

BYDUREON injektioneste, depotsuspensio, esitäytetty kynä 2 mg
Vaikuttavat aineet ja niiden määrät
Yksi esitäytetty kynä sisältää eksenatidia 2 mg / 0,85 ml.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, depotsuspensio, esitäytetty kynä (BCise).
Kliiniset tiedot
Käyttöaiheet
Bydureon on tarkoitettu tyypin 2 diabetesta sairastaville aikuisille, nuorille ja vähintään 10-vuotiaille lapsille parantamaan glukoositasapainoa yhdistettynä muihin verensokeria alentaviin lääkevalmisteisiin, perusinsuliini mukaan lukien, kun niiden käytöllä ei yhdessä ruokavalion ja liikunnan kanssa saavuteta riittävää glukoositasapainoa.
Katso kohdista Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakodynamiikka tutkimustulokset, jotka koskevat yhdistelmiä, vaikutuksia glukoositasapainoon ja sydän- ja verisuonitapahtumiin sekä tutkittuja potilasryhmiä.
Annostus ja antotapa
Annostus
Suositeltu annos on 2 mg eksenatidia kerran viikossa.
Potilaiden, jotka siirtyvät eksenatidia välittömästi vapauttavasta valmisteesta (Byetta) depotmuotoiseen eksenatidivalmisteeseen (Bydureon tai Bydureon BCise), veren glukoositaso voi ohimenevästi kohota, mutta se yleensä tasaantuu neljän ensimmäisen viikon aikana hoidon aloittamisesta. Potilaat voivat vaihtaa hoitoa depotmuotoisten eksenatidivalmisteiden (Bydureon tai Bydureon BCise) välillä eikä vaihtamisen odoteta vaikuttavan merkittävästi veren glukoosipitoisuuksiin.
Kun depotmuotoinen eksenatidivalmiste lisätään käynnissä olevaan metformiini- ja/tai tiatsolidiinidionihoitoon, metformiini- ja/tai tiatsolidiinidioniannosta ei tarvitse muuttaa. Kun valmiste lisätään sulfonyyliureahoitoon, sulfonyyliurea-annoksen pienentämistä on harkittava hypoglykemiariskin vähentämiseksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Yhdistelmähoitoa tiatsolidiinidionin kanssa on tutkittu vain aikuisilla.
Depotmuotoinen eksenatidivalmiste pistetään kerran viikossa samana viikonpäivänä. Tarvittaessa viikottaista pistospäivää voidaan muuttaa edellyttäen, että edellinen lääkeannos pistettiin vähintään kolme vuorokautta aikaisemmin. Depotmuotoinen eksenatidivalmiste voidaan pistää mihin tahansa vuorokauden aikaan riippumatta ruokailusta.
Jos lääkeannos jää ottamatta, se on otettava niin pian kuin mahdollista, kunhan seuraava annosteluaikataulun mukainen pistos on vähintään kolmen päivän kuluttua. Sen jälkeen potilas voi palata normaaliin annosteluaikataulun mukaiseen kerran viikossa annosteluun. Jos annos on unohtunut ja seuraava annosteluaikataulun mukainen annos on yhden tai kahden päivän kuluttua, potilaan ei pidä ottaa unohtunutta annosta vaan palata depotmuotoisen eksenatidivalmisteen normaaliin annosteluaikatauluun seuraavana annosteluaikataulun mukaisena päivänä.
Tämän lääkevalmisteen käyttö ei vaadi ylimääräistä omaseurantaa. Verensokerin omaseuranta on välttämätöntä sulfonyyliurean ja insuliinin annoksen säätämiseksi erityisesti, kun hoito pitkävaikutteisella eksenatidilla aloitetaan ja insuliiniannosta pienennetään. Insuliiniannoksen pienentämiseen suositellaan asteittaista lähestymistapaa.
Jos toinen glukoosipitoisuutta pienentävä hoito aloitetaan depotmuotoisen eksenatidihoidon lopettamisen jälkeen, on otettava huomioon lääkevalmisteen pitkävaikutteisuus (ks. kohta Farmakokinetiikka).
Erityisryhmät
Iäkkäät
Annoksen säätäminen ei ole tarpeen iän perusteella. Koska munuaistoiminta yleensä heikkenee iän myötä, on kuitenkin kiinnitettävä huomiota potilaan munuaistoimintaan (ks. kohta Munuaisten vajaatoiminta) (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen säätäminen ei ole tarpeen hoidettaessa potilaita, joilla on lievä tai keskivaikea munuaisten vajaatoiminta.
Depotmuotoista eksenatidivalmistetta ei suositella, jos potilaalla on loppuvaiheen munuaissairaus tai vaikea munuaisten vajaatoiminta (glomerulusten suodatusnopeus [GFR] < 30 ml/min) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksan vajaatoiminta
Annoksen säätäminen ei ole tarpeen maksan vajaatoimintaa sairastaville potilaille (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Annoksen säätäminen ei ole tarpeen vähintään 10-vuotiailla lapsilla ja nuorilla. Tietoja alle 10-vuotiaista lapsista ei ole saatavilla (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Antotapa
Ihon alle
Potilas pistää itse depotmuotoisen eksenatidilääkkeen. Kukin kertakäyttöinen lääkekynä on tarkoitettu vain yhden henkilön käyttöön.
On erittäin suositeltavaa, että terveydenhuollon ammattilainen neuvoo potilaalle ja huoltajalle depotmuotoisen eksenatidivalmisteen oikean käytön ennen hoidon aloittamista. Lääkepakkauksessa on käyttäjän opas, jota täytyy huolellisesti noudattaa.
Lääkeannos pistetään ihon alle vatsaan, reiteen tai olkavarren takaosaan välittömästi lääkevalmisteen valmiiksi sekoittamisen jälkeen.
Kun käytetään samanaikaisesti insuliinia, depotmuotoinen eksenatidivalmiste ja insuliini täytyy antaa kahtena erillisenä pistoksena.
Ohjeet lääkevalmisteen valmistukseen ennen käyttöä on esitetty kohdassa Käyttö- ja käsittelyohjeet. ja käyttäjän oppaassa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Depotmuotoista eksenatidivalmistetta ei pidä käyttää tyypin 1 diabeteksen eikä diabeettisen ketoasidoosin hoitoon.
Pitkävaikutteinen eksenatidi ei korvaa insuliinia. Diabeettista ketoasidoosia on ilmoitettu esiintyneen insuliinista riippuvaisilla potilailla insuliinin käytön nopean keskeyttämisen tai annoksen pienentämisen jälkeen (ks. kohta Annostus ja antotapa).
Depotmuotoista eksenatidivalmistetta ei saa pistää laskimoon tai lihakseen.
Munuaisten vajaatoiminta
Eksenatidia välittömästi vapauttavan valmisteen kerta-annokset lisäsivät ruuansulatuskanavan haittavaikutusten yleisyyttä ja vakavuutta potilailla, joilla oli loppuvaiheen munuaissairaus ja jotka saivat dialyysihoitoa. Näin ollen depotmuotoisia eksenatidivalmisteita ei suositella potilaille, joilla on loppuvaiheen munuaissairaus tai vaikea munuaisten vajaatoiminta (GFR < 30 ml/min).
Eksenatidin käytön yhteydessä on ilmoitettu melko harvinaisina haittavaikutuksina munuaistoiminnan muutoksia, kuten suurentunutta seerumin kreatiniiniarvoa, munuaistoiminnan heikentymistä, kroonisen munuaisten vajaatoiminnan pahenemista ja akuuttia munuaisten vajaatoimintaa, jotka saattavat edellyttää hemodialyysia. Jotkin näistä haittavaikutuksista ilmenivät potilailla, joilla oli samanaikaisesti mahdollisesti nestetasapainoon vaikuttavia haittoja, kuten pahoinvointia, oksentelua ja/tai ripulia, ja/tai potilas sai lääkettä, jonka tiedetään vaikuttavan munuaisten toimintaan tai nestetasapainoon. Potilaiden samanaikaiseen lääkitykseen sisältyi ACE:n estäjiä, angiotensiini II ‑reseptorin salpaajia, tulehduskipulääkkeitä ja diureetteja. Munuaistoiminnan muutosten on havaittu palautuneen, kun potilas sai tukihoitoa ja kun munuaistoimintaan mahdollisesti vaikuttava lääkehoito, kuten eksenatidihoito, lopetettiin.
Vaikea ruuansulatuskanavan sairaus
Depotmuotoista eksenatidivalmistetta ei ole tutkittu potilailla, joilla on vaikea ruuansulatuskanavan sairaus, kuten gastropareesi. Valmisteen käyttö aiheuttaa usein ruuansulatuskanavaan kohdistuvia haittavaikutuksia, kuten pahoinvointia, oksentelua ja ripulia. Siksi tätä lääkevalmistetta ei suositella potilaille, joilla on vaikea ruuansulatuskanavan sairaus.
Akuutti haimatulehdus
GLP-1-reseptorin agonistien käyttöön on liittynyt akuutin haimatulehduksen riski. Bydureon BCise ‑valmisteella tehdyissä kliinisissä tutkimuksissa ilmeni akuuttia haimatulehdusta 0,4 %:lla potilaista. Depotmuotoisen eksenatidivalmisteen käytön yhteydessä on spontaanisti ilmoitettu äkillistä haimatulehdusta. Haimatulehdusten on havaittu paranevan tukihoidolla, mutta hyvin harvoin on ilmoitettu nekrotisoivaa tai hemorragista haimatulehdusta ja/tai kuolemia. Potilaalle on kerrottava äkillisen haimatulehduksen tyypillisistä oireista, kuten jatkuvasta kovasta vatsakivusta. Jos epäillään haimatulehdusta, hoito tällä lääkevalmisteella on lopetettava. Jos akuutin haimatulehduksen diagnoosi varmistuu, lääkitystä ei pidä aloittaa uudelleen. Varovaisuutta on noudatettava, kun hoidetaan potilaita, joilla on aiemmin ollut haimatulehdus.
Muut samanaikaisesti käytetyt lääkevalmisteet
Depotmuotoisten eksenatidivalmisteiden samanaikaista käyttöä D‑fenyylialaniinijohdosten (meglitinidien), alfa-glukosidaasin estäjien, dipeptidyylipeptidaasi‑4:n estäjien tai muiden GLP‑1-reseptoriagonistien kanssa ei ole tutkittu. Depotmuotoisen eksenatidivalmisteen käyttöä eksenatidia välittömästi vapauttavan valmisteen kanssa ei ole tutkittu eikä sitä suositella.
Lääkevasta-aineista johtuva hoidon tehottomuus pediatrisilla potilailla
Pediatrisilla potilailla korkeiden lääkevasta-ainetitterien kehittymistodennäköisyys on mahdollisesti suurempi kuin aikuisilla (ks. kohta Haittavaikutukset). Potilailla, joilla on korkeammat vasta-ainetitterit, saattaa olla heikentynyt HbA1c-vaste.
Lääkevasta-aineiden kaupallisia tutkimuksia ei ole saatavilla. Jos haluttua veren glukoosin hoitotavoitetta ei saavuteta, vaikka hoitomyöntyvyys on vahvistetusti hyvä, lääkärien on hoidon tehottomuuden syystä riippumatta harkittava jotakin muuta diabeteksen hoitovaihtoehtoa.
Yhteisvaikutukset varfariinin kanssa
On ilmoitettu spontaanisti tapauksia, joissa INR-arvo (International Normalized Ratio) on suurentunut ja joihin on liittynyt verenvuotoa, kun varfariinia ja eksenatidia on käytetty samanaikaisesti (ks. kohta Yhteisvaikutukset).
