
TAGRISSO tabletti, kalvopäällysteinen 40 mg, 80 mg
Vaikuttavat aineet ja niiden määrät
TAGRISSO 40 mg tabletti
Yksi tabletti sisältää osimertinibimesylaattia määrän, joka vastaa 40 mg osimertinibia.
TAGRISSO 80 mg tabletti
Yksi tabletti sisältää osimertinibimesylaattia määrän, joka vastaa 80 mg osimertinibia.
Apuaine, jonka vaikutus tunnetaan
TAGRISSO 40 mg tabletit
Yksi tabletti sisältää 0,3 mg natriumia.
TAGRISSO 80 mg tabletit
Yksi tabletti sisältää 0,6 mg natriumia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen.
Kliiniset tiedot
Käyttöaiheet
TAGRISSO on tarkoitettu monoterapiana:
-
liitännäishoitoon kasvaimen täydellisen resektion jälkeen aikuispotilaille, joilla on levinneisyysasteen IB–IIIA ei-pienisoluinen keuhkosyöpä (NSCLC), jossa on epidermaalisen kasvutekijän reseptorin (EGFR:n) eksonin 19 deleetioita tai eksonin 21 (L858R) substituutiomutaatioita (ks. kohta Farmakodynamiikka)
-
sellaisten aikuispotilaiden hoitoon, joilla on paikallisesti edennyt, leikkaukseen soveltumaton NSCLC, jossa on EGFR:n eksonin 19 deleetioita tai eksonin 21 (L858R) substituutiomutaatioita, ja joiden tauti ei ole edennyt platinapohjaisen kemosädehoidon aikana tai sen jälkeen.
-
ensilinjan hoitoon aikuispotilaille, joilla on paikallisesti edennyt tai metastaattinen NSCLC, jossa on aktivoivia EGFR:n mutaatioita
- sellaisten aikuispotilaiden hoitoon, joilla on paikallisesti edennyt tai metastaattinen NSCLC, jossa on EGFR:n T790M-mutaatio.
TAGRISSO on tarkoitettu yhdistelmänä:
- pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa ensilinjan hoitoon aikuispotilaille, joilla on pitkälle edennyt NSCLC, jossa on EGFR:n eksonin 19 deleetioita tai eksonin 21 (L858R) substituutiomutaatioita.
Ehto
Ainoastaan syöpälääkkeiden antoon perehtyneen lääkärin tulee aloittaa hoito.
Annostus ja antotapa
TAGRISSO-hoidon aloittaa syöpähoitojen käyttöön perehtynyt lääkäri.
Kun harkitaan TAGRISSO-valmisteen käyttöä, EGFR-mutaatiostatus on määritettävä (liitännäishoidon tai paikallisesti edenneiden, leikkaukseen soveltumattomien kasvainten yhteydessä kasvainnäytteistä ja paikallisesti edenneen tai metastaattisen taudin yhteydessä kasvain- tai plasmanäytteistä) validoidulla testimenetelmällä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Monoterapia
Suositeltu annos on 80 mg osimertinibia kerran vuorokaudessa.
Yhdistelmähoito
TAGRISSO-valmisteen suositeltu annos on 80 mg osimertinibia kerran vuorokaudessa pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa.
Katso pemetreksedin ja sisplatiinin tai karboplatiinin valmisteyhteenvedoista niiden annostusta koskevat tiedot.
Jos potilas saa valmistetta liitännäishoitona, hoitoa tulisi jatkaa, kunnes tauti uusiutuu tai toksisuus ei ole hyväksyttävissä. Yli 3 vuoden pituista hoitoa ei ole tutkittu.
Jos potilaalla on paikallisesti edennyt tai metastaattinen keuhkosyöpä, TAGRISSO-hoitoa tulisi jatkaa, kunnes tauti etenee tai toksisuus ei ole hyväksyttävissä.
Jos TAGRISSO-valmisteen annos unohtuu, se tulee ottaa heti muistettaessa. Jos seuraavan annoksen ottamiseen on alle 12 tuntia, annos jätetään väliin.
TAGRISSO voidaan ottaa ruoan kanssa tai ilman ruokaa, samaan aikaan joka päivä.
Annoksen muuttaminen
Hoidon keskeyttäminen ja/tai annoksen pienentäminen saattaa olla tarpeen perustuen potilaan turvallisuuteen ja lääkityksen siedettävyyteen. Jos annoksen pienentäminen on tarpeen, annos on pienennettävä 40 mg:aan kerran vuorokaudessa.
Suositukset annoksen pienentämisestä toksisuudesta johtuvien haittavaikutusten vuoksi on esitetty taulukossa 1.
Taulukko 1: TAGRISSO-valmisteen suositellut annoksen muutokset
Kohde-elin | Haittavaikutusa | Annoksen muuttaminen |
Keuhkotb | Interstitiaalinen keuhkosairaus (ILD) / pneumoniittic | TAGRISSO-hoito on lopetettava pysyvästi |
| Asteen 1 sädepneumoniitti | TAGRISSO-hoidon keskeyttämistä on harkittava tai hoitoa on jatkettava kliinisen tarpeen mukaan |
| Asteen 2 sädepneumoniitti | TAGRISSO-hoito on keskeytettävä, kunnes oireet häviävät. TAGRISSO-hoito voidaan aloittaa uudelleen. Hoito on lopetettava pysyvästi, jos oireet eivät ole hävinneet 4 viikon kuluttua tai asteen 2 sädepneumoniitti uusiutuu |
| Asteen 3 tai 4 sädepneumoniitti | TAGRISSO-hoito on lopetettava pysyvästi |
Sydänb | QTc-aika yli 500 ms ainakin kahdessa erillisessä EKG-tutkimuksessa | TAGRISSO-hoito on keskeytettävä, kunnes QTc-aika on alle 481 ms tai QTc-aika on palautunut lähtötasolle, jos lähtötason QTc-aika on vähintään 481 ms. Hoito aloitetaan uudelleen pienemmällä annoksella (40 mg) |
QTc-ajan piteneminen, johon liittyy vakavan arytmian merkkejä tai oireita | TAGRISSO-hoito on lopetettava pysyvästi | |
Ihob | Stevens–Johnsonin oireyhtymä ja toksinen epidermaalinen nekrolyysi | TAGRISSO-hoito on lopetettava pysyvästi |
Veri ja imukudosb | Aplastinen anemia | TAGRISSO-hoito on lopetettava pysyvästi |
Muu | Vähintään asteen 3 haittavaikutus | TAGRISSO-hoito on keskeytettävä korkeintaan kolmeksi viikoksi |
Jos vähintään asteen 3 haittavaikutus lievenee asteeseen 0–2 TAGRISSO-hoidon oltua keskeytettynä korkeintaan kolmen viikon ajan | TAGRISSO-hoito voidaan aloittaa uudelleen samalla annoksella (80 mg) tai pienemmällä annoksella (40 mg) | |
Vähintään asteen 3 haittavaikutus, joka ei lievene asteeseen 0–2 hoidon oltua keskeytettynä korkeintaan kolmen viikon ajan | TAGRISSO-hoito on lopetettava pysyvästi |
a Huom.: Kliinisten haittatapahtumien voimakkuus on luokiteltu National Cancer Instituten (NCI) haittavaikutusten CTCAE-luokituksen (Common Terminology Criteria for Adverse Events) kriteerien version 5.0. mukaisesti.
b Ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
c ILD/pneumoniitti sisältää: definitiivisen platinapohjaisen kemosädehoidon jälkeinen ILD/pneumoniitti.
EKG: Elektrokardiogrammi; QTc: QT-väli korjattuna sydämen syketiheyden suhteen
Yhdistelmähoito
Kun TAGRISSO-valmistetta käytetään yhdistelmänä, minkä tahansa yhdistelmähoitoon kuuluvan lääkkeen annosta on tarvittaessa muutettava. Katso TAGRISSO-valmisteen annoksen muuttamista koskevat ohjeet taulukosta 1. Pemetreksedin, sisplatiinin tai karboplatiinin annoksia muutetaan niiden valmisteyhteenvedoissa annettujen ohjeiden mukaisesti. Sisplatiinia ja/tai karboplatiinia saa käyttää enintään 4 hoitosyklin ajan.
Erityisryhmät
Annoksen muuttaminen ei ole tarpeen potilaan iän, painon, sukupuolen, etnisen taustan tai tupakoinnin vuoksi (ks. kohta Farmakokinetiikka).
Maksan vajaatoiminta
Kliinisten tutkimusten perusteella annoksen muuttaminen ei ole tarpeen potilaille, joilla on lievä maksan vajaatoiminta (Child–Pugh-luokka A) tai kohtalainen maksan vajaatoiminta (Child–Pugh-luokka B). Populaatiofarmakokineettisen analyysin perusteella annoksen muuttamista ei myöskään suositella potilaille, joilla on lievä maksan vajaatoiminta (kokonaisbilirubiini ≤ viitearvon yläraja [ULN] ja aspartaattiaminotransferaasi ASAT > ULN tai kokonaisbilirubiini > 1,0–1,5 x ULN ja mikä tahansa ASAT‑arvo) tai kohtalainen maksan vajaatoiminta (kokonaisbilirubiini 1,5–3 x ULN ja mikä tahansa ASAT‑arvo). Tämän valmisteen tehoa ja turvallisuutta ei tiedetä potilailla, joilla on vaikea maksan vajaatoiminta. Käyttöä potilaille, joilla on vaikea maksan vajaatoiminta, ei suositella ennen kuin saadaan lisää tietoa (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Kliinisten tutkimusten ja populaatiofarmakokineettisen analyysin perusteella annoksen muuttaminen ei ole tarpeen potilaille, joilla on lievä, kohtalainen tai vaikea munuaisten vajaatoiminta. Tämän lääkevalmisteen tehoa ja turvallisuutta ei tiedetä potilailla, joilla on loppuvaiheen munuaissairaus (kreatiniinipuhdistuma alle 15 ml/min Cockcroftin ja Gaultin yhtälöllä laskettuna) tai jotka saavat dialyysihoitoa. Varovaisuutta on noudatettava hoidettaessa potilaita, joilla on vaikea ja loppuvaiheen munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
TAGRISSO-valmisteen turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Tämä lääkevalmiste otetaan suun kautta. Tabletti niellään kokonaisena veden kanssa. Tablettia ei saa murskata, jakaa tai pureskella.
Jos potilas ei pysty nielemään tablettia, se voidaan ensin liuottaa 50 ml:aan hiilihapotonta vettä. Tabletti pudotetaan veteen murskaamatta sitä, sekoitetaan kunnes se on liuennut ja liuos juodaan välittömästi. Lasiin lisätään vielä puolen lasin verran vettä, jotta lasiin ei jää yhtään lääkettä, ja juodaan välittömästi. Muita nesteitä ei pidä käyttää.
Jos valmiste on annettava nenä-mahaletkulla, noudatetaan samaa menettelytapaa kuin edellä, mutta ensimmäiseen liuottamiseen käytetään 15 ml nestettä ja jäämien huuhtelemiseen 15 ml. Saatu 30 ml nestettä annetaan nenä-mahaletkun valmistajan ohjeiden mukaan asianmukaisin vesihuuhteluin. Liuos ja lääkejäämät on annettava 30 minuutin kuluessa tablettien lisäämisestä veteen.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Mäkikuismaa sisältäviä valmisteita ei tule käyttää samanaikaisesti TAGRISSO-valmisteen kanssa (ks. kohta Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
EGFR-mutaatiostatuksen määrittäminen
Kun harkitaan TAGRISSO-valmisteen käyttöä liitännäishoitona NSCLC-kasvaimen täydellisen resektion jälkeen, positiivinen EGFR-mutaatiostatus (eksonin 19 deleetiot [Ex19del] tai eksonin 21 L858R-substituutiomutaatiot [L858R]) viittaa hoidon soveltuvuuteen. Validoitu testi on tehtävä kliinisessä laboratoriossa käyttämällä biopsianäytteestä tai leikkauspreparaatista saatua kasvainkudoksen DNA:ta.
Kun harkitaan TAGRISSO-valmisteen käyttöä potilaille, joilla on paikallisesti edennyt, leikkaukseen soveltumaton NSCLC ja joiden tauti ei ole edennyt platinapohjaisen kemosädehoidon aikana tai sen jälkeen, positiivinen EGFR-mutaatiostatus (eksonin 19 deleetiot tai eksonin 21 [L858R] substituutiomutaatiot) viittaa hoitoon soveltuvuuteen. Validoitu testi on tehtävä kliinisessä laboratoriossa käyttämällä biopsianäytteestä saatua kasvainkudoksen DNA:ta.
Harkittaessa TAGRISSO-valmisteen käyttöä paikallisesti edenneen tai metastaattisen NSCLC:n hoitoon on tärkeää, että EGFR-mutaatiostatusmäärityksen tulos on positiivinen. Validoitu testi on tehtävä käyttämällä joko kudosnäytteestä eristettyä kasvaimen DNA:ta tai plasmanäytteestä eristettyä kiertävää kasvain-DNA:ta (ctDNA).
Joko kudos- tai plasmanäytteestä tehdyllä testillä määritetty positiivinen EGFR-mutaatiostatus (aktivoivien EGFR-mutaatioiden suhteen, jos kyseessä on ensilinjan hoito, tai eksonin 19 deleetion tai eksonin 21 (L858R) substituutiomutaatioiden suhteen, kun TAGRISSO-valmistetta annetaan yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, jos kyseessä on ensilinjan hoito, tai T790M-mutaatioiden suhteen, jos tauti on edennyt EGFR-toimintaan kohdennetun tyrosiinikinaasin estäjähoidon aikana tai sen jälkeen) osoittaa, että potilas soveltuu saamaan TAGRISSO-hoitoa. Mikäli plasmapohjaisen ctDNA-testin tulos on negatiivinen, suositellaan kuitenkin mahdollisuuksien mukaan jatkotutkimusta kudostestillä, sillä plasmapohjainen testi saattaa antaa virheellisen negatiivisen tuloksen.
On käytettävä ainoastaan varmoja, luotettavia ja herkkiä testejä, joiden käyttökelpoisuus EGFR–mutaatiostatuksen määrittämisessä on osoitettu.
Interstitiaalinen keuhkosairaus (ILD)
Kliinisissä tutkimuksissa, myös definitiivisen platinapohjaisen kemosädehoidon jälkeen, TAGRISSO-hoitoa saaneilla potilailla on todettu vaikeaa, hengenvaarallista tai kuolemaan johtavaa ILD:tä tai ILD:n kaltaisia haittavaikutuksia (kuten pneumoniittia). Useimmat tapaukset lievenivät tai paranivat itsestään hoidon keskeyttämisen jälkeen. Potilaat, joilla oli ollut aiemmassa anamneesissa ILD, lääkkeen aiheuttama ILD tai steroidihoitoa edellyttänyt sädepneumoniitti tai joilla havaittiin viitteitä kliinisesti aktiivisesta ILD:stä, suljettiin pois kliinisistä tutkimuksista (ks. kohta Haittavaikutukset).
Kaikki potilaat, joilla esiintyy äkillisesti alkavia hengityselinoireita (kuten hengenahdistus, yskä, kuume) ja/tai tällaisten oireiden selittämätöntä pahenemista, on huolellisesti tutkittava ILD:n varalta. Hoito tällä lääkevalmisteella on keskeytettävä oireiden selvittämisen ajaksi. Jos ILD todetaan, TAGRISSO‑hoito on lopetettava ja asiaan kuuluva hoito on aloitettava tarpeen mukaan. TAGRISSO-hoito voidaan aloittaa uudelleen vain huolellisen harkinnan jälkeen ottaen huomioon potilaan hoidosta saamat hyödyt ja sen riskit.
Sädepneumoniitti
Sädepneumoniittitapauksia todetaan keuhkoihin kohdistuneen sädehoidon jälkeen yleensä enintään vuoden ajan. Ohjeet TAGRISSO-annoksen muuttamisesta definitiivisen platinapohjaisen kemosädehoidon jälkeen ilmenneen sädepneumoniitin vuoksi, ks. kohta Annostus ja antotapa.
Lääkeaineen aiheuttamat vaikeat ihoreaktiot
Stevens‑Johnsonin oireyhtymää (SJS) on raportoitu esiintymistiheydellä ”harvinainen” ja toksista epidermaalista nekrolyysiä (TEN) esiintymistiheydellä ”tuntematon” TAGRISSO-hoidon yhteydessä. Ennen hoidon aloittamista potilaille on kerrottava SJS:n ja TEN:n merkeistä ja oireista. Jos SJS:ään tai TEN:iin viittaavia merkkejä ja oireita ilmenee, TAGRISSO-hoito on keskeytettävä. TAGRISSO-hoito on lopetettava välittömästi, jos potilaalla todetaan SJS tai TEN.
