
KEPPRA koncentrat till infusionsvätska, lösning 100 mg/ml
Kvalitativ och kvantitativ sammansättning
Varje ml innehåller 100 mg levetiracetam.
Varje 5 ml-flaska innehåller 500 mg levetiracetam.
Hjälpämne med känd effekt:
Varje flaska innehåller 19 mg natrium.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Koncentrat till infusionsvätska, lösning (sterilt koncentrat).
Kliniska uppgifter
Terapeutiska indikationer
Keppra är indicerat som monoterapi vid partiella anfall med eller utan sekundär generalisering hos vuxna och ungdomar från 16 år med nydiagnostiserad epilepsi.
Keppra är indicerat som tilläggsbehandling
- vid partiella anfall med eller utan sekundär generalisering hos vuxna, ungdomar och barn från fyra år med epilepsi.
- vid myokloniska anfall hos vuxna och ungdomar från 12 år med juvenil myoklonisk epilepsi.
- vid primärt generaliserade tonisk-kloniska anfall hos vuxna och ungdomar från 12 år med idiopatisk generaliserad epilepsi.
Keppra koncentrat är ett alternativ för patienter när oral administrering tillfälligt inte är möjlig.
Dosering och administreringssätt
Dosering
Behandling med Keppra kan påbörjas med antingen intravenös eller oral administrering.
Övergång till eller från oral till intravenös administrering kan göras direkt utan titrering. Den totala dagliga dosen och administreringsfrekvensen bör bibehållas.
Partiella anfall
Den rekommenderade dosen för monoterapi (från 16 år) och tilläggsbehandling är densamma och i enlighet med vad som anges nedan.
Samtliga indikationer
Vuxna (≥18 år) och ungdomar (12 till 17 år) som väger 50 kg eller mer
Den initiala terapeutiska dosen är 500 mg två gånger dagligen. Denna dos kan insättas från första behandlingsdagen. En lägre initial dos om 250 mg två gånger dagligen kan emellertid ges baserat på läkarens bedömning av behovet av att minska anfall kontra potentiella biverkningar. Denna dos kan ökas till 500 mg två gånger dagligen efter två veckor.
Den dagliga dosen kan ökas upp till 1500 mg två gånger dagligen beroende på klinisk respons och tolerabilitet. Dosjustering kan ske med ökningar och minskningar om 250 mg eller 500 mg två gånger dagligen varannan till var fjärde vecka.
Ungdomar (12 till 17 år) som väger mindre än 50 kg och barn från 4 års ålder
Läkaren bör förskriva den bäst lämpade läkemedelsformen, förpackningsstorleken och styrkan utifrån vikt, ålder och dos. Se avsnittet Pediatrisk population för dosjusteringar utifrån vikt.
Behandlingstid
Det finns ingen erfarenhet av administrering av intravenös levetiracetam under perioder längre än 4 dagar.
Avslutande av behandling
Om levetiracetam-behandlingen måste avbrytas rekommenderas en gradvis utsättning (t ex till vuxna och ungdomar som väger mer än 50 kg: en dosminskning med 500 mg två gånger dagligen varannan till var fjärde vecka; till barn och ungdomar som väger mindre än 50 kg: dosminskningar bör inte överstiga 10 mg/kg två gånger dagligen varannan vecka).
Särskilda patientgrupper
Äldre (65 år och äldre)
Dosjustering rekommenderas till äldre patienter med nedsatt njurfunktion (se ”Nedsatt njurfunktion” nedan).
Nedsatt njurfunktion
Den dagliga dosen måste justeras individuellt med hänsyn till njurfunktion.
För vuxna patienter, se tabellen nedan och justera dosen enligt denna. För att använda denna doseringstabell måste patientens kreatininclearance (CLcr) ml/min uppskattas. CLcr ml/min kan värderas genom bestämning av serumkreatinin (mg/dl), för vuxna och ungdomar som väger 50 kg eller mer genom att använda följande formel:
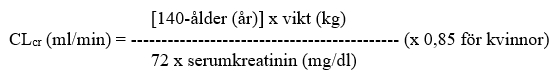
Därefter justeras CLcr för kroppens ytarea (BSA; body surface area) enligt följande:
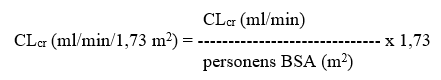
Dosjustering för vuxna och ungdomar som väger mer än 50 kg med nedsatt njurfunktion:
| Grupp | Kreatininclearance (ml/min/1,73 m2) | Dos och frekvens |
Normal Lätt Måttlig Svår Patienter med njursjukdom i slutstadiet som genomgår dialys (1) | ≥ 80 50-79 30-49 < 30 - | 500 till 1500 mg två gånger per dag 500 till 1000 mg två gånger per dag 250 till 750 mg två gånger per dag 250 till 500 mg två gånger per dag 500 till 1000 mg en gång per dag (2) |
(1) En startdos om 750 mg rekommenderas första behandlingsdagen med levetiracetam.
(2) Efter dialys rekommenderas en tilläggsdos om 250 till 500 mg.
För barn med nedsatt njurfunktion måste levetiracetamdosen justeras efter njurfunktionen eftersom clearance av levetiracetam är beroende av njurfunktionen. Denna rekommendation är baserad på en studie på vuxna patienter med nedsatt njurfunktion.
CLcr ml/min/1,73 m2 kan värderas genom bestämning av serumkreatinin (mg/dl), för yngre ungdomar och barn, genom att använda följande formel (Schwartz formel):
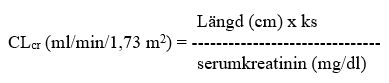
ks=0,55 hos barn yngre än 13 år och ungdomar (flickor); ks=0,7 hos ungdomar (pojkar).