Hypoglykemia
Kliinisissä tutkimuksissa hypoglykemian riski suureni, kun depotmuotoista eksenatidivalmistetta käytettiin yhdessä sulfonyyliurean kanssa. Lisäksi kliinisissä tutkimuksissa todettiin sulfonyyliureayhdistelmähoidossa enemmän hypoglykemiakohtauksia lievää munuaisten vajaatoimintaa sairastavilla potilailla kuin potilailla, joiden munuaiset toimivat normaalisti. Sulfonyyliurea-annoksen pienentämistä on harkittava sen käyttöön liittyvän hypoglykemiariskin pienentämiseksi.
Nopea painon lasku
Eksenatidin käytön yhteydessä on ilmoitettu nopeaa painon laskua, yli 1,5 kg viikossa. Näin nopealla painon laskulla saattaa olla haitallisia seurauksia. Potilaita, joiden paino laskee nopeasti, on tarkkailtava sappikivitaudin merkkien ja oireiden varalta.
Hoidon lopettaminen
Depotmuotoisen eksenatidihoidon lopettamisen jälkeen sen vaikutus saattaa jatkua 10 viikkoa plasman eksenatidipitoisuuksien pienentyessä. Tämä on otettava huomioon harkittaessa muita lääkkeitä ja niiden annoksia, sillä haittavaikutukset ja lääkkeen teho saattavat ainakin osittain jatkua, kunnes eksenatidipitoisuudet pienenevät.
Aspiraatio yleisanestesian tai syvän sedaation yhteydessä
GLP-1-reseptoriagonisteja käyttävillä potilailla on ilmoitettu aspiraatiotapauksia yleisanestesian tai syvän sedaation yhteydessä. Siksi on otettava huomioon lisääntynyt riski mahaan jääneestä sisällöstä viivästyneen tyhjentymisen takia (ks. Haittavaikutukset) ennen yleisanestesian tai syvän sedaation aikana suoritettavia toimenpiteitä.
Yhteisvaikutukset
Sulfonyyliureat
Sulfonyyliurean annos saattaa vaatia säätämistä siihen liittyvän lisääntyneen hypoglykemiariskin vuoksi (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Mahalaukun tyhjentyminen
Eksenatidin vaikutusta mahalaukun tyhjentymiseen tutkittiin käyttämällä parasetamolia merkkiaineena. Tutkimustulokset viittaavat siihen, että depotmuotoisen eksenatidivalmisteen mahalaukun tyhjentymistä hidastava vaikutus on vähäinen eikä sen odoteta kliinisesti merkittävästi pienentävän samanaikaisesti suun kautta annettujen lääkkeiden imeytymisnopeutta tai ‑määrää. Siksi mahalaukun hidastuneelle tyhjentymiselle herkkien lääkevalmisteiden annosta ei tarvitse säätää.
Kun yhden gramman parasetamolitabletteja annettiin joko ruuan kanssa tai ilman ruokaa 14 viikon depotmuotoisen eksenatidihoidon jälkeen, parasetamolin pitoisuuskäyrän (AUC) alle jäävässä pinta-alassa ei havaittu merkittäviä muutoksia kontrollijaksoon verrattuna. Parasetamolin maksimipitoisuus (Cmax) pieneni paastotilanteessa 16 % ja aterian jälkeen 5 %. Aika huippupitoisuuteen (tmax) oli kontrollijakson aikana noin 1 tunti, kun Bydureon-hoidon jälkeen tmax oli paastotilanteessa 1,4 tuntia ja aterian jälkeen 1,3 tuntia.
Seuraavat yhteisvaikutustutkimukset on tehty annoksella 10 mikrog välittömästi vapautuvaa eksenatidia, mutta ei depotmuotoisilla eksenatidivalmisteilla.
Varfariini
Kun varfariini annettiin 35 minuuttia välittömästi vapautuvan eksenatidin jälkeen, varfariinin tmax-arvoissa havaittiin noin kahden tunnin viive. Kliinisesti merkittäviä muutoksia ei havaittu Cmax-arvossa eikä plasman lääkeainepitoisuuskäyrän alle jäävässä pinta-alassa (AUC). INR-arvon (International Normalized Ratio) suurenemisia on ilmoitettu spontaanisti varfariinin ja depotmuotoisen eksenatidivalmisteen samanaikaisen käytön yhteydessä. INR-arvoja on seurattava aloitettaessa depotmuotoinen eksenatidi hoito potilaille, jotka käyttävät varfariinia ja/tai kumarolijohdoksia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
HMG-CoA-reduktaasin estäjät
Kun välittömästi vapautuvan eksenatidin kanssa annettiin samanaikaisesti kerta-annos lovastatiinia (40 mg), lovastatiinin AUC pieneni noin 40 %, Cmax noin 28 %, ja tmax piteni noin 4 tuntia verrattuna pelkällä lovastatiinilla saatuihin vastaaviin arvoihin. Välittömästi vapautuvan eksenatidin 30 viikkoa kestäneissä lumekontrolloiduissa kliinisissä tutkimuksissa eksenatidin ja HMG-CoA-reduktaasin estäjien samanaikaisen käytön ei havaittu aiheuttavan lipidiprofiileissa johdonmukaisia muutoksia (ks. kohta Farmakodynamiikka). Vaikka annosmuutos ei ole tarpeen, lipidiarvoja on seurattava säännöllisesti.
Digoksiini ja lisinopriili
Välittömästi vapautuvan eksenatidin, digoksiinin ja lisinopriilin samanaikaisessa käytössä kliinisesti merkittäviä muutoksia ei havaittu Cmax-arvossa eikä plasman lääkeainepitoisuuskäyrän alle jäävässä pinta-alassa (AUC), mutta tmax-arvossa havaittiin 2 tunnin viive.
Etinyyliestradioli ja levonorgestreeli
Kun yhdistelmäehkäisyvalmistetta (30 mikrog etinyyliestradiolia + 150 mikrog levonorgestreelia) otettiin suun kautta tuntia ennen välittömästi vapautuvaa eksenatidia, etinyyliestradiolin ja levonorgestreelin pitoisuuskäyrä (AUC), huippupitoisuudet (Cmax) ja minimipitoisuudet (Cmin) eivät muuttuneet. Kun ehkäisyvalmistetta otettiin suun kautta 35 minuuttia eksenatidin jälkeen, etinyyliestradiolin ja levonorgestreelin AUC-arvot eivät muuttuneet, mutta etinyyliestradiolin huippupitoisuus (Cmax) pieneni 45 % ja levonorgestreelin 27−41 %. Tmax viivästyi 2−4 tuntia, koska mahan tyhjeneminen hidastui. Huippupitoisuuksien (Cmax) pienenemisen kliininen merkitys on vähäinen eikä suun kautta otetun ehkäisyvalmisteen annosta tarvitse muuttaa.
Pediatriset potilaat
Eksenatidin yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Koska depotmuotoisella eksenatidivalmisteella on pitkä puhdistuma-aika, naisten, jotka voivat tulla raskaaksi, on käytettävä ehkäisyä depotmuotoisen eksenatidilääkityksen aikana. Tämä lääkehoito on lopetettava ainakin 3 kuukautta ennen suunniteltua raskaaksi tuloa.
Raskaus
Depotmuotoisen eksenatidivalmisteen käytöstä raskauden aikana ei ole riittävästi tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Mahdollista riskiä ihmiselle ei tiedetä. Depotmuotoista eksenatidivalmistetta ei pidä käyttää raskauden aikana, jolloin suositellaan insuliinia.
Imetys
Ei tiedetä, erittyykö eksenatidi ihmisillä äidinmaitoon. Depotmuotoista eksenatidivalmistetta ei pidä käyttää imetyksen aikana.
Hedelmällisyys
Vaikutuksia ihmisen hedelmällisyyteen ei ole tutkittu.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Depotmuotoisella eksenatidivalmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Kun valmistetta käytetään yhdessä sulfonyyliurean kanssa, potilasta on neuvottava noudattamaan varotoimia hypoglykemian välttämiseksi ajaessaan autoa ja käyttäessään koneita.
Haittavaikutukset
Turvallisuusprofiili
Kliinisten tutkimusten aikana ilmenneet yleisimmät haittavaikutukset aikuisilla olivat ruuansulatuskanavan oireet (pääasiassa pahoinvointi (8 %), jolla oli taipumus kadota, kun hoitoa jatkettiin), päänsärky (4 %) ja pistoskohdan reaktiot, kuten injektiokohdan kutina (3 %) ja injektiokohdan punoitus (2 %). Lisäksi hyvin yleinen haittavaikutus sulfonyyliurean kanssa oli hypoglykemia (ks. Tarkempia tietoja haittavaikutuksista jäljempänä). Useimmat haittavaikutukset olivat lieviä tai kohtalaisia.
Haittavaikutustaulukko
Taulukossa 1 esitetään yhteenveto haittavaikutuksista, jotka on todettu Bydureon BCise ‑valmisteelle aikuisilla tehdyissä kliinisissä tutkimuksissa.
Bydureon BCise ‑valmistetta koskevat yhdistetyt kliinisten tutkimusten tiedot sisältävät kaksi aikuisilla tehtyä vaiheen 3 verrokkikontrolloitua tutkimusta, jotka kestivät 6–12 kuukautta. Tutkimusten seurantavaiheet ja jatkotutkimukset sisältyvät yhdistettyihin tietoihin. Taustahoitoja olivat pelkkä ruokavalio ja liikunta tai lisäksi metformiini, sulfonyyliurea, tiatsolidiinidioni tai suun kautta otettavien glukoosipitoisuutta pienentävien lääkevalmistiden yhdistelmä. Taulukossa 1 on esitetty myös haittavaikutukset, joita havaittiin depotmuotoisen eksenatidivalmisteen käytön yhteydessä, mutta ei Bydureon BCise ‑valmisteella tehdyissä kliinisissä tutkimuksissa.
Depotmuotoisella eksenatidivalmisteella tehdyissä kliinisissä tutkimuksissa taustahoitoja olivat ruokavalio ja liikunta, metformiini, sulfonyyliurea, tiatsolidiinidioni, suun kautta otettavien glukoosipitoisuutta pienentävien lääkkeiden yhdistelmä tai perusinsuliini.
