2-salpaajilla tai protonipumpun estäjillä (esim. famotidiini ja omepratsoli) vähentää todennäköisesti dasatinibialtistusta. Kerta-annostutkimuksessa terveillä koehenkilöillä famotidiinin anto 10 tuntia ennen dasatinibin kerta-annosta pienensi dasatinibialtistusta 61 %. Tutkimuksessa, jossa 14 terveelle koehenkilölle annettiin dasatinibia 100 mg:n kerta-annos 22 tuntia sen jälkeen, kun koehenkilöille oli saavutettu neljän päivän 40 mg:n omepratsoliannoksen jälkeen vakaa tila, dasatinibin AUC-arvo pieneni 43 % ja Cmax-arvo 42 %. Dasatinibihoitoa saaville potilaille tulisi harkita antasidien antoa H2-salpaajien ja protonipumpun estäjien sijaan (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Antasidit
Ei-kliiniset tiedot osoittavat, että dasatinibin liukoisuus riippuu pH:sta. Kun terveille koehenkilöille annettiin samanaikaisesti alumiinihydroksidi/magnesiumhydroksidi-antasideja ja kerta-annos dasatinibia, pieneni AUC 55 % ja Cmax 58 %. Kun antasidit annettiin 2 tuntia ennen dasatinibin kerta-annosta, ei havaittu oleellisia dasatinibin pitoisuuden tai altistuksen muutoksia. Siten antasideja voidaan antaa vähintään 2 tuntia ennen dasatinibia tai 2 tuntia dasatinibin jälkeen (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Lääkeaineet, joiden pitoisuutta plasmassa dasatinibi saattaa muuttaa
Dasatinibin ja CYP3A4:n substraatin samanaikainen käyttö saattaa lisätä altistusta CYP3A4:n substraatille. Terveillä koehenkilöillä tehdyssä tutkimuksessa dasatinibin 100 mg:n kerta-annos suurensi simvastatiinin, tunnetun CYP3A4:n substraatin, AUC-arvoa 20 % ja Cmax-arvoa 37 %. Vaikutus voi olla suurempi toistuvien dasatinibiannosten jälkeen. Siksi CYP3A4:n substraatteja, joilla on kapea terapeuttinen alue (esim. astemitsoli, terfenadiini, sisapridi, pimotsidi, kinidiini, bepridiili tai ergotalkaloidit [ergotamiini, dihydroergotamiini]) tulee antaa varoen potilaille, jotka saavat dasatinibia (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
In vitro ‑tutkimusten perusteella interaktiot CYP2C8:n substraattien, kuten glitatsonien, kanssa ovat mahdollisia.
Pediatriset potilaat
Yhteisvaikutustutkimuksia on tehty ainoastaan aikuisille.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / ehkäisy miehille ja naisille
Seksuaalisesti aktiivisten miesten ja hedelmällisessä iässä olevien naisten tulee käyttää tehokasta ehkäisyä hoidon aikana.
Raskaus
Ihmisissä havaittuihin vaikutuksiin perustuen dasatinibin epäillään aiheuttavan synnynnäisiä epämuodostumia, mukaan lukien hermostoputken sulkeutumishäiriöitä ja sikiöön kohdistuvia farmakologisia haittavaikutuksia, jos sitä käytetään raskauden aikana. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. Kohta Prekliiniset tiedot turvallisuudesta).
Dasatinib Krka ‑valmistetta ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa dasatinibilla. Jos Dasatinib Krka ‑valmistetta käytetään raskauden aikana, potilaalle tulee kertoa mahdollisesta sikiöön kohdistuvasta riskistä.
Imetys
Tiedot dasatinibin erittymisestä ihmisen tai eläimen maitoon ovat riittämättömät/rajalliset. Fysikaalis-kemialliset ja saatavilla olevat farmakodynaamiset/toksikologiset tiedot dasatinibista viittaavat siihen, että dasatinibi erittyy rintamaitoon eikä riskiä imeväiselle voida poissulkea.
Imetys on lopetettava Dasatinib Krka ‑hoidon ajaksi.
Hedelmällisyys
Eläinkokeissa dasatinibihoidolla ei todettu vaikutusta uros- tai naarasrottien hedelmällisyyteen (ks. Kohta Prekliiniset tiedot turvallisuudesta). Lääkäreiden ja muiden terveydenhuollon ammattilaisten on kerrottava soveltuvan ikäisille miehille Dasatinib Krka ‑valmisteen mahdollisista vaikutuksista hedelmällisyyteen ja siemennesteen tallettamisen mahdollisuudesta.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Dasatinib Krka ‑valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaille tulee kertoa, että heillä saattaa ilmetä haittavaikutuksia, kuten huimausta tai näön sumenemista, dasatinibihoidon aikana. Sen takia varovaisuutta suositellaan noudatettavaksi ajettaessa autoa tai käytettäessä koneita.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Alla esitetyt tiedot koskevat kaikkia dasatinibiannoksia yksilääkehoitoina, joita on testattu kliinisissä tutkimuksissa (N = 2 900), mukaan lukien 324 vastadiagnosoidun kroonisen vaiheen KML-aikuispotilasta, 2 388 imatinibille resistenttiä tai intoleranttia kroonisen tai edenneen vaiheen KML- tai Ph+ ALL -aikuispotilasta sekä 188 pediatrista potilasta.
Niillä 2 712 aikuispotilaalla, joilla oli kroonisen vaiheen KML, edenneen vaiheen KML tai Ph+ ALL, hoidon keston mediaani oli 19,2 kuukautta (vaihteluväli 0–93,2 kk). Satunnaistetussa tutkimuksessa vastadiagnosoidun kroonisen vaiheen KML-potilaille annetun hoidon keston mediaani oli noin 60 kuukautta. Niillä 1 618 aikuispotilaalla, joilla oli kroonisen vaiheen KML, hoidon keston mediaani oli 29 kuukautta (vaihteluväli 0–92,9 kk). Niillä 1 094 aikuispotilaalla, joilla oli edenneen vaiheen KML tai Ph+ ALL, hoidon keston mediaani oli 6,2 kuukautta (vaihteluväli 0–93,2 kk). Pediatrisissa tutkimuksissa 188 potilaalla hoidon keston mediaani oli 26,3 kuukautta (vaihteluväli 0–99,6 kk). Potilasryhmässä, jossa oli 130 dasatinibia saanutta pediatrista potilasta, joilla oli kroonisen vaiheen KML, hoidon keston mediaani oli 42,3 kuukautta (vaihteluväli 0,1–99,6 kuukautta).
Suurin osa dasatinibilla hoidetuista potilaista sai haittavaikutuksia jossain vaiheessa. Dasatinibia saaneista yhteensä 2 712 aikuispotilaasta 520:lla (19 %) ilmeni hoidon lopettamiseen johtaneita haittavaikutuksia.
Dasatinibin turvallisuusprofiili oli pediatrisilla Ph+ CP-KML -potilailla lääkemuodosta riippumatta samankaltainen kuin aikuisilla. Pediatrisilla potilailla ei kuitenkaan ilmoitettu perikardiaalista effuusiota, pleuraeffuusiota, keuhkoedeemaa eikä keuhkoverenpainetautia. Dasatinibia saaneista 130 pediatrisesta potilaasta, joilla oli CP-KML, 2:lla (1,5 %) ilmeni hoidon lopettamiseen johtaneita haittavaikutuksia.
Taulukko haittavaikutuksista
Seuraavat haittavaikutukset, lukuun ottamatta laboratoriotestipoikkeavuuksia, raportoitiin dasatinibilla yksilääkehoitona hoidetuilla potilailla kliinisissä tutkimuksissa ja markkinoille tulon jälkeen (taulukko 5). Haittavaikutukset on esitetty elinjärjestelmittäin ja esiintyvyyden mukaan. Esiintyvyydet on luokiteltu seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000), tuntematon (koska saatavilla oleva markkinoille tulon jälkeinen tieto ei riitä arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 5: Yhteenvetotaulukko haittavaikutuksista
| Infektiot | |
| Hyvin yleinen | infektio (mukaan lukien bakteeri-, virus-, sieni- ja määrittämätön infektio) |
| Yleinen | pneumonia (myös bakteeri-, virus- ja sieniperäinen), ylähengitysteiden infektio/tulehdus, herpesvirusinfektio (mukaan luettuna sytomegalovirus CMV), enterokoliitti, sepsis (mukaan lukien melko harvinaiset kuolemaan johtaneet tapaukset) |
| Tuntematon | hepatiitti B:n uudelleen aktivoituminen |
| Veri ja imukudos | |
| Hyvin yleinen | myelosuppressio (myös anemia, neutropenia, trombosytopenia) |
| Yleinen | kuumeinen neutropenia |
| Melko harvinainen | lymfadenopatia, lymfosytopenia |
| Harvinainen | puhdas punasoluaplasia |
| Immuunijärjestelmä | |
| Melko harvinainen | yliherkkyys (myös kyhmyruusu) |
| Harvinainen | anafylaktinen sokki |
| Umpieritys | |
| Melko harvinainen | kilpirauhasen vajaatoiminta |
| Harvinainen | kilpirauhasen liikatoiminta, kilpirauhastulehdus |
| Aineenvaihdunta ja ravitsemus | |
| Yleinen | ruokahalun häiriöta, hyperurikemia |
| Melko harvinainen | tuumorilyysioireyhtymä, elimistön kuivuminen, hypoalbuminemia, hyperkolesterolemia |
| Harvinainen | diabetes mellitus |
| Psyykkiset häiriöt | |
| Yleinen | depressio, unettomuus |
| Melko harvinainen | ahdistuneisuus, sekavuustila, mielialan ailahtelu, heikentynyt libido |
| Hermosto | |
| Hyvin yleinen | päänsärky |
| Yleinen | neuropatia (mukaan lukien perifeerinen neuropatia), huimaus, makuhäiriö, uneliaisuus |
| Melko harvinainen | keskushermoston verenvuoto*b, pyörtyminen, vapina, amnesia, tasapainohäiriö |
| Harvinainen | aivoverenkierron häiriöt, ohimenevät aivoverenkiertohäiriöt (TIA), kouristuskohtaus, näköhermon tulehdus, kasvohermon halvaus, dementia, ataksia |
| Silmät | |
| Yleinen | näön häiriöt (mukaan lukien näköhäiriö, näön samentuminen ja näön tarkkuuden alentuminen), kuivasilmäisyys |
| Melko harvinainen | näön heikkeneminen, konjunktiviitti, valonarkuus, kyynelvuodon lisääntyminen |
| Kuulo ja tasapainoelin | |
| Yleinen | tinnitus |
| Melko harvinainen | kuulonmenetys, vertigo |
| Sydän | |
| Yleinen | sydämen kongestiivinen vajaatoiminta / sydämen toimintahäiriö*c, perikardiaalinen effuusio*, rytmihäiriöt (myös takykardia), palpitaatiot |
| Melko harvinainen | sydäninfarkti (mukaan lukien kuolemaan johtaneet)*, pidentynyt QT-aika elektrokardiogrammissa*, perikardiitti, kammioarytmia (myös kammiotakykardia), angina pectoris, kardiomegalia, epänormaali T-aalto elektrokardiogrammissa, kohonnut troponiiniarvo |
| Harvinainen | cor pulmonale, sydänlihastulehdus, akuutti sepelvaltimotautikohtaus, sydämenpysähdys, PR-välin pidentyminen elektrokardiogrammissa, sepelvaltimotauti, pleuroperikardiitti |
| Tuntematon | eteisvärinä/eteislepatus |
| Verisuonisto | |
| Hyvin yleinen | verenvuoto*d |
| Yleinen | hypertensio, punastuminen |
| Melko harvinainen | hypotensio, tromboflebiitti, tromboosi |
| Harvinainen | syvä laskimotukos, embolia, livedo reticularis (sinikalpeus) |
| Tuntematon | tromboottinen mikroangiopatia |
| Hengityselimet, rintakehä ja välikarsina | |
| Hyvin yleinen | pleuraeffuusio*, hengenahdistus |
| Yleinen | keuhkoedeema*, pulmonaalinen hypertensio*, keuhkoinfiltraatio, pneumoniitti, yskä |
| Melko harvinainen | keuhkovaltimoiden verenpainetauti, bronkospasmi, astma, kylothorax* |
| Harvinainen | keuhkoembolia, akuutti hengitysvaikeusoireyhtymä |
| Tuntematon | interstitiaalinen keuhkosairaus |
| Ruoansulatuselimistö | |
| Hyvin yleinen | ripuli, oksentelu, pahoinvointi, mahakipu |
| Yleinen | maha-suolistoverenvuoto*, koliitti (myös neutropeeninen koliitti), gastriitti, limakalvotulehdus (mukaan lukien mukosiitti / stomatiitti), dyspepsia, vatsan pingottuminen, ummetus, suun pehmytkudossairaus |
| Melko harvinainen | haimatulehdus (myös akuutti haimatulehdus), ylemmän maha-suolikanavan haavauma, esofagiitti, askites*, peräaukon haavauma, dysfagia, gastroesofageaalinen refluksitauti |
| Harvinainen | proteiinia menettävä gastroenteropatia, ileus, peräaukon fisteli |
| Tuntematon | kuolemaan johtava maha-suolikanavan verenvuoto* |
| Maksa ja sappi | |
| Melko harvinainen | hepatiitti, kolekystiitti, kolestaasi |
| Iho ja ihonalainen kudos | |
| Hyvin yleinen | ihottumae |
| Yleinen | hiustenlähtö, dermatiitti (myös ekseema), kutina, akne, kuiva iho, urtikaria, liikahikoilu |
| Melko harvinainen | neutrofiilinen dermatoosi, valoyliherkkyys, pigmenttihäiriö, pannikuliitti, ihohaavauma, rakkulaihottumat, kynnen rakennehäiriöt, käsi-jalkaoireyhtymä (kämmenten ja jalkapohjien erytrodysestesia), hiushäiriö |
| Harvinainen | leukosytoklastinen verisuonitulehdus, ihon sidekudostuminen |
| Tuntematon | Stevens–Johnsonin oireyhtymäf |
| Luusto, lihakset ja sidekudos | |
| Hyvin yleinen | lihas- ja luustokipug |
| Yleinen | nivelkipu, myalgia, lihasheikkous, tuki- ja liikuntaelinten jäykkyys, lihasspasmi |
| Melko harvinainen | rabdomyolyysi, luukuolio, lihastulehdus, jännetulehdus, niveltulehdus |
| Harvinainen | epifyysin luutumisen hidastuminenh, kasvuhäiriöh |
| Munuaiset ja virtsatiet | |
| Melko harvinainen | munuaistoiminnan heikkeneminen (myös munuaisten vajaatoiminta), tiheä virtsaamistarve, proteinuria |
| Tuntematon | nefroottinen oireyhtymä |
| Raskauteen, synnytykseen ja perinataalikauteen liittyvät haitat | |
| Harvinainen | keskenmeno |
| Sukupuolielimet ja rinnat | |
| Melko harvinainen | gynekomastia, kuukautishäiriö |
| Yleisoireet ja antopaikassa todettavat haitat | |
| Hyvin yleinen | perifeerinen edeemai, väsymys, kuume, kasvoedeemaj |
| Yleinen | voimattomuus, kipu, rintakipu, yleistynyt edeema*k, vilunväristykset |
| Melko harvinainen | huonovointisuus, muu pinnallinen edeemal |
| Harvinainen | kävelyhäiriö |
| Tutkimukset | |
| Yleinen | painonlasku, painonnousu |
| Melko harvinainen | kohonnut veren kreatiinikinaasiarvo, kohonnut glutamyylitransferaasiarvo |
| Vammat, myrkytykset ja hoitokomplikaatiot | |
| Yleinen | ruhjeet |
a Kattaa seuraavat: ruokahalun heikkeneminen, varhainen kylläisyydentunne, ruokahalun lisääntyminen.
