
KYPROLIS pulver till infusionsvätska, lösning 10 mg, 30 mg, 60 mg
Kvalitativ och kvantitativ sammansättning
Kyprolis 10 mg pulver till infusionsvätska, lösning
En injektionsflaska innehåller 10 mg carfilzomib.
Hjälpämne med känd effekt
En injektionsflaska innehåller 37 mg natrium.
En injektionsflaska innehåller 500 mg cyklodextrin (sulfobutylbetadexnatrium).
Kyprolis 30 mg pulver till infusionsvätska, lösning
En injektionsflaska innehåller 30 mg carfilzomib.
Hjälpämne med känd effekt
En injektionsflaska innehåller 109 mg natrium.
En injektionsflaska innehåller 1 500 mg cyklodextrin (sulfobutylbetadexnatrium).
Kyprolis 60 mg pulver till infusionsvätska, lösning
En injektionsflaska innehåller 60 mg carfilzomib.
Hjälpämne med känd effekt
En injektionsflaska innehåller 216 mg natrium.
En injektionsflaska innehåller 3 000 mg cyklodextrin (sulfobutylbetadexnatrium).
Efter rekonstituering innehåller 1 ml lösning 2 mg carfilzomib.
För fullständig förteckning över hjälpämnen, se avsnitt Förteckning över hjälpämnen.
Läkemedelsform
Pulver till infusionsvätska, lösning.
Kliniska uppgifter
Terapeutiska indikationer
Kyprolis i kombination med daratumumab och dexametason, med lenalidomid och dexametason eller med enbart dexametason är indicerat för behandling av multipelt myelom hos vuxna patienter som tidigare har fått minst en behandling (se avsnitt Farmakodynamiska egenskaper).
Villkor
Valmiste annetaan syövän lääkehoitoon perehtyneen lääkärin valvonnassa.
Dosering och administreringssätt
Behandling med Kyprolis ska ske under övervakning av en läkare med erfarenhet av cancerbehandling.
Dosering
Dosen beräknas utifrån patientens kroppsyta vid behandlingsstart. Patienter som har en större kroppsyta än 2,2 m2 ska få en dos som baseras på en kroppsyta på 2,2 m2. Det krävs inga dosjusteringar för viktförändringar som är mindre än eller lika med 20 %.
Kyprolis i kombination med lenalidomid och dexametason
När Kyprolis ges i kombination med lenalidomid och dexametason administreras det intravenöst som en 10 minuter lång infusion, under två på varandra följande dagar, varje vecka i tre veckor (dag 1, 2, 8, 9, 15 och 16), vilket följs av en 12 dagars återhämtningsperiod (dag 17 till 28) såsom visas i tabell 1. Varje 28‑dagarsperiod betraktas som en behandlingscykel.
Kyprolis administreras med en startdos på 20 mg/m2 (maxdos 44 mg) dag 1 och 2 i cykel 1. Om denna dos tolereras ökas dosen från och med dag 8 i cykel 1 till 27 mg/m2 (maxdos 60 mg). Från och med cykel 13 ska Kyprolis‑doserna på dag 8 och 9 uteslutas.
Behandlingen kan fortgå tills sjukdomen progredierar eller tills patienten drabbas av oacceptabel toxicitet.
Behandling med Kyprolis i kombination med lenalidomid och dexametason i mer än 18 cykler ska grundas på en individuell bedömning av nyttan mot den eventuella risken med fortsatt behandling, eftersom data avseende carfilzomibs tolerabilitet och toxicitet efter 18 cykler är begränsad (se avsnitt Farmakodynamiska egenskaper).
I kombination med Kyprolis administreras lenalidomid 25 mg peroralt på dag 1‑21 och dexametason 40 mg peroralt eller intravenöst på dag 1, 8, 15 och 22 i 28‑dagarscyklerna. Lämpliga dosminskningar av startdosen lenalidomid ska göras i enlighet med rekommendationerna i den aktuella produktresumén för lenalidomid, till exempel för patienter med nedsatt njurfunktion vid behandlingsstart. Dexametason ska administreras 30 minuter till 4 timmar före Kyprolis.
Tabell 1. Kyprolis i kombination med lenalidomid och dexametason
Cykel 1 | |||||||||||
Vecka 1 | Vecka 2 | Vecka 3 | Vecka 4 | ||||||||
Dag 1 | Dag 2 | Dag 3‑7 | Dag 8 | Dag 9 | Dag 10‑14 | Dag 15 | Dag 16 | Dag 17‑21 | Dag 22 | Dag 23‑28 | |
Kyprolis (mg/m2)a | 20 | 20 | - | 27 | 27 | - | 27 | 27 | - | - | - |
Dexametason (mg) | 40 | - | - | 40 | - | - | 40 | - | - | 40 | - |
Lenalidomid | 25 mg dagligen | - | - | ||||||||
Cykel 2‑12 | |||||||||||
Vecka 1 | Vecka 2 | Vecka 3 | Vecka 4 | ||||||||
Dag 1 | Dag 2 | Dag 3‑7 | Dag 8 | Dag 9 | Dag 10‑14 | Dag 15 | Dag 16 | Dag 17‑21 | Dag 22 | Dag 23‑28 | |
Kyprolis (mg/m2)a | 27 | 27 | - | 27 | 27 | - | 27 | 27 | - | - | - |
Dexametason (mg) | 40 | - | - | 40 | - | - | 40 | - | - | 40 | - |
Lenalidomid | 25 mg dagligen | - | - | ||||||||
Cykel 13 och därefter | |||||||||||
Vecka 1 | Vecka 2 | Vecka 3 | Vecka 4 | ||||||||
Dag 1 | Dag 2 | Dag 3‑7 | Dag 8 | Dag 9 | Dag 10‑14 | Dag 15 | Dag 16 | Dag 17‑21 | Dag 22 | Dag 23‑28 | |
Kyprolis (mg/m2)a | 27 | 27 | - | - | - | - | 27 | 27 | - | - | - |
Dexametason (mg) | 40 | - | - | 40 | - | - | 40 | - | - | 40 | - |
Lenalidomid | 25 mg dagligen | - | - | ||||||||
| a. Infusionstiden är 10 minuter och förblir densamma under behandlingsregimen | |||||||||||
Kyprolis i kombination med dexametason
När Kyprolis ges i kombination med dexametason administreras det intravenöst som en 30 minuter lång infusion, under två på varandra följande dagar, varje vecka i tre veckor (dag 1, 2, 8, 9, 15 och 16), vilket följs av en 12 dagars återhämtningsperiod (dag 17 till 28) såsom visas i tabell 2. Varje 28‑dagarsperiod betraktas som en behandlingscykel.
Kyprolis administreras med en startdos på 20 mg/m2 (maxdos 44 mg) dag 1 och 2 i cykel 1. Om denna dos tolereras ökas dosen från och med dag 8 i cykel 1 till 56 mg/m2 (maxdos 123 mg).
Behandlingen kan fortgå tills sjukdomen progredierar eller tills patienten drabbas av oacceptabel toxicitet.
När Kyprolis administreras i kombination med dexametason, administreras dexametason 20 mg peroralt eller intravenöst på dag 1, 2, 8, 9, 15, 16, 22 och 23 i 28‑dagarscyklerna. Dexametason ska administreras 30 minuter till 4 timmar före Kyprolis.
Tabell 2. Kyprolis i kombination med enbart dexametason
Cykel 1 | |||||||||||||
Vecka 1 | Vecka 2 | Vecka 3 | Vecka 4 | ||||||||||
Dag 1 | Dag 2 | Dag 3‑7 | Dag 8 | Dag 9 | Dag 10‑14 | Dag 15 | Dag 16 | Dag 17‑21 | Dag 22 | Dag 23 | Dag 24‑28 | ||
Kyprolis (mg/m2)a | 20 | 20 | - | 56 | 56 | - | 56 | 56 | - | - | - | - | |
Dexametason (mg) | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | |
Cykel 2 och alla efterföljande cykler | |||||||||||||
Vecka 1 | Vecka 2 | Vecka 3 | Vecka 4 | ||||||||||
Dag 1 | Dag 2 | Dag 3‑7 | Dag 8 | Dag 9 | Dag 10‑14 | Dag 15 | Dag 16 | Dag 17‑21 | Dag 22 | Dag 23 | Dag 24‑28 | ||
Kyprolis (mg/m2)a | 56 | 56 | - | 56 | 56 | - | 56 | 56 | - | - | - | - | |
Dexametason (mg) | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | |
| a. Infusionstiden är 30 minuter och förblir densamma under behandlingsregimen | |||||||||||||
Kyprolis i kombination med daratumumab och dexametason
När Kyprolis ges i kombination med daratumumab och dexametason administreras det intravenöst som en 30 minuter lång infusion, under två på varandra följande dagar, varje vecka i tre veckor (dag 1, 2, 8, 9, 15 och 16), vilket följs av en 12‑dagars återhämtningsperiod (dag 17 till 28) såsom visas i tabell 3. Varje 28‑dagarsperiod betraktas som en behandlingscykel.
Kyprolis administreras med en startdos på 20 mg/m2 (maxdos 44 mg) dag 1 och 2 i cykel 1. Om denna dos tolereras ökas dosen från och med dag 8 i cykel 1 till 56 mg/m2 (maxdos 123 mg).
Behandlingen kan fortgå tills sjukdomen progredierar eller tills patienten drabbas av oacceptabel toxicitet.
En dos på 20 mg dexametason administreras peroralt eller intravenöst på dagarna 1, 2, 8, 9, 15 och 16, följt av 40 mg peroralt eller intravenöst på dag 22 i varje 28-dagarscykel. För patienter > 75 år administreras 20 mg dexametason peroralt eller intravenöst en gång i veckan efter den första veckan. Dexametason ska administreras 30 minuter till 4 timmar före Kyprolis.
Daratumumab kan administreras intravenöst eller subkutant.
Vid intravenös administrering ges daratumumab med en dos på 16 mg/kg (faktisk kroppsvikt), en gång per vecka, dock med den första dosen uppdelad på 8 mg/kg på dag 1 och 2 i cykel 1. Därefter administreras daratumumab 16 mg/kg en gång per vecka på dag 8, 15 och 22 i cykel 1 och på dag 1, 8, 15 och 22 i cykel 2, sedan varannan vecka under 4 cykler (cykel 3 till 6) och sedan var fjärde vecka under de återstående cyklerna eller till sjukdomsprogression.
Alternativt kan daratumumab ges subkutant med en dos på 1 800 mg på dag 1, 8, 15 och 22 i cykel 1, på dag 1, 8, 15 och 22 i cykel 2, sedan varannan vecka under 4 cykler (cykel 3 till 6) och sedan var fjärde vecka under de återstående cyklerna eller till sjukdomsprogression.
Se produktresumén för daratumumab för ytterligare information rörande användningen av den subkutana beredningen.
De dagar då fler än ett av dessa läkemedel administreras är den rekommenderade administreringsordningen följande: dexametason, läkemedel inför daratumumabinfusion (se avsnittet Samtidig läkemedelsbehandling), carfilzomib, daratumumab och läkemedel efter daratumumabinfusion (se avsnittet Samtidig läkemedelsbehandling).
Se produktresuméerna för daratumumab och dexametason för ytterligare information om administrering.
Tabell 3. Kyprolis i kombination med dexametason och daratumumab
| Cykel 1 | |||||||||||
Vecka 1 | Vecka 2 | Vecka 3 | Vecka 4 | |||||||||
Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | |
Kyprolis (mg/m2)a | 20 | 20 | - | 56 | 56 | - | 56 | 56 | - | - | - | - |
Dexametason (mg)b | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 40 | - | - |
Daratumumab (intravenöst ELLER subkutant) | ||||||||||||
Intravenös administrering (mg/kg) | 8 | 8 | - | 16 | - | - | 16 | - | - | 16 | - | - |
Subkutan administrering (mg) | 1 800 | - | - | 1 800 | - | - | 1 800 | - | - | 1 800 | - | - |
| Cykel 2 | |||||||||||
Vecka 1 | Vecka 2 | Vecka 3 | Vecka 4 | |||||||||
Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | |
Kyprolis (mg/m2)a | 56 | 56 | - | 56 | 56 | - | 56 | 56 | - | - | - | - |
Dexametason (mg)b | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 40 | - | - |
Daratumumab (intravenöst ELLER subkutant) | ||||||||||||
Intravenös administrering (mg/kg) | 16 | - | - | 16 | - | - | 16 | - | - | 16 | - | - |
Subkutan administrering (mg) | 1 800 | - | - | 1 800 | - | - | 1 800 | - | - | 1 800 | - | - |
| Cykel 3–6 | |||||||||||
Vecka 1 | Vecka 2 | Vecka 3 | Vecka 4 | |||||||||
Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | |
Kyprolis (mg/m2)a | 56 | 56 | - | 56 | 56 | - | 56 | 56 | - | - | - | - |
Dexametason (mg)b | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 40 | - | - |
Daratumumab (intravenöst ELLER subkutant) | ||||||||||||
Intravenös administrering (mg/kg) | 16 | - | - | - | - | - | 16 | - | - | - | - | - |
Subkutan administrering (mg) | 1 800 | - | - | - | - | - | 1 800 | - | - | - | - | - |
| Cykel 7 och alla efterföljande cykler | |||||||||||
Vecka 1 | Vecka 2 | Vecka 3 | Vecka 4 | |||||||||
Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | Dag | |
Kyprolis (mg/m2)a | 56 | 56 | - | 56 | 56 | - | 56 | 56 | - | - | - | - |
Dexametason (mg)b | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 40 | - | - |
Daratumumab (intravenöst ELLER subkutant) | ||||||||||||
Intravenös administrering (mg/kg) | 16 | - | - | - | - | - | - | - | - | - | - | - |
Subkutan administrering (mg) | 1 800 | - | - | - | - | - | - | - | - | - | - | - |
a. Infusionstiden är 30 minuter och förblir densamma under behandlingsregimen b. Till patienter > 75 år administreras 20 mg dexametason oralt eller intravenöst varje vecka efter den första veckan. | ||||||||||||
Samtidig läkemedelsbehandling
Profylaktisk antiviral behandling ska övervägas för patienter som behandlas med Kyprolis för att minska risken för reaktivering av herpes zoster‑infektion (se avsnitt Biverkningar).
