
COMETRIQ kapsel, hård 20 mg + 80 mg
Kvalitativ och kvantitativ sammansättning
En hård kapsel innehåller kabozantinib (S)-malat motsvarande 20 mg eller 80 mg kabozantinib.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Hård kapsel.
Kliniska uppgifter
Terapeutiska indikationer
COMETRIQ är indicerat för behandling av vuxna patienter med progressiv, icke-resektabel lokalt avancerad eller metastaserad medullär tyreoideacancer.
Vid ställningstagande till individuell behandling för patienter som är RET- (rearranged during transfection) mutationsnegativ eller där status inte är känt ska man beakta att nyttan av behandlingen kan vara lägre (se viktig information i avsnitt Farmakodynamiska egenskaper).
Villkor
Hoito tulee aloittaa syövän hoitoon tarkoitettujen lääkevalmisteiden antamiseen perehtyneen lääkärin valvonnassa.
Dosering och administreringssätt
Förskrivning av COMETRIQ ska göras av en läkare med erfarenhet av administrering av cancerläkemedel.
Dosering
COMETRIQ (kabozantinib) kapslar och CABOMETYX (kabozantinib) tabletter är inte bioekvivalenta och får inte bytas ut mot varandra (se avsnitt Farmakokinetiska egenskaper).
Den rekommenderade doseringen av COMETRIQ är 140 mg dagligen, i form av en 80 mg orange kapsel och tre 20 mg grå kapslar. Behandlingen bör fortsätta tills patienten inte längre har någon klinisk nytta av behandlingen eller tills oacceptabel toxicitet uppstår.
Det bör förväntas att majoriteten av de patienter som behandlas med COMETRIQ kommer att behöva en eller fler dosjusteringar (reduktion och/eller avbrott) på grund av toxicitet. Patienter bör därför övevakas noggrant under behandlingens åtta första veckor (se avsnitt Varningar och försiktighet).
Tillfällig utsättning och/eller dossänkning av COMETRIQ kan krävas för att hantera misstänkta biverkningar. Vid nödvändig dossänkning rekommenderas en minskning till 100 mg dagligen i form av en 80 mg orange kapsel och en 20 mg grå kapsel, och vidare ned till 60 mg dagligen, i form av tre 20 mg grå kapslar.
Utsättning rekommenderas för att hantera toxicitet av CTCAE (Common Terminology Criteria for Adverse Events) grad 3 eller högre eller intolerabel toxicitet av grad 2.
Dossänkning rekommenderas för händelser som, om ihållande, kan bli allvarliga eller oacceptabla.
Då de flesta händelser kan inträffa i ett tidigt skede av behandlingen bör läkaren bedöma patienten noggrant under de första åtta veckorna för att avgöra om dosjusteringar är motiverade. Händelser som i allmänhet har tidig debut inkluderar hypokalcemi, hypokalemi, trombocytopeni, hypertension, palmar-plantar erytrodysestesi (PPES) och gastrointestinala biverkningar (buk- eller munsmärta, slemhinneinflammation, förstoppning, diarré och kräkningar).
Förekomsten av vissa allvarliga biverkningar (som GI-fistel) kan vara beroende av den kumulativa dosen och uppstå först i ett senare skede i behandlingen.
Om en patient missar en dos ska den missade dosen inte tas om det är mindre än 12 timmar kvar tills nästa dos.
Samtidig behandling med andra läkemedel
Samtidig behandling med läkemedel som är starka hämmare av CYP3A4 bör användas med försiktighet, och kronisk användning av samtidigt administrerade läkemedel som är starka inducerare av CYP3A4 bör undvikas (se avsnitt Varningar och försiktighet och Interaktioner).
Val av ett annat läkemedel för samtidig administrering som har låg eller obefintlig potential att inducera eller hämma CYP3A4 bör övervägas.
Äldre patienter
Ingen specifik dosjustering för användning av kabozantinib i äldre (≥ 65 år) rekommenderas. Emellertid har en trend av ökad frekvens av allvarliga biverkningar i patienter äldre än 75 år observerats.
Etnicitet
Erfarenheten av kabozantinib i icke-vita patienter är begränsad.
Nedsatt njurfunktion
Kabozantinib ska användas med försiktighet till patienter med lätt eller måttligt nedsatt njurfunktion.
Kabozantinib rekommenderas inte till patienter med gravt nedsatt njurfunktion eftersom säkerhet och effekt inte har fastställts i denna population.
Nedsatt leverfunktion
För patienter med lätt eller måttligt nedsatt leverfunktion är den rekommenderade dosen av kabozantinib 60 mg en gång dagligen. Noggrann övervakning av den övergripande säkerheten rekommenderas för dessa patienter (se avsnitt Farmakokinetiska egenskaper), eftersom dosjustering eller behandlingsavbrott kan bli nödvändigt. Kabozantinib rekommenderas inte till patienter med gravt nedsatt leverfunktion eftersom säkerhet och effekt inte har fastställts i denna population.
Nedsatt hjärtfunktion
Det finns begränsade data från patienter med nedsatt hjärtfunktion. Inga specifika doseringsrekommendationer kan ges.
Pediatrisk population
Säkerhet och effekt hos barn under 18 år har inte fastställts. Inga data finns tillgängliga.
Administreringssätt
COMETRIQ är för oral användning. Kapslarna ska sväljas hela och får inte öppnas. Patienterna ska instrueras att avstå från föda minst 2 timmar före och 1 timme efter att ha tagit COMETRIQ.
Kontraindikationer
Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
Varningar och försiktighet
Dosreducering och avbrott i behandlingen förekom hos 79 % respektive 72 % av de patienter som behandlades med kabozantinib i den pivotala kliniska studien. Hos 41 % av patienterna krävdes två dosreduceringar. Mediantiden till den första dosreduceringen var 43 dagar, och till första avbrottet 33 dagar. Noggrann övervakning av patienter rekommenderas därför under behandlingens åtta första veckor (se avsnitt Dosering och administreringssätt).