Dosjustering för barn och ungdomar som väger mindre än 50 kg med nedsatt njurfunktion:
| Grupp | Kreatinin-clearance (ml/min/1,73m2) | Dos och frekvens |
| Barn från 4 år och ungdomar som väger mindre än 50 kg | ||
| Normal | ≥ 80 | 10 till 30 mg/kg (0,10 till 0,30 ml/kg) två gånger per dag |
| Lätt | 50-79 | 10 till 20 mg/kg (0,10 till 0,20 ml/kg) två gånger per dag |
| Måttlig | 30-49 | 5 till 15 mg/kg (0,05 till 0,15 ml/kg) två gånger per dag |
| Svår | < 30 | 5 till 10 mg/kg (0,05 till 0,10 ml/kg) två gånger per dag |
| Patienter med njursjukdom i slutstadiet som genomgår dialys | -- | 10 till 20 mg/kg (0,10 till 0,20 ml/kg) en gång per dag (1) (2) |
(1) 15 mg/kg (0,15 ml/kg) som initialdos rekommenderas på behandlingens första dag med levetiracetam.
(2) Efter dialys rekommenderas en tilläggsdos om 5 till 10 mg/kg (0,05 till 0,10 ml/kg).
Nedsatt leverfunktion
Det behövs ingen dosjustering hos patienter med lätt till måttligt nedsatt leverfunktion. Hos patienter med svårt nedsatt leverfunktion kan kreatininclearance ge en underskattning av njurinsufficiensen. Därför rekommenderas en 50%:ig reduktion av den dagliga dosen när kreatininclearance är <60 ml/min/1,73 m2.
Pediatrisk population
Läkaren bör förskriva den bäst lämpade läkemedelsformen, förpackningsstorleken och styrkan utifrån ålder, vikt och dos.
Monoterapi
Säkerhet och effekt med Keppra som monoterapi har inte säkerställts för barn och ungdomar under 16 år.
Data saknas.
Ungdomar (16 och 17 år) som väger 50 kg eller mer med partiella anfall med eller utan sekundär generalisering och nydiagnostiserad epilepsi
Se ovanstående avsnitt om Vuxna (≥18 år) och ungdomar (12 till 17 år) som väger 50 kg eller mer.
Tilläggsterapi för barn 4 till 11 år och ungdomar (12 till 17 år) som väger mindre än 50 kg.
Den initiala terapeutiska dosen är 10 mg/kg två gånger dagligen.
Den dagliga dosen kan ökas upp till 30 mg/kg två gånger dagligen beroende på klinisk respons och tolerabilitet. Dosjusteringar bör inte överstiga ökningar eller minskningar med 10 mg/kg två gånger dagligen varannan vecka. Lägsta effektiva dos ska användas för samtliga indikationer.
Dosen för barn som väger 50 kg eller mer är densamma som för vuxna för samtliga indikationer.
Se ovanstående avsnitt om Vuxna (≥18 år) och ungdomar (12 till 17 år) som väger 50 kg eller mer beträffande samtliga indikationer.
Dosrekommendationer för barn och ungdomar:
| Vikt | Startdos: 10 mg/kg två gånger dagligen | Maxdos: 30 mg/kg två gånger dagligen |
| 15 kg (1) | 150 mg två gånger dagligen | 450 mg två gånger dagligen |
| 20 kg (1) | 200 mg två gånger dagligen | 600 mg två gånger dagligen |
| 25 kg | 250 mg två gånger dagligen | 750 mg två gånger dagligen |
| Från 50 kg (2) | 500 mg två gånger dagligen | 1500 mg två gånger dagligen |
(1) Barn som väger 25 kg eller mindre bör helst starta behandlingen med Keppra 100 mg/ml oral lösning.
(2) Dos till barn och ungdomar som väger 50 kg eller mer är samma som till vuxna.
Tilläggsterapi för spädbarn och barn yngre än 4 år
Säkerhet och effekt av Keppra koncentrat till infusionsvätska, lösning, har inte säkerställts för barn under 4 år.
Tillgängliga data beskrivs i avsnitt Biverkningar, Farmakodynamiska egenskaper och Farmakokinetiska egenskaper men rekommendation angående dosering kan inte göras.
Administreringssätt
Keppra koncentrat ska bara användas för intravenöst bruk och den rekommenderade dosen måste spädas i åtminstone 100 ml kompatibel spädningsvätska och administreras intravenöst som en 15‑minuters intravenös infusion (se avsnitt Särskilda anvisningar för destruktion och övrig hantering).
Kontraindikationer
Överkänslighet mot den aktiva substansen eller andra pyrrolidonderivat eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Nedsatt njurfunktion
Administrering av levetiracetam till patienter med nedsatt njurfunktion kan kräva dosjustering. Hos patienter med svårt nedsatt leverfunktion rekommenderas utredning av njurfunktionen före fastställande av dosen (se avsnitt Dosering och administreringssätt).
Akut njurskada
Användning av levetiracetam har i mycket sällsynta fall associerats med akut njurskada, med debut efter några dagar till flera månader.
Cytopenier
Sällsynta fall av cytopenier (neutropeni, agranulocytos, leukopeni, trombocytopeni och pancytopeni) har förekommit i samband med administrering av levetiracetam, vanligtvis i början av behandlingen. Fullständig blodstatus rekommenderas hos patienter som upplever betydande svaghet, pyrexi, återkommande infektioner eller koagulationsrubbningar (se avsnitt Biverkningar).
Självmord
Självmord, självmordsförsök, suicidtankar och självmordsbeteende har rapporterats hos patienter som behandlats med antiepileptika (inklusive levetiracetam). En metaanalys av randomiserade placebokontrollerade studier med antiepileptika har också visat en liten ökad risk för suicidtankar och självmordsbeteende. Mekanismen för denna risk är inte känd.
Därför ska patienter övervakas för tecken på depression och/eller suicidtankar och självmordsbeteende och lämplig behandling bör övervägas. Patienter (och deras vårdgivare) bör rådas till att uppsöka medicinsk rådgivning om tecken på depression och/eller suicidtankar och självmordsbeteende uppstår.
Onormalt och aggressivt uppförande
Levetiracetam kan orsaka psykotiska symtom och avvikande beteende, inklusive irritabilitet och aggressivitet. Patienter som behandlas med levetiracetam ska övervakas med avseende på utveckling av psykiatriska tecken som tyder på betydande förändringar av sinnesstämning och/eller personlighet. Om sådana beteenden observeras ska anpassning av behandlingen eller en gradvis utsättning av behandlingen övervägas. Om man överväger utsättning, se avsnitt Dosering och administreringssätt.