Trombosprofylax rekommenderas för patienter som behandlas med Kyprolis i kombination med daratumumab och dexametason, med lenalidomid och dexametason eller med enbart dexametason och ska baseras på en bedömning av patientens underliggande risker och kliniska status. För andra läkemedel som kan behövas samtidigt, till exempel förebyggande behandling med syraneutraliserande medel (antacida), hänvisas till de aktuella produktresuméerna för lenalidomid och dexametason.
Patienter som behandlas med Kyprolis i kombination med daratumumab och dexametason ska få läkemedel före infusion (premedicinering) för att minska risken för infusionsrelaterade reaktioner av daratumumab.
Se produktresumén för daratumumab för ytterligare information om samtidig läkemedelsbehandling, bl.a. läkemedel före och efter infusion.
Hydrering, vätske‑ och elektrolytkontroller
Adekvat uppvätskning krävs innan den första dosen administreras i cykel 1, i synnerhet hos patienter som löper hög risk att drabbas av tumörlyssyndrom eller njurtoxicitet. Alla patienter ska övervakas för tecken på övervätskning och behovet av extra vätska ska anpassas efter den enskilda patientens behov. Justering av den totala vätskevolymen ska baseras på klinisk status vid behandlingsstart hos patienter med hjärtsvikt eller som löper risk att drabbas av hjärtsvikt (se avsnitt Varningar och försiktighet).
Rekommenderad uppvätskning innefattar både perorala vätskor (30 ml/kg/dag under 48 timmar före dag 1 i cykel 1) och intravenösa vätskor (250 ml till 500 ml lämpliga intravenösa vätskor före varje dos i cykel 1). Ge ytterligare 250 ml till 500 ml intravenösa vätskor vid behov efter administrering av Kyprolis i cykel 1. Peroral och/eller intravenös uppvätskning ska fortsatt ges efter behov i de efterföljande cyklerna.
När Kyprolis ges i kombination med intravenöst daratumumab, krävs inte oral eller intravenös uppvätskning de dagar då intravenöst daratumumab administreras.
Kaliumnivåerna i serum ska kontrolleras varje månad eller oftare under behandling med Kyprolis beroende på patientens kliniska status. Kontrollfrekvensen avgörs av kaliumnivåerna före behandlingsstarten, samtidigt användning av andra läkemedel (t.ex. läkemedel med känd ökad risk för hypokalemi) och medföljande komorbiditeter.
Rekommenderade dosjusteringar
Doseringen av Kyprolis ska modifieras efter eventuell behandlingsrelaterad toxicitet. Rekommenderade åtgärder och dosjusteringar beskrivs i tabell 4. Dosnivåminskningar visas i tabell 5.
Tabell 4. Dosjusteringar vid behandling med Kyprolis
Hematologisk toxicitet | Rekommenderad åtgärd |
|
|
|
|
|
|
Icke‑hematologisk toxicitet (njurrelaterad) | Rekommenderad åtgärd |
|
|
Övrig icke‑hematologisk toxicitet | Rekommenderad åtgärd |
|
|
| a. Se tabell 5 för dosnivåminskningar | |
Tabell 5. Dosnivåminskningar för Kyprolis
Behandlingsregim | Kyprolisdos | Första minskningen av Kyprolisdosen | Andra minskningen av Kyprolisdosen | Tredje minskningen av Kyprolisdosen |
Kyprolis, lenalidomid och dexametason | 27 mg/m2 | 20 mg/m2 | 15 mg/m2 a | — |
Kyprolis och dexametason | 56 mg/m2 | 45 mg/m2 | 36 mg/m2 | 27 mg/m2 a |
Kyprolis, daratumumab och dexametason | 56 mg/m2 | 45 mg/m2 | 36 mg/m2 | 27 mg/m2 a |
Obs! Infusionstiden för Kyprolis ändras inte under dosminskningarna a. Om symtomen inte går tillbaka ska behandlingen med Kyprolis avbrytas | ||||
Särskilda populationer
Nedsatt njurfunktion
Patienter med måttligt eller gravt nedsatt njurfunktion rekryterades till kombinationsstudierna med Kyprolis och dexametason, men fick ej delta i kombinationsstudierna med Kyprolis och lenalidomid. Data är därför begränsad för Kyprolis i kombination med lenalidomid och dexametason hos patienter med kreatininclearance (CrCL) < 50 ml/min. Lämpliga dossänkningar av startdosen av lenalidomid bör beaktas för patienter med nedsatt njurfunktion vid baslinjen, i enlighet med de rekommendationer som anges i lenalidomids produktresumé.
Baserat på tillgängliga farmakokinetiska data rekommenderas inga justeringar av startdosen av Kyprolis för patienter med lindrigt, måttligt eller gravt nedsatt njurfunktion vid behandlingsstart och inte heller för patienter i dialys (se avsnitt Farmakokinetiska egenskaper). I kliniska fas 3‑studier var dock incidensen av biverkningen akut njursvikt högre hos patienter med lägre kreatininclearance vid baslinjen än bland patienter med högre kreatininclearance vid baslinjen.
Njurfunktionen ska kontrolleras vid behandlingsstarten och därefter minst en gång per månad eller i enlighet med godtagen klinisk praxis, i synnerhet bland patienter med lägre kreatininclearance vid baslinjen (CrCL < 30 ml/min). Lämpliga dosjusteringar baserade på uppkommen toxicitet ska göras (se tabell 4). Det finns begränsad data över effekt och säkerhet hos patienter med kreatininclearance < 30 ml/min vid baslinjen.
Eftersom Kyprolisclearance vid dialys inte har studerats ska läkemedlet administreras efter dialystillfället.
Nedsatt leverfunktion
Patienter med måttligt eller gravt nedsatt leverfunktion fick ej delta i studier där Kyprolis gavs i kombination med antingen lenalidomid och dexametason eller med enbart dexametason.
Kyprolis farmakokinetik har inte utvärderats hos patienter med gravt nedsatt leverfunktion. Baserat på tillgängliga farmakokinetiska data rekommenderas inga justeringar av startdosen för patienter med lindrigt eller måttligt nedsatt leverfunktion. Dock har högre förekomst av avvikande leverfunktion, biverkningar ≥ grad 3 och allvarliga biverkningar rapporterats hos patienter med lindrigt till måttligt nedsatt leverfunktion vid behandlingsstart jämfört med patienter med normal leverfunktion (se avsnitt Varningar och försiktighet och Farmakokinetiska egenskaper). Leverenzymer och bilirubin ska kontrolleras vid behandlingens start och därefter en gång per månad under behandlingen med carfilzomib, oavsett värdet vid behandlingsstart, och lämpliga dosjusteringar ska göras baserat på uppkommen toxicitet (se tabell 4). Patienter med måttligt eller gravt nedsatt leverfunktion ska övervakas extra noga, eftersom data över effekt och säkerhet är begränsad för denna patientgrupp.
Äldre patienter
I kliniska studier var förekomsten av vissa biverkningar (däribland hjärtsvikt) totalt sett högre hos patienter som var ≥ 75 år jämfört med patienter som var < 75 år (se avsnitt Varningar och försiktighet).
Pediatrisk population
Säkerhet och effekt för Kyprolis för pediatriska patienter har inte fastställts. Inga data finns tillgängliga.
Administreringssätt
Kyprolis ska administreras via intravenös infusion. Dosen 20/27 mg/m2 administreras under 10 minuter. Dosen 20/56 mg/m2 ska administreras under 30 minuter.
Kyprolis får inte administreras som en intravenös stötdos eller bolusinjektion.
Den intravenösa administreringsslangen ska genomspolas med fysiologisk koksaltlösning eller 5 % glukoslösning för injektion omedelbart före och efter administreringen av Kyprolis.
Kyprolis ska inte blandas med eller administreras som infusion tillsammans med andra läkemedel.
Anvisningar om beredning av läkemedlet före administrering finns i avsnitt Särskilda anvisningar för destruktion och övrig hantering.
Kontraindikationer
- Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt Förteckning över hjälpämnen.
- Kvinnor som ammar (se avsnitt Fertilitet, graviditet och amning).
Eftersom Kyprolis administreras i kombination med andra läkemedel ska produktresuméerna för dessa läkemedel läsas för information om ytterligare kontraindikationer.
Varningar och försiktighet
Eftersom Kyprolis administreras i kombination med andra läkemedel ska produktresuméerna för dessa läkemedel läsas innan behandlingen med Kyprolis sätts in. Eftersom lenalidomid kan användas i kombination med Kyprolis måste i synnerhet kraven på graviditetstestning och eventuell användning av lämplig preventivmetod i samband med lenalidomidbehandling beaktas (se avsnitt Fertilitet, graviditet och amning).
Hjärtat
Nyuppkommen eller förvärrad hjärtsvikt (t.ex. hjärtinsufficiens, lungödem, sänkt ejektionsfraktion), myokardischemi samt -infarkt har förekommit efter administrering av Kyprolis. Dödsfall på grund av hjärtstillestånd har förekommit inom en dag från administreringen av Kyprolis och fall med dödlig utgång har rapporterats för hjärtsvikt och myokardinfarkt. För potentiella dosrelaterade effekter, se avsnitt Biverkningar.
Adekvat uppvätskning är ett krav före dosering i cykel 1 och samtliga patienter ska kontrolleras för tecken på övervätskning, i synnerhet patienter som löper risk att drabbas av hjärtsvikt. Justering av den totala vätskevolymen ska baseras på klinisk status vid behandlingsstart hos patienter med hjärtsvikt eller som löper risk att drabbas av hjärtsvikt (se avsnitt Dosering och administreringssätt).
Avbryt behandlingen med Kyprolis vid hjärthändelser av grad 3 eller 4 tills dessa har gått tillbaka och överväg att återuppta behandlingen med en dossänkning med 1 dosnivå baserat på en individuell bedömning av nytta och risk för patienten (se avsnitt Dosering och administreringssätt).
Risken för hjärtsvikt är högre hos äldre patienter (≥ 75 år). Risken för hjärtsvikt är också ökad hos patienter med asiatiskt ursprung.
En noggrann bedömning av kardiovaskulära riskfaktorer rekommenderas innan behandling påbörjas.
Patienter med hjärtsvikt i NYHA‑(New York Heart Association)‑klass III eller IV, som nyligen hade haft myokardinfarkt eller med rytmstörningar där ingen effekt erhölls av läkemedel, var uteslutna från de kliniska studierna. Dessa patienter kan löpa högre risk för hjärtkomplikationer. Patienter med tecken eller symtom på hjärtsvikt i NYHA‑klass III eller IV, som nyligen har haft myokardinfarkt (under de senaste 4 månaderna) och patienter med okontrollerad angina eller arytmier, ska genomgå en omfattande kardiologisk utvärdering innan behandling med Kyprolis påbörjas. Detta för att kunna optimera patientens status, med särskilt fokus på blodtryckskontroll och vätskebalans. Patienterna ska därefter behandlas med försiktighet och övervakas noggrant.
EKG‑förändringar
I kliniska studier och efter godkännande för försäljning har fall av förlängt QT‑intervall rapporterats. Fall av kammartakykardi har rapporterats hos patienter som får Kyprolis.
Lungtoxicitet
Akut andnödssyndrom (ARDS), akut andningssvikt och akut diffus infiltrativ lungsjukdom, såsom pneumonit och interstitiell lungsjukdom, har förekommit hos patienter som får Kyprolis. Några av dessa fall har haft dödlig utgång. Utvärdera och avbryt behandlingen med Kyprolis tills tillstånden har gått tillbaka och överväg om behandlingen med Kyprolis ska återupptas baserat på en individuell bedömning av nytta och risk (se avsnitt Dosering och administreringssätt).
Pulmonell hypertoni
Pulmonell hypertoni har rapporterats hos patienter som behandlas med Kyprolis. Några av dessa fall har haft dödlig utgång. Gör en bedömning på lämpligt sätt. Avbryt behandlingen med Kyprolis tills den pulmonella hypertonin har gått tillbaka och överväg därefter om behandlingen med Kyprolis ska återupptas eller inte baserat på en individuell bedömning av nytta och risk (se avsnitt Dosering och administreringssätt).
Andnöd
Andnöd har rapporterats hos patienter som behandlas med Kyprolis. Gör en bedömning av andnöden för att utesluta kardiopulmonella tillstånd, däribland hjärtsvikt och olika lungtillstånd. Avbryt behandlingen med Kyprolis vid andnöd av grad 3 och 4 och vänta tills den har gått tillbaka. Överväg därefter om behandling med Kyprolis ska återupptas baserat på en individuell bedömning av nytta och risk (se avsnitt Dosering och administreringssätt och Biverkningar).
Hypertoni
Hypertoni, däribland hypertensiv kris, har observerats vid behandling av Kyprolis. Några av dessa fall har haft dödlig utgång. Hypertoni rapporterades oftare hos patienter som fick Kyprolis i kombination med daratumumab i studie 20160275. Innan behandlingen påbörjas och under behandlingen rekommenderas att högt blodtryck är under kontroll. Under behandlingen med Kyprolis ska samtliga patienter utvärderas rutinmässigt med avseende på blodtrycket och vid behov behandlas. Om hypertonin inte kan hållas under kontroll ska Kyprolisdosen sänkas. Vid en hypertensiv kris ska behandlingen med Kyprolis avbrytas tills hypertonin har gått tillbaka. Överväg därefter om behandlingen med Kyprolis ska återupptas baserat på en individuell bedömning av nytta och risk (se avsnitt Dosering och administreringssätt).