Levertoxicitet
Avvikelser i leverfunktionstester (ökning av alaninaminotransferas (ALAT), aspartataminotransferas (ASAT) och bilirubin) har ofta observerats hos patienter som behandlas med kabozantinib. Det rekommenderas att utföra leverfunktionstester (ALAT, ASAT och bilirubin) innan behandling med kabozantinib påbörjas och noggrann övervakning under behandlingen. För patienter där försämrat resultat på leverfunktionstest anses relaterat till kabozantinibbehandling (där ingen alternativ orsak är uppenbar) bör dosen sänkas eller behandlingen avbrytas enligt rekommendationerna i avsnitt Dosering och administreringssätt.
Perforeringar, fistlar och intraabdominella abscesser
Allvarliga gastrointestinala perforeringar och fistlar, ibland med dödlig utgång, och intraabdominella abscesser har observerats med kabozantinib. Patienter som nyligen har genomgått strålbehandling, har inflammatorisk tarmsjukdom (t.ex. Crohns sjukdom, ulcerös kolit, peritonit eller divertikulit), har tumörinfiltration i luftstrupen, bronkerna eller matstrupen, har komplikationer från tidigare gastrointestinal kirurgi (särskilt när detta är förenat med fördröjd eller ofullständig läkning), har komplikationer från strålbehandling av brösthålan (inklusive mediastinum) bör noggrant utvärderas före insättande av kabozantinib och bör därefter kontrolleras noggrant för symtom på perforeringar och fistlar. Icke-gastrointestinala fistlar bör vid behov uteslutas när mukosit uppstår efter behandlingsstart. Kabozantinib ska sättas ut vid GI-perforation, eller en GI- eller icke-GI-fistel.
Tromboemboliska biverkningar
Fall av venös tromboembolism, inklusive lungembolism och arteriell tromboembolism, ibland dödlig, har observerats med kabozantinib. Kabozantinib bör användas med försiktighet i patienter som löper risk för eller som har en historia av sådana biverkningar. Kabozantinib ska sättas ut i patienter som utvecklar en akut hjärtinfarkt eller någon annan kliniskt signifikant arteriell tromboembolisk komplikation.
Blödning
Svår blödning, ibland dödlig, har observerats med kabozantinib. Patienter med tecken på involvering av tumör i luftstrupen eller bronkerna eller en historia av hemoptys före behandlingsstart bör noggrant utvärderas före insättande av kabozantinib. Kabozantinib bör inte ges till patienter med allvarlig blödning eller nyligen inträffad hemoptys.
Aneurysmer och arteriella dissektioner
Användning av VEGF-hämmare till patienter med eller utan hypertoni kan främja bildningen av aneurysmer och/eller arteriella dissektioner. Denna risk ska noga övervägas innan kabozantinib sätts in hos patienter med riskfaktorer såsom hypertoni eller tidigare aneurysm.
Gastrointestinala (GI) besvär
Diarré, illamående/kräkningar, nedsatt aptit och stomatit/oral smärta var några av de vanligast rapporterade GI-biverkningarna (se avsnitt Biverkningar). Snabb medicinsk hantering, inklusive stödjande vård med antiemetika, antidiarroika eller antacida, bör sättas in för att förhindra uttorkning, obalans i elektrolyterna och viktminskning. Behandlingsavbrott, dosreducering eller permanent utsättning av kabozantinib bör beaktas vid bestående eller återkommande betydande GI-biverkningar (se avsnitt Dosering och administreringssätt).
Sårkomplikationer
Sårkomplikationer har observerats med kabozantinib. Om möjligt ska behandlingen avbrytas minst 28 dagar före planerad kirurgi, inklusive tandkirurgiska ingrepp eller invasiva tandingrepp. Beslut om återupptagande av behandling efter kirurgi ska baseras på klinisk bedömning av adekvat sårläkning. Kabozantinib ska avbrytas i patienter med sårläkningskomplikationer som kräver läkarvård.
Hypertension
Hypertension, inklusive hypertensiv kris, har observerats med kabozantinib. Blodtrycket ska vara välkontrollerat innan behandling med kabozantinib påbörjas. Efter påbörjad behandling med kabozantinib ska blodtrycket monitoreras tidigt och regelbundet och behandlas med blodtryckssänkande terapi efter behov. Vid bestående hypertension trots användning av blodtryckssänkande läkemedel, ska behandling med kabozantinib avbrytas tills dess att blodtrycket är kontrollerat varpå kabozantinibbehandlingen kan återupptas med reducerad dos. Kabozantinib ska sättas ut vid allvarlig och ihållande hypertension trots blodtryckssänkande behandling och dosreducering av kabozantinib. Vid hypertensiv kris ska kabozantinib sättas ut.
Hjärtsvikt
Kabozantinib har förknippats med en ökad risk för hjärtsvikt. Denna risk kan förvärras av vanliga biverkningar av kabozantinib (till exempel hypertoni, hypotyreos och arteriella trombotiska händelser) som kan leda till hjärtsvikt. Patienterna ska övervakas för tecken och symtom på hjärtsvikt genom hela behandlingen. Dessa biverkningar ska hanteras omgående, behandlingsavbrott och/eller dosjustering bör övervägas om nödvändigt (se avsnitt Dosering och administreringssätt) och behandling med TKI ska avbrytas hos patienter som utvecklar svårt hjärtsvikt.
Osteonekros
Fall av osteonekros i käken har observerats med kabozantinib. En munundersökning bör genomföras före insättning av kabozantinib och regelbundet under behandlingen. Patienterna bör instrueras i god munhygien. Behandlingen med kabozantinib bör om möjligt avbrytas minst 28 dagar före planerad tandkirurgi eller invasiva tandingrepp. Försiktighet är indicerat i patienter som får preparat förknippade med osteonekros i käken, såsom bifosfonater. Kabozantinib bör sättas ut för patienter som får osteonekros i käken.
Palmar-plantar erytrodysestesi
Palmar-plantar erytrodysestesi har observerats med kabozantinib. Vid allvarlig PPES bör man överväga att avbryta behandlingen med kabozantinib. Kabozantinibbehandlingen bör återupptas med en lägre dos när PPES har åtgärdats till grad 1.
