
ALECENSA kapseli, kova 150 mg
Vaikuttavat aineet ja niiden määrät
Yksi kova kapseli sisältää alektinibihydrokloridia määrän, joka vastaa 150 mg:aa alektinibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kova kapseli sisältää 33,7 mg laktoosia (monohydraattina) ja 6 mg natriumia (natriumlauryylisulfaattina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kova kapseli.
Kliiniset tiedot
Käyttöaiheet
Resekoidun ei-pienisoluisen keuhkosyövän (NSCLC) adjuvanttihoito
Alecensa on tarkoitettu monoterapiana suuren uusiutumisriskin ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavien aikuispotilaiden adjuvanttihoitoon kasvaimen täydellisen resektion jälkeen (ks. valintakriteerit kohdasta Farmakodynamiikka).
Edenneen ei-pienisoluisen keuhkosyövän hoito
Alecensa on tarkoitettu monoterapiana edennyttä ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavien aikuispotilaiden ensilinjan hoitoon.
Alecensa on tarkoitettu monoterapiana edennyttä ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavien aikuispotilaiden hoitoon, kun potilasta on aiemmin hoidettu kritsotinibilla.
Ehto
Hoidon aloittavan ja hoitoa seuraavan lääkärin tulee olla perehtynyt syöpälääkkeiden käyttöön.
Annostus ja antotapa
Syöpälääkkeiden käyttöön perehtyneen lääkärin on aloitettava Alecensa-hoito ja valvottava sen toteuttamista.
ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavien potilaiden soveltuvuus hoitoon on varmistettava validoidulla ALK-määrityksellä. Ei-pienisoluista keuhkosyöpää sairastavien potilaiden ALK-positiivisuus pitää selvittää ennen Alecensa-hoidon aloittamista.
Annostus
Suositeltu Alecensa-annos on 600 mg (neljä 150 mg:n kapselia) kahdesti vuorokaudessa aterian yhteydessä (kokonaisvuorokausiannos on 1200 mg).
Jos potilaalla on perussairautena vaikea maksan vajaatoiminta (Child–Pugh C), aloitusannos on 450 mg kahdesti vuorokaudessa aterian yhteydessä (kokonaisvuorokausiannos on 900 mg).
Hoidon kesto
Resekoidun ei-pienisoluisen keuhkosyövän adjuvanttihoito
Alecensa-hoitoa jatketaan, kunnes tauti uusiutuu, ilmaantuu kestämättömiä haittavaikutuksia tai kunnes hoito on jatkunut 2 vuotta.
Edenneen ei-pienisoluisen keuhkosyövän hoito
Alecensa-hoitoa jatketaan, kunnes tauti etenee tai ilmaantuu kestämättömiä haittavaikutuksia.
Annosten viivästyminen tai ottamatta jääminen
Jos suunniteltu Alecensa-annos jää ottamatta, potilas voi ottaa annoksen myöhässä, jos seuraavaan annokseen ottamisajankohtaan on yli 6 tuntia. Potilas ei saa ottaa kahta annosta samanaikaisesti korvatakseen ottamatta jääneen annoksen. Jos potilas oksentaa Alecensa‑annoksen ottamisen jälkeen, potilaan pitää ottaa seuraava annos hoitoaikataulun mukaisena ajankohtana.
Annoksen muuttaminen
Haittavaikutusten hoito saattaa edellyttää Alecensa-annoksen pienentämistä, hoidon keskeyttämistä tilapäisesti tai hoidon lopettamista pysyvästi. Alecensa-annosta pitää pienentää 150 mg:n annoksina kaksi kertaa vuorokaudessa siedettävyyden mukaan. Alecensa-hoito pitää lopettaa pysyvästi, jos potilas ei siedä 300 mg:n annosta kaksi kertaa vuorokaudessa.
Ohjeet annosmuutoksiin esitetään seuraavissa taulukoissa 1 ja 2.
Taulukko 1. Annoksen pienentäminen
| Annoksen pienentäminen | Annostus |
| Annos | 600 mg kaksi kertaa vuorokaudessa |
| Ensimmäinen annoksen pienentämiskerta | 450 mg kaksi kertaa vuorokaudessa |
| Toinen annoksen pienentämiskerta | 300 mg kaksi kertaa vuorokaudessa |
Taulukko 2. Ohjeet annoksen pienentämiseen tiettyjen haittavaikutusten yhteydessä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset)
| CTCAE-luokka | Alecensa-hoito |
| Minkä tahansa vaikeusasteen interstitiaalinen keuhkosairaus tai pneumoniitti | Keskeytä Alecensa-hoito heti ja lopeta se pysyvästi, jos interstitiaaliseen keuhkosairauteen tai pneumoniittiin ei tunnisteta muita mahdollisia syitä. |
| ALAT- tai ASAT-pitoisuuden kohoaminen > viisinkertaiseksi normaaliarvojen ylärajaan nähden (upper limit of normal, ULN) ja kokonaisbilirubiinipitoisuus ≤ kaksinkertainen normaaliarvojen ylärajaan nähden | Keskeytä hoito tilapäisesti, kunnes pitoisuudet palautuvat lähtötilanteeseen tai ≤ kolminkertaiseksi normaaliarvojen ylärajaan nähden (ULN), jatka hoitoa sitten pienemmällä annoksella (ks. taulukko 1). |
| ALAT- tai ASAT-pitoisuuden kohoaminen > kolminkertaiseksi normaaliarvojen ylärajaan nähden (ULN) ja kokonaisbilirubiinipitoisuus > kaksinkertainen normaaliarvojen ylärajaan nähden, kun potilaalla ei ole kolestaasia tai hemolyysiä | Lopeta Alecensa-hoito pysyvästi. |
| Bradykardiaa, vaikeusaste 2 tai vaikeusaste 3 (oireinen, saattaa olla vaikea-asteinen ja kliinisesti merkityksellinen, lääkärinhoito aiheellista) | Keskeytä hoito tilapäisesti, kunnes bradykardia lievenee vaikeusasteeseen ≤ 1 (oireeton) tai sydämen syke nousee ≥ 60 lyöntiin minuutissa. Selvitä muiden sellaisten lääkevalmisteiden käyttö, joiden tiedetään aiheuttavan bradykardiaa, sekä verenpainelääkevalmisteiden käyttö. Jos potilaan todetaan käyttävän samanaikaisesti muita bradykardiaa aiheuttavia lääkevalmisteita ja niiden käyttö lopetetaan tai annosta muutetaan, jatka hoitoa aiemmalla annoksella, kun bradykardia on lieventynyt vaikeusasteeseen ≤ 1 (oireeton) tai sydämen syke on noussut ≥ 60 lyöntiin minuutissa. Jos muita bradykardiaa aiheuttavia lääkevalmisteita ei ole todettu olevan käytössä tai jos bradykardiaa aiheuttavien lääkevalmisteiden käyttöä ei lopeteta eikä annosta muuteta, jatka hoitoa pienemmällä annoksella (ks. taulukko 1), kun bradykardia on lieventynyt vaikeusasteeseen ≤ 1 (oireeton) tai sydämen syke on noussut ≥ 60 lyöntiin minuutissa. |
| Bradykardiaa, vaikeusaste 4 (hengenvaaralliset seuraukset, kiireellinen hoito aiheellista) | Lopeta hoito pysyvästi, jos muita bradykardiaa aiheuttavia lääkevalmisteita ei ole todettu olevan käytössä. Jos potilaan todetaan käyttävän samanaikaisesti muita bradykardiaa aiheuttavia lääkevalmisteita ja niiden käyttö lopetetaan tai annosta muutetaan, jatka hoitoa pienemmällä annoksella (ks. taulukko 1), kun bradykardia on lieventynyt vaikeusasteeseen ≤ 1 (oireeton) tai sydämen syke on noussut ≥ 60 lyöntiin minuutissa; potilasta on tarkkailtava tiheästi kliinisen tarpeen mukaan. Lopeta hoito pysyvästi, jos oireet uusiutuvat. |
| Kreatiinikinaasipitoisuuden kohoaminen > viisinkertaiseksi normaaliarvojen ylärajaan (ULN) nähden | Keskeytä hoito tilapäisesti, kunnes arvot palautuvat lähtötasolle tai ≤ 2,5‑kertaisiksi normaaliarvojen ylärajaan (ULN) nähden, jatka hoitoa sitten samalla annoksella. |
| Kreatiinikinaasipitoisuuden kohoaminen > kymmenkertaiseksi normaaliarvojen ylärajaan (ULN) nähden tai kreatiinikinaasipitoisuuden kohoaminen toisen kerran > viisinkertaiseksi normaaliarvojen ylärajaan (ULN) nähden | Keskeytä hoito tilapäisesti, kunnes arvot palautuvat lähtötasolle tai ≤ 2,5‑kertaisiksi normaaliarvojen ylärajaan (ULN) nähden, jatka hoitoa sitten pienemmällä annoksella taulukko 1:n mukaisesti. |
| Hemolyyttinen anemia, johon liittyy hemoglobiinipitoisuus < 100 g/l (vaikeusaste ≥ 2) | Keskeytä hoito tilapäisesti, kunnes anemia korjaantuu. Jatka hoitoa sitten pienennetyllä annoksella (taulukko 1). |
ALAT = alaniiniaminotransferaasi; ASAT = aspartaattiaminotransferaasi; CTCAE = USA:n kansallisen syöpäinstituutin yleisten haittavaikutusten luokitus (NCI Common Terminology Criteria for Adverse Events)
a Sydämen syke alle 60 lyöntiä minuutissa.
Erityiset potilasryhmät
Maksan vajaatoiminta
Lievää (Child–Pugh A) tai keskivaikeaa (Child–Pugh B) maksan vajaatoimintaa perussairautena sairastavien potilaiden aloitusannosta ei tarvitse muuttaa. Jos potilaalla on perussairautena vaikea maksan vajaatoiminta (Child–Pugh C), aloitusannos on 450 mg kahdesti vuorokaudessa (kokonaisvuorokausiannos on 900 mg) (ks. kohta Farmakokinetiikka). Kaikkia maksan vajaatoimintaa sairastavia potilaita kehotetaan seuraamaan asianmukaisesti (esim. maksan toimintaa osoittavat merkkiaineet), ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Munuaisten vajaatoiminta
Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa. Alecensa-valmistetta ei ole tutkittu vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla. Alektinibin eliminaatio munuaisten kautta on hyvin vähäistä, joten vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Iäkkäät potilaat (≥ 65-vuotiaat)
Vähäiset tiedot Alecensa-valmisteen turvallisuudesta ja tehosta 65-vuotiaille ja sitä vanhemmille potilaille eivät viittaa siihen, että Alecensa-annosta olisi tarpeen muuttaa iäkkäitä potilaita hoidettaessa (ks. kohta Farmakokinetiikka). Yli 80-vuotiaita potilaita koskevaa tietoa ei ole saatavilla.
Pediatriset potilaat
Alecensa-valmisteen turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Vaikeasti lihavat potilaat (> 130 kg)
Alecensa-valmistetta koskevat farmakokineettiset simulaatiot eivät osoita, että altistus lääkkeelle olisi vaikeasti lihavilla (eli > 130 kg:n painoisilla) potilailla pieni, mutta alektinibi jakautuu laajasti, ja alektinibilla tehdyissä kliinisissä tutkimuksissa mukana olleiden potilaiden paino oli 36,9−123 kg. Yli 130 kg:n painoisista potilaista ei ole tietoja saatavissa.
Antotapa
Alecensa otetaan suun kautta. Kovat kapselit pitää niellä kokonaisina eikä niitä saa avata eikä liuottaa. Kapselit pitää ottaa ruokailun yhteydessä (ks. kohta Farmakokinetiikka).