Akut njursvikt
Fall av akut njursvikt har rapporterats hos patienter som får Kyprolis. Några av dessa fall har haft dödlig utgång. Akut njursvikt var vanligare hos patienter med långt framskridet recidiverande och refraktärt multipelt myelom som fick Kyprolis i monoterapi. I kliniska fas 3‑studier var incidensen av biverkningen akut njursvikt högre hos försökspersoner med lägre kreatininclearance vid behandlingsstart än bland försökspersoner med högre kreatininclearance vid behandlingsstart. För större delen av patienterna var kreatininclearance stabilt över tid. Njurfunktionen ska övervakas minst en gång per månad eller i enlighet med god klinisk praxis, i synnerhet bland patienter med lägre kreatininclearance vid behandlingsstart. Minska eller avbryt doseringen vid behov (se avsnitt Dosering och administreringssätt).
Tumörlyssyndrom
Fall av tumörlyssyndrom, däribland fall med dödlig utgång, har rapporterat hos patienter som får Kyprolis. Patienter med hög tumörbörda ska anses löpa högre risk att drabbas av tumörlyssyndrom. Säkerställ god uppvätskning av patienten före administreringen av Kyprolis i cykel 1 samt vid behov även i efterföljande cykler (se avsnitt Dosering och administreringssätt). Läkemedel som sänker urinsyranivåerna ska övervägas för patienter som löper hög risk att drabbas av tumörlyssyndrom. Under behandlingen ska förekomst av tumörlyssyndrom övervakas, med bl.a. regelbundna mätningar av serumelektrolyter, och behandlas utan dröjsmål. Avbryt behandlingen med Kyprolis tills tumörlyssyndromet har gått tillbaka (se avsnitt Dosering och administreringssätt).
Infusionsreaktioner
Infusionsreaktioner, däribland livshotande reaktioner, har rapporterats hos patienter som får Kyprolis. Symtomen kan vara feber, frossbrytningar, artralgi, myalgi, ansiktsrodnad, ansiktsödem, kräkningar, svaghet, andnöd, hypotoni, synkope, bradykardi, trånghetskänsla i bröstet eller angina. Dessa reaktioner kan förekomma omedelbart efter eller i upp till 24 timmar efter administreringen av Kyprolis. Dexametason ska administreras före Kyprolis för att minska förekomsten och svårighetsgraden av reaktionerna (se avsnitt Dosering och administreringssätt).
Blödning och trombocytopeni
Fall av blödning (t.ex. gastrointestinala, pulmonella och intrakraniella blödningar) har rapporterats hos patienter som behandlats med Kyprolis, ofta tillsammans med trombocytopeni. Några av dessa fall har haft dödlig utgång (se avsnitt Biverkningar).
Kyprolis orsakar trombocytopeni, där de lägsta trombocytnivåerna observeras på dag 8 eller dag 15 i varje 28‑dagarscykel och där nivåerna har återhämtat sig till utgångsvärdet vid starten av nästa cykel (se avsnitt Biverkningar). Regelbundna kontroller av trombocytantalet ska göras under behandlingen med Kyprolis. Minska eller avbryt doseringen vid behov (se avsnitt Dosering och administreringssätt).
Venösa tromboemboliska händelser
Fall av ventrombos, inklusive djup ventrombos samt fall av lungemboli med dödlig utgång, har rapporterats hos patienter som fått Kyprolis.
Patienter med kända riskfaktorer för tromboemboli, inklusive tidigare förekomst av trombos, ska övervakas noggrant. Vidtag åtgärder för att försöka minimera alla påverkbara riskfaktorer (t.ex. rökning, hypertoni och hyperlipidemi). Iakttag försiktighet vid samtidig administrering av andra ämnen som kan öka risken för trombos (t.ex. erytropoetiska medel eller hormonersättning). Patienter och läkare uppmanas att vara uppmärksamma på tecken och symtom på tromboemboli. Patienterna ska uppmanas att söka läkarvård om de drabbas av symtom såsom andnöd, bröstsmärtor, blodiga upphostningar eller svullnad eller smärta i armar och/eller ben.
Tromboprofylax ska övervägas baserat på en individuell nytta‑riskbedömning.
Levertoxicitet
Fall av levertoxicitet, däribland fall med dödlig utgång, har rapporterats. Kyprolis kan orsaka förhöjda nivåer av serumtransaminaser (se avsnitt Biverkningar). Minska eller avbryt doseringen vid behov (se avsnitt Dosering och administreringssätt). Leverenzymer och bilirubin ska kontrolleras vid behandlingsstart och därefter en gång per månad under behandlingen med carfilzomib, oavsett värdet vid behandlingsstart.
Trombotisk mikroangiopati
Fall av trombotisk mikroangiopati, däribland trombotisk trombocytopen purpura och hemolytiskt uremiskt syndrom (TTP/HUS), har rapporterats hos patienter som får Kyprolis. Några av dessa fall har haft dödlig utgång. Tecken och symtom på TTP/HUS ska övervakas. Om en sådan diagnos misstänks ska behandlingen med Kyprolis avbrytas och patienten utvärderas för eventuell TTP/HUS. Om diagnosen TTP/HUS utesluts kan behandlingen med Kyprolis återupptas. Säkerheten då Kyprolis‑behandlingen återupptas hos patienter som tidigare har drabbats av TTP/HUS är inte känd.
Posteriort reversibelt encefalopatisyndrom
Fall av posteriort reversibelt encefalopatisyndrom (PRES) har rapporterats hos patienter som får Kyprolis. PRES, som tidigare benämndes reversibelt bakre leukoencefalopatisyndrom (RPLS), är ett sällsynt neurologiskt tillstånd som kan ge symtom som krampanfall, huvudvärk, letargi, förvirring, blindhet, förändrad medvetandegrad samt andra synstörningar och neurologiska störningar, tillsammans med hypertoni. Diagnosen bekräftas med neurologiska röntgentekniker. Behandlingen med Kyprolis ska avbrytas vid misstänkt PRES. Säkerheten då Kyprolis behandlingen återupptas hos patienter som tidigare har drabbats av PRES är inte känd.
Reaktivering av hepatit B‑virus (HBV)
Fall av reaktivering av hepatit B‑virus (HBV) har rapporterats hos patienter som får carfilzomib.
Alla patienter ska screenas för HBV innan behandling med carfilzomib inleds. För patienter med positiv HBV‑serologi ska profylax med antivirala medel övervägas. De ska övervakas för kliniska och laboratoriska tecken på HBV‑reaktivering under och efter behandling. Experter inom behandling av HBV‑infektion ska konsulteras vid behov. Säkerheten vid fortsatt behandling med carfilzomib, efter att HBV‑reaktiveringen har kontrollerats, är inte känd. Därför ska återupptagande av behandling diskuteras med experter på HBV‑vård.
Progressiv multifokal leukoencefalopati
Fall av progressiv multifokal leukoencefalopati (PML) har rapporterats hos patienter som får carfilzomib och som tidigare fått eller samtidigt får immunhämmande behandling.
Patienter som får carfilzomib ska övervakas för nya eller försämrade neurologiska, kognitiva eller beteenderelaterade tecken och symtom som kan tyda på PML som en del av en differentialdiagnos av CNS‑sjukdomar.
Vid misstänkt PML ska administrering pausas tills PML har uteslutits av en specialist genom lämplig diagnostisk testning. Vid bekräftad PML ska behandling med carfilzomib avbrytas.
Kontraception
Kvinnliga patienter i fertil ålder (och/eller deras partner) måste använda effektiva preventivmetoder under och i en månad efter behandlingen. Manliga patienter måste använda effektiva preventivmetoder under och i 3 månader efter behandlingen om deras partner är gravid eller är i fertil ålder och inte använder en effektiv preventivmetod (se avsnitt Fertilitet, graviditet och amning). Carfilzomib kan minska effekten av p‑piller (se avsnitt Interaktioner).
Natriuminnehåll
Kyprolis 10 mg pulver till infusionsvätska, lösning
Detta läkemedel innehåller 37 mg natrium per 10 mg injektionsflaska, motsvarande 1,9 % av WHOs högsta rekommenderat dagligt intag (2 gram natrium för vuxna).
Kyprolis 30 mg pulver till infusionsvätska, lösning
Detta läkemedel innehåller 109 mg natrium per 30 mg injektionsflaska, motsvarande 5,5 % av WHOs högsta rekommenderat dagligt intag (2 gram natrium för vuxna).
Kyprolis 60 mg pulver till infusionsvätska, lösning
Detta läkemedel innehåller 216 mg natrium per 60 mg injektionsflaska, motsvarande 11 % av WHOs högsta rekommenderat dagligt intag (2 gram natrium för vuxna).
Cyklodextrininnehåll
Kyprolis 10 mg pulver till infusionsvätska, lösning
Detta läkemedel innehåller 500 mg cyklodextrin (sulfobutylbetadexnatrium) per 10 mg injektionsflaska, motsvarande 88 mg/kg för en vuxen person som väger 70 kg.
Kyprolis 30 mg pulver till infusionsvätska, lösning
Detta läkemedel innehåller 1 500 mg cyklodextrin (sulfobutylbetadexnatrium) per 30 mg injektionsflaska, motsvarande 88 mg/kg för en vuxen person som väger 70 kg.
Kyprolis 60 mg pulver till infusionsvätska, lösning
Detta läkemedel innehåller 3 000 mg cyklodextrin (sulfobutylbetadexnatrium) per 60 mg injektionsflaska, motsvarande 88 mg/kg för en vuxen person som väger 70 kg.
Interaktioner
Carfilzomib metaboliseras primärt via peptidas- och epoxidhydrolasaktivitet och det är därför inte sannolikt att dess farmakokinetiska profil påverkas av samtidig administrering av cytokrom P450‑hämmare och -inducerare.
In vitro‑studier har visat att carfilzomib inte inducerar humant CYP3A4 i odlade humana hepatocyter. I en klinisk studie där peroralt midazolam användes som CYP3A‑prob och man studerade carfilzomib i en dos om 27 mg/m2 (2‑10 minuter lång infusion) visades att midazolams farmakokinetik inte påverkades av samtidig administrering av carfilzomib, vilket tyder på att carfilzomib inte förväntas hämma metabolismen av CYP3A4/5‑substrat och inte är en CYP3A4‑inducerare hos människor. Det utfördes inte någon klinisk studie som undersökte dosen 56 mg/m2. Det är dock inte känt om carfilzomib inducerar CYP1A2, 2C8, 2C9, 2C19 eller 2B6 vid terapeutiska koncentrationer. Försiktighet ska iakttas när carfilzomib kombineras med läkemedel som är substrat till dessa enzymer, t.ex. p‑piller. Effektiva preventivmetoder ska användas (se avsnitt Fertilitet, graviditet och amning och även den aktuella produktresumén för lenalidomid). Om patienten använder p‑piller ska hon byta till en alternativ effektiv preventivmetod.
Carfilzomib hämmar inte CYP1A2, 2B6, 2C8, 2C9, 2C19 eller 2D6 in vitro och förväntas därför inte påverka exponeringen för läkemedel som är substrat till dessa enzymer, som ett resultat av hämning.
Carfilzomib är ett substrat till P‑glykoprotein (P‑gp) men inte till BCRP. Med tanke på att Kyprolis administreras intravenöst och metaboliseras i omfattande grad är det dock inte sannolikt att carfilzomibs farmakokinetiska profil påverkas av P‑gp- eller BCRP‑hämmare eller -inducerare. In vitro hämmar carfilzomib utflödestransport av digoxin, ett P‑gp‑substrat, med 25 % vid koncentrationer (3 µM) lägre än de som förväntas vid terapeutiska doser. Försiktighet ska iakttas när carfilzomib kombineras med substrat till P‑gp (t.ex. digoxin, kolchicin).
Carfilzomib hämmar OATP1B1 in vitro, med IC50 = 2,01 µM, men det är inte känt om carfilzomib hämmar andra transportörer som OATP1B3, OAT1, OAT3, OCT2 och BSEP, vid den systemiska nivån. Carfilzomib hämmar inte humant UGT2B7 men hämmar humant UGT1A1 med ett IC50 på 5,5 µM. Med tanke på att carfilzomib elimineras så snabbt, med särskilt en snabb sänkning av den systemiska koncentrationen 5 minuter efter avslutad infusion, är risken för kliniskt relevanta interaktioner med substrat till OATP1B1 och UGT1A1 troligen låg.
Fertilitet, graviditet och amning
Kvinnor i fertil ålder/Kontraception hos män och kvinnor
Kvinnliga patienter i fertil ålder som behandlas med Kyprolis (och/eller deras partner) måste använda effektiva preventivmetoder under och i en månad efter behandlingen.
Det kan inte uteslutas att effekten av p‑piller avtar under behandlingen med carfilzomib (se avsnitt Interaktioner). På grund av att carfilzomib är förknippat med ökad risk för ventromboemboli ska dessutom kvinnor undvika hormonella preventivmetoder som är förknippade med en risk för trombos under behandlingen med carfilzomib (se avsnitt Varningar och försiktighet och Biverkningar). Patienter som använder p‑piller eller en hormonell preventivmetod som är förknippad med en risk för trombos ska byta till en alternativ effektiv preventivmetod.
Manliga patienter måste använda effektiva preventivmetoder under och i 3 månader efter behandlingen om deras partner är gravid eller är i fertil ålder och inte använder en effektiv preventivmetod.
Graviditet
Det finns inga data från användningen av carfilzomib i gravida kvinnor.
Djurstudier har visat reproduktionstoxikologiska effekter (se avsnitt Prekliniska säkerhetsuppgifter).
Baserat på dess verkningsmekanism och resultaten från djurstudier kan Kyprolis ge upphov till fosterskador när det administreras till gravida kvinnor. Kyprolis ska inte användas under graviditet om inte den potentiella nyttan för kvinnan är större än de potentiella riskerna för fostret. Om Kyprolis används under graviditeten eller om patienten blir gravid under behandlingen med detta läkemedel ska hon informeras om den potentiella faran för fostret.