Försämring av anfall
Liksom med andra typer av antiepileptika kan levetiracetam i sällsynta fall förvärra anfallsfrekvensen eller anfallens allvarlighetsgrad. Denna paradoxala effekt har oftast rapporterats inom den första månaden efter initiering av levetiracetam eller ökning av dosen. Effekten har varit reversibel vid utsättande av läkemedlet eller minskning av dosen. Patienten ska uppmanas att omedelbart kontakta sin läkare i händelse av förvärrad epilepsi.
Brist på effekt eller försämring av anfall har till exempel rapporterats hos patienter med epilepsi förknippad med mutationer på spänningsstyrda natriumkanalers alfa-8-subenhet (SCN8A).
Förlängt QT‑intervall på EKG
Förlängt QT‑intervall har i sällsynta fall observerats på EKG under övervakningen efter godkännandet för försäljning. Levetiracetam ska användas med försiktighet hos patienter med QTc‑intervallförlängning hos patienter som får samtidig behandling med läkemedel som påverkar QTc‑intervallet eller hos patienter med relevant befintlig hjärtsjukdom eller elektrolytstörningar.
Pediatrisk population
Tillgängliga data på barn tyder inte på någon påverkan på tillväxt och pubertet. Långtidseffekter på inlärning, intelligens, tillväxt, endokrina funktioner, pubertet och förmåga att få barn är fortfarande okända hos barn.
Hjälpämnen
Detta läkemedel innehåller 2,5 mmol (eller 57 mg) natrium per maximal engångsdos (0,8 mmol (eller 19 mg) per flaska), motsvarande 2,85 % av WHOs högsta rekommenderat dagligt intag (2 gram natrium för vuxna). Detta bör beaktas av patienter som ordinerats saltfattig kost.
Interaktioner
Antiepileptika
Data från kliniska studier före marknadsföring genomförda på vuxna indikerar att levetiracetam inte påverkar andra antiepileptikas serumkoncentrationer (fenytoin, karbamazepin, valproatsyra, fenobarbital, lamotrigin, gabapentin och primidon) och att dessa antiepileptika inte påverkar levetiracetams farmakokinetik.
Liksom hos vuxna finns inga belägg för kliniskt signifikanta läkemedelsinteraktioner hos pediatriska patienter som fått upp till 60 mg/kg/dag av levetiracetam.
En retrospektiv utvärdering av farmakokinetiska interaktioner hos barn och ungdomar med epilepsi (4 till 17 år) bekräftade att tilläggsbehandling med oralt administrerad levetiracetam inte påverkade serumkoncentrationer vid steady state av samtidigt administrerad karbamazepin och valproat. Data tydde dock på ett 20% högre clearance av levetiracetam hos barn som tar enzyminducerande antiepileptika. Dosjusteringar krävs inte.
Probenecid
Probenecid (500 mg fyra gånger dagligen), ett ämne som blockerar njurarnas tubulära sekretion, har visats hämma renal utsöndring av den primära metaboliten men inte av levetiracetam. Koncentrationen av denna metabolit förblir emellertid låg.
Metotrexat
Samtidig administrering av levetiracetam och metotrexat har rapporterats minska clearance för metotrexat, vilket resulterar i högre/förlängd blodkoncentration av metotrexat till potentiellt toxiska nivåer. Nivåerna av metotrexat och levetiracetam i blod bör övervakas noga hos patienter som behandlas samtidigt med de två läkemedlen.
Perorala preventivmedel och andra farmakokinetiska interaktioner
Levetiracetam 1000 mg dagligen påverkade inte farmakokinetiken hos perorala preventivmedel (etinyl-estradiol och levonorgestrel); endokrina parametrar (luteiniseringshormon och progesteron) ändrades inte. Levetiracetam 2000 mg dagligen påverkade inte farmakokinetiken hos digoxin och warfarin; protrombintiden ändrades inte. Samtidig administrering med digoxin, perorala preventivmedel och warfarin påverkade inte levetiracetams farmakokinetik.
Alkohol
Det finns inga data rörande interaktion mellan levetiracetam och alkohol.
Fertilitet, graviditet och amning
Kvinnor i fertil ålder
Kvinnor i fertil ålder bör få råd från en specialist. Behandling med levetiracetam bör omprövas när en kvinna planerar att bli gravid. Som med alla antiepileptika ska plötslig utsättning av levetiracetam undvikas, eftersom detta kan leda till anfall med allvarliga följder för både kvinnan och det ofödda barnet. Monoterapi är om möjligt alltid att föredra eftersom behandling med flera antiepileptika kan vara förenad med en större risk för medfödda missbildningar än monoterapi, beroende på vilka antiepileptika som används.
Graviditet
En stor mängd data från gravida kvinnor, som exponerats för levetiracetam som monoterapi (över 1 800, där exponeringen skedde under den 1:a trimestern hos över 1 500 av dem), har dokumenterats efter marknadsgodkännandet och tyder inte på någon ökning av risken för allvarliga medfödda missbildningar. Det finns begränsad evidens tillgänglig för den neurologiska utvecklingen hos barn som exponerats för levetiracetam monoterapi in utero. Data från två observationella populationsbaserade registerstudier som genomförts i stort sett i samma dataset från de nordiska länderna och som omfattade mer än 1000 barn födda av kvinnor med epilepsi som prenatalt exponerats för levetiracetam monoterapi tyder inte på någon ökad risk för autismspektrumtillstånd eller intellektuell funktionsnedsättning jämfört med barn födda av kvinnor med epilepsi som inte exponerats för ett antiepileptiskt läkemedel in utero. Den genomsnittliga uppföljningstiden för barn i levetiracetamgruppen var kortare än för gruppen av barn som inte exponerats för något antiepileptiskt läkemedel (t.ex. 4,4 år jämfört med 6,8 år i en av studierna).
Levetiracetam kan användas under graviditet om det efter noggrant övervägande anses vara kliniskt nödvändigt. I sådana fall rekommenderas den lägsta effektiva dosen.