b Kattaa seuraavat: keskushermoston verenvuoto, aivojen verenpurkauma, aivoverenvuoto, kovakalvon ulkopuolinen verenpurkauma, kallonsisäinen verenvuoto, hemorraginen aivohalvaus, lukinkalvonalainen verenvuoto, kovakalvonalainen verenpurkauma ja kovakalvonalainen verenvuoto.
c Kattaa seuraavat: aivojen natriureettisen peptidipitoisuuden suureneminen, sydämen kammion toimintahäiriö, sydämen vasemman kammion toimintahäiriö, sydämen oikean kammion toimintahäiriö, sydämen vajaatoiminta, akuutti sydämen vajaatoiminta, krooninen sydämen vajaatoiminta, kongestiivinen sydämen vajaatoiminta, kardiomyopatia, kongestiivinen kardiomyopatia, diastolinen toimintahäiriö, ejektiofraktion pienentyminen ja ventrikulaarinen vajaatoiminta, sydämen vasemman kammion vajaatoiminta, sydämen oikean kammion vajaatoiminta ja ventrikulaarinen hypokinesia.
d Ei kata seuraavia: ruoansulatuskanavan verenvuoto ja keskushermoston verenvuoto; nämä haittavaikutukset on esitetty kohdissa ruoansulatuselimistö ja hermosto.
e Kattaa seuraavat: lääkeaineihottuma, punoitus, erythema multiforme, erytroosi, hilseilevä ihottuma, yleistynyt punoitus, genitaalialueen ihottuma, kuumuuden laukaisema ihottuma, milia (luufinnit), miliaria (hikirakkulatauti), märkärakkulainen psoriaasi, ihottuma, punoittava ihottuma, follikulaarinen ihottuma, yleistynyt ihottuma, makulaarinen ihottuma, makulopapulaarinen ihottuma, papulaarinen ihottuma, kutiava ihottuma, pustuloosinen ihottuma, rakkulaihottuma, ihoärsytys, toksinen ihottuma, rakkulainen nokkosihottuma ja verisuonitulehduksen aiheuttama ihottuma.
f Markkinoille saattamisen jälkeen on raportoitu yksittäisiä Stevens-Johnsonin oireyhtymätapauksia. Ei voitu määritellä, olivatko havaitut ihon ja limakalvon haittavaikutukset yhteydessä dasatinibiin vai muihin samanaikaisesti käytettyihin lääkevalmisteisiin.
g Lihas- ja luustokipua on ilmoitettu hoidon aikana tai sen lopettamisen jälkeen.
h Pediatrisissa tutkimuksissa esiintymistiheydeksi on ilmoitettu yleinen.
i Gravitaatioedeema, paikallistunut edeema, perifeerinen edeema.
j Silmän sidekalvon edeema, silmien edeema, silmien turvotus, silmäluomien edeema, kasvojen edeema, huulten edeema, makulaarinen edeema, suun edeema, orbitaalinen edeema, periorbitaalinen edeema, kasvojen turvotus.
k Nesteylimäärä, nesteretentio, maha-suolikanavan edeema, yleistynyt edeema, perifeerinen turvotus, edeema, sydäntaudista johtuva edeema, perinefriittinen effuusio, toimenpiteen jälkeinen edeema, viskeraalinen edeema.
l Sukuelinten turvotus, leikkaushaavan edeema, sukuelinten edeema, siittimen edeema, siittimen turvotus, kivespussin edeema, ihoturvotus, kivesten turvotus, vulvovaginaalinen turvotus.
* Lisätietoja, ks. Kohta ”Valikoitujen haittavaikutusten kuvaus”
Valikoitujen haittavaikutusten kuvaus
Myelosuppressio
Dasatinib Krka ‑hoitoon liittyy anemiaa, neutropeniaa ja trombosytopeniaa. Näitä ilmenee aikaisemmin ja useammin potilailla, joilla on edenneen vaiheen KML tai Ph+ ALL, kuin potilailla, joilla on kroonisen vaiheen KML (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Verenvuoto
Dasatinibia saavilla potilailla raportoitiin lääkkeen käyttöön liittyviä verenvuotoa koskevia haittavaikutuksia, jotka vaihtelivat pienistä verenpurkaumista ja nenäverenvuodosta 3. ja 4. asteen ruoansulatuskanavan ja keskushermoston verenvuotoihin (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Nesteretentio
Termillä ”nesteretentio” voidaan yhteisesti kuvailla monenlaisia haittavaikutuksia kuten pleuraeffuusio, askites, keuhkoedeema ja perikardiaalinen effuusio, johon voi liittyä pinnallista edeemaa. Vastadiagnosoitua kroonisen vaiheen KML:aa koskeneessa tutkimuksessa todettiin seuraavia dasatinibiin liittyneitä nesteretentiota koskevia haittavaikutuksia, kun seuranta oli kestänyt vähintään 60 kuukautta: pleuraeffuusio (28 %), pinnallinen edeema (14 %), keuhkoverenpainetauti (5 %), yleistynyt edeema (4 %) ja perikardiaalinen effuusio (4 %). Potilaista < 2 %:lla raportoitiin kongestiivinen sydämen vajaatoiminta / sydämen toimintahäiriö ja keuhkoedeema.
Dasatinibiin liittyneen pleuraeffuusion (kaikki vaikeusasteet) kumulatiivinen esiintymistiheys oli 10 % 12 kuukauden kohdalla, 14 % 24 kuukauden kohdalla, 19 % 36 kuukauden kohdalla, 24 % 48 kuukauden kohdalla ja 28 % 60 kuukauden kohdalla. Pleuraeffuusio oli uusiutuva yhteensä 46 dasatinibilla hoidetulla potilaalla. Potilaista 17:llä ilmeni 2 erillistä pleuraeffuusioon liittyvää haittavaikutusta, 6:lla 3 haittavaikutusta, 18:lla 4–8 haittavaikutusta ja 5:lla > 8 haittavaikutusta.
Dasatinibiin liittynyt 1. tai 2. asteen pleuraeffuusio ilmeni ensimmäisen kerran 114 viikon (mediaani, vaihteluväli 4–299 viikkoa) kuluttua. Dasatinibiin liittyneet pleuraeffuusiot olivat vaikeita (3. tai 4. aste) alle 10 %:lla kaikista pleuraeffuusion saaneista potilaista. Dasatinibiin liittynyt ≥ 3. asteen pleuraeffuusio ilmeni ensimmäisen kerran 175 viikon (mediaani, vaihteluväli 114–274 viikkoa) kuluttua. Dasatinibiin liittyneen pleuraeffuusion (kaikki vaikeusasteet) keston mediaani oli 283 päivää (~ 40 viikkoa).
Pleuraeffuusio oli tavallisesti palautuva, ja se hoidettiin keskeyttämällä dasatinibihoito ja käyttämällä diureetteja tai muita asianmukaisia tukihoitotoimia (ks. Kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Dasatinibihoitoon liittynyt pleuraeffuusio ilmeni 73 potilaalla, joista 45:n (62 %) hoito keskeytettiin ja 30:n (41 %) annosta pienennettiin. Lisäksi 34:lle (47 %) potilaalle annettiin diureetteja, 23:lle (32 %) kortikosteroideja ja 20:lle (27 %) sekä kortikosteroideja että diureetteja. Yhdeksälle (12 %) potilaalle tehtiin hoidollinen pleurapunktio.
Dasatinibihoitoa saaneista 6 %:lla hoito keskeytettiin lääkkeeseen liittyneen pleuraeffuusion vuoksi. Pleuraeffuusio ei heikentänyt potilaiden kykyä saavuttaa vaste. Pleuraeffuusion saaneista dasatinibiryhmän potilaista 96 % saavutti täydellisen sytogeneettisen vasteen (cCCyR), 82 % saavutti merkittävän molekulaarisen vasteen (MMR) ja 50 % molekulaarisen vasteen 4,5 login aleneman (MR4,5) hoidon keskeytyksistä tai annosmuutoksista huolimatta.
Kohdassa Varoitukset ja käyttöön liittyvät varotoimet on lisätietoja potilaista, joilla on kroonisen vaiheen KML, edenneen vaiheen KML tai Ph+ ALL.
Kylothorax-tapauksia on raportoitu potilailla, joilla on pleuraeffuusio. Jotkin kylothorax-tapaukset poistuivat, kun dasatinibin anto lopetettiin, keskeytettiin tai kun annosta pienennettiin, mutta suurimmassa osassa tapauksista tarvittiin lisähoitoa.
Keuhkovaltimoiden verenpainetauti (pulmonaaliarteriahypertensio, PAH)
Dasatinibialtistuksen yhteydessä on ilmoitettu haittavaikutuksena PAHia (prekapillaarista pulmonaaliarteriahypertensiota, joka on vahvistettu sydämen oikean puolen katetrisaatiolla). Ilmoitusten mukaan PAH on ilmennyt dasatinibihoidon aloittamisen jälkeen, yli vuodenkin hoidon jälkeen. Potilaat, joilla on ilmoitettu PAH dasatinibihoidon aikana, ovat usein käyttäneet muita lääkevalmisteita samanaikaisisesti tai heillä on ollut muita sairauksia pahanlaatuisen perussairautensa lisäksi. Kun dasatinibihoito on lopetettu, hemodynaamisten ja kliinisten tutkimusten tulokset ovat joillakin PAH-potilailla parantuneet.
QT-ajan pidentyminen
Faasin III tutkimuksessa vastadiagnosoidun kroonisen vaiheen KML-potilailla, yhdellä potilaalla (< 1 %) dasatinibihoitoa saaneista oli QTcF > 500 millisekuntia, kun seuranta oli kestänyt vähintään 12 kuukautta (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet). Kun seuranta oli kestänyt vähintään 60 kuukautta, yhdelläkään muulla potilaalla ei ollut raportoitu > 500 millisekunnin QTcF-aikoja.
Viidessä faasin II kliinisessä tutkimuksessa potilailla, joilla aikaisempi imatinibihoito ei tuottanut tulosta tai potilaat eivät sietäneet sitä, oli mukana 865 potilasta, jotka saivat dasatinibia 70 mg kahdesti vuorokaudessa. Toistetut EKG:t otettiin ennalta määrättyinä ajankohtina ennen hoitoa ja hoidon aikana. EKG:t tulkittiin keskitetysti. QT-aika suhteutettiin sydämen sykkeeseen Friderician menetelmällä. Päivänä 8 kaikkina annosten ottamisen jälkeisinä ajankohtina keskimääräiset muutokset lähtötason QTcF-ajasta olivat 4-6 millisekuntia; (95 %:n luottamusvälin yläraja oli < 7 millisekuntia). 2 182 potilaalla, joilla aikaisempi imatinibihoito ei tuottanut tulosta tai potilaat eivät sietäneet sitä ja jotka saivat dasatinibihoitoa, 15 (1 %) QTc-ajan pidentymistapausta raportoitiin haittavaikutuksena. Kahdellakymmenelläyhdellä potilaalla (1 %) QTcF oli yli 500 millisekuntia (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sydämeen liittyvät haittavaikutukset
Potilaita, joilla on riskitekijöitä tai aikaisempi sydänsairaushistoria, on huolellisesti seurattava sydämen toimintahäiriöön liittyvien kliinisten merkkien tai oireiden varalta sekä tarvittaessa hoidettava asianmukaisesti (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Hepatiitti B:n uudelleen aktivoituminen
Hepatiitti B:n uudelleen aktivoitumista on ilmoitettu BCR‑ABL-tyrosiinikinaasin estäjien käytön yhteydessä. Tämä aiheutti joissakin tapauksissa maksan vajaatoimintaa tai fulminanttia hepatiittia, joka johti maksansiirtoon tai kuolemaan (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annoksen optimointia koskeneessa faasin III tutkimuksessa potilailla, joilla oli kroonisen vaiheen KML, ja joilla aikaisempi imatinibihoito ei tuottanut tulosta tai potilaat eivät sietäneet sitä, (hoidon mediaanikesto 30 kuukautta), pleuraeffuusiota ja sydämen kongestiivista vajaatoimintaa / sydämen toimintahäiriötä esiintyi vähemmän, kun dasatinibiannostus oli 100 mg kerran vuorokaudessa, kuin dasatinibiannostuksen ollessa 70 mg kahdesti vuorokaudessa.
Myös myelosuppressiota esiintyi harvemmin hoitoryhmässä, jossa annostus oli 100 mg kerran vuorokaudessa (ks. Alla Poikkeavuudet laboratoriotesteissä). Hoidon keston mediaani oli 37 kuukautta (vaihteluväli 1–91 kuukautta) ryhmässä, jonka annostus oli 100 mg kerran vuorokaudessa. Taulukossa 6a on esitetty valikoitujen haittavaikutusten kumulatiiviset esiintymistiheydet, jotka raportoitiin ryhmässä, joka sai suositeltua aloitusannosta 100 mg kerran vuorokaudessa.
Taulukko 6a: Valikoidut haittavaikutukset, joita raportoitiin faasin III annoksen optimointitutkimuksessa (imatinibille intolerantti tai resistentti kroonisen vaiheen KML)a
Seuranta vähintään 2 vuotta | Seuranta vähintään 5 vuotta | Seuranta vähintään 7 vuotta | |||||||||
Kaikki asteet | Aste 3/4 | Kaikki asteet | Aste 3/4 | Kaikki asteet | Aste 3/4 | ||||||
| Suositeltu termi | Prosenttia (%) potilaista | ||||||||||
Ripuli Nesteretentio Pinnallinen edeema Pleuraeffuusio Yleistynyt edeema Perikardiaalinen effuusio Keuhkoverenpainetauti Verenvuoto Maha-suolikanavan verenvuoto | 27 34 18 18 3 2 0 11 2 | 2 4 0 2 0 1 0 1 1 | 28 42 21 24 4 2 0 11 2 | 2 6 0 4 0 1 0 1 1 | 28 48 22 28 4 3 2 12 2 | 2 7 0 5 0 1 1 1 1 | |||||
a Tulokset faasin III annoksen optimointitutkimuksesta, jossa käytettiin suositeltua aloitusannosta 100 mg kerran vuorokaudessa (n = 165)
Annoksen optimointia koskeneessa faasin III tutkimuksessa potilailla, joilla oli edenneen vaiheen KML ja Ph+ ALL, hoidon mediaanikesto oli 14 kuukautta akseleraatiovaiheen KML-potilailla, 3 kuukautta myelooisen blastikriisivaiheen KML-potilailla, 4 kuukautta lymfaattisen blastikriisivaiheen KML-potilailla ja 3 kuukautta Ph+ ALL ‑potilailla. Taulukossa 6b on esitetty valikoidut haittavaikutukset, joita raportoitiin käytettäessä suositeltua aloitusannosta 140 mg kerran vuorokaudessa. Tutkimuksessa arvioitiin myös hoitoa annostuksella 70 mg kahdesti vuorokaudessa. Hoito 140 mg:lla kerran vuorokaudessa oli yhtä tehokas kuin 70 mg:lla kahdesti vuorokaudessa, mutta ensin mainitun hoito-ohjelman turvallisuusprofiili oli edullisempi.