Haittavaikutukset luetellaan seuraavassa taulukossa MedDRA-terminologian mukaisesti elinjärjestelmän ja absoluuttisen esiintymistiheyden perusteella. Esiintymistiheyden määritelmät ovat: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10) ja melko harvinainen (≥ 1 / 1 000, < 1/100), harvinainen (≥ 1 / 10 000, < 1 / 1 000), hyvin harvinainen (< 1 / 10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1: Aikuisilla tehdyissä kliinisissä tutkimuksissa ja spontaanien raporttien perusteella Bydureon BCise ‑valmisteella todetut haittavaikutukset
| Elinjärjestelmä/ haittavaikutukset | Esiintyvyys | |||||
| Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Hyvin harvinainen | Tuntematon | |
| Veri ja imukudos | ||||||
| Lääkeaineen aiheuttama trombosytopenia9 | X | |||||
| Maksa ja sappi | ||||||
| sappirakkotulehdus11 | X | |||||
| sappikivitauti | X | |||||
| Immuunijärjestelmä | ||||||
| anafylaktinen reaktio2 | X | |||||
| Aineenvaihdunta ja ravitsemus | ||||||
| hypoglykemia (sulfonyyliurean kanssa)5,6,7 | X | |||||
| hypoglykemia (ilman sulfonyyliureaa)5,6,7 | X | |||||
| hypoglykemia (insuliinin kanssa)3,4,5 | X | |||||
| pienentynyt ruokahalu | X | |||||
| dehydraatio | X | |||||
| Hermosto | ||||||
| päänsärky | X | |||||
| heitehuimaus | X | |||||
| makuhäiriö | X | |||||
| uneliaisuus2 | X | |||||
| Ruoansulatuselimistö | ||||||
| pahoinvointi5 | X | |||||
| ripuli | X | |||||
| oksentelu | X | |||||
| ummetus | X | |||||
| dyspepsia | X | |||||
| gastroesofageaalinen refluksi | X | |||||
| vatsan turvotus | X | |||||
| vatsakipu | X | |||||
| ilmavaivat | X | |||||
| äkillinen haimatulehdus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | X | |||||
| röyhtäily2 | X | |||||
| suolitukos2 | X | |||||
| hidastunut mahan tyhjeneminen10 | X | |||||
| Iho ja ihonalainen kudos | ||||||
| nokkosihottuma | X | |||||
| liikahikoilu | X | |||||
| makulaarinen ja papulaarinen ihottuma | X | |||||
| kutina | X | |||||
| hiustenlähtö2 | X | |||||
| angioedeema9 | X | |||||
| pistoskohdan absessit ja selluliitti9 | X | |||||
| Munuaiset ja virtsatiet | ||||||
| muuttunut munuaistoiminta8 | X | |||||
| Yleisoireet ja antopaikassa todettavat haitat | ||||||
| pistoskohdan kutina5 | X | |||||
| pistoskohdan punoitus5 | X | |||||
| väsymys | X | |||||
| pistoskohdan reaktiot5 | X | |||||
| voimattomuus | X | |||||
| pistoskohdan ihottuma5 | X | |||||
| hermostuneisuuden tunne2 | X | |||||
| Tutkimukset | ||||||
| INR-arvon (International Normalized Ratio) suureneminen9 (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | X | |||||
1 Esiintyvyys perustuu päättyneisiin tehoa ja turvallisuutta arvioineisiin pitkäaikaistutkimuksiin (n = 526), ellei toisin ole ilmoitettu. Sisältää seurannan 70 päivän ajalta viimeisen annoksen saamisen jälkeen ja jatkovaiheen.
2 Esiintyvyys perustuu kahteentoista depotmuotoisella eksenatidivalmisteella tehtyyn päättyneeseen tehoa ja turvallisuutta arvioineeseen pitkäaikaistutkimukseen, n = 2 868 (yhteensä).
3 Perustuu hypoglykemiatapahtumiin, jotka 1. johtavat tajunnanmenetykseen, kouristuskohtaukseen tai koomaan, joka päättyy glukagonin tai glukoosin antamisen jälkeen, TAI 2. korjaantuakseen edellyttävät apua kolmannelta osapuolelta tajunnan tai käyttäytymisen heikkenemisen vuoksi ja jolloin glukoosiarvo on < 54 mg/dl (3 mmol/l) TAI 3. aiheuttavat hypoglykemiaan viittaavia oireita, joihin liittyy glukoosiarvo, joka on < 54 mg/dl (3 mmol/l) ennen hoitoa.
4 Esiintymistiheys, joka ilmoitettiin 28 viikkoa kestäneen kontrolloidun hoitojakson perusteella tutkimuksessa, jossa arvioitiin depotmuotoista eksenatidivalmistetta glargiini-insuliinin lisälääkkeenä (N = 231).
5 Ks. Tarkempia tietoja haittavaikutuksista jäljempänä.
6 Yhdistetyissä tiedoissa ilmoitetut esiintymistiheydet saatiin kahden vaiheen 3 kliinisen tutkimuksen kontrolloiduista vaiheista (n = 410).
7 Perustuu hypoglykemiatapahtumiin, joissa on hypoglykemiaan viittaavia oireita, joihin liittyy glukoosiarvo, joka on < 54 mg/ml (3 mmol/l) ennen hoitoa.
8 Sisältää akuutin munuaisten vajaatoiminnan, kroonisen munuaisten vajaatoiminnan pahenemisen, munuaisten toiminnan heikentymisen ja suurentuneen seerumin kreatiniiniarvon. Ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
9 Esiintyvyys perustuu depotmuotoisella eksenatidivalmisteella tehtyjen spontaanien ilmoitusten tietoihin (käyttäjien lukumäärää ei tiedetä).
10 Esiintyvyys perustuu kuuteentoista depotmuotoisella eksenatidivalmisteella tehtyyn päättyneeseen tehoa ja turvallisuutta arvioineeseen pitkäaikaistutkimukseen, n = 4 086.
11 Esiintyvyys perustuu Bydureon-valmisteella tehtyihin päättyneisiin turvallisuutta ja tehoa arvioineisiin tutkimuksiin (yhteensä n = 3 560); mukaan lukien DURATION 7- ja DURATION 8 ‑tutkimukset.
Tarkempia tietoja haittavaikutuksista
Lääkeaineen aiheuttama trombosytopenia
Myyntiluvan myöntämisen jälkeen aikuisilla on ilmoitettu lääkeaineen aiheuttamaa trombosytopeniaa, johon liittyy eksenatidista riippuvaisten verihiutalevasta-aineiden muodostuminen. Lääkeaineen aiheuttama trombosytopenia on lääkeaineesta riippuvaisten verihiutalevasta-aineiden aiheuttama immuunivälitteinen reaktio. Herkistävän lääkeaineen läsnä ollessa nämä vasta-aineet tuhoavat verihiutaleita.
Hypoglykemia
Bydureon BCise ‑valmisteen käytön yhteydessä ei ilmennyt merkittäviä hypoglykemiatapahtumia aikuisilla tehdyissä kliinisissä tutkimuksissa. Vähäisen hypoglykemian kokonaisesiintyvyys oli 6,3 %. Kun valmistetta käytettiin yhdessä sulfonyyliurean kanssa, hypoglykemiakohtausten esiintyvyys suureni (26,1 %) verrattuna käyttöön ilman sulfonyyliureaa (0,9 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Sulfonyyliurean käyttöön liittyvän hypoglykemiariskin pienentämiseksi voidaan harkita sulfonyyliurean annoksen pienentämistä (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Kun perusinsuliiniin lisättiin depotmuotoinen eksenatidivalmiste, insuliinin annosta ei tarvinnut muuttaa. Kliinisesti merkittäviä eroja hypoglykemiakohtausten ilmaantuvuudessa insuliiniin verrattuna ei todettu, kun depotmuotoista eksenatidivalmistetta käytettiin yhdessä perusinsuliinin kanssa. Depotmuotoista eksenatidivalmistetta yhdessä insuliinin kanssa saaneiden ryhmässä ei ilmennyt yhtään vaikeaa hypoglykemiakohtausta.
Pahoinvointi
Yleisimmin ilmoitettu ruuansulatuskanavaan kohdistuva haittavaikutus aikuisilla oli pahoinvointi. Kliinisen tutkimuksen kontrolloidun vaiheen aikana, jossa verrattiin Bydureon BCise ‑valmistetta välittömästi vapautuvaan eksenatidiin, pahoinvointia ilmoitettiin 9,6 %:lla Bydureon BCise ‑valmistetta saaneista potilaista ja 20,5 %:lla välittömästi vapautuvaa eksenatidia saaneista potilaista. Molempien kliinisten tutkimusten kontrolloidun vaiheen aikana pahoinvointia ilmoitettiin yhteensä 9,3 %:lla potilaista, jotka olivat saaneet Bydureon BCise ‑valmistetta. Pahoinvointi oli useimmiten lievää tai kohtalaista, sitä ilmeni hoidon alussa ja se väheni ajan myötä.
Pistoskohdan reaktiot
Aikuisilla tehtyjen kliinisten tutkimusten kontrolloidun vaiheen aikana pistoskohdan reaktioita havaittiin yleisemmin Bydureon BCise ‑valmistetta saaneilla potilailla (24 %) kuin vertailuvalmistetta saaneilla potilailla (4 %, välittömästi vapautuva eksenatidi). Nämä pistoskohdan reaktiot olivat tavallisesti lieviä eivätkä yleensä johtaneet tutkimushoidon keskeyttämiseen. Potilaiden oireita voidaan lievittää lääkityksen jatkuessa. Myöhemmät injektiot on annettava eri kohtiin joka viikko. Myyntiluvan myöntämisen jälkeen on raportoitu depotmuotoiselle eksenatidivalmisteelle pistoskohdan märkäpesäke- ja selluliittitapauksia.
Kliinisissä tutkimuksissa havaittiin usein ihonalaisia injektiokohdan nystyröitä yhdenmukaisesti poly(D,L‑laktidi‑ko‑glykolidi)polymeerimikropartikkelilääkemuotojen tunnettujen ominaisuuksien kanssa. Useimmat yksittäiset nystyrät eivät vaikuttaneet tutkimukseen osallistumiseen ja hävisivät ajan kuluessa.
Immunogeenisuus
Proteiini- ja peptidilääkkeillä saattaa olla immunogeenisia ominaisuuksia, joten potilaille saattaa kehittyä eksenatidivasta-aineita depotmuotoisen eksenatidihoidon aikana.
Noin 42 %:lla potilaista kehittyi matala eksenatidivasta-ainetitteri ja 32 %:lla potilaista kehittyi korkea vasta-ainetitteri aikuisilla tehtyjen tutkimusten missä tahansa vaiheessa. Näiden positiivisen vasta-ainetitterin ja erityisesti korkean titterin omaavien tutkittavien prosentuaalinen osuus oli suurimmillaan noin 8.–16. annostusviikoilla ja pieneni sen jälkeen ajan myötä. Tutkimuksen päätemuuttujan kohdalla noin 43 %:lla potilaista oli matala eksenatidivasta-ainetitteri ja 14 %:lla potilaista oli korkea vasta-ainetitteri. Glukoositasapaino (HbA1c) oli Bydureon BCise ‑valmistetta saaneilla potilailla, joiden vasta-ainetitteri oli viimeisellä käynnillä matala (‑1,1 % – ‑1,5 %), yleisesti samaa luokkaa kuin potilailla, joilla ei havaittu vasta-ainetittereitä (‑1,1 % – ‑1,4 %). Vaikka potilailla, joilla oli viimeisellä käynnillä korkea vasta-ainetitteri, HbA1c-vaste oli heikentynyt, HbA1c-arvojen pieneneminen oli näillä potilailla kliinisesti merkittävää (‑0,6 % – ‑0,7 %).
Bydureon BCise ‑valmistetta saaneilla aikuisilla potilailla, joilta voitiin määrittää vasta-aineet (N = 393), mahdollisesti immunogeenisten pistoskohdan reaktioiden (useimmiten pistoskohdan nystyröiden) ilmaantuvuus oli kahdessa tutkimuksessa noin 20 %. Näitä reaktioita havaittiin harvemmin vasta-ainenegatiivisilla potilailla (16 %) ja potilailla, joilla oli matala vasta-ainetitteri (16 %), kuin potilailla, joilla oli korkea vasta-ainetitteri (27 %).
Nopea painonlasku
30 viikkoa kestäneessä aikuisilla tehdyssä tutkimuksessa noin 3 %:lla (n = 4/148) depotmuotoista eksenatidivalmistetta saaneista potilaista esiintyi ainakin yksi jakso, jolloin paino laski nopeasti (kahden peräkkäisen tutkimuskäynnin välillä kirjattu painon lasku oli yli 1,5 kg viikossa).