Lenalidomid är strukturellt besläktat med talidomid. Talidomid är en substans med känd teratogen effekt hos människor och orsakar svåra livshotande missbildningar. Om lenalidomid används under graviditeten förväntas det orsaka en teratogen effekt hos människor. Villkoren i lenalidomids program för förebyggande av graviditet måste uppfyllas av samtliga patienter om det inte finns tillförlitliga belägg för att patienten är infertil. Läs den aktuella produktresumén för lenalidomid.
Amning
Det är okänt om carfilzomib eller dess metaboliter utsöndras i bröstmjölk. Baserat på dess farmakologiska egenskaper kan en risk för det ammade barnet/spädbarnet inte uteslutas. Som en försiktighetsåtgärd är därför amning kontraindicerat under och i minst 2 dagar efter behandlingen med Kyprolis.
Fertilitet
Inga fertilitetsstudier har utförts på djur (se avsnitt Prekliniska säkerhetsuppgifter).
Effekter på förmågan att framföra fordon och använda maskiner
Kyprolis har mindre effekt på förmågan att framföra fordon och använda maskiner.
Trötthet, yrsel, svimning, dimsyn, sömnighet och/eller blodtrycksfall har observerats i kliniska studier. Patienter som behandlas med Kyprolis ska rådas att inte framföra fordon eller använda maskiner om de drabbas av något av dessa symtom.
Biverkningar
Sammanfattning av säkerhetsprofilen
Allvarliga biverkningar som kan förekomma vid behandling med Kyprolis är: hjärtsvikt, myokardinfarkt, hjärtstillestånd, myokardischemi, interstitiell lungsjukdom, pneumonit, akut andnödssyndrom, akut andningssvikt, pulmonell hypertoni, andnöd, hypertoni inklusive hypertensiv kris, akut njurskada, tumörlyssyndrom, infusionsrelaterade reaktioner, gastrointestinal blödning, intrakraniell blödning, pulmonell blödning, trombocytopeni, leversvikt, reaktivering av hepatit B‑virus, posteriort reversibelt encefalopatisyndrom (PRES), trombotisk mikroangiopati och trombotisk trombocytopen purpura (TTP)/hemolytiskt uremiskt syndrom (HUS). I kliniska studier med Kyprolis förekom hjärttoxicitet och andnöd tidigt under behandlingen med Kyprolis (se avsnitt Varningar och försiktighet). De vanligaste biverkningarna (förekom hos > 20 % av försökspersonerna) var: anemi, trötthet, trombocytopeni, illamående, diarré, feber, andnöd, luftvägsinfektion, hosta och neutropeni.
Efter inledande carfilzomibdoser på 20 mg/m2 ökades dosen till 27 mg/m2 i studie PX‑171‑009 och till 56 mg/m2 i studie 2011‑003 (se avsnitt Farmakodynamiska egenskaper). En jämförelse av biverkningarna i Kd‑armen (Kyprolis och dexametason) i studie 2011‑003 kontra biverkningarna i KRd‑armen (Kyprolis, lenalidomid och dexametason) i studie PX‑171‑009 tyder på att det kan finnas ett potentiellt dossamband för följande biverkningar: hjärtsvikt (Kd 8,2 %, KRd 6,4 %), andnöd (Kd 30,9 %, KRd 22,7 %), hypertoni (Kd 25,9 %, KRd 15,8 %) och pulmonell hypertoni (Kd 1,3 %, KRd 0,8 %).
I studie 20160275 (se avsnitt Farmakodynamiska egenskaper), där administrering av Kyprolis i kombination med daratumumab och dexametason (KdD) jämfördes med Kyprolis i kombination med dexametason (Kd), inträffade dödsfall pga. biverkningar inom 30 dagar efter den sista dosen av all studiebehandling för 10 % av patienterna i KdD‑armen jämfört med 5 % av patienterna i Kd‑armen. Den vanligaste dödsorsaken för patienter i båda armarna (KdD kontra Kd) var infektioner (5 % kontra 3 %). Risken för dödliga behandlingsrelaterade biverkningar var högre bland försökspersoner ≥ 65 år. Allvarliga biverkningar rapporterades hos 56 % av patienterna i KdD‑armen och hos 46 % av patienterna i Kd‑armen. De vanligaste allvarliga biverkningarna som rapporterats i KdD‑armen jämfört med Kd‑armen var anemi (2 % kontra 1 %), diarré (2 % kontra 0 %), feber (4 % kontra 2 %), lunginflammation (12 % kontra 9 %), influensa (4 % kontra 1 %), sepsis (4 % kontra 1 %) och bronkit (2 % kontra 0 %).
Tabell över biverkningar
Biverkningarna anges nedan efter organsystem och frekvenskategori (se tabell 6). Frekvenskategorierna bestämdes på basis av rådata för den incidens som rapporterades för varje biverkning i ett dataset av poolade kliniska studier (N = 3 878). Inom varje organsystem och frekvenskategori presenteras biverkningarna efter fallande allvarlighetsgrad.
Tabell 6. Tabell över biverkningar
MedDRA:s klassificering av organsystem | Mycket vanliga (≥ 1/10) | Vanliga (≥ 1/100, < 1/10) | Mindre vanliga (≥ 1/1 000, < 1/100) | Sällsynta (≥ 1/10 000, < 1/1 000) |
Infektioner och infestationer | Lunginflammation Luftvägsinfektioner | Sepsis Lunginfektion Influensa Herpes zoster* Urinvägsinfektion Bronkit Gastroenterit Virusinfektion Nasofaryngit Rinit | Kolit orsakad av Clostridium difficile Cytomegalovirusinfektion Reaktivering av hepatit B‑virus | |
Immunsystemet | Överkänslighet mot läkemedel | |||
Blodet och lymfsystemet | Trombocytopeni Neutropeni Anemi Lymfopeni Leukopeni | Febril neutropeni | HUS TTP | Trombotisk mikroangiopati |
Metabolism och nutrition | Hypokalemi Minskad aptit | Uttorkning Hyperkalemi Hypomagnesemi Hyponatremi Hyperkalcemi Hypokalcemi Hypofosfatemi Hyperurikemi Hypoalbuminemi Hyperglykemi | Tumörlyssyndrom | |
Psykiska störningar | Sömnsvårigheter | Ångest Förvirringstillstånd | ||
Centrala och perifera nervsystemet | Yrsel Perifer neuropati Huvudvärk | Parestesi Hypoestesi | Intrakraniella blödningar Cerebrovaskulära händelser PRES | |
Ögon | Katarakt Dimsyn | |||
Öron och balansorgan | Tinnitus | |||
Hjärtat | Hjärtsvikt Myokardinfarkt Förmaksflimmer Takykardi Sänkt ejektionsfraktion Hjärtklappning | Hjärtstillestånd Kardiomyopati Myokardischemi Perikardit Perikardiell utgjutning Kammartakykardi | ||
Blodkärl | Hypertoni | Djup ventrombos Hypotoni Blodvallningar | Hypertensiv kris Blödningar | Hypertensiv nödsituation |
Andningsvägar, bröstkorg och mediastinum | Andnöd Hosta | Lungemboli Lungödem Näsblödningar Orofarynxsmärta Dysfoni Väsande andning Pulmonell hypertoni | ARDS Akut andningssvikt Pulmonella blödningar Interstitiell lungsjukdom Pneumonit | |
Magtarmkanalen | Kräkningar Diarré Förstoppning Buksmärta Illamående | Gastrointestinala blödningar Dyspepsi Tandvärk | Gastrointestinal perforation Akut pankreatit | |
Lever och gallvägar | Förhöjt alaninaminotransferas Förhöjt aspartataminotransferas Förhöjt gamma-glutamyltransferas Hyperbilirubinemi | Leversvikt Kolestas | ||
Hud och subkutan vävnad | Utslag Klåda Erytem Hyperhidros | Angioödem | ||
Muskuloskeletala systemet och bindväv | Ryggsmärta Artralgi Smärta i armar och ben Muskelspasmer | Muskuloskeletal smärta Muskuloskeletal bröstsmärta Skelettsmärta Myalgi Muskelsvaghet | ||
Njurar och urinvägar | Förhöjt blodkreatinin | Akut njurskada Njursvikt Nedsatt njurfunktion Sänkt renalt kreatininclearance | ||
Allmänna symtom och/eller symtom vid administreringsstället | Feber Perifert ödem Asteni Trötthet Frossa | Bröstsmärta Smärta Reaktioner vid infusionsstället Influensaliknande sjukdom Sjukdomskänsla | Multiorgan-dysfunktion-syndrom | |
Undersökningar | Förhöjt C‑reaktivt protein Förhöjd blodurinsyra | |||
Skador och förgiftningar och behandlingskomplikationer | Infusionsrelaterade reaktioner | |||
| * Frekvensen är beräknad med hjälp av data från kliniska studier i vilka de flesta patienter fick profylaktisk behandling | ||||
Beskrivning av valda biverkningar
Hjärtsvikt, myokardinfarkt och myokardischemi
I kliniska studier med Kyprolis rapporterades hjärtsvikt hos ca 5 % av försökspersonerna (ca 3 % av försökspersonerna hade ≥ grad 3), myokardinfarkt rapporterades hos ca 1 % av försökspersonerna (ca 1 % av försökspersonerna hade ≥ grad 3) och myokardischemi rapporterades hos < 1 % av försökspersonerna (< 1 % av försökspersonerna hade ≥ grad 3). Dessa biverkningar uppträdde vanligen tidigt under Kyprolis‑behandlingen (< 5 cykler).
I studie 20160275 var den totala incidensen av hjärtsjukdomar (alla typer och av alla grader) i undergruppen av patienter med kärlsjukdomar vid baslinjen eller hypertoni vid baslinjen 29,9 % kontra 19,8 % (KdD kontra Kd) respektive 30,6 % kontra 18,1 %. För hjärthändelser med dödlig utgång var incidensen 1,9 % kontra 0,0 % (KdD kontra Kd) respektive 1,5 % kontra 0,0 %. Ingen enskild typ av hjärthändelse svarade för skillnaden som rapporterades mellan KdD‑ och Kd‑armarna i undergruppen av patienter med kärlsjukdomar vid baslinjen eller hypertoni vid baslinjen.
För klinisk behandling av hjärtrelaterade tillstånd under behandlingen med Kyprolis, se avsnitt Varningar och försiktighet.
Andnöd
Andnöd rapporterades hos omkring 24 % av försökspersonerna i kliniska studier med Kyprolis. Majoriteten av fallen av andnödbiverkningar var inte allvarliga (< 5 % av försökspersonerna hade biverkningar ≥ grad 3), gick tillbaka, ledde sällan till behov av behandlingsavbrott och uppträdde tidigt under studien (< 3 cykler). För klinisk behandling av andnöd under behandlingen med Kyprolis, se avsnitt Varningar och försiktighet.
Hypertoni inklusive hypertensiv kris
Hypertensiv kris har förekommit efter administrering av Kyprolis. Några av dessa fall har haft dödlig utgång. I kliniska studier förekom hypertonibiverkningar hos omkring 21 % av försökspersonerna och 8 % av försökspersonerna hade biverkningar ≥ grad 3, men hypertensiv kris förekom hos < 0,5 % av försökspersonerna. Förekomsten av hypertonibiverkningar var ungefär densamma hos patienter med eller utan en tidigare anamnes på hypertoni. För klinisk behandling av hypertoni under behandlingen med Kyprolis, se avsnitt Varningar och försiktighet.
Trombocytopeni
Trombocytopeni rapporterades hos omkring 33 % av försökspersonerna i kliniska studier med Kyprolis och ca 20 % av försökspersonerna hade biverkningar ≥ grad 3. I studie 20160275 var förekomsten av trombocytopeni av grad ≥ 3 24,4 % i KdD‑armen och 16,3 % i Kd‑armen. Kyprolis orsakar trombocytopeni genom att avknoppningen av trombocyter från megakaryocyter hämmas, vilket leder till en klassisk cyklisk trombocytopeni där de lägsta trombocytkoncentrationerna förekommer på dag 8 eller 15 i varje 28‑dagarscykel och där koncentrationerna vanligtvis har gått tillbaka till utgångsvärdet till starten av nästa cykel. För klinisk behandling av trombocytopeni under behandlingen med Kyprolis, se avsnitt Varningar och försiktighet.
Venösa tromboemboliska händelser
Fall av ventrombos, inklusive djup ventrombos och lungemboli med dödlig utgång, har rapporterats hos patienter som fått Kyprolis (se avsnitt Varningar och försiktighet). Den totala förekomsten av ventromboemboli var högre i de grupper som fick Kyprolis i tre fas 3‑studier. I studie PX‑171‑009 var förekomsten av ventromboemboli 15,6 % i KRd‑gruppen och 9,0 % i Rd‑gruppen. Tromboemboli av grad ≥ 3 rapporterades hos 5,6 % av patienterna i KRd‑armen och hos 3,9 % av patienterna i Rd‑armen. I studie 2011‑003 var förekomsten av ventromboemboli 12,5 % i Kd‑gruppen och 3,3 % i gruppen som fick bortezomib och dexametason (Vd). Tromboemboli av grad ≥ 3 rapporterades hos 3,5 % av patienterna i Kd‑armen och 1,8 % av patienterna i Vd‑armen. I studie 20160275 var förekomsten av ventromboemboli 6,2 % i KdD‑armen och 11,1 % i Kd‑armen. Ventromboemboli av grad ≥ 3 rapporterades hos 1,9 % av patienterna i KdD‑armen och 6,5 % av patienterna i Kd‑armen.
Leversvikt
Fall av leversvikt, däribland fall med dödlig utgång, har rapporterats hos < 1 % av försökspersonerna i kliniska studier med Kyprolis. För klinisk behandling av levertoxicitet under behandlingen med Kyprolis, se avsnitt Varningar och försiktighet.
