
IMATINIB KRKA D.D. tabletti, kalvopäällysteinen 100 mg
Vaikuttavat aineet ja niiden määrät
100 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 100 mg imatinibia (mesilaattina).
Apuaine, jonka vaikutus tunnetaan:
Yksi kalvopäällysteinen tabletti sisältää 114 mg laktoosia (monohydraattina).
400 mg kalvopäällysteiset tabletit
Yksi kalvopäällysteinen tabletti sisältää 400 mg imatinibia (mesilaattina).
Apuaine, jonka vaikutus tunnetaan:
Yksi kalvopäällysteinen tabletti sisältää 456 mg laktoosia (monohydraattina).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kalvopäällysteinen tabletti (tabletti)
Kliiniset tiedot
Käyttöaiheet
Imatinib Krka d.d. on tarkoitettu
- Philadelphia-kromosomi (bcr-abl)-positiivisen (Ph+) kroonisen myelooisen leukemian (KML) hoitoon aikuis- ja lapsipotilaille, joiden sairaus on vasta diagnosoitu mutta joille luuytimensiirtoa ei katsota ensisijaiseksi hoitomuodoksi.
- Philadelphia-kromosomipositiivisen kroonisen myelooisen leukemian hoitoon aikuis- ja lapsipotilaille alfa-interferonihoidon epäonnistuttua kroonisessa vaiheessa tai taudin ollessa blastikriisivaiheessa tai akseleraatiovaiheessa.
- kemoterapian osana aikuis- ja lapsipotilaille, joilla on vasta diagnosoitu Philadelphia-kromosomipositiivinen akuutti lymfaattinen leukemia (Ph+ ALL).
- monoterapiana aikuispotilaille, joilla on uusiutunut tai vaikeahoitoinen Ph+ ALL.
- aikuispotilaille, joilla on myelodysplastinen oireyhtymä tai myeloproliferatiivinen sairaus (MDS/MPD), johon liittyy verihiutalekasvutekijäreseptorigeenien (PDGFR) uudelleenjärjestäytymistä.
- aikuispotilaille, joilla on pitkälle edennyt hypereosinofiilinen oireyhtymä (HES) ja/tai krooninen eosinofiilinen leukemia (CEL), johon liittyy FIP1L1-PDGFRα:n uudelleenjärjestäytymistä.
Imatinib Krka d.d. ‑valmisteen vaikutusta luuytimensiirron lopputulokseen ei ole selvitetty.
Imatinib Krka d.d. on tarkoitettu
- pahanlaatuista Kit (CD 117)-positiivista ruuansulatuskanavan stroomakasvainta (GIST) sairastavien aikuisten potilaiden hoitoon silloin, kun kasvainta ei voida leikata ja/tai kun kasvain on metastasoitunut.
- Kit (CD 117)-positiivisen GIST:in resektion jälkeiseen liitännäishoitoon aikuisille potilaille, joiden taudin uusiutumisriski on huomattava. Liitännäishoitoa ei tulisi antaa potilaille, joiden uusiutumisriski on matala tai erittäin matala.
- aikuisten dermatofibrosarcoma protuberans- (DFSP-) potilaiden hoitoon, kun kasvainta ei voida leikata ja aikuisille potilaille, joilla on uusiutunut ja/tai metastasoitunut DFSP, jota ei voi leikata.
Näyttö imatinibin tehosta aikuis- ja lapsipotilaiden KML:ssa perustuu hematologisten ja sytogeneettisten vasteiden määrään ja havaittuun aikaan ilman merkkejä taudin etenemisestä, Ph+ ALL:issa ja MDS/MPD:ssä hematologiseen ja sytogeneettiseen vasteeseen, HES:ssä/CEL:ssä hematologisten vasteiden määrään, aikuispotilaiden GIST:issä, jota ei voida leikata ja/tai joka on metastasoitunut ja DFSP:ssä objektiivisten vasteiden määrään, sekä liitännäishoidetussa GIST:issä uusiutumisvapaaseen eloonjäämiseen. Imatinibin käytöstä on hyvin vähän kokemusta potilailla, joilla on myelodysplastinen oireyhtymä / myeloproliferatiivinen sairaus ja siihen liittyvää PDGFR-geenin uudelleenjärjestäytymistä (ks. kohta Farmakodynamiikka). Juuri diagnosoitua kroonisessa vaiheessa olevaa KML:aa lukuun ottamatta kontrolloituja tutkimuksia, jotka osoittaisivat kliinistä tehoa tai eloonjäämisetua näissä taudeissa, ei ole tehty.
Ehto
Hoito tulee aloittaa tilanteesta riippuen hematologista syöpäsairautta tai pahanlaatuista sarkoomaa sairastavien potilaiden hoitoon perehtyneen lääkärin toimesta.
Annostus ja antotapa
Annostus
Hoito tulee aloittaa tilanteesta riippuen hematologista syöpäsairautta tai pahanlaatuista sarkoomaa sairastavien potilaiden hoitoon perehtyneen lääkärin toimesta.
400 mg:n ja sitä suurempia annoksia varten (katso annossuositus alla) on saatavana 400 mg:n tabletti (ei jakouurteellinen).
Muita kuin 400 mg:n ja 800 mg:n annoksia varten (katso annossuositus alla) on saatavana 100 mg:n jakouurteellinen tabletti.
Määrätty annos tulee antaa suun kautta aterian yhteydessä ja ison vesilasillisen kera ruuansulatuskanavan ärsytyksen riskin minimoimiseksi. Annokset 400 mg ja 600 mg pitää antaa kerran vuorokaudessa. Sen sijaan 800 mg annos pitää antaa kahdesti vuorokaudessa 400 mg annoksena aamulla ja illalla.
Jos potilas ei pysty nielemään kalvopäällysteistä tablettia, voidaan tabletti sekoittaa lasilliseen hiilihapotonta vettä tai omenamehua. Vaadittava määrä tabletteja laitetaan sopivaan määrään nestettä (noin 50 ml yhtä 100 mg:n tablettia kohti ja noin 200 ml yhtä 400 mg:n tablettia kohti), ja sekoitetaan lusikalla. Suspensio annetaan välittömästi tabletin/tablettien täydellisen hajoamisen jälkeen.
Annostus aikuisille kroonisessa myelooisessa leukemiassa (KML)
Kroonisessa vaiheessa oleville aikuisille KML-potilaille suositeltu Imatinib Krka d.d. -annos on 400 mg/vrk. KML on kroonisessa vaiheessa, kun kaikki seuraavat edellytykset täyttyvät: veressä ja luuytimessä blasteja < 15 %, perifeerisessä veressä basofiilejä < 20 %, verihiutaleita > 100 x 109/l.
Akseleraatiovaiheessa oleville aikuispotilaille suositeltu Imatinib Krka d.d. -annos on 600 mg/vrk. KML on akseleraatiovaiheessa, mikäli jokin seuraavista edellytyksistä täyttyy: veressä tai luuytimessä on blasteja ≥ 15 % mutta < 30 %, veressä tai luuytimessä on ≥ 30 % blasteja ja promyelosyyttejä yhteensä (olettaen että blasteja < 30 %), perifeerisessä veressä on basofiilejä ≥ 20 %, verihiutaleita on (ei hoidosta johtuen) < 100 x 109/l.
Blastikriisissä oleville aikuispotilaille suositeltu Imatinib Krka d.d. ‑annos on 600 mg/vrk. Blastikriisissä veressä tai luuytimessä on ≥ 30 % blasteja tai potilaalla on luuytimen ulkopuolinen sairaus pois lukien hepatosplenomegalia.
Hoidon kesto: Kliinisissä tutkimuksissa imatinibihoitoa jatkettiin, kunnes tauti alkoi edetä. Hoidon keskeyttämisen vaikutusta sen jälkeen, kun täydellinen sytogeneettinen vaste on saavutettu, ei ole tutkittu.
Annoksen suurentamista 400 mg:sta 600 mg:aan tai 800 mg:aan taudin kroonisessa vaiheessa tai 600 mg:sta suurimpaan annokseen 800 mg (400 mg annosteltuna kahdesti vuorokaudessa) akseleraatiovaiheessa tai blastikriisissä oleville potilaille voidaan harkita seuraavissa tapauksissa edellyttäen, ettei vaikeita haittavaikutuksia ja vaikeaa leukemiaan liittymätöntä neutropeniaa tai trombosytopeniaa ole esiintynyt: jos tauti etenee (ajankohdasta riippumatta); jos vähintään 3 kuukauden hoito ei ole tuottanut tyydyttävää hematologista vastetta; jos 12 kuukauden hoito ei ole tuottanut sytogeneettistä vastetta tai jos aiemmin saavutettu hematologinen ja/tai sytogeneettinen vaste häviää. Potilaita on seurattava huolellisesti, kun annosta nostetaan, koska korkeammilla annoksilla voi esiintyä enemmän haittavaikutuksia.
Annostus lapsille kroonisessa myelooisessa leukemiassa (KML)
Annoksen määrittämisen lapsille ja nuorille tulee perustua kehon pinta-alaan (mg/m2). Kroonisen vaiheen KML:a ja pitkälle edennyttä KML:a sairastaville lapsille ja nuorille suositellaan annosta 340 mg/m2 vuorokaudessa (kokonaisannos ei saa olla yli 800 mg). Lääke voidaan antaa kerran vuorokaudessa tai vaihtoehtoisesti vuorokausiannos voidaan jakaa kahteen annostelukertaan – yksi annos aamulla ja yksi illalla. Tämänhetkinen annossuositus perustuu kokemukseen pienellä joukolla lapsipotilaita (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Alle 2-vuotiaiden lasten hoidosta ei ole kokemusta.
Annoksen suurentamista 340 mg/m2:stä vuorokaudessa 570 mg/m2:een vuorokaudessa (kokonaisannos ei saa olla yli 800 mg) lapsille ja nuorille voidaan harkita seuraavissa tapauksissa edellyttäen, ettei vaikeita haittavaikutuksia ja vaikeaa leukemiaan liittymätöntä neutropeniaa tai trombosytopeniaa ole esiintynyt: jos tauti etenee (ajankohdasta riippumatta); jos vähintään 3 kuukauden hoito ei ole tuottanut tyydyttävää hematologista vastetta; jos 12 kuukauden hoito ei ole tuottanut sytogeneettistä vastetta tai jos aiemmin saavutettu hematologinen ja/tai sytogeneettinen vaste häviää. Potilaita on seurattava huolellisesti, kun annosta nostetaan, koska korkeammilla annoksilla voi esiintyä enemmän haittavaikutuksia.
Annostus aikuisille Philadelphia-kromosomipositiivisessa akuutissa lymfaattisessa leukemiassa (Ph+ ALL)
Aikuispotilaille, joilla on Ph+ ALL, suositeltu Imatinib Krka d.d. ‑annos on 600 mg/vrk. Hoidon kaikkien vaiheiden tulee tapahtua kyseisen sairauden hoitoon perehtyneiden hematologian asiantuntijoiden valvonnassa.
Hoitojen ajoitus: Nykytietojen perusteella imatinibin on osoitettu olevan tehokas ja turvallinen, kun sitä käytetään annoksella 600 mg/vrk yhdessä muiden syöpälääkkeiden kanssa kemoterapian induktiovaiheessa, konsolidaatiovaiheessa ja ylläpitovaiheessa (ks. kohta Farmakodynamiikka) aikuispotilailla, joilla on äskettäin todettu Ph+ ALL. Imatinibihoidon kesto voi vaihdella valitun hoito-ohjelman mukaan, mutta pitempiaikainen altistus imatinibille on yleensä tuottanut parempia tuloksia.
Imatinibimonoterapia annoksella 600 mg/vrk on tehokas ja turvallinen hoito aikuispotilaille, joilla on uusiutunut tai vaikeahoitoinen Ph+ ALL, ja hoitoa voidaan jatkaa, kunnes tauti etenee.
Annostus lapsille Philadelphia-kromosomipositiivisessa akuutissa lymfaattisessa leukemiassa (Ph+ ALL)
Lasten annostus perustuu kehon pinta-alaan (mg/m2). Ph+ ALL ‑lapsipotilaille suositeltu annos on 340 mg/m2/vrk (600 mg kokonaisannos ei saa ylittyä).
Annostus myelodysplastisessa oireyhtymässä tai myeloproliferatiivisissa sairauksissa (MDS/MPD)
Aikuispotilaille, joilla on MDS/MPD, suositeltu Imatinib Krka d.d. ‑annos on 400 mg/vrk.
Hoidon kesto: Toistaiseksi ainoassa tätä aihetta selvittäneessä kliinisessä tutkimuksessa imatinibihoitoa jatkettiin taudin etenemiseen asti (ks. kohta Farmakodynamiikka). Hoidon keston keskiarvo oli analyysihetkellä 47 kuukautta (24 päivää – 60 kuukautta).
Annostus hypereosinofiilisessä oireyhtymässä (HES)/kroonisessa eosinofiilisessä leukemiassa (CEL)
Aikuispotilaille, joilla on HES/CEL, suositeltu Imatinib Krka d.d. ‑annos on 100 mg/vrk.
Annoksen suurentamista 100 mg:sta 400 mg:aan voidaan harkita, mikäli hoitovaste ei ole riittävä eikä potilaalle ole kehittynyt haittavaikutuksia.
Hoitoa tulisi jatkaa niin kauan kuin potilas hyötyy siitä.
Annostus ruuansulatuskanavan stroomakasvaimissa (GIST)
Aikuispotilaille, joilla on pahanlaatuinen ruuansulatuskanavan stroomakasvain (GIST), jota ei voida leikata ja/tai joka on metastasoitunut, suositeltu Imatinib Krka d.d. -annos on 400 mg/vrk.
Annoksen suurentamisen 400 mg:sta 600 mg:aan tai 800 mg:aan vaikutuksesta potilaille, joiden tauti etenee pienemmällä annoksella, on vain vähän tietoa (ks. kohta Farmakodynamiikka).
Hoidon kesto: Kliinisessä GIST-tutkimuksessa imatinibihoitoa jatkettiin, kunnes tauti eteni. Tulosten analysoinnin aikaan hoidon keston mediaani oli 7 kuukautta (7 päivää - 13 kuukautta). Hoidon keskeyttämisen vaikutusta vasteen saavuttamisen jälkeen ei ole tutkittu.
Aikuispotilaille, GIST:in resektion jälkeiseen liitännäishoitoon, suositeltu Imatinib Krka d.d. -annos on 400 mg päivässä. Optimaalinen hoitoaika ei ole vielä vakiintunut. Tämän indikaation tueksi tehdyssä kliinisessä tutkimuksessa hoitoaika oli 36 kuukautta (ks. kohta Farmakodynamiikka).
Annostus dermatofibrosarcoma protuberansissa (DFSP)
Aikuispotilaille, joilla on DFSP, suositeltu Imatinib Krka d.d. ‑annos on 800 mg/vrk.
Annoksen muuttaminen haittavaikutusten vuoksi
Muut kuin hematologiset haittavaikutukset
Jos imatinibin käytön yhteydessä ilmenee vaikea muu kuin hematologinen haittavaikutus, hoito on keskeytettävä, kunnes tilanne on normalisoitunut. Sen jälkeen hoito voidaan aloittaa uudelleen, kun se on tarkoituksenmukaista haittavaikutuksen vakavuuteen nähden.
Mikäli bilirubiini nousee > 3 x viitearvon normaalin ylärajan tai mikäli transaminaasit nousevat > 5 x yli viitearvon normaalin ylärajan, imatinibihoito tulisi keskeyttää, kunnes bilirubiinitaso on laskenut < 1,5 x viitearvon ylärajan ja transaminaasitasot laskeneet < 2,5 x viitearvon ylärajan. Imatinibihoitoa voidaan tämän jälkeen jatkaa alennetuilla vuorokausiannoksilla. Aikuisilla annosta tulee pienentää 400 mg:sta 300 mg:aan tai 600 mg:sta 400 mg:aan tai 800 mg:sta 600 mg:aan vuorokaudessa, ja lapsilla 340 mg:sta 260 mg:aan per m2/vrk.
Hematologiset haittavaikutukset
Annoksen pienentämistä tai hoidon keskeyttämistä vaikean neutropenian ja trombosytopenian vuoksi suositellaan oheisen taulukon mukaisesti.
Annoksen muuttaminen neutropenian ja trombosytopenian vuoksi:
HES/CEL (aloitusannos 100 mg) | ANC < 1,0 x 109/l ja/tai trombosyyttiarvo < 50 x 109/l | 1. Imatinibihoito lopetetaan, kunnes ANC on ≥ 1,5 x 109/l ja trombosyyttiarvo ≥ 75 x 109/l. 2. Hoito aloitetaan uudelleen imatinibilla aikaisemmalla annoksella (jota käytettiin ennen vakavaa haittavaikutusta). |
Kroonisen myelooisen leukemian krooninen vaihe, MDS/MPD ja GIST (aloitusannos 400 mg) HES/CEL (annos 400 mg) | ANC < 1,0 x 109/l ja/tai trombosyyttiarvo < 50 x 109/l | 1. Imatinibihoito lopetetaan, kunnes ANC on ≥ 1,5 x 109/l ja trombosyyttiarvo ≥ 75 x 109/l. 2. Hoito aloitetaan uudelleen imatinibilla aikaisemmalla annoksella (jota käytettiin ennen vakavaa haittavaikutusta). 3. Jos ANC palaa tasolle < 1,0 x 109/l ja/tai trombosyyttiarvo tasolle < 50 x 109/l, toistetaan vaihe 1 ja aloitetaan imatinibihoito uudelleen pienennetyllä 300 mg annoksella. |
Lasten kroonisen myelooisen leukemian krooninen vaihe (annos 340 mg/m2) | ANC < 1,0 x 109/l ja/tai trombosyyttiarvo < 50 x 109/l | 1. Imatinibihoito lopetetaan, kunnes ANC on ≥ 1,5 x 109/l ja trombosyyttiarvo ≥ 75 x 109/l. 2. Hoito aloitetaan uudelleen imatinibilla aikaisemmalla annoksella (jota käytettiin ennen vakavaa haittavaikutusta). 3. Jos ANC palaa tasolle < 1,0 x109/l ja/tai trombosyyttiarvo tasolle < 50 x 109/l, toistetaan vaihe 1 ja aloitetaan imatinibihoito uudelleen pienennetyllä 260 mg/m2 annoksella. |
Kroonisen myelooisen leukemian akseleraatiovaihe ja blastikriisi ja Ph+ ALL (aloitusannos 600 mg) | aANC < 0,5 x 109/l ja/tai trombosyyttiarvo < 10 x 109/l | 1. Tarkistetaan, liittyykö sytopenia leukemiaan (luuydinaspiraatti tai biopsia). 2. Jos sytopenia ei liity leukemiaan, imatinibiannos pienennetään 400 mg:aan. 3. Jos sytopenia jatkuu 2 viikon ajan, annos pienennetään vielä 300 mg:aan. 4. Jos sytopenia jatkuu 4 viikon ajan eikä vieläkään liity leukemiaan, imatinibihoito lopetetaan, kunnes ANC on ≥ 1 x 109/l ja trombosyyttiarvo ≥ 20 x 109/l, ja sen jälkeen hoito aloitetaan uudelleen 300 mg:n annoksella. |
Lasten kroonisen myelooisen leukemian akseleraatiovaihe ja blastikriisi (aloitusannos 340 mg/m2) | aANC < 0,5 x 109/l ja/tai trombosyyttiarvo < 10 x 109/l | 1. Tarkistetaan, liittyykö sytopenia leukemiaan (luuydinaspiraatti tai biopsia). 2. Jos sytopenia ei liity leukemiaan, imatinibiannos pienennetään 260 mg/m2:een. 3. Jos sytopenia jatkuu 2 viikon ajan, annos pienennetään vielä 200 mg/m2:een. 4. Jos sytopenia jatkuu 4 viikon ajan eikä vieläkään liity leukemiaan, imatinibihoito lopetetaan, kunnes ANC on ≥ 1 x 109/l ja trombosyyttiarvo ≥ 20 x 109/l, ja sen jälkeen hoito aloitetaan uudelleen 200 mg/m2:n annoksella. |
Dermatofibrosarcoma protuberans (annos 800 mg) | ANC < 1,0 x 109/l ja/tai trombosyyttiarvo < 50 x 109/l | 1. Imatinibihoito lopetetaan, kunnes ANC on ≥ 1,5 x 109/l ja trombosyyttiarvo ≥ 75 x 109/l. 2. Imatinibihoito aloitetaan uudelleen annoksella 600 mg. 3. Jos ANC palaa tasolle < 1,0 x 109/l ja/tai trombosyyttiarvo tasolle < 50 x 109/l, toistetaan vaihe 1 ja aloitetaan imatinibihoito uudelleen pienennetyllä 400 mg annoksella. |
ANC = neutrofiilien absoluuttinen määrä a joka ilmenee vähintään 1 kuukautta kestäneen hoidon jälkeen | ||
Erityisryhmät
Pediatriset potilaat
Tietoja käytöstä KML:aa sairastaville alle 2-vuotiaille lapsille ja alle 1-vuotiaille Philadelphia-kromosomipositiivista akuuttia lymfaattista leukemiaa sairastaville lapsille ei ole (ks. kohta Farmakodynamiikka). Lapsista, joilla on myelodysplastinen oireyhtymä / myeloproliferatiivinen tauti, dermatofibrosarcoma protuberans, GIST ja hypereosinofiilinen oireyhtymä/krooninen eosinofiilinen leukemia, on hyvin vähän kokemusta.
Imatinibin turvallisuutta ja tehoa alle 18-vuotiaiden, myelodysplastista oireyhtymää / myeloproliferatiivista tautia, dermatofibrosarcoma protuberansia, GIST:iä ja HES/CEL:iä sairastavien lasten ja nuorten hoidossa ei ole osoitettu kliinisissä tutkimuksissa. Saatavissa olevan julkaistun tiedon perusteella, joka on kuvattu kohdassa Farmakodynamiikka, ei voida antaa suosituksia annostuksesta.
Maksan vajaatoiminta
Imatinibi metaboloituu pääosin maksan välityksellä. Potilaille, joilla on lievä, kohtalainen tai vaikea maksan vajaatoiminta pitää antaa pienintä suositettua annosta 400 mg vuorokaudessa. Annosta voidaan pienentää, jos se ei ole siedetty (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset ja Farmakokinetiikka).
Maksan vajaatoiminnan luokittelu:
Maksan vajaatoiminta | Maksan toimintakokeen arvot |
Lievä | kokonaisbilirubiini: = 1,5 ULN ASAT: > ULN (voi olla normaali tai < ULN, jos kokonaisbilirubiini on > ULN) |
Kohtalainen | kokonaisbilirubiini: > 1,5–3,0 ULN ASAT: mikä tahansa arvo |
Vaikea | kokonaisbilirubiini: > 3–10 ULN ASAT: mikä tahansa arvo |
ULN = normaaliarvon yläraja laitoksessa
ASAT = aspartaattiaminotransferaasi
Munuaisten vajaatoiminta
Potilaille, joilla on munuaisten vajaatoiminta tai jotka saavat dialyysihoitoa, tulee antaa aloitusannoksena pienin suositeltu vuorokausiannos eli 400 mg. Näiden potilaiden kohdalla suositellaan kuitenkin varovaisuutta. Annosta voidaan pienentää, jos potilas ei siedä sitä. Jos annos on siedetty, sitä voidaan suurentaa, ellei teho ole riittävä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Iäkkäät henkilöt
Imatinibin farmakokinetiikkaa ei ole erityisesti tutkittu iäkkäillä henkilöillä. Kliinisissä tutkimuksissa aikuisilla potilailla, joista yli 20 % oli 65-vuotiaita tai vanhempia, ei havaittu iästä merkittävästi riippuvia muutoksia farmakokinetiikassa. Erityinen annossuositus iäkkäille henkilöille ei ole tarpeen.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Yhteisvaikutuksia voi esiintyä annettaessa imatinibia yhdessä muiden lääkkeiden kanssa. Varovaisuutta on noudatettava, kun imatinibin kanssa käytetään samanaikaisesti proteaasin estäjiä, atsoliryhmään kuuluvia sienilääkkeitä, tiettyjä makrolidiantibiootteja (ks. kohta Yhteisvaikutukset), CYP3A4-substraatteja, joilla on pieni terapeuttinen leveys (esim. siklosporiini, pimotsidi, takrolimuusi, sirolimuusi, ergotamiini, diergotamiini, fentanyyli, alfentaniili, terfenadiini, bortetsomibi, dosetakseli, kinidiini), tai varfariinia ja muita kumariinijohdoksia (ks. kohta Yhteisvaikutukset).
Imatinibin käyttö samanaikaisesti CYP3A4 indusoivien lääkkeiden (esim. deksametasonin, fenytoiinin, karbamatsepiinin, rifampisiinin, fenobarbitaalin tai mäkikuisman (Hypericum perforatum)) kanssa saattaa vähentää merkitsevästi imatinibialtistusta, mahdollisesti lisäten hoidon epäonnistumisen riskiä. Siksi voimakkaiden CYP3A4-indusoijien ja imatinibin yhteiskäyttöä tulee välttää (ks. kohta Yhteisvaikutukset).
Hypotyreoosi
Potilailla, joiden kilpirauhanen on poistettu ja jotka saavat levotyroksiinikorvaushoitoa, on ilmoitettu kliinistä hypotyreoosia imatinibihoidon aikana (ks. kohta Yhteisvaikutukset). Näiden potilaiden kohdalla tyreotropiini-arvoja (TSH-arvoja) on seurattava huolellisesti.
Maksatoksisuus
Imatinibi metaboloituu pääosin maksassa ja vain 13 % erittyy munuaisten kautta. Potilailla, joilla on maksan vajaatoiminta (lievä, kohtalainen tai vaikea), perifeeristä verenkuvaa ja maksaentsyymejä on seurattava huolellisesti (ks. kohdat Annostus ja antotapa, Haittavaikutukset ja Farmakokinetiikka). On huomioitava, että GIST-potilailla saattaa olla maksan metastaaseja, mikä voi johtaa maksan vajaatoimintaan.
Imatinibin käytön yhteydessä on raportoitu maksavauriotapauksia, mukaan lukien maksan vajaatoiminta ja maksanekroosi. Kun imatinibi yhdistettiin suuriannoksisiin kemoterapiahoitoihin, havaittiin vakavien maksavaikutusten lisääntymistä. Maksan toimintaa tulee seurata huolellisesti, jos imatinibi yhdistetään kemoterapiahoitoihin, joiden tiedetään voivan aiheuttaa maksan toimintahäiriöitä (ks. kohdat Yhteisvaikutukset ja Haittavaikutukset).
Nesteretentio
Vaikeita nestekertymiä (pleuraeffuusio, ödeema, keuhkoödeema, askites, pinnallinen ödeema) on ilmoitettu esiintyneen noin 2,5 %:lla imatinibia saaneista vasta diagnosoiduista KML-potilaista. Siksi potilaiden säännöllistä punnitsemista suositellaan voimakkaasti. Odottamattoman nopean painonnousun syy on selvitettävä tarkasti ja tarvittaessa on ryhdyttävä asianmukaisiin tuki- ja hoitotoimiin. Kliinisissä tutkimuksissa näitä tapahtumia havaittiin useammin iäkkäillä henkilöillä ja potilailla, joilla oli aiemmin ollut sydänsairauksia. Siksi varovaisuutta on noudatettava potilaiden kohdalla, joilla on sydämen toimintahäiriö.
Potilaat, joilla on sydänsairaus
Jos potilaalla on jokin sydänsairaus, sydämen vajaatoiminnan riskitekijöitä tai jos hänellä on aiemmin ollut munuaisten vajaatoiminta, häntä tulee seurata huolellisesti. Kaikki potilaat, joille kehittyy sydämen tai munuaisten vajaatoimintaan viittaavia merkkejä tai oireita, tulee arvioida ja hoitaa.
Potilailla, joilla on hypereosinofiilinen oireyhtymä (HES), jossa esiintyy HES-solujen piilevää infiltraatiota sydänlihakseen, on imatinibihoidon aloittamisen jälkeiseen HES-solujen degranulaatioon liittynyt yksittäistapauksina kardiogeenista sokkia ja vasemman kammion toimintahäiriöitä. Tapaukset ovat olleet ohimeneviä, kun potilaille on annettu systeemisiä steroideja, verenkiertoa on tuettu ja imatinibihoito on väliaikaisesti keskeytetty. Imatinibihoidon yhteydessä on toisinaan ilmoitettu sydämeen kohdistuvia haittatapahtumia, joten imatinibihoidon hyötyjä ja riskejä tulee punnita huolellisesti ennen hoidon aloittamista, jos potilaalla on hypereosinofiilinen oireyhtymä/krooninen eosinofiilinen leukemia.
Myelodysplastisen oireyhtymän ja myeloproliferatiivisten sairauksien ja PDGFR-geenin uudelleenjärjestäytymisen yhteydessä voi esiintyä korkeita eosinofiiliarvoja. Potilaille, joilla on hypereosinofiilinen oireyhtymä/krooninen eosinofiilinen leukemia, ja potilaille, joilla on myelodysplastinen oireyhtymä / myeloproliferatiivinen sairaus ja korkeat eosinofiiliarvot, tulee harkita kardiologin tutkimusta, sydämen ultraäänitutkimusta ja seerumin troponiinin määritystä ennen imatinibihoidon aloittamista. Jos tutkimustuloksissa on poikkeavuuksia, kardiologin on ehkä syytä seurata potilaan tilaa, ja systeemisten steroidien antamista estohoitona (1–2 mg/kg) samanaikaisesti imatinibin kanssa ensimmäisten 1–2 viikon ajan tulee harkita.
Ruuansulatuskanavan verenvuoto
GIST-tutkimuksessa, kun potilaiden kasvainta ei voida leikata ja/tai kun kasvain on metastasoitunut, raportoitiin sekä ruuansulatuskanavan verenvuotoa että verenvuotoja kasvaimien alueella (ks. kohta Haittavaikutukset). Käytettävissä olevan tiedon perusteella ei ole tunnistettu altistavia tekijöitä (kuten kasvaimen koko, sijainti tai häiriö hyytymistekijöissä), jotka lisäisivät GIST-potilaan riskiä saada kummankaan tyyppinen verenvuoto. Koska lisääntynyt verisuonitus ja taipumus verenvuotoon kuuluu osana GIST-tautiin, on verenvuodot pyrittävä tavanomaisia tutkimus- ja hoitomenetelmiä käyttäen havaitsemaan ja hoitamaan kaikilla potilailla.
Markkinoille tulon jälkeisessä käytössä KML-, ALL- sekä muilla potilailla on raportoitu lisäksi mahalaukun antraalista vaskulaarista ektasiaa, joka on ruuansulatuskanavan verenvuodon harvinainen aiheuttaja (ks. kohta Haittavaikutukset). Tarvittaessa voidaan harkita imatinibihoidon keskeyttämistä.
Tuumorilyysisyndrooma
Kliinisesti merkittävän nestehukan korjaaminen ja korkeiden virtsahappotasojen alentaminen on suositeltavaa ennen imatinibihoidon aloittamista mahdollisen tuumorilyysisyndrooman (TLS) ilmenemisen vuoksi (ks. kohta Haittavaikutukset).
Hepatiitti B:n uudelleen aktivoituminen
Hepatiitti B:n uudelleen aktivoitumista on tapahtunut kyseisen viruksen pysyvillä kantajilla sen jälkeen, kun potilas on saanut BCR-ABL-tyrosiinikinaasin estäjiä. Tämä aiheutti joissakin tapauksissa akuuttia maksan vajaatoimintaa tai fulminanttia hepatiittia, joka johti maksansiirtoon tai kuolemaan.
Potilaat on testattava hepatiitti B -viruksen varalta ennen Imatinib Krka d.d. -hoidon aloittamista. Maksasairauksien ja hepatiitti B:n hoitoon perehtyneitä asiantuntijoita on kuultava ennen hoidon aloittamista, jos potilaan hepatiitti B -serologia on positiivinen (mukaan lukien potilaat, joilla sairaus on aktiivinen) ja jos potilas saa positiivisen hepatiitti B -testituloksen hoidon aikana. Hepatiitti B -viruksen kantajia, jotka tarvitsevat Imatinib Krka d.d. -hoitoa, on seurattava tarkasti aktiivisen hepatiitti B-virusinfektion oireiden varalta koko hoidon ajan ja useita kuukausia hoidon jälkeen (ks. kohta Haittavaikutukset).
Fototoksisuus
Suoraa auringonvaloa on vältettävä tai altistumisen on oltava mahdollisimman vähäistä, sillä imatinibihoitoon liittyy fototoksisuusriski. Potilaita on ohjattava käyttämään suojautumiskeinoja kuten suojaavaa vaatetusta ja aurinkosuojaa, jossa on korkea suojakerroin (SPF).
Tromboottinen mikroangiopatia
BCR-ABL-tyrosiinikinaasin estäjien käyttöön on liittynyt tromboottista mikroangiopatiaa, myös yksittäisiä tapauskertomuksia Imatinib Krka d.d. -hoidon yhteydessä (ks. kohta Haittavaikutukset). Jos Imatinib Krka d.d. -hoitoa saavalla potilaalla havaitaan tromboottiseen mikroangiopatiaan liittyviä laboratorio- tai kliinisiä löydöksiä, hoito on keskeytettävä ja tromboottisesta mikroangiopatiasta on tehtävä perusteellinen arvio, johon sisältyy ADAMTS13-aktiivisuuden ja ADAMTS13-vasta-aineiden määritys. Jos ADAMTS13-vasta-aineet ovat koholla ja ADAMTS13-aktiivisuus on samanaikaisesti alentunut, Imatinib Krka d.d. -hoitoa ei pidä aloittaa uudelleen.
Laboratoriokokeet
Täydellinen verenkuva on määritettävä säännöllisesti imatinibihoidon aikana. Kroonista myelooista leukemiaa sairastavien potilaiden imatinibihoitoon on liittynyt neutropeniaa tai trombosytopeniaa. Näiden sytopenioiden esiintyminen liittyy todennäköisesti kuitenkin hoidettavan taudin vaiheeseen, ja ne olivat tavallisempia kroonisen myelooisen leukemian akseleraatiovaihetta tai blastikriisiä sairastavilla kuin kroonista vaihetta sairastavilla potilailla. Imatinibihoito voidaan keskeyttää tai annosta pienentää kohdan Annostus ja antotapa suositusten mukaan.
Imatinibihoitoa saavien potilaiden maksan toimintaa (transaminaasit, bilirubiini, alkalinen fosfataasi) pitää seurata säännöllisesti.
Potilailla, joiden munuaistoiminta on heikentynyt, plasman imatinibialtistus vaikuttaa olevan suurempi kuin potilailla, joiden munuaistoiminta on normaali. Tämä johtuu todennäköisesti siitä, että imatinibia sitovan happaman alfa-1-glykoproteiinin (AGP) pitoisuus munuaisten vajaatoimintapotilaiden plasmassa on kohonnut. Munuaisten vajaatoimintapotilaille tulee antaa pienin mahdollinen aloitusannos. Vaikeaa munuaisten vajaatoimintaa sairastavia potilaita on hoidettava varoen. Annosta voidaan pienentää, jos siedettävyysongelmia ilmenee (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Pitkäkestoiseen imatinibihoitoon voi liittyä kliinisesti merkitsevää munuaisten toiminnan heikkenemistä. Tämän vuoksi munuaisten toiminta tulee arvioida ennen imatinibihoidon aloitusta ja sitä tulee seurata tarkasti hoidon aikana, kiinnittäen erityistä huomiota potilaisiin, joilla on munuaisten vajaatoiminnan riskitekijöitä. Jos munuaisten vajaatoimintaa havaitaan, tulee aloittaa tarkoituksenmukainen hoito tavanomaisten hoitokäytäntöjen mukaisesti.
Pediatriset potilaat
Kasvun hidastumista on ilmoitettu imatinibia saaneilla lapsilla ja nuorilla ennen murrosikää. Pediatrisilla KML-potilailla tehdyssä havainnoivassa tutkimuksessa raportoitiin tilastollisesti merkitsevää (mutta kliiniseltä merkitykseltään epävarmaa) pituuden keskiarvon keskihajontapisteiden laskua 12 ja 24 kuukauden hoidon jälkeen kahdessa pienessä alaryhmässä murrosiän vaiheesta ja sukupuolesta riippumatta. Vastaavia tuloksia on havaittu pediatrisilla ALL-potilailla tehdyssä havainnoivassa tutkimuksessa. Imatinibihoitoa saavien lasten kasvun tarkkaa seurantaa suositellaan (ks. kohta Haittavaikutukset).
Imatinib Krka d.d. sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei tule käyttää tätä lääkettä.
Yhteisvaikutukset
Vaikuttavat aineet, jotka voivat suurentaaimatinibin plasmapitoisuuksia:
Sytokromi P450 isoentsyymi CYP3A4:n toimintaa estävät lääkkeet (esim. proteaasin estäjät, kuten indinaviiri, lopinaviiri/ritonaviiri, ritonaviiri, sakinaviiri, telapreviiri, nelfinaviiri, bosepreviiri; atsoliryhmään kuuluvat sienilääkkeet, mukaan lukien ketokonatsoli, itrakonatsoli, posakonatsoli, vorikonatsoli; tietyt makrolidiryhmän antibiootit, kuten erytromysiini, klaritromysiini ja telitromysiini) saattavat heikentää imatinibin metaboliaa ja suurentaa imatinibipitoisuuksia. Imatinibialtistus voimistui merkitsevästi (imatinibin Cmax-keskiarvo suureni 26 % ja AUC-keskiarvo 40 %) terveillä koehenkilöillä, kun sitä annettiin samanaikaisesti ketokonatsolin (CYP3A4-estäjä) kerta-annoksen kanssa. Varovaisuus on tarpeen annettaessa imatinibia samanaikaisesti CYP3A4-estäjäryhmään kuuluvien lääkkeiden kanssa.
Vaikuttavat aineet, jotka voivat pienentääimatinibin plasmapitoisuuksia:
CYP3A4:n toimintaa indusoivat aineet (esim. deksametasoni, fenytoiini, karbamatsepiini, rifampisiini, fenobarbitaali, fosfenytoiini, primidoni tai mäkikuisma (Hypericum perforatum)) saattavat vähentää merkitsevästi imatinibialtistusta, mahdollisesti lisäten hoidon epäonnistumisen riskiä. Esihoito useilla rifampisiiniannoksilla (600 mg), joita seurasi 400 mg:n kerta-annos imatinibia, pienensi Cmax-arvoa vähintään 54 % ja AUC(0-∞)-arvoa vähintään 74 %:lla vastaaviin ilman rifampisiinihoitoa saatuihin arvoihin verrattuna. Samanlaisia tuloksia saatiin myös potilailla, jotka saivat imatinibihoitoa pahanlaatuisten glioomien hoitoon ja käyttivät samanaikaisesti entsyymejä indusoivia epilepsialääkkeitä kuten karbamatsepiinia, okskarbatsepiinia ja fenytoiinia. Imatinibin AUC-arvo plasmassa pieneni 73 % verrattuna potilaisiin, jotka eivät käyttäneet entsyymejä indusoivia epilepsialääkkeitä. Imatinibin yhteiskäyttöä rifampisiinin tai muiden voimakkaiden CYP3A4-indusoijien kanssa tulee välttää.
Vaikuttavat aineet, joiden plasmapitoisuus voi muuttua imatinibin vaikutuksesta
Imatinibi suurentaa simvastatiinin (CYP3A4-substraatti) Cmax-keskiarvot kaksinkertaisiksi ja AUC-keskiarvot 3,5-kertaisiksi, mikä osoittaa imatinibin estävän CYP3A4:ää. Siksi suositellaan noudattamaan varovaisuutta käytettäessä imatinibia samanaikaisesti kapean terapeuttisen leveyden omaavien CYP3A4-substraattien (esim. syklosporiini, pimotsidi, takrolimuusi, sirolimuusi, ergotamiini, diergotamiini, fentanyyli, alfentaniili, terfenadiini, bortetsomibi, dosetakseli ja kinidiini) kanssa. Imatinibi saattaa suurentaa muiden CYP3A4:n metaboloimien lääkkeiden plasmapitoisuuksia (esim. triatsolibentsodiatsepiinien, dihydropyridiini kalsiumkanavan salpaajien, tiettyjen HMG-CoA reduktaasin estäjien, eli statiinien; jne.).
Koska imatinibin käyttöön liittyy tunnetusti lisääntynyt riski verenvuodoille (esim. hemorragia), antikoagulanttihoitoa tarvitseville potilaille pitää käyttää pienimolekyylipainoista tai tavanomaista hepariinia kumariinijohdannaisten (esim. varfariinin) sijaan.
In vitro imatinibi estää sytokromi P450 isoentsyymi CYP2D6:n toimintaa pitoisuuksina, jotka ovat samanlaisia kuin CYP3A4:n toimintaan vaikuttavat pitoisuudet. Imatinibi annoksella 400 mg kahdesti vuorokaudessa esti CYP2D6-välitteisen metoprololin metaboliaa, metoprololin Cmax ja AUC-arvo kohosivat noin 23 % (90 % luottamusväli [1,16–1,30]). Annoksen muuttaminen ei ole tarpeen, kun imatinibia annetaan samanaikaisesti CYP2D6-substraattien kanssa. Varovaisuutta on kuitenkin noudatettava imatinibin ja kapean terapeuttisen leveyden omaavien CYP2D6-substraattien, kuten metoprololin yhteydessä. Metoprololia saavien potilaiden seurantaa tulee harkita.
In vitro, imatinibi estää parasetamolin O-glukuronidaatiota Ki-arvolla 58,5 μmol/l. Tätä estoa ei ole havaittu in vivo 400 mg imatinibi- ja 1000 mg parasetamoliannoksen jälkeen. Suurempia imatinibi- ja parasetamoliannoksia ei ole tutkittu.
Siksi tulee noudattaa varovaisuutta, kun suuria annoksia imatinibia ja parasetamolia annetaan samanaikaisesti.
Jos potilaan kilpirauhanen on poistettu ja häntä hoidetaan levotyroksiinilla, samanaikainen imatinibihoito saattaa pienentää plasman levotyroksiinipitoisuuksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Varovaisuutta on siis syytä noudattaa. Tämän yhteisvaikutuksen syntymekanismia ei kuitenkaan vielä tunneta.
Imatinibin käytöstä samanaikaisesti kemoterapian kanssa potilailla, joilla on Ph+ ALL, on kliinistä kokemusta (ks. kohta Farmakodynamiikka), mutta imatinibin ja kemoterapiahoitojen välisiä yhteisvaikutuksia ei tunneta täysin. Imatinibiin liittyvät haittatapahtumat (maksatoksisuus, myelosuppressio tai muut) saattavat lisääntyä, ja on ilmoitettu, että valmisteen samanaikaiseen käyttöön L-asparaginaasin kanssa saattaa liittyä maksatoksisuuden lisääntymistä (ks. kohta Haittavaikutukset). Tästä syystä imatinibin käyttö yhdistelmähoidossa vaatii erityistä varovaisuutta.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset
Hedelmällisessä iässä olevia naisia on kehotettava käyttämään tehokasta ehkäisyä hoidon aikana ja vähintään 15 päivän ajan imatinibihoidon lopettamisen jälkeen.
Raskaus
On vain vähän tietoja imatinibin käytöstä raskaana oleville naisille. Markkinoilletulon jälkeen imitinibihoitoa saaneilla naisilla on raportoitu keskenmenoja ja lapsilla synnynnäisiä epämuodostumia. Eläinkokeissa on kuitenkin havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta), eikä mahdollista riskiä sikiölle tunneta. Imatinibia ei pidä käyttää raskauden aikana, ellei käyttö ole selvästi välttämätöntä. Jos sitä käytetään raskauden aikana, potilaalle on kerrottava sikiöön mahdollisesti kohdistuvasta riskistä.
Imetys
Imatinibin jakaantumisesta äidinmaitoon on rajallista tietoja. Kahden imettävän naisen tutkimuksessa paljastui, että sekä imatinibi että sen aktiivi metaboliitti voi jakaantua äidinmaitoon. Maidon plasmasuhde, jota tutkittiin yhdellä potilaalla, oli imatinibille määritettynä 0,5 ja sen metaboliitille määritettynä 0,9, joka viittaa metaboliitin suurempaan jakaantumiseen maitoon. Ottaen huomioon imatinibin ja sen metaboliitin yhdistetty pitoisuus sekä imeväisten suurin mahdollinen päivittäinen maitomäärä, kokonaisaltistus oletetaan olevan matala (~10 % terapeuttisesta annoksesta). Koska vaikutukset matala-annoksiselle imatinibi-altistukselle imeväiselle ovat tuntemattomia, imatinibia käyttävien naisten ei tule kuitenkaan imettää hoidon aikana, eikä vähintään 15 päivään imatinibihoidon lopettamisen jälkeen.
Hedelmällisyys
Prekliinisissä tutkimuksissa lääke ei vaikuttanut uros- eikä naarasrottien hedelmällisyyteen, vaikka vaikutuksia reproduktiivisiin parametreihin havaittiin (ks. kohta Prekliiniset tiedot turvallisuudesta). Imatinibilääkitystä saavilla potilailla ei ole suoritettu tutkimuksia, ja tutkimuksia lääkkeen mahdollisista vaikutuksista hedelmällisyyteen ja gametogeneesiin ei ole tehty. Jos potilas on huolissaan imatinibihoidon vaikutuksesta hedelmällisyyteensä, hänen tulee keskustella asiasta lääkärin kanssa.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Potilaille pitää kertoa, että he saattavat kokea haittavaikutuksia kuten huimausta, näön hämärtymistä tai uneliaisuutta imatinibihoidon aikana. Sen vuoksi autolla ajettaessa ja koneita käytettäessä on noudatettava varovaisuutta.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Potilailla, joilla on pitkälle edennyt syöpäsairaus, voi olla useita muita sairauksia, jotka vaikeuttavat haittavaikutusten syysuhteiden arviointia erilaisten oireiden vuoksi, jotka liittyvät perussairauteen, perussairauden etenemiseen ja monien lääkkeiden yhteiskäyttöön.
Kliinisissä KML-tutkimuksissa lääkkeen käytön lopettaminen lääkkeisiin liittyvien haittavaikutusten vuoksi havaittiin 2,4 %:lla juuri diagnosoiduista potilaista, 4 %:lla taudin myöhäisen kroonisen vaiheen potilaista interferonihoidon epäonnistuttua, 4 %:lla akseleraatiovaiheen potilaista interferonihoidon epäonnistuttua ja 5 %:lla blastikriisipotilaista interferonihoidon epäonnistuttua. GIST-tutkimuksessa lääkkeen käyttö lopetettiin lääkkeeseen liittyvien haittavaikutusten vuoksi 4 %:lla potilaista.
Kahta poikkeusta lukuun ottamatta haittavaikutukset olivat samanlaisia kaikissa käyttöaiheissa. KML-potilailla havaittiin enemmän myelosuppressiota kuin GIST-potilailla, mikä johtuu todennäköisesti perustaudista. GIST-tutkimuksessa, kun potilaiden kasvainta ei voida leikata ja/tai kun kasvain on metastasoitunut 7 potilaalla (5 %) oli 3/4 asteen (CTC, Yleinen Toxicity Criteria) verenvuoto: ruuansulatuskanavan verenvuoto (3 potilasta); verenvuotoja kasvaimien alueella (3 potilasta) tai molemmat (1 potilas). Ruuansulatuskanavan verenvuodot saattavat olla lähtöisin ruuansulatuskanavan alueella sijaitsevista kasvaimista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Ruuansulatuskanavan ja kasvaimen verenvuodot voivat olla vakavia ja joskus kuolemaan johtavia. Yleisimpiä (≥ 10 %) lääkkeisiin liittyviä haittavaikutuksia molemmissa potilasryhmissä olivat lievä pahoinvointi, oksentelu, ripuli, vatsakipu, väsymys, lihassärky, lihaskouristukset ja ihottuma. Pinnallinen turvotus oli yleinen löydös kaikissa tutkimuksissa, ja sitä kuvattiin lähinnä periorbitaaliseksi turvotukseksi ja alaraajaturvotukseksi. Turvotus oli kuitenkin vain harvoin vaikeaa, ja se voidaan hoitaa diureeteilla, muilla tukitoimilla tai imatinibiannosta pienentämällä.
Kun imatinibi yhdistettiin suuriannoksiseen kemoterapiaan Ph+ ALL-potilailla, todettiin ohimenevää maksatoksisuutta, joka ilmeni transaminaasiarvojen nousuna ja hyperbilirubinemiana. Ottaen huomioon turvallisuustietokannan rajallisuuden, lapsilla tähän mennessä ilmoitetut haittatapahtumat vastaavat Ph+ ALL -aikuispotilaiden tunnettua turvallisuusprofiilia. Ph+ ALL -lapsipotilaita koskeva turvallisuustietokanta on hyvin rajallinen, mutta uusia turvallisuusriskejä ei ole tunnistettu.
Sekalaisia haittavaikutuksia kuten pleuraeffuusio, askites, keuhkoödeema ja nopea painonnousu, johon voi liittyä pinnallista turvotusta, voidaan kuvata kollektiivisesti ”nestekertymiksi”. Nämä voidaan yleensä hoitaa keskeyttämällä imatinibihoito väliaikaisesti ja diureeteilla ja muilla asianmukaisilla tukitoimilla. Jotkut näistä haittavaikutuksista saattavat kuitenkin olla vakavia tai hengenvaarallisia, ja useita blastikriisipotilaita, joilla oli todettu pleuraeffuusio, kongestiivinen sydämen vajaatoiminta ja munuaisten vajaatoiminta, on kuollut. Kliinisissä tutkimuksissa ei lapsipotilailla havaittu erityisiä turvallisuuteen liittyviä löydöksiä.
Haittavaikutukset
Haittavaikutukset, joita on raportoitu useampia kuin yksittäinen tapaus, on lueteltu alla elinryhmittäin ja esiintymistiheyden mukaan luokiteltuina. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen esiintymistiheyden mukaan yleisimmistä alkaen.
Taulukossa 1 on lueteltu haittavaikutukset ja niiden esiintymistiheydet.
Taulukko 1 Taulukoitu yhteenveto haittavaikutuksista
Infektiot | |
Melko harvinainen: | Herpes zoster, herpes simplex, nasofaryngiitti, keuhkokuume1, sinuiitti, selluliitti, ylähengitystieinfektiot, influenssa, virtsatieinfektiot, gastroenteriitti, sepsis |
Harvinainen: | Sieni-infektio |
Tuntematon: | Hepatiitti B:n uudelleen aktivoituminen* |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | |
Harvinainen: | Tuumorilyysisyndrooma |
Tuntematon: | Kasvaimen verenvuoto/kasvaimen nekroosi* |
Immuunijärjestelmä | |
Tuntematon: | Anafylaktinen sokki* |
Veri ja imukudos | |
Hyvin yleinen: | Neutropenia, trombosytopenia, anemia |
Yleinen: | Pansytopenia, kuumeinen neutropenia |
Melko harvinainen: | Trombosytoosi, lymfopenia, luuydinsuppressio, eosinofilia, lymfadenopatia |
Harvinainen: | Hemolyyttinen anemia, tromboottinen mikroanginopatia |
Aineenvaihdunta ja ravitsemus | |
Yleinen: | Ruokahaluttomuus |
Melko harvinainen: | Hypokalemia, ruokahalun lisääntyminen, hypofosfatemia, ruokahalun vähentyminen, dehydraatio, kihti, hyperurikemia, hyperkalsemia, hyperglykemia, hyponatremia |
Harvinainen: | Hyperkalemia, hypomagnesemia |
Psyykkiset häiriöt | |
Yleinen: | Unettomuus |
Melko harvinainen: | Masennus, sukupuolivietin heikentyminen, ahdistuneisuus |
Harvinainen: | Sekavuustila |
Hermosto | |
Hyvin yleinen: | Päänsärky2 |
Yleinen: | Huimaus, tuntohäiriöt, makuaistin häiriöt, heikentynyt tunto |
Melko harvinainen: | Migreeni, uneliaisuus, pyörtyminen, perifeerinen neuropatia, muistihäiriöt, iskias, levottomien jalkojen oireyhtymä, vapina, aivoverenvuoto |
Harvinainen: | Kohonnut aivopaine, kouristukset, näköhermotulehdus |
Tuntematon: | Aivoturvotus* |
Silmät | |
Yleinen: | Silmäluomien turvotus, lisääntynyt kyyneleritys, sidekalvon verenvuoto, sidekalvotulehdus, silmien kuivuminen, näön hämärtyminen |
Melko harvinainen: | Silmien ärsytys, silmäkipu, silmäkuopan turvotus, kovakalvon verenvuoto, verkkokalvon verenvuoto, silmäluomitulehdus, makulaturvotus |
Harvinainen: | Kaihi, glaukooma, papillan turvotus |
Tuntematon: | Lasiaisen verenvuoto* |
Kuulo ja tasapainoelin | |
Melko harvinainen: | Kiertohuimaus, tinnitus, kuulon heikkeneminen |
Sydän | |
Melko harvinainen: | Sydämentykytys, takykardia, kongestiivinen sydämen vajaatoiminta3, keuhkoödeema |
Harvinainen: | Sydämen rytmihäiriöt, eteisvärinä, sydänpysähdys, sydäninfarkti, angina pectoris, perikardiumeffuusio |
Tuntematon: | Perikardiitti*, sydäntamponaatio* |
Verisuonisto4 | |
Yleinen: | Punastuminen, verenvuoto |
Melko harvinainen: | Hypertensio, hematoomat, subduraalihematooma, ääreisosien kylmyys, hypotensio, Raynaudin ilmiö |
Tuntematon: | Tromboosi/embolia* |
Hengityselimet, rintakehä ja välikarsina | |
Yleinen: | Hengenahdistus, nenäverenvuoto, yskä |
Melko harvinainen: | Pleuraeffuusio5, nielun ja kurkunpään kipu, nielutulehdus |
Harvinainen: | Pleurakipu, keuhkofibroosi, keuhkohypertensio, keuhkoverenvuoto |
Tuntematon: | Akuutti hengitysvajaus11*, interstitielli keuhkosairaus* |
Ruoansulatuselimistö | |
Hyvin yleinen: | Pahoinvointi, ripuli, oksentelu, dyspepsia, vatsakipu6 |
Yleinen: | Ilmavaivat, vatsan pullotus, gastroesofageaalinen refluksi, ummetus, suun kuivuminen, gastriitti |
Melko harvinainen: | Suutulehdus, suun haavaumat, ruoansulatuskanavan verenvuoto7, röyhtäily, veriripuli, ruokatorvitulehdus, askites, mahahaava, verioksennukset, huulitulehdus, nielemishäiriö, haimatulehdus |
Harvinainen: | Koliitti, ileus, tulehduksellinen suolistosairaus |
Tuntematon: | Ileus/suolentukkeuma*, ruoansulatuskanavan perforaatio*, divertikuliitti*, mahalaukun antraalinen vaskulaarinen ektasia (GAVE)* |
Maksa ja sappi | |
Yleinen: | Kohonneet maksaentsyymiarvot |
Melko harvinainen: | Hyperbilirubinemia, hepatiitti, ikterus |
Harvinainen: | Maksan vajaatoiminta8, maksanekroosi |
Iho ja ihonalainen kudos | |
Hyvin yleinen: | Turvotus silmäkuopan ympärillä, ihotulehdus/ekseema/ihottuma |
Yleinen: | Kutina, kasvojen turvotus, ihon kuivuminen, punoitus, hiustenlähtö, yöhikoilu, valoherkkyysreaktiot |
Melko harvinainen: | Märkärakkulainen ihottuma, ruhjeet, lisääntynyt hikoilu, nokkosihottuma, mustelmat, tavallista suurempi alttius mustelmille, niukka karvaisuus, ihon hypopigmentaatio, eksfoliatiivinen dermatiitti, kynsien murtuminen, follikuliitti, petekiat, psoriaasi, purppura, ihon hyperpigmentaatio, rakkulaiset ihomuutokset, pannikuliitti12 |
Harvinainen: | Akuutti kuumeinen neutrofiilinen dermatoosi (Sweetin oireyhtymä), kynsien värimuutokset, angioödeema, vesirakkulainen ihottuma, erythema multiforme, leukosytoklastinen vaskuliitti, Stevens-Johnsonin oireyhtymä, akuutti yleistynyt eksanteemainen pustuloosi ihottuma (AGEP), pemfigus* |
Tuntematon: | Palmoplantaarinen erytrodysestesiaoireyhtymä*, likenoidinen keratoosi*, punajäkälä*, toksinen epidermaalinen nekrolyysi*, lääkeihottuma, johon liittyy eosinofilia ja systeemisiä oireita (DRESS)*, pseudoporfyria* |
Luusto, lihakset ja sidekudos | |
Hyvin yleinen: | Lihasspasmit ja -krampit, luusto- ja lihaskipu (mm. lihaskipu9, nivelkipu, luukipu10) |
Yleinen: | Nivelten turvotus |
Melko harvinainen: | Nivelten ja lihasten jäykkyys, osteonekroosi* |
Harvinainen: | Lihasheikkous, niveltulehdus, rabdomyolyysi/myopatia |
Tuntematon: | Kasvun hidastuminen lapsilla* |
Munuaiset ja virtsatiet | |
Melko harvinainen: | Munuaiskipu, verivirtsaisuus, akuutti munuaisten vajaatoiminta, tavallista suurempi virtsaamistiheys |
Tuntematon: | Krooninen munuaisten vajaatoiminta |
Sukupuolielimet ja rinnat | |
Melko harvinainen: | Gynekomastia, erektiohäiriöt, runsaat kuukautiset, kuukautisten epäsäännöllisyys, sukupuolitoimintojen häiriöt, nännien kipu, rintojen turpoaminen, kivespussin turvotus |
Harvinainen: | Hemorraaginen keltarauhanen/ munasarjakysta |
Yleisoireet ja antopaikassa todettavat haitat | |
Hyvin yleinen: | Nesteen kertyminen elimistöön ja turvotus, väsymys |
Yleinen: | Heikkous, kuume, yleistynyt voimakas turvotus (anasarca), vilunväreet, jäykkyys |
Melko harvinainen: | Rintakipu, huonovointisuus |
Tutkimukset | |
Hyvin yleinen: | Painon nousu |
Yleinen: | Painon lasku |
Melko harvinainen: | Kohonneet veren kreatiniiniarvot, kohonneet veren kreatiinifosfokinaasiarvot, kohonneet veren laktaattidehydrogenaasiarvot, kohonneet veren alkalisen fosfataasin arvot |
Harvinainen: | Kohonneet veren amylaasiarvot |
* Tämäntyyppisiä reaktioita on ilmoitettu lähinnä imatinibin markkinoille tulon jälkeen. Tiedot perustuvat sekä spontaaneihin tapausraportteihin että vakaviin haittatapahtumiin, joita on todettu meneillään olevissa tutkimuksissa, laajennetun saatavuuden tutkimuksissa, kliinisissä farmakologisissa tutkimuksissa ja eksploratiivisissa tutkimuksissa toistaiseksi hyväksymättömillä käyttöaiheilla. Koska ilmoitetut reaktiot on todettu populaatiossa, jonka kokoa ei tiedetä, niiden esiintymistiheyttä ja mahdollista syy-yhteyttä imatinibialtistuksen kanssa ei välttämättä pystytä arvioimaan luotettavasti.
1 Keuhkokuumetta ilmoitettiin yleisimmin potilailla, joilla oli GIST tai akseleraatio- tai blastikriisivaiheessa oleva KML.
2 Päänsärky oli yleisintä GIST-potilailla.
3 Sydämeen kohdistuneita haittatapahtumia kuten kongestiivista sydämen vajaatoimintaa todettiin potilasvuosiin nähden yleisemmin potilailla, joilla oli akseleraatio- tai blastikriisivaiheessa oleva KML, kuin potilailla, joilla oli kroonisessa vaiheessa oleva KML.
4 Punastuminen oli yleisintä GIST-potilailla, kun taas verenvuodot (verenpurkaumat, verenvuoto) olivat yleisimpiä potilailla, joilla oli GIST tai akseleraatio- tai blastikriisivaiheessa oleva KML.
5 Pleuraeffuusiota ilmoitettiin yleisemmin potilailla, joilla oli GIST tai akseleraatio- tai blastikriisivaiheessa oleva KML, kuin potilailla, joilla oli kroonisessa vaiheessa oleva KML.
6+7 Vatsakipua ja ruoansulatuskanavan verenvuotoa esiintyi yleisimmin GIST-potilailla.
8 Joitakin kuolemaan johtaneita maksan vajaatoimintatapauksia ja maksanekroositapauksia on ilmoitettu.
9 Imatinibin markkinoille tulon jälkeen on todettu luusto- ja lihaskipua imatinibihoidon aikana tai sen päättymisen jälkeen.
10 Luusto- ja lihaskipua ja siihen liittyviä tapahtumia todettiin yleisemmin KML-potilailla kuin GIST-potilailla.
11 Kuolemaan johtaneita tapauksia on raportoitu potilailla, joilla on ollut pitkälle edennyt tauti, vaikeita infektioita, vaikea neutropenia ja muita vakavia samanaikaisia kliinisiä tiloja.
12 Mukaan lukien kyhmyruusu.
Laboratoriokoearvojen poikkeavuudet
Hematologia
KML-potilailla sytopeniat, etenkin neutropenia ja trombosytopenia, ovat olleet yhdenmukainen löydös kaikissa tutkimuksissa, ja tiedot viittaavat siihen, että esiintymistiheys on suurempi suuria ≥ 750 mg:n annoksia käytettäessä (I vaiheen tutkimus). Sytopenioiden esiintyminen riippui kuitenkin selvästi myös taudin vaiheesta. Kolmannen ja neljännen asteen neutropenioiden (ANC < 1,0 x 109/l) ja trombosytopenioiden (trombosyyttiarvo < 50 x 109/l) esiintymistiheys oli 4–6 kertaa suurempi blastikriisissä ja akseleraatiovaiheessa olevilla potilailla (neutropenia 59–64 % ja trombosytopenia 44–63 %) verrattuna kroonisen myelooisen leukemian kroonisessa vaiheessa oleviin potilaisiin, joiden sairaus oli vasta diagnosoitu (neutropenia 16,7 % ja trombosytopenia 8,9 %). Vasta diagnosoidussa kroonisen myelooisen leukemian kroonisessa vaiheessa 4. asteen neutropeniaa (ANC < 0,5 x 109/l) havaittiin 3,6 %:lla potilaista ja trombosytopeniaa (trombosyyttiarvo < 10 x 109/l) < 1 %:lla potilaista. Neutropeniajaksojen mediaanikesto oli yleensä 2–3 viikkoa ja trombosytopeniajaksojen yleensä 3−4 viikkoa. Nämä tapahtumat voidaan yleensä hoitaa joko pienentämällä imatinibiannosta tai keskeyttämällä hoito, mutta ne voivat harvoissa tapauksissa johtaa hoidon pysyvään keskeyttämiseen. KML:aa sairastavilla lapsipotilailla yleisimmin todettuja toksisia vaikutuksia olivat 3. ja 4. asteen sytopeniat, kuten neutropenia, trombosytopenia ja anemia. Niitä esiintyy yleensä ensimmäisten hoitokuukausien aikana.
GIST-tutkimuksessa, kun potilaiden kasvainta ei voida leikata ja/tai kun kasvain on metastasoitunut, 3. asteen anemiaa raportoitiin 5,4 %:lla ja 4. asteen anemiaa 0,7 %:lla potilaista. Tähän ovat, ainakin joillain potilaista, voineet vaikuttaa ruuansulatuskanavan tai kasvaimensisäiset verenvuodot. Kolmannen asteen neutropeniaa havaittiin 7,5 %:lla; 4. asteen neutropeniaa 2,7 %:lla ja 3. asteen trombosytopeniaa 0,7 %:lla potilaista. Yhdelläkään potilaalla ei havaittu 4. asteen trombosytopeniaa. Valkosolujen ja neutrofiilien määrät laskivat pääasiassa hoidon ensimmäisten kuuden viikon aikana, minkä jälkeen arvot pysyivät suhteellisen vakiona.
Biokemia
KML-potilailla havaittiin huomattavaa transaminaasi- (< 5 %) tai bilirubiiniarvojen (< 1 %) nousua ja se saatiin yleensä hoidettua pienentämällä annosta tai keskeyttämällä hoito (näiden jaksojen mediaanikesto oli noin yksi viikko). Hoito lopetettiin pysyvästi poikkeavien maksa-arvojen vuoksi alle 1 %:lla KML-potilaista. GIST-potilaista (tutkimus B2222) 6,8 %:lla havaittiin kolmannen tai neljännen asteen ALAT:in (alaniiniaminotransferaasin) pitoisuuden nousu ja 4,8 %:lla kolmannen tai neljännen asteen ASAT:in (aspartaattiaminotransferaasin) nousu. Bilirubiinin nousua havaittiin alle 3 %:lla.
Sytolyyttistä ja kolestaattista maksatulehdusta ja maksan vajaatoimintaa on havaittu, jotka joissain tapauksissa ovat johtaneet kuolemaan, kuten yhdellä suuren parasetamoliannoksen saaneella potilaalla.
Valikoitujen haittavaikutusten kuvaus:
Hepatiitti B:n uudelleen aktivoituminen
Hepatiitti B:n uudelleen aktivoitumista on ilmoitettu BCR-ABL-tyrosiinikinaasin estäjien käytön yhteydessä. Tämä aiheutti joissakin tapauksissa akuuttia maksan vajaatoimintaa tai fulminanttia hepatiittia, joka johti maksansiirtoon tai kuolemaan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kokemusta suositellun terapeuttisen annostuksen ylittämisestä on vain rajoitetusti. Yksittäisiä imatinibiyliannostustapauksia on raportoitu spontaanisti ja kirjallisuudessa. Yliannostustapauksissa potilasta tulee seurata ja asianmukainen oireellinen hoito antaa. Useimmiten raportoitu tulos näissä tapauksissa oli ”kohentunut” tai ”toipunut”. Tapauksia, jotka on raportoitu muissa annosväleissä:
Aikuiset
1200–1600 mg (kestäen vaihtelevasti 1–10 päivän välillä): Pahoinvointia, oksentelua, ripulia, ihottuma, eryteema, edeema, turvotus, heikotus, lihasnykäyksiä, trombosytopenia, pansytopenia, vatsakipua, päänsärkyä, vähentynyt ruokahalu.
1800–3200 mg (jopa 3200 mg päivässä 6 päivän ajan): Voimattomuus, myalgia, suurentunut kreatiniini fosfokinaasi, suurentunut bilirubiini, ruuansulatuskanavan kipu.
6400 mg (kerta-annos): Yksi kirjallisuusraportti yhdestä potilaasta, joka koki pahoinvointia, oksentelua, vatsakipua, kuumetta, kasvojen turvotusta, alentunut neutrofiililuku, kohonneita transaminaasilukuja.
8–10 g (kerta-annos): Oksentelua ja ruuansulatuskanavan kipua on raportoitu.
Pediatriset potilaat
Yksi 3-vuotias poika, joka altistui 400 mg kerta-annokselle, koki oksentelua, ripulia sekä anoreksiaa, ja toinen 3-vuotias poika, joka altistui 980 mg kerta-annokselle, koki veren valkosolumäärän alenemista sekä ripulia.
Yliannostustapauksissa potilasta tulee tarkkailla ja antaa asiaankuuluvaa tukihoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, BCR-ABL-tyrosiinikinaasin estäjät, ATC-koodi: L01EA01.
Vaikutusmekanismi
Imatinibi on pienimolekyylinen tyrosiinikinaasin estäjä, joka estää voimakkaasti Bcr-Abl-tyrosiinikinaasin toimintaa ja useita reseptorityrosiinikinaaseja (c-Kit-proto-onkogeenin koodaama kantasolutekijän [SCF] Kit-reseptori, DDR1- ja DDR2-reseptorit, kasvutekijäreseptori CSF-1R ja verihiutalekasvutekijäreseptorit alfa ja beeta [PDGFR-alfa ja PDGFR-beeta]). Imatinibi voi myös estää näiden reseptorikinaasien aktivaation välittämiä solutason tapahtumia.
Farmakodynaamiset vaikutukset
Imatinibi on proteiinityrosiinikinaasin estäjä, joka estää voimakkaasti Bcr-Abl-tyrosiinikinaasia in vitro-, solu- ja in vivo ‑pitoisuuksina. Aine estää selektiivisesti proliferaatiota ja indusoi apoptoosia Bcr-Abl-positiivisissa solulinjoissa sekä tuoreissa leukemiasoluissa, jotka ovat peräisin Philadelphia-kromosomin suhteen positiivisilta kroonista myelooista leukemiaa sairastavilta potilailta sekä akuuttia lymfoblastista leukemiaa sairastavilta potilailta.
Aineella on kasvaimia estävä vaikutus in vivo, kun sitä annetaan ainoana aineena eläinmalleissa, joissa käytetään Bcr-Abl-positiivisia kasvainsoluja.
Imatinibi on myös verihiutalekasvutekijän (PDGF) reseptorityrosiinikinaasin, PDGF-R, ja kantasolutekijän (SCF) reseptorityrosiinikinaasin, c-Kit, estäjä ja se estää PDGF- ja SCF-välitteisiä solutapahtumia. In vitro, ruuansulatuskanavan stroomakasvainsoluissa, joissa aktivaattorina toimii kit-mutaatio, imatinibi estää soluproliferaatiota ja aiheuttaa apoptoosia. PDGF:n konstitutiivisen tuotannon tai PDGF-reseptorin tai Abl-proteiinityrosiinikinaasien konstitutiivisen aktivaation (jonka syynä on niiden yhdistyminen eri proteiineihin) on arveltu osallistuvan MDS:n, MPD:n, HES:n/CEL:n ja DFSP:n patogeneesiin. Imatinibi estää viestinkulkua ja proliferaatiota soluissa, joiden PDGFR- ja Abl-kinaasitoiminta on häiriintynyt.
Kliiniset tutkimukset kroonisessa myelooisessa leukemiassa (KML)
Näyttö imatinibin tehosta perustuu hematologisten ja sytogeneettisten vasteiden määrään ja aikaan ilman taudin etenemistä. Juuri diagnosoitua kroonisessa vaiheessa olevaa KML:aa lukuun ottamatta kontrolloituja tutkimuksia, jotka osoittaisivat kliinistä tehoa, kuten sairauteen kuuluvien oireiden vähenemistä tai eloonjäämisetua, ei ole.
Kolme suurta kansainvälistä avointa kontrolloimatonta II vaiheen tutkimusta tehtiin potilaille, jotka sairastivat kroonisen myelooisen leukemian Philadelphia-kromosomipositiivista (Ph+) pitkälle edennyttä vaihetta, blasti- tai akseleraatiovaihetta tai muita Ph+-leukemiatyyppejä tai kroonisen myelooisen leukemian kroonista vaihetta, johon aiempi alfainterferonihoito ei ollut tehonnut. Potilaille, joilla on vasta diagnosoitu Philadelphia-kromosomin suhteen positiivinen krooninen myelooinen leukemia, on tehty yksi suuri, avoin, kansainvälinen, satunnaistettu III vaiheen monikeskustutkimus. Lisäksi lapsia ja nuoria on hoidettu kahdessa I vaiheen tutkimuksessa ja yhdessä II vaiheen tutkimuksessa.
Kaikissa kliinisissä tutkimuksissa 38-40 % potilaista oli ≥ 60-vuotiaita ja 10-12 % ≥ 70-vuotiaita.
Krooninen vaihe, vasta diagnosoidut potilaat
III vaiheen tutkimuksessa aikuispotilailla verrattiin pelkkää imatinibihoitoa yhdistelmähoitoon, jossa potilaat saivat alfa-interferonia ja sytarabiinia. Potilailla oli mahdollisuus vaihtaa toiseen hoitoryhmään, jos heillä ei saavutettu lainkaan hoitovastetta (täydellisen hematologisen vasteen puuttuminen kuuden kuukauden kohdalla, valkosolujen määrän kasvu, huomattavan sytogeneettisen vasteen puuttuminen 24 kuukauden kohdalla), jos hoitovaste hävisi (täydellisen hematologisen tai huomattavan sytogeneettisen vasteen häviäminen) tai jos he sietivät hoidon huonosti. Imatinibiryhmässä potilaita hoidettiin 400 mg:n vuorokausiannoksella. Alfa-interferoniryhmässä potilaat saivat alfa-interferonia subkutaanisesti tavoiteannoksen 5 MIU/m2 /vrk, sekä subkutaanisesti sytarabiinia 20 mg/m2 /vrk 10 päivän ajan kuukaudessa.
Kaikkiaan 1 106 potilasta satunnaistettiin, 553 potilasta, molempiin hoitoryhmiin. Potilaiden ominaisuudet lähtötilanteessa olivat hyvin samankaltaiset molemmissa hoitoryhmissä. Potilaiden iän mediaani oli 51 vuotta (vaihteluväli 18–70 vuotta) ja 21,9 % potilaista oli ≥ 60 vuotiaita. Potilaista 59 % oli miehiä ja 41 % naisia; 89,9 % valkoihoisia ja 4,7 % tummaihoisia. Seitsemän vuotta viimeisen potilaan rekrytoinnin jälkeen ensisijaishoidon mediaanikesto imatinibiryhmässä oli 82 kk ja alfainterferoniryhmässä 8 kk. Toissijaisen imatinibihoidon mediaanikesto oli 64 kk. Imatinibia ensisijaishoitona saaneiden potilaiden saama keskimääräinen vuorokausiannos oli 406 ± 76 mg. Tutkimuksen ensisijainen tehokkuuspäätemuuttuja oli havaittu aika ilman merkkejä taudin etenemisestä. Taudin etenemisellä tarkoitettiin mitä tahansa seuraavista tapahtumista: taudin eteneminen akseleraatiovaiheeseen tai blastikriisiin; kuolema; täydellisen hematologisen vasteen tai huomattavan sytogeneettisen vasteen häviäminen; tai valkosolujen lukumäärän kasvu asianmukaisesta hoidosta huolimatta potilailla, joilla ei ole saavutettu täydellistä hematologista vastetta. Pääasiallisia toissijaisia päätemuuttujia olivat huomattava sytogeneettinen vaste, hematologinen vaste, molekulaarinen vaste (arvioitu minimaalinen jäljellä oleva tauti), taudin etenemiseen akseleraatiovaiheeseen tai blastikriisiin kulunut aika, sekä eloonjääminen. Vastetta koskevat tiedot on esitetty Taulukossa 2.
Taulukko 2 Vasteet kliinisessä tutkimuksessa koskien vasta diagnosoitua kroonista myelooista leukemiaa (84 kuukauden tiedot)
| (Parhaat vasteet) | Imatinibi n=553 | Alfainterferoni + sytarabiini n=553 |
| Hematologinen vaste | ||
| Täydellisten hematologisten vasteiden määrä n (%) | 534 (96,6 %)* | 313 (56,6 %)* |
| [95 %:n luottamusväli] | [94,7 %; 97,9 %] | [52,4 %; 60,8 %] |
| Sytogeneettinen vaste | ||
| Huomattava vaste n (%) | 490 (88,6 %)* | 129 (23,3 %)* |
| [95 %:n luottamusväli] | [85,7 %, 91,1 %] | [19,9 %, 27,1 %] |
| Täydellinen sytogeneettinen vaste n (%) | 456 (82,5 %)* | 64 (11,6 %)* |
| Osittainen sytogeneettinen vaste n (%) | 34 (6,1 %) | 65 (11,8 %) |
| Molekulaarinen vaste** | ||
| Huomattava vaste 12 kuukauden kohdalla (%) | 153/305=50,2 % | 8/83=9,6 % |
| Huomattava vaste 24 kuukauden kohdalla (%) | 73/104=70,2 % | 3/12=25 % |
| Huomattava vaste 84 kuukauden kohdalla (%) | 102/116=87,9 % | 3/4=75 % |
* p<0,001, Fisherin eksakti testi ** molekulaarinen vaste prosentteina perustuvat saatavilla oleviin näytteisiin Hematologisen vasteen kriteerit (kaikki vasteet varmistetaan ≥ 4 viikon jälkeen): Valkosolumäärä < 10 x 109 /l, verihiutaleiden määrä < 450 x 109 /l, myelosyyttejä + metamyelosyyttejä < 5 % veressä, ei blasteja eikä promyelosyyttejä veressä, basofiileja < 20 %, ei luuytimenulkoista sairautta. Sytogeneettisen vasteen kriteerit: täydellinen (0 % Ph+ -metafaaseja), osittainen (1–35 %), vähäinen (36–65 %) tai minimaalinen (66–95 %). Huomattava vaste (0–35 %) kattaa sekä täydellisen että osittaisen vasteen. Huomattavan molekulaarisen vasteen kriteerit: perifeerisessä veressä ≥ 3 logaritmin väheneminen Bcr-Abl transkriptien määrässä (mitattuna reaaliaikaisella kvantitatiivisella käänteiskopioijaentsyymi-PCR-menetelmällä) standardoituun lähtötilanteeseen verrattuna. | ||
Ensisijaishoidon aikaansaamat täydelliset hematologiset vasteet, huomattavat sytogeneettiset vasteet ja täydelliset sytogeneettiset vasteet arvioitiin Kaplan-Meierin menetelmällä, jossa hoitoon vastaamattomat potilaat jätettiin pois laskuista viimeisenä tutkimuspäivänä. Tällä menetelmällä saadut arviot ensisijaishoitona käytetyn imatinibin aikaansaamista kumulatiivisista vasteista paranivat 12 ja 84 hoitokuukauden välisenä aikana seuraavasti: täydelliset hematologiset vasteet 96,4 %:sta 98,4 %:iin ja täydelliset sytogeneettiset vasteet 69,5 %:sta 87,2 %:iin.
Seitsemän vuoden seurannassa taudin etenemiseen liittyviä tapahtumia esiintyi 93:lla (16,8 %) imatinibiryhmän potilaista: akseleraatiovaiheeseen/blastikriisivaiheeseen siirtyi 37 potilasta (6,7 %), huomattava sytogeneettinen vaste menetettiin 31 potilaalla (5,6 %), täydellinen hematologinen vaste menetettiin tai valkosolumäärä nousi 15 potilaalla (2,7 %), ja 10 potilasta (1,8 %) menehtyi jonkin muun syyn kuin KML:n takia. Sitä vastoin interferonia ja sytarabiinia saaneiden potilaiden ryhmässä esiintyi 165 tapahtumaa (29,8 %), ja näistä tapahtumista 130 ilmaantui, kun alfainterferonia ja sytarabiinia käytettiin ensisijaishoitona.
Alfainterferoniryhmään verrattuna imatinibiryhmässä oli 84 kuukauden jälkeen merkitsevästi enemmän potilaita, joiden tauti ei ollut edennyt akseleraatiovaiheeseen tai blastikriisivaiheeseen (92,5 % vs. 85,1 %; p < 0,001). Taudin vuotuiset etenemisprosentit akseleraatiovaiheeseen tai blastikriisivaiheeseen pienenivät hoidon jatkuessa, ja neljäntenä ja viidentenä hoitovuotena vuotuinen etenemisprosentti oli alle 1 %. Arvioiden mukaan 84 kuukauden elossaolo ilman taudin etenemistä oli imatinibiryhmässä 81,2 % ja verrokkiryhmässä 60,6 % (p < 0,001). Taudin vuotuiset etenemisprosentit pienenivät imatinibiryhmässä ajan mittaan taudin etenemistyypistä riippumatta.
Imatinibihoitoa saaneista potilaista menehtyi yhteensä 71 (12,8 %) ja alfainterferonia ja sytarabiinia saaneista potilaista 85 (15,4 %). Arvioitu kokonaiselossaolo 84 kuukauden kohdalla oli imatinibihoitoon satunnaistetussa ryhmässä 86,4 % (83, 90) ja alfainterferoni- ja sytarabiinihoitoon satunnaistetussa ryhmässä 83,3 % (80, 87) (p = 0,073, log rank -testi). Tähän lopputapahtumaan (tapahtumaan kulunut aika) vaikuttaa voimakkaasti se, että niin monet alfainterferoni- ja sytarabiinihoitoon satunnaistetut potilaat siirtyivät imatinibihoitoon. Imatinibihoidon vaikutusta elossaoloon vasta diagnosoidussa kroonisen vaiheen KML:ssa on tutkittu tarkemmin yllä raportoitujen imatinibitietojen retrospektiivisessä analyysissa, kun mukaan otettiin myös primaariset tiedot toisesta vaiheen III tutkimuksesta, jossa interferonin ja sytarabiinin yhdistelmää (n = 325) annettiin täysin samalla tavalla. Tässä retrospektiivisessä analyysissa imatinibi todettiin interferonin ja sytarabiinin yhdistelmää paremmaksi (p < 0,001); 42 kuukauden kuluttua 47 (8,5 %) imatinibia saaneista potilaista ja 63 (19,4 %) interferonia ja sytarabiinia saaneista potilaista oli menehtynyt.
Sytogeneettisen ja molekulaarisen vasteen voimakkuudella oli selkeä vaikutus imatinibia saaneiden potilaiden pitkäaikaistuloksiin. Arvioiden mukaan täydellisen sytogeneettisen vasteen (osittaisen sytogeneettisen vasteen) 12 kuukauden kohdalla saavuttaneista potilaista 96 %:lla (93 %:lla) tauti ei ollut edennyt 84 kuukauden kohdalla akseleraatiovaiheeseen/blastikriisivaiheeseen. Potilaista, jotka eivät olleet saavuttaneet huomattavaa sytogeneettistä vastetta 12 kuukauden kohdalla, vain 81 %:n tauti ei ollut muuttunut 84 kuukauden kohdalla pitkälle edenneeksi KML:ksi (p < 0,001 yhteensä, p = 0,25 täydellinen vs. osittainen sytogeneettinen vaste). Jos potilaalla todettiin 12 kuukauden kohdalla vähintään 3 logaritmin väheneminen Bcr-Abl-transkriptien määrässä, todennäköisyys, että hänen tautinsa ei ollut edennyt akseleraatiovaiheeseen tai blastikriisivaiheeseen 84 kuukauden kohdalla, oli 99 %. Myös 18 kuukauden kohdalla tehty analyysi antoi samansuuntaista näyttöä.
Tässä tutkimuksessa oli sallittu annoksen suurentaminen 400 mg:sta vuorokaudessa 600 mg:aan ja sen jälkeen 600 mg:sta 800 mg:aan vuorokaudessa. 42 kuukauden seurannan jälkeen 11 potilaalla varmistettiin (4 viikon sisällä) sytogeneettisen vasteen häviäminen. Näistä 11 potilaasta 4:llä annosta suurennettiin 800 mg:aan vuorokaudessa, joista kaksi sai sytogeneettisen vasteen takaisin (1 osittainen ja 1 täydellinen, jolla myös havaittiin molekulaarinen vaste). Niillä 7 potilaalla, joilla annosta ei suurennettu, ainoastaan yksi sai takaisin täydellisen sytogeneettisen vasteen. Eräiden haittavaikutusten osuus oli suurempi niillä 40 potilailla, joilla annosta suurennettiin 800 mg:aan, verrattuna potilasjoukkoon ennen annoksen suurentamista (n = 551). Yleisempiä haittavaikutuksia olivat ruuansulatuskanavan verenvuodot, sidekalvotulehdukset ja transaminaasi- ja bilirubiiniarvojen kohoaminen. Muita haittavaikutuksia raportoitiin joko vähemmän tai yhtä yleisesti.
Krooninen vaihe, epäonnistunut interferonihoito
532 aikuispotilasta hoidettiin 400 mg aloitusannoksena. Potilaat jakautuivat kolmeen pääryhmään: hematologinen epäonnistuminen (29 %), sytogeneettinen epäonnistuminen (35 %) tai interferoni-intoleranssi (36 %). Potilaat olivat saaneet aiemmin interferonihoitoa, jonka mediaanikesto oli 14 kuukautta annoksilla ≥ 25 x 106 IU/viikko; kaikkien sairaus oli myöhäisessä kroonisessa vaiheessa, ja taudin toteamisesta kulunut mediaaniaika oli 32 kuukautta. Tutkimuksen ensisijainen tehokkuusmuuttuja oli huomattavan sytogeneettisen vasteen osuus (täydellinen + osittainen vaste, 0‑35 % Ph+-metafaaseja luuytimessä).
Tässä tutkimuksessa 65 % potilaista saavutti huomattavan sytogeneettisen vasteen, joka oli täydellinen 53 %:lla (varmistettu 43 %:lla) potilaista (Taulukko 3). Täydellinen hematologinen vaste saavutettiin 95 %:lla potilaista.
Akseleraatiovaihe
Tutkimukseen otettiin 235 akseleraatiovaiheen aikuispotilasta. Ensimmäisten 77 potilaan aloitusannos oli 400 mg; sittemmin tutkimussuunnitelmaa muutettiin suuremman annoksen sallivaksi, ja loppujen 158 potilaan aloitusannos oli 600 mg.
Ensisijainen tehokkuusmuuttuja oli hematologisen vasteen osuus. Hematologinen vaste ilmoitettiin joko täydellisenä hematologisena vasteena, leukemian olemassaoloa koskevan näytön puuttumisena (esim. blastien poistuminen luuytimestä ja verestä siten, ettei täyttä ääreisveren toipumista kuitenkaan saavutettu kuten täydellisessä vasteessa) tai paluuna kroonisen myelooisen leukemian krooniseen vaiheeseen. Varmistettu hematologinen vaste saavutettiin 71,5 %:lla potilaista (Taulukko 3). Tärkeää on se, että 27,7 % potilaista saavutti myös huomattavan sytogeneettisen vasteen, joka oli täydellinen 20,4 %:lla (varmistettu 16 %:lla) potilaista. 600 mg:n annosta saaneiden potilaiden osalta arvioidut tämänhetkiset mediaani elossaoloajat olivat seuraavat: elossa ilman taudin etenemistä 22,9 kuukautta ja kaikkiaan 42,5 kuukautta.
Myelooinen blastikriisi
Tutkimukseen otettiin 260 myelooista blastikriisiä sairastavaa potilasta. Heistä 95 (37 %) oli saanut aiemmin kemoterapiaa joko akseleraatiovaiheen tai blastikriisin hoitona (”aiemmin hoidetut potilaat”), kun taas 165 (63 %) ei ollut saanut aiempaa hoitoa (”hoitamattomat potilaat”). Ensimmäisten 37 potilaan aloitusannos oli 400 mg; sittemmin tutkimussuunnitelmaa muutettiin suuremman annoksen sallivaksi, ja loppujen 223 potilaan aloitusannos oli 600 mg.
Ensisijainen tehokkuusmuuttuja oli hematologisen vasteen osuus. Hematologinen vaste ilmoitettiin joko täydellisenä hematologisena vasteena, leukemian olemassaoloa koskevan näytön puuttumisena tai paluuna kroonisen myelooisen leukemian krooniseen vaiheeseen käyttäen samoja kriteerejä kuin akseleraatiovaihetta koskevissa tutkimuksissa. Tässä tutkimuksessa hematologisen vasteen saavutti 31 % potilaista (36 % aiemmin hoitamattomista potilaista ja 22 % aiemmin hoidetuista potilaista) (Taulukko 3). Lisäksi vaste oli yleisempi 600 mg:n annosta saaneilla potilailla (33 %) kuin 400 mg:n annosta saaneilla potilailla (16 %, p = 0,0220). Senhetkinen arvio potilaiden mediaanielossaolosta oli aiemmin hoitamattomien potilaiden osalta 7,7 kuukautta ja hoidettujen potilaiden osalta 4,7 kuukautta.
Lymfaattinen blastikriisi
I vaiheen tutkimuksiin otettiin rajoitettu määrä potilaita (n = 10). Hematologisen vasteen osuus oli 70 % ja kesto 2–3 kuukautta.
Taulukko 3 Vasteet kliinisissä tutkimuksissa koskien kroonista myelooista leukemiaa aikuisilla
Tutkimus 0110 37 kuukauden tiedot Krooninen vaihe, epäonnistunut interferonihoito (n=532) | Tutkimus 0109 40,5 kuukauden tiedot Akseleraatiovaihe (n=235) | Tutkimus 0102 38 kuukauden tiedot Myelooinen blastikriisi (n=260) | |
| % potilaista (95 %:n luottamusväli) | |||
Hematologinen vaste1 Täydellinen hematologinen vaste (CHR) Ei näyttöä leukemiasta (NEL) Paluu krooniseen vaiheeseen (RTC) | 95 % (92,3–96,3) 95 % Ei sovellettavissa Ei sovellettavissa | 71 % (65,3–77,2) 42 % 12 % 17 % | 31 % (25,2–36,8) 8 % 5 % 18 % |
Huomattava sytogeneettinen vaste2 Täydellinen (Varmistettu3) [95 %:n luottamusväli] Osittainen | 65 % (61,2–69,5) 53 % (43 %) [38,6–47,2] 12 % | 28 % (22,0–33,9) 20 % (16 %) [11,3–21,0] 7 % | 15 % (11,2–20,4) 7 % (2 %) [0,6–4,4] 8 % |
1Hematologisen vasteen kriteerit (kaikki vasteet varmistetaan ≥ 4 viikon jälkeen): Täydellinen hematologinen vaste (CHR): Tutkimus 0110 [valkosolumäärä < 10 x 109 /l, trombosyyttiarvo < 450 x 109 /l, myelosyyttejä + metamyelosyyttejä < 5 % veressä, ei blasteja eikä promyelosyyttejä veressä, basofiileja < 20 %, ei luuytimenulkoista sairautta] ja tutkimuksissa 0102 ja 0109 [ANC ≥ 1,5 x 109/l, trombosyyttiarvo ≥ 100 x 109/l, ei blasteja veressä, luuydinblasteja < 5 % eikä luuytimenulkoista sairautta] Ei näyttöä leukemiasta (NEL): Samat kriteerit kuin täydellisessä hematologisesssa vasteessa, mutta ANC ≥ 1 x 109/l ja trombosyyttiarvo ≥ 20 x 109/l (vain 0102 ja 0109). Paluu krooniseen vaiheeseen (RTC): luuydin- ja ääreisveriblasteja < 15 %, luuytimessä ja ääreisveressä blasteja + promyelosyyttejä < 30 %, ääreisveressä basofiilejä < 20 %, ei luuytimenulkoista sairautta lukuun ottamatta pernaa ja maksaa (vain 0102 ja 0109). 2Sytogeneettisen vasteen kriteerit: Huomattavassa vasteessa yhdistyvät sekä täydellinen että osittainen vaste: Täydellinen (0 % Ph+-metafaaseja), osittainen (1–35 %) 3Täydellinen sytogeneettinen vaste, joka varmistettiin toisella luuytimen sytogeneettisellä tutkimuksella aikaisintaan kuukauden päästä ensimmäisestä luuytimen tutkimuksesta. | |||
Lapsipotilaat
I faasin suurenevin annoksin tehtyyn tutkimukseen osallistui yhteensä 26 alle 18-vuotiasta lasta, joilla oli joko kroonisen vaiheen KML (n = 11) tai blastikriisissä oleva KML tai Philadelphia-kromosomipositiivinen akuutti leukemia (n = 15). Tutkimukseen osallistuneita potilaita oli ennen tutkimusta hoidettu tehokkaasti. 46 %:lle potilaista oli tehty luuydinsiirto ja 73 %:lle oli annettu kemoterapiaa useilla eri lääkeaineilla. Potilaat saivat imatinibia 260 mg/m2/vrk (n = 5), 340 mg/m2/vrk (n = 9), 440 mg/m2/vrk (n = 7) ja 570 mg/m2/vrk (n = 5). 9 kroonisen vaiheen KML:aa sairastavasta potilaasta, joiden sytogeneettiset tiedot ovat käytettävissä, 4 potilasta (44 %) saavutti täydellisen sytogeneettisen vasteen ja 3 potilasta (33 %) osittaisen sytogeneettisen vasteen. Huomattavan sytogeneettisen vasteen saavutti siis 77 % potilaista.
II faasin avoimeen, yhdellä hoitoryhmällä toteutettavaan monikeskustutkimukseen osallistui yhteensä 51 lasta, joilla oli äskettäin diagnosoitu ja hoitamaton kroonisen vaiheen KML. Potilaat saivat imatinibia 340 mg/m2/vrk, ja hoitoa annettiin keskeytyksettä, ellei annosta rajoittavaa toksisuutta ilmennyt. Imatinibihoito sai aikaan nopean hoitovasteen lapsilla, joiden KML oli äskettäin diagnosoitu, ja täydellinen hematologinen vaste saavutettiin 78 %:lla potilaista 8 viikon hoidon jälkeen. Huomattavaan täydelliseen hematologiseen vasteprosenttiin liittyi myös täydellisen sytogeneettisen vasteen kehittyminen 65 %:lle potilaista, mikä vastaa aikuisilla saatuja tuloksia. Lisäksi osittainen sytogeneettinen vaste todettiin 16 %:lla potilaista, ja huomattava sytogeneettinen vaste saavutettiin 81 %:lla potilaista. Suurimmalla osalla potilaista, joilla todettiin täydellinen sytogeneettinen vaste, se kehittyi 3–10 kuukaudessa. Vasteen saavuttamiseen kuluneen ajan mediaani oli 5,6 kuukautta Kaplan-Meierin estimaattiin perustuen.
Kliiniset tutkimukset Philadelphia-kromosomipositiivisessa akuutissa lymfaattisessa leukemiassa (Ph+ ALL)
Vasta diagnosoitu Ph+ ALL
Kontrolloidussa tutkimuksessa (ADE10), jossa verrattiin imatinibihoitoa ja kemoterapiainduktiota 55 vasta diagnosoidulla, 55-vuotiaalla tai sitä vanhemmalla potilaalla, monoterapiana annetulla imatinibilla saavutettiin merkitsevästi useammin täydellinen hematologinen vaste kuin kemoterapialla (imatinibi 96,3 %, kemoterapia 50 %; p = 0,0001). Kun kemoterapiapotilaille, joilla ei saavutettu vastetta tai saavutettiin vain heikko vaste, annettiin imatinibia pelastavana hoitona, täydellinen hematologinen vaste saavutettiin 9 potilaalla 11:sta (81,8 %). Tämän kliinisen vaikutuksen yhteydessä bcr-abl-transkriptien määrä väheni enemmän imatinibihoitoa saaneilla potilailla kuin kemoterapiapotilailla 2 hoitoviikon jälkeen (p = 0,02). Kaikki potilaat saivat imatinibia ja vakauttavaa kemoterapiaa (ks. Taulukko 4) induktion jälkeen, ja 8 viikon kohdalla bcr-abl-transkriptien määrä oli sama molemmissa hoitoryhmissä. Kuten tutkimusasetelman perusteella oletettiinkin, remission pituudessa, tautivapaassa elossaoloajassa tai kokonaiseloonjäämisessä ei havaittu eroja. Potilailla, joilla saavutettiin täydellinen molekulaarinen vaste ja minimaalinen jäännöstauti, saavutettiin kuitenkin parempia tuloksia sekä remission kestossa (p = 0,01) että tautivapaassa elossaoloajassa (p = 0,02).
211 vasta diagnosoidun Ph+ ALL-potilaan populaatiossa tehdyissä neljässä kontrolloimattomassa kliinisessä tutkimuksessa (AAU02, ADE04, AJP01 ja AUS01) saadut tulokset ovat yhdenmukaisia edellä kuvattujen tulosten kanssa. Kun imatinibi yhdistettiin kemoterapiainduktioon (ks. Taulukko 4), saavutettiin täydellinen hematologinen vaste 93 %:lla potilaista (147 potilaalla 158 arviointikelpoisesta potilaasta) ja huomattava sytogeneettinen vaste 90 %:lla (19 potilaalla 21 arviointikelpoisesta potilaasta). Täydellinen molekulaarinen vaste saavutettiin 48 %:lla (49 potilasta 102 arviointikelpoisesta potilaasta). Tautivapaa elossaoloaika ja kokonaiseloonjääminen olivat johdonmukaisesti yli 1 vuoden ja parempia kuin historiallisessa verrokkiryhmässä (tautivapaa elossaoloaika, p < 0,001; kokonaiseloonjääminen, p < 0,0001) kahdessa tutkimuksessa (AJP01 ja AUS01).
Taulukko 4 Yhdistelmähoitona imatinibin kanssa käytetty kemoterapiahoito
| Tutkimus ADE10 | |
| Esivaihe | DEX 10 mg/m2 suun kautta, päivät 1–5; CP 200 mg/m2 laskimoon, päivät 3, 4, 5; MTX 12 mg intratekaalisesti, päivä 1 |
| Remission induktio | DEX 10 mg/m2 suun kautta, päivät 6–7, 13–16; VCR 1 mg laskimoon, päivät 7, 14; IDA 8 mg/m2 laskimoon (0,5 h), päivät 7, 8, 14, 15; CP 500 mg/m2 laskimoon (1 h) päivä 1; Ara-C 60 mg/m2 laskimoon, päivät 22–25, 29–32 |
| Konsolidaatiohoidot I, III, V | MTX 500 mg/m2 laskimoon (24 h), päivät 1, 15; 6-MP 25 mg/m2 suun kautta, päivät 1–20 |
| Konsolidaatiohoidot II, IV | Ara-C 75 mg/m2 laskimoon (1 h), päivät 1–5; VM26 60 mg/m2 laskimoon (1 h), päivät 1–5 |
| Tutkimus AAU02 | |
| Induktiohoito (de novo Ph+ ALL) | Daunorubisiini 30 mg/m2 laskimoon, päivät 1–3, 15–16; VCR 2 mg kokonaisannos laskimoon, päivät 1, 8, 15, 22; CP 750 mg/m2 laskimoon, päivät 1, 8; prednisoni 60 mg/m2 suun kautta, päivät 1–7, 15–21; IDA 9 mg/m2 suun kautta, päivät 1–28; MTX 15 mg intratekaalisesti, päivät 1, 8, 15, 22; Ara-C 40 mg intratekaalisesti, päivät 1, 8, 15, 22; metyyliprednisoloni 40 mg intratekaalisesti, päivät 1, 8, 15, 22 |
| Konsolidaatio (de novo Ph+ ALL) | Ara-C 1 000 mg/m2/12 h laskimoon (3 h), päivät 1–4; mitoksantroni 10 mg/m2 laskimoon, päivät 3–5; MTX 15 mg intratekaalisesti, päivä 1; metyyliprednisoloni 40 mg intratekaalisesti, päivä 1 |
| Tutkimus ADE04 | |
| Esivaihe | DEX 10 mg/m2 suun kautta, päivät 1–5; CP 200 mg/m2 laskimoon, päivät 3–5; MTX 15 mg intratekaalisesti, päivä 1 |
| Induktiohoito I | DEX 10 mg/m2 suun kautta, päivät 1–5; VCR 2 mg laskimoon, päivät 6, 13, 20; daunorubisiini 45 mg/m2 laskimoon, päivät 6–7, 13–14 |
| Induktiohoito II | CP 1 g/m2 laskimoon (1 h), päivät 26, 46; Ara-C 75 mg/m2 laskimoon (1 h), päivät 28–31, 35–38, 42–45; 6-MP 60 mg/m2 suun kautta, päivät 26−46 |
| Konsolidaatiohoito | DEX 10 mg/m2 suun kautta, päivät 1–5; vindesiini 3 mg/m2 laskimoon, päivä 1; MTX 1,5 g/m2 laskimoon (24 h), päivä 1; etoposidi 250 mg/m2 laskimoon (1 h) päivät 4–5; Ara-C 2x 2 g/m2 laskimoon (3 h, 12 h välein), päivä 5 |
| Tutkimus AJP01 | |
| Induktiohoito | CP 1,2 g/m2 laskimoon (3 h), päivä 1; daunorubisiini 60 mg/m2 laskimoon (1 h), päivät 1-3; vinkristiini 1,3 mg/m2 laskimoon, päivät 1, 8, 15, 21; prednisoloni 60 mg/m2/vrk suun kautta |
| Konsolidaatiohoito | Vuorottainen syöpälääkitys: suuriannoksinen kemoterapia, jossa MTX 1 g/m2 laskimoon (24 h), päivä 1, ja Ara-C 2 g/m2 laskimoon (12 h välein), päivät 2–3, 4 hoitojaksoa |
| Ylläpitohoito | VCR 1,3 g/m2 laskimoon, päivä 1; prednisoloni 60 mg/m2 suun kautta, päivät 1–5 |
| Tutkimus AUS01 | |
| Induktio- ja konsolidaatiohoito | Hyper-CVAD-lääkitys: CP 300 mg/m2 laskimoon (3 h, 12 h välein), päivät 1–3; vinkristiini 2 mg laskimoon, päivät 4, 11; doksorubisiini 50 mg/m2 laskimoon (24 h), päivä 4; DEX 40 mg/vrk päivinä 1–4 ja 11−14, vuorottain seuraavan lääkityksen kanssa: MTX 1 g/m2 laskimoon (24 h), päivä 1, Ara-C 1 g/m2 laskimoon (2 h, 12 h välein), päivät 2–3 (yhteensä 8 hoitojaksoa) |
| Ylläpitohoito | VCR 2 mg laskimoon kerran kuukaudessa 13 kk ajan; prednisoloni 200 mg suun kautta, 5 vrk/kk 13 kk ajan |
| Kaikkiin hoitoihin kuului steroidien anto keskushermostoprofylaksiaa varten. | |
| Ara-C: sytosiiniarabinosidi; CP: syklofosfamidi; DEX: deksametasoni; MTX: metotreksaatti; 6-MP: 6-merkaptopuriini; VM26: teniposidi; VCR: vinkristiini; IDA: idarubisiini | |
Pediatriset potilaat
I2301-tutkimuksessa avoimeen, sekventiaalisilla kohorteilla toteutettuun, satunnaistamattomaan, vaiheen III monikeskustutkimukseen otettiin mukaan yhteensä 93 lasta, nuorta ja nuorta aikuista (ikä 1–22 v), joilla oli Ph+ ALL. Potilaat saivat imatinibin (340 mg/m2/vrk) ja intensiivisen kemoterapian yhdistelmähoitoa induktiohoidon jälkeen. Imatinibihoitoa annettiin jaksottaisesti kohorteissa 1–5. Imatinibihoidon kestoa pidennettiin ja aloittamista aikaistettiin kohorteittain: kohortissa 1 imatinibihoito oli vähiten intensiivistä ja kohortissa 5 intensiivisintä (ts. jatkuvan, päivittäisen imatinibihoidon kesto päivinä ensimmäisten kemoterapiahoitojaksojen aikana oli pisin). Kohortin 5 potilailla (n = 50) jatkuva, päivittäinen imatinibialtistus hoidon alkuvaiheessa yhdessä kemoterapian kanssa paransi 4 v elossaoloa ilman tapahtumia (69,6 %) verrattuna historiallisiin verrokkeihin (n = 120), jotka saivat tavanomaista kemoterapiaa ilman imatinibihoitoa (31,6 %). Arvioitu 4 v kokonaiselossaolo kohortin 5 potilailla oli 83,6 % verrattuna historiallisiin verrokkeihin (44,8 %). 20 potilasta 50:stä (40 %) sai hematopoieettisen kantasolusiirron kohortissa 5.
Taulukko 5 Yhdistelmähoitona imatinibin kanssa käytetty kemoterapiahoito I2301-tutkimuksessa
Konsolidaatiojakso 1 (3 viikkoa) | Etoposidi (100 mg/m2/vrk, laskimoon): päivät 1–5 Ifosfamidi (1,8 g/m2/vrk, laskimoon): päivät 1–5 MESNA (360 mg/m2/annos 3 h välein, 8 annosta/vrk, laskimoon): päivät 1–5 Granulosyyttikasvutekijä (5 μg/kg, ihon alle): päivät 6–15 tai kunnes ANC (absoluuttinen neutrofiiliarvo) > 1 500 neutropenian jälkeen Intratekaalinen metotreksaatti (mukautettu iän mukaan): VAIN päivä 1 Intratekaalinen kolmoishoito (mukautettu iän mukaan): päivä 8, 15 |
Konsolidaatiojakso 2 (3 viikkoa) | Metotreksaatti (5 g/m2 24 tunnin aikana, laskimoon): päivä 1 Foliinihappo (75 mg/m2 36 tunnin kohdalla, laskimoon; 15 mg/m2 laskimoon tai suun kautta 6 h välein, 6 annosta)iii: päivät 2 ja 3 Intratekaalinen kolmoishoito (mukautettu iän mukaan): päivä 1 Sytarabiini (3 g/m2/annos 12 h välein x 4, laskimoon): päivät 2 ja 3 Granulosyyttikasvutekijä (5 μg/kg, ihon alle): päivät 4–13 tai kunnes ANC > 1 500 neutropenian jälkeen |
Uudelleeninduktiojakso 1 (3 viikkoa) | Vinkristiini (1,5 mg/m2/vrk, laskimoon): päivät 1, 8, ja 15 Daunorubisiini (45 mg/m2/vrk boluksena, laskimoon): päivät 1 ja 2 Syklofosfamidi (250 mg/m2/annos 12 h välein, 4 annosta, laskimoon): päivät 3 ja 4 PEG-asparaginaasi (2 500 ky/m2, lihakseen): päivä 4 Granulosyyttikasvutekijä (5 μg/kg, ihon alle): päivät 5–14 tai kunnes ANC > 1 500 neutropenian jälkeen Intratekaalinen kolmoishoito (mukautettu iän mukaan): päivät 1 ja 15 Deksametasoni (6 mg/m2/vrk, suun kautta): päivät 1–7 ja 15–21 |
Tehostusjakso 1 (9 viikkoa) | Metotreksaatti (5 g/m2 24 tunnin aikana, laskimoon): päivät 1 ja 15 Foliinihappo (75 mg/m2 36 tunnin kohdalla, laskimoon; 15 mg/m2 laskimoon tai suun kautta 6 h välein, 6 annosta)iii: päivät 2, 3, 16 ja 17 Intratekaalinen kolmoishoito (mukautettu iän mukaan): päivät 1 ja 22 Etoposidi (100 mg/m2/vrk, laskimoon): päivät 22–26 Syklofosfamidi (300 mg/m2/vrk, laskimoon): päivät 22–26 MESNA (150 mg/m2/vrk, laskimoon): päivät 22–26 Granulosyyttikasvutekijä (5 μg/kg, ihon alle): päivät 27–36 tai kunnes ANC > 1 500 neutropenian jälkeen Sytarabiini (3 g/m2, 12 h välein, laskimoon): päivät 43, 44 L-asparaginaasi (6 000 ky/m2, lihakseen): päivä 44 |
Uudelleeninduktiojakso 2 (3 viikkoa) | Vinkristiini (1,5 mg/m2/vrk, laskimoon): päivät 1, 8 ja 15 Daunorubisiini (45 mg/m2/vrk boluksena, laskimoon): päivät 1 ja 2 Syklofosfamidi (250 mg/m2/annos 12 h välein, 4 annosta, laskimoon): päivät 3 ja 4 PEG-asparaginaasi (2500 ky/m2, lihakseen): päivä 4 Granulosyyttikasvutekijä (5 μg/kg, ihon alle): päivät 5–14 tai kunnes ANC > 1 500 neutropenian jälkeen Intratekaalinen kolmoishoito (mukautettu iän mukaan): päivät 1 ja 15 Deksametasoni (6 mg/m2/vrk, suun kautta): päivät 1–7 ja 15–21 |
Tehostusjakso 2 (9 viikkoa) | Metotreksaatti (5 g/m2 24 tunnin aikana, laskimoon): päivät 1 ja 15 Foliinihappo (75 mg/m2 36 tunnin kohdalla, laskimoon; 15 mg/m2 laskimoon tai suun kautta 6 h välein, 6 annosta)iii: päivät 2, 3, 16 ja 17 Intratekaalinen kolmoishoito (mukautettu iän mukaan): päivät 1 ja 22 Etoposidi (100 mg/m2/vrk, laskimoon): päivät 22–26 Syklofosfamidi (300 mg/m2/vrk, laskimoon): päivät 22–26 MESNA (150 mg/m2/vrk, laskimoon): päivät 22–26 Granulosyyttikasvutekijä (5 μg/kg, ihon alle): päivät 27–36 tai kunnes ANC > 1 500 neutropenian jälkeen Sytarabiini (3 g/m2, 12 h välein, laskimoon): päivät 43, 44 L-asparaginaasi (6 000 ky/m2, lihakseen): päivä 44 |
Ylläpito (8 viikon hoitojaksot) Hoitojaksot 1–4 | Metotreksaatti (5 g/m2 24 tunnin aikana, laskimoon): päivä 1 Foliinihappo (75 mg/m2 36 tunnin kohdalla, laskimoon; 15 mg/m2 laskimoon tai suun kautta 6 h välein, 6 annosta)iii: päivät 2 ja 3 Intratekaalinen kolmoishoito (mukautettu iän mukaan): päivät 1, 29 Vinkristiini (1,5 mg/m2, laskimoon): päivät 1, 29 Deksametasoni (6 mg/m2/vrk, suun kautta): päivät 1–5; 29–33 6-merkaptopuriini (75 mg/m2/vrk, suun kautta): päivät 8–28 Metotreksaatti (20 mg/m2/viikko, suun kautta): päivät 8, 15, 22 Etoposidi (100 mg/m2, laskimoon): päivät 29–33 Syklofosfamidi (300 mg/m2, laskimoon): päivät 29–33 MESNA laskimoon päivät 29–33 Granulosyyttikasvutekijä (5 μg/kg, ihon alle): päivät 34-43 |
Ylläpito (8 viikon hoitojaksot) Hoitojakso 5 | Pään sädehoito (vain jakso 5) 12 Gy 8 fraktiossa kaikille potilaille, joiden tila toteamishetkellä CNS1 tai CNS2 18 Gy 10 fraktiossa potilaille, joiden tila toteamishetkellä CNS3 Vinkristiini (1,5 mg/m2/vrk, laskimoon): päivät 1, 29 Deksametasoni (6 mg/m2/vrk, suun kautta): päivät 1–5; 29–33 6-merkaptopuriini (75 mg/m2/vrk, suun kautta): päivät 11–56 (6-merkaptopuriini tauotetaan 6–10 päivän pään sädehoidon ajaksi alkaen hoitojakson 5 päivästä 1. 6-merkpatopuriini aloitetaan ensimmäisenä päivänä pään sädehoidon päättymisen jälkeen.) Metotreksaatti (20 mg/m2/viikko, suun kautta): päivät 8, 15, 22, 29, 36, 43, 50 |
Ylläpito (8 viikon hoitojaksot) Hoitojaksot 6–12 | Vinkristiini (1,5 mg/m2/vrk, laskimoon): päivät 1, 29 Deksametasoni (6 mg/m2/vrk, suun kautta): päivät 1–5; 29–33 6-MP (75 mg/m2/vrk, suun kautta): päivät 1–56 Metotreksaatti (20 mg/m2/viikko, suun kautta): päivät 1, 8, 15, 22, 29, 36, 43, 50 |
MESNA = 2-merkaptoetaanisulfonaattinatrium, iii = tai kunnes metotreksaattipitoisuus on < 0,1 μmol, Gy = gray
AIT07-tutkimus oli avoin, satunnaistettu, vaiheen II/III monikeskustutkimus, johon osallistui 128 potilasta (1 – < 18 v), jotka saivat imatinibin ja kemoterapian yhdistelmähoitoa. Tutkimuksen turvallisuustiedot näyttävät olevan yhdenmukaiset imatinibin turvallisuusprofiilin kanssa Ph+ ALL -potilailla.
Uusiutunut/vaikeahoitoinen Ph+ ALL
Kun imatinibia annettiin monoterapiana potilaille, joilla oli uusiutunut/vaikeahoitoinen Ph+ ALL, saavutettiin hematologinen vaste 30 %:lla (täydellinen vaste, 9 %) ja huomattava sytogeneettinen vaste 23 %:lla. Kaikkiaan 53 potilasta 411:sta oli arviointikelpoisia. 353 potilaalta ei kerätty ensisijaisia vastetietoja (an expanded access program). Koko potilasjoukossa (411 potilasta, joilla oli uusiutunut/vaikeahoitoinen Ph+ ALL), taudin etenemiseen kulunut mediaaniaika vaihteli 2,6 kuukaudesta 3,1 kuukauteen, ja kokonaiselossaoloajan mediaani 401 arviointikelpoisella potilaalla vaihteli 4,9 kuukaudesta 9 kuukauteen. Tiedot olivat samanlaiset, kun analyysi tehtiin uudelleen ja mukaan otettiin vain 55-vuotiaat ja sitä vanhemmat potilaat.
Kliiniset tutkimukset myelodysplastisessa oireyhtymässä / myeloproliferatiivisissa sairauksissa (MDS/MPD)
Imatinibin käytöstä tässä käyttöaiheessa on hyvin vähän kokemusta, ja se perustuu hematologisten ja sytogeneettisten vasteiden määrään. Kliinistä hyötyä tai elinajan pitenemistä ei ole osoitettu yhdessäkään kliinisessä tutkimuksessa. Yhdessä avoimessa vaiheen II kliinisessä monikeskustutkimuksessa (tutkimus B2225) imatinibia annettiin monentyyppisille potilaille, joilla oli hengenvaarallisia sairauksia liittyneenä Abl-, Kit- tai PDGFR-proteiinityrosiinikinaaseihin. Tähän tutkimukseen osallistui myös 7 potilasta, joilla oli myelodysplastinen oireyhtymä / myeloproliferatiivinen tauti. He saivat imatinibia annoksena 400 mg/vrk. Kolme potilasta saavutti täydellisen hematologisen vasteen ja yksi potilas osittaisen hematologisen vasteen. Alkuperäisen analyysin tekemisvaiheessa neljällä potilaalla todettiin PDGFR-geenin uudelleenjärjestäytymistä, ja näistä potilaista kolme saavutti hematologisen vasteen (2 täydellisen ja 1 osittaisen hematologisen vasteen). Nämä potilaat olivat 20–72-vuotiaita.
Hoidon pitkäaikaisturvallisuus- ja tehotietojen keräämiseksi toteutettiin havainnoiva rekisteritutkimus (tutkimus L2401). Siihen otettiin imatinibihoitoa saavia potilaita, joilla oli myeloproliferatiivisia kasvaimia ja PDGFR-β-geenin uudelleenjärjestymä. Rekisteri sisälsi 23 potilasta, joiden imatinibivuorokausiannoksen mediaani oli 264 mg (vaihteluväli 100–400 mg) ja hoidon mediaanikesto 7,2 v (vaihteluväli 0,1–12,7 v). Koska kyseessä oli havainnoiva rekisteri, oli hematologisia arviointeja saatavilla 22, sytogeneettisiä arviointeja 9 ja molekulaarisia arviointeja 17 tutkimukseen otetuista 23 potilaasta. Jos konservatiivisesti oletetaan, että ne potilaat joilta tietoja ei ollut saatavilla eivät saavuttaneet vastetta, niin 20 potilasta 23:sta (87 %) saavutti täydellisen hematologisen vasteen, 9 potilasta 23:sta (39,1 %) saavutti täydellisen sytogeneettisen vasteen ja 11 potilasta 23:sta (47,8 %) saavutti molekulaarisen vasteen. Jos vasteprosentit lasketaan potilaista, joilta on tiedossa vähintään yksi validi arviointitulos, täydellisen hematologisen vasteen saavutti 20 potilasta 22:sta (90,9 %), täydellisen sytogeneettisen vasteen 9 potilasta 9:stä (100 %) ja molekulaarisen vasteen 11 potilasta 17:stä (64,7 %).
Lisäksi 13 julkaisussa on annettu tietoa 24 potilaasta, joilla oli myelodysplastinen oireyhtymä / myeloproliferatiivinen tauti. 21 potilasta sai imatinibia annoksena 400 mg/vrk, ja loput kolme saivat pienempiä annoksia. Yhdellätoista potilaalla todettiin PDGFR-geenin uudelleenjärjestäytymistä. Heistä 9 saavutti täydellisen hematologisen vasteen ja 1 osittaisen hematologisen vasteen. Nämä potilaat olivat 2–79-vuotiaita. Eräässä julkaisussa annettiin äskettäin uutta tietoa kuudesta näistä 11 potilaasta, ja tietojen mukaan kaikki kuusi ovat edelleen sytogeneettisessä remissiossa (vaihteluväli 32–38 kuukautta). Samassa julkaisussa raportoitiin pitkäaikaisseurantatietoja 12 potilaasta, joilla oli myelodysplastinen oireyhtymä / myeloproliferatiivinen tauti ja PDGFR-geenin uudelleenjärjestäytymistä (5 potilasta B2225-tutkimuksesta). Näillä potilailla imatinibihoidon mediaanikesto oli 47 kuukautta (vaihteluväli 24 vuorokautta – 60 kuukautta). Näistä potilaista kuutta on nyt seurattu yli 4 vuoden ajan. Yksitoista potilasta saavutti nopeasti täydellisen hematologisen vasteen. Kymmenellä potilaalla sytogeneettiset poikkeavuudet korjaantuivat täysin ja RT-PCR-tutkimuksella määritettävät fuusiotranskriptit joko vähenivät tai hävisivät täysin. Hematologisten vasteiden mediaanikesto on ollut 49 kuukautta (vaihteluväli 19–60 kuukautta) ja sytogeneettisten vasteiden 47 kuukautta (vaihteluväli 16−59 kuukautta). Kokonaiselinaika on 65 kuukautta diagnoosista (vaihteluväli 25–234 kuukautta). Jos potilaalla ei ole todettu tätä geenin translokaatiota, ei imatinibihoidon antamisesta ole hyötyä.
Pediatrisilla potilailla, joilla on myelodysplastinen oireyhtymä / myeloproliferatiivinen sairaus, ei ole tehty kontrolloituja kliinisiä tutkimuksia. Viisi MDS/MPD-tapausta, joihin liittyi PDGFR-geenin uudelleenjärjestäytymistä, on raportoitu neljässä eri julkaisussa. Nämä potilaat olivat iältään 3 kk − 4 vuotta ja imatinibia annettiin annoksin 50 mg/vrk tai 92,5–340 mg/m2/vrk. Kaikki potilaat saavuttivat täydellisen hematologisen vasteen, sytogeneettisen vasteen ja/tai kliinisen vasteen.
Kliiniset tutkimukset koskien hypereosinofiilista oireyhtymää (HES)/kroonista eosinofiilistä leukemiaa (CEL)
Yhdessä avoimessa vaiheen II kliinisessä monikeskustutkimuksessa (tutkimus B2225) imatinibia annettiin monentyyppisille potilaille, joilla oli hengenvaarallisia sairauksia liittyneenä Abl-, Kit- tai PDGFR-proteiinityrosiinikinaaseihin. Tässä tutkimuksessa 14 potilasta, joilla oli hypereosinofiilinen oireyhtymä/krooninen eosinofiilinen leukemia, sai imatinibia annoksena 100–1 000 mg/vrk. Lisäksi 35 julkaistussa tapauskertomuksessa ja tapaussarjassa on annettu tietoa 162 potilaasta, joilla oli hypereosinofiilinen oireyhtymä / krooninen eosinofiilinen leukemia ja jotka saivat imatinibia annoksena 75–800 mg/vrk. Sytogeneettiset poikkeavuudet arvioitiin tässä 176 potilaan kokonaispopulaatiossa 117 potilaalta. Näistä 117 potilaasta 61 todettiin FIP1L1-PDGFRα-fuusiokinaasipositiivisiksi. Kolmessa muussa julkaistussa raportissa on kuvattu vielä 4 hypereosinofiilista oireyhtymää sairastavaa potilasta, jotka todettiin FIP1L1-PDGFRα-positiivisiksi. Kaikki 65 FIP1L1-PDGFRα-fuusiokinaasipositiivista potilasta saavuttivat täydellisen hematologisen vasteen, joka säilyi kuukausien ajan (vaihteluväli 1+ – 44+ kuukautta raportointivaiheeseen mennessä). Erään tuoreen julkaisun tietojen mukaan näistä 65 potilaasta 21 saavutti myös täydellisen molekulaarisen remission. Seuranta-ajan mediaani oli 28 kuukautta (vaihteluväli 13–67 kuukautta). Nämä potilaat olivat 25–72-vuotiaita. Tutkijat ovat raportoineet tapauskertomuksissa myös oireiston ja elinten toimintahäiriöiden paranemista. Paranemista raportoitiin seuraavissa elinjärjestelmissä: sydän, hermosto, iho/ihonalaiskudokset, hengityselimet/rintakehä/välikarsina, tuki- ja liikuntaelimistö/sidekudokset/verisuonet sekä ruoansulatuskanava.
Kontrolloituja tutkimuksia hypereosinofiilista oireyhtymää tai kroonista eosinofiilista leukemiaa sairastavilla pediatrisilla potilailla ei ole tehty. Kolme potilasta, joilla oli hypereosinofiilinen oireyhtymä ja krooninen eosinofiilinen leukemia, joihin liittyi PDGFR-geenin uudelleenjärjestäytymistä, on raportoitu kolmessa julkaisussa. Nämä potilaat olivat 2–16-vuotiaita ja imatinibia annettiin annoksin 300 mg/m2/vrk tai 200–400 mg/vrk. Kaikki potilaat saavuttivat täydellisen hematologisen vasteen, täydellisen sytogeneettisen vasteen ja/tai täydellisen molekulaarisen vasteen.
Kliininen tutkimus koskien ruuansulatuskanavan stroomakasvaimia (GIST), jota ei voida leikata ja/tai joka on metastasoitunut
Pahanlaatuista ruuansulatuskanavan stroomakasvainta (GIST), jota ei voida leikata ja/tai joka on metastasoitunut, sairastavilla potilailla suoritettiin yksi II vaiheen, avoin, kontrolloimaton, satunnaistettu, kansainvälinen tutkimus. Tutkimukseen otettiin 147 potilasta, jotka satunnaistettiin saamaan suun kautta joko 400 mg tai 600 mg imatinibia kerran vuorokaudessa 36 kuukauden ajan. Potilaat olivat 18–83-vuotiaita ja heillä oli histologisesti todettu Kit-positiivinen pahanlaatuinen GIST, jota ei voitu leikata ja/tai joka oli metastasoitunut. Immunohistokemiallinen määritys suoritettiin rutiininomaisesti Kit vasta-aineella (A-4502, kaniinin polyklonaalinen antiseerumi, 1:100; DAKO Corporation, Carpinteria, CA). Antigeenin eristämisen jälkeinen analyysi perustui avidiini-biotiiniperoksidaasi-kompleksi-menetelmään.
Ensisijainen tehon osoitus perustui objektiivisten vasteiden määrään. Kasvainten piti olla mitattavissa ainakin yhdellä kasvainalueella ja vasteen luokittelu perustui SWOG-kriteeriin (Southwestern Oncology Group). Tulokset on esitetty Taulukossa 6.
Taulukko 6 Paras hoitovaste (kasvaimiin) GIST-tutkimuksessa STIB2222
| Paras hoitovaste | Kaikki annokset (n=147) 400 mg (n=73) 600 mg (n=74) n (%) |
| Täydellinen vaste | 1(0,7) |
| Osittainen vaste | 98 (66,7) |
| Vakiintunut sairaus | 23 (15,6) |
| Etenevä sairaus | 18 (12,2) |
| Ei mitattavissa | 5 (3,4) |
| Tuntematon | 2 (1,4) |
Kahden annosryhmän välillä ei ollut eroja vasteiden määrissä. Huomattava osa potilaista, joilla oli vakiintunut sairaus välianalyysin aikaan, saavuttivat osittaisen vasteen pidemmällä hoidolla (mediaani seuranta-aika 31 kuukautta). Mediaaniaika vasteeseen oli 13 viikkoa (95 %:n luottamusväli 12–23). Mediaaniaika hoidon epäonnistumiseen hoitoon vastanneilla potilailla oli 122 viikkoa (95 %:n luottamusväli 106–147), kun se koko tutkimusjoukossa oli 84 viikkoa (95 %:n luottamusväli 71–109). Mediaania kokonaiskuolleisuutta ei ole vielä saavutettu. Kaplan-Meier estimaatti elossaololle 36 kuukauden seurannan jälkeen on 68 %.
Kahdessa kliinisessä tutkimuksessa (tutkimus B2222 ja ryhmän sisäinen tutkimus S0033) imatinibin päivittäinen annos suurennettiin 800 mg:aan niillä potilailla, joilla tauti eteni 400 mg:n tai 600 mg:n annoksilla. Päivittäinen annos suurennettiin 800 mg:aan kaiken kaikkiaan 103 potilaalla. Näistä 6 potilasta saavutti osittaisen vasteen ja 21:lla tauti vakiintui annoksen suurennuksen jälkeen; kliinistä hyötyä oli 26 %:lla. Käytettävissä olevan turvallisuustiedon perusteella annoksen suurentaminen 800 mg:aan vuorokaudessa potilailla, joiden tauti etenee 400 mg:n ja 600 mg:n annoksilla, ei näytä vaikuttavan imatinibin turvallisuusprofiiliin.
Kliiniset tutkimukset liitännäishoidon GISTistä
Liitännäishoitotilanteessa imatinibia tutkittiin kaksoissokkoutetussa, pitkäaikaisessa, plasebokontrolloidussa faasin III monikeskustutkimuksessa (Z9001) 773 potilaalla. Potilaiden ikä vaihteli 18 vuodesta 91 vuoteen. Potilailla, joilla oli kudosnäytteeseen perustuva diagnoosi ensivaiheen GIST:istä, jossa oli immunokemiallisesti osoitettu Kit-proteiini sekä kasvaimen maksimikoko ≥ 3 cm, ja täydellinen resektio ensivaiheen GIST:istä tehtynä 14-70 päivää ennen rekisteröintiä otettiin mukaan. Ensivaiheen GIST:in poistoleikkauksen jälkeen potilaat satunnaistettiin yhteen kahdesta tutkimushaarasta: imatinibi 400 mg/päivä tai vastaavanlainen plasebo yhden vuoden ajan.
Ensisijainen päätetapahtuma tutkimuksessa oli uusiutumisvapaa eloonjääminen (recurrence-free survival [RFS]), joka määriteltiin satunnaistamisen ajankohdasta uusiutumisen ajankohtaan tai kuolemaan, mistä tahansa syystä.
Imatinibi pitkitti huomattavasti RFS:ää, 75 % potilaista olivat uusiutumisvapaita 38 kuukauden jälkeen imatinibi-ryhmässä ja 20 kuukauden jälkeen plaseboryhmässä (95 %:n luottamusväli, [30 – ei laskettavissa], [14 – ei laskettavissa], vastaavasti), (riskisuhde = 0,398 [0,259-0,610], p < 0,0001). Yhden vuoden kohdalla kokonais RFS oli huomattavasti parempi imatinibihoidolla (97,7 %) kun plasebolla (82,3 %), (p < 0,0001). Uusiutumisen riski pieneni siten noin 89 % verrattuna plaseboon (riskisuhde = 0,113 [0,049-0,264]).
Potilaiden primaari GIST:in uusiutumisriskiä leikkauksen jälkeen arvioitiin takautuvasti seuraaviin ennustetekijöihin perustuen: kasvaimen kokoon, mitoosi-indeksiin, sijaintiin. Mitoosi-indeksi -tietoja oli saatavilla 556 potilaalta 713 potilaasta lähtöryhmien mukaisesta populaatiosta (ITT, intention-to- treat). Alaryhmän analyysitulokset United States National Institutes of Health (NIH) ja Armed Forces Institute of Pathology (AFIP) -riskiluokitukseen perustuen ovat esitetty Taulukossa 7. Etua ei voitu osoittaa matalan ja erittäin matalan riskin ryhmissä. Kokonaiseloonjäämisetua ei ollut havaittavissa.
Taulukko 7 Tutkimuksen Z9001 yhteenveto RFS analyysi NIH ja AFIP –riskiluokitukseen perustuen
| Riskiperuste | Riskitaso | Potilaiden osuus % | Tapahtumi-en määrä / potilaiden määrä | Kokonais riskisuhde (95% luottamusväli)* | RFS arviot (%) | |
| 12 kuu-kautta | 24 kuu-kautta | |||||
| imatinibi vs placebo | imatinibi vs placebo | imatinibi vs placebo | ||||
| NIH | Matala | 29,5 | 0/86 vs. 2/90 | E.A. | 100 vs. 98,7 | 100 vs. 95,5 |
| Keskiasteen | 25,7 | 4/75 vs. 6/78 | 0,59 (0,17; 2,10) | 100 vs. 94,8 | 97,8 vs. 89,5 | |
| Korkea | 44,8 | 21/140 vs. 51/127 | 0,29 (0,18; 0,49) | 94,8 vs. 64,0 | 80,7 vs. 46,6 | |
| AFIP | Erittäin matala | 20,7 | 0/52 vs. 2/63 | E.A. | 100 vs. 98,1 | 100 vs. 93,0 |
| Matala | 25,0 | 2/70 vs. 0/69 | E.A. | 100 vs. 100 | 97,8 vs. 100 | |
| Kohtuullinen | 24,6 | 2/70 vs. 11/67 | 0,16 (0,03; 0,70) | 97,9 vs. 90,8 | 97,9 vs. 73,3 | |
| Korkea | 29,7 | 16/84 vs. 39/81 | 0,27 (0,15; 0,48) | 98,7 vs. 56,1 | 79,9 vs. 41,5 | |
* Koko seuranta-aika; E.A. – Ei arvioitavissa
Toisessa avoimessa vaiheen III monikeskustutkimuksessa (SSG XVIII/AIO) verrattiin 12 kk ja 36 kk kestäneitä imatinibihoitoja (400 mg/vrk) potilailla, joille oli tehty GIST-kasvaimen resektio ja joita koski jokin seuraavista: kasvaimen halkaisija > 5 cm ja mitoosiaktiivisuus > 5/50 HPF-kenttä (high power field); kasvaimen halkaisija > 10 cm ja mikä tahansa mitoosiaktiivisuus; tai minkä tahansa kokoinen kasvain ja mitoosiaktiivisuus > 10/50 HPF tai kasvain revennyt vatsaonteloon. Tutkimukseen otettiin mukaan ja satunnaistettiin yhteensä 397 potilasta (199 potilasta 12 kuukauden ryhmään ja 198 potilasta 36 kuukauden ryhmään). Mediaani-ikä oli 61 vuotta (jakauma 22–84 vuotta). Seuranta-ajan mediaani oli 54 kk (satunnaistamisajankohdasta tiedonkeruun päättymiseen). Ensimmäisen potilaan satunnaistamisesta tiedonkeruun päättymiseen kului yhteensä 83 kk.
Tutkimuksen ensisijainen päätetapahtuma oli relapsiton elossaolo (RFS), joka määriteltiin ajaksi satunnaistamisajankohdasta uusiutumisajankohtaan tai mistä tahansa syystä johtuneeseen kuolemaan.
36 kk imatinibihoito pidensi RFS:ää merkitsevästi verrattuna 12 kk imatinibihoitoon (kokonaisriskisuhde [HR] = 0,46 [0,32, 0,65], p < 0,0001) (Taulukko 8, Kuva 1).
36 kk imatinibihoito pidensi merkitsevästi myös kokonaiselossaoloa verrattuna 12 kk imatinibihoitoon (HR = 0,45 [0,22, 0,89], p = 0,0187) (Taulukko 8, Kuva 2).
Pidempi hoidon kesto (> 36 kuukautta) voi viivästyttää uusien relapsien ilmaantumisajankohtaa; tämän löydöksen vaikutus kokonaiselossaoloon jää kuitenkin tuntemattomaksi.
Kuolemantapauksia oli 12 kk hoitoryhmässä yhteensä 25 ja 36 kk ryhmässä 12.
36 kuukauden imatinibihoito oli 12 kuukauden hoitoa parempi ITT-analyysissä, joka sisälsi koko tutkimuspopulaation. Suunnitellussa mutaatiotyypin mukaisessa alaryhmäanalyysissä 36 kuukauden hoidossa potilailla, joilla oli mutaatio eksonissa 11, RFS:n HR oli 0,35 [95 %:n luottamusväli: 0,22, 0,56]. Muille harvinaisemmille mutaatioalaryhmille ei voida vetää johtopäätöksiä johtuen havaittujen tapahtumien vähäisestä määrästä.
Taulukko 8 12 kk ja 36 kk imatinibihoito (SSGXVIII/AIO-tutkimus)
| 12 kk hoitoryhmä | 36 kk hoitoryhmä | |
| RFS | % (luottamusväli) | % (luottamusväli) |
| 12 kk | 93,7 (89,2-96,4) | 95,9 (91,9-97,9) |
| 24 kk | 75,4 (68,6-81,0) | 90,7 (85,6-94,0) |
| 36 kk | 60,1 (52,5-66,9) | 86,6 (80,8-90,8) |
| 48 kk | 52,3 (44,0-59,8) | 78,3 (70,8-84,1) |
| 60 kk | 47,9 (39,0-56,3) | 65,6 (56,1-73,4) |
| Elossaolo | ||
| 36 kk | 94,0 (89,5-96,7) | 96,3 (92,4-98,2) |
| 48 kk | 87,9 (81,1-92,3) | 95,6 (91,2-97,8) |
| 60 kk | 81,7 (73,0-87,8) | 92,0 (85,3-95,7) |
Kuva 1 Ensisijaisen päätetapahtuman (relapsiton elossaolo, RFS) Kaplan–Meier-estimaatit (ITT-populaatio)
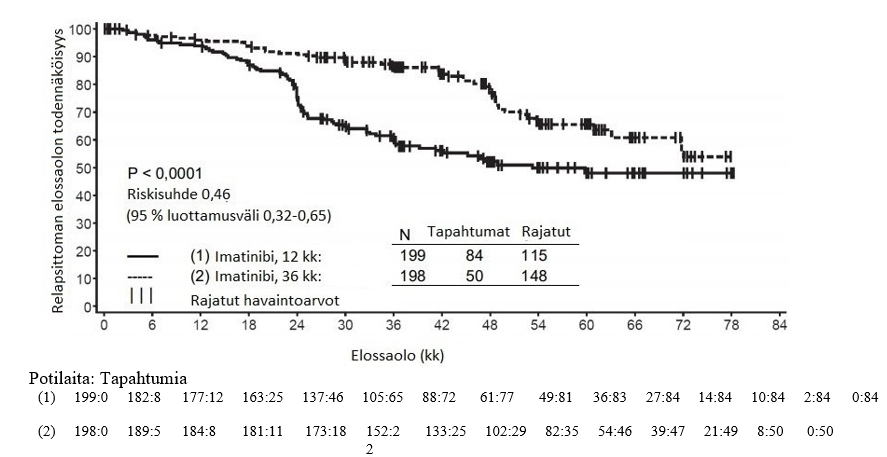
Kuva 2 Kokonaiselossaolon Kaplan–Meier-estimaatit (ITT-populaatio)
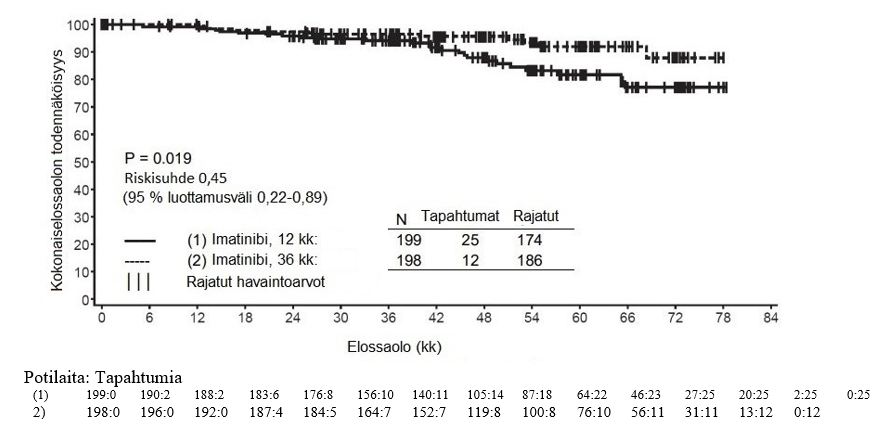
C-Kit-positiivista ruuansulatuskanavan stroomakasvainta sairastavilla pediatrisilla potilailla ei ole tehty kontrolloituja tutkimuksia. Seitsemässä eri julkaisussa on raportoitu 17 GIST:iä (Kit- ja PDGFR- mutaatioiden kera tai ilman) sairastavaa potilaasta. Nämä potilaat olivat 8-18-vuotiaita ja imatinibia annettiin sekä liitännäishoitona että metastasoituneen taudin hoidossa. Annokset vaihtelivat välillä 300-800 mg/vrk. Suurimmassa osassa tapauksista pediatrisilta GIST-potilailta puuttuivat tiedot c-Kit- tai PDGFR-mutaatiostatuksesta, mikä saattoi olla vaihtelevien kliinisten hoitotulosten syynä.
Kliiniset tutkimukset dermatofibrosarcoma protuberansin (DFSP) hoidossa
Vaiheen II avoimeen kliiniseen monikeskustutkimukseen (tutkimus B2225) osallistui 12 potilasta, joilla oli dermatofibrosarcoma protuberans (DFSP) ja jotka saivat imatinibia annoksella 800 mg/vrk. DFSP-potilaiden ikä oli 23–75 vuotta; sairaus oli metastaattinen ja uusiutunut paikallisesti alkuvaiheen resektioleikkauksen jälkeen, eikä soveltunut uuteen resektioleikkaukseen tutkimukseenottoajankohtana. Hoidon vaikutusta arvioitiin ensisijaisesti objektiivisten vasteprosenttien perusteella. Yhdeksällä tutkimukseen otetuista 12 potilaasta saavutettiin vaste (1 täydellinen ja 8 osittaista vastetta). Kolme osittaisen vasteen saavuttanutta potilasta parantui myöhemmin kokonaan leikkauksella. B2225-tutkimuksessa hoidon keston mediaani oli 6,2 kk ja pisin hoitoaika 24,3 kk. Viidessä julkaistussa tapausselostuksessa on kuvattu kuusi muuta imatinibihoitoa saanutta DFSP-potilasta, joiden ikävaihtelu oli 18 kuukaudesta 49 vuoteen. Kirjallisuudessa mainitut aikuispotilaat saivat joko 400 mg (4 potilasta) tai 800 mg (1 potilas) imatinibia päivässä. Viidellä (5) potilaalla saavutettiin vaste (3 täydellistä ja 2 osittaista vastetta). Julkaistussa kirjallisuudessa hoidon keston mediaani vaihteli 4 viikosta yli 20 kuukauteen. Lähes kaikilla potilailla, joilla saavutettiin vaste imatinibihoitoon, oli translokaatio t(17:22)[(q22:q13)] tai sen geenituotetta.
Dermatofibrosarcoma protuberansia sairastavilla pediatrisilla potilailla ei ole tehty kontrolloituja tutkimuksia. Kolmessa julkaisussa on raportoitu viisi potilasta, joilla oli DFSP ja siihen liittyvä PDGFR-geenin uudelleenjärjestäytyminen. Näiden potilaiden iät vaihtelivat vastasyntyneestä neljääntoista vuoteen. Imatinibia annettiin annoksin 50 mg/vrk tai 400–520 mg/m2/vrk. Kaikki potilaat saavuttivat osittaisen ja/tai täydellisen vasteen.
Farmakokinetiikka
Imatinibin farmakokinetiikka
Imatinibin farmakokinetiikkaa on arvioitu 25–1 000 mg:n annoksilla. Plasman farmakokineettiset profiilit analysoitiin ensimmäisenä päivänä sekä päivänä 7 tai 28, johon mennessä plasman pitoisuudet olivat saavuttaneet vakaan tilan.
Imeytyminen
Imatinibin absoluuttinen biologinen keskimääräinen hyötyosuus on 98 %. Imatinibin plasman AUC-arvot vaihtelevat paljon potilaiden välillä, kun lääkettä otetaan suun kautta. Kun imatinibi otetaan rasvaisen aterian kera, imeytyminen heikkeni vain vähän (Cmax väheni 11 % ja tmax piteni 1,5 h) ja AUC väheni hieman (7,4 %), verrattuna paasto-olosuhteisiin. Aikaisemman ruuansulatuskanavan leikkauksen vaikutusta lääkkeen imeytymiseen ei ole tutkittu.
Jakautuminen
Kliinisesti merkittävällä konsentraatiolla imatinibi sitoutui plasman proteiineihin noin 95 %:sti in vitro ‑tutkimuksen perusteella, lähinnä albumiiniin ja alfa-happo-glykoproteiiniin ja vähäisessä määrin lipoproteiiniin.
Biotransformaatio
Tärkein ihmisen veressä todettavista metaboliiteista on N-demetyloitunut piperatsiinijohdos, jolla on samanlainen in vitro teho kuin vaikuttavalla aineella. Tämän metaboliitin plasma-AUC-arvo on vain 16 % imatinibin AUC-arvosta. N-demetyloituneen metaboliitin sitoutuminen plasman proteiineihin on samansuuruista kuin vaikuttavan aineen.
Imatinibi ja N-demetyloitunut metaboliitti vastasivat yhdessä noin 65 %:sta verenkierrossa havaitusta radioaktiivisuudesta (AUC(0-48h)). Loppuosa verenkierrossa havaitusta radioaktiivisuudesta koostui useista vähäisistä metaboliiteista.
In vitro ‑tutkimukset osoittivat, että CYP3A4 oli merkittävin ihmisen P450 entsyymi, joka katalysoi imatinibin biotransformaatiota. Joukosta mahdollisia yhtä aikaa annettavia lääkkeitä (parasetamoli, asikloviiri, allopurinoli, amfoterisiini, sytarabiini, erytromysiini, flukonatsoli, hydroksiurea, norfloksasiini, V-penisilliini) ainoastaan erytromysiini (IC50 50 μmol/l) ja flukonatsoli (IC50 118 μmol/l) estivät imatinibin metaboliaa niin, että sillä voi olla kliinistä merkitystä.
Imatinibin osoitettiin in vitro ‑tutkimuksissa olevan kilpaileva estäjä CYP2C9, CYP2D6 ja CYP3A4/5 substraateille. Ki-arvot ihmisen maksan mikrosomeissa olivat vastaavasti 27; 7,5 ja 7,9 μmol/l. Imatinibin suurimmat plasmapitoisuudet potilailla ovat 2–4 μmol/l, mistä syystä CYP2D6 ja/tai CYP3A4/5 välityksellä tapahtuva, samaan aikaan annetun lääkkeen, metabolian estyminen on mahdollista. Imatinibi ei vaikuttanut 5-fluorourasiilin biotransformaatioon mutta se esti paklitakselin metaboliaa, johtuen CYP2C8 kilpailevasta estosta (Ki = 34,7 μmol/l). Tämä Ki-arvo on huomattavasti suurempi kuin imatinibin odotetut plasmapitoisuudet potilailla, mistä johtuen yhteisvaikutuksia ei odoteta annettaessa yhdessä imatinibia ja 5-fluorourasiilia tai paklitakselia.
Eliminaatio
Suun kautta annetun 14C-merkityn imatinibiannoksen poistumisanalyysin perusteella noin 81 % annoksesta poistui 7 päivän kuluessa ulosteisiin (68 % annoksesta) ja virtsaan (13 % annoksesta). Muuttumattomana imatinibista poistui 25 % annoksesta (5 % virtsaan, 20 % ulosteisiin), ja loppuosa oli metaboliitteja.
Plasmafarmakokinetiikka
Suun kautta terveille vapaaehtoisille koehenkilöille annetun imatinibin puoliintumisaika (t½) oli noin 18 h, minkä perusteella kerran vuorokaudessa tapahtuva annostelu on riittävä. Suun kautta annetun imatinibin AUC-keskiarvo suureni lineaarisesti ja suhteessa annokseen suurennettaessa annoksia alueella 25–1 000 mg. Imatinibin kinetiikka ei muuttunut toistuvassa annostelussa, ja kumuloituminen oli 1,5–2,5-kertaista vakaassa tilassa, kun annos otettiin kerran vuorokaudessa.
Farmakokinetiikka GIST-potilailla
Käytettäessä samaa annosta (400 mg vuorokaudessa), GIST-potilailla vakaan tilan altistus oli 1,5‑kertaa suurempi kuin KML-potilailla havaittu. Alustavan GIST-potilaille tehdyn populaatiofarmakokinetiikka-analyysin mukaan kolme muuttujaa (albumiini, valkosolumäärä, bilirubiini) vaikuttivat tilastollisesti merkitsevästi imatinibin farmakokinetiikkaan. Pienentynyt albumiinin määrä vähensi puhdistumaa (CL/f); ja valkosolujen suurempi määrä vähensi CL/f:tä. Nämä riippuvuudet eivät kuitenkaan ole niin voimakkaita, että annostusta täytyisi muuttaa. Tässä potilasryhmässä maksan metastaasien esiintyminen voi mahdollisesti johtaa maksan vajaatoimintaan ja metabolian heikkenemiseen.
Populaatiofarmakokinetiikka
Populaatiofarmakokinetiikkaa KML-potilailla koskevan analyysin perusteella ikä vaikutti vähäisessä määrin jakautumistilavuuteen (suureni 12 %, yli 65-vuotiailla potilailla). Tätä muutosta ei pidetä kliinisesti merkitsevänä. Painon vaikutus imatinibin puhdistumaan on sellainen, että 50 kg painavalla potilaalla odotettavissa oleva keskipuhdistuma on 8,5 l/h, mutta 100 kg painavalla potilaalla puhdistuma suurenee arvoon 11,8 l/h. Näitä muutoksia ei katsota riittäviksi, jotta annosta tulisi painon perusteella muuttaa. Sukupuoli ei vaikuta imatinibin kinetiikkaan.
Farmakokinetiikka lapsilla ja nuorilla
Kuten aikuisilla potilailla, lapsipotilailla, sekä I vaiheen että II vaiheen tutkimuksissa, imatinibi imeytyi nopeasti suun kautta annettaessa. Annokset 260 ja 340 mg/m2/vrk lapsilla ja nuorilla aiheuttivat samankaltaisen altistuksen kuin annokset 400 ja 600 mg aikuisilla. AUC(0-24)-arvojen vertailu päivinä yksi ja kahdeksan käytettäessä annosta 340 mg/m2/vrk paljasti lääkeaineen 1,7‑kertaisen kumuloitumisen toistuvan kerran vuorokaudessa ‑annostelun yhteydessä.
Hematologista sairautta (KML, Ph+ ALL tai muu imatinibilla hoidettava hematologinen sairaus) sairastavilla pediatrisilla potilailla toteutetun poolatun populaatiofarmakokinetiikan analyysin perusteella imatinibin puhdistuma suurenee kehon pinta-alan suurentuessa. Kehon pinta-alan vaikutuksen suhteen tehtyjen korjausten jälkeen muilla demografisilla tekijöillä kuten iällä, painolla ja painoindeksillä ei ollut kliinisesti merkitseviä vaikutuksia imatinibialtistukseen. Analyysi vahvisti, että imatinibialtistus oli samaa luokkaa pediatrisilla potilailla, jotka saivat 260 mg/m2 kerran vuorokaudessa (enintään 400 mg kerran vuorokaudessa) tai 340 mg/m2 kerran vuorokaudessa (enintään 600 mg kerran vuorokaudessa), ja aikuispotilailla, jotka saivat imatinibia 400 mg tai 600 mg kerran vuorokaudessa.
Elintoimintojen heikkeneminen
Imatinibi ja sen metaboliitit eivät erity merkittävässä määrin munuaisten kautta. Potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta, vaikuttaa olevan suurempi imatinibialtistus plasmassa kuin potilailla, joiden munuaistoiminta on normaali. Altistus suurenee noin 1,5–2-kertaiseksi, mikä vastaa imatinibia voimakkaasti sitovan proteiinin, AGP:n, pitoisuuden suurenemista plasmassa 1,5-kertaiseksi. Vapaan imatinibin puhdistuma on todennäköisesti samanlainen munuaisten vajaatoimintapotilailla ja potilailla, joiden munuaistoiminta on normaali, sillä imatinibi eliminoituu vain vähäisessä määrin munuaisten kautta (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Vaikka farmakokineettisen analyysin tulokset osoittivat, että henkilöiden välillä on suurta vaihtelua, keskimääräinen altistus imatinibille ei suurentunut potilailla, joilla oli eriasteinen maksan vajaatoiminta, verrattuna potilaisiin, joilla oli normaali maksan toiminta (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Prekliiniset tiedot turvallisuudesta
Imatinibin prekliininen turvallisuusprofiili arvioitiin rotilla, koirilla, apinoilla ja kaneilla.
Toistuvan annostelun toksisuuskokeissa huomattiin lieviä tai kohtalaisia hematologisia muutoksia rotilla, koirilla ja apinoilla. Rotilla ja koirilla niihin liittyi luuydinmuutoksia.
Rotilla ja koirilla maksa oli toksisuuden kohde-elin. Molemmilla eläinlajeilla havaittiin lievää tai kohtalaista transaminaasiarvojen nousua ja lievää kolesteroli-, triglyseridi-, kokonaisproteiini- ja albumiiniarvojen laskua. Rotan maksassa ei havaittu histopatologisia muutoksia. Kaksi viikkoa hoidetuilla koirilla havaittiin vakavaa maksatoksisuutta, johon liittyi maksaentsyymiarvojen kohoamista, hepatosellulaarista nekroosia, sappitiehyeiden nekroosia ja sappitiehyeiden hyperplasiaa.
Kaksi viikkoa hoidetuilla apinoilla havaittiin munuaistoksisuutta, johon liittyi pesäkemäistä mineralisaatiota ja munuaistiehyeiden laajenemista ja tubulusten nefroosia. Usealla apinalla havaittiin veren ureatypen (BUN) ja kreatiniinin nousu. Rotilla 13 viikkoa kestäneessä tutkimuksessa ≥ 6 mg/kg annoksilla havaittiin munuaisnystyn ja virtsarakon välimuotoisen epiteelin (transitional epithelium) hyperplasiaa, johon ei liittynyt muutoksia seerumi- tai virtsa-arvoissa. Imatinibin pitkäaikaisessa annostuksessa havaittiin opportunististen infektioiden määrän kasvu.
39 viikkoa kestäneessä apinakokeessa ei saatu määritettyä haittavaikutuksetonta tasoa (NOAEL) pienimmällä 15 mg/kg annoksella, joka on noin kolmannes ihmiselle tarkoitetusta 800 mg maksimiannoksesta perustuen kehon pinta-alaan. Hoito johti normaalisti oireettomana olevan malariainfektion pahenemiseen näillä eläimillä.
Imatinibi ei ollut genotoksinen in vitro bakteereilla tehdyssä solutestissä (Ames-testi), in vitro nisäkässolutestissä (hiiren lymfooma) eikä in vivo rotan mikrotumatestissä. Imatinibilla havaittiin genotoksista vaikutusta in vitro nisäkässolutestissä (kiinalaisen hamsterin munasarja) klastogeenisuuden (kromosomien poikkeavuus) esiintyessä metabolisen aktivaation yhteydessä. Kaksi valmistusprosessin välituotetta, jotka esiintyvät myös lopullisessa valmisteessa, ovat mutageenejä Ames-testillä mitattuna. Toinen näistä välituotteista antoi positiivisen tuloksen myös hiiren lymfoomatestissä.
Urosrotilla suoritetussa hedelmällisyystutkimuksessa 70 päivää ennen parittelua kestäneellä annostuksella, annoksella 60 mg/kg (vastaten noin 800 mg/vrk maksimiannosta ihmiselle perustuen kehon pinta-alaan) havaittiin kivesten ja lisäkivesten painon pienenemistä ja liikkuvan siemennesteen osuuden vähenemistä. Annoksilla ≤ 20 mg/kg ei havaittu vastaavaa. Lievää tai kohtalaista spermatogeneesin vähenemistä havaittiin myös koirilla annettaessa suun kautta ≥ 30 mg/kg annoksia. Kun naarasrotille annettiin ennen parittelua 14 vuorokauden ajan imatinibia ja annostusta jatkettiin 6:nteen gestaatiopäivään asti, ei parittelussa eikä tiineiden naaraiden määrässä havaittu poikkeavuutta. Annoksella 60 mg/kg, naarasrotilla implantaation jälkeinen alkionmenetys oli merkittävä ja elävien sikiöiden määrä väheni. Annoksilla ≤ 20 mg/kg ei havaittu vastaavaa.
Rotilla tehdyssä tutkimuksessa, jossa selvitettiin lääkkeen oraalisen annostelun vaikutuksia pre- ja postnataalikehitykseen, havaittiin punaista emätineritettä tiineyden 14. tai 15. päivänä 45 mg/kg/vrk annosta saaneiden ryhmässä. Samalla annoksella kuolleena syntyneiden poikasten ja synnytyksen jälkeisinä päivinä 0–4 kuolleiden poikasten määrä oli suurentunut. Käytettäessä samaa annosta ensimmäisen polven jälkeläisten keskimääräinen ruumiinpaino oli alentunut syntymästä lopettamiseen saakka. Preputiaalisen separaation vaatimukset saavuttavien poikueiden määrä oli hieman alentunut. Lääke ei vaikuttanut ensimmäisen polven fertiliteettiin, kun taas annoksella 45 mg/kg/vrk havaittiin resorptioiden määrän kasvua ja elinkykyisten sikiöiden määrän laskua. Ei havaittavia vaikutuksia aiheuttava annos (NOEL) sekä naarasrotilla että ensimmäisen polven jälkeläisillä oli 15 mg/kg/vrk (neljäsosa ihmisen maksimiannoksesta, 800 mg).
Rotilla imatinibi oli teratogeeninen, kun sitä annettiin organogeneesin aikana ≥ 100 mg/kg annoksilla, vastaten noin 800 mg/vrk maksimiannosta ihmiselle perustuen kehon pinta-alaan. Teratogeenisiä vaikutuksia olivat eksenkefalia tai enkefaloseele, puuttuvat/pienentyneet otsaluut ja puuttuvat päälakiluut. Annoksilla ≤ 30 mg/kg ei havaittu vastaavaa.
Nuorilla rotilla (päivät 10–70 syntymän jälkeen) suoritetussa kehitystoksisuutta koskevassa tutkimuksessa ei todettu uusia kohde-elimiä niiden tunnettujen kohde-elinten lisäksi, jotka on todettu aikuisilla rotilla. Nuorilla eläimillä suoritetussa toksisuustutkimuksessa todettiin vaikutuksia eläinten kasvuun sekä hidastunutta emättimen avautumista ja esinahan eriytymistä annoksilla, jotka tuottivat noin 0,3–2 kertaa suuremman altistuksen kuin mitä suurin suositeltu annos, 340 mg/m2, keskimäärin aikaansaa pediatrisilla potilailla. Lisäksi kuolleisuutta todettiin nuorilla eläimillä (vieroitusvaiheen aikoihin) noin 2 kertaa suuremmalla altistuksella kuin mitä suurin suositeltu annos, 340 mg/m2, keskimäärin aikaansaa pediatrisilla potilailla.
Kaksi vuotta kestäneessä annoksilla 15, 30 ja 60 mg/kg/vrk rotilla suoritetussa karsinogeenisuustutkimuksessa pitkäikäisyys lyheni tilastollisesti merkitsevästi uroksilla annoksella 60 mg/kg/vrk ja naarailla annoksilla ≥ 30 mg/kg/vrk. Kuolleiden eläinten histopatologisissa tutkimuksissa havaittiin pääasiallisina kuolinsyinä tai lopettamisen syinä sydänlihassairaus (molemmat sukupuolet), krooninen etenevä munuaissairaus (naaraat) ja esinahkarauhasten papillooma. Kasvainmuutosten kohde-elimet olivat munuaiset, virtsarakko, virtsaputki, esinahka- ja häpykielirauhaset, ohutsuoli, lisäkilpirauhaset, lisämunuaiset ja rauhasista vapaa osa mahalaukusta.
Esinahka- ja häpykielialueilla havaittiin papillooma/karsinooma annoksilla 30 mg/kg/vrk tai yli, mikä vastaa ihmisellä noin 0,5- tai 0,3-kertaista altistusta (perustuen AUC:hen) 400 mg:n ja 800 mg:n vuorokausiannoksella. Lapsilla tämä vastaa 0,4-kertaista altistusta (perustuen AUC:hen) vuorokausiannoksella 340 mg/m2/vrk. Korkein altistumistaso, jolla haitallista vaikutusta ei voitu havaita (NOEL), oli 15 mg/kg/vrk. Munuaisten adenooma/karsinooma, virtsarakon ja virtsaputken papilloomat, ohutsuolen adenokarsinoomat, lisäkilpirauhasten adenoomat, hyvän- ja pahanlaatuiset lisämunuaisytimen kasvaimet, mahalaukun rauhasista vapaan osan papilloomat/karsinoomat havaittiin annoksella 60 mg/kg/vrk, joka vastaa noin 1,7- tai 1-kertaista ihmisen päivittäistä altistumista (AUC:n perusteella) annostasolla 400 mg/vrk tai 800 mg/vrk ja 1,2-kertaista päivittäistä altistumista lapsilla ja nuorilla (AUC:n perusteella) annostasolla 340 mg/m2/vrk. Korkein altistumistaso, jolla haitallista vaikutusta ei voitu havaita (NOEL), oli 30 mg/kg/vrk.
Rottien karsinogeenisuustutkimusten löydösten mekanismia ja merkitystä ihmiselle ei ole vielä selvitetty.
Aiemmissa prekliinisissä tutkimuksissa ei kartoitettu ei-neoplastisia vaurioita kardiovaskulaarisessa järjestelmässä, haimassa, endokriinisissä elimissä eikä hampaissa. Tärkeimpiä muutoksia olivat sydänlihaksen liikakasvu ja sydämen laajentuma, jotka johtivat sydämen vajaatoimintaoireisiin joillakin eläimillä.
Vaikuttava aine, imatinibi, on ympäristöriski pohjaeläimistölle.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin:
Laktoosimonohydraatti
Maissitärkkelys
Hydroksipropyyliselluloosa
Mikrokiteinen selluloosa (E460)
Krospovidoni
Kolloidinen vedetön piidioksidi
Magnesiumstearaatti (E470b)
Kalvopäällyste:
Poly(vinyylialkoholi)
Titaanidioksidi (E171)
Makrogoli 3000
Talkki
Punainen rautaoksidi (E172)
Keltainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
IMATINIB KRKA D.D. tabletti, kalvopäällysteinen
100 mg (J) (L:kyllä) 120 fol (148,14 €)
PF-selosteen tieto
100 mg kalvopäällysteiset tabletit:
Läpipainopakkaus (PVC/PE/PVDC-kalvo//alumiinifolio): 20, 30, 60, 90, 120 tai 180 kalvopäällysteistä tablettia pahvikotelossa.
400 mg kalvopäällysteiset tabletit:
Läpipainopakkaus (PVC/PE/PVDC-kalvo//alumiinifolio): 10, 30, 60 tai 90 kalvopäällysteistä tablettia pahvikotelossa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
100 mg: Oranssinruskea, pyöreä (halkaisija 11 mm), hieman kaksoiskupera viistoreunainen kalvopäällysteinen tabletti, jossa on toisella puolella jakouurre. Tabletin voi jakaa yhtä suuriin annoksiin.
400 mg: Oranssinruskea, soikea (mitat: 22 mm x 9 mm), kaksoiskupera kalvopäällysteinen tabletti.
Käyttö- ja käsittelyohjeet
Ei erityisvaatimuksia hävittämisen suhteen.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
IMATINIB KRKA D.D. tabletti, kalvopäällysteinen
100 mg 120 fol
- Ylempi erityiskorvaus (100 %). Leukemiat, muut pahanlaatuiset veri- ja luuydintaudit sekä pahanlaatuiset imukudostaudit (117), Pahanlaatuiset kasvaimet, joita ei ole edellä erikseen mainittu (130).
- Peruskorvaus (40 %).
ATC-koodi
L01EA01
Valmisteyhteenvedon muuttamispäivämäärä
19.03.2024
Yhteystiedot
Tekniikantie 14
02150 Espoo
Suomi
020-7545330
www.krka.biz
info.fi@krka.biz