Nopeutunut sydämen syke
Bydureon BCise ‑valmisteella aikuisilla tehtyjen kliinisten tutkimusten kontrolloidussa vaiheessa havaittiin sydämen sykkeen nopeutuneen lähtötilanteesta (74 lyöntiä minuutissa) keskimäärin 2,4 lyöntiä minuutissa. 15 %:lla depotmuotoista eksenatidihoitoa saaneista potilaista sydämen syke nopeutui keskimäärin ≥ 10 lyöntiä minuutissa. Suunnilleen 5−10 %:lla muiden hoitoryhmien potilaista sydämen syke nopeutui keskimäärin ≥ 10 lyöntiä minuutissa.
Pediatriset potilaat
Kliinisessä tutkimuksessa vähintään 10-vuotiailla lapsilla ja nuorilla havaittu eksenatidin turvallisuusprofiili (ks. kohta Farmakodynamiikka) oli samankaltainen kuin aikuisilla tehdyissä tutkimuksissa on todettu.
Pediatrisessa tutkimuksessa ei ilmennyt yhtään vaikeaa hypoglykemiatapahtumaa.
24 viikkoa kestäneen kaksoissokkoutetun hoitojakson aikana yhdellä potilaalla (1,7 %) depotmuotoista eksenatidivalmistetta saaneessa ryhmässä ja yhdellä potilaalla (4,3 %) lumeryhmässä ilmeni lievää hypoglykemiaa (lievä hypoglykemia määriteltiin ei-vaikeaksi hypoglykemiatapahtumaksi, johon kuului hypoglykemiaan viittaavia oireita ja glukoosiarvo < 3 mmol/l [54 mg/dl] ennen hoitoa). Molemmat potilaat saivat insuliinia taustahoitona.
Muita hypoglykemiatapahtumia, jotka eivät täyttäneet vaikean eivätkä lievän tapahtuman kriteereitä, ilmoitettiin tutkijan mukaan 8 potilaalla (13,6 %) depotmuotoista eksenatidivalmistetta saaneessa ryhmässä ja 1 potilaalla (4,3 %) lumeryhmässä. Heistä 6 potilasta depotmuotoista eksenatidivalmistetta saaneessa ryhmässä ja 1 potilas lumeryhmässä sai insuliinia taustahoitona.
Pediatrisessa tutkimuksessa suurin missä tahansa vaiheessa tutkimuksen aikana ilmoitettu vasta-aineiden titteri oli matala (< 625) noin 29,3 %:lla potilaista ja korkea (≥ 625) noin 63,8 %:lla potilaista. Positiivisen vasta-ainetitterin omaavien potilaiden prosentuaalinen osuus oli suurimmillaan noin viikolla 12. Kun tutkimusta jatkettiin viikkoon 52 asti, korkean vasta-ainetitterin omaavien potilaiden prosentuaalinen osuus pieneni (30,4 %:iin) ja matalan vasta-ainetitterin omaavien potilaiden prosentuaalinen osuus suureni (41,3 %:iin). Potilailla, joilla on korkeammat vasta-ainetitterit, saattaa olla heikompi HbA1c-vaste (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostuksen merkkejä ja oireita (välittömästi vapautuvalla eksenatidilla tehtyjen kliinisten tutkimusten perusteella) ovat voimakas pahoinvointi, voimakas oksentelu ja veren glukoosipitoisuuden nopea lasku. Yliannostustapauksissa on aloitettava asianmukainen tukihoito potilaan kliinisten merkkien ja oireiden perusteella.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Diabeteslääkkeet, glukagonin kaltaiset peptidi-1 (GLP-1) analogit. ATC-koodi: A10BJ01.
Vaikutusmekanismi
Eksenatidi on glukagonin kaltaisen peptidin 1 (GLP‑1:n) reseptoriagonisti, jonka useat vaikutukset ovat glukagonin kaltaisen peptidin 1 (GLP‑1) verensokeria alentavien vaikutusten kaltaisia. Eksenatidin ja ihmisen GLP‑1:n aminohappojärjestykset ovat osittain päällekkäiset. Eksenatidin on osoitettu sitoutuvan tunnettuun ihmisen GLP‑1-reseptoriin ja aktivoivan sen in vitro. Syklinen AMP ja/tai muut solunsisäiset signaalinvälitysreitit toimivat sen vaikutusmekanismin välittäjinä.
Eksenatidi lisää haiman beetasoluissa tapahtuvaa insuliinin eritystä glukoosista riippuvaisella tavalla. Kun verenglukoosi laskee, insuliinin eritys vähenee. Kun eksenatidia käytettiin metformiinin ja/tai tiatsolidiinidionin kanssa, hypoglykemian ilmaantuvuuden ei todettu olevan suurempi kuin lumelääkkeellä metformiinin ja/tai tiatsolidiinidionin kanssa, mikä saattaa johtua tästä glukoosista riippuvaisesta insulinotrooppisesta mekanismista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Eksenatidi estää glukagonin eritystä, minkä tiedetään olevan tyypin 2 diabeteksessa liian runsasta. Kun glukagonipitoisuudet pienenevät, maksa tuottaa vähemmän glukoosia. Eksenatidi ei kuitenkaan heikennä normaalia glukagonivastetta eikä muitakaan hypoglykemian aiheuttamia hormonivasteita.
Eksenatidi hidastaa mahan tyhjenemistä ja siten aterioista peräisin olevan glukoosin pääsyä verenkiertoon.
Eksenatidin on osoitettu vähentävän syömistä, mikä johtuu heikentyneestä ruokahalusta ja lisääntyneestä kylläisyyden tunteesta.
Farmakodynaamiset vaikutukset
Eksenatidi parantaa glukoositasapainoa alentamalla pitkäkestoisesti tyypin 2 diabetesta sairastavilla potilailla sekä aterianjälkeistä että paastoverenglukoosia. Farmakokineettisten ja farmakodynaamisten ominaisuuksiensa ansiosta depotmuotoinen eksenatidivalmiste soveltuu annettavaksi kerran viikossa toisin kuin luonnollinen GLP‑1.
Eksenatidin farmakodynaamisessa tutkimuksessa todettiin tyypin 2 diabetesta sairastavilla potilailla (n = 13) ensivaiheen insuliinierityksen palautuminen ja toisen vaiheen insuliinierityksen paraneminen vasteena laskimonsisäiseen glukoosibolukseen.
Kliininen teho ja turvallisuus
Alla esitetään Bydureon BCise ‑valmisteella tehtyjen kahden tutkimuksen ja depotmuotoisella eksenatidivalmisteella tehtyjen kuuden kliinisen pitkäaikaistutkimuksen tulokset. Näihin tutkimuksiin osallistui 1 766 aikuista tutkittavaa (joista 556 sai Bydureon BCise ‑valmistetta), joista 53 % oli miehiä ja 47 % naisia. Yli 65-vuotiaita oli 304 (17 %).
Lisäksi kaksoissokkoutettuun, lumekontrolloituun, kardiovaskulaarisia tuloksia arvioivaan tutkimukseen (EXSCEL) osallistui 14 752 aikuista tutkittavaa, joilla oli tyypin 2 diabetes ja minkä tahansa suuruinen kardiovaskulaarinen riski, kun valmiste lisättiin heidän senhetkiseen tavanomaiseen hoitoonsa.
Glukoositasapaino
Bydureon BCise
28 viikkoa kestäneessä aikuisilla tehdyssä avoimessa tutkimuksessa verrattiin Bydureon BCise ‑valmistetta välittömästi vapautuvaan eksenatidiin tutkittavilla, jotka noudattivat pelkkää ruokavalio- ja liikuntaohjelmaa tai jotka saivat suun kautta otettavaa glukoosipitoisuutta pienentävää vakaata lääkehoitoa. HbA1c-arvo pieneni lähtötilanteesta molemmissa hoitoryhmissä. Bydureon BCise ‑valmiste pienensi HbA1c-arvoja enemmän kuin välittömästi vapautuva eksenatidi lähtötilanteesta viikkoon 28 mennessä (taulukko 2). Tutkimuksen 28 viikon pituista verrokkikontrolloitua vaihetta seurasi 24 viikon pituinen jatkovaihe, jonka aikana kaikki tutkimukseen osallistuvat tutkittavat saivat hoitoa tällä lääkevalmisteella. Vaikutus HbA1c-arvoihin säilyi kliinisesti merkittävänä 52 viikon ajan, mutta pieneni osittain ajan myötä ryhmässä, jossa tutkittavat olivat aluksi saaneet Bydureon BCise ‑valmistetta.
Sekä Bydureon BCise ‑valmistetta että välittömästi vapautuvaa eksenatidia saaneilla potilailla paino oli laskenut viikolla 28 lähtötilanteeseen verrattuna (taulukko 2). Ero näiden kahden hoitoryhmän välillä ei ollut merkitsevä. Painon lasku oli säilynyt viikolla 52.
Taulukko 2: Tulokset yhdestä 28 viikkoa kestäneestä tutkimuksesta, jossa verrattiin Bydureon BCise valmistetta välittömästi vapautuvaan eksenatidiin yhdistettynä pelkkään ruokavalioon ja liikuntaan tai suun kautta otettavaan glukoosipitoisuutta pienentävään vakaaseen lääkehoitoon (modifioidut intent-to-treat‑potilaat1)
| Bydureon BCise, 2 mg kerran viikossa | Välittömästi vapautuva eksenatidi 10 mikrog kaksi kertaa vuorokaudessa | |
| N | 229 | 146 |
| HbA1c (keskiarvo, %) | ||
| Lähtötaso | 8,5 | 8,5 |
| Muutos lähtötasosta (± SE)2 | -1,4 (± 0,1) | -1,0 (± 0,1) |
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna välittömästi vapautuvaan eksenatidiin (95 % CI)2 | -0,37* (-0,63, -0,10) | |
| HbA1c-arvon < 7 % saavuttaneiden potilaiden prosenttiosuus3 | 49 | 43 |
| Keskimääräinen paino (kg) | ||
| Lähtötaso | 97 | 97 |
| Muutos lähtötasosta (± SE)2 | -1,5 (± 0,3) | -1,9 (± 0,4) |
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna välittömästi vapautuvaan eksenatidiin (95 % CI)2 | + 0,40 (-0,48, 1,28) | |
| Plasman paastoglukoosin keskimääräinen muutos lähtötasosta (mmol/l) (± SE)2 | -1,8 (± 0,2) | -1,3 (± 0,3) |
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna välittömästi vapautuvaan eksenatidiin (95 % CI)2 | -0,56 (-1,20, 0,08) | |
N = potilaiden määrä hoitoryhmässä, SE = keskivirhe, CI = luottamusväli.
*p-arvo < 0,01.
1 Kaikki satunnaistetut potilaat, jotka saivat ainakin yhden annoksen tutkimuslääkettä.
2 Pienimmän neliösumman keskiarvot.
3 Viimeisestä havainnosta laskettu arvio (Last Observation Carried Forward, LOCF).
28 viikkoa kestäneessä avoimessa tutkimuksessa (suun kautta annettava lääkitys sokkoutettu) Bydureon BCise ‑valmistetta verrattiin sitagliptiiniin ja lumelääkkeeseen tutkittavilla, jotka saivat myös metformiinia vähintään 1 500 mg vuorokaudessa. Bydureon BCise ‑valmiste pienensi HbA1c-arvoa lähtötilanteesta viikkoon 28 mennessä enemmän kuin sitagliptiini ja lumelääke (taulukko 3).