Perifer neuropati
I en randomiserad, öppen multicenterstudie med patienter som fick Kyprolis 20/56 mg/m2 som en 30 minuter lång infusion i kombination med dexametason (Kd, n = 464) jämfört med bortezomib plus dexametason (Vd, n = 465) rapporterades fall av perifer neuropati av grad 2 och högre hos 7 % av patienterna med recidiverande multipelt myelom i Kd‑armen jämfört med 35 % i Vd‑armen vid tidpunkten för den i förväg planerade analysen av total överlevnad (OS). I studie 20160275 rapporterades fall av perifer neuropati av grad 2 och högre hos 10,1 % av patienterna med recidiverande multipelt myelom i KdD‑armen jämfört med 3,9 % i Kd‑armen.
Infusionsreaktioner
I studie 20160275 förelåg en högre risk för infusionsreaktion när carfilzomib administrerades med daratumumab.
Luftvägsinfektioner
I studie 20160275 rapporterades luftvägsinfektioner som allvarliga biverkningar i varje behandlingsgrupp (27,6 % i KdD‑armen och 15,0 % i Kd‑armen). I studie 20160275 rapporterades lunginflammation som allvarlig biverkning i varje behandlingsgrupp (15,3 % i KdD‑armen och 9,8 % i Kd‑armen). 1,3 % och 0 % av biverkningarna hade dödlig utgång i KdD‑armen respektive Kd‑armen.
Sekundära primära maligniteter
I studie 20160275 rapporterades sekundära primära maligniteter i varje behandlingsgrupp (1,9 % i KdD‑armen och 1,3 % i Kd‑armen).
Opportunistiska infektioner
I studie 20160275 rapporterades opportunistiska infektioner i varje behandlingsgrupp (9,4 % i KdD‑armen och 3,9 % i Kd‑armen). Opportunistiska infektioner som förekom hos ≥ 1 % av patienterna i KdD‑armen var herpes zoster, oral candidos, oral herpes och herpes simplex.
Reaktivering av hepatit B
I studie 20160275 var förekomsten av hepatit B‑reaktivering 0,6 % i KdD‑armen kontra 0 % i Kd‑armen.
Andra särskilda populationer
Äldre patienter
I kliniska studier med Kyprolis var förekomsten av vissa biverkningar (däribland hjärtarytmier och hjärtsvikt (se avsnitt Varningar och försiktighet), andnöd, leukopeni och trombocytopeni) totalt sett högre hos patienter som var ≥ 75 år jämfört med patienter som var < 75 år.
I studie 20160275 var 47 % av de 308 patienterna som fick KdD 20/56 mg/m2 två gånger per vecka ≥ 65 år. I KdD‑armen av studien inträffade behandlingsrelaterade biverkningar med dödlig utgång för 6 % av patienterna < 65 år och 14 % av patienterna ≥ 65 år. I Kd‑armen inträffade sådana händelser för 8 % av patienterna < 65 år och 3 % av patienterna ≥ 65 år.
Rapportering av misstänkta biverkningar
Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta‑riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning via det nationella rapporteringssystemet: www.fimea.fi.
Överdosering
Informationen är i dagsläget otillräcklig för att det ska gå att dra slutsatser om säkerheten för doser som är högre än de som har utvärderats i kliniska studier. Akut uppkomst av frossbrytningar, hypotoni, njurinsufficiens, trombocytopeni och lymfopeni rapporterades efter det att en dos på 200 mg Kyprolis hade administrerats av misstag.
Det finns ingen känd specifik antidot till överdosering av carfilzomib. I händelse av en överdos ska patienten övervakas, i synnerhet för de Kyprolis‑biverkningar som anges i avsnitt Biverkningar.
Farmakologiska egenskaper
Farmakodynamiska egenskaper
Farmakoterapeutisk grupp: Antineoplastiska medel, andra antineoplastiska medel, ATC‑kod: L01XG02
Verkningsmekanism
Carfilzomib är en proteasominhibitor (tetrapeptid) av epoxyketontyp som selektivt och irreversibelt binder till N‑terminala treonininnehållande aktiva ytor hos 20S proteasom, den proteolytiska kärnpartikeln i 26S proteasom, och uppvisar liten till ingen aktivitet mot andra proteasklasser. Carfilzomib hade antiproliferativa och proapoptotiska aktiviteter i prekliniska modeller av hematologiska tumörer. I djurstudier hämmade carfilzomib proteasomaktiviteten i blod och vävnad och fördröjde tumörtillväxt i modeller av multipelt myelom. In vitro hade carfilzomib minimal neurotoxicitet och uppvisade minimala reaktioner mot icke‑proteasomproteaser.
Farmakodynamisk effekt
Intravenös administrering av carfilzomib ledde till suppression av proteasomkymotrypsinliknande (CT‑L) aktivitet när det mättes i blodet 1 timme efter den första dosen. Doser på ≥ 15 mg/m2 inducerade konsekvent (≥ 80 %) hämning av CT‑L‑aktiviteten hos proteasom. Dessutom ledde administrering av carfilzomib till hämning av latent membranprotein 2 (LMP2) och multikatalytiskt endopeptidaskomplex‑liknande 1 (MECL1) subenheter i immunproteasom i intervallet från 26 % till 32 % respektive 41 % till 49 % vid 20 mg/m2. Proteasomhämningen varade i ≥ 48 timmar efter den första carfilzomibdosen i varje doseringsvecka. Kombinationsdosering med lenalidomid och dexametason påverkade inte proteasomhämning.
Vid den högre dosen på 56 mg/m2 observerades inte bara en kraftigare hämning av CT‑L‑subenheterna (≥ 90 %) jämfört med doser på 15 till 20 mg/m2, utan dessutom kraftigare hämning av andra proteasomsubenheter (LMP7, MECL1 och LMP2). Hämningen av LMP7‑, MECL1- och LMP2‑subenheterna var omkring 8 %, 23 % respektive 34 % högre vid en dos på 56 mg/m2 jämfört med doser på 15 till 20 mg/m2. Liknande proteasomhämning med carfilzomib uppnåddes med infusioner på 2 till 10 samt 30 minuter vid de 2 dosnivåer (20 och 36 mg/m2) som undersöktes.
Klinisk effekt och säkerhet
Kyprolis i kombination med lenalidomid och dexametason för behandling av patienter med recidiverande multipelt myelom – studie PX‑171‑009 (ASPIRE)
Säkerhet och effekt för Kyprolis utvärderades i en randomiserad, öppen multicenterstudie på 792 patienter med recidiverande multipelt myelom, där kombinationen Kyprolis, lenalidomid och dexametason utvärderades i förhållande till behandling med enbart lenalidomid och dexametason, med randomiseringen 1:1.
I denna studie utvärderades Kyprolis vid en initial dos på 20 mg/m2, vilken ökades till 27 mg/m2 på dag 8 i cykel 1 och administrerades som en 10 minuter lång infusion två gånger per vecka i 3 av 4 veckor. Behandling med Kyprolis administrerades i högst 18 cykler om den inte avbröts tidigare på grund av sjukdomsprogression eller oacceptabel toxicitet. Administrering av lenalidomid och dexametason kunde fortgå fram till progression eller oacceptabel toxicitet.
Patienter med följande uteslöts från studien: kreatininclearance < 50 ml/min, hjärtsvikt i NYHA‑klass III till IV eller myokardinfarkt inom de senaste 4 månaderna, sjukdomsprogression under behandling med en bortezomibinnehållande regim, eller progression under de första 3 månaderna efter insättning av behandlingen med lenalidomid och dexametason eller progression någon gång under behandlingen med lenalidomid och dexametason om detta var försökspersonens senaste behandlingslinje. Studiens urvalskriterier tillät rekrytering av ett litet antal patienter med myelom refraktärt mot bortezomib (N = 118) eller lenalidomid (N = 57). Rekryterade patienter definierades som refraktära mot en behandling om de uppfyllde något av följande 3 kriterier: svarade inte (< minimal respons) på någon behandlingsregim, progression under någon behandlingsregim eller progression inom 60 dagar från fullbordan av någon behandlingsregim. Denna studie utvärderade inte nytta‑riskförhållandet i den bredare refraktära populationen.
Sjukdomsstatus och andra parametrar vid studiestart var välmatchade mellan de två armarna, inklusive ålder (64 år, intervall 31‑91 år), kön (56 % män), ECOG‑funktionsstatus (48 % med funktionsstatus 1), genetiska högriskmutationer innefattande de genetiska subtyperna t(4;14), t(14;16) eller deletion 17p i ≥ 60 % av plasmacellerna (13 %), genetiska mutationer av okänd risk, vilket innefattade försökspersoner med resultat som inte samlades in eller inte analyserades (47 %) samt sjukdom i ISS‑stadium III vid studiestart (20 %). Försökspersonerna hade tidigare genomgått 1 till 3 behandlingslinjer (median var 2), däribland tidigare behandling med bortezomib (66 %), talidomid (44 %) och lenalidomid (20 %).
Resultaten från studie PX‑171‑009 sammanfattas i tabell 7 samt i figur 1 och figur 2.
Tabell 7. Sammanfattning av effektanalysen i studie PX‑171‑009 på recidiverande multipelt myelom
| KRd kombinationsbehandling | ||
| KRd‑arma (N = 396) | Rd‑arma (N = 396) | |
| PFS månader median (95 % CI) | 26,3 (23,3; 30,5) | 17,6 (15,0; 20,6) |
| HR (95 % CI); ensidigt p‑värdeb | 0,69 (0,57; 0,83); < 0,0001 | |
| OS månader median (95 % CI) | 48,3 (42,4; 52,8) | 40,4 (33,6; 44,4) |
| HR (95 % CI); ensidigt p‑värdeb | 0,79 (0,67; 0,95); 0,0045 | |
| ORR, n (%) | 345 (87,1) | 264 (66,7) |
| sCR | 56 (14,1) | 17 (4,3) |
| CR | 70 (17,7) | 20 (5,1) |
| VGPR | 151 (38,1) | 123 (31,1) |
| PR | 68 (17,2) | 104 (26,3) |
| 95 % CI av ORR | 83,4; 90,3 | 61,8; 71,3 |
| Ensidigt p‑värde | < 0,0001 | |
KRd = Kyprolis, lenalidomid och dexametason; Rd = lenalidomid och dexametason; PFS = progressionsfri överlevnad; HR = riskkvot; CI = konfidensintervall; OS = total överlevnad; ORR = total responsfrekvens; sCR = stringent komplett respons; CR = komplett respons; VGPR = mycket bra partiell respons; PR = partiell respons; IMWG = International Myeloma Working Group; EBMT = European society for blood and marrow transplantation a. Fastställdes av en oberoende granskningskommitté med standardiserade objektiva IMWG/EBMT‑responskriterier. b. Statistiskt signifikant. | ||
Patienter som fick Kyprolis, lenalidomid och dexametason (KRd) uppvisade bättre progressionsfri överlevnad (PFS) jämfört med dem som fick lenalidomid och dexametason (Rd), (HR = 0,69, med ensidigt p‑värde < 0,0001), vilket motsvarar 45 % förbättring av PFS eller 31 % riskminskning, fastställt med standardiserade objektiva IMWG/EBMT‑responskriterier av en oberoende granskningskommitté (IRC).
PFS‑nytta med KRd observerades konsekvent i samtliga undergrupper, vilket innefattar patienter ≥ 75 år (N = 96), patienter med genetiska mutationer med hög risk (N = 100) eller okänd risk (N = 375) och patienter med kreatininclearance vid studiestart på 30 ‑ < 50 ml/min (N = 56).
Figur 1. Kaplan‑Meier‑kurva över progressionsfri överlevnad vid recidiverande multipelt myeloma
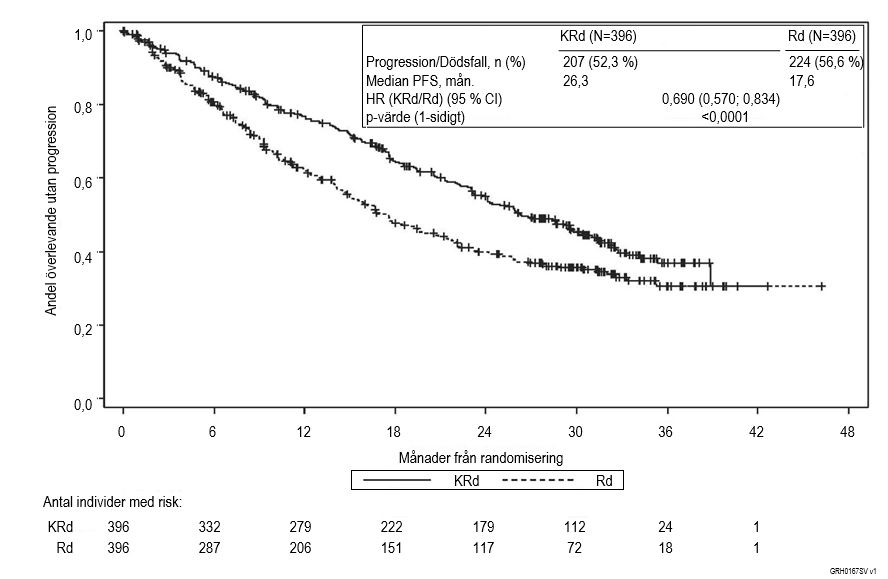
KRd = Kyprolis, lenalidomid och dexametason; Rd = lenalidomid och dexametason; PFS = progressionsfri överlevnad; HR = riskkvot; CI = konfidensintervall; IMWG = International Myeloma Working Group; EBMT = European society for blood and marrow transplantation; mån. = månader
Obs! Utfall för respons och progression fastställdes med hjälp av standardiserade objektiva IMWG/EBMT‑responskriterier.
a. Studie PX‑171‑009
En planerad analys av total överlevnad (OS) utfördes efter 246 dödsfall i KRd‑armen och 267 dödsfall i Rd‑armen. Mediantiden för uppföljningen var omkring 67 månader. En statistiskt signifikant fördel med avseende på total överlevnad observerades hos patienter i KRd‑armen jämfört med patienterna i Rd‑armen. Patienterna i KRd‑armen uppvisade 21 % sänkt risk för dödsfall jämfört med dem i Rd‑armen (HR = 0,79; 95 % CI: 0,67, 0,95; p‑värde = 0,0045). Median OS var 7,9 månader längre för patienter i KRd‑armen jämfört med dem i Rd‑armen (se tabell 7 och figur 2).