Taulukko 6b: Valikoidut haittavaikutukset, joita raportoitiin faasin III annoksen optimointitutkimuksessa:edenneen vaiheen KML ja Ph+ ALLa
140 mg kerran vuorokaudessa n = 304 | ||
| Kaikki asteet | Aste 3/4 | |
| Suositeltu termi | Prosenttia (%) potilaista | |
Ripuli Nesteretentio Pinnallinen edeema Pleuraeffuusio Yleistynyt edeema Kongestiivinen sydämen vajaatoiminta / sydämen toimintahäiriöb Perikardiaalinen effuusio Keuhkoedeema Verenvuoto Maha-suolikanavan verenvuoto | 28 33 15 20 2 1 2 1 23 8 | 3 7 < 1 6 0 0 1 1 8 6 |
a Faasin III annoksen optimointitutkimuksen viimeisellä, 2 vuoden seurantakäynnillä saadut tulokset potilaista, jotka saivat suositeltua aloitusannosta 140 mg kerran vuorokaudessa (n = 304).
b Mukaan lukien kammion toimintahäiriö, sydämen vajaatoiminta, kongestiivinen sydämen vajaatoiminta, kardiomyopatia, kongestiivinen kardiomyopatia, diastolinen toimintahäiriö, ejektiofraktion pieneneminen ja kammion vajaatoiminta.
Lisäksi kahdessa tutkimuksessa tutkittiin yhteensä 161:tä pediatrista Ph+ ALL ‑potilasta, jotka saivat dasatinibihoitoa yhdessä kemoterapian kanssa. Pivotaalitutkimuksessa oli 106 pediatrista potilasta, jotka saivat dasatinibihoitoa yhdessä kemoterapian kanssa jatkuvalla annosteluohjelmalla. Tukitutkimuksessa oli 55 pediatrista potilasta, joista 35 sai dasatinibihoitoa yhdessä kemoterapian kanssa ei-jatkuvalla annosteluohjelmalla (kaksi viikkoa hoitoa, minkä jälkeen 1–2 viikkoa ilman hoitoa) ja 20 sai dasatinibihoitoa yhdessä kemoterapian kanssa jatkuvalla annosteluohjelmalla. Niiden 126 pediatrisen Ph+ ALL -potilaan, joita hoidettiin dasatinibilla jatkuvalla annosteluohjelmalla, hoidon keston mediaani oli 23,6 kuukautta (vaihteluväli 1,4–33 kuukautta).
Jatkuvalla annosteluohjelmalla hoitoa saaneista 126:sta pediatrisesta Ph+ ALL -potilaasta 2:lla (1,6 %) ilmeni hoidon lopettamiseen johtaneita haittavaikutuksia. Taulukossa 7 on esitetty haittavaikutukset, joita raportoitiin näissä kahdessa tutkimuksessa ≥10 %:n esiintymistiheydellä niillä pediatrisilla potilailla, jotka saivat hoitoa jatkuvalla annosteluohjelmalla. Huomattavaa on, että pleuraeffuusiota raportoitiin 7 potilaalla (5,6 %) tässä ryhmässä, eikä sitä siksi ole otettu mukaan taulukkoon.
Taulukko 7: Haittavaikutukset, joita raportoitiin ≥ 10 %:lla pediatrisista Ph+ ALL ‑potilaista, jotka saivat dasatinibihoitoa yhdessä kemoterapian kanssa jatkuvalla annosteluohjelmalla (N=126)a
| Prosenttia (%) potilaista | ||
| Haittavaikutus | Kaikki asteet | Aste 3/4 |
| Kuumeinen neutropenia | 27,0 | 26,2 |
| Pahoinvointi | 20,6 | 5,6 |
| Oksentelu | 20,6 | 4,8 |
| Vatsakipu | 14,3 | 3,2 |
| Ripuli | 12,7 | 4,8 |
| Kuume | 12,7 | 5,6 |
| Päänsärky | 11,1 | 4,8 |
| Ruokahalun heikkeneminen | 10,3 | 4,8 |
| Väsymys | 10,3 | 0 |
a Pivotaalitutkimuksessa oli yhteensä 106 potilasta, joista 24 sai oraalisuspensiota vähintään kerran, ja 8 potilasta näistä 24:stä sai ainoastaan oraalisuspensiota.
Poikkeavuudet laboratoriotesteissä
Hematologia
Faasin III tutkimuksessa vastadiagnosoidun kroonisen vaiheen KML-potilailla, jotka saivat dasatinibihoitoa, raportoitiin seuraavia 3. ja 4. asteen poikkeavuuksia laboratoriotesteissä, kun seuranta oli kestänyt vähintään 12 kuukautta: neutropenia (21 %), trombosytopenia (19 %) ja anemia (10 %). Kun seuranta oli kestänyt vähintään 60 kuukautta, neutropenian kumulatiivinen esiintymistiheys oli 29 %, trombosytopenian 22 % ja anemian 13 %.
Dasatinibihoitoa saaneilla vastadiagnosoidun kroonisen vaiheen KML-potilailla, jotka kokivat 3. tai 4. asteen myelosuppression, paraneminen tapahtui yleensä hoidon lyhyen keskeyttämisen jälkeen ja/tai annoksen pienentämisen seurauksena tai hoidon pysyvän keskeyttämisen jälkeen (1,6 % potilaista), kun seuranta oli jatkunut vähintään 12 kuukautta. Kun seuranta oli jatkunut vähintään 60 kuukautta, 3. tai 4. asteen myelosuppression vuoksi pysyvästi hoidon lopettaneiden potilaiden kumulatiivinen osuus oli 2,3 %.
KML-potilailla, joilla aikaisempi imatinibihoito ei tuottanut tulosta, tai potilaat eivät sietäneet hoitoa, sytopeniat (trombosytopenia, neutropenia ja anemia) olivat yleinen löydös. Sytopenioiden esiintyvyys riippui kuitenkin myös selkeästi sairauden vaiheesta. Asteen 3 tai 4 hematologiset poikkeavuudet esitetään taulukossa 8.
Taulukko 8: CTC-asteiden 3/4 hematologiset laboratoriotulosten poikkeavuudet kliinisissä tutkimuksissa potilailla, joilla aikaisempi imatinibihoito ei tuottanut tulosta, tai potilaat eivät sietäneet sitäa
Krooninen vaihe (n = 165)b | Akseleraatiovaihe (n = 157)c | Myelooinen blastikriisivaihe (n = 74)c | Lymfaattinen blastikriisivaihe ja Ph+ ALL (n = 168)c | |
| Prosenttia (%) potilaista | ||||
| Hematologiset parametrit | ||||
| Neutropenia | 36 | 58 | 77 | 76 |
| Trombosytopenia | 23 | 63 | 78 | 74 |
| Anemia | 13 | 47 | 74 | 44 |
a Faasin III annoksen optimointitutkimuksen tulokset 2 vuoden seurannan kohdalla.
b CA180-034-tutkimuksen tulokset suositellulla aloitusannoksella 100 mg kerran vuorokaudessa.
c CA180-035-tutkimuksen tulokset suositellulla aloitusannoksella 140 mg kerran vuorokaudessa.
CTC-asteet: neutropenia (aste 3 ≥ 0,5 – < 1,0 × 109/l, aste 4 < 0,5 × 109/l); trombosytopenia (aste 3 ≥ 25 – < 50 × 109/l,
aste 4 < 25 × 109/l); anemia (hemoglobiini aste 3 ≥ 65 – < 80 g/l, aste 4 < 65 g/l).
Asteen 3 tai 4 sytopeniaa ilmeni 100 mg:n päivittäisen kerta-annoksen ryhmässä 2 ja 5 vuoden kuluttua kumulatiivisesti samassa määrin: neutropenia (35 % vs 36 %), trombosytopenia (23 % vs 24 %) ja anemia (13 % vs 13 %).
Asteen 3 ja 4 myelosuppression saaneet potilaat toipuivat yleensä lyhyen annostelun keskeyttämisen ja/tai annoksen pienentämisen jälkeen; pysyvästi hoito lopetettiin 5 %:lla potilaista. Suurin osa potilaista jatkoi hoitoa ilman myelosuppression uudelleen ilmaantumista.
Biokemia
Vastadiagnosoidun kroonisen vaiheen KML-potilailla, jotka saivat dasatinibihoitoa, raportoitiin 3. tai 4. asteen hypofosfatemiaa 4 %:lla potilaista ja ≤ 1 %:lla potilaista raportoitiin 3. tai 4. asteen transaminaasi-, kreatiniini- tai bilirubiinipitoisuuksien nousua, kun seuranta oli kestänyt vähintään 12 kuukautta. Kun seuranta oli kestänyt vähintään 60 kuukautta, 3. tai 4. asteen hypofosfatemian kumulatiivinen esiintymistiheys oli 7 %, 3. tai 4. asteen kreatiniini- ja bilirubiinipitoisuuksien nousun 1 % ja 3. tai 4. asteen transaminaasipitoisuuksien nousun 1 %. Dasatinibihoitoa ei keskeytetty näiden biokemiallisten laboratorioparametrien vuoksi.
2 vuoden seuranta
Asteen 3 ja 4 transaminaasi- tai bilirubiinipitoisuuksien suurenemista raportoitiin 1 %:lla kroonisen vaiheen KML-potilaista (imatinibihoito ei tuottanut tulosta, tai potilaat eivät sietäneet sitä), mutta edenneen vaiheen KML-potilailla ja Ph+ ALL -potilailla suurenemista raportoitiin 1–7 %:lla. Se hoidettiin yleensä annosta pienentämällä tai hoidon lopettamisella. Annoksen optimointia koskeneessa faasin III tutkimuksessa kroonisen vaiheen KML:ssa asteen 3 tai 4 transaminaasi- tai bilirubiiniarvojen nousua todettiin ≤ 1 %:lla potilaista, ja se oli yhtä harvinaista kaikissa neljässä hoitoryhmässä. Faasin III annoksen optimointia koskeneessa tutkimuksessa edenneen vaiheen KML:ssa ja Ph+ ALL:ssa asteen 3 tai 4 transaminaasi- tai bilirubiiniarvojen nousua todettiin 1–5 %:lla potilaista kaikissa hoitoryhmissä.
Keskimäärin 5 %:lla dasatinibia saaneista potilaista, joilla oli normaali lähtöarvo, ilmeni asteen 3 tai 4 ohimenevää hypokalsemiaa jossain vaiheessa tutkimusta. Vähentyneeseen kalsiumin määrään ei yleensä liittynyt kliinisiä oireita. Asteen 3 tai 4 hypokalsemia korjaantui yleensä suun kautta otettavalla kalsiumin korvaushoidolla.
Asteen 3 tai 4 hypokalsemiaa, hypokalemiaa ja hypofosfatemiaa todettiin kaikissa KML:n vaiheissa, mutta ne olivat yleisempiä myelooisen tai lymfaattisen blastikriisivaiheen KML:aa ja Ph+ ALL:aa sairastavilla potilailla. Asteen 3 tai 4 kreatiniinipitoisuuden nousua esiintyi < 1 %:lla kroonisen vaiheen KML-potilaista ja yleisemmin, 1–4 %:lla, edenneen vaiheen KML-potilaista.
Pediatriset potilaat
Dasatinibin turvallisuusprofiili oli pediatrisilla Ph+ CP-KML -potilailla, jotka saivat dasatinibia yksilääkehoitona, samankaltainen kuin aikuisilla. Dasatinibin turvallisuusprofiili oli pediatrisilla Ph+ ALL ‑potilailla, jotka saivat dasatinibia yhdessä kemoterapian kanssa, verrannollinen dasatinibin tunnettuun turvallisuusprofiiliin aikuisilla, ja vastasi kemoterapian odotettuja haittavaikutuksia. Poikkeuksena oli pleuraeffuusion vähäisempi esiintyvyys pediatrisilla potilailla aikuisiin verrattuna.
Pediatrisissa KML-tutkimuksissa havaitut laboratorioarvojen poikkeavuuksien määrät olivat verrannollisia aikuisilla havaittuihin laboratoriotutkimusten löydöksiin.
Pediatrisissa ALL-tutkimuksissa havaitut laboratorioarvojen poikkeavuuksien määrät olivat verrannollisia aikuisilla havaittuihin laboratoriotutkimusten löydöksiin tilanteessa, jossa akuuttia leukemiaa sairastava potilas saa kemoterapiaa.
Erityisryhmät
Vaikka dasatinibin turvallisuusprofiili iäkkäämmillä henkilöillä on samanlainen kuin nuoremmilla henkilöillä, vähintään 65-vuotiailla ilmenee nuorempia todennäköisemmin yleisesti raportoituja haittavaikutuksia, kuten väsymystä, pleuraeffuusiota, dyspneaa, yskää, maha-suolikanavan alaosan verenvuotoja ja ruokahalun häiriöitä, sekä harvemmin raportoituja haittavaikutuksia, kuten vatsan pingotusta, huimausta, perikardiaalista effuusiota, kongestiivista sydämen vajaatoimintaa ja painonlaskua. Siksi tällaisia potilaita tulee seurata tarkasti (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa saadut kokemukset dasatinibiyliannostuksesta rajoittuvat yksittäisiin tapauksiin. Suurin yliannostus, 280 mg vuorokaudessa viikon ajan, raportoitiin kahdella potilaalla, ja molemmilla havaittiin merkittävä trombosyyttimäärän pienentyminen. Dasatinibihoitoon voi liittyä 3. tai 4. asteen myelosuppressiota (ks. Kohta Varoitukset ja käyttöön liittyvät varotoimet), ja siksi potilaiden tilaa on seurattava tarkoin myelosuppression varalta, jos suositeltu annos ylitetään, ja heille on annettava asianmukaista tukihoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, proteiinikinaasin estäjät, ATC-koodi: L01EA02
Farmakodynaamiset vaikutukset
Dasatinibi ehkäisee BCR-ABL-kinaasin ja SRC-kinaasiperheeseen kuuluvien kinaasien aktiivisuutta sekä myös lukuisten muiden onkogeenisten kinaasien, mukaan lukien c-KIT, efriini (EPH) ‑reseptorikinaasit ja PDGFβ-reseptori, aktiivisuutta. Dasatinibi on voimakas BCR-ABL-kinaasin subnanomolaarinen estäjä 0,6–0,8 nM:n pitoisuuksina. Dasatinibi sitoutuu sekä inaktiivisessa että aktiivisessa muodossa olevaan BCR-ABL-entsyymiin.
Vaikutusmekanismi
Dasatinibi tehoaa in vitro leukeemisiin solulinjoihin, jotka ovat imatinibille herkkiä tai resistenttejä. Ei-kliiniset tutkimukset osoittavat, että dasatinibi voi tehota imatinibiresistenssiin, joka on seurausta BCR-ABL:n yli-ilmentymästä, BCR-ABL-kinaasidomeenien mutaatioista, vaihtoehtoisten signaalireittien, joihin liittyvät SRC-perheen kinaasit (LYN, HCK), aktivoitumisesta ja monilääkeresistenssiä aiheuttavasta geeniyli-ilmentymästä. Dasatinibi estää SRC-perheen kinaaseja subnanomolaarisina pitoisuuksina.
Erillisissä in vivo -tutkimuksissa, joissa käytettiin KML:n hiirimalleja, dasatinibi esti kroonisen vaiheen KML:n etenemisen blastikriisivaiheeseen ja lisäsi elinaikaa hiirillä, joihin oli siirretty potilaiden eri elimistön alueilta, myös keskushermostosta, saatuja KML-solulinjoja.
