

CIMZIA säiliö annostelulaitteeseen (ava), 200 mg injektioneste, liuos
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Potilas
Vaikuttavat aineet ja niiden määrät
Yksi säiliö annostelulaitteeseen sisältää 200 mg sertolitsumabipegolia yhdessä ml:ssa.
Sertolitsumabipegoli on rekombinantti, humanisoitu Fab’-vasta-ainefragmentti tuumorinekroositekijä alfaa (TNF‑α) vastaan. Fab’-fragmentti tuotetaan Escherichia coli ‑bakteerissa ja konjugoidaan polyetyleeniglykoliin (PEG).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste).
Kliiniset tiedot
Käyttöaiheet
Nivelreuma
Cimzia yhdessä metotreksaatin kanssa on tarkoitettu
- aikuispotilaiden keskivaikean tai vaikean aktiivisen nivelreuman hoitoon, kun vaste taudinkulkua muuttaviin reumalääkkeisiin (disease-modifying antirheumatic drugs, DMARD), kuten metotreksaattiin, ei ole ollut riittävä. Cimzia voidaan antaa monoterapiana, jos potilas ei siedä metotreksaattia tai jos metotreksaattihoidon jatkaminen ei sovi potilaalle.
- aikuisten vaikean, aktiivisen ja etenevän nivelreuman hoitoon, silloin kun potilas ei ole saanut aiempaa hoitoa metotreksaatilla tai muilla taudinkulkua muuttavilla reumalääkkeillä.
Cimzian, kun sitä on annettu yhdistelmänä metotreksaatin kanssa, on osoitettu hidastavan nivelvaurioiden radiografista etenemistä ja parantavan fyysistä toimintakykyä.
Aksiaalinen spondylartriitti
Cimzia on tarkoitettu aikuispotilaiden vaikeiden, aktiivisten aksiaalisten spondylartriittien hoitoon, ja näitä ovat:
Selkärankareuma (eli röntgenpositiivinen aksiaalinen spondylartriitti)
Cimzia on tarkoitettu aikuispotilaiden vaikean, aktiivisen selkärankareuman hoitoon, kun tulehduskipulääkkeillä (NSAIDit) ei ole saatu riittävää hoitovastetta tai kun potilas ei siedä niitä.
Aksiaalinen spondylartriitti ilman radiografista näyttöä selkärankareumasta (eli röntgennegatiivinen aksiaalinen spondylartriitti)
Cimzia on tarkoitettu niiden aikuispotilaiden vaikean, aktiivisen röntgennegatiivisen aksiaalisen spondylartriitin hoitoon, joilla on objektiivisia tulehduksen merkkejä magneettikuvissa (MRI) ja/tai suurentunut C-reaktiivisen proteiinin (CRP) arvo, silloin kun tulehduskipulääkkeillä (NSAIDit) ei ole saatu riittävää hoitovastetta tai kun potilas ei siedä niitä.
Nivelpsoriaasi
Cimzia yhdessä metotreksaatin kanssa on tarkoitettu aikuispotilaiden aktiivisen nivelpsoriaasin hoitoon, kun vaste aiemmin käytettyyn taudinkulkua muuttavaan reumalääkehoitoon (DMARD) ei ole ollut riittävä.
Cimzia voidaan antaa monoterapiana, jos potilas ei siedä metotreksaattia tai jos metotreksaattihoidon jatkaminen ei sovi potilaalle.
Läiskäpsoriaasi
Cimzia on tarkoitettu keskivaikeaan tai vaikeaan läiskäpsoriaasiin aikuisille, joille voidaan antaa systeemistä hoitoa.
Tarkemmat tiedot hoitovaikutuksista, ks. kohta Farmakodynamiikka.
Ehto
Hoito on aloitettava ja toteutettava käyttöaiheissa mainittujen sairauksien diagnosointiin ja hoitoon perehtyneen erikoislääkärin seurannassa.
Annostus ja antotapa
Hoito on aloitettava ja toteutettava Cimzian käyttöaiheissa mainittujen sairauksien diagnosointiin ja hoitoon perehtyneen erikoislääkärin seurannassa. Potilaille on annettava erityinen muistutuskortti.
Annostus
Nivelreuma, nivelpsoriaasi, aksiaalinen spondylartriitti, läiskäpsoriaasi
Aloitusannos
Cimzia-hoidon suositeltu aloitusannos aikuispotilaille on 400 mg (annetaan kahtena 200 mg:n injektiona ihon alle) viikoilla 0, 2 ja 4. Nivelreumassa ja nivelpsoriaasissa metotreksaattihoitoa jatketaan tarvittaessa Cimzia-hoidon aikana.
Ylläpitoannos
Nivelreuma
Aloitusannoksen jälkeen Cimzia-hoidon suositeltu ylläpitoannos nivelreumaa sairastaville aikuisille on 200 mg kahden viikon välein. Kliinisen vasteen vahvistuttua voidaan harkita vaihtoehtoista ylläpitoannostusta 400 mg neljän viikon välein. Metotreksaattihoitoa jatketaan tarvittaessa Cimzia-hoidon aikana.
Aksiaalinen spondylartriitti
Aloitusannoksen jälkeen Cimzia-hoidon suositeltu ylläpitoannos aksiaalista spondylartriittia sairastaville aikuisille on 200 mg kahden viikon välein tai 400 mg neljän viikon välein. Kun Cimzia-hoito on jatkunut vähintään vuoden, voidaan harkita ylläpitoannoksen pienentämistä tasolle 200 mg neljän viikon välein, mikäli potilas on saavuttanut pitkäkestoisen remission (ks. kohta Farmakodynamiikka).
Nivelpsoriaasi
Aloitusannoksen jälkeen Cimzia-hoidon suositeltu ylläpitoannos nivelpsoriaasia sairastaville aikuisille on 200 mg kahden viikon välein. Kliinisen vasteen vahvistuttua voidaan harkita vaihtoehtoista ylläpitoannostusta 400 mg neljän viikon välein. Metotreksaattihoitoa jatketaan tarvittaessa Cimzia-hoidon aikana.
Käytettävissä olevat tiedot viittaavat siihen, että kliininen vaste edellä mainituissa käyttöaiheissa saavutetaan tavallisesti 12 hoitoviikon kuluessa. Jos potilaalla ei todeta hyötyä hoidosta ensimmäisten 12 hoitoviikon aikana, hoidon jatkamista on harkittava tarkoin uudelleen.
Läiskäpsoriaasi
Aloitusannoksen jälkeen Cimzia-annoksen ylläpitoannos läiskäpsoriaasia sairastaville aikuisille on 200 mg kahden viikon välein. Vaihtoehtoisesti voidaan antaa 400 mg kahden viikon välein, jos potilaalla on riittämätön vaste (ks. kohta Farmakodynamiikka).
Läiskäpsoriaasia sairastavista potilaista käytettävissä olevat tiedot viittaavat siihen, että kliininen hoitovaste saavutetaan tavallisesti 16 hoitoviikon kuluessa. Jos potilaalla ei todeta hyötyä hoidosta ensimmäisten 16 hoitoviikon aikana, hoidon jatkamista on harkittava tarkoin uudelleen. Joidenkin hoidon alussa osittaista hoitovastetta osoittaneiden potilaiden hoitovaste voi myöhemmin parantua, jos hoito jatkuu yli 16 viikon ajan.
Ottamatta jäänyt annos
Jos potilaalta jää annos ottamatta, häntä on neuvottava pistämään seuraava Cimzia-annos heti, kun hän huomaa unohtaneensa sen ja jatkamaan sen jälkeen annosten pistämistä annetun ohjeen mukaisesti.
Erityisryhmät
Pediatriset potilaat (< 18-vuotiaat)
Cimzian turvallisuutta ja tehoa lasten ja alle 18‑vuotiaiden nuorten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Iäkkäät potilaat (≥ 65‑vuotiaat)
Annosta ei tarvitse muuttaa. Populaatiofarmakokineettiset analyysit eivät viitanneet siihen, että iällä olisi vaikutusta (ks. kohta Farmakokinetiikka).
Munuaisten ja maksan vajaatoiminta
Cimzia-injektionestettä ei ole tutkittu näillä potilasryhmillä. Annostussuosituksia ei voida antaa (ks. kohta Farmakokinetiikka).
Antotapa
Annostelulaitteen säiliön kokonaissisällön (1 ml) saa antaa vain ihonalaisena injektiona sähkömekaanisella ava-injektiolaitteella. Sopivia pistoskohtia ovat reisi tai vatsa.
Cimzia-injektioneste, liuos, säiliö annostelulaitteeseen on tarkoitettu kertakäyttöön sähkömekaanisella injektiolaitteella, jonka nimi on ava. Kun potilas on saanut riittävän opastuksen injektiotekniikkaan, hän voi pistää pistoksen itse käyttämällä sähkömekaanista ava-injektiolaitetta ja kertakäyttöistä säiliötä annostelulaitteeseen, jos lääkäri arvioi sen asianmukaiseksi ja potilas on tarvittavassa lääkärin seurannassa. Lääkärin pitää keskustella potilaan kanssa siitä, mikä vaihtoehtoisista injektiolaitteista on sopivin.
Alustava Ava-injektiolaitteen ensimmäinen versio ei tue kahden viikon välein annettavaa 400 mg:n ylläpitoannosta (läiskäpsoriaasi) tai pienempää 200 mg:n ylläpitoannosta neljän viikon välein (aksiaalinen spondylartriitti). Jos potilasta hoidetaan näillä ylläpitoannoksilla, lääkäriä neuvotaan käyttämään Ava Connect -versiota ava-injektiolaitteen ava Connect -versiota tai muita menetelmiä.
Lääkkeen annostelussa on noudatettava pakkausselosteen lopussa ja sähkömekaanisen ava-injektiolaitteen ohjekirjassa olevia käyttöohjeita.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Aktiivisessa vaiheessa oleva tuberkuloosi tai muu vaikea infektio, kuten sepsis tai opportunisti-infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Keskivaikea tai vaikea sydämen vajaatoiminta (NYHA-luokat III tai IV) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Infektiot
Potilaiden tilaa on seurattava tarkoin infektioiden, kuten tuberkuloosin, oireiden ja löydösten havaitsemiseksi ennen Cimzia-hoitoa, sen aikana ja jälkeen. Koska sertolitsumabipegolin eliminaatio voi kestää jopa 5 kuukautta, seurantaa on jatkettava koko tämän ajan (ks. kohta Vasta-aiheet).
Cimzia-hoitoa ei saa aloittaa potilaille, joilla on kliinisesti merkittävä aktiivisessa vaiheessa oleva infektio, krooninen tai paikallinen infektio mukaan lukien, ennen kuin infektio on saatu hallintaan (ks. kohta Vasta-aiheet).
Jos potilaalle kehittyy uusi infektio Cimzia-hoidon aikana, hänen tilaansa on seurattava tarkoin. Jos potilaalle kehittyy uusi vakava infektio, Cimzian anto on lopetettava siihen saakka, kunnes infektio on saatu hallintaan. Lääkärin on oltava varovainen harkitessaan Cimzia-hoitoa potilaalle, jolla on aiemmin ollut toistuvia tai opportunistisia infektioita tai taustalla oleva sairaus, joka saattaa altistaa hänet infektioille, immunosuppressiivisten lääkkeiden samanaikainen käyttö mukaan lukien.
Nivelreumaa sairastavilla potilailla ei välttämättä esiinny heidän sairautensa ja samanaikaisten lääkitysten vuoksi infektion tyypillisiä oireita, kuten kuumetta. Infektion, etenkin vakavan infektion epätyypillisten kliinisten ilmenemismuotojen, varhainen havaitseminen on siksi erittäin tärkeää, jotta diagnoosin ja hoidon aloittamisen viive voidaan minimoida.
Cimzia-hoitoa saaneilla potilailla on raportoitu vakavia infektioita, kuten sepsistä ja tuberkuloosia (miliaarituberkuloosi, hajapesäkkeinen tuberkuloosi ja ekstrapulmonaalinen tuberkuloosi mukaan lukien) sekä opportunisti-infektioita (esim. histoplasmoosia, nokardiaa, kandidiaasia). Osa näistä tapauksista on johtanut kuolemaan.
Tuberkuloosi
Kaikki potilaat on tutkittava aktiivisen ja inaktiivisen (piilevän) tuberkuloosi-infektion varalta ennen Cimzia-hoidon aloittamista. Tutkimuksessa on selvitettävä tarkoin potilaan aiemmat terveystiedot, kuten potilaan aiemmin sairastama tuberkuloosi tai aiempi mahdollinen kontakti aktiivista tuberkuloosia sairastavan kanssa ja potilaan aiemmin ja/tai parhaillaan saama immunosuppressiivinen hoito. Kaikille potilaille on tehtävä asianmukaiset seulontatestit, esim. ihoon tehtävä tuberkuliinikoe ja keuhkokuvaus (mahdollisesti paikallisten suositusten mukaisesti). Näiden testien tekeminen suositellaan kirjaamaan muistutuskorttiin. Lääkettä määrääviä muistutetaan väärän negatiivisen tuberkuliinikokeen vaarasta, etenkin vaikeasti sairailla tai immuunipuutteisilla potilailla.
Jos potilaalla todetaan aktiivinen tuberkuloosi ennen Cimzia-hoitoa tai sen aikana, hoitoa ei saa aloittaa, tai se on keskeytettävä (ks. kohta Vasta-aiheet).
Jos potilaalla epäillään piilevää tuberkuloosia, on syytä konsultoida tuberkuloosin hoitoon perehtynyttä lääkäriä. Kaikissa alla mainituissa tilanteissa Cimzia-hoidon hyöty‒riski-suhde on arvioitava hyvin tarkoin.
Jos todetaan piilevä tuberkuloosi, asianmukainen tuberkuloosihoito on aloitettava paikallisten suositusten mukaisesti ennen Cimzia-hoidon aloittamista.
Tuberkuloosilääkitystä on harkittava ennen Cimzia-hoidon aloittamista myös silloin, jos potilaalla on aiemmin ollut piilevä tai aktiivinen tuberkuloosi eikä riittävää hoitoa voida varmistaa, sekä silloin, jos potilaalla on merkittäviä tuberkuloosin riskitekijöitä huolimatta negatiivisesta testituloksesta piilevän tuberkuloosin suhteen. Biologisten kokeiden tekemistä tuberkuloosin seulomiseksi on harkittava ennen Cimzia-hoidon aloittamista, jos piilevä tuberkuloosi on mahdollinen BCG‑rokotuksesta huolimatta.
TNF:n estäjällä, myös Cimzialla, hoidetuilla potilailla on ilmennyt aktiivista tuberkuloosia aiemmin tai samanaikaisesti annetusta tuberkuloosin estohoidosta huolimatta. Joillekin potilaille, joiden aktiivinen tuberkuloosi on hoidettu tuloksellisesti, on Cimzia-hoidon aikana kehittynyt tuberkuloosi uudestaan.
Potilaita on kehotettava kääntymään lääkärin puoleen, jos heille ilmaantuu Cimzia-hoidon aikana tai sen jälkeen tuberkuloosiin viittaavia löydöksiä/oireita (esim. pitkittyvää yskää, kuihtumista/painon laskua, lievää kuumeilua, haluttomuutta).
Hepatiitti B -viruksen (HBV) reaktivaatio
Hepatiitti B:n reaktivaatiota on ilmennyt potilailla, jotka ovat saaneet TNF:n estäjää, mukaan lukien sertolitsumabipegolia, ja jotka ovat tämän viruksen kroonisia kantajia (ts. pinta-antigeenipositiivisia). Jotkut tapauksista ovat johtaneet kuolemaan.
Potilailta pitää testata HBV‑infektio ennen Cimzia-hoidon aloittamista. Jos HBV‑infektiotestin tulos on positiivinen, on suositeltavaa konsultoida hepatiitti B:n hoitoon erikoistunutta lääkäriä.
HBV:n kantajia, joille Cimzia-hoito on tarpeen, pitää tarkkailla huolellisesti aktiivisen HBV‑infektion merkkien ja oireiden varalta koko hoidon ajan sekä useita kuukausia hoidon lopettamisen jälkeen. Ei ole olemassa riittäviä tietoja HBV:tä kantavien potilaiden HBV‑reaktivaation estosta käytettäessä viruslääkkeitä samanaikaisesti TNF:n estäjähoidon kanssa. Jos potilaalle kehittyy HBV‑infektion reaktivaatio, Cimzia-hoito lopetetaan ja potilaalle aloitetaan tehokas antiviraalinen lääkitys sekä asianmukainen supportiivinen hoito.
Pahanlaatuiset kasvaimet ja lymfoproliferatiiviset sairaudet
TNF:n estäjien mahdollista osuutta pahanlaatuisten kasvainten kehittymiseen ei tiedetä. Varovaisuutta on noudatettava, kun harkitaan hoitoa TNF:n estäjillä potilaille, joilla on aiemmin ollut pahanlaatuisia kasvaimia, tai kun harkitaan hoidon jatkamista, kun potilaalle on kehittynyt pahanlaatuinen kasvain.
TNF:n estäjiä saaneilla potilailla lymfoomien, leukemian tai muiden pahanlaatuisten kasvainten mahdollista riskiä ei tämänhetkisen tietämyksen perusteella voida sulkea pois.
Cimzialla ja muilla TNF:n estäjillä tehdyissä kliinisissä lääketutkimuksissa lymfoomaa ja muita pahanlaatuisia kasvaimia on havaittu useammin TNF:n estäjiä saaneilla potilailla kuin lumelääkettä saaneilla verrokeilla (ks. kohta Haittavaikutukset). Potilailla, jotka ovat saaneet TNF:n estäjiä niiden markkinoille tulon jälkeen, on raportoitu leukemiatapauksia. Pitkään hyvin aktiivista tulehdussairautta sairastaneilla nivelreumapotilailla on suurentunut lymfooma- ja leukemiariski, mikä vaikeuttaa riskin arviointia.
Tutkimuksia ei ole tehty potilailla, joilla on ollut aiemmin pahanlaatuisia kasvaimia, tai potilailla, joiden hoitoa jatketaan Cimzia-lääkityksen aikana ilmenneestä pahanlaatuisesta kasvaimesta huolimatta.
Ihosyövät
Melanoomaa ja merkelinsolukarsinoomaa on raportoitu TNF:n estäjillä hoidetuilla potilailla, sertolitsumabipegoli mukaan luettuna (ks. kohta Haittavaikutukset). Säännöllisin väliajoin toistuvaa ihon tutkimista suositellaan etenkin potilailta, joilla on ihosyövän riskitekijöitä.
Pahanlaatuiset kasvaimet lapsipotilailla
Pahanlaatuisia kasvaimia on raportoitu lapsilla, nuorilla ja nuorilla aikuisilla (≤ 22‑vuotiailla), jotka ovat saaneet TNF:n estäjiä (hoidon alkaessa ikä ollut ≤ 18 vuotta) näiden markkinoille tulon jälkeen. Osa näistä tapauksista on johtanut potilaan kuolemaan. Noin puolet raportoiduista pahanlaatuisista kasvaimista oli lymfoomia. Muissa tapauksissa esiintyi monia erilaisia pahanlaatuisia kasvaimia, myös harvinaisia pahanlaatuisia kasvaimia, jotka liittyvät tavallisesti immuunisupressioon. TNF:n estäjiä saaneilla lapsilla ja nuorilla pahanlaatuisten kasvainten riskiä ei voida sulkea pois.