QTc‑ajan piteneminen
QTc‑ajan pitenemistä on todettu TAGRISSO‑hoitoa saaneilla potilailla. QTc‑ajan piteneminen saattaa lisätä kammioperäisten takyarytmioiden (kuten kääntyvien kärkien takykardian, torsades de pointes) tai äkillisen kuoleman riskiä. ADAURA-, LAURA‑, FLAURA-, FLAURA2- tai AURA‑tutkimuksissa ei ilmoitettu QTc-aikaan liittyviä rytmihäiriöitä (ks. kohta Haittavaikutukset). Potilaat, joilla oli kliinisesti merkittäviä rytmi‑ ja johtumishäiriöitä levossa otetussa elektrokardiogrammissa (EKG) (esimerkiksi QTc‑aika yli 470 ms), suljettiin pois näistä tutkimuksista (ks. kohta Haittavaikutukset).
TAGRISSO-valmisteen käyttöä tulisi mahdollisuuksien mukaan välttää potilailla, joilla on synnynnäinen pitkä QT ‑oireyhtymä. EKG:n ja elektrolyyttien säännöllistä seurantaa on harkittava, jos potilaalla on kongestiivinen sydämen vajaatoiminta, elektrolyyttihäiriöitä tai jos potilas käyttää lääkkeitä, joiden tiedetään pidentävän QTc‑aikaa. Hoito tulisi keskeyttää, jos potilaan QTc‑aika on yli 500 ms vähintään kahdessa erillisessä EKG:ssä, kunnes QTc‑aika on alle 481 ms tai kunnes QTc‑aika on palautunut lähtötasolle, jos lähtötason QTc‑aika on vähintään 481 ms. TAGRISSO-hoitoa voidaan sitten jatkaa pienemmällä annoksella, kuten on kuvattu taulukossa 1. TAGRISSO-hoito tulisi lopettaa pysyvästi, jos potilaalla ilmenee QTc‑ajan pitenemistä ja samanaikaisesti jokin seuraavista: kääntyvien kärkien takykardia (torsades de pointes), monimuotoinen kammiotakykardia, vakavan arytmian merkkejä/oireita.
Muutokset sydämen supistumisvireydessä
Kaikissa kliinisissä tutkimuksissa vasemman kammion ejektiofraktio pieneni vähintään 10 prosenttiyksikköä ja laski alle 50 %:iin 4,2 %:lla (65/1 557) TAGRISSO-monoterapiaa saaneista potilaista, joiden vasemman kammion ejektiofraktio oli arvioitu lähtötilanteessa ja ainakin kerran sen jälkeen. Potilailla, joilla on sydämeen liittyviä riskitekijöitä tai sairaus, joka voi vaikuttaa vasemman kammion ejektiofraktioon, on harkittava sydämen toiminnan seurantaa, joka sisältää vasemman kammion ejektiofraktion arvioinnin lähtötilanteessa ja hoidon aikana. Jos potilaalle kehittyy merkittäviä sydämeen liittyviä oireita tai merkkejä hoidon aikana, on harkittava sydämen toiminnan seurantaa, mukaan lukien vasemman kammion ejektiofraktion arviointi. Liitännäishoitoa arvioineessa lumekontrolloidussa tutkimuksessa (ADAURA) 1,5 %:lla (5/325) TAGRISSO-hoitoa saaneista potilaista ja 1,5 %:lla (5/331) lumehoitoa saaneista potilaista todettiin vasemman kammion ejektiofraktion pienentyneen vähintään 10 prosenttiyksikköä ja laskeneen alle 50 %:iin. LAURA-tutkimuksessa platinapohjaisen kemosädehoidon jälkeen 3,0 %:lla (4/135) TAGRISSO-valmistetta saaneista potilaista, joiden vasemman kammion ejektiofraktio oli arvioitu sekä lähtötilanteessa että sen jälkeen, todettiin vasemman kammion ejektiofraktion pienentyneen vähintään 10 prosenttiyksikköä ja laskeneen alle 50 %:iin. Tätä vaikutusta ei havaittu yhdelläkään lumelääkettä saaneella potilaalla. FLAURA2-tutkimuksessa 8,0 %:lla (21/262) TAGRISSO-valmistetta yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa saaneista potilaista, joiden vasemman kammion ejektiofraktio oli arvioitu lähtötilanteessa ja ainakin kerran sen jälkeen, todettiin vasemman kammion ejektiofraktion pienentyneen vähintään 10 prosenttiyksikköä ja laskeneen alle 50 %:iin.
Sarveiskalvotulehdus
Sarveiskalvotulehdusta ilmoitettiin 0,6 %:lla (n = 11) 1 956:sta TAGRISSO-monoterapiaa ADAURA-, FLAURA-, FLAURA2-, LAURA‑ ja AURA-tutkimuksissa saaneesta potilaasta. Potilaat, joilla ilmenee sarveiskalvotulehdukseen viittaavia oireita tai merkkejä, kuten akuutti tai paheneva silmätulehdus, kyynelnesteen eritys, valoherkkyys, näön hämärtyminen, silmäkipu ja/tai silmien punoitus, on ohjattava pikaisesti silmätautien erikoislääkärille (ks. kohta Annostus ja antotapa, taulukko 1).
Aplastinen anemia
TAGRISSO-hoidon yhteydessä on ilmoitettu harvinaisina tapauksina aplastista anemiaa, ja jotkin tapahtumat ovat johtaneet kuolemaan. Ennen hoidon aloittamista potilaille on kerrottava aplastisen anemian merkeistä ja oireista, joita voivat olla esimerkiksi pitkäkestoinen kuume, mustelmat, verenvuodot, ihon kalpeus, infektiot ja väsymys. Jos potilaalle kehittyy aplastiseen anemiaan viittaavia merkkejä ja oireita, on harkittava potilaan tarkkaa seurantaa ja TAGRISSO-hoidon keskeyttämistä tai lopettamista. TAGRISSO-hoito on lopetettava, jos potilaalla on varmistettu aplastinen anemia (ks. kohta Annostus ja antotapa).
Ikä ja paino
Iäkkäillä (yli 65‑vuotiailla) potilailla tai potilailla, joiden paino on pieni (alle 50 kg), vähintään asteen 3 haittatapahtumien riski saattaa olla suurentunut. Tällaisten potilaiden kohdalla suositellaan tarkkaa seurantaa (ks. kohta Haittavaikutukset).
Hepatiitti B -viruksen (HBV:n) uudelleenaktivoituminen
TAGRISSO-hoitoa saaneilla potilailla voi esiintyä hepatiitti B -viruksen uudelleenaktivoitumista, mikä voi joissakin tapauksissa johtaa fulminanttiin hepatiittiin, maksan vajaatoimintaan ja kuolemaan. Potilaita, jotka on serologisesti todettu HBV-positiivisiksi, on seurattava TAGRISSO-hoidon aikana HBV:n uudelleenaktivoitumisen kliinisesti ja laboratoriokokein havaittavien merkkien varalta. Jos hepatiitti B -virus aktivoituu uudelleen TAGRISSO-hoidon aikana, TAGRISSO-hoito on keskeytettävä, ja potilasta on hoidettava paikallisten hoitosuositusten mukaisesti.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol (23 mg) natriumia per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Farmakokineettiset yhteisvaikutukset
Voimakkaat CYP3A4‑induktorit voivat vähentää altistusta osimertinibille. Osimertinibi saattaa lisätä altistusta rintasyövän resistenssiproteiinin (BCRP:n) ja P-glykoproteiinin (P‑gp:n) substraateille.
Vaikuttavat aineet, jotka saattavat suurentaa osimertinibin pitoisuutta plasmassa
In vitro ‑tutkimukset ovat osoittaneet, että osimertinibin vaiheen I metabolia tapahtuu pääasiassa CYP3A4:n ja CYP3A5:n välityksellä. Kliinisessä farmakokineettisessä tutkimuksessa potilaille annettiin osimertinibia sekä samanaikaisesti 200 mg:n itrakonatsoliannos (voimakas CYP3A4:n estäjä) kahdesti päivässä. Tällä ei ollut kliinisesti merkittävää vaikutusta osimertinibialtistukseen (käyrän alla oleva pinta-ala [AUC] suureni 24 % ja Cmax pieneni 20 %). CYP3A4:n estäjät eivät siten todennäköisesti vaikuta osimertinibialtistukseen. Muita katalysoivia entsyymejä ei ole tunnistettu.
Vaikuttavat aineet, jotka saattavat pienentää osimertinibin pitoisuutta plasmassa
Kliinisessä farmakokineettisessä tutkimuksessa osimertinibin vakaan tilan AUC pieneni potilailla 78 %, kun sitä annettiin samanaikaisesti rifampisiinin kanssa (600 mg vuorokaudessa 21 vuorokauden ajan). Vastaavasti altistus metaboliitille AZ5104 pieneni; AUC pieneni 82 % ja Cmax 78 %. TAGRISSO‑valmisteen samanaikaista käyttöä voimakkaiden CYP3A‑induktorien (kuten fenytoiinin, rifampisiinin ja karbamatsepiinin) kanssa tulisi välttää. Keskivahvat CYP3A4-induktorit (kuten bosentaani, efavirentsi, etraviriini ja modafiniili) saattavat myös pienentää osimertinibialtistusta, ja niiden käytössä tulisi noudattaa varovaisuutta tai välttää käyttöä mahdollisuuksien mukaan. Ei ole olemassa kliinistä tietoa, jonka perusteella voitaisiin suositella TAGRISSO-valmisteen annoksen muuttamista. Samanaikainen mäkikuisman käyttö on vasta-aiheista (ks. kohta Vasta-aiheet).
Mahahapon eritystä vähentävien aineiden vaikutus osimertinibiin
Kliinisessä farmakokineettisessä tutkimuksessa omepratsolin samanaikainen antaminen ei aiheuttanut kliinisesti merkittäviä muutoksia osimertinibialtistuksiin. Mahalaukun pH‑arvoon vaikuttavia lääkkeitä voidaan rajoituksetta käyttää samanaikaisesti TAGRISSO‑valmisteen kanssa.
Vaikuttavat aineet, joiden pitoisuuteen plasmassa TAGRISSO saattaa vaikuttaa
In vitro ‑tutkimusten mukaan osimertinibi on kompetitiivinen BCRP‑kuljettajaproteiinien inhibiittori.
Kliinisessä farmakokineettisessä tutkimuksessa TAGRISSO‑valmisteen samanaikainen antaminen rosuvastatiinin (sensitiivinen BCRP:n substraatti) kanssa suurensi rosuvastatiinin AUC‑arvoa 35 % ja Cmax‑arvoa 72 %. Potilaita, jotka käyttävät samanaikaisesti lääkevalmisteita, jotka ovat osittain riippuvaisia BCRP:stä ja joilla on kapea terapeuttinen indeksi, on seurattava huolellisesti samanaikaisesti käytettävän lääkkeen siedettävyyden muuttumiseen viittaavien merkkien varalta, sillä altistus saattaa suurentua TAGRISSO‑hoidon aikana (ks. kohta Farmakokinetiikka).
Kliinisessä farmakokineettisessä tutkimuksessa TAGRISSO‑valmisteen samanaikainen annostelu simvastatiinin (herkkä CYP3A4:n substraatti) kanssa pienensi simvastatiinin AUC-arvoa 9 % ja Cmax-arvoa 23 %. Nämä muutokset olivat pieniä, eikä niillä todennäköisesti ole kliinistä merkitystä. Kliiniset farmakokineettiset yhteisvaikutukset CYP3A4:n substraattien kanssa ovat epätodennäköisiä. Riskiä hormonaalisten ehkäisyvalmisteiden altistuksen pienenemiselle ei voida poissulkea.
Kliinisessä tutkimuksessa, jossa arvioitiin pregnaani X ‑reseptoriin (PXR) liittyviä yhteisvaikutuksia, TAGRISSO-valmisteen samanaikainen antaminen feksofenadiinin (P‑gp:n substraatti) kanssa suurensi feksofenadiinin AUC-arvoa 56 % (90 %:n luottamusväli 35, 79) ja Cmax-arvoa 76 % (90 %:n luottamusväli 49, 108) kerta-annoksen antamisen jälkeen sekä AUC-arvoa 27 % (90 %:n luottamusväli 11, 46) ja Cmax-arvoa 25 % (90 %:n luottamusväli 6, 48) vakaassa tilassa. Potilaita, jotka käyttävät samanaikaisesti lääkkeitä, jotka ovat osittain riippuvaisia P‑gp:stä ja joilla on kapea terapeuttinen indeksi, kuten digoksiinia, dabigatraania tai aliskireenia, on seurattava huolellisesti samanaikaisesti käytettävän lääkkeen siedettävyyden muuttumiseen viittaavien merkkien varalta, sillä altistus saattaa suurentua TAGRISSO-hoidon aikana (ks. kohta Farmakokinetiikka).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisy miehillä ja naisilla
Naisia, jotka voivat tulla raskaaksi, on kehotettava välttämään raskaaksi tulemista TAGRISSO‑hoidon aikana. Potilaita kehotetaan käyttämään tehokasta ehkäisyä tämän lääkehoidon lopettamisen jälkeen seuraavasti: naiset vähintään kaksi kuukautta ja miehet vähintään 4 kuukautta. Riskiä hormonaalisten ehkäisyvalmisteiden altistuksen pienenemiselle ei voida sulkea pois.
Raskaus
Osimertinibin käytöstä raskaana oleville naisille ei ole olemassa tietoja tai on vain vähän tietoja. Eläimillä tehdyissä tutkimuksissa on havaittu lisääntymistoksisuutta (alkiokuolleisuutta, sikiön kasvun hidastumista ja vastasyntyneiden kuolemia, ks. kohta Prekliiniset tiedot turvallisuudesta). Vaikutusmekanismiin ja prekliinisiin tietoihin perustuen osimertinibi saattaa vahingoittaa sikiötä, jos sitä annetaan raskaana olevalle naiselle. TAGRISSO-valmistetta ei pidä käyttää raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa osimertinibilla.
Imetys
Ei tiedetä, erittyvätkö osimertinibi ja/tai sen metaboliitit ihmisillä äidinmaitoon. Ei ole riittävästi tietoa osimertinibin ja/tai sen metaboliittien erittymisestä maitoon koe-eläimillä. Osimertinibia ja sen metaboliitteja on kuitenkin havaittu imeväisikäisissä poikasissa, ja poikasten heikentynyttä kasvua ja eloonjäämistä on todettu (ks. kohta Prekliiniset tiedot turvallisuudesta). Imetettävään vauvaan kohdistuvia riskejä ei voida poissulkea. Imetys on lopetettava TAGRISSO-hoidon ajaksi.
Hedelmällisyys
TAGRISSO-valmisteen vaikutuksesta ihmisen hedelmällisyyteen ei ole tietoa. Eläinkokeet ovat osoittaneet, että osimertinibi vaikuttaa urosten ja naaraiden lisääntymiselimiin ja saattaa heikentää hedelmällisyyttä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
TAGRISSO‑valmisteella ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Tutkimukset EGFR-mutaatiopositiivisilla NSCLC-potilailla
Tiedot monoterapiana käytetyn TAGRISSO-valmisteen turvallisuudesta perustuvat yhdistettyihin tietoihin 1 956 potilaasta, joilla oli EGFR-mutaatiopositiivinen ei-pienisoluinen keuhkosyöpä. Nämä potilaat saivat TAGRISSO-valmistetta 80 mg:n vuorokausiannoksella viidessä satunnaistetussa faasin 3 tutkimuksessa (ADAURA, liitännäishoito; FLAURA ja FLAURA2 (monoterapiahaara), ensilinjan hoito; LAURA (platinapohjaisen kemosädehoidon jälkeinen hoito) ja AURA3, ainoastaan toisen linjan hoito), kahdessa faasin 2 yksihaaraisessa tutkimuksessa (AURAex ja AURA2, toisen linjan tai sitä myöhemmän linjan hoito) ja yhdessä faasin 1 tutkimuksessa (AURA1, ensilinjan tai sitä myöhemmän linjan hoito) (ks. kohta Farmakodynamiikka). Suurin osa haittavaikutuksista oli vaikeusastetta 1 tai 2. Yleisimmin ilmoitetut lääkkeen haittavaikutukset olivat ripuli (46 %), ihottuma (45 %), kynsivallintulehdus (33 %), kuiva iho (31 %) ja suutulehdus (23 %). Näissä tutkimuksissa vaikeusasteen 3 haittavaikutuksia oli 11 % ja vaikeusasteen 4 haittavaikutuksia 0,2 %. Niiden potilaiden joukosta, jotka saivat 80 mg TAGRISSO-valmistetta kerran vuorokaudessa, annosta pienennettiin 3,4 %:lla potilaista haittavaikutusten vuoksi. Haittavaikutusten takia hoito keskeytettiin 5,5 %:lla potilaista.