Kliininen teho ja turvallisuus
Faasin I tutkimuksessa havaittiin hematologinen ja sytogeneettinen vaste kaikissa KML:n vaiheissa ja Ph+ ALL:ssa ensimmäisillä 84 potilaalla, joita hoidettiin ja tutkittiin 27 kuukauden ajan. Vaste säilyi kaikkien KML- ja Ph+ ALL -tautivaiheiden ajan.
Neljässä yksihaaraisessa, kontrolloimattomassa, avoimessa faasin II kliinisessä tutkimuksessa selvitettiin dasatinibin turvallisuutta ja tehoa KML:n kroonista, akseleraatio- tai myelooista blastikriisivaihetta sairastavilla potilailla, jotka olivat imatinibiresistenttejä tai -intolerantteja. Yksi satunnaistettu ei-vertaileva tutkimus tehtiin kroonisen vaiheen potilailla, joiden hoito 400 tai 600 mg:lla imatinibia vuorokaudessa oli epäonnistunut. Dasatinibin aloitusannos oli 70 mg kahdesti vuorokaudessa. Annosmuutokset hyväksyttiin tehon parantamiseksi tai toksisuuden hoitamiseksi (ks. Kohta Annostus ja antotapa).
Kahdessa satunnaistetussa, avoimessa faasin III tutkimuksessa on arvioitu dasatinibihoidon tehoa kerran tai kahdesti vuorokaudessa annosteltuna. Lisäksi on tehty yksi avoin, satunnaistettu, faasin III vertailututkimus, johon otetuilla aikuispotilailla oli vastadiagnosoitu kroonisen vaiheen KML.
Dasatinibin tehokkuus perustuu hematologiseen ja sytogeneettiseen vasteeseen.
Hoitovasteen kesto ja arvioitu eloonjäämisaste tuovat lisänäyttöä dasatinibin kliinisistä eduista.
Kliinisissä tutkimuksissa on arvioitu yhteensä 2 712 potilasta, joista 23 % oli ≥ 65-vuotiaita ja 5 % oli
≥ 75-vuotiaita.
Kroonisen vaiheen KML – vastadiagnosoitu
Kansainvälisessä, avoimessa, satunnaistetussa ja vertailevassa faasin III monikeskustutkimuksessa on tutkittu aikuispotilaita, joilla oli vastadiagnosoitu kroonisen vaiheen KML. Potilaat satunnaistettiin saamaan joko dasatinibia 100 mg kerran vuorokaudessa tai imatinibia 400 mg kerran vuorokaudessa. Primaarinen päätetapahtuma oli 12 kuukauden kuluessa saavutettava vahvistettu täydellinen sytogeneettinen vaste (cCCyR). Sekundaariset päätetapahtumat olivat täydellisen sytogeneettisen vasteen kesto (vasteen kestävyyden mittari), aika vahvistetun täydellisen sytogeneettisen vasteen saavuttamiseen, merkittävä molekulaarinen vaste (MMR), aika merkittävän molekulaarisen vasteen saavuttamiseen, etenemisvapaa elinaika (PFS) ja kokonaiselinaika (OS). Muita merkityksellisiä tehoon liittyviä tuloksia olivat täydellinen sytogeneettinen vaste (CcyR) ja täydellinen molekulaarinen vaste (CMR). Tutkimus on kesken.
Yhteensä 519 potilasta satunnaistettiin hoitoryhmään: 259 dasatinibiryhmään ja 260 imatinibiryhmään. Hoitoryhmät olivat lähtötilanteessa keskenään hyvin tasapainossa iän, sukupuolen ja rodun suhteen: Iän mediaani oli 46 vuotta dasatinibiryhmässä (10 % vähintään 65-vuotiaita) ja 49 vuotta imatinibiryhmässä (11 % vähintään 65-vuotiaita). Naisten osuus dasatinibiryhmässä oli 44 % ja imatinibiryhmässä 37 %. Valkoihoisia oli dasatinibiryhmässä 51 % ja imatinibiryhmässä 55 %, ja aasialaisia oli dasatinibiryhmässä 42 % ja imatinibiryhmässä 37 %. Hasford-pistemäärän (Hasford scores) jakauma oli samankaltainen dasatinibiryhmässä ja imatinibiryhmässä (pieni riski 33 % vs 34 %; kohtalainen riski 48 % vs 47 %; suuri riski 19 % vs 19 %).
Seuranta kesti vähintään 12 kuukautta, ja tänä aikana 85 % dasatinibiryhmään satunnaistetuista potilaista ja 81 % imatinibiryhmään satunnaistetuista potilaista sai yhä ensilinjan hoitoa.
Hoito keskeytettiin 12 kuukauden aikana sairauden etenemisen vuoksi dasatinibiryhmässä 3 prosentilla potilaista ja imatinibiryhmässä 5 prosentilla potilaista.
Seuranta kesti vähintään 60 kuukautta, ja tänä aikana 60 % dasatinibiryhmään satunnaistetuista potilaista ja 63 % imatinibiryhmään satunnaistetuista potilaista sai yhä ensilinjan hoitoa. Hoito keskeytettiin 60 kuukauden aikana sairauden etenemisen vuoksi dasatinibiryhmässä 11 prosentilla potilaista ja imatinibiryhmässä 14 prosentilla potilaista.
Tehoa koskevat tulokset ovat taulukossa 9. Tilastollisesti merkitsevästi suurempi osuus dasatinibiryhmän potilaista kuin imatinibiryhmän potilaista saavutti varmistetun täydellisen sytogeneettisen vasteen (cCCyR) ensimmäisen 12 hoitokuukauden aikana. Dasatinibin teho osoitettiin yhdenmukaisesti kaikissa eri alaryhmissä, mukaan lukien ikä, sukupuoli ja Hasford-lähtöpisteet.
Taulukko 9: Faasin III tutkimuksen tehoa koskevat tulokset vastadiagnosoidun kroonisen vaiheen KML-potilailla
dasatinibi n = 259 | imatinibi n = 260 | p-arvo | |
| Hoitoon vastanneet (95 %:n luottamusväli) | |||
Sytogeneettinen vaste 12 kuukauden aikana | |||
| cCCyRa | 76,8 % (71,2–81,8) | 66,2 % (60,1–71,9) | p < 0,007* |
| CcyRb | 85,3 % (80,4–89,4) | 73,5 % (67,7-78,7) | ⎯ |
| 24 kuukauden aikana | |||
| cCCyRa | 80,3 % | 74,2 % | ⎯ |
| CcyRb | 87,3 % | 82,3 % | ⎯ |
| 36 kuukauden aikana | |||
| cCCyRa | 82,6 % | 77,3 % | ⎯ |
| CcyRb | 88,0 % | 83,5 % | ⎯ |
| 48 kuukauden aikana | |||
| cCCyRa | 82,6 % | 78,5 % | ⎯ |
| CcyRb | 87,6 % | 83,8 % | ⎯ |
| 60 kuukauden aikana | |||
| cCCyRa | 83,0 % | 78,5 % | ⎯ |
| CcyRb | 88,0 % | 83,8 % | ⎯ |
| Merkittävä molekulaarinen vastec | |||
| 12 kuukautta | 52,1 % (45,9–58,3) | 33,8 % (28,1–39,9) | p < 0,00003* |
| 24 kuukautta | 64,5 % (58,3–70,3) | 50 % (43,8–56,2) | ⎯ |
| 36 kuukautta | 69,1 % (63,1–74,7) | 56,2 % (49,9–62,3) | ⎯ |
| 48 kuukautta | 75,7 % (70,0–80,8) | 62,7 % (56,5–68,6) | ⎯ |
| 60 kuukautta | 76,4 % (70,8–81,5) | 64,2 % (58,1–70,1) | p = 0,0021 |
| Riskitiheyssuhde (HR) | |||
12 kuukauden aikana (99,99 %:n luottamusväli) | |||
| Aika cCCyR:n saavuttamiseen | 1,55 (1,0–2,3) | p < 0,0001* | |
| Aika MMR:n saavuttamiseen | 2,01 (1,2–3,4) | p < 0,0001* | |
| cCCyR:n säilyminen | 0,7 (0,4–1,4) | p < 0,035 | |
24 kuukauden aikana (95 %:n luottamusväli) | |||
| Aika cCCyR:n saavuttamiseen | 1,49 (1,22–1,82) | ⎯ | |
| Aika MMR:n saavuttamiseen | 1,69 (1,34–2,12) | ⎯ | |
| cCCyR:n säilyminen | 0,77 (0,55–1,10) | ⎯ | |
36 kuukauden aikana (95 %:n luottamusväli) | |||
| Aika cCCyR:n saavuttamiseen | 1,48 (1,22–1,80) | ⎯ | |
| Aika MMR:n saavuttamiseen | 1,59 (1,28–1,99) | ⎯ | |
| cCCyR:n säilyminen | 0,77 (0,53–1,11) | ⎯ | |
48 kuukauden aikana (95 %:n luottamusväli) | |||
| Aika cCCyR:n saavuttamiseen | 1,45 (1,20–1,77) | ⎯ | |
| Aika MMR:n saavuttamiseen | 1,55 (1,26–1,91) | ⎯ | |
| cCCyR:n säilyminen | 0,81 (0,56–1,17) | ⎯ | |
60 kuukauden aikana (95 %:n luottamusväli) | |||
| Aika cCCyR:n saavuttamiseen | 1,46 (1,20–1,77) | p = 0,0001 | |
| Aika MMR:n saavuttamiseen | 1,54 (1,25–1,89) | p < 0,0001 | |
| cCCyR:n säilyminen | 0,79 (0,55–1,13) | p = 0,1983 | |
a Varmistettu täydellinen sytogeneettinen vaste (cCCyR) määritellään vasteeksi, joka on otettu kahdesti peräkkäin (toteamiskertojen välillä vähintään 28 päivää).
b Täydellinen sytogeneettinen vaste (CcyR) perustuu yksittäiseen sytogeneettiseen luuydintutkimukseen.
c Merkittävä molekulaarinen vaste = BCR-ABL-transkripti / kontrollitranskripti ≤ 0,1 % RQ-PCR-menetelmällä määritettynä perifeerisistä verinäytteistä, jotka on vakioitu kansainvälisen asteikon mukaisesti. Nämä ovat kumulatiivisia lukuja määritellyn aikavälin vähimmäisseurannasta.
* Mukautettu Hasford-pistemäärään ja osoitti tilastollista merkitsevyyttä ennalta määritellyn nominaaliasteikollisen merkitsevyystason mukaan.
60 kuukauden seurannan jälkeen mediaaniaika varmistetun täydellisen sytogeneettisen vasteen (cCCYR) saavuttamiseen oli 3,1 kuukautta dasatinibiryhmässä ja 5,8 kuukautta imatinibiryhmässä niillä potilailla, joilla täydellinen sytogeneettinen vaste (CcyR) varmistettiin. 60 kuukauden seurannan jälkeen mediaaniaika merkittävän molekulaarisen vasteen (MMR) saavuttamiseen oli 9,3 kuukautta dasatinibiryhmässä ja 15,0 kuukautta imatinibiryhmässä niillä potilailla, joilla merkittävä molekulaarinen vaste saavutettiin. Nämä tulokset ovat yhteneväisiä 12, 24 ja 36 kuukauden kohdalla saatujen tulosten kanssa.
Aika merkittävän molekulaarisen vasteen (MMR) saavuttamiseen on esitetty graafisesti kuvassa 1. Aika MMR:n saavuttamiseen oli yhdenmukaisesti lyhyempi dasatinibihoitoa saaneilla potilailla kuin imatinibihoitoa saaneilla potilailla.
Kuva 1: Kaplan-Meier-arvio: Aika merkittävän molekulaarisen vasteen (MMR) saavuttamiseen
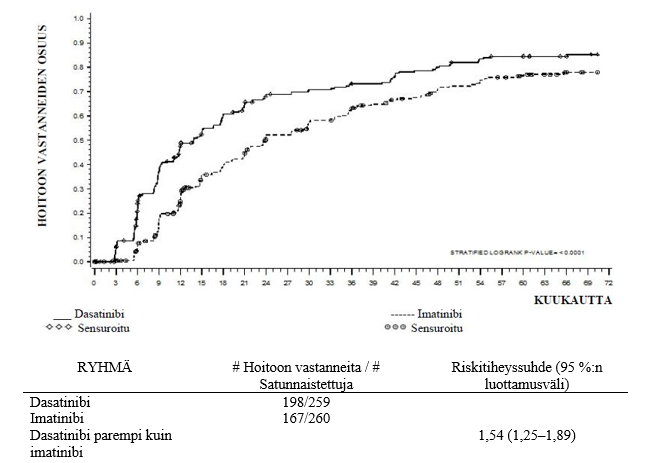
Varmistettujen täydellisten sytogeneettisten vasteiden (cCCyR) osuus oli 3 kuukauden kuluttua dasatinibiryhmässä 54 % ja imatinibiryhmässä 30 %; 6 kuukauden kuluttua vastaavat luvut olivat 70 % ja 56 %; 9 kuukauden kuluttua 75 % ja 63 %; 24 kuukauden kuluttua 80 % ja 74 %; 36 kuukauden kuluttua 83 % ja 77 %; 48 kuukauden kuluttua 83 % ja 79 % ja 60 kuukauden kuluttua 83 % ja 79 %. Nämä tulokset olivat yhdenmukaisia primaarisen päätetapahtuman kanssa. Vastaavat MMR-osuudet dasatinibiryhmässä ja imatinibiryhmässä olivat 3 kuukauden kuluttua 8 % ja 0,4 %; 6 kuukauden kuluttua 27 % ja 8 %; 9 kuukauden kuluttua 39 % ja 18 %; 12 kuukauden kuluttua 46 % ja 28 %; 24 kuukauden kuluttua 64 % ja 46 %; 36 kuukauden kuluttua 67 % ja 55 %; 48 kuukauden kuluttua 73 % ja 60 % ja 60 kuukauden kuluttua 76 % ja 64 %. Myös nämä tulokset olivat yhdenmukaisia primaarisen päätetapahtuman kanssa.
Kuvassa 2 on esitetty graafisesti merkittävän molekulaarisen vasteen (MMR) saavuttaneiden määrät tiettyinä ajankohtina. MMR:n saavuttaneita oli yhdenmukaisesti enemmän dasatinibiryhmässä kuin imatinibiryhmässä.
Kuva 2: MMR:n saavuttaneet eri ajankohtina – kaikki satunnaistetut potilaat faasin III tutkimuksessa, joka koski vastadiagnosoitua kroonisen vaiheen KML:aa
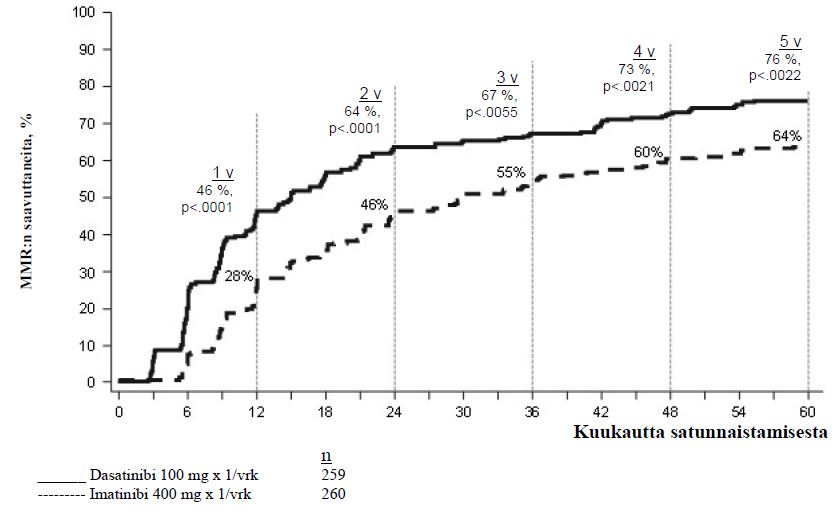
Niiden potilaiden osuus, joilla saavutettiin tutkimuksen missä tahansa vaiheessa BCR-ABL-suhde ≤ 0,01 % (4 login alenema), oli suurempi dasatinibiryhmässä (54,1 %) kuin imatinibiryhmässä (45 %). Niiden potilaiden osuus, joilla saavutettiin tutkimuksen missä tahansa vaiheessa BCR-ABL-suhde ≤ 0,0032 % (4,5 login alenema), oli suurempi dasatinibiryhmässä (44 %) kuin imatinibiryhmässä (34 %).