Vasta-aiheet
Yliherkkyys alektinibille tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Interstitiaalinen keuhkosairaus ja pneumoniitti
Alecensa-valmisteella tehdyissä kliinisissä tutkimuksissa on raportoitu interstitiaalista keuhkosairautta ja pneumoniittia (ks. kohta Haittavaikutukset). Potilaita pitää tarkkailla pneumoniittiin viittaavien keuhko-oireiden havaitsemiseksi. Alecensa-hoito pitää keskeyttää heti, jos potilaalla todetaan interstitiaalinen keuhkosairaus tai pneumoniitti, ja hoito pitää lopettaa pysyvästi, jos interstitiaaliseen keuhkosairauteen tai pneumoniittiin ei ole tunnistettu muita mahdollisia syitä (ks. kohta Annostus ja antotapa).
Maksatoksisuus
Alecensa-valmisteella tehdyissä kliinisissä pivotaalitutkimuksissa esiintyi alaniiniaminotransferaasi- (ALAT) ja aspartaattiaminotransferaasipitoisuuksien (ASAT) kohoamista yli viisinkertaiseksi normaaliarvojen ylärajaan nähden (ULN) sekä bilirubiinipitoisuuksien kohoamista yli kolminkertaisiksi normaaliarvojen ylärajaan nähden (ks. kohta Haittavaikutukset). Suurin osa näistä tapahtumista ilmeni kolmen ensimmäisen hoitokuukauden aikana. Alecensa-valmisteella tehdyissä kliinisissä pivotaalitutkimuksissa raportoitiin, että kolmella potilaalla, joiden ASAT/ALAT-pitoisuuden nousu oli vaikeusasteeltaan 3–4, oli lääkeaineen aiheuttama maksavaurio. Alecensa-valmisteella tehdyissä kliinisissä tutkimuksissa yhdellä potilaalla ilmeni ALAT- tai ASAT-pitoisuuksien kohoamista kolminkertaiseksi tai yli kolminkertaiseksi normaaliarvojen ylärajaan nähden ja samaan aikaan kokonaisbilirubiinipitoisuuden kohoamista kaksinkertaiseksi tai yli kaksinkertaiseksi normaaliarvojen ylärajaan nähden alkalisen fosfataasin pitoisuuden säilyessä normaaleina.
Maksan toimintaa, ALAT-, ASAT- ja kokonaisbilirubiinipitoisuus mukaan lukien, pitää seurata lähtötilanteessa ja sen jälkeen kolmen ensimmäisen hoitokuukauden aikana kahden viikon välein. Tämän jälkeen pitoisuuksia pitää seurata määräajoin, koska tapahtumia voi ilmetä myöhemmin kuin kolmen kuukauden kuluessa. Jos potilaan aminotransferaasi- ja bilirubiinipitoisuudet kohoavat, ne on määritettävä tiheämmin. Alecensa-hoito pitää haittavaikutuksen vaikeusasteesta riippuen joko keskeyttää ja jatkaa sen jälkeen aiempaa pienemmällä annoksella tai lopettaa pysyvästi, kuten taulukossa 2 esitetään (ks. kohta Annostus ja antotapa).
Vaikea lihassärky ja kreatiinikinaasipitoisuuden kohoaminen
Alecensa-valmisteella tehdyissä pivotaalitutkimuksissa raportoitiin lihassärkyä tai muskuloskeletaalista kipua, mukaan lukien vaikeusasteen 3 tapahtumia (ks. kohta Haittavaikutukset).
Alecensa-valmisteella tehdyissä pivotaalitutkimuksissa esiintyi kreatiinikinaasipitoisuuden kohoamista, mukaan lukien vaikeusasteen 3 tapahtumia (ks. kohta Haittavaikutukset). Kaikissa kliinisissä tutkimuksissa (BO40336, BO28984, NP28761, NP28673) mediaaniaika vaikeusasteen ≥ 3 kreatiinikinaasipitoisuuden kohoamiseen oli 15 päivää.
Potilaita tulee kehottaa ilmoittamaan kaikesta selittämättömästä lihassärystä, -arkuudesta tai ‑heikkoudesta. Kreatiinikinaasipitoisuudet pitää määrittää kahden viikon välein ensimmäisen hoitokuukauden ajan sekä kliinisen tarpeen mukaan potilailta, jotka ilmoittavat oireista. Kreatiinikinaasipitoisuuksien kohoamisen vaikeusasteen mukaisesti Alecensa-hoito pitää keskeyttää ja jatkaa myöhemmin, tai annosta pitää pienentää (ks. kohta Annostus ja antotapa).
Bradykardia
Alecensa-hoidon yhteydessä voi esiintyä oireista bradykardiaa (ks. kohta Haittavaikutukset). Sydämen syketaajuutta ja verenpainetta pitää seurata kliinisen tarpeen mukaan. Oireettoman bradykardian yhteydessä annosmuutokset eivät ole tarpeen (ks. kohta Annostus ja antotapa). Jos potilaalla on oireista bradykardiaa tai hengenvaarallisia tapahtumia, bradykardiaa tunnetusti aiheuttavien lääkevalmisteiden, samoin kuin verenpainelääkevalmisteiden, samanaikaista käyttöä pitää arvioida ja muuttaa Alecensa-hoitoa taulukossa 2 kuvatulla tavalla (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset, P‑gp:n substraatit ja BCRP:n substraatit).
Hemolyyttinen anemia
Alecensa-hoidon yhteydessä on raportoitu hemolyyttistä anemiaa (ks. kohta Haittavaikutukset). Jos hemoglobiinipitoisuus on alle 100 g/l ja hemolyyttistä anemiaa epäillään, keskeytä Alecensa-hoito ja aloita asianmukaiset laboratoriotutkimukset. Jos hemolyyttinen anemia varmistuu, jatka Alecensa-hoitoa pienennetyllä annoksella, kun hemolyyttinen anemia on korjaantunut, kuten taulukossa 2 kuvataan (ks. kohta Annostus ja antotapa).
Maha-suolikanavan perforaatio
Alektinibihoitoa saaneilla potilailla on raportoitu maha-suolikanavan perforaatioita, kun potilailla on ollut niiden suurentunut riski (esim. aiempi divertikuliitti, etäpesäkkeitä maha-suolikanavassa tai sellaisten lääkevalmisteiden samanaikainen käyttö, joihin tiedetään liittyvän maha-suolikanavan perforaatioiden riski). Jos potilaalle kehittyy maha-suolikanavan perforaatio, Alecensa-hoidon lopettamista pitää harkita. Potilaille pitää kertoa maha-suolikanavan perforaatioiden oireista ja löydöksistä ja heitä pitää kehottaa oireiden ilmaantuessa ottamaan nopeasti yhteyttä lääkäriin.
Valoyliherkkyys
Alecensa-valmisteen käytön yhteydessä on raportoitu valoyliherkkyyttä auringonvalolle (ks. kohta Haittavaikutukset). Potilaita pitää neuvoa välttämään pitkäkestoista altistumista auringonvalolle Alecensa-hoidon aikana ja vähintään 7 vuorokauden ajan hoidon päättymisen jälkeen. Potilaita pitää myös neuvoa suojaamaan iho palamiselta käyttämällä laajakirjoista ultravioletti A- (UVA) ja ultravioletti B (UVB) ‑säteilyltä suojaavaa auringonsuojavoidetta ja huulivoidetta (suojakerroin ≥ 50).
Alkio-sikiötoksisuus
Raskaana olevalle naiselle annettu Alecensa saattaa vahingoittaa sikiötä. Naispotilaiden, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisymenetelmää hoidon aikana ja vähintään viiden viikon ajan viimeisen Alecensa-annoksen jälkeen (ks. kohdat Yhteisvaikutukset, Raskaus ja imetys ja Prekliiniset tiedot turvallisuudesta). Miespotilaiden, joiden naispuolinen kumppani voi tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisymenetelmää hoidon aikana ja vähintään kolmen kuukauden ajan viimeisen Alecensa-annoksen jälkeen (ks. kohdat Raskaus ja imetys ja Prekliiniset tiedot turvallisuudesta).
Laktoosi-intoleranssi
Tämä lääkevalmiste sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, synnynnäinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei tule käyttää tätä lääkettä.
Natriumsisältö
Tämä lääkevalmiste sisältää 48 mg natriumia per vuorokausiannos (1200 mg), joka vastaa 2,4 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset alektinibiin
CYP3A4 on in vitro ‑tietojen perusteella pääasiallinen sekä alektinibin että sen aktiivisen metaboliitin M4:n metaboliaa välittävä entsyymi, ja CYP3A:n osuus koko maksametaboliasta on 40−50 %. M4:n tehon ja aktiivisuuden in vitro anaplastisen lymfoomakinaasin (ALK) suhteen on osoitettu olevan samankaltainen.
CYP3A:n indusorit
Useiden 600 mg:n rifampisiiniannosten (rifampisiini on voimakas CYP3A:n indusori) ottaminen suun kautta kerran vuorokaudessa samaan aikaan suun kautta otettavan 600 mg:n alektinibikerta-annoksen kanssa pienensi alektinibin Cmax-arvoa 51 % ja AUCinf-arvoa 73 % sekä suurensi M4:n Cmax-arvon 2,20-kertaiseksi ja AUCinf-arvon 1,79-kertaiseksi. Vaikutus alektinibin ja M4:n yhdistettyyn altistukseen oli vähäinen, sillä Cmax-arvo pieneni 4 % ja AUCinf-arvo pieneni 18 %. Alektinibin ja M4:n yhdistettyyn altistukseen kohdistuvien vaikutusten perusteella annosta ei tarvitse muuttaa, kun Alecensa-valmistetta käytetään samaan aikaan CYP3A:n indusorien kanssa. Voimakkaita CYP3A:n indusoreita (mm. karbamatsepiinia, fenobarbitaalia, fenytoiinia, rifabutiinia, rifampisiinia ja mäkikuismaa [Hypericum perforatum]) samanaikaisesti käyttävien potilaiden asianmukaista seurantaa suositellaan.
CYP3A:n estäjät
Useiden 400 mg:n posakonatsoliannosten (posakonatsoli on voimakas CYP3A:n estäjä) ottaminen suun kautta kaksi kertaa vuorokaudessa samaan aikaan suun kautta otettavan 300 mg:n alektinibikerta-annoksen kanssa lisäsi alektinibialtistusta siten, että Cmax-arvo suureni 1,18-kertaiseksi ja AUCinf-arvo suureni 1,75-kertaiseksi, kun taas M4-altistus väheni siten, että Cmax-arvo pieneni 71 % ja AUCinf-arvo pieneni 25 %. Vaikutus alektinibin ja M4:n yhdistettyyn altistukseen oli vähäinen, sillä Cmax-arvo pieneni 7 % ja AUCinf-arvo suureni 1,36-kertaiseksi. Alektinibin ja M4:n yhdistettyyn altistukseen kohdistuvien vaikutusten perusteella annosta ei tarvitse muuttaa, kun Alecensa-valmistetta käytetään samaan aikaan CYP3A:n estäjien kanssa. Voimakkaita CYP3A:n estäjiä (mm. ritonaviiria, sakinaviiria, telitromysiiniä, ketokonatsolia, itrakonatsolia, vorikonatsolia, posakonatsolia, nefatsodonia, greippihedelmää tai pomeranssia) samanaikaisesti käyttävien potilaiden asianmukaista seurantaa suositellaan.