TNF:n estäjillä hoidetuilla potilailla on markkinoilletulon jälkeen raportoitu hepatospleenistä T‑solulymfoomaa (HSTCL). Tämä harvinainen T-solulymfooma on taudinkuvaltaan hyvin aggressiivinen ja tavallisesti fataali. Suurin osa TNF:n estäjillä raportoiduista tapauksista ilmeni Crohnin tautia tai haavaista paksusuolitulehdusta sairastavilla nuorilla tai nuorilla aikuisilla miehillä. Lähes kaikkia näistä potilaista oli hoidettu immunosuppressanteilla ‒ atsatiopriinilla ja/tai 6‑merkaptopuriinilla ‒ samanaikaisesti TNF:n estäjän kanssa joko diagnoosihetkellä tai sitä ennen. Hepatospleenisen T‑solulymfooman kehittymisen riskiä ei voida poissulkea potilailla, joita hoidetaan Cimzialla.
Krooninen keuhkoahtaumatauti (COPD)
Toisen TNF:n estäjän, infliksimabin, käyttöä arvioivassa eksploratiivisessa kliinisessä tutkimuksessa keskivaikeaa tai vaikeaa kroonista keuhkoahtaumatautia (COPD) sairastavilla potilailla raportoitiin pahanlaatuisia kasvaimia, lähinnä keuhkojen tai pään ja kaulan kasvaimia, useammin infliksimabihoitoa saaneilla potilailla kuin verrokeilla. Kaikki potilaat olivat aiemmin tupakoineet runsaasti. TNF:n estäjien käytössä on siksi oltava varovainen hoidettaessa kroonista keuhkoahtaumatautia sairastavia potilaita sekä potilaita, joilla pahanlaatuisten kasvainten riski on suurentunut runsaan tupakoinnin vuoksi.
Kongestiivinen sydämen vajaatoiminta
Cimzia on vasta-aiheinen keskivaikeaa tai vaikeaa sydämen vajaatoimintaa sairastaville (ks. kohta Vasta-aiheet). Toisella TNF:n estäjällä tehdyssä kliinisessä lääketutkimuksessa havaittiin kongestiivisen sydämen vajaatoiminnan pahenemista ja kongestiivisesta sydämen vajaatoiminnasta johtuvan kuolleisuuden lisääntymistä. Kongestiivista sydämen vajaatoimintaa on ilmoitettu myös Cimzia-hoitoa saaneilla nivelreumaa sairastavilla potilailla. Cimzia-valmistetta on annettava varoen lievää sydämen vajaatoimintaa (NYHA-luokka I tai II) sairastaville potilaille. Jos potilaalle kehittyy uusia kongestiivisen sydämen vajaatoiminnan oireita tai aiemmat oireet pahenevat, Cimzia-hoito on lopetettava.
Hematologiset reaktiot
TNF:n estäjien käytön yhteydessä on ilmoitettu harvinaisina tapauksina pansytopeniaa, mukaan lukien aplastista anemiaa. Vereen kohdistuvia haittavaikutuksia, kuten potilaan hoidon kannalta merkittäviä sytopenioita (esim. leukopeniaa, pansytopeniaa, trombosytopeniaa) on ilmoitettu Cimzia-hoidon yhteydessä (ks. kohta Haittavaikutukset). Kaikkia potilaita on neuvottava hakeutumaan heti lääkärin hoitoon, jos heille kehittyy veren dyskrasiaan tai infektioon viittaavia löydöksiä tai oireita (esim. pitkittynyttä kuumetta, mustelmia, verenvuotoa, kalpeutta) Cimzia-hoidon aikana. Potilaan Cimzia-hoidon keskeyttämistä on harkittava, jos veriarvojen merkittävät poikkeavuudet varmistuvat.
Neurologiset vaikutukset
TNF:n estäjien käyttöön on liittynyt harvinaisina tapauksina uusien kliinisten oireiden ilmaantumista tai aiempien kliinisten oireiden pahenemista ja/tai kuvantamalla todettu demyelinaatiosairaus, multippeliskleroosi mukaan lukien. Jos potilaalla on jo ennestään olemassa oleva tai vasta äskettäin ilmennyt demyelinaatiosairaus, TNF:n estäjähoidon hyötyjä ja riskejä on harkittava tarkoin ennen Cimzia-hoidon aloittamista. Cimzia-hoitoa saaneilla on ilmoitettu harvinaisina tapauksina neurologisia häiriöitä, kuten kouristuskohtauksia, hermotulehduksia ja perifeeristä neuropatiaa.
Yliherkkyys
Cimzia-valmisteen antamisen jälkeen harvinaisina haittavaikutuksina on raportoitu vaikeita yliherkkyysreaktioita. Osa näistä reaktioista ilmaantui ensimmäisen Cimzia-injektion jälkeen. Jos vaikeita reaktioita ilmenee, Cimzia-valmisteen antaminen on lopetettava heti ja aloitettava asianmukainen hoito.
Cimzian käytöstä potilaille, joilla on esiintynyt vaikeita yliherkkyysreaktioita muille TNF:n estäjille, on vähän tietoja, joten tällaisten potilaiden hoidossa on oltava varovainen.
Herkkyys lateksille
Cimzia-annostelulaitteen säiliön irrotettavan korkin sisällä oleva neulan suojus sisältää luonnonkumin (lateksin) johdannaista (ks. kohta Pakkaukset ja valmisteen kuvaus). Luonnonkumin kanssa kosketuksiin joutuminen voi aiheuttaa vaikeita allergisia reaktioita lateksille herkistyneille henkilöille. Cimzia-annostelulaitteen irrotettavassa korkissa ei ole tähän mennessä havaittu antigeenistä lateksiproteiinia. Yliherkkyysreaktioiden mahdollista riskiä ei kuitenkaan voida täysin sulkea pois lateksille herkistyneillä henkilöillä.
Immunosuppressio
Koska tuumorinekroositekijä (TNF) välittää tulehdusreaktiota ja muuntaa solujen immuunivastetta, TNF:n estäjiin, myös Cimzia-hoitoon, liittyy mahdollisesti immunosuppressiota, joka vaikuttaa elimistön vastustuskykyyn infektioita ja pahanlaatuisia kasvaimia vastaan.
Autoimmuniteetti
Cimzia-hoito voi johtaa tumavasta-aineiden muodostumiseen ja melko harvinaisissa tilanteissa SLE‑tyyppisen oireyhtymän kehittymiseen (ks. kohta Haittavaikutukset). Pitkäaikaisen Cimzia-hoidon vaikutusta autoimmuunisairauksien kehittymiseen ei tiedetä. Jos potilaalle kehittyy SLE‑tyyppiseen oireyhtymään viittaavia oireita Cimzia-hoidon jälkeen, hoito on lopetettava. Cimzia-hoitoa ei ole tutkittu erityisesti SLE‑potilasryhmässä (ks. kohta Haittavaikutukset).
Rokotukset
Cimzia-hoitoa saaville potilaille voidaan antaa rokotuksia, ei kuitenkaan eläviä taudinaiheuttajia sisältäviä rokotteita. Cimzia-hoitoa saaneiden potilaiden vasteesta eläviä taudinaiheuttajia sisältäville rokotteille tai infektion toissijaisesta tarttumisesta elävästä rokotteesta ei ole tietoa. Cimzia-hoidon aikana ei saa antaa samanaikaisesti eläviä taudinaiheuttajia sisältäviä rokotteita.
Lumekontrolloidussa kliinisessä tutkimuksessa reumapotilailla todettiin samanlainen vasta-ainevaste Cimzia- ja lumeryhmän välillä, kun pneumokokkipolysakkaridirokotetta ja influenssarokotetta annettiin samanaikaisesti Cimzian kanssa. Kuitenkin Cimziaa ja metotreksaattia saavilla potilailla oli matalampi humoraalinen vaste verrattuna pelkästään Cimziaa saaviin potilaihin. Tämän kliinistä merkitystä ei tunneta.
Muiden biologisten valmisteiden samanaikainen käyttö
Kliinisissä lääketutkimuksissa havaittiin vaikeita infektioita ja neutropeniaa anakinran (interleukiini 1 ‑reseptoriantagonisti) tai abataseptin (CD28‑modulaattori) ja toisen TNF:n estäjän, etanerseptin, samanaikaisen käytön yhteydessä, eikä hoidosta havaittu olevan lisähyötyä pelkällä TNF:n estäjällä annettuun hoitoon nähden. Muun TNF:n estäjän ja joko abataseptin tai anakinran yhdistelmäkäytön yhteydessä havaittujen haittavaikutusten luonteen vuoksi myös anakinran tai abataseptin ja muiden TNF:n estäjien käytöstä yhdistelmänä voi aiheutua samankaltaisia haittoja. Tämän vuoksi sertolitsumabipegolin käyttöä yhdistelmänä anakinran tai abataseptin kanssa ei suositella (ks. kohta Yhteisvaikutukset).
Leikkaukset
Kirurgisten toimenpiteiden turvallisuudesta Cimzia-hoitoa saaville potilaille on vähän kokemusta. Jos potilaalle suunnitellaan kirurgista toimenpidettä, sertolitsumabipegolin 14 vuorokauden pituinen puoliintumisaika on otettava huomioon. Jos potilaalle on tehtävä leikkaus Cimzia-hoidon aikana, infektioiden ilmaantumista on seurattava tarkoin ja ryhdyttävä asianmukaisiin toimenpiteisiin.
Aktivoidun partiaalisen tromboplastiiniajan (aPTT) määritys
Cimzia-hoitoa saaneilla potilailla on havaittu häiriöitä tiettyjen veren hyytymistä mittaavien kokeiden yhteydessä. Cimzia saattaa aiheuttaa virheellisiä suurentuneita aPTT‑tuloksia potilailla, joilla ei ole hyytymishäiriöitä. Tällaisia vaikutuksia on havaittu seuraavien kokeiden yhteydessä: Diagnostica Stagon testit PTT‑Lupus Anticoagulant (LA) test ja Standard Target Activated Partial Thromboplastin time (STA‑PTT) Automate tests sekä Instrumentation Laboratoriesin testit HemosIL APTT‑SP liquid ja HemosIL lyophilized silica test. Hoito saattaa vaikuttaa myös muihin aPTT‑määrityksiin. Ei ole näyttöä siitä, että Cimzia-hoito vaikuttaisi veren hyytymiseen in vivo. Kun potilas on saanut Cimzia-hoitoa, veren hyytymiskokeiden poikkeavien tulosten tulkintaan on kiinnitettävä erityistä huomiota. Trombiiniajan (TT) ja protrombiiniajan (PT) määrityksiin kohdistuvia häiriöitä ei ole havaittu.
Iäkkäät potilaat
Kliinisissä tutkimuksissa infektioiden esiintymistiheys näytti olevan suurempi ≥ 65‑vuotiailla kuin nuoremmilla, mutta kokemusta iän vaikutuksesta on vähän. Iäkkäiden potilaiden hoidossa on oltava varovainen, ja infektioiden ilmaantumiseen on kiinnitettävä erityistä huomiota.
Yhteisvaikutukset
Samanaikainen hoito metotreksaatin, kortikosteroidien, ei-steroidisten tulehduskipulääkkeiden (NSAIDit) ja kipulääkkeiden kanssa ei populaatiofarmakokineettisen analyysin perusteella näyttänyt vaikuttavan sertolitsumabipegolin farmakokineettisiin ominaisuuksiin.
Sertolitsumabipegolin ja anakinran tai abataseptin käyttöä yhdistelmänä ei suositella (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Cimzian ja metotreksaatin samanaikainen anto ei vaikuttanut merkittävästi metotreksaatin farmakokinetiikkaan. Tutkimusten välisessä vertailussa sertolitsumabipegolin farmakokinetiikka vaikutti olevan samankaltainen kuin terveillä tutkimushenkilöillä oli aiemmin havaittu.
Raskaus ja imetys
Hedelmällisessä iässä olevat naiset
Naisten, jotka voivat tulla raskaaksi, on huomioitava riittävän ehkäisyn käyttö. Jos nainen suunnittelee raskautta, on arvioitava, onko Cimzia-hoidon jatkaminen kliinisesti tarpeen. Jos Cimzia-valmisteen päätetään antaa poistua elimistöstä ennen hedelmöittymistä, ehkäisyä pitää jatkaa 5 kuukauden ajan viimeisen Cimzia-annoksen jälkeen (ks. kohta Farmakokinetiikka).
Raskaus
Ihmisiä koskevat tiedot
Laajat tiedot prospektiivisesti raportoiduista raskauksista (yli 1 500:sta Cimzia-valmisteelle ensimmäisen raskauskolmanneksen aikana altistuneesta raskaudesta), joiden lopputulos on tiedossa, eivät viittaa siihen, että valmisteella olisi epämuodostumia aiheuttavaa tai fetaalista/neonataalista toksisuutta. Jatkuva tiedonkeruu, mukaan lukien lääketurvaan liittyvien tapahtumien raportointi ja raskausrekisteri, on käynnissä.
Raskausrekisterissä (OTIS-tutkimus) vakavien syntymävikojen osuus elävänä syntyneillä vauvoilla oli 15/132 (11,4 %) naisilla, jotka olivat saaneet Cimzia-hoitoa ainakin ensimmäisen raskauskolmanneksen aikana, ja 8/126 (6,3 %) naisilla, joilla oli samoja käyttöaiheiden mukaisia sairauksia, mutta jotka eivät olleet saaneet Cimzia-hoitoa (suhteellinen riski 1,85; 95 %:n luottamusväli 0,74–4,60). Vastaavanlainen yhteys havaittiin, kun Cimzia-hoitoa saaneita naisia verrattiin naisiin, joilla ei ollut Cimzia-valmisteen hyväksyttyjen käyttöaiheiden mukaista sairautta (suhteellinen osuus 10/126 [7,9 %] ja suhteellinen riski 1,65; 95 %:n luottamusväli 0,75–3,64). Vakavien tai lievempien syntymävikojen suhteen ei tunnistettu mitään tiettyä piirrettä.
Cimzia-hoitoa saaneen ryhmän ja kummankaan vertailuryhmän välillä ei ollut selkeitä eroja spontaaneissa keskenmenoissa, vakavissa tai opportunistisissa infektioissa, sairaalahoidossa tai rokotteen haittavaikutuksissa lapsilla, joita seurattiin enintään viiden vuoden ajan. Kuolleena syntyneitä tai raskauden keskeytymisiä ei raportoitu lainkaan Cimzia-ryhmässä, kun taas ryhmässä, jossa tutkittavilla oli sairauksia, mutta he eivät olleet altistuneet Cimzia-valmisteelle, raportoitiin kaksi kuolleena syntynyttä ja kolme raskauden keskeytymistä. Tietojen tulkintaan voivat vaikuttaa tutkimusmenetelmään liittyvät rajoitukset, mukaan lukien pieni otoskoko ja ei‑satunnaistettu tutkimusasetelma.
Kliinisessä tutkimuksessa, joka tehtiin 21:llä Cimzia-valmistetta raskauden aikana saaneella naisella, plasman sertolitsumabipegolipitoisuudet olivat ei‑raskaana olevilla aikuispotilailla havaitulla pitoisuuksien vaihteluvälillä (ks. kohta Farmakokinetiikka).
Kliinisessä tutkimuksessa 16:tta raskaana olevaa naista hoidettiin sertolitsumabipegolilla (200 mg joka toinen viikko tai 400 mg joka neljäs viikko). Syntymän yhteydessä 14 imeväiseltä mitatut plasman sertolitsumabipegolipitoisuudet olivat 13 näytteessä alle määritysrajan (Below the Limit of Quantification, BLQ); yhdessä näytteessä pitoisuus oli 0,042 mikrog/ml. Lapsen ja äidin plasman sertolitsumabipegolipitoisuuksien suhdeluku oli syntymähetkellä 0,09 %.Viikolla 4 ja viikolla 8 kaikkien imeväisten pitoisuudet olivat alle määritysrajan. Pienten sertolitsumabipegolipitoisuuksien kliinistä merkitystä imeväisille ei tunneta. Ennen eläviä tai heikennettyjä eläviä taudinaiheuttajia sisältävien rokotteiden (esim. BCG-rokotteen) antamista on suositeltavaa odottaa vähintään viisi kuukautta siitä, kun äidille on annettu viimeisen kerran Cimzia-valmistetta raskauden aikana, paitsi jos rokotuksen hyödyt ovat selvästi suuremmat kuin teoreettinen riski, joka aiheutuu elävää tai heikennettyä elävää taudinaiheuttajaa sisältävän rokotteen antamisesta imeväisille.
Eläimiä koskevat tiedot
Eläinkokeet jyrsijän (rotan) TNF‑α:n estäjällä eivät tuoneet esiin näyttöä hedelmällisyyden heikkenemisestä eivätkä sikiölle aiheutuvasta haitasta. Tutkimukset ovat kuitenkin olleet riittämättömiä ihmisen reproduktiivisen toksisuuden kannalta (ks. kohta Prekliiniset tiedot turvallisuudesta). Raskauden aikana annettu Cimzia saattaa vaikuttaa vastasyntyneen normaaliin immuunivasteeseen TNF‑α:n estymisen vuoksi.
Non-kliiniset tutkimukset viittaavat lievään tai merkityksettömään sertolitsumabipegolin homologisen Fab-fragmentin siirtymiseen istukan kautta (ei Fc‑alue) (ks. kohta Prekliiniset tiedot turvallisuudesta).
Cimzia-valmistetta saa käyttää raskauden aikana vain, jos se on kliinisesti välttämätöntä. Annoksen muuttaminen ei ole tarpeen.
Imetys
Cimzia-valmistetta voidaan käyttää imetyksen aikana.
Cimzia-hoitoa saaneilla 17 imettävällä naisella tehdyssä kliinisessä tutkimuksessa osoitettiin, että sertolitsumabipegolin siirtyminen plasmasta rintamaitoon on minimaalista. Äidin sertolitsumabipegoliannoksesta 24 tunnin aikana imeväiseen kulkeutuneeksi prosenttiosuudeksi arvioitiin 0,04–0,30 %. Sertolitsumabipegoli on lisäksi proteiini, joka suun kautta otettuna hajoaa maha-suolikanavassa, joten absoluuttinen biologinen hyötyosuus rintaruokituilla imeväisillä on oletettavasti hyvin pieni.
Hedelmällisyys
Urospuolisilla jyrsijöillä on havaittu vaikutuksia siittiöiden liikkuvuutta osoittavissa mittareissa ja siittiömäärän vähenemistä, mutta ei ilmeisiä vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Kliinisessä tutkimuksessa arvioitiin sertolitsumabipegolin vaikutusta siemennesteen laatuparametreihin. 20 tervettä miestä satunnaistettiin saamaan ihonalaisesti joko 400 mg:n kerta-annos sertolitsumabipegolia tai lumelääkettä. 14 viikon seurannan aikana ei sertolitsumabipegolilla todettu vaikutusta siemennesteen laatuparametreihin verrattuna lumelääkkeeseen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Cimzia saattaa vaikuttaa vähäisessä määrin ajokykyyn ja kykyyn käyttää koneita. Huimausta (kiertohuimaus, näköhäiriöt ja väsymys mukaan lukien) saattaa esiintyä Cimzian annon jälkeen (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin tiivistelmä
Nivelreuma
Cimziaa tutkittiin kontrolloiduissa ja avoimissa tutkimuksissa 4 049 nivelreumapotilaalla 92 kuukauden ajan.
Lumekontrolloiduissa tutkimuksissa Cimzia-hoitoa saaneiden potilaiden altistuksen kesto oli noin nelinkertainen lumeryhmään verrattuna. Tämä ero altistuksessa johtuu pääasiassa siitä, että lumelääkettä saaneet potilaat keskeyttivät todennäköisemmin tutkimukseen osallistumisensa aikaisin. Lisäksi tutkimuksissa RA‑I ja RA‑II potilaiden, joilla ei esiintynyt vastetta viikolla 16, oli keskeytettävä tutkimukseen osallistuminen, ja suurin osa näistä potilaista sai lumelääkettä.