Kuvassa 3 on esitetty graafisesti MR4,5-vasteen saavuttaneet eri ajankohtina. MR4,5-vasteen saavuttaneita oli ajan myötä yhdenmukaisesti enemmän dasatinibiryhmässä kuin imatinibiryhmässä.
Kuva 3: MR4,5-vasteen saavuttaneet eri ajankohtina – kaikki satunnaistetut potilaat faasin III tutkimuksessa, joka koski vastadiagnosoitua kroonisen vaiheen KML:aa
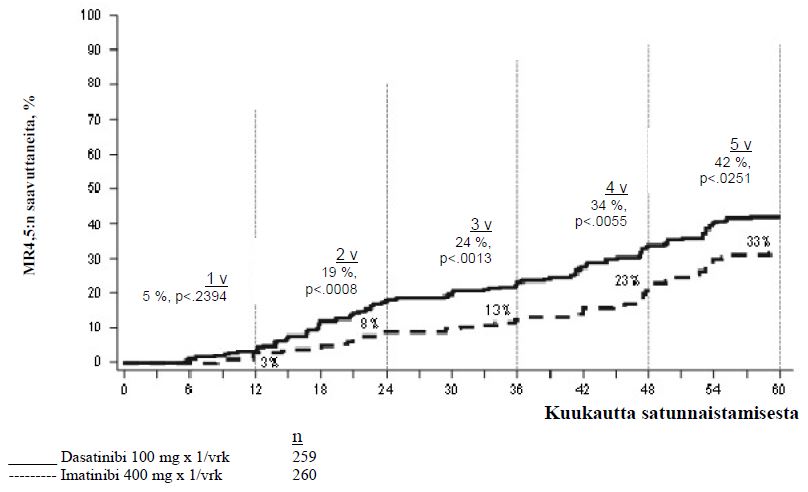
Niiden potilaiden osuus, jotka Hasford-pistemäärän perusteella saavuttivat tutkimuksen missä tahansa vaiheessa merkittävän molekulaarisen vasteen (MMR), oli suurempi dasatinibiryhmässä kuin imatinibiryhmässä (pieni riski: 90 % ja 69 %; kohtalainen riski 71 % ja 65 %; suuri riski: 67 % ja 54 %).
Lisäanalyysissa varhainen molekulaarinen vaste (määritelmä: BCR-ABL-pitoisuus ≤ 10 % 3 kk:n kohdalla) saavutettiin suuremmalla osalla potilaista dasatinibiryhmässä (84 %) kuin imatinibiryhmässä (64 %). Kuten taulukosta 10 käy ilmi, varhaisen molekulaarisen vasteen saavuttaneilla potilailla taudin etenemisriski oli pienempi, suuremmalla osalla potilaista tauti ei ollut edennyt ja elossa olleiden osuus oli myös suurempi.
Taulukko 10: Dasatinibia saaneet potilaat, joiden BCR-ABL oli ≤ 10 % ja > 10 % 3 kuukauden kohdalla
| Dasatinibi, n = 235 | Potilaat, joiden BCR-ABL ≤ 10 % 3 kk:n kohdalla | Potilaat, joiden BCR-ABL > 10 % 3 kk:n kohdalla |
| Potilaita, lkm (%) | 198 (84,3) | 37 (15,7) |
| Potilaat, joilla ilmeni transformaatio, 60 kk, n/N (%) | 6/198 (3,0) | 5/37 (13,5) |
| PFS, 60 kk (95 %:n luottamusväli) | 92,0 % (89,6–95,2) | 73,8 % (52,0–86,8) |
| OS, 60 kk (95 %:n luottamusväli) | 93,8 % (89,3–96,4) | 80,6 % (63,5–90,2) |
Kuvassa 4 on esitetty graafisesti elossa olleiden osuus (OS) tiettyinä ajankohtina. Elossa olleiden osuus oli yhdenmukaisesti suurempi siinä ryhmässä, jossa potilaat saivat dasatinibia ja jotka saavuttivat BCR-ABL-pitoisuuden ≤ 10 % 3 kk:n kohdalla, kuin niillä, jotka sitä eivät saavuttaneet.
Kuva 4: Laatikko-janakuvio: Merkittävä elossa olleiden osuus (OS) dasatinibille mitattuna BCR-ABL-pitoisuutena (≤ 10 % tai > 10 %) 3 kuukauden kohdalla faasin III tutkimuksessa, joka koski vastadiagnosoitua kroonisen vaiheen KML:aa
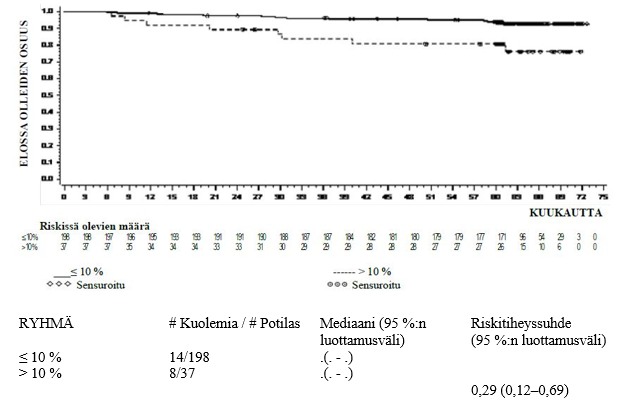
Sairauden etenemiseksi määritettiin valkosolujen lisääntyminen asianmukaisesta hoidosta huolimatta, täydellisen hematologisen vasteen (CHR), osittaisen sytogeneettisen vasteen (CyR) tai täydellisen sytogeneettisen vasteen (CcyR) menettäminen, taudin eteneminen akseleraatio- tai blastikriisivaiheisiin tai kuolema. Niiden potilaiden osuus, joiden tauti ei ollut edennyt 60 kuukauden kohdalla, oli 88,9 % sekä dasatinibiryhmässä että imatinibiryhmässä (luottamusväli 84–92,4 %). 60 kuukauden kohdalla tauti siirtyi akseleraatio- tai blastikriisivaiheisiin pienemmällä osalla dasatinibihoitoa saaneista (n = 8; 3 %) kuin imatinibihoitoa saaneista potilaista (n = 15; 5,8 %). Arvioitu 60 kuukauden eloonjääminen oli dasatinibiryhmässä 90,9 % (luottamusväli: 86,6–93,8 %) ja imatinibiryhmässä 89,6 % (luottamusväli: 85,2–92,8 %). Dasatinibi ja imatinibi eivät eronneet toisistaan elossa olleiden osuuden (OS) (riskitiheyssuhde 1,01; 95 %:n luottamusväli: 0,58–1,73, p = 0,9800) tai PFS:n (riskitiheyssuhde 1,00; 95 %:n luottamusväli: 0,58–1,72, p = 0,9998) suhteen.
BCR-ABL-sekvensointi tehtiin niiden potilaiden verinäytteistä, joilla raportoitiin taudin eteneminen tai jotka lopettivat dasatinibi- tai imatinibihoidon ja joiden verinäyte oli käytettävissä. Mutaatioita esiintyi samassa määrin molemmissa hoitoryhmissä. Dasatinibihoitoa saaneilla potilailla todetut mutaatiot olivat T315I, F317I/L ja V299L. Imatinibihoitoryhmässä mutaatioiden kirjo oli erilainen. In vitro -tutkimusten perusteella dasatinibi ei näytä tehoavan T315I-mutaatioon.
Kroonisen vaiheen KML – resistenssi tai intoleranssi aiempaan imatinibihoitoon
Kaksi kliinistä tutkimusta tehtiin potilailla, jotka olivat imatinibiresistenttejä tai -intolerantteja; ensisijainen hoitotehon päätetapahtuma näissä tutkimuksissa oli ”merkittävä sytogeneettinen vaste” (McyR).
Tutkimus 1
Avoin, satunnaistettu, ei-vertaileva monikeskustutkimus tehtiin potilailla, joiden hoito 400 mg:lla tai 600 mg:lla imatinibia epäonnistui. Potilaat satunnaistettiin (2:1) saamaan dasatinibia (70 mg kahdesti vuorokaudessa) tai imatinibia (400 mg kahdesti vuorokaudessa). Siirtyminen vaihtoehtoiseen hoitoryhmään sallittiin, jos potilailla havaittiin taudin etenemistä tai intoleranssia, jota ei voitu hoitaa annosta muuttamalla. Primaari päätetapahtuma oli merkittävä sytogeneettinen vaste (McyR) 12 viikon kohdalla. Tuloksia on saatavilla 150 potilaasta: 101 satunnaistettiin dasatinibihoitoon ja 49 imatinibihoitoon (kaikki imatinibiresistenttejä). Mediaaniaika diagnoosista satunnaistamiseen oli 64 kuukautta dasatinibiryhmässä ja 52 kuukautta imatinibiryhmässä.
Kaikki potilaat olivat saaneet runsaasti aikaisempia hoitoja. Täydellisen hematologisen vasteen imatinibille oli aikaisemmin saavuttanut 93 % koko potilaspopulaatiosta. Merkittävän sytogeneettisen vasteen imatinibille oli aikaisemmin saavuttanut 28 % dasatinibiryhmän potilaista ja 29 % imatinibiryhmän potilaista.
Hoidon keston mediaani dasatinibilla oli 23 kuukautta (44 %:a potilaista hoidettiin > 24 kuukautta) ja imatinibilla 3 kuukautta (10 %:a potilaista > 24 kuukautta). Dasatinibiryhmässä 93 % potilaista saavutti täydellisen hematologisen vasteen ennen siirtymistä toiseen hoitoon ja 82 % imatinibiryhmän potilaista saavutti täydellisen hematologisen vasteen ennen toiseen hoitoon siirtymistä.
Kolmen kuukauden seurannassa merkittävä sytogeneettinen vaste saavutettiin useammin dasatinibiryhmässä (36 %) kuin imatinibiryhmässä (29 %). 22 % dasatinibiryhmän potilaista sai täydellisen sytogeneettisen vasteen, kun taas vain 8 % imatinibiryhmän potilaista saavutti täydellisen sytogeneettisen vasteen (CcyR). Pidemmän hoitojakson ja seurannan aikana (hoidon mediaani 24 kk) dasatinibihoitoa saaneista potilaista 53 %:lla saavutettiin merkittävä sytogeneettinen vaste (44 %:lla täydellinen sytogeneettinen vaste, CcyR) ja imatinibilla hoidetuista potilaista 33 %:lla saavutettiin merkittävä sytogeneettinen vaste (18 %:lla täydellinen sytogeneettinen vaste, CcyR) ennen siirtymistä toiseen hoitoon. Dasatinibihoitoa saaneista potilaista, jotka olivat saaneet 400 mg imatinibia ennen tutkimukseen osallistumista, 61 %:lla saavutettiin merkittävä sytogeneettinen vaste dasatinibitutkimushaarassa ja 50 %:lla imatinibihaarassa.
Kaplan–Meier-arvion perusteella merkittävä sytogeneettinen vaste säilyi vuoden ajan 92 %:lla (95 %:n luottamusväli: 85–100 %) dasatinibiryhmässä (CcyR 97 %, 95 %:n luottamusväli: 92–100 %) ja 74 %:lla (95 %:n luottamusväli: 49–100 %) imatinibiryhmässä (CcyR 100 %). Merkittävä sytogeneettinen vaste säilyi 18 kuukautta 90 %:lla (95 %:n luottamusväli: 82–98 %) dasatinibiryhmässä (CcyR 94 %, 95 %:n luottamusväli: 87–100 %) ja 74 %:lla (95 %:n luottamusväli: 49–100 %) imatinibiryhmässä (CcyR 100 %).
Kaplan–Meier-arvion perusteella vuoden kohdalla niiden potilaiden osuus, joiden tauti ei ollut edennyt, oli 91 % (95 %:n luottamusväli: 85–97 %) dasatinibiryhmässä ja 73 % (95 %:n luottamusväli: 54–91 %) imatinibiryhmässä. 2 vuoden kohdalla niiden potilaiden osuus, joiden tauti ei ollut edennyt, oli 86 % (95 %:n luottamusväli: 78–93 %) dasatinibiryhmässä ja 65 % (95 %:n luottamusväli: 43–87 %) imatinibiryhmässä.
Dasatinibiryhmän potilaista 43 %:lla ja imatinibiryhmän potilaista 82 %:lla hoito epäonnistui. Epäonnistumiseksi määritettiin sairauden eteneminen tai siirtyminen toiseen hoitoon (vasteen puute, tutkimuslääkeintoleranssi yms.).
Merkittävä molekulaarisen vasteentaso (määritetty BCR-ABL/kontrollitranskriptio ≤ 0,1 % RQ-PCR ääreisveren näytteistä) oli 29 % dasatinibiryhmässä ja 12 % imatinibiryhmässä ennen siirtymistä toiseen hoitoon.
Tutkimus 2
Avoin, yksihaarainen monikeskustutkimus tehtiin imatinibiresistenteillä tai -intoleranteilla potilailla (potilaat, joilla oli merkittäviä haittavaikutuksia aikaisemman imatinibihoidon aikana).
387 potilasta sai dasatinibia 70 mg kahdesti vuorokaudessa (288 resistenttiä ja 99 intoleranttia). Mediaaniaika diagnoosista hoidon aloittamiseen oli 61 kuukautta. Suurin osa potilaista (53 %) oli saanut aikaisemmin imatinibihoitoa yli 3 vuoden ajan. Resistenteimmät potilaat (72 %) olivat saaneet > 600 mg imatinibia vuorokaudessa. 35 % potilaista oli saanut imatinibin lisäksi aikaisemmin sytotoksista kemoterapiaa, 65 % interferonia ja 10 %:lle oli aikaisemmin tehty kantasolujensiirto. 38 %:lla potilaista oli lähtötilanteessa mutaatioita, joiden tiedetään vaikuttavan imatinibiresistenssiin. Dasatinibihoidon keston mediaani oli 24 kuukautta ja 51 %:a potilaista hoidettiin > 24kuukautta. Tulokset hoitotehosta on esitetty taulukossa 11. Merkittävä sytogeneettinen vaste (McyR) saavutettiin 55 % imatinibiresistentillä potilaalla ja 82 % imatinibi-intolerantilla potilaalla. Vähintään 24 kuukauden seurantatutkimuksessa tauti oli edennyt vain 21 potilaalla 240 potilaasta, jotka olivat saavuttaneet merkittävän sytogeneettisen vasteen, eikä merkittävän sytogeneettisen vasteen mediaanikestoa saavutettu.