Tiedot TAGRISSO-valmisteen turvallisuudesta, kun sitä annetaan yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, perustuvat tietoihin 276 potilaasta, joilla oli EGFR-mutaatiopositiivinen ei-pienisoluinen keuhkosyöpä, ja ne vastasivat TAGRISSO-monoterapian turvallisuustietoja sekä pemetreksedin ja platinapohjaisen solunsalpaajahoidon tunnettuja turvallisuusprofiileja. Yleisimmin ilmoitetut lääkkeen haittavaikutukset, kun TAGRISSO-valmistetta annettiin yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, olivat ihottuma (49 %), ripuli (43 %), ruokahalun huonontuminen (31 %), suutulehdus (31 %), kynsivallintulehdus (27 %) ja kuiva iho (24 %). Kun TAGRISSO-valmistetta annetaan yhdistelmähoitona, tutustu kyseiseen yhdistelmähoitoon kuuluvien lääkkeiden valmisteyhteenvetoihin ennen hoidon aloittamista.
Potilaat, joilla oli aiemmin ollut interstitiaalinen keuhkosairaus (ILD), lääkkeen aiheuttama ILD tai steroidihoitoa edellyttänyt sädepneumoniitti tai joilla havaittiin viitteitä kliinisesti aktiivisesta ILD:stä, suljettiin pois kliinisistä tutkimuksista. Potilaat, joilla oli kliinisesti merkittäviä rytmi‑ ja johtumishäiriöitä levossa otetussa elektrokardiogrammissa (EKG) (esimerkiksi QTc‑aika yli 470 ms), suljettiin pois näistä tutkimuksista. Potilaiden vasemman kammion ejektiofraktio arvioitiin tutkimuksen sisäänottovaiheessa ja 12 viikon välein sen jälkeen.
Haittavaikutustaulukko
Taulukossa 2 haittavaikutukset on mahdollisuuksien mukaan luokiteltu esiintymistiheyden mukaan sen perusteella, mikä vastaavien haittatapahtumaraporttien ilmaantuvuus oli yhdistetyissä tutkimustiedoissa. Tutkimukset, joista tieto on kerätty, koskevat kaiken kaikkiaan 1 956:a EGFR-mutaatiopositiivista NSCLC-potilasta, jotka ovat saaneet TAGRISSO-monoterapiaa 80 mg:n vuorokausiannoksella ADAURA-, FLAURA‑, FLAURA2‑, LAURA‑, AURA3‑, AURAex‑, AURA2- ja AURA1-tutkimuksissa, sekä 276:ta potilasta, jotka ovat saaneet TAGRISSO-valmistetta yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa FLAURA2-tutkimuksessa.
Haittavaikutukset on lueteltu MedDRA:n elinjärjestelmäluokituksen (SOC) mukaan. Kunkin elinjärjestelmän haittavaikutukset on lueteltu esiintymistiheyden mukaan yleisimmistä alkaen. Haittavaikutukset on esitetty kussakin yleisyysluokassa haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä. Lisäksi kukin haittavaikutus on luokiteltu esiintymistiheyden mukaan käyttämällä CIOMS III ‑luokitusta seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 2: ADAURA-, LAURA-, FLAURA-, FLAURA2- ja AURA-tutkimuksissa ilmoitetut haittavaikutukset
MedDRA-elinjärjestelmä ja MedDRA-termi | TAGRISSOa | TAGRISSO pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssab | ||
CIOMS-yleisyysluokat/ kokonaisesiintymistiheys (kaikki CTCAE-asteet)c | CTCAE-asteen 3 tai tätä vaikeampien haittavaikutusten esiintymistiheysc | CIOMS-yleisyysluokat / kokonaisesiintymistiheys (kaikki CTCAE-asteet)c | CTCAE-asteen 3 tai tätä vaikeampien haittavaikutusten esiintymistiheysc | |
Infektiot | ||||
Hepatiitti B:n uudelleenaktivoituminend | Tuntematon | 0 % | 0 % | |
Veri ja imukudos | ||||
Aplastinen anemia | Harvinainen (0,05 %) | 0,05 % | 0 % | 0 % |
Trombosytopenia | Yleinen (7 %) | 0,6 % | Hyvin yleinen (18,5 %) | 6,9 % |
Neutropenia | Yleinen (6 %) | 0,9 % | Hyvin yleinen (24,6 %) | 13,4 % |
Leukopenia | Yleinen (5 %) | 0,4 % | Hyvin yleinen (12,7 %) | 2,9 % |
Lymfopenia | Yleinen (1,6 %) | 0,3 % | Yleinen (2,5 %) | 1,1 % |
Aineenvaihdunta ja ravitsemus | ||||
Ruokahalun huonontuminen | Hyvin yleinen (19 %) | 1,1 % | Hyvin yleinen (31 %) | 2,9 % |
Silmät | ||||
Sarveiskalvotulehduse | Melko harvinainen (0,6 %) | 0,05 % | Melko harvinainen (0,7 %) | 0 % |
Sydän | ||||
Sydämen vajaatoiminta | Melko harvinainen (0,5 %) | 0,2 % | Yleinen (1,8 %) | 1,1 %f |
Hengityselimet, rintakehä ja välikarsina | ||||
Nenäverenvuoto | Yleinen (6 %) | 0 % | Yleinen (7 %) | 0,4 % |
Interstitiaalinen keuhkosairaus | Yleinen (4,3 %)g | 1,4 %h | Yleinen (3,3 %)i | 0,7 %j |
Sädepneumoniittik | Yleinen (4 %) | 0,2 % | 0 % | 0 % |
Eosinofiilinen pneumoniad | Tuntematon | 0 % | 0 % | |
Ruoansulatuselimistö | ||||
Ripuli | Hyvin yleinen (46 %) | 1,5 % | Hyvin yleinen (43 %) | 2,9 % |
Suutulehdusl | Hyvin yleinen (23 %) | 0,4 % | Hyvin yleinen (31 %) | 0,4 % |
Iho ja ihonalainen kudos | ||||
Ihottumam | Hyvin yleinen (45 %) | 0,8 % | Hyvin yleinen (49 %) | 2,5 % |
Kynsivallintulehdusn | Hyvin yleinen (33 %) | 0,4 % | Hyvin yleinen (27 %) | 0,7 % |
Kuiva ihoo | Hyvin yleinen (31 %) | 0,2 % | Hyvin yleinen (24 %) | 0 % |
Kutinap | Hyvin yleinen (17 %) | 0,05 % | Yleinen (8 %) | 0 % |
Hiustenlähtö | Yleinen (5 %) | 0 % | Yleinen (9 %) | 0 % |
Käsi-jalkaoireyhtymä | Yleinen (1,9 %) | 0 % | Yleinen (5 %) | 0 % |
Nokkosihottuma | Yleinen (1,9 %) | 0,1 % | Yleinen (1,4 %) | 0,4 % |
Ihon hyperpigmentaatioq | Melko harvinainen (0,9 %) | 0 % | Yleinen (2,5 %) | 0 % |
Erythema multiformer | Melko harvinainen (0,3 %) | 0 % | Yleinen (1,4 %) | 0,7 % |
Ihovaskuliittis | Melko harvinainen (0,2 %) | 0 % | 0 % | |
Stevens–Johnsonin oireyhtymät | Harvinainen (0,02 %) | 0 % | 0 % | |
Toksinen epidermaalinen nekrolyysid | Tuntematon | 0 % | 0 % | |
Tutkimukset | ||||
Pienentynyt vasemman kammion ejektiofraktiou,v | Yleinen (4,1 %) | Yleinen (8,0 %) | ||
Suurentunut veren kreatiinifosfokinaasipitoisuus | Yleinen (2 %) | 0,4 % | Yleinen (3,3 %) | 1,1 % |
QTc-ajan piteneminenw | Yleinen (1,1 %) | Yleinen (1,8 %) | ||
Tutkimukset (koetuloksiin perustuvat löydökset CTCAE-asteiden muutoksina) | ||||
Alentunut leukosyyttien määräu | Hyvin yleinen (65 %) | 1,9 % | Hyvin yleinen (88 %) | 20 % |
Alentunut lymfosyyttien määräu | Hyvin yleinen (64 %) | 8 % | Hyvin yleinen (78 %) | 16 % |
Alentunut verihiutaleiden määräu | Hyvin yleinen (53 %) | 1,3 % | Hyvin yleinen (85 %) | 16 % |
Alentunut neutrofiilien määräu | Hyvin yleinen (36 %) | 4,0 % | Hyvin yleinen (85 %) | 36 % |
Suurentunut veren kreatiniinipitoisuus | Yleinen (5,6 %) | 0,05 % | Hyvin yleinen (22 %) | 0,4 % |
Luusto, lihakset ja sidekudos | ||||
Lihastulehdus | Melko harvinainen (0,2 %) | 0 % | 0 % | 0 % |
a Tiedot ovat yhdistettyjä tietoja ADAURA-, FLAURA‑, FLAURA2- (monoterapiahaara), LAURA‑ ja AURA-tutkimuksista (AURA3, AURAex, AURA2 ja AURA1); yhteenvedossa ovat mukana tapahtumat ainoastaan potilailla, jotka ovat saaneet vähintään yhden annoksen TAGRISSO‑valmistetta satunnaistettuna hoitona.
b Tiedot ovat peräisin FLAURA2-tutkimuksen yhdistelmähoitohaarasta; yhteenvedossa ovat mukana tapahtumat ainoastaan potilailla, jotka ovat saaneet vähintään yhden annoksen tutkimuksessa käytettyä yhdistelmää (TAGRISSO, pemetreksedi ja sisplatiini tai karboplatiini) satunnaistettuna hoitona. Tutkimushoidon keston mediaani oli 22,3 kuukautta hoitohaarassa, jossa potilaat saivat TAGRISSO-valmistetta pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa.
c National Cancer Instituten (NCI) haittavaikutusten CTCAE‑luokitus (Common Terminology Criteria for Adverse Events), versio 5.0.
d Raportoitu markkinoilletulon jälkeisessä käytössä.
e Sisältää: sarveiskalvon epiteelin vioittuma, sarveiskalvon eroosio, sarveiskalvotulehdus, pisteinen sarveiskalvotulehdus.
f Kaksi CTCAE-luokituksen asteen 5 tapahtumaa (jotka johtivat kuolemaan) ilmoitettiin.
g Sisältää: interstitiaalinen keuhkosairaus (1,8 %), keuhkotulehdus (2,2 %), keuhkofibroosi (0,2 %), organisoituva keuhkokuume (0,1 %).
h Kahdeksan CTCAE-luokituksen asteen 5 tapahtumaa (jotka johtivat kuolemaan) ilmoitettiin.
i Sisältää: interstitiaalinen keuhkosairaus (1,8 %), keuhkotulehdus (1,1 %), organisoituva keuhkokuume (0,4 %).
j Yksi CTCAE-luokituksen asteen 5 tapahtuma (joka johti kuolemaan) ilmoitettiin.
k Sisältää: keuhkojen sädefibroosi (0,05 %).
l Sisältää: suun haavaumat, suutulehdus.
m Sisältää: akne, ihotulehdus, aknetyyppinen ihottuma, lääkeaineihottuma, ihon punoitus, karvatupen tulehdus, märkärakkula, ihottuma, punoittava ihottuma, follikulaarinen ihottuma, täpläinen ihottuma, täpläinen ja näppyläinen ihottuma, näppyläinen ihottuma, märkärakkulainen ihottuma, kutiava ihottuma, rakkulainen ihottuma, ihon pinnallinen haavauma.
n Sisältää: kynsipedin sairaudet, kynsipedin infektio, kynsipedin tulehdus, kynsien värjäytyminen, kynsisairaus, kynsien surkastuminen, kynsitulehdus, kynsien pigmentaatio, harjanteet kynsissä, kynsitoksisuus, kynsikipu, kynsien lohkeilu, kynnen irtoaminen, kaikkien kynsien irtoaminen, kynsien pehmeneminen, kynsivallintulehdus.
o Sisältää: kuiva iho, ekseema, ihon halkeamat, kseroderma, kseroosi.
p Sisältää: silmäluomien kutina, kutina.
q Erythema dyschromicum perstans -tapauksia on raportoitu markkinoilletulon jälkeen.
r ADAURA-, FLAURA‑, FLAURA2- (monoterapiahaara), LAURA‑ ja AURA-tutkimuksissa kuusi potilasta 1 956 potilaasta ilmoitti erythema multiformea. Markkinoilletulon jälkeen on myös raportoitu erythema multiformea, mukaan lukien 7 raporttia markkinoilletulon jälkeisestä seurantatutkimuksesta (N = 3 578).
s Arvioitu esiintymistiheys. Piste-estimaatin 95 %:n luottamusvälin yläraja on 3 / 1 956 (0,4 %).
t Yksi tapahtuma raportoitiin markkinoilletulon jälkeisessä tutkimuksessa, ja esiintymistiheys perustuu ADAURA-, FLAURA‑, FLAURA2- (monoterapiahaara), LAURA‑ ja AURA-tutkimuksiin ja markkinoilletulon jälkeiseen tutkimukseen (N = 5 534).
u Kuvaa laboratoriolöydösten, ei ilmoitettujen haittavaikutusten, ilmaantuvuutta.
v Kuvaa pienenemistä vähintään 10 prosenttiyksiköllä ja laskua alle 50 %:iin.
w Kuvaa niiden potilaiden ilmaantuvuutta, joilla QTcF-aika on pidentynyt yli 500 ms:iin.
Valikoitujen haittavaikutusten kuvaus
Interstitiaalinen keuhkosairaus (ILD)
ADAURA-, FLAURA-, FLAURA2- (monoterapiahaara) AURA‑ ja LAURA-tutkimuksissa ILD:tä tai ILD:n kaltaisia haittavaikutuksia ilmoitettiin 4,3 %:lla 1 956 potilaasta. Kahdeksan kuolemaan johtanutta tapausta ilmoitettiin. Liitännäishoitoasetelmassa ei ilmoitettu kuolemaan johtaneita tapauksia. ILD:n esiintyvyys oli 10,4 % etniseltä taustaltaan japanilaisilla potilailla, 2,8 % etniseltä taustaltaan aasialaisilla, mutta ei japanilaisilla potilailla ja 3,2 % muilla kuin aasialaisilla potilailla. Mediaaniaika ensimmäisen annoksen saamisesta ILD:n tai ILD:n kaltaisten haittavaikutusten puhkeamiseen oli 85 päivää (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
ILD:tä tai ILD:n kaltaisia haittavaikutuksia ilmoitettiin 7,7 %:lla 143 potilaasta, jotka saivat LAURA-tutkimuksessa TAGRISSO-valmistetta definitiivisen platinapohjaisen kemosädehoidon jälkeen. 0,7 %:lla potilaista (n = 1) tällaiset haittavaikutukset johtivat kuolemaan. ILD:n ilmaantuvuus oli etniseltä taustaltaan aasialaisilla mutta ei japanilaisilla potilailla 6,6 % ja muilla kuin aasialaisilla potilailla 17,2 %. Yhdelläkään japanilaisella potilaalla ei ilmennyt ILD-tapahtumaa. Mediaaniaika ensimmäisen annoksen saamisesta ILD:n tai ILD:n kaltaisten haittavaikutusten puhkeamiseen oli 1,9 kuukautta. Mediaaniaika viimeisen sädehoitoannoksen saamisesta ILD:n tai ILD:n kaltaisten haittavaikutusten puhkeamiseen oli 3,0 kuukautta.
ILD:tä tai ILD:n kaltaisia haittavaikutuksia ilmoitettiin 3,3 %:lla 276 potilaasta, jotka saivat FLAURA2-tutkimuksessa TAGRISSO-valmistetta yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa. 0,4 %:lla potilaista (n = 1) tällaiset haittavaikutukset johtivat kuolemaan. ILD:n ilmaantuvuus oli japanilaisilla potilailla 14,9 % ja muilla kuin aasialaisilla potilailla 1,7 %. Yhdelläkään etniseltä taustaltaan aasialaisella mutta ei japanilaisella potilaalla ei ilmennyt ILD-tapahtumaa FLAURA2-tutkimuksen yhdistelmähoitohaarassa. Mediaaniaika ensimmäisen annoksen saamisesta ILD:n tai ILD:n kaltaisten haittavaikutusten puhkeamiseen oli 161 päivää.