Proteinuri
Proteinuri har observerats med kabozantinib. Övervaka urinprotein regelbundet under behandling med kabozantinib. Kabozantinib bör sättas ut i patienter som utvecklar nefrotiskt syndrom.
Posteriort reversibelt encefalopatisyndrom
Posteriort reversibelt encefalopatisyndrom (PRES), har observerats med kabozantinib. PRES bör beaktas för alla patienter med symtom som tyder på denna diagnos, inklusive krampanfall, huvudvärk, synstörningar, förvirring eller förändrad mental funktion. Behandlingen med kabozantinib bör sättas ut för patienter med PRES.
Förlängning av QT-intervall
Kabozantinib bör användas med försiktighet till patienter med en historia av förlängt QT-intervall, patienter som behandlas med antiarytmika och patienter med relevant tidigare hjärtsjukdom, bradykardi eller störningar i elektrolytbalansen. Vid användning av kabozantinib ska regelbunden övervakning med on-treatment EKG och elektrolyter (serumkalcium, serumkalium, serummagnesium) övervägas. Samtidig behandling med starka hämmare av CYP3A4, som kan öka plasmanivåerna av kabozantinib, ska användas med försiktighet.
CYP3A4-inducerare och -hämmare
Kabozantinib är ett CYP3A4-substrat. Samtidig administrering av kabozantinib med den potenta CYP3A4-hämmaren ketokonazol medförde en ökning av plasmaexponeringen av kabozantinib. Försiktighet krävs vid samtidig administrering av kabozantinib tillsammans med potenta CYP3A4-hämmare. Samtidig administrering av kabozantinib med den starka CYP3A4-induceraren rifampicin medförde en minskning av plasmaexponeringen av kabozantinib. Därför bör kronisk administrering av ämnen med starkt CYP3A4-inducerande effekt tillsammans med kabozantinib undvikas (se avsnitt Dosering och administreringssätt och Interaktioner)
P-glykoproteinsubstrat
Kabozantinib var en hämmare (IC50 = 7,0 μM) men inte ett substrat av P‑glykoprotein (P‑gp)-transportaktiviteter i ett dubbelriktat analyssystem med MDCK‑MDR1-celler. Därför kan kabozantinib potentiellt öka plasmakoncentrationerna av samtidigt administrerade substrat av P‑gp. Patienterna ska varnas för att ta ett P‑gp-substrat (t.ex. fexofenadin, aliskiren, ambrisentan, dabigatranetexilat, digoxin, kolchicin, maravirok, posakonazol, ranolazin, saxagliptin, sitagliptin, talinolol, tolvaptan) samtidigt med kabozantinib.
MRP2-hämmare
Administration av MRP2-hämmare kan resultera i ökningar av plasmakoncentrationerna av kabozantinib. Därför ska försiktighet iakttas vid samtidig användning av MRP2-hämmare (t.ex. ciklosporin, efavirenz, emtricitabin).
Hjälpämne
Natrium
Detta läkemedel innehåller mindre än 1 mmol natrium (23 mg) per kapsel, d.v.s är näst intill ”natriumfritt”.
Interaktioner
Effekt av andra läkemedel på kabozantinib
CYP3A4-hämmare och inducerare
Administrering av den potenta CYP3A4-hämmaren ketokonazol (400 mg dagligen i 27 dagar) till friska frivilliga försökspersoner minskade kabozantinibclearance (med 29 %) och ökade plasmaexponeringen vid engångsdos av kabozantinib (AUC) med 38 %. Därför ska samtidig administrering av potenta CYP3A4-hämmare (t.ex. ritonavir, itrakonazol, erytromycin, klaritromycin, grapefruktjuice) med kabozantinib hanteras med försiktighet.
Administrering av den potenta CYP3A4-induceraren rifampicin (600 mg dagligen i 31 dagar) till friska frivilliga försökspersoner ökade kabozantinibs clearance (4,3 gånger) och minskade plasmaexponeringen vid engångsdos av kabozantinib (AUC) med 77 %. Kronisk samtidig administrering av potenta CYP3A4-inducerare (t.ex. fenytoin, karbamazepin, rifampicin, fenobarbital eller naturläkemedel som innehåller johannesört [Hypericum perforatum]) med kabozantinib ska därför undvikas.
Medel som förändrar pH i magsäcken
Samtidig administrering av protonpumpshämmaren esomeprazol (40 mg dagligen under 6 dagar) och en enkel dos på 100 mg kabozantinib till friska frivilliga resulterade inte i några kliniskt signifikanta effekter på plasmaexponeringen av kabozantinib (AUC). Ingen dosjustering är indicerad när medel som förändrar pH i magsäcken (bl.a. protonpumpshämmare, H2-receptorantagonister och antacida) ges samtidigt med kabozantinib.
MRP2-hämmare
In vitro -data visar att kabozantinib är ett substrat av MRP2. Därför kan administration av MRP2-hämmare resultera i ökningar av plasmakoncentrationerna av kabozantinib.
Sekvestreringsmedel för gallsalt
Sekvestreringsmedel för gallsalt såsom kolestyramin och cholestagel kan interagera med kabozantinib och kan påverka absorptionen (eller reabsorptionen) vilket medför potentiellt minskad exponering (se avsnitt Farmakokinetiska egenskaper). Den kliniska betydelsen av dessa potentiella interaktioner är okänd.
Effekten av kabozantinib på andra läkemedel
Effekten av kabozantinib på farmakokinetiken hos kontraceptiva steroider har inte undersökts. Eftersom opåverkad kontraceptiv effekt inte kan garanteras rekommenderas en ytterligare kontraceptiv metod, exempelvis en barriärmetod.
På grund av kabozantinibs höga plasmaproteinbindningsgrad (avsnitt Farmakokinetiska egenskaper) kan interaktion med warfarin vara möjlig genom bortträngning från bindningsställen (”displacement”). I händelse av en sådan kombination, bör INR-värdet övervakas.