Fysiologiska förändringar under graviditet kan påverka levetiracetam-koncentrationen. Minskad levetiracetam-koncentration i plasma har observerats under graviditet. Denna minskning är mer uttalad under tredje trimestern (upp till 60% av utgångsvärdet före graviditet). Lämplig klinisk behandling ska säkerställas för gravida kvinnor som behandlas med levetiracetam.
Amning
Levetiracetam utsöndras i human bröstmjölk. Därför rekommenderas inte amning.
Om levetiracetam-behandling är nödvändig under amning, ska nyttan/risken med behandling dock vägas mot vikten av amning.
Fertilitet
I djurstudier upptäcktes ingen effekt på fertilitet (se avsnitt Prekliniska säkerhetsuppgifter). Inga kliniska data finns, eventuell risk för människa är okänd.
Effekter på förmågan att framföra fordon och använda maskiner
Levetiracetam har liten eller måttlig effekt på förmågan att framföra fordon och använda maskiner. Då känsligheten kan variera mellan individer, kan vissa patienter uppleva somnolens eller andra symtom relaterade till centrala nervsystemet, särskilt i början av behandlingen eller efter dosökning. Därför rekommenderas försiktighet hos dessa patienter vid aktiviteter som kräver skärpt uppmärksamhet, t ex framförande av fordon eller handhavande av maskinell utrustning. Patienter rekommenderas att inte framföra fordon eller använda maskiner tills det är fastställt att deras förmåga att utföra sådana aktiviteter inte påverkas.
Biverkningar
Sammanfattning av säkerhetsprofilen
De oftast rapporterade biverkningarna var nasofaryngit, somnolens, huvudvärk, utmattning och yrsel. Säkerhetsprofilen nedan baseras på den sammanlagda säkerhetsanalysen av placebokontrollerade kliniska studier avseende alla indikationer, med totalt 3416 patienter behandlade med levetiracetam. Dessa data är kompletterade med användning av levetiracetam i öppna fortsättningsstudier samt med erfarenhet efter marknadsföring. Levetiracetams säkerhetsprofil är i allmänhet densamma i alla åldersgrupper (vuxna och pediatriska patienter) och för alla godkända epilepsi-indikationer.
Lista över biverkningar
Biverkningar som rapporterats från kliniska studier (vuxna, ungdomar, barn och spädbarn >1 månad) och efter marknadsföring listas i följande tabell efter organklass och frekvens. Biverkningarna presenteras i fallande allvarlighetsgrad och deras frekvens är definierad på följande sätt: mycket vanliga (≥1/10); vanliga (≥1/100, <1/10); mindre vanliga (≥1/1 000, <1/100); sällsynta (≥1/10 000, <1/1 000); och mycket sällsynta (<1/10 000).
| MedDRA organklass | Frekvens | ||||
| Mycket vanliga | Vanliga | Mindre vanliga | Sällsynta | Mycket sällsynta | |
| Infektioner och infestationer | Nasofaryngit | Infektion | |||
| Blodet och lymfsystemet | Trombocytopeni, leukopeni | Pancytopeni, neutropeni, agranulocytos | |||
| Immunsystemet | Läkemedelsutlösta utslag med eosinofili och systemiska symtom (DRESS)(1), hypersensitivitet (inklusive angioödem och anafylaxi) | ||||
| Metabolism och nutrition | Anorexi | Viktminskning, viktökning | Hyponatremi | ||
| Psykiska störningar | Depression, fientlighet/ aggression, ångest, insomni, nervositet/ irritabilitet | Självmordsförsök, självmordstankar, psykotisk störning, onormalt uppförande, hallucination, ilska, förvirring, panikattack, emotionell labilitet/ humörsvängningar, agitation | Självmord, personlighets-störningar, onormalt tänkande, delirium | Tvångssyndrom(2) | |
| Centrala och perifera nervsystemet | Somnolens, huvudvärk | Konvulsion, balansrubbning, yrsel, letargi, tremor | Amnesi, försämring av minnet, onormal koordination/ataxi, parestesi, störning i uppmärksamheten | Koreoatetos, dyskinesi, hyperkinesi, gångrubbning, encefalopati, försämring av anfall, malignt neuroleptikasyndrom(3) | |
| Ögon | Diplopi, dimsyn | ||||
| Öron och balansorgan | Vertigo | ||||
| Hjärtat | Förlängt QT‑intervall på EKG | ||||
| Andningsvägar, bröstkorg och mediastinum | Hosta | ||||
| Magtarmkanalen | Buksmärta, diarré, dyspepsi, kräkningar, illamående | Pankreatit | |||
| Lever och gallvägar | Onormalt leverfunktionstest | Leversvikt, hepatit | |||
| Hud och subkutan vävnad | Utslag | Alopeci, eksem, klåda | Toxisk epidermal nekrolys, Stevens-Johnsons syndrom, erythema multiforme | ||
| Muskuloskeletala systemet och bindväv | Muskelsvaghet, myalgi | Rabdomyolys och förhöjt kreatinfosfokinas i blodet(3) | |||
| Njurar och urinvägar | Akut njurskada | ||||
| Allmänna symtom och/eller symtom vid administreringsstället | Asteni/ utmattning | ||||
| Skador och förgiftningar och behandlings-komplikationer | Skada | ||||
(1) Se Beskrivning av utvalda biverkningar.
(2) Mycket sällsynta fall där tvångssyndrom (OCD) utvecklats hos patienter med underliggande anamnes av OCD eller psykisk störning har observerats vid övervakning efter godkännande för försäljning.
(3) Prevalensen är signifikant högre hos japanska patienter jämfört med hos icke-japanska patienter.
Beskrivning av utvalda biverkningar
Överkänslighetsreaktioner som påverkar flera organ
Överkänslighetsreaktioner som påverkar flera organ (även kallade DRESS, Drug Reaction with Eosinophilia and Systemic Symptoms) har rapporterats i sällsynta fall hos patienter som behandlats med levetiracetam. Kliniska manifestationer kan utvecklas 2 till 8 veckor efter påbörjad behandling. Dessa reaktioner varierar i uttryck, men orsakar vanligtvis feber, utslag, ansiktsödem, lymfadenopati, hematologiska avvikelser och kan vara förknippade med påverkan i olika organsystem, främst levern. Vid misstanke om överkänslighetsreaktion i flera organ ska levetiracetam sättas ut.
