
VIRAMUNE oraalisuspensio 50 mg/5 ml
Vaikuttavat aineet ja niiden määrät
Yksi ml oraalisuspensiota sisältää 10 mg nevirapiinia (hemihydraattina).
Yksi pullo sisältää 2,4 g nevirapiinia (hemihydraattina) 240 ml:ssa Viramune oraalisuspensiota.
Apuaineet, joiden vaikutus tunnetaan
Yksi ml oraalisuspensiota sisältää 150 mg sakkaroosia, 162 mg sorbitolia, 1,8 mg metyyliparahydroksibentsoaattia ja 0,24 mg propyyliparahydroksibentsoaattia.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Oraalisuspensio
Kliiniset tiedot
Käyttöaiheet
Viramune on tarkoitettu yhdessä muiden antiretroviraalisten lääkevalmisteiden kanssa HIV-1 infektoituneiden aikuisten, nuorten ja minkä tahansa ikäisten lasten hoitoon (ks. kohta Annostus ja antotapa).
Viramune-valmistetta on eniten tutkittu yhdessä nukleosidianalogikäänteiskopioijaentsyymin estäjien kanssa käytettynä. Viramune-hoidon jälkeisen lääkevalinnan tulisi perustua kliiniseen kokemukseen ja resistenssitesteihin (ks. kohta Farmakodynamiikka).
Ehto
Hoidon saa määrätä vain HIV-infektion hoitoon perehtynyt lääkäri.
Annostus ja antotapa
Viramune-hoidon saa määrätä vain HIV-infektion hoitoon perehtynyt lääkäri.
Annostus
16-vuotiaat ja sitä vanhemmat potilaat
Suositeltava annos on 20 ml (200 mg) Viramune oraalisuspensiota kerran vuorokaudessa ensimmäiset 14 päivää (tätä aloitusjaksoa tulisi noudattaa, koska sen on havaittu vähentävän ihottuman esiintyvyyttä), ja sen jälkeen 20 ml (200 mg) oraalisuspensiota kahdesti vuorokaudessa yhdessä ainakin kahden muun antiretroviraalisen lääkeaineen kanssa.
Viramune-valmistetta on saatavissa myös 200 mg tabletteina yli 16-vuotiaille potilaille tai vanhemmille lapsille, erityisesti niille, jotka painavat vähintään 50 kg tai joiden kehon pinta-ala on yli 1,25 m2.
Jos annoksen huomataan unohtuneen 8 tunnin kuluessa tavallisesta ottoajankohdasta, potilaan pitää ottaa unohtunut annos niin pian kuin mahdollista. Jos annos unohtuu yli 8 tuntia tavallisesta ottoajankohdasta, potilaan pitää ottaa seuraava annos tavalliseen aikaan.
Annostuksessa huomioitavaa
Potilaille, joilla ilmenee ihottumaa 14 päivän aloitusjakson aikana 200 mg:n vuorokausiannoksella (4 mg/kg/vrk tai 150 mg/m2 lapsipotilaille), Viramune annosta ei saa lisätä ennen kuin ihottuma on hävinnyt. Ihottumaa ilman muita oireita pitää seurata tarkasti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Annostusta 200 mg kerran vuorokaudessa ei pidä jatkaa yli 28 päivän ajan, vaan siinä vaiheessa pitää etsiä vaihtoehtoista hoitoa mahdollisesti liian pieneksi jäävän annoksen ja resistenssin riskien vuoksi.
Potilaiden, jotka keskeyttävät nevirapiinin käytön pidemmäksi ajaksi kuin 7 päiväksi, pitää aloittaa uudelleen suositellulla annostuksella noudattaen kahden viikon aloitusjaksoa.
On toksisia vaikutuksia, jotka vaativat Viramune hoidon keskeyttämistä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Iäkkäät
Nevirapiinia ei ole erityisesti tutkittu yli 65-vuotiailla potilailla.
Munuaisten toimintahäiriö
Potilaille, joilla on dialyysia vaativa munuaisten toimintahäiriö, suositellaan 200 mg:n lisäannosta aina dialyysin jälkeen. Potilailla, joilla glomerulussuodosnopeus on ≥ 20 ml/min, ei annoksen muuttaminen ole tarpeen, ks. kohta Farmakokinetiikka.
Maksan toimintahäiriö
Nevirapiinia ei tule käyttää potilailla, joilla on vaikea maksan toimintahäiriö (Child–Pugh C, ks. kohta Vasta-aiheet). Annosta ei tarvitse säätää potilailla, joilla on lievä tai keskivaikea maksan toimintahäiriö (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Kokonaisvuorokausiannos ei saa kenelläkään potilaalla ylittää 400 mg:aa. Viramune-valmistetta voidaan annostella lapsipotilaille joko kehon pinta-alan tai painon mukaan seuraavasti: Mostellerin kaavan mukaisesti lasketun kehon pinta-alan mukaan annosteltuna suositeltu annos suun kautta kaikenikäisille lapsipotilaille on 150 mg/m2 kerran vuorokaudessa kahden viikon ajan ja sen jälkeen 150 mg/m2 kaksi kertaa vuorokaudessa.
Lapsipotilaille tarvittavan Viramune oraalisuspension (50 mg/5 ml) määrän laskeminen kehon pinta-alan perusteella annoksella 150 mg/m2:
| Kehon pinta-ala (m2) | Määrä (ml) |
| 0,08 – 0,25 | 2,5 |
| 0,25 – 0,42 | 5 |
| 0,42 – 0,58 | 7,5 |
| 0,58 – 0,75 | 10 |
| 0,75 – 0,92 | 12,5 |
| 0,92 – 1,08 | 15 |
| 1,08 – 1,25 | 17,5 |
| 1,25+ | 20 |
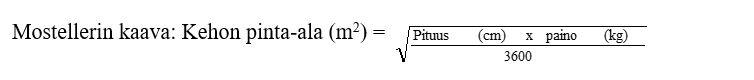
Painon mukaan suositeltu annos suun kautta alle 8-vuotiaille lapsipotilaille on 4 mg/kg kerran vuorokaudessa kahden viikon ajan ja sen jälkeen 7 mg/kg kahdesti vuorokaudessa. 8-vuotiaille ja sitä vanhemmille lapsille suositeltu annos on 4 mg/kg kerran vuorokaudessa kahden viikon ajan ja sen jälkeen 4 mg/kg kahdesti vuorokaudessa.
Lapsipotilaille tarvittavan Viramune oraalisuspension (50 mg/5 ml) määrän laskeminen kahden viikon aloitusjakson jälkeen.
| Paino (kg) < 8-vuotiaille potilaille, jotka saavat painon mukaan 7 mg/kg. | Paino (kg) ≥ 8-vuotiaille potilaille, jotka saavat painon mukaan 4 mg/kg. | Määrä (ml) |
| 1,79 – 5,36 | 3,13 – 9,38 | 2,5 |
| 5,36 – 8,93 | 9,38 – 15,63 | 5 |
| 8,93 – 12,50 | 15,63 – 21,88 | 7,5 |
| 12,50 – 16,07 | 21,88 – 28,12 | 10 |
| 16,07 – 19,64 | 28,12 – 34,37 | 12,5 |
| 19,64 – 23,21 | 34,37 – 40,62 | 15 |
| 23,21 – 26,79 | 40,62– 46,88 | 17,5 |
| 26,79+ | 46,88+ | 20 |
Kaikkien alle 16-vuotiaiden Viramune oraalisuspensiota saavien potilaiden painoa tai kehon pinta-alaa pitäisi seurata säännöllisesti, jotta voidaan arvioida annoksen säätämisen tarve.
Antotapa
On tärkeää käyttää mitattu Viramune oraalisuspensioannos kokonaisuudessaan. Käyttöä helpottaa annosteluruisku. Jos käytetään jotakin muuta mittavälinettä (esim. mittamukia tai lusikkaa suurehkoja annoksia varten), se on huuhdeltava perusteellisesti vedellä ja myös huuhteluvesi on annettava potilaalle. Viramune voidaan ottaa ruoan kanssa tai ilman ruokaa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Hoidon aloittaminen uudestaan potilaille, joiden hoito on jouduttu lopettamaan pysyvästi vaikean ihottuman, ihottuman johon liittyy yleisoireita, yliherkkyysreaktioiden tai nevirapiinin aiheuttaman kliinisen hepatiitin vuoksi.
Potilaat, joilla maksan toiminta on vakavasti heikentynyt (Child–Pugh luokka C) tai joilla ennen hoidon aloittamista ASAT- tai ALAT-arvot ovat yli viisinkertaiset normaalin ylärajaan verrattuna ennen kuin ASAT/ALAT-lähtöarvot ovat vakiintuneet alle viisinkertaisiksi normaalin ylärajaan verrattuna.
Hoidon aloittaminen uudestaan potilaille, joiden ASAT- tai ALAT-arvot olivat aikaisemmin nevirapiinin käytön aikana yli viisinkertaiset verrattuna normaalin ylärajaan ja joilla maksan toimintakokeiden arvot kohosivat uudestaan, kun nevirapiinin käyttö aloitettiin uudestaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Samanaikainen mäkikuismaa (Hypericum perforatum) sisältävien rohdosvalmisteiden käyttö, koska se saattaa alentaa plasman nevirapiinipitoisuutta sekä vähentää nevirapiinin kliinistä vaikutusta (ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Viramune-valmistetta pitää käyttää vähintään kahden muun antiretroviraalisen lääkeaineen kanssa (ks. kohta Farmakodynamiikka).
Viramune-valmistetta ei pidä käyttää ainoana antiretroviraalisena lääkkeenä, koska minkä tahansa antiretroviraalisen monoterapian on osoitettu johtavan virusresistenssiin.
Ensimmäiset 18 viikkoa nevirapiinihoidossa ovat kriittistä aikaa, jolloin potilaita pitää seurata tarkasti, mahdollisesti ilmaantuvien vakavien ja hengenvaarallisten ihoreaktioiden (mukaan lukien Stevens–Johnsonin syndrooma (SJS) ja toksinen epidermaalinen nekrolyysi (TEN)) sekä vakavan hepatiitin/maksan vajaatoiminnan varalta. Maksa- ja ihoreaktioiden riski on suurin 6 ensimmäisen hoitoviikon aikana. Minkä tahansa maksatapahtuman riski säilyy kuitenkin tämän ajanjakson jälkeen ja seurannan pitää jatkua säännöllisin väliajoin. Naissukupuoli ja nevirapiinihoidon aloituksen yhteydessä korkeampi CD4+ -solujen määrä (> 250/mm3 aikuisilla naispotilailla ja > 400/mm3 aikuisilla miespotilailla) on liitetty suurempaan riskiin saada maksahaittavaikutus, jos potilaalla on mitattavissa oleva plasman HIV-1 RNA – eli pitoisuus on ≥ 50 kopiota/ml nevirapiinihoitoa aloitettaessa. Koska vakavaa ja hengenvaarallista maksatoksisuutta on havaittu kontrolloiduissa ja kontrolloimattomissa tutkimuksissa lähinnä potilailla, joiden plasman HIV-1 viruskuorma on 50 kopiota/ml tai enemmän, nevirapiinihoitoa ei saa aloittaa aikuisille naispotilaille, joiden CD4+ -solujen määrä on yli 250 solua/mm3 tai aikuisille miespotilaille, joiden CD4+ -solujen määrä on yli 400 solua/mm3, ja joilla on mitattavissa oleva plasman HIV-1 RNA, ellei hoidon hyöty ole riskiä suurempi. Joissakin tapauksissa maksavaurio on edennyt hoidon lopettamisesta huolimatta. Potilaiden, joille kehittyy hepatiitin, vaikean ihoreaktion tai yliherkkyysreaktioiden merkkejä tai oireita, pitää lopettaa nevirapiini ja hakeutua välittömästi lääkärin hoitoon. Nevirapiinia ei saa aloittaa uudelleen vaikeiden maksa-, iho- tai yliherkkyysreaktioiden jälkeen (ks. kohta Vasta-aiheet). Annosta pitää noudattaa tarkasti, erityisesti 14 päivän aloitusjaksoa (ks. kohta Annostus ja antotapa). |
Ihoreaktiot
Vakavia ja hengenvaarallisia ihoreaktioita, mukaan lukien kuolemaan johtaneita tapauksia on havaittu nevirapiinilla hoidetuilla potilailla pääasiassa hoidon ensimmäisten 6 viikon aikana. Tällaisia haittavaikutuksia ovat olleet Stevens–Johnsonin syndrooma, toksinen epidermaalinen nekrolyysi ja yliherkkyysreaktioina ihottuma, johon liittyy monia yleisoireita sekä sisäelimiin liittyviä löydöksiä. Potilaita pitää seurata tehokkaasti ensimmäisten 18 viikon aikana. Potilaita pitää seurata tarkasti, jos kehittyy ihottuma ilman muita oireita. Nevirapiinin käyttö pitää keskeyttää pysyvästi kaikilla potilailla, joille kehittyy vaikea ihottuma tai ihottuma, johon liittyy yleisoireita (kuten kuumetta, rakkuloita, suun leesioita, sidekalvontulehdusta, kasvojen turvotusta, lihas- tai nivelkipuja tai huonovointisuutta), mukaan lukien Stevens–Johnsonin syndrooma tai toksinen epidermaalinen nekrolyysi. Nevirapiinin käyttö pitää keskeyttää pysyvästi kaikilla potilailla, joille kehittyy yliherkkyysreaktio (ihottuma, jolle on ominaista yleisoireet, sekä sisäelimiin liittyvät löydökset kuten hepatiitti, eosinofilia, granulosytopenia ja munuaisten vajaatoiminta) ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Nevirapiinin annostelu suositeltua suuremmalla annoksella saattaa lisätä ihoreaktioiden esiintyvyyttä ja vakavuutta (esimerkiksi Stevens–Johnsonin syndrooma ja toksinen epidermaalinen nekrolyysi).