P-glykoproteinsubstrat
Kabozantinib var en hämmare (IC50 = 7,0 μM) men inte ett substrat av P‑glykoprotein (P‑gp)-transportaktiviteter i ett dubbelriktat analyssystem med MDCK‑MDR1-celler. Därför kan kabozantinib potentiellt öka plasmakoncentrationerna av samtidigt administrerade substrat av P‑gp. Patienterna ska varnas för att ta ett P‑gp-substrat (t.ex. fexofenadin, aliskiren, ambrisentan, dabigatranetexilat, digoxin, kolchicin, maravirok, posakonazol, ranolazin, saxagliptin, sitagliptin, talinolol, tolvaptan) samtidigt med kabozantinib.
Fertilitet, graviditet och amning
Kvinnor i fertil ålder/Preventivmedel för män och kvinnor
Kvinnor i fertil ålder måste tillrådas att undvika graviditet medan de tar kabozantinib. Kvinnliga partner till manliga patienter som tar kabozantinib måste också undvika graviditet. Effektiva preventivmetoder bör användas av både manliga och kvinnliga patienter och deras partner under behandling och i minst 4 månader efter avslutad behandling. Eftersom orala preventivmedel möjligen inte anses vara ett ”effektivt preventivmedel”, bör de användas tillsammans med en annan metod, exempelvis en barriärmetod (se avsnitt Interaktioner).
Graviditet
Inga studier av kabozantinib under graviditet har utförts. Djurstudier har uppvisat embryofetala och teratogena effekter (se avsnitt Prekliniska säkerhetsuppgifter). Den potentiella risken för människor är okänd. Kabozantinib bör inte användas under graviditet om inte kvinnans kliniska tillstånd kräver behandling med kabozantinib.
Amning
Det är okänt om kabozantinib och/eller dess metaboliter utsöndras i modersmjölk. På grund av den potentiella risken för skada på barnet ska mödrar avbryta amningen vid behandling med kabozantinib och undvika amning i minst 4 månader efter att behandlingen har avslutats.
Fertilitet
Data på human fertilitet saknas. Baserat på icke-kliniska säkerhetsdata kan manlig och kvinnlig fertilitet nedsättas av behandling med kabozantinib (se avsnitt Prekliniska säkerhetsuppgifter). Både män och kvinnor bör rådas att söka rådgivning och överväga fertilitetsbevarande åtgärder före behandling.
Effekter på förmågan att framföra fordon och använda maskiner
Kabozantinib har mindre effekt på förmågan att framföra fordon och använda maskiner. Biverkningar som trötthet och svaghet har förknippats med kabozantinib. Därför ska patienter rekommenderas att iaktta försiktighet vid framförande av fordon och användning av maskiner.
Biverkningar
Sammanfattning av säkerhetsprofil
De vanligaste allvarliga biverkningarna förknippade med kabozantinib är lunginflammation, slemhinneinflammation, hypokalcemi, dysfagi, uttorkning, lungemboli och hypertoni. De vanligaste biverkningarna oavsett grad (som upplevdes av minst 20 % av patienterna) inkluderade diarré, PPES, viktnedgång, minskad aptit, illamående, trötthet, smakförändringar, förändrad hårfärg, hypertoni, stomatit, förstoppning, kräkningar, slemhinneinflammation, asteni, hypokalcemi och dysfoni.
De vanligaste laboratorieavvikelserna var ökat aspartataminotransferas (ASAT), ökat alaninaminotransferas (ALAT), ökat alkaliskt fosfatas (ALP), lymfopeni, hypokalcemi, neutropeni, trombocytopeni, hypofosfatemi, hyperbilirubinemi, hypomagnesemi och hypokalemi.
Biverkningstabell
Biverkningarna är listade i Tabell 1 efter organsystemklass i MedDRA-systemet och efter frekvenskategori. Frekvenserna baseras på samtliga grader och definieras som mycket vanliga (≥ 1/10), vanliga (≥ 1/100 till < 1/10); mindre vanliga (≥ 1/1,000 till < 1/100), ingen känd frekvens (kan inte beräknas från tillgängliga data). Inom varje frekvensområde presenteras biverkningarna efter fallande allvarlighetsgrad.
Tabell 1: Biverkningar som har rapporterats med kabozantinib
| Infektioner och infestationer | |
| Vanliga | abscess* (inklusive invärtes, hud, tänder), lunginflammation, follikulit, svampinfektion (inklusive hud, oral, genital) |
| Mindre vanliga | Aspergillom |
| Endokrina systemet | |
| Vanliga | Hypotyreos |
| Metabolism och nutrition | |
| Mycket vanliga | minskad aptit, hypokalcemic, hypokalemic, hypomagnesemic |
| Vanliga | dehydrering*, hypoalbuminemic, hyperbilirubinemid, hypofosfatemic |
| Psykiska störningar | |
| Vanliga | ångest, depression, förvirringstillstånd |
| Mindre vanliga | onormala drömmar, delirium |
| Centrala och perifera nervsystemet | |
| Mycket vanliga | dysgeusi, huvudvärk, yrsel |
| Vanliga | cerebrovaskulär händelse*, perifer neuropati, parestesi, ageusi, tremor |
| Mindre vanliga | ataxi, störd uppmärksamhet, hepatisk encefalopati, medvetslöshet, talsvårigheter, posteriort reversibelt encefalopatisyndrom* |
| Ögon | |
| Vanliga | Dimsyn |
| Mindre vanliga | katarakt, konjunktivit |
| Öron och balansorgan | |
| Vanliga | öronvärk, tinnitus |
| Mindre vanliga | Hörselnedsättning |
| Hjärtat | |
| Vanliga | Förmaksflimmer, hjärtsvikt |
| Mindre vanliga | angina pectoris, supraventrikulär takykardi |
| Ingen känd frekvens | Hjärtinfarkt |
| Blodkärl | |
| Mycket vanliga | hypertoni*f |
| Vanliga | hypotonig, djup ventrombos*, venös trombos*, arteriell trombos*, blekhet, kalla händer och fötter |
| Mindre vanliga | Hypertensiv krish, artäremboli |
| Ingen känd frekvens | aneurysmer och arteriella dissektioner |
| Andningsvägar, bröstkorg och mediastinum | |
| Mycket vanliga | dysfoni, orofaryngeal smärta |
| Vanliga | icke-gastrointestinala fistlar* (inklusive i luftrören, i pneumomediastinum och trakeo-esofagala), lungemboli*, blödningar i andningsvägarna* (inklusive pulmonella, bronkiella, trakeala), aspirationspneumoni |
