
ULTOMIRIS infuusiokonsentraatti, liuosta varten 300 mg/3 ml, 1100 mg/11 ml
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Lääkäri
Potilas
Opas potilaalle, vanhemmille ja lailliselle huoltajalle
Vaikuttavat aineet ja niiden määrät
Ultomiris on ravulitsumabivalmiste, joka on valmistettu kiinanhamsterin munasarjan (CHO) soluviljelmässä yhdistelmä-DNA-tekniikalla.
Ultomiris 300 mg/3 ml infuusiokonsentraatti, liuosta varten
Yksi 3 ml:n injektiopullo sisältää 300 mg ravulitsumabia (100 mg/ml).
Laimentamisen jälkeen infusoitavan liuoksen lopullinen pitoisuus on 50 mg/ml.
Apuaine(et), joiden vaikutus tunnetaan:
Natrium (4,6 mg 3 ml:n injektiopulloa kohden), polysorbaatti 80 (1,5 mg injektiopulloa kohden)
Ultomiris 1 100 mg/11 ml infuusiokonsentraatti, liuosta varten
Yksi 11 ml:n injektiopullo sisältää 1 100 mg ravulitsumabia (100 mg/ml).
Laimentamisen jälkeen infusoitavan liuoksen lopullinen pitoisuus on 50 mg/ml.
Apuaine(et), joiden vaikutus tunnetaan:
Natrium (16,8 mg 11 ml:n injektiopulloa kohden), polysorbaatti 80 (5,5 mg injektiopulloa kohden)
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti)
Kliiniset tiedot
Käyttöaiheet
Paroksysmaalinen nokturnaalinen hemoglobinuria (PNH)
Ultomiris on tarkoitettu aikuisten ja vähintään 10 kg painavien pediatristen potilaiden PNH:n hoitoon:
- hemolyysipotilailla, joilla on hyvin aktiiviseen sairauteen viittaavia kliinisiä oireita
- potilailla, jotka ovat kliiniseltä tilaltaan vakaita saatuaan hoitoa ekulitsumabilla vähintään viimeisten 6 kuukauden ajan
Epätyypillinen hemolyyttis-ureeminen oireyhtymä (aHUS)
Ultomiris on tarkoitettu aHUSin hoitoon aikuisille ja pediatrisille potilaille, jotka painavat vähintään 10 kg ja jotka eivät aikaisemmin ole saaneet komplementin estäjähoitoa tai jotka ovat saaneet ekulitsumabia vähintään 3 kuukauden ajan ja joilla on näyttöä vasteesta ekulitsumabille.
Yleistynyt myasthenia gravis (gMG)
Ultomiris on tarkoitettu gMG:n tavanomaisen hoidon lisähoidoksi gMG:tä sairastaville aikuisille, jotka ovat positiivisia asetyylikoliinireseptorin (AChR) vasta-aineille.
Neuromyelitis optica -kirjon häiriö (NMOSD)
Ultomiris on tarkoitettu aikuisille potilaille, joilla on todettu akvaporiini 4:n (AQP4) vasta-aineita (ks. kohta Farmakodynamiikka).
Ehto
Valmistetta saa antaa vain terveydenhuollon ammattilainen veri-, munuais-, neuromuskulaaristen tai neuroinflammatoristen tautien hoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Ravulitsumabia saa antaa vain terveydenhuollon ammattilainen veri-, munuais-, neuromuskulaaristen tai neuroinflammatoristen tautien hoitoon perehtyneen lääkärin valvonnassa.
Annostus
Aikuiset PNH-, aHUS-, gMG- ja NMOSD-potilaat
Suositeltu annostusohjelma käsittää latausannoksen ja ylläpitoannoksia, jotka annetaan infuusiona laskimoon. Annosten suuruus määräytyy potilaan painon mukaan, taulukon 1 mukaisesti. Ylläpitoannokset annetaan aikuispotilaille (≥ 18-vuotiaille) 8 viikon välein. Ensimmäinen ylläpitoannos annetaan 2 viikon kuluttua latausannoksesta.
Annoksen antoajankohta voi silloin tällöin poiketa enintään ±7 vuorokautta aikataulun mukaisesta infusointipäivästä (lukuun ottamatta ensimmäistä ravulitsumabin ylläpitoannosta), mutta sitä seuraava annos on annettava alkuperäisen aikataulun mukaisesti.
Taulukko 1: Painoon perustuva ravulitsumabinannostus vähintään 40 kg:n painoisille aikuispotilaille
| Painoalue (kg) | Latausannos (mg) | Ylläpitoannos (mg)* | Annosväli |
| ≥ 40, <60 | 2 400 | 3 000 | 8 viikon välein |
| ≥ 60, <100 | 2 700 | 3 300 | 8 viikon välein |
| ≥ 100 | 3 000 | 3 600 | 8 viikon välein |
*Ensimmäinen ylläpitoannos annetaan 2 viikon kuluttua latausannoksesta.
Ohjeet hoidon aloittamisesta potilaille, jotka eivät ole aikaisemmin saaneet komplementin estäjähoitoa tai jotka vaihtavat hoitoa ekulitsumabin, on annettu taulukossa 2.
Taulukko 2: Ravulitsumabihoidon aloitusohjeet
| Potilasryhmä | Ravulitsumabin painoon perustuva latausannos | Ravulitsumabin ensimmäinen painoon perustuva ylläpitoannos |
|---|---|---|
| Ei saa tällä hetkellä hoitoa ravulitsumabilla tai ekulitsumabilla | Hoidon alkaessa | 2 viikkoa ravulitsumabin latausannoksen jälkeen |
| Saa tällä hetkellä hoitoa ekulitsumabilla | Seuraavan aikataulun mukaisen ekulitsumabiannoksen antoajankohtana | 2 viikkoa ravulitsumabin latausannoksen jälkeen |
Pediatriset potilaat, joiden paino on ≥ 40 kg
Näitä potilaita on hoidettava aikuisten annostussuositusten mukaisesti (taulukko 1).
Pediatriset potilaat, joiden paino on ≥ 10 kg – < 40 kg
Painoon perustuvat annokset ja annosvälit pediatrisille potilaille, joiden paino on ≥ 10 kg – < 40 kg, on esitetty taulukossa 3.
Potilaille, jotka vaihtavat ekulitsumabista ravulitsumabiin, ravulitsumabin latausannos tulee antaa 2 viikon kuluttua viimeisestä ekulitsumabi-infuusiosta, minkä jälkeen ylläpitoannokset tulee antaa taulukossa 3 esitetyn painoon perustuvan annostuksen mukaisesti, alkaen 2 viikon kuluttua latausannoksesta.
Taulukko 3: Painoon perustuva ravulitsumabin annostus alle 40 kg painaville pediatrisille PNH- tai aHUS-potilaille
| Painoalue (kg) | Latausannos (mg) | Ylläpitoannos(mg)* | Annosväli |
| ≥ 10, < 20 | 600 | 600 | 4 viikon välein |
| ≥ 20, < 30 | 900 | 2 100 | 8 viikon välein |
| ≥ 30, < 40 | 1 200 | 2 700 | 8 viikon välein |
*Ensimmäinen ylläpitoannos annetaan 2 viikon kuluttua latausannoksesta.
Ravulitsumabia ei ole tutkittu pediatrisilla PNH-potilailla, jotka painavat alle 30 kg. Näiden potilaiden ravulitsumabiannostus perustuu pediatrisille aHUS-potilaille käytettyyn annostukseen ja ravulitsumabihoitoa saaneiden pediatristen aHUS- ja PNH-potilaiden farmakokineettisiin/farmakodynaamisiin saatavilla oleviin tietoihin.
PNH on krooninen sairaus, ja ravulitsumabihoitoa on suositeltavaa jatkaa potilaan eliniän ajan, jollei ravulitsumabihoidon lopettaminen ole kliinisesti aiheellista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
aHUS-potilailla tromboottisten mikroangiopatiatapahtumien (TMA) hoitoa ravulitsumabilla pitää jatkaa vähintään 6 kuukauden ajan. Tämän jälkeen hoidon kestoa on harkittava yksilöllisesti jokaisen potilaan kohdalla. Pitkäkestoinen hoito voi olla tarpeen, jos potilaalla on hoitavan lääkärin arvion mukaan suurentunut TMA:n uusiutumisriski (tai mikäli se on kliinisesti aiheellista) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Aikuisilla gMG- ja NMOSD-potilailla ravulitsumabihoitoa on tutkittu vain pitkäaikaisessa annossa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ravulitsumabia ei ole tutkittu gMG-potilailla, joiden MGFA (Myasthenia Gravis Foundation of America) -luokka on V.
Lisäannokset plasmanvaihdon, plasmafereesin tai laskimonsisäisen immunoglobuliinihoidon (IVIg-hoidon) jälkeen
Plasmanvaihdon, plasmafereesin ja IVIg-hoidon on osoitettu pienentävän ravulitsumabin pitoisuutta seerumissa. Plasmanvaihdon, plasmafereesin ja IVIg-hoidon yhteydessä on tarpeen antaa lisäannos ravulitsumabia (taulukko 4).
Taulukko 4: Ravulitsumabin lisäannos plasmafereesin, plasmanvaihdon tai IVIg-hoidon jälkeen
| Painoalue (kg) | Viimeisin ravulitsumabiannos (mg) | Lisäannos (mg) kunkin plasmanvaihto- tai plasmafereesi-intervention jälkeen | Lisäannos (mg) IVIg-hoitosyklin päättymisen jälkeen |
|---|---|---|---|
| ≥ 40 – < 60 | 2 400 | 1 200 | 600 |
| 3 000 | 1 500 | ||
| ≥ 60 – < 100 | 2 700 | 1 500 | 600 |
| 3 300 | 1 800 | ||
| ≥ 100 | 3 000 | 1 500 | 600 |
| 3 600 | 1 800 | ||
| Ravulitsumabin lisäannoksen ajoitus | 4 tunnin kuluessa kustakin plasmanvaihto- tai plasmafereesi-interventiosta | 4 tunnin kuluessa IVIg-hoitosyklin päättymisestä | |
Lyhenteet: IVIg = laskimonsisäinen immunoglobuliini, kg = kilogrammaa
Erityisryhmät
Iäkkäät potilaat
Annoksen säätäminen ei ole tarpeen 65-vuotiaille ja sitä vanhemmille PNH-, aHUS-, gMG- ja NMOSD-potilaille. Ei ole olemassa näyttöä siitä, että iäkkäiden potilaiden hoito vaatisi erityisiä varotoimia, joskin ravulitsumabin käytöstä iäkkäille PNH-, aHUS- ja NMOSD-kliinisissä tutkimuksissa on niukasti kokemusta.
Munuaisten vajaatoiminta
Annoksen säätäminen ei ole tarpeen munuaisten vajaatoimintaa sairastaville potilaille ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Ravulitsumabin turvallisuutta ja tehoa maksan vajaatoimintaa sairastavien potilaiden hoidossa ei ole tutkittu. Farmakokineettiset tiedot kuitenkin viittaavat siihen, ettei annoksen säätäminen ole tarpeen maksan vajaatoimintaa sairastaville potilaille.
Pediatriset potilaat
Ravulitsumabin turvallisuutta ja tehoa alle 10 kg:n painoisten pediatristen PNH- ja aHUS-potilaiden hoidossa ei ole varmistettu. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdassa Haittavaikutukset, ei voida antaa suosituksia annostuksesta.
Ravulitsumabin turvallisuutta ja tehoa pediatristen gMG- ja NMOSD-potilaiden hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Vain infuusiona laskimoon.
Tämä lääkevalmiste on annettava 0,2 µm:n suuruisen suodattimen läpi, eikä sitä saa antaa nopeana infuusiona eikä boluksena laskimoon. Ultomiris-valmisteen annon jälkeen koko letku huuhdellaan 0,9-prosenttisella natriumkloridi-injektionesteellä, USP.
3 ml:n ja 11 ml:n injektiopulloissa oleva Ultomiris-infuusiokonsentraatti on laimennettava 50 mg/ml:n lopulliseen pitoisuuteen. Laimentamisen jälkeen Ultomiris on annettava ruiskutyyppistä pumppua tai infuusiopumppua käyttäen vähintään 0,17–1,3 tunnin (10–75 minuutin) kestoisena infuusiona laskimoon. Infuusion minimikesto riippuu potilaan painosta (ks. taulukot 5 ja 6 alla).
Taulukko 5: Ultomiris annoksen antonopeus
| Painoalue (kg)a | Latausannos (mg) | Infuusion minimikesto minuutteina (tunteina) | Ylläpitoannos (mg) | Infuusion minimikesto minuutteina (tunteina) |
| ≥ 10, < 20b | 600 | 45 (0,8) | 600 | 45 (0,8) |
| ≥ 20, < 30b | 900 | 35 (0,6) | 2 100 | 75 (1,3) |
| ≥ 30, < 40b | 1 200 | 31 (0,5) | 2 700 | 65 (1,1) |
| ≥40, <60 | 2 400 | 45 (0,8) | 3 000 | 55 (0,9) |
| ≥60, <100 | 2 700 | 35 (0,6) | 3 300 | 40 (0,7) |
| ≥100 | 3 000 | 25 (0,4) | 3 600 | 30 (0,5) |
a Paino hoidon aikana.
b Vain PNH:n ja aHUSin käyttöaiheisiin.
Taulukko 6: Ultomiris lisäannoksen antonopeus
| Painoalue (kg)a | Lisäannosb (mg) | Infuusion minimikesto minuutteina (tunteina) |
≥ 40 – < 60
| 600 | 15 (0,25) |
| 1 200 | 25 (0,42) | |
| 1 500 | 30 (0,5) | |
| ≥ 60 – < 100 | 600 | 12 (0,20) |
| 1 500 | 22 (0,36) | |
| 1 800 | 25 (0,42) | |
| ≥ 100 | 600 | 10 (0,17) |
| 1 500 | 15 (0,25) | |
| 1 800 | 17 (0,28) |
a Paino hoidon aikana.
b Ravulitsumabin lisäannoksen valitseminen, ks. taulukko 4.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Potilaat, joilla on hoitamaton Neisseria meningitidis -infektio hoitoa aloitettaessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
- Potilaat, joilla ei ole voimassa olevaa rokotesuojaa Neisseria meningitidis -infektiota vastaan, elleivät he saa profylaktista hoitoa asianmukaisilla antibiooteilla siihen saakka, kunnes rokotuksesta on kulunut 2 viikkoa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Vakava meningokokki-infektio
Vaikutusmekanisminsa vuoksi ravulitsumabi lisää potilaan alttiutta meningokokki-infektiolle/-sepsikselle (Neisseria meningitidis). Minkä tahansa seroryhmän aiheuttamia meningokokki-infektioita saattaa esiintyä (ks. kohta Haittavaikutukset). Tämän infektioriskin pienentämiseksi kaikki potilaat on rokotettava meningokokki-infektioita vastaan vähintään 2 viikkoa ennen ravulitsumabihoidon aloittamista, ellei ravulitsumabihoidon viivästyttämiseen liittyvä riski ole suurempi kuin meningokokki-infektion riski. Jos potilaan ravulitsumabihoito aloitetaan ennen kuin meningokokkirokotteen saamisesta on kulunut 2 viikkoa, potilaan on saatava asianmukaista profylaktista antibioottihoitoa siihen saakka, kunnes rokotuksesta on kulunut 2 viikkoa. Yleisesti patogeenisten seroryhmien aiheuttamien meningokokki-infektioiden ehkäisemiseksi suositellaan rokotusta kaikkia seroryhmiä vastaan, joille rokote on saatavissa, mukaan lukien A, C, Y, W135 ja B. Potilaat on rokotettava ja uudelleen rokotettava voimassa olevien kansallisten rokotussuositusten mukaisesti. Jos potilaan hoito vaihdetaan ekulitsumabista, lääkärin tulee varmistaa, että potilaalla on kansallisten rokotussuositusten mukainen voimassa oleva rokotesuoja meningokokki-infektiota vastaan.