Mahan pH-arvoa suurentavat lääkevalmisteet
Useiden kerran vuorokaudessa annettujen 40 mg:n esomepratsoliannosten (esomepratsoli on protonipumpun estäjä) ei osoitettu vaikuttavan kliinisesti oleellisesti alektinibin ja M4:n yhdistettyyn altistukseen. Annosta ei näin ollen tarvitse muuttaa, kun Alecensa-valmistetta käytetään samaan aikaan protonipumpun estäjien tai muiden mahan pH-arvoa suurentavien lääkevalmisteiden (esim. H2-reseptorin antagonistien tai antasidien) kanssa.
Kuljettajaproteiinien vaikutus alektinibialtistukseen
M4 on P-glykoproteiinin (P-gp) substraatti. Koska alektinibi estää P-gp:tä, samanaikainen käyttö P‑gp:n estäjien kanssa ei oletettavasti vaikuta oleellisesti M4-altistukseen.
Alektinibin vaikutukset muihin lääkevalmisteisiin
CYP:n substraatit
In vitro alektinibilla ja M4:llä on heikko CYP3A4:ää aikariippuvasti estävä vaikutus ja alektinibi indusoi kliinisinä pitoisuuksina heikosti CYP3A4:ää ja CYP2B6:ta.
Useat 600 mg:n alektinibiannokset eivät vaikuttaneet herkän CYP3A:n substraatin midatsolaamin (2 mg) altistukseen. Samanaikaisesti käytettyjen CYP3A:n substraattien annosta ei siksi tarvitse muuttaa.
CYP2B6:n ja pregnaani X ‑reseptorin (PXR) säätelemien entsyymien indusoitumisen riskiä ei CYP3A4:ää lukuun ottamatta voida kokonaan sulkea pois. Samanaikaisesti annosteltavien suun kautta otettavien ehkäisyvalmisteiden teho voi heikentyä.
P‑gp:n substraatit
Alektinibi ja sen pääasiallinen aktiivinen metaboliitti M4 ovat in vitro effluksiproteiini P‑gp:n estäjiä. Alektinibi ja M4 saattavat siksi suurentaa samanaikaisesti annettujen P‑gp:n substraattien pitoisuutta plasmassa. Kun alektinibia annetaan samanaikaisesti P‑gp:n substraattien kanssa (esim. digoksiini, dabigatraanieteksilaatti, topotekaani, sirolimuusi, everolimuusi, nilotinibi ja lapatinibi), suositellaan asianmukaista seurantaa.
Rintasyövän resistenssiproteiinin (BCRP) substraatit
Alektinibi ja M4 ovat in vitro effluksiproteiini BCRP:n estäjiä. Alektinibi ja M4 saattavat siksi suurentaa samanaikaisesti annettujen BCRP:n substraattien pitoisuutta plasmassa. Kun alektinibia annetaan samanaikaisesti BCRP:n substraattien kanssa (esim. metotreksaatti, mitoksantroni, topotekaani ja lapatinibi), suositellaan asianmukaista seurantaa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisia, jotka voivat tulla raskaaksi, on kehotettava välttämään raskaaksi tuloa Alecensa-hoidon aikana (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ehkäisy naispotilaille
Alecensa-hoitoa saavien naispotilaiden, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisymenetelmää sekä hoidon aikana että vähintään viiden viikon ajan viimeisen Alecensa-annoksen jälkeen (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Ehkäisy miespotilaille
Miespotilaiden, joiden naispuolinen kumppani voi tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisymenetelmää hoidon aikana ja vähintään kolmen kuukauden ajan viimeisen Alecensa-annoksen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Raskaus
Ei ole olemassa tietoja tai on vain vähän tietoja alektinibin käytöstä raskaana oleville naisille. Alektinibi saattaa vaikutusmekanisminsa perusteella vahingoittaa sikiötä, jos alektinibia käytetään raskauden aikana. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Jos naispotilas tulee raskaaksi Alecensa-hoidon aikana tai viiden viikon aikana viimeisen Alecensa-annoksen jälkeen, hänen on otettava yhteyttä hoitavaan lääkäriin ja hänelle on kerrottava sikiölle mahdollisesti aiheutuneesta haitasta.
Miespotilaan, jonka naispuolinen kumppani tulee raskaaksi miespotilaan Alecensa-hoidon aikana tai kolmen kuukauden kuluessa viimeisen Alecensa-annoksen ottamisesta, on otettava yhteyttä lääkäriin, ja hänen naispuolisen kumppaninsa on käännyttävä lääkärin puoleen valmisteen aneugeenisuudesta sikiölle mahdollisesti aiheutuvan haitan vuoksi (ks. kohta Prekliiniset tiedot turvallisuudesta).
Imetys
Ei tiedetä, erittyvätkö alektinibi ja/tai sen metaboliitit ihmisen rintamaitoon. Vastasyntyneeseen/imeväiseen kohdistuvaa riskiä ei voida poissulkea. Äitejä on kehotettava olemaan imettämättä Alecensa-hoidon aikana.
Hedelmällisyys
Eläimillä ei ole tehty erityisiä hedelmällisyyttä koskevia tutkimuksia alektinibin vaikutusten selvittämiseksi. Yleistä toksisuutta koskeneissa tutkimuksissa ei havaittu haitallisia vaikutuksia urosten tai naaraiden lisääntymiselimiin (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Alecensa-valmisteella on vähäinen vaikutus ajokykyyn ja koneiden käyttökykyyn. Ajaessa tai koneita käyttäessä on noudatettava varovaisuutta, koska potilaille saattaa tulla oireista bradykardiaa (esim. pyörtyminen, heitehuimaus, hypotensio) tai näköhäiriöitä Alecensa-hoidon aikana (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Jäljempänä esitetyt tiedot kuvastavat Alecensa-altistusta 533:lla resekoitua tai edennyttä ALK-positiivista ei-pienisoluista keuhkosyöpää (NSCLC) sairastavalla potilaalla. Nämä potilaat saivat kliinisissä pivotaalitutkimuksissa Alecensa-hoitoa suositeltuna annoksena 600 mg kaksi kertaa päivässä resekoidun ei-pienisoluisen keuhkosyövän adjuvanttihoitoon (BO40336, ALINA) tai edenneen ei-pienisoluisen keuhkosyövän hoitoon (BO28984, ALEX; NP28761; NP28673). Ks. lisätietoja kliinisen tutkimuksen tutkittavista kohdasta Farmakodynamiikka.
BO40336-tutkimuksessa (ALINA; N = 128) Alecensa-altistuksen keston mediaani oli 23,9 kuukautta. Tutkimuksessa BO28984 (ALEX; N = 152) Alecensa-altistuksen keston mediaani oli 28,1 kuukautta. Vaiheen II kliinisissä tutkimuksissa (NP28761, NP28673; N = 253) Alecensa-altistuksen keston mediaani oli 11,2 kuukautta.
Yleisimpiä haittavaikutuksia (≥ 20 %) olivat ummetus, lihassärky, turvotus, suurentunut bilirubiinipitoisuus, suurentunut ASAT-arvo, anemia, ihottuma ja suurentunut ALAT-arvo.
Haittavaikutustaulukko
Taulukossa 3 luetellaan haittavaikutukset, joita esiintyi kliinisissä tutkimuksissa (BO40336, BO28984, NP28761, NP28673) Alecensa-hoitoa saaneilla potilailla.
Taulukossa 3 luetellut haittavaikutukset esitetään elinjärjestelmittäin ja esiintyvyysluokittain seuraavan esitystavan mukaisesti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100 – < 1/10), melko harvinainen (≥ 1/1 000 – < 1/100), harvinainen (≥ 1/10 000 – < 1/1 000), hyvin harvinainen (< 1/10 000). Haittavaikutukset on esitetty kussakin elinjärjestelmäluokassa haittavaikutusten yleisyyden ja vaikeusasteen mukaan alenevassa järjestyksessä. Haittavaikutukset on esitetty samassa esiintyvyys- ja vaikeusasteluokassa vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 3. Alecensa-valmisteen kliinisissä tutkimuksissa (BO40336, BO28984, NP28761, NP28673; N = 533) raportoidut haittavaikutukset
Elinjärjestelmäluokka Haittavaikutus (MedDRA-luokka) | Alecensa N = 533 | |
| Esiintyvyysluokka (kaikki vaikeusasteet) | Esiintyvyysluokka (vaikeusasteet 3–4) | |
| Veri ja imukudos | ||
| Anemia1) | Hyvin yleinen | Yleinen |
| Hemolyyttinen anemia2) | Yleinen | -* |
| Hermosto | ||
| Makuaistin häiriö 3) | Yleinen | Melko harvinainen |
| Silmät | ||
| Näköhäiriöt4) | Yleinen | -* |
| Sydän | ||
| Bradykardia5) | Hyvin yleinen | -* |
| Hengityselimet, rintakehä ja välikarsina | ||
| Interstitiaalinen keuhkosairaus / pneumoniitti | Yleinen | Melko harvinainen |
| Ruoansulatuselimistö | ||
| Ripuli | Hyvin yleinen | Yleinen |
| Oksentelu | Hyvin yleinen | Melko harvinainen |
| Ummetus | Hyvin yleinen | Melko harvinainen |
| Pahoinvointi | Hyvin yleinen | Melko harvinainen |
| Stomatiitti6) | Yleinen | Melko harvinainen |
| Maksa ja sappi | ||
| Suurentunut ASAT-arvo | Hyvin yleinen | Yleinen |
| Suurentunut ALAT-arvo | Hyvin yleinen | Yleinen |
| Suurentunut bilirubiinipitoisuus7) | Hyvin yleinen | Yleinen |
| Suurentunut alkalisen fosfataasin arvo | Hyvin yleinen | Melko harvinainen |
| Lääkeaineen aiheuttama maksavaurio8) | Melko harvinainen | Melko harvinainen |
| Iho ja ihonalainen kudos | ||
| Ihottuma9) | Hyvin yleinen | Yleinen |
| Valoyliherkkyys | Yleinen | Melko harvinainen |
| Luusto, lihakset ja sidekudos | ||
| Lihassärky10) | Hyvin yleinen | Melko harvinainen |
| Suurentunut kreatiinikinaasipitoisuus | Hyvin yleinen | Yleinen |
| Munuaiset ja virtsatiet | ||
| Suurentunut veren kreatiniinipitoisuus | Hyvin yleinen | Melko harvinainen** |
| Akuutti munuaisvaurio | Yleinen | Melko harvinainen** |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Turvotus11) | Hyvin yleinen | Melko harvinainen |
| Tutkimukset | ||
| Painon nousu | Hyvin yleinen | Melko harvinainen |
| Aineenvaihdunta ja ravitsemus | ||
| Hyperurikemia12) | Yleinen | -* |
*Vaikeusasteen 3–4 haittavaikutuksia ei havaittu.
**Sisältää yhden vaikeusasteen 5 tapahtuman (havaittiin edenneen ei-pienisoluisen keuhkosyövän yhteydessä).
1) sisältää anemian, pienentyneen hemoglobiinipitoisuuden ja normokromisen normosyyttisen anemian.