Sädepneumoniitti
LAURA-tutkimuksessa definitiivisen platinapohjaisen kemosädehoidon jälkeen sädepneumoniittia ilmoitettiin 48 %:lla TAGRISSO-hoitoa saaneista 143 potilaasta ja 38 %:lla lumelääkettä saaneista 73 potilaasta. Kolmella potilaalla (2,1 %) ilmeni asteen 3 tapahtumia, kaikki TAGRISSO-haarassa. Yhtään asteen 4 tai 5 tapahtumaa ei ilmoitettu kummassakaan haarassa. Mediaaniaika ensimmäisen annoksen saamisesta sädepneumoniitin puhkeamiseen oli TAGRISSO-haarassa 1,7 kuukautta ja lumelääkehaarassa 1,8 kuukautta. Mediaaniaika viimeisen sädehoitoannoksen saamisesta sädepneumoniitin puhkeamiseen oli TAGRISSO-haarassa 2,5 kuukautta ja lumelääkehaarassa 2,6 kuukautta.
QTc-ajan piteneminen
ADAURA-, FLAURA-, FLAURA2-, LAURA- ja AURA-tutkimuksiin osallistuneista 1 956 potilaasta, jotka saivat TAGRISSO-monoterapiaa (80 mg), 1,1 %:lla potilaista (n = 21) todettu QTc-aika oli yli 500 ms, ja 4 %:lla potilaista (n = 79) QTc‑aika pidentyi enemmän kuin 60 ms lähtötasosta. TAGRISSO‑valmisteen farmakokineettinen ja farmakodynaaminen analyysi ennusti pitoisuudesta riippuvaisen QTc‑ajan pitenemisen. ADAURA-, LAURA-, FLAURA-, FLAURA2- tai AURA-tutkimuksissa ei ilmoitettu QTc‑ajan pitenemiseen liittyviä rytmihäiriöitä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka). Kun tarkasteltiin potilaita, jotka saivat TAGRISSO-valmistetta yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, niiden potilaiden osuus, joilla QTc-aika piteni yli 500 ms:iin ja enemmän kuin 60 ms lähtötasosta, oli pieni (1,8 %) ja vastaavanlainen kuin monoterapian yhteydessä (1,5 %).
Vaikutukset ruoansulatuselimistöön
ADAURA-, FLAURA-, FLAURA2-, LAURA‑ ja AURA-tutkimuksissa (TAGRISSO-monoterapia; N = 1 956) 46 %:lla potilaista ilmoitettiin ripulia, joista 36 % oli 1. vaikeusasteen, 8,1 % 2. asteen ja 1,5 % 3. asteen tapahtumia. Yhtään asteen 4 tai 5 haittatapahtumaa ei raportoitu. Annosta jouduttiin pienentämään 0,6 %:lla potilaista ja hoitoa tauottamaan 1,9 %:lla potilaista. Neljässä tapauksessa (0,2 %) tämä johti hoidon keskeyttämiseen. ADAURA-tutkimuksessa mediaaniaika haittavaikutuksen ilmaantumiseen oli 22 päivää, FLAURA-tutkimuksessa 19 päivää, FLAURA2-tutkimuksessa (monoterapiahaara) 22 päivää ja AURA3-tutkimuksessa 22 päivää. Asteen 2 haittatapahtuman keston mediaani oli ADAURA-tutkimuksessa 11 päivää, FLAURA-tutkimuksessa 19 päivää, FLAURA2-tutkimuksessa (monoterapiahaara) 17 päivää ja AURA3-tutkimuksessa 6 päivää. Kun tarkasteltiin potilaita, jotka saivat TAGRISSO-valmistetta yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, ripulia ilmoitettiin 43 %:lla potilaista, kun monoterapiaa saaneilla potilailla vastaava osuus oli 41 %. Useimmat näistä ripulitapahtumista olivat asteen 1 ja asteen 2 haittatapahtumia.
Hematologiset tapahtumat
TAGRISSO-valmistetta saaneilla potilailla leukosyyttien, lymfosyyttien, neutrofiilien ja verihiutaleiden laboratorioarvojen mediaanien havaittiin alenevan varhaisessa vaiheessa. Arvot vakiintuivat ajan myötä ja pysyivät sitten viitealueen alarajan yläpuolella. Haittatapahtumina ilmoitettiin leukopeniaa, lymfopeniaa, neutropeniaa ja trombosytopeniaa. Suurin osa näistä tapahtumista oli vaikeusasteeltaan lieviä tai kohtalaisia, eivätkä ne johtaneet hoidon keskeyttämiseen. TAGRISSO-hoidon yhteydessä on ilmoitettu harvinaisina tapauksina aplastista anemiaa, ja jotkin tapahtumat ovat johtaneet kuolemaan. TAGRISSO-hoito on lopetettava, jos potilaalla on varmistettu aplastinen anemia (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Iäkkäät
ADAURA-, FLAURA-, FLAURA2-, LAURA- ja AURA3‑tutkimuksissa (TAGRISSO-monoterapia; n = 1 956) 43 % potilaista oli vähintään 65‑vuotiaita ja 11 % oli vähintään 75‑vuotiaita. Nuorempiin potilaisiin (alle 65‑vuotiaisiin) verrattuna vähintään 65-vuotiaat raportoivat enemmän tutkimuslääkkeen annoksen muuttamiseen (hoidon keskeyttämiseen tai annoksen pienentämiseen) johtaneita haittavaikutuksia (vähintään 65-vuotiaat: 18 %, alle 65-vuotiaat: 13 %). Ilmoitetut haittatapahtumat olivat tyypiltään samankaltaisia iästä riippumatta. Vanhemmat potilaat raportoivat enemmän vähintään asteen 3 haittavaikutuksia (14 %) verrattuna nuorempiin potilaisiin (9 %). Hoidon tehossa ei kaiken kaikkiaan havaittu eroa näiden vanhempien ja nuorempien potilaiden välillä. Nämä tulokset ovat linjassa faasin 2 AURA-tutkimusten turvallisuutta ja tehoa koskevien tulosten kanssa. 276 potilaasta, jotka saivat TAGRISSO-valmistetta yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, 104 potilasta oli vähintään 65‑vuotiaita ja 23 potilasta oli vähintään 75‑vuotiaita. Vanhemmat (vähintään 65‑vuotiaat) potilaat raportoivat saman verran vähintään asteen 3 haittavaikutuksia (36 %) verrattuna alle 65‑vuotiaisiin potilaisiin (36 %). Annoksen muuttamista haittavaikutusten vuoksi raportoitiin suuremmalla osalla vähintään 65‑vuotiaista potilaista (34 %) verrattuna alle 65‑vuotiaisiin (20 %).
Pienipainoiset
Potilailla, jotka saivat TAGRISSO-monoterapiaa (80 mg; N = 1 956) ja joiden paino oli pieni (alle 50 kg), ilmoitettiin enemmän vähintään asteen 3 haittavaikutuksia (20 %:lla) ja QTc-ajan pidentymistä (12 %:lla) kuin potilailla, joiden paino oli suurempi (vähintään 50 kg) (vähintään asteen 3 haittavaikutuksia 10 %:lla ja QTc-ajan pidentymistä 6 %:lla). Potilailla, jotka saivat TAGRISSO-valmistetta yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa ja joiden paino oli pieni (alle 50 kg), ilmoitettiin vähintään asteen 3 haittavaikutuksia samankaltaisilla esiintymistiheyksillä (32 %) kuin potilailla, joiden paino oli suurempi (vähintään 50 kg) (37 %). Sitä vastoin kuivaa ihoa ja suutulehdusta raportoitiin suuremmilla esiintymistiheyksillä potilailla, joiden paino oli pieni (alle 50 kg) (kuivaa ihoa 34 %:lla ja suutulehdusta 40 %:lla), kuin potilailla, joiden paino oli suurempi (vähintään 50 kg) (kuivaa ihoa 22 %:lla ja suutulehdusta 30 %:lla).
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
TAGRISSO-valmisteen kliinisissä tutkimuksissa pienelle määrälle potilaita annettiin korkeintaan 240 mg:n vuorokausiannoksia, eikä annosta rajoittavaa toksisuutta havaittu. Näissä tutkimuksissa potilailla, jotka saivat TAGRISSO‑valmistetta 160 mg:n ja 240 mg:n vuorokausiannoksella, ilmeni useita tyypillisiä EGFR-TKI:n aiheuttamia haittavaikutuksia (pääasiassa ripulia ja ihottumaa) useammin ja vaikeampina kuin 80 mg:n annosta saaneilla. Vahingossa tapahtuneista yliannostuksista ihmisillä on vain vähän tietoa. Kaikki olivat yksittäisiä tapauksia, joissa potilas oli erehdyksessä ottanut ylimääräisen TAGRISSO‑valmisteen vuorokausiannoksen, eikä niillä ollut kliinisiä seurauksia.
TAGRISSO‑valmisteen yliannostukseen ei ole olemassa erityistä hoitoa. Epäiltäessä yliannostusta TAGRISSO‑hoito on keskeytettävä ja aloitettava oireiden mukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, proteiinikinaasin estäjät. ATC-koodi: L01EB04
Vaikutusmekanismi
Osimertinibi on tyrosiinikinaasin inhibiittori (TKI). Se estää irreversiibelisti sellaisia EGFR:iä, joissa on herkistäviä mutaatioita (EGFRm) ja TKI-resistenssimutaatio T790M.
Farmakodynaamiset vaikutukset
In vitro ‑tutkimuksissa on osoitettu, että osimertinibilla on suuri teho ja kyky inhiboida EGFR:ää (näennäinen IC50 6–54 nM fosfo-EGFR:ää vastaan) kaikissa kliinisesti merkittävissä NSCLC-solulinjoissa, joissa on herkistävä EGFR‑mutaatio ja T790M‑mutaatio. Tämä johtaa solujen kasvun estymiseen, kun taas vaikutus villityypin EGFR:ää vastaan solulinjoissa on huomattavasti pienempi (näennäinen IC50 480 nM – 1,8 μM fosfo-EGFR:ää vastaan). In vivo ‑tutkimuksissa osimertinibin antaminen suun kautta pienensi kasvainta EGFRm‑ ja T790M‑NSCLC‑vieraslajisiirre- ja siirtogeenisten hiirten keuhkokasvainmalleissa.
Sydämen elektrofysiologia
TAGRISSO‑valmisteen QTc‑aikaa pidentävää vaikutusta arvioitiin 210 potilaalla, jotka saivat AURA2‑tutkimuksessa 80 mg osimertinibia vuorokaudessa. Osimertinibin vaikutusta QTc‑aikoihin arvioitiin tekemällä sarja EKG-tutkimuksia kerta-annoksen jälkeen sekä vakaassa tilassa. Farmakokineettisen ja farmakodynaamisen analyysin perusteella lääkkeen 80 mg:n annokseen liittyvä ennustettu QTc-ajan piteneminen on 14 ms, ylärajan ollessa 16 ms (90 %:n luottamusväli).
Kliininen teho ja turvallisuus
EGFR-mutaatiopositiivisen ei-pienisoluisen keuhkosyövän (NSCLC) liitännäishoito potilailla, joista osa oli saanut aiemmin liitännäissolunsalpaajahoitoa – ADAURA
TAGRISSO-valmisteen teho ja turvallisuus EGFR-mutaatiopositiivisen (Ex19del tai L858R) ei-pienisoluisen keuhkosyövän liitännäishoitona potilailla, joille oli tehty täydellinen kasvaimen resektio ja mahdollisesti annettu myös aiempaa liitännäissolunsalpaajahoitoa, osoitettiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa (ADAURA).
Tutkimukseen soveltuvilla potilailla oli oltava leikattavissa oleva levinneisyysasteen IB–IIIA kasvain (American Joint Commission on Cancer ‑organisaation [AJCC] 7. painoksen mukaisesti) ja EGFR-mutaatio (Ex19del tai L858R), joka oli todettu prospektiivisesti keskuslaboratoriossa Cobas-EGFR-mutaatiotestillä joko biopsianäytteestä tai leikkauspreparaatista.
Potilaat satunnaistettiin suhteessa 1:1 saamaan TAGRISSO-valmistetta (n = 339, 80 mg suun kautta kerran vuorokaudessa) tai lumelääkettä (n = 343) heidän toivuttuaan leikkauksesta ja tavanomaisesta liitännäissolunsalpaajahoidosta, jos sitä oli annettu. Potilaat, jotka eivät saaneet solunsalpaajia liitännäishoitona, satunnaistettiin 10 viikon kuluessa leikkauksesta ja potilaat, jotka saivat solunsalpaajia liitännäishoitona, satunnaistettiin 26 viikon kuluessa leikkauksesta. Satunnaistamisessa käytettiin stratifikaatiotekijöinä EGFR-mutaation tyyppiä (Ex19del tai L858R), etnistä taustaa (aasialainen tai muu kuin aasialainen) ja levinneisyysastetta, joka määriteltiin pTNM-luokituksella (pathological tumor-node-metastasis; levinneisyysaste IB tai II tai IIIA) AJCC:n 7. painoksen perusteella. Hoitoa jatkettiin, kunnes tauti uusiutui, potilaalla ilmeni toksisuutta, jota ei voitu hyväksyä, tai 3 vuotta täyttyi.
Tärkein tehoa kuvaava päätetapahtuma oli tutkijalääkärin arvioima tautivapaa elossaoloaika (DFS) potilailla, joiden syövän levinneisyysaste oli II–IIIA. Tehoa kuvaavana päätetapahtumana oli myös tutkijalääkärin arvioima DFS potilailla, joiden syövän levinneisyysaste oli IB–IIIA (koko populaatio). Muita tehoa kuvaavia päätetapahtumia olivat tautivapaan elossaolon osuus, kokonaiselossaoloaika (OS), kokonaiselossaolo-osuus ja aika terveyteen liittyvän elämänlaadun (HRQoL) huononemiseen SF-36-mittarilla mitattuna.
Koko tutkimuspopulaation demografiset tiedot ja tautitiedot lähtötilanteessa olivat: mediaani-ikä 63 vuotta (vaihteluväli 30–86 vuotta), vähintään 75-vuotiaita 11 %, naisia 70 % ja aasialaisia 64 %. 72 % potilaista ei ollut koskaan tupakoinut. WHO:n mukainen suorituskykyluokitus oli 0 (64 %:lla potilaista) tai 1 (36 %:lla). Levinneisyysaste oli 31 %:lla IB, 34 %:lla II ja 35 %:lla IIIA. Mitä tulee EGFR-mutaatiostatukseen, 55 %:lla oli eksonin 19 deleetio ja 45 %:lla oli eksonin 21 L858R-substituutiomutaatio; 9 potilaalla (1 %) oli samanaikaisesti myös de novo T790M-mutaatio. Useimmat potilaat (60 %) saivat liitännäishoitona solunsalpaajia ennen satunnaistamista (IB: 26 %; IIA: 71 %; IIB: 73 %; IIIA: 80 %). DFS-analyysin ajankohtana 205 potilasta (61 %) sai edelleen aktiivista hoitoa; niistä 73 potilaasta (11 %), joilla oli mahdollisuus suorittaa 3 vuoden hoitojakso loppuun, 40 potilasta kuului osimertinibihaaraan (12 %) ja 33 potilasta lumehaaraan (10 %).
Tauti uusiutui 37 potilaalla TAGRISSO-hoidon aikana. Yleisimmin ilmoitettuja uusiutumispaikkoja olivat keuhkot (19 potilasta), imusolmukkeet (10 potilasta) ja keskushermosto (5 potilasta). Tauti uusiutui 157 potilaalla lumehoidon aikana. Yleisimmin ilmoitettuja uusiutumispaikkoja olivat keuhkot (61 potilasta), imusolmukkeet (48 potilasta) ja keskushermosto (34 potilasta).