Rokotus ei välttämättä riitä ehkäisemään meningokokki-infektiota. Bakteerilääkkeiden asianmukaista käyttöä koskevat viralliset suositukset tulee huomioida. Vakavia tai kuolemaan johtaneita meningokokki-infektio/-sepsistapauksia on raportoitu potilailla, jotka ovat saaneet hoitoa ravulitsumabilla, ja potilailla, jotka ovat saaneet hoitoa muilla komplementin estäjillä. Kaikkia potilaita on seurattava meningokokki-infektion ja ‑sepsiksen varhaisten merkkien varalta, ja infektiota epäiltäessä potilas on tutkittava välittömästi ja hoito asianmukaisilla antibiooteilla aloitettava. Potilaille tulee kertoa näistä merkeistä ja oireista, ja heitä on neuvottava hakeutumaan lääkärinhoitoon välittömästi, jos niitä esiintyy. Lääkärien tulee antaa potilaille potilasopas ja potilaskortti.
Immunisaatio
On suositeltavaa, että potilaiden immunisaatio aloitetaan voimassa olevien rokotussuositusten mukaisesti ennen ravulitsumabihoidon aloittamista.
Rokotus saattaa tehostaa komplementin aktivoitumista edelleen. Tällöin komplementtivälitteisiä tauteja sairastavien potilaiden perussairauden merkit ja oireet saattavat lisääntyä. Tämän vuoksi potilaita tulee suositellun rokotuksen jälkeen seurata huolellisesti sairauden oireiden varalta.
Alle 18-vuotiaat potilaat on rokotettava Haemophilus influenzae- ja pneumokokki-infektioita vastaan, ja rokotuksissa on noudatettava tarkoin kutakin ikäryhmää koskevia kansallisia rokotussuosituksia.
Muut systeemiset infektiot
Ravulitsumabi-hoito on annettava varoen potilaille, joilla on aktiivisia systeemisiä infektioita. Ravulitsumabi estää komplementin terminaalisen osan aktivaation, minkä vuoksi potilaat saattavat olla tavallista alttiimpia Neisseria-bakteerilajien ja kapselillisten bakteerien aiheuttamille infektioille. Vakavia Neisseria-bakteerilajien (muiden kuin Neisseriameningitidis -lajin) aiheuttamia infektioita, mukaan lukien disseminoituneita gonokokki-infektioita, on raportoitu.
Potilaita on informoitava pakkausselosteen sisältämistä tiedoista mahdollisten vakavien infektioiden ja niiden merkkien ja oireiden tunnistamisen parantamiseksi. Lääkärien tulee neuvoa potilaita tippurin ehkäisyssä.
Infuusioon liittyvät reaktiot
Ravulitsumabin anto saattaa aiheuttaa systeemisiä infuusioon liittyviä reaktioita ja allergisia tai yliherkkyysreaktioita, mukaan lukien anafylaksiaa (ks. kohta Haittavaikutukset).
Systeemiseen infuusioon liittyvän reaktion tapauksessa, jossa potilaalla esiintyy merkkejä kardiovaskulaarisen tilan epävakaudesta tai hengityselinten toiminnan heikentymisestä, ravulitsumabin anto on keskeytettävä ja potilaalle on annettava asianmukaista elintoimintoja tukevaa hoitoa.
PNH-potilaan hoidon lopettaminen
Jos PNH-potilaan ravulitsumabihoito lopetetaan, potilasta on seurattava huolellisesti vakavan intravaskulaarisen hemolyysin merkkien ja oireiden varalta, joita ovat LDH (laktaattidehydrogenaasi) -pitoisuuden kohoaminen ja äkillinen PNH-kloonin koon tai hemoglobiiniarvon pienentyminen sekä esimerkiksi seuraavien oireiden uusiutuminen: väsymys, hemoglobinuria, vatsakipu, hengenahdistus, vakava verisuonitapahtuma (mukaan lukien tromboosi), nielemishäiriö tai erektiohäiriö. Potilaita, jotka lopettavat ravulitsumabihoidon, tulee seurata vähintään 16 viikon ajan hemolyysin ja muiden reaktioiden havaitsemiseksi. Jos hemolyysin merkkejä ja oireita, mukaan lukien LDH-pitoisuuden kohoamista, ilmaantuu lopettamisen jälkeen, ravulitsumabihoidon uudelleen aloittamista tulee harkita.
aHUS-potilaan hoidon lopettaminen
Ravulitsumabihoidon lopettamisesta ei ole spesifisiä tietoja. Pitkäkestoisessa prospektiivisessa havainnointitutkimuksessa C5-komplementin estäjähoidon (ekulitsumabin) lopettamisen jälkeen TMA uusiutui 13,5 kertaa useammin ja potilaiden munuaistoiminta heikkeni suuntauksenomaisesti verrattuna potilaisiin, jotka jatkoivat hoitoa.
Jos ravulitsumabihoito on lopetettava, potilasta on seurattava huolellisesti ja jatkuvasti TMA:n oireiden ja löydösten varalta. Seuranta ei kuitenkaan välttämättä riitä vaikeiden TMA-komplikaatioiden ennustamiseen tai ehkäisyyn.
Hoidon lopettamisen jälkeen esiintyvät TMA-komplikaatiot voidaan tunnistaa, jos potilaalla todetaan jokin seuraavista:
- vähintään 2 seuraavista laboratoriotuloksista samanaikaisesti: trombosyyttiarvon pieneneminen vähintään 25 %:lla lähtötilanteen arvosta tai ravulitsumabihoidon aikana todetusta suurimmasta trombosyyttiarvosta; seerumin kreatiniiniarvon suureneminen vähintään 25 %:lla lähtötilanteen arvosta tai ravulitsumabihoidon aikana todetusta pienimmästä arvosta; tai seerumin LDH-arvon suureneminen vähintään 25 %:lla lähtötilanteen arvosta tai ravulitsumabihoidon aikana todetusta pienimmästä arvosta (tulokset on vahvistettava toistamalla mittaus)
tai
-
mikä tahansa seuraavista TMA-oireista: mielentilan muutos tai epileptiset kohtaukset tai muut munuaisten ulkopuoliset TMA-oireet, kuten sydämen ja verisuoniston poikkeavuudet, perikardiitti, ruoansulatuskanavan oireet / ripuli; tai verisuonitukos.
Jos TMA-komplikaatioita ilmaantuu ravulitsumabihoidon lopettamisen jälkeen, on harkittava ravulitsumabihoidon aloittamista uudelleen lataus- ja ylläpitoannoksilla (ks. kohta Annostus ja antotapa.)
gMG-potilaan hoidon lopettaminen
Koska gMG on krooninen sairaus, ravulitsumabihoidosta hyötyviä potilaita, jotka lopettavat hoidon, on seurattava perussairauden oireiden varalta. Jos gMG:n oireita esiintyy lopettamisen jälkeen, ravulitsumabihoidon uudelleen aloittamista on harkittava.
NMOSD-potilaan hoidon lopettaminen
Koska NMOSD on krooninen sairaus, ravulitsumabihoidosta hyötyviä potilaita, jotka lopettavat hoidon, on seurattava NMOSD:n relapsin oireiden varalta. Jos NMOSD:n relapsin oireita esiintyy lopettamisen jälkeen, ravulitsumabihoidon uudelleen aloittamista on harkittava.
Vaihtaminen ekulitsumabista ravulitsumabiin
Ravulitsumabihoitoa ei suositella gMG-potilaille, jotka eivät reagoi ekulitsumabihoitoon hyväksytyllä annostuksella.
Natriumpitoisuus
0,9 prosenttisella (9 mg/ml) natriumkloridi-injektionesteellä laimennettuna tämä lääkevalmiste sisältää 0,18 g natriumia per 72 ml enimmäisannoksella, joka vastaa 9,1 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Polysorbaatti 80 -pitoisuus
Tämä lääkevalmiste sisältää 1,5 mg polysorbaattia 80 per 3 ml:n injektiopullo ja 5,5 mg per 11 ml:n injektiopullo, joka vastaa enintään 0,53 mg/kg aikuisille potilaille ja yli 10 kg:n painoisille pediatrisille potilaille annettavalla enimmäisannoksella. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Yhteisvaikutustutkimuksia ei ole tehty. Koska ravulitsumabi saattaa estää rituksimabin aiheuttamaa komplementtiriippuvaista sytotoksisuutta, ravulitsumabi saattaa heikentää rituksimabin odotettuja farmakodynaamisia vaikutuksia.
Pitkäaikainen laskimonsisäinen ihmisen immunoglobuliinihoito (IVIg-hoito) saattaa häiritä monoklonaalisten vasta-aineiden kuten ravulitsumabin kierrätysmekanismia endosomien neonataalivastasyntyneen Fc-reseptoreissa (FcRn) ja siten pienentää ravulitsumabin pitoisuutta seerumissa.
Samanaikaisen plasmanvaihdon, plasmafereesin tai laskimonsisäisen immunoglobuliinihoidon (IVIg-hoidon) tapauksessa ks. ohjeet kohdasta Annostus ja antotapa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta ehkäisyä hoidon aikana ja 8 kuukautta hoidon päättymisen jälkeen.
Raskaus
Ei ole olemassa kliinisiä tietoja ravulitsumabin käytöstä raskaana oleville naisille.
Lisääntymistoksisuutta koskevissa eläinkokeissa ei käytetty ravulitsumabia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Lisääntymistoksisuustutkimukset tehtiin hiirillä käyttämällä hiiren korvikemolekyyliä BB5.1, jonka avulla arvioitiin C5-proteiinin eston vaikutusta sukupuolielimiin. Näissä tutkimuksissa ei havaittu erityisiä testattavaan aineeseen liittyviä toksisuuksia. Ihmisen immunoglobuliini G:n (IgG) tiedetään läpäisevän ihmisen veri-istukkaesteen, joten on mahdollista, että ravulitsumabilla on komplementin terminaalista osaa estävä vaikutus sikiön verenkierrossa.
Ei ole tehty riittäviä eläinkokeita lisääntymistoksisuuden selvittämiseksi (ks. kohta Prekliiniset tiedot turvallisuudesta).
Ravulitsumabin käyttöä raskaana oleville naisille voidaan harkita hyöty-riskiarvion jälkeen.
Imetys
Ei tiedetä, erittyykö ravulitsumabi ihmisen rintamaitoon. Hiirillä tehdyissä ei-kliinisissä lisääntymistoksisuustutkimuksissa, joissa käytettiin hiiren korvikemolekyyliä BB5.1, ei havaittu haittavaikutuksia hoitoa saaneiden emojen imettämissä poikasissa.
Imeväiseen kohdistuvia riskejä ei voida poissulkea.
Koska monet lääkevalmisteet ja immunoglobuliinit erittyvät ihmisen rintamaitoon, ja koska vakavia haittavaikutuksia imeväisille ei voida poissulkea, rintaruokinta on lopetettava ravulitsumabihoidon ajaksi ja 8 kuukaudeksi hoidon päättymisen jälkeen.
Hedelmällisyys
Ravulitsumabilla ei ole tehty erityisiä hedelmällisyyttä koskevia konventionaalisia tutkimuksia.
Hiirillä tehdyissä konventionaalisissa lisääntymistoksisuutta koskevissa tutkimuksissa, joissa käytettiin hiiren korvikemolekyyliä BB5.1, hoitoa saaneilla naarailla ja uroksilla ei havaittu hedelmällisyyteen kohdistuvia haittavaikutuksia.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ultomiris-valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Ravulitsumabin yleisimmät haittavaikutukset ovat päänsärky (30,6 %), ylähengitystieinfektio (21,6 %), nenänielutulehdus (20,4 %), ripuli (18,7 %), kuume (17,7 %), pahoinvointi (15 %), nivelkipu (14,4 %), selkäkipu (13,6 %), uupumus (13,3 %), vatsakipu (12,3 %), huimaus (10,7 %) ja virtsatieinfektio (10,7 %). Vakavimmat haittavaikutukset ovat meningokokki-infektio (0,7 %) mukaan lukien meningokokkisepsis, meningokokkiaivokalvotulehdus, meningokokkiaivotulehdus ja meningokokki-infektio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) sekä disseminoitunut gonokokki-infektio (0,2 %), mukaan lukien disseminoitunut gonokokki-infektio ja gonokokki-infektio.
Haittavaikutusten taulukkomuotoinen luettelo
Taulukossa 7 on lueteltu kliinisissä tutkimuksissa ja markkinoille tulon jälkeen havaitut haittavaikutukset.
Haittavaikutukset on lueteltu MedDRA:n elinjärjestelmäluokkien ja seuraavien yleisyysluokkien mukaan: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on kussakin yleisyysluokassa esitetty vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 7: Haittavaikutukset kliinisissä tutkimuksissa ja markkinoille tulon jälkeen
| MedDRA-elinjärjestelmäluokka | Hyvin yleinen (≥ 1/10) | Yleinen (≥ 1/100, < 1/10) | Melko harvinainen (≥ 1/1 000, < 1/100) |
|---|---|---|---|
| Infektiot | Virtsatieinfektioa Ylähengitystieinfektio, Nenänielutulehdus | Meningokokki-infektiob Disseminoitunut gonokokki-infektiod | |
| Immuunijärjestelmä | Yliherkkyyse | Anafylaktinen reaktiod
| |
| Hermosto | Päänsärky, Huimaus | ||
| Ruoansulatuselimistö | Ripuli, Pahoinvointi, Vatsakipu | Oksentelu, Dyspepsia | |
| Iho ja ihonalainen kudos | Nokkosihottuma, Kutina, Ihottuma
| ||
| Luusto, lihakset ja sidekudos | Nivelkipu, Selkäkipu | Lihaskipu, Lihaskouristukset | |
| Yleisoireet ja antopaikassa todettavat haitat | Kuume, Uupumus | Influenssan kaltainen sairaus, Vilunväristykset, Voimattomuus
| |
| Vammat, myrkytykset ja hoitokomplikaatiot | Infuusioon liittyvä reaktio |
aVirtsatieinfektio on yleistermi, joka käsittää suositellut termit (Preferred Terms) virtsatieinfektio, virtsateiden bakteeri-infektio, enterokokki-virtsatieinfektio ja Escherichia-virtsatieinfektio.
bMeningokokki-infektio käsittää suositellut termit (Preferred Terms) meningokokki-infektio, meningokokkisepsis, meningokokkiaivokalvotulehdus ja meningokokkiaivotulehdus.
c Disseminoitunut gonokokki-infektio käsittää suositellut termit (Preferred Terms) disseminoitunut gonokokki-infektio ja gonokokki-infektio.
d Arvioitu markkinoille tulon jälkeisen kokemuksen perusteella.
e Yliherkkyys käsittää suositellut termit (Preferred Terms) lääkeyliherkkyys, johon liittyy syy-yhteys, ja yliherkkyys.