Haittavaikutusten vuoksi hoidon keskeyttäneiden potilaiden osuus oli kontrolloitujen tutkimusten aikana 4,4 % Cimzia-hoitoa saaneista ja 2,7 % lumehoitoa saaneista.
Yleisimmin esiintyneet haittavaikutukset kuuluivat Infektiot-ryhmään, ja niitä raportoitiin 14,4 %:lla Cimzia-hoitoa saaneista ja 8,0 %:lla lumehoitoa saaneista, Yleisoireet ja antopaikassa todettavat haitat ‑ryhmään, joissa niitä raportoitiin 8,8 %:lla Cimzia-hoitoa saaneista ja 7,4 %:lla lumehoitoa saaneista tai Iho ja ihonalainen kudos ‑ryhmään, joissa niitä raportoitiin 7,0 %:lla Cimzia-hoitoa saaneista ja 2,4 %:lla lumehoitoa saaneista.
Aksiaalinen spondylartriitti
Cimziaa tutkittiin aluksi 325:llä aktiivista aksiaalista spondylartriittia (mukaan lukien selkärankareuma ja röngennegatiivinen aksiaalinen spondylartriitti) sairastaneella potilaalla pisimmillään 4 vuoden ajan kliinisessä AS001‑tutkimuksessa, jossa 24 viikon lumekontrolloitua vaihetta seurasi annoksen suhteen sokkoutettu 24 viikon jakso ja 156 viikon avoin hoitojakso. Tämän jälkeen Cimziaa on tutkittu myös 317 potilaalla, jolla oli röntgennegatiivinen aksiaalinen spondylartriitti, lumekontrolloidussa tutkimuksessa 52 viikon ajan (AS0006). Cimziaa on tutkittu myös aksiaalista spondylartriittia (mukaan lukien selkärankareuma ja röntgennegatiivinen aksiaalinen spondylartriitti) sairastavilla potilailla kliinisessä tutkimuksessa enintään 96 viikon ajan. Tässä tutkimuksessa 48 viikon avointa sisäänottovaihetta (N = 736) seurasi 48 viikon lumekontrolloitu vaihe (N = 313) niiden potilaiden osalta, joiden remissio oli säilynyt (C-OPTIMISE). Cimziaa tutkittiin myös 96 viikon pituisessa avoimessa tutkimuksessa 89:llä aksiaalista spondylartriittia sairastavalla potilaalla, joilla oli aiemmin ollut dokumentoituja anteriorisen uveiitin pahenemisvaiheita. Kaikissa neljässä tutkimuksessa Cimzian turvallisuusprofiili näillä potilailla oli yhdenmukainen nivelreuman hoidossa todetun turvallisuusprofiilin ja aiempien Cimzia-kokemusten kanssa.
Nivelpsoriaasi
Cimziaa on tutkittu 409:llä nivelpsoriaasia sairastaneella potilaalla pisimmillään 4 vuoden ajan kliinisessä PsA001‑tutkimuksessa, jossa 24 viikon lumekontrolloitua vaihetta seurasi annoksen suhteen sokkoutettu 24 viikon jakso ja 168 viikon avoin hoitojakso. Cimzian turvallisuusprofiili nivelpsoriaasin hoidossa oli yhdenmukainen nivelreuman hoidossa todetun turvallisuusprofiilin ja aiempien Cimzia-kokemusten kanssa.
Läiskäpsoriaasi
Cimzia-valmistetta on tutkittu 1 112 psoriaasipotilaalla kontrolloiduissa ja avoimissa tutkimuksissa enintään kolmen vuoden ajan. Vaiheen III ohjelmassa aloitus- ja ylläpitovaiheita seurasi 96 viikon avoin hoitojakso (ks. kohta Farmakodynamiikka). Cimzia-valmisteen pitkän aikavälin turvallisuusprofiili annostuksilla 400 mg ja 200 mg kahden viikon välein oli yleisesti ottaen samankaltainen ja yhdenmukainen aiempien Cimzia-kokemusten kanssa.
Kontrolloiduissa kliinisissä tutkimuksissa viikon 16 loppuun mennessä vakavia haittatapahtumia saaneita potilaita oli Cimzia-ryhmässä 3,5 % ja lumelääkeryhmässä 3,7 %.
Kontrolloiduissa kliinisissä tutkimuksissa haittatapahtumien vuoksi hoidon keskeyttäneitä potilaita oli Cimzia-ryhmässä 1,5 % ja lumelääkeryhmässä 1,4 %.
Viikon 16 loppuun mennessä ilmoitetut yleisimmät haittavaikutukset kuuluivat elinjärjestelmäluokkaan Infektiot, ja niitä ilmoitettiin 6,1 %:lla Cimzia-potilaista ja 7 %:lla lumelääkettä saaneista potilaista. Yleisoireita ja antopaikassa todettavia haittoja ilmoitettiin 4,1 %:lla Cimzia-potilaista ja 2,3 %:lla lumelääkettä saaneista potilaista, ihon ja ihonalaisen kudoksen häiriöitä ilmoitettiin 3,5 %:lla Cimzia-potilaista ja 2,8 %:lla lumelääkettä saaneista potilaista.
Haittavaikutustaulukko
Haittavaikutukset, jotka perustuvat ensisijaisesti kokemuksiin lumekontrolloituista kliinisistä tutkimuksista ja valmisteen markkinoilletulon jälkeisiin tapauksiin, jotka ainakin mahdollisesti liittyivät Cimzia-hoitoon, on lueteltu seuraavassa taulukossa 1 esiintymistiheyden ja elinjärjestelmäluokan mukaan. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥1/10), yleinen (≥1/100, <1/10), melko harvinainen (≥1/1 000, <1/100), harvinainen (≥1/10 000, <1/1 000), hyvin harvinainen (<1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 1 Haittavaikutukset kliinisissä lääketutkimuksissa ja valmisteen markkinoilletulon jälkeen
Elinjärjestelmä | Yleisyys | Haittavaikutukset |
Infektiot | Yleinen | bakteeri-infektiot (märkäpesäkkeet mukaan lukien), virusinfektiot (herpes zoster, papilloomavirus, influenssa mukaan lukien) |
Melko harvinainen | sepsis (usean elimen toimintahäiriöt, septinen sokki mukaan lukien), tuberkuloosi (miliaari-, hajapesäkkeinen ja ekstrapulmonaalinen tuberkuloosi mukaan lukien), sieni-infektiot (opportunisti-infektiot mukaan lukien) | |
Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | Melko harvinainen | veren ja imukudoksen syövät (lymfooma ja leukemia mukaan lukien), elinten kiinteät kasvaimet, ei-melanooma ihosyöpä, syövän esiasteet (suun leukoplakia, melanosyyttiluomi mukaan lukien), hyvänlaatuiset kasvaimet ja kystat (ihon papillooma mukaan lukien) |
Harvinainen | ruoansulatuselimistön kasvaimet, melanooma | |
Tuntematon | merkelinsolukarsinooma*, Kaposin sarkooma | |
Veri ja imukudos | Yleinen | eosinofiiliset häiriöt, leukopenia (neutropenia, lymfopenia mukaan lukien) |
Melko harvinainen | anemia, lymfadenopatia, trombosytopenia, trombosytoosi | |
Harvinainen | pansytopenia, suurentunut perna, erytrosytoosi, veren valkosolujen morfologiset poikkeavuudet | |
Immuunijärjestelmä | Melko harvinainen | suonitulehdukset, lupus erythematosus, lääkeaineyliherkkyys (anafylaktinen sokki mukaan lukien), allergiset häiriöt, autovasta-ainepositiivisuus |
Harvinainen | angioneuroottinen edeema, sarkoidoosi, seerumitauti, ihonalaisen rasvakerroksen tulehdus (erythema nodosum mukaan lukien), dermatomyosiitin oireiden paheneminen** | |
Umpieritys | Harvinainen | kilpirauhashäiriöt |
Aineenvaihdunta ja ravitsemus | Melko harvinainen | elektrolyyttitasapainon häiriöt, dyslipidemia, ruokahalun häiriöt, painon muutos |
Harvinainen | hemosideroosi | |
Psyykkiset häiriöt | Melko harvinainen | ahdistuneisuus ja mielialahäiriöt (näihin liittyvät oireet mukaan lukien) |
Harvinainen | itsemurhayritys, delirium, mielenterveyshäiriö | |
Hermosto | Yleinen | päänsärky (migreeni mukaan lukien), aistien poikkeavuudet |
Melko harvinainen | perifeeriset neuropatiat, heitehuimaus, vapina | |
Harvinainen | kouristuskohtaukset, aivohermotulehdus, koordinaatiokyvyn tai tasapainon heikkeneminen | |
Tuntematon | multippeliskleroosi*, Guillain-Barrén oireyhtymä* | |
Silmät | Melko harvinainen | näköhäiriöt (näkökyvyn heikkeneminen mukaan lukien), silmän ja silmäluomen tulehdus, kyynelerityksen häiriö |
Kuulo ja tasapainoelin | Melko harvinainen | tinnitus, kiertohuimaus (vertigo) |
Sydän | Melko harvinainen | sydänlihassairaudet (sydämen vajaatoiminta mukaan lukien), iskeemiset sepelvaltimosairaudet, sydämen rytmihäiriöt (eteisvärinä mukaan lukien), sydämentykytys |
Harvinainen | sydänpussitulehdus, eteis-kammiokatkos | |
Verisuonisto | Yleinen | hypertensio |
Melko harvinainen | verenvuoto (mistä tahansa kohdasta), hyperkoagulaatio (laskimotukkotulehdus, keuhkoembolia mukaan lukien), pyörtyminen, edeema (perifeerinen, kasvojen edeema mukaan lukien), ekkymoosit (hematooma, petekiat mukaan lukien) | |
Harvinainen | aivoverenkiertohäiriö, arterioskleroosi, Raynaud’n oireyhtymä, livedo reticularis, telangiektasia | |
Hengityselimet, rintakehä ja välikarsina | Melko harvinainen | astma ja siihen liittyvät oireet, pleuraeffuusio ja sen oireet, hengitystiekongestio ja ‑tulehdus, yskä |
Harvinainen | interstitiaalinen keuhkosairaus, keuhkotulehdus | |
Ruoansulatuselimistö | Yleinen | pahoinvointi |
Melko harvinainen | askites, ruoansulatuselimistön haavauma ja perforaatio, maha-suolikanavan tulehdus (missä tahansa kohdassa), suutulehdus, ruoansulatushäiriöt, vatsan pingottuneisuus, suun ja nielun kuivuus | |
Harvinainen | nielemiskipu, hypermotiliteetti | |
Maksa ja sappi | Yleinen | hepatiitti (suurentuneet maksaentsyymiarvot mukaan lukien) |
Melko harvinainen | maksasairaus (kirroosi mukaan lukien), kolestaasi, suurentuneet veren bilirubiiniarvot | |
Harvinainen | kolelitiaasi | |
Iho ja ihonalainen kudos | Yleinen | ihottuma |
Melko harvinainen | alopesia, psoriaasin (myös palmoplantaaripustuloosin) ja siihen liittyvien sairauksien ilmeneminen tai paheneminen, dermatiitti ja ekseema, hikirauhasten häiriöt, ihon haavaumat, valoherkkyys, akne, ihon värimuutos, ihon kuivuus, kynsien ja kynsipedin häiriöt | |
Harvinainen | ihon hilseily ja kesiminen, rakkulaiset sairaudet, hiusten koostumuksen häiriöt, Stevens-Johnsonin oireyhtymä**, erythema multiforme**, jäkälää muistuttavat reaktiot | |
Luusto, lihakset ja sidekudos | Melko harvinainen | lihasten häiriöt, suurentunut veren kreatiinikinaasipitoisuus |
Munuaiset ja virtsatiet | Melko harvinainen | munuaisten vajaatoiminta, verivirtsaisuus, virtsarakon ja virtsaputken oireet |
Harvinainen | munuaissairaus (nefriitti mukaan lukien) | |
Sukupuolielimet ja rinnat | Melko harvinainen | kuukautiskierron ja kuukautisvuodon häiriöt (kuukautisten puuttuminen mukaan lukien), rintojen häiriöt |
Harvinainen | sukupuolitoimintojen häiriöt | |
Yleisoireet ja antopaikassa todettavat haitat | Yleinen | kuume, kipu (missä tahansa kohdassa), voimattomuus, kutina (missä tahansa kohdassa), pistoskohdan reaktiot |
Melko harvinainen | vilunväristykset, influenssankaltainen sairaus, muuttunut käsitys ruumiinlämmöstä, yöhikoilu, flush-oire | |
Harvinainen | fistelit (missä tahansa) | |
Tutkimukset | Melko harvinainen | suurentunut veren alkalisen fosfataasin pitoisuus, pidentynyt koagulaatioaika |
Harvinainen | suurentunut veren virtsahappopitoisuus | |
Vammat ja myrkytykset | Melko harvinainen | ihovauriot, paranemisen heikkeneminen |
*Nämä haitat ovat liittyneet TNF:n estäjien lääkeryhmään, mutta esiintyvyyttä sertolitsumabipegolihoidon yhteydessä ei tiedetä.
**Nämä haitat ovat liittyneet TNF:n estäjien lääkeryhmään.
Lisäksi seuraavia haittavaikutuksia on havaittu melko harvoin käytettäessä Cimzia-valmistetta muihin käyttöaiheisiin: ruoansulatuselimistön ahtaumat ja tukkeumat, yleisen terveydentilan heikkeneminen, keskenmeno ja atsoospermia.
Valikoitujen haittavaikutusten kuvaus
Infektiot
Uusien infektioiden ilmaantuvuusaste nivelreumapotilaille oli lumekontrolloiduissa kliinisissä lääketutkimuksissa 1,03 tapausta potilasvuotta kohden kaikilla Cimzia-hoitoa saaneilla potilailla ja 0,92 tapausta potilasvuotta kohden lumehoitoa saaneilla. Infektiot olivat pääasiassa ylempien hengitysteiden infektioita, virtsatieinfektioita, alempien hengitysteiden infektioita ja herpes-infektioita (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet).
Lumekontrolloiduissa kliinisissä nivelreumatutkimuksissa uusia vakavia infektioita esiintyi useammin Cimzia-hoitoa saaneissa ryhmissä (0,07 tapausta potilasvuotta kohden kaikkien annostusten yhteydessä) kuin lumelääkettä saaneilla (0,02 tapausta potilasvuotta kohden). Yleisimmät vakavat infektiot olivat keuhkokuume, tuberkuloosi-infektiot. Vakavia infektioita olivat myös invasiiviset opportunisti-infektiot (esim. pneumokystoosi, sieniesofagiitti, nokardioosi ja disseminoitunut vyöruusu). Infektioriskin suurentumisesta ei ole näyttöä altistuksen jatkuessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Uusien infektiotapausten ilmaantuvuus lumekontrolloiduissa kliinisissä psoriaasitutkimuksissa oli 1,37 potilasvuotta kohden kaikilla Cimzia-hoidetuilla potilailla ja 1,59 potilasvuotta kohden lumelääkettä saaneilla potilailla. Infektiot olivat pääasiassa ylempien hengitysteiden infektioita ja virusinfektioita (mukaan lukien herpesinfektiot). Vakavien infektioiden ilmaantuvuus Cimzia-hoitoa saaneilla potilailla oli 0,02 potilasvuotta kohden. Lumelääkepotilailla ei ilmoitettu vakavia infektioita. Infektioriskin suurentumisesta ei ole näyttöä altistuksen jatkuessa.
Pahanlaatuiset kasvaimet ja lymfoproliferatiiviset sairaudet
Nivelreumapotilailla tehdyissä kliinisissä Cimzia-tutkimuksissa, joissa hoitoa sai yhteensä 4 049 potilasta (vastaa 9 277 potilasvuotta), havaittiin ei-melanooma ihosyöpää lukuun ottamatta 121 pahanlaatuista kasvainta, joista viisi oli lymfoomia. Lymfoomatapausten esiintymistiheys oli 0,05 tapausta 100 potilasvuotta kohden ja melanooman esiintymistiheys oli 0,08 tapausta 100 potilasvuotta kohden Cimzia-valmisteella tehdyissä nivelreumaan liittyvissä kliinisissä lääketutkimuksissa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Myös nivelpsoriaasia koskeneessa kliinisessä faasin III tutkimuksessa havaittiin yksi lymfoomatapaus.
Lukuun ottamatta ihosyöpätapauksia, jotka eivät ole melanoomaa, psoriaasia koskeneissa kliinisissä Cimzia-tutkimuksissa havaittiin 11 pahanlaatuista kasvainta, mukaan lukien yksi lymfoomatapaus. Tutkimuksissa annettiin hoitoa yhteensä 1 112 potilaalle, mikä vastaa 2 300:aa potilasvuotta.
Autoimmuniteetti
Nivelreuman pivotaalitutkimuksissa yhteensä 16,7 %:lle Cimzia-hoitoa saaneista, lähtötilanteessa tumavasta-ainenegatiivisista potilaista kehittyi positiivinen tumavasta-ainetiitteri verrattuna 12,0 %:iin lumeryhmän potilaista. Kaikkiaan 2,2 %:lle Cimzia-hoitoa saaneista, lähtötilanteessa anti-dsDNA -vasta-ainenegatiivisista potilaista kehittyi positiivinen anti-dsDNA-vasta-ainetiitteri verrattuna lumeryhmän 1,0 %:iin. Nivelreumapotilailla tehdyissä sekä lumekontrolloiduissa että avoimissa kliinisissä seurantatutkimuksissa raportoitiin melko harvoin SLE‑tyyppistä oireyhtymää. Muita immuunivälitteisiä sairauksia on raportoitu harvoin eikä syy-yhteyttä Cimzia-hoitoon tiedetä. Pitkäaikaisen Cimzia-hoidon vaikutusta autoimmuunisairauksien kehittymiseen ei tiedetä.
Pistoskohdan reaktiot
Lumekontrolloiduissa kliinisissä nivelreumatutkimuksissa 5,8 %:lle Cimzia-hoitoa saaneista potilaista kehittyi pistoskohdan reaktioita kuten punoitusta, kutinaa, hematooma, kipua, turvotusta tai mustelmia verrattuna 4,8 %:iin lumelääkettä saaneista potilaista. Pistoskohdan kipua esiintyi 1,5 %:lla Cimzia-hoitoa saaneista potilaista, mutta yksikään tapaus ei johtanut potilaan vetäytymiseen tutkimuksesta.
Kreatiinikinaasiarvon suureneminen
Kreatiinikinaasin (CK) arvo suureni yleensä useammin aksiaalista spondylartriittia sairastaneilla kuin nivelreumaa sairastaneilla. Esiintymistiheys suureni sekä lumelääkkeellä hoidetuilla potilailla (2,8 %:lla aksiaalista spondylartriittia sairastaneista vs. 0,4 %:lla nivelreumaa sairastaneista) että Cimzialla hoidetuilla potilailla (4,7 %:lla aksiaalista spondylartriittia sairastaneista vs. 0,8 %:lla nivelreumaa sairastaneista). CK‑arvo suureni aksiaalista spondylartriittia koskeneessa tutkimuksessa enimmäkseen lievästi tai kohtalaisesti ja ohimenevästi. CK‑arvon suurenemisen kliinistä merkitystä ei tiedetä, eikä hoitoa tarvinnut yhdessäkään tapauksessa lopettaa arvon suurenemisen vuoksi.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‒haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisten tutkimusten yhteydessä ei havaittu annosta rajoittavaa toksisuutta. Valmistetta on annettu toistuvina enintään 800 mg:n annoksina ihon alle ja toistuvina 20 mg/kg annoksina laskimoon. Yliannostuksen yhteydessä suositellaan potilaiden tilan tarkkaa seurantaa haittavaikutusten ja -tapahtumien varalta. Asianmukainen oireenmukainen hoito on aloitettava heti.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Immunosuppressantit, tuumorinekroositekijä alfan (TNF‑α) estäjät, ATC-koodi: L04AB05
Vaikutusmekanismi
Cimzialla on suuri affiniteetti (KD = 90 pM) ihmisen TNF‑α:aan, johon se sitoutuu. TNF‑α on keskeinen tulehdusta edistävä sytokiini ja sillä on keskeinen merkitys tulehdusprosessissa. Cimzia neutraloi selektiivisesti TNF‑α:aa (ihmisen TNF‑α:n estävä pitoisuus (IC90) saatiin L929 hiiren fibrosarkooman sytotoksisuusmäärityksessä in vitro annoksella 4 ng/ml), mutta se ei neutraloi lymfotoksiini α:aa (TNF‑β).