Rabdomyolyysia on havaittu potilailla, jotka ovat saaneet nevirapiinin käyttöön liittyviä iho- ja/tai maksareaktioita.
Prednisonin samanaikaisen käytön (40 mg/vuorokausi Viramune-valmisteen annon ensimmäisten 14 päivän aikana) ei ole osoitettu vähentävän nevirapiiniin liittyvän ihottuman esiintymistä, ja siihen saattaa liittyä ihottuman esiintymistiheyden ja vakavuuden lisääntyminen nevirapiinihoidon ensimmäisen 6 viikon aikana.
Joitain riskitekijöitä, jotka altistavat vaikeille ihoreaktioille, on tunnistettu. Niitä ovat epäonnistuminen aloitusannoksen 200 mg vuorokaudessa (4 mg/kg/vrk tai 150 mg/m2 lapsipotilaille) noudattamisessa aloitusjakson aikana ja pitkä viive ensioireiden ja lääkärikäynnin välillä. Ihottuman kehittymisen riski näyttää olevan naisilla suurempi kuin miehillä, olipa kyseessä nevirapiinia sisältävä tai sisältämätön hoito-ohjelma.
Potilaille pitää kertoa, että yksi nevirapiinin merkittävimmistä haittavaikutuksista on ihottuma. Heitä pitää neuvoa ilmoittamaan heti lääkärilleen mistä tahansa ihottumasta ja välttämään viivettä ensioireiden ja lääkärikäynnin välillä. Nevirapiiniin liittyvistä ihottumista suurin osa ilmenee ensimmäisen kuuden hoitoviikon aikana, minä aikana potilaita pitää tarkkailla huolellisesti ihottuman havaitsemiseksi. Potilaille pitää kertoa, että mikäli mitä tahansa ihottumaa esiintyy kahden viikon aloitusjakson aikana, lääkeannosta ei saa nostaa, ennen kuin ihottuma on hävinnyt. Annostusta 200 mg kerran vuorokaudessa ei pidä jatkaa yli 28 päivän ajan, vaan siinä vaiheessa pitää etsiä vaihtoehtoista hoitoa mahdollisesti liian pieneksi jäävän altistumisen ja resistenssiriskin vuoksi. Erityisesti lapsipotilaiden huolellinen tarkkailu on taattava, etenkin kahdeksantoista ensimmäisen hoitoviikon aikana, koska ihoreaktioiden havaitseminen itse ja niistä kertominen voi olla lapsipotilailla epätodennäköisempää kuin aikuisilla.
Jokaisen potilaan, joka saa vaikean ihottuman tai ihottuman, johon liittyy yleisoireita, kuten kuumetta, rakkuloita, suun leesioita, sidekalvontulehdusta, kasvojen turvotusta, lihas- ja nivelkipuja tai huonovointisuutta, pitää keskeyttää lääkevalmisteen käyttö ja hakeutua välittömästi lääkärin hoitoon. Näillä potilailla nevirapiinihoitoa ei saa aloittaa uudestaan. Jos potilailla esiintyy mahdollisesti nevirapiiniin liittyvä ihottuma, maksan toimintakokeet pitää tehdä. Potilaiden, joiden maksa-arvot ovat kohtalaisesti tai voimakkaasti nousseet (ASAT tai ALAT yli viisinkertainen verrattuna normaalin ylärajaan), nevirapiinihoito pitää lopettaa pysyvästi. Jos ilmenee yliherkkyysreaktio, jolle on ominaista ihottuma ja yleisoireina kuume, nivelkivut, lihaskivut ja imusolmukkeiden suurentuminen sekä sisäelimiin liittyviä löydöksiä kuten hepatiitti, eosinofilia, granulosytopenia ja munuaisten vajaatoiminta, nevirapiinin käyttö pitää pysyvästi lopettaa eikä käyttöä saa aloittaa uudelleen (ks. kohta Vasta-aiheet). |
Maksareaktiot
Vakavaa ja hengenvaarallista maksatoksisuutta, mukaan luettuna äkillinen ja voimakasoireinen kuolemaan johtava hepatiitti, on havaittu nevirapiinilla hoidetuilla potilailla. Ensimmäiset 18 viikkoa ovat kriittistä aikaa, joka vaatii tarkkaa seurantaa. Maksareaktioiden riski on suurimmillaan kuuden ensimmäisen hoitoviikon aikana. Riski säilyy kuitenkin tämän ajanjakson jälkeen ja tarkkailun pitää jatkua säännöllisin väliajoin koko hoidon keston ajan.
Rabdomyolyysia on havaittu potilailla, jotka ovat saaneet nevirapiinin käyttöön liittyviä iho- ja/tai maksareaktioita.
Antiretroviraalihoitoa aloitettaessa yli 2,5-kertaiset ASAT- tai ALAT-arvot ja/tai samanaikainen hepatiitti-infektio (B- ja/tai C-hepatiitti) ovat yhteydessä suurempaan riskiin saada maksahaittavaikutuksia yleisesti antiretroviraalihoidon aikana, nevirapiinia sisältävät hoito-ohjelmat mukaan lukien.
Naissukupuoleen sekä aiemmin hoitamattomiin potilaisiin, joilla on korkeampi CD4+ -solujen määrä nevirapiinihoitoa aloitettaessa, on liitetty suurentunut riski saada maksahaittavaikutuksia. Naisilla riski saada oireellinen maksatapahtuma, johon liittyy usein ihottuma, on kolminkertainen miehiin nähden (5,8 % verrattuna 2,2 %:iin). Aiemmin hoitamattomilla potilailla, joilla on mitattavissa oleva määrä plasman HIV-1 RNA:ta ja korkeampi CD4+ -solujen määrä nevirapiinihoitoa aloitettaessa, on sukupuolesta riippumatta suurempi riski saada oireellinen maksatapahtuma. Retrospektiivisessä tarkastelussa, jossa potilaista, joilla valtaosalla plasman HIV-1 viruskuorma oli 50 kopiota/ml tai enemmän, naisilla, joilla CD4+ -solujen määrä oli yli 250 solua/mm3, oli kaksitoistakertainen riski saada oireellinen maksahaittavaikutus verrattuna naisiin, joilla CD4+ -solujen määrä oli alle 250 solua/mm3 (11,0 % verrattuna 0,9 %:iin). Miehillä, joilla oli mitattavissa oleva määrä plasman HIV-1 RNA:ta ja CD4+ -solujen määrä oli yli 400 solua/mm3, havaittiin suurentunut riski verrattuna miehiin, joilla CD4+ -solujen määrä oli alle 400 solua/mm3 (6,3 % verrattuna 1,2 %:iin). Tiettyihin CD4+ -solujen määriin perustuvaa suurentunutta toksisuusriskiä ei ole havaittu potilailla, joiden viruskuorma oli alle mittausrajan (eli < 50 kopiota/ml).
Potilaille pitää kertoa, että maksareaktiot ovat merkittävä nevirapiinin haittavaikutus, joka vaatii tarkkaa seurantaa ensimmäisen 18 viikon aikana. Heille pitää kertoa, että hepatiittiin viittaavien oireiden ilmaantuessa pitää nevirapiinihoito keskeyttää ja hakeutua välittömästi lääkärin tutkimuksiin, joiden tulisi sisältää maksan toimintakokeet.
Maksa-arvojen seuranta
Laboratoriotutkimukset, joihin kuuluvat maksan toimintakokeet, pitää tehdä ennen nevirapiinihoidon aloittamista ja sopivin väliajoin hoidon aikana.
Maksan toimintakokeissa on raportoitu poikkeavia arvoja nevirapiinin käytön yhteydessä, joissain tapauksissa ensimmäisten hoitoviikkojen aikana.
Maksaentsyymien oireetonta nousua on kuvattu usein ja se ei ole välttämättä vasta-aihe nevirapiinin käytölle. Oireeton GT:n nousu ei ole vasta-aihe käytön jatkamiselle.
Maksan toimintakokeita pitää seurata kahden viikon välein hoidon kahden ensimmäisen kuukauden aikana, kolmannen kuukauden kohdalla ja sen jälkeen säännöllisesti. Maksa-arvoja pitää seurata potilailta, joilla on hepatiittiin ja/tai yliherkkyyteen viittaavia merkkejä tai oireita.
Jos ASAT tai ALAT on ennen hoitoa tai hoidon aikana yli 2,5-kertainen verrattuna normaalin ylärajaan, maksa-arvoja pitää seurata useammin lääkärikäyntien yhteydessä. Nevirapiinia ei saa antaa potilaille, joilla ASAT tai ALAT on yli viisinkertainen normaalin ylärajaan verrattuna ennen kuin ASAT- ja ALAT-lähtöarvot ovat vakiintuneet alle viisinkertaisen normaalin ylärajan (ks. kohta Vasta-aiheet).
Lääkärin ja potilaan pitää olla varuillaan hepatiitin esioireiden tai löydösten, kuten anoreksian, pahoinvoinnin, keltaisuuden, bilirubinurian, akolisten ulosteiden, maksan suurenemisen tai maksan kosketusarkuuden suhteen. Potilaita pitää neuvoa hakeutumaan viipymättä lääkärin hoitoon, mikäli näitä oireita ilmenee. Jos ASAT tai ALAT kohoaa yli viisinkertaiseksi hoidon aikana verrattuna normaalin ylärajaan, nevirapiinin käyttö pitää välittömästi keskeyttää. Jos ASAT ja ALAT palautuvat lähtötasolle, ja jos potilaalla ei ole hepatiitin kliinisiä merkkejä tai oireita, ihottumaa, yleisoireita tai muita löydöksiä, jotka viittaavat elinten toimintahäiriöön, on ehkä mahdollista, tapauskohtaisesti, aloittaa nevirapiini uudelleen käyttäen alkuannoksena 200 mg/vrk 14 päivän ajan, jonka jälkeen annoksena käytetään 400 mg/vrk. Näissä tapauksissa vaaditaan tiheämpää maksan toiminnan seurantaa. Jos maksan toimintakokeiden arvot kohoavat nopeasti uudestaan, nevirapiinin käyttö pitää lopettaa pysyvästi. Jos kliinistä hepatiittia, jolle on ominaista anoreksia, pahoinvointi, oksentelu, keltaisuus SEKÄ laboratoriolöydökset (kuten kohtalaisesti tai voimakkaasti kohonneet maksan toimintakokeiden arvot (lukuun ottamatta GT:n kohoamista), esiintyy, nevirapiinin käyttö pitää lopettaa pysyvästi. Viramune-valmistetta ei saa aloittaa uudestaan potilaille, joilta nevirapiinin käyttö lopetettiin pysyvästi sen aiheuttaman kliinisen hepatiitin vuoksi. |
Maksasairaus
Viramune-valmisteen turvallisuutta ja tehoa ei ole varmistettu potilailla, joilla on taustalla merkittäviä maksan toiminnan häiriöitä. Viramune on vasta-aiheinen potilaille, joilla on vaikea maksan vajaatoiminta (Child–Pugh luokka C, ks. kohta Vasta-aiheet). Farmakokineettiset tulokset viittaavat siihen, että varovaisuutta pitää noudattaa annettaessa nevirapiinia potilaille, joilla on keskivaikea maksan toimintahäiriö (Child–Pugh B). Niillä antiretroviraalihoitoa saavilla potilailla, joilla on krooninen hepatiitti B tai C, on suurempi riski saada vakavia, mahdollisesti fataaleja maksahaittavaikutuksia. Jos hepatiitin B tai C hoitoon annetaan samanaikaisesti muita antiviraalisia lääkkeitä, tutustu myös näiden valmisteiden tuoteselosteisiin.
Niillä antiretroviraalista yhdistelmähoitoa saavilla potilailla, joilla on aikaisemmin ollut maksan vajaatoimintaa, mukaan lukien krooninen aktiivinen hepatiitti, esiintyy useammin maksan toiminnan poikkeavuuksia. Heitä on seurattava tavanomaisen käytännön mukaisesti. Jos tällaisten potilaiden maksasairauden pahenemista todetaan, on harkittava hoidon keskeyttämistä tai lopettamista.
Muut varoitukset
Altistuksen jälkeinen profylaksi: Vakavaa maksatoksisuutta, maksan siirtoa vaativa maksan vajaatoiminta mukaan lukien, on raportoitu HIV-infektoitumattomilla henkilöillä, jotka ovat saaneet hyväksytyn käyttöaiheen vastaisesti kerrannaisannoksia Viramune-valmistetta profylaktisesti altistuksen jälkeen (PEP). Viramune-valmisteen käyttöä ei ole arvioitu erityisessä PEP-tutkimuksessa, etenkään hoidon keston suhteen, ja siksi se on erittäin epäsuotavaa.
Nevirapiinia sisältävä yhdistelmähoito ei ole parantava hoito HIV-1 infektoituneille potilaille; potilaille saattaa edelleen kehittyä edenneeseen HIV-1 infektioon liittyviä sairauksia, kuten opportunistisia infektioita.
Hormonaalisia ehkäisymenetelmiä, depomedroksiprogesteroniasetaattia (DMPA) lukuun ottamatta, ei saa käyttää ainoana ehkäisymenetelmänä Viramune-valmistetta saavilla naisilla, koska nevirapiini saattaa pienentää näiden lääkevalmisteiden plasmapitoisuuksia. Tästä syystä ja HIVin tartuntavaaran vuoksi suositellaan muita ehkäisykeinoja (esim. kondomeja). Lisäksi jos menopaussin jälkeistä hormonihoitoa käytetään yhtä aikaa nevirapiinin kanssa, pitää sen terapeuttista vaikutusta seurata.
Paino ja metaboliset parametrit
Antiretroviraalisen hoidon aikana saattaa ilmetä painon nousua sekä veren lipidi- ja glukoosiarvojen nousua. Tällaiset muutokset saattavat osittain liittyä hoitotasapainoon ja elämäntapaan. Lipidien kohdalla on joissain tapauksissa näyttöä siitä, että syynä on lääkehoito, kun taas vahvaa näyttöä minkään tietyn hoidon vaikutuksesta painon nousuun ei ole. Veren lipidi- ja glukoosiarvojen seurannan osalta viitataan HIV-infektion hoitosuosituksiin. Rasva-aineenvaihdunnan häiriöitä on hoidettava kliinisen käytännön mukaisesti.
Kliinisissä tutkimuksissa Viramune-valmisteen käyttöön on liittynyt HDL-kolesterolin nousu ja kokonaiskolesterolin ja HDL-kolesterolin suhteen kokonaisparaneminen. Tästä ei kuitenkaan ole tehty erityisiä tutkimuksia, joten näiden havaintojen kliinistä merkitystä ei tunneta. Viramune-valmisteen ei myöskään ole osoitettu aiheuttaneen glukoositasapainon häiriöitä.
Osteonekroosi: osteonekroositapauksia on esiintynyt erityisesti edenneen HIV-infektion ja/tai pitkäaikaisen antiretroviraalisen yhdistelmähoidon (CART) yhteydessä, vaikkakin syitä tapauksille on ollut useita (mukaan lukien kortikosteroidihoito, alkoholin käyttö, vaikea immuunisuppressio, korkea painoindeksi). Potilaita pitää neuvoa ottamaan yhteyttä lääkäriin, jos heillä esiintyy nivelsärkyä ja -kipua, nivelten jäykkyyttä tai liikkumisvaikeuksia.
Immuunireaktivaatio-oireyhtymä: Antiretroviraalisen yhdistelmähoidon (CART) aloitus voi vaikeaa immuunikatoa sairastavilla HIV-infektoituneilla potilailla laukaista tulehdusreaktion. Opportunististen patogeenien aiheuttama latentti infektio voi muuttua oireiseksi aiheuttaen vakavia kliinisiä oireita tai oireiden lisääntymistä. Tällaisia oireita on havaittu erityisesti yhdistelmähoidon ensimmäisinä viikkoina tai kuukausina. Esimerkkejä tulehduksista ovat sytomegaloviruksen aiheuttama retiniitti, yleistynyt ja/tai paikallinen mykobakteeri-infektio ja Pneumocystis jiroveciin aiheuttama keuhkokuume. Kaikkia tulehdusoireita pitää seurata ja tarvittaessa aloittaa niiden hoito. Immuunireaktivaation yhteydessä on raportoitu myös autoimmuunisairauksia (kuten Basedowin tauti ja autoimmuunihepatiitti). Taudin puhkeamiseen kuluvan ajan on raportoitu kuitenkin olevan vaihteleva, ja näitä tapahtumia voi ilmaantua useita kuukausia hoidon aloittamisen jälkeen.
Saatavana olevat farmakokineettiset tiedot viittaavat siihen, että rifampisiinin ja nevirapiinin samanaikaista käyttöä ei suositella. Lisäksi seuraavien yhdisteiden käyttämistä Viramune-valmisteen kanssa samanaikaisesti ei suositella: efavirentsi, ketokonatsoli, etraviriini, rilpiviriini, elvitegraviiri (yhdessä kobisistaatin kanssa), atatsanaviiri (yhdessä ritonaviirin kanssa), fosamprenaviiri (ellei sitä annostella yhdessä matala-annoksisen ritonaviirin kanssa) (ks. kohta Yhteisvaikutukset).
Tsidovudiiniin liittyy yleisesti granulosytopeniaa. Tämän vuoksi potilailla, jotka saavat nevirapiinia ja tsidovudiinia samanaikaisesti ja erityisesti pediatriset potilaat ja potilaat, jotka saavat suurempia tsidovudiiniannoksia tai potilaat, joilla on huonot luuydinvarastot ja näistä etenkin ne, joilla on pitkälle edennyt HIV-sairaus, on suurentunut granulosytopenian riski. Näiden potilaiden hematologisia arvoja pitää monitoroida tarkasti.
Yliherkkyys
Sakkaroosi: Viramune oraalisuspensio sisältää 150 mg/ml sakkaroosia. Potilaiden, joilla on harvinainen perinnöllinen fruktoosi-intoleranssi, glukoosi-galaktoosi imeytymishäiriö tai sakkaroosi-isomaltaasin vajaatoiminta, ei tule käyttää tätä lääkettä.
Sorbitoli: Viramune oraalisuspensio sisältää 162 mg/ml sorbitolia. Tätä lääkevalmistetta ei pidä antaa potilaille, joilla on perinnöllinen fruktoosi-intoleranssi (HFI).
Metyyli- ja propyyliparahydroksibentsoaatti: Viramune oraalisuspensio sisältää metyyliparahydroksibentsoaattia ja propyyliparahydroksibentsoaattia, jotka voivat aiheuttaa allergisia reaktioita (mahdollisesti viivästyneitä).
Yhteisvaikutukset
Nevirapiini indusoi CYP3A:ta ja mahdollisesti CYP2B6:ta. Maksimaalinen induktio tapahtuu 2-4 viikon kuluessa lääkityksen aloittamisesta ylläpitoannoksella.
Tätä metaboliareittiä käyttävien lääkeaineiden pitoisuudet plasmassa saattavat pienentyä käytettäessä yhdessä nevirapiinin kanssa. P450-reitin kautta metaboloituvien lääkevalmisteiden terapeuttisen tehon huolellista tarkkailua suositellaan käytettäessä yhdessä nevirapiinin kanssa.
Ruoka, antasidit tai emäksistä puskuria sisältävät lääkevalmisteet eivät vaikuta nevirapiinin imeytymiseen.
Yhteisvaikutustiedot esitetään geometrisinä keskiarvoina 90 %:n luottamusvälillä (90 % CI) aina kun nämä tiedot ovat saatavilla. ND = ei määritelty, ↑ = suurenee ↓ = pienenee, ↔ ei vaikutusta
Lääkevalmisteet terapeuttisen alueen mukaan | Yhteisvaikutus | Yhteiskäyttöä koskevat suositukset |
INFEKTIOLÄÄKKEET | ||
ANTIRETROVIRAALISET LÄÄKEAINEET | ||
Nukleosidianalogit (NRTI:t) | ||
Didanosiini 100-150 mg kahdesti vuorokaudessa | Didanosiini AUC ↔ 1,08 (0,92-1,27) | Didanosiinia ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. |
Emtrisitabiini | Emtrisitabiini ei ole ihmisen CYP 450 ‑entsyymien estäjä. | Viramune-valmistetta ja emtrisitabiinia voidaan käyttää ilman annosten muuttamista. |
Abakaviiri | Abakaviiri ei estä sytokromi P450 ‑isoformeja ihmisen maksan mikrosomeissa. | Viramune-valmistetta ja abakaviiria voidaan käyttää ilman annosten muuttamista. |
Lamivudiini 150 mg kahdesti vuorokaudessa | Ei muutoksia lamivudiinin puhdistumassa tai jakautumistilavuudessa. Tulokset viittaavat siihen, että nevirapiinilla ei ole indusoivaa vaikutusta lamivudiinin puhdistumaan. | Lamivudiinia ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. |
Stavudiini: 30/40 mg kahdesti vuorokaudessa | Stavudiini AUC ↔ 0,96 (0,89-1,03) Nevirapiini: pitoisuuksissa ei tapahtunut muutoksia aikaisempaan aineistoon verrattuna. | Stavudiinia ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. |
Tenofoviiri 300 mg kerran vuorokaudessa | Tenofoviirin pitoisuus plasmassa pysyi muuttumattomana annettaessa yhdessä nevirapiinin kanssa. | Tenofoviiria ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. |
Tsidovudiini 100-200 mg kolmesti vuorokaudessa | Tsidovudiini AUC ↓ 0,72 (0,60-0,96) Nevirapiini: Tsidovudiini ei vaikuttanut nevirapiinin farmakokinetiikkaan. | Tsidovudiinia ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. Tsidovudiiniin liittyy yleisesti granulosytopeniaa. Tämän vuoksi potilailla, jotka saavat nevirapiinia ja tsidovudiinia samanaikaisesti ja erityisesti pediatriset potilaat ja potilaat, jotka saavat suurempia tsidovudiiniannoksia tai potilaat, joilla on huonot luuydinvarastot ja näistä etenkin ne, joilla on pitkälle edennyt HIV-sairaus, on suurentunut granulosytopenian riski. Näiden potilaiden hematologisia arvoja pitää monitoroida tarkasti. |
Ei-nukleosidianalogit (NNRTI:t) | ||
Efavirentsi 600 mg kerran vuorokaudessa | Efavirentsi AUC ↓ 0,72 (0,66-0,86) | Efavirentsin ja Viramune-valmisteen yhteiskäyttöä ei suositella lisääntyneen toksisuuden takia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Lisäksi yhteiskäyttö ei lisää tehoa verrattuna jommankumman NNRTI:n käyttöön yksin (2NN-tutkimuksen tulokset, ks. kohta Farmakodynamiikka). |
Etraviriini | Etraviriinin ja nevirapiinin samanaikainen käyttö voi vähentää merkittävästi etraviriinin pitoisuutta plasmassa sekä sen terapeuttista vaikutusta. | Viramune-valmisteen ja NNRTI:n samanaikaista käyttöä ei suositella. |
Rilpiviriini | Yhteisvaikutusta ei ole tutkittu. | Viramune-valmisteen ja NNRTI:n samanaikaista käyttöä ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). |
Proteaasin estäjät (PI:t) | |||
Atatsanaviiri/ ritonaviiri 300/100 mg kerran vuorokaudessa 400/100 mg kerran vuorokaudessa | Atatsanaviiri/r 300/100 mg: Atatsanaviiri/r 400/100 mg Nevirapiini AUC ↑ 1,25 (1,17-1,34) | Atatsanaviirin/ritonaviirin ja Viramune-valmisteen yhteiskäyttöä ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). | |
Darunaviiri/ritonaviiri 400/100 mg kahdesti vuorokaudessa | Darunaviiri AUC ↑ 1,24 (0,97-1,57) Nevirapiini AUC ↑ 1,27 (1,12-1,44) | Darunaviiria ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. | |
Fosamprenaviiri 1400 mg kahdesti vuorokaudessa | Amprenaviiri AUC ↓ 0,67 (0,55-0,80) Nevirapiini AUC ↑ 1,29 (1,19-1,40) | Fosamprenaviirin ja Viramune-valmisteen yhteiskäyttöä ei suositella ilman että samanaikaisesti annetaan ritonaviiria (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). | |
Fosamprenaviiri/ ritonaviiri 700/100 mg kahdesti vuorokaudessa | Amprenaviiri AUC ↔ 0,89 (0,77-1,03) Nevirapiini AUC ↑ 1,14 (1,05-1,24) | Fosamprenaviiri/ritonaviiria ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. | |
Lopinaviiri/ritonaviiri (kapselit) 400/100 mg kahdesti vuorokaudessa | Aikuispotilaat: | Lopinaviiri/ritonaviiri annoksen nostoa 533/133 mg:aan (4 kapseliin) tai 500/125 mg (5 tablettia, joissa jokaisessa 100/25 mg) kahdesti vuorokaudessa ruoan kanssa suositellaan käytettäessä yhdessä Viramune-valmisteen kanssa. Viramune-annoksen muuttamista ei tarvita annettaessa yhdessä lopinaviirin kanssa. | |
Lopinaviiri/ritonaviiri (oraaliliuos) 300/75 mg/m2 kahdesti vuorokaudessa | Lapsipotilaat: | Lapsilla lopinaviiri/ritonaviirin annoksen nostoa 300/75 mg/m2:aan kahdesti vuorokaudessa ruoan kanssa pitää harkita käytettäessä yhdessä Viramune-valmisteen kanssa, etenkin potilailla, joilla epäillään alentunutta herkkyyttä lopinaviiri/ritonaviirille. | |
Ritonaviiri 600 mg kahdesti vuorokaudessa | Ritonaviiri AUC ↔ 0,92 (0,79-1,07) Nevirapiini: Yhteiskäyttö ritonaviirin kanssa ei johda kliinisesti merkitsevään muutokseen nevirapiinin pitoisuudessa plasmassa. | Ritonaviiria ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. | |
Sakinaviiri/ritonaviiri | Rajoitettu tieto ritonaviirilla tehostetun sakinaviirin (pehmeät kapselit) käytöstä ei viittaa kliinisesti merkitseviin yhteisvaikutuksiin ritonaviirilla tehostetun sakinaviirin ja nevirapiinin välillä. | Sakinaviiri/ritonaviiria ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. | |
Tipranaviiri/ritonaviiri 500/200 mg kahdesti vuorokaudessa | Erityisiä yhteisvaikutustutkimuksia ei ole tehty. Saatavilla oleva rajallinen tieto faasi IIa-tutkimuksesta HIV-infektoiduilla potilailla on osoittanut kliinisesti merkityksettömän 20 % pienenemisen tipranaviirin Cmin-arvossa. | Tipranaviiria ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. | |
FUUSION ESTÄJÄT | |||
Enfuvirtidi | Metaboliareitin vuoksi kliinisesti merkitseviä farmakokineettisiä yhteisvaikutuksia ei ole odotettavissa enfuvirtidin ja nevirapiinin välillä. | Enfuvirtidia ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. | |
Maraviroki 300 mg kerran vuorokaudessa | Maraviroki AUC ↔ 1,01 (0,6 -1,55) Nevirapiinin pitoisuuksia ei ole mitattu, yhteisvaikutusta ei ole odotettavissa. | Maravirokia ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. | |
INTEGRAASIN ESTÄJÄT | |||
Elvitegraviiri/ kobisistaatti | Yhteisvaikutusta ei ole tutkittu. Kobisistaatti on sytokromi P450 3A ‑estäjä ja se estää merkittävästi sekä maksaentsyymejä että muita metaboliareittejä. Sen vuoksi yhtäaikainen annostelu todennäköisesti muuttaisi sekä kobisistaatin että Viramune-valmisteen pitoisuuksia plasmassa. | Viramune-valmisteen ja elvitegraviirin ja kobisistaatin yhdistelmän samanaikaista annostelua ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). | |
Raltegraviiri 400 mg kahdesti vuorokaudessa | Kliinistä tietoa ei ole saatavilla. Raltegraviirin metaboliareitin vuoksi yhteisvaikutuksia ei ole odotettavissa. | Raltegraviiria ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. | |
ANTIBIOOTIT | |||
Klaritromysiini 500 mg kahdesti vuorokaudessa | Klaritromysiini AUC ↓ 0,69 (0,62-0,76) Metaboliitti 14-OH klaritromysiini Nevirapiini AUC ↑ 1,26 | Klaritromysiinialtistus pieneni merkitsevästi ja 14 OH-metaboliittialtistus kasvoi. Koska klaritromysiinin aktiivisen metaboliitin aktiivisuus solunsisäistä Mycobacterium avium -kompleksia vastaan on alentunut, saattaa yleinen aktiivisuus tätä patogeeniä kohtaan muuttua. Vaihtoehtoa klaritromysiinille, kuten esim. atsitromysiiniä, pitää harkita. Maksan poikkeavuuksia suositellaan seurattavan tarkasti. | |
Rifabutiini 150 tai 300 mg kerran vuorokaudessa | Rifabutiini AUC ↑ 1,17 (0,98-1,40) Metaboliitti 25-O-desasetyylirifabutiini AUC ↑ 1,24 (0,84-1,84) Nevirapiinin puhdistuman raportoitiin kasvavan (9 %) aikaisempaan tietoon verrattuna. Muutos ei ollut kliinisesti merkitsevä. | Ei havaittavissa merkittävää vaikutusta rifabutiinin tai Viramune-valmisteen keskimääräisiin farmakokineettisiin parametreihin. Rifabutiinia ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. Suuren yksilöiden välisen vaihtelevuuden vuoksi joillakin potilailla rifabutiinialtistus saattaa kuitenkin kasvaa huomattavasti ja heillä saattaa olla suurempi riski rifabutiinin toksisuudelle. Sen vuoksi varovaisuutta on noudatettava samanaikaisessa annostelussa. | |
Rifampisiini 600 mg kerran vuorokaudessa | Rifampisiini AUC ↔ 1,11 (0,96-1,28) Nevirapiini AUC ↓ 0,42 | Rifampisiinin ja Viramune-valmisteen samanaikaista käyttöä ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Lääkärit, jotka hoitavat samanaikaisesti tuberkuloosia sairastavia potilaita, joilla on Viramune-valmistetta sisältävä hoito-ohjelma, voivat harkita sen sijaan rifabutiinin yhteiskäyttöä. | |
SIENILÄÄKKEET | |||
Flukonatsoli 200 mg kerran vuorokaudessa | Flukonatsoli AUC ↔ 0,94 (0,88-1,01) Nevirapiini: altistus: ↑100 % verrattuna aikaisempaan tietoon annettaessa pelkästään nevirapiinia. | Kohonneen Viramune-altistuksen riskin vuoksi varovaisuutta on noudatettava annettaessa lääkevalmisteita samanaikaisesti ja potilaita on valvottava tarkoin. | |
Itrakonatsoli 200 mg kerran vuorokaudessa | Itrakonatsoli AUC ↓ 0,39 Nevirapiini: ei merkitseviä muutoksia nevirapiinin farmakokineettisissä parametreissa. | Itrakonatsolin annoksen suurentamista pitää harkita annettaessa näitä kahta lääkeainetta samanaikaisesti. | |
Ketokonatsoli 400 mg kerran vuorokaudessa | Ketokonatsoli AUC ↓ 0,28 (0,20-0,40) Nevirapiini: plasmapitoisuudet: ↑ 1,15-1,28 verrattuna aikaisempiin kontrolleihin. | Ketokonatsolin ja Viramune-valmisteen samanaikaista käyttöä ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). | |
VIRUSLÄÄKKEET KROONISEN B- JA C-HEPATIITIN HOITOON | |||
Adefoviiri | Tulokset in vitro -tutkimuksista osoittivat adefoviirin antagonisoivan nevirapiinin vaikutusta heikosti (ks. kohta Farmakodynamiikka). Tätä ei ole vahvistettu kliinisissä tutkimuksissa, eikä tehon heikkenemistä ole odotettavissa. Adefoviiri ei vaikuttanut yleisimpiin CYP-isoformeihin, joiden tiedetään olevan osallisena ihmisen lääkeainemetaboliassa, ja se erittyy munuaisten kautta. Kliinisesti merkitsevää lääke-lääke-yhteisvaikutusta ei ole odotettavissa. | Adefoviiria ja Viramune-valmistetta voidaan käyttää yhdessä ilman annosten muuttamista. | |
Entekaviiri | Entekaviiri ei ole sytokromi P450 ‑entsyymien (CYP 450) substraatti, indusoija tai estäjä. Entekaviirin metaboliareitin johdosta kliinisesti merkitsevää lääke-lääke-yhteisvaikutusta ei ole odotettavissa. | Entekaviiria ja Viramune-valmistetta voidaan käyttää yhdessä ilman annosten muuttamista. | |
Interferonit (pegyloitu interferonialfa-2a ja –alfa-2b) | Interferoneilla ei tiedetä olevan vaikutusta CYP 3A4:ään tai 2B6:een. Kliinisesti merkitsevää lääke-lääke-yhteisvaikutusta ei ole odotettavissa. | Interferoneja ja Viramune-valmistetta voidaan käyttää yhdessä ilman annosten muuttamista. | |
Ribaviriini | Tulokset in vitro -tutkimuksista osoittivat ribaviriinin antagonisoivan nevirapiinin vaikutusta heikosti (ks. kohta Farmakodynamiikka). Tätä ei ole vahvistettu kliinisissä tutkimuksissa, eikä tehon heikkenemistä ole odotettavissa. Ribaviriini ei estä sytokromi P450 ‑entsyymejä, eikä toksisuustutkimuksissa ole saatu näyttöä ribaviriinin maksaentsyymejä indusoivasta vaikutuksesta. Kliinisesti merkitsevää lääke-lääke-yhteisvaikutusta ei ole odotettavissa. | Ribaviriinia ja Viramune-valmistetta voidaan käyttää yhdessä ilman annosten muuttamista. | |
Telbivudiini | Telbivudiini ei ole sytokromi P450-entsyymijärjestelmän (CYP 450) substraatti, indusoija tai estäjä. Telbivudiinin metaboliareitin johdosta kliinisesti merkitsevää lääke-lääke-yhteisvaikutusta ei ole odotettavissa. | Telbivudiinia ja Viramune-valmistetta voidaan antaa yhdessä ilman annosten muuttamista. | |
ANTASIDIT | |||
Simetidiini | Simetidiini: ei merkitsevää muutosta havaittavissa simetidiinin farmakokineettisissä parametreissä. Nevirapiini Cmin ↑ 1,07 | Simetidiiniä ja Viramune-valmistetta voidaan käyttää ilman annosten muuttamista. | |
ANTITROMBOOTIT | |||
Varfariini | Nevirapiinin ja antitromboottisen aineen, varfariinin, yhteisvaikutus on monimutkainen, ja koagulaatioaika saattaa joko pidentyä tai lyhentyä käytettäessä näitä aineita samanaikaisesti. | Antikoagulaation tasoa on seurattava tarkoin. | |
EHKÄISYVALMISTEET | |||
Depo-medroksiprogesteroni-asetaatti (DMPA) 150 mg joka 3. kuukausi | DMPA AUC ↔ Nevirapiini AUC ↑ 1,20 | Viramune-valmisteen yhteiskäyttö ei muuttanut DMPA:n ovulaatiota estävää vaikutusta. Annoksen muuttamista ei tarvita käytettäessä DMPA:ta ja Viramune-valmistetta samanaikaisesti. | |
Etinyyliestradioli (EE) 0,035 mg | EE AUC ↓ 0,80 (0,67 - 0,97) | Suun kautta otettavia hormonaalisia ehkäisyvalmisteita ei tule käyttää ainoana ehkäisynä Viramune-valmistetta käyttävillä naisilla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Tehon ja turvallisuuden kannalta sopivia annoksia muille hormonaalisille ehkäisyvalmisteille (oraalinen tai muu käyttötapa) kuin DMPA ei ole osoitettu yhdessä Viramune-valmisteen kanssa. | |
Noretisteroni (NET) 1,0 mg kerran vuorokaudessa | NET AUC ↓ 0,81 (0,70 - 0,93) | ||
KIPULÄÄKKEET/OPIOIDIT | |||
Metadoni, potilaan yksilöllinen annostus | Metadoni AUC ↓ 0,40 (0,31 - 0,51) | Kun metadonia saaville potilaille aloitetaan Viramune-hoito, heitä pitää tarkkailla mahdollisesti ilmenevien vieroitusoireiden vuoksi ja metadoniannosta muuttaa vastaavasti. | |
ROHDOSVALMISTEET | |||
Mäkikuisma | Nevirapiinin pitoisuus plasmassa saattaa alentua käytettäessä samanaikaisesti mäkikuismaa (Hypericum perforatum) sisältäviä rohdosvalmisteita. Tämä johtuu mäkikuisman lääkevalmistemetaboliaa ja/tai kuljetusproteiineja indusoivasta vaikutuksesta. | Mäkikuismaa sisältäviä rohdosvalmisteita ja Viramune-valmistetta ei saa käyttää samanaikaisesti (ks. kohta Vasta-aiheet). Jos potilas on jo käyttänyt mäkikuismaa, tarkista nevirapiinin pitoisuus plasmassa sekä mahdollisesti myös virustasot ja lopeta mäkikuisman käyttö. Nevirapiinipitoisuus saattaa nousta mäkikuisman lopettamisen jälkeen. Viramune-valmisteen annosta saatetaan joutua säätämään. Indusoiva vaikutus saattaa säilyä ainakin 2 viikkoa mäkikuisman käytön lopettamisen jälkeen. | |
Muu informaatio:
Nevirapiinimetaboliitit: Tutkimukset käyttäen ihmisen maksan mikrosomeja osoittivat, että dapsoni, rifabutiini, rifampisiini ja trimetopriimi/sulfametoksatsoli eivät vaikuttaneet nevirapiinin hydroksyloituneiden metaboliittien muodostumiseen. Sen sijaan ketokonatsoli ja erytromysiini estivät merkitsevästi nevirapiinin hydroksyloituneiden metaboliittien muodostumista.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset / Ehkäisy miehille ja naisille
Hedelmällisessä iässä olevien naisten ei tule käyttää suun kautta otettavia ehkäisyvalmisteita ainoana ehkäisymenetelmänä, koska nevirapiini saattaa alentaa näiden lääkevalmisteiden pitoisuutta plasmassa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Raskaus
Tällä hetkellä saatavilla oleva tieto ei viittaa epämuodostumiin tai sikiön/vastasyntyneen toksisuuteen raskaana olevilla naisilla. Toistaiseksi tarjolla ei myöskään ole muuta tarkkaa epidemiologista tietoa. Tiineenä olevilla rotilla ja kaneilla tehdyissä lisääntymistutkimuksissa ei ole havaittu (merkittävää) teratogeenisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Riittäviä ja hyvin kontrolloituja tutkimuksia ei ole tehty raskaana olevilla naisilla. Varovaisuutta pitää noudattaa määrättäessä nevirapiinia raskaana oleville naisille (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Koska maksatoksisuus on yleisempää naisilla, joiden CD4+ -solujen määrä on yli 250 solua/mm3 ja plasman HIV-1 RNA määrä on mitattavissa (50 kopiota/ml tai enemmän), on terveydentila otettava huomioon hoitopäätöstä tehtäessä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Aiemmin hoitoa saaneilla ja nevirapiinihoidon aloittaneilla naisilla, joilla oli alle mittausrajan oleva viruskuorma (plasman HIV-1:stä vähemmän kuin 50 kopiota/ml) ja CD4+ -solujen määrä yli 250 solua/mm3, ei ole suurentunutta toksisuusriskiä. Vastaavasta ei ole näyttöä raskaana oleville naisille. Kaikki satunnaistetut tutkimukset, jotka käsittelivät erityisesti tätä asiaa, jättivät raskaana olevat naiset pois tutkimuksesta ja raskaana olevat naiset olivat aliedustettuina sekä kohortti-tutkimuksissa että meta-analyyseissä.
Imetys
On suositeltavaa, että HIV-infektion saaneet naiset eivät imetä lapsiaan HIV-tartunnan välttämiseksi.
Hedelmällisyys
Lisääntymistutkimuksissa nevirapiini vähensi rottien hedelmällisyyttä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Valmisteen vaikutuksesta ajokykyyn ja koneidenkäyttökykyyn ei ole erityisiä tutkimuksia. Potilaille on kuitenkin kerrottava, että nevirapiinihoidon aikana saattaa esiintyä haittavaikutuksia kuten väsymystä. Autolla ajettaessa ja koneita käytettäessä on siis noudatettava varovaisuutta. Jos potilaalla on väsymystä, hänen on vältettävä vaarallisia toimia, kuten ajamista tai koneiden käyttöä.
Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Useimmin ilmoitetut Viramune hoitoon liittyvät haittavaikutukset kaikissa kliinisissä tutkimuksissa olivat ihottuma, allergiset reaktiot, hepatiitti, epänormaalit arvot maksan toimintakokeissa, pahoinvointi, oksentelu, ripuli, vatsakipu, uupumus, kuume, päänsärky ja lihassärky.
| Markkinoille tulon jälkeen saatu kokemus on osoittanut, että vakavimmat haittavaikutukset ovat Stevens–Johnsonin syndrooma/toksinen epidermaalinen nekrolyysi, vakava hepatiitti/maksan vajaatoiminta, sekä lääkereaktio, johon liittyy eosinofiliaa ja systeemisiä oireita, joille on ominaista ihottuma ja yleisoireina kuume, nivelkivut, lihaskivut ja imusolmukkeiden suurentuminen sekä sisäelimiin liittyviä löydöksiä, kuten hepatiitti, eosinofilia, granulosytopenia ja munuaisten vajaatoiminta. Ensimmäiset 18 viikkoa on kriittinen ajanjakso, jolloin vaaditaan tarkkaa seurantaa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). |
Taulukoitu yhteenveto haittavaikutuksista
Seuraavia haittavaikutuksia, joilla saattaa olla syy-yhteys Viramune-valmisteen antoon, on raportoitu. Arvioidut esiintymistiheydet perustuvat kliinisistä tutkimuksista koottuihin tietoihin haittavaikutuksista, joiden arvioitiin liittyvän Viramune-hoitoon.
Esiintymistiheydet on luokiteltu seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, <1/10); melko harvinainen (≥ 1/1,000, < 1/100); harvinainen (≥1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000)
Veri ja imukudos
Yleinen: granulosytopenia
Melko harvinainen: anemia
Immuunijärjestelmä
Yleinen: yliherkkyys (mukaan lukien anafylaksia, angioedeema, nokkosihottuma)
Melko harvinainen: anafylaktinen reaktio
Harvinainen: lääkereaktio, johon liittyy eosinofiliaa ja systeemisiä oireita
Hermosto
Yleinen: päänsärky
Ruoansulatuselimistö
Yleinen: pahoinvointi, oksentelu, vatsakipu, ripuli
Maksa ja sappi
Yleinen: hepatiitti (myös vaikea ja hengenvaarallinen maksatoksisuus) (1,9 %)
Melko harvinainen: keltaisuus
Harvinainen: fulminantti hepatiitti (joka voi johtaa kuolemaan)
Iho ja ihonalainen kudos
Hyvin yleinen: ihottuma (12,5 %)
Melko harvinainen: Stevens–Johnsonin syndrooma/toksinen epidermaalinen nekrolyysi (joka voi johtaa kuolemaan) (0,2 %), angioedeema, nokkosihottuma
Luusto, lihakset ja sidekudos
Melko harvinainen: nivelkipu, lihaskipu
Yleisoireet ja antopaikassa todettavat haitat
Yleinen: kuume, uupumus
Tutkimukset
Yleinen: epänormaalit arvot maksan toimintakokeissa (alaniiniaminotransferaasi koholla; transaminaasit koholla; aspartaattiaminotransferaasi koholla; gammaglutamyylitransferaasi koholla; maksaentsyymi koholla; hypertransaminasemia)
Melko harvinainen: veren fosforipitoisuus alentunut; kohonnut verenpaine
Kuvaus valikoiduista haittavaikutuksista
Tutkimuksessa 1100.1090, josta saatiin suurin osa (n=28) haittavaikutuksista, lumelääkettä saaneilla potilailla esiintyi useammin granulosytopeniaa (3,3 %) kuin nevirapiinia saaneilla potilailla (2,5 %).
Anafylaktinen reaktio tunnistettiin markkinoille tulon jälkeisessä valvonnassa, mutta sitä ei havaittu satunnaistetuissa kontrolloiduissa kliinisissä tutkimuksissa. Yleisyysluokka arvioitiin satunnaistetussa kontrolloidussa kliinisessä tutkimuksessa nevirapiinia saaneiden potilaiden kokonaismäärään perustuvasta tilastollisesta laskelmasta (n=2718).
Alentunutta veren fosforipitoisuutta ja koholla olevaa verenpainetta oli havaittavissa kliinisissä tutkimuksissa, joissa annettiin lisäksi tenofoviiria/emtrisitabiinia.
Metaboliset parametrit
Paino sekä veren lipidi- ja glukoosiarvot saattavat nousta antiretroviraalisen hoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kun nevirapiinia on käytetty yhdistelmähoitona muiden antiretroviraalisten lääkeaineiden kanssa, seuraavia haittavaikutuksia on ilmoitettu: haimatulehdus, perifeerinen neuropatia ja trombosytopenia. Nämä haittavaikutukset ovat yleensä liittyneet muihin antiretroviraalisiin lääkeaineisiin ja voidaan odottaa ilmaantuvaksi, kun nevirapiinia käytetään yhdessä muiden lääkeaineiden kanssa. On kuitenkin epätodennäköistä, että nämä haittavaikutukset liittyisivät nevirapiinihoitoon. Maksan ja munuaisten vajaatoiminta-oireyhtymää on raportoitu harvoin.
Vaikeaa immuunikatoa sairastavilla HIV-infektoituneilla potilailla voi antiretroviraalisen yhdistelmähoidon (CART) aloitus laukaista piilevän opportunisti-infektion. Autoimmuunisairauksia (kuten Basedowin tauti ja autoimmuunihepatiitti) on myös raportoitu. Taudin puhkeamiseen kuluvan ajan on raportoitu kuitenkin olevan vaihteleva, ja näitä tapahtumia voi ilmaantua useita kuukausia hoidon aloittamisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Osteonekroositapauksia on esiintynyt erityisesti potilailla, joilla on yleisesti tunnettuja riskitekijöitä, edennyt HIV-infektio tai pitkäaikainen antiretroviraalinen yhdistelmähoito (CART). Tapausten esiintymistiheyttä ei tunneta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Iho ja ihonalaiset kudokset
Nevirapiinin yleisin haittavaikutus on ihottuma, jota ilmeni 12,5 %:lla potilaista käytettäessä yhdistelmähoitoa kontrolloiduissa tutkimuksissa.
Ihottumat ovat tavallisesti lieviä tai kohtalaisia. Vartalolla, kasvoissa ja raajoissa esiintyy makulopapulaarisena, kutiavana tai kutiamattomana, punoittavana kylvönä näppylöitä. Yliherkkyyttä (anafylaktinen reaktio, angioödeemaa ja nokkosihottumaa) on raportoitu. Ihottumat esiintyvät yksinään tai lääkereaktiona, johon liittyy eosinofiliaa ja systeemisiä oireita, joille on ominaista ihottumaan liittyvinä yleisoireina kuume, nivelkivut, lihaskivut ja imusolmukkeiden suurentuminen sekä sisäelimiin liittyviä löydöksiä, kuten hepatiitti, eosinofilia, granulosytopenia ja munuaisten toimintahäiriö.
Vaikeita ja hengenvaarallisia ihoreaktioita on esiintynyt nevirapiinilla hoidetuilla potilailla, mukaan luettuna Stevens–Johnsonin syndrooma ja toksinen epidermaalinen nekrolyysi. Kuolemaan johtaneita Stevens–Johnsonin syndroomatapauksia, toksisia epidermaalisia nekrolyysitapauksia ja lääkereaktioita, joihin liittyy eosinofiliaa ja systeemisiä oireita, on raportoitu. Suurin osa vaikeista ihottumista ilmaantui ensimmäisen 6 viikon aikana. Jotkut näistävaativat sairaalahoitoa ja yksi potilas tarvitsi kirurgista hoitoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Maksa-sappi
Useimmin havaitut poikkeavuudet laboratoriokokeissa ovat kohonneet arvot maksan toimintakokeissa, mukaan lukien ALAT, ASAT, GT, kokonaisbilirubiini ja alkalinen fosfataasi. Oireeton GT-tason nousu on yleisin. Keltaisuutta on raportoitu. Hepatiittitapauksia (vakava ja hengenvaarallinen maksatoksisuus, mukaan luettuna äkillinen ja voimakasoireinen kuolemaan johtava hepatiitti) on raportoitu nevirapiinilla hoidetuilla potilailla. Vakavan maksahaittatapahtuman paras ennustaja oli kohonneet arvot maksan toimintakokeissa lähtötasolla. Ensimmäiset 18 viikkoa on kriittinen ajanjakso, jolloin vaaditaan tarkkaa seurantaa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Kliinisiin tutkimuksiin osallistuneen 361 lapsipotilaan, joista suurin osa sai yhdistelmähoitoa tsidovudiinin ja/tai didanosiinin kanssa, hoidosta saadun kokemuksen perusteella nevirapiinihoitoon liittyvät yleisimmät haittavaikutukset olivat samankaltaisia kuin aikuisilla todetut haittavaikutukset. Granulosytopeniaa todettiin yleisemmin lapsilla. Avoimessa kliinisessä tutkimuksessa (ACTG 180) lääkevalmisteesta johtuvaksi arvioitua granulosytopeniaa esiintyi 5/37 (13,5 %) potilaassa. Kaksoissokkoutetussa lumekontrolloidussa ACTG 245-tutkimuksessa vakavan lääkevalmisteesta johtuvan granulosytopenian esiintyvyys oli 5/305 (1,6 %). Tässä ryhmässä on raportoitu yksittäisiä Stevens–Johnsonin syndroomatapauksia ja Stevens–Johnson/toksinen epidermaalinen nekrolyysi välimuoto-oireyhtymätapauksia.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Nevirapiiniyliannostukseen ei ole antidoottia. Viramune-valmisteen yliannostustapauksia on raportoitu, annokset vaihtelivat 800–6000 mg vuorokaudessa aina 15 vrk:n ajan. Potilailla esiintyi turvotusta, kyhmyruusua, väsymystä, kuumetta, päänsärkyä, unettomuutta, pahoinvointia, keuhkoinfiltraatteja, ihottumaa, huimausta, oksentelua, transaminaasiarvojen kohoamista ja painon alenemista. Kaikki nämä oireet väistyivät, kun nevirapiinin anto keskeytettiin.
Pediatriset potilaat
Yksi vakava vahingossa tapahtunut yliannostustapaus vastasyntyneellä on raportoitu. Nielty annos oli 40 kertaa suositeltua annosta 2 mg/kg/vrk suurempi. Potilaalla havaittiin lievä isoloitunut neutropenia ja hyperlaktatemia, jotka hävisivät spontaanisti yhden viikon sisällä ilman mitään kliinisiä komplikaatioita. Vuotta myöhemmin lapsen kehitys oli pysynyt normaalina.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Systeemiset viruslääkkeet, käänteiskopioijaentsyymin estäjät, muut kuin nukleosidirakenteiset, ATC-koodi: J05AG01.
Vaikutusmekanismi
Nevirapiini on HIV-1:n ei-nukleosidinen käänteiskopioijaentsyymin estäjä. Nevirapiini on ei-kilpaileva HIV-1:n käänteiskopioijaentsyymin estäjä, mutta se ei biologisesti merkittävästi estä HIV-2 käänteiskopioijaentsyymiä tai eukaryoottisia DNA polymeraaseja α, β, γ, δ.
Antiviraalinen aktiivisuusin vitro:
Nevirapiinin keskimääräinen EC50-arvo (50 % estävä pitoisuus) ryhmäpaneelin M HIV-1-isolaattien tyyppejä A, B, C, D, F, G ja H ja kiertäviä rekombinanttimuotoja (CRF), CRF01_AE, CRF02_AG ja CRF12_BF vastaan ihmisen alkion munuaisten 293‑soluissa oli 63 nM. Paneelissa, jossa oli 2923 vallitsevasti alatyypiltään B HIV-1-isolaattia, keskimääräinen EC50-arvo oli 90 nM. Samanlaisia EC50-arvoja saadaan, kun nevirapiinin antiviraalista aktiivisuutta mitataan perifeerisillä veren mononukleaarisoluilla, monosyyteistä johdetuilla makrofageilla ja lymfoblastoidisilla solulinjoilla. Nevirapiinilla ei ollut antiviraalista vaikutusta soluviljelmässä ryhmän O HIV-1- ja HIV-2-isolaatteja vastaan.
Nevirapiini yhdessä efavirentsin kanssa osoitti voimakasta antagonistista anti-HIV-1-vaikutusta in vitro (ks. kohta Yhteisvaikutukset) ja sillä oli additiivisesta antagonistiseen vaikutusta proteaasin estäjä ritonaviirin tai fuusionestäjä enfuvirtidin kanssa. Nevirapiini osoitti additiivisesta synergistiseen anti-HIV-1-vaikutusta yhdessä proteaasin estäjien amprenaviirin, atatsanaviirin, indinaviirin, lopinaviirin, sakinaviirin ja tipranaviirin sekä nukleosidianalogien abakaviirin, didanosiinin, emitrisitabiinin, lamivudiinin, stavudiinin, tenofoviirin ja tsidovudiinin kanssa. Anti-HBV-lääkevalmiste adefoviiri ja anti-HCV-lääkevalmiste ribaviriini antagonisoivat nevirapiinin anti-HIV-1-vaikutusta in vitro.
Resistenssi
HIV-1-isolaatteja, joiden herkkyys nevirapiinille on pienentynyt (100‑250-kertaisesti), tavataan soluviljelmissä. Genotyypin tutkimuksessa todettiin mutaatioita HIV-1 RT geenissä Y181C ja/tai V106A riippuen viruskannasta ja käytetystä solulinjasta. Nevirapiiniresistenssin ilmaantumiseen kuluva aika soluviljelmässä ei muuttunut, kun valikoimassa oli nevirapiini yhdistettynä useiden muiden ei-nukleosidianalogien kanssa.
Genotyypin tutkimus isolaateissa potilailla, jotka eivät olleet aiemmin saaneet antiretroviraalista lääkehoitoa ja saivat nevirapiinia kerran vuorokaudessa (n=25) tai kaksi kertaa vuorokaudessa (n=46) yhdessä lamivudiinin ja stavudiinin kanssa 48 viikon ajan ja joiden viraalinen hoito epäonnistui (n=71), osoitti, että 8/25 ja 23/46 potilaalla oli isolaatti, jossa oli yksi tai useampi seuraavista ei-nukleosisanalogi resistenssiin liittyvistä substituutioista:
Y181C, K101E, G190A/S, K103N, V106A/M, V108I, Y188C/L, A98G, F227L ja M230L.
Ristiresistenssi
Ei-nukleosidianalogeille ristiresistenttien HIV-kantojen nopea ilmeneminen on havaittu in vitro tutkimuksissa. Ristiresistenssi efavirentsille on odotettavissa nevirapiinihoidon virologisen epäonnistumisen jälkeen. Riippuen resistenssitestin tuloksista etraviriinia sisältävää hoitoa voidaan käyttää tämän jälkeen. Ristiresistenssi nevirapiinin ja joko HIV-proteaasin estäjien, HIV-integraasinestäjien tai HIV-fuusionestäjien välillä on epätodennäköistä, koska lääkeaineilla on eri kohde-entsyymit. Samaten mahdollisuudet ristiresistenssiin nevirapiinin ja NRTI:n välillä ovat vähäisiä, koska molekyylit sitoutuvat eri kohtiin käänteiskopioijaentsyymissä.
Nevirapiini-resistentit HIV-1 kliiniset isolaatit olivat ristiresistenttejä NNRTI-lääkkeille delavirdiinille ja efavirentsille. Niistä aminohappojen substituutioista, jotka ilmaantuivat potilailla, jotka eivät olleet aiemmin saaneet lääkehoitoa, ja joita hoidettiin nevirapiinilla, substituutiot Y181C ja M230L ovat yksittäin liitetty vähentyneeseen kliiniseen vasteeseen etraviriinille, ja G190A/S, K101E, A98G on liitetty myös vähentyneeseen kliiniseen vasteeseen etraviriinille.
Kliiniset tulokset
Viramune-valmistetta on tutkittu aikaisemmin hoitamattomilla ja hoidetuilla potilailla.
Tutkimukset potilailla, jotka eivät ole aiemmin saaneet lääkehoitoa
2NN tutkimus
Kaksois- ei-nukleosidirakenteinen tutkimus 2 NN oli satunnaistettu, avoin prospektiivinen monikeskustutkimus, jossa vertailtiin ei-nukleosidianalogeja nevirapiinia, efavirentsia ja molempia lääkevalmisteita yhdessä annettuna.
1216 potilaalle, jotka eivät olleet aiemmin saaneet antiretroviraalista lääkehoitoa ja joiden HIV-1 RNA –pitoisuus plasmassa oli lähtötilanteessa yli 5000 kopiota/ml, annettiin joko Viramune-valmistetta 400 mg kerran vuorokaudessa, Viramune-valmistetta 200 mg kaksi kertaa vuorokaudessa, efavirentsia 600 mg kerran vuorokaudessa tai Viramune-valmistetta (400 mg) ja efavirentsia (800 mg) kerran vuorokaudessa sekä stavudiinia ja lamivudiinia 48 viikon ajan.
Ensisijainen päätetapahtuma oli hoidon epäonnistuminen, joka määritettiin seuraavasti: pienempi kuin 1 log10 lasku HIV-1 RNA-pitoisuudessa plasmassa 12 ensimmäisen viikon aikana tai viikosta 24 eteenpäin kaksi peräkkäistä mittausta, joissa yli 50 kopiota/ml, tai taudin eteneminen.
Mediaani-ikä oli 34 vuotta ja noin 64 % potilaista oli miehiä, mediaani CD4+ -solujen määrä oli 170 ja 190 solua/mm3 potilasryhmissä, jotka saivat Viramune-valmistetta kahdesti vuorokaudessa tai vastaavasti efavirentsia. Hoitoryhmien välillä ei ollut merkittäviä eroja demografisissa tai lähtötilanteen ominaisuuksissa.
Ennalta määritelty ensisijainen tehovertailu tehtiin Viramune-valmisteen kahdesti vuorokaudessa ja efavirentsin hoitoryhmien välillä.
Neviirapiinihoito kahdesti vuorokaudessa ja efavirentsihoito eivät eronnet merkittävästi tehon suhteen (p= 0.091) hoidon epäonnistumisella tai minkään hoidon osatekijän epäonnistumisella, mukaan lukien virologinen epäonnistuminen, mitattuna.
Nevirapiinin (400 mg) + efavirentsin (800 mg) samanaikaiseen käyttöön liittyi eniten kliinisiä haittavaikutuksia ja hoidon epäonnistumisia (53.1 %). Tätä hoitoa ei suositella, koska nevirapiinin ja efavirentsin käyttö yhdessä ei tuonut lisätehoa ja aiheutti enemmän haittavaikutuksia kuin kumpikin lääkevalmiste erikseen käytettynä.
Kahdellakymmenellä prosentilla nevirapiinia saaneista ja 18 %:lla efavirentsiä saaneista potilaista oli ainakin yksi tason 3 tai 4 kliininen haittavaikutus. Kliiniseksi haittavaikutukseksi raportoitu kliininen hepatiitti ilmaantui kymmenelle (2,6 %) nevirapiinia kahdesti vuorokaudessa saaneelle potilaalle ja kahdelle (0,5 %) efavirentsia saaneelle potilaalle. Niiden potilaiden osuus, joilla oli ainakin yksi maksaan liittyvä tason 3 tai 4 toksisuuslöydös laboratoriotestissä oli 8,3 % nevirapiinia kahdesti vuorokaudessa saaneilla ja 4,5 % efavirentsia saaneilla. Näistä potilaista nevirapiinia kahdesti vuorokaudessa saaneista potilaista 6,7 %:lle ilmaantui lisäksi hepatiitti B ja 20,0 %:lle hepatiitti C. Efavirentsia saaneista potilaista hepatiitti B ilmaantui 5,6 %:lle ja hepatiitti C 11,1 %:lle.
2NN kolmen vuoden seurantatutkimus
Tämä on retrospektiivinen monikeskustutkimus, jossa vertaillaan Viramune-valmisteen ja efavirentsin antiviraalista tehoa 3 vuoden aikana yhdessä stavudiinin ja lamivudiinin kanssa 2 NN-potilailla viikosta 49 viikkoon 144. Potilaita, jotka osallistuivat 2NN tutkimukseen ja olivat vielä aktiivisessa seurannassa viikolla 48, kun tutkimus lopetettiin ja joita yhä hoidettiin tutkimusklinikassa, pyydettiin osallistumaan tähän tutkimukseen. Tutkimuksen ensisijaiset päätetapahtumat (niiden potilaiden prosentuaalinen osuus, joilla hoito epäonnistui) ja toissijaiset päätetapahtumat ja myös muu lääkitys olivat samanlaiset kuin alkuperäisessä 2NN-tutkimuksessa.
Tässä tutkimuksessa dokumentoitiin pysyvä vaste Viramune-valmisteelle vähintään 3 vuoden ajaksi ja Viramune-valmisteen 200 mg kahdesti vuorokaudessa ja efavirentsin välillä todettiin samanarvoisuus 10 % rajojen sisällä hoidon epäonnistumisen osalta. Ensisijaisessa (p = 0.92) eikä toissijaisessa päätetapahtumassa ollut tilastollisesti merkittävää eroa efavirentsin ja Viramune-valmisteen 200 mg kahdesti vuorokaudessa välillä.
Aikaisemmin lääkehoitoa saaneilla potilailla tehdyt tutkimukset
NEFA-tutkimus
NEFA-tutkimus on kontrolloitu prospektiivinen satunnaistettu tutkimus, jossa arvioitiin hoitovaihtoehtoja potilaille, joilla viruskuorma oli mittauskynnyksen alapuolella ja jotka vaihtoivat proteaasin estäjään perustuvasta hoidosta joko Viramune-valmisteeseen, efavirentsiin tai abakaviiriin. Tutkimukseen satunnaistettiin 460 aikuista, jotka käyttivät kahta nukleosidianalogia ja ainakin yhtä proteaasin estäjää ja joiden HIV-1 RNA -pitoisuus plasmassa oli ollut alle 200 kopiota/ml ainakin edeltävien kuuden kuukauden ajan ennen proteaasin estäjän vaihtoa Viramune-valmisteeseen (155 potilasta), efavirentsiin (156) tai abakaviiriin (149). Ensisijainen päätetapahtuma oli kuolema, taudin eteneminen AIDS:ksi tai HIV-1 RNA pitoisuuden nousu vähintään yli 200 kopioon/ml.
12 kuukauden kohdalla Kaplan–Meier estimaatit päätetapahtuman saavuttamisen todennäköisyydestä olivat 10 % Viramune-ryhmässä, 6 % efavirentsiryhmässä ja 13 % abakaviiriryhmässä (P=0.10 intention-to-treat -analyysin mukaan).
Haittavaikutusten kokonaisesiintyvyys oli huomattavasti alhaisempaa abakaviiriryhmässä (61 potilasta tai 41 %) verrattuna nevirapiiniryhmään (83 potilasta tai 54 %) tai efavirentsiryhmään (89 potilasta tai 57 %). Huomattavasti pienempi osa abakaviiriryhmän potilaista (9 potilasta tai 6 %) verrattuna nevirapiiniryhmän (26 potilasta tai 17 %) tai efavirentsiryhmän (27 potilasta tai 17 %) potilaisiin keskeytti lääkevalmisteen haittavaikutusten takia
Perinataali-tartunta
Viramune-valmisteen käyttöä suhteessa perinataaliin tartuntaan on selvitetty useissa tutkimuksissa, etenkin HIVNET 012 -tutkimuksessa. Tämä tutkimus osoitti merkitsevän vähennyksen tartunnassa annettaessa nevirapiinia kerta-annoksena Viramune-ryhmässä (13,1 % (n = 310)) ultralyhyeen tsidovudiinihoitoon verrattuna (25,1 % (n=308), p = 0.00063). Viramune-monoterapiaan on liittynyt NNRTI-resistenssin kehittyminen. Jos nevirapiinia annetaan äideille ja vastasyntyneille kerta-annoksena, nevirapiinia sisältävän HIV-hoidon teho saattaa laskea, jos HIV-hoito aloitetaan näillä potilailla myöhemmin ja kerta-annoshoidon saamisesta on kulunut kuusi kuukautta tai vähemmän. Muiden antiretroviraalisten valmisteiden yhdistäminen nevirapiinin kerta-annokseen vaimentaa nevirapiiniresistenssin esilletuloa. Silloin kun muita antiretroviraalisia lääkeineita on saatavilla, kerta-annoksena annettava Viramune pitäisi yhdistää muihin tehokkaisiin antiretroviraalisiin lääkkeisiin, kuten kansainvälisesti tunnustetuissa ohjeissa on suositeltu.
Näiden tulosten kliinistä merkitystä eurooppalaisessa väestössä ei ole osoitettu. Lisäksi jos Viramune-valmistetta annetaan kerta-annoksena HIV-1 infektion vertikaalisen tartunnan estoon, ei maksatoksisuuden riskiä äidille ja lapselle voida sulkea pois.
Pediatriset potilaat
Tulokset eteläafrikkalaisen tutkimuksen BI 1100.1368 48 viikon analyysistä vahvistavat, että 4/7 mg/kg ja 150 mg/m2 nevirapiiniannosryhmät olivat hyvin siedettyjä ja tehokkaita hoidettaessa lapsipotilaita, jotka eivät olleet saaneet aiemmin antiretroviraalista hoitoa. Huomattava paraneminen CD4+ -solujen osuudessa havaittiin viikkoon 48 mennessä molemmissa annosryhmissä. Molemmat annokset alensivat tehokkaasti viruskuormaa. Tässä 48 viikon tutkimuksessa ei kummassakaan ryhmässä todettu odottamattomia turvallisuuteen liittyviä löydöksiä.
Farmakokinetiikka
Viramune tablettien ja oraalisuspension on todettu olevan hyötyosuudeltaan samankaltaisia ja ne ovat keskenään vaihdettavissa, kun annos on enintään 200 mg.
Imeytyminen: Terveillä vapaaehtoisilla ja HIV-1 infektoituneilla aikuisilla nevirapiini imeytyy hyvin (> 90 %) suun kautta annettuna. Kahdellatoista terveellä aikuisella nevirapiinin kerta-annoksen hyötyosuus oli 93 ± 9 % (keskimääräinen keskihajonta) 50 mg tabletilla ja 91 ± 8 % oraaliliuoksella. Käytettäessä 200 mg kerta-annosta nevirapiinin huippupitoisuus plasmassa oli 2 ± 0,4 µg/ml (7,5 mikroM) 4 tunnissa. Toistuvasti annosteltuna nevirapiinin huippupitoisuudet lisääntyivät lineaarisesti annosalueella 200‑400 mg vuorokaudessa. Kirjallisuudessa raportoidut 20 HIV-infektoituneen potilaan tiedot viittaavat siihen, että vakaan tilan huippupitoisuus on 5,74 μg/ml (5,00-7,44), pienin pitoisuus 3,73 μg/ml (3,20 – 5,08) ja AUC 109 h*μg/ml (96,0-143,5) potilailla, jotka saivat nevirapiinia 200 mg kaksi kertaa vuorokaudessa. Muu julkaistu tieto tukee tätä päätelmää. Pitkäaikaisteho on todennäköisin potilailla, joilla nevirapiinin pienimmät pitoisuudet ovat yli 3,5 μg/ml.
Jakautuminen: Nevirapiini on rasvaliukoinen ja fysiologisessa pH:ssa se on pääasiallisesti ionisoitumattomassa muodossa. Käytettäessä laskimonsisäistä annostelua terveillä aikuisilla, nevirapiinin jakaantumistilavuus (Vd) oli 1,2 ± 0,09 l/kg viitaten siihen, että nevirapiini jakaantuu ihmisissä laajasti kudoksiin. Nevirapiini läpäisee helposti istukan ja erittyy äidinmaitoon. Nevirapiinista on sitoutuneena n. 60 % plasman proteiineihin plasmapitoisuuden ollessa 1-10 µg/ml. Nevirapiinin pitoisuudet ihmisellä aivo-selkäydinnesteessä (n = 6) olivat 45 % (± 5 %) plasman pitoisuuksista; tämä suhde on lähes yhtä suuri plasman proteiineihin sitoutumattoman osan kanssa.
Biotransformaatio ja eliminaatio: In vivo tutkimukset ihmisillä ja in vitro tutkimukset ihmisen maksan mikrosomeilla ovat osoittaneet, että nevirapiini metaboloituu pääasiassa sytokromi P450:n kautta useiksi hydroksyloituneiksi metaboliiteiksi. In vitro tutkimukset ihmisen maksan mikrosomeilla osoittavat, että nevirapiinin oksidatiivinen metabolia välittyy pääasiallisesti CYP3A-isoentsyymien kautta, vaikka vähäistä metaboliaa saattaa tapahtua muiden isoentsyymien kautta. Erittymistutkimuksessa vakaassa tilassa annettiin kahdeksalle terveelle miespuoliselle vapaaehtoiselle nevirapiinia 200 mg kahdesti vuorokaudessa ja sen jälkeen yksi yksittäinen 50 mg:n annos radioaktiivisesti merkittyä 14C- nevirapiinia, josta jäljitettiin 91,4 ± 10,5 %. Virtsaan erittyi 81,3 ± 11,1 % ja ulosteisiin 10,1 ± 1,5 %. Enemmän kuin 80 % virtsan radioaktiivisuudesta oli peräisin hydroksyloituneiden metaboliittien glukuronidikonjugaateista. Näin sytokromi P450 metabolia, glukuronidikonjugaatio ja glukuronidimetaboliittien erittyminen virtsaan osoittautuvat nevirapiinin metaboloitumisen ja eliminaation ensisijaiseksi reitiksi ihmisillä. Vain pieni osa (< 5 %) virtsan radioaktiivisuudesta oli kanta-ainetta (< 3 % kokonaisannoksesta), joten erittymisellä muuttumattomana virtsaan on vain vähäinen merkitys nevirapiinin eliminaatiossa.
Nevirapiini indusoi maksan sytokromi P450 järjestelmän entsyymejä. Kun siirrytään kerta-annoksesta annostukseen 200–400 mg/päivä kahden–neljän viikon ajaksi, suun kautta otetun nevirapiinin puhdistuma kasvaa n. 1,5–2-kertaiseksi oman metabolian induktion vuoksi. Induktiosta seuraa myös, että nevirapiinin terminaalifaasin puoliintumisaika plasmassa pienenee n. 45 tunnista (yksittäinen annos) n. 25–30 tuntiin käytettäessä jatkuvaa 200–400 mg vuorokausiannosta.
Munuaisten toimintahäiriö: Nevirapiinin kerta-annoksen kinetiikkaa on verrattu 23 potilaalla, joilla oli joko lievä (glomerulussuodosnopeus ≥ 50 ml/min, mutta alle 80 ml/min), kohtalainen (glomerulussuodosnopeus ≥ 30 ml/min, mutta alle 50 ml/min) tai vaikea (glomerulussuodosnopeus alle 30 ml/min) munuaisten toimintahäiriö, munuaisten vajaatoiminta tai dialyysihoitoa vaativa loppuvaiheen munuaissairaus, sekä 8 potilaalla, joilla munuaisten toiminta oli normaali (glomerulussuodosnopeus yli 80 ml/min). Munuaisten toimintahäiriö (lievä, kohtalainen tai vaikea) ei aiheuttanut merkittäviä muutoksia nevirapiinin farmakokinetiikassa. Loppuvaiheen munuaissairautta sairastavilla dialyysihoitoa saavilla potilailla havaittiin kuitenkin 43,5 %:n pieneneminen nevirapiinin AUC:ssa viikon kestävän altistuksen aikana. Nevirapiinin hydroksimetaboliittien kumuloitumista plasmaan havaittiin myös. Tulokset viittaavat siihen, että nevirapiinihoidon täydentäminen 200 mg:n Viramune-lisäannoksella aina dialyysihoidon jälkeen auttaisi kompensoimaan dialyysin vaikutuksia nevirapiinin puhdistumaan. Muutoin potilaiden, joiden glomerulussuodosnopeus on ≥ 20 ml/min, nevirapiiniannosta ei tarvitse säätää.
Maksan toimintahäiriö: Tehtiin vakaan tilan tutkimus, jossa verrattiin 46 potilasta, joilla oli
lievä (n=17: Ishak-pisteet 1-2),
kohtalainen (n=20; Ishak-pisteet 3-4),
tai vaikea (n=9; Ishak-pisteet 5-6, Child–Pugh luokka A 8 potilaalla, yhdellä potilaalla Child–Pugh pisteet eivät sovellettavissa)
maksafibroosi, jota käytettiin maksan toimintahäiriön mittarina.
Tutkitut potilaat saivat 200 mg Viramune-valmistetta sisältävää antiretroviraalista hoitoa kahdesti vuorokaudessa vähintään 6 viikkoa ennen farmakokineettistä näytteenottoa. Hoidon mediaanikesto oli 3,4 vuotta. Tässä tutkimuksessa ei nevirapiinin eikä sen viiden oksidatiivisen metaboliitin moniannostelun farmakokineettinen luonne muuttunut.
Kuitenkin noin 15 prosentilla potilaista, joilla oli maksafibroosi, nevirapiinin matalimmat (trough) pitoisuudet olivat yli 9 000 ng/ml (kaksinkertaiset verrattuna tavalliseen keskimääräiseen matalimpaan pitoisuuteen). Potilaita, joilla on maksan toimintahäiriö, pitäisi tarkkailla huolellisesti, jotta lääkevalmisteen aiheuttama toksisuus havaitaan.
200 mg nevirapiinikerta-annoksen farmakokineettisessä tutkimuksessa HIV-negatiivisilla potilailla, joilla oli lievä tai kohtalainen maksan toimintahäiriö (Child–Pugh luokka A, n=6; Child–Pugh luokka B, n=4), nevirapiinin AUC:n merkittävää nousua havaittiin yhdellä Child–Pugh luokan B potilaalla, jolla oli askites. Tämä viittaa siihen, että potilailla, joilla on heikentyvä maksan toiminta ja askites, saattaa olla riski nevirapiinin kertymiselle systeemiseen verenkiertoon. Koska nevirapiini indusoi omaa metaboliaansa moniannostelussa, tämä kerta-annostutkimus ei välttämättä kuvasta maksan toimintahäiriön vaikutusta moniannostelun farmakokinetiikkaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sukupuoli ja iäkkäät
Monikansallisessa 2NN tutkimuksessa tehtiin 1077 potilaan farmakokineettinen populaatio-alatutkimus, jossa 391 potilaista oli naisia. Naispuolisilla potilailla nevirapiinin puhdistuma oli 13,8 % pienempi kuin miespotilailla. Tätä eroa ei pidetä kliinisesti merkitsevänä. Koska paino tai painoindeksi ei vaikuttanut nevirapiinin puhdistumaan, sukupuolen vaikutusta ei voi selittää kehon koolla. Iällä (19‑68 vuotta) tai syntyperällä (mustat, eteläamerikkalaiset ja valkoihoiset) ei ole vaikutusta nevirapiinin farmakokinetiikkaan HIV-1 infektoituneilla aikuisilla. Nevirapiinia ei ole erityisesti tutkittu yli 65-vuotiailla potilailla.
Pediatriset potilaat
Nevirapiinin farmakokinetiikkaa koskevia tietoja on saatu kahdesta päälähteestä: 48 viikon tutkimuksesta lapsipotilailla Etelä-Afrikassa (BI 1100.1368), jossa oli mukana 123 HIV-1-positiivista potilasta, jotka eivät olleet saaneet aiemmin antiretroviraalista hoitoa ja jotka olivat iältään 3 kuukaudesta 16 vuoteen, ja yhdistetystä analyysistä viidestä PACTG (Paediatric AIDS Clinical Trials Group) -protokollasta, joka käsitti 495 potilasta iältään 14 päivää – 19 vuotta.
Tiheän näytteenoton ryhmässä 33 potilaan (ikä 0,77 – 13,7 vuotta) farmakokineettiset tiedot osoittivat, että nevirapiinin puhdistuma lisääntyy iän myötä tavalla, joka on suhteessa kehon pinta-alan kasvuun. Annettaessa nevirapiinia 150 mg/m2 kahdesti vuorokaudessa (kahden viikon aloitusjakson 150 mg/m2 kerran vuorokaudessa jälkeen) saatiin nevirapiinin jäännöspitoisuuden geometriseksi keskiarvoksi tai keskiarvoksi 4-6 μg/ml (johon pyrittiin aikuisista saatujen tietojen perusteella). Lisäksi havaitut nevirapiinin jäännöspitoisuudet olivat keskenään vertailukelpoisia molemmilla tavoilla mitattuna.
Yhdistetty analyysi PACTG (Paediatric AIDS Clinical Trials Group) -protokollista 245, 356, 366, 377 ja 403 mahdollistivat näissä tutkimuksissa olleiden alle 3 kuukauden ikäisten lapsipotilaiden (n=17) arvioinnin. Havaitut plasman nevirapiinipitoisuudet olivat aikuisilla ja muilla lapsipotilailla havaittujen pitoisuuksien vaihtelualueella, mutta vaihtelu potilaiden välillä oli suurempaa, etenkin toisen ikäkuukauden aikana.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta ja genotoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille, lukuunottamatta kliinisissä tutkimuksissa havaittuja seikkoja. Karsinogeenisuustutkimuksissa nevirapiini aiheutti maksan kasvaimia rotilla ja hiirillä. Nämä löydökset johtuvat erittäin todennäköisesti nevirapiinin voimakkaasta maksaentsyymejä indusoivasta vaikutuksesta eikä sen genotoksisuudesta.
Farmaseuttiset tiedot
Apuaineet
Karbomeeri
Metyyliparahydroksibentsoaatti (E218)
Propyyliparahydroksibentsoaatti (E216)
Sorbitoli
Sakkaroosi
Polysorbaatti 80
Natriumhydroksidi (pH:n säätöön)
Puhdistettu vesi
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Lääkevalmiste pitää käyttää 6 kuukauden kuluessa pullon avaamisesta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VIRAMUNE oraalisuspensio
50 mg/5 ml (L:ei) 240 ml (124,01 €)
PF-selosteen tieto
Valkoinen korkeapainepolyetyleeni (HDPE) -pullo, jossa on kaksiosainen turvakorkki (ulkopinta valkoista polyetyleeniä, sisäpinta polypropyleeniä) ja valkoinen polyetyleenitiiviste. Jokaisessa pullossa on 240 ml oraalisuspensiota.
Valmisteen kuvaus:
Valkoinen tai melkein valkoinen homogeeninen suspensio.
Käyttö- ja käsittelyohjeet
Käyttöohjeet:
Viramune-oraalisuspensiota on ravistettava varovasti ennen antamista. Tarvittavat annosmäärät pitää mitata käyttämällä annosruiskua. Viramune oraalisuspensio pitää käyttää 6 kuukauden kuluessa pullon avaamisesta.
Hävittäminen:
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VIRAMUNE oraalisuspensio
50 mg/5 ml 240 ml
- Ei korvausta.
ATC-koodi
J05AG01
Valmisteyhteenvedon muuttamispäivämäärä
15.10.2024
Yhteystiedot
 BOEHRINGER INGELHEIM FINLAND KY
BOEHRINGER INGELHEIM FINLAND KY Tammasaarenkatu 5
00180 Helsinki
010 310 2800
www.boehringer-ingelheim.fi
medinfo.finland@boehringer-ingelheim.com