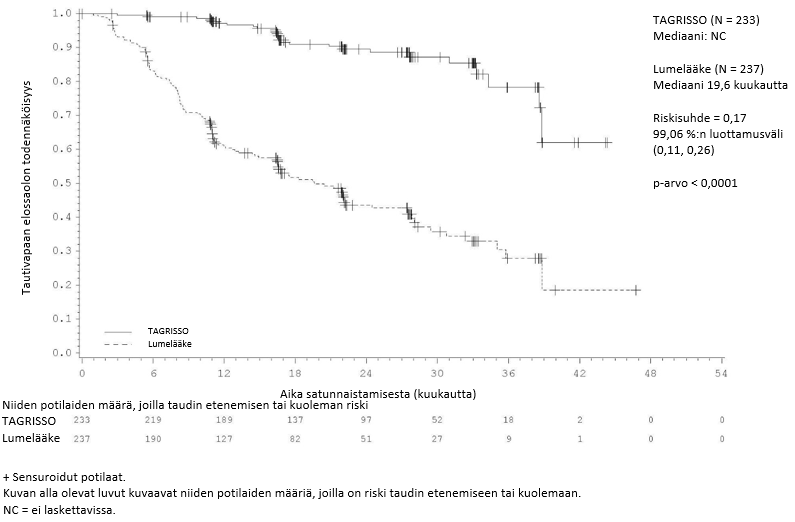
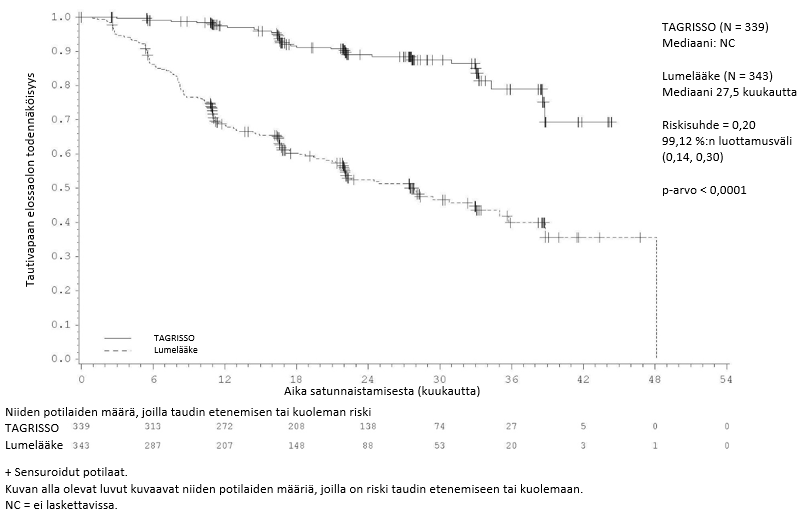
ADAURA-tutkimuksessa todettiin, että taudin uusiutumisen tai kuoleman riski oli TAGRISSO-haarassa tilastollisesti merkitsevästi pienempi kuin lumehaarassa, kun syövän levinneisyysaste oli II–IIIA. Vastaavia tuloksia todettiin potilailla, joiden syövän levinneisyysaste oli IB–IIIA.
Taulukossa 3 esitetään yhteenveto ADAURA-tutkimuksen tehoa koskevista tuloksista tutkijalääkärin arvion mukaisesti.
Taulukko 3. ADAURA-tutkimuksen tehoa koskevat tulokset (tutkijalääkärin arvio)
| Potilaat, joiden syövän levinneisyysaste oli II–IIIA | Potilaat, joiden syövän levinneisyysaste oli IB–IIIA | ||
Tehoa kuvaava muuttuja | TAGRISSO | Lumelääke | TAGRISSO | Lumelääke |
Tautivapaa elossaoloaika (DFS) | ||||
Tapahtumien lukumäärä (%) | 26 (11) | 130 (55) | 37 (11) | 159 (46) |
Uusiutunut tauti (%) | 26 (11) | 129 (54) | 37 (11) | 157 (46) |
Kuolemat (%) | 0 | 1 (0,4) | 0 | 2 (0,6) |
Tautivapaan elossaolon (DFS) mediaani, kuukautta (95 %:n luottamusväli) | NC (38,8, NC) | 19,6 (16,6, 24,5) | NC (NC, NC) | 27,5 (22,0, 35,0) |
HR (99,06 %:n luottamusväli); p-arvo | 0,17 (0,11, 0,26); p < 0,0001a | 0,20 (0,14, 0,30); p < 0,0001b | ||
Tautivapaan elossaolon (DFS) osuus 12 kuukauden kohdalla (%) (95 %:n luottamusväli) | 97 (94, 99) | 61 (54, 67) | 97 (95, 99) | 69 (63, 73) |
Tautivapaan elossaolon (DFS) osuus 24 kuukauden kohdalla (%) (95 %:n luottamusväli) | 90 (84, 93) | 44 (37, 51) | 89 (85, 92) | 52 (46, 58) |
Tautivapaan elossaolon (DFS) osuus 36 kuukauden kohdalla (%) (95 %:n luottamusväli)c, d | 78 (65, 87) | 28 (19, 38) | 79 (69, 86) | 40 (32, 48) |
HR = riskisuhde; NC = ei laskettavissa
DFS-tulokset perustuvat tutkijalääkärin arvioon.
Riskisuhde < 1 suosii TAGRISSO-valmistetta.
DFS-tulosten osalta seuranta-ajan mediaani oli 22,1 kuukautta TAGRISSO-valmistetta saaneilla, 14,9 kuukautta lumelääkettä saaneilla (kun syövän levinneisyysaste oli II–IIIA) ja 16,6 kuukautta lumelääkettä saaneilla (kun syövän levinneisyysaste oli IB–IIIA).
DFS-tulokset perustuvat ensisijaiseen analyysiin (17.1.2020).
a Korjattu välianalyysin suhteen (maturiteetti 33 %), tilastollisen merkitsevyyden saavuttaminen edellytti p-arvoa < 0,0094.
b Korjattu välianalyysin suhteen (maturiteetti 29 %), tilastollisen merkitsevyyden saavuttaminen edellytti p-arvoa < 0,0088.
c Potilaita, joilla oli taudin etenemisen tai kuoleman riski 36 kuukauden kohdalla, oli TAGRISSO-haarassa 18 ja lumelääkehaarassa 9 (potilaat, joiden syövän levinneisyysaste oli II–IIIA).
d Potilaita, joilla oli taudin etenemisen tai kuoleman riski 36 kuukauden kohdalla, oli TAGRISSO-haarassa 27 ja lumelääkehaarassa 20 (potilaat, joiden syövän levinneisyysaste oli IB–IIIA).
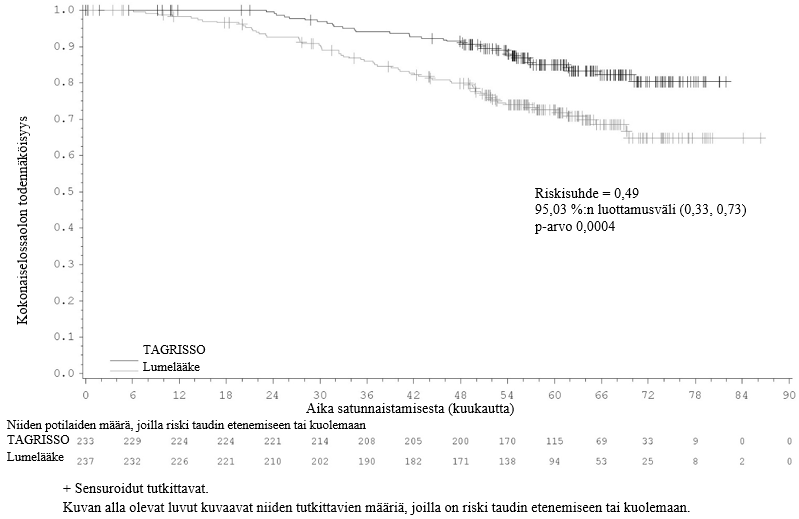
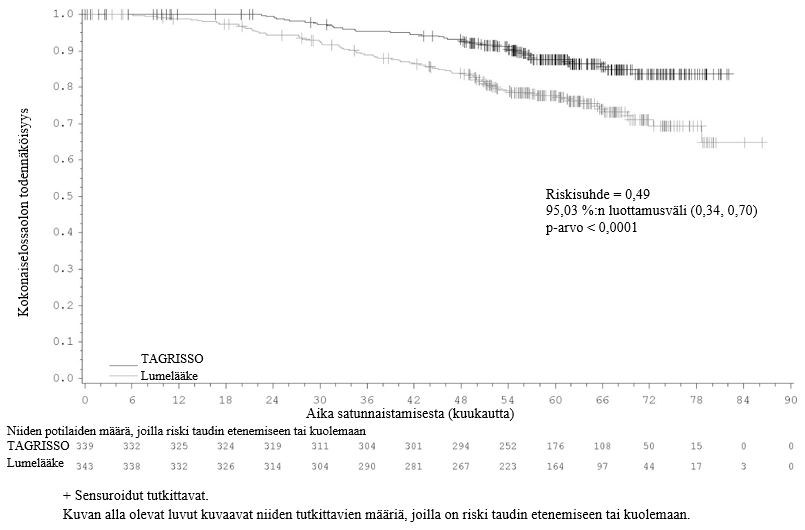
Lopullisessa kokonaiselossaoloaika-analyysissa (tiedonkeruun katkaisupiste 27.1.2023) todettiin, että kokonaiselossaolotulokset olivat tilastollisesti merkitsevästi paremmat TAGRISSO-hoitoa saaneilla potilailla kuin lumelääkettä saaneilla sekä niiden potilaiden joukossa, joiden taudin levinneisyysaste oli II–IIIA (100 kokonaiselossaolotapahtumaa [maturiteetti 21 %]; riskisuhde [HR] = 0,49; 95,03 %:n luottamusväli: 0,33, 0,73; p-arvo = 0,0004), että koko populaatiossa (IB–IIIA; 124 kokonaiselossaolotapahtumaa [maturiteetti 18 %]; riskisuhde = 0,49; 95,03 %:n luottamusväli: 0,34, 0,70; p-arvo < 0,0001). Kummassakaan potilasryhmässä kokonaiselossaoloajan mediaania ei saavutettu kummassakaan hoitohaarassa eivätkä 95 %:n luottamusvälit olleet laskettavissa. Kokonaiselossaoloajan seuranta-ajan mediaanikesto kaikilla potilailla oli TAGRISSO-haarassa 59,9 kuukautta (potilailla, joiden taudin levinneisyysaste oli II–IIIA) ja 60,4 kuukautta (potilailla, joiden taudin levinneisyysaste oli IB–IIIA) ja lumelääkehaarassa 56,2 kuukautta (potilailla, joiden taudin levinneisyysaste oli II–IIIA) ja 59,4 kuukautta (potilailla, joiden taudin levinneisyysaste oli IB–IIIA).
Kuva 1. Tautivapaata elossaoloaikaa kuvaava Kaplan–Meier-käyrä potilailla, joiden taudin levinneisyysaste oli II–IIIA (tutkijalääkärin arvio)
Kuva 2. Tautivapaata elossaoloaikaa kuvaava Kaplan–Meier-käyrä potilailla, joiden taudin levinneisyysaste oli IB–IIIA (koko populaatio) (tutkijalääkärin arvio)
TAGRISSO-hoito paransi johdonmukaisesti tautivapaata elossaoloaikaa verrattuna lumelääkkeeseen kaikissa analysoiduissa ennalta määritellyissä alaryhmissä, joiden määrittelyperusteina olivat mm. etninen tausta, ikä, sukupuoli ja EGFR-mutaation tyyppi (Ex19Del tai L858R).
Kuva 3. Kokonaiselossaoloaikaa kuvaava Kaplan–Meier-käyrä potilailla, joiden taudin levinneisyysaste oli II–IIIA
Kuva 4. Kokonaiselossaoloaikaa kuvaava Kaplan–Meier-käyrä potilailla, joiden taudin levinneisyysaste oli IB–IIIA (koko populaatio)
Keskushermoston tautivapaan elossaoloajan eksploratiivisessa analyysissä (aika taudin etenemiseen keskushermostossa tai kuolemaan), jossa TAGRISSO-hoitoa saaneita potilaita verrattiin lumelääkettä saaneisiin, riskisuhde oli 0,18 (95 %:n luottamusväli 0,10, 0,33; p < 0,0001) koko populaatiossa (levinneisyysasteet IB–IIIA).
Potilaan ilmoittamat vaikutukset (Patient Reported Outcomes, PRO)
ADAURA-tutkimuksessa terveyteen liittyvää elämänlaatua arvioitiin SF-36v2-kyselylomakkeella [Short Form (36) Health Survey, versio 2]. SF-36v2-lomake täytettiin 12 viikon kohdalla, 24 viikon kohdalla ja tämän jälkeen 24 viikon välein satunnaistamisesta laskettuna aina hoidon päättymiseen tai keskeyttämiseen asti. Kokonaisuutena arvioiden terveyteen liittyvä elämänlaatu säilyi molemmissa haaroissa hyvänä 30 kuukauteen asti: vähintään 70 %:lla potilaista, joiden syövän levinneisyysaste oli II–IIIA, ei todettu SF-36-lomakkeen fyysisen osion tulosten kliinisesti merkittävää huononemista eikä kuolemaa (70 % TAGRISSO-haarassa ja 76 % lumelääkehaarassa) eikä SF-36-lomakkeen psyykkisen osion tulosten kliinisesti merkittävää huononemista eikä kuolemaa (70 % TAGRISSO-haarassa ja 71 % lumelääkehaarassa).
Paikallisesti edennyt, leikkaukseen soveltumaton EGFR‑mutaatiopositiivinen NSCLC – LAURA
TAGRISSO-valmisteen tehoa ja turvallisuutta arvioitiin satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa (LAURA) sellaisten EGFR-mutaatiopositiivista, paikallisesti edennyttä, leikkaukseen soveltumatonta NSCLC:tä sairastavien potilaiden hoidossa, joiden tauti ei ollut edennyt definitiivisen platinapohjaisen kemosädehoidon aikana tai sen jälkeen. Potilaat määrättiin saamaan joko samanaikaisesti annettavaa kemosädehoitoa (CCRT) tai peräkkäin annettavaa kemosädehoitoa (SCRT). Näihin hoito-ohjelmiin kuului, että potilaat olivat saaneet korkeintaan 6 viikkoa ennen satunnaistamista platinapohjaista solunsalpaajaa ainakin kahden hoitosyklin ajan tai vähintään viisi kerran viikossa annettavaa annosta sekä sädehoitoa kokonaisannoksella 60 Gy ± 10 % (54–66 Gy). Edellytyksenä oli, että potilaan kasvainkudosnäytteistä oli todettu EGFR:n eksonin 19 deleetio tai eksonin 21 L858R-mutaatio keskitetyllä tai paikallisella määrityksellä, jossa käytettiin sertifioitua/hyväksyttyä testiä.
Potilaat satunnaistettiin (2:1) saamaan joko 80 mg TAGRISSO-valmistetta suun kautta kerran vuorokaudessa (n = 143) tai lumelääkettä (n = 73). Satunnaistaminen stratifioitiin aiemman kemosädehoitostrategian (CCRT vs. SCRT), kasvaimen kemosädehoitoa edeltävän levinneisyysasteen (IIIA vs. IIIB/IIIC) ja Kiinan kohortin mukaan. Tutkimushoitoa jatkettiin, kunnes potilas ei enää sietänyt hoitoa tai taudin eteneminen vahvistettiin. Siirtyminen lumelääkkeen käytöstä TAGRISSO-hoitoon sallittiin, jos potilaan tauti eteni.
Ensisijainen tehoa kuvaava päätetapahtuma oli sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) arvioima etenemisvapaa elossaoloaika (PFS). Muita tehoa kuvaavia päätetapahtumia olivat kokonaiselossaoloaika (OS) ja keskushermostoon liittyvä etenemisvapaa elossaoloaika, jonka arvioi neuroradiologiaan keskittyvä BICR-arvioijataho.
Koko tutkimuspopulaation demografiset tiedot ja tautitiedot lähtötilanteessa olivat: mediaani-ikä 63 vuotta (vaihteluväli 36–84 vuotta), vähintään 75-vuotiaita 13 %, naisia 61 %, aasialaisia 82 % ja valkoihoisia 14 %. 70 % potilaista ei ollut koskaan tupakoinut. WHO:n mukainen suorituskykyluokitus lähtötilanteessa oli 0 (51 %:lla potilaista) tai 1 (49 %:lla potilaista). NSCLC:n levinneisyysaste oli 35 %:lla potilaista IIIA, 49 %:lla potilaista IIIB ja 16 %:lla potilaista IIIC. EGFR-mutaatiostatuksen osalta 54 %:lla oli eksonin 19 deleetioita ja 45 %:lla eksonin 21 L858R-mutaatioita. Ennen satunnaistamista 89 % potilasta oli saanut CCRT-hoitoa ja 11 % SCRT-hoitoa. Kaikki potilaat saivat platinapohjaista solunsalpaajahoitoa (55 % sai karboplatiinipohjaista ja 44 % sisplatiinipohjaista solunsalpaajahoitoa). Kokonaissäteilyannoksen mediaani oli 60 Gy kummassakin hoitohaarassa.