Cimzian osoitettiin neutraloivan annoksesta riippuvaisesti solukalvoon kiinnittynyttä ja liukoista ihmisen TNF‑α:aa. Inkuboitaessa ihmisen monosyyttejä sertolitsumabipegolilla todettiin annoksesta riippuvainen lipopolysakkaridien indusoiman TNF‑α:n ja IL‑1β:n tuotannon estyminen.
Cimzia ei sisällä Fc‑fragmenttia, jonka täydellinen vasta-aine tavallisesti sisältää, eikä se sen vuoksi sido komplementtia eikä aiheuta vasta-aineriippuvaista soluvälitteistä sytotoksisuutta in vitro. Se ei saa aikaan apoptoosia in vitro ihmisen ääreisveren monosyyteissä eikä lymfosyyteissä, eikä se saa myöskään aikaan neutrofiilien degranulaatiota.
Kliininen teho
Nivelreuma
Cimzian tehoa ja turvallisuutta on arvioitu kahdessa satunnaistetussa, lumekontrolloidussa, kaksoissokkoutetussa kliinisessä tutkimuksessa RA‑I (RAPID 1) ja RA‑II (RAPID 2) ≥ 18‑vuotiailla aktiivista nivelreumaa sairastaneilla potilailla, joiden nivelreuma oli diagnosoitu American College of Rheumatology (ACR) ‑kriteerien mukaan. Jokaisella potilaalla oli ≥ 9 turvonnutta ja arkaa niveltä ja heillä oli ollut aktiivinen nivelreuma vähintään 6 kuukauden ajan ennen lähtötilannetta. Cimzia-valmistetta annettiin kummassakin tutkimuksessa ihon alle yhdessä suun kautta annettavan metotreksaatin kanssa vähintään 6 kuukauden ajan, jolloin annostus säilyi samana, eli vähintään 10 mg viikoittain, kahden kuukauden ajan. Cimzian käytöstä yhdistelmänä muiden taudinkulkua muuttavien reumalääkkeiden kanssa metotreksaattia lukuun ottamatta ei ole kokemusta.
Cimzian tehoa ja turvallisuutta on arvioitu satunnaistetussa, lumekontrolloidussa, kaksoissokkoutetussa kliinisessä tutkimuksessa (C‑EARLY) aktiivista nivelreumaa sairastaneilla aikuispotilailla, jotka eivät olleet aiemmin saaneet taudinkulkua muuttavia reumalääkkeitä. C‑EARLY‑tutkimukseen otetut potilaat olivat ≥ 18‑vuotiaita, jokaisella oli ≥ 4 turvonnutta ja arkaa niveltä, ja keskivaikea tai vaikea aktiivinen ja etenevä nivelreuma piti olla diagnosoitu kuluneen vuoden sisällä (vuoden 2010 ACR / European League Against Rheumatism [EULAR] ‑luokituskriteerien mukaan). Potilaat olivat saaneet diagnoosin keskimäärin 2,9 kuukautta ennen lähtötilannetta, eivätkä he olleet saaneet taudinkulkua muuttavia reumalääkkeitä (eivät myöskään metotreksaattia). Hoito metotreksaatilla (10 mg/viikko) aloitettiin sekä Cimzia- että lumelääkeryhmässä viikosta 0 alkaen, sen annos titrattiin suurimpaan siedettyyn annokseen viikkoon 8 mennessä (sallittu annos viikkoa kohti oli vähintään 15 mg ja enintään 25 mg), ja hoitoa jatkettiin koko tutkimuksen ajan (keskimääräinen viikoittainen metotreksaattiannos oli viikon 8 jälkeen lumeryhmässä 22,3 mg ja Cimzia-ryhmässä 21,1 mg).
Taulukko 2 Kliinisen tutkimuksen kuvaus
| Tutkimus-numero | Potilas-määrä | Aktiivilääkkeen annostus | Tutkimuksen tavoite |
| RA‑I (52 viikkoa) | 982 | 400 mg (viikoilla 0, 2, 4) + metotreksaatti 200 mg tai 400 mg 2 viikon välein + metotreksaatti | Oireiden ja löydösten hoidon arviointi ja rakenteellisten vaurioiden estäminen. Ensisijaiset päätetapahtumat: ACR 20 viikolla 24 ja mTSS:n muutos lähtötilanteesta viikolla 52 |
RA‑II (24 viikkoa) | 619 | 400 mg (viikoilla 0, 2, 4) + metotreksaatti 200 mg tai 400 mg 2 viikon välein + metotreksaatti | Oireiden ja löydösten hoidon arviointi ja rakenteellisten vaurioiden estäminen. Ensisijainen päätetapahtuma: ACR 20 viikolla 24. |
C-EARLY (viikkoon 52 asti) | 879 | 400 mg (viikoilla 0, 2, 4) + metotreksaatti 200 mg 2 viikon välein + metotreksaatti | Oireiden ja löydösten hoidon arviointi ja rakenteellisten vaurioiden estäminen potilailla, jotka eivät olleet saaneet taudinkulkua muuttavia reumalääkkeitä. Ensisijainen päätetapahtuma: potilaat (%), joilla saavutettu pitkäkestoinen remissio* viikolla 52 |
mTSS: modified Total Sharp Score *Pitkäkestoinen remissio viikolla 52, määritelmä: DAS28 (ESR) < 2,6 sekä viikolla 40 että viikolla 52. | |||
Oireet ja löydökset
Kliinisten tutkimusten RA‑I ja RA‑II tulokset on esitetty taulukossa 3. ACR‑vaste 20 saavutettiin kummassakin kliinisessä tutkimuksessa tilastollisesti merkitsevästi yleisemmin viikosta 1 lähtien ja ACR‑vaste 50 viikosta 2 lähtien lumeryhmään verrattuna. Vasteet säilyivät viikkoon 52 (RA‑I) ja 24 (RA‑II) saakka. RA‑I-tutkimuksen alussa aktiivista hoitoa saamaan satunnaistetuista 783 potilaasta 508 oli tutkimuksessa mukana koko 52 viikkoa kestäneen lumekontrolloidun jakson ajan ja jatkoi avoimessa jatkotutkimuksessa. Heistä 427 oli mukana avoimessa jatkotutkimuksessa sen koko 2 vuoden kestoajan, joten heidän kokonaisaltistusaikansa Cimzia-hoidolle oli 148 viikkoa. Tänä ajankohtana ACR‑vaste 20 todettiin 91 %:lla potilaista. DAS28 (ESR) ‑indeksin pieneneminen lähtötilanteesta oli myös merkitsevästi suurempaa (p < 0,001) viikolla 52 (RA‑I) ja viikolla 24 (RA‑II) lumelääkkeeseen verrattuna, ja se säilyi 2 vuoden ajan RA‑I-tutkimuksen avoimessa jatkotutkimuksessa.
Taulukko 3 ACR-vasteet kliinisissä tutkimuksissa RA‑I ja RA‑II
Tutkimus RA‑I Yhdistelmähoito metotreksaatin kanssa (24 ja 52 viikkoa) | Tutkimus RA‑II Yhdistelmähoito metotreksaatin kanssa (24 viikkoa) | |||
| Vaste | Lumelääke + metotreksaatti N = 199 | Cimzia 200 mg + metotreksaatti kahden viikon välein N = 393 | Lumelääke + metotreksaatti N = 127 | Cimzia 200 mg + metotreksaatti kahden viikon välein N = 246 |
| ACR 20 | ||||
| Viikko 24 | 14 % | 59 %** | 9 % | 57 %** |
| Viikko 52 | 13 % | 53 %** | N/A | N/A |
| ACR 50 | ||||
| Viikko 24 | 8 % | 37 %** | 3 % | 33 %** |
| Viikko 52 | 8 % | 38 %** | N/A | N/A |
| ACR 70 | ||||
| Viikko 24 | 3 % | 21 %** | 1 % | 16 %* |
| Viikko 52 | 4 % | 21 %** | N/A | N/A |
| Suurin kliininen vastea. | 1 % | 13 %** | ||
Cimzia vs. lumelääke: * p ≤ 0,01, ** p < 0,001
a. Suurin kliininen vaste on määritelty vasteen ACR 70 saavuttamiseksi jokaisen arviointikerran yhteydessä koko 6 kuukauden mittaisen jakson ajan.
Waldin testin p‑arvo on esitetty hoitojen vertailemiseksi logistisen regression avulla, jossa faktorit ovat hoito ja alue.
Prosentuaaliset vasteet, jotka perustuvat niiden potilaiden lukumäärään, joista tietoja on saatu (n) kyseiseen päätepisteeseen ja ajankohtaan, saattavat olla erilaiset kuin N.
C‑EARLY-tutkimuksessa ensisijainen ja tärkein toissijainen päätetapahtuma saavutettiin. Tutkimuksen päätulokset on esitetty taulukossa 4.
Taulukko 4 C‑EARLY-tutkimus: Potilaat (%), joilla pitkäkestoinen remissio ja pitkäkestoinen vähäinen tautiaktiivisuus saavutettu viikolla 52
| Vaste | Lume + metotreksaatti N = 213 | Cimzia 200 mg + metotreksaatti N = 655 |
Pitkäkestoinen remissio* (DAS28 [ESR] < 2,6 sekä viikolla 40 että viikolla 52) | 15,0 % | 28,9 %** |
Pitkäkestoinen vähäinen tautiaktiivisuus (DAS28 [ESR] ≤ 3,2 sekä viikolla 40 että viikolla 52) | 28,6 % | 43,8 %** |
*C-EARLY-tutkimuksen ensisijainen päätetapahtuma (viikkoon 52 asti) Koko analyysijoukko, puuttuvat tiedot korvattiin non-responder imputation (NRI) ‑menetelmällä. **Cimzia + metotreksaatti vs. lumelääke + metotreksaatti: p < 0,001 p-arvo on arvioitu logistisella regressiomallilla, jossa faktoreina olivat hoitoryhmä, alue ja aika nivelreuman diagnoosista lähtötilanteeseen (≤ 4 kk vs. > 4 kk) | ||
DAS28 (ESR) -indeksi pieneni lähtötilanteesta enemmän Cimzian ja metotreksaatin yhdistelmää saaneilla potilailla kuin lumelääkkeen ja metotreksaatin yhdistelmää saaneilla potilailla jo viikosta 2 alkaen aina viikon 52 loppuun asti (p < 0,001 jokaisella käynnillä). Käynneillä tehdyt arviot remissiosta (DAS28 [ESR] < 2,6), tautiaktiivisuuden vähäisyydestä (DAS28 [ESR] ≤ 3,2) sekä ACR50‑ ja ACR70‑vasteesta osoittivat, että Cimzian ja metotreksaatin yhdistelmä tuotti nopeamman ja suuremman vasteen kuin lumelääkkeen ja metotreksaatin yhdistelmä. Nämä tulokset säilyivät 52 hoitoviikon ajan tutkittavilla, jotka eivät olleet saaneet taudinkulkua muuttavia reumalääkkeitä.
Radiografinen vaste
Tutkimuksessa RA‑I rakenteellisia nivelvaurioita arvioitiin röntgenkuvin ja ne ilmaistiin mTSS:n ja sen osa-alueiden, eroosiopisteiden ja nivelraon ahtautumista (joint space narrowing, JSN) kuvaavien pisteiden muutoksena viikolla 52 lähtötilanteeseen verrattuna. Cimzia-hoitoa saaneessa ryhmässä esiintyi lumeryhmään verrattuna huomattavasti vähemmän röntgenkuvin todettavaa etenemistä viikolla 24 ja viikolla 52 (ks. taulukko 5). Lumeryhmässä 52 %:lla potilaista ei esiintynyt röntgenkuvin todettavaa etenemistä (mTSS ≤ 0,0) viikolla 52 verrattuna 69 %:iin 200 mg:n Cimzia-annoksia saaneessa ryhmässä.
Taulukko 5 Muutokset 12 kuukauden aikana tutkimuksessa RA‑I
Lumelääke + metotreksaatti N = 199 Keskiarvo (keskihajonta) | Cimzia 200 mg + metotreksaatti N = 393 Keskiarvo (keskihajonta) | Cimzia 200 mg + metotreksaatti – lumelääke + metotreksaatti Keskimääräinen ero | |
| mTSS | |||
| Viikko 52 | 2,8 (7,8) | 0,4 (5,7) | −2,4 |
| Eroosiopisteet | |||
| Viikko 52 | 1,5 (4,3) | 0,1 (2,5) | −1,4 |
| JSN-pisteet | |||
| Viikko 52 | 1,4 (5,0) | 0,4 (4,2) | −1,0 |
p‑arvot olivat < 0,001 sekä mTSS:n että eroosiopisteiden osalta ja ≤ 0,01 JSN‑pisteiden osalta. ANCOVA perustui kunkin mittaustuloksen osalta järjestykseen asetettuihin (ordinary scale) muutoksiin lähtötilanteesta siten, että faktoreina olivat alue ja hoitoryhmä, ja kovariaattina mittaustuloksen järjestysluku lähtötilanteessa.
RA‑I-tutkimuksen alussa aktiivista hoitoa saamaan satunnaistetuista 783 potilaasta 508 oli tutkimuksessa mukana koko 52 viikkoa kestäneen lumekontrolloidun jakson ajan ja jatkoi avoimessa jatkotutkimuksessa. Rakenteellisten vaurioiden etenemisen osoitettiin estyneen pitkäkestoisesti 449 potilaalla siinä potilasjoukossa, jotka saivat Cimzia-hoitoa vähintään 2 vuoden ajan (RA‑I ja avoin jatkotutkimus) ja joista oli kahden vuoden päättyessä arvioitavissa olevaa tietoa.
C‑EARLY-tutkimuksessa Cimzian ja metotreksaatin yhdistelmä esti röntgenkuvin todettavan taudin etenemisen verrattuna lumelääkkeen ja metotreksaatin yhdistelmään viikolla 52 (ks. taulukko 6). Röntgenkuvin todettavaa taudin etenemistä ei todettu viikolla 52 lumelääkkeen ja metotreksaatin yhdistelmää saaneista potilaista 49,7 %:lla (mTSS-muutos ≤ 0,5) verrattuna 70,3 %:iin Cimzian ja metotreksaatin yhdistelmää saaneista (p < 0,001).
Taulukko 6 Röntgenkuvissa todettu muutos C‑EARLY-tutkimuksen viikolla 52
Lumelääke + metotreksaatti N = 163 Keskiarvo (keskihajonta) | Cimzia 200 mg + metotreksaatti N = 528 Keskiarvo (keskihajonta) | Cimzia 200 mg + metotreksaatti – Lumelääke + metotreksaatti Ero* | |
mTSS Viikko 52 | 1,8 (4,3) | 0,2 (3,2)** | −0,978 (−1,005 – −0,500) |
Eroosiopisteet Viikko 52 | 1,1 (3,0) | 0,1 (2,1)** | −0,500 (−0,508 – −0,366) |
JSN-pisteet Viikko 52 | 0,7 (2,3) | 0,1 (1,7)** | 0,000 (0,000 – 0,000) |
Tutkittavat, joiden röntgenkuvat arvioitiin, lineaarinen ekstrapolaatio. *Muutosta kuvaava Hodges-Lehmannin piste-estimointi (point estimate of shift) ja 95 %:n asymptomaattinen (Moses) luottamusväli. **Cimzia + metotreksaatti vs. lumelääke + metotreksaatti, p < 0,001. p-arvo on arvioitu ANCOVA-mallilla, jossa faktorit olivat hoitoryhmä, alue, aika nivelreuman diagnoosista lähtötilanteeseen (≤ 4 kk vs. > 4 kk) ja kovariaattina lähtötilanne. | |||
Vaste fyysiseen toimintakykyyn ja terveyteen liittyvä hoitotulos
Tutkimuksissa RA‑I ja RA‑II Cimzia-hoitoa saaneet potilaat raportoivat merkittävää parantumista lumelääkkeeseen nähden arvioitaessa fyysistä toimintakykyä Health Assessment Questionnaire – Disability Index (HAQ‑DI) ‑kyselyllä sekä väsymystä (uupuneisuutta) Fatigue Assessment Scale (FAS) ‑pisteytyksellä viikosta 1 lähtien tutkimusten loppuun saakka. Cimzia-hoitoa saaneilla potilailla raportoitiin kummassakin kliinisessä tutkimuksessa merkitsevästi suurempaa paranemista SF‑36-asteikon fyysisen ja mielenterveysosion yhteenvetopisteiden ja kaikkien osioiden pisteiden osalta. Fyysisen toimintakyvyn ja HRQoL-pisteiden paraneminen säilyi RA‑I-tutkimuksen koko kahden vuoden mittaisen avoimen jatkotutkimuksen ajan. Cimzia-hoitoa saaneet potilaat raportoivat tilastollisesti merkitsevää paranemista lumelääkkeeseen verrattuna Work Productivity Survey ‑kyselyllä arvioituna.
C‑EARLY-tutkimuksessa Cimzian ja metotreksaatin yhdistelmää saaneet potilaat raportoivat merkittävää parantumista viikolla 52 verrattuna lumelääkkeen ja metotreksaatin yhdistelmää saaneisiin potilaisiin (-48,5 vs -44,0 [pienimmän neliösumman keskiarvo, p < 0,05]) arvioitaessa kipua Patient Assessment of Arthritis Pain (PAAP) ‑kyselyllä.
Kliininen DoseFlex-tutkimus
Kahden Cimzia-annostuksen (200 mg kahden viikon välein ja 400 mg neljän viikon välein) tehoa ja turvallisuutta on verrattu lumelääkkeeseen kliinisessä kaksoissokkotutkimuksessa, jossa oli ensin 18 viikon avoin esivaihe (run-in) ja sitten 16 viikon satunnaistettu ja lumekontrolloitu vaihe. Tutkimukseen osallistuneilla aikuispotilailla oli ACR‑kriteerien mukaan diagnosoitu aktiivinen nivelreuma, jonka vaste metotreksaattihoitoon ei ollut riittävä.
Potilaat saivat aloitusannoksena 400 mg Cimziaa viikoilla 0, 2 ja 4 ja tämän jälkeen 200 mg Cimziaa kahden viikon välein tutkimuksen avoimessa aloitusvaiheessa. Viikolla 16 hoitoon vastanneet (ACR 20 ‑vaste saavutettu) satunnaistettiin viikolla 18 saamaan joko 200 mg Cimziaa kahden viikon välein, 400 mg Cimziaa neljän viikon välein tai lumelääkettä yhdessä metotreksaatin kanssa vielä 16 viikon ajan (tutkimuksen kokonaiskesto 34 viikkoa). Nämä kolme hoitoryhmää olivat kliinisen vasteen suhteen hyvin tasapainossa aktiivisen esivaiheen jälkeen (ACR 20: 83–84 % viikolla 18).