Valittujen haittavaikutusten kuvaus
Meningokokki-infektio/-sepsis/-aivotulehdus
Rokotus pienentää meningokokki-infektioiden riskiä, muttei poista riskiä kokonaan. Kliinisiin tutkimuksiin osallistuneista potilaista < 1 %:lle kehittyi vakava meningokokki-infektio ravulitsumabihoidon aikana. Kaikki nämä potilaat olivat aikuisia PNH- tai NMOSD-potilaita, jotka oli rokotettu. Ks. kohdasta Varoitukset ja käyttöön liittyvät varotoimet tietoja meningokokki-infektiosta ja sen ehkäisystä sekä epäillyn meningokokki-infektion hoidosta. Ravulitsumabihoitoa saaneilla potilailla meningokokki-infektiot ovat ilmenneet meningokokkisepsiksenä ja meningokokkiaivotulehduksena. Potilaille tulee kertoa meningokokki-infektion merkeistä ja oireista, ja heitä on neuvottava hakeutumaan lääkärinhoitoon välittömästi, jos niitä esiintyy
Infuusioon liittyvät reaktiot
Infuusioon liittyvät reaktiot olivat yleisiä (≥ 1 %) kliinisissä tutkimuksissa. Nämä tapahtumat olivat vaikeusasteeltaan lieviä tai keskivaikeita sekä ohimeneviä, ja niitä olivat selkäkipu, vatsakipu, lihaskouristukset, verenpaineen lasku, verenpaineen nousu, vilunväristykset, epämukavuuden tunne raajoissa, lääkeyliherkkyys (allerginen reaktio), dysgeusia (paha maku suussa) ja uneliaisuus. Nämä reaktiot eivät vaatineet ravulitsumabihoidon lopettamista.
Immunogeenisuus
Aikuisilla PNH-potilailla tehdyissä tutkimuksissa (N = 475), pediatrisilla PNH-potilailla tehdyssä tutkimuksessa (N = 13), aHUS-potilailla tehdyissä tutkimuksissa (N = 89) gMG-potilailla tehdyssä tutkimuksessa (N = 86) ja NMOSD-potilailla tehdyssä tutkimuksessa (N = 58) havaittiin kaksi (0,3 %) tapausta, joissa potilaalle kehittyi hoidon aikana lääkevasta-aineita ravulitsumabille (1 aikuinen PNH-potilas ja 1 aikuinen aHUS-potilas). Nämä lääkevasta-aineet olivat ohimeneviä ja titteriltään pieniä, eivätkä ne korreloituneet kliinisen vasteen tai haittatapahtumien kanssa.
Pediatriset potilaat
Paroksysmaalinen nokturnaalinen hemoglobinuria (PNH)
Pediatriseen PNH-tutkimukseen (ALXN1210‑PNH‑304) otetuilla potilailla (N = 13, ikä 9–17 vuotta) ravulitsumabin turvallisuusprofiili vaikutti samankaltaiselta kuin aikuisilla PNH-potilailla. Yleisimmät pediatrisilla PNH-potilailla raportoidut haittavaikutukset olivat vatsakipu, pahoinvointi, nenänielutulehdus ja päänsärky, joita esiintyi kolmella potilaalla (23,1 %).
Epätyypillinen hemolyyttis-ureeminen oireyhtymä (aHUS)
ALXN1210‑aHUS‑312-tutkimukseen otetuilla pediatrisilla potilailla (N = 34, ikä vähintään 10 kuukautta ja alle 18 vuotta), joilla oli näyttöä aHUS-tapahtumista, ravulitsumabin turvallisuusprofiili vaikutti samankaltaiselta kuin aikuispotilailla, joilla oli näyttöä aHUS-tapahtumista. Turvallisuusprofiilit eri ikäisten pediatristen potilaiden alaryhmissä vaikuttavat samankaltaisilta. Tiedot turvallisuudesta alle 2‑vuotiaille potilaille rajoittuvat neljään potilaaseen. Yleisimmät (> 20 %) pediatrisilla potilailla raportoidut haittavaikutukset olivat kuume, oksentelu, ripuli, päänsärky, nenänielutulehdus, ylähengitystieinfektio ja vatsakipu.
Yleistynyt myasthenia gravis (gMG)
Ravulitsumabia ei ole tutkittu pediatrisilla gMG-potilailla.
Neuromyelitis optica -kirjon häiriö (NMOSD)
Ravulitsumabia ei ole tutkittu pediatrisilla NMOSD-potilailla.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Jos potilas saa yliannoksen, infuusio on keskeytettävä välittömästi ja potilasta on seurattava huolellisesti haittavaikutusten merkkien tai oireiden varalta. Asianmukainen oireenmukainen hoito on aloitettava.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, komplementin estäjät, ATC-koodi: L04A J02
Vaikutusmekanismi
Ravulitsumabi on monoklonaalinen IgG2/4K-vasta-aine, joka sitoutuu spesifisesti komplementtiproteiiniin C5, estäen sitä pilkkoutumasta C5a:ksi (proinflammatorinen anafylatoksiini) ja C5b:ksi (solukalvoa tuhoavan kompleksin [MAC tai C5b‑9] käynnistävä alayksikkö) ja estäen siten C5b-9:ää muodostumasta. Ravulitsumabi säilyttää komplementin aktivaation varhaiset komponentit, jotka ovat välttämättömiä mikro-organismien opsonisaatiolle ja immuunikompleksien poistamiselle.
Farmakodynaamiset vaikutukset
Faasin 3 tutkimuksissa ravulitsumabihoidolla havaittiin sekä komplementin estäjiä aiemmin saamattomilla aikuisilla ja pediatrisilla potilailla että ekulitsumabia aiemmin saaneilla PNH-potilailla olevan välitön, täydellinen ja pitkäkestoinen seerumin vapaata C5-proteiinia estävä vaikutus (pitoisuus <0,5 µg/ml) ensimmäisen infuusion päättymiseen mennessä, ja tämä vaikutus säilyi koko 26-viikkoisen hoitojakson ajan kaikilla potilailla. Välitön ja täydellinen seerumin vapaata C5-proteiinia estävä vaikutus havaittiin myös aikuisilla ja pediatrisilla aHUS-potilailla, aikuisilla gMG-potilailla ja aikuisilla NMOSD-potilailla ensimmäisen infuusion päättymiseen mennessä, ja tämä vaikutus säilyi koko ensisijaisen hoitojakson ajan.
PNH-, aHUS-, gMG- ja NMOSD-potilaiden farmakodynaamisen vasteen laajuus ja kesto riippuivat ravulitsumabialtistuksesta. Alle 0,5 µg/ml:n vapaan C5-proteiinin pitoisuus korreloitui maksimaalisen intravaskulaarisen hemolyysin hallinnan ja täydellisen komplementin terminaalisen osan eston kanssa. gMG:ssä komplementin terminaalisen osan aktivoituminen johtaa solukalvoa tuhoavan kompleksin kertymiseen hermo-lihasliitoksessa ja hermo-lihasjohtumisen heikentymiseen. NMOSD-potilailla komplementin terminaalisen osan aktivaatio johtaa MAC:n muodostukseen sekä C5a:sta riippuvaiseen tulehdukseen, astrosyyttien nekroosiin ja ympäröivien gliasolujen ja neuronien vaurioitumiseen.
Kliininen teho ja turvallisuus
Paroksysmaalinen nokturnaalinen hemoglobinuria (PNH)
Ravulitsumabin turvallisuutta ja tehoa aikuisilla PNH-potilailla arvioitiin kahdessa avoimessa, satunnaistetussa, aktiivikontrolloidussa faasin 3 tutkimuksessa:
- tutkimuksessa, johon osallistui aikuisia PNH-potilaita, jotka eivät olleet aiemmin saaneet hoitoa komplementin estäjillä,
- tutkimuksessa, johon osallistui aikuisia PNH-potilaita, jotka olivat kliiniseltä tilaltaan vakaita saatuaan ekulitsumabihoitoa vähintään edellisten 6 kuukauden ajan.
Ravulitsumabia annettiin kohdassa Annostus ja antotapa kuvatun suositellun annostusohjelman mukaisesti (4 ravulitsumabi-infuusiota 26 viikon aikana) ja ekulitsumabia ekulitsumabin suositellun annostusohjelman mukaisesti eli 600 mg kerran viikossa ensimmäisten 4 viikon ajan ja sen jälkeen 900 mg 2 viikon välein (15 infuusiota 26 viikon aikana).
Potilaat rokotettiin meningokokki-infektiota vastaan ennen ravulitsumabi- tai ekulitsumabihoidon aloittamista tai aloittamisen yhteydessä tai heille annettiin profylaktista hoitoa asianmukaisilla antibiooteilla siihen saakka, kunnes rokotuksesta oli kulunut 2 viikkoa.
Demografiset ja lähtötilanteen ominaisuudet ravulitsumabi- ja ekulitsumabihoitoryhmien välillä eivät eronneet merkittävästi toisistaan kummassakaan faasin 3 tutkimuksessa. Edellisten 12 kuukauden aikana saatujen verensiirtojen lukumäärä oli samankaltainen ravulitsumabi- ja ekulitsumabihoitoryhmien välillä kummassakin faasin 3 tutkimuksessa.
Tutkimus aikuisilla PNH-potilailla, jotka eivät olleet aiemmin saaneet hoitoa komplementin estäjillä (ALXN1210-PNH-301)
Tutkimus komplementin estäjiä aiemmin saamattomilla potilailla oli 26 viikon pituinen, avoin, satunnaistettu, aktiivikontrolloitu faasin 3 monikeskustutkimus, johon osallistui 246 potilasta. Sitä seurasi pitkäkestoinen jatkovaihe, jonka aikana kaikki potilaat saivat ravulitsumabia. Tutkimukseen otettiin potilaita, joiden sairaus oli hyvin aktiivinen, määritelmänä lähtötilanteen LDH-pitoisuus ≥1,5 × viitealueen yläraja seulonnassa ja vähintään yksi seuraavista PNH:n merkeistä tai oireista 3 kuukauden kuluessa seulonnasta: väsymys, hemoglobinuria, vatsakipu, hengenahdistus, anemia (hemoglobiini < 100 g/l), vakava verisuonitapahtuma (mukaan lukien tromboosi), nielemishäiriö tai erektiohäiriö, tai aiempi punasolusiirto PNH:n vuoksi.
Yli 80 % potilaista kummassakin hoitoryhmässä oli saanut verensiirron tutkimuksen alkamista edeltävien 12 kuukauden aikana. Suurin osa potilaista tutkimuksessa komplementin estäjiä aiemmin saamattomista potilaista oli erittäin hemolyyttisiä lähtötilanteessa; 86,2 %:lla mukaan otetuista potilaista LDH-pitoisuus oli ≥3 × viitealueen yläraja. LDH-pitoisuus on suora intravaskulaarisen hemolyysin mittari PNH:ssa.
Taulukossa 8 on esitetty komplementin estäjiä aiemmin saamattomilla potilailla tehtyyn tutkimukseen otettujen PNH:ta sairastavien potilaiden lähtötilanteen ominaisuudet, joissa ei havaittu selviä kliinisesti merkittäviä eroja hoitoryhmien välillä.
Taulukko 8: Lähtötilanteen ominaisuudet komplementin estäjiä aiemmin saamattomilla potilailla tehdyssä tutkimuksessa
| Muuttuja | Tunnusluku | Ravulitsumabi(N = 125) | Ekulitsumabi(N = 121) |
| Ikä (vuotta) PNH-diagnoosin yhteydessä | Keskiarvo (keskihajonta) | 37,9 (14,90) | 39,6 (16,65) |
| Mediaani | 34,0 | 36,5 | |
| Min., maks. | 15, 81 | 13, 82 | |
| Ikä (vuotta) tutkimuksen ensimmäisen infuusion yhteydessä | Keskiarvo (keskihajonta) | 44,8 (15,16) | 46,2 (16,24) |
| Mediaani | 43,0 | 45,0 | |
| Min., maks. | 18, 83 | 18, 86 | |
| Sukupuoli (n, %) | Mies | 65 (52,0) | 69 (57,0) |
| Nainen | 60 (48,0) | 52 (43,0) | |
| Hoitoa edeltävä LDH-pitoisuus | Keskiarvo (keskihajonta) | 1 633,5 (778,75) | 1 578.3 (727,06) |
| Mediaani | 1 513,5 | 1 445,0 | |
| Potilaat, jotka saivat punasolusiirron ensimmäistä annosta edeltävien 12 kuukauden aikana | n (%) | 103 (82,4) | 100 (82,6) |
| Siirrettyjen punasoluyksiköiden lukumäärä ensimmäistä annosta edeltävien 12 kuukauden aikana | Yhteensä | 925 | 861 |
| Keskiarvo (keskihajonta) | 9,0 (7,74) | 8,6 (7,90) | |
| Mediaani | 6,0 | 6,0 | |
| PNH-punasolukloonin koko | Mediaani | 33,6 | 34,2 |
| PNH-granulosyyttikloonin koko | Mediaani | 93,8 | 92,4 |
| Potilaat, joilla oli PNH:hon liittyviä sairaustilojaa ennen tietoon perustuvaa suostumusta | n (%) | 121 (96,8) | 120 (99,2) |
| Anemia | 103 (82,4) | 105 (86,8) | |
| Hematuria tai hemoglobinuria | 81 (64,8) | 75 (62,0) | |
| Aplastinen anemia | 41 (32,8) | 38 (31,4) | |
| Munuaisten vajaatoiminta | 19 (15,2) | 11 (9,1) | |
| Myelodysplastinen oireyhtymä | 7 (5,6) | 6 (5,0) | |
| Raskauskomplikaatio | 3 (2,4) | 4 (3,3) | |
| Muub | 27 (21,6) | 13 (10,7) |
a Potilaskertomuksen perusteella.
b Raportointilomakkeessa olevaan ”Muu”-kategoriaan kuului trombosytopenia, krooninen munuaissairaus, pansytopenia ja useita muita sairaustiloja.