Sekä Bydureon BCise ‑valmistetta että sitagliptiinia saaneilla potilailla paino oli laskenut viikolla 28 lähtötilanteeseen verrattuna (taulukko 3). Ero näiden kahden hoitoryhmän välillä ei ollut merkitsevä.
Taulukko 3: Tulokset yhdestä 28 viikkoa kestäneestä tutkimuksesta, jossa verrattiin Bydureon BCise ‑valmistetta sitagliptiiniin ja lumelääkkeeseen yhdistelmänä metformiinin kanssa (modifioidut intent-to-treat‑potilaat1)
| Bydureon BCise 2 mg kerran viikossa | Sitagliptiini 100 mg kerran vuorokaudessa | Lumelääke kerran vuorokaudessa | |
| N | 181 | 122 | 61 |
| HbA1c (keskiarvo, %) | |||
| Lähtötaso | 8,4 | 8,5 | 8,5 |
| Muutos lähtötasosta (± SE)2 | -1,1 (± 0,1) | -0,8 (± 0,1) | -0,4 (± 0,2) |
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna sitagliptiiniin (95 % CI)2 | -0,38* (-0,70, -0,06) | ||
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna lumelääkkeeseen (95 % CI)2 | -0,72** (-1,15, -0,30) | ||
| HbA1c-arvon < 7 % saavuttaneiden potilaiden prosenttiosuus3 | 43* | 32 | 25 |
| Keskimääräinen paino (kg) | |||
| Lähtötaso | 89 | 88 | 89 |
| Muutos lähtötasosta (± SE)2 | -1,1 (± 0,3) | -1,2 (± 0,3) | +0,2 (± 0,5) |
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna sitagliptiiniin (95 % CI)2 | +0,07 (-0,73, 0,87) | ||
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna lumelääkkeeseen (95 % CI)2 | -1,27# (-2,34, -0,20) | ||
| Plasman paastoglukoosin keskimääräinen muutos lähtötasosta (mmol/l) (± SE)2 | -1,2 (± 0,2) | -0,6 (± 0,3) | +0,5 (± 0,4) |
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna sitagliptiiniin (95 % CI)2 | -0,56 (-1,21, 0,09) | ||
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna lumelääkkeeseen (95 % CI)2 | -1,71§ (-2,59, -0,83) |
N = potilaiden määrä hoitoryhmässä, SE = keskivirhe, CI = luottamusväli.
*p-arvo < 0,05, **p-arvo < 0,01, #nimellinen p‑arvo < 0,05, §nimellinen p‑arvo < 0,001.
1 Kaikki satunnaistetut potilaat, jotka saivat ainakin yhden annoksen tutkimuslääkettä.
2 Pienimmän neliösumman keskiarvot.
3 Viimeisestä havainnosta laskettu arvio (Last Observation Carried Forward, LOCF).
Depotmuotoinen eksenatidivalmiste
Kahdessa aikuisilla tehdyssä tutkimuksessa verrattiin depotmuotoista eksenatidivalmistetta annoksella 2 mg kerran viikossa välittömästi vapautuvaan eksenatidiin annoksella 5 mikrogrammaa annettuna kaksi kertaa vuorokaudessa 4 viikon ajan, minkä jälkeen välittömästi vapautuvan eksenatidin annos suurennettiin 10 mikrogrammaan annettuna kaksi kertaa vuorokaudessa. Toinen tutkimus kesti 24 viikkoa (n = 252) ja toinen 30 viikkoa (n = 295), minkä jälkeisessä avoimessa jatkovaiheessa kaikki potilaat saivat depotmuotoista eksenatidivalmistetta 2 mg kerran viikossa seuraavat 7 vuotta (n = 258). Molemmissa tutkimuksissa HbA1c-arvon lasku oli ilmeinen kummassakin hoitoryhmässä jo ensimmäisessä HbA1c-mittauksessa hoidon aloittamisen jälkeen (4 tai 6 viikon kohdalla).
Depotmuotoinen eksenatidivalmiste laski HbA1c-arvoa tilastollisesti merkitsevästi verrattuna välittömästi vapautuvaan eksenatidiin (taulukko 4).
Depotmuotoisella eksenatidivalmisteella ja välittömästi vapautuvalla eksenatidilla havaittiin molemmissa tutkimuksissa kliinisesti merkittävä vaikutus HbA1c-arvoon riippumatta taustalla olevasta diabeteslääkityksestä.
Depotmuotoista eksenatidivalmistetta saaneista tutkittavista kliinisesti merkityksellisesti ja tilastollisesti merkitsevästi suuremmalla osalla HbA1c-arvo pieneni näissä kahdessa tutkimuksessa alle tason ≤ 7 % tai < 7 % verrattuna välittömästi vapautuvaa eksenatidia saaneisiin (p < 0,05 ja p < 0,0001).
Sekä depotmuotoinen että välittömästi vapautuva eksenatidi laskivat potilaan painoa lähtötasoon nähden, vaikka ero näiden kahden hoitohaaran välillä ei ollut merkitsevä.
Kontrolloimattomassa jatkotutkimuksessa arviointikelpoisilla potilailla (n = 121), jotka siirtyivät välittömästi vapautuvasta eksenatidista depotmuotoiseen eksenatidivalmisteeseen viikolla 30, HbA1c‑arvo oli parantunut viikolla 52 samalle tasolle (pienenemä -2 %) lähtötilanteeseen verrattuna kuin depotmuotoista eksenatidivalmistetta saaneilla potilailla. Kaikilla potilailla (258 potilaasta osallistui jatkotutkimukseen 122), jotka jatkoivat 7 vuoden kontrolloimattoman jatkotutkimuksen loppuun saakka, HbA1c‑arvo suureni vähitellen viikosta 52 lähtien, mutta oli edelleen pienempi 7 vuoden jälkeen lähtötilanteeseen verrattuna (-1,5 %). Näillä potilailla painon lasku säilyi yli 7 vuotta.
Taulukko 4: Tulokset kahdesta tutkimuksesta, depotmuotoinen vs. välittömästi vapautuva eksenatidi yhdistettynä pelkkään ruokavalioon ja liikuntaan, yhdistettynä metformiiniin ja/tai sulfonyyliureaan ja yhdistettynä metformiiniin ja/tai tiatsolidiinidioniin (intent-to-treat‑potilaat)
| 24 viikkoa kestänyt tutkimus | Depotmuotoinen eksenatidivalmiste 2 mg | Välittömästi vapautuva eksenatidi 10 mikrog kaksi kertaa vuorokaudessa |
| N | 129 | 123 |
| HbA1c (keskiarvo, %) | ||
| Lähtötaso | 8,5 | 8,4 |
| Muutos lähtötasosta (± SE) | -1,6 (±0,1)** | -0,9 (±0,1) |
| Hoitojen välinen keskimääräinen ero muutoksessa lähtötasosta (95 % CI) | -0,67 (-0,94, -0,39)** | |
| HbA1c-arvon < 7,0 % saavuttaneiden potilaiden prosenttiosuus | 58 | 30 |
| Plasman paastoglukoosin muutos (mmol/l)(± SE) | -1,4 (±0,2) | -0,3 (±0,2) |
| Keskimääräinen paino (kg) | ||
| Lähtötaso | 97 | 94 |
| Muutos lähtötasosta (± SE) | -2,3 (±0,4) | -1,4 (±0,4) |
| Hoitojen välinen keskimääräinen ero muutoksessa lähtötasosta (95 % CI) | -0,95 (-1,91, 0,01) | |
| 30 viikkoa kestänyt tutkimus | ||
| N | 148 | 147 |
| HbA1c (keskiarvo, %) | ||
| Lähtötaso | 8,3 | 8,3 |
| Muutos lähtötasosta (± SE) | ‑1,9 (±0,1)* | ‑1,5 (±0,1) |
| Hoitojen välinen keskimääräinen ero muutoksessa lähtötasosta (95 % CI) | -0,33 (-0.54, -0,12)* | |
| HbA1c-arvon < 7,0 % saavuttaneiden potilaiden prosenttiosuus | 73 | 57 |
| Plasman paastoglukoosin muutos (mmol/l) (± SE) | -2,3 (±0,2) | -1,4 (±0,2) |
| Keskimääräinen paino (kg) | ||
| Lähtötaso | 102 | 102 |
| Muutos lähtötasosta (± SE) | ‑3,7 (±0,5) | ‑3,6 (±0,5) |
| Hoitojen välinen keskimääräinen ero muutoksessa lähtötasosta (95 % CI) | -0,08 (-1,29, 1,12) | |
SE = keskivirhe, CI = luottamusväli, *p < 0,05, **p < 0,0001
26 viikkoa kestäneessä aikuisilla tehdyssä tutkimuksessa depotmuotoista eksenatidivalmistetta 2 mg verrattiin glargiini-insuliiniin kerran päivässä annettuna. Glargiini-insuliiniin verrattuna depotmuotoisella eksenatidivalmisteella saatiin parempi HbA1c-arvon muutos, se laski keskimääräistä painoa merkittävästi enemmän ja siihen liittyi vähemmän hypoglykemiatapahtumia (taulukko 5).
Taulukko 5: Tulokset 26 viikkoa kestäneestä tutkimuksesta depotmuotoinen eksenatidivalmiste vs. glargiini-insuliini yhdistettynä pelkkään metformiiniin tai metformiinin ja sulfonyyliurean yhdistelmään (intent-to-treat‑potilaat)
| Depotmuotoinen eksenatidivalmiste 2 mg | Glargiini-insuliini1 | |
| N | 233 | 223 |
| HbA1c (keskiarvo, %) | ||
| Lähtötaso | 8,3 | 8,3 |
| Muutos lähtötasosta (± SE) | ‑1,5 (±0,1)* | ‑1,3 (±0,1)* |
| Hoitojen välinen keskimääräinen ero muutoksessa lähtötasosta (95 % CI) | -0,16 (-0,29, -0,03)* | |
| HbA1c-arvon < 7,0 % saavuttaneiden potilaiden prosenttiosuus | 62 | 54 |
| Plasman paastoglukoosin muutos (mmol/l) (± SE) | -2,1 (±0,2) | -2,8 (±0,2) |
| Keskimääräinen paino (kg) | ||
| Lähtötaso | 91 | 91 |
| Muutos lähtötasosta (± SE) | -2,6 (±0,2) | +1,4 (±0,2) |
| Hoitojen välinen keskimääräinen ero muutoksessa lähtötasosta (95 % CI) | -4,05 (-4,57, -3,52) * | |
SE = keskivirhe, CI = luottamusväli, *p < 0,05
1Glargiini-insuliinin annoksella pyrittiin glukoositasoon 4,0−5,5 mmol/l (72−100 mg/dl). Keskimääräinen glargiini-insuliiniannos oli tutkimuksen alussa 10,1 IU/vrk ja se nousi tasolle 31,1 IU/vrk glargiini-insuliinia saaneilla potilailla.
Tulokset viikolla 156 olivat yhdenmukaiset aiemmin viikon 26 kohdalla tehdyn väliraportin kanssa. Glargiini-insuliiniin verrattuna depotmuotoinen eksenatidihoito paransi jatkuvasti ja merkittävästi glukoositasopainoa ja painonhallintaa. Turvallisuuslöydökset viikolla 156 olivat yhdenmukaiset viikolla 26 raportoitujen tulosten kanssa.