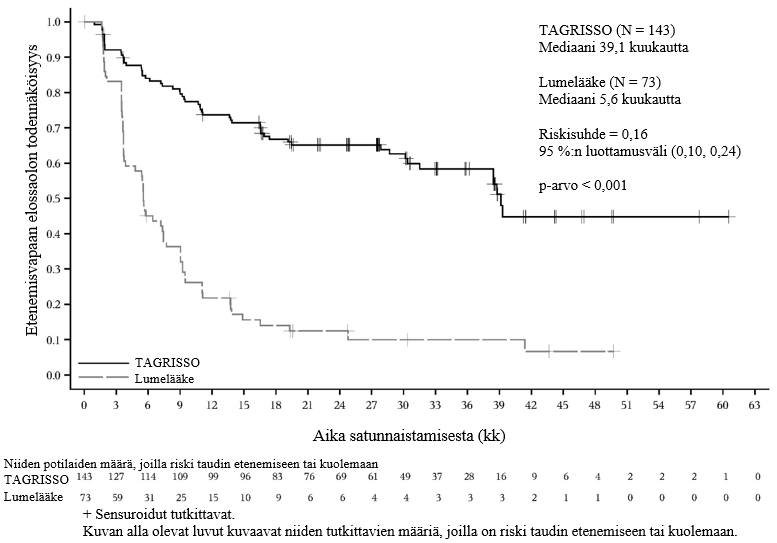
TAGRISSO-hoito platinapohjaisen kemosädehoidon jälkeen johti tilastollisesti merkitsevään etenemisvapaan elossaoloajan pitenemiseen lumelääkkeen käyttöön verrattuna (maturiteetti 56 %; riskisuhde [HR] = 0,16; 95 %:n luottamusväli: 0,10, 0,24; p < 0,001, mediaani TAGRISSO-hoidolla 39,1 kuukautta ja lumelääkehoidolla 5,6 kuukautta).
Tutkimussuunnitelman mukaisesti kaikille potilaille tehtiin lähtötilanteessa aivojen magneettikuvaus. Kaikille paitsi yhdelle potilaalle tehtiin suunnitellusti aivojen magneettikuvaus hoidon aikana. Uusia keskushermostoleesioita ilmeni neuroradiologisesti arvioituna pienemmällä osalla TAGRISSO-haaran potilaista kuin lumelääkehaaran potilaista (17/143 [12 %] vs. 26/73 [36 %]).
Kokonaiselossaoloajan välianalyysin ajankohtana (tiedonkeruun katkaisupiste 5.1.2024) ei ollut saavutettu tilastollista merkitsevyyttä. 50 potilasta lumelääkehaaran 62 potilaasta (80,6 %) sai BICR:n vahvistaman taudin etenemisen jälkeen TAGRISSO-hoitoa.
LAURA-tutkimuksen tehoa koskevat tulokset esitetään yhteenvetona taulukossa 4, ja Kaplan–Meier-käyrä etenemisvapaasta elossaoloajasta on esitetty kuvassa 5.
Taulukko 4. LAURA-tutkimuksen tehoa koskevat tulokset
Tehoa kuvaava muuttuja | TAGRISSO | Lumelääke |
Etenemisvapaa elossaoloaika (PFS)a | ||
Tapahtumien lukumäärä (%) | 57 (40) | 63 (86) |
Mediaani PFS, kuukautta (95 %:n luottamusväli) | 39,1 (31,5, NC) | 5,6 (3,7, 7,4) |
HR (95 %:n luottamusväli); p-arvo | 0,16 (0,10, 0,24); p < 0,001 | |
Kokonaiselossaoloaika (OS) | ||
Kuolemien lukumäärä (%) | 28 (20) | 15 (21) |
Mediaani OS, kuukautta (95 %:n luottamusväli) | 54,0 (46,5, NC) | NC (42,1, NC) |
HR (95 %:n luottamusväli); p-arvo | 0,81 (0,42, 1,56); p = 0,530a | |
HR = riskisuhde; NC = ei laskettavissa
PFS-tulokset ovat BICR:n arvioimat.
Kaikkien potilaiden PFS-tulosten osalta seuranta-ajan mediaani oli 22,0 kuukautta TAGRISSO-hoitohaarassa ja 5,6 kuukautta lumelääkehaarassa.
a Korjattu välianalyysin suhteen (maturiteetti 20 %), tilastollisen merkitsevyyden saavuttaminen edellytti p-arvoa < 0,000036.
Kuva 5. LAURA-tutkimuksen etenemisvapaata elossaoloaikaa kuvaavat Kaplan–Meier-käyrät (BICR-arvioinnin mukaan)
Aiemmin hoitamaton EGFR-mutaatiopositiivinen paikallisesti edennyt tai metastaattinen ei-pienisoluinen keuhkosyöpä (NSCLC)
FLAURA – monoterapia
TAGRISSO-valmisteen teho ja turvallisuus osoitettiin satunnaistetussa, kaksoissokkoutetussa, aktiivikontrolloidussa tutkimuksessa (FLAURA) sellaisten potilaiden hoidossa, joilla oli EGFR-mutaatiopositiivinen, paikallisesti edennyt, kuratiiviseen leikkaukseen tai sädehoitoon soveltumaton, tai metastaattinen NSCLC ja jotka eivät olleet aiemmin saaneet systeemistä hoitoa edenneeseen tautiin. Edellytyksenä oli, että potilaan kasvainkudosnäytteistä oli todettu paikallisella tai keskitetyllä määrityksellä toinen kahdesta yleisestä EGFR-mutaatiosta, joiden tiedetään liittyvän EGFR-TKI-herkkyyteen (Ex19del tai L858R).
Potilaat satunnaistettiin suhteessa 1:1 saamaan joko TAGRISSO-valmistetta (n = 279, 80 mg suun kautta kerran vuorokaudessa) tai EGFR-TKI-vertailuvalmistetta (n = 277; gefitinibi 250 mg suun kautta kerran vuorokaudessa tai erlotinibi 150 mg suun kautta kerran vuorokaudessa). Satunnaistamisen yhteydessä tehty stratifiointi perustui EGFR-mutaatiotyyppiin (Ex19del tai L858R) ja etniseen taustaan (aasialaiset tai muut kuin aasialaiset). Potilas sai tutkimushoitoa, kunnes hänelle kehittyi intoleranssi hoidolle tai tutkijalääkäri katsoi, että potilas ei enää kliinisesti hyötynyt hoidosta. EGFR-TKI-vertailuvalmistetta saaneet potilaat saivat taudin etenemisen jälkeen vaihtaa avoimeen TAGRISSO-hoitoon, jos kasvainnäytteiden T790M-mutaatiotestin tulos oli positiivinen. Ensisijainen tehoa kuvaava päätetapahtuma oli tutkijalääkärin arvioima etenemisvapaa elossaoloaika (PFS).
Koko tutkimuspopulaation demografiset tiedot ja tautitiedot lähtötilanteessa olivat: mediaani-ikä 64 vuotta (vaihteluväli 26–93 vuotta), vähintään 75‑vuotiaita 14 %, naisia 63 %, valkoihoisia 36 %, aasialaisia 62 %, 64 % tutkittavista ei ollut koskaan tupakoinut, 100 %:lla potilaista WHO:n mukainen suorituskykyluokitus oli 0 tai 1. 36 %:lla potilaista oli metastaattinen luusairaus, 35 %:lla potilaista oli rintakehän ulkopuolisia metastaaseja sisäelimissä ja 21 %:lla potilaista oli keskushermostometastaaseja (todettu keskushermostossa sijaitseva leesio lähtötilanteessa, todettu aiemmin sairaushistoriassa ja/tai aiemman leikkauksen ja/tai keskushermostometastaaseihin aiemmin annetun sädehoidon perusteella).
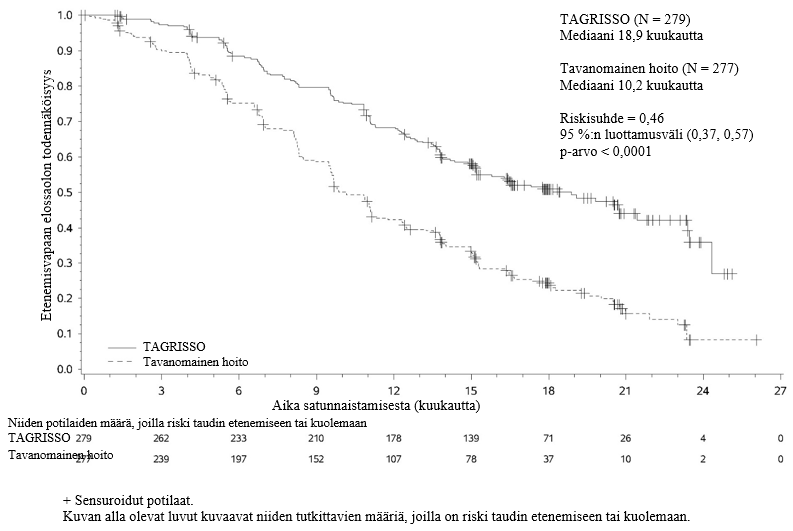
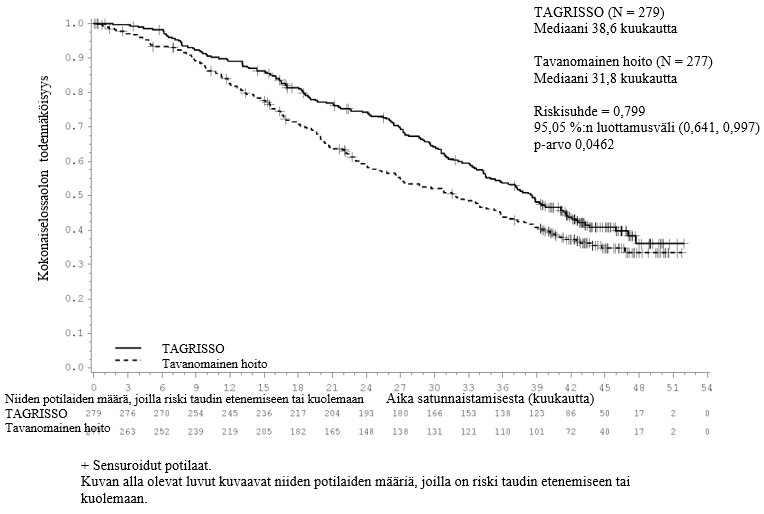
TAGRISSO-valmisteen osoitettiin pidentävän etenemisvapaata elossaoloaikaa (PFS) kliinisesti merkittävästi ja tilastollisesti merkitsevästi verrattuna EGFR-TKI-vertailuvalmisteeseen (mediaani PFS TAGRISSO-valmisteella 18,9 kuukautta ja EGFR-TKI-vertailuvalmisteella 10,2 kuukautta, riskisuhde = 0,46, 95 %:n luottamusväli: 0,37, 0,57; p < 0,0001). Tutkijalääkärin arvioimista FLAURA-tutkimuksen tehoa koskevista tuloksista on esitetty yhteenveto taulukossa 5, ja Kaplan–Meier-käyrä etenemisvapaasta elossaoloajasta on esitetty kuvassa 6. Kokonaiselossaolotietojen lopullisen analyysin kohdalla (maturiteetti 58 %) potilailla, jotka oli satunnaistettu saamaan TAGRISSO-valmistetta, osoitettiin tilastollisesti merkitsevästi pidempi kokonaiselossaoloaika riskisuhteella 0,799 (95,05 %:n luottamusväli: 0,641, 0,997) ja kliinisesti merkittävä pidempi elossaoloajan mediaani verrattuna EGFR-TKI-vertailuvalmisteeseen (taulukko 5 ja kuva 7). TAGRISSO-valmistetta saaneista potilaista oli elossa suurempi osuus 12 kuukauden kohdalla (89 %), 18 kuukauden kohdalla (81 %), 24 kuukauden kohdalla (74 %) ja 36 kuukauden kohdalla (54 %) kuin EGFR-TKI-vertailuvalmistetta saaneista potilaista (12 kuukauden kohdalla 83 %, 18 kuukauden kohdalla 71 %, 24 kuukauden kohdalla 59 % ja 36 kuukauden kohdalla 44 %). Taudin etenemisen jälkeen todettujen päätetapahtumien analyysi osoitti, että etenemisvapaaseen elossaoloaikaan liittyvä hyöty säilyi seuraavien linjojen hoitojen aikana.
Taulukko 5. FLAURA-tutkimuksen tehoa koskevat tulokset (tutkijalääkärin arvio)
Tehoa kuvaava muuttuja | TAGRISSO | EGFR-TKI-vertailuhoito (gefitinibi tai erlotinibi) |
Etenemisvapaa elossaoloaika (PFS) | ||
Tapahtumien lukumäärä (maturiteetti 62 %) | 136 (49) | 206 (74) |
Mediaani PFS, kuukautta (95 %:n luottamusväli) | 18,9 (15,2, 21,4) | 10,2 (9,6, 11,1) |
HR (95 %:n luottamusväli); p‑arvo | 0,46 (0,37, 0,57); p < 0,0001 | |
Kokonaiselossaoloaika (OS) | ||
Kuolemien lukumäärä (maturiteetti 58 %) | 155 (56) | 166 (60) |
Mediaani OS, kuukautta (95 %:n luottamusväli) | 38,6 (34,5, 41,8) | 31,8 (26,6, 36,0) |
HR (95,05 %:n luottamusväli); p‑arvo | 0,799 (0,641, 0,997); p = 0,0462a | |
Objektiivinen hoitovaste (ORR)*b | ||
Vasteiden määrä (n), hoitovaste (95 %:n luottamusväli) | 223 80 % (75, 85) | 210 76 % (70, 81) |
Ristitulosuhde (95 %:n luottamusväli); p‑arvo | 1,3 (0,9, 1,9); p = 0,2421 | |
Vasteen kesto (DoR)* | ||
Vasteen keston mediaani, kuukautta (95 %:n luottamusväli) | 17,2 (13,8, 22,0) | 8,5 (7,3, 9,8) |
Toinen PFS ensimmäisen seuraavan hoidon aloittamisen jälkeen (PFS2) | ||
Niiden potilaiden lukumäärä, joilla tauti eteni toisen kerran (%) | 73 (26) | 106 (38) |
Mediaani PFS2, kuukautta (95 %:n luottamusväli) | NC (23,7, NC) | 20,0 (18,0, NC) |
HR (95 %:n luottamusväli); p‑arvo | 0,58 (0,44, 0,78); p = 0,0004 | |
Aika satunnaistamisesta ensimmäiseen seuraavaan hoitoon tai kuolemaan (TFST) | ||
Niiden potilaiden lukumäärä, jotka saivat ensimmäistä seuraavaa hoitoa tai kuolivat (%) | 115 (41) | 175 (63) |
Mediaani TFST, kuukautta (95 %:n luottamusväli) | 23,5 (22,0, NC) | 13,8 (12,3, 15,7) |
HR (95 %:n luottamusväli); p‑arvo | 0,51 (0,40, 0,64); p < 0,0001 | |
Aika satunnaistamisesta toiseen seuraavaan hoitoon tai kuolemaan (TSST) | ||
Niiden potilaiden lukumäärä, jotka saivat toista seuraavaa hoitoa tai kuolivat (%) | 75 (27) | 110 (40) |
Mediaani TSST, kuukautta (95 %:n luottamusväli) | NC (NC, NC) | 25,9 (20,0, NC) |
HR (95 %:n luottamusväli); p‑arvo | 0,60 (0,45, 0,80); p = 0,0005 | |
HR = riskisuhde; NC = ei laskettavissa
PFS-, ORR-, DoR- ja PFS2-tulokset perustuvat tutkijalääkärin arvioon (RECIST).
* Perustuu vahvistamattomaan vasteeseen.
Seuranta-ajan mediaani oli 15,0 kuukautta TAGRISSO-valmistetta saaneilla ja 9,7 kuukautta EGFR-TKI-vertailuvalmistetta saaneilla potilailla.
Elossaolon seuranta-ajan mediaani oli 35,8 kuukautta TAGRISSO-valmistetta saaneilla ja 27,0 kuukautta EGFR-TKI-vertailuvalmistetta saaneilla potilailla.
PFS-, ORR-, DoR-, PFS2-, TFST- ja TSST-tulokset ovat tiedonkeruun katkaisupisteeltä 12.6.2017. Kokonaiselossaoloaikaa koskevat tulokset ovat tiedonkeruun katkaisupisteeltä 25.6.2019.
Riskisuhde < 1 suosii TAGRISSO-valmistetta, ristitulosuhde (odds ratio) > 1 suosii TAGRISSO-valmistetta.
a Korjattu välianalyysin suhteen (maturiteetti 25 %), tilastollisen merkitsevyyden saavuttaminen edellytti p‑arvoa < 0,0495.
b BICR:n arvioimat ORR-tulokset vastasivat tutkijalääkärin raportoimia tuloksia; BICR-arvion mukaan ORR oli TAGRISSO-valmisteella 78 % (95 %:n luottamusväli: 73, 83) ja EGFR-TKI-vertailuvalmisteella 70 % (95 %:n luottamusväli: 65, 76).