Ensisijaiset päätetapahtumat olivat verensiirron tarpeelta välttyminen sekä hemolyysi, joka mitattiin suoraan LDH-pitoisuuden normalisoitumisena (LDH-pitoisuus ≤1 × viitealueen yläraja, joka on 246 U/l). Tärkeimmät toissijaiset päätetapahtumat olivat LDH-pitoisuuden prosenttimuutos lähtötilanteesta, elämänlaadun muutos (väsymystä mittaavissa FACIT-Fatigue-pisteissä), niiden potilaiden osuus, joilla esiintyi läpilyöntihemolyysiä, ja niiden potilaiden osuus, joiden hemoglobiini stabilisoitui.
Ravulitsumabi oli vertailukelpoinen ekulitsumabiin nähden molempien ensisijaisten päätetapahtumien, eli punasolusiirron tarpeelta välttymisen tutkimussuunnitelmassa määriteltyjen suositusten mukaisesti ja LDH-pitoisuuden normalisoitumisen päivästä 29 päivään 183, suhteen sekä kaikkien neljän tärkeimmän toissijaisen päätetapahtuman suhteen (kuva 1).
Kuva 1: Ensisijaisten ja toissijaisten päätetapahtumien analyysi – koko analyysijoukko (komplementin estäjiä aiemmin saamattomilla potilailla tehty tutkimus)
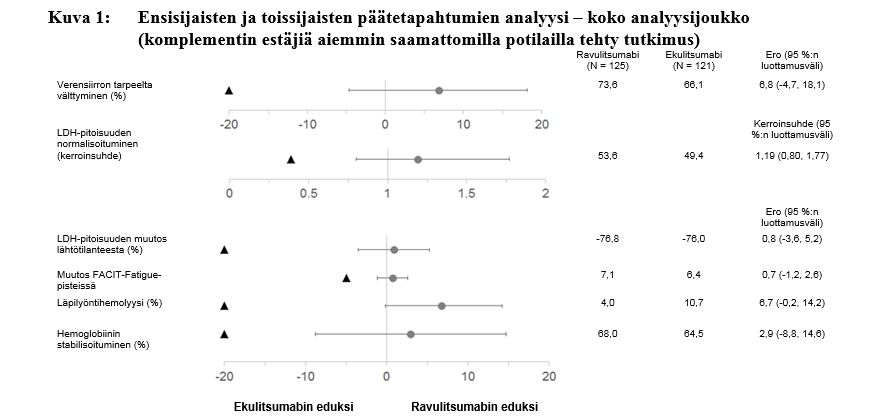
Huom: Mustat kolmiot osoittavat vertailukelpoisuusmarginaalit ja harmaat pisteet piste-estimaatit.
Huom: LDH = laktaattidehydrogenaasi; FACIT = kroonisen sairauden hoidon funktionaalinen arviointi (Functional Assessment of Chronic Illness Therapy).
Tutkimuksen lopulliseen tehokkuusanalyysiin kuuluivat kaikki ravulitsumabihoitoa milloin tahansa saaneet potilaat (n = 244). Näiden potilaiden hoidon mediaanikesto oli 1 423 päivää. Lopullisella analyysillä vahvistettiin, että ensisijaisen arviointijakson aikana havaitut ravulitsumabihoidon vasteet säilyivät koko tutkimuksen ajan.
Tutkimus aikuisilla PNH-potilailla, jotka olivat aiemmin saaneet hoitoa ekulitsumabilla (ALXN1210-PNH-302)
Tutkimus ekulitsumabia aiemmin saaneilla potilailla oli 26 viikon pituinen, avoin, satunnaistettu, aktiivikontrolloitu faasin 3 monikeskustutkimus, johon osallistui 195 PNH-potilasta, jotka olivat kliiniseltä tilaltaan vakaita (LDH-pitoisuus ≤1,5 × viitealueen yläraja) saatuaan hoitoa ekulitsumabilla vähintään edellisten 6 kuukauden ajan. Sitä seurasi pitkäkestoinen jatkovaihe, jonka aikana kaikki potilaat saivat ravulitsumabia.
PNH-sairauskertomus oli samankaltainen ravulitsumabi- ja ekulitsumabihoitoryhmien välillä. Edellisten 12 kuukauden aikana saatujen verensiirtojen lukumäärä oli samankaltainen ravulitsumabi- ja ekulitsumabihoitoryhmien välillä, ja yli 87 % potilaista kummassakin hoitoryhmässä ei ollut saanut verensiirtoa tutkimuksen alkamista edeltävien 12 kuukauden aikana. Keskimääräinen PNH-punasolukloonin koko oli 60,05 %, keskimääräinen PNH-granulosyyttikloonin koko oli 83,30 % ja keskimääräinen PNH-monosyyttikloonin koko oli 85,86 %.
Taulukossa 9 on esitetty ekulitsumabia aiemmin saaneilla potilailla tehtyyn tutkimukseen otettujen PNH-potilaiden lähtötilanteen ominaisuudet, joissa ei havaittu selviä kliinisesti merkittäviä eroja hoitoryhmien välillä.
Taulukko 9: Lähtötilanteen ominaisuudet ekulitsumabia aiemmin saaneilla potilailla tehdyssä tutkimuksessa
| Muuttuja | Tunnusluku | Ravulitsumabi(N = 97) | Ekulitsumabi(N = 98) |
| Ikä (vuotta) PNH-diagnoosin yhteydessä | Keskiarvo (keskihajonta) | 34,1 (14,41) | 36,8 (14,14) |
| Mediaani | 32,0 | 35,0 | |
| Min., maks. | 6, 73 | 11, 74 | |
| Ikä (vuotta) tutkimuksen ensimmäisen infuusion yhteydessä | Keskiarvo (keskihajonta) | 46,6 (14,41) | 48,8 (13,97) |
| Mediaani | 45,0 | 49,0 | |
| Min., maks. | 18, 79 | 23, 77 | |
| Sukupuoli (n, %) | Mies | 50 (51,5) | 48 (49,0) |
| Nainen | 47 (48,5) | 50 (51,0) | |
| Hoitoa edeltävä LDH-pitoisuus | Keskiarvo (keskihajonta) | 228,0 (48,71) | 235,2 (49,71) |
| Mediaani | 224,0 | 234,0 | |
| Potilaat, jotka saivat punasolujen/kokoveren siirtoja ensimmäistä annosta edeltävien 12 kuukauden aikana | n (%) | 13 (13,4) | 12 (12,2) |
| Siirrettyjen punasolu-/kokoveriyksiköiden lukumäärä ensimmäistä annosta edeltävien 12 kuukauden aikana | Yhteensä | 103 | 50 |
| Keskiarvo (keskihajonta) | 7,9 (8,78) | 4,2 (3,83) | |
| Mediaani | 4,0 | 2,5 | |
| Potilaat, joilla oli PNH:hon liittyviä sairaustilojaa ennen tietoon perustuvaa suostumusta | n (%) | 90 (92,8) | 96 (98,0) |
| Anemia | 64 (66,0) | 67 (68,4) | |
| Hematuria tai hemoglobinuria | 47 (48,5) | 48 (49,0) | |
| Aplastinen anemia | 34 (35,1) | 39 (39,8) | |
| Munuaisten vajaatoiminta | 11 (11,3) | 7 (7,1) | |
| Myelodysplastinen oireyhtymä | 3 (3,1) | 6 (6,1) | |
| Raskauskomplikaatio | 4 (4,1) | 9 (9,2) | |
| Muub | 14 (14,4) | 14 (14,3) |
a Potilaskertomuksen perusteella.
b Raportointilomakkeessa olevaan ”Muu”-kategoriaan kuului neutropenia, krooninen munuaissairaus, trombosytopenia ja useita muita sairaustiloja.
Ensisijainen päätetapahtuma oli hemolyysi, joka mitattiin LDH-pitoisuuden prosenttimuutoksena lähtötilanteesta. Toissijaiset päätetapahtumat olivat niiden potilaiden osuus, joilla esiintyi läpilyöntihemolyysiä, elämänlaadun muutos (väsymystä mittaavissa FACIT-Fatigue-pisteissä), verensiirron tarpeelta välttyminen ja niiden potilaiden osuus, joiden hemoglobiini stabilisoitui.
Ravulitsumabi oli vertailukelpoinen ekulitsumabiin nähden ensisijaisen päätetapahtuman, eli LDH-pitoisuuden prosenttimuutoksen lähtötilanteesta päivään 183, suhteen sekä kaikkien neljän tärkeimmän toissijaisen päätetapahtuman suhteen (kuva 2).
Kuva 2: Ensisijaisten ja toissijaisten päätetapahtumien analyysi – koko analyysijoukko (ekulitsumabia aiemmin saaneilla potilailla tehty tutkimus)
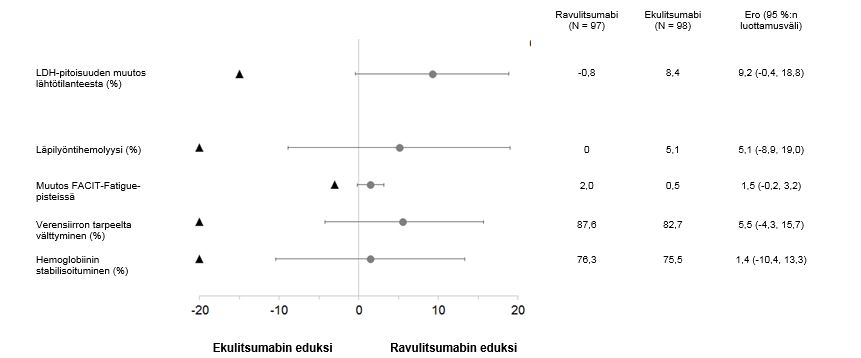
Huom: Mustat kolmiot osoittavat vertailukelpoisuusmarginaalit ja harmaat pisteet piste-estimaatit.
LDH = laktaattidehydrogenaasi.
Tutkimuksen lopulliseen tehokkuusanalyysiin kuuluivat kaikki ravulitsumabihoitoa milloin tahansa saaneet potilaat (n=192). Näiden potilaiden hoidon mediaanikesto oli 968 päivää. Lopullisella analyysilla vahvistettiin, että ensisijaisen arviointijakson aikana havaitut ravulitsumabihoidon vasteet säilyivät koko tutkimuksen ajan.
Epätyypillinen hemolyyttis-ureeminen oireyhtymä (aHUS)
Tutkimus aikuisilla aHUS-potilailla (ALXN1210-aHUS-311)
Aikuisilla tehty tutkimus oli yhdellä hoitoryhmällä toteutettu faasin 3 monikeskustutkimus, johon otetuilla potilailla oli dokumentoitu aHUS, ei ollut aiempia komplementin estäjähoitoja ennen tutkimukseen osallistumista sekä oli näyttöä tromboottisesta mikroangiopatiasta (TMA). Tutkimus koostui 26 viikon pituisesta alkuarviointivaiheesta, jonka jälkeen potilailla oli mahdollisuus osallistua enintään 4,5 vuotta kestävään jatkovaiheeseen.
Tutkimukseen otettiin yhteensä 58 potilasta, joilla oli dokumentoitu aHUS. Tutkimuksen mukaanottokriteerit sulkivat pois potilaat, joilla oli ADAMTS13:n (disintegriini ja metalloproteinaasi, jossa on trombospondiini 13 rakennekuvioita) puutoksesta johtuva TMA tai Shiga-toksiinia tuottavan Escherichia colin aiheuttama hemolyyttis-ureeminen oireyhtymä (STEC-HUS) ja kobalamiini C metabolian geneettinen häiriö. Kaksi potilasta suljettiin pois koko analyysipopulaatiosta STEC-HUS -diagnoosin varmistumisen takia. Potilaista 93 %:lla oli lähtötilanteessa munuaisten ulkopuolisia löydöksiä (sydän ja verisuonisto, keuhkot, keskushermosto, ruoansulatuskanava, iho, luustolihakset) tai aHUS-oireita.
Taulukossa 10 on esitetty ALXN1210‑aHUS‑311-tutkimukseen otettujen, koko analyysipopulaation muodostaneiden 56 aikuispotilaan demografiset tiedot ja lähtötilanteen ominaisuudet.
Taulukko 10: Lähtötilanteen ominaisuudet aikuisilla tehdyssä tutkimuksessa
| Muuttuja | Tunnusluku | Ravulitsumabi (N = 56) |
| Ikä (vuotta) ensimmäisen infuusion ajankohtana | Keskiarvo (keskihajonta) Min., maks. | 42,2 (14,98) 19,5; 76,6 |
Sukupuoli Mies |
n (%) |
19 (33,9) |
Rotu Aasialainen Valkoihoinen Tuntematon/muu | n (%) |
15 (26,8) 29 (51,8) 12 (21,4) |
| Aiempi elinsiirto | n (%) | 8 (14,3) |
Veren trombosyytit (109/l)
| n Mediaani (min., maks.) | 56 95,25 (18; 473) |
Veren hemoglobiini (g/l)
| n Mediaani (min., maks.) | 56 85,00 (60,5; 140) |
Seerumin LDH (U/l)
| n Mediaani (min., maks.) | 56 508,00 (229,5; 3 249) |
eGFR (ml/min/1,73 m2)
| n Mediaani (min., maks.) | 55 10,00 (4; 80) |
| Dialyysihoitoa saavat potilaat | N (%) | 29(51,8) |
| Äskettäin synnyttäneet potilaat | N (%) | 8 (14,3) |
Huom.: prosenttiosuudet perustuvat potilaiden kokonaismäärään.
Lyhenteet: eGFR = arvioitu glomerulusten suodatusnopeus; LDH = laktaattidehydrogenaasi; maks. = suurin; min. = pienin.
Ensisijainen päätetapahtuma oli täydellinen TMA-vaste 26 viikon pituisen alkuarviointivaiheen aikana eli hematologisten muuttujien normalisoituminen (trombosyyttiarvo ≥ 150 x 109/l ja LDH ≤ 246 U/l) sekä seerumin kreatiniiniarvon paraneminen ≥ 25 %:lla lähtötilanteen arvosta. Potilaiden oli täytettävä kaikki täydellisen TMA-vasteen kriteerit kahdessa erillisessä arvioinnissa, joiden välissä oli vähintään 4 viikon (28 vuorokauden) tauko, sekä kaikissa näiden arvioiden välillä mahdollisesti tehdyissä mittauksissa.
Täydellinen TMA-vaste todettiin 30 potilaalla 56:sta (53,6 %) 26 viikon pituisen alkuarviointivaiheen aikana, kuten taulukossa 11 on esitetty.
