
FEBUXOSTAT KRKA tabletti, kalvopäällysteinen 120 mg
Vaikuttavat aineet ja niiden määrät
Yksi kalvopäällysteinen tabletti sisältää 120 mg febuksostaattia.
Apuaine(et), joiden vaikutus tunnetaan:
- laktoosi (monohydraattina): 109 mg
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Febuxostat Krka on tarkoitettu kroonisen hyperurikemian hoitoon silloin, kun uraattikiteitä on jo päässyt muodostumaan (mm. kun potilaalla on tai on aiemmin ollut kihtikyhmyjä ja/tai kihtiartriitti).
Febuxostat Krka on tarkoitettu hyperurikemian ehkäisyyn ja hoitoon aikuispotilaille, jotka saavat solunsalpaajahoitoa hematologisen syöpäsairauden vuoksi ja joiden tuumorilyysioireyhtymän (TLS) riski on kohtalainen tai suuri.
Febuxostat Krka on tarkoitettu aikuisille.
Annostus ja antotapa
Annostus
Kihti: Febuxostat Krka ‑tablettien suositusannos on 80 mg kerran vuorokaudessa suun kautta. Lääkkeen voi ottaa aterioista riippumatta. Jos seerumin virtsahappopitoisuus on 2–4 viikon kuluttua > 6 mg/dl (357 μmol/l), voidaan harkita Febuxostat Krka ‑annoksen suurentamista 120 mg:aan kerran vuorokaudessa.
Febuxostat Krka vaikuttaa niin nopeasti, että seerumin virtsahappopitoisuus voidaan tarkistaa 2 viikon kuluttua. Hoidon tavoitteena on saada seerumin virtsahappopitoisuus laskemaan alle arvon 6 mg/dl (357 μmol/l) ja myös pysymään sen alla.
Kihtikohtausten estohoitoa suositellaan jatkettavaksi vähintään 6 kuukauden ajan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tuumorilyysioireyhtymä:
Febuxostat Krka ‑tablettien suositusannos on 120 mg kerran vuorokaudessa suun kautta. Lääkkeen voi ottaa aterioista riippumatta.
Febuxostat Krka ‑hoito on aloitettava kaksi vuorokautta ennen solunsalpaajahoitoa ja sitä on jatkettava vähintään 7 vuorokautta. Hoitoa voidaan kuitenkin lääkärin harkinnan mukaan pidentää 9 vuorokauteen solunsalpaajahoidon keston mukaan.
Iäkkäät
Iäkkäiden potilaiden annostusta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Tämän lääkevalmisteen tehoa ja turvallisuutta vaikean munuaisten vajaatoiminnan yhteydessä (kreatiniinipuhdistuma < 30 ml/min, ks. kohta Farmakokinetiikka) ei ole täysin selvitetty.
Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annosta ei tarvitse muuttaa.
Maksan vajaatoiminta
Febuksostaatin tehoa ja turvallisuutta ei ole tutkittu vaikeaa maksan vajaatoimintaa (Child–Pugh-luokka C) sairastavilla potilailla.
Kihti: Suositusannos lievää maksan vajaatoimintaa sairastaville potilaille on 80 mg. Keskivaikeaa maksan vajaatoimintaa sairastavista potilaista on niukasti tietoa.
Tuumorilyysioireyhtymä: Vain vaikeaa maksan vajaatoimintaa sairastavat potilaat suljettiin pois keskeisestä vaiheen 3 tutkimuksesta (FLORENCE). Tutkimukseen osallistuneiden potilaiden annoksen muuttaminen ei ollut tarpeen maksan toiminnan perusteella.
Pediatriset potilaat
Febuxostat Krka ‑tablettien turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Suun kautta.
Febuxostat Krka otetaan suun kautta ja voidaan ottaa ruokailun yhteydessä tai tyhjään mahaan.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille (ks. myös kohta Haittavaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Sydän ja verisuonisto
Kroonisen hyperurikemian hoito
Potilailla, joilla oli todettu merkittävä sydän- ja verisuonitauti (kuten sydäninfarkti, aivohalvaus tai epävakaa sepelvaltimotauti), havaittiin valmisteen kehitystyön aikana ja yhdessä rekisteröinnin jälkeisessä tutkimuksessa (CARES) enemmän kuolemaan johtaneita sydän- ja verisuonitapahtumia febuksostaatin käytön yhteydessä verrattuna allopurinoliin.
Seuraavassa rekisteröinnin jälkeisessä tutkimuksessa (FAST) febuksostaatti oli kuitenkin vähintään samanveroinen kuin allopurinoli sekä kuolemaan johtaneiden että kuolemaan johtamattomien sydän- ja verisuonitapahtumien ilmaantuvuuden suhteen.
Tämän potilasryhmän hoidossa on noudatettava varovaisuutta, ja tämän ryhmän potilaita on seurattava säännöllisesti.
Katso lisätietoja febuksostaatin kardiovaskulaarisesta turvallisuudesta kohdista Haittavaikutukset ja Farmakodynamiikka.
Hyperurikemian ehkäisy ja hoito potilailla, joilla on riski saada tuumorilyysioireyhtymä
Solunsalpaajahoitoa hematologisen syöpäsairauden vuoksi saavien ja kohtalaisen tai suuren tuumorilyysioireyhtymän (TLS) riskin omaavien potilaiden on oltava kliinisesti asianmukaisessa sydänvalvonnassa febuksostaattihoidon aikana.
Lääkeaineallergia / yliherkkyys
Vaikuttavan aineen markkinoille tulon jälkeisen käyttökokemuksen yhteydessä on raportoitu harvoin vakavia allergisia/yliherkkyysreaktioita, hengenvaarallinen Stevens–Johnsonin oireyhtymä, toksinen epidermaalinen nekrolyysi mukaan lukien, sekä akuutteja anafylaktisia reaktioita/sokkeja. Nämä reaktiot ilmaantuivat useimmiten febuksostaattihoidon ensimmäisen kuukauden aikana. Osalla, mutta ei kaikilla, näistä potilaista raportoitiin munuaisten vajaatoimintaa ja/tai aiempaa yliherkkyyttä allopurinolille. Vaikeisiin yliherkkyysreaktioihin, myös lääkeaineihottumaan, johon liittyy eosinofiliaa ja systeemioireita (drug reaction with eosinophilia and systemic symptoms, DRESS), liittyi joissakin tapauksissa kuumetta, hematologisia vaikutuksia munuais- tai maksa-affisio.
Potilaalle on kerrottava allergisten/yliherkkyysreaktioiden oireista ja löydöksistä ja häntä on kehotettava tarkkailemaan niiden ilmaantumista (ks. kohta Haittavaikutukset). Jos vakava allerginen/yliherkkyysreaktio, Stevens–Johnsonin oireyhtymä mukaan lukien, ilmaantuu, febuksostaattihoito on lopetettava välittömästi, sillä hoidon varhaiseen lopettamiseen liittyy parempi ennuste. Jos potilaalle on kehittynyt allerginen/yliherkkyysreaktio, Stevens–Johnsonin oireyhtymä ja akuutti anafylaktinen reaktio/sokki mukaan lukien, tälle potilaalle ei enää missään vaiheessa saa aloittaa uudelleen febuksostaattihoitoa.
Akuutit kihtikohtaukset (kihdin paheneminen)
Febuksostaattihoitoa ei saa aloittaa ennen kuin akuutti kihtikohtaus on mennyt kokonaan ohi. Febuksostaatti voi aiheuttaa akuutteja kihtikohtauksia hoidon alkuvaiheessa. Tämä johtuu seerumin virtsahappopitoisuuden muutoksista ja siitä aiheutuvasta kudoksiin kertyneen uraatin vapautumisesta (ks. kohdat Haittavaikutukset ja Farmakodynamiikka). Febuksostaattihoitoa aloitettaessa potilaalle on suositeltavaa määrätä jotakin ei-steroidaalista tulehduskipulääkettä (NSAIDia) tai kolkisiinia kihtikohtausten estoon vähintään 6 kuukauden ajaksi (ks. kohta Annostus ja antotapa).
Febuksostaattihoitoa ei saa keskeyttää, jos potilas saa hoidon aikana kihtikohtauksen. Kohtaus hoidetaan potilaalle sopivalla tavalla varsinaista hoitoa keskeyttämättä. Febuksostaattihoidon jatkaminen keskeytyksettä vähentää myöhempien kihtikohtausten esiintymistä ja voimakkuutta.
Ksantiinikiteet
Febuksostaatti saattaa harvinaisissa tapauksissa suurentaa virtsan absoluuttista ksantiinipitoisuutta niin paljon, että virtsateihin muodostuu ksantiinikiteitä. Tämä koskee potilaita, joiden uraattituotanto on huomattavasti normaalia voimakkaampaa (esim. syöpä, syöpähoidot, Lesch–Nyhanin oireyhtymä). Ksantiinikiteiden muodostumista virtsateihin ei havaittu Febuxostat Krka ‑valmisteella tehdyssä keskeisessä kliinisessä tutkimuksessa potilailla, joilla oli tuumorilyysioireyhtymä. Febuksostaattihoidosta ei ole kokemusta potilailla, joilla on Lesch-Nyhanin oireyhtymä, joten sitä ei myöskään suositella.
Merkaptopuriini / atsatiopriini
Febuksostaatin määräämistä potilaille, jotka saavat samanaikaisesti merkaptopuriinia/atsatiopriinia, ei suositella, koska febuksostaatin aikaansaama ksantiinioksidaasin estyminen saattaa suurentaa merkaptopuriinin/atsatiopriinin pitoisuuksia plasmassa, mikä voi aiheuttaa vaikeaa toksisuutta.