| Mindre vanliga | atelektas, svalgödem, pneumonit, pneumothorax |
| Magtarmkanalen | |
| Mycket vanliga | diarré*, illamående*, stomatit, förstoppning, kräkningar*, buksmärtae, dyspepsi, dysfagi, glossodyni |
| Vanliga | gastrointestinal perforation*, gastrointestinal fistel*, gastrointestinal blödning*, pankreatit, hemorrojder, analfissur, anal inflammation, keilit |
| Mindre vanliga | Esofagit |
| Lever och gallvägar | |
| Vanliga | Kolelitias |
| Hud och subkutan vävnad | |
| Mycket vanliga | palmar-plantar erytrodysestesi*, förändrad hårfärg, hudutslag, hudtorrhet, alopeci, erytem |
| Vanliga | hyperkeratos, acne, blåsor, onormal hårväxt, exfoliation, hypopigmentering |
| Mindre vanliga | hudsår, telangiektasi |
| Ingen känd frekvens | kutan vaskulit |
| Muskuloskeletala systemet och bindväv | |
| Mycket vanliga | artralgi, muskelspasmer, smärta i extremitet |
| Vanliga | muskuloskeletal bröstsmärta, osteonekros i käken* |
| Mindre vanliga | Rabdomyolys |
| Njurar och urinvägar | |
| Vanliga | proteinuri*, dysuri, hematuri |
| Mindre vanliga | akut njursvikt |
| Reproduktionsorgan och bröstkörtel | |
| Mindre vanliga | amenorré, vaginal blödning |
| Allmänna symtom och/eller symtom vid administreringsstället | |
| Mycket vanliga | trötthet, slemhinneinflammation, asteni |
| Vanliga | försämrad sårläkning*, frossa, ansiktsödem |
| Mindre vanliga | cysta, ansiktssmärta, lokaliserat ödem |
| Undersökningar | |
| Mycket vanliga | viktnedgång, förhöjda serumkoncentrationer av ALAT, ASAT, och ALP, förhöjt LDH och TSH i blod*d, trombocytopenia |
| Vanliga | förhöjt kreatinin i blod, lymfopenia, neutropenia, förhöjt lipas |
| Mindre vanliga | förkortad aktiverad partiell tromboplastintid, förhöjt antal eosinofilerb, förhöjt antal trombocyterb |
* Se avsnitt Biverkningar Beskrivning av utvalda biverkningar för ytterligare karaktärisering.
Följande termer har slagits samman för att erhålla en lämplig frekvenskategori:
a Sänkta hematologiparametrar: lymfopeni och minskat antal lymfocyter; neutropeni och minskat antal neutrofiler; trombocytopeni och minskat antal blodplättar.
b Förhöjda hematologiparametrar: förhöjt antal eosinofiler och eosinofili; förhöjt antal trombocyter och trombocytos.
c Sänkta biokemiparametrar: hypoalbuminemi och minskat albumin i blodet; hypokalcemi och minskat kalcium i blodet; hypokalemi och minskat kalium i blodet; hypomagnesemi och minskat magnesium i blodet; hypofosfatemi och minskat fosfor i blodet.
d Förhöjda biokemiparametrar: hyperbilirubinemi och förhöjt bilirubin i blodet; hypotyreoidism och förhöjt tyreoideastimulerande hormon i blodet.
e Buksmärta, obehag i buken, övre buksmärta och nedre buksmärta.
f Hypertoni och förhöjt blodtryck.
g Hypotoni och sänkt blodtryck.
h Inga fall av hypertensiv kris har rapporterats i kliniska studier med Cometriq, frekvensen baseras på samlad data från kabozantinib (inklusive data från Cabometyx 60 mg, tabletter).
Beskrivning av utvalda biverkningar
Ett TSH (tyreoideastimulerande hormon)-värde över det normala efter första dosen observerades hos 57 % av patienterna på kabozantinib kontra 19 % av patienterna på placebo (oavsett utgångsvärden). Nittiotvå procent av patienterna i kabozantinibarmen hade tyreoidektomi i anamnesen och 89 % tog sköldkörtelhormoner före den första dosen.
En ökning från baseline i korrigerat QT-intervall enligt Fridericia (QTcF) på 10 ‑ 15 ms på dag 29 (men inte på dag 1) efter påbörjad kabozantinibbehandling (med en dos på 140 mg dagligen) observerades i en kontrollerad klinisk studie i cancerpatienter (se avsnitt Varningar och försiktighet). Denna effekt var inte förknippad med en förändring i hjärtvågens form eller nya rytmer. Inga patienter som behandlades med kabozantinib hade ett QTcF‑intervall > 500 ms.
Se avsnitt Varningar och försiktighet för rekommendationer angående övervakning och hantering av följande biverkningar: perforationer, fistlar och intraabdominella abcesser, tromboemboliska biverkningar, blödning, aneurysmer och arteriella dissektioner, gastrointestinala besvär, sårkomplikationer, hypertoni, osteonekros, palmar-plantar erytrodysestesi, proteinuri och posteriort reversibelt encefalopatisyndrom.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till
webbplats: www.fimea.fi
Säkerhets- och utvecklingscentret för läkemedelsområdet Fimea
Biverkningsregistret
PB 55
00034 FIMEA
Överdosering
Det finns ingen specifik behandling tillgänglig i händelse av överdosering av kabozantinib och möjliga överdoseringssymtom har inte fastställts.
I händelse av misstänkt överdos ska kabozantinib sättas ut och understödjande behandling sättas in. Metabola kliniska laboratorievärden bör kontrolleras minst en gång i veckan eller när så bedöms kliniskt lämpligt för att bedöma eventuella trendförändringar. Biverkningar som förknippas med överdos ska behandlas symtomatiskt.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: cytostatikum, proteinkinashämmare, ATC-kod: L01EX07
Verkningsmekanism
Kabozantinib är en liten molekyl som hämmar flera receptortyrosinkinaser (RTK) inblandade i tumörtillväxt och angiogenes, patologisk benremodellering och metastasutveckling av cancer. Kabozantinib utvärderades med avseende på dess hämmande aktivitet mot en mängd olika kinaser och identifierades som en hämmare av MET- (hepatocyttillväxtfaktor) och VEGF- (vaskulär endotel tillväxtfaktor) receptorerna. Dessutom hämmar kabozantinib andra tyrosinkinaser, inklusive RET, GAS6‑receptorn (AXL), stamcellsfaktorreceptorn (KIT) och Fms‑liknande tyrosinkinas‑3 (FLT3).