Kuva 6. FLAURA-tutkimuksen etenemisvapaata elossaoloaikaa kuvaavat Kaplan–Meier-käyrät (tutkijalääkärin arvio)
Kuva 7. FLAURA-tutkimuksen kokonaiselossaoloaikaa kuvaavat Kaplan–Meier-käyrät
Etenemisvapaaseen elossaoloaikaan liittyvä TAGRISSO-valmisteella saavutettu hyöty EGFR-TKI-vertailuvalmisteeseen verrattuna oli yhdenmukainen kaikissa ennalta analysoitaviksi määritellyissä alaryhmissä, mukaan lukien etniseen taustaan, ikään, sukupuoleen, tupakointitaustaan, lähtötilanteessa todettuun keskushermostometastaasistatukseen ja EGFR-mutaatiotyyppiin (eksonin 19 deleetio tai L858R) perustuvat alaryhmät.
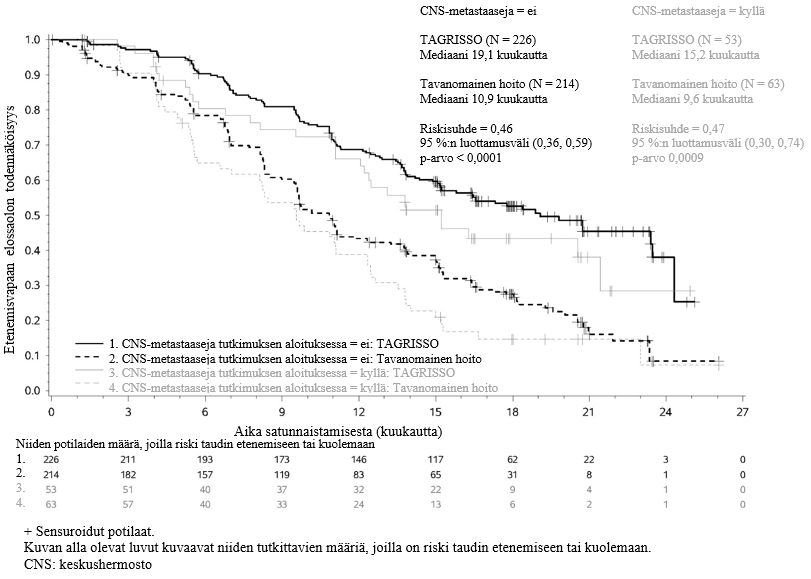
Keskushermostometastaaseihin liittyvät tehoa koskevat tulokset FLAURA-tutkimuksessa
FLAURA-tutkimukseen voitiin satunnaistaa mukaan potilaita, joilla oli keskushermostometastaaseja, joita ei ollut hoidettu steroideilla, ja joiden neurologinen status oli ollut vakaa vähintään kahden viikon ajan definitiivisen hoidon ja steroidihoidon päättymisen jälkeen. 556 potilaasta 200 potilaalle oli tehty lähtötilanteessa aivokuvaus. Sokkoutettu riippumaton keskitetty arvioijataho (BICR) arvioi nämä kuvaukset, ja arvioinnin tuloksena oli alaryhmä, jossa 128/556 (23 %) potilaalla oli todettu keskushermostometastaaseja, ja näistä tuloksista on esitetty yhteenveto taulukossa 6. FLAURA-tutkimuksessa RECIST v1.1 ‑kriteereillä arvioitu keskushermostoon liittyvä teho osoitti, että keskushermostoon liittyvän etenemisvapaan elossaoloajan tilastollinen merkitsevyys parani (riskisuhde = 0,48, 95 %:n luottamusväli: 0,26, 0,86; p = 0,014).
Taulukko 6. Keskushermostoon liittyvä teho BICR-arvioinnin mukaan potilailla, joilla oli keskushermostometastaaseja lähtötilanteen aivokuvauksessa FLAURA-tutkimuksessa
Tehoa kuvaava muuttuja | TAGRISSO | EGFR-TKI-vertailuvalmiste |
Keskushermostoon liittyvä etenemisvapaa elossaoloaikaa |
|
|
Tapahtumien lukumäärä (%) | 18 (30) | 30 (45) |
Keskushermostoon liittyvän etenemisvapaan elossaoloajan mediaani, kuukautta | NC (16,5, NC) | 13,9 (8,3, NC) |
HR (95 %:n luottamusväli); p-arvo | 0,48 (0,26, 0,86); p = 0,014 | |
Potilaat, joilla ei ollut tapahtunut keskushermostoon liittyvää taudin etenemistä ja jotka olivat elossa 6 kuukauden kohdalla (%) | 87 (74, 94) | 71 (57, 81) |
Potilaat, joilla ei ollut tapahtunut keskushermostoon liittyvää taudin etenemistä ja jotka olivat elossa 12 kuukauden kohdalla (%) | 77 (62, 86) | 56 (42, 68) |
HR = riskisuhde; NC = ei laskettavissa
Riskisuhde < 1 suosii TAGRISSO-valmistetta, ristitulosuhde (odds ratio) > 1 suosii TAGRISSO-valmistetta.
a Keskushermostoon liittyvä etenemisvapaa elossaoloaika määritettiin RECIST v1.1 ‑kriteereillä sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) tekemällä keskushermoston arvioinnilla (BICR:n lähtötilanteessa toteamat keskushermoston mitattavissa ja ei-mitattavissa olevat leesiot); n = 61 TAGRISSO-valmisteelle ja n = 67 EGFR-TKI-vertailuvalmisteelle; vasteet ovat vahvistamattomia.
FLAURA-tutkimuksessa arvioitiin ennalta määritetty etenemisvapaan elossaoloajan alaryhmä, joka perustui keskushermostometastaasistatukseen (todettu keskushermostossa sijaitseva leesio lähtötilanteessa, todettu aiemmin sairaushistoriassa ja/tai aiemman leikkauksen ja/tai keskushermostometastaaseihin aiemmin annetun sädehoidon perusteella) tutkimuksessa aloittamisen hetkellä, ja arvioinnin tulokset on esitetty kuvassa 8. Keskushermostoleesiostatuksesta tutkimuksen sisäänottovaiheessa riippumatta TAGRISSO-haaran potilailla osoitettiin tehoon liittyvä hyöty verrattuna EGFR-TKI-vertailuhaaraan, ja TAGRISSO-haarassa oli vähemmän potilaita, joilla oli uusia keskushermostoleesioita (11/279, 3,9 %), kuin EGFR-TKI-vertailuhaarassa (34/277, 12,3 %). Niiden potilaiden alaryhmässä, joilla ei ollut keskushermostoleesioita lähtötilanteessa, uusia keskushermostoleesioita oli TAGRISSO-haarassa vähemmän (7/226, 3,1 %) kuin EGFR-TKI-vertailuhaarassa (15/214, 7,0 %).
Kuva 8. Etenemisvapaa elossaoloaika (tutkijan arvio) keskushermostometastaasistatuksen mukaan tutkimuksessa aloittamisen hetkellä, Kaplan–Meier-käyrä (koko analyysisarja) FLAURA-tutkimuksessa
Potilaan ilmoittamat vaikutukset (Patient Reported Outcomes, PRO)
Potilaiden ilmoittamat oireet ja terveyteen liittyvää elämänlaatua koskevat tiedot kerättiin sähköisesti EORTC QLQ‑C30:llä ja sen keuhkosyöpäosiolla (EORTC QLQ‑LC13). LC13 täytettiin aluksi kerran viikossa ensimmäisten 6 viikon ajan ja sen jälkeen 3 viikon välein ennen taudin etenemistä ja sen jälkeen. C30 arvioitiin 6 viikon välein ennen taudin etenemistä ja sen jälkeen. Lähtötilanteessa ei havaittu eroja potilaiden ilmoittamissa oireissa, toimintakyvyssä tai terveyteen liittyvässä elämänlaadussa TAGRISSO-haaran ja EGFR-TKI-vertailuhaaran (gefitinibi tai erlotinibi) välillä. Hoitomyöntyvyys oli ensimmäisten 9 kuukauden aikana yleisesti hyvä (≥ 70 %) ja samankaltainen molemmissa haaroissa.
Keskeisten keuhkosyövän oireiden analyysi
Lähtötilanteesta kuukauteen 9 asti kerätyt tiedot osoittivat, että TAGRISSO-ryhmässä ja EGFR-TKI-vertailuryhmässä oli tapahtunut samanlaisia parannuksia potilaan ilmoittamien viiden ennalta määrätyn keskeisimmän oireen (yskä, hengenahdistus, rintakipu, väsymys ja ruokahaluttomuus) osalta, ja yskän osalta todettu parannus saavutti vahvistetun kliinisen merkittävyyden raja-arvon. TAGRISSO- ja EGFR-TKI-vertailuryhmissä potilaan ilmoittamien oireiden osalta ei todettu kliinisesti merkittäviä eroja (vähintään 10 pisteen eroja) kuukauteen 9 mennessä.
Terveyteen liittyvän elämänlaadun ja fyysisen toimintakyvyn paranemisen analyysi
Molemmilla ryhmillä ilmoitettiin samankaltaisia parannuksia useimmissa toimintakykyyn liittyvissä domeeneissa ja yleisessä terveydentilassa / terveyteen liittyvässä elämänlaadussa, mikä osoittaa potilaiden kokonaisterveydentilan parantuneen. Kuukauteen 9 mennessä TAGRISSO-ryhmän ja EGFR-TKI-vertailuryhmän välillä ei havaittu kliinisesti merkittäviä eroja toimintakyvyn tai terveyteen liittyvän elämänlaadun suhteen.
FLAURA2 – yhdistelmähoito
Yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa käytetyn TAGRISSO-valmisteen teho ja turvallisuus osoitettiin satunnaistetussa, avoimessa, aktiivikontrolloidussa tutkimuksessa (FLAURA2) sellaisten potilaiden hoidossa, joilla oli EGFR-mutaatiopositiivinen, paikallisesti edennyt tai metastaattinen NSCLC ja jotka eivät olleet aiemmin saaneet systeemistä hoitoa edenneeseen tautiin. Edellytyksenä oli, että potilaan kasvainkudosnäytteistä oli todettu paikallisella tai keskitetyllä määrityksellä toinen kahdesta yleisestä EGFR-mutaatiosta, joiden tiedetään liittyvän EGFR-TKI-herkkyyteen (Ex19del tai L858R).
Potilaat satunnaistettiin suhteessa 1:1 jompaankumpaan seuraavista hoitohaaroista:
-
TAGRISSO (80 mg) suun kautta kerran vuorokaudessa sekä pemetreksedi (500 mg/m2) ja tutkijalääkärin valinnan mukaan sisplatiini (75 mg/m2) tai karboplatiini (AUC5) laskimoon kunkin 21 päivän mittaisen hoitosyklin päivänä 1 yhteensä 4 hoitosyklin ajan ja sen jälkeen TAGRISSO (80 mg) suun kautta kerran vuorokaudessa ja pemetreksedi (500 mg/m2) laskimoon 3 viikon välein (n = 279)
-
TAGRISSO (80 mg) suun kautta kerran vuorokaudessa (n = 278).
Satunnaistamisen yhteydessä tehty stratifiointi perustui etniseen taustaan (kiinalaiset/aasialaiset, ei-kiinalaiset/aasialaiset tai ei-aasialaiset), WHO:n mukaiseen suorituskykyluokitukseen (0 tai 1) sekä kudosmääritysmenetelmään (keskitetty tai paikallinen). Potilas sai tutkimushoitoa, kunnes hänelle kehittyi intoleranssi hoidolle tai tutkijalääkäri katsoi, että potilas ei enää kliinisesti hyötynyt hoidosta.
Ensisijainen tehoa kuvaava päätetapahtuma oli tutkijalääkärin RECIST 1.1 ‑kriteereillä arvioima etenemisvapaa elossaoloaika (PFS), ja keskeinen toissijainen tehoa kuvaava päätetapahtuma oli kokonaiselossaoloaika.
Koko tutkimuspopulaation demografiset tiedot ja tautitiedot lähtötilanteessa olivat: mediaani-ikä 61 vuotta (vaihteluväli 26–85 vuotta), vähintään 75‑vuotiaita 8 %, naisia 61 %, aasialaisia 64 % ja valkoihoisia 28 %. 66 % potilaista ei ollut koskaan tupakoinut. WHO:n mukainen suorituskykyluokitus lähtötilanteessa oli 0 (37 %:lla potilaista) tai 1 (63 %:lla potilaista). 98,7 %:lla potilaista histologinen tyyppi oli pääasiassa adenokarsinooma. Potilaista, joiden tauti oli metastaattinen, 49 %:lla oli metastaattinen luusairaus, 53 %:lla oli rintakehän ulkopuolisia metastaaseja ja 20 %:lla oli maksametastaaseja. 41 %:lla potilaista oli keskushermostometastaaseja (jotka tutkijalääkäri totesi lähtötilanteessa keskushermostossa sijaitsevan leesion, sairaushistorian ja/tai aiemman leikkauksen ja/tai keskushermostometastaaseihin aiemmin annetun sädehoidon perusteella). Kasvaimen EGFR-mutaatiotyyppi satunnaistamisen aikaan oli 60,5 %:lla potilaista eksonin 19 deleetio ja 38,2 %:lla eksonin 21 L858R-mutaatio. Potilaista 0,7 %:lla oli kasvaimia, joissa oli sekä eksonin 19 deleetio että eksonin 21 L858R-mutaatio.
Yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa käytetyn TAGRISSO-valmisteen osoitettiin pidentävän etenemisvapaata elossaoloaikaa (PFS) tilastollisesti merkitsevästi verrattuna TAGRISSO-monoterapiaan. Etenemisvapaaseen elossaoloaikaan liittyvä hyöty oli yhdenmukainen kaikissa analysoiduissa alaryhmissä. Lopullisessa kokonaiselossaoloaika-analyysissa todettiin, että kokonaiselossaolotulokset olivat tilastollisesti merkitsevästi paremmat potilailla, jotka oli satunnaistettu saamaan TAGRISSO-valmistetta yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa verrattuna TAGRISSO-monoterapiaan.
Taulukossa 7 on esitetty yhteenveto FLAURA2-tutkimuksen tehoa koskevista tuloksista. Kaplan–Meier-käyrä etenemisvapaasta elossaoloajasta on esitetty kuvassa 9, ja Kaplan–Meier-käyrä kokonaiselossaoloajasta on esitetty kuvassa 10.
Taulukko 7. FLAURA2-tutkimuksen tehoa koskevat tulokset (tutkijalääkärin arvio)
Tehoa kuvaava muuttuja | TAGRISSO pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa (N = 279) | TAGRISSO |
Etenemisvapaa elossaoloaika (PFS) | ||
Tapahtumien lukumäärä (%) | 120 (43) | 166 (60) |
Mediaani PFS, kuukautta (95 %:n luottamusväli)a | 25,5 (24,7, NC) | 16,7 (14,1, 21,3) |
HR (95 %:n luottamusväli); p‑arvo | 0,62 (0,49, 0,79); p < 0,0001 | |
Kokonaiselossaoloaika (OS) | ||
Kuolemien lukumäärä (%) | 144 (52) | 171 (62) |
Mediaani OS, kuukautta (95 %:n luottamusväli) | 47,5 (41,0, NC) | 37,6 (33,2, 43,2) |
HR (95 %:n luottamusväli); p‑arvo | 0,77 (0,61, 0,96); p = 0,0202b | |
HR = riskisuhde; NC = ei laskettavissa
PFS-tulokset perustuvat tutkijalääkärin arvioon (RECIST).
Kaikkien potilaiden PFS-tulosten osalta seuranta-ajan mediaani oli 19,5 kuukautta hoitohaarassa, jossa potilaat saivat TAGRISSO-valmistetta pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, ja 16,5 kuukautta hoitohaarassa, jossa potilaat saivat TAGRISSO-valmistetta monoterapiana.
PFS-tulokset ovat tiedonkeruun katkaisupisteeltä 3.4.2023 (maturiteetti 51 %). OS-tulokset ovat tiedonkeruun katkaisupisteeltä 12.6.2025 (maturiteetti 57 %).
a Sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) PFS-tulokset vastasivat tutkijalääkärin raportoimia tuloksia.
b Korjattu välianalyysien suhteen, tilastollisen merkitsevyyden saavuttaminen lopullisessa kokonaiselossaoloaika-analyysissa edellytti p‑arvoa < 0,04953.