Kaplan–Meier-arvion perusteella merkittävä sytogeneettinen vaste säilyi vuoden ajan 95 %:lla (95 %:n luottamusväli: 92–98 %) potilaista vuoden ajan ja 88 %:lla (95 %:n luottamusväli: 83–93 %) potilaista 2 vuotta. Täydellinen sytogeneettinen vaste säilyi 97 %:lla (95 %:n luottamusväli: 94–99 %) potilaista vuoden ajan ja 90 %:lla potilaista (95 %:n luottamusväli: 86–95 %) potilaista 2 vuotta. 42 %:lla imatinibiresistenteistä potilaista, joilla aiemmin ei saavutettu merkittävää sytogeneettistä vastetta imatinibille (n = 188), saavutettiin merkittävä sytogeneettinen vaste dasatinibille.
38 %:lla potilaista, jotka osallistuivat tähän tutkimukseen, oli 45 erilaista BCR-ABL-mutaatiota. Täydellinen hematologinen vaste tai merkittävä sytogeneettinen vaste saavutettiin potilailla, joilla oli erilaisia imatinibiresistenssiin liittyviä BCR-ABL-mutaatioita lukuun ottamatta T315I. 2 vuoden kohdalla potilailla oli samanlainen merkittävän sytogeneettisen vasteen taso riippumatta, jos heillä lähtötilanteessa oli BCR-ABL-mutaatio (63 %), P-loopin mutaatio (61 %) tai ei mutaatioita (62 %).
Arvioitu osuus imatinibiresistenssipotilaista, joiden tauti ei ollut edennyt vuoden kuluttua, oli 88 % (95 %:n luottamusväli: 84–92 %) ja 2 vuoden kuluttua 75 % (95 %:n luottamusväli: 69–81 %). Arvioitu osuus imatinibi-intoleranteista potilaista, joiden tauti ei ollut edennyt vuoden kuluttua, oli 98 % (95 %:n luottamusväli: 95–100 %) ja 2 vuoden kuluttua 94 % (95 %:n luottamusväli: 88–99 %).
Merkittävän molekulaarisen vasteen taso 24 kuukauden kohdalla oli 45 % (35 % imatinibiresistenteille potilaille ja 74 % imatinibi-intoleranteille potilaille).
Akseleraatiovaiheen KML
Avoin, yksihaarainen monikeskustutkimus tehtiin imatinibi-intoleranteilla tai resistenteillä potilailla. 174 potilasta sai dasatinibia 70 mg kahdesti vuorokaudessa (161 imatinibiresistenttiä ja 13 imatinibi-intoleranttia). Mediaaniaika diagnoosista hoidon aloittamiseen oli 82 kuukautta. Dasatinibihoidon mediaanikesto oli 14 kuukautta ja 31 %:a potilaista hoidettiin > 24 kuukautta. Merkittäviä molekulaarisia vasteita (arvioitiin 41 potilaalla, joilla oli täydellinen sytogeneettinen vaste, CcyR) 24 kuukauden kohdalla oli 46 %. Lisää tehotuloksia on raportoitu taulukossa 11.
Myelooisen blastikriisivaiheen KML
Avoin, yksihaarainen monikeskustutkimus tehtiin imatinibi-intoleranteilla tai -resistenteillä potilailla. 109 potilasta sai dasatinibia 70 mg kahdesti vuorokaudessa (99 imatinibiresistenttiä ja 10 imatinibi-intoleranttia). Mediaaniaika diagnoosista hoidon aloittamiseen oli 48 kuukautta. Dasatinibihoidon mediaanikesto oli 3,5 kuukautta ja 12 %:a potilaista hoidettiin > 24 kuukautta. Merkittäviä molekulaarisia vasteita (arvioitiin 19 potilaalla, joilla oli täydellinen sytogeneettinen vaste, CcyR) 24 kuukauden kohdalla oli 68 %. Lisää tehotuloksia on raportoitu taulukossa 11.
Lymfaattinen blastikriisivaiheen KML ja Ph+ ALL
Avoin, yksihaarainen monikeskustutkimus tehtiin lymfaattisen blastikriisivaiheen KML-potilailla tai Ph+ ALL -potilailla, jotka olivat resistenttejä tai intolerantteja aikaisemmalle imatinibihoidolle. 48 lymfaattisen blastikriisivaiheen KML-potilasta sai dasatinibia 70 mg kahdesti vuorokaudessa (42 imatinibiresistenttiä ja 6 imatinibi-intoleranttia). Mediaaniaika diagnoosista hoidon aloittamiseen oli 28 kuukautta. Dasatinibihoidon keston mediaani oli 3 kuukautta ja 2 %:a potilaista hoidettiin > 24 kuukautta. Merkittäviä molekulaarisia vasteita (kaikilla hoitoa saaneilla 22 potilaalla oli täydellinen sytogeneettinen vaste, CcyR) 24 kuukauden kohdalla oli 50 %. Lisäksi 46 Ph+ ALL ‑potilasta sai dasatinibia 70 mg kahdesti vuorokaudessa (44 imatinibiresistenttiä ja 2 imatinibi-intoleranttia). Mediaaniaika diagnoosista hoidon aloittamiseen oli 18 kuukautta. Dasatinibihoidon mediaanikesto oli 3 kuukautta ja 7 %:a potilaista hoidettiin > 24 kuukautta. Merkittäviä molekulaarisia vasteita (kaikilla hoitoa saaneilla 25 potilaalla oli täydellinen sytogeneettinen vaste, CcyR) 24 kuukauden kohdalla oli 52 %. Lisää tehotuloksia on raportoitu taulukossa 11. Huomattavaa on, että merkittävät hematologiset vasteet (MaHR) saavutettiin nopeasti (suurin osa 35 päivän sisällä ensimmäisestä dasatinibiannoksesta lymfaattisen blastikriisivaiheen KML-potilailla ja 55 päivän sisällä Ph+ ALL -potilailla).
Taulukko 11: Hoitoteho dasatinibin faasin II yksihaaraisissa kliinisissä kokeissaa
Krooninen (n = 387) | Akseleraatio (n = 174) | Myelooinen blastikriisi (n = 109) | Lymfaattinen blastikriisi (n = 48) | Ph+ ALL (n = 46) | ||
| Hematologinen vasteb (%) | ||||||
| MaHR (95 % CI) | n/a | 64 % (57–72) | 33 % (24–43) | 35 % (22–51) | 41 % (27–57) | |
| CHR (95 % CI) | 91 % (88–94) | 50 % (42–58) | 26 % (18–35) | 29 % (17–44) | 35 % (21–50) | |
| NEL (95 % CI) | n/a | 14 % (10–21) | 7 % (3–14) | 6 % (1–17) | 7 % (1–18) | |
| MaHR kesto (%; Kaplan–Meier arvio) | ||||||
| 1 vuosi | n/a | 79 % (71–87) | 71 % (55–87) | 29 % (3–56) | 32 % (8–56) | |
| 2 vuotta | n/a | 60 % (50–70) | 41 % (21–60) | 10 % (0–28) | 24 % (2–47) | |
| Sytogeneettinen vaste c (%) | ||||||
| McyR (95 % CI) | 62 % (57–67) | 40 % (33–48) | 34 % (25–44) | 52 % (37–67) | 57 % (41–71) | |
| CcyR (95 % CI) | 54 % (48–59) | 33 % (26–41) | 27 % (19–36) | 46 % (31–61) | 54 % (39–69) | |
| Eloonjääminen (%; Kaplan–Meier arvio) | ||||||
Elossa olleiden osuus ilman taudin etenemistä 1 vuosi | 91 % (88–94) | 64 % (57–72) | 35 % (25–45) | 14 % (3–25) | 21 % (9–34) | |
| 2 vuotta | 80 % (75–84) | 46 % (38–54) | 20 % (11–29) | 5 % (0–13) | 12 % (2–23) | |
| Elossa olleiden osuus | ||||||
| 1 vuosi | 97 % (95–99) | 83 % (77–89) | 48 % (38–59) | 30 % (14–47) | 35 % (20–51) | |
| 2 vuotta | 94 % (91–97) | 72 % (64–79) | 38 % (27–50) | 26 % (10–42) | 31 % (16–47) | |
Tässä taulukossa esitetyt tiedot ovat tutkimuksista, joissa aloitusannoksena käytettiin 70 mg kaksi kertaa vuorokaudessa. Katso kohdasta Annostus ja antotapa suositeltu aloitusannos.
a Lihavoidut numerot ovat primäärin päätetapahtuman tulokset.
b Hematologisen vasteen kriteerit (kaikki vasteet vahvistettu 4 viikon jälkeen): merkittävä hematologinen vaste (MaHR) = täydellinen hematologinen vaste (CHR) + ei merkkejä leukemiasta (NEL).
CHR (kroonisen vaiheen KML): valkosoluja ≤ normaalin yläraja, verihiutaleita < 450 000/mm3, ei blasteja tai promyelosyyttejä ääreisverenkierrossa, < 5 % myelosyyttejä + metamyelosyyttejä ääreisverenkierrossa, basofiileja ääreisverenkierrossa < 20 %, eikä ekstramedullaarista sairautta.
CHR (edennyt KML/Ph+ ALL): valkosoluja ≤ normaalin yläraja, absoluuttinen neutrofiilien määrä ≥ 1 000/mm3, verihiutaleiden ≥ 100 000/mm3, ei blasteja tai promyelosyyttejä ääreisverenkierrossa, luuydinblasteja ≤ 5 %, < 5 % myelosyyttejä + metamyelosyyttejä ääreisverenkierrossa, basofiileja ääreisverenkierrossa < 20 %, eikä ekstramedullaarista sairautta.
NEL: samat kriteerit kuin CHR:ssa paitsi absoluuttinen neutrofiilien määrä ≥ 500/mm3 ja < 1 000/mm3 tai verihiutaleita ≥ 20 000/mm3 ja ≤ 100 000/mm3.
c Sytogeneettisen vasteen kriteerit: täydellinen (0 % Ph+-metafaaseja) tai osittainen (> 0–35 %). Merkittävässä sytogeneettisessa vasteessa, McyR (0–35 %) yhdistyvät sekä täydellinen että osittainen vaste.
n/a = ei saatavilla; CI = luottamusväli; ULN = normaaliarvon yläraja (upper limit of normal range).
Dasatinibihoidon jälkeen luuytimensiirron saaneiden potilaiden lopullista hoitotulosta ei ole arvioitu.
Faasin III kliiniset tutkimukset KML-potilailla, joilla oli krooninen vaihe, akseleraatiovaihe tai myelooinen blastikriisivaihe, ja Ph+ ALL -vaiheen potilailla, jotka olivat resistenttejä tai intolerantteja imatinibille
Kahdessa avoimessa satunnaistetussa tutkimuksessa verrattiin kerran vuorokaudessa annetun dasatinibihoidon tehoa kahdesti vuorokaudessa annetun dasatinibihoidon tehoon. Alla esitetyt tulokset perustuvat vähintään 2 vuoden ja 7 vuoden seuranta-aikaan dasatinibihoidon alkamisesta lukien.
Tutkimus 1
Tutkimuksessa, jossa oli mukana kroonisen vaiheen KML:aa sairastavia potilaita, ensisijainen päätetapahtuma oli imatinibille resistenttien potilaiden merkittävä sytogeneettinen vaste (McyR). Tärkein toissijainen päätetapahtuma oli imatinibille resistenttien potilaiden merkittävä sytogeneettinen vaste (McyR) suhteessa kokonaisvuorokausiannokseen. Muita toissijaisia päätetapahtumia olivat merkittävän sytogeneettisen vasteen kesto, PFS ja kokonaiselinaika (OS). Yhteensä 670 potilasta, joista 497 oli imatinibiresistenttejä, jaettiin satunnaistetusti ryhmiin, jotka saivat dasatinibia 100 mg kerran vuorokaudessa, 140 mg kerran vuorokaudessa, 50 mg kahdesti vuorokaudessa tai 70 mg kahdesti vuorokaudessa. Hoidon keston mediaani oli vähintään 59 kuukautta (vaihteluväli 28–66 kuukautta) kaikille niille potilaille, jotka yhä saivat hoitoa ja joiden seuranta-aika oli vähintään 5 vuotta (n = 205). Hoidon keston mediaani oli kaikille potilaille 7 vuoden seurannan kohdalla 29,8 kuukautta (vaihteluväli < 1–92,9 kuukautta).
Hoito todettiin tehokkaaksi kaikissa dasatinibia saaneissa hoitoryhmissä, ja ensisijaiseen tehoa mittaavaan päätetapahtumaan perustuva teho oli yhdenvertainen (non-inferiority) kerran vuorokaudessa ja kahdesti vuorokaudessa hoitoa saaneissa ryhmissä (merkittävän sytogeneettisen vasteen ero 1,9 %, 95 %:n luottamusväli -6,8 – 10,6 %). Hoidon turvallisuus ja siedettävyys osoitettiin kuitenkin muita paremmaksi annostuksella 100 mg:lla kerran vuorokaudessa. Tulokset hoidon tehosta esitellään taulukoissa 12 ja 13.
Taulukko 12: Dasatinibihoidon teho annoksen optimointia koskeneessa faasin III tutkimuksessa: imatinibille resistentti tai intolerantti kroonisen vaiheen KML (2 vuoden tulokset)a
| Kaikki potilaat | n = 167 | |
| Imatinibille resistentit potilaat | n = 124 | |
| Hematologinen vaste b (%) (95 % CI) | ||
| CHR | 92 % (86–95) | |
| Sytogeneettinen vaste c (%) (95 % CI) | ||
McyR Kaikki potilaat Imatinibille resistentit potilaat CcyR Kaikki potilaat Imatinibille resistentit potilaat | 63 % (56–71) 59 % (50–68) 50 % (42–58) 44 % (35–53) | |
| Merkittävä molekulaarinen vaste CcyR:n saavuttaneillad (%) (95 % CI) | ||
| Kaikki potilaat | 69 % (58–79) | |
| Imatinibille resistentit potilaat | 72 % (58–83) | |
a Tulokset raportoitu suositellulla aloitusannoksella 100 mg kerran vuorokaudessa.
b Hematologisen vasteen kriteerit (kaikki vasteet vahvistettu 4 viikon kuluttua): Täydellinen hematologinen vaste (CHR)
(krooninen KML): valkosolut ≤ normaalin yläraja, verihiutaleet < 450 000/mm3, ei blasteja tai promyelosyyttejä ääreisverenkierrossa, < 5 % myelosyyttejä + metamyelosyyttejä ääreisverenkierrossa, basofiileja ääreisverenkierrossa < 20 %, eikä ekstramedullaarista sairautta.
c Sytogeneettisen vasteen kriteerit: täydellinen (0 % Ph+ metafaasit) tai osittainen (> 0–35 %). Merkittävässä sytogeneettisessä vasteessa (McyR) yhdistyvät sekä täydellinen että osittainen (> 0–35 %) vaste.
d Merkittävän molekulaarisen vasteen kriteerit: määritelmän mukaan BCR-ABL-transkripti / kontrollitranskripti ≤ 0,1 % RQ-PCR-menetelmällä määritettynä perifeerisistä verinäytteistä.