Tutkimuksen ensisijainen päätetapahtuma oli ACR 20 ‑vasteen saavuttaneiden osuus viikolla 34. Viikon 34 tulokset on esitetty taulukossa 7. Molemmilla Cimzia-annostuksilla saavutettiin pitkäkestoinen kliininen vaste, joka oli lumelääkkeeseen verrattuna tilastollisesti merkitsevä viikolla 34. ACR 20 ‑päätetapahtuman saavuttivat sekä 200 mg Cimziaa kahden viikon välein saaneet että 400 mg Cimziaa neljän viikon välein saaneet.
Taulukko 7 ACR‑vaste kliinisessä DoseFlex-tutkimuksessa viikolla 34
| Hoitoviikot 0–16 | Cimzia 400 mg + metotreksaatti viikoilla 0, 2 and 4, sitten Cimzia 200 mg + metotreksaatti kahden viikon välein | ||
| Satunnaistettu kaksoissokkohoito viikoilla 18‒34 | Lumelääke + metotreksaatti N = 69 | Cimzia 200 mg + metotreksaatti kahden viikon välein N = 70 | Cimzia 400 mg + metotreksaatti neljän viikon välein N = 69 |
ACR 20 p‑arvo* | 45 % N/A | 67 % 0,009 | 65 % 0,017 |
ACR 50 p‑arvo* | 30 % N/A | 50 % 0,020 | 52 % 0,010 |
ACR 70 p‑arvo* | 16 % N/A | 30 % 0,052 | 38 % 0,005 |
N/A: ei saatavana. *Waldin testin p‑arvot on arvioitu hoitojen (Cimzia 200 mg vs. lumelääke ja Cimzia 400 mg vs. lumelääke) vertailemiseksi logistisen regressiomallin avulla, jossa on hoitoon liittyviä faktoreja. | |||
Aksiaalinen spondylartriitti (röntgennegatiivisen aksiaalisen spondylartriitin ja selkärankareuman osajoukot)
AS001
Cimzian tehoa ja turvallisuutta on arvioitu yhdessä satunnaistetussa, kaksoissokkoutetussa ja lumekontrolloidussa monikeskustutkimuksessa (AS001) 325:llä ≥ 18‑vuotiaalla potilaalla, joiden aikuisiällä alkanut aktiivinen aksiaalinen spondylartriitti oli kestänyt vähintään 3 kuukautta aksiaalisen spondylartriitin ASAS (Assessment of Spondyloarthritis International Society) ‑luokittelukriteerien mukaan. Osalla näistä aksiaalista spondylartriittia sairastaneista potilaista näkyi röntgenkuvissa selkärankareuman (eli röntgenpositiivisen aksiaalisen spondylartriitin) muutoksia, osalla ei (röntgennegatiivinen aksiaalinen spondylartriitti). BASDAI-indeksillä (Bath Ankylosing Spondylitis Disease Activity Index) määriteltynä sairaus oli aktiivinen (indeksi ≥ 4), rankakivun vaikeusaste oli ≥ 4 numeroasteikolla 0–10 ja CRP‑arvo oli suurentunut tai magneettikuvissa näkyi tutkimushetkellä näyttöä sakroiliitista. Tutkimukseen otettiin vain potilaita, joilla vähintään yksi NSAID-hoitokokeilu oli epäonnistunut potilaan intoleranssin tai riittämättömän hoitovasteen vuoksi. Potilaista yhteensä 16 % oli altistunut aiemmin TNF:n estäjälle. Potilaat saivat joko 400 mg Cimziaa aloitusannoksena viikoilla 0, 2 ja 4 (kummassakin hoitoryhmässä) tai lumelääkettä ja sen jälkeen joko 200 mg Cimziaa kahden viikon välein, 400 mg Cimziaa neljän viikon välein tai lumelääkettä. Potilaista 87,7 % sai samanaikaisesti NSAID-hoitoa. Tehon ensisijainen päätetapahtuma oli ASAS20‑vasteen saaneiden osuus viikolla 12. Tutkimuksessa 24 viikon kaksoissokkoutettua ja lumekontrolloitua hoitojaksoa seurasi annoksen suhteen sokkoutettu 24 viikon hoitojakso ja 156 viikon avoin hoitojakso. Tutkimus kesti pisimmillään 204 viikkoa. Kaikki potilaat saivat Cimziaa sekä annoksen suhteen sokkoutetun että avoimen seurantajakson aikana. Yhteensä 199 potilasta (61,2 % satunnaistetuista potilaista) pysyi mukana tutkimuksessa viikon 204 loppuun asti.
Keskeiset hoidon tehoa koskevat tulokset
Kliinisessä AS001‑tutkimuksessa ASAS20‑vaste saavutettiin viikolla 12 58 %:lla potilaista, jotka saivat Cimziaa 200 mg kahden viikon välein, ja 64 %:lla potilaista, jotka saivat Cimziaa 400 mg neljän viikon välein verrattuna 38 %:iin lumelääkettä saaneista potilaista (p < 0,01). Koko tutkimusjoukossa ASAS20‑vasteen saavuttaneiden prosenttiosuus oli jokaisella tutkimuskäynnillä viikosta 1 viikon 24 loppuun saakka kliinisesti merkityksellinen ja merkitsevästi lumeryhmää suurempi (p ≤ 0,001 joka käynnillä) niissä kahdessa ryhmässä, joista toinen sai Cimziaa 200 mg kahden viikon välein ja toinen 400 mg neljän viikon välein. Viikoilla 12 ja 24 ASAS40‑vasteen saavuttaneiden potilaiden prosenttiosuus oli suurempi Cimzia-hoitoryhmissä kuin lumelääkeryhmässä.
Saavutetut tulokset olivat samankaltaisia sekä selkärankareumaa että röntgennegatiivista aksiaalista spondylartriittia sairastaneiden alaryhmissä. Naispotilailla ASAS20‑vaste ei eronnut tilastollisesti merkitsevästi lumelääkkeestä ennen kuin viikon 12 jälkeen.
ASAS 5/6 ‑vaste, osittainen remissio ja BASDAI‑50‑vaste paranivat tilastollisesti merkitsevästi viikoilla 12 ja 24, ja nämä tulokset säilyivät viikkoon 48 asti sekä koko tutkimusjoukossa että alaryhmissä. Kliinisestä AS001‑tutkimuksesta saadut keskeiset hoidon tehoa koskevat tulokset on esitetty taulukossa 8.
Kaikkien edellä mainittujen keskeisten hoidon tehoa koskevien tulosten paraneminen säilyi tutkimuksessa mukana pysyneillä potilailla viikon 204 loppuun asti sekä koko tutkimusjoukossa että alaryhmissä.
Taulukko 8 Kliinisestä AS001‑tutkimuksesta saadut keskeiset hoidon tehoa koskevat tulokset (potilaiden prosenttiosuudet)
| Parametrit | Selkärankareuma | Röntgennegatiivinen aksiaalinen spondylartriitti | Aksiaalinen spondylartriitti Koko tutkimusjoukko | |||
Lume-lääke N = 57 | Kaikki Cimzia- annostukset (a) N = 121 | Lume-lääke N = 50 | Kaikki Cimzia- annostukset(a) N = 97 | Lumelääke N = 107 | Kaikki Cimzia- annostukset (a) N = 218 | |
ASAS20(b,c) Viikko 12 Viikko 24 | 37 % 33 % | 60 %* 69 %** | 40 % 24 % | 61 %* 68 %** | 38 % 29 % | 61 %** 68%** |
ASAS40(c,d) Viikko 12 Viikko 24 | 19 % 16 % | 45 %** 53 %** | 16 % 14 % | 47 %** 51 %** | 18 % 15 % | 46 %** 52 %** |
ASAS 5/6(c,d) Viikko 12 Viikko 24 | 9 % 5 % | 42 %** 40 %** | 8 % 4 % | 44 %** 45 %** | 8 % 5 % | 43 %** 42 %** |
Osittainen remissio(c,d) Viikko 12 Viikko 24 | 2 % 7 % | 20 %** 28 %** | 6 % 10 % | 29 %** 33 %** | 4 % 9 % | 24 %** 30 %** |
BASDAI 50(c,d) Viikko 12 Viikko 24 | 11 % 16 % | 41 %** 49 %** | 16 % 20 % | 49 %** 57 %** | 13 % 18 % | 45 %** 52 %** |
(a) Kaikki Cimzia-annostukset = tiedot kahdesta ryhmästä, joista toinen sai Cimziaa 200 mg kahden viikon välein ja sitä ennen 400 mg:n aloitusannoksen viikoilla 0, 2 ja 4, ja toinen sai Cimziaa 400 mg neljän viikon välein ja sitä ennen 400 mg:n aloitusannoksen viikoilla 0, 2 ja 4
(b) Tulokset ovat satunnaistetusta joukosta
(c) Waldin testin p‑arvot on esitetty hoitojen vertailemiseksi logistisella regressiomallilla, jossa faktorit ovat hoito ja alue
(d) Koko analyysijoukko
N/A = ei saatavana
*p ≤ 0,05 Cimzia vs. lumelääke
**p < 0,001 Cimzia vs. lumelääke
Selkärangan liikkuvuus
Selkärangan liikkuvuutta arvioitiin kaksoissokkoutetun ja lumekontrolloidun jakson aikana BASMI-mittarin avulla useina ajankohtina, mukaan lukien lähtötilanteessa ja viikoilla 12 ja 24. Lumelääkettä ja Cimziaa saaneiden välillä osoitettiin jokaisella lähtötilanteen jälkeisellä tutkimuskäynnillä kliinisesti merkityksellisiä ja tilastollisesti merkitseviä eroja. Ero lumelääkeryhmään verrattuna oli yleensä suurempi röntgennegatiivista aksiaalista spondylartriittia sairastaneiden ryhmässä kuin selkärankareumaa sairastaneiden ryhmässä. Tämä saattaa johtua siitä, että ensin mainitussa ryhmässä rakennevauriot eivät olleet yhtä pitkäaikaisia kuin jälkimmäisenä mainitussa ryhmässä.
Viikolla 24 saavutettu lineaarisen BASMI-pistemäärän paraneminen säilyi tutkimuksessa mukana pysyneillä potilailla viikon 204 loppuun asti.
Vaste fyysiseen toimintakykyyn ja terveyteen liittyvä hoitotulos
Kliinisessä AS001‑tutkimuksessa Cimzialla hoidetuilla potilailla raportoitiin lumelääkettä saaneisiin verrattuna fyysisen toimintakyvyn merkitsevää paranemista BASFI-mittarilla arvioituna sekä kivun lievittymistä arvioituna kokonaiskivun ja yöllisen selkäkivun numeroasteikoilla. Cimzialla hoidetuilla potilailla raportoitiin lumelääkettä saaneisiin verrattuna väsymyksen (uupuneisuuden) merkittävää lievittymistä arvioituna BASDAI-mittarin fatigue-uupumusosiolla ja lisäksi terveyteen liittyvien elämänlaatupisteiden (HRQoL) paranemista arvioituna selkärankareumapotilaiden elämänlaatumittarilla (ASQoL) ja SF‑36‑kyselylomakkeen fyysisen ja mielenterveysosion yhteenvetopisteillä ja kaikkien osioiden pisteillä. Lumelääkettä saaneisiin verrattuna Cimzialla hoidetut potilaat raportoivat aksiaaliseen spondylartriittiin liittyvää merkittävää tuottavuuden paranemista sekä töissä että kotona Work Productivity Survey ‑kyselyn perusteella. Kaikki edellä mainitut parantuneet tulokset säilyivät tutkimuksessa vielä mukana olleilla potilailla pääosin viikon 204 loppuun asti.
Tulehduksen estyminen magneettikuvissa
Kuvantamisen alatutkimuksessa arvioitiin magneettikuvissa näkyviä tulehduksen merkkejä 153 potilaalla viikolla 12 SPARCC (Spondyloarthritis Research Consortium of Canada) ‑pistemäärän muutoksena lähtötilanteesta SI‑nivelten osalta ja ASspiMRI‑a-pistemäärän (Berlin-modifikaatiot) muutoksena lähtötilanteesta selkärangan osalta. Cimzialla (kaikki annostusryhmät) hoidetuilla potilailla havaittiin viikolla 12 tulehduksen merkkien merkittävää estymistä sekä SI‑nivelissä että selkärangassa tarkasteltaessa aksiaalisen spondylartriittipotilaiden koko joukkoa ja selkärankareuman ja röntgennegatiivisen aksiaalisen spondylartriitin alaryhmiä. Niillä tutkimuksessa vielä mukana olleilla potilailla, joista oli olemassa sekä lähtötilanteen että viikon 204 arvot, tulehduksen merkkien estyminen sekä SI‑nivelissä (n = 72) että selkärangassa (n = 82) säilyi pääosin viikon 204 loppuun asti sekä aksiaalisen spondylartriittipotilaiden koko joukossa että selkärankareuman ja röntgennegatiivisen aksiaalisen spondylartriitin alaryhmissä.
C-OPTIMISE
Annoksen pienentämisen ja hoidon lopettamisen tehoa ja turvallisuutta potilailla, jotka olivat saavuttaneet pitkäkestoisen remission, arvioitiin varhaisvaiheen aktiivista aksiaalista spondylartriittia (oireet kestäneet alle 5 vuotta) sairastavilla aikuispotilailla (18–45-vuotiailla), joiden ASDAS-pisteet olivat ≥ 2,1 (ja tautiin liittyvät tutkimukseenottokriteerit olivat samanlaiset kuin AS001-tutkimuksessa) ja joiden vaste vähintään kahdelle NSAID-lääkkeelle oli ollut riittämätön tai jotka eivät sietäneet NSAID-lääkkeitä tai joiden kohdalla niiden käyttö oli vasta-aiheista. Tutkimukseen osallistui aksiaalisen spondylartriitin molempiin alaryhmiin (selkärankareuma ja röntgennegatiivinen aksiaalinen spondylartriitti) kuuluvia potilaita. Potilaat osallistuivat ensin 48 viikkoa kestäneeseen avoimeen sisäänottovaiheeseen (osa A), jonka aikana he kaikki saivat kolme 400 mg:n latausannosta Cimziaa viikoilla 0, 2 ja 4 ja sen jälkeen 200 mg Cimziaa kahden viikon välein viikolta 6 viikolle 46.
Potilaat, jotka saavuttivat pitkäkestoisen remission (määriteltiin inaktiiviseksi taudiksi [ASDAS < 1,3] vähintään 12 viikon ajan) ja jotka olivat edelleen remissiossa viikolla 48, satunnaistettiin tutkimuksen osaan B, jossa he saivat joko 200 mg Cimziaa kahden viikon välein (N = 104), 200 mg Cimziaa neljän viikon välein (annoksen pienentäminen, N = 105) tai lumelääkettä (hoidon lopettaminen, N = 104) 48 viikon ajan.
Ensisijainen tehomuuttuja oli niiden potilaiden prosenttiosuus, joilla ei esiintynyt taudin aktivoitumista osan B aikana.
Potilaat, joiden tauti aktivoitui osan B aikana, eli joiden ASDAS-pisteet olivat ≥ 2,1 kahdella peräkkäisellä käynnillä tai > 3,5 millä tahansa osan B käynnillä, saivat toisiolääkityksenä 200 mg Cimziaa kahden viikon välein vähintään 12 viikon ajan (lumehoitoa saaneet potilaat saivat 400 mg:n latausannoksen Cimziaa viikoilla 0, 2 ja 4).
Kliininen vaste
Niiden potilaiden prosenttiosuus, jotka saavuttivat pitkäkestoisen remission osan A viikolla 48, oli aksiaalista spondylartriittia sairastavien potilaiden kokonaisjoukossa 43,9 % ja samankaltainen röntgennegatiivista aksiaalista spondylartriittia (45,3 %) ja selkärankareumaa (42,8 %) sairastavien potilaiden alaryhmissä.
Osaan B satunnaistetuista potilaista (N = 313) tilastollisesti merkitsevästi (p < 0,001, NRI) suuremman osan tauti ei aktivoitunut, kun Cimzia-hoitoa jatkettiin annoksella 200 mg kahden viikon välein (83,7 %) tai annoksella 200 mg neljän viikon välein (79,0 %) verrattuna potilaisiin, joiden hoito keskeytettiin (20,2 %).
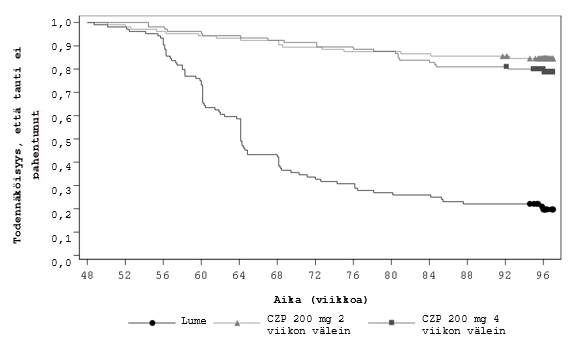
Hoidon lopettaneiden potilaiden ryhmän ja molempien Cimzia-ryhmien välinen ero taudin aktivoitumiseen kuluneessa ajassa oli tilastollisesti merkitsevä (molempien vertailujen p < 0,001) ja kliinisesti merkittävä. Lumeryhmässä taudin aktivoitumisvaiheet alkoivat noin 8 viikon kuluttua Cimzian lopettamisesta, ja suurin osa aktivoitumisvaiheista ilmeni 24 viikon kuluessa hoidon lopettamisesta (kuva 1).
Kuva 1 Kaplan-Meierin käyrä taudin aktivoitumiseen kuluneesta ajasta
Non-responder imputation (NRI) ‑menetelmä; tulokset ovat satunnaistetusta joukosta
Huomautus: taudin aktivoitumiseen kulunut aika määriteltiin satunnaistamispäivän ja taudin aktivoitumispäivän väliseksi ajaksi. Niiden tutkittavien osalta, joilla taudin aktivoitumista ei ilmennyt, taudin aktivoitumiseen kulunut aika sensuroitiin viikon 96 käynnin järjestämispäivänä.
Kaplan-Meierin käyrä katkaistiin 97 viikon kohdalta, kun tutkimus oli < 5 %:lla osallistujista vielä kesken.
Tutkimuksen osan B tulokset esitetään taulukossa 9.
Taulukko 9 Kliinisen vasteen pysyvyys tutkimuksen osassa B viikolla 96
| Päätetapahtumat | Lumelääke (hoidon lopetus) N = 104 | CIMZIA 200 mg kahden viikon välein N = 104 | CIMZIA 200 mg neljän viikon välein N = 105 |
| ASDAS-MI, n (%)1 | |||
| Osan B lähtötilanne (viikko 48) | 84 (80,8) | 90 (86,5) | 89 (84,8) |
| Viikko 96 | 11 (10,6) | 70 (67,3)* | 61 (58,1)* |
| ASAS40, n (%)1 | |||
| Osan B lähtötilanne (viikko 48) | 101 (97,1) | 103 (99,0) | 101 (96,2) |
| Viikko 96 | 22 (21,2) | 88 (84,6)* | 77 (73,3)* |
| BASDAI, muutos osan B lähtötilanteesta (viikko 48), pienimmän neliösumman keskiarvo (SE)2 | |||
| Viikko 96 | 3,02 (0,226) | 0,56 (0,176)* | 0,78 (0,176)* |
| ASDAS, muutos osan B lähtötilanteesta (viikko 48), pienimmän neliösumman keskiarvo (SE)2 | |||
| Viikko 96 | 1,66 (0,110) | 0,24 (0,077)* | 0,45 (0,077)* |
1 Non-responder imputation (NRI) ‑menetelmä; tulokset ovat satunnaistetusta joukosta
2 Toistomittausten sekamalli (MMRM); tulokset ovat satunnaistetusta joukosta
ASDAS-MI = selkärankareuman tautiaktiivisuus – merkittävä parannus; ASAS: Assessment of Sponyloarthritis international Society; ASAS40= ASAS 40 % vastekriteerit; SE = keskivirhe;
Huomautus: ASDAS-pisteissä tapahtunut merkittävä parannus määritellään > 2,0 pisteen laskuksi lähtötilanteesta.