Farmakodynamisk effekt
Kabozantinib uppvisade dosrelaterad inhibering av tumörtillväxt, regression, och/eller inhiberad metastasering i ett brett spektrum av prekliniska tumörmodeller.
Kabozantinib befanns vara effektivt i patienter med medullär tyreoideacancer med vildtyp- eller RET-mutation.
Kliniska data i medullär tyreoideacancer
En randomiserad, dubbelblind multicenterstudie som jämförde kabozantinib (N = 219) med placebo (N = 111) genomfördes i patienter med inoperabel lokalt avancerad eller metastaserande MTC och dokumenterad radiologisk sjukdomsprogression inom 14 månader före inträdet i studien. Det primära målet var att jämföra progressionsfri överlevnad (PFS) i patienter som fick kabozantinib kontra patienter som fick placebo. De sekundära målen var att jämföra total objektiv svarsfrekvens (ORR) och total överlevnad (OS). Centraliserad, oberoende, blindad granskning av bilddata användes vid utvärderingen av PFS och ORR. Patienterna behandlades till sjukdomsprogression eller till oacceptabel toxicitet.
Resultaten från PFS-analysen baserad på RECIST-bedömningen i den centrala granskningen visade en statistiskt signifikant skillnad i PFS-duration med kabozantinib jämfört med placebo: mediandurationen var 11,2 månader för patienter i kabozantinibarmen kontra 4,0 månader för patienter i placeboarmen (stratified hazard ratio [HR] = 0,28; 95 % CI: 0,19, 0,40; p < 0,0001; Figur 1). PFS-resultaten var konsekventa över alla baseline- och demografiska subgrupper som utvärderades, inklusive tidigare behandling med tyrosinkinashämmare (som kan ha bestått av ämnen med riktad verkan mot banor som motverkar angiogenes), RET-mutationsstatus (inklusive patienter som dokumenterats inte lida av RET-mutationer), tidigare cancerbehandling eller strålbehandling eller befintliga skelettmetastaser.
ORR var 27,9 % och 0 % för patienter i kabozantinibarmen respektive placeboarmen (p < 0,0001; Tabell 2). Mediandurationen för objektiv respons var 14,6 månader (95 % CI: 11,1, 17,5) för patienter i kabozantinibarmen.
Figur 1: Kaplan Meier-diagram över progressionsfri överlevnad
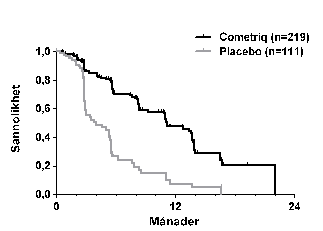
| Antal patienter i riskzonen | ||||||||
| Månad | 0 | 3 | 6 | 9 | 12 | 15 | 18 | 21 |
| Cometriq | 219 | 121 | 78 | 55 | 31 | 12 | 2 | 1 |
| Placebo | 111 | 35 | 11 | 6 | 3 | 2 | 0 | 0 |
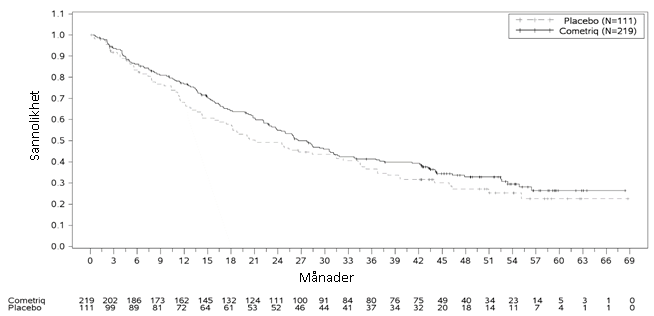
Den slutliga analysen av OS utfördes efter att 218 händelser (dödsfall) inträffat och visar på en trend av ökad medianöverlevnad på 5,5 månader i kabozantinibarmen: median (månader) 26,6 för kabozantinib jämfört med 21,1 för placebo (HR = 0,85 [95 % konfidensintervall: 0,64, 1,12], p‑värde = 0,2409).
Figur 2: Kaplan Meier-diagram över total överlevnad
Tabell 2: Sammanfattning av de viktigaste effektresultaten
| Kabozantinib | Placebo | |
| Medianvärde, progressionsfri överlevnad | 11,2 månader | 4,0 månader |
| HR: 0,28 (0,19, 0,40) p < 0,0001 | ||
| Medianvärde, total överlevnad | 26,6 månader | 21,1 månader |
| HR: 0,85 (0,64, 1,12) p = 0,2409 | ||
| Total objektiv svarsfrekvensa (95 % CI) | 27,9 % (21,9 %, 34,5 %) | 0 % |
| p < 0,0001 | ||
| Svarsduration; median (95 % CI) | 14,6 månader 11,1, 17,5) | - |
| Sjukdomskontrollb(95 % CI) | 55,3 % (48,3 %, 62,2 %) | 13,5 % (7,6 %, 21,6 %) |
| Kalcitoninresponsa | 47 % (49/104)c | 3 % (1/40)c |
| CEA (carcinoembryonalt antigen)-responsa | 33 % (47/143)c | 2 % (1/55)c |
a Respons = CR + PR
b Sjukdomskontroll = SD+ ORR
c Avser de patienter som gick att utvärdera avseende respons
RET-mutationsstatus
Av de 215 patienterna med tillräckliga data för att bedöma mutationsstatus klassificerades 78,6 % (n = 169) som RET-mutationspositiva (av vilka 126 var positiva för M918T-mutationen), och 21,4 % (n = 46) som RET-mutationsnegativa. För ytterligare 115 patienter gick inte RET-mutationsstatus att fastställa eller var oklar. Alla tre subgrupperna uppvisade förhöjt PFS I kabozantinibarmen jämfört med placeboarmen (HR på 0,23, 0,53 respektive 0,30 för den RET-mutationspositiva, -negativa och okända subgruppen). Objektiva svarsfrekvenser i dessa subgrupper var generellt konsekventa med PFS-resultaten: den RET-positiva, RET-negativa och okända subgruppen uppvisade tumörsvarsfrekvenser på 32 %, 22 % respektive 25 %.