Risken för anorexi är högre när levetiracetam administreras samtidigt med topiramat.
I flera fall av alopeci sågs återhämtning när Keppra sattes ut.
Benmärgssuppression identifierades i några av fallen av pancytopeni.
Fall med encefalopati inträffade vanligen i början av behandlingen (några dagar till några månader) och var reversibla efter avslutad behandling.
Pediatrisk population
Hos patienter i åldern 1 månad till yngre än 4 år har totalt 190 patienter behandlats med levetiracetam i placebokontrollerade studier och öppna fortsättningsstudier. Sextio av dessa patienter behandlades med levetiracetam i placebokontrollerade studier. Hos patienter i åldern 4-16 år har totalt 645 patienter behandlats med levetiracetam i placebokontrollerade studier och öppna fortsättningsstudier. 233 av dessa patienter behandlades med levetiracetam i placebokontrollerade studier. I båda dessa åldersgrupper är data kompletterade med erfarenhet av levetiracetamanvändning efter marknadsföringen.
Dessutom exponerades 101 spädbarn yngre än 12 månader i en säkerhetsstudie efter marknadsföringen. Inga nya säkerhetsrisker för levetiracetam identifierades för spädbarn yngre än 12 månader med epilepsi.
Levetiracetams biverkningsprofil är i allmänhet densamma i alla åldersgrupper och för alla godkända epilepsi-indikationer. Resultat av säkerheten hos pediatriska patienter i placebokontrollerade studier överensstämde med levetiracetams säkerhetsprofil hos vuxna utom för beteende- och psykiatriska biverkningar som var vanligare hos barn än hos vuxna. Hos barn och ungdomar i åldern 4-16 år rapporterades kräkning (mycket vanlig, 11,2%), agitation (vanlig, 3,4%), humörsvängningar (vanlig, 2,1%), emotionell labilitet (vanlig 1,7%), aggression (vanlig, 8,2%),onormalt uppförande (vanlig, 5,6%) och letargi (vanlig, 3,9%) oftare än i andra åldersgrupper eller i den totala säkerhetsprofilen. Hos spädbarn och barn i åldern 1 månad till mindre än 4 år rapporterades irritabilitet (mycket vanlig, 11,7%) och onormal koordination (vanlig, 3,3%) oftare än i andra åldersgrupper eller i den totala säkerhetsprofilen.
I en dubbelblind, placebokontrollerad pediatrisk säkerhetsstudie med ”non-inferiority”-design har kognitiva och neuropsykologiska effekter av levetiracetam utvärderats hos barn 4-16 år med partiella anfall. Man kom fram till att Keppra inte skilde sig (var ”non-inferior”) från placebo när det gällde förändring från baslinjen beträffande poäng i Leiter-R Attention och Memory, Memory Screen Composite i per protokoll-populationen. Resultat relaterade till beteende och känslofunktioner tydde på en försämring hos levetiracetam-behandlade patienter avseende aggressiva beteenden mätt på ett standardiserat och systematiskt sätt genom användning av ett validerat verktyg (CBCL – Achenbach Child Behaviour Checklist). Emellertid upplevde patienter som tog levetiracetam i den uppföljande, öppna, långtidsstudien ingen försämring, i genomsnitt, av sina beteenden eller känslofunktioner; specifikt var mätningar av aggressivt beteende inte sämre än utgångsvärdet.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Symtom
Somnolens, agitation, aggressivitet, medvetandesänkning, andningsdepression och koma observerades vid överdosering med Keppra.
Hantering av överdosering
Det finns ingen specifik antidot mot levetiracetam. Behandling av en överdos är symtomatisk och kan inkludera hemodialys. Effektiviteten vid dialysutsöndringen är 60% för levetiracetam och 74% för den primära metaboliten.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: antiepileptika, övriga antiepileptika, ATC kod: N03AX14.
Den aktiva substansen levetiracetam är ett pyrrolidonderivat (S-enantiomer av α-etyl-2-oxo-1-pyrrolidin acetamid), kemiskt obesläktad till existerande antiepileptiska aktiva substanser.
Verkningsmekanism
Verkningsmekanismen för levetiracetam är ännu inte helt klarlagd. In vitro- och in vivo-experiment tyder på att levetiracetam inte påverkar cellernas basala egenskaper eller normal neurotransmission.
In vitro-studier visar att levetiracetam påverkar intraneuronala Ca2+-nivåer genom partiell hämning av Ca2+-strömmar av N-typ och genom att reducera frisläppandet av Ca2+ från intraneuronala lager. Dessutom upphäver levetiracetam delvis reduktionen av GABA- och glycin-medierade strömmar inducerad av zink och β-karboliner. Vidare har levetiracetam i in vitro-studier visats binda till ett specifikt bindningsställe i hjärnvävnad hos gnagare. Detta bindningsställe är det synaptiska vesikelproteinet 2A, som förmodas vara involverat i vesikelfusion och exocytos av neurotransmittorer. Levetiracetam och besläktade analoger visar en rangordning av affinitet för bindning till det synaptiska vesikelproteinet 2A som korrelerar till styrkan av deras anfallsskydd i den audiogena epilepsimodellen hos mus. Detta fynd tyder på att interaktionen mellan levetiracetam och det synaptiska vesikelproteinet 2A verkar bidraga till läkemedlets antiepileptiska verkningsmekanism.
Farmakodynamiska effekter
Levetiracetam visar anfallsskydd i ett brett urval av djurmodeller av partiella och primärt generaliserade anfall utan att ha pro-konvulsiv effekt. Den primära metaboliten är inaktiv.
Hos människa har en aktivitet i både partiella och generaliserade epileptiska tillstånd (epileptiform urladdning/fotoparoxysmal respons) bekräftat den breda farmakologiska profilen hos levetiracetam.