Figur 2. Kaplan‑Meier‑kurva av en analys av total överlevnad vid recidiverande multipelt myeloma
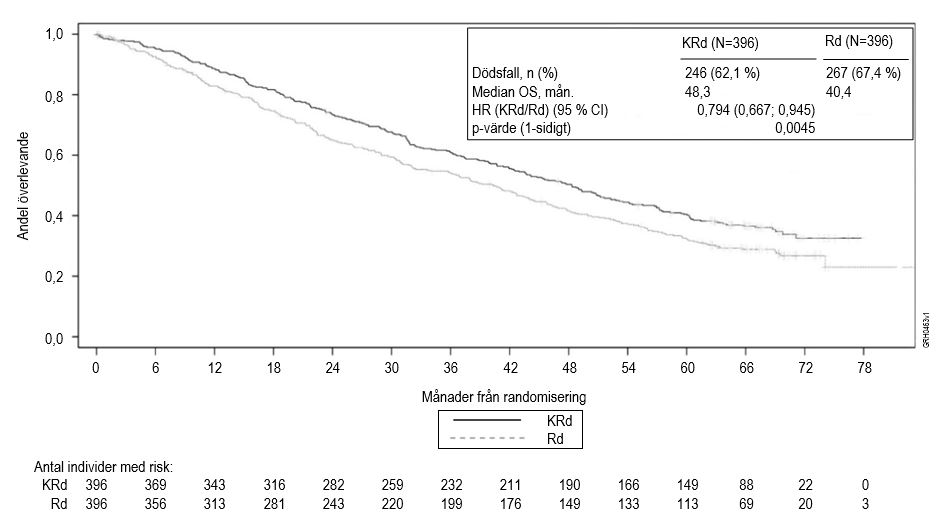
KRd = Kyprolis, lenalidomid och dexametason; Rd = lenalidomid och dexametason; OS = total överlevnad; HR = riskkvot; CI = konfidensintervall; mån. = månader
a. Studie PX‑171‑009
Patienter som behandlades med KRd rapporterade bättre övergripande hälsostatus med högre poäng för övergripande hälsostatus/livskvalitet (QoL) jämfört med Rd under 18 behandlingscykler (ej multiplicitetsjusterat ensidigt p‑värde = 0,0001) mätt med EORTC QLQ‑C30, ett validerat mätinstrument för multipelt myelom.
Kyprolis i kombination med dexametason för behandling av patienter med recidiverande multipelt myelom – studie 2011‑003 (ENDEAVOR)
Säkerhet och effekt för Kyprolis utvärderades i en randomiserad, öppen multicenterstudie i fas 3 av Kyprolis plus dexametason (Kd) jämfört med bortezomib plus dexametason (Vd). Totalt 929 patienter med recidiverande eller refraktärt multipelt myelom som tidigare hade genomgått 1 till 3 behandlingslinjer rekryterades och randomiserades (464 i Kd‑armen, 465 i Vd‑armen).
Studien utvärderade Kyprolis vid en initial dos på 20 mg/m2, vilken ökades till 56 mg/m2 på dag 8 i cykel 1 och administrerades som en 30 minuter lång infusion två gånger per vecka i 3 av 4 veckor fram till sjukdomsprogression eller oacceptabel toxicitet.
Patienterna som randomiserades till Vd‑armen fick bortezomib antingen intravenöst (n = 108) eller subkutant (n = 357). Patienter med följande uteslöts från studien: kreatininclearance < 15 ml/min, hjärtsvikt i NYHA‑klass III till IV, myokardinfarkt inom de senaste 4 månaderna eller de med vänsterkammar‑ejektionsfraktion (LVEF) < 40 %. Studiens urvalskriterier tillät rekrytering av patienter som tidigare hade behandlats med carfilzomib (n = 3) eller bortezomib (n = 502) så länge som patienterna hade uppvisat åtminstone partiell respons (PR) vid den tidigare behandlingen med proteasomhämmare, inte hade avbrutit behandlingen med proteasomhämmare på grund av toxicitet och som inte hade behandlats med proteasomhämmare inom de senaste 6 månaderna.
Demografi och parametrar vid studiestart för studie 2011‑003 var välmatchade mellan de två armarna, inklusive tidigare behandling med bortezomib (54 %), tidigare behandling med lenalidomid (38 %), lenalidomidrefraktära (25 %) ålder (65 år, intervall 30‑89 år), kön (51 % män), ECOG‑funktionsstatus (45 % med funktionsstatus 1), genetiska högriskmutationer innefattande de genetiska subtyperna t(4;14) eller t(14;16) hos 10 % eller mer av de screenade plasmacellerna, eller deletion 17p i ≥ 20 % av plasmacellerna (23 %), genetiska mutationer av okänd risk, vilket innefattade försökspersoner med resultat som inte samlades in eller inte analyserades (9 %) samt sjukdom i ISS‑stadium III vid studiestart (24 %).
Resultaten från studie 2011‑003 sammanfattas i tabell 8.
Tabell 8. Sammanfattning av effektanalysen i studie 2011‑003 på recidiverande multipelt myelom
| Kd‑arm (N = 464) | Vd‑arm (N = 465) | |
| PFS månader median (95 % CI)a | 18,7 (15,6; NE) | 9,4 (8,4; 10,4) |
| HR (95 % CI); ensidigt p‑värdeb | 0,533 (0,44; 0,65); < 0,0001 | |
| Total överlevnad månader median (95 % CI) | 47,6 (42,5; NE) | 40,0 (32,6; 42,3) |
| HR (95 % CI); ensidigt p‑värdeb | 0,791 (0,65; 0,96); 0,010 | |
| ORR n (%)a, c | 357 (76,9) | 291 (62,6) |
| ≥ CRd | 58 (12,5) | 29 (6,2) |
| ≥ VGPRe | 252 (54,3) | 133 (28,6) |
| 95 % CI av ORR | 72,8, 80,7 | 58,0, 67,0 |
| Ensidigt p‑värdeb | < 0,0001 | |
Kd = Kyprolis plus dexametason; Vd = bortezomib och dexametason; CI = konfidensintervall; NE = ej uppskattningsbart; HR = Riskkvot; ORR = total responsfrekvens; CR = komplett respons; VGPR = mycket bra partiell respons a. Dessa effektmått fastställdes av en oberoende granskningskommitté b. Statistiskt signifikant. c. Total respons definieras som att uppnå bästa totala respons för PR, VGPR, CR eller sCR d. Statistiskt signifikant, ensidigt p‑värde = 0,0005 e. Statistiskt signifikant, ensidigt p‑värde = 0,0001 | ||
Studien visade signifikant förbättring av PFS för patienter i Kd‑armen jämfört med dem i Vd‑armen (HR: 0,53; 95 % CI: 0,44; 0,65 [p‑värde < 0,0001]) (se figur 3).
Liknande PFS‑resultat observerades för patienter som tidigare hade behandlats med bortezomib (HR 0,56; 95 % CI: 0,44; 0,73) och patienter som inte hade behandlats med bortezomib (HR 0,48; 95 % CI: 0,36; 0,66).
De positiva PFS‑resultaten med Kd observerades konsekvent i samtliga subgrupper, inklusive patienter ≥ 75 år (n = 143), patienter med genetiska högriskmutationer (n = 210) och patienter med kreatininclearance på 30 ‑ < 50 ml/min vid behandlingsstarten (n = 128).
Hos patienter som tidigare hade behandlats med bortezomib (54 %) var median‑PFS 15,6 månader i Kd‑armen jämfört med 8,1 månader i Vd‑armen (HR = 0,56; 95 % CI: 0,44; 0,73), ORR var 71,2 % jämfört med 60,3 %.
Hos patienter som tidigare hade behandlats med lenalidomid (38 %) var median‑PFS 12,9 månader i Kd‑armen jämfört med 7,3 månader i Vd‑armen (HR = 0,69; 95 % CI: 0,52; 0,92), ORR var 70,1 % jämfört med 59,3 %. Hos patienter som var refraktära mot lenalidomid (25 %) var median‑PFS 8,6 månader i Kd‑armen jämfört med 6,6 månader i Vd‑armen (HR = 0,80; 95 % CI: 0,57; 1,11), ORR var 61,9 % jämfört med 54,9 %.
Figur 3. Kaplan‑Meier‑kurva över progressionsfri överlevnad fastställd av den oberoende granskningskommittén (ITT‑population) studie 2011‑003
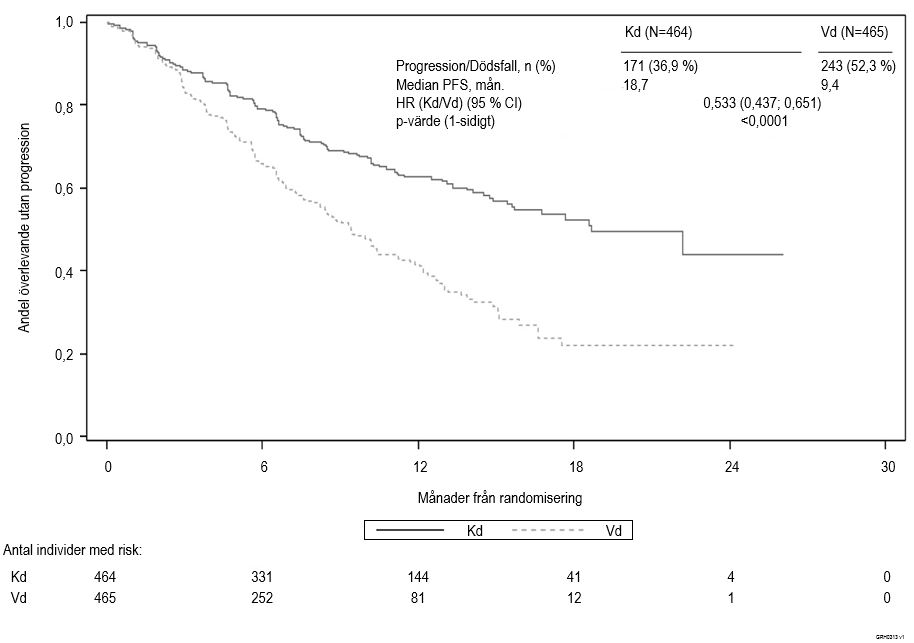
Kd = Kyprolis plus dexametason; Vd = bortezomib plus dexametason; PFS = progressionsfri överlevnad; mån. = månader; HR = riskkvot; CI = konfidensintervall
En i förväg planerad andra interimsanalys över total överlevnad (OS) utfördes efter 189 dödsfall i Kd‑armen och 209 dödsfall i Vd‑armen. Vid tidpunkten för analysen hade 80 % av de fall som krävdes för slutanalysen registrerats. Mediantiden för uppföljningen var ungefär 37 månader. En statistiskt signifikant fördel i total överlevnad observerades för patienter i Kd-armen jämfört med patienterna i Vd-armen (HR = 0,791; 95 % CI; 0,65, 0,96; p‑värde = 0,010) (se figur 4).
Figur 4. Kaplan‑Meier‑kurva över total överlevnad i studie 2011‑003 på recidiverande multipelt myelom
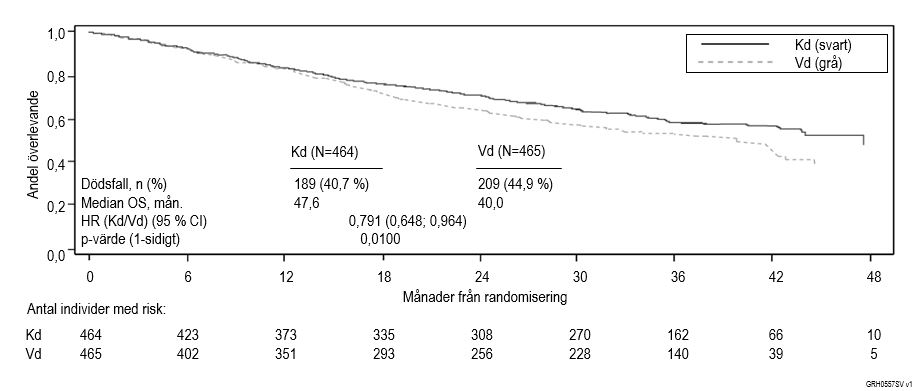
Kd = Kyprolis plus dexametason; Vd = bortezomib plus dexametason; OS = total överlevnad; mån. = månader; HR = riskkvot; CI = konfidensintervall
Kyprolis i kombination med daratumumab och dexametason för behandling av patienter med recidiverande eller refraktärt multipelt myelom – studie 20160275 (CANDOR)
Säkerhet och effekt för Kyprolis utvärderades i en randomiserad, öppen multicenter‑ och överlägsenhetsstudie i fas 3 av Kyprolis med daratumumab plus dexametason (KdD) jämfört med Kyprolis plus dexametason (Kd). Totalt 466 patienter med recidiverande eller refraktärt multipelt myelom som tidigare hade genomgått 1 till 3 behandlingslinjer rekryterades och randomiserades i en 2:1‑randomisering (312 i KdD‑armen och 154 i Kd‑armen).
I KdD‑ och Kd‑armarna utvärderades Kyprolis vid en startdos på 20 mg/m2, vilken ökades till 56 mg/m2 på dag 8 i cykel 1 och administrerades som en 30 minuter lång infusion två gånger per vecka i 3 av 4 veckor.
Patienter som hade följande undantogs från studien: känd måttlig eller svår ihållande astma under de senaste 2 åren, känd kronisk obstruktiv lungsjukdom (KOL) med en FEV1 < 50 % av predikterat normalvärde och aktiv hjärtsvikt.