Taulukko 13: Dasatinibihoidon pitkäaikaisteho faasin III annoksen optimointitutkimuksessa: imatinibille resistentti tai intolerantti kroonisen vaiheen KMLa
| Seuranta-aika vähintään | ||||
| 1 vuosi | 2 vuotta | 5 vuotta | 7 vuotta | |
| Merkittävä molekulaarinen vaste | ||||
| Kaikki potilaat | NA | 37 % (57/154) | 44 % (71/160) | 46 % (73/160) |
| Imatinibille resistentit potilaat | NA | 35 % (41/117) | 42 % (50/120) | 43 % (51/120) |
| Imatinibille intolerantit potilaat | NA | 43 % (16/37) | 53 % (21/40) | 55 % (22/40) |
| Elossa olleiden osuus ilman taudin etenemistäb | ||||
| Kaikki potilaat | 90 % (86–95) | 80 % (73–87) | 51 % (41–60) | 42 % (33–51) |
| Imatinibille resistentit potilaat | 88 % (82–94) | 77 % (68–85) | 49 % (39–59) | 39 % (29–49) |
| Imatinibille intolerantit potilaat | 97 % (92–100) | 87 % (76–99) | 56 % (37–76) | 51 % (32–67) |
| Elossa olleiden osuus (OS) | ||||
| Kaikki potilaat | 96 % (93–99) | 91 % (86–96) | 78 % (72–85) | 65 % (56–72) |
| Imatinibille resistentit potilaat | 94 % (90–98) | 89 % (84–95) | 77 % (69–85) | 63 % (53–71) |
| Imatinibille intolerantit potilaat | 100 % (100–100) | 95 % (88–100) | 82 % (70–94) | 70 % (52–82) |
a Tulokset raportoitu suositellulla aloitusannoksella 100 mg kerran vuorokaudessa.
b Etenemisen määritelmä: suureneva valkosolumäärä, CHR:n tai McyR:n menetys, Ph+-metafaasien lisääntyminen ≥ 30 %, vahvistettu akseleraatiovaihe tai blastikriisivaihe tai kuolema. Etenemisvapaa elinaika analysoitiin hoitoaikomusperiaatteen mukaisesti ja potilaita seurattiin tapahtumien ja niiden hoidon suhteen.
Kaplan–Meier-arvion perusteella merkittävä sytogeneettinen vaste (McyR) säilyi 18 kuukautta 93 %:lla (95 %:n luottamusväli: 88–98 %) potilaista, jotka saivat 100 mg dasatinibia kerran vuorokaudessa.
Teho arvioitiin myös imatinibi-intoleranteilla potilailla. Tässä potilasryhmässä 100 mg kerran vuorokaudessa saaneilla potilailla merkittävä sytogeneettinen vaste (McyR) saavutettiin 77 %:lla ja täydellinen sytogeneettinen vaste (CcyR) 67 %:lla.
Tutkimus 2
Tutkimuksessa, jossa oli mukana edenneen vaiheen KML:aa ja Ph+ ALL:aa sairastavia potilaita, ensisijainen päätetapahtuma oli merkittävä hematologinen vaste (MaHR). Yhteensä 611 potilasta jaettiin satunnaistetusti ryhmiin, jotka saivat dasatinibihoitoa joko 140 mg kerran vuorokaudessa tai 70 mg kahdesti vuorokaudessa. Hoidon keston mediaani oli noin 6 kuukautta (vaihteluväli 0,03–31 kuukautta).
Ensisijaiseen tehoa mittaavaan päätetapahtumaan perustuva teho oli yhdenvertainen (non-inferiority) kerran vuorokaudessa ja kahdesti vuorokaudessa hoitoa saaneissa ryhmissä (merkittävän hematologisen vasteen ero 0,8 %, 95 %:n luottamusväli −7,1 – 8,7 %). Hoidon turvallisuus ja siedettävyys osoitettiin kuitenkin paremmaksi annostuksella 140 mg:lla kerran vuorokaudessa. Hoitovasteet esitellään taulukossa 14.
Taulukko 14: Dasatinibihoidon teho annoksen optimointia koskeneessa faasin III tutkimuksessa: edenneen vaiheen KML ja Ph+ ALL (2 vuoden tulokset)a
Akseleraatiovaihe (n = 158) | Myelooinen blastikriisivaihe (n = 75) | Lymfaattinen blastikriisivaihe (n = 33) | Ph+ ALL (n = 40) | |
MaHRb (95 % CI) | 66 % (59–74) | 28 % (18–40) | 42 % (26–61) | 38 % (23–54) |
CHRb (95 % CI) | 47 % (40–56) | 17 % (10–28) | 21 % (9–39) | 33 % (19–49) |
NELb (95 % CI) | 19 % (13–26) | 11 % (5–20) | 21 % (9–39) | 5 % (1–17) |
McyRc (95 % CI) | 39 % (31–47) | 28 % (18–40) | 52 % (34–69) | 70 % (54–83) |
CcyR (95 % CI) | 32 % (25–40) | 17 % (10–28) | 39 % (23–58) | 50 % (34–66) |
a Tulokset raportoitu suositellulla aloitusannoksella 140 mg kerran vuorokaudessa (ks. Kohta Annostus ja antotapa).
b Hematologisen vasteen kriteerit (kaikki vasteet vahvistettu 4 viikon jälkeen): merkittävä hematologinen vaste (MaHR) = täydellinen hematologinen vaste (CHR) + ei merkkejä leukemiasta (NEL).
CHR: valkosoluja ≤ normaalin yläraja (ULN), absoluuttinen neutrofiilien määrä ≥ 1 000/mm3, verihiutaleita ≥ 100 000/mm3, ei blasteja tai promyelosyyttejä ääreisverenkierrossa, blasteja luuytimessä ≤ 5 %, < 5 % myelosyyttejä + metamyelosyyttejä ääreisverenkierrossa, basofiileja ääreisverenkierrossa < 20 %, eikä ekstramedullaarista sairautta.
NEL: samat kriteerit kuin CHR:ssa paitsi absoluuttinen neutrofiilien määrä ≥ 500/mm3 ja < 1 000/mm3 tai verihiutaleita ≥ 20 000/mm3 ja ≤ 100 000/mm3
c Merkittävässä sytogeneettisessä vasteessa (McyR) yhdistyvät sekä täydellinen (0 % Ph+-metafaaseja) että osittainen (> 0–35 %) vaste.
CI = luottamusväli, ULN = normaaliarvon yläraja (upper limit normal range).
Akseleraatiovaiheen (KML) potilailla, joiden hoitoannos oli 140 mg kerran vuorokaudessa, ei saavutettu merkittävän hematologisen vasteen (MaHR) mediaanikestoa eikä kokonaiselinajan (OS) mediaania, ja heillä mediaani etenemisvapaa elinaika (PFS) oli 25 kuukautta.
Myelooisen blastikriisivaiheen potilailla (KML), joiden hoitoannos oli 140 mg kerran vuorokaudessa, merkittävän hematologisen vasteen mediaanikesto oli 8 kuukautta, mediaani etenemisvapaa elinaika 4 kuukautta ja kokonaiselinajan (OS) mediaani 8 kuukautta. Lymfaattisen blastikriisivaiheen potilailla (KML), joiden hoitoannos oli 140 mg kerran vuorokaudessa, merkittävän hematologisen vasteen (MaHR) mediaanikesto oli 5 kuukautta, mediaani etenemisvapaa elinaika 5 kuukautta ja kokonaiselinajan (OS) mediaani 11 kuukautta.
Ph+ ALL -potilailla, joiden hoitoannos oli 140 mg kerran vuorokaudessa, merkittävän hematologisen vasteen (MaHR) mediaanikesto oli 5 kuukautta, mediaani etenemisvapaa elinaika 4 kuukautta ja kokonaiselinajan (OS) mediaani 7 kuukautta.
Pediatriset potilaat
Pediatriset potilaat, joilla on KML
130 potilaasta, jolla oli kroonisen vaiheen KML (CP-KML) ja joita hoidettiin kahdessa pediatrisessa tutkimuksessa, avoimessa faasin I satunnaistamattomassa vaihtelevin annoksin tehdyssä tutkimuksessa ja avoimessa faasin II satunnaistamattomassa tutkimuksessa, 84 potilaalla (vain faasin II tutkimuksessa) oli vastadiagnosoitu CP-KML ja 46 potilaalla (17:llä faasin I tutkimuksessa ja 29:llä faasin II tutkimuksessa) aikaisempi imatinibihoito ei tuottanut tulosta tai potilaat eivät sietäneet sitä. Yhdeksänkymmentäseitsemän pediatrista CP-KML-potilasta 130:stä sai dasatinibitabletteja 60 mg/m2 kerran vuorokaudessa (potilailla, joiden kehon pinta-ala oli suuri, suurin vuorokausiannos oli 100 mg kerran vuorokaudessa). Potilaat saivat hoitoa, kunnes sairaus alkoi edetä tai hoidon toksisuus kasvoi liian suureksi.
Tehon tärkeimmät päätetapahtumat olivat täydellinen sytogeneettinen vaste (CcyR), merkittävä sytogeneettinen vaste (McyR) ja merkittävä molekulaarinen vaste (MMR). Tulokset hoitotehosta on esitetty taulukossa 15.
Taulukko 15: Dasatinibihoidon teho pediatrisilla potilailla, joilla on CP-KML
Kumulatiivinen vaste eri ajankohtina lyhimmän seurantajakson mukaan
| 3 kuukautta | 6 kuukautta | 12 kuukautta | 24 kuukautta | |
| CcyR | ||||
| (95 %:n luottamusväli) | ||||
| Vastadiagnosoitu | 43,1 % | 66,7 % | 96,1 % | 96,1 % |
| (N = 51)a | (29,3–57,8) | (52,1–79,2) | (86,5–99,5) | (86,5–99,5) |
| Aiempi imatinibihoito | 45,7 % | 71,7 % | 78,3 % | 82,6 % |
| (N = 46)b | (30,9–61,0) | (56,5–84,0) | (63,6–89,1) | (68,6–92,2) |
| McyR | ||||
| (95 %:n luottamusväli) | ||||
| Vastadiagnosoitu | 60,8 % | 90,2 % | 98,0 % | 98,0 % |
| (N = 51)a | (46,1–74,2) | (78,6–96,7) | (89,6–100) | (89,6–100) |
| Aiempi imatinibihoito | 60,9 % | 82,6 % | 89,1 % | 89,1 % |
| (N = 46)b | (45,4–74,9) | (68,6–92,2) | (76,4–96,4) | (76,4–96,4) |
| MMR | ||||
| (95 %:n luottamusväli) | ||||
| Vastadiagnosoitu | 7,8 % | 31,4 % | 56,9 % | 74,5 % |
| (N = 51)a | (2,2–18,9) | (19,1–45,9) | (42,2–70,7) | (60,4–85,7) |
| Aiempi imatinibihoito | 15,2 % | 26,1 % | 39,1 % | 52,2 % |
| (N = 46)b | (6,3–28,9) | (14,3–41,1) | (25,1–54,6) | (36,9–67,1) |
a Potilaat faasin II pediatrisesta tutkimuksesta, jossa potilailla vastadiagnosoitu CP-KML ja jossa valmistetta annettiin suun kautta tablettina
b Potilaat faasin I ja faasin II pediatrisista tutkimuksista, joissa potilailla imatinibille resistentti tai intolerantti CP‑KML ja joissa valmistetta annettiin suun kautta tablettina
Faasin I pediatrisessa tutkimuksessa vähintään seitsemän vuoden seurannan jälkeen 17 potilaalla, joilla oli imatinibille resistentti tai intolerantti CP-KML, mediaani etenemisvapaa elinaika oli 53,6 kuukautta ja elossa olleiden osuus (OS) 82,4 %.
Faasin II pediatrisessa tutkimuksessa tabletteja saaneiden 51 potilaan, joilla oli vastadiagnosoitu CP‑KML, arvioitu elossa olevien potilaiden osuus ilman taudin etenemistä 24 kuukauden kohdalla oli 94,0 % (82,6–98,0) ja 29 potilaan, joilla oli imatinibille resistentti tai intolerantti CP-KML, 81,7 % (61,4–92,0). 24 kuukauden seurannan jälkeen elossa olleiden osuus (OS) oli vastadiagnosoiduilla potilailla 100 % ja imatinibille resistenteillä tai intoleranteilla potilailla 96,6 %.
Faasin II pediatrisessa tutkimuksessa yhdellä vastadiagnosoidulla potilaalla ja kahdella imatinibille resistentillä tai intolerantilla potilaalla tauti eteni blastikriisivaiheen KML:ksi.
Tutkimuksessa 33 vastadiagnosoitua pediatrista potilasta, joilla oli CP-KML, sai dasatinibijauhetta oraalisuspensiota varten 72 mg/m2:n annoksena. Tällä annoksella altistus on 30 % pienempi kuin suositellulla annoksella. Näillä potilailla 12 kuukauden kohdalla CcyR oli 87,9 % (95 %:n luottamusväli: 71,8–96,6) ja MMR 45,5 % (95 %:n luottamusväli: 28,1–63,6).
Dasatinibilla hoidetuilla pediatrisilla potilailla, joilla oli CP-KML ja jotka olivat aiemmin saaneet imatinibihoitoa, hoidon päätyttyä todetut mutaatiot olivat T315A, E255K ja F317L. Mutaatiot E255K ja F317L todettiin kuitenkin myös jo ennen hoitoa. Potilailla, joilla oli vastadiagnosoitu CP-KML, ei todettu mutaatioita hoidon päätyttyä.
Pediatriset potilaat, joilla on ALL
Dasatinibihoidon tehoa yhdessä kemoterapian kanssa arvioitiin pivotaalitutkimuksessa, jossa tutkittiin yli 1-vuotiaita pediatrisia potilaita, joilla oli vastadiagnosoitu Ph+ ALL.
Tutkimus oli faasin II historiallisesti kontrolloitu monikeskustutkimus, jossa tutkittiin dasatinibia tavanomaisen kemoterapian lisänä 106 pediatrisella potilaalla, joilla oli vastadiagnosoitu Ph+ ALL. Näistä 106 potilaasta 104:llä oli vahvistettu Ph+ ALL. Potilaat saivat dasatinibia päivittäisannoksella 60 mg/m2 jatkuvalla annosteluohjelmalla enintään 24 kuukauden ajan yhdessä kemoterapian kanssa. Kahdeksankymmentäkaksi potilasta sai ainoastaan dasatinibitabletteja, ja 24 potilasta sai dasatanibia oraalisuspensiona vähintään kerran. Näistä 24:stä 8 sai ainoastaan oraalisuspensiota. Kemoterapiahoito-ohjelma oli sama kuin AIEOP‑BFM ALL 2000 ‑tutkimuksessa (tavanomainen kemoterapeuttinen monen lääkeaineen kemoterapiaohjelma). Ensisijainen tehoa mittaava päätetapahtuma oli 3 vuoden tapahtumavapaa elossaolo-osuus (EFS), joka oli 65,5 % (55,5, 73,7).
Minimaalisen jäännöstaudin (MRD) suhteen negatiivisten potilaiden osuus, joka arvioitiin Ig/TCR-uudelleenjärjestymällä, oli 71,7 % kaikista hoidetuista potilaista konsolidaatiovaiheen päättymisen jälkeen. Kun osuuden arvio perustui 85 potilaan arvioitavissa oleviin Ig/TCR-tuloksiin, negatiivisten osuus oli 89,4 %. Minimaalisen jäännöstaudin suhteen negatiivisten osuudet olivat virtaussytometrialla mitattuina induktiovaiheen lopussa 66,0 % ja konsolidaatiovaiheen lopussa 84 %.
Farmakokinetiikka
Dasatinibin farmakokinetiikkaa tutkittiin 229 terveellä aikuisella koehenkilöllä sekä 84 potilaalla.
Imeytyminen
Dasatinibi imeytyy nopeasti potilailla oraalisen annon jälkeen ja huippupitoisuudet saavutetaan 0,5–3 tunnissa. Oraalisen annon jälkeen keskimääräisen altistuksen lisääntyminen (AUCτ) on suunnilleen suhteessa annoslisäyksiin, kun annos on 25–120 mg kaksi kertaa vuorokaudessa. Dasatinibin keskimääräinen terminaalinen puoliintumisaika potilailla on noin 5–6 tuntia.