26 viikkoa kestäneessä kaksoissokkotutkimuksessa depotmuotoista eksenatidivalmistetta verrattiin sitagliptiinin ja pioglitatsonin enimmäisvuorokausiannoksiin aikuisilla tutkittavilla, jotka saivat myös metformiinia. Kaikissa hoitoryhmissä HbA1c-arvo pieneni merkittävästi lähtötilanteeseen nähden. Depotmuotoinen eksenatidivalmiste oli sekä sitagliptiinia että pioglitatsonia tehokkaampi HbA1c-arvon muutoksen suhteen lähtötilanteeseen nähden.
Depotmuotoinen eksenatidivalmiste laski painoa merkittävästi enemmän kuin sitagliptiini. Pioglitatsonia saaneiden potilaiden paino nousi (taulukko 6).
Taulukko 6: Tulokset 26 viikkoa kestäneestä tutkimuksesta depotmuotoinen eksenatidi vs. sitagliptiini vs. pioglitatsoni, yhdessä metformiinin kanssa (intent-to-treat‑potilaat)
| Depotmuotoinen eksenatidivalmiste 2 mg | Sitagliptiini 100 mg | Pioglitatsoni 45 mg | |
| N | 160 | 166 | 165 |
| HbA1c (keskiarvo, %) | |||
| Lähtötaso | 8,6 | 8,5 | 8,5 |
| Muutos lähtötasosta (± SE) | -1,6 (±0,1)* | -0,9 (±0,1)* | -1,2 (±0,1)* |
| Hoitojen välinen keskimääräinen ero muutoksessa lähtötasosta (95 % CI) vs. sitagliptiini | -0,63 (-0,89, -0,37)** | ||
| Hoitojen välinen keskimääräinen ero muutoksessa lähtötasosta (95 % CI) vs. pioglitatsoni | -0,32 (-0,57, -0,06,)* | ||
| HbA1c-arvon < 7,0 % saavuttaneiden potilaiden prosenttiosuus | 62 | 36 | 49 |
| Plasman paastoglukoosin muutos (mmol/l)(± SE) | -1,8 (±0,2) | -0,9 (±0,2) | -1,5 (±0,2) |
| Keskimääräinen paino (kg) | |||
| Lähtötaso | 89 | 87 | 88 |
| Muutos lähtötasosta (± SE) | -2,3 (±0,3) | -0,8 (±0,3) | +2,8 (±0,3) |
| Hoitojen välinen keskimääräinen ero muutoksessa lähtötasosta (95 % CI) vs. sitagliptiini | -1,54 (-2,35, -0,72)* | ||
| Hoitojen välinen keskimääräinen ero muutoksessa lähtötasosta (95 % CI) vs. pioglitatsoni | -5,10 (-5,91, -4,28)** | ||
SE = keskivirhe, CI = luottamusväli, *p < 0,05, **p < 0,0001
28 viikkoa kestäneessä aikuisilla tehdyssä kaksoissokkoutetussa tutkimuksessa depotmuotoisen eksenatidin ja dapagliflotsiinin yhdistelmää verrattiin pelkkään depotmuotoiseen eksenatidiin ja pelkkään dapagliflotsiiniin tutkittavilla, jotka käyttivät myös metformiinia. HbA1c-arvo pieneni kaikissa hoitoryhmissä lähtötasoon verrattuna. Depotmuotoista eksenatidia ja dapagliflotsiinia saaneiden ryhmässä HbA1c‑arvo pieneni lähtötasosta enemmän kuin pelkkää depotmuotoista eksenatidia tai pelkkää dapagliflotsiinia saaneilla (taulukko 5).
Depotmuotoisen eksenatidin ja dapagliflotsiinin yhdistelmän käytön yhteydessä paino laski huomattavasti enemmän kuin kumpaakaan lääkevalmistetta yksinään käytettäessä (taulukko 7).
Taulukko 7: Tulokset yhdestä 28 viikkoa kestäneestä tutkimuksesta, jossa verrattiin depotmuotoista eksenatidia ja dapagliflotsiinia pelkkään depotmuotoiseen eksenatidiin ja pelkkään dapagliflotsiiniin yhdistelmänä metformiinin kanssa (intent-to-treat–potilaat)
Depotmuotoinen eksenatidi 2 mg kerran viikossa + dapagliflotsiini 10 mg kerran vuorokaudessa | Depotmuotoinen eksenatidi 2 mg kerran viikossa + lumelääke kerran vuorokaudessa | Dapagliflotsiini 10 mg kerran vuorokaudessa + lumelääke kerran viikossa | |
| N | 228 | 227 | 230 |
| HbA1c (keskiarvo, %) | |||
| Lähtötaso | 9,3 | 9,3 | 9,3 |
| Muutos lähtötasosta (± SE)a | -2,0 (± 0,1) | -1,6 (± 0,1) | -1,4 (± 0,1) |
| Keskimääräinen ero muutoksessa lähtötasosta yhdistelmän ja yksittäisen vaikuttavan lääkevalmisteen välillä (95 %:n luottamusväli) | -0,38* (-0,63, -0,13) | -0,59** (-0,84, -0,34) | |
| HbA1c-arvon < 7,0 % saavuttaneiden potilaiden osuus (%) | 45 | 27 | 19 |
| Plasman paastoglukoosin keskimääräinen muutos lähtötasosta (mmol/l)(± SE)a | -3,7 (± 0,2) | -2,5 (± 0,2) | -2,7 (± 0,2) |
| Keskimääräinen ero muutoksessa lähtötasosta yhdistelmän ja yksittäisen vaikuttavan lääkevalmisteen välillä (95 %:n luottamusväli) | -1,12** (-1,55, -0,68) | -0,92** (-1,36, -0,49) | |
| 2 tuntia aterian jälkeen mitatun plasman glukoosipitoisuuden keskimääräinen muutos lähtötasosta (mmol/l) (± SE)a | -4,9 (± 0,2) | -3,3 (± 0,2) | -3,4 (± 0,2) |
| Keskimääräinen ero muutoksessa lähtötasosta yhdistelmän ja yksittäisen vaikuttavan lääkevalmisteen välillä (95 %:n luottamusväli) | -1,54** (-2,10, -0,98) | -1,49** (-2,04, -0,93) | |
| Keskimääräinen paino (kg) | |||
| Lähtötaso | 92 | 89 | 91 |
| Muutos lähtötasosta (± SE)a | -3,6 (± 0,3) | -1,6 (± 0,3) | -2,2 (± 0,3) |
| Keskimääräinen ero muutoksessa lähtötasosta yhdistelmän ja yksittäisen vaikuttavan lääkevalmisteen välillä (95 %:n luottamusväli) | -2,00** (-2,79, -1,20) | -1,33** (-2,12, -0,55) | |
SE = keskivirhe, N = potilaiden määrä
a Korjatut pienimmän neliösumman keskiarvot ja tutkimusryhmien ero(t) lähtötasossa todetuista arvoista tapahtuneen muutoksen suhteen viikolla 28 malllinnettiin käyttämällä toistettujen mittausten sekamallia. Malli sisälsi kiinteinä tekijöinä hoidon, alueen, lähtötilanteen HbA1c-ositteen (< 9,0 % tai ≥ 9,0 %), viikon sekä hoitoon liittyvät yhteisvaikutukset viikoittain. Kovariaattina oli lähtötason arvo.
* p < 0,01, ** p < 0,001.
Kaikki p-arvot ovat kerrannaisuuden suhteen korjattuja p-arvoja.
Analyysit eivät sisällä mittauksia, jotka on tehty hätälääkityksen käytön jälkeen tai tutkimuslääkkeen käytön ennenaikaisen lopettamisen jälkeen.
28 viikkoa kestäneessä aikuisilla tehdyssä kaksoissokkoutetussa tutkimuksessa depotmuotoista eksenatidia pelkän glargiini-insuliinin lisälääkkeenä tai glargiini-insuliinin ja metformiinin lisälääkkeenä verrattiin lumelääkkeeseen pelkän glargiini-insuliinin lisälääkkeenä tai glargiini-insuliinin ja metformiinin lisälääkkeenä. Glargiini-insuliinin annoksella pyrittiin plasman paastoglukoosipitoisuuteen, joka oli 4,0–5,5 mmol/l (72–99 mg/dl). Depotmuotoinen eksenatidivalmiste pienensi HbA1c-arvoa lähtötilanteesta viikkoon 28 mennessä enemmän kuin lumelääke (taulukko 8).
Depotmuotoisen eksenatidin käytön yhteydessä paino laski viikkoon 28 mennessä enemmän kuin lumelääkettä käytettäessä (taulukko 8).
Taulukko 8: Tulokset yhdestä 28 viikkoa kestäneestä tutkimuksesta, jossa verrattiin depotmuotoista eksenatidia lumelääkkeeseen yhdistelmänä pelkän glargiini-insuliinin tai glargiini-insuliinin ja metformiinin kanssa (intent-to-treat‑potilaat)
Depotmuotoinen eksenatidi 2 mg + glargiini-insuliinia | Lumelääke + glargiini-insuliinia | |
| N | 230 | 228 |
| HbA1c (keskiarvo, %) | ||
| Lähtötaso | 8,5 | 8,5 |
| Muutos lähtötasosta (± SE)b | ‑1,0 (± 0,1) | ‑0,2 (± 0,1) |
| Keskimääräinen hoitojen välinen ero muutoksessa lähtötasosta (95 %:n luottamusväli) | ‑0,74* (‑0,94, ‑0,54) | |
| HbA1c-arvon ≤ 7 % saavuttaneiden potilaiden osuus (%)c | 33* | 7 |
| Keskimääräinen paino (kg) | ||
| Lähtötaso | 94 | 94 |
| Muutos lähtötasosta (± SE)b | ‑1,0 (± 0,3) | 0,5 (± 0,3) |
| Keskimääräinen hoitojen välinen ero muutoksessa lähtötasosta (95 %:n luottamusväli) | ‑1,52* (‑2,19, ‑0,85) | |
| 2 tuntia aterian jälkeen mitatun plasman glukoosipitoisuuden muutos lähtötasosta (mmol/l) (± SE)b,d | ‑1,6 (± 0,3) | ‑0,1 (± 0,3) |
| Keskimääräinen hoitojen välinen ero muutoksessa lähtötasosta (95 %:n luottamusväli) | ‑1,54* (‑2,17, ‑0,91) | |
N = potilaiden määrä kussakin hoitoryhmässä, SE = keskivirhe, *p-arvo < 0,001 (korjattu kerrannaisuuden suhteen).
a. Pienimmän neliösumman keskiarvojen muutos insuliinin keskimääräisessä vuorokausiannoksessa oli depotmuotoista eksenatidivalmistetta saaneiden ryhmässä 1,6 yksikköä ja lumeryhmässä 3,5 yksikköä.
b. Korjatut pienimmän neliösumman keskiarvot ja tutkimusryhmien ero(t) lähtötasossa todetuista arvoista tapahtuneen muutoksen suhteen viikolla 28 mallinnettiin käyttämällä toistettujen mittausten sekamallia. Malli sisälsi kiinteinä tekijöinä hoidon, alueen, lähtötilanteen HbA1c-tason (< 9,0 % tai ≥ 9,0 %), lähtötason sulfonyyliurean käytön (käyttö vs. ei käyttöä), viikon sekä hoitoon liittyvät yhteisvaikutukset viikoittain. Kovariaattina oli lähtötason arvo. Kaksi tuntia aterian jälkeen mitatun plasman glukoosipitoisuuden absoluuttinen muutos viikolla 28 mallinnettiin samalla tavalla käyttämällä kovarianssianalyysiä (ANCOVA).
c. Kaikki potilaat, joista ei ollut saatavilla päätemuuttujaa koskevia tietoja, tulkittiin potilaiksi, jotka eivät saaneet vastetta.
d. Ateriarasituskokeen jälkeen.