Klinisk effekt och säkerhet
Tilläggsbehandling vid partiella anfall med eller utan sekundär generalisering hos vuxna, ungdomar och barn från fyra år med epilepsi.
Effekten av levetiracetam hos vuxna har visats i tre dubbelblinda, placebokontrollerade studier med dagliga doser på 1000 mg, 2000 mg eller 3000 mg, administrerade som två separata doser, med en behandlingsduration på upp till 18 veckor. I en poolad analys var procentandelen av patienterna som uppnådde en minskning på 50% eller mer från baslinjen av frekvensen av partiella anfall per vecka vid en stadigvarande dos (12/14 veckor) 27,7%, 31,6% respektive 41,3% av patienterna som behandlades med 1000, 2000 respektive 3000 mg levetiracetam och 12,6% av patienterna i placebogruppen.
Pediatrisk population
Hos pediatriska patienter (4 till 16 år) fastställdes effekten av levetiracetam i en dubbelblind, placebokontrollerad 14-veckors studie som inkluderade 198 patienter. I studien erhöll patienterna en fast dos av levetiracetam, 60 mg/kg/dag, (administrerad som två doser per dag).
44,6% av patienterna som behandlades med levetiracetam och 19,6% av patienterna i placebogruppen fick en minskning av frekvensen av partiella anfall per vecka med 50% eller mer från baslinjen. Vid fortsatt långtidsbehandling var 11,4% av patienterna anfallsfria under minst 6 månader och 7,2% var anfallsfria under minst 1 år. 35 spädbarn yngre än 1 år med partiella anfall har exponerats i placebokontrollerade kliniska studier varav endast 13 var <6 månader.
Monoterapi vid partiella anfall med eller utan sekundär generalisering hos patienter från 16 år med nydiagnostiserad epilepsi.
Effekt av levetiracetam som monoterapi har visats i en dubbelblind, parallellgrupps-, ”non-inferiority”-, jämförande studie med en depotberedning av karbamazepin hos 576 patienter som var 16 år eller äldre och som hade nydiagnostiserad epilepsi. Patienterna hade uppvisat oprovocerade partiella anfall eller enbart generaliserade tonisk-kloniska anfall. Patienterna randomiserades till en depotberedning av karbamazepin 400-1200 mg/dag eller levetiracetam 1000-3000 mg/dag och behandlingsperioden var upp till 121 veckor beroende på behandlingssvaret.
Sex månaders anfallsfrihet uppnåddes hos 73,0% av patienterna som behandlades med levetiracetam och hos 72,8% av patienterna som behandlades med en depotberedning av karbamazepin; den justerade absoluta skillnaden mellan behandlingarna var 0,2% (95% konfidensintervall: -7.8 8.2). Mer än hälften av patienterna förblev anfallsfria i 12 månader (56,6% och 58,5% för patienter behandlade med levetiracetam respektive en depotberedning av karbamazepin).
I en studie som avspeglar klinisk praxis visades att annan samtidig antiepileptisk behandling kunde sättas ut för ett begränsat antal patienter som svarat på tilläggsbehandling med levetiracetam (36 av 69 vuxna patienter).
Tilläggsbehandling vid myokloniska anfall hos vuxna och ungdomar från 12 år med juvenil myoklonisk epilepsi.
Effekten av levetiracetam fastställdes i en dubbelblind, placebokontrollerad 16 veckors studie hos patienter 12 år eller äldre med idiopatisk generaliserad epilepsi med myokloniska anfall i olika syndrom. Majoriteten av patienterna hade juvenil myoklonisk epilepsi.
I denna studie var dosen levetiracetam 3000 mg/dag, administrerad som två separata doser.
58,3% av patienterna som behandlades med levetiracetam och 23,3% av patienterna i placebogruppen fick en minskning av antalet dagar med myokloniska anfall per vecka på minst 50%. Vid fortsatt långtidsbehandling var 28,6% av patienterna fria från myokloniska anfall under minst 6 månader och 21,0% var fria från myokloniska anfall under minst 1 år.
Tilläggsbehandling vid primärt generaliserade tonisk-kloniska anfall hos vuxna och ungdomar från 12 år med idiopatisk generaliserad epilepsi.
Effekten av levetiracetam fastställdes i en 24-veckors dubbelblind, placebokontrollerad studie som inkluderade vuxna, ungdomar och ett begränsat antal barn med idiopatisk generaliserad epilepsi med primärt generaliserade tonisk-kloniska (PGTC) anfall i olika syndrom (juvenil myoklonisk epilepsi, juvenil absensepilepsi, absensepilepsi hos barn eller epilepsi med grand mal-anfall vid uppvaknandet). I denna studie var doserna av levetiracetam 3000 mg/dag för vuxna och ungdomar respektive 60 mg/kg/dag för barn, administrerade som två separata doser.
72,2% av patienterna som behandlades med levetiracetam och 45,2% av patienterna i placebogruppen fick en minskning av frekvensen av PGTC-anfall per vecka på 50% eller mer. Vid fortsatt långtidsbehandling var 47,4% av patienterna fria från tonisk-kloniska anfall under minst 6 månader och 31,5% var fria från tonisk-kloniska anfall under minst 1 år.
Farmakokinetiska egenskaper
Den farmakokinetiska profilen har karakteriserats efter oral administrering. En engångsdos på 1500 mg levetiracetam löst i 100 ml av en kompatibel spädningsvätska och infunderat intravenöst under 15 minuter är bioekvivalent med 1500 mg levetiracetam intaget oralt, givet som tre 500 mg tabletter.
Intravenös administrering av doser upp till 4000 mg lösta i 100 ml 0,9% natriumklorid infunderade under 15 minuter och doser upp till 2500 mg lösta i 100 ml 0,9% natriumklorid infunderade under 5 minuter utvärderades. Inga problem gällande säkerhet identifierades av farmakokinetik- eller säkerhetsprofilen.
Levetiracetam är en lättlöslig och permeabel förening. Den farmakokinetiska profilen är linjär med låg intra- och inter-individuell variabilitet. Clearance ändras inte efter upprepad administrering. Levetiracetams tidsoberoende farmakokinetikprofil bekräftades också efter intravenös infusion av 1500 mg under 4 dagar med dosering två gånger dagligen.