Demografi och parametrar vid studiestart var i allmänhet likvärdiga mellan de två armarna, däribland kön (57,5 % män), ras (78,5 % vita försökspersoner), ålder (64 år, intervall 29–84 år), tidigare behandling med bortezomib (90 %), bortezomibrefraktäritet (29 %), genetiska högriskmutationer innefattande de genetiska subtyperna t(4; 14), t(14; 16) eller deletion 17p (16 %) och genetiska mutationer av okänd risk som innefattade försökspersoner utan färdiga resultat, med misslyckade resultat eller med resultat av otillräcklig kvantitet (51 %). En mindre andel försökspersoner var ≥ 75 år i KdD‑armen (9,0 %) än i Kd‑armen (14,3 %). Patienterna hade i median erhållit 2,0 tidigare behandlingslinjer (intervall 1 till 4). En högre andel av patienterna i KdD‑armen var tidigare transplanterade (62,5 %) jämfört med Kd‑armen (48,7 %). Endast 1 patient i KdD‑armen hade fått tidigare behandling med monoklonala antikroppar riktade mot CD38.
Resultaten från den primära analysen av studie 20160275 sammanfattas i tabell 9 samt i figur 5 och figur 6.
Tabell 9. Sammanfattning av effekt i studie 20160275 vid primäranalys
KdD‑arm (n = 312) | Kd‑arm (n = 154) | |
| PFS månader median (95 % CI)a | NE (NE; NE) | 15,8 (12,1; NE) |
| HR (95 % CI); ensidigt p‑värdeb | 0,630 (0,464; 0,854); 0,0014 | |
| ORR (%) (95 % CI)a, c | 84,3 (79,8; 88,1) | 74,7 (67,0; 81,3) |
| Responskategori, n (%) | ||
| n med respons | 263 | 115 |
| CR | 89 (28,5) | 16 (10,4) |
| MRD[–]CR | 43 (13,8) | 5 (3,2) |
| VGPR | 127 (40,7) | 59 (38,3) |
| PR | 47 (15,1) | 40 (26,0) |
| Oddskvot | 1,925 (1,184; 3,129) | |
| Ensidigt p‑värdeb | 0,0040 | |
| MRD[–]CR vid 12 månader | 12,5 (9,0; 16,7) | 1,3 (0,2; 4,6) |
| Oddskvot | 11,329 (2,703; 47,476) | |
| Ensidigt p‑värdeb | < 0,0001 | |
KdD = Kyprolis plus dexametason och daratumumab; Kd = Kyprolis plus dexametason; CI = konfidensintervall; NE = ej uppskattningsbart; HR = riskkvot; ORR = total responsfrekvens; CR = komplett respons; VGPR = mycket bra partiell respons; MRD[–]CR = komplett respons och ingen minimal kvarvarande sjukdom (MRD negativ) a. Dessa effektmått fastställdes av en oberoende granskningskommitté med hjälp av IMWG‑responskriterierna. b. Statistiskt signifikant. c. Total respons (ORR) definieras som att uppnå bästa totala respons för PR, VGPR och CR, eller bättre. | ||
Vid tidpunkten för den primära PFS analysen under prövningen påvisades en förbättring av PFS i KdD‑armen jämfört med Kd‑armen (riskkvot [HR] = 0,630; 95 % CI: 0,464, 0,854; p = 0,0014), vilket representerar en 37-procentig minskning av risken för sjukdomsprogression eller dödsfall för patienterna som behandlades med KdD. Det gick inte att beräkna median‑PFS för KdD‑armen. Median‑PFS för Kd‑armen var 15,8 månader.
Hos patienter som tidigare hade behandlats med lenalidomid (42,3 %) var median‑PFS NE i KdD‑armen jämfört med 12,1 månader i Kd‑armen (HR = 0,52; 95 % CI: 0,34; 0,80), ORR var 78,9 % kontra 74,3 % (OR = 1,29; 95 % CI: 0,65; 2,54) och MRD[–]CR vid 12 månader var 11,4 % kontra 0,0 % (OR = NE, 95 % CI: NE; NE). Hos patienter som var refraktära mot lenalidomid (33 %) var median‑PFS NE i KdD‑armen jämfört med 11,1 månader i Kd‑armen (HR = 0,45; 95 % CI: 0,28; 0,74), ORR var 79,8 % jämfört med 72,7 % (OR = 1,48; 95 % CI: 0,69; 3,20) och MRD[–]CR vid 12 månader 13,1 % kontra 0,0 % (OR = NE, 95 % CI: NE; NE).
Begränsade data finns tillgängliga för äldre patienter (≥ 75 år). Totalt 43 patienter över 75 år rekryterades till studie 20160275 (25 patienter i KdD‑armen och 18 patienter i Kd‑armen). En HR på 1,459 (95 % CI: 0,504; 4,223) i PFS observerades. Risken för dödliga behandlingsrelaterade biverkningar var högre bland försökspersoner ≥ 65 år (se avsnitt Biverkningar). KdD ska användas med försiktighet hos patienter ≥ 75 år efter noggrann övervägning av den potentiella nyttan/risken på individuell basis.
Figur 5. Kaplan‑Meier‑kurva över progressionsfri överlevnad (ITT‑population) fastställd med IRC‑studie 20160275
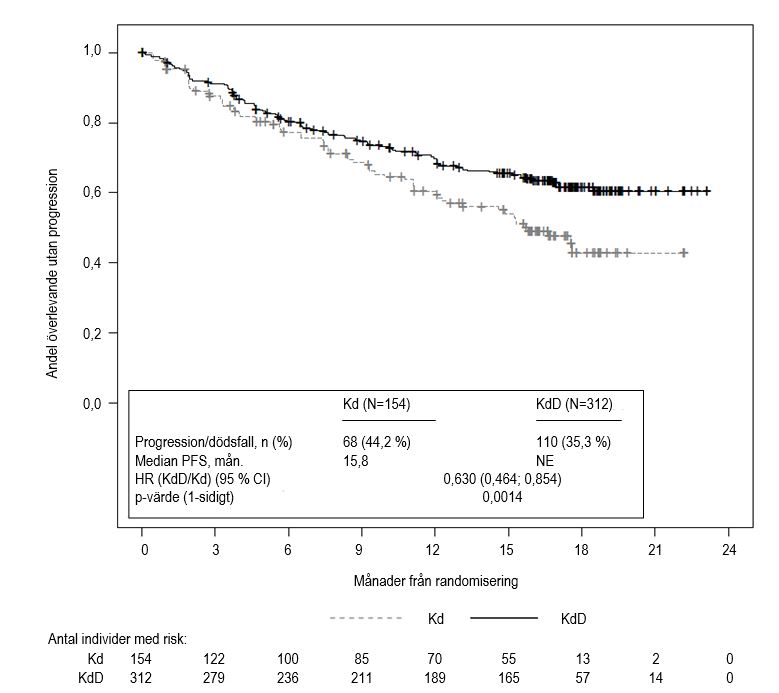
ORR var 84,3 % för patienterna i KdD‑armen och 74,7 % i Kd‑armen (se tabell 9). Mediantiden till respons gick inte att beräkna för KdD‑armen och var 16,6 månader (13,9; NE) för Kd‑armen. Mediantid till respons var 1,0 (1; 14) månader för KdD‑armen och 1,0 (1; 10) månader för Kd‑armen.
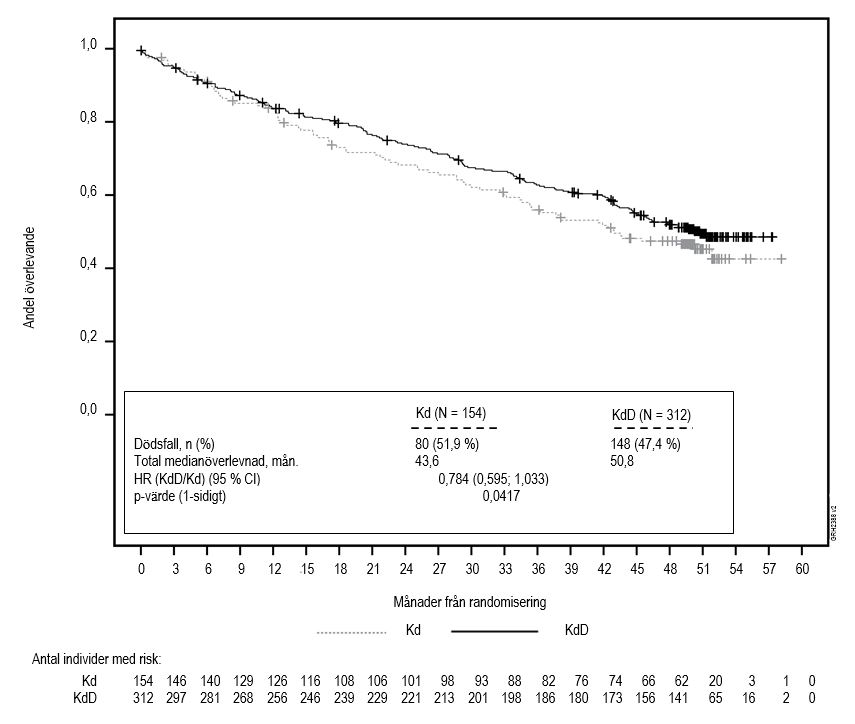
Vid tidpunkten för den slutliga analysen hade 148 försökspersoner (47,4 %) i KdD‑gruppen och 80 försökspersoner (51,9 %) i Kd‑gruppen dött. Median OS (95 % CI) var 50,8 (44,7, NE) månader för KdD‑gruppen och 43,6 (35,3, NE) månader för Kd‑gruppen, med en HR (KdD/Kd) på 0,784 (95 % CI: 0,595, 1,033; 1‑sidigt p = 0,0417). Detta ensidiga p‑värde uppfyllde inte den statistiska signifikansnivån 0,021 för den här slutliga analysen. Median uppföljningstid var 50,6 månader i KdD‑gruppen och 50,1 månader i Kd‑gruppen.
Figur 6. Kaplan‑Meier‑kurva över total överlevnad i studie 20160275
Kyprolis som monoterapi hos patienter med recidiverande och refraktärt multipelt myelom
Ytterligare klinisk erfarenhet har erhållits från Kyprolis i monoterapi hos patienter med recidiverande och refraktärt multipelt myelom. Studie PX‑171‑011 var en öppen, randomiserad fas 3‑studie (N = 315; exponering för ≥ 3 tidigare behandlingsregimer krävdes). Patienterna som rekryterades till studie PX‑171‑011 hade tidigare behandlats i större omfattning och hade lägre organ- och benmärgsfunktion jämfört med de som rekryterades till studie PX‑171‑009. PX‑171‑011 utvärderade Kyprolis i monoterapi jämfört med en kontrollarm (kortikosteroider och cyklofosfamid). Studien uppfyllde inte det primära effektmåttet, som var att visa att Kyprolis i monoterapi var överlägset den aktiva kontrollarmen vad avser total överlevnad (HR = 0,975 [95 % CI: 0,760; 1,249]). PX‑171‑003A1 var en enarmad fas 2‑studie (N = 266; exponering för ≥ 2 tidigare behandlingsregimer krävdes), vilken uppfyllde det primära effektmåttet avseende IRC‑bedömd ORR (22,9 %).
Hjärtats elektrofysiologi
En utvärdering av möjliga effekter av carfilzomib på hjärtats funktion utfördes genom analys medelst blindad centraliserad granskning av EKG‑tripletter från 154 försökspersoner med långt framskridna tumörsjukdomar, däribland multipelt myelom. Effekten av carfilzomib på hjärtats repolarisering mätt med QT‑intervall med Fridericias korrigering (QTcF‑intervall) och analys av förhållandet mellan koncentration och QTc ger inga tydliga signaler om eventuella dosrelaterade effekter. Den övre gränsen för det ensidiga 95 %‑konfidensintervallet (CI) för förutsagd effekt på QTcF vid Cmax var 4,8 msek. Med Bazetts korrigering (QTcB‑intervall) var det övre gränsvärdet 5,9 msek för ett ensidigt 95 %‑konfidensintervall (CI) för förutsagd effekt på QTcB vid Cmax.
Pediatrisk population
Europeiska läkemedelsmyndigheten har beviljat undantag från kravet att skicka in studieresultat för Kyprolis för alla grupper av den pediatriska populationen för multipelt myelom (information om pediatrisk användning finns i avsnitt Dosering och administreringssätt).
Farmakokinetiska egenskaper
Absorption
Cmax och AUC efter en 2 till 10 minuter lång intravenös infusion på 27 mg/m2 var 4 232 ng/ml respektive 379 ng•h/ml. Efter upprepad dosering av Kyprolis vid 15 och 20 mg/m2 var den systemiska exponeringen (AUC) och halveringstiden ungefär densamma på dag 1 och 15 eller 16 i cykel 1, vilket tyder på att det inte förekom någon systemisk ackumulering av carfilzomib. Vid doser mellan 20 och 56 mg/m2 förelåg en dosberoende ökning av exponeringen.
En 30 minuter lång infusion ledde till en liknande halveringstid och AUC men till 2 till 3 gånger lägre Cmax jämfört med det som observerades med en 2 till 10 minuter lång infusion av samma dos. Efter en 30 minuters infusion av dosen 56 mg/m2 var AUC (948 ng•h/ml) omkring 2,5 gånger den som observerades vid dosnivån 27 mg/m2 och Cmax (2 079 ng/ml) var lägre än den för 27 mg/m2 givet som en 2 till 10 minuter lång infusion.
Distribution
Den genomsnittliga distributionsvolymen vid jämvikt för en dos på 20 mg/m2 carfilzomib var 28 l. När bindningen av carfilzomib till humana plasmaproteiner testades in vitro var den i genomsnitt 97 % över koncentrationsintervallet 0,4 till 4 mikromolar.