Huomautus: tutkimuksen osan A lähtötilannetta käytettiin viitearvona kliinistä parannusta kuvaavien ASDAS-muuttujien ja ASAS-muuttujien määrittämisessä
* Nimellinen p < 0,001, CIMZIA vs. lumelääke
Tulehduksen estyminen magneettikuvissa
Tutkimuksen osassa B arvioitiin magneettikuvissa näkyviä tulehduksen merkkejä viikolla 48 ja viikolla 96, ja tulokset esitettiin SPARCC‑pistemäärän muutoksena lähtötilanteesta SI‑nivelten osalta ja ASspiMRI‑a-pistemäärän (Berlin-modifikaatiot) muutoksena lähtötilanteesta. Potilailla, jotka olivat saavuttaneet pitkäkestoisen remission viikolla 48, ei joko todettu lainkaan tulehdusta tai sitä todettiin hyvin vähän, eikä tulehduksen merkittävää lisääntymistä havaittu viikolla 96 hoitoryhmästä riippumatta.
Uusintahoito potilailla, joiden tauti aktivoitui
Tutkimuksen osassa B tauti aktivoitui 70 %:lla (73/104) lumehoitoa saaneista potilaista, 14 %:lla (15/105) 200 mg Cimziaa neljän viikon välein saaneista potilaista ja 6,7 %:lla (7/104) 200 mg Cimziaa kahden viikon välein saaneista potilaista. Taudin pahenemisen jälkeen näille potilaille annettiin Cimzia-hoitoa annoksella 200 mg kahden viikon välein.
Ryhmässä, joka oli määrätty saamaan 200 mg Cimziaa neljän viikon välein, kaikki 15 potilasta, joiden tauti aktivoitui, saivat täydet 12 viikkoa toisiolääkitystä Cimzialla. Näiden potilaiden ASDAS-tietojen perusteella 12 potilaalla (80 %) tautiaktiivisuus oli ASDAS-pisteiden mukaan vähäistä tai tauti oli inaktiivinen (eli kaikki ASDAS-pisteet < 2,1) 12 viikkoa avoimen hoidon uudelleenaloittamisen jälkeen.
Hoidon lopettaneiden ryhmässä niistä 73 potilaasta, joiden tauti aktivoitui, 71 potilasta sai täydet 12 viikkoa toisiolääkitystä Cimzialla. Näiden potilaiden ASDAS-tietojen perusteella 64 potilaalla (90 %) tautiaktiivisuus oli ASDAS-pisteiden mukaan vähäistä tai tauti oli inaktiivinen (eli kaikki ASDAS-pisteet < 2,1) 12 viikkoa avoimen hoidon uudelleenaloittamisen jälkeen.
C-OPTIMISE-tutkimuksen tulosten perusteella annoksen pienentämistä voidaan harkita vuoden kestäneen Cimzia-hoidon jälkeen, mikäli potilas on saavuttanut pitkäkestoisen remission (ks. kohta Annostus ja antotapa). Cimzia-hoidon lopettamiseen liittyy suuri taudin aktivoitumisen riski.
Röntgennegatiivinen aksiaalinen spondylartriitti
Cimzian tehoa ja turvallisuutta arvioitiin 52 viikon pituisessa satunnaistetussa, kaksoissokkoutetussa monikeskustutkimuksessa (AS0006) 317:llä vähintään 18-vuotiaalla potilaalla, joilla oli aikuisiällä alkanut aksiaalinen spondylartriitti ja selkäkipua vähintään 12 kuukauden ajan. Potilaiden piti täyttää röntgennegatiivisen aksiaalisen spondylartriitin ASAS-kriteerit (ei suvussa esiintymistä eikä hyvää vastetta ei-steroidisille tulehduskipulääkkeille), ja heillä piti olla objektiivisia tulehduksen merkkejä siten, että C-reaktiivisen proteiinin (CRP) pitoisuudet olivat yli viitealueen ylärajan ja/tai magneettikuvauksessa oli havaittavissa sakroiliittia, mikä viittaa tulehdukselliseen sairauteen [positiivinen CRP (yli viitealueen ylärajan) ja/tai positiivinen magneettikuvauksen tulos] mutta ei yksiselitteistä radiografista näyttöä sakroiliaalisten nivelten rakenteellisista vaurioista. BASDAI-indeksillä määriteltynä sairaus oli aktiivinen (indeksi ≥ 4), ja rankakivun vaikeusaste oli ≥ 4 numeroasteikolla 0–10. Tutkimukseen otettiin vain potilaita, joilla vähintään kaksi NSAID-hoitokokeilua oli epäonnistunut potilaan intoleranssin tai riittämättömän hoitovasteen vuoksi. Potilaat saivat joko lumelääkettä tai 400 mg Cimziaa aloitusannoksena viikoilla 0, 2 tai 4 ja sen jälkeen 200 mg Cimziaa kahden viikon välein. Normaalin lääkityksen (esim. ei-steroidiset tulehduskipulääkkeet, taudin kulkua muuttavat reumalääkkeet, kortikosteroidit, kipulääkkeet) käyttö ja annoksen muuttaminen oli sallittua milloin tahansa. Ensisijaisena tehomuuttujana oli Ankylosing Spondylitis Disease Activity Score -pistemäärän merkittävänä kohentumisena (ASDAS-MI) mitattu vaste viikolla 52. ASDAS-MI-vaste määriteltiin ASDAS-pistemäärän vähenemiseksi (kohentumiseksi) vähintään 2,0 pisteellä lähtötilanteeseen nähden tai pienimmän mahdollisen pistemäärän saavuttamiseksi. ASAS 40 oli toissijaisena päätetapahtumana.
Lähtötilanteessa 37 %:lla CIMZIA-ryhmän potilaista ja 41 %:lla lumelääkeryhmän potilaista oli korkean taudin aktiivisuus (ASDAS ≥ 2,1, ≤ 3,5). 62 %:lla CIMZIA-ryhmän potilaista ja 58 %:lla lumelääkeryhmän potilaista oli erittäin korkea taudin aktiivisuus (ASDAS > 3,5).
Kliininen vaste
Tutkimus AS0006, johon osallistuneilla tutkittavilla ei ollut radiografisia tulehduksen merkkejä SI-nivelissä, vahvisti aiemmin AS001-tutkimuksessa todetun tehon tässä potilaiden osajoukossa.
Viikolla 52 tilastollisesti merkitsevästi suurempi osa Cimzia-hoitoa saaneista potilaista saavutti ADAS-MI-vasteen lumelääkehoitoa saaneisiin potilaisiin verrattuna. Cimzia-hoitoa saaneilla potilailla oli myös kohentumista lumelääkkeeseen verrattuna useissa aksiaalisen spondylartriitin aktiivisuuden komponenteissa, mukaan lukien CRP. Sekä viikolla 12 että viikolla 52 ASAS 40 ‑vasteita oli merkitsevästi enemmän Cimzia-hoitoa saaneilla kuin lumelääkettä saaneilla. Keskeiset tulokset on esitetty taulukossa 10.
Taulukko 10 ASDAS-MI- ja ASAS 40 -vasteet tutkimuksessa AS0006 (potilaiden prosenttiosuus)
| Parametrit | Lumelääke N=158 | Cimziaa 200 mg 2 viikon välein N=159 |
ASDAS-MI Viikko 52 | 7 % | 47 %* |
ASAS 40 Viikko 12 Viikko 52 | 11 % 16 % | 48 %* 57 %* |
a Cimzia 2 viikon välein annettuna, mitä ennen annettiin 400 mg:n aloitusannos viikoilla 0, 2 ja 4
* p < 0,001
Kaikki prosenttiosuudet edustavat niiden potilaiden suhteellista osuutta, jotka saivat vasteeen täydessä analyysijoukossa.
Viikolla 52 niiden potilaiden prosenttiosuus, jotka saavuttivat ASDAS-asteikon inaktiivisen taudin tilanteen (ASDAS < 1,3), oli 36,4 % Cimzia-ryhmässä ja 11,8 % lumelääkeryhmässä.
Viikolla 52 Cimzia-hoitoa saaneilla potilailla havaittiin kliinisesti merkittävä kohentuminen MASES -asteikolla mitattuna lumelääkkeeseen verrattuna (pienimmän neliösumman keskimääräinen muutos lähtötilanteesta oli -2,4 Cimzia-ryhmässä ja -0,2 lumelääkeryhmässä).
Nivelpsoriaasi
Cimzian tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa ja lumekontrolloidussa kliinisessä monikeskustutkimuksessa (PsA001), johon osallistuneilla 409:llä ≥ 18‑vuotiaalla potilaalla oli aikuisiällä alkanut aktiivinen nivelpsoriaasi. Potilailla oli ollut CASPAR (Classification Criteria for Psoriatic Arthritis) ‑kriteerit täyttävä nivelpsoriaasi vähintään 6 kuukautta. Potilailla oli ≥ 3 turvonnutta ja aristavaa niveltä ja suurentuneet akuutin vaiheen reaktanttiarvot. Potilailla oli myös aktiivisia psoriaattisia iholeesioita tai anamneesissa dokumentoitu psoriaasi sekä yksi tai useampi epäonnistunut hoitokokeilu taudinkulkua muuttavilla reumalääkkeillä (DMARD). Potilailla sai olla anamneesissa yksi aiempi TNF:n estäjähoito, ja 20 % potilaista oli aiemmin altistunut TNF:n estäjille. Potilaat saivat ensin aloitusannoksena 400 mg Cimziaa viikoilla 0, 2 ja 4 (molemmissa hoitoryhmissä) tai lumelääkettä ja tämän jälkeen joko 200 mg Cimziaa kahden viikon välein, 400 mg Cimziaa neljän viikon välein tai lumelääkettä kahden viikon välein. Potilaista 72,6 % sai samanaikaisesti tulehduskipulääkkeitä (NSAIDeja) ja 70,2 % tavanomaisia taudinkulkua muuttavia reumalääkkeitä (DMARD). Ensisijaisia päätetapahtumia oli kaksi: viikolla 12 ACR 20 ‑vasteen saavuttaneiden potilaiden prosenttiosuus ja mTSS–pistemäärän muutos lähtötilanteesta viikolle 24. Cimzian tehoa ja turvallisuutta ei analysoitu erikseen sellaisilla nivelpsoriaasipotilailla, joiden vallitseva oire oli sakroiliitti tai aksiaalinen spondylartriitti.
Tutkimuksen 24 viikon lumekontrolloitua ja kaksoissokkoutettua hoitojaksoa seurasi annoksen suhteen sokkoutettu 24 viikon hoitojakso ja 168 viikon avoin hoitojakso. Tutkimus kesti pisimmillään 216 viikkoa. Kaikki potilaat saivat Cimziaa sekä annoksen suhteen sokkoutetun jakson että avoimen seurantajakson aikana. Yhteensä 264 potilasta (64,5 %) pysyi tutkimuksessa viikon 216 loppuun asti.
ACR‑vaste
ACR 20 -vaste saavutettiin viikoilla 12 ja 24 tilastollisesti merkitsevästi (p < 0,001) useammin Cimzialla hoidetuilla kuin lumelääkkeellä hoidetuilla potilailla. Lumelääkkeeseen verrattuna ACR 20 ‑vasteen saavuttaneiden potilaiden prosenttiosuus oli lähtötilanteesta viikon 24 loppuun saakka kliinisesti merkityksellinen jokaisella seurantakäynnillä hoitoryhmissä, joista toinen sai 200 mg Cimziaa kahden viikon välein ja toinen 400 mg Cimziaa neljän viikon välein (nominaalinen p‑arvo ≤ 0,001 jokaisella käynnillä). Lisäksi ACR 50‑ ja 70‑vasteen saavuttaneiden prosenttiosuudet Cimzia-hoitoryhmissä paranivat merkitsevästi. Nivelpsoriaasille tyypillistä perifeeristä aktiivisuutta mittaavat parametrit (esim. turvonneiden nivelten lukumäärä, kipeiden/aristavien nivelten lukumäärä, daktyliitti ja entesiitti) paranivat Cimzialla hoidetuilla potilailla viikoilla 12 ja 24 (nominaalinen p‑arvo < 0,01).
Taulukossa 11 on esitetty kliinisestä PsA001‑tutkimuksesta saadut keskeiset tehoa koskevat tulokset.
Taulukko 11 Kliinisestä PsA001‑tutkimuksesta saadut keskeiset hoidon tehoa koskevat tulokset (potilaiden prosenttiosuudet)
| Vaste | Lumelääke N = 136 | Cimzia(a) 200 mg kahden viikon välein N = 138 | Cimzia(b) 400 mg neljän viikon välein N = 135 |
ACR 20 Viikko 12 Viikko 24 | 24 % 24 % | 58 %** 64 %** | 52 %** 56 %** |
ACR 50 Viikko 12 Viikko 24 | 11 % 13 % | 36 %** 44 %** | 33 %** 40 %** |
ACR 70 Viikko 12 Viikko 24 | 3 % 4 % | 25 %** 28 %** | 13 %* 24 %** |
| Vaste | Lumelääke N = 86 | Cimzia(a) 200 mg kahden viikon välein N = 90 | Cimzia(b) 400 mg neljän viikon välein N = 76 |
PASI 75 (c) Viikko 12 Viikko 24 Viikko 48 | 14 % 15 % N/A | 47 %*** 62 %*** 67 % | 47 %*** 61 %*** 62 % |
(a) Cimziaa kahden viikon välein, sitä ennen aloitusannoksena 400 mg viikoilla 0, 2 ja 4
(b) Cimziaa neljän viikon välein, sitä ennen aloitusannoksena 400 mg viikoilla 0, 2 ja 4
(c) Tutkittavat, joilla psoriaasi peitti vähintään 3 % ihoalueesta lähtötilanteessa
*p < 0,01, Cimzia vs. lumelääke
**p < 0,001, Cimzia vs. lumelääke
***p < 0,001 (nominaalinen), Cimzia vs. lumelääke
Tulokset ovat satunnaistetusta joukosta. Hoitoero: Cimzia 200 mg ‒ lumelääke ja 400 mg ‒ lumelääke ‑vertailua (ja vastaavat 95 %:n luottamusvälit ja p‑arvot) estimoitiin kaksisuuntaisella Waldin asymptoottisten keskivirheiden testillä. Puuttuvat tiedot korvattiin non-responder imputation (NRI) ‑menetelmällä niiden potilaiden osalta, jotka poistuivat ennenaikaisesti hoidosta tai joiden hoidosta ei muuten saatu kaikkea tarvittavaa tietoa.
Niistä 273 potilaasta, jotka alussa satunnaistettiin saamaan joko 200 mg Cimziaa kahden viikon välein tai 400 mg Cimziaa neljän viikon välein, 237 (86,8 %) sai edelleen samaa hoitoa viikolla 48. Niistä 138 potilaasta, jotka satunnaistettiin saamaan 200 mg Cimziaa kahden viikon välein, 92 saavutti ACR 20 ‑vasteen, 68 ACR 50 ‑vasteen ja 48 ACR 70 ‑vasteen viikolla 48. Niistä 135 potilaasta, jotka alussa satunnaistettiin saamaan 400 mg Cimziaa neljän viikon välein, 89 saavutti ACR 20 ‑vasteen, 62 ACR 50 ‑vasteen ja 41 ACR 70 ‑vasteen. Tutkimuksessa mukana pysyneiden potilaiden ACR 20‑, ACR 50‑ ja ACR 70 ‑vasteet säilyivät viikon 216 loppuun asti. Näin kävi myös perifeerisen aktiivisuuden muille parametreille (esim. turvonneiden nivelten lukumäärä, kipeiden/aristavien nivelten lukumäärä, daktyliitti ja entesiitti).
Radiografinen vaste
Kliinisessä PsA001‑tutkimuksessa rakenteellisten nivelvaurioiden etenemisen estoa arvioitiin röntgenkuvin ja tätä ilmaistiin mTSS:n ja sen osa-alueiden, eroosiopisteiden (erosion score, ES) ja nivelraon kaventumista (joint space narrowing, JSN) kuvaavien pisteiden, muutoksena lähtötilanteesta viikolle 24. mTSS‑arviointi muutettiin nivelpsoriaasiin sopivaksi lisäämällä arviointiin sormen kärkinivelet (DIP‑nivelet). Lumehoitoon verrattuna Cimzia-hoito esti vaurion radiografisen etenemisen viikolla 24 mitattuna mTSS‑pistemäärän muutoksena lähtötilanteesta (pienimmän neliösumman keskiarvo [±SE] oli lumelääkeryhmässä 0,28 [±0,07] vs. 0,06 [±0,06] yhdistetyssä Cimzia-annosryhmässä; p = 0,007). Radiografinen eteneminen estyi Cimzia-hoidossa viikolle 48 asti niillä potilailla, joilla radiografisen etenemisen riski oli muita suurempi (eli potilailla, joiden mTSS-lähtöpisteet > 6). Tutkimuksessa mukana pysyneillä potilailla radiografinen eteneminen estyi edelleen viikolle 216.
Vaste fyysiseen toimintakykyyn ja terveyteen liittyvä hoitotulos
Kliinisessä PsA001‑tutkimuksessa Cimzia-hoitoa saaneet potilaat raportoivat merkittävää parantumista lumelääkkeeseen verrattuna arvioitaessa fyysistä toimintakykyä Health Assessment Questionnaire – Disability Index (HAQ‑DI) ‑kyselyllä, kipua PAAP‑kyselyllä sekä väsymystä (uupuneisuutta) Fatigue Assessment Scale (FAS) ‑pisteytyksellä. Cimzia-hoitoa saaneet potilaat raportoivat merkittävää paranemista lumelääkettä saaneisiin verrattuna arvioitaessa terveyteen liittyvää elämänlaatua nivelpsoriaasipotilaiden elämänlaatukyselyllä (PsAQoL) ja SF‑36-kyselyn fyysisen toimintakyvyn ja mielenterveyden osioilla sekä arvioitaessa nivelpsoriaasiin liittyvää tuottavuutta palkka- ja kotitöissä Work Productivity Survey ‑kyselyllä. Kaikkien edellä mainitut parantuneet tulokset säilyivät viikon 216 loppuun asti.
Läiskäpsoriaasi
Cimzia-valmisteen tehoa ja turvallisuutta arvioitiin kahdessa lumekontrolloidussa tutkimuksessa (CIMPASI-1 ja CIMPASI-2) ja yhdessä lumelääkkeellä ja vaikuttavalla aineella kontrolloidussa tutkimuksessa (CIMPACT) vähintään 18-vuotiailla potilailla, joilla oli ollut keskivaikea tai vaikea krooninen läiskäpsoriaasi vähintään 6 kuukautta. Potilaiden psoriaasin vaikeusastetta kuvaava PASI (Psoriasis Area ja Severity Index) -indeksi oli ≥ 12, psoriaasin levinneisyyttä kuvaava BSA (Body Surface Area) ‑arvo ≥ 10 %, lääkärin yleisarvio (Physician Global Assessment, PGA) ≥ 3, ja potilaita pidettiin sopivina systeemiseen hoitoon ja/tai valohoitoon ja/tai fotokemoterapiaan. Faasin III tutkimukseen (CIMPASI-1, CIMPASI-2 ja CIMPACT) ei otettu mukaan potilaita, joilla ei ollut saavutettu ensisijaista vastetta mihinkään aikaisempaan biologiseen hoitoon (määritelmä: ei vastetta ensimmäisten 12 hoitoviikon aikana). CIMPACT-tutkimuksessa Cimzian tehoa ja turvallisuutta verrattiin etarneseptiin.