Det finns inga tecken på någon relevant köns-, ras- eller dygnsvariabilitet. Den farmakokinetiska profilen är jämförbar mellan friska frivilliga försökspersoner och patienter med epilepsi.
Vuxna och ungdomar
Distribution
Maximal plasmakoncentration (Cmax) observerad hos 17 försökspersoner efter en intravenös engångsdos på 1500 mg infunderad under 15 minuter var 51 ± 19 µg/ml (aritmetiskt medelvärde ± standardavvikelse).
Det finns inga data beträffande vävnadsdistribution hos människa.
Varken levetiracetam eller dess primära metabolit är signifikant bundet till plasmaproteiner (<10%).
Levetiracetams distributionsvolym är ca. 0,5 till 0,7 l/kg, ett värde som ligger nära den totala kroppsvattenvolymen.
Metabolism
Levetiracetam metaboliseras i låg omfattning hos människor. Den huvudsakliga metabola vägen (24% av dosen) är en enzymatisk hydrolys av acetamid-gruppen. Produktionen av den primära metaboliten, ucb L057, stöds inte av lever cytokrom P450 isoformer. Hydrolys av acetamid-gruppen var mätbar i ett stort antal vävnader inklusive blodceller. Metaboliten ucb L057 är farmakologiskt inaktiv.
Två mindre metaboliter identifierades också. En erhölls genom hydroxylering av pyrrolidonringen (1,6% av dosen) och den andra genom öppnandet av pyrrolidonringen (0,9% av dosen). Andra oidentifierade komponenter stod för endast 0,6% av dosen.
Ingen omvandling mellan enantiomerer påvisades in vivo för levetiracetam eller dess primära metabolit.
In vitro har levetiracetam och dess primära metabolit visat att de inte hämmar de viktigaste humana cytokrom P450 isoformerna i lever (CYP3A4, 2A6, 2C9, 2C19, 2D6, 2E1 och 1A2), glukoronyl transferas (UGT1A1 och UGT1A6) och epoxidhydroxylas aktiviteter. Vidare påverkar levetiracetam inte in vitro glukuronidering av valproatsyra.
I odlade humana hepatocyter hade levetiracetam liten eller ingen effekt på CYP1A2, SULT1E1 eller UGT1A1. Levetiracetam orsakade mild induktion av CYP2B6 och CYP3A4. Data in vitro och interaktionsdata in vivo för orala preventivmedel, digoxin och warfarin indikerar att ingen signifikant enzyminduktion förväntas in vivo. Därför är det inte troligt att Keppra interagerar med andra läkemedel eller vice versa.
Eliminering
Halveringstiden i plasma hos vuxna var 7 ±1 timmar och varierade varken med dos, administreringsväg eller upprepad dosering. Den genomsnittliga totala kroppseliminationen var 0,96 ml/min/kg.
Den huvudsakliga utsöndringen var via urin, vilken i genomsnitt stod för 95% av dosen (ca 93% av dosen var utsöndrad inom 48 timmar). Utsöndring via faeces stod för endast 0,3% av dosen.
Den kumulativa urinutsöndringen av levetiracetam och dess primära metabolit stod för 66% respektive 24% av dosen under de första 48 timmarna.
Renal utsöndring av levetiracetam och ucb L057 är 0,6 respektive 4,2 ml/min/kg vilket tyder på att levetiracetam utsöndras genom glomerulär filtration med efterföljande tubulär reabsorption och att den primära metaboliten också utsöndras genom aktiv tubulär sekretion tillsammans med glomerulär filtration. Levetiracetams eliminering är korrelerad till kreatininclearance.
Äldre
Hos äldre ökas halveringstiden med ca 40% (10 till 11 timmar). Detta relateras till försämrad njurfunktion hos denna grupp (se avsnitt Dosering och administreringssätt).
Nedsatt njurfunktion
Apparent clearance av både levetiracetam och dess primära metabolit är korrelerad till kreatininclearance. Därför rekommenderas justering av den dagliga dosen av Keppra med hänsyn till kreatininclearance hos patienter med måttligt till kraftigt nedsatt njurfunktion (se avsnitt Dosering och administreringssätt)
Hos anuriska vuxna patienter med njursjukdom i slutstadiet var halveringstiden ca 25 timmar under perioder mellan dialys respektive 3,1 timmar under dialys.
Den fraktionella elimineringen av levetiracetam var 51% under en typisk 4-timmars dialys.
Nedsatt leverfunktion
Hos personer med lätt och måttligt nedsatt leverfunktion förekom ingen relevant ändring av clearance av levetiracetam. Hos de flesta försökspersonerna med kraftigt nedsatt leverfunktion reducerades clearance av levetiracetam med mer än 50% beroende på en samtidigt nedsatt njurfunktion (se avsnitt Dosering och administreringssätt).
Pediatrisk population
Barn (4 till 12 år)
Farmakokinetiken hos barn har inte undersökts efter intravenös administrering. Baserat på levetiracetams farmokokinetiska egenskaper, farmakokinetiken hos vuxna efter intravenös administrering och farmakokinetiken hos barn efter oral administrering förväntas dock exponeringen (AUC) för levetiracetam vara liknande hos barn i åldern 4 till 12 år efter intravenös och oral administrering.
Efter administrering av en engångsdos (20 mg/kg) till barn med epilepsi (6 till 12 år) var levetiracetams halveringstid 6 timmar. Apparent viktjusterad clearance var ca 30% högre än hos vuxna med epilepsi.
Levetiracetam absorberades snabbt efter upprepad administrering av oral dos (20 till 60 mg/kg/dag) till barn med epilepsi (4 till 12 år). Maximal plasmakoncentration observerades 0,5 till 1,0 timme efter dosering. Linjära och dosproportionella ökningar observerades för maximala plasmakoncentrationer och area under kurvan. Halveringstiden för eliminering var cirka 5 timmar. Skenbart kroppsclearance var 1,1 ml/min/kg.
Prekliniska säkerhetsuppgifter
Ickekliniska data visade ingen speciell risk för människor baserat på gängse studier av farmakologisk säkerhet, genotoxicitet och carcinogen potential.