Jos tämän yhdistelmän käyttöä ei voida välttää, merkaptopuriinin/atsatiopriinin annosta suositellaan pienentämään enintään 20 %:iin aiemmin määrätystä annoksesta, jotta voidaan välttää mahdolliset hematologiset vaikutukset (ks. kohdat Yhteisvaikutukset ja Prekliiniset tiedot turvallisuudesta).
Potilasta on seurattava tarkoin ja merkaptopuriinin/atsatiopriinin annosta on sen jälkeen säädettävä hoitovasteen arvioinnin ja mahdollisten toksisten vaikutusten ilmaantumisen mukaan.
Elinsiirtopotilaat
Febuksostaatin käyttöä elinsiirtopotilaille ei suositella, sillä tämän potilasryhmän hoidosta tällä valmisteella ei ole kokemusta (ks. kohta Farmakodynamiikka).
Teofylliini
Farmakokineettisiä yhteisvaikutuksia ei havaittu, kun febuksostaattia (80 mg) ja teofylliiniä (400 mg kerta-annos) annettiin samanaikaisesti terveille tutkimushenkilöille (ks. kohta Yhteisvaikutukset). Febuksostaattia (80 mg) voidaan käyttää samanaikaisesti teofylliinin kanssa ilman plasman teofylliinipitoisuuksien suurenemisen riskiä. 120 mg febuksostaattihoitoa koskevia tietoja ei ole saatavilla.
Maksa
Vaiheen 3 yhdistetyissä kliinisissä tutkimuksissa febuksostaattia saaneilla potilailla (5,0 %) todettiin lieviä maksan toiminnan poikkeavuuksia. Maksan toimintakokeiden tekemistä suositellaan ennen febuksostaattihoidon aloittamista ja sen jälkeen määräajoin lääkärin tekemän kliinisen arvion mukaan (ks. kohta Farmakodynamiikka).
Kilpirauhanen
Pitkäkestoisissa avoimissa jatkotutkimuksissa TSH-arvojen nousua (> 5,5 μIU/ml) todettiin pitkäkestoista febuksostaattihoitoa saaneilla potilailla (5,5 %). Febuksostaattia tulisi antaa varoen potilaille, joilla on todettu muutoksia kilpirauhasen toiminnassa (ks. kohta Farmakodynamiikka).
Apuaineet
Febuxostat Krka sisältää laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Merkaptopuriini/atsatiopriini
Febuksostaatilla on ksantiinioksidaasia estävä vaikutus, joten näiden lääkevalmisteiden samanaikaista käyttöä ei suositella. Febuksostaatin ksantiinioksidaasia estävä vaikutus saattaa suurentaa näiden lääkkeiden pitoisuutta plasmassa ja aiheuttaa siten luuydintoksisuutta.
Merkaptopuriinin/atsatiopriinin annos pitää pienentää enintään 20 %:iin aiemmin määrätystä annoksesta, jos niitä annetaan samanaikaisesti febuksostaatin kanssa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Prekliiniset tiedot turvallisuudesta).
Ehdotettu annosmuutos perustui rotilla saaduista prekliinisistä tiedoista tehtyyn mallintamiseen ja simulaatioanalyysiin. Muutoksen riittävyys varmistettiin tuloksilla kliinisestä yhteisvaikutustutkimuksesta, jossa terveet vapaaehtoiset saivat pelkästään atsatiopriinia 100 mg tai atsatiopriinia pienennetyllä annoksella (25 mg) yhdistelmänä febuksostaatin (40 tai 120 mg) kanssa.
Febuksostaatin ja muiden solunsalpaajien välisiä yhteisvaikutuksia ei ole tutkittu.
Tuumorilyysioireyhtymän keskeisessä tutkimuksessa annettiin febuksostaattia 120 mg vuorokaudessa potilaille, joilla oli useita solunsalpaajahoitoja, monoklonaaliset vasta-aineet mukaan lukien. Lääkkeiden välisiä tai lääkkeiden ja taudin välisiä yhteisvaikutuksia ei kuitenkaan selvitetty tämän tutkimuksen aikana. Siksi mahdollisia yhteisvaikutuksia toisen samanaikaisesti annettavan solunsalpaajan kanssa ei voida sulkea pois.
Rosiglitatsoni/CYP2C8:n substraatit
Febuksostaatin on osoitettu olevan heikko CYP2C8:n estäjä in vitro. Tutkimuksessa terveillä tutkimushenkilöillä febuksostaatin (120 mg x 1) ja rosiglitatsonin (4 mg kerta-annos suun kautta) samanaikainen käyttö ei vaikuttanut rosiglitatsonin eikä sen metaboliitin N-desmetyylirosiglitatsonin farmakokinetiikkaan. Tämä viittaa siihen, että febuksostaatti ei estä CYP2C8-entsyymiä in vivo. Näin ollen febuksostaatin samanaikainen käyttö rosiglitatsonin tai muiden CYP2C8:n substraattien kanssa ei todennäköisesti edellytä kyseisten lääkkeiden annoksen muuttamista.
Teofylliini
Terveillä tutkimushenkilöillä on tehty febuksostaattia koskeva yhteisvaikutustutkimus, jossa arvioitiin, suurentaako ksantiinioksidaasin esto veren teofylliinipitoisuuksia kuten muiden ksantiinioksidaasin estäjien yhteydessä on ilmoitettu. Tutkimustulokset osoittivat, että febuksostaatin (80 mg x 1) samanaikainen käyttö teofylliinin (400 mg kerta-annos) kanssa ei vaikuttanut teofylliinin farmakokinetiikkaan eikä turvallisuuteen. Näin ollen febuksostaatin (80 mg) ja teofylliinin samanaikainen käyttö ei vaadi erityistä varovaisuutta. 120 mg febuksostaattihoitoa koskevia tietoja ei ole.
Naprokseeni ja muut glukuronidaation estäjät
Febuksostaatin metabolia riippuu UDP-glukuronyylitransferaasista (UGT-entsyymeistä). Glukuronidaatiota estävät lääkkeet, kuten ei-steroidaaliset tulehduskipulääkkeet (NSAIDit) ja probenesidi, voivat teoriassa vaikuttaa febuksostaatin eliminaatioon. Terveillä tutkimushenkilöillä febuksostaatin ja naprokseenin (250 mg kahdesti vuorokaudessa) samanaikainen käyttö suurensi febuksostaattialtistusta (Cmax 28 %, AUC 41 % ja t1/2 26 %). Kliinisissä tutkimuksissa naprokseenin tai muiden ei-steroidaalisten tulehduskipulääkkeiden/COX-2-estäjien käyttöön ei liittynyt kliinisesti merkityksellistä haittatapahtumien ilmaantuvuuden suurenemista.
Febuksostaattia ja naprokseenia saa käyttää samanaikaisesti eikä kummankaan lääkkeen annostusta tarvitse muuttaa.
Glukuronidaation indusorit
UGT-entsyymien toimintaa voimakkaasti indusoivat lääkkeet saattavat kiihdyttää febuksostaatin metaboliaa ja heikentää sen tehoa. Siksi seerumin virtsahappopitoisuuksia suositellaan seuraamaan 1‑2 viikon ajan glukuronidaatiota voimakkaasti indusoivien lääkkeiden käytön aloittamisen jälkeen. Glukuronidaation indusorien käytön lopettaminen voi puolestaan suurentaa febuksostaatin pitoisuuksia plasmassa.
Kolkisiini/indometasiini/hydroklooritiatsidi/varfariini
Febuksostaattia saa käyttää samanaikaisesti kolkisiinin tai indometasiinin kanssa. Annosmuutokset eivät ole tarpeen.
Samanaikainen hydroklooritiatsidihoito ei vaadi febuksostaattiannoksen muuttamista.
Febuksostaatin ja varfariinin yhteiskäyttö ei vaadi varfariiniannoksen muuttamista. Febuksostaatin (80 mg tai 120 mg kerran päivässä) antaminen varfariinin kanssa ei vaikuttanut varfariinin farmakokinetiikkaan terveillä tutkimushenkilöillä. Febuksostaatin yhteiskäyttö ei vaikuttanut INR-arvoon eikä hyytymistekijä VII:n aktiivisuuteen.
Desipramiini/CYP2D6:n substraatit
Febuksostaatin on osoitettu olevan heikko CYP2D6:n estäjä in vitro. Terveillä tutkimushenkilöillä tehdyssä tutkimuksessa febuksostaatti 120 mg kerran vuorokaudessa suurensi desipramiinin (CYP2D6:n substraatti) AUC-arvoa keskimäärin 22 %. Tämä viittaa siihen, että febuksostaatti saattaa olla heikko CYP2D6-entsyymin estäjä myös in vivo. Febuksostaatin ja muiden CYP2D6:n substraattien yhteiskäytön ei odoteta vaativan näiden lääkeaineiden annosmuutoksia.
Antasidit
Magnesiumhydroksidia ja alumiinihydroksidia sisältävien antasidien on osoitettu hidastavan samanaikaisesti annetun febuksostaatin imeytymistä (noin 1 tunnilla) ja pienentävän sen huippupitoisuutta (Cmax) noin 32 %, mutta AUC-arvossa ei todettu merkitseviä muutoksia. Febuksostaattia saa siten käyttää samanaikaisesti antasidien kanssa.
Raskaus ja imetys
Raskaus
Tiedot hyvin rajallisesta määrästä valmisteelle altistuneita raskauksia eivät viittaa febuksostaatin haitallisiin vaikutuksiin raskauteen tai sikiön/vastasyntyneen terveyteen. Eläinkokeissa ei ole havaittu suoria tai epäsuoria vaikutuksia raskauteen, alkion/sikiön kehitykseen tai synnytykseen (ks. kohta Prekliiniset tiedot turvallisuudesta).Mahdollista riskiä ihmisille ei tunneta. Febuksostaattia ei pidä käyttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö febuksostaatti ihmisen rintamaitoon. Eläinkokeissa tämän lääkeaineen on havaittu erittyvän maitoon ja vaikuttavan haitallisesti poikasten kehitykseen. Imeväiseen kohdistuvia riskejä ei voida poissulkea. Febuksostaattia ei pidä käyttää rintaruokinnan aikana.
Hedelmällisyys
Eläimillä annoksiin 48 mg/kg/vrk saakka tehdyt reproduktiotutkimukset eivät viitanneet hedelmällisyyteen kohdistuviin annosriippuvaisiin haittavaikutuksiin (ks. kohta Prekliiniset tiedot turvallisuudesta). Febuxostat Krka ‑valmisteen vaikutusta ihmisen hedelmällisyyteen ei tiedetä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Febuksostaatin käytön yhteydessä on raportoitu uneliaisuutta, heitehuimausta, parestesioita ja näön sumenemista. Ajaminen, koneiden käyttö ja muut vaaralliset toimet edellyttävät varovaisuutta, kunnes potilas on kohtuullisen varma, ettei Febuxostat Krka heikennä hänen suorituskykyään.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kliinisissä tutkimuksissa (4 072 vähintään yhden 10–300 mg:n annoksen saanutta tutkimuspotilasta) , myyntiluvan myöntämisen jälkeen toteutetuissa turvallisuustutkimuksissa (FAST-tutkimus: 3 001 vähintään yhden 80–120 mg:n annoksen saanutta tutkimuspotilasta) ja vaikuttavan aineen markkinoille tulon jälkeisen käyttökokemuksen yhteydessä kihtipotilailla yleisimmin ilmoitettuja haittavaikutuksia olivat kihtikohtaukset, maksan toiminnan poikkeavuudet, ripuli, pahoinvointi, päänsärky, huimaus, hengenahdistus, ihottuma, kutina, nivelkipu, lihaskipu, raajakipu, turvotus ja väsymys. Nämä haittavaikutukset olivat vaikeusasteeltaan pääasiassa lieviä tai keskivaikeita. Harvinaisia vakavia yliherkkyysreaktioita febuksostaatille, joista osaan liittyi systeemisiä oireita, ja harvinaisia äkillisiä sydänperäisiä kuolemia on esiintynyt valmisteen markkinoille tulon jälkeen.
Haittavaikutustaulukko
Seuraavassa taulukossa on lueteltu ne febuksostaattia saaneilla potilailla todetut yleiset (≥ 1/100 ja < 1/10), melko harvinaiset (≥ 1/1 000 ja < 1/100) ja harvinaiset (≥ 1/10 000 ja < 1/1 000) haittavaikutukset, joita esiintyi febuksostaattihoitoa saaneilla potilailla.
Esiintymistiheydet perustuvat tutkimuksiin sekä valmisteen markkinoille tulon jälkeiseen käyttökokemukseen kihtipotilailla.
Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1: Haittavaikutukset pitkäkestoisissa vaiheen 3 yhdistetyissä jatkotutkimuksissa, myyntiluvan myöntämisen jälkeen toteutetuissa turvallisuustutkimuksissa ja valmisteen markkinoille tulon jälkeenkihtipotilailla.
Veri ja imukudos | Harvinaiset Pansytopenia, trombosytopenia, agranulosytoosi*, anemia# |
Immuunijärjestelmä | Harvinaiset Anafylaktinen reaktio*, lääkeaineyliherkkyys* |
Umpieritys | Melko harvinaiset Suurentunut veren tyreotropiinipitoisuus, kilpirauhasen vajaatoiminta# |
Silmät | Melko harvinaiset Näön sumeneminen Harvinaiset Verkkokalvon valtimotukos# |
Aineenvaihdunta ja ravitsemus | Yleiset*** Kihtikohtaukset Melko harvinaiset Diabetes mellitus, hyperlipidemia, vähentynyt ruokahalu, painon nousu Harvinaiset Painon lasku, lisääntynyt ruokahalu, ruokahaluttomuus |
Psyykkiset häiriöt | Melko harvinaiset Sukupuolisen halun heikentyminen, unettomuus Harvinaiset Hermostuneisuus, masentuneisuus#, unihäiriöt# |
Hermosto | Yleiset Päänsärky, huimaus Melko harvinaiset Parestesiat, hemipareesi, uneliaisuus, letargia#, makuhäiriöt, hypestesia, hyposmia Harvinaiset Makuaistin menettäminen#, polttava tunne# |
Kuulo ja tasapainoelin | Melko harvinaiset Tinnitus Harvinaiset Pyörrytys# |
Sydän | Melko harvinaiset Eteisvärinä, sydämentykytys, EKG:n poikkeavuudet, vasemman puolen haarakatkos (ks. kohta Tuumorilyysioireyhtymä), sinustakykardia (ks. kohta Tuumorilyysioireyhtymä), rytmihäiriöt# Harvinaiset Äkillinen sydänperäinen kuolema* |
Verisuonisto | Melko harvinaiset Verenpaineen nousu, punastelu, kuumat aallot, verenvuoto (ks. kohta Tuumorilyysioireyhtymä) Harvinaiset Verenkiertokollapsi# |
Hengityselimet | Yleiset Hengenahdistus Melko harvinaiset Keuhkoputkitulehdus, ylähengitystieinfektio, alahengitystieinfektio#, yskä, voimakas, vetinen nuha# Harvinaiset Keuhkokuume# |
Ruoansulatuselimistö | Yleiset Ripuli**, pahoinvointi Melko harvinaiset: Vatsakipu, ylävatsakipu#, vatsan pingottuneisuus, gastroesofageaalinen refluksitauti, oksentelu, suun kuivuminen, ruoansulatushäiriöt, ummetus, tihentynyt ulostamistarve, ilmavaivat, epämiellyttävä tunne maha-suolikanavassa, suun haavaumat, huulten turvotus#, haimatulehdus Harvinaiset Maha-suolikanavan perforaatio#, suutulehdus# |
Maksa ja sappi | Yleiset Maksan toiminnan poikkeavuudet** Melko harvinaiset Sappikivitauti Harvinaiset Hepatiitti, ikterus*, maksavaurio*, sappirakkotulehdus# |
Iho ja ihonalainen kudos | Yleiset Ihottuma (mukaan lukien harvemmin raportoidut erityyppiset ihottumat, ks. jäljempänä), kutina Melko harvinaiset Dermatiitti, nokkosihottuma, ihon värimuutos, ihovaurio, petekiat, makulaarinen ihottuma, makulopapulaarinen ihottuma, papulaarinen ihottuma, liikahikoilu, alopesia, ekseema#, punoitus, yöhikoilu#, psoriaasi#, kutiseva ihottuma# Harvinaiset Toksinen epidermaalinen nekrolyysi*, Stevens–Johnsonin oireyhtymä*, angioedeema*, lääkeaineihottuma, johon liittyy eosinofiliaa ja systeemioireita (drug reaction with eosinophilia and systemic symptoms, DRESS)*, yleistynyt ihottuma (vakava)*, kesivä ihottuma, follikulaarinen ihottuma, rakkulainen ihottuma, märkärakkulainen ihottuma, erytematoottinen ihottuma, tuhkarokkotyyppinen ihottuma |
Luusto, lihakset ja sidekudos | Yleiset Nivelkipu, lihaskipu, raajakipu# Melko harvinaiset Niveltulehdus, tuki- ja liikuntaelimistön kipu, lihasheikkous, lihaskouristukset, lihasten kireys, limapussitulehdus, nivelten turvotus#, selkäkipu#, tuki- ja liikuntaelimistön jäykkyys#, nivelten jäykkyys Harvinaiset Rabdomyolyysi*, kiertäjäkalvosinoireyhtymä#, polymyalgia rheumatica# |
Munuaiset ja virtsatiet | Melko harvinaiset Munuaisten vajaatoiminta, munuaiskivitauti, verivirtsaisuus, tihentynyt virtsaamistarve, proteinuria, virtsaamispakko, virtsatieinfektio# Harvinaiset Tubulointerstitiaalinen nefriitti* |
Sukupuolielimet ja rinnat | Melko harvinaiset Erektiohäiriö |
Yleisoireet ja antopaikassa todettavat haitat | Yleiset Turvotus, väsymys Melko harvinaiset Rintakipu, epämiellyttävä tunne rinnassa, kipu#, huonovointisuus# Harvinaiset Jano, kuumuuden tunne# |
Tutkimukset | Melko harvinaiset Veren amylaasipitoisuuden suureneminen, trombosyyttipitoisuuden pieneneminen, veren valkosolumäärän väheneminen, veren lymfosyyttimäärän väheneminen, veren kreatiinipitoisuuden suureneminen, veren kreatiniinipitoisuuden suureneminen, hemoglobiinipitoisuuden pieneneminen, veren ureapitoisuuden suureneminen, veren triglyseridipitoisuuden suureneminen, veren kolesterolipitoisuuden suureneminen, hematokriitin pieneneminen, veren laktaattidehydrogenaasipitoisuuden suureneminen, veren kaliumpitoisuuden suureneminen, INR-arvon suureneminen# Harvinaiset Veren glukoosipitoisuuden suureneminen, aktivoituneen partiaalisen tromboplastiiniajan piteneminen, veren punasolumäärän pieneneminen, veren alkalisen fosfataasipitoisuuden suureneminen, veren kreatiinifosfokinaasipitoisuuden suureneminen* |
Vammat, myrkytykset ja hoitokomplikaatiot | Melko harvinaiset Kontuusio# |
* Valmisteen markkinoille tulon jälkeiseen käyttökokemukseen perustuvat hoitoon liittyvät haittavaikutukset
** Hoidosta aiheutuva ei-infektiivinen ripuli ja vaiheen 3 yhdistetyissä tutkimuksissa havaitut maksan toiminnan poikkeavuudet ovat yleisempiä potilailla, jotka käyttävät samanaikaisesti kolkisiinia.
*** Ks. kohdasta Farmakodynamiikka kihtikohtausten ilmaantuvuus kussakin vaiheen 3 satunnaistetussa, kontrolloidussa tutkimuksessa.
# Myyntiluvan myöntämisen jälkeen toteutetuissa turvallisuustutkimuksissa havaitut haittavaikutukset
Joidenkin haittavaikutusten kuvaus
Valmisteen markkinoille tulon jälkeen on esiintynyt harvoin vakavia yliherkkyysreaktioita febuksostaatille, Stevens–Johnsonin oireyhtymä, toksinen epidermaalinen nekrolyysi ja anafylaktinen reaktio/sokki mukaan lukien. Stevens–Johnsonin oireyhtymälle ja toksiselle epidermaaliselle nekrolyysille tyypillistä on etenevä ihottuma, johon liittyy rakkuloita tai limakalvovaurioita ja silmä-ärsytystä. Yliherkkyysreaktioihin febuksostaatille voi liittyä seuraavia oireita: ihoreaktioita, joille on tyypillistä infiltroitunut makulopapulaarinen ihottuma, yleistynyt tai hilseilevä ihottuma, mutta myös ihomuutoksia, kasvojen turvotusta, kuumetta, hematologisia poikkeavuuksia, kuten trombosytopeniaa ja eosinofiliaa, ja yhteen tai useaan elimeen (maksaan ja munuaisiin, tubulointerstitiaalinen nefriitti mukaan lukien) liittyviä poikkeavuuksia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kihtikohtauksia havaittiin yleisesti pian hoidon aloittamisen jälkeen ja ensimmäisten hoitokuukausien aikana. Tämän jälkeen kihtikohtausten esiintyvyys vähenee ajasta riippuvaisesti. Kihtikohtausten estohoitoa suositellaan (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Tuumorilyysioireyhtymä
Turvallisuusprofiilin yhteenveto
Satunnaistetussa, kaksoissokkoutetussa vaiheen 3 keskeisessä FLORENCE (FLO-01) -tutkimuksessa verrattiin febuksostaattia allopurinoliin (346 potilasta, jotka saivat solunsalpaajahoitoa hematologisen syöpäsairauden vuoksi ja joiden tuumorilyysioireyhtymän (TLS) riski oli kohtalainen tai suuri). Yhteensä vain 22 potilaalla (6,4 %) ilmeni haittavaikutuksia, 11 (6,4 %) potilaalla kummassakin hoitoryhmässä. Suurin osa haittavaikutuksista oli joko lieviä tai kohtalaisia.
Kaiken kaikkiaan FLORENCE-tutkimuksessa ei noussut esiin mitään erityistä turvallisuusriskiä aiemman kihtipotilailla havaitun febuksostaattikäyttökokemuksen lisäksi, lukuun ottamatta seuraavia kolmea haittavaikutusta (lueteltu edellä taulukossa 1).
Sydän:
Melko harvinaiset: vasemman puolen haarakatkos, sinustakykardia
Verisuonisto:
Melko harvinaiset: verenvuoto
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Yliannostustapauksessa potilaalle tulee antaa oireenmukaista ja elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Kihtilääkkeet, virtsahapon muodostumista estävät valmisteet, ATC-koodi: M04AA03
Vaikutusmekanismi
Virtsahappo on ihmiselimistössä puriinien metabolian lopputuote, jota muodostuu kun hypoksantiini muuttuu ksantiiniksi ja ksantiini virtsahapoksi. Ksantiinioksidaasi (XO) katalysoi tämän aineenvaihduntaprosessin molempia vaiheita. Febuksostaatti on 2-aryylitiatsolijohdannainen, jonka terapeuttinen vaikutus perustuu ksantiinioksidaasin selektiiviseen estoon ja tämän kautta seerumin virtsahappopitoisuuden pienentämiseen. Febuksostaatti on voimakas ei-puriiniselektiivinen ksantiinioksidaasin estäjä (NP-SIXO), jonka estovaikutuksen Ki-arvo on alle 1 nM in vitro. Febuksostaatin on osoitettu estävän voimakkaasti ksantiinioksidaasin sekä hapettunutta että pelkistynyttä muotoa. Febuksostaatti ei terapeuttisina pitoisuuksina estä muita puriini- tai pyrimidiinimetaboliaan osallistuvia entsyymejä, joita ovat guaniinideaminaasi, hypoksantiiniguaniinifosforibosyylitransferaasi, orotaattifosforibosyylitransferaasi, orotidiinimonofosfaattidekarboksylaasi ja puriininukleosidifosforylaasi.
Kliininen teho ja turvallisuus
Kihti
Febuksostaattitablettien teho osoitettiin kolmessa vaiheen 3 pivotaalitutkimuksessa (kaksi pivotaalitutkimusta, APEX ja FACT, sekä lisätutkimus CONFIRMS on kuvattu jäljempänä). Näihin tutkimuksiin osallistui 4 101 potilasta, jotka sairastivat hyperurikemiaa ja kihtiä. Jokaisessa vaiheen 3 pivotaalitutkimuksessa febuksostaatti pienensi seerumin virtsahappopitoisuutta ja piti sen saavutetussa pitoisuudessa allopurinolia paremmin. APEX- ja FACT-tutkimuksissa ensisijaisena päätetapahtumana oli niiden potilaiden osuus, joiden seerumin virtsahappopitoisuus oli < 6,0 mg/dl (357 µmol/l) tutkimuksen kolmena viimeisenä kuukautena. Lisäksi tehdyssä vaiheen 3 CONFIRMS-tutkimuksessa, jonka tulokset on saatu sen jälkeen, kun febuksostaattivalmisteelle on myönnetty myyntilupa, ensisijainen tehon päätetapahtuma oli niiden potilaiden osuus, joiden seerumin virtsahappopitoisuus oli loppukäynnillä < 6,0 mg/dl. Näihin tutkimuksiin ei otettu elinsiirtopotilaita (ks. kohta Annostus ja antotapa).
APEX-tutkimus: APEX-tutkimus (the Allopurinol and Placebo-Controlled Efficacy Study of Febuxostat) oli 28 viikon pituinen vaiheen 3 satunnaistettu, kaksoissokkoutettu monikeskustutkimus. Tässä tutkimuksessa 1 072 potilasta satunnaistettiin saamaan joko lumelääkettä (n = 134), febuksostaattiannosta 80 mg kerran vuorokaudessa (n = 267), febuksostaattiannosta 120 mg kerran vuorokaudessa (n = 269), febuksostaattiannosta 240 mg kerran vuorokaudessa (n = 134) tai allopurinolia (300 mg kerran vuorokaudessa [n = 258], jos potilaan seerumin kreatiniinipitoisuus oli lähtötilanteessa ≤ 1,5 mg/dl, tai 100 mg kerran vuorokaudessa [n = 10], jos potilaan seerumin kreatiniinipitoisuus oli lähtötilanteessa > 1,5 mg/dl ja ≤ 2,0 mg/dl). Febuksostaattiannosta 240 mg (kaksi kertaa suositeltavan enimmäisannoksen suuruinen annos) käytettiin lääkkeen turvallisuuden arviointiin.
APEX-tutkimus osoitti, että febuksostaatti annoksina 80 mg kerran vuorokaudessa ja 120 mg kerran vuorokaudessa pienensi seerumin virtsahappopitoisuuden alle arvon 6 mg/dl (357 μmol/l) tilastollisesti merkitsevästi paremmin kuin tavanomaiset allopurinoliannokset 300 mg (n = 258) ja 100 mg (n = 10) (ks. taulukko 2 ja kuva 1).
FACT-tutkimus: FACT-tutkimus (the Febuxostat Allopurinol Controlled Trial) oli 52 viikon pituinen vaiheen 3 satunnaistettu, kaksoissokkoutettu monikeskustutkimus. Tässä tutkimuksessa 760 potilasta satunnaistettiin saamaan febuksostaattiannosta 80 mg kerran vuorokaudessa (n = 256), febuksostaattiannosta 120 mg kerran vuorokaudessa (n = 251) tai allopurinolia 300 mg kerran vuorokaudessa (n = 253).
FACT-tutkimus osoitti, että febuksostaatti annoksina 80 mg kerran vuorokaudessa ja 120 mg kerran vuorokaudessa pienensi seerumin virtsahappopitoisuuden alle arvon 6 mg/dl (357 µmol/l) ja piti sen saavutetussa pitoisuudessa tilastollisesti merkitsevästi paremmin kuin tavanomainen allopurinoliannos 300 mg.
Ensisijaisen tehon päätetapahtuman tulokset on esitetty yhteenvetona taulukossa 2:
Taulukko 2
Potilaat, joiden seerumin virtsahappopitoisuus oli < 6,0 mg/dl (< 357 µmol/l) kolmella viimeisellä kuukausikäynnillä
| Tutkimus | Febuksostaatti 80 mg kerran vuorokaudessa | Febuksostaatti 120 mg kerran vuorokaudessa | Allopurinoli 300 / 100 mg kerran vuorokaudessa1 |
APEX (28 viikkoa) | 48 % * (n = 262) | 65 % *, # (n = 269) | 22 % (n = 268) |
FACT (52 viikkoa) | 53 %* (n = 255) | 62 %* (n = 250) | 21 % (n = 251) |
Yhdistetyt tulokset | 51 %* (n = 517) | 63 %*, # (n = 519) | 22 % (n = 519) |
1 tulokset potilaista, jotka saivat allopurinolia joko 100 mg kerran vuorokaudessa (n = 10: potilaat, joiden seerumin kreatiniinipitoisuus oli > 1,5 ja ≤ 2,0 mg/dl) tai 300 mg kerran vuorokaudessa (n = 509) yhdistettiin analyysejä varten. * p < 0,001 vs allopurinoli, # p < 0,001 vs 80 mg | |||
Febuksostaatti pienensi seerumin virtsahappopitoisuutta nopeasti ja pitkäkestoisesti. Seerumin virtsahappopitoisuuden havaittiin pienentyneen tasolle < 6,0 mg/dl (357 μmol/l) viikon 2 käynnillä, ja se pysyi tällä tasolla koko hoitojakson ajan. Kuvassa 1 esitetään kahdesta vaiheen 3 pivotaalitutkimuksesta saadut hoitoryhmäkohtaiset keskimääräiset seerumin virtsahappopitoisuudet ajan funktiona.
Kuva 1: Keskimääräiset seerumin virtsahappopitoisuudet vaiheen 3 yhdistetyissä pivotaalitutkimuksissa
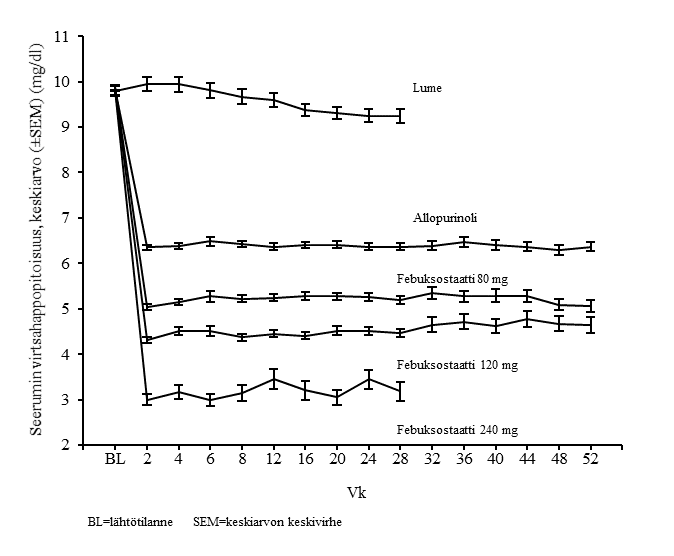
Huom.: 509 potilasta sai allopurinolia annoksena 300 mg kerran vuorokaudessa; 10 potilasta, joiden seerumin kreatiniinipitoisuus oli > 1,5 ja ≤ 2,0 mg/dl, saivat 100 mg kerran vuorokaudessa. (APEX-tutkimuksessa 10 potilasta 268:sta).
Febuksostaattiannosta 240 mg (kaksi kertaa suositellun enimmäisannoksen suuruinen annos) käytettiin febuksostaatin turvallisuuden arviointiin.
CONFIRMS-tutkimus: CONFIRMS-tutkimus oli 26 viikon pituinen vaiheen 3 satunnaistettu, kontrolloitu, tutkimus, jossa arvioitiin 40 mg:n ja 80 mg:n febuksostaattiannosten tehoa verrattuna 300 mg:n ja 200 mg:n allopurinoliannoksiin potilailla, joilla oli kihti ja hyperurikemia. Tässä tutkimuksessa 2269 potilasta satunnaistettiin saamaan febuksostaattiannosta 40 mg kerran vuorokaudessa (n = 757), febuksostaattiannosta 80 mg kerran vuorokaudessa (n = 756) tai allopurinolia 300/200 mg kerran vuorokaudessa (n = 756). Potilaista vähintään 65 %:lla oli lievä tai keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma 30–89 ml/min). Kihtikohtausten estohoito oli pakollinen 26 viikon mittaisen jakson ajan.
Niiden potilaiden osuus, joiden seerumin virtsahappopitoisuus oli loppukäynnillä < 6,0 mg/dl (357 µmol/l), oli 45 % febuksostaattia 40 mg:n annoksina saaneista, 67 % febuksostaattia 80 mg:n annoksina saaneista ja 42 % allopurinolia 300/200 mg:n annoksina saaneista.
Ensisijainen päätetapahtuma munuaisten vajaatoimintaa sairastavien potilaiden alaryhmässä
APEX-tutkimuksessa tehoa arvioitiin 40:llä munuaisten vajaatoimintaa sairastavalla potilaalla (seerumin kreatiniinipitoisuus lähtötilanteessa > 1,5 mg/dl ja ≤ 2,0 mg/dl). Allopurinoliryhmään satunnaistetut munuaisten vajaatoimintapotilaat saivat allopurinolia enintään 100 mg kerran vuorokaudessa. Ensisijainen tehon päätetapahtuma saavutettiin 44 %:lla febuksostaattitabletteja 80 mg kerran vuorokaudessa saaneista potilaista, 45 %:lla febuksostaattitabletteja 120 mg kerran vuorokaudessa saaneista potilaista ja 60 %:lla febuksostaattitabletteja 240 mg kerran vuorokaudessa saaneista potilaista, mutta 0 %:lla potilaista, jotka saivat allopurinolia 100 mg kerran vuorokaudessa tai lumelääkettä.
Terveiden vapaaehtoisten seerumin virtsahappopitoisuuden prosentuaalisessa laskussa ei todettu kliinisesti merkitseviä eroja heidän munuaistensa toiminnasta riippumatta (58 % potilailla, joiden munuaiset toimivat normaalisti, ja 55 % potilailla, joilla oli vaikeita munuaisten toimintahäiriöitä).
CONFIRMS-tutkimuksessa määriteltiin prospektiivisesti analyysi potilaista, joilla oli kihti ja munuaisten vajaatoimintaa. Analyysi osoitti, että febuksostaatti pienensi seerumin virtsahappopitoisuuden alle arvon 6 mg/dl huomattavasti tehokkaammin kuin 300 mg/200 mg allopurinolia, kun potilaalla oli kihti ja lievä tai keskivaikea munuaisten vajaatoiminta (65 % tutkituista potilaista).
Ensisijainen päätetapahtuma alaryhmässä, jossa potilaiden seerumin virtsahappopitoisuus oli ≥ 10 mg/dl
Noin 40 %:lla potilaista (sekä APEX- että FACT-tutkimuksissa) seerumin virtsahappopitoisuus oli lähtötilanteessa ≥ 10 mg/dl. Tässä alaryhmässä ensisijainen tehon päätetapahtuma (seerumin virtsahappopitoisuus 3 viimeisellä käynnillä < 6,0 mg/dl) saavutettiin 41 %:lla febuksostaattitabletteja 80 mg kerran vuorokaudessa saaneista potilaista, 48 %:lla febuksostaattitabletteja 120 mg kerran vuorokaudessa saaneista potilaista ja 66 %:lla febuksostaattitabletteja 240 mg kerran vuorokaudessa saaneista potilaista, mutta 9 %:lla potilaista, jotka saivat allopurinolia 300 mg/100 mg kerran vuorokaudessa, ja 0 %:lla potilaista, jotka saivat lumelääkettä.
CONFIRMS-tutkimuksessa niiden potilaiden osuus, jotka saavuttivat ensisijaisen tehon päätetapahtuman (seerumin virtsahappopitoisuus viimeisellä käynnillä < 6,0 mg/dl), kun potilaan seerumin virtsahappopitoisuus oli lähtötilanteessa ≥ 10 mg/dl, oli 27 % febuksostaattia 40 mg:n annoksina kerran vuorokaudessa saaneista potilaista (66/249), 49 % febuksostaattia 80 mg:n annoksina kerran vuorokaudessa saaneista potilaista (125/254) ja 31 % allopurinolia 300 mg/200 mg kerran vuorokaudessa saaneista potilaista (72/230).
Kliiniset tulokset: potilaat, jotka tarvitsivat hoitoa kihtikohtauksiin
APEX-tutkimus: 8 viikkoa kestäneen estohoitojakson aikana suurempi osa tutkimuspotilaita 120 mg febuksostaattia saaneessa ryhmässä (36 %) verrattuna 80 mg febuksostaattia (28 %), 300 mg allopurinolia (23 %) ja lumelääkettä (20 %) saaneisiin ryhmiin tarvitsi hoitoa kihtikohtauksiin. Kohtaukset lisääntyivät estohoitojakson jälkeen ja vähenivät vähitellen ajan mittaan. Viikkojen 8–28 aikana kihtikohtauksiin sai hoitoa 46–55 % tutkimuspotilaista. Tutkimusten neljän viimeisen viikon aikana (viikot 24–28) kihtikohtauksia havaittiin 15 %:lla (80 mg tai 120 mg febuksostaattia saaneista), 14 %:lla (300 mg allopurinolia saaneista) ja 20 %:lla (lumelääkettä saaneista) potilaista.
FACT-tutkimus: 8 viikkoa kestäneen estohoitojakson aikana suurempi osa tutkimuspotilaita 120 mg febuksostaattia saaneessa ryhmässä (36 %) verrattuna 80 mg febuksostaattia (22 %) ja 300 mg allopurinolia (21 %) saaneisiin ryhmiin tarvitsi hoitoa kihtikohtauksiin. Kihtikohtausten ilmaantuvuus suureni 8 viikon estohoitojakson jälkeen ja pieneni vähitellen ajan mittaan (64 % ja 70 % tutkimuspotilaista sai hoitoa kihtikohtauksiin viikkoina 8–52). Tutkimusten neljän viimeisen viikon aikana (viikot 49–52) kihtikohtauksia havaittiin 6–8 %:lla (80 mg tai 120 mg febuksostaattia saaneista) ja 11 %:lla (300 mg allopurinolia saaneista) potilaista.
APEX- ja FACT-tutkimuksissa kihtikohtauksiin hoitoa tarvinneiden potilaiden osuus oli hoitovaiheen viimeisten 32 viikon aikana (viikkojen 20–24 ja viikkojen 49–52 välisellä jaksolla) lukumääräisesti pienempi niissä ryhmissä, joissa seerumin keskimääräinen uraattipitoisuus oli lähtötilanteen jälkeen < 6,0 mg/dl, < 5,0 mg/dl tai < 4,0 mg/dl verrattuna ryhmiin, joissa se oli ≥ 6,0 mg/dl.
CONFIRMS-tutkimuksen aikana (ensimmäisestä päivästä 6 kuukauteen saakka) kihtikohtauksiin hoitoa tarvinneiden potilaiden prosenttiosuus oli 31 % febuksostaattia 80 mg:n annoksina saaneista ja 25 % allopurinoliryhmässä. Kihtikohtauksiin hoitoa tarvinneiden potilaiden osuudessa ei havaittu eroa 40 mg tai 80 mg febuksostaattia saaneilla potilailla.
Pitkäkestoiset, avoimet jatkotutkimukset
EXCEL-tutkimus (C02-021): Excel-tutkimus oli kolmen vuoden pituinen vaiheen 3, avoin, satunnaistettu, allopurinolikontrolloitu, turvallisuutta selvittävä monikeskustutkimuksena toteutettu jatkotutkimus, johon osallistuneet potilaat olivat olleet mukana vaiheen 3 pivotaalitutkimuksissa (Apex tai FACT). Tutkimukseen otettiin mukaan yhteensä 1086 potilasta, jotka saivat: febuksostaattiannosta 80 mg kerran vuorokaudessa (n = 649), febuksostaattiannosta 120 mg kerran vuorokaudessa (n = 292) tai allopurinolia 300/100 mg kerran vuorokaudessa (n = 145). Noin 69 % potilaista ei tarvinnut hoitoonsa muutoksia lopullisen vakaan hoidon saavuttamiseksi. Jos potilaan seerumin virtsahappopitoisuus oli kolmella peräkkäisellä kerralla > 6,0 mg/dl, hänen osallistumisensa tutkimukseen keskeytettiin. Seerumin virtsahappopitoisuudet pysyivät ajan kuluessa ennallaan (eli 91 %:lla 80 mg febuksostaattia ja 93 %:lla 120 mg febuksostaattia aluksi saaneista potilaista seerumin virtsahappopitoisuus oli < 6 mg/dl 36 kuukauden kohdalla).
Kolmen vuoden tiedot osoittivat kihtikohtausten ilmaantuvuuden pienentyneen niin, että alle 4 % potilaista tarvitsi hoitoa kihtikohtauksen vuoksi (ts. yli 96 % potilaista ei tarvinnut hoitoa kihtikohtaukseen) kuukausien 16–24 ja 30–36 aikana.
Primaari palpoitavissa ollut kihtikyhmy hävisi täysin lähtötilanteesta tutkimuksen loppukäyntiin mennessä 46 %:lla 80 mg febuksostaattia kerran vuorokaudessa saaneista potilaista ja 38 %:lla 120 mg febuksostaattia kerran vuorokaudessa saaneista potilaista.
Tutkimus TMX-01-005 (FOCUS) oli 5 vuoden mittainen vaiheen 2, avoin, turvallisuutta selvittävä monikeskustutkimuksena toteutettu jatkotutkimus potilailla, jotka olivat olleet mukana febuksostaatilla tehdyssä 4 viikkoa kestäneessä kaksoissokkoutetulla annostuksella toteutetussa tutkimuksessa TMX-00-004. Tutkimukseen otettiin mukaan 116 potilasta, jotka saivat aluksi 80 mg febuksostaattia kerran vuorokaudessa. Potilaista 62 % ei tarvinnut annosmuutoksia, jotta seerumin virtsahappopitoisuus pysyi alle arvon 6 mg/dl, mutta 38 %:lla potilaista annosta oli muutettava lopullisen vakaan annoksen saavuttamiseksi.
Niiden potilaiden osuus, joiden seerumin virtsahappopitoisuus oli loppukäynnillä < 6,0 mg/dl (357 µmol/l), oli kaikilla febuksostaattiannoksilla yli 80 % (81–100 %).
Vaiheen 3 kliinisissä tutkimuksissa febuksostaattihoitoa saaneilla potilailla todettiin lieviä poikkeamia maksan toiminnassa (5,0 %). Nämä prosenttiosuudet olivat samaa luokkaa kuin allopurinolihoidon aikana ilmoitetut prosenttiosuudet (4,2 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Pitkäaikaisten avointen jatkotutkimusten yhteydessä todettiin TSH-arvojen kohoamista (> 5,5 µIU/ml) 5,5 %:lla pitkäaikaista febuksostaattihoitoa saavista potilaista ja 5,8 %:lla pitkäaikaista allopurinolihoitoa saavista potilaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pitkäkestoiset tutkimukset valmisteen markkinoille tulon jälkeen
CARES-tutkimus oli satunnaistettu, kaksoissokkoutettu, vertailukelpoisuutta arvioinut monikeskustutkimus, jossa verrattiin febuksostaattihoidon kardiovaskulaarisia tuloksia allopurinolihoitoon kihtipotilailla, joilla oli aiemmin todettu merkittävä sydän- ja verisuonitauti, kuten sydäninfarkti, sairaalahoitoa vaatinut epävakaa sepelvaltimotauti, sepelvaltimoiden tai aivojen revaskularisaatiotoimenpide, aivohalvaus, sairaalahoitoa edellyttänyt ohimenevä aivoverenkiertohäiriö, ääreisverisuonien sairaus tai diabetes mellitus, johon liittyi näyttöä mikrovaskulaarisesta tai makrovaskulaarisesta sairaudesta. Jotta saavutettiin alle 6 mg/dl:n virtsahappopitoisuus seerumissa, febuksostaattiannos titrattiin 40 mg:sta enintään 80 mg:aan (munuaisten toiminnasta riippumatta) ja allopurinoliannos titrattiin 100 mg:n lisäyksin 300–600 mg:aan potilailla, joiden munuaiset toimivat normaalisti tai joilla oli lievä munuaisten vajaatoiminta, ja 200–400 mg:aan potilailla, joilla oli keskivaikea munuaisten vajaatoiminta.
Ensisijainen päätetapahtuma CARES-tutkimuksessa oli aika merkittävän sydän- ja verisuonitapahtuman (MACE) ensimmäiseen ilmaantumiseen, ja se oli yhdistelmäpäätetapahtuma, joka sisälsi kuolemaan johtamattoman sydäninfarktin, kuolemaan johtamattoman aivohalvauksen, sydän- ja verisuoniperäisen kuoleman ja epävakaan sepelvaltimotaudin, johon liittyi kiireellinen sepelvaltimon revaskularisaatio.
Päätetapahtumat (ensisijaiset ja toissijaiset) analysoitiin käyttämällä hoitoaikeen mukaista analyysiä (ITT), jossa olivat mukana kaikki satunnaistetut tutkimushenkilöt, jotka olivat saaneet ainakin yhden annoksen kaksoissokkoutettua tutkimuslääkettä.
Yhteensä 56,6 % potilaista lopetti tutkimuslääkkeen käytön ennenaikaisesti, ja 45 % potilaista ei tullut kaikille tutkimuskäynneille.
Yhteensä 6 190 potilasta käsittävän seurannan keston mediaani oli 32 kuukautta, ja altistuksen keston mediaani oli febuksostaattiryhmän potilailla 728 päivää (n = 3 098) ja allopurinoliryhmän potilailla 719 päivää (n = 3 092).
Ensisijaisia merkittävän sydän- ja verisuonitapahtuman päätetapahtumia (MACE, yhdistetty) ilmeni febuksostaatti- ja allopurinolihoitoryhmissä saman verran (febuksostaattiryhmässä 10,8 %:lla ja allopurinoliryhmässä 10,4 %:lla potilaista; riskisuhde 1,03, kaksitahoinen toistettu 95 %:n luottamusväli 0,89–1,21).
Merkittävien sydän- ja verisuonitapahtumien yksittäisten komponenttien analyysissä sydän- ja verisuoniperäisiä kuolemantapauksia oli febuksostaattiryhmässä enemmän kuin allopurinoliryhmässä (febuksostaattiryhmässä 4,3 %:lla ja allopurinoliryhmässä 3,2 %:lla potilaista; riskisuhde 1,34, 95 %:n luottamusväli 1,03–1,73). Muiden merkittävien sydän- ja verisuonitapahtumien ilmaantuvuudet olivat samanlaisia febuksostaatti- ja allopurinoliryhmissä: kuolemaan johtamatonta sydäninfarktia ilmeni febuksostaattiryhmässä 3,6 %:lla ja allopurinoliryhmässä 3,8 %:lla potilaista (riskisuhde 0,93, 95 %:n luottamusväli 0,72–1,21) ja kuolemaan johtamatonta aivohalvausta febuksostaattiryhmässä 2,3 %:lla ja allopurinoliryhmässä 2,3 %:lla potilaista (riskisuhde 1,01, 95 %:n luottamusväli 0,73–1,41), ja kiireellinen revaskularisaatiotoimenpide epävakaan sepelvaltimotaudin vuoksi tehtiin 1,6 %:lle febuksostaattiryhmän potilaista ja 1,8 %:lle allopurinoliryhmän potilaista (riskisuhde 0,86, 95 %:n luottamusväli 0,59–1,26). Kaikki kuolinsyyt kattava kuolleisuus oli myös febuksostaattiryhmässä suurempi kuin allopurinoliryhmässä (febuksostaattiryhmässä 7,8 % ja allopurinoliryhmässä 6,4 % potilaista; riskisuhde 1,22, 95 %:n luottamusväli 1,01–1,47), mikä johtui pääasiassa suuremmasta sydän- ja verisuoniperäisten kuolemantapausten määrästä febuksostaattiryhmässä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vahvistetut määrät, jotka koskivat sairaalahoitoa sydämen vajaatoiminnan tai iskemiaan liittymättömän rytmihäiriön vuoksi, laskimoperäisiä tromboembolisia tapahtumia ja sairaalahoitoa vaativia ohimeneviä aivoverenkiertohäiriöitä, olivat vastaavia febuksaattia ja allopurinolia saaneilla potilailla.
FAST-tutkimus oli prospektiivinen, satunnaistettu, avoin, päätetapahtuman osalta sokkoutettu tutkimus, jossa verrattiin febuksostaattihoidon ja allopurinolihoidon kardiovaskulaarisia turvallisuusprofiileja potilailla, joilla oli krooninen hyperurikemia (kun uraattikiteitä oli jo päässyt muodostumaan) ja kardiovaskulaarisia riskitekijöitä (eli vähintään 60‑vuotiailla potilailla, joilla oli ainakin yksi muu kardiovaskulaarinen riskitekijä). Sisäänottokriteerit täyttävät potilaat saivat allopurinolihoitoa ennen satunnaistamista, ja annosta muutettiin tarvittaessa kliinisen harkinnan, EULAR-suositusten ja hyväksytyn annostuksen mukaan. Allopurinolilla toteutetun aloitusvaiheen lopussa potilaat, joilla seerumin virtsahappopitoisuus oli < 0,36 mmol/l (< 6 mg/dl) tai jotka saivat suurinta siedettyä tai suurinta hyväksyttyä allopurinoliannosta, satunnaistettiin suhteessa 1:1 saamaan joko febuksostaatti- tai allopurinolihoitoa. FAST-tutkimuksen ensisijainen päätetapahtuma oli aika minkä tahansa sellaisen tapahtuman ensimmäiseen ilmaantumiseen, joka sisältyi yhdistettyyn APTC-päätetapahtumaan (yhdistettyyn Antiplatelet Trialists’ Collaborative ‑päätetapahtumaan), joka käsitti i) sairaalahoidon kuolemaan johtamattoman sydäninfarktin tai biomarkkeripositiivisen äkillisen sepelvaltimo-oireyhtymän (ACS) vuoksi; ii) kuolemaan johtamattoman aivohalvauksen; iii) sydän- ja verisuonitapahtumasta johtuvan kuoleman. Primaarianalyysi perustui hoitoa saaneiden potilaiden analyysiin.
Satunnaistettuja potilaita oli yhteensä 6 128, joista 3 063 satunnaistettiin saamaan febuksostaattia ja 3 065 allopurinolia.
Hoitoa saaneiden potilaiden primaarianalyysissä febuksostaatti oli vähintään samanveroinen kuin allopurinoli ensisijaisen päätetapahtuman ilmaantuvuuden suhteen. Ensisijainen päätetapahtuma ilmeni 172:lla febuksostaattia saaneella potilaalla (1,72/100 potilasvuotta) ja 241:llä allopurinolia saaneella potilalla (2,05/100 potilasvuotta), ja korjattu riskisuhde oli 0,85 (95 %:n luottamusväli 0,70‑1,03), p < 0,001. Ensisijaista päätetapahtumaa koskeneessa hoitoa saaneiden potilaiden analyysissä sellaisten potilaiden alaryhmästä, joilla oli aiemmin todettu sydäninfarkti, aivohalvaus tai äkillinen sepelvaltimo-oireyhtymä, ei todettu merkitseviä eroja hoitoryhmien välillä: febuksostaattiryhmässä tapahtumia todettiin 65 potilaalla (9,5 %) ja allopurinoliryhmässä 83 potilaalla (11,8 %); korjattu riskisuhde oli 1,02 (95 %:n luottamusväli 0,74–1,42); p = 0,202.
Febuksostaattihoitoon ei liittynyt sydän- ja verisuoniperäisten kuolemantapausten tai mistä tahansa syystä johtuneiden kuolemantapausten ilmaantuvuuden suurenemista koko potilaspopulaatiossa tai sellaisten potilaiden alaryhmässä, joilla oli lähtötilanteessa aiemmin todettu sydäninfarkti, aivohalvaus tai äkillinen sepelvaltimo-oireyhtymä. Febuksostaattiryhmässä oli kaiken kaikkiaan vähemmän kuolemantapauksia (62 sydän- ja verisuoniperäistä kuolemantapausta ja 108 mistä tahansa syystä johtunutta kuolemantapausta) kuin allopurinoliryhmässä (82 sydän- ja verisuoniperäistä kuolemantapausta ja 174 mistä tahansa syystä johtunutta kuolemantapausta).
Virtsahappopitoisuudet pienenivät febuksostaattihoitoa saaneilla enemmän kuin allopurinolihoitoa saaneilla.
Tuumorilyysioireyhtymä
FLORENCE (FLO-01) -tutkimuksessa arvioitiin febuksostaatin tehoa ja turvallisuutta tuumorilyysioireyhtymän ehkäisyssä ja hoidossa. Febuksostaatti vähensi uraatin määrää selvästi paremmin ja nopeammin kuin allopurinoli.
Keskeisessä, satunnaistetussa (1:1), kaksoissokkoutetussa, vaiheen 3 FLORENCE-tutkimuksessa verrattiin febuksostaattia 120 mg kerran vuorokaudessa saaneita potilaita allopurinolia 200−600 mg vuorokaudessa saaneisiin potilaisiin (allopurinolin keskimääräinen vuorokausiannos [± keskihajonta]: 349,7 ± 112,90 mg) seerumin virtsahappopitoisuuden sääntelyn suhteen. Sisäänottokriteerit täyttävien potilaiden piti soveltua allopurinolihoitoon tai heillä ei saanut olla mahdollisuutta käyttää rasburikaasia. Ensisijaiset päätetapahtumat olivat käyrän alle jäävä seerumin virtsahappopitoisuus (AUC sUA1-8) ja seerumin kreatiniinipitoisuuden (sC) muutos, molemmat mitattuina lähtötilanteesta päivään 8.
Tutkimukseen osallistui yhteensä 364 hematologista syöpäsairautta sairastavaa ja solunsalpaajahoitoa saavaa potilasta, joilla oli kohtalainen/suuri tuumorilyysioireyhtymän riski. Keskimääräinen AUC sUA1‑8 (mgxh/dl) oli merkittävästi pienempi febuksostaattia saaneilla potilailla (514,0 ± 225,71, allopurinoliryhmässä 708,0 ± 234,42; pienimmän neliösumman keskiarvojen ero: -196,794 [95 %:n luottamusväli: -238,600; -154,988]; p < 0,0001). Lisäksi seerumin keskimääräinen virtsahappopitoisuus oli merkittävästi pienempi febuksostaattia saaneilla potilailla ensimmäisten 24 tunnin hoidon aikana ja kaikissa mittauspisteissä tämän jälkeen. Keskimääräisessä seerumin kreatiniinipitoisuuden muutoksessa (%) ei havaittu merkittävää eroa febuksostaatin ja allopurinolin välillä (febuksostaatti: -0,83 ± 26,98, allopurinoli: -4,92 ± 16,70; pienimmän neliösumman keskiarvojen ero: 4,0970 [95 %:n luottamusväli: -0,6467; 8,8406]; p = 0,0903). Toissijaisten päätetapahtumien osalta ei havaittu merkittävää muutosta laboratoriokokein varmistetun TLS:n (febuksostaattiryhmä: 8,1 %, allopurinoliryhmä: 9,2 %; riskisuhde: 0,875 [95 %:n luottamusväli: 0,4408; 1,7369]; p = 0,8488) eikä kliinisesti todetun TLS:n esiintyvyydessä (febuksostaattiryhmä: 1,7 %, allopurinoliryhmä: 1,2 %; riskisuhde: 0,994 [95 %:n luottamusväli: 0,9691; 1,0199]; p = 1,0000). Kaikkien hoidon aikana ilmenneiden merkkien ja oireiden esiintyvyys oli febuksostaattiryhmässä 67,6 % ja allopurinoliryhmässä 64,7 %. Kaikkien hoidon aikana ilmenneiden haittavaikutusten esiintyvyys oli sekä febuksostaatti- että allopurinoliryhmässä 6,4 %. FLORENCE-tutkimuksessa seerumin virtsahappopitoisuuden sääntely toimi selvästi paremmin febuksostaatilla allopurinoliin verrattuna potilailla, joiden oli tarkoitus saada allopurinolia. Tietoja ei tällä hetkellä ole saatavilla febuksostaatin vertailusta rasburikaasiin. Febuksostaatin tehoa ja turvallisuutta ei ole osoitettu potilailla, joilla on akuutti vaikea TLS, esim. potilailla, joilla muut uraattipitoisuutta pienentävät hoidot ovat epäonnistuneet.
Farmakokinetiikka
Terveillä vapaaehtoisilla febuksostaatin AUC-arvo ja huippupitoisuudet plasmassa (Cmax) suurenivat annosriippuvaisesti 10–120 mg:n suuruisten kerta-annosten jälkeen ja jatkuvassa annostelussa. Febuksostaatin AUC-arvo suureni enemmän kuin suhteessa annokseen, kun annokset olivat suuruudeltaan 120–300 mg. Febuksostaatti ei juuri kerry elimistöön, kun 10–240 mg:n suuruisia annoksia annetaan 24 tunnin välein. Febuksostaatin näennäinen terminaalinen eliminaation puoliintumisaika (t1/2) on keskimäärin n. 5–8 tuntia.
Populaatiofarmakokineettiset/farmakodynaamiset analyysit tehtiin 211 potilaalla, joilla oli hyperurikemia ja kihti ja jotka saivat febuksostaattihoitoa annoksena 40–240 mg kerran vuorokaudessa. Näistä analyyseistä saadut arviot febuksostaatin farmakokineettisistä parametreista ovat yleisesti ottaen verrattavissa terveillä tutkimushenkilöillä todettuihin parametreihin, mikä viittaa siihen, että terveistä tutkimushenkilöillä saadut tiedot sopivat farmakokineettisten/farmakodynaamisten arviointien tekemiseen kihtiä sairastavilla potilailla.
Imeytyminen
Febuksostaatti imeytyy nopeasti (tmax 1,0–1,5 h) ja hyvin (vähintään 84 %). Kun potilaat saivat suun kautta 80 mg febuksostaattia kerta-annoksena tai kerran vuorokaudessa useiden päivien ajan, Cmax oli noin 2,8–3,2 µg/ml. Annoksella 120 mg Cmax oli vastaavasti noin 5,0–5,3 µg/ml. Febuksostaattitablettien absoluuttista biologista hyötyosuutta ei ole tutkittu.
Kun potilaille annettiin runsaasti rasvaa sisältävän aterian yhteydessä suun kautta 80 mg febuksostaattia kerran vuorokaudessa useiden päivien ajan, febuksostaatin Cmax-arvo pieneni 49 % ja AUC-arvo 18 %. Kun potilaille annettiin runsaasti rasvaa sisältävän aterian yhteydessä 120 mg:n kerta-annos febuksostaattia, Cmax pieneni 38 % ja AUC-arvo 16 %. Seerumin virtsahappopitoisuuden prosentuaalisessa pienenemisessä ei kuitenkaan todettu kliinisesti merkitseviä muutoksia (kun potilaat saivat 80 mg:n annoksia useiden päivien ajan). Febuksostaattitabletit voi siis ottaa aterioista riippumatta.
Jakautuminen
Vakaassa tilassa febuksostaatin näennäinen jakautumistilavuus (Vss/F) on 10–300 mg:n suuruisten oraalisten annosten jälkeen 29–75 l. Febuksostaatti sitoutuu plasman proteiineihin (pääasiassa albumiiniin) noin 99,2-prosenttisesti. Proteiineihin sitoutumisaste on vakio pitoisuusalueella, joka saavutetaan 80–120 mg:n annoksilla. Aktiivisten metaboliittien sitoutumisaste plasman proteiineihin on noin 82–91 %.
Biotransformaatio
Febuksostaatti metaboloituu suuressa määrin UDP-glukuronyylitransferaasivälitteisen konjugaation ja sytokromi (CYP) P450 -välitteisen oksidaation kautta. Febuksostaatilla on todettu neljä farmakologisesti aktiivista hydroksyylimetaboliittia, joista kolmea löytyy ihmisen plasmasta. In vitro -tutkimukset ihmisen maksan mikrosomeilla osoittivat, että näitä oksidatiivisia metaboliitteja muodostui pääasiassa CYP1A1-, CYP1A2-, CYP2C8- ja CYP2C9-entsyymien vaikutuksesta ja febuksostaattiglukuronidia pääasiassa UGT 1A1-, 1A8- ja 1A9-entsyymien vaikutuksesta.
Eliminaatio
Febuksostaatti eliminoituu sekä maksa- että munuaisteitse. 80 mg:n suuruisen oraalisen 14C-merkityn febuksostaattiannoksen jälkeen noin 49 % annoksesta erittyi virtsaan muuttumattomana febuksostaattina (3 %), vaikuttavan aineen asyyliglukuronidina (30 %), tunnettuina oksidatiivisina metaboliitteina ja niiden konjugaatteina (13 %) sekä muina tuntemattomina metaboliitteina (3 %). Virtsaan erittymisen lisäksi noin 45 % annoksesta erittyi ulosteeseen muuttumattomana febuksostaattina (12 %), vaikuttavan aineen asyyliglukuronidina (1 %), tunnettuina oksidatiivisina metaboliitteina ja niiden konjugaatteina (25 %) sekä muina tuntemattomina metaboliitteina (7 %).
Munuaisten vajaatoiminta
Kun lievää, keskivaikeaa tai vaikeaa munuaisten vajaatoimintaa sairastaville potilaille annettiin useita 80 mg:n suuruisia febuksostaattiannoksia, ei febuksostaatin Cmax-arvossa todettu merkitseviä eroja sellaisiin tutkimushenkilöihin nähden, joiden munuaiset toimivat normaalisti. Febuksostaatin keskimääräinen kokonais-AUC suureni noin 1,8-kertaiseksi (7,5 μg⋅h/ml normaalin munuaistoiminnan yhteydessä ja 13,2 μg⋅h/ml vaikean munuaisten vajaatoiminnan yhteydessä). Aktiivisten metaboliittien Cmax suureni enintään 2-kertaiseksi ja AUC enintään 4-kertaiseksi. Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden annostusta ei kuitenkaan tarvitse muuttaa.
Maksan vajaatoiminta
Kun lievää (Child–Pugh-luokka A) tai keskivaikeaa (Child–Pugh-luokka B) maksan vajaatoimintaa sairastaville potilaille annettiin useita 80 mg:n suuruisia febuksostaattiannoksia, ei febuksostaatin tai sen metaboliittien Cmax- ja AUC-arvoissa todettu merkitseviä eroja sellaisiin tutkimushenkilöihin nähden, joiden maksa toimi normaalisti. Vaikeaa maksan vajaatoimintaa (Child–Pugh-luokka C) sairastavilla potilailla ei ole tehty tutkimuksia.
Ikä
Kun iäkkäille tutkimushenkilöille annettiin suun kautta useita febuksostaattiannoksia, ei febuksostaatin tai sen metaboliittien AUC-arvoissa todettu merkitseviä eroja nuorempiin terveisiin tutkimushenkilöihin nähden.
Sukupuoli
Useiden oraalisten febuksostaattiannosten jälkeen febuksostaatin Cmax-arvo oli naisilla 24 % suurempi kuin miehillä. AUC-arvo oli naisilla 12 % suurempi kuin miehillä. Painoon suhteutetuissa Cmax- ja AUC-arvoissa ei kuitenkaan ollut sukupuolten välisiä eroja. Annosta ei tarvitse muuttaa sukupuolen perusteella.
Prekliiniset tiedot turvallisuudesta
Haittoja on koe-eläimissä todettu yleensä, kun on käytetty altistusta, joka ylittää suurimman ihmisille käytettävän annostuksen.
Rottia koskevien tietojen farmakokineettinen mallinnus ja simulaatio viittaavat siihen, että merkaptopuriinin/atsatiopriinin kliininen annos pitää pienentää enintään 20 %:iin aiemmin määrätystä annoksesta, jos niitä käytetään samanaikaisesti febuksostaatin kanssa, jotta voidaan välttää mahdolliset hematologiset vaikutukset (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Karsinogeenisuus, mutageenisuus, hedelmällisyyden heikkeneminen
Virtsarakon kasvainten (transitiosellulaaristen papilloomien ja karsinoomien) ilmaantuvuuden lisääntymistä todettiin vain suurinta annosta (noin 11-kertainen ihmisten altistukseen verrattuna) saavilla urosrotilla, joilla oli myös ksantiinikiviä. Uros- tai naaraspuolisilla hiirillä tai rotilla ei todettu minkään muun kasvaintyypin ilmaantuvuuden lisääntymistä. Näiden löydösten katsotaan johtuvan puriinien metabolian ja virtsan koostumuksen lajikohtaisista ominaispiirteistä eikä niillä ole kliinistä merkitystä.
Febuksostaatilla tehdyt tavanomaiset geenitoksisuustestisarjat eivät paljastaneet mitään biologisesti oleellisia geenitoksisia vaikutuksia.
Suun kautta annettu febuksostaatti (enimmäisannos 48 mg/kg/vrk) ei vaikuttanut uros- eikä naarasrottien hedelmällisyyteen eikä lisääntymistoimintoihin.
Febuksostaatin ei ole havaittu heikentävän hedelmällisyyttä. Sillä ei myöskään ole todettu teratogeenisiä eikä sikiöön kohdistuvia haitallisia vaikutuksia. Suuret annokset (noin 4,3-kertaiset ihmisten altistukseen verrattuna) olivat toksisia rottaemoille, pienensivät vieroitusindeksiä ja häiritsivät poikasten kehitystä. Tiineillä rotilla (annokset noin 4,3-kertaisia ihmisten altistukseen verrattuna) ja tiineillä kaniineilla (annokset noin 13-kertaisia ihmisten altistukseen verrattuna) tehdyt teratologiset tutkimukset eivät paljastaneet mitään teratogeenisiä vaikutuksia.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Laktoosimonohydraatti
Selluloosa, mikrokiteinen
Hydroksipropyyliselluloosa
Kroskarmelloosinatrium
Piidioksidi, kolloidinen, hydratoitu
Magnesiumstearaatti
Tabletin päällyste
Poly(vinyylialkoholi)
Makrogoli 3350
Titaanidioksidi (E 171)
Talkki
Keltainen rautaoksidi (E 172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
FEBUXOSTAT KRKA tabletti, kalvopäällysteinen
120 mg (L:kyllä) 28 fol (21,36 €)
PF-selosteen tieto
Läpipainopakkaus (PVC/PVDC/PVC//Al): 14, 28, 56 tai 84 kalvopäällysteistä tablettia kotelossa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Ruskeankeltainen, lievästi kaksoiskupera kapselin muotoinen kalvopäällysteinen tabletti, jossa on molemmilla puolilla jakouurre. Tabletin mitat: noin 19 mm x 8 mm. Jakouurre on tarkoitettu vain nielemisen helpottamiseksi eikä jakamiseksi yhtä suuriin annoksiin.
Käyttö- ja käsittelyohjeet
Ei erityisvaatimuksia hävittämisen suhteen.
Korvattavuus
FEBUXOSTAT KRKA tabletti, kalvopäällysteinen
120 mg 28 fol
- Alempi erityiskorvaus (65 %). Febuksostaatti: Kihdin hoito erityisin edellytyksin (288).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Febuksostaatti: Vaikeaa kroonista hyperurikemiaa sairastavien potilaiden hoito erityisin edellytyksin (349).
ATC-koodi
M04AA03
Valmisteyhteenvedon muuttamispäivämäärä
26.03.2025
Yhteystiedot
Tekniikantie 14
02150 Espoo
Suomi
020-7545330
www.krka.biz
info.fi@krka.biz