Ytterligare genetisk analys påvisade somatiska tumörmutationer i HRAS, KRAS eller NRAS hos en liten del av patienterna. Dessa patienter (n = 16) uppvisade signifikant förlängning av PFS (HR 0,15) och en objektiv svarsfrekvens på 31 %. RET-mutationsnegativa patienter utan tecken på RAS-mutation (n = 33) uppvisade en minskad PFS-nytta på kabozantinib (HR på 0,87) och en lägre svarsfrekvens på 18 % jämfört med andra mutationssubgrupper.
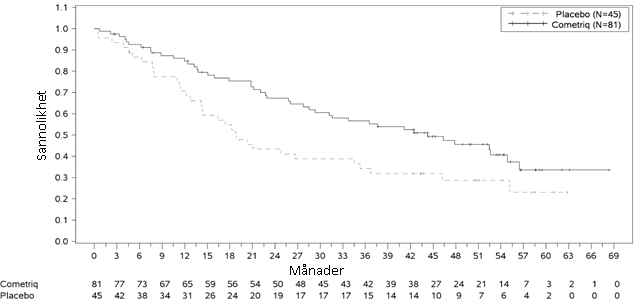
En signifikant förbättring av OS observerades i undergruppen RET M918T-mutationspositiva patienter (n=81/219 kabozantinibarmen): 44,3 månader i kabozantinibarmen jämfört med 18,9 månader i placeboarmen (HR = 0,60, p = 0,0255). Ingen förbättring av OS förekom i de RET M918T-negativa och okända undergrupperna.
Figur 3: Kaplan Meier-analys av OS hos patienter med en RET M918T-mutation
Pediatrisk population
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för kabozantinib i en eller flera grupper av den pediatriska populationen för behandling av maligna solida tumörer (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Absorption
Vid oral administrering av kabozantinib nås maximal plasmakoncentration vid 2 till 5 timmar efter dosering. Plasma-koncentrationstidsprofiler visar en andra absorptionstopp ungefär 24 timmar efter administrering, vilket tyder på att kabozantinib kan genomgå enterohepatisk recirkulation.
Upprepad daglig dosering av 140 mg kabozantinib i 19 dagar resulterade i en ungefärlig 4‑ till 5‑faldig ackumulering av kabozantinib (baserat på AUC) jämfört med administrering av en enda dos. Steady state uppnås från ungefär dag 15.
En fettrik måltid ökade måttligt Cmax och AUC (41 % respektive 57 %) jämfört med fastande mage i friska frivilliga försökspersoner som tog en enda peroral dos 140 mg kabozantinib. Det finns ingen information om den exakta effekten av matintag 1 timme efter administrering av kabozantinib.
Bioekvivalens kunde inte visas mellan kabozantinib som kapsel respektive tablett efter en engångsdos på 140 mg till friska försökspersoner. En ökning på 19 % av Cmax för tablettformuleringen (CABOMETYX) jämfört med kapselformuleringen (COMETRIQ) observerades. En skillnad på mindre än 10 % i AUC observerades mellan kabozantinib formulerat som tablett (CABOMETYX) och kapsel (COMETRIQ).
Distribution
Kabozantinib är starkt proteinbundet in vitro i humanplasma (≥ 99,7 %). Baserat på populations-farmakokinetiska (PK) modellen, är distributionsvolymen (V/F) cirka 349 l (SE: ± 2,73 %). Proteinbindningen var oförändrad hos patienter med lätt eller måttligt nedsatt njur- eller leverfunktion.
Metabolism
Kabozantinib metaboliserades in vivo. Fyra metaboliter observerades i plasma vid exponeringar (AUC) större än 10 % av moderföreningen: XL184‑N‑oxid, XL184 amid‑spjälkningsprodukt, XL184‑monohydroxisulfat, och 6‑desmetylamid-spjälkningsprodukt-sulfat. Två icke-konjugerade metaboliter (XL184‑N‑oxid och XL184 amid-spjälkningsprodukt) som har < 1 % av moderföreningen kabozantinibs målriktade kinasinhiberingspotens står för vardera < 10 % av den totala läkemedelsrelaterade plasmaexponeringen.
Kabozantinib är ett substrat för CYP3A4-metabolism in vitro i egenskap av en neutraliserande antikropp mot CYP3A4-inhiberad bildning av metabolit XL184 N‑oxid med > 80 % i en NADPH-katalyserad humanlevermikrosomal (HLM) inkubering. Däremot hade neutraliserande antikroppar mot CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6 och CYP2E1 ingen effekt på bildningen av kabozantinibs metaboliter. En neutraliserande antikropp mot CYP2C9 uppvisade en minimal effekt på kabozantinibs metabolitbildning (dvs en minskning på < 20 %).
Eliminering
Plasmahalveringstiden för kabozantinib i engångsdosstudier i friska frivilliga försökspersoner är cirka 120 timmar. Genomsnittlig clearance (CL/F) vid steady-state i cancerpatienter uppskattades till 4,4 l/h i en populations PK-analys. Inom en samlingsperiod på 48 dagar efter en engångsdos 14C‑kabozantinib i friska frivilliga försökspersoner utsöndrades cirka 81 % av den totala administrerade radioaktiviteten, varav 54 % i avföring och 27 % i urin.