Biverkningar som inte observerats i kliniska studier men som observerats hos råtta och i mindre utsträckning hos mus vid exponeringsnivåer liknande humana exponeringsnivåer och med möjlig relevans för klinisk användning var leverförändringar, som indikerar en adapterande respons såsom ökad vikt och centrilobular hypertrofi, fettinfiltration och ökade leverenzymer i plasma.
Inga oönskade effekter på fertiliteten observerades hos han- eller honråttor vid doser upp till 1800 mg/kg/dag (6 gånger den maximala rekommenderade humana dosen [MRHD] på basis av mg/m2 eller exponering) hos föräldrar eller F1-generationen.
Två utvecklingsstudier på embryo/foster (embryo-foetal development [EFD] studies) utfördes på råttor vid 400, 1200 och 3600 mg/kg/dag. Vid 3600 mg/kg/dag visades i endast den ena av de 2 EFD-studierna en liten minskning i fostervikt som förknippades med en marginell ökning i skelettvariation/mindre anomalier. Ingen effekt sågs på embryodödlighet och ingen ökning i incidensen av missbildningar. NOAEL (No Observed Adverse Effect Level) var 3600 mg/kg/dag för dräktiga honråttor (12 gånger MRHD på mg/m2-basis) och 1200 mg/kg/dag för foster.
Fyra utvecklingsstudier på embryo/foster utfördes på kaniner med doser om 200, 600, 800, 1200 och 1800 mg/kg/dag. Dosnivån 1800 mg/kg/dag medförde en markant maternell toxicitet och en minskning i fostervikt förknippad med en ökning i incidensen av foster med kardiovaskulära anomalier/skelettanomalier. NOAEL var <200 mg/kg/dag för mödrarna och 200 mg/kg/dag för fostren (likvärdigt med MRHD på mg/m2-basis).
En peri-och postnatal utvecklingsstudie utfördes på råttor med levetiracetamdoser om 70, 350 och 1800 mg/kg/dag. NOAEL var ≥1800 mg/kg/dag för F0-honorna och för överlevnad, tillväxt och utveckling av F1-avkomman fram till avvänjning (6 gånger MRHD på mg/m2-basis).
Studier på neonatala och juvenila råttor och hundar visade att det inte fanns några negativa effekter i någon av standardmätpunkterna för utveckling och mognad vid doser upp till 1800 mg/kg/dag (6-17 gånger MRHD på mg/m2-basis).
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Natriumacetat
Koncentrerad ättiksyra
Natriumklorid
Vatten för injektionsvätskor
Inkompatibiliteter
Detta läkemedel får inte blandas med andra läkemedel förutom de som nämns under avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Hållbarhet
3 år
Ur ett mikrobiologiskt perspektiv ska produkten användas direkt efter spädning. Om den inte används omedelbart ligger ansvaret för hållbarhetstider och förvaring, före och under användning, på användaren. Denna förvaring ska normalt inte vara längre än 24 timmar vid +2 - +8 °C, såvida inte spädningen har utförts under kontrollerade och validerade aseptiska förhållanden.
Särskilda förvaringsanvisningar
Inga särskilda förvaringsanvisningar.
För förvaringsanvisningar för färdigberedd produkt, se avsnitt Hållbarhet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
KEPPRA infuusiokonsentraatti, liuosta varten
100 mg/ml (L:ei) 10 x 5 ml (403,30 €)
PF-selosteen tieto
5 ml injektionsflaska av glas (typ 1) försluten med en grå bromobutylgummipropp (ej överdragen) och förseglad med en avknäppbar kapsyl av aluminium/polypropen.
Varje kartong innehåller 10 flaskor.
Läkemedlets utseende:
Klar, färglös vätska.
Särskilda anvisningar för destruktion och övrig hantering
Se tabell 1 för rekommenderad beredning och administrering av Keppra koncentrat till infusionsvätska, lösning för att uppnå en total daglig dos på 500 mg, 1000 mg, 2000 mg, eller 3000 mg uppdelat på två doser.
Tabell 1. Beredning och administrering av Keppra koncentrat till infusionsvätska, lösning
Dos | Uppdragningsvolym | Volym spädningsvätska | Infusions-tid | Administrerings-frekvens | Total daglig dos |
250 mg | 2,5 ml (halv 5 ml flaska) | 100 ml | 15 minuter | Två gånger dagligen | 500 mg/dag |
500 mg | 5 ml (en 5 ml flaska) | 100 ml | 15 minuter | Två gånger dagligen | 1000 mg/dag |
1000 mg | 10 ml (två 5 ml flaskor) | 100 ml | 15 minuter | Två gånger dagligen | 2000 mg/dag |
1500 mg | 15 ml (tre 5 ml flaskor) | 100 ml | 15 minuter | Två gånger dagligen | 3000 mg/dag |
Detta läkemedel är endast för engångsanvändning, all oanvänd lösning ska kasseras.
Keppra koncentrat till infusionsvätska, lösning visades vara fysikaliskt kompatibelt och kemiskt stabilt under minst 24 timmar när det blandades med följande spädningsvätskor och förvarades i PVC-påsar vid kontrollerad rumstemperatur 15-25°C.
Spädningsvätskor:
- Natriumklorid 9 mg/ml (0,9%) injektionsvätska, lösning
- Ringer-laktat injektionsvätska, lösning
- Dextros 50 mg/ml (5%) injektionsvätska, lösning
Läkemedel med partiklar eller missfärgning ska inte användas.
Allt oanvänt läkemedel ska kasseras enligt lokala riktlinjer.
Ersättning
KEPPRA infuusiokonsentraatti, liuosta varten
100 mg/ml 10 x 5 ml
- Ei korvausta.
- Epilepsian hoidossa lääkevaihto vain saman kauppanimen valmisteeseen.
Atc-kod
N03AX14
Datum för översyn av produktresumén
13.10.2025
Yhteystiedot
Bertel Jungin aukio 5, 6 krs.
02600 Espoo
+358 9 2514 4221
etunimi.sukunimi@ucb.com