Analyysit eivät sisällä mittauksia, jotka on tehty hätälääkityksen käytön jälkeen tai tutkimuslääkkeen käytön ennenaikaisen lopettamisen jälkeen.
Kardiovaskulaarinen arviointi
EXSCEL oli pragmaattinen kardiovaskulaarisia tuloksia arvioinut tutkimus, johon osallistuneilla aikuisilla potilailla oli tyypin 2 diabetes ja minkä tahansa suuruinen kardiovaskulaarinen riski. Yhteensä 14 752 potilasta satunnaistettiin suhteessa 1:1 saamaan joko depotmuotoista eksenatidivalmistetta 2 mg kerran viikossa tai lumelääkettä, jotka lisättiin potilaan senhetkiseen tavanomaiseen hoitoon, johon saattoi sisältyä SGLT2:n estäjiä. Tavanomaisen kliinisen käytännön mukaisen potilaiden seurannan mediaani oli 38,7 kuukautta ja hoidon keston mediaani oli 27,8 kuukautta. Tutkimuksen päättyessä elossaolotietoja oli saatavilla potilaista seuraavasti: 98,9 %:sta depotmuotoista eksenatidivalmistetta saaneiden ryhmässä ja 98,8 %:sta lumeryhmässä. Keskimääräinen ikä tutkimuksessa aloittamisen hetkellä oli 62 vuotta (8,5 % potilaista oli vähintään 75‑vuotiaita). Noin 62 % potilaista oli miehiä. Keskimääräinen painoindeksi oli 32,7 kg/m2 ja potilaat olivat sairastaneet diabetesta keskimäärin 13,1 vuoden ajan. Keskimääräinen HbA1c oli 8,1 %. Noin 49,3 %:lla potilaista oli lievä munuaisten vajaatoiminta (glomerulusten laskennallinen suodatusnopeus [eGFR] ≥ 60 − ≤ 89 ml/min/1,73 m2) ja 21,6 %:lla keskivaikea munuaisten vajaatoiminta (eGFR ≥ 30 − ≤ 59 ml/min/1,73 m2). Yhteensä 26,9 %:lla potilaista ei ollut aiemmin ollut yhtään kardiovaskulaarista tapahtumaa ja 73,1 %:lla oli ollut aiemmin ainakin yksi kardiovaskulaarinen tapahtuma.
EXSCEL-tutkimuksen ensisijainen turvallisuutta (vähintään samanveroisuutta) ja tehoa (paremmuutta) koskeva päätemuuttuja oli aika ensimmäiseen vahvistettuun merkittävään sydänperäiseen haittatapahtumaan (Major Adverse Cardiac Event, MACE): kardiovaskulaarisyistä johtuva kuolema, ei-fataaliin sydäninfarktiin tai ei-fataaliin aivohalvaukseen. Kaikista syistä johtuva kuolleisuus oli ensimmäinen arvioitu toissijainen päätemuuttuja.
Potilaan senhetkiseen tavanomaiseen hoitoon lisätty depotmuotoinen eksenatidi ei suurentanut kardiovaskulaarista riskiä tyypin 2 diabetesta sairastavilla potilailla lumelääkkeeseen verrattuna (riskisuhde: 0,91; 95 %:n luottamusväli: 0,832, 1,004; p < 0,001 vähintään samanveroisuudelle), ks. kuva 1. EXSCEL-tutkimuksen ennalta määritellyssä alaryhmäanalyysissä MACE-tapahtuman riskisuhde oli 0,86 (95 %:n luottamusväli: 0,77, 0,97) potilailla, joilla lähtötilanteen eGFR oli ≥ 60 ml/min/1,73 m2, ja 1,01 (95 %:n luottamusväli: 0,86, 1,19) potilailla, joilla lähtötilanteen eGFR oli < 60 ml/min/1,73 m2. Ensisijaista yhdistettyä ja toissijaisia kardiovaskulaarisia päätemuuttujia koskevat tulokset on esitetty kuvassa 2.
Kuva 1: Aika ensimmäiseen arvioituun MACE-tapahtumaan (intent-to-treat‑potilaat)
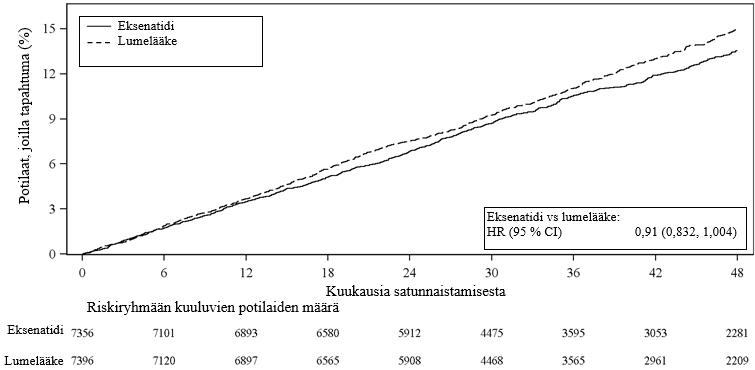
HR = riskisuhde, CI = luottamusväli
Kuva 2: Forest-kuvaaja: ensisijaisen ja toissijaisten päätemuuttujien analyysi (intent-to-treat‑potilaat)
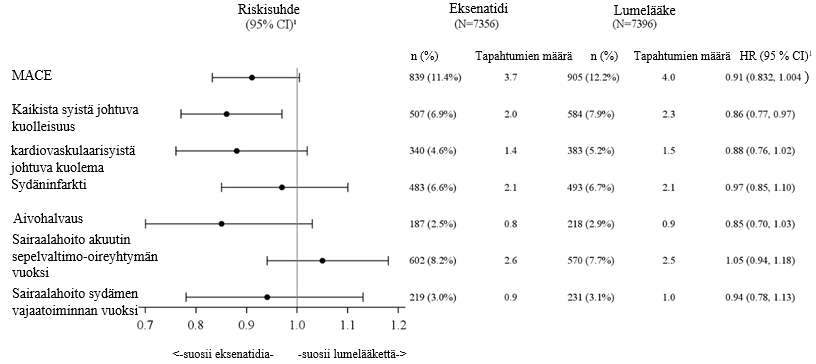
CI = luottamusväli; HR = riskisuhde; MACE = merkittävä sydänperäinen haittatapahtuma; n = niiden potilaiden määrä, joilla todettiin tapahtuma; N = hoitoryhmän potilaiden määrä.
1 Riskisuhde (vaikuttava aine / lumelääke) ja luottamusväli perustuvat Coxin suhteellisen riskiregressioanalyysimalliin , joka oli stratifioitu aiemman kardiovaskulaarisen tapahtuman mukaan ja jossa käytettiin hoitoryhmää vain selittävänä muuttujana.
Depotmuotoista eksenatidia saaneiden ryhmässä verensokeria alentavan lisälääkityksen tarve pieneni 33 % (altistuksen suhteen vakioitu ilmaantuvuus 100 potilasvuotta kohti 10,5) lumeryhmään verrattuna (altistuksen suhteen vakioitu ilmaantuvuus 100 potilasvuotta kohti 15,7). HbA1c-arvojen havaittiin laskeneen tutkimuksen aikana niin, että hoitojen välinen kokonaisero oli ‑0,53 % (depotmuotoinen eksenatidi verrattuna lumelääkkeeseen).
Potilaan paino
Potilaan paino laski depotmuotoisilla eksenatidivalmisteilla tehdyissä tutkimuksissa lähtötilanteeseen nähden. Tämä painon lasku todettiin riippumatta pahoinvoinnin esiintymisestä, joskin paino laski enemmän ryhmässä, jossa potilailla ilmeni pahoinvointia (keskimääräinen painon lasku potilailla, joilla ilmeni pahoinvointia, oli ‑1,9 − ‑5,2 kg vs. ‑1,0 − ‑2,9 kg ilman pahoinvointia).
Plasman/seerumin glukoosi
Depotmuotoinen eksenatidihoito pienensi merkittävästi plasman/seerumin paastoglukoosiarvoja. Pienenemä havaittiin jo 4 viikon jälkeen. Glargiini-insuliinilla tehdyssä lumekontrolloidussa tutkimuksessa plasman paastoglukoosiarvo muuttui lähtötilanteesta viikkoon 28 mennessä depotmuotoista eksenatidivalmistetta saaneiden ryhmässä ‑0,7 mmol/l ja lumeryhmässä ‑0,1 mmol/l. Lisäksi havaittiin aterianjälkeisten glukoosiarvojen pieneneminen.
Molempien depotmuotoisten eksenatidivalmisteiden kohdalla plasman paastoglukoosiarvojen paraneminen säilyi viikkoon 52 saakka.
Beetasolujen toiminta
Depotmuotoisilla eksenatidivalmisteilla tehdyissä kliinisissä tutkimuksissa on osoitettu beetasolujen toiminnan paranevan, kun mittareina on käytetty esim. beetasolujen toiminnan homeostaasimallimääritystä (HOMA-B). Vaikutus beetasolujen toimintaan säilyi viikkoon 52 saakka.
Verenpaine
Depotmuotoisilla eksenatidivalmisteilla tehdyissä tutkimuksissa havaittiin systolisen verenpaineen laskua (0,8 mmHg – 4,7 mmHg). Vertailevassa 30 viikkoa kestäneessä eksenatidia välittömästi vapauttavaa lääkemuotoa verrokkina käyttäneessä tutkimuksessa sekä depotmuotoinen että välittömästi vapautuva eksenatidi laskivat merkittävästi systolista verenpainetta lähtötilanteeseen verrattuna (depotmuotoinen eksenatidi -4,7 ± 1,1 mmHg ja välittömästi vapautuva eksenatidi ‑3,4 ± 1,1 mmHg). Lääkitysten välinen ero ei ollut merkitsevä. Verenpaineen lasku säilyi viikolle 52.
Glargiini-insuliinin kanssa tehdyssä lumekontrolloidussa tutkimuksessa systolinen verenpaine muuttui lähtötilanteesta viikkoon 28 mennessä depotmuotoista eksenatidivalmistetta saaneiden ryhmässä ‑2,6 mmHg ja lumeryhmässä ‑0,7 mmHg.
Yhdistelmähoito depotmuotoisella eksenatidilla ja dapagliflotsiinilla oli laskenut keskimääräistä systolista verenpainetta viikolla 28 huomattavasti (-4,3 ± 0,8 mmHg) verrattuna pelkkään depotmuotoiseen eksenatidiin (-1,2 ± 0,8 mmHg, p < 0,01) tai pelkkään dapagliflotsiiniin (‑1,8 ± 0,8 mmHg, p < 0,05).
Paastolipidit
Depotmuotoisilla eksenatidivalmisteilla ei ole todettu lipidiparametreihin kohdistuvia haitallisia vaikutuksia.