CIMPASI-1- ja CIMPASI-2-tutkimuksissa ensisijaiset tehon päätetapahtumat olivat niiden potilaiden osuudet, joilla saavutettiin PASI‑75‑vaste ja PGA-taso "täysin parantunut " tai "lähes parantunut" (lähtötaso parantunut vähintään 2 pisteellä) viikolla 16. CIMPACT-tutkimuksessa ensisijainen tehon päätetapahtuma oli niiden potilaiden osuus, joilla saavutettiin PASI‑75‑vaste viikolla 12. Tärkeimmät toissijaiset päätetapahtumat olivat PASI‑75 ja PGA viikolla 16. Kaikissa kolmessa tutkimuksessa tärkein toissijainen päätetapahtuma oli PASI‑90 viikolla 16.
CIMPASI-1-tutkimuksessa arvioitiin 234 potilasta ja CIMPASI-2-tutkimuksessa 227. Molemmissa tutkimuksissa potilaat satunnaistettiin saamaan lumelääkettä tai 200 mg Cimziaa kahden viikon välein (viikoilla 0, 2 ja 4 annetun 400 mg:n Cimzia-aloitusannoksen jälkeen) tai 400 mg Cimziaa kahden viikon välein. Ne Cimzia-valmistetta käyttämään satunnaistetut potilaat, jotka saavuttivat PASI‑50‑vasteen viikolla 16, jatkoivat Cimzia-hoitoa enintään viikkoon 48 asti samalla satunnaistetulla annoksella. Potilaat, jotka oli alun perin satunnaistettu lumelääkeryhmään ja jotka saavuttivat PASI‑50‑vasteen, mutta eivät PASI‑75‑vastetta viikolla 16, saivat 200 mg Cimziaa kahden viikon välein (400 mg:n Cimzia-aloitusannos viikoilla 16, 18 ja 20). Potilaat, joiden vaste viikolla 16 oli riittämätön (PASI‑50 potilailla, joilla ei saavutettu vastetta), saivat 400 mg Cimziaa kahden viikon välein avoimessa hoitojaksossa enintään 128 viikon ajan.
CIMPACT-tutkimuksessa arvioitiin 559 potilasta. Potilaat satunnaistettiin saamaan lumelääkettä tai 200 mg Cimziaa kahden viikon välein (viikoilla 0, 2 ja 4 annetun 400 mg:n Cimzia-aloitusannoksen jälkeen) tai 400 mg Cimziaa kahden viikon välein enintään viikolle 16 asti tai etanerseptiä 50 mg kahdesti viikossa enintään viikolle 12 asti.
Potilaat, jotka alun perin oli satunnaistettu saamaan Cimziaa ja jotka saavuttivat PASI‑75‑vasteen viikolla 16, satunnaistettiin uudelleen alkuperäisen annostusohjelmansa perusteella. Potilaat, jotka saivat 200 mg Cimziaa kahden viikon välein, satunnaistettiin uudelleen saamaan joko 200 mg Cimziaa kahden viikon välein, 400 mg Cimziaa neljän viikon välein tai lumelääkettä. Potilaat, jotka saivat 400 mg Cimziaa kahden viikon välein, satunnaistettiin uudelleen saamaan joko 400 mg Cimziaa kahden viikon välein, 200 mg Cimziaa kahden viikon välein tai lumelääkettä. Potilaita arvioitiin kaksoissokkoutetusti ja lumekontrolloidusti viikon 48 loppuun asti. Kaikki ne tutkittavat, jotka eivät saavuttaneet PASI‑75‑vastetta viikolla 16, siirtyivät toisiohoitoryhmään, jossa hoitona oli 400 mg Cimziaa kahden viikon välein avoimessa hoitojaksossa enintään 128 viikon ajan.
Kaikissa kolmessa tutkimuksessa sokkoutettua 48 viikon ylläpitojaksoa seurasi 96 viikon avoin hoitojakso potilaille, jotka olivat saaneet PASI 50 -vasteen viikkoon 48 mennessä. Kaikki nämä potilaat, mukaan lukien ne, jotka saivat 400 mg Cimziaa kahden viikon välein, aloittivat avoimen jakson Cimzia-annoksella 200 mg kahden viikon välein.
Potilaat olivat pääsääntöisesti miehiä (64 %) ja valkoihoisia (94 %). Heidän keski-ikänsä oli 45,7 vuotta (18–80 vuotta); 7,2 % oli ≥ 65-vuotiaita. Niistä 850 satunnaistetusta potilaasta, jotka saivat joko lumelääkettä tai Cimziaa näissä lumekontrolloiduissa tutkimuksissa, 29 % ei ollut saanut aiempaa systeemistä hoitoa psoriaasiin. Potilaista 47% oli saanut aiempaa valohoitoa tai fotokemoterapiaa, ja 30 % oli saanut aiempaa biologista hoitoa psoriaasiin. Näistä 850 potilaasta 14 % oli saanut vähintään yhtä TNF:n estäjää, 13 % oli saanut IL-17:n estäjää ja 5 % oli saanut IL‑12/23:n estäjää. 18 % potilaista oli lähtötilanteessa ilmoittanut sairastaneensa aiemmin nivelpsoriaasia. Keskimääräinen PASI-indeksi lähtötilanteessa oli 20, ja vaihteluväli oli 12–69. Lähtötilanteen PGA-taso vaihteli keskivaikeasta (70 %) vaikeaan (30 %). Keskimääräinen BSA-lähtöarvo oli 25 %, ja vaihteluväli oli 10–96 %.
Kliininen vaste viikoilla 16 ja 48
CIMPASI-1- ja CIMPASI-2-tutkimusten keskeiset tulokset on esitetty taulukossa 12.
Taulukko 12 Kliininen vaste CIMPASI-1- ja CIMPASI-2-tutkimuksissa viikoilla 16 ja 48
| Viikko 16 | Viikko 48 | ||||
| CIMPASI-1 | |||||
Lumelääke N = 51 | Cimzia 200 mg Q2Wa) N = 95 | Cimzia 400 mg Q2W N = 88 | Cimzia 200 mg Q2W N = 95 | Cimzia 400 mg Q2W N = 88 | |
| PGA ”täysin parantunut” tai ”lähes parantunut”b) | 4,2 % | 47,0 %* | 57,9 %* | 52,7 % | 69,5 % |
| PASI‑75 | 6,5 % | 66,5 %* | 75,8 %* | 67,2 % | 87,1 % |
| PASI‑90 | 0,4 % | 35,8 %* | 43,6 %* | 42,8 % | 60,2 % |
| CIMPASI-2 | |||||
Lumelääke N = 49 | Cimzia 200 mg Q2Wa) N = 91 | Cimzia 400 mg Q2W N = 87 | Cimzia 200 mg Q2W N = 91 | Cimzia 400 mg Q2W N = 87 | |
| PGA ”täysin parantunut” tai ”lähes parantunut”b | 2,0 % | 66,8 %* | 71,6 %* | 72,6 % | 66,6 % |
| PASI‑75 | 11,6 % | 81,4 %* | 82,6 %* | 78,7 % | 81,3 % |
| PASI‑90 | 4,5 % | 52,6 %* | 55,4 %* | 59,6 % | 62,0 % |
a) 400 mg:n Cimzia-aloitusannos viikoilla 0, 2 ja 4, minkä jälkeen 200 mg kahden viikon välein.
b) Viisiportainen PGA‑arviointiasteikko. Hoidon onnistumistaso "täysin parantunut" (0) tai "lähes parantunut" (1) tarkoittaa, että psoriaasin merkkejä ei ole tai että leesioiden väri on normaali tai vaaleanpunainen, plakit eivät ole paksuuntuneet ja ihon paikoittainen hilseily on hyvin vähäistä tai sitä ei ole lainkaan.
* Cimzia verrattuna lumelääkkeeseen: p < 0,0001.
Hoitoon vastanneiden määriä ja p-arvoja PASI:n ja PGA:n suhteen arvioitiin logistisella regressiomallilla, jossa puuttuvat tiedot laskettiin moninkertaista imputointia käyttävällä MCMC-menetelmällä. Tutkittavia, jotka siirrettiin toisiohoitoon tai jotka keskeyttivät osallistumisensa (koska eivät saavuttaneet PASI‑50‑vastetta), käsiteltiin tutkittavina, joilla ei saavutettu hoitovastetta viikolla 48.
Tulokset koskevat satunnaistettujen tutkittavien joukkoa.
Tärkeimmät tulokset CIMPACT-tutkimuksesta on esitetty taulukossa 13.
Taulukko 13 Kliininen vaste CIMPACT-tutkimuksessa viikoilla 12 ja 16
| Viikko 12 | Viikko 16 | ||||||
Lumelääke N = 57 | Cimzia 200 mg Q2Wa) N = 165 | Cimzia 400 mg Q2W N = 167 | Etanersepti 50 mg BiW N = 170 | Lumelääke N = 57 | Cimzia 200 mg Q2W N = 165 | Cimzia 400 mg Q2W N = 167 | |
| PASI‑75 | 5 % | 61,3 %*,§ | 66,7 %*,§§ | 53,3 % | 3,8 % | 68,2 %* | 74,7 %* |
| PASI‑90 | 0,2 % | 31,2 %* | 34,0 %* | 27,1 % | 0,3 % | 39,8 %* | 49,1 %* |
| PGA ”täysin parantunut” tai ”lähes parantunut”b) | 1,9 % | 39,8 %** | 50,3 %* | 39,2 % | 3,4 % | 48,3 %* | 58,4 %* |
a) 400 mg:n Cimzia-aloitusannos viikoilla 0, 2 ja 4, minkä jälkeen 200 mg kahden viikon välein.
b) Viisiportainen PGA‑arviointiasteikko. Hoidon onnistumistaso "täysin parantunut" (0) tai "lähes parantunut" (1) tarkoittaa, että psoriaasin merkkejä ei ole tai että leesioiden väri on normaali tai vaaleanpunainen, plakit eivät ole paksuuntuneet ja ihon paikoittainen hilseily on hyvin vähäistä tai sitä ei ole lainkaan.
* Cimzia verrattuna lumelääkkeeseen: p < 0,0001.
§ Cimzia 200 mg kahden viikon välein verrattuna etanerseptiin, 50 mg kahdesti viikossa, osoittautui vertailukelpoiseksi (ero etarneseptin ja kahden viikon välein annetun 200 mg:n Cimzia-annoksen välillä oli 8,0 %, 95 %:n luottamusväli: ‑2,9, 18,9, ennalta määritetyn 10 %:n vertailukelpoisuusrajan perusteella).
§§ Cimzia 400 mg kahden viikon välein verrattuna etanerseptiin, 50 mg kahdesti viikossa, osoittautui paremmaksi (p < 0,05).
** Cimzia verrattuna lumelääkkeeseen: p < 0,001. Hoitoon vastanneiden määrät ja p-arvot perustuvat logistiseen regressiomalliin. Puuttuvat tiedot laskettiin moninkertaista imputointia käyttävällä MCMC-menetelmällä.
Tulokset koskevat satunnaistettujen tutkittavien joukkoa.
Kaikissa kolmessa tutkimuksessa PASI‑75‑vaste saavutettiin merkitsevästi suuremmalla osalla Cimzia-hoidossa kuin lumelääkehoidossa viikosta 4 lähtien.
Molemmilla Cimzia-annoksilla osoitettiin teho verrattuna lumelääkkeeseen potilaan iästä, sukupuolesta, ruumiinpainosta, BMI:stä, psoriaasin kestosta tai potilaan aikaisemmista systeemisistä tai biologisista hoidoista riippumatta.
Vasteen säilyminen
Niistä CIMPASI-1‑ ja CIMPASI-2‑tutkimusten yhteisanalyysissa mukana olleista potilaista, joilla saavutettiin PASI‑75‑vaste viikolla 16 ja jotka saivat Cimziaa joko 400 mg (N = 134 [175:stä satunnaistetusta tutkittavasta]) tai 200 mg (N = 132 [186:sta satunnaistetusta tutkittavasta]) kahden viikon välein, vasteen säilyttäneitä oli viikolla 48 ensin mainitussa hoitoryhmässä 98,0 % ja toisena mainitussa hoitoryhmässä 87,5 %. Vastaavasti niistä potilaista, joiden PGA-taso oli ”täysin parantunut” tai ”lähes parantunut” viikolla 16 ja jotka saivat Cimziaa joko 400 mg (N = 103/175) tai 200 (N = 95/186) mg kahden viikon välein, vasteen säilyttäneitä oli viikolla 48 ensin mainitussa hoitoryhmässä 85,9 % ja toisena mainitussa hoitoryhmässä vastaavasti 84,3 %.
Vasteen säilymistä arvioitiin 96 viikon avoimen lisähoitojakson jälkeen (viikko 144). Kaksikymmentäyksi prosenttia kaikista satunnaistetuista tutkittavista katosi seurannasta ennen viikkoa 144. Noin 27 %:lla tutkittavista, jotka pysyivät tutkimuksessa mukana loppuun saakka ja aloittivat avoimen hoitojakson (viikot 48–144) annoksella 200 mg Cimziaa kahden viikon välein, annosta suurennettiin 400 mg:aan Cimziaa kahden viikon välein vasteen säilyttämiseksi. Analyysissa, jossa kaikkia potilaita, joiden hoito oli epäonnistunut, pidettiin hoitoon vastaamattomina, 200 mg Cimziaa kahden viikon välein saaneessa hoitoryhmässä vasteen säilyttäneitä oli 96 viikkoa kestäneen avoimen lisähoitojakson jälkeen 84,5 % tutkittavista, jotka saavuttivat PASI 75 ‑vasteen viikolla 16 ja 78,4 % tutkittavista, joiden PGA-taso oli viikolla 16 ”täysin parantunut” tai ”lähes parantunut”. Cimziaa 400 mg kahden viikon välein saaneessa ryhmässä, joka sai avoimen hoitojakson alussa Cimziaa 200 mg kahden viikon välein, vasteen säilyttäneitä oli 84,7 % tutkittavista, jotka saavuttivat PASI 75 ‑vasteen viikolla 16, ja 73,1 % tutkittavista, joiden PGA-taso oli viikolla 16 ”täysin parantunut” tai ”lähes parantunut”.
Hoitoon vastanneiden määrät perustuvat logistiseen regressiomalliin, jossa puuttuvat tiedot laskettiin 48 tai 144 viikon ajalta moninkertaisella imputoinnilla (MCMC-menetelmä) yhdistettynä hoidon epäonnistumisen osalta NRI-menetelmään.
CIMPACT-tutkimuksessa viikolla 16 PASI‑75‑vasteen saaneilla potilailla, jotka saivat 400 mg Cimziaa kahden viikon välein ja jotka satunnaistettiin uudelleen saamaan joko Cimziaa 400 mg tai 200 mg kahden viikon välein tai lumelääkettä, Cimziaa saaneissa ryhmissä oli prosentuaalisesti enemmän PASI‑75‑vasteen saavuttaneita viikolla 48 kuin lumelääkeryhmässä (98,0 %, 80,0 % ja 36,0 % ryhmittäin). Viikolla 16 PASI‑75‑vasteen saaneista, 200 mg Cimziaa kahden viikon välein saaneista potilaista, jotka oli uudelleen satunnaistettu saamaan joko 400 mg Cimziaa neljän viikon välein, 200 mg Cimziaa kahden viikon välein tai lumelääkettä, Cimzia-ryhmissä oli myös suurempi prosenttiosuus PASI‑75‑vasteen saaneita viikolla 48 kuin lumelääkeryhmässä (88,6 %, 79,5 % ja 45,5 % ryhmittäin). Puuttuvat tiedot korvattiin non-responder imputation (NRI) -menetelmällä.
Elämänlaatu / potilaiden ilmoittamat hoitotulokset
Tilastollisesti merkitsevät parannukset lumelääkkeeseen verrattuna viikolla 16 lähtötilanteesta alkaen (CIMPASI-1 ja CIMPASI-2) havaittiin DLQI-indeksissä (Dermatology Life Quality Index). Keskimääräinen väheneminen (parantuminen) DLQI-indeksissä lähtötilanteesta alkaen oli -8,9…-11,1, kun Cimzia-annos oli 200 mg kahden viikon välein, ja -9,6…-10,0, kun Cimzia-annos oli 400 mg kahden viikon välein verrattuna lumelääkeryhmään, jossa vastaavat arvot olivat -2,9…-3,3 viikolla 16.
Lisäksi viikolla 16 suurempi osa Cimziaa saaneista potilaista sai DLQI-indeksin 0 tai 1 (45,5 % ja 50,6 % ryhmässä 400 mg Cimziaa kahden viikon välein; 47,4 % ja 46,2 % ryhmässä 200 mg Cimziaa kahden viikon välein; 5,9 % ja 8,2 % lumelääkeryhmässä).
Parannus DLQI-indeksissä säilyi viikkoon 144 saakka tai pieneni hiukan.
Lumelääkettä saaneisiin verrattuna Cimzia-hoitoa saaneiden potilaiden ilmoittama paraneminen oli Hospital Anxiety and Depression Scale (HADS)-D ‑asteikolla suurempi.
Immunogeenisuus
Seuraavat tiedot kuvaavat niiden potilaiden prosenttiosuutta, joiden sertolitsumabipegolin vasta-aineiden testitulosten katsottiin olevan positiivisia ELISA-määrityksessä ja myöhemmin vielä herkemmässä määrityksessä. Nämä tulokset ovat hyvin riippuvaisia määrityksen herkkyydestä ja spesifisyydestä. Määrityksissä havaittu vasta-aineiden (neutraloivat vasta-aineet mukaan lukien) ilmaantuvuus on hyvin riippuvainen monista eri tekijöistä, joita ovat esimerkiksi määrityksen herkkyys ja spesifisyys, testimenetelmä, näytteiden käsittely, näytteiden ottoajankohta, samanaikaiset lääkitykset ja perussairaudet. Näistä syistä alla kuvatuissa tutkimuksissa havaitun sertolitsumabipegolin vasta-aineiden ilmaantuvuuden vertaaminen vasta-aineiden ilmaantuvuuteen muissa tutkimuksissa tai muiden valmisteiden käytön yhteydessä voi antaa harhaanjohtavia tuloksia.
Nivelreuma
Niiden potilaiden kokonaisprosenttiosuus, joilla havaittiin vähintään kerran vasta-aineita Cimzia-valmisteelle, oli 9,6 % nivelreumaa koskevissa lumekontrolloiduissa tutkimuksissa. Noin kolmanneksella vasta-ainepositiivisista potilaista vasta-aineet olivat neutraloivia in vitro. Samanaikaista immunosuppressiivista hoitoa (metotreksaatti) saaneille potilaille muodostui vasta-aineita harvemmin kuin potilaille, jotka eivät saaneet immunosuppressiivista hoitoa lähtötilanteesta. Vasta-aineiden muodostumiseen liittyi lääkeaineen pitoisuuden pienenemistä plasmassa ja osalla potilaista tehon heikkenemistä.