Kuva 9. FLAURA2-tutkimuksen etenemisvapaata elossaoloaikaa kuvaavat Kaplan–Meier-käyrät (tutkijalääkärin arvio)
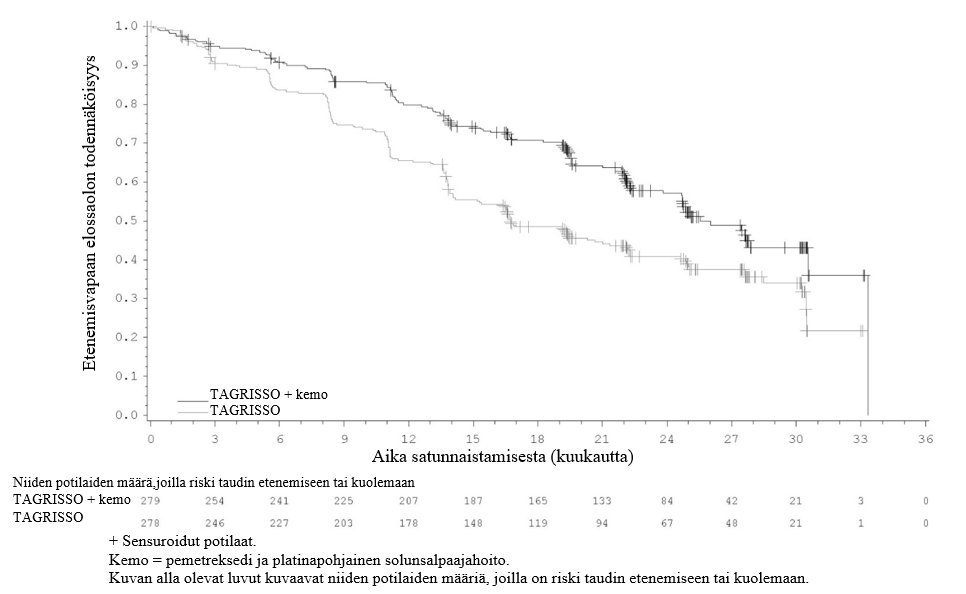
Kuva 10. FLAURA2-tutkimuksen kokonaiselossaoloaikaa kuvaavat Kaplan–Meier-käyrät
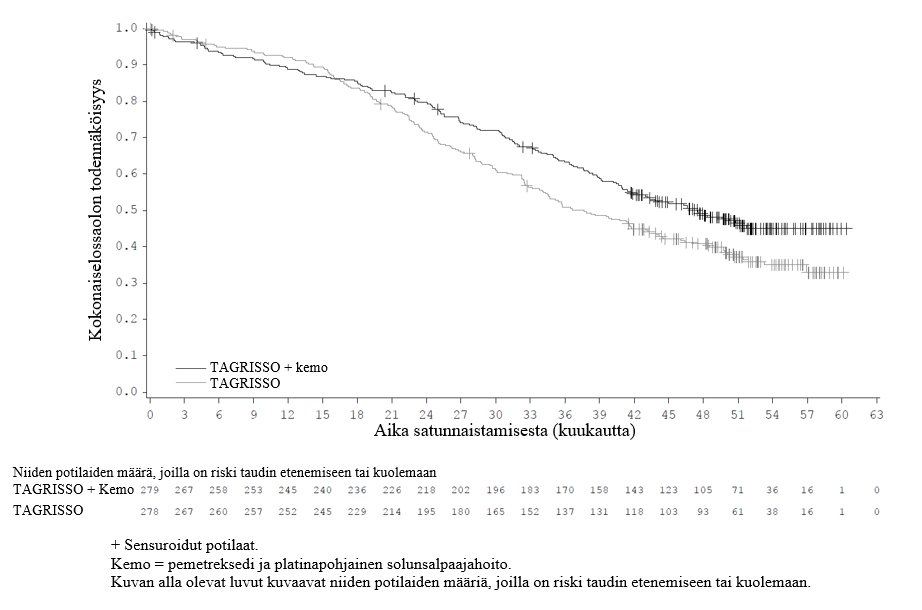
Keskushermostometastaaseihin liittyvät tehoa koskevat tulokset FLAURA2-tutkimuksessa
FLAURA2-tutkimukseen voitiin satunnaistaa mukaan potilaita, joilla oli oireettomia keskushermostometastaaseja, joita ei ollut hoidettu steroideilla ja joiden neurologinen status oli ollut vakaa vähintään kahden viikon ajan definitiivisen hoidon ja steroidihoidon päättymisen jälkeen. Kaikkien potilaiden lähtötilanteen aivokuvaustulokset olivat saatavilla. BICR arvioi nämä kuvaukset käyttämällä muokattuja RECIST-kriteerejä, ja arvioinnin tuloksena oli alaryhmä, jossa 222 potilaalla 557:stä (40 %) oli todettu mitattavissa ja/tai ei-mitattavissa olevia keskushermostoleesioita (cFAS), sekä toinen alaryhmä, jossa 78 potilaalla 557:stä (14 %) oli todettu mitattavissa olevia keskushermostoleesioita (cEFR). Eksploratiivisen analyysin perusteella keskushermostoon liittyvän vasteen saavuttaneiden osuus oli molemmissa hoitohaaroissa > 65 %, ja hoitohaarassa, jossa potilaat saivat TAGRISSO-valmistetta pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, täydellisen vasteen saavuttaneiden osuus oli suurempi (59,3 % potilaista) kuin hoitohaarassa, jossa potilaat saivat TAGRISSO-valmistetta monoterapiana (43,3 % potilaista). Vasteen keston mediaania ei saavutettu hoitohaarassa, jossa potilaat saivat TAGRISSO-valmistetta pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, ja se oli 26,2 kuukautta hoitohaarassa, jossa potilaat saivat TAGRISSO-valmistetta monoterapiana. cEFR-alaryhmässä keskushermostoon liittyvän täydellisen vasteen saavuttaneiden osuus oli 47,5 % hoitohaarassa, jossa potilaat saivat TAGRISSO-valmistetta pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, ja 15,8 % hoitohaarassa, jossa potilaat saivat TAGRISSO-valmistetta monoterapiana.
Aiempaa hoitoa saaneet T790M-positiiviset NSCLC-potilaat – AURA3
TAGRISSO-hoidon turvallisuus ja teho osoitettiin satunnaistetussa, avoimessa, aktiivikontrolloidussa faasin 3 tutkimuksessa (AURA3) sellaisten potilaiden hoidossa, joilla on paikallisesti edennyt tai metastaattinen ei-pienisoluinen keuhkosyöpä (NSCLC), jossa on EGFR:n T790M-mutaatio, ja joiden tauti on edennyt EGFR-TKI-hoidon aikana tai sen jälkeen. Tutkimuksessa edellytettiin, että kaikilla siihen osallistuvilla potilailla on ennen satunnaistamista keskuslaboratoriossa Cobas-EGFR‑mutaatiotestillä määritetty EGFR T790M-mutaatiopositiivinen NSCLC. T790M-mutaatiostatus arvioitiin myös käyttämällä ctDNA:ta, joka oli eristetty tutkimuksen sisäänottovaiheen aikana otetusta plasmanäytteestä. Ensisijainen tehoa kuvaava päätetapahtuma oli tutkijalääkärin arvioima etenemisvapaa elossaoloaika (Progression-Free Survival, PFS). Muita tehoa kuvaavia päätetapahtumia olivat tutkijalääkärin arvioimat objektiivinen hoitovaste (ORR), vasteen kesto (DoR) ja kokonaiselossaoloaika (OS).
Potilaat satunnaistettiin suhteessa 2:1 (TAGRISSO: platinapohjainen kahden valmisteen yhdistelmäkemoterapia) saamaan TAGRISSO-valmistetta (n = 279) tai platinapohjaista kahden valmisteen yhdistelmäkemoterapiaa (n = 140). Satunnaistamisen yhteydessä tehty stratifiointi perustui etniseen taustaan (aasialaiset ja muut kuin aasialaiset). TAGRISSO-haaran potilaat saivat TAGRISSO-valmistetta 80 mg suun kautta kerran vuorokaudessa, kunnes potilaalle kehittyi intoleranssi hoidolle tai tutkijalääkäri katsoi, että potilas ei enää kliinisesti hyötynyt hoidosta. Solunsalpaajahoito koostui pemetreksedista (500 mg/m2) ja karboplatiinista (AUC5) tai pemetreksedista (500 mg/m2) ja sisplatiinista (75 mg/m2), jotka annettiin kunkin 21 päivän hoitosyklin päivänä 1 enintään 6 hoitosyklin ajan. Potilaat, joiden tauti ei edennyt platinapohjaisen solunsalpaajahoidon neljän hoitosyklin jälkeen, saattoivat saada pemetreksediylläpitohoitoa (pemetreksedi 500 mg/m2 kunkin 21 päivän hoitosyklin päivänä 1). Solunsalpaajahaaran tutkittavat, joiden tauti oli edennyt objektiivisen radiologisen arvioinnin mukaan (arvion teki tutkijalääkäri ja arvion vahvisti riippumaton keskitetty kuvantamiseen erikoistunut arvioijataho), saivat mahdollisuuden aloittaa TAGRISSO-hoidon.
Koko tutkimuspopulaation demografiset tiedot ja tautitiedot lähtötilanteessa olivat: mediaani-ikä 62, vähintään 75-vuotiaita 15 %, naisia 64 %, valkoihoisia 32 %, aasialaisia 65 %, 68 % tutkittavista ei ollut koskaan tupakoinut, 100 %:lla potilaista WHO:n mukainen suorituskykyluokitus oli 0 tai 1. 54 %:lla potilaista oli rintakehän ulkopuolisia metastaaseja sisäelimissä, mukaan lukien 34 % potilaita, joilla oli keskushermostometastaaseja (todettu keskushermostossa sijaitseva leesio lähtötilanteessa, todettu aiemmin sairaushistoriassa ja/tai aiemman leikkauksen ja/tai keskushermostometastaaseihin aiemmin annetun sädehoidon perusteella), ja 23 % potilaita, joilla oli maksametastaaseja. 42 %:lla potilaista oli metastaattinen luusairaus.
AURA3-tutkimuksessa osoitettiin, että etenemisvapaa elossaoloaika piteni tilastollisesti merkitsevästi TAGRISSO-hoitoa saaneilla potilailla solunsalpaajahoitoon verrattuna. Tutkijalääkärin arvioimista AURA3-tutkimuksen tehoa koskevista tuloksista on esitetty yhteenveto taulukossa 8, ja Kaplan–Meier-käyrä etenemisvapaasta elossaoloajasta on esitetty kuvassa 11. Lopullisessa kokonaiselossaoloaika-analyysissa ei havaittu tilastollisesti merkitseviä eroja hoitohaarojen välillä.
Taulukko 8. AURA3‑tutkimuksen tehoa koskevat tulokset (tutkijalääkärin arvio)
Tehoa kuvaava muuttuja | TAGRISSO | Solunsalpaajahoito (pemetreksedi/sisplatiini tai pemetreksedi/karboplatiini) | |
Etenemisvapaa elossaoloaika (PFS) | |||
Tapahtumien lukumäärä (maturiteetti, %) | 140 (50) | 110 (79) | |
Mediaani PFS, kuukautta (95 %:n luottamusväli) | 10,1 (8,3, 12,3) | 4,4 (4,2, 5,6) | |
HR (95 %:n luottamusväli); p-arvo | 0,30 (0,23, 0,41); p < 0,001 | ||
Kokonaiselossaoloaika (OS)a | |||
Kuolemien lukumäärä (maturiteetti, %) | 188 (67,4) | 93 (66,4) | |
Mediaani OS, kuukautta (95 %:n luottamusväli) | 26,8 (23,5, 31,5) | 22,5 (20,2, 28,8) | |
HR (95,56 %:n luottamusväli); p-arvo | 0,87 (0,67, 1,13); p = 0,277 | ||
Objektiivinen hoitovasteb | |||
Vasteiden määrä, hoitovaste (95 %:n luottamusväli) | 197 | 44 | |
Ristitulosuhde (odds ratio) (95 %:n luottamusväli); p-arvo | 5,4 (3,5, 8,5); p < 0,001 | ||
Vasteen kesto (DoR)b | |||
Vasteen keston mediaani, kuukautta (95 %:n luottamusväli) | 9,7 (8,3, 11,16) | 4,1 (3,0, 5,6) | |
HR = riskisuhde; NC = ei laskettavissa; OS = kokonaiselossaoloaika
Kaikki tehoa koskevat tulokset perustuvat tutkijalääkärin arvioon (RECIST).
a Lopullinen kokonaiselossaoloajan (OS) analyysi tehtiin, kun maturiteetti oli 67 %. Riskisuhteen luottamusväli on korjattu aiempien välianalyysien suhteen. Kokonaiselossaoloajan analyysiä ei korjattu cross-overista johtuvien mahdollisesti sekoittavien vaikutusten suhteen (99 [71 %] solunsalpaajahaaran potilasta sai seuraavaksi osimertinibihoitoa).
b Tutkijalääkärin arvioimat ORR- ja DoR-tulokset vastasivat sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) raportoimia tuloksia; BICR-arvion mukaan ORR oli osimertinibilla 64,9 % [95 %:n luottamusväli: 59,0, 70,5] ja solunsalpaajahoidolla 34,3 % [95 %:n luottamusväli: 25,6, 42,8]; BICR-arvion mukaan DoR oli osimertinibilla 11,2 kuukautta (95 %:n luottamusväli: 8,3, NC) ja solunsalpaajahoidolla 3,1 kuukautta (95 %:n luottamusväli: 2,9, 4,3).
Kuva 11. AURA3-tutkimuksen etenemisvapaata elossaoloaikaa kuvaavat Kaplan–Meier-käyrät (tutkijalääkärin arvio)
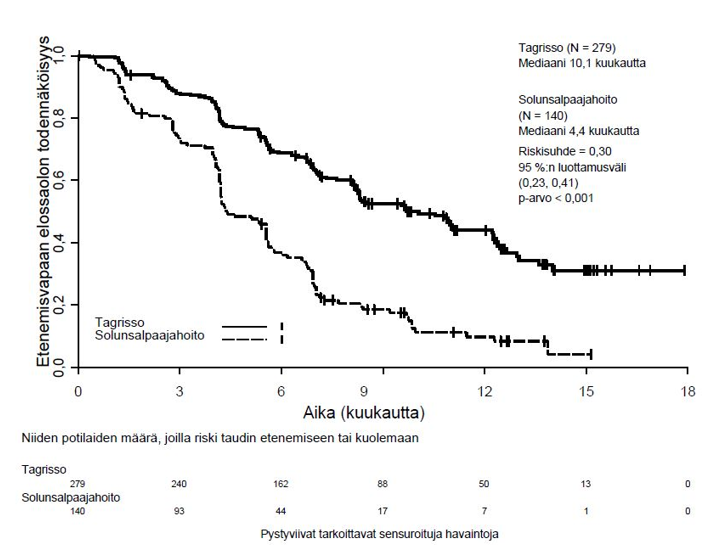
Sokkoutettu riippumaton keskitetty arvioijataho (BICR) suoritti etenemisvapaan elossaoloajan herkkyysanalyysin, jonka mukaan etenemisvapaan elossaoloajan mediaani oli TAGRISSO-hoidolla 11,0 kuukautta ja solunsalpaajahoidolla 4,2 kuukautta. Tämä analyysi osoitti hoitovaikutuksen (riskisuhde 0,28, 95 %:n luottamusväli: 0,20, 0,38), joka oli yhdenmukainen tutkijalääkärin arvioinnissa havaitun hoitovaikutuksen kanssa.
Kaikissa ennalta määritellyissä analysoiduissa alaryhmissä, joita olivat etniseen taustaan, ikään, sukupuoleen, tupakointitaustaan ja EGFR-mutaatioon (eksonin 19 deleetio ja L858R) perustuvat alaryhmät, havaittiin johdonmukaisesti kliinisesti merkityksellisiä parannuksia etenemisvapaassa elossaoloajassa riskisuhteilla, jotka olivat alle 0,50 TAGRISSO-hoitoa saaneiden potilaiden eduksi verrattuna solunsalpaajahoitoa saaneisiin.
Keskushermostometastaaseihin liittyvät tehoa koskevat tulokset AURA3-tutkimuksessa
Tutkimukseen voitiin randomoida mukaan potilaita, joilla oli oireettomia stabiileja aivometastaaseja, joita ei ollut hoidettu steroideilla ainakaan 4 viikkoon ennen tutkimushoidon alkamista. Taulukossa 9 on esitetty yhteenveto keskushermostoon liittyvän tehon BICR-arvioinnista RECIST v1.1 ‑kriteereillä alaryhmässä, jonka 116/419 (28 %) potilaalla oli todettu keskushermostometastaaseja lähtötilanteen aivokuvauksessa.
Taulukko 9. Keskushermostoon liittyvä teho BICR-arvion mukaan potilailla, joilla oli keskushermostometastaaseja AURA3-tutkimuksen lähtötilanteen aivokuvauksessa