Metabolism
Carfilzomib metaboliserades snabbt och i hög grad. De övervägande metaboliterna som identifierades i human plasma och urin, och som genererades in vitro av humana hepatocyter, var peptidfragment och diolen av carfilzomib, vilket tyder på att peptidasklyvning och epoxidhydrolys var de huvudsakliga metabolismvägarna. Cytokrom P450‑medierade mekanismer hade endast en mindre roll i den totala carfilzomibmetabolismen. Metaboliterna har ingen känd biologisk aktivitet.
Eliminering
Efter intravenös administrering av doser ≥ 15 mg/m2 utsöndrades carfilzomib snabbt från den systemiska cirkulationen med en halveringstid på ≤ 1 timme på dag 1 i cykel 1. Systemiskt clearance varierade från 151 till 263 l/timme och överskred det hepatiska blodflödet, vilket tyder på att carfilzomib i huvudsak utsöndrades extrahepatiskt. Carfilzomib elimineras primärt via metabolism med efterföljande utsöndring av metaboliterna i urin.
Särskilda populationer
Farmakokinetiska populationsanalyser tyder på att ålder, kön och etnicitet inte har någon effekt på carfilzomibs farmakokinetik.
Nedsatt leverfunktion
I en farmakokinetisk studie utvärderades 33 patienter med recidiverande eller progredierande långt framskridna tumörsjukdomar (fasta tumörer; n = 31, eller hematologiska tumörsjukdomar; n = 2) med normal leverfunktion (bilirubin ≤ övre normala gränsvärdet [ULN]; aspartataminotransferas [ASAT] ≤ ULN, n = 10), lindrigt nedsatt leverfunktion (bilirubin > 1‑1,5 × ULN eller ASAT > ULN, men bilirubin ≤ ULN, n = 14), eller måttligt nedsatt leverfunktion (bilirubin > 1,5‑3 × ULN; oavsett ASAT, n = 9). Carfilzomibs farmakokinetik har inte studerats hos patienter med gravt nedsatt leverfunktion (bilirubin > 3 × ULN, oavsett ASAT). Kyprolis, som monoterapi, administrerades intravenöst under 30 minuter vid 20 mg/m2 på dag 1 och 2, samt vid 27 mg/m2 på dag 8, 9, 15 och 16 i cykel 1. Om patienterna tolererade dosen fick de 56 mg/m2 från och med cykel 2. Leverfunktionen vid baslinjen hade inte någon markant effekt på den totala systemiska exponeringen (AUClast) för carfilzomib efter en bolusdos eller upprepad dosering (geometrisk medelkvot för AUClast vid dosen 27 mg/m2 i cykel 1, dag 16, för patienter med lindrigt och måttligt nedsatt leverfunktion jämfört med patienter med normal leverfunktion var 144,4 % respektive 126,1 %; vid dosen 56 mg/m2 i cykel 2, dag 1, var den 144,7 % respektive 121,1 %). Hos patienter med lindrigt eller måttligt nedsatt leverfunktion vid baslinjen, vilka samtliga hade fasta tumörer, var incidensen för avvikande leverfunktion, biverkningar ≥ grad 3 och allvarliga biverkningar högre jämfört med patienter med normal leverfunktion (se avsnitt Dosering och administreringssätt).
Nedsatt njurfunktion
Carfilzomibs farmakokinetik studerades i två studier med syfte att undersöka effekten av nedsatt njurfunktion.
Den första studien utfördes på 50 patienter med multipelt myelom och normal njurfunktion (CrCL > 80 ml/min, n = 12), lindrigt (CrCL 50‑80 ml/min, n = 12), måttligt (CrCL 30‑49 ml/min, n = 10) eller gravt (CrCL < 30 ml/min, n = 8) nedsatt njurfunktion samt patienter i dialys (n = 8). Kyprolis, som monoterapi, administrerades intravenöst under 2 till 10 minuter vid doser på upp till 20 mg/m2. Farmakokinetiska data samlades in från patienterna efter dosen på 15 mg/m2 i cykel 1 och efter dosen på 20 mg/m2 i cykel 2. Den andra studien utfördes på 23 patienter med recidiverande multipelt myelom och kreatininclearance ≥ 75 ml/min (n = 13) samt patienter med terminal njursjukdom (ESRD) i dialys (n = 10). Farmakokinetiska data samlades in från patienterna efter en dos på 27 mg/m2 som en 30 minuter lång infusion på dag 16 i cykel 1 och efter dosen på 56 mg/m2 dag 1 i cykel 2.
Resultaten från båda studierna visar att njurfunktionen inte hade någon markant effekt på exponeringen av carfilzomib efter en singeldos eller efter upprepad dosering. Den geometriska medelkvoten för AUClast vid dosen 15 mg/m2 på dag 1 i cykel 1 till patienter med lindrig, måttlig eller gravt nedsatt njurfunktion samt patienter i dialys jämfört med patienter med normal njurfunktion var 124,36 %, 111,07 %, 84,73 % respektive 121,72 %. Den geometriska medelkvoten för AUClast vid doserna 27 mg/m2 på dag 16 i cykel 1 och 56 mg/m2 på dag 1 i cykel 2 till ESRD‑patienter jämfört med patienter med normal njurfunktion var 139,72 % respektive 132,75 %. I den första studien var halten M14‑metabolit, ett peptidfragment och den vanligaste metaboliten i blodcirkulationen, 2 till 3 gånger högre hos patienter med måttligt respektive gravt nedsatt njurfunktion, och 7 gånger högre hos patienter som krävde dialys (baserat på AUClast). I den andra studien var exponeringen för M14 högre (ungefär 4‑faldig) hos patienter med ESRD än hos patienter med normal njurfunktion. Denna metabolit har ingen känd biologisk aktivitet. Allvarliga biverkningar relaterade till försämrad njurfunktion var vanligare hos patienter med njurdysfunktion vid baslinjen (se avsnitt Dosering och administreringssätt).
Prekliniska säkerhetsuppgifter
Carfilzomib orsakade kromosomskador i ett in vitro‑test på lymfocyter från perifert blod. Carfilzomib var inte mutagent in vitro i Ames test och orsakade inte kromosomskada in vivo i ett mikrokärntest i musbenmärg.
Apor som fick en enstaka intravenös bolusinjektion carfilzomib vid 3 mg/kg (vilket motsvarar 36 mg/m2 och liknar den rekommenderade dosen till människa på 27 mg/m2 baserat på kroppsyta) drabbades av hypotoni, ökad hjärtfrekvens och förhöjda serumnivåer av troponin T. Upprepad administrering av intravenösa bolusinjektioner av carfilzomib vid ≥ 2 mg/kg/dos till råttor och 2 mg/kg/dos till apor med dosscheman som liknar de som används kliniskt ledde till mortalitet på grund av toxicitet i följande system: hjärta‑kärl (hjärtsvikt, hjärtfibros, vätskeansamling i hjärtsäcken, hjärtblödning/degeneration), magtarmkanalen (nekros/blödning), njurarna (glomerulonefropati, tubulärnekros, dysfunktion), och lungorna (blödning/inflammation). Dosen på 2 mg/kg/dos till råttor motsvarar omkring halva den rekommenderade dosen till människa på 27 mg/m2 baserat på kroppsyta. Den högsta icke‑allvarligt toxiska dosen på 0,5 mg/kg hos apor ledde till interstitiell njurinflammation tillsammans med tendenser till glomerulopati och lindrig hjärtinflammation. Dessa resultat rapporterades vid 6 mg/m2, vilket är lägre än den rekommenderade dosen på 27 mg/m2 till människa.
Inga fertilitetsstudier med carfilzomib har utförts. Det observerades inga reproduktionseffekter under 28 dagars upprepad dosering i toxicitetsstudier på råtta och apor och inte heller i 6 månader (råtta) och 9 månader (apor) långa allmäntoxicitetsstudier. Carfilzomib orsakade toxicitet hos embryon och foster hos dräktiga kaniner vid doser som var lägre än hos patienter som fick den rekommenderade dosen. Carfilzomib som administrerades till dräktiga råttor under organogenesen var inte teratogent vid doser upp till 2 mg/kg/dag, vilket är ungefär hälften av den rekommenderade dosen till människa på 27 mg/m2 baserat på kroppsyta.
Farmaceutiska uppgifter
Förteckning över hjälpämnen
Betadex‑sulfobutyleter‑natrium, vattenfri citronsyra (E330), natriumhydroxid (för pH‑justering).
Inkompatibiliteter
Då blandbarhetsstudier saknas får detta läkemedel inte blandas med andra läkemedel.
Kyprolis pulver till infusionsvätska, lösning, får inte blandas med natriumkloridlösning 9 mg/ml (0,9 %) för injektion.
Hållbarhet
Pulver i injektionsflaska (oöppnad)
3 år.
Rekonstituerad lösning
Kemisk och fysikalisk stabilitet för den rekonstituerade lösningen i injektionsflaskan, sprutan eller infusionspåsen har visats i 24 timmar vid 2 °C till 8 °C eller i 4 timmar vid 25 °C. Tiden från rekonstituering till administrering får inte överstiga 24 timmar.
Med tanke på risken för mikrobiologisk tillväxt ska produkten användas omedelbart. Om den inte används omedelbart är förvaringstiden och -förhållandena för den färdigberedda lösningen användarens ansvar och får inte vara längre än 24 timmar vid 2 °C‑8 °C.
Särskilda förvaringsanvisningar
Förvaras i kylskåp (2 °C‑8 °C).
Får ej frysas.
Förvaras i originalförpackningen. Ljuskänsligt.
Förvaringsanvisningar för läkemedlet efter berdning finns i avsnitt Hållbarhet.
Förpackningstyp och innehåll
Markkinoilla olevat pakkaukset
Resepti
KYPROLIS infuusiokuiva-aine, liuosta varten
10 mg (L:ei) 1 kpl (305,58 €)
30 mg (L:ei) 1 kpl (874,62 €)
60 mg (L:ei) 1 kpl (1704,85 €)
PF-selosteen tieto
Kyprolis 10 mg pulver till infusionsvätska, lösning
10 ml injektionsflaska av klart typ I‑glas, försluten med en fluorpolymerlaminerad elastomerpropp och en aluminiumförsegling med ett ljusblått snäpplock av plast.
Kyprolis 30 mg pulver till infusionsvätska, lösning
30 ml injektionsflaska av klart typ I‑glas, försluten med en fluorpolymerlaminerad elastomerpropp och en aluminiumförsegling med ett orange snäpplock av plast.
Kyprolis 60 mg pulver till infusionsvätska, lösning
50 ml injektionsflaska av klart typ I‑glas, försluten med en fluorpolymerlaminerad elastomerpropp och en aluminiumförsegling med ett lila snäpplock av plast.
Förpackningsstorlekar med en injektionsflaska.
Läkemedlets utseende:
Vitt till benvitt frystorkat pulver.
Särskilda anvisningar för destruktion och övrig hantering
Allmänna försiktighetsåtgärder
Carfilzomib är ett cytotoxiskt ämne. Därför ska försiktighet iakttas vid hantering och beredning av Kyprolis. Användning av handskar och annan skyddsutrustning rekommenderas.
Rekonstituering och förberedelser för intravenös administrering
Injektionsflaskorna med Kyprolis innehåller inga antimikrobiella konserveringsmedel och de är endast avsedda för engångsbruk. Korrekt aseptisk teknik måste iakttas.
Den rekonstituerade lösningen innehåller carfilzomib i en koncentration av 2 mg/ml. Läs de fullständiga beredningsanvisningarna före rekonstitueringen:
- Beräkna dosen (mg/m2) och antalet Kyprolis‑injektionsflaskor som krävs med utgångspunkt från patientens kroppsyta vid behandlingsstart. Patienter som har en större kroppsyta än 2,2 m2 ska få en dos som baseras på en kroppsyta på 2,2 m2. Det krävs inga dosjusteringar för viktförändringar ≤ 20 %.
- Ta fram injektionsflaskan från kylen precis innan den ska användas.
- Använd endast en 21‑gauge‑nål eller en nål med högre gauge‑tal (utvändig diameter på 0,8 mm eller mindre). Rekonstituera varje injektionsflaska aseptiskt genom att långsamt injicera 5 ml (för 10 mg injektionsflaska), 15 ml (för 30 mg injektionsflaska) eller 29 ml (för 60 mg injektionsflaska) sterilt vatten för injektion genom proppen och rikta vätskan mot INSIDAN AV INJEKTIONSFLASKANS VÄGG för att minimera skumbildning.
- Snurra flaskan försiktigt och/eller vänd den långsamt upp och ned i omkring 1 minut eller tills pulvret är helt upplöst. FÅR EJ SKAKAS. Om det bildas skum ska injektionsflaskan få stå tills skummet har lagt sig (omkring 5 minuter) och lösningen är klar.
- Kontrollera lösningen visuellt för partiklar och missfärgning innan den ska administreras. Den rekonstituerade produkten ska vara klar, färglös till något gulaktig och ska inte administreras om någon missfärgning eller partiklar observeras.
- Kassera allt oanvänt läkemedel som finns kvar i injektionsflaskan.
- Kyprolis kan administreras direkt via en intravenös infusion eller alternativt administreras via en infusionspåse. Det får inte administreras som en intravenös stötdos eller bolusinjektion.
- När administreringen ska göras via en infusionspåse, använd endast en 21‑gauge‑nål eller en nål med högre gauge‑tal (utvändig diameter på 0,8 mm eller mindre). Dra upp den beräknade dosen från injektionsflaskan och späd i en 50 eller 100 ml infusionspåse som innehåller 5 % glukoslösning för injektion.
Destruktion
Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar.
Ersättning
KYPROLIS infuusiokuiva-aine, liuosta varten
10 mg 1 kpl
30 mg 1 kpl
60 mg 1 kpl
- Ei korvausta.
Atc-kod
L01XG02
Datum för översyn av produktresumén
14.12.2023
Yhteystiedot
Keilaranta 10, PL 86
02101 Espoo
09 5490 0500