Farmakokinetik i speciella patientgrupper
Nedsatt njurfunktion
Resultat från en studie på patienter med nedsatt njurfunktion tyder på att förhållandet mellan geometriska LS-medelvärden för plasma-kabozantinib, Cmax och AUC0-inf var 19 % och 30 % högre för patienter med lätt nedsatt njurfunktion (90 % CI för Cmax 91,60 % till 155,51 %, AUC0-inf 98,79 % till 171,26 %) och 2 % och 6-7 % högre (90 % CI för Cmax 78,64 % till 133,52 %; AUC0-inf 79,61 % till 140,11 %), för patienter med måttligt nedsatt njurfunktion jämfört med personer med normal njurfunktion. Patienter med gravt nedsatt njurfunktion har inte studerats.
Nedsatt leverfunktion
Resultat från en studie på patienter med nedsatt leverfunktion visar att exponeringen (AUC0-inf) ökade med 81 % och 63 % hos patienter med lätt till måttligt nedsatt leverfunktion, respektive (90 % CI för AUC0-inf: 121,44 % till 270,34 % för lätt och 107,37 % till 246,67 % måttligt). Patienter med gravt nedsatt leverfunktion har inte studerats.
Etnicitet
Inga data finns tillgängliga för att avgöra eventuella etnicitetsbaserade skillnader i PK.
Prekliniska säkerhetsuppgifter
Följande effekter har inte setts hos människa, men har setts i exponeringar nära klinisk exponering och bedöms därför ha möjlig klinisk relevans:
I upp till 6 månader långa studier av toxicitet med upprepad dosering i råttor och hundar var målorganen magtarmkanalen, benmärgen, lymfoida vävnader, njure, binjure och vävnader i reproduktionsorganen. NOAEL (nivå utan observerade skadliga effekter) för dessa fynd var lägre än mänskliga kliniska exponeringsnivåer vid avsedd terapeutisk dos.
Kabozantinib har inte uppvisat någon mutagen eller klastogen potential i en standarduppsättning genotoxiska tester. Kabozantinibs cancerogena potential har utvärderats i två arter: rasH2-transgena möss och Sprague-Dawleyråttor. I den 2 år långa karcinogenicitetsstudien på råttor bestod de kabozantinibrelaterade neoplastiska fynden av en ökad incidens av godartad feokromocytom, ensamt eller i kombination med malign feokromocytom/komplex malign feokromocytom, i binjuremärgen hos båda könen vid exponeringar långt under den avsedda exponeringen hos människa. Den kliniska relevansen av de neoplastiska förändringarna som observerats i råttor är osäker, men är troligen låg. Kabozantinib var inte cancerogen i rasH2 musmodell vid en något högre exponering än den avsedda humana terapeutiska exponeringen.
I fertilitetsstudier i råttor observerades minskad fertilitet i både hanar och honor. Vidare observerades hypospermatogenes hos hanhundar vid exponeringsnivåer under kliniska exponeringsnivåer för människa vid avsedd terapeutisk dos.
Embryofetala utvecklingsstudier utfördes på råtta och kanin. Hos råttor orsakade kabozantinib postimplantationsförlust, fetalt ödem, gomspalt/kluven läpp, dermal aplasi och böjd eller rudimentär svans. Hos kaniner gav kabozantinib upphov till fetala mjukdelsförändringar (minskad mjältstorlek, liten eller saknad mellanliggande lunglob) och ökade den totala förekomsten av fostermissbildningar. NOAEL för embryo-fetal toxicitet och teratogena fynd understeg de kliniska exponeringsnivåerna för människa vid avsedd terapeutisk dos.
Juvenila råttor (jämförbart med en > 2 år gammal pediatrisk population) som gavs kabozantinib uppvisade ökade WBC-parametrar, minskad hematopoes, pubescent/omoget kvinnligt reproduktionssystem (utan fördröjd vaginal öppning), tandabnormaliteter, minskat benmineralinnehåll och täthet, leverpigmentering och gallgångshyperplasi. Fynden i livmoder/äggstockar och minskad hematopoes tycks vara övergående, medan effekterna på benparametrar samt leverpigmentering kvarstod. Bedömning i juvenila råttor (jämförbart med en < 2 år gammal pediatrisk population) har inte utförts.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Kapselinnehåll
Mikrokristallin cellulosa
Kroskarmellosnatrium
Natriumstärkelseglykolat
Kolloidal vattenfri kiseldioxid
Stearinsyra
Kapselhölje
Gelatin
Svart järnoxid (E172) (endast 20 mg kapslar)
Röd järnoxid (E172) (endast 80 mg kapslar)
Titandioxid (E171)
Tryckfärg
Shellack
Svart järnoxid (E172)
Propylenglykol
Inkompatibiliteter
Ej relevant.
Hållbarhet
3 år.
Särskilda förvaringsanvisningar
Förvaras vid högst 25°C.
Förvaras i originalförpackningen fuktkänsligt.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
COMETRIQ kapseli, kova
20 mg + 80 mg (L:ei) 84+28 fol (4652,50 €)
PF-selosteen tieto
PVC/PE/PCTFE-aluminiumblister med baksida av aluminiumfolie, förseglad i en värmeförseglad förpackning av kartong.
Varje blisterkarta innehåller:
21 kapslar om 20 mg och 7 kapslar om 80 mg (140 mg daglig dos ger 7 dagar).
28-dagarsförpackning:
112 kapslar (4 blisterkartor med 21 x 20 mg och 7 x 80 mg) (140 mg daglig dos ger 28 dagar).
Läkemedlets utseende:
Cometriq 20 mg hård kapsel:
De hårda kapslarna är grå med “XL184 20mg” tryckt i svart på huvuddelen av kapseln. Kapseln innehåller ett benvitt till vitt pulver.
Cometriq 80 mg hård kapsel:
De hårda kapslarna är orange med “XL184 80mg” tryckt i svart på huvuddelen av kapseln. Kapseln innehåller ett benvitt till vitt pulver.
Särskilda anvisningar för destruktion och övrig hantering
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
COMETRIQ kapseli, kova
20 mg + 80 mg 84+28 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Kabotsantinibi: Munuaissyövän ja kilpirauhassyövän hoito erityisin edellytyksin (3012).
Atc-kod
L01EX07
Datum för översyn av produktresumén
15.01.2026
Yhteystiedot
Kista Science Tower, Färögatan 33
SE-164 51 Kista
Sweden
+46 8 451 60 00
www.ipsen.com
info.se@ipsen.com