Taulukko 11: Täydellinen TMA-vaste ja täydellisen TMA-vasteen osa-alueiden analyysi 26 viikon pituisessa alkuarviointivaiheessa (ALXN1210-aHUS-311)
| Yhteensä | Vasteen saavuttaneet | ||
| n | Osuus (95 %:n luottamusväli)a | ||
| Täydellinen TMA-vaste | 56 | 30 | 0,536 (0,396; 0,675) |
| Täydellisen TMA-vasteen osa-alueet | |||
| Trombosyyttiarvon normalisoituminen | 56 | 47 | 0,839 (0,734; 0,944) |
| LDH-arvon normalisoituminen | 56 | 43 | 0,768 (0,648; 0,887) |
| Seerumin kreatiniiniarvon paraneminen ≥ 25 %:lla lähtötilanteeseen nähden | 56 | 33 | 0,589 (0,452; 0,727) |
| Veriarvojen normalisoituminen | 56 | 41 | 0,732 (0,607; 0,857) |
a 95 %:n luottamusvälit osuuksille perustuvat jatkuvuuskorjattuun asymptoottiseen Gaussin likiarvomenetelmään.
Lyhenteet: LDH = laktaattidehydrogenaasi; TMA = tromboottinen mikroangiopatia.
Täydellinen TMA-vaste todettiin lisäksi kuudella muulla potilaalla jatkovaiheen päivinä 169, 302, 401, 407, 1 247 ja 1 359 eli täydellinen TMA-vaste saavutettiin yhteensä 36 potilaalla 56:sta (64,3 %; 95 %:n luottamusväli: 50,8 %; 77,7 %) tutkimuksen loppuun mennessä. Yksittäisillä osa-alueilla vasteen saavuttaneiden määrä suureni trombosyyttiarvon normalisoitumisen kohdalla 48 potilaaseen (85,7 %; 95 %:n luottamusväli: 75,7 %; 95,8 %), LDH-arvon normalisoitumisen kohdalla 49 potilaaseen (87,5 %; 95 %:n luottamusväli: 77,9 %; 97,1 %) ja munuaistoiminnan paranemisen kohdalla 37 potilaaseen (66,1 %; 95 %:n luottamusväli: 52,8 %; 79,4 %).
Mediaaniaika täydellisen TMA-vasteen saavuttamiseen oli 86 vuorokautta (7–1 359 vuorokautta). Keskimääräisen trombosyyttiarvon nopeaa nousua havaittiin pian ravulitsumabihoidon aloittamisen jälkeen; arvo nousi lähtötilanteen arvosta 118,52 × 109/l arvoon 243,54 × 109/l päivänä 8 ja pysyi arvoa 227 × 109/l suurempana kaikilla alkuarviointivaiheen (26 viikkoa) myöhemmillä käynneillä. Keskimääräinen LDH-arvo laski vastaavasti lähtötilanteen arvosta kahden ensimmäisen hoitokuukauden aikana ja säilyi sen jälkeen samanlaisena alkuarviointivaiheen (26 viikkoa) loppuun asti.
Yli kahdella kolmasosalla potilaspopulaatiosta, jolla oli lähtötilanteessa CKD-asteen 4 tai 5 krooninen munuaissairaus, CKD-aste pieneni vähintään yhdellä asteella tutkimuksen päivään 743 mennessä. eGFR-arvona mitattu munuaistoiminnan paraneminen pysyi vakaana tutkimuksen loppuun asti. Monilla potilailla (19/30) kroonisen munuaissairauden asteen pieneneminen jatkui 26 viikon pituisen alkuarviointivaiheen aikana täydellisen TMA-vasteen saavuttamisen jälkeen.
Niistä 27 potilaasta, jotka eivät tarvinneet dialyysihoitoa tutkimukseenottovaiheessa, 19 potilasta pärjäsi koko tutkimusjakson ilman dialyysihoitoa. Dialyysihoito aloitettiin tutkimuksen aikana 8 potilaalle, joista kaksi lopetti dialyysihoidon tutkimuksen aikana. Yksi dialyysihoidon tutkimuksen jatkovaiheen aikana lopettaneista potilaista aloitti dialyysihoidon uudelleen ja jatkoi sitä tutkimuksen päättymiseen asti.
Taulukko 12: Toissijaiset tehotulokset ALXN1210‑aHUS‑311-tutkimuksen 26 viikon pituisessa alkuarviointivaiheessa
| Muuttujat | ALXN1210‑aHUS‑311-tutkimus (N = 56) | |
Hematologiset TMA-muuttujat, Veren trombosyytit (109/l) Seerumin LDH (U/l) | Todettu arvo (n = 48) 237,96 (73,528) 232,00 194,46 (58,099) 176,50 | Muutos lähtötilanteesta (n = 48) 114,79 (105,568) 125,00 -519,83 (572,467) -310,75 |
Hemoglobiiniarvon nousu ≥ 20 g/l lähtötilanteesta, vahvistettu alkuarviointivaiheen aikana n/m |
40/56 | |
Kroonisen munuaissairauden asteen muutos lähtötilanteesta, päivä 183 Parania Pahenib |
32/47
| |
eGFR (ml/min/1,73 m2), päivä 183
Keskiarvo (keskihajonta) | Todettu arvo (n = 48) 51,83 (39,162) 40,00 | Muutos lähtötilanteesta (n = 47) 34,80 (35,454) 29,00 |
Huom: n: niiden potilaiden lukumäärä, joista oli saatavana tietoa spesifistä arviointia varten päivän 183 käynnillä. m: spesifiset kriteerit täyttäneiden potilaiden lukumäärä. Kroonisen munuaissairauden (CKD) aste määritetään National Kidney Foundation -säätiön Chronic Kidney Disease Stage -asteikolla. Aste 5 on huonoin ja aste 1 paras luokitus. Lähtötilanteen arvo johdetaan viimeisestä saatavana olevasta eGFR-arvosta, joka on mitattu ennen hoidon aloittamista. Parani/paheni: verrattuna kroonisen munuaissairauden asteeseen lähtötilanteessa. *95 %:n luottamusvälit perustuvat eksakteihin luottamusrajoihin, jotka on määritetty Clopper–Pearsonin menetelmällä.a Lukuun ottamatta potilaita, joilla on lähtötilanteessa asteen 1 krooninen munuaissairaus, koska siinä ei voi tapahtua paranemista. b Lukuun ottamatta potilaita, joilla on lähtötilanteessa asteen 5 sairaus, koska siinä ei voi tapahtua pahenemista.
Lyhenteet: eGFR = arvioitu glomerulusten suodatusnopeus; LDH = laktaattidehydrogenaasi; TMA = tromboottinen mikroangiopatia.
Tutkimuksen lopullinen tehoanalyysi kaikista potilaista, jotka saivat ravulitsumabia hoidon mediaanikeston ollessa 130,36 viikkoa, vahvisti, että ensisijaisen arviointijakson aikana havaitut ravulitsumabihoidon vasteet säilyivät koko tutkimuksen ajan.
Yleistynyt myasthenia gravis (gMG)
Tutkimus aikuisilla gMG-potilailla
Ravulitsumabin tehoa ja turvallisuutta aikuisilla gMG-potilailla arvioitiin faasin 3 satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa (ALXN1210-MG-306). Tutkimuksen jälkeen siihen osallistuneilla potilailla oli mahdollisuus osallistua avoimeen jatkohoitojaksoon, jossa kaikki potilaat saivat ravulitsumabia.
Tutkimukseen otetuilla gMG-potilailla (diagnoosista kulunut vähintään 6 kuukautta) oli positiivinen tulos serologisesta asetyylikoliinireseptori (AChR) -vasta-ainetestistä, kliininen MGFA (Myasthenia Gravis Foundation of America) -luokka II–IV sekä jäljellä olevia oireita, joiden määritelmänä oli MG-ADL (Myasthenia Gravis Activities of Daily Living) -kokonaispistemäärä ≥ 6. Potilaat satunnaistettiin saamaan joko ravulitsumabia (N = 86) tai lumelääkettä (N = 89). Immunosuppressanttihoitoa (kortikosteroideja, atsatiopriinia, syklofosfamidia, siklosporiinia, metotreksaattia, mykofenolaattimofetiilia tai takrolimuusia) saavien potilaiden annettiin jatkaa hoitoaan koko tutkimuksen ajan. Myös varahoito (suuret kortikosteroidiannokset, plasmanvaihto/plasmafereesi tai IVIg-hoito) sallittiin, jos potilaalla esiintyi tutkimussuunnitelman määritelmän mukaista kliinistä pahenemista.
Yhteensä 162 (92,6 %) potilasta oli mukana ALXN1210-MG-306-tutkimuksen 26 viikon pituisen satunnaiskontrolloidun hoitojakson loppuun asti. Potilaiden lähtötilanteen ominaisuudet on esitetty taulukossa 13. Suurin osa (97 %) tutkimukseen otetuista potilaista oli saanut vähintään yhtä seuraavista immunomoduloivista hoidoista tutkimukseen ottamista edeltävien 2 vuoden aikana: immunosuppressanttihoito, plasmanvaihto/plasmafereesi, IVIg-hoito.
Taulukko 13: Sairauteen liittyvät ominaisuudet tutkimuksen ALXN1210-MG-306 lähtötilanteessa
| Muuttuja | Tunnusluku | Lumelääke (N = 89) | Ravulitsumabi (N = 86) |
| Sukupuoli Mies Nainen | n (%) | 44 (49,4) 45 (50,6) | 42 (48,8) 44 (51,2) |
| Ikä (vuotta) ensimmäisen annoksen yhteydessä | Keskiarvo (keskihajonta) (min., maks.) |
53,3 (16,05) |
58,0 (13,82) |
| Iäkkäät (≥ 65 vuoden ikäiset) potilaat tutkimuksen alkaessa | n (%) | 24 (27,0) | 30 (34,9) |
| MG:n kesto diagnoosista laskien (vuotta) | Keskiarvo (keskihajonta) (min., maks.) |
10,0 (8,90) |
9,8 (9,68) |
| Lähtötilanteen MG-ADL-pisteet | Keskiarvo (keskihajonta) (min., maks.) |
8,9 (2,30) |
9,1 (2,62) |
| Lähtötilanteen QMG-pisteet | Keskiarvo (keskihajonta) (min., maks.) |
14,5 (5,26) (2,0; 27,0) |
14,8 (5,21) (6,0; 39,0) |
| Lähtötilanteen MGFA-luokka Luokka II (lievä heikkous) Luokka III (keskivaikea heikkous) Luokka IV (vaikea heikkous) | n (%) |
45 (51) 5 (6) |
41 (48) 6 (7) |
| Potilaat, joille tehty aiempi intubaatio diagnoosin jälkeen (MGFA-luokka V) | n (%) | 9 (10,1) | 8 (9,3) |
| Potilaat, joilla ollut aiempi myasteeninen kriisi diagnoosin jälkeena | n (%) | 17 (19,1) | 21 (24,4) |
Vakaiden immunosuppressanttihoitojen lukumääräb tutkimukseen otettaessa 0 1 ≥ 2 | n (%) | 8 (9,0) 34 (38,2) 47 (52,8) | 10 (11,6) 40 (46,5) 36 (41,9) |
a Tiedot aiemmista myasteenisista kriiseistä kerättiin sairauskertomuksesta, eikä niitä arvioitu kliinisen tutkimussuunnitelman määritelmän perusteella.
b Immunosuppressanttihoidot käsittivät kortikosteroidit, atsatiopriinin, syklofosfamidin, siklosporiinin, metotreksaatin, mykofenolaattimofetiilin ja takrolimuusin.
Lyhenteet: maks. = maksimi; min. = minimi; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGFA = Myasthenia Gravis Foundation of America; QMG = Quantitative Myasthenia Gravis
Ensisijainen päätetapahtuma oli MG-ADL-kokonaispisteiden muutos lähtötilanteesta viikolle 26.
Toissijaisia päätetapahtumia, joilla arvioitiin niin ikään muutosta lähtötilanteesta viikolle 26, olivat muutos QMG (Quantitative Myasthenia Gravis) -kokonaispisteissä, niiden potilaiden osuus, joiden QMG-kokonaispisteet paranivat vähintään 5 pisteellä ja MG-ADL-kokonaispisteet vähintään 3 pisteellä, sekä muutokset elämänlaatua mittaavissa arvioinneissa.
Ravulitsumabi paransi MG-ADL-kokonaispisteitä tilastollisesti merkitsevästi verrattuna lumelääkkeeseen. Ensisijaisen päätetapahtuman ja toissijaisten päätetapahtumien tulokset on esitetty taulukossa 14.
Taulukko 14: Ensisijaisen päätetapahtuman ja toissijaisten päätetapahtumien analyysi
| Tehon päätepahtumat viikon 26 aikapisteessä | Lumelääke (N = 89) LS-keskiarvo (SEM) | Ravulitsumabi (N = 86) LS-keskiarvo (SEM) | Vertailun tunnusluku | Hoitovaikutus (95 %:n CI) | p-arvo (toistettujen mittausten sekamallia käytettäessä) |
| MG-ADL | -1,4 (0,37) | -3,1 (0,38) | Ero muutoksessa lähtötilanteesta | -1,6 (-2,6; -0,7) | 0,0009 |
| QMG | -0,8 (0,45) | -2,8 (0,46) | Ero muutoksessa lähtötilanteesta | -2,0 (-3,2; -0,8) | 0,0009 |
| MG-QoL15r | -1,6 (0,70) | -3,3 (0,71) | Ero muutoksessa lähtötilanteesta | -1,7 (-3,4; 0,1) | 0,0636 |
| Neuro‑QoL‑fatigue | -4,8 (1,87) | -7,0 (1,92) | Ero muutoksessa lähtötilanteesta | -2,2 (-6,9; 2,6) | 0,3734 a |
a Päätetapahtumaa ei testattu muodollisesti tilastollisen merkitsevyyden suhteen; nimellinen p-arvo raportoitiin.
Lyhenteet: CI = luottamusväli; LS = pienimmän neliösumman; MG-ADL = Myasthenia Gravis Activities of Daily Living; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15‑item scale; Neuro-QoL-fatigue = Neurological Quality of Life Fatigue; QMG = Quantitative Myasthenia Gravis; SEM = keskiarvon keskivirhe.
Kliinisen vasteen saavuttaminen MG-ADL-kokonaispisteiden osalta määriteltiin tutkimuksessa ALXN1210-MG-306 vähintään 3 pisteen parannuksena. Tämän kliinisen vasteen saavuttaneiden osuus viikon 26 aikapisteessä oli 56,7 % ravulitsumabiryhmässä ja 34,1 % lumelääkeryhmässä (nimellinen p = 0,0049). Kliinisen vasteen saavuttaminen QMG-kokonaispisteiden osalta määriteltiin vähintään 5 pisteen parannuksena. Tämän kliinisen vasteen saavuttaneiden osuus viikon 26 aikapisteessä oli 30,0 % ravulitsumabiryhmässä ja 11,3 % lumelääkeryhmässä (p = 0,0052).
Taulukosta 15 käy ilmi, kuinka monella potilaalla havaittiin kliinistä pahenemista ja kuinka monta potilasta tarvitsi varahoitoa 26 viikon pituisen satunnaiskontrolloidun hoitojakson aikana.