Pediatriset potilaat
Depotmuotoisen eksenatidivalmisteen (2 mg kerran viikossa) tai lumelääkkeen tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa rinnakkaisryhmätutkimuksessa vähintään 10-vuotiailla lapsilla ja nuorilla, joiden tyypin 2 diabeteksen hoitona oli vain ruokavalio ja liikunta tai myös vakaa-annoksinen suun kautta otettava diabeteslääkitys ja/tai insuliini. Depotmuotoinen eksenatidivalmiste pienensi HbA1c‑arvoa enemmän kuin lumelääke 24 viikon jälkeen (taulukko 9).
Taulukko 9: Tulokset yhdestä 24 viikkoa kestäneestä tutkimuksesta, jossa depotmuotoista eksenatidivalmistetta verrattiin lumelääkkeeseen vähintään 10-vuotiailla pediatrisilla ja nuorilla potilailla (intent‑to‑treat-potilaat)
| Depotmuotoinen eksenatidivalmiste, 2 mg kerran viikossa | Lumelääke kerran viikossa | |
|---|---|---|
| Intent-to-treat-joukko (N) | 58 | 24 |
| HbA1c (keskiarvo, %) | ||
| Lähtötaso | 8,11 | 8,22 |
| Muutos lähtötasosta (± SE) | -0,36 (0,18) | 0,49 (0,27) |
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna lumelääkkeeseen (95 % CI)a | ‑0,85 (‑1,51, ‑0,19)* | |
| Plasman paastoglukoosi (mmol/l) | ||
| Lähtötaso | 9,24 | 9,08 |
| Muutos lähtötasosta (± SE) | ‑0,29 (0,424) | 0,91 (0,63) |
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna lumelääkkeeseen (95 % CI)b | ‑1,2 (‑2,72, 0,32) | |
| Keskimääräinen paino (kg) | ||
| Lähtötaso | 100,33 | 96,96 |
| Muutos lähtötasosta (± SE) | ‑0,59 (0,67) | 0,63 (0,98) |
| Keskimääräinen ero muutoksessa lähtötasosta verrattuna lumelääkkeeseen (95 % CI)b | ‑1,22 (‑3,59, 1,15) | |
| HbA1c-arvon < 7,0 % saavuttaneiden potilaiden osuus | 31,0 % | 8,3 % |
| HbA1c-arvon ≤ 6,5 % saavuttaneiden potilaiden osuus | 19,0 % | 4,2 % |
| HbA1c-arvon < 6,5 % saavuttaneiden potilaiden osuus | 19,0 % | 4,2 % |
*p = 0,012
a Korjatut pienimmän neliösumman keskiarvot ja tutkimusryhmien erot lähtötasossa todetuista arvoista tapahtuneen muutoksen suhteen kullakin käynnillä mallinnettiin käyttämällä toistettujen mittausten sekamallia. Malli sisälsi kiinteinä tekijöinä hoitoryhmän, alueen, käynnin, hoitoryhmän ja käynnin yhteisvaikutuksen, lähtötilanteen HbA1c-arvon sekä lähtötilanteen HbA1c-arvon ja käynnin yhteisvaikutuksen, ja siinä käytettiin strukturoimatonta kovarianssimatriisia.
b Korjatut pienimmän neliösumman keskiarvot ja tutkimusryhmien erot lähtötasossa todetuista arvoista tapahtuneen muutoksen suhteen kullakin käynnillä mallinnettiin käyttämällä toistettujen mittausten sekamallia. Malli sisälsi kiinteinä tekijöinä hoitoryhmän, alueen, käynnin, hoitoryhmän ja käynnin yhteisvaikutuksen, lähtötilanteen arvon, seulonta-ajankohdan HbA1c-arvon (< 9,0 % tai ≥ 9,0 %) sekä lähtötilanteen arvon ja käynnin yhteisvaikutuksen, ja siinä käytettiin strukturoimatonta kovarianssimatriisia.
Farmakokinetiikka
Eksenatidin imeytymisominaisuuksien vuoksi depotmuotoinen eksenatidivalmiste vapauttaa hitaasti vaikuttavaa ainetta. Kun eksenatidi on imeytynyt verenkiertoon, se jakautuu ja eliminoituu tunnettujen systeemisten farmakokineettisten ominaisuuksiensa mukaisesti (kuvattu tässä kappaleessa).
Imeytyminen
Kun Bydureon BCise ‑valmistetta annettiin 2 mg:n annoksella kerran viikossa, eksenatidin plasman keskiarvo ylitti tehokkaan annoksen minimipitoisuuden (~ 50 pg/ml) 2 viikossa. Keskimääräiset pitoisuudet plasmassa nousivat asteittain 8 viikkoon saakka, minkä jälkeen eksenatidipitoisuus pysyi tasolla noin 153‑208 pg/ml, mikä osoitti vakaan tilan saavuttamisen. Eksenatidin vakaan tilan pitoisuus säilyy, kun annosväli on yksi viikko, ja tämän keskimääräisen terapeuttisen pitoisuuden vaihtelu huippupitoisuudesta minimipitoisuuteen on hyvin vähäistä.
Jakautuminen
Kun ihon alle annetaan kerta-annos eksenatidia, sen keskimääräinen näennäinen jakautumistilavuus on 28 l.
Biotransformaatio ja eliminaatio
Prekliiniset tutkimukset ovat osoittaneet, että eksenatidi eliminoituu pääasiallisesti glomerulussuodatuksella ja tämän jälkeen proteolyyttisen hajoamisen kautta. Eksenatidin keskimääräinen näennäispuhdistuma on 9 l/h. Nämä eksenatidin farmakokineettiset ominaisuudet eivät ole annoksesta riippuvaisia. Noin 10 viikkoa depotmuotoisen eksenatidilääkityksen lopettamisen jälkeen plasman keskimääräiset eksenatidipitoisuudet olivat alle pienimmän mitattavissa olevan pitoisuuden.
Erityisryhmät
Munuaisten vajaatoiminta
Eksenatidin vakaan tilan pitoisuuksissa tai siedettävyydessä ei havaittu kliinisesti merkittäviä eroja potilailla, joilla oli lievä tai keskivaikea munuaisten vajaatoiminta (eGFR 30–89 ml/min/1,73 m2) ja jotka saivat Bydureon BCise ‑valmistetta, verrattuna potilaisiin, joiden munuaiset toimivat normaalisti.
Maksan vajaatoiminta
Farmakokineettisiä tutkimuksia ei ole tehty potilailla, joilla on maksan vajaatoiminta. Eksenatidi eliminoituu pääasiassa munuaisten kautta, joten maksan toimintahäiriön ei odoteta vaikuttavan veren eksenatidipitoisuuksiin.
Sukupuoli, rotu ja paino
Sukupuolella, rodulla tai painolla ei ole kliinisesti merkittävää vaikutusta eksenatidin farmakokinetiikkaan.
Iäkkäät
Iäkkäistä henkilöistä on saatavilla niukasti tietoa, mutta näyttää siltä, ettei ikä aina noin 75 vuoteen asti muuta sanottavasti eksenatidialtistusta.
Farmakokineettisessä tutkimuksessa välittömästi vapautuvan eksenatidin annoksella 10 mikrog iäkkäiden, 75−85-vuotiaiden tyypin 2 diabetesta sairastavien potilaiden (N = 15), saama altistus (AUC) suureni keskimäärin 36 % verrattuna 45−65-vuotiaiden (N = 15) altistukseen. Altistuksen suureneminen iäkkäissä johtui todennäköisesti heikentyneestä munuaistoiminnasta (ks. kohta Annostus ja antotapa).
Pediatriset potilaat
Populaatiofarmakokineettinen analyysi tyypin 2 diabetesta sairastavilla vähintään 10-vuotiailla lapsilla ja nuorilla, joilla oli matalat lääkevasta-ainetitterit, osoitti, että depotmuotoisen eksenatidivalmisteen (2 mg) anto johti samankaltaiseen altistukseen kuin aikuisilla on todettu.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta ja genotoksisuutta koskevien konventionaalisten tutkimusten tulokset välittömästi vapautuvalla eksenatidilla tai depotmuotoisilla eksenatidivalmisteilla eivät viittaa erityiseen vaaraan ihmisille.
Rotilla ja hiirillä on todettu kilpirauhasen kasvaimia pitkävaikutteisten GLP-1-reseptoriagonistien käytön yhteydessä. Depotmuotoisella eksenatidivalmisteella rotilla tehdyssä kaksi vuotta kestäneessä karsinogeenisuustutkimuksessa havaittiin C-soluadenoomien ja C-solukarsinoomien ilmaantuvuuden suureneminen annoksilla, jotka olivat AUC-arvojen perusteella vähintään kaksinkertaisia verrattuna systeemiseen altistukseen ihmisillä. Näiden löydösten kliinistä merkitystä ei tällä hetkellä tunneta.
Eksenatidilla tehdyt eläinkokeet eivät osoittaneet haitallisia vaikutuksia hedelmällisyyteen. Suurilla annoksilla oli vaikutuksia luustoon ja ne hidastivat sikiön ja vastasyntyneen kasvua.
Farmaseuttiset tiedot
Apuaineet
Injektiokuiva-aine
poly(D,L-laktidi-ko-glykolidi)
sakkaroosi
Vehikkeli
Keskipitkäketjuiset triglyseridit
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Kyniä voidaan säilyttää ennen käyttöä 4 viikkoa alle 30 °C:n lämpötilassa.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Kyniä täytyy säilyttää vaakatasossa.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
BYDUREON injektioneste, depotsuspensio, esitäytetty kynä
2 mg (L:ei) 4 x 1 annos (BCise, 2 mg/0,85 ml) (106,90 €)
PF-selosteen tieto
Suspensio on pakattu 2 ml:n tyypin I lasista valmistettuun sylinteriampulliin, joka on suljettu toisesta päästä kumisella (bromibutyyli) sinetti/tulppayhdistelmällä (combiseal) ja toisesta päästä kumisella (bromibutyyli) männällä. Valmis lääkevalmiste koostuu suspensiolla täytetystä sylinteriampullista, joka on asennettu kynään. Kynässä on integroitu neula.
Pakkauksessa on neljä esitäytettyä kerta-annoskynää (BCise) tai monipakkaus joka sisältää 12 (3 x 4) esitäytettyä kerta-annoskynää (BCise).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen tai melkein valkoinen läpinäkymätön suspensio.
Käyttö- ja käsittelyohjeet
Esitäytetty kynä on tarkoitettu vain yhtä käyttökertaa varten.
Terveydenhuollon ammattilaisten on koulutettava potilaat ja huoltajat.
BCise-kynä täytyy ottaa jääkaapista ja sen täytyy antaa olla vaakatasossa vähintään 15 minuutin ajan ennen pistoksen antamista. Suspensio täytyy sekoittaa ravistamalla voimakkaasti vähintään 15 sekunnin ajan. Suspensio on tarkastettava silmämääräisesti ennen käyttöä. Suspension saa käyttää vain, jos se on tasaisesti sekoittunut, valkoista tai melkein valkoista ja sameaa eikä kynän ikkunan seinämässä, pohjalla tai yläosassa näy valkoista lääkeainetta. Kun suspensio on sekoitettu valmiiksi, valmisteluvaiheet täytyy suorittaa välittömästi ja suspensio pistää ihon alle. Lue pakkausselosteesta ja käyttäjän oppaasta lisätietoja suspensiosta ja antamisesta.
Potilaalle on neuvottava, miten kynä hävitetään turvallisesti käytön jälkeen.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
BYDUREON injektioneste, depotsuspensio, esitäytetty kynä
2 mg 4 x 1 annos
- Ei korvausta.
ATC-koodi
A10BJ01
Valmisteyhteenvedon muuttamispäivämäärä
20.11.2024
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi