

ZIEXTENZO injektioneste, liuos, esitäytetty ruisku 6 mg
Vaikuttavat aineet ja niiden määrät
Yksi esitäytetty ruisku sisältää 6 mg pegfilgrastiimia* (pegfilgrastimum) 0,6 ml:ssa injektionestettä. Pelkkään proteiiniin perustuva pitoisuus on 10 mg/ml.**
*Tuotettu Escherichia coli -soluissa yhdistelmä-DNA-tekniikalla ja konjugoitu sen jälkeen polyetyleeniglykoliin (PEG).
**Pitoisuus on 20 mg/ml, jos PEG-osa lasketaan mukaan.
Tämän valmisteen voimakkuutta ei pidä verrata minkään muun samaan lääkeaineryhmään kuuluvan pegyloidun tai pegyloimattoman proteiinin voimakkuuteen. Lisätietoja, ks. kohta Farmakodynamiikka.
Apuaineet, joiden vaikutus tunnetaan
Yksi esitäytetty ruisku sisältää 30 mg sorbitolia (E 420).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste).
Kliiniset tiedot
Käyttöaiheet
Neutropenian keston lyhentäminen ja kuumeisen neutropenian esiintymistiheyden vähentäminen aikuisilla potilailla, jotka saavat solunsalpaajia syövän hoitoon (lukuun ottamatta kroonista myelooista leukemiaa ja myelodysplastisia oireyhtymiä).
Ehto
Hoidon aloittavat onkologiaan ja/tai hematologiaan perehtyneet lääkärit ja hoito toteutetaan heidän valvonnassaan.
Annostus ja antotapa
Suositellaan, että Ziextenzo-hoidon aloittavat onkologiaan ja/tai hematologiaan perehtyneet lääkärit ja hoito toteutetaan heidän valvonnassaan.
Annostus
Ziextenzon suositeltu annostus on 6 mg (yksi esitäytetty ruisku) kutakin solunsalpaajasykliä kohti vähintään 24 tuntia solunsalpaajalääkityksen jälkeen.
Erityiset potilasryhmät
Pediatriset potilaat
Pegfilgrastiimin turvallisuutta ja tehoa lasten hoidossa ei ole vielä varmistettu. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta.
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa munuaisten vajaatoimintaa sairastaville potilaille, ei myöskään potilaille, joilla on loppuvaiheen munuaissairaus.
Antotapa
Ziextenzo annetaan ihon alle.
Injektiot annetaan reiteen, vatsaan tai olkavarteen. Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen käsittelystä ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yleiset varoitukset ja varotoimet
Rajalliset kliiniset tutkimustulokset viittaavat siihen, että pegfilgrastiimilla on vastaavanlainen vaikutus potilaiden toipumisaikaan vaikeasta neutropeniasta kuin filgrastiimilla de novo akuutissa myelooisessa leukemiassa (AML) (ks. kohta Farmakodynamiikka). Pegfilgrastiimin pitkäaikaisvaikutuksia ei kuitenkaan ole osoitettu AML:n hoidossa, joten sen käytössä on noudatettava varovaisuutta tässä potilasryhmässä.
Granulosyyttikasvutekijä (G‑CSF) voi edistää myeloidisten solujen kasvua in vitro, ja samankaltaisia vaikutuksia saattaa esiintyä myös joissakin ei-myeloidisissa soluissa in vitro.
Pegfilgrastiimin tehoa ja turvallisuutta ei ole tutkittu myelodysplastista oireyhtymää, kroonista myelogeenista leukemiaa eikä sekundaarista AML:ää sairastavien potilaiden hoidossa, eikä sitä pitäisi antaa näille potilaille. Erityistä huomiota on kiinnitettävä kroonisen myelooisen leukemian blastitransformaation erottamiseen AML:stä.
Pegfilgrastiimin tehoa ja turvallisuutta ei ole osoitettu alle 55-vuotiaiden potilaiden hoidossa de novo akuutissa myelooisessa leukemiassa, johon liittyy sytogenetiikka t(15;17).
Pegfilgrastiimin tehoa ja turvallisuutta ei ole tutkittu suuriannoksista solunsalpaajahoitoa saavilla potilailla. Tätä lääkevalmistetta ei saa käyttää solunsalpaajien vakiintuneiden annossuositusten ylittämiseen.
Keuhkoihin liittyvät haittatapahtumat
Granulosyyttikasvutekijöiden (G‑CSF) antamisen jälkeen on raportoitu keuhkoihin kohdistuneita haittavaikutuksia, erityisesti interstitiaalista pneumoniaa. Näiden vaikutusten vaara saattaa olla suurempi potilailla, joilla on esiintynyt hiljattain keuhkoinfiltraatteja tai keuhkokuume (ks. kohta Haittavaikutukset).
Keuhko-oireiden, kuten yskän, kuumeen ja hengenahdistuksen, ilmaantuminen samanaikaisesti radiologisten infiltraattien kanssa sekä keuhkofunktioiden heikkeneminen neutrofiilien määrän samalla lisääntyessä saattavat olla äkillisen hengitysvajausoireyhtymän (ARDS) esioireita. Tällaisessa tilanteessa pegfilgrastiimilääkitys tulisi keskeyttää lääkärin harkinnan mukaan ja antaa asianmukaista hoitoa (ks. kohta Haittavaikutukset).
Munuaiskerästulehdus (glomerulonefriitti)
Filgrastiimia ja pegfilgrastiimia saavilla potilailla on raportoitu munuaiskerästulehdusta. Munuaiskerästulehdus parani yleensä filgrastiimi- tai pegfilgrastiimiannoksen pienentämisen tai hoidon lopettamisen jälkeen. Virtsatutkimuksia suositellaan tehtäväksi säännöllisin välein.
Kapillaarivuoto-oireyhtymä
Kapillaarivuoto-oireyhtymää on raportoitu granulosyyttikasvutekijöiden antamisen jälkeen. Sen tyypillisiä oireita ovat hypotensio, hypoalbuminemia, turvotus ja hemokonsentraatio. Jos potilaalle kehittyy kapillaarivuoto-oireyhtymän oireita, hänen tilaansa on seurattava tarkoin ja annettava oireenmukaista hoitoa, tarvittaessa myös tehohoitoa (ks. kohta Haittavaikutukset).
Splenomegalia ja pernan repeämä
Pegfilgrastiimin antamisen jälkeen on esiintynyt splenomegaliaa, joka on kuitenkin yleensä ollut oireetonta, ja pernan repeämiä, jotka ovat joissakin tapauksissa johtaneet kuolemaan (ks. kohta Haittavaikutukset). Pernan kokoa on sen vuoksi seurattava tarkoin (esim. tunnustelu, ultraäänitutkimus). Pernan repeämän mahdollisuus on otettava huomioon, jos potilaalla esiintyy kipua vasemmalla ylävatsassa tai olkapään kärjessä.
Trombosytopenia ja anemia
Pegfilgrastiimihoito yksinään ei estä trombosytopeniaa ja anemiaa, koska luuydintä lamaavaa solunsalpaajahoitoa jatketaan täysin annoksin hoito-ohjelman mukaisin välein. Trombosyytti- ja hematokriittiarvoja on seurattava säännöllisin välein. Erityistä varovaisuutta on noudatettava käytettäessä yksittäisiä solunsalpaajia tai solunsalpaajien yhdistelmiä, joiden tiedetään aiheuttavan vaikeaa trombosytopeniaa.
Myelodysplastinen oireyhtymä ja akuutti myelooinen leukemia rinta- ja keuhkosyöpäpotilailla
Markkinoille tulon jälkeisessä havainnoivassa tutkimuksessa pegfilgrastiimi annettuna samanaikaisesti solunsalpaajien ja/tai sädehoidon kanssa on yhdistetty myelodysplastisen oireyhtymän (MDS) ja akuutin myelooisen leukemian (AML) kehittymiseen rinta- ja keuhkosyöpäpotilailla (katso kohta Haittavaikutukset). Rinta- ja keuhkosyöpäpotilaita on seurattava myelodysplastisen oireyhtymän ja akuutin myelooisen leukemian merkkien ja oireiden varalta.
Sirppisoluanemia
Sirppisolukriisejä on esiintynyt pegfilgrastiimin käytön aikana potilailla, joilla on sirppisolupoikkeavuus tai sirppisolutauti (ks. kohta Haittavaikutukset). Varovaisuutta on noudatettava määrättäessä pegfilgrastiimia potilaille, joilla on sirppisolupoikkeavuus tai sirppisolutauti, ja asianmukaisia kliinisiä parametrejä ja laboratorioarvoja on seurattava tarkoin ja tarkkailtava erityisesti tämän lääkkeen mahdollista yhteyttä pernan suurentumiseen ja vaso-okklusiiviseen kriisiin.
Leukosytoosi
Alle 1 %:lla pegfilgrastiimihoitoa saaneista potilaista on havaittu valkosoluarvoja, jotka ovat 100 × 109/l tai suurempia. Tämänasteiseen leukosytoosiin suoranaisesti liittyviä haittatapahtumia ei ole raportoitu. Tällainen valkosoluarvon nousu on ohimenevä, se todetaan yleensä 24–48 tunnin kuluttua lääkkeen antamisesta ja se on tämän lääkkeen farmakodynaamisten vaikutusten mukainen. Kliinisten vaikutusten ja mahdollisen leukosytoosin vuoksi valkosoluarvoa on seurattava säännöllisin välein hoidon aikana. Jos valkosoluarvo ylittää tason 50 × 109/l sen jälkeen, kun odotettu pohjalukema on saavutettu, tämän lääkkeen käyttö on lopetettava heti.
Yliherkkyys
Pegfilgrastiimia saavilla potilailla on raportoitu yliherkkyysoireita, myös anafylaktisia reaktioita, ensimmäisen tai myöhempien hoitojaksojen yhteydessä. Pegfilgrastiimihoito on lopetettava pysyvästi, jos potilaalla havaitaan kliinisesti merkittävää yliherkkyyttä. Pegfilgrastiimia ei saa antaa potilaille, joilla on aikaisemmin esiintynyt pegfilgrastiimi- tai filgrastiimiyliherkkyyttä. Mahdolliset vakavat allergiset reaktiot on hoidettava asianmukaisesti, ja potilaan tilaa on seurattava tarkoin useiden vuorokausien ajan.
Stevens–Johnsonin oireyhtymä
Stevens–Johnsonin oireyhtymää, joka voi olla hengenvaarallinen tai johtaa kuolemaan, on raportoitu pegfilgrastiimihoidon yhteydessä harvoin. Jos potilaalle on kehittynyt Stevens–Johnsonin oireyhtymä pegfilgrastiimin käytön yhteydessä, potilaalle ei saa enää koskaan antaa pegfilgrastiimihoitoa.
Immunogeenisuus
Immunogeenisuuden mahdollisuus on olemassa, kuten kaikkia proteiinilääkkeitä käytettäessä. Pegfilgrastiimin vasta-aineiden muodostuminen on yleensä vähäistä. Sitoutuvia vasta-aineita esiintyy, kuten on odotettavissa kaikkia biologisia lääkkeitä käytettäessä, mutta toistaiseksi niillä ei ole havaittu olevan neutraloivaa vaikutusta.
Aortiitti
Aortiittia on raportoitu granulosyyttikasvutekijöiden (G-CSF) antamisen jälkeen terveillä henkilöillä ja syöpäpotilailla. Oireita ovat olleet muun muassa kuume, vatsakipu, huonovointisuus, selkäkipu ja tulehdusmarkkereiden kohoaminen (esim. C-reaktiivisen proteiinin ja valkoisten verisolujen arvot). Aortiitti diagnosoitiin useimmissa tapauksissa CT-kuvauksella, ja se parani yleensä, kun G-CSF:n antaminen lopetettiin. Katso myös kohta Haittavaikutukset.
Muut varoitukset
Pegfilgrastiimihoidon tehoa ja turvallisuutta veren kantasolujen mobilisaatiossa ei ole tutkittu riittävästi potilailla eikä terveillä luovuttajilla.
Kasvutekijähoidosta aiheutuvaan luuytimen hematopoieettisen aktiivisuuden lisääntymiseen on liittynyt ohimeneviä positiivisia löydöksiä luuston kuvantamistutkimuksissa. Tämä on otettava huomioon luuston kuvantamistuloksia tulkittaessa.
Apuaineet
Tämä lääkevalmiste sisältää 30 mg sorbitolia per esitäytetty ruisku, joka vastaa 50 mg/ml. Sorbitolia (tai fruktoosia) sisältävien muiden valmisteiden samanaikaisen annon sekä ravinnosta saatavan sorbitolin (tai fruktoosin) additiivinen vaikutus on huomioitava.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per 6 mg:n annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Koska nopeasti jakautuvat myeloidiset solut saattavat olla herkkiä solunsalpaajille, pegfilgrastiimi tulisi antaa vähintään 24 tuntia solunsalpaajien jälkeen. Kliinisissä tutkimuksissa pegfilgrastiimia on annettu turvallisesti 14 päivää ennen solunsalpaajalääkitystä. Pegfilgrastiimin samanaikaista käyttöä minkään solunsalpaajan kanssa ei ole tutkittu potilaiden hoidossa. Eläinkoemalleissa pegfilgrastiimin ja 5‑fluorourasiilin (5‑FU) tai muiden antimetaboliittien samanaikaisen käytön on todettu voimistavan luuydinlamaa.
Kliinisissä tutkimuksissa ei ole tutkittu erityisesti mahdollisia yhteisvaikutuksia muiden hematopoieettisten kasvutekijöiden ja sytokiinien kanssa.
Yhteisvaikutuksen mahdollisuutta litiumin kanssa, joka myös edistää neutrofiilien vapautumista, ei ole erityisesti tutkittu. Viitteitä tällaisen yhteisvaikutuksen haitallisuudesta ei ole saatu.
Pegfilgrastiimin tehoa ja turvallisuutta ei ole tutkittu potilailla, jotka saavat nitrosoureoita tai muita viivästynyttä luuydinlamaa aiheuttavia solunsalpaajia.
Erityisiä interaktio- tai metaboliatutkimuksia ei ole tehty, mutta kliinisissä tutkimuksissa pegfilgrastiimilla ei ole havaittu yhteisvaikutuksia minkään muun lääkevalmisteen kanssa.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja pegfilgrastiimin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Pegfilgrastiimin käyttöä ei suositella raskauden aikana eikä sellaisten naisten hoitoon, jotka voivat tulla raskaaksi ja jotka eivät käytä ehkäisyä.
Imetys
Ei ole riittävästi tietoa pegfilgrastiimin/metaboliittien erittymisestä ihmisen rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea. On päätettävä lopetetaanko rintaruokinta vai lopetetaanko pegfilgrastiimihoito ottaen huomioon rintaruokinnasta aiheutuvat hyödyt lapselle ja hoidosta koituvat hyödyt äidille.
Hedelmällisyys
Pegfilgrastiimi ei vaikuttanut uros- eikä naarasrottien lisääntymistoimintoihin eikä hedelmällisyyteen kerran viikossa annettuina kumulatiivisina annoksina, jotka olivat noin 6–9 kertaa suurempia kuin ihmisille suositeltu annos (kehon pinta-alan perusteella) (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Pegfilgrastiimilla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Tiivistelmä turvallisuustiedoista
Useimmin raportoidut haittavaikutukset olivat luukipu (hyvin yleinen [≥ 1/10]) ja lihas- ja luustokipu (yleinen [≥ 1/100, < 1/10]). Luukipu oli yleensä lievää tai kohtalaista ja ohimenevää, ja se saatiin useimmiten hallintaan tavallisilla kipulääkkeillä.
Yliherkkyysreaktion tyyppisiä oireita, kuten ihottumaa, nokkosihottumaa, angioedeemaa, hengenahdistusta, ihon punoitusta, kasvojen ja kaulan punoitusta ja hypotensiota, on esiintynyt ensimmäisellä tai myöhemmillä hoitokerroilla pegfilgrastiimin yhteydessä (melko harvinainen [≥ 1/1 000, < 1/100]). Pegfilgrastiimihoitoa saavilla potilailla voi esiintyä vakavia allergisia reaktioita, myös anafylaksiaa (melko harvinainen) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Solunsalpaajahoidossa olevilla syöpäpotilailla on raportoitu granulosyyttikasvutekijöiden antamisen jälkeen melko harvoin (≥ 1/ 1 000, < 1/100) kapillaarivuoto-oireyhtymää, joka voi olla hengenvaarallinen, jos hoito viivästyy, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja jäljempänä oleva kappale Tärkeimpien haittavaikutusten kuvaus.
Splenomegaliaa, joka on yleensä oireetonta, esiintyy melko harvoin.
Pegfilgrastiimin antamisen jälkeen on raportoitu melko harvoin pernan repeämiä, jotka ovat joissakin tapauksissa johtaneet kuolemaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Keuhkoihin kohdistuneita haittavaikutuksia, kuten interstitiaalista pneumoniaa, keuhkoedeemaa, keuhkoinfiltraatteja ja keuhkofibroosia, on raportoitu melko harvoin. Nämä ovat johtaneet melko harvoin hengitysvajaukseen tai äkilliseen hengitysvajausoireyhtymään (ARDS), jotka voivat johtaa kuolemaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Yksittäisiä sirppisolukriisejä on raportoitu potilailla, joilla on sirppisolupoikkeavuus tai sirppisolutauti (melko harvinainen sirppisolupotilailla) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Alla olevan taulukon tiedot perustuvat kliinisissä tutkimuksissa raportoituihin haittavaikutuksiin ja spontaaneihin haittavaikutusilmoituksiin. Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1. Haittavaikutukset
| Elinjärjestelmä (MedDRA) | Haittavaikutukset | |||
Hyvin yleinen (≥ 1/10) | Yleinen (≥ 1/100, < 1/10) | Melko harvinainen (≥ 1/1 000, < 1/100) | Harvinainen (≥ 1/10 000, < 1/1 000) | |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Myelodysplastinen oireyhtymä1 Akuutti myelooinen leukemia1 | |||
| Veri ja imukudos | Trombosytopenia1 Leukosytoosi1 | Sirppisoluanemiaan liittyvä kriisi2 Splenomegalia2 Pernan repeämä2 | ||
| Immuunijärjestelmä | Yliherkkyysreaktiot Anafylaksia | |||
| Aineenvaihdunta ja ravitsemus | Virtsahappoarvon kohoaminen | |||
| Hermosto | Päänsärky1 | |||
| Verisuonisto | Kapillaarivuotooireyhtymä1 | Aorttatulehdus | ||
| Hengityselimet, rintakehä ja välikarsina | Äkillinen hengitysvajausoireyhtymä2 Keuhkoihin kohdistuvat haittavaikutukset (interstitiaalinen pneumonia, keuhkoedeema, keuhkoinfiltraatit ja keuhkofibroosi) Hemoptyysi | Keuhkoverenvuoto | ||
| Ruoansulatuselimistö | Pahoinvointi1 | |||
| Iho ja ihonalainen kudos | Sweetin oireyhtymä (akuutti kuumeinen neutrofiilinen dermatoosi)1,2 Ihon vaskuliitti1,2 | Stevens–Johnsonin oireyhtymä | ||
| Luusto, lihakset ja sidekudos | Luukipu | Lihas- ja luustokipu (lihaskipu, nivelkipu, raajakipu, selkäkipu, lihas- ja luustokipu, niskakipu) | ||
| Munuaiset ja virtsatiet | Munuaiskerästulehdus2 | |||
| Yleisoireet ja antopaikassa todettavat haitat | Injektiokohdan kipu1 Muu kuin sydänperäinen rintakipu | Injektiokohdan reaktiot2 | ||
| Tutkimukset | Laktaatti-dehydrogenaasiarvon ja alkalisen fosfataasiarvon kohoaminen1 ALAT- tai ASAT-arvon ohimenevä kohoaminen maksan toimintakokeissa1 | |||
1 Ks. jäljempänä oleva kappale Tärkeimpien haittavaikutusten kuvaus.
2 Tämä haittavaikutus todettiin markkinoille tulon jälkeisessä seurannassa, mutta sitä ei havaittu aikuispotilaiden satunnaistetuissa kliinisissä vertailututkimuksissa. Yleisyysluokitus perustui tilastolliseen laskelmaan, jossa olivat mukana yhdeksässä satunnaistetussa kliinisessä tutkimuksessa pegfilgrastiimia saaneiden 1 576 potilaan tiedot.
Tärkeimpien haittavaikutusten kuvaus
Sweetin oireyhtymää on raportoitu melko harvoin, ja joissakin tapauksissa taustalla olevat pahanlaatuiset verisairaudet ovat voineet vaikuttaa sen kehittymiseen.
Pegfilgrastiimihoitoa saaneilla potilailla on raportoitu melko harvoin ihon vaskuliittia. Vaskuliitin syntymekanismia näillä potilailla ei tunneta.
Injektiokohdan reaktioita, kuten injektiokohdan punoitusta (melko harvinainen) sekä injektiokohdan kipua (yleinen), on esiintynyt ensimmäisen tai myöhempien pegfilgrastiimihoitokertojen yhteydessä.
Leukosytoosia (valkosoluarvo > 100 × 109/l) on raportoitu yleisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Korjautuvaa lievää tai kohtalaista virtsahappoarvon ja alkalisen fosfataasiarvon nousua esiintyi melko harvoin ja korjautuvaa lievää tai kohtalaista laktaattidehydrogenaasiarvon nousua melko harvoin, kun pegfilgrastiimihoitoa annettiin solunsalpaajalääkityksen jälkeen. Arvojen kohoamiseen ei liittynyt kliinisiä oireita.
Pahoinvointia ja päänsärkyä esiintyi hyvin yleisesti solunsalpaajahoitoa saaneilla potilailla.
Maksan toimintakokeissa on todettu melko harvoin kohonneita alaniiniaminotransferaasiarvoja (ALAT) tai aspartaattiaminotransferaasiarvoja (ASAT), kun potilaat ovat saaneet pegfilgrastiimia solunsalpaajahoidon jälkeen. Nämä ovat ohimeneviä muutoksia, ja arvot palautuvat lähtötasolle.
Rinta- ja keuhkosyöpäpotilailla tehdyssä epidemiologisessa tutkimuksessa on havaittu lisääntynyt myelodysplastisen oireyhtymän ja akuutin myelooisen leukemian riski Ziextenzo-hoidon ja samanaikaisesti annettujen solunsalpaajien ja/tai sädehoidon jälkeen (katso kohta Varoitukset ja käyttöön liittyvät varotoimet).
Trombosytopeniaa on raportoitu yleisesti.
Lääkkeen markkinoille tulon jälkeen granulosyyttikasvutekijöiden käytön yhteydessä on raportoitu kapillaarivuoto-oireyhtymää. Sitä on esiintynyt yleensä potilailla, joilla on pitkälle edennyt pahanlaatuinen sairaus tai sepsis tai jotka saavat useita solunsalpaajia tai joille on tehty afereesi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Lääkkeen käytöstä lapsipotilaiden hoidossa on vain vähän kokemuksia. Vakavia haittavaikutuksia on todettu useammin 0–5‑vuotiailla nuoremmilla lapsilla (92 %) kuin 6–11‑vuotiailla (80 %) ja 12–21‑vuotiailla (67 %) vanhemmilla lapsilla ja aikuisilla. Yleisin raportoitu haittavaikutus oli luukipu (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Vakavia haittavaikutuksia ei havaittu, kun muutamille terveille tutkittaville ja ei-pienisoluista keuhkosyöpää sairastaville potilaille annettiin ihonalaisina kerta-annoksina 300 mikrog/kg. Haittatapahtumat olivat samanlaisia kuin pienempiä pegfilgrastiimiannoksia saaneilla potilailla.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: immunostimulantit, kasvutekijät; ATC-koodi: L03AA13
Ziextenzo on ns. biosimilaari lääkevalmiste. Yksityiskohtaisempaa tietoa on saatavilla Euroopan lääkeviraston verkkosivulta: http://www.ema.europa.eu.
Ihmisen granulosyyttiryhmiä stimuloiva kasvutekijä (G‑CSF) on glykoproteiini, joka säätelee neutrofiilien muodostumista ja vapautumista luuytimestä. Pegfilgrastiimissa yhdistelmä-DNA-tekniikalla tuotettu ihmisen G‑CSF (r‑metHuG‑CSF) on kovalenttisesti konjugoitunut yhteen 20 kilodaltonin kokoiseen polyetyleeniglykolimolekyyliin (PEG). Pegfilgrastiimi on filgrastiimin pitkävaikutteinen muoto, jonka pitempi vaikutuksen kesto perustuu vähäisempään munuaispuhdistumaan.
Pegfilgrastiimilla ja filgrastiimilla on todettu olevan samanlainen vaikutusmekanismi, joka suurentaa huomattavasti perifeerisen veren neutrofiilien määrää 24 tunnin kuluessa ja vain vähän monosyyttien ja/tai lymfosyyttien määrää. Kuten filgrastiimin myös pegfilgrastiimin avulla muodostuneet neutrofiilit toimivat normaalisti tai normaalia tehokkaammin, mikä on osoitettu kemotaksista ja fagosytoosia mittaavilla testeillä. G‑CSF:llä, kuten muillakin hematopoieettisilla kasvutekijöillä, on todettu olevan ihmisen endoteelisoluja stimuloivia ominaisuuksia in vitro. G‑CSF voi edistää myeloidisten, myös pahanlaatuisten, solujen kasvua in vitro, ja samankaltaisia vaikutuksia saattaa esiintyä myös joissakin ei‑myeloidisissa soluissa in vitro.
Kahdessa keskeisessä satunnaistetussa kaksoissokkotutkimuksessa, jossa suuren riskin II‑IV asteen rintasyöpää sairastavat potilaat saivat doksorubisiinia ja dosetakselia sisältävää luuydintä lamaavaa solunsalpaajahoitoa, yksi pegfilgrastiimiannos solunsalpaajasykliä kohti lyhensi neutropenian kestoa ja vähensi kuumeisen neutropenian esiintymistä samassa määrin kuin todettiin annettaessa filgrastiimia päivittäin (kerran päivässä annettujen annosten lukumäärä 11 (mediaani)). Ilman kasvutekijätukea tämän hoito-ohjelman yhteydessä raportoidun 4. asteen neutropenian kesto (keskiarvo) on ollut 5–7 päivää ja kuumeisen neutropenian ilmaantuvuus 30‑40 %. Tutkimuksessa (n = 157), jossa käytettiin pegfilgrastiimia 6 mg:n vakioannoksena, 4. asteen neutropenian kesto (keskiarvo) oli pegfilgrastiimiryhmässä 1,8 vuorokautta ja filgrastiimiryhmässä 1,6 vuorokautta (ero 0,23 vuorokautta, 95 %:n luottamusväli ‑0,15, 0,63). Kuumeista neutropeniaa esiintyi koko tutkimusjakson aikana pegfilgrastiimia saaneessa ryhmässä 13 %:lla ja filgrastiimia saaneessa ryhmässä 20 %:lla potilaista (ero 7 %, 95 %:n luottamusväli ‑19 %, 5 %). Toisessa tutkimuksessa (n = 310), jossa käytettiin painonmukaista annosta (100 mikrog/kg), 4. asteen neutropenian kesto (keskiarvo) oli pegfilgrastiimiryhmässä 1,7 vuorokautta ja filgrastiimiryhmässä 1,8 vuorokautta (ero 0,03 vuorokautta, 95 %:n luottamusväli ‑0,36, 0,30). Kuumeisen neutropenian kokonaisesiintyvyys oli pegfilgrastiimia saaneiden potilaiden ryhmässä 9 % ja filgrastiimia saaneiden ryhmässä 18 % (ero 9 %, 95 %:n luottamusväli ‑16,8 %, ‑1,1 %).
Rintasyöpäpotilaiden lumekontrolloidussa kaksoissokkotutkimuksessa arvioitiin pegfilgrastiimin vaikutusta kuumeisen neutropenian ilmaantuvuuteen sellaisen solunsalpaajahoidon jälkeen, jossa kuumeisen neutropenian esiintyvyys on yleensä 10‑20 % (dosetakseli 100 mg/m2 3 viikon välein 4 syklin ajan). Potilaita oli yhteensä 928, ja he saivat satunnaistetusti joko pegfilgrastiimia tai lumevalmistetta kerta-annoksena noin 24 tunnin kuluttua solunsalpaaja-annoksesta (2. päivänä) jokaisen syklin aikana. Kuumeista neutropeniaa esiintyi pegfilgrastiimiryhmään satunnaistetuilla potilailla vähemmän (1 %) kuin lumeryhmän potilailla (17 %, p < 0,001). Kliinisesti diagnosoituun kuumeiseen neutropeniaan liittyvä sairaalahoidon ja laskimonsisäisen mikrobilääkityksen tarve oli pegfilgrastiimiryhmässä vähäisempi kuin lumeryhmässä (1 % ja 14 %, p < 0,001; ja 2 % ja 10 %, p < 0,001).
Suppeassa (n = 83) 2. vaiheen satunnaistetussa kaksoissokkotutkimuksessa, jossa potilaat saivat solunsalpaajahoitoa de novo akuuttiin myelooiseen leukemiaan, pegfilgrastiimia (6 mg kerta-annoksena) verrattiin filgrastiimiin annosteltuna induktiohoidon aikana. Toipumisajan vakavasta neutropeniasta arvioitiin olevan molemmissa hoitoryhmissä 22 vuorokautta (mediaani). Pitkäaikaista hoitotulosta ei tutkittu (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sarkoomaa sairastavien lapsipotilaiden 2. vaiheen (n = 37) satunnaistetussa avoimessa monikeskustutkimuksessa, jossa pegfilgrastiimia (100 mikrog/kg) annettiin vinkristiiniä, doksorubisiinia ja syklofosfamidia (VAdriaC/IE) sisältävän solunsalpaajahoidon 1. syklin jälkeen, vaikean neutropenian (neutrofiilimäärä < 0,5 × 109/l) kesto oli pitempi 0–5‑vuotiailla nuoremmilla lapsilla (8,9 vrk) kuin 6–11‑vuotiailla (6 vrk) ja 12–21‑vuotiailla (3,7 vrk) vanhemmilla lapsilla ja aikuisilla. Lisäksi kuumeista neutropeniaa esiintyi enemmän 0–5‑vuotiailla nuoremmilla lapsilla (75 %) kuin 6–11‑vuotiailla (70 %) ja 12–21‑vuotiailla (33 %) vanhemmilla lapsilla ja aikuisilla (ks. kohdat Haittavaikutukset ja Farmakokinetiikka).
Farmakokinetiikka
Ihonalaisen kerta-annoksen jälkeen pegfilgrastiimin huippupitoisuus seerumissa saavutetaan 16–120 tunnin kuluttua annoksesta ja pegfilgrastiimin pitoisuudet seerumissa säilyvät luuydintä lamaavan solunsalpaajahoidon jälkeisen neutropenian keston ajan. Pegfilgrastiimi eliminoituu epälineaarisesti suhteessa annokseen; pegfilgrastiimin seerumipuhdistuma vähenee annoksen suurentuessa. Pegfilgrastiimi näyttää eliminoituvan pääasiassa neutrofiilivälitteisen puhdistuman kautta, ja tämä mekanismi saturoituu suurempia annoksia käytettäessä. Itsesäätelevän puhdistumamekanismin mukaisesti pegfilgrastiimin pitoisuus seerumissa pienenee nopeasti neutrofiilimäärän alkaessa suurentua (ks. kuva 1).
Kuva 1. Seerumin pegfilgrastiimipitoisuuden ja absoluuttisen neutrofiilimäärän (ANC) mediaaniarvojen profiili 6 mg:n kertainjektion jälkeen potilailla, jotka ovat saaneet solunsalpaajahoitoa
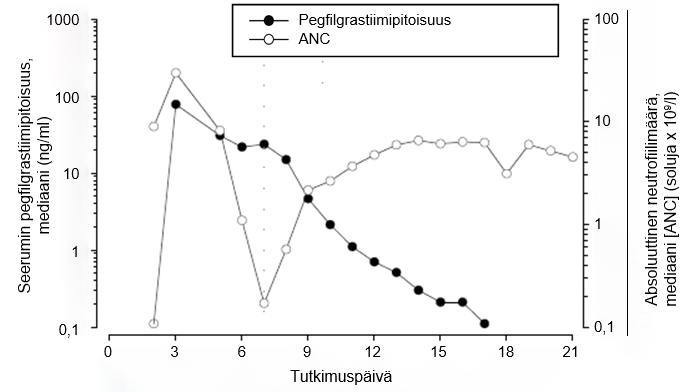
Neutrofiilivälitteisen puhdistumamekanismin vuoksi munuaisten tai maksan vajaatoiminnan ei odoteta vaikuttavan pegfilgrastiimin farmakokinetiikkaan. Eriasteinen munuaisten vajaatoiminta, mukaan lukien loppuvaiheen munuaissairaus, ei vaikuttanut pegfilgrastiimin farmakokinetiikkaan avoimessa kerta-annostutkimuksessa (n = 31).
Iäkkäät
Rajalliset tutkimustulokset viittaavat siihen, että pegfilgrastiimin farmakokinetiikka on iäkkäillä (> 65‑vuotiailla) samanlainen kuin muillakin aikuisilla.
Pediatriset potilaat
Pegfilgrastiimin farmakokinetiikkaa tutkittiin 37:llä sarkoomaa sairastavalla lapsipotilaalla, jotka saivat pegfilgrastiimia 100 mikrog/kg VAdriaC/IE-solunsalpaajahoidon päättymisen jälkeen. Pegfilgrastiimialtistuksen (AUC) keskiarvo (± keskihajonta) oli nuorimmassa ikäryhmässä (0–5‑vuotiailla) suurempi (47,9 ± 22,5 mikrog·hr/ml) kuin 6–11‑vuotiailla (22,0 ± 13,1 mikrog·hr/ml) ja 12–21‑vuotiailla (29,3 ± 23,2 mikrog·hr/ml) vanhemmilla lapsilla (ks. kohta Farmakodynamiikka). Nuorinta ikäryhmää (0–5‑vuotiaita) lukuun ottamatta AUC:n keskiarvo näytti olevan lapsilla samanlainen kuin suuren riskin II‑IV asteen rintasyöpää sairastavilla aikuisilla, jotka saivat pegfilgrastiimia 100 mikrog/kg doksorubisiini-/dosetakselihoidon päättymisen jälkeen (ks. kohdat Haittavaikutukset ja Farmakodynamiikka).
Prekliiniset tiedot turvallisuudesta
Tavanomaisista toistuvilla annoksilla tehdyistä toksisuustutkimuksista saadut prekliiniset tiedot toivat esiin odotettuja farmakologisia vaikutuksia, joita olivat valkosolumäärän suureneminen, myeloidinen hyperplasia luuytimessä, ekstramedullaarinen hematopoieesi ja pernan suureneminen.
Jälkeläisillä ei havaittu haittavaikutuksia, kun tiineille rotille annettiin pegfilgrastiimia ihon alle, mutta kaniineilla pegfilgrastiimin on havaittu aiheuttavan alkio-/sikiötoksisuutta (alkionmenetyksiä), kun kumulatiiviset annokset olivat noin 4-kertaisia verrattuna ihmisille suositeltuun annokseen. Näitä vaikutuksia ei havaittu, kun tiineille kaniineille annettiin annoksia, jotka vastasivat ihmisille suositeltua annosta. Rotilla tehdyt tutkimukset ovat osoittaneet, että pegfilgrastiimi voi läpäistä istukan. Tutkimukset rotilla osoittivat, ettei ihon alle annettu pegfilgrastiimi vaikuttanut lisääntymistoimintoihin, hedelmällisyyteen, kiimakiertoon, pariuttamisen ja parittelun väliseen aikaan eikä sikiön elossaoloaikaan kohdussa. Näiden löydösten merkitystä ihmisen kannalta ei tunneta.
Farmaseuttiset tiedot
Apuaineet
Väkevä etikkahappo
Sorbitoli (E 420)
Polysorbaatti 20
Natriumhydroksidi (pH-arvon säätöön)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden, varsinkaan natriumkloridiliuosten, kanssa.
Kestoaika
3 vuotta.
Säilytys
Säilytä jääkaapissa (2 °C ‑ 8 °C).
Ziextenzo voidaan ottaa huoneenlämpöön (ei yli 35 °C) yhden kerran enintään 120 tunnin ajaksi. Ziextenzo on hävitettävä, jos se on ollut huoneenlämmössä kauemmin kuin 120 tuntia.
Ei saa jäätyä. Alle 24 tuntia kestänyt tahaton altistuminen pakkaselle (yhden kerran) ei vaikuta haitallisesti Ziextenzon säilyvyyteen.
Pidä pakkaus ulkopakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ZIEXTENZO injektioneste, liuos, esitäytetty ruisku
6 mg (L:ei) 0,6 ml (automaattinen turvamekanismi, 6 mg/0,6 ml (10 mg/ml)) (270,84 €)
PF-selosteen tieto
Esitäytetty ruisku (tyypin I lasia), jossa on kuminen männän pysäytin (bromobutyylikumi, lateksiton), mäntä, ruostumattomasta teräksestä valmistettu koon 29 neula sekä kuminen neulansuojus (termoelastomeeri, lateksiton), jossa on automaattinen turvamekanismi.
Yksi esitäytetty ruisku sisältää 0,6 ml injektionestettä.
Pakkauksessa 1 esitäytetty ruisku muovikotelossa.
Valmisteen kuvaus:
Kirkas väritön tai hieman kellertävä injektioneste, liuos.
Käyttö- ja käsittelyohjeet
Ennen käyttöä on tarkastettava silmämääräisesti, ettei Ziextenzo-liuoksessa ole hiukkasia. Vain kirkasta ja väritöntä tai hieman kellertävää liuosta saa antaa injektiona.
Voimakas ravistaminen voi aiheuttaa pegfilgrastiimin aggregaation, jolloin se muuttuu biologisesti tehottomaksi.
Anna esitäytetyn ruiskun lämmetä huoneenlämpöiseksi 15-30 minuutin ajan ennen ruiskun käyttöä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ZIEXTENZO injektioneste, liuos, esitäytetty ruisku
6 mg 0,6 ml
- Ylempi erityiskorvaus (100 %). Rintasyöpä (115), Eturauhassyöpä (116), Leukemiat, muut pahanlaatuiset veri- ja luuydintaudit sekä pahanlaatuiset imukudostaudit (117), Aplastinen anemia (122), Gynekologiset syövät (128), Itsenäinen verihiutaleiden tai granulosyyttien niukkuus (129), Pahanlaatuiset kasvaimet, joita ei ole edellä erikseen mainittu (130).
- Peruskorvaus (40 %).
ATC-koodi
L03AA13
Valmisteyhteenvedon muuttamispäivämäärä
30.11.2023
Yhteystiedot
Edvard Thomsens Vej 14
København S København
Danmark
010 6133 415
info.suomi@sandoz.com