Kahdessa pitkäaikaisessa (altistus pisimmillään 5 vuotta) avoimessa tutkimuksessa niiden potilaiden kokonaisprosenttiosuus, joilla havaittiin vasta-aineita Cimzia-valmisteelle vähintään kerran, oli 13 % (Cimzia-vasta-aineiden muodostus oli tilapäistä 8,4 prosentilla ja pysyvää 4,7 prosentilla kaikista potilaista). Niiden potilaiden kokonaisprosenttiosuus, jotka olivat vasta-ainepositiivisia ja joilla plasman lääkeainepitoisuus oli laskenut pysyvästi, oli 9,1 %. Kuten lumekontrolloiduissa tutkimuksissa, vasta-ainepositiivisuuteen liittyi osalla potilaista tehon heikkenemistä.
Faasin III tutkimustietoihin perustuva farmakodynaaminen malli ennustaa, että noin 15 %:lle potilaista kehittyy vasta-aineita kuuden kuukauden kuluessa, kun heidän hoitonsa toteutetaan suositellulla annoksella (aloitusannoksen jälkeen 200 mg kahden viikon välein) ilman samanaikaista metotreksaattihoitoa. Tämä potilasosuus pienenee, kun samanaikaisen metotreksaattihoidon annosta suurennetaan. Nämä tulokset ovat melko samansuuntaiset havaittujen tietojen kanssa.
Nivelpsoriaasi
Niiden potilaiden kokonaisprosenttiosuus, joilla havaittiin vähintään kerran vasta-aineita Cimzia-valmisteelle viikolle 24 asti, oli 11,7 % faasin III lumekontrolloidussa tutkimuksessa, johon osallistui nivelpsoriaasipotilaita. Vasta-aineiden muodostumiseen liittyi lääkeaineen pitoisuuden pienenemistä plasmassa.
Koko tutkimuksen (pisimmillään 4 vuoden altistuksen) aikana niiden potilaiden kokonaisprosenttiosuus, joilla havaittiin vähintään kerran vasta-aineita Cimzia-valmisteelle, oli 17,3 % (Cimzia-vasta-aineiden muodostus oli tilapäistä 8,7 %:lla ja pysyvää 8,7 %:lla). Niiden potilaiden kokonaisprosenttiosuus, jotka olivat vasta-ainepositiivisia ja joilla plasman lääkeainepitoisuus oli laskenut pysyvästi, oli 11,5 %.
Läiskäpsoriaasi
Lumelääkkeellä ja vaikuttavalla aineella kontrolloiduissa faasin III tutkimuksissa Cimzia-vasta-aineille vähintään kerran positiivisia potilaita oli enintään 48 viikon hoidon aikana 8,3 % (22/265) ryhmässä, joka sai Cimziaa 400 mg kahden viikon välein, ja 19,2 % (54/281) ryhmässä, joka sai Cimziaa 200 mg kahden viikon välein. CIMPASI-1- ja CIMPASI-2-tutkimuksissa kuusikymmentä potilasta oli vasta-aine-positiivisia; 27:ltä näistä potilaista voitiin määrittää neutraloivat vasta-aineet ja tulos oli positiivinen. Vasta-ainepositiivisuutta havaittiin ensimmäisen kerran avoimella hoitojaksolla 2,8 %:lla (19/668) potilaista. Vasta-ainepositiivisuuteen liittyi pienentynyt lääkepitoisuus plasmassa ja joillakin potilailla lääkkeen heikentynyt teho.
Aksiaalinen spondylartriitti
AS001
Aksiaalista spondylartriittia (alaryhminä selkärankareuma ja röntgennegatiivinen aksiaalinen spondylartriitti) sairastavilla potilailla tehdyssä faasin III lumekontrolloidussa AS001-tutkimuksessa niiden potilaiden kokonaisprosenttiosuus, joilla havaittiin vähintään kerran vasta-aineita Cimzia-valmisteelle viikkoon 24 mennessä, oli 4,4 %. Vasta-aineiden muodostumiseen liittyi lääkeaineen pitoisuuden pienenemistä plasmassa.
Niiden potilaiden kokonaisprosenttiosuus, joilla havaittiin vähintään kerran vasta-aineita Cimzia-valmisteelle koko tutkimuksen aikana (kesto enintään 192 viikkoa), oli 9,6 % (Cimzia-vasta-aineiden muodostus oli tilapäistä 4,8 %:lla ja pysyvää 4,8 %:lla). Niiden potilaiden kokonaisprosenttiosuus, jotka olivat vasta-ainepositiivisia ja joilla plasman lääkeainepitoisuus oli laskenut pysyvästi, oli arviolta 6,8 %.
AS0006 ja C-OPTIMISE
Herkempää ja lääketolerantimpaa määritystä käytettiin ensimmäisen kerran AS0006-tutkimuksessa (ja myöhemmin myös C-OPTIMISE-tutkimuksessa). Tämän seurauksena suuremmassa osassa näytteitä todettiin mitattavissa olevia Cimzia-vasta-ainepitoisuuksia, joten suurempi määrä potilaita luokiteltiin vasta-ainepositiivisiksi. AS0006-tutkimuksessa Cimzia-vasta-ainepositiivisten potilaiden kokonaismäärä oli 97 % (248/255 potilasta) enintään 52 viikon pituisen hoidon jälkeen. Vain suurimmat titterit olivat yhteydessä pienentyneisiin Cimzia-pitoisuuksiin plasmassa. Tehoon kohdistuvaa vaikutusta ei kuitenkaan havaittu. C-OPTIMISE-tutkimuksesta saatiin Cimzia-vasta-aineiden osalta samankaltaisia tuloksia. C-OPTIMISE-tutkimuksen tulokset myös osoittivat, että Cimzia-annoksen pienentäminen tasolle 200 mg neljän viikon välein ei vaikuttanut immunogeenisuustuloksiin.
Noin 22 %:lla (54/248) AS0006-tutkimukseen osallistuneista potilaista, jotka olivat Cimzia-vasta-ainepositiivisia, oli neutraloiviksi luokiteltuja vasta-aineita. Neutraloivia vasta-aineita ei arvioitu C-OPTIMISE-tutkimuksessa.
Farmakokinetiikka
Plasman sertolitsumabipegolipitoisuudet olivat yleisesti suhteessa annokseen. Nivelreuma- ja psoriaasipotilaiden farmakokinetiikan havaittiin olevan samankaltainen kuin terveiden tutkimushenkilöiden.
Imeytyminen
Kun sertolitsumabipegolia annetaan ihon alle, plasman huippupitoisuudet saavutetaan 54−171 tunnissa injektion jälkeen. Sertolitsumabipegolin biologinen hyötyosuus (F) on ihon alle annettuna noin 80 % (vaihteluväli 76–88 %) verrattuna valmisteen antamiseen laskimoon.
Jakautuminen
Populaatiofarmakokineettisessä analyysissa näennäiseksi jakautumistilavuudeksi (V/F) arvioitiin 8,01 litraa nivelreumaa sairastavilla potilailla ja 4,71 litraa läiskäpsoriaasipotilailla.
Biotransformaatio ja eliminaatio
Pegylaatio, PEG-polymeerien kovalenttinen kiinnittyminen peptideihin, viivästyttää näiden partikkelien eliminaatiota verenkierrosta useilla mekanismeilla, kuten munuaispuhdistuman heikkenemisellä, proteolyysin vähenemisellä ja immunogeenisuuden vähenemisellä. Tämän mukaisesti sertolitsumabipegoli on polyetyleeniglykoliin konjugoitu Fab’-vasta-ainefragmentti ja tarkoituksena on pidentää Fab’-fragmentin terminaalisen eliminaation puoliintumisaika plasmassa vastaavaksi kuin koko vasta-ainevalmisteen puoliintumisaika. Terminaalisen eliminaatiovaiheen puoliintumisaika (t1/2) oli kaikilla tutkituilla annoksilla noin 14 vuorokautta.
Puhdistuman arvioitiin nivelreumapotilaiden populaatiofarmakokineettisessä analyysissa olleen ihon alle annetun annoksen jälkeen 21,0 ml/h, jolloin yksilöiden välinen vaihtelu on 30,8 % (CV) ja antokertojen välinen vaihtelu 22,0 %. Aiemmin käytetyllä ELISA-menetelmällä arvioituna vasta-aineiden muodostuminen sertolitsumabipegolia vastaan johti puhdistuman suurenemiseen noin kolminkertaiseksi. Yksittäisen 40 kg:n painoisen nivelreumapotilaan puhdistuma oli 29 % pienempi ja 120 kg:n painoisen nivelreumapotilaan puhdistuma oli 38 % suurempi 70 kg:n painoiseen henkilöön verrattuna. Puhdistuma psoriaasipotilailla oli ihon alle annetun annoksen jälkeen 14 ml/h, jolloin yksilöiden välinen vaihtelu oli 22,2 % (CV).
Fab’-fragmentti sisältää proteiiniyhdisteitä ja sen oletetaan hajoavan proteolyysin kautta peptideiksi ja aminohapoiksi. Dekonjugoitunut PEG-osa eliminoituu plasmasta nopeasti ja erittyy tuntemattomassa määrin munuaisten kautta.
Erityispotilasryhmät
Munuaisten vajaatoiminta
Erityisiä kliinisiä tutkimuksia, joilla arvioitaisiin munuaisten vajaatoiminnan vaikutusta sertolitsumabipegolin tai sen PEG-osan farmakokinetiikkaan, ei ole tehty. Lievää munuaisten vajaatoimintaa sairastaviin potilaisiin perustuva populaatiofarmakokineettinen analyysi ei kuitenkaan viitannut kreatiniinipuhdistumaan kohdistuviin vaikutuksiin. Annossuositusten antamiseen keskivaikeaa ja vaikeaa munuaisten vajaatoimintaa sairastavien hoidossa ei ole riittävästi tietoa. Sertolitsumabipegolin PEG-osan farmakokinetiikan oletetaan olevan riippuvainen munuaisten toiminnasta, mutta sitä ei ole tutkittu munuaisten vajaatoimintaa sairastavilla potilailla.
Maksan vajaatoiminta
Erityisiä kliinisiä tutkimuksia, joilla arvioitaisiin maksan vajaatoiminnan vaikutusta sertolitsumabipegolin farmakokinetiikkaan, ei ole tehty.
Iäkkäät potilaat (≥ 65‑vuotiaat)
Iäkkäillä potilailla ei ole tehty erityisiä kliinisiä tutkimuksia, mutta ikään liittyviä vaikutuksia ei havaittu nivelreumapotilailla tehdyssä populaatiofarmakokineettisessä analyysissä, jossa 78 tutkittavista (13,2 % potilaista) oli vähintään 65‑vuotiaita ja iäkkäin tutkimuspotilas 83‑vuotias. Ikään liittyviä vaikutuksia ei havaittu aikuisilla läiskäpsoriaasipotilailla tehdyssä populaatiofarmakokineettisessä analyyseissa.
Raskaus
Kliinisessä tutkimuksessa 21 naista sai Cimzia-valmistetta ylläpitoannoksena 200 mg tai 400 mg kahden viikon välein tai 400 mg neljän viikon välein raskauden aikana ja vähintään 13 viikkoa synnytyksen jälkeen (ks. kohta Raskaus ja imetys).
Populaatiofarmakokineettisen mallinnuksen perusteella arvioitiin, että tutkituilla annostusohjelmilla Cimzia-valmisteen aikaan saaman systeemisen altistuksen mediaani oli raskauden aikana 22 % (AUC) ja 36 % (Cmin) pienempi (eniten altistuksen vähenemistä havaittiin kolmannen raskauskolmanneksen aikana) kuin synnytyksen jälkeen tai ei‑raskaana olevilla henkilöillä.
Vaikka plasman sertolitsumabipegolipitoisuudet olivat raskauden aikana pienempiä kuin synnytyksen jälkeen, ne olivat silti ei‑raskaana olevilla psoriaasia, aksiaalista spondylartriittia tai nivelreumaa sairastavilla potilailla havaitulla pitoisuuksien vaihteluvälillä.
Sukupuoli
Sukupuoli ei vaikuttanut sertolitsumabipegolin farmakokineettisiin ominaisuuksiin. Koska puhdistuma on pienempi, jos paino on alhaisempi, naisten systeeminen altistus sertolitsumabipegolille saattaa yleensä olla jonkin verran suurempi kuin miehillä.
Farmakokineettiset/farmakodynaamiset suhteet
Faasien II ja III kliinisten tutkimustietojen perusteella nivelreumapotilailla määriteltiin populaation altistus‒vastesuhde plasman annosvälinaikaisen keskimääräisen sertolitsumabipegolipitoisuuden (Cavg) ja tehon (ACR 20 vasteen saaneen määritelmä) välillä. Tyypillinen annosvälin aikainen pitoisuus plasmassa (Cavg), joka sai aikaan puolet ACR 20 ‑vasteen suurimmasta todennäköisyydestä (EC50), oli 17 µg/ml (95 %:n luottamusväli: 10–23 µg/ml). Vastaavasti faasin III kliinisten tutkimustietojen perusteella psoriaasipotilailla määriteltiin populaation altistus‒vastesuhde plasman sertolitsumabipegolipitoisuuden ja PASI-indeksin välillä: EC90 oli 11,1 µg/ml.
Prekliiniset tiedot turvallisuudesta
Valmisteen turvallisuutta selvittänyt ei-kliininen pivotaalitutkimus tehtiin cynomolgus-apinoilla. Histopatologinen tutkimus paljasti rotilla ja apinoilla ihmiselle annettavia annoksia suuremmilla annoksilla solujen vakuolisaatiota, jota esiintyi useissa elimissä lähinnä makrofageissa (imusolmukkeissa, injektiokohdassa, pernassa, lisämunuaisissa, kohdussa, kohdunkaulassa, aivokammion suonipunoksessa ja suonipunoksen epiteelisoluissa). Tämä löydös johtui todennäköisesti polyetyleeniglykoliosan soluunotosta. Ihmisen vakuoleja sisältävien makrofagien toiminnalliset tutkimukset in vitro osoittivat, että kaikki tutkitut toiminnot olivat säilyneet. Rotilla tehdyt tutkimukset osoittivat, että yli 90 % annetusta polyetyleeniglykolista eliminoitui kerta-annoksen jälkeisten kolmen kuukauden aikana, ja virtsa oli tällöin tärkein eliminaatioreitti.
Sertolitsumabipegoli ei ristireagoi jyrsijän TNF:n kanssa. Toksikologisia reproduktiotutkimuksia on siksi tehty rotan TNF:n tunnistavalla homologisella reagenssilla. Näiden tietojen merkitys ihmiseen kohdistuvan riskin arvioinnin kannalta saattaa olla vähäinen. Rotilla ei havaittu emon hyvinvointiin tai naaraan hedelmällisyyteen, alkioon tai sikiöön, syntymän aikaisiin tai sen jälkeisiin lisääntymisindekseihin kohdistuvia haittoja käytettäessä jyrsijän anti-rotta-TNF‑α:n PEGyloitua Fab'-fragmenttia (cTN3 PF) pitkäkestoisen TNF‑α-suppression jälkeen. Urosrotilla havaittiin siittiöiden liikkuvuuden vähenemistä ja tendenssimäisesti siittiömäärän vähenemistä.
Jakautumistutkimukset ovat osoittaneet, että cTN3 PF:n erittyminen istukan ja maidon kautta sikiöön ja vastasyntyneen verenkiertoon on vähäistä. Sertolitsumabipegoli ei sitoudu vastasyntyneen Fc-reseptoriin (FcRn). Tiedot istukan läpäisevyysmallista ex vivo viittaavat lievään tai merkityksettömään sertolitsumabipegolin siirtymiseen sikiötilaan. Lisäksi on tehty FcRn-välitteisiä transsytoosikokeita soluilla, jotka on transfektoitu ihmisen FcRn-reseptorilla. Transsytoosin havaittiin näissä kokeissa olevan merkityksetöntä (ks. kohta Raskaus ja imetys).
Prekliinisissä tutkimuksissa ei osoitettu mutageenisuutta eikä klastogeenisuutta. Sertolitsumabipegolilla ei ole tehty karsinogeenisuustutkimuksia.
Farmaseuttiset tiedot
Apuaineet
Natriumasetaatti
Natriumkloridi
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopimattomuustutkimuksia ei ole tehty, lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
2 vuotta.
Katso myös kohdasta Säilytys käyttöaika säilytettäessä huoneenlämmössä (enintään 25 °C).
Säilytys
Säilytä jääkaapissa (2–8 ºC).
Ei saa jäätyä.
Pidä annostelulaitteen säiliö ulkopakkauksessa. Herkkä valolle.
Annostelulaitteita voidaan säilyttää huoneenlämmössä (enintään 25 °C) valolta suojattuna yhden enintään 10 vuorokauden mittaisen ajanjakson ajan, jonka jälkeen annostelulaitteet on käytettävä tai hävitettävä.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
CIMZIA injektioneste, liuos, säiliö annostelulaitteeseen
200 mg (L:ei) 2 x 1 ml (876,35 €)
PF-selosteen tieto
Yhden ml:n säiliö annostelulaitteeseen, joka sisältää esitäytetyn ruiskun (tyypin I lasia), jossa on männän pysäytin (bromobutyylikumia). Esitäytetty ruisku sisältää 200 mg sertolitsumabipegolia. Neulan suojus on styreenibutadieenikumia ja sisältää luonnonkumin (lateksin) johdannaista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pakkaus, jossa 2 säiliötä annostelulaitteeseen ja 2 alkoholipyyhettä.
Kerrannaispakkaus, jossa 6 (3 x 2) säiliötä annostelulaitteeseen ja 6 (3 x 2) alkoholipyyhettä.
Kerrannaispakkaus, jossa 10 (5 x 2) säiliötä annostelulaitteeseen ja 10 (5 x 2) alkoholipyyhettä.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai opaalinhohtoinen, väritön tai keltainen liuos. Liuoksen pH on noin 4,7.
Käyttö- ja käsittelyohjeet
Pakkausselosteessa ja sähkömekaanisen ava-injektiolaitteen ohjekirjassa on annettu tarkat tiedot säiliöön annostelulaitetta varten pakatun Cimzia-valmisteen valmistelusta ja annosta.
Tämä lääkevalmiste on yhtä käyttökertaa varten. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
CIMZIA injektioneste, liuos, säiliö annostelulaitteeseen
200 mg 2 x 1 ml
- Alempi erityiskorvaus (65 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli ja tosilitsumabi (tulehdukselliset reumasairaudet): Nivelreuman, juveniilin polyartriitin, psoriaasiin liittyvän niveltulehduksen, selkärankareuman tai edellä mainittuja niveltulehduksia läheisesti muistuttavan niveltulehduksen hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman hoito erityisin edellytyksin (281).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abatasepti, adalimumabi, bimekitsumabi, etanersepti, golimumabi, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sarilumabi, sekukinumabi, sertolitsumabipegoli, tosilitsumabi ja ustekinumabi (tulehdukselliset reumasairaudet): Eräiden reumasairauksien hoito erityisin edellytyksin / Adalimumabi: Uveiitin hoito erityisin edellytyksin / Tosilitsumabi: Aktiivisen yleisoireisen lastenreuman ja jättisoluarteriitin hoito erityisin edellytyksin (313), Adalimumabi, bimekitsumabi, brodalumabi, etanersepti, guselkumabi, iksekitsumabi, infliksimabi, risankitsumabi, sekukinumabi, sertolitsumabipegoli, tildrakitsumabi ja ustekinumabi (ihopsoriaasi): Vaikean kroonisen ihopsoriaasin hoito erityisin edellytyksin (319).
ATC-koodi
L04AB05
Valmisteyhteenvedon muuttamispäivämäärä
30.01.2025
Yhteystiedot
Bertel Jungin aukio 5, 6 krs.
02600 Espoo
+358 9 2514 4221
etunimi.sukunimi@ucb.com