2) tapaukset raportoitu BO40336-tutkimuksessa (N = 128)
3) sisältää makuaistin häiriöt, heikentyneen makuaistin ja makuhäiriöt
4) sisältää näön sumenemisen, näkökyvyn heikkenemisen, lasiaiskellujat, heikentyneen näkötarkkuuden, astenopian, diplopian, valonarkuuden ja fotopsian
5) sisältää bradykardian ja sinusbradykardian
6) sisältää stomatiitin ja suun haavaumat
7) sisältää suurentuneen veren bilirubiinipitoisuuden, hyperbilirubinemian, suurentuneen konjugoituneen bilirubiinin pitoisuuden ja suurentuneen veren konjugoitumattoman bilirubiinipitoisuuden
8) sisältää kaksi potilasta, joilla raportoitiin MedDRA-termi lääkeaineen aiheuttama maksavaurio, sekä yhden potilaan, jolla raportoitiin vaikeusasteen 4 ASAT- ja ALAT-pitoisuuksien suureneminen ja jolla oli maksabiopsialla vahvistettu lääkeaineen aiheuttama maksavaurio
9) sisältää ihottuman, makulopapulaarisen ihottuman, dermatiitin, aknetyyppisen ihottuman, eryteeman, papulaarisen ihottuman, kutisevan ihottuman, makulaarisen ihottuman, eksfoliatiivisen ihottuman ja erytematoottisen ihottuman
10) sisältää lihassäryn, muskuloskeletaalisen kivun ja nivelkivun
11) sisältää perifeerisen turvotuksen, turvotuksen, yleistyneen turvotuksen, silmäluomien turvotuksen ja periorbitaalisen turvotuksen, kasvojen turvotuksen, paikallisen turvotuksen, raajojen turpoamisen, kasvojen turpoamisen, huulten turpoamisen, turpoamisen, nivelten turpoamisen ja silmäluomien turpoamisen
12) sisältää hyperurikemiatapauksia ja suurentuneen veren virtsahappopitoisuuden.
Valikoitujen haittavaikutuksien kuvaus
Interstitiaalinen keuhkosairaus / pneumoniitti
Kliinisissä tutkimuksissa 1,7 %:lla Alecensa-hoitoa saaneista potilaista esiintyi interstitiaalista keuhkosairautta / pneumoniittia. Näistä 0,4 % oli vaikeusasteen 3 tapauksia. Hoidon lopetti interstitiaalisen keuhkosairauden / pneumoniitin vuoksi 1,1 % potilaista, ja 0,4 %:lla potilaista tapahtuman vuoksi tehtiin annosmuutoksia. Vaiheen III kliinisessä tutkimuksessa BO28984 vaikeusasteen 3 tai 4 interstitiaalista keuhkosairautta / pneumoniittia ei havaittu Alecensa-hoitoa saaneilla potilailla, mutta havaittiin 2,0 %:lla kritsotinibia saaneista potilaista. Interstitiaalinen keuhkosairaus ei johtanut näissä kliinisissä tutkimuksissa yhdenkään potilaan kuolemaan. Potilaita pitää tarkkailla pneumoniittiin viittaavien keuhko-oireiden havaitsemiseksi (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Maksatoksisuus
Kliinisissä tutkimuksissa kolmella potilaalla oli dokumentoitu lääkeaineen aiheuttama maksavaurio (mukaan lukien kaksi potilasta, joilla raportoitiin lääkeaineen aiheuttama maksavaurio, ja yksi potilas, jolla raportoitiin vaikeusasteen 4 suurentunut ASAT- ja ALAT-pitoisuus ja jolla oli maksabiopsian avulla dokumentoitu lääkeaineen aiheuttama maksavaurio). Alecensa-hoitoa saaneilla potilailla raportoitiin kaikissa kliinisissä tutkimuksissa haittavaikutuksina suurentuneita ASAT- ja ALAT-arvoja (suurentuneita ASAT-arvoja 23,6 %:lla potilaista ja suurentuneita ALAT-arvoja 20,5 %:lla potilaista). Suurin osa näistä tapahtumista oli vaikeusasteeltaan luokkaa 1 tai 2, ja vaikeusasteen ≥ 3 suurentuneita ASAT-arvoja raportoitiin 3,0 %:lla potilaista ja ALAT-arvoja 3,2 %:lla potilaista. Tapahtumat ilmaantuivat yleensä kolmen ensimmäisen hoitokuukauden aikana, ne olivat tavallisesti ohimeneviä ja hävisivät, kun Alecensa-hoito keskeytettiin tilapäisesti (ASAT-arvojen osalta 2,3 %:lla potilaista ja ALAT-arvojen osalta 3,6 %:lla potilaista) tai annosta pienennettiin (ASAT-arvojen osalta 1,7 %:lla potilaista ja ALAT-arvojen osalta 1,5 %:lla potilaista). Alecensa-hoito piti lopettaa suurentuneiden ASAT-arvojen vuoksi 1,3 %:lla potilaista ja suurentuneiden ALAT-arvojen vuoksi 1,5 %:lla potilaista. Vaiheen III kliinisessä tutkimuksessa BO28984 vaikeusasteen 3 tai 4 suurentuneita ALAT-arvoja havaittiin 4,6 %:lla ja ASAT-arvoja 5,3 %:lla Alecensa-hoitoa saavista potilaista verrattuna 16,6 %:iin (ALAT-arvoja) ja 10,6 %:iin (ASAT-arvoja) kritsotinibihoitoa saavista potilaista.
Alecensa-hoitoa kaikissa kliinisissä tutkimuksissa saaneista potilaista 25,9 %:lla raportoitiin haittavaikutuksena suurentuneita bilirubiinipitoisuuksia. Suurin osa näistä tapahtumista oli vaikeusasteeltaan luokkaa 1 tai 2, ja vaikeusasteen ≥ 3 tapahtumia raportoitiin 3,9 %:lla potilaista. Tapahtumat ilmaantuivat yleensä kolmen ensimmäisen hoitokuukauden aikana, ne olivat tavallisesti ohimeneviä, ja valtaosa hävisi, kun annosta muutettiin. Suurentunut bilirubiinipitoisuus johti 8,3 %:lla potilaista annosmuutoksiin, ja 2,1 %:lla potilaista Alecensa-hoito piti lopettaa suurentuneiden bilirubiinipitoisuuksien vuoksi. Vaiheen III kliinisessä tutkimuksessa BO28984 vaikeusasteen 3 tai 4 suurentuneita bilirubiinipitoisuuksia todettiin 5,9 %:lla Alecensa-hoitoa saavista potilaista, kun taas kritsotinibihoidossa niitä ei todettu yhdelläkään potilaalla.
ALAT- tai ASAT-arvojen suurenemista kolminkertaisiksi tai yli kolminkertaisiksi normaaliarvojen ylärajaan nähden ja samaan aikaan kokonaisbilirubiiniarvojen suurenemista kaksinkertaisiksi tai yli kaksinkertaisiksi normaaliarvojen ylärajaan nähden alkalisen fosfataasin pitoisuuksien säilyessä normaaleina ilmeni yhdellä potilaalla (0,2 %) Alecensa-valmisteen kliinisissä tutkimuksissa.
Potilaiden maksan toimintaa sekä ALAT-, ASAT- ja kokonaisbilirubiinipitoisuuksia pitää seurata kohdassa Varoitukset ja käyttöön liittyvät varotoimet. esitetyllä tavalla ja hoitaa kohdassa Annostus ja antotapa annettujen suositusten mukaisesti.
Bradykardia
Alecensa-hoitoa kaikissa kliinisissä tutkimuksissa saaneilla potilailla on raportoitu vaikeusasteeltaan luokan 1 tai 2 bradykardiaa (11,3 %). Yhdelläkään potilaalla ei ollut vaikeusasteen ≥ 3 tapahtumia. 102 potilaalla kaikkiaan 521 Alecensa-hoitoa saaneesta potilaasta (19,6 %), joista oli EKG-rekisteröintisarja saatavissa, sydämen syketiheys oli annoksen ottamisen jälkeen alle 50 lyöntiä minuutissa. Vaiheen III kliinisessä tutkimuksessa BO28984 Alecensa-hoitoa saaneista potilaista 12,4 %:lla sydämen syketiheys oli annoksen ottamisen jälkeen alle 50 lyöntiä minuutissa verrattuna 17,6 %:iin kritsotinibia saaneista potilaista. Potilaat, joille kehittyy oireista bradykardiaa, pitää hoitaa kohdissa Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet annettujen suositusten mukaisesti. Bradykardia ei johtanut Alecensa-hoidon keskeyttämiseen.
Vaikea lihassärky ja kreatiinikinaasiarvojen kohoaminen
Alecensa-hoitoa kaikissa kliinisissä tutkimuksissa saaneilla potilailla on raportoitu lihassärkyä (35,3 %), mukaan lukien lihassärkytapahtumia (24,2 %), nivelkipua (16,3 %) ja muskuloskeletaalista kipua (0,8 %). Suurin osa tapahtumista oli vaikeusasteeltaan luokkaa 1 tai 2, ja viiden potilaan (0,9 %) tapahtuma oli vaikeusasteeltaan luokkaa 3. Alecensa-annosta oli tarpeen muuttaa näiden haittavaikutusten vuoksi yhdeksällä potilaalla (1,7 %). Lihassärky ei johtanut Alecensa-hoidon keskeyttämiseen. Kreatiinikinaasiarvojen kohoamista ilmeni Alecensa-valmisteen kaikissa kliinisissä tutkimuksissa 56,2 %:lla 491 potilaasta, joiden kreatiinikinaasin laboratoriotulokset olivat saatavilla. Vaikeusasteen ≥ 3 kreatiinikinaasin kohoamisen ilmaantuvuus oli 5,5 %. Mediaaniaika vaikeusasteen ≥ 3 kreatiinikinaasiarvojen nousuun oli kaikissa tutkimuksissa 15 päivää. Kreatiinikinaasiarvojen noususta johtuvia annosmuutoksia tehtiin 5,4 %:lle potilaista, mutta kreatiinikinaasiarvojen nousu ei johtanut Alecensa-hoidon keskeyttämiseen. Kliinisessä tutkimuksessa BO28984 vaikea-asteista nivelkipua havaittiin yhdellä potilaalla (0,7 %) alektinibihoitoa saaneilla ja kahdella potilaalla (1,3 %) kritsotinibihoitoa saaneilla. Vaikeusasteen ≥ 3 kreatiinikinaasiarvojen kohoamista raportoitiin 3,3 %:lla Alecensa-hoitoa saaneista potilaista ja 4,6 %:lla kritsotinibihoitoa saaneista potilaista.
Hemolyyttinen anemia
Hemolyyttistä anemiaa on havaittu kliinisissä tutkimuksissa 3,1 %:lla Alecensa-hoitoa saaneista potilaista. Kyse oli vaikeusasteen 1 tai 2 tapauksista (ei vakavia), ja ne eivät johtaneet hoidon lopettamiseen (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Vaikutukset ruoansulatuskanavaan
Ummetus (39,6 %), ripuli (18,8 %), pahoinvointi (17,6 %) ja oksentelu (12,4 %) olivat yleisimmin raportoidut ruoansulatuskanavan haittavaikutukset. Suurin osa tapahtumista oli vaikeusasteeltaan lieviä tai kohtalaisia; vaikeusasteen 3 tapahtumia raportoitiin ripulin (1,1 %), pahoinvoinnin (0,4 %), ummetuksen (0,4 %) ja oksentelun (0,2 %) osalta. Nämä tapahtumat eivät johtaneet Alecensa-hoidon keskeyttämiseen. Mediaaniaika ummetuksen, pahoinvoinnin, ripulin ja/tai oksentelun alkamiseen kaikissa kliinisissä tutkimuksissa oli 21 päivää. Tapahtumien esiintymistiheys harveni ensimmäisen hoitokuukauden jälkeen. Vaiheen III kliinisessä tutkimuksessa BO28984 alektinibihaarassa vaikeusasteen 3 ja 4 pahoinvointia ja ummetusta raportoitiin kumpaakin yhdellä potilaalla (0,7 %) ja ripulia raportoitiin 2 potilaalla (1,3 %); kritsotinibihaarassa vaikeusasteen 3 ja 4 pahoinvoinnin ilmaantuvuus oli 3,3 %, oksentelun ilmaantuvuus 3,3 % ja ripulin ilmaantuvuus 2,0 %.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannoksen saanutta potilasta pitää tarkkailla huolellisesti, ja yleinen elintoimintoja tukeva hoito on aloitettava. Alecensa-yliannoksen hoitoon ei ole spesifistä vasta-ainetta.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset aineet, proteiinikinaasin estäjät, ATC-koodi: L01ED03.
Vaikutusmekanismi
Alektinibi on erittäin selektiivinen ja voimakas ALK- ja RET (rearranged during transfection) ‑tyrosiinikinaasin estäjä. ALK-tyrosiinikinaasin aktiivisuuden estyminen johti prekliinisissä tutkimuksissa signaalireittien, STAT 3 (signal transducer and activator of transcription 3) ja PI3K (fosfoinositidi-3-kinaasi) / AKT (proteiinikinaasi B) mukaan lukien, alavirran estymiseen ja solukuoleman (apoptoosin) induktioon.
Alektinibilla osoitettiin aktiivisuutta in vitro ja in vivo ALK-entsyymin mutatoituneita muotoja vastaan, kritsotinibiresistenssiä aiheuttavat mutaatiot mukaan lukien. Alektinibin pääasiallisella metaboliitilla (M4) on todettu in vitro samankaltainen vaikutus ja aktiivisuus.
Alektinibi ei prekliinisten tietojen perusteella ole P-gp:n eikä BCRP:n substraatti. P-glykoproteiini ja BCRP ovat kumpikin uloskuljettajia (effluksiproteiineja) veri-aivoesteessä, ja alektinibi kykenee siksi jakautumaan keskushermostoon ja säilymään siellä.
Kliininen teho ja turvallisuus
Resekoidun ALK-positiivisen ei-pienisoluisen keuhkosyövän adjuvanttihoito
Alecensa-valmisteen tehoa ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavien potilaiden hoitoon kasvaimen täydellisen poistoleikkauksen jälkeen tutkittiin maailmanlaajuisessa vaiheen III satunnaistetussa, avoimessa kliinisessä tutkimuksessa (BO40336; ALINA). Tutkimukseen mukaan soveltuneilla potilailla edellytettiin olevan levinneisyysasteen IB (≥ 4 cm:n kasvaimia) – levinneisyysasteen IIIA ei-pienisoluinen keuhkosyöpä UICC/AJCC (Union for International Cancer Control/American Joint Committee on Cancer) ‑levinneisyysluokituksen (7th Edition) mukaan, kun ALK-positiivinen sairaus on tunnistettu paikallisesti tehdyllä CE-merkityllä ALK-testillä tai keskitetysti tehdyllä Ventana ALK (D5F3) ‑immunohistokemiallisella (IHC) menetelmällä.
Seuraavat valintakriteerit määrittelevät potilaat, jolla on suuri uusiutumisriski ja joita käyttöaihe koskee. Lisäksi ne kuvastavat potilasjoukkoa, joilla on UICC/AJCC:n (7. painos) levinneisyyskriteerien mukainen levinneisyysasteen IB (≥ 4 cm) – IIIA ei-pienisoluinen keuhkosyöpä:
Kasvaimen koko ≥ 4 cm tai minkä tahansa kokoinen kasvain, johon liittyy joko N1- tai N2-status tai rintakehän rakenteisiin (keuhkopussinlehteen, rintakehän seinämään, palleaan, palleahermoon, välikarsinanmyötäiseen keuhkopussiin, parietaaliseen sydänpussinlehteen, välikarsinaan, sydämeen, suuriin verisuoniin, henkitorveen, palaavaan kurkunpäähermoon, ruokatorveen, nikamansolmuun, henkitorven harjuun suoraan tunkeutuneita) levinneitä kasvaimia tai pääkeuhkoputken < 2 cm distaalisesti henkitorven harjusta affisioivia kasvaimia, jotka eivät kuitenkaan affisioi henkitorven harjua, tai kasvaimia, joihin liittyy atelektaasi tai koko keuhkon obstruktiivinen pneumoniitti, tai kasvaimia, joissa on primaarisena erillinen nodulus / primaarisina erillisiä noduluksia samassa lohkossa tai toisessa samanpuoleisessa lohkossa.
Tutkimukseen ei otettu mukaan N2-statuksen potilaita, joiden kasvaimet olivat tunkeutuneet myös välikarsinaan, sydämeen, suuriin verisuoniin, henkitorveen, palaavaan kurkunpäähermoon, ruokatorveen, nikamansolmuun, henkitorven harjuun tai joilla oli erillinen kasvainnodulus / erillisiä kasvainnoduluksia toisessa samanpuoleisessa lohkossa.
Potilaat satunnaistettiin (1:1) saamaan kasvaimen resektion jälkeen Alecensa-valmistetta tai platinapohjaista solunsalpaajaa. Satunnaistaminen ositettiin etnisen taustan (aasialainen ja muu kuin aasialainen) ja sairauden levinneisyysasteen (IB, II ja IIIA) perusteella. Alecensa-valmistetta annettiin suun kautta suositeltuna annoksena 600 mg kaksi kertaa päivässä yhteensä 2 vuoden ajan tai kunnes sairaus uusiutui tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Platinapohjaista solunsalpaajaa annettiin laskimoon neljän 21 päivän pituisen hoitosyklin ajan noudattaen jotakin seuraavista hoito-ohjelmista:
75 mg/m2 sisplatiinia päivänä 1 ja 25 mg/m2 vinorelbiinia päivinä 1 ja 8
75 mg/m2 sisplatiinia päivänä 1 ja 1250 mg/m2 gemsitabiinia päivinä 1 ja 8
75 mg/m2 sisplatiinia päivänä 1 ja 500 mg/m2 pemetreksediä päivänä 1.
Jos potilas ei sietänyt sisplatiiniin perustuvaa hoito-ohjelmaa, sisplatiinin sijaan annettiin karboplatiinia edellä mainittuina yhdistelminä annoksena, jolla plasmassa olevan vapaan karboplatiinin pitoisuus-aikakuvaajan pinta-ala (AUC) oli 5 mg/ml/min tai AUC-arvo oli 6 mg/ml/min.
Ensisijainen tehon päätetapahtuma oli tutkijan arvioima tauditon elossaoloaika (disease-free survival, DFS). Tauditon elossaoloaika määriteltiin satunnaistamispäivämäärästä minkä tahansa seuraavien ilmaantumispäivämäärään kuluneeksi ajaksi: sairauden ensimmäinen dokumentoitu uusiutuminen, uusi primaari ei-pienisoluinen keuhkosyöpä tai mistä tahansa syystä aiheutunut kuolema sen mukaan, mikä näistä tapahtui ensimmäisenä. Toissijaiset ja eksploratiiviset tehon päätetapahtumat olivat kokonaiselinaika (overall survival, OS) ja sairauden uusiutumiseen keskushermostossa tai tutkittavan kuolemaan (CNS-DFS) kulunut aika.
Yhteensä 257:ää potilasta tutkittiin: 130 potilasta satunnaistettiin Alecensa-haaraan ja 127 potilasta satunnaistettiin solunsalpaajahoitohaaraan. Kaiken kaikkiaan iän mediaani oli 56 vuotta (vaihteluväli: 26–87), ja 24 % oli ≥ 65‑vuotiaita, 52 % oli naisia, 56 % oli aasialaisia, 60 % ei ollut koskaan tupakoinut, 53 %:lla ECOG-suorituskykypisteet olivat 0; 10 %:lla potilaista oli levinneisyysasteen IB sairaus, 36 %:lla oli levinneisyysasteen II sairaus ja 54 %:lla oli levinneisyysasteen IIIA sairaus.
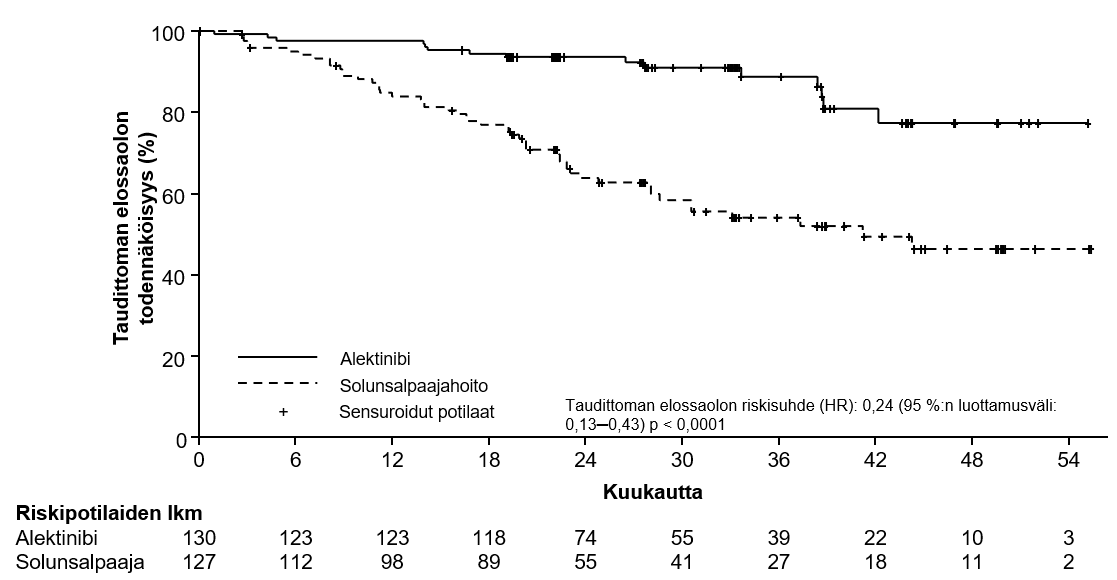
ALINA-tutkimuksessa osoitettiin, että levinneisyysasteen II-IIIA ja levinneisyysasteen IB (≥ 4 cm) -IIIA (ITT-potilasjoukko) potilasjoukoissa Alecensa-hoitoa saaneiden potilaiden tauditon elossaoloaika piteni tilastollisesti merkitsevästi verrattuna solunsalpaajahoitoa saaneisiin potilaisiin. Kokonaiselinaikaa koskevat tiedot olivat keskeneräiset taudittoman elossaoloajan analyysiajankohtana, jolloin kaikkiaan oli raportoitu 2,3 % kuolemista. Elossaolon seurannan keston mediaani oli Alecensa-haarassa 27,8 kuukautta ja solunsalpaajahoitohaarassa 28,4 kuukautta.
Tauditonta elossaoloaikaa koskevien tehon tulosten yhteenveto on taulukossa 4 ja kuvassa 1.
Taulukko 4 Tutkijan arvioon perustuvaa tauditonta elossaoloaikaa koskevat ALINA-tutkimuksen tulokset
| Tehoa koskeva parametri | Levinneisyysaste II-IIIA | ITT-potilasjoukko | ||
| Alecensa n = 116 | Solunsalpaaja-hoito n = 115 | Alecensa n = 130 | Solunsalpaaja-hoito n = 127 | |
| Tauditonta elossaoloa koskevien tapahtumien lukumäärä (%) | 14 (12,1) | 45 (39,1) | 15 (11,5) | 50 (39,4) |
| Taudittoman elossaoloajan mediaani, kuukautta (95 %:n luottamusväli) | NE (NE–NE) | 44,4 (27,8–NE) | NE (NE–NE) | 41,3 (28,5–NE) |
| Ositettu riskitiheyksien suhde (HR) (95 %:n luottamusväli)* | 0,24 (0,13–0,45) | 0,24 (0,13–0,43) | ||
| p-arvo (log-rank)* | < 0,0001 | < 0,0001 | ||
ITT = hoitoaikeen mukainen (Intent-to-Treat); NE = ei arvioitavissa (Not Estimable)
*Ositettu levinneisyysasteen II-IIIA sairautta sairastavien potilaiden etnisen taustan mukaan, ositettu etnisen taustan ja levinneisyysasteen IB-IIIA mukaisen levinneisyysasteen mukaan.
Kuva 1. Tutkijan ITT-potilasjoukossa arvioiman taudittoman elossaolon Kaplan–Meier-käyrä
Edenneen ALK-positiivisen ei-pienisoluisen keuhkosyövän hoito
Aiemmin hoitamattomat potilaat
Alecensa-valmisteen turvallisuutta ja tehoa tutkittiin maailmanlaajuisessa vaiheen III satunnaistetussa, avoimessa kliinisessä tutkimuksessa (BO28984, ALEX) ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla, jotka eivät olleet aiemmin saaneet hoitoa. Kaikilta potilailta edellytettiin ennen tutkimukseen satunnaistamista keskitetysti tehty ALK-proteiinin ilmentymisen testaus kudosnäytteestä Ventana anti‑ALK (D5F3) ‑immunohistokemiallisella menetelmällä.
Vaiheen III tutkimukseen otettiin mukaan yhteensä 303 potilasta, joista 151 potilasta satunnaistettiin kritsotinibihaaraan ja 152 potilasta satunnaistettiin Alecensa-haaraan. Alecensa-haarassa potilaat saivat Alecensa-valmistetta suun kautta suositeltuina annoksina 600 mg kaksi kertaa vuorokaudessa.
Satunnaistamisen ositustekijöitä olivat ECOG (Eastern Cooperative Oncology Group) ‑suorituskykypisteet (0/1 vs. 2), etninen tausta (aasialainen vs. muu kuin aasialainen) ja lähtötilanteessa olleet etäpesäkkeet keskushermostossa (kyllä vs. ei). Tutkimuksen ensisijainen päätetapahtuma oli Alecensa-hoidon paremmuuden osoittaminen kritsotinibiin verrattuna, mitä mitattiin tutkijan RECIST (Response Evaluation Criteria in Solid Tumors) ‑version 1.1 kriteerein arvioiman etenemisvapaan ajan (PFS) perusteella. Alecensa-haaran lähtötilanteen demografiset ja sairautta koskevat ominaisuudet olivat iän mediaani 58 vuotta (kritsotinibihaarassa 54 vuotta), 55 % naisia (kritsotinibihaarassa 58 %), 55 % muita kuin aasialaistaustaisia (kritsotinibihaarassa 54 %), 61 % tupakoimattomia (kritsotinibihaarassa 65 %), 93 %:lla ECOG-suorituskykypisteet 0 tai 1 (kritsotinibihaarassa 93 %), 97 %:lla levinneisyysasteen IV tauti (kritsotinibihaarassa 96 %), 90 %:lla adenokarsinooma histologia (kritsotinibihaarassa 94 %), 40 %:lla lähtötilanteessa etäpesäkkeitä keskushermostossa (kritsotinibihaarassa 38 %) ja 17 % saanut aiemmin keskushermoston sädehoitoa (kritsotinibihaarassa 14 %).
Tutkimuksessa saavutettiin sen ensisijainen päätetapahtuma primaarianalyysissa, jossa osoitettiin tutkijan arvion perusteella tilastollisesti merkitsevä paraneminen taudin etenemisvapaassa ajassa. Yhteenveto tehoa koskevista tiedoista esitetään taulukossa 5, ja Kaplan–Meier-käyrä tutkijan arvioimasta taudin etenemisvapaasta ajasta esitetään kuvassa 2. Lisäksi kuvassa 3 esitetään kokonaiselinajan loppuanalyysiin perustuva kokonaiselinajan Kaplan–Meier-käyrä.
Taulukko 5 Yhteenveto tutkimuksen BO28984 (ALEX) tehoa koskevista tuloksista
Kritsotinibi n = 151 | Alecensa n = 152 | |
| Seurannan keston mediaani (kuukautta)‡ | 23,3 (vaihteluväli 0,3–123,5) | 53,5 (vaihteluväli 0,5–126,8) |
| Ensisijainen tehoa koskeva parametri | ||
Etenemisvapaa aika (PFS) (tutkijan arvio)† Niiden potilaiden lukumäärä, joilla todettiin tapahtumia n (%) Mediaani (kuukautta) [95 %:n luottamusväli] | 102 (68 %) 11,1 [9,1–13,1] | 62 (41 %) NE [17,7–NE] |
Riskitiheyksien suhde (HR) [95 %:n luottamusväli] Ositetun log-rank-testin p-arvo | 0,47 [0,34–0,65] p < 0,0001 | |
| Toissijainen tehoa koskeva parametri | ||
Etenemisvapaa aika (PFS) (riippumattoman arviointikomitean arvio)*, † Niiden potilaiden lukumäärä, joilla todettiin tapahtumia n (%) Mediaani (kuukautta) [95 %:n luottamusväli] | 92 (61 %) 10,4 [7,7–14,6] | 63 (41 %) 25,7 [19,9–NE] |
Riskitiheyksien suhde (HR) [95 %:n luottamusväli] Ositetun log-rank-testin p-arvo | 0,50 [0,36–0,70] p < 0,0001 | |
Aika keskushermostoon etenemiseen (riippumattoman arviointikomitean arvio)*, **, † Niiden potilaiden lukumäärä, joilla todettiin tapahtumia n (%) | 68 (45 %) | 18 (12 %) |
Syyspesifinen riskitiheyksien suhde (HR) [95 %:n luottamusväli] Ositetun log-rank-testin p-arvo | 0,16 [0,10–0,28] p < 0,0001 | |
Keskushermostoon etenemisen 12 kuukauden kumulatiivinen ilmaantuvuus (riippumattoman arviointikomitean arvio) [95 %:n luottamusväli] | 41,4 % [33,2–49,4] | 9,4 % [5,4–14,7] |
Kokonaisvaste (ORR) (tutkijan arvio)*, ***, † Vasteen saaneita n (%) [95 %:n luottamusväli] | 114 (75,5 %) [67,8–82,1] | 126 (82,9 %) [76,0–88,5] |
Kokonaiselinaika*, ‡ Niiden potilaiden lukumäärä, joilla todettiin tapahtumia n (%) Mediaani (kuukautta) [95 %:n luottamusväli] | 73 (48,3 %) 54,2 [34,6–75,6] | 76 (50,0 %) 81,1 [62,3–NE] |
Riskitiheyksien suhde (HR) [95 %:n luottamusväli] | 0,78 [0,56–1,08] | |
Vasteen kesto (DOR) (tutkijan arvio)‡ Mediaani (kuukautta) [95 %:n luottamusväli] | n = 115 11,1 [7,9–13,0] | n = 126 42,3 [31,3–51,3] |
Keskushermoston kokonaisvaste (CNS-ORR) potilailla, joilla oli lähtötilanteessa mitattavissa olleita etäpesäkkeitä keskushermostossa† Vasteen keskushermostossa saaneita n (%) [95 %:n luottamusväli] Kokonaisvasteen keskushermostossa saaneita n (%) Vasteen kesto keskushermostossa, mediaani (kuukautta) [95 %:n luottamusväli] | n = 22 11 (50,0 %) [28,2–71,8] 1 (5 %) 5,5 [2,1–17,3] | n = 21 17 (81,0 %) [58,1–94,6] 8 (38 %) 17,3 [14,8–NE] |
Keskushermoston kokonaisvaste (CNS-ORR) potilailla, joilla oli lähtötilanteessa keskushermostossa etäpesäkkeitä, jotka olivat ja eivät olleet mitattavissa (riippumattoman arviointikomitean arvio)† Vasteen keskushermostossa saaneita n (%) [95 %:n luottamusväli] Täydellisen vasteen (CR) keskushermostossa saaneita n (%) Vasteen kesto keskushermostossa (CNS-DOR), mediaani (kuukautta) [95 %:n luottamusväli] | n = 58 15 (25,9 %) [15,3–39,0] 5 (9 %) 3,7 [3,2–6,8] | n = 64 38 (59,4 %) [46,4–71,5] 29 (45 %) NE [17,3–NE] |
* Hierarkkisen testauksen keskeisten toissijaisten päätetapahtumien osio
** Kilpaileva riskianalyysi etenemisestä keskushermostossa, systeemisestä etenemisestä ja kuolemasta kilpailevina tapahtumina
*** 2 potilaalla kritsotinibihaarassa ja 6 potilaalla alektinibihaarassa oli täydellinen vaste
† Tiedot primaarianalyysista
‡ Tiedot kokonaiselinajan loppuanalyysista, joka tehtiin 149 kuoleman jälkeen.
NE = ei arvioitavissa (not estimable), CNS= central nervous system, PFS= Progression Free Survival (etenemisvapaa aika), ORR= Objective Response Rate (kokonaisvaste)
DOR=Duration Of Response (vasteen kesto), HR = hazard ratio (riskitiheyksien suhde)
Taudin etenemisvapaa aika potilailla, joilla oli lähtötilanteessa etäpesäkkeitä keskushermostossa (riskitiheyksien suhde [HR] = 0,40, 95 %:n luottamusväli: 0,25–0,64, etenemisvapaan ajan mediaani Alecensa-hoitohaarassa = ei arvioitavissa [NE], 95 %:n luottamusväli: 9,2–NE, etenemisvapaan ajan mediaani kritsotinibihoitohaarassa = 7,4 kuukautta, 95 %:n luottamusväli: 6,6–9,6) ja joilla ei ollut lähtötilanteessa etäpesäkkeitä keskushermostossa (riskitiheyksien suhde [HR] = 0,51, 95 %:n luottamusväli: 0,33–0,80, etenemisvapaan ajan mediaani Alecensa-hoitohaarassa = NE, 95 %:n luottamusväli: NE–NE, etenemisvapaan ajan mediaani kritsotinibihoitohaarassa = 14,8 kuukautta, 95 %:n luottamusväli:10,8–20,3), osoitti kummassakin osajoukossa, että Alecensa-hoidon hyöty oli kritsotinibihoitoa suurempi.
Kuva 2. Kaplan–Meierin kuvaaja tutkijan arvioimasta etenemisvapaasta ajasta tutkimuksessa BO28984 (ALEX)

Kuva 3: Kaplan–Meierin kuvaaja kokonaiselinajasta tutkimuksessaBO28984 (ALEX)

Kritsotinibihoitoa aiemmin saaneet potilaat
Alecensan turvallisuutta ja tehoa ALK-positiivista ei-pienisoluista keuhkosyöpää sairastaville potilaille, jotka olivat aiemmin saaneet kritsotinibihoitoa, tutkittiin kahdessa vaiheen I/II kliinisessä tutkimuksessa (NP28673 ja NP28761).
NP28673
Tutkimus NP28673 oli vaiheen I/II yhden hoitoryhmän monikeskustutkimus potilailla, joilla oli pitkälle edennyt ALK-positiivinen ei-pienisoluinen keuhkosyöpä (NSCLC) ja joiden syöpä oli aiemmin edennyt kritsotinibihoidon aikana. Potilaat olivat mahdollisesti saaneet aiemmin kritsotinibin lisäksi myös solunsalpaajahoitoa. Tutkimuksen vaiheen II osassa oli mukana yhteensä 138 potilasta, jotka saivat Alecensa-valmistetta suun kautta suositusannoksina 600 mg kaksi kertaa vuorokaudessa.
Ensisijainen päätetapahtuma oli arvioida Alecensa-hoidon tehoa kokonaisvasteella (Objective Response Rate, ORR), joka perustui riippumattoman arviointikomitean (Independent Review Committee, IRC) keskitettyyn arvioon koko potilasjoukosta (sytotoksisilla solunsalpaajilla hoidetuilla ja hoitamattomilla). Arviointiin käytettiin kiinteissä kasvaimissa todetun vasteen arviointiin tarkoitettujen RECIST-kriteerien versiota 1.1. Toinen ensisijainen päätetapahtuma oli riippumattoman arviointikomitean RECIST 1.1 ‑kriteereitä käyttäen tekemä keskitetty arvio kokonaisvasteesta potilailla, jotka olivat aiemmin saaneet hoitoa sytotoksisilla solunsalpaajilla. Tilastollisesti merkitsevä tulos saavutettaisiin, jos arvioidun ORR:n luottamusvälin alaraja ylittäisi etukäteen määritellyn 35 %:n kynnyksen.
Potilaiden demografiset ominaisuudet olivat yhdenmukaiset ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavan potilasjoukon kanssa. Koko tutkimuspotilasjoukon demografiset ominaisuudet olivat: valkoihoisia 67 %, aasialaisia 26 %, naisia 56 % ja iän mediaani 52 vuotta. Suurimmalla osalla potilaista (70 %) ei ollut tupakointitaustaa. ECOG‑suorituskykyluokka oli lähtötilanteessa 0 tai 1 90,6 %:lla potilaista ja 2 9,4 %:lla potilaista. Potilaiden tullessa mukaan tutkimukseen 99 %:lla potilaista oli graduksen IV syöpä, 61 %:lla oli etäpesäkkeitä aivoissa ja 96 %:lla potilaista oli adenokarsinoomaksi luokiteltuja kasvaimia. Tutkimukseen mukaan otetuista potilaista 20 %:lla syöpä oli edennyt pelkän kritsotinibihoidon aikana ja 80 %:lla syöpä oli edennyt kritsotinibista ja vähintään yhdestä solunsalpaajasta koostuneen hoidon aikana.
Tutkimus NP28761
Tutkimus NP28761 oli vaiheen I/II yhden hoitoryhmän monikeskustutkimus potilailla, joilla oli pitkälle edennyt ALK-positiivinen ei-pienisoluinen keuhkosyöpä (NSCLC) ja joiden syöpä oli aiemmin edennyt kritsotinibihoidon aikana. Potilaat olivat mahdollisesti saaneet aiemmin kritsotinibin lisäksi myös solunsalpaajahoitoa. Tutkimuksen vaiheen II osassa oli mukana yhteensä 87 potilasta, jotka saivat Alecensa-valmistetta suun kautta suositusannoksina 600 mg kaksi kertaa vuorokaudessa.
Ensisijainen päätetapahtuma oli arvioida Alecensa-hoidon tehoa kokonaisvasteella (ORR), joka perustui riippumattoman arviointikomitean RECIST-versiolla 1.1 tekemään keskitettyyn arvioon. Tilastollisesti merkitsevä tulos saavutettaisiin, jos arvioidun ORR:n luottamusvälin alaraja ylittäisi etukäteen määritellyn 35 %:n kynnyksen.
Potilaiden demografiset ominaisuudet olivat yhdenmukaiset ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavan potilasjoukon kanssa. Koko tutkimuspotilasjoukon demografiset ominaisuudet olivat: valkoihoisia 84 %, aasialaisia 8 %, naisia 55 %. Iän mediaani oli 54 vuotta. Suurimmalla osalla potilaista (62 %) ei ollut tupakointitaustaa. ECOG-suorituskykyluokka oli lähtötilanteessa 0 tai 1 89,7 %:lla potilaista ja 2 10,3 %:lla potilaista. Potilaiden tullessa mukaan tutkimukseen 99 %:lla potilaista oli graduksen IV syöpä, 60 %:lla oli etäpesäkkeitä aivoissa ja 94 %:lla potilaista oli adenokarsinoomaksi luokiteltuja kasvaimia. Tutkimukseen mukaan otetuista potilaista 26 %:lla syöpä oli edennyt pelkän kritsotinibihoidon aikana ja 74 %:lla syöpä oli edennyt kritsotinibista ja vähintään yhdestä solunsalpaajasta koostuneen hoidon aikana.
Yhteenveto tutkimusten NP28673 ja NP28761 tärkeimmistä tehoa koskevista tuloksista esitetään taulukossa 6. Yhteenveto keskushermostoa koskevien päätetapahtumien yhdistetystä analyysista esitetään taulukossa 7.
Taulukko 6. Tehoa koskevat tulokset tutkimuksista NP28673 ja NP28761
NP28673 Alecensa 600 mg kaksi kertaa vuorokaudessa | NP28761 Alecensa 600 mg kaksi kertaa vuorokaudessa | |
| Seurannan keston mediaani (kuukautta) | 21 (vaihteluväli 1–30) | 17 (vaihteluväli 1–29) |
| Ensisijaiset tehon parametrit | ||
Kokonaisvaste (ORR, riippumattoman arviointikomitean arvio) potilasjoukossa, jossa vaste oli arvioitavissa Vasteen saaneita n (%) [95 %:n luottamusväli] | n = 122a 62 (50,8 %) [41,6–60,0 %] | n = 67b 35 (52,2 %) [39,7–64,6 %] |
Kokonaisvaste (ORR, riippumattoman arviointikomitean arvio) potilailla, joita oli aiemmin hoidettu kemoterapialla Vasteen saaneita n (%) [95 %:n luottamusväli] | n = 96 43 (44,8 %) [34,6–55,3 %] | |
| Toissijaiset tehon parametrit | ||
Vasteen kesto (DOR, riippumattoman arviointikomitean arvio) Niiden potilaiden lukumäärä, joilla todettiin tapahtumia n (%) Mediaani (kuukautta) [95 %:n luottamusväli] | n = 62 36 (58,1 %) 15,2 [11,2–24,9] | n = 35 20 (57,1 %) 14,9 [6,9–NE] |
Etenemisvapaa aika (PFS, riippumattoman arviointikomitean arvio) Niiden potilaiden lukumäärä, joilla todettiin tapahtumia n (%) Keston mediaani (kuukautta) [95 % luottamusväli] | n = 138 98 (71,0 %) 8,9 [5,6–12,8] | n = 87 58 (66,7 %) 8,2 [6,3–12,6] |
NE = ei arvioitavissa (not estimable); kokonaisvaste = ORR, Objective Response Rate; vasteen kesto = DOR, Duration Of Response; etenemisvapaa aika = PFS, Progression Free Survival.
a 16 potilaalla ei ollut riippumattoman arviointikomitean mukaan mitattavissa sairautta lähtötilanteessa, eikä näitä potilaita sisällytetty potilasjoukkoon, jonka vaste oli arvioitavissa.
b 20 potilaalla ei ollut riippumattoman arviointikomitean mukaan mitattavissa olevaa sairautta lähtötilanteessa, eikä näitä potilaita sisällytetty potilasjoukkoon, jonka vaste oli arvioitavissa.
Tutkimusten NP28673 ja NP28761 kokonaisvastetta osoittavat tulokset olivat yhdenmukaiset kaikissa potilaiden lähtötilanteen ominaisuuksiin (kuten ikä, sukupuoli, rotu, ECOG-suorituskykyluokka, keskushermoston etäpesäkkeet ja aiempi solunsalpaajahoito) perustuvissa osajoukoissa etenkin kun huomioidaan, että joissakin osajoukoissa potilaita oli vähän.
Taulukko 7. Yhteenveto tutkimusten NP28673 ja NP28761 keskushermostoa koskevien päätetapahtumien yhdistetystä analyysista
| Keskushermostoa koskevat parametrit (NP28673 ja NP28761) | Alecensa 600 mg kaksi kertaa vuorokaudessa |
| Potilaat, joilla oli lähtötilanteessa mitattavissa olleita keskushermoston leesioita | n = 50 |
Keskushermostoa koskeva kokonaisvaste (CNS ORR, riippumattoman arviointikomitean arvio) Vasteen saaneita (%) [95 %:n luottamusväli] Täydellinen vaste Osittainen vaste |
32 (64,0 %) [49,2–77,1 %] 11 (22,0 %) 21 (42,0 %) |
Keskushermoston leesioissa todetun vasteen kesto (CNS DOR, riippumattoman arviointikomitean arvio) Niiden potilaiden lukumäärä, joilla todettiin tapahtumia (%) Mediaani (kuukautta) [95 %:n luottamusväli] | n = 32 18 (56,3 %) 11,1 [7,6–NE] |
Keskushermosto = CNS, Central Nervous System; kokonaisvaste = ORR, Objective Response Rate; vasteen kesto =DOR, Duration of Response; NE = ei arvioitavissa (not estimable)
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Alecensa-valmisteen käytöstä keuhkosyövän (pienisoluisen ja ei-pienisoluisen syövän) hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Alektinibin ja sen pääasiallisen aktiivisen metaboliitin (M4) farmakokineettisiä parametreja on tutkittu ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavilla potilailla ja terveillä tutkittavilla. Populaatiofarmakokineettisen analyysin perusteella alektinibin vakaan tilan geometriset keskiarvot (variaatiokerroin %) olivat Cmax n. 665 ng/ml (44,3 %), Cmin n. 572 ng/ml (47,8 %) ja AUC0-12hr n. 7 430 ng*h/ml (45,7 %). M4:n vakaan tilan geometriset keskiarvot olivat Cmax n. 246 ng/ml (45,4 %), Cmin n. 222 ng/ml (46,6 %) ja AUC0-12h n. 2 810 ng*h/ml (45,9 %).
Imeytyminen
ALK-positiivista ei-pienisoluista keuhkosyöpää sairastaville potilaille aterian jälkeen suun kautta kaksi kertaa vuorokaudessa annettu 600 mg:n alektinibiannos imeytyi ja Tmax saavutettiin noin 4–6 tunnissa.
Alektinibin vakaa tila saavutetaan 600 mg kaksi kertaa päivässä jatkuvasti annettavilla annoksilla 7 päivän kuluessa. Kaksi kertaa vuorokaudessa otettujen 600 mg:n annosten kertymissuhde oli noin kuusinkertainen. Populaatiofarmakokineettinen analyysi tukee alektinibin suhdetta annokseen koko annosalueella 300–900 mg aterian jälkeen otettuna.
Alektinibikapseleiden absoluuttinen biologinen hyötyosuus oli terveillä tutkittavilla aterian jälkeen 36,9 % (90 %:n luottamusväli: 33,9–40,3 %).
Suun kautta runsasrasvaisen, runsaskalorisen aterian yhteydessä otetun kerta-annoksen 600 mg jälkeen alektinibin ja M4:n altistus suureni noin kolminkertaiseksi paastotilaan verrattuna (ks. kohta Annostus ja antotapa).
Jakautuminen
Alektinibi ja sen pääasiallinen metaboliitti M4 sitoutuvat vaikuttavan aineen pitoisuudesta riippumatta voimakkaasti ihmisen plasman proteiineihin (> 99 %). Ihmisen veressä ja plasmassa olevien pitoisuuksien suhteen keskiarvo in vitro on kliinisesti oleellisilla pitoisuuksilla alektinibin osalta 2,64 ja M4:n osalta 2,50.
Laskimoon annetun alektinibin vakaan tilan jakautumistilavuuden geometrinen keskiarvo (Vss) oli 475 l, mikä viittaa siihen, että alektinibi jakautuu laajasti kudoksiin.
In vitro -tietojen perusteella alektinibi ei ole P-gp:n substraatti. Alektinibi ja M4 eivät ole BCRP:n tai orgaanisten anionien kuljettajapolypeptidien (organic anion-transporting polypeptide, OATP) 1B1/B3:n substraatteja.
Biotransformaatio
Metaboliatutkimukset in vitro osoittivat, että CYP3A4 on alektinibin ja sen päämetaboliitin M4:n metaboliaa välittävä pääasiallinen CYP-isoentsyymi. Sen arvioidaan vastaavan 40–50 %:sta alektinibin metaboliaa. Ihmisellä tehdyn massatasetutkimuksen tulokset osoittivat, että alektinibi ja M4 olivat pääasialliset plasmassa kiertävät fraktiot ja että ne käsittivät yhdessä noin 76 % plasmassa olevasta kokonaisradioaktiivisuudesta. Metaboliitin ja kanta-aineen suhteen geometrinen keskiarvo vakaassa tilassa on 0,399.
Metaboliitti M1b:tä havaittiin vähäisempänä metaboliittina in vitro ja terveiden tutkittavien plasmasta. Metaboliitti M1b:n ja sen vähäisemmän isomeerin M1a:n muodostumista katalysoivat todennäköisesti CYP-isoentsyymit (muut isoentsyymit kuin CYP3A) ja aldehydidehydrogenaasi (ALDH) yhdessä.
In vitro -tutkimukset osoittavat, ettei alektinibi eikä sen aktiivinen pääasiallinen metaboliitti (M4) estä CYP1A2:ta, CYP2B6:ta, CYP2C9:ää, CYP2C19:ää tai CYP2D6:ta kliinisesti merkityksellisillä pitoisuuksilla. Alektinibi ei estänyt OATP1B1:tä/ OATP1B3:a, OAT1:tä, OAT3:a eikä OCT2:ta kliinisesti oleellisina pitoisuuksina in vitro.
Eliminaatio
Terveille tutkittaville suun kautta annetun 14C-leimatun alektinibikerta-annoksen jälkeen suurin osa radioaktiivisuudesta erittyi ulosteeseen (keskimäärin todettu määrä 97,8 %), ja vain pieni osa erittyi virtsaan (keskimäärin todettu määrä 0,46 %). Ulosteeseen erittyneestä osuudesta 84 % oli muuttumatonta alektinibia ja 5,8 % oli M4:ää.
Alektinibin näennäinen puhdistuma (Cl/F) oli populaatiofarmakokineettisen analyysin perusteella 81,9 l/tunti. Alektinibin yksilöllisen eliminaation puoliintumisajan estimaattien geometrinen keskiarvo oli 32,5 tuntia. M4-metaboliitin vastaavat arvot olivat 217 l/tunti ja 30,7 tuntia.
Farmakokinetiikka erityisryhmissä
Munuaisten vajaatoiminta
Muuttumattomana aineena virtsaan erittyneen alektinibin ja aktiivisen metaboliitin M4:n määrä on hyvin pieni (< 0,2 % annoksesta). Alektinibin ja M4:n altistukset olivat populaatiofarmakokineettisen analyysin perusteella samankaltaiset sekä lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla että potilailla, joiden munuaisten toiminta oli normaali. Alektinibin farmakokinetiikkaa ei ole tutkittu vaikeaa munuaisten vajaatoimintaa sairastavilla potilailla.
Maksan vajaatoiminta
Alektinibin eliminaatio tapahtuu pääasiassa maksassa metaboloitumalla, joten maksan vajaatoiminta saattaa suurentaa alektinibin ja/tai sen pääasiallisen metaboliitin M4:n pitoisuuksia plasmassa. Alektinibin ja M4:n altistukset olivat populaatiofarmakokineettisen analyysin perusteella samankaltaiset lievää maksan vajaatoimintaa sairastavilla potilailla ja potilailla, joiden maksan toiminta oli normaali.
Vaikeaa (Child–Pugh C) maksan vajaatoimintaa sairastaville tutkittaville suun kautta annetun 300 mg:n alektinibikerta-annoksen jälkeen alektinibin Cmax-arvo oli sama ja AUCinf-arvo suureni 2,2‑kertaiseksi kaltaistettujen terveiden tutkittavien näihin parametreihin verrattuna. M4:n Cmax-arvo oli 39 % ja AUCinf-arvo oli 34 % pienempi, minkä seurauksena yhdistetty altistus alektinibille ja M4:lle (AUCinf) oli vaikeaa maksan vajaatoimintaa sairastavilla potilailla 1,8‑kertainen kaltaistettuihin terveisiin tutkittaviin verrattuna.
Maksan vajaatoimintaa koskeneessa tutkimuksessa oli mukana myös keskivaikeaa (Child–Pugh B) maksan vajaatoimintaa sairastanut ryhmä. Altistuksen alektinibille havaittiin olleen tässä ryhmässä hieman suurempi kaltaistettuihin terveisiin tutkittaviin verrattuna. Child–Pugh B ‑ryhmän tutkittavilla ei kuitenkaan yleensä todettu poikkeavuuksia bilirubiini- ja albumiinipitoisuudessa tai protrombiiniajassa. Tämä osoittaa, että he eivät välttämättä täysin edustaneet keskivaikeaa maksan vajaatoimintaa sairastavia tutkittavia, joiden metabolia on heikentynyt.
Iän, painon, rodun ja sukupuolen vaikutus
Iällä, painolla, rodulla ja sukupuolella ei ollut kliinisesti merkityksellistä vaikutusta alektinibin ja M4:n systeemiseen altistukseen. Kliinisiin tutkimuksiin mukaan otettujen potilaiden paino oli 36,9−123 kg. Vaikeasti lihavista (> 130 kg) potilaista ei ole tietoja saatavissa (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Karsinogeenisuus
Alektinibin karsinogeenisuuden selvittämiseksi ei ole tehty karsinogeenisuustutkimuksia.
Mutageenisuus
Alektinibi ei ollut mutageeninen in vitro bakteerien käänteismutaatiotestissä (Amesin testi). Se kuitenkin lisäsi hieman poikkeavuuksien lukumäärää in vitro kiinanhamsterin keuhkosoluissa tehdyssä sytogeneettisessä testissä, johon liittyi metabolian aktivaatio, samoin kuin rotan luuytimen mikrotumissa tehdyssä mikrotumakokeessa. Mikrotumainduktion mekanismi oli poikkeava kromosomin segregaatio (aneugeenisuus) eikä klastogeeninen vaikutus kromosomeihin.
Hedelmällisyyden heikkeneminen
Eläimillä ei ole tehty erityisiä hedelmällisyyttä koskevia tutkimuksia alektinibin vaikutusten selvittämiseksi. Yleistä toksisuutta koskeneissa tutkimuksissa ei havaittu haitallisia vaikutuksia urosten tai naaraiden lisääntymiselimiin. Nämä tutkimukset tehtiin rotalla ja apinalla altistuksilla, jotka olivat rotalla vähintään 2,6-kertaiset ja apinalla vähintään 0,5-kertaiset ihmisen altistukseen nähden suositusannoksen 600 mg kaksi kertaa vuorokaudessa pitoisuus-aikakuvaajan pinta-alan (AUC) arvolla mitattuna.
Teratogeenisuus
Alektinibi aiheutti alkio-sikiötoksisuutta tiineille rotille ja kaniineille. Tiineille rotille alektinibi aiheutti täydellisiä alkioiden ja sikiöiden menetyksiä (keskenmenoja) altistuksilla, jotka olivat 4,5-kertaiset ihmisen AUC-altistukseen nähden, ja sikiöiden pienikokoisuutta, mihin liittyi luutumisen viivästymistä ja vähäisiä elinten poikkeavuuksia altistuksilla, jotka olivat 2,7-kertaiset ihmisen AUC-altistukseen nähden. Tiineille kaniineille alektinibi aiheutti alkioiden ja sikiöiden menetyksiä, sikiöiden pienikokoisuutta ja lisääntynyttä luustomuutosten esiintymistä altistuksilla, jotka olivat 2,9-kertaiset ihmisen suositusannosten AUC-altistukseen nähden.
Muut
Alektinibi absorboi 200–400 nm:n ultraviolettivaloa (UV-valoa) ja sillä osoitettiin fototoksisia vaikutuksia valoa koskevassa turvallisuuskokeessa in vitro, joka tehtiin UVA-säteilytetyssä hiiren fibroblastiviljelmässä.
Toistetuilla annoksilla tehdyissä toksikologisissa tutkimuksissa sekä rotan että apinan kohde-elimiä kliinisesti oleellisilla altistuksilla olivat mm. erytroidinen järjestelmä, maha-suolikanava sekä maksaan ja sappeen liittyvä järjestelmä.
Kun altistukset olivat suositusannoksen AUC-arvon perusteella vähintään 10–60 % ihmisen altistuksesta, havaittiin erytrosyyttien morfologisia poikkeavuuksia. Kun altistukset olivat suositusannoksen AUC-arvon perusteella vähintään 20–120 % ihmisen altistuksesta, kummallakin lajilla havaittiin maha-suolikanavan limakalvon proliferatiivisen alueen laajenemista. Kun rotan ja/tai apinan altistukset olivat suositusannoksen AUC-arvon perusteella vähintään 20–30 % ihmisen altistuksesta, havaittiin suurentuneita maksan alkalisen fosfataasin (AFOS) ja suoran bilirubiinin pitoisuuksia sekä sapenjohtimen epiteelin vakuolisaatiota/rappeutumista/nekroosia ja hepatosyyttien suurenemista/fokaalista nekroosia.
Apinoilla on havaittu suunnilleen kliinisesti oleellisilla altistuksilla lievää hypotensiivistä vaikutusta.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Laktoosimonohydraatti
Hydroksipropyyliselluloosa
Natriumlauryylisulfaatti
Magnesiumstearaatti
Karmelloosikalsium
Kapselin kuori
Hypromelloosi
Karrageeni
Kaliumkloridi
Titaanidioksidi (E 171)
Maissitärkkelys
Karnaubavaha
Painomuste
Punainen rautaoksidi (E 172)
Keltainen rautaoksidi (E 172)
Indigokarmiinialumiinilakka (E 132)
Karnaubavaha
Valkoinen shellakka
Glyserolimono-oleaatti
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
5 vuotta.
Säilytys
Läpipainopakkaukset
Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Purkit
Säilytä alkuperäispakkauksessa, ja pidä purkki tiiviisti suljettuna. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ALECENSA kapseli, kova
150 mg (L:kyllä) 224 fol (4x56) (4652,81 €)
PF-selosteen tieto
Alumiini/alumiiniläpipainopakkaukset (PA/Al/PVC/Al), jotka sisältävät 8 kovaa kapselia.
Pakkauskoko: 224 kovaa kapselia (neljä 56 kovan kapselin pakkausta).
HDPE-purkki, jossa turvasuljin ja kiinteä kuivausaine.
Pakkauskoko: 240 kovaa kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Valkoinen kova 19,2 mm:n pituinen kapseli, jonka kansiosaan on painettu mustalla musteella ˮALEˮ ja jonka runko-osaan on painettu mustalla musteella ˮ150 mgˮ.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ALECENSA kapseli, kova
150 mg 224 fol
- Ylempi erityiskorvaus (100 %). Alektinibi: Monoterapiana ALK-positiivista ei-pienisoluista keuhkosyöpää sairastavien aikuispotilaiden hoito erityisin edellytyksin (1504).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Alektinibi: Keuhkosyövän hoito erityisin edellytyksin (3001).
ATC-koodi
L01ED03
Valmisteyhteenvedon muuttamispäivämäärä
29.01.2026
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com