Taulukko 15: Kliininen pahentuminen ja varahoito
| Muuttuja | Tunnusluku | Lumelääke (N = 89) | Ravulitsumabi (N = 86) |
| Potilaat, joilla havaittiin kliinistä pahentumista | n (%) | 15 (16,9) | 8 (9,3) |
| Potilaat, jotka tarvitsivat varahoitoaa | n (%) | 14 (15,7) | 8 (9,3) |
a Varahoito käsitti suuret kortikosteroidiannokset, plasmanvaihdon/plasmafereesin tai IVIg-hoidon.
Potilailla, jotka saivat aluksi Ultomiris-hoitoa satunnaiskontrolloidussa hoitojaksossa ja jotka jatkoivat sen jälkeen Ultomiris-hoidon saamista enintään 164 viikon ajan avoimessa jatkohoitojaksossa, hoitovaikutus säilyi (kuva 3). Potilailla, jotka saivat aluksi lumelääkettä 26 viikon pituisessa satunnaiskontrolloidussa hoitojaksossa ja sen jälkeen Ultomiris-hoitoa avoimessa jatkohoitojaksossa, hoitovaste saavutettiin jatkohoitojaksossa nopeasti ja sen havaittiin säilyvän kaikkien päätetapahtumien osalta, mukaan lukien MG-ADL ja QMG (kuva 3). Hoidon mediaanikesto oli noin 2 vuotta.
Kuva 3: MG-ADL-kokonaispisteiden (A) ja QMG-kokonaispisteiden (B) muutos satunnaiskontrolloidun hoitojakson lähtötilanteesta enintään viikolle 164 (keskiarvo ja 95 %:n CI)
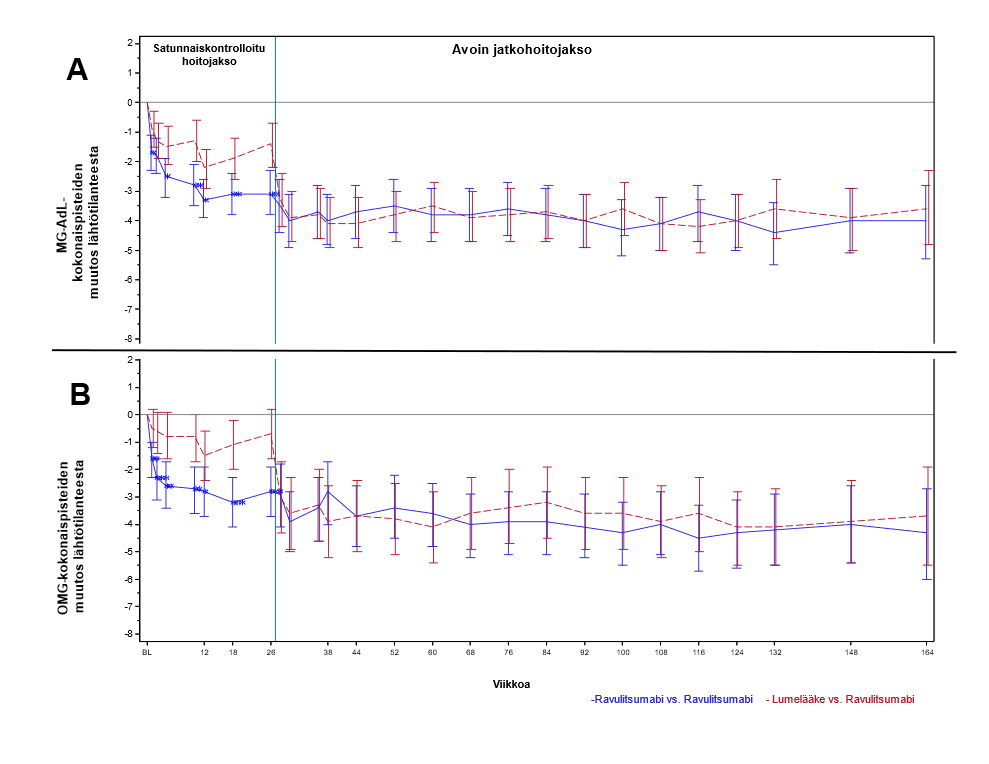
Huom.:
Satunnaistetun ja kontrolloidun jakson luvut perustuvat 175 potilaasta saatuihin tietoihin. Avoimen jatkohoitojakson luvut perustuvat 161 potilaasta saatuihin tietoihin.
Lyhenteet: CI = luottamusväli; MG-ADL = Myasthenia Gravis Activities of Daily Living; QMG = Quantitative Myasthenia Gravis
Tutkimuksen avoimessa jatkohoitojaksossa kliinikoilla oli mahdollisuus säätää immunosuppressanttihoitoja. Avoimen jatkohoitojakson lopussa (Ultomiris-hoidon mediaanikesto sekä satunnaistetun ja kontrolloidun jakson että avoimen jatkohoitojakson aikana oli 759 vuorokautta) 30,1 % potilaista pienensi kortikosteroidihoidon vuorokausiannostaan ja 12,4 % lopetti kortikosteroidihoidon. Yleisin syy kortikosteroidihoitojen muutoksiin oli MG-oireiden lievittyminen ravulitsumabihoidon aikana.
Neuromyelitis optica -kirjon häiriö (NMOSD)
Tutkimus aikuisilla NMOSD-potilailla
Ravulitsumabin tehoa AQP4-vasta-ainepositiivisilla NMOSD-potilailla arvioitiin maailmanlaajuisessa avoimessa kliinisessä tutkimuksessa (ALXN1210-NMO-307).
Tutkimukseen ALXN1210-NMO-307 otettiin 58 aikuista NMOSD-potilasta, joilla oli positiivinen serologinen testitulos AQP4-vasta-aineille, vähintään 1 relapsi seulontavaihetta edeltävien 12 kuukauden aikana ja pistemäärä ≤ 7 laajennetulla toimintakyvyn rajoittuneisuusasteikolla (EDSS). Aiempi hoito immunosuppressanteilla ei ollut mukaan oton vaatimuksena, ja 53,4 % potilaista sai ravulitsumabimonoterapiaa. Valittuja immunosuppressanttihoitoja (kortikosteroidit, atsatiopriini, mykofenolaattimofetiili, takrolimuusi) saavien potilaiden sallittiin jatkaa hoitoaan yhdessä ravulitsumabin kanssa, edellyttäen, että annostus oli vakaa tutkimusviikolle 106 asti. Lisäksi akuutti relapsin hoito (suuret kortikosteroidiannokset, plasmanvaihto/plasmafereesi tai IVIg-hoito) sallittiin, jos potilaalla esiintyi relapsi tutkimuksen aikana.
Tutkimukseen otettujen potilaiden keski-ikä oli 47,4 vuotta (vaihteluväli 18–74 vuotta), ja suurin osa potilaista oli naisia (90 %). Potilaiden mediaani-ikä NMOSD:n ensimmäisen kliinisen ilmenemisen yhteydessä oli 42,5 vuotta (vaihteluväli 16–73 vuotta). Lähtötilanteen sairausominaisuudet on esitetty taulukossa 16.
Taulukko 16: Potilaiden sairaushistoria ja lähtötilanteen ominaisuudet tutkimuksessa
ALXN1210-NMO-307
| Muuttuja | Tunnusluku | ALXN1210-NMO-307 Ravulitsumabi |
| Aika NMOSD:n ensimmäisestä kliinisestä ilmenemisestä ensimmäiseen tutkimuslääkeannokseen (vuotta) | Keskiarvo (SD) | 5,2 (6,38) |
| Mediaani | 2,0 | |
| Min., maks. | 0,19; 24,49 | |
| Vuotuisten relapsien määrä seulontaa edeltävien 24 kuukauden aikana | Keskiarvo (SD) | 1,87 (1,59) |
| Mediaani | 1,44 | |
| Min., maks. | 0,5; 6,9 | |
| Lähtötilanteen HAI-pisteet | Keskiarvo (SD) | 1,2 (1,42) |
| Mediaani | 1,0 | |
| Min., maks. | 0, 7 | |
| Lähtötilanteen EDSS-pisteet | Keskiarvo (SD) | 3,30 (1,58) |
| Mediaani | 3,25 | |
| Min., maks. | 0,0; 7,0 | |
| Potilaat, jotka olivat milloin tahansa aiemmin saaneet rituksimabia | n (%) | 21 (36,2) |
| Potilaat, jotka saivat pelkkää vakaa-annoksista kortikosteroidihoitoa tutkimukseen otettaessa | n (%) | 11 (19,0) |
| Potilaat, jotka eivät saaneet mitään immunosuppressanttihoitoa tutkimukseen otettaessa | n (%) | 31 (53,4) |
Lyhenteet: EDSS = laajennettu toimintakyvyn rajoittuneisuusasteikko (Expanded Disability Status Scale); HAI = Hauserin liikkumisindeksi (Hauser Ambulation Index); Maks. = maksimi; Min. = minimi; NMOSD = neuromyelitis optica -kirjon häiriö; SD = keskihajonta.
Tutkimuksen ALXN1210-NMO-307 ensisijainen päätetapahtuma oli aika ensimmäiseen tutkimuksen aikana esiintyneeseen, riippumattoman arviointikomitean vahvistamaan relapsiin. Ravulitsumabihoitoa saavilla potilailla ei esiintynyt vahvistettuja relapseja ensisijaisen hoitojakson aikana. Kellään ravulitsumabia saavista potilaista ei esiintynyt relapseja seurantavaiheessa, jonka mediaanikesto oli 90,93 viikkoa. Ravulitsumabihoitoa saavien potilaiden päätetapahtuman tulos oli yhdenmukainen relapsittomuus riippumatta siitä, saivatko potilaat samanaikaista immunosuppressanttihoitoa vai ei.
Lopullisessa tehoanalyysissä, jossa seurantavaiheen mediaani oli 170,29 viikkoa, ravulitsumabihoitoa saavilla potilailla ei esiintynyt vahvistettuja relapseja tutkimuksen loppuun asti. Ensisijaisen arviointijakson aikana havaitut ravulitsumabihoidon vasteet säilyivät koko tutkimuksen ajan. Lisäksi niiden 27 potilaan joukossa, jotka saivat lähtötilanteessa immunosuppressanttihoitoa, 17:lla (63 %) vähintään yksi immunosuppressanttihoito väheni tai se lopetettiin ravulitsumabihoidon aikana.
Ravulitsumabia ei ole tutkittu NMOSD-potilaiden relapsien akuutissa hoidossa.
Pediatriset potilaat
Paroksysmaalinen nokturnaalinen hemoglobinuria (PNH)
Tutkimus pediatrisilla PNH-potilailla (ALXN1210-PNH-304)
Pediatrinen tutkimus (ALXN1210-PNH-304) on avoin faasin 3 monikeskustutkimus ekulitsumabia aiemmin saaneilla ja komplementin estäjiä aiemmin saamattomilla pediatrisilla PNH-potilailla. Välianalyysin tuloksissa yhteensä 13 pediatrista PNH-potilasta päätti ALXN1210-PNH-304-tutkimuksen ensisijaisen arviointijakson (26 viikkoa) aikana annetun ravulitsumabihoidon. Näistä 13 potilaasta viisi ei ollut aiemmin saanut komplementin estäjiä ja kahdeksan sai ekulitsumabia ennen tutkimuksen alkamista.
Useimmat potilaista olivat iältään 12–17 vuotta ensimmäisen infuusion aikana (keski-ikä 14,4 vuotta), ja kaksi potilasta oli alle 12 vuotta (11 vuotta ja 9 vuotta). Kahdeksan 13:sta potilaasta oli tyttöjä. Potilaiden keskipaino lähtötilanteessa oli 56 kg (vaihteluväli 37–72 kg). Taulukossa 17 on esitetty tutkimukseen ALXN1210‑PNH-304 otettujen pediatristen potilaiden sairaushistoria ja ominaisuudet lähtötilanteessa.
Taulukko 17: Sairaushistoria ja ominaisuudet lähtötilanteessa (koko analyysijoukko)
| Muuttuja | Komplementin estäjiä aiemmin saamattomat potilaat (N = 5) | Ekulitsumabia aiemmin saaneet potilaat (N = 8) |
PNH-punasolukloonin koko (%) Mediaani (min., maks.) | (N = 4) 40,05 (6,9; 68,1) | (N = 6) 71,15 (21,2; 85,4) |
PNH-granulosyyttikloonin koko (%) Mediaani (min., maks.) | 78,30 (36,8; 99,0) | 91,60 (20,3; 97,6) |
| Potilaat, jotka saivat punasolujen/kokoveren siirtoja ensimmäistä annosta edeltävien 12 kuukauden aikana, n (%) | 2 (40,0) | 2 (25,0) |
Punasolujen/kokoveren siirtojen lukumäärä ensimmäistä annosta edeltävien 12 kuukauden aikana Yhteensä |
10 |
2 |
Siirrettyjen punasolu-/kokoveriyksiköiden lukumäärä ensimmäistä annosta edeltävien 12 kuukauden aikana Yhteensä | 14 7,0 (3; 11) | 2 2,0 (2; 2) |
| Potilaat, joilla oli PNH:hon liittyviä sairaustiloja ennen tietoon perustuvaa suostumusta, n (%) | 5 (100) | 8 (100) |
| Anemia | 2 (40,0) | 5 (62,5) |
| Hematuria tai hemoglobinuria | 2 (40,0) | 5 (62,5) |
| Aplastinen anemia | 3 (60,0) | 1 (12,5) |
| Munuaisten vajaatoiminta | 2 (40,0) | 2 (25,0) |
| Muua | 0 | 1 (12,5) |
Hoitoa edeltävä LDH-pitoisuus (U/l) Mediaani (min., maks.) |
588,50 (444; 2 269,7) |
251,50 (140,5; 487) |
a Muina PNH:hon liittyvinä sairaustiloina raportoitiin ”munuais- ja haimainfarkteja” ja ”useita emboliseen prosessiin mahdollisesti viittaavia leesioita”.
Huom: prosenttiosuudet perustuivat potilaiden kokonaismäärään kussakin kohortissa.
Lyhenteet: LDH = laktaattidehydrogenaasi; maks. = maksimi; min. = minimi; PNH = paroksysmaalinen nokturnaalinen hemoglobinuria.
Potilaat saivat kehonpainonsa mukaisen latausannoksen ravulitsumabia päivänä 1 ja kehonpainonsa mukaisen ylläpitoannoksen päivänä 15 ja sen jälkeen 8 viikon välein, jos potilaan paino oli ≥ 20 kg, tai 4 viikon välein, jos potilaan paino oli < 20 kg. Potilailla, jotka saivat ekulitsumabihoitoa tutkimukseen otettaessa, tutkimushoidon päivän 1 oli määrä olla 2 viikkoa potilaan viimeisen ekulitsumabiannoksen jälkeen.
Ravulitsumabin painoon perustuvalla annostuksella oli välitön, täydellinen ja pitkäkestoinen komplementin terminaalista osaa estävä vaikutus, joka säilyi koko 26 viikon pituisen ensisijaisen arviointijakson ajan riippumatta siitä, oliko potilas aiemmin saanut ekulitsumabia vai ei. Ravulitsumabihoidon aloittamisen jälkeen seerumin terapeuttinen vakaan tilan ravulitsumabipitoisuus saavutettiin välittömästi ensimmäisen annoksen jälkeen, ja se säilyi koko 26 viikon pituisen ensisijaisen arviointijakson ajan kummassakin kohortissa. Tutkimuksessa ei esiintynyt läpilyöntihemolyysitapahtumia, eikä yhdenkään potilaan vapaan C5-proteiinin pitoisuus ollut yli 0,5 µg/ml lähtötilanteen jälkeen.
LDH-pitoisuuden keskimääräinen prosenttimuutos lähtötilanteesta päivänä 183 oli -47,91 % komplementin estäjiä aiemmin saamattomien potilaiden kohortissa. Ekulitsumabia aiemmin saaneiden potilaiden kohortissa pitoisuus säilyi vakaana 26 viikon pituisen ensisijaisen arviointijakson ajan. Hemoglobiini stabilisoitui viikkoon 26 mennessä 60 %:lla (3/5) komplementin estäjiä aiemmin saamattomista potilaista ja 75 %:lla (6/8) ekulitsumabia aiemmin saaneista potilaista. Potilaista 84,6 % (11/13) välttyi verensiirroilta 26 viikon pituisen ensisijaisen arviointijakson aikana.
Välianalyysin tehoa koskevat tulokset on esitetty alla olevassa taulukossa 18.
Taulukko 18: Tehoa koskevat tulokset pediatrisilla PNH-potilailla tehtävästä tutkimuksesta (ALXN1210-PNH-304) – 26 viikon pituinen ensisijainen arviointijakso
| Päätetapahtuma | Ravulitsumabi (ei aiempaa hoitoa, N = 5) | Ravulitsumabi (hoidon vaihto, N = 8) |
|---|---|---|
LDH-pitoisuuden prosenttimuutos lähtötilanteesta Keskiarvo (keskihajonta) |
-47,91 (52,716) |
4,65 (44,702) |
Verensiirron tarpeelta välttyminen Prosenttiosuus (95 %:n luottamusväli) |
60,0 (14,66; 94,73) |
100,0 (63,06; 100,00) |
Hemoglobiinin stabilisoituminen Prosenttiosuus (95 %:n luottamusväli) |
60,0 (14,66; 94,73) |
75 (34,91; 96,81) |
| Läpilyöntihemolyysi (%) | 0 | 0 |
Lyhenteet: LDH = laktaattidehydrogenaasi
Pitkäkestoista tehoa koskevat tulokset, jotka ulottuivat tutkimuksen loppuun asti ja joissa hoidon mediaanikesto oli 915 päivää, osoittivat pitkäkestoisen hoitovasteen pediatrisilla PNH-potilailla.
Näiden välianalyysin tulosten perusteella ravulitsumabin teho pediatrisilla PNH-potilailla vaikuttaa olevan samanlainen kuin aikuisilla PNH-potilailla.
Epätyypillinen hemolyyttis-ureeminen oireyhtymä (aHUS)
Näyttö, joka tukee Ultomiris-valmisteen käyttöä pediatristen aHUS-potilaiden hoidossa, on saatu yhdestä pediatrisilla potilailla tehdystä kliinisestä tutkimuksesta (johon otettiin yhteensä 31 potilasta, joilla oli dokumentoitu aHUS. Koko analyysipopulaatioon otettiin 28 potilasta, joiden ikä vaihteli 10 kuukaudesta 17 vuoteen.).
Tutkimus pediatrisilla aHUS-potilailla (ALXN1210-aHUS-312)
Pediatrinen tutkimus oli 26 viikon pituinen, yhdellä hoitoryhmällä toteutettava faasin 3 monikeskustutkimus pediatrisilla potilailla. Potilailla oli mahdollisuus osallistua enintään 4,5 vuotta kestävään jatkovaiheeseen.
Tutkimukseen otettiin yhteensä 24 potilasta, joilla oli dokumentoitu aHUS-diagnoosi ja näyttöä TMA-tapahtumista, ja jotka eivät olleet aiemmin saaneet ekulitsumabia. Näistä potilasta 20 oli mukana koko analyysipopulaatiossa. Tutkimuksen mukaanottokriteerit sulkivat pois potilaat, joilla oli ADAMTS13:n (disintegriini ja metalloproteinaasi, jossa on trombospondiini 13 rakennekuvioita) puutoksesta johtuva TMA, STEC-HUS ja kobalamiini C metabolian geneettinen häiriö. Neljä potilasta sai 1–2 annosta lääkettä ennen tutkimuksesta vetäytymistä; nämä potilaat suljettiin pois koko analyysipopulaatiosta, koska soveltuvuutta aHUS:n suhteen ei ollut vahvistettu. Koko populaatiossa keskimääräinen paino lähtötilanteessa oli 21,2 kg; suurin osa potilaista oli lähtötilanteessa painoalueella ≥ 10 – < 20 kg. Suurimmalla osalla potilaista (70,0 %:lla) oli ennen hoidon aloittamista munuaisten ulkopuolisia löydöksiä (sydän ja verisuonisto, keuhkot, keskushermosto, ruoansulatuskanava, iho, luustolihakset) tai aHUS-oireita lähtötilanteessa. Lähtötilanteessa 35,0 %:lla (n = 7) potilaista oli asteen 5 krooninen munuaissairaus.
Tutkimukseen otettiin yhteensä 10 potilasta, jotka vaihtoivat ekulitsumabihoidon ravulitsumabihoitoon ja joilla oli dokumentoitu aHUS-diagnoosi ja näyttöä TMA-tapahtumista. Potilaiden oli täytynyt saavuttaa kliininen vaste ekulitsumabihoidolle ennen tutkimukseen osallistumista (so. LDH < 1,5 x viitealueen yläraja, trombosyyttiarvo ≥ 150 000/μl ja eGFR-arvo > 30 ml/min/1,73 m2). Näin ollen tietoa ei ole ravulitsumabin käytöstä potilailla, joilla ekulitsumabi on tehotonta.
Taulukossa 19 on esitetty ALXN1210-aHUS-312-tutkimukseen otettujen pediatristen potilaiden ominaisuudet lähtötilanteessa.
Taulukko 19: Demografiset tiedot ja lähtötilanteen ominaisuudet ALXN1210‑aHUS‑312-tutkimuksessa
| Muuttuja | Tunnusluku | Ravulitsumabi (ei aiempaa hoitoa, N = 20) | Ravulitsumabi (hoidon vaihto, N = 10) |
Ikäryhmä (vuotta) ensimmäisen infuusion ajankohtana Vastasyntynyt – < 2 vuotta | n (%) |
4 (20,0) |
1 (10,0)
|
Sukupuoli Mies | n (%) |
8 (40,0) |
9 (90,0) |
Rotua Intiaani tai Alaskan alkuperäiskansaa | n (%) |
1 (5,0) |
0 (0,0) |
| Aiempi elinsiirre | n (%) | 1 (5,6) | 1 (10,0) |
| Veren trombosyyttiarvo (109/l) | Mediaani (min., maks.) | 51,25 (14; 125) | 281,75 (207; 415,5) |
| Hemoglobiini (g/l) | Mediaani (min., maks.) | 74,25 (32; 106) | 132,0 (114,5; 148) |
| LDH (U/l) | Mediaani (min., maks.) | 1 963,0 (772; 4 985) | 206,5 (138,5; 356) |
| eGFR (ml/min/1,73 m2) | Mediaani (min., maks.) | 22,0 (10; 84) | 99,75 (54; 136,5) |
| Tarvitsi dialyysihoitoa lähtötilanteessa | n (%) | 7 (35,0) | 0 (0,0) |
Huom.: Prosenttiosuudet perustuvat potilaiden kokonaismäärään.
a Potilaille saatettiin ilmoittaa enemmän kuin yksi rotu.
Lyhenteet: eGFR = arvioitu glomerulusten suodatusnopeus; LDH = laktaattidehydrogenaasi; maks. = suurin; min. = pienin.
Ensisijainen päätetapahtuma oli täydellinen TMA-vaste 26 viikon pituisen alkuarviointivaiheen aikana eli hematologisten muuttujien normalisoituminen (trombosyyttiarvo ≥ 150 x 109/l ja LDH ≤ 246 U/l) sekä seerumin kreatiniiniarvon paraneminen ≥ 25 %:lla lähtötilanteen arvosta potilailla, jotka eivät olleet saaneet aiemmin ekulitsumabia. Potilaiden oli täytettävä kaikki täydellisen TMA-vasteen kriteerit kahdessa erillisessä arvioinnissa, joiden välissä oli vähintään 4 viikon (28 vuorokauden) tauko, sekä kaikissa näiden arvioiden välillä mahdollisesti tehdyissä mittauksissa.
Täydellinen TMA-vaste todettiin 26 viikon pituisen alkuarviointivaiheen aikana 15 potilaalla niistä 20 potilaasta, jotka eivät olleet saaneet aiempia hoitoja (75,0 %), kuten taulukossa 20 on esitetty.
Taulukko 20: Täydellinen TMA-vaste ja täydellisen TMA-vasteen osa-alueiden analyysi 26 viikon pituisen alkuarviointivaiheen aikana (ALXN1210-aHUS-312)
| Yhteensä | Vasteen saavuttaneet | ||
| n | Osuus (95 %:n luottamusväli)a | ||
| Täydellinen TMA-vaste | 20 | 15 | 0,750 (0,509; 0,913) |
| Täydellisen TMA-vasteen osa-alueet | |||
| Trombosyyttiarvon normalisoituminen | 20 | 19 | 0,950 (0,751; 0,999) |
| LDH-arvon normalisoituminen | 20 | 18 | 0,900 (0,683; 0,988) |
| Seerumin kreatiniiniarvon paraneminen ≥ 25 %:lla lähtötilanteen arvosta | 20 | 16 | 0,800 (0,563; 0,943) |
| Veriarvojen normalisoituminen | 20 | 18 | 0,900 (0,683; 0,988) |
a 95 %:n luottamusvälit osuuksille perustuvat jatkuvuuskorjattuun asymptoottiseen Gaussin likiarvomenetelmään.
Lyhenteet: LDH = laktaattidehydrogenaasi; TMA = tromboottinen mikroangiopatia
Alkuarviointivaiheen aikana täydellisen TMA-vasteen saavuttamiseen kuluneen ajan mediaani oli 30 vuorokautta (15–99 vuorokautta). Täydellinen TMA-vaste säilyi kaikilla sen saavuttaneilla potilailla koko alkuarviointivaiheen ajan, ja heidän munuaistoiminnassaan tapahtui jatkuvaa paranemista. Keskimääräisessä trombosyyttiarvossa havaittiin nousua pian ravulitsumabihoidon aloittamisen jälkeen; arvo nousi lähtötilanteen arvosta 71,70 × 109/l arvoon 302,41 × 109/l päivänä 8 ja pysyi arvoa 304 × 109/l suurempana kaikilla alkuarviointivaiheen (26 viikkoa) myöhemmillä käynneillä päivän 22 jälkeen.
Täydellinen TMA-vaste todettiin lisäksi kolmella muulla potilaalla jatkovaiheen päivinä 295 (2 potilasta) ja 351 (1 potilas), eli täydellinen TMA-vaste saavutettiin 18 pediatrisella potilaalla 20:sta (90 %; 95 %:n luottamusväli: 68,3 %; 98,8 %) tutkimuksen loppuun mennessä. Yksittäisillä osa-alueilla vasteen saavuttaneiden määrä suureni 19 potilaaseen 20:sta trombosyyttiarvon normalisoitumisen kohdalla (95,0 %; 95 %:n luottamusväli: 75,1 %; 99,9 %), 19 potilaaseen 20:sta LDH-arvon normalisoitumisen kohdalla (95,0 %; 95 %:n luottamusväli: 75,1 %; 99,9 %) ja 18 potilaaseen 20:sta munuaistoiminnan paranemisen kohdalla (90,0 %; 95 %:n luottamusväli: 68,3 %; 98,8 %).
Kaikki seitsemän potilasta, jotka tarvitsivat dialyysihoitoa tutkimukseenottovaiheessa, pystyivät lopettamaan dialyysihoidon. Näistä potilaista kuudella dialyysihoito oli lopetettu jo päivään 36 mennessä. Yhdellekään potilaalle ei aloitettu tai aloitettu uudelleen dialyysihoitoa tutkimuksen aikana. Niistä 16 potilaasta, joista oli saatavilla lähtötilanteen ja viikon 52 (päivän 351) tietoja, 16 potilaan kroonisen munuaissairauden (CKD) asteessa oli tapahtunut paranemista lähtötilanteeseen verrattuna. Potilailla, joista oli saatavilla tietoja tutkimuksen loppuun asti, CKD-aste joko parani entisestään tai ei muuttunut. eGFR-arvona mitattu munuaistoiminnan paraneminen pysyi vakaana tutkimuksen loppuun asti. Taulukossa 21 on esitetty yhteenveto ALXN1210 aHUS 312 tutkimuksen toissijaisista tehotuloksista.
Taulukko 21: Toissijaiset tehotulokset ALXN1210‑aHUS‑312-tutkimuksen 26 viikon pituisessa alkuarviointivaiheessa
| Muuttujat | ALXN1210‑aHUS‑312-tutkimus (N = 20) | |
|---|---|---|
Hematologiset TMA-muuttujat, Veren trombosyytit (109/l) Seerumin LDH (U/l) | Todettu arvo (n = 17) 304,94 (75,711) 318,00 262,41 (59,995) 247,00 | Muutos lähtötilanteesta (n = 17) 245,59 (91,827) 247,00 -2 044,13 (1 328,059) -1 851,50 |
| Hemoglobiiniarvon nousu ≥ 20 g/l lähtötilanteesta, vahvistettu alkuarviointivaiheen aikana n/m osuus (95 %:n luottamusväli)** | 17/20 0,850 (0,621; 0,968) | |
Kroonisen munuaissairauden asteen muutos lähtötilanteesta, päivä 183 Parania Pahenib |
| |
eGFR (ml/min/1,73 m2), päivä 183
Keskiarvo (keskihajonta) | Todettu arvo (n = 17) 108,5 (56,87) 108,0 | Muutos lähtötilanteesta (n = 17) 85,4 (54,33) 80,0 |
Huom.: n: niiden potilaiden lukumäärä, joista oli saatavana tietoa spesifistä arviointia varten päivän 183 käynnillä. m: spesifiset kriteerit täyttäneiden potilaiden lukumäärä. Kroonisen munuaissairauden (CKD) aste määritetään National Kidney Foundation -säätiön Chronic Kidney Disease Stage -asteikolla. Aste 1 on paras ja aste 5 huonoin luokitus. Lähtötilanteen arvo johdetaan viimeisestä saatavana olevasta eGFR-arvosta, joka on mitattu ennen hoidon aloittamista.
Parani/paheni: verrattuna kroonisen munuaissairauden asteeseen lähtötilanteessa.
*95 %:n luottamusvälit perustuvat eksakteihin luottamusrajoihin, jotka on määritetty Clopper–Pearsonin menetelmällä.a Lukuun ottamatta potilaita, joilla on lähtötilanteessa asteen 1 sairaus, koska siinä ei voi tapahtua paranemista. b Lukuun ottamatta potilaita, joilla on lähtötilanteessa asteen 5 sairaus, koska siinä ei voi tapahtua pahenemista.
Lyhenteet: eGFR = arvioitu glomerulusten suodatusnopeus; LDH = laktaattidehydrogenaasi; TMA = tromboottinen mikroangiopatia.
Aiemmin ekulitsumabia saaneiden potilaiden sairaus pysyi hallinnassa heidän siirryttyään ravulitsumabihoitoon, sillä heidän veriarvonsa ja munuaisarvonsa pysyivät vakaina, eikä mitään selkeitä vaikutuksia turvallisuuteen havaittu.
Ravulitsumabin teho aHUSin hoidossa vaikuttaa olevan samanlainen pediatrisilla potilailla ja aikuispotilailla. Tutkimuksen lopullinen tehoanalyysi kaikista pediatrisista potilaista, jotka saivat ravulitsumabia hoidon mediaanikeston ollessa 130,60 viikkoa, vahvisti, että ensisijaisen arviointijakson aikana havaitut ravulitsumabihoidon vasteet säilyivät koko tutkimuksen ajan.
Yleistynyt myasthenia gravis (gMG)
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Ultomiris-valmisteen käytöstä myasthenia graviksen hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä. Ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Neuromyelitis optica -kirjon häiriö (NMOSD)
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Ultomiris-valmisteen käytöstä NMOSD:n hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä. Ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Farmakokinetiikka
Imeytyminen
Koska ravulitsumabi annetaan laskimoinfuusiona ja lääkeannos on liuos, 100 % annetusta ravulitsumabiannoksesta katsotaan biologisesti hyödynnettäväksi. Aika suurimpaan havaittuun pitoisuuteen (tmax) on odotettavissa infuusion lopussa tai pian sen jälkeen. Terapeuttiset vakaan tilan lääkepitoisuudet saavutetaan ensimmäisen annoksen jälkeen.
Jakautuminen
Keskimääräinen (keskihajonta) keskustilan tilavuus ja vakaan tilan jakautumistilavuus aikuisilla ja pediatrisilla PNH- ja aHUS-potilailla sekä aikuisilla gMG- ja NMOSD-potilailla on esitetty taulukossa 22.
Biotransformaatio ja eliminaatio
Monoklonaalisena immunoglobuliini G (IgG) -vasta-aineena ravulitsumabin odotetaan metaboloituvan samalla tavalla kuin mikä tahansa endogeeninen IgG-vasta-aine (hajoamalla pieniksi peptideiksi ja aminohapoiksi katabolisten reittien kautta), ja myös eliminoituvan samalla tavalla. Ravulitsumabi koostuu ainoastaan luonnollisesti esiintyvistä aminohapoista, ja sillä ei ole tunnettuja aktiivisia metaboliitteja. Ravulitsumabin keskimääräinen (keskihajonta) terminaalinen eliminaation puoliintumisaika ja puhdistuma aikuisilla ja pediatrisilla PNH-potilailla, aikuisilla ja pediatrisilla aHUS-potilailla sekä aikuisilla gMG- ja NMOSD-potilailla on esitetty taulukossa 22.
Taulukko 22: Arvioidun keskustilan tilavuuden, jakautumistilavuuden, biotransformaation ja eliminaation parametrit ravulitsumabin annon jälkeen
| Aikuiset ja pediatriset PNH-potilaat | Aikuiset ja pediatriset aHUS-potilaat | Aikuiset gMG-potilaat | Aikuiset NMOSD-potilaat | |
| Arvioitu keskustilan tilavuus (litraa) Keskiarvo (SD) | Aikuiset: 3,44 (0,66) Pediatriset: 2,87 (0,60) | Aikuiset: 3,25 (0,61) Pediatriset: 1,14 (0,51) | 3,42 (0,756) | 2,91 (0,571) |
| Vakaan tilan jakautumistilavuus (litraa) Keskiarvo (SD) | 5,30 (0,9) | 5,22 (1,85) | 5,74 (1,16) | 4,77 (0,819) |
| Terminaalinen eliminaation puoliintumisaika (vuorokautta) Keskiarvo (SD) | 49,6 (9,1) | 51,8 (16,2) | 56,6 (8,36) | 64,3 (11,0) |
| Puhdistuma (litraa/vrk) Keskiarvo (SD) | 0,08 (0,022) | 0,08 (0,04) | 0,08 (0,02) | 0,05 (0,016) |
Lyhenteet: aHUS = epätyypillinen hemolyyttis-ureeminen oireyhtymä; gMG = yleistynyt myasthenia gravis; NMOSD = neuromyelitis optica -kirjon häiriö; PNH = paroksysmaalinen nokturnaalinen hemoglobinuria; SD = keskihajonta.
Lineaarisuus/ei-lineaarisuus
Tutkitulla annosvälillä ja annostusohjelmalla ravulitsumabin farmakokinetiikka muuttui suhteessa annokseen ja oli ajan suhteen lineaarista.
Erityisryhmät
Paino
Paino on PNH-, aHUS-, gMG- ja NMOSD-potilailla merkittävä kovariaatti, ja enemmän painavien potilaiden altistukset ovat pienempiä. Painoon perustuvat annostussuositukset on esitetty kohdassa Annostus ja antotapa olevissa taulukoissa 1, 3 ja 4.
Muodollisia tutkimuksia sukupuolen, rodun, iän (iäkkäät potilaat) tai maksan tai munuaisten vajaatoiminnan vaikutuksesta ravulitsumabin farmakokinetiikkaan ei tehty. Populaatiofarmakokineettisessä arvioinnissa sukupuolen, iän, rodun ja maksan tai munuaisten vajaatoiminnan ei kuitenkaan havaittu vaikuttavan ravulitsumabin farmakokinetiikkaan terveillä vapaaehtoisilla eikä PNH-, aHUS-, gMG- ja NMOSD-potilailla, eikä annostuksen säätämistä näin ollen pidetä tarpeellisena.
Ravulitsumabin farmakokinetiikkaa on tutkittu aHUS-potilailla, jotka sairastivat eriasteista munuaisten vajaatoimintaa, ja myös dialyysihoitoa saavilla potilailla. Näissä potilasalaryhmissä (mukaan lukien potilaat, joilla on proteinuria) todetuissa farmakokineettisissä parametreissa ei ole havaittu eroja.
Prekliiniset tiedot turvallisuudesta
Lisääntymistoksisuutta arvioivissa eläinkokeissa ei käytetty ravulitsumabia vaan komplementtia estävää hiiren korvikevasta-ainetta BB5.1, jota annettiin hiirille. Näissä tutkimuksissa ei havaittu selviä hoitoon liittyviä vaikutuksia tai haittavaikutuksia. Kun emot altistettiin vasta-aineelle organogeneesin aikana, suuremman vasta-aineannoksen (noin neljä kertaa ihmisen suurin suositeltu ravulitsumabiannos kehonpainojen vertailun perusteella) saaneiden emojen synnyttämien 230 jälkeläisen joukossa havaittiin kaksi verkkokalvon dysplasiatapausta ja yksi napatyrätapaus. Altistus ei kuitenkaan lisännyt sikiöiden menetyksiä tai vastasyntyneiden poikasten kuolemia.
Eläinkokeita ravulitsumabin mahdollisen genotoksisuuden ja karsinogeenisuuden arvioimiseksi ei ole tehty.
Tulokset hiirillä tehdyistä konventionaalisista tutkimuksista, joissa käytettiin hiiren korvikemolekyyliä BB5.1, eivät viittaa erityiseen vaaraan ihmisille.
Farmaseuttiset tiedot
Apuaineet
Dinatriumvetyfosfaattiheptahydraatti (E 339), natriumdivetyfosfaattimonohydraatti (E 339), polysorbaatti 80 (E 433), arginiini, sakkaroosi, injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Laimentamiseen saa käyttää laimentimena ainoastaan 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionestettä.
Kestoaika
18 kuukautta.
Lääkevalmiste on käytettävä heti laimentamisen jälkeen. Laimennetun valmisteen kemiallisen ja fysikaalisen säilyvyyden on kuitenkin osoitettu olevan enintään 24 tuntia 2–8 °C:n lämpötilassa ja enintään 4 tuntia huoneenlämmössä.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
ULTOMIRIS infuusiokonsentraatti, liuosta varten
300 mg/3 ml (L:ei) 3 ml (100 mg/ml) (5876,24 €)
1100 mg/11 ml (L:ei) 11 ml (100 mg/ml) (21064,59 €)
PF-selosteen tieto
Pakkauskoko: yksi injektiopullo.
Ultomiris 300 mg/3 ml infuusiokonsentraatti, liuosta varten
3 ml steriiliä konsentraattia injektiopullossa (tyypin I lasia), jossa on tulppa ja sinetti.
Ultomiris 1 100 mg/11 ml infuusiokonsentraatti, liuosta varten
11 ml steriiliä konsentraattia injektiopullossa (tyypin I lasia), jossa on tulppa ja sinetti.
Valmisteen kuvaus:
Läpikuultava, kirkas tai kellertävä liuos, jonka pH-arvo on 7,4 ja osmolaliteetti noin 250–350 mOsm/kg.
Käyttö- ja käsittelyohjeet
Injektiopullot ovat kertakäyttöisiä.
Tämä lääkevalmiste on laimennettava 50 mg/ml:n lopulliseen pitoisuuteen.
Aseptista tekniikkaa on käytettävä.
Ultomiris-infuusiokonsentraatti valmistellaan seuraavasti:
- Laimennettavien injektiopullojen lukumäärä perustuu potilaan painoon ja potilaalle määrättyyn annokseen (ks. kohta Annostus ja antotapa).
- Injektiopullot tulee tarkastaa silmämääräisesti ennen laimentamista; liuoksessa ei saa näkyä hiukkasia tai merkkejä saostumisesta. Älä käytä liuosta, jos siinä näkyy hiukkasia tai merkkejä saostumisesta.
- Laskettu määrä lääkevalmistetta vedetään asianmukaisesta määrästä injektiopulloja ja laimennetaan infuusiopussissa käyttämällä laimentimena 0,9-prosenttista (9 mg/ml) natriumkloridi-injektionestettä. Ks. annosten antamisen viitetaulukot jäljempänä. Valmiste on sekoitettava varovasti. Infuusiopussia ei saa ravistaa.
- Laimentamisen jälkeen infusoitavan liuoksen lopullinen pitoisuus on 50 mg/ml
- Liuos tulee käyttää heti laimentamisen jälkeen, ellei sitä ole säilytetty 2–8 °C:n lämpötilassa. Jos liuosta on säilytetty 2–8 °C:n lämpötilassa, anna laimennetun liuoksen lämmetä huoneenlämpöiseksi ennen antoa. Älä anna nopeana infuusiona tai boluksena laskimoon. Ks. infuusioiden minimikestot taulukoista 5 ja 6. Infuusiot on annettava 0,2 µm:n suuruisen suodattimen läpi. Ultomiris-valmisteen annon jälkeen koko letku huuhdellaan 0,9-prosenttisella natriumkloridi-injektionesteellä, USP.
- Jos lääkevalmistetta ei käytetä heti laimentamisen jälkeen, säilytysaika ei saa ylittää 24 tuntia 2–8 °C:n lämpötilassa tai 4 tuntia huoneenlämmössä odotettu infuusioaika mukaan luettuna.
Taulukko 23: Ultomiris latausannoksen antamisen viitetaulukko
| Painoalue (kg)a | Latausannos (mg) | Ultomiris-valmisteen määrä (ml) | NaCl-laimentimen määräb (ml) | Kokonaismäärä (ml) |
| ≥ 10, < 20 | 600 | 6 | 6 | 12 |
| ≥ 20, < 30 | 900 | 9 | 9 | 18 |
| ≥ 30, < 40 | 1 200 | 12 | 12 | 24 |
| ≥40, <60 | 2 400 | 24 | 24 | 48 |
| ≥60, <100 | 2 700 | 27 | 27 | 54 |
| ≥100 | 3 000 | 30 | 30 | 60 |
a Paino hoidon aikana.
b Ultomiris-valmisteen saa laimentaa ainoastaan 0,9 prosenttisella (9 mg/ml) natriumkloridi-injektionesteellä.
Taulukko 24: Ultomiris ylläpitoannosten antamisen viitetaulukko
| Painoalue (kg)a | Ylläpitoannos (mg) | Ultomiris-valmisteen määrä (ml) | NaCl-laimentimen määräb (ml) | Kokonaismäärä (ml) |
| ≥ 10, < 20 | 600 | 6 | 6 | 12 |
| ≥ 20, < 30 | 2 100 | 21 | 21 | 42 |
| ≥ 30, < 40 | 2 700 | 27 | 27 | 54 |
| ≥40, <60 | 3 000 | 30 | 30 | 60 |
| ≥60, <100 | 3 300 | 33 | 33 | 66 |
| ≥100 | 3 600 | 36 | 36 | 72 |
a Paino hoidon aikana.
b Ultomiris-valmisteen saa laimentaa ainoastaan 0,9 prosenttisella (9 mg/ml) natriumkloridi-injektionesteellä.
Taulukko 25: Ultomiris lisäannosten antamisen viitetaulukko
| Painoalue (kg)a | Lisäannos (mg) | Ultomiris-valmisteen määrä (ml) | NaCl-laimentimen määräb (ml) | Kokonaismäärä (ml) |
≥ 40, < 60
| 600 | 6 | 6 | 12 |
| 1 200 | 12 | 12 | 24 | |
| 1 500 | 15 | 15 | 30 | |
| ≥ 60, < 100 | 600 | 6 | 6 | 12 |
| 1 500 | 15 | 15 | 30 | |
| 1 800 | 18 | 18 | 36 | |
| ≥ 100 | 600 | 6 | 6 | 12 |
| 1 500 | 15 | 15 | 30 | |
| 1 800 | 18 | 18 | 36 |
a Paino hoidon aikana.
b Ultomiris-valmisteen saa laimentaa ainoastaan 0,9‑prosenttisella (9 mg/ml) natriumkloridi-injektionesteellä.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
ULTOMIRIS infuusiokonsentraatti, liuosta varten
300 mg/3 ml 3 ml
1100 mg/11 ml 11 ml
- Ei korvausta.
ATC-koodi
L04AJ02
Valmisteyhteenvedon muuttamispäivämäärä
11.09.2025
Yhteystiedot
Forskaren, Hagaplan 4
SE-113 68 Tukholma/Stockholm
Ruotsi/Sverige
+46 (0)8 557 727 50
alexion.nordics@alexion.com