Terveillä koehenkilöillä, joille annettiin 100 mg:n kerta-annos dasatinibia 30 minuuttia runsasrasvaisen aterian jälkeen, dasatinibin keskimääräinen AUC suureni 14 %. Kun 30 minuuttia ennen dasatinibin antoa annettiin vähärasvainen ateria, kasvoi dasatinibin keskimääräinen AUC 21 %. Havaitut ruoan vaikutukset altistukseen eivät ole kliinisesti merkittäviä. Dasatinibialtistuksen vaihtelu on suurempaa paastoavilla potilailla (47 % CV) verrattuna vähärasvaisen aterian saaneisiin potilaisiin (39 % CV) ja runsasrasvaisen aterian saaneisiin potilaisiin (32 % CV).
Populaatiofarmakokineettisen analyysin perusteella dasatinibialtistuksen vaihtelun syynä arvioitiin olevan pääasiassa hyötyosuuden toteamiskertojen välinen vaihtelu (44 % CV) ja vähäisemmässä määrin hyötyosuuden yksilöiden välinen vaihtelu (30 % CV) ja puhdistuman yksilöiden välinen vaihtelu (32 % CV). Satunnaisen toteamiskertojen välisen altistuksen vaihtelun ei odoteta vaikuttavan kumulatiiviseen altistukseen, tehokkuuteen tai turvallisuuteen.
Jakautuminen
Potilailla dasatinibin näennäinen jakautumistilavuus on suuri (2 505 l), variaatiokerroin (CV % 93 %), mikä viittaa siihen, että lääkevalmiste kulkeutuu hyvin suuressa määrin verisuonien ulkopuolelle. In vitro ‑tutkimusten perusteella dasatinibi sitoutui noin 96-prosenttisesti plasman proteiineihin kliinisesti merkittävinä pitoisuuksina.
Biotransformaatio
Dasatinibi metaboloituu merkittävästi ihmisillä useiden entsyymien vaikutuksesta. Terveillä koehenkilöillä, joille annettiin 100 mg [14C]-merkittyä dasatinibia, muuttumaton dasatinibi edusti 29 % plasman radioaktiivisuudesta. Pitoisuudet plasmassa ja mitattu in vitro ‑aktiivisuus osoittavat, että dasatinibin metaboliiteilla ei todennäköisesti ole merkitystä lääkkeen farmakologiassa. CYP3A4 on tärkein dasatinibia metaboloiva entsyymi.
Eliminaatio
Dasatinibin keskimääräinen terminaalinen puoliintumisaika on 3–5 tuntia. Keskimääräinen ilmeinen oraalinen puhdistuma on 363,8 l/h (CV % 81,3 %).
Eliminaatio tapahtuu pääasiassa ulosteeseen, enimmäkseen metaboliitteina. [14C]-merkityn dasatinibin oraalisen kerta-annoksen jälkeen noin 89 % annoksesta eliminoitui 10 päivässä ja radioaktiivisuudesta 4 % päätyi virtsaan ja 85 % ulosteeseen. Virtsaan erittyneestä annoksesta 0,1 % ja ulosteeseen erittyneestä annoksesta 19 % oli muuttumatonta dasatinibia ja loput metaboliitteja.
Maksan ja munuaisten vajaatoiminta
Maksan vajaatoiminnan vaikutusta dasatinibin kerta-annoksen farmakokinetiikkaan tutkittiin kahdeksalla lievää maksan vajaatoimintaa sairastavalla tutkittavalla, jotka saivat annoksen 50 mg, ja viidellä vaikeaa maksan vajaatoimintaa sairastavalla tutkittavalla, jotka saivat annoksen 20 mg, ja näitä verrattiin terveisiin verrokkeihin, jotka saivat annoksen 70 mg dasatinibia. Dasatinibin keskimääräinen Cmax-arvo ja keskimääräinen AUC-arvo asetettiin 70 mg:n annoksen perusteella, ja arvot pienenivät 47 % ja 8 %, tässä järjestyksessä, lievää maksan vajaatoimintaa sairastavilla potilailla verrattuna tutkittaviin, joilla maksan toiminta oli normaali. Vaikeaa maksan vajaatoimintaa sairastavilla potilailla keskimääräinen Cmax-arvo ja keskimääräinen AUC-arvo, jotka asetettiin 70 mg:n annoksen perusteella, pienenivät 43 % ja 28 %, tässä järjestyksessä, verrattuna tutkittaviin, joilla maksan toiminta oli normaali (ks. Kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Dasatinibi ja sen metaboliitit erittyvät vähäisessä määrin munuaisten kautta.
Pediatriset potilaat
Dasatinibin farmakokinetiikkaa tutkittiin 104 pediatrisella potilaalla, joilla oli leukemia tai kiinteitä kasvaimia (72 sai tabletteja ja 32 jauhetta oraalisuspensiota varten).
Pediatrisia potilaita tutkineessa farmakokineettisessa tutkimuksessa annoksen suhteen normalisoitu dasatinibialtistus (Cavg, Cmin ja Cmax) näyttää olevan samanlainen 21:n CP-KML-potilaan ja 16:n Ph+ ALL -potilaan välillä.
Tablettimuodossa annetun dasatinibin farmakokinetiikkaa tutkittiin 72 pediatrisella potilaalla, joilla oli uusiutunut tai hoitoon huonosti reagoiva leukemia tai kiinteitä kasvaimia. Suun kautta otetut annokset olivat 60–120 mg/m2 kerran vuorokaudessa ja 50–110 mg/m2 kahdesti vuorokaudessa. Kummankin tutkimuksen tiedot yhdistettiin, ja ne osoittivat, että dasatinibi imeytyi nopeasti. Kaikilla annoksilla ja kaikissa ikäryhmissä keskimääräinen Tmax oli 0,5–6 tuntia ja keskimääräinen puoliintumisaika 2–5 tuntia. Dasatinibin farmakokinetiikka osoitti annosriippuvuutta, sillä pediatrisilla potilailla altistuksen havaittiin kasvavan suhteessa annokseen. Dasatinibin farmakokinetiikassa ei ollut merkittävää eroa lasten ja nuorten välillä. Dasatinibin annoksen suhteen normalisoidut Cmax:n, AUC (0-t):n ja AUC (inf):n geometriset keskiarvot näyttivät olevan samankaltaiset lapsilla ja nuorilla eri annoksilla. Populaatiofarmakokineettiseen malliin perustuva simulaatio ennusti, että kohdassa Annostus ja antotapa kuvatulta kehonpainon mukaan porrastetulta tabletin annossuositukselta odotetaan samaa altistusta kuin tablettina annetulta 60 mg/m2:n annokselta. Nämä tiedot on otettava huomioon, jos potilas vaihtaa tabletista oraalisuspensioon tai toisin päin.
Prekliiniset tiedot turvallisuudesta
Dasatinibin ei-kliinistä turvallisuusprofiilia arvioitiin in vitro- ja in vivo ‑tutkimussarjalla hiirillä, rotilla, apinoilla ja kaneilla.
Tärkeimmät toksisuudet esiintyivät ruoansulatuskanavassa ja hematopoieettisessa tai lymfaattisessa järjestelmässä. Gastrointestinaalitoksisuus oli annosta rajoittava tekijä rotilla ja apinoilla. Rotilla vähäisiin tai pieniin punasoluparametrien laskuihin liittyi luuydinmuutoksia; samanlaisia muutoksia esiintyi apinoilla, mutta niitä esiintyi harvemmin. Lymfaattisen järjestelmän toksisuuteen kuului rotilla imusolmukkeiden, pernan ja kateenkorvan imukudostoiminnan heikkeneminen sekä lymfaattisen järjestelmän elinten painon pieneneminen. Muutokset ruoansulatuskanavassa sekä hematopoieettisen ja lymfaattisen järjestelmän muutokset palautuivat hoidon keskeyttämisen jälkeen.
Munuaismuutokset apinoilla, joita hoidettiin enimmillään 9 kuukautta, rajoittuivat munuaisten normaalin mineralisaation lisääntymiseen. Ihoverenvuotoa esiintyi akuuteissa oraalisissa kerta-annostutkimuksissa apinoilla, mutta ei toistuvan annon tutkimuksissa apinoilla eikä rotilla. Rotilla dasatinibi ehkäisi verihiutaleiden aggregaatiota in vitro ja pitkitti orvaskeden verenvuotoaikaa in vivo, mutta ei aiheuttanut spontaania verenvuotoa.
Dasatinibin aktiivisuus in vitro hERG- ja Purkinjen säikeillä suoritetuissa tutkimuksissa viittasi siihen, että sydänkammion repolarisaation piteneminen (QT-aika) on mahdollista. Kuitenkaan in vivo ‑kerta-annostutkimuksissa tajuissaan olevilla, telemetrian avulla seuratuilla apinoilla ei esiintynyt QT-ajan tai EKG:n muutoksia.
Dasatinibi ei ollut mutageeninen in vitro ‑bakteerisolututkimuksissa (Amesin testi) eikä genotoksinen in vivo ‑mikronukleuskokeessa rotalla. Dasatinibi oli klastogeeninen in vitro kiinanhamsterin jakaantuvissa munasoluissa (CHO).
Dasatinibi ei vaikuttanut urosten ja naaraiden hedelmällisyyteen tavanomaisissa rotilla tehdyissä hedelmällisyystutkimuksissa ja varhaisen vaiheen sikiön kehitystä koskevissa tutkimuksissa, mutta indusoi alkiokuolleisuutta annostasoilla, jotka vastasivat suurin piirtein kliinistä altistusta ihmisillä. Alkion- ja sikiönkehitystä koskevissa tutkimuksissa dasatinibi samoin indusoi alkiokuolleisuutta, johon liittyi rottapoikueen koon pienenemistä sekä sikiön luuston muutoksia rotilla ja kaneilla. Nämä muutokset ilmenivät annoksilla, jotka eivät aiheuttaneet toksisuutta emoille, mikä viittaa siihen, että dasatinibi on selektiivinen organogeneesin aikana vaikuttava lisääntymistoksinen yhdiste.
Dasatinibi indusoi immunosuppressiota hiirillä. Vaikutus oli annoksesta riippuvainen ja saatiin hyvin hallintaan dasatinibin annosta pienentämällä ja/tai annosaikataulua muuttamalla. Dasatinibilla oli fototoksisia vaikutuksia in vitro ‑fototoksisuusanalyysissä (neutral red uptake) hiiren fibroblasteissa. Dasatinibi ei ollut fototoksinen in vivo kun sitä annettiin kerta-annoksena suun kautta karvattomille naarashiirille jopa 3-kertainen annos ihmisten altistukseen verrattuna suositellun terapeuttisen annoksen (perustuu AUC:hen) jälkeen.
Rotille annettiin kaksivuotisessa karsinogeenisuustutkimuksessa 0,3, 1 ja 3 mg/kg/vrk dasatinibia suun kautta. Suurimmalla annoksella saavutettiin plasmassa altistus (AUC), joka vastasi yleisesti ottaen altistusta, joka ihmisellä saavutetaan suositellulla aloitusannoksella, 100–140 mg päivittäin. Kohdun ja kohdunkaulan okasolusyöpien ja papilloomien ilmaantuvuus suuria annoksia saaneilla naarailla ja eturauhasen adenoomien ilmaantuvuus pieniä annoksia saaneilla uroksilla suureni yhteen laskettuna tilastollisesti merkitsevästi. Näiden rotan karsinogeenisuustutkimuksen löydösten merkitystä ihmiselle ei tunneta.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Laktoosimonohydraatti (200)
Mikrokiteinen selluloosa (101 ja 102)
Kroskarmelloosinatrium
Hydroksipropyyliselluloosa (MW 80 000)
Magnesiumstearaatti
Kalvopäällyste
Laktoosimonohydraatti
Hypromelloosi (15 mPas)
Titaanidioksidi (E171)
Triasetiini
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
DASATINIB KRKA tabletti, kalvopäällysteinen
20 mg (L:kyllä) 60 fol (266,12 €)
50 mg (L:kyllä) 60 fol (526,48 €)
70 mg (L:kyllä) 60 fol (311,01 €)
PF-selosteen tieto
Läpipainopakkaus (OPA/alumiini/PVC//alumiinifolio): 30 tai 60 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Dasatinib Krka 20 mg kalvopäällysteiset tabletit
Valkoinen tai luonnonvalkoinen, kaksoiskupera, pyöreä kalvopäällysteinen tabletti, jonka halkaisija on noin 5,6 mm. Tabletin toiselle puolelle on painettu ”D7SB” ja toiselle puolelle ”20”.
Dasatinib Krka 50 mg kalvopäällysteiset tabletit
Valkoinen tai luonnonvalkoinen, kaksoiskupera, soikea kalvopäällysteinen tabletti, jonka pituus on noin 11,0 mm ja leveys noin 6,0 mm. Tabletin toiselle puolelle on painettu ”D7SB” ja toiselle puolelle ”50”.
Dasatinib Krka 70 mg kalvopäällysteiset tabletit
Valkoinen tai luonnonvalkoinen, kaksoiskupera, pyöreä kalvopäällysteinen tabletti, jonka halkaisija on noin 9,1 mm. Tabletin toiselle puolelle on painettu ”D7SB” ja toiselle puolelle ”70”.
Dasatinib Krka 80 mg kalvopäällysteiset tabletit
Valkoinen tai luonnonvalkoinen, kaksoiskupera, kolmionmuotoinen kalvopäällysteinen tabletti, jonka pituus on noin 10,4 mm ja leveys noin 10,6 mm. Tabletin toiselle puolelle on painettu ”D7SB” ja toiselle puolelle ”80”.
Dasatinib Krka 100 mg kalvopäällysteiset tabletit
Valkoinen tai luonnonvalkoinen, kaksoiskupera, soikea kalvopäällysteinen tabletti, jonka pituus on noin 15,1 mm ja leveys noin 7,1 mm. Tabletin toiselle puolelle on painettu ”D7SB” ja toiselle puolelle ”100”.
Dasatinib Krka 140 mg kalvopäällysteiset tabletit
Valkoinen tai luonnonvalkoinen, kaksoiskupera, pyöreä kalvopäällysteinen tabletti, jonka halkaisija on noin 11,7 mm. Tabletin toiselle puolelle on painettu ”D7SB” ja toiselle puolelle ”140”.
Käyttö- ja käsittelyohjeet
Kalvopäällysteiset tabletit koostuvat tablettiytimestä sekä sitä peittävästä kalvopäällysteestä, jonka tarkoitus on ehkäistä terveydenhuoltohenkilöstön altistus vaikuttavalle aineelle. Jos tabletit murskautuvat tai hajoavat vahingossa, lääkkeen hävittämisessä suositellaan käytettävän lateksi- tai nitriilikäsineitä ihoaltistusriskin minimoimiseksi.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
DASATINIB KRKA tabletti, kalvopäällysteinen
20 mg 60 fol
50 mg 60 fol
70 mg 60 fol
- Ylempi erityiskorvaus (100 %). Leukemiat, muut pahanlaatuiset veri- ja luuydintaudit sekä pahanlaatuiset imukudostaudit (117).
- Peruskorvaus (40 %).
ATC-koodi
L01EA02
Valmisteyhteenvedon muuttamispäivämäärä
12.04.2023
Yhteystiedot
Tekniikantie 14
02150 Espoo
Suomi
020-7545330
www.krka.biz
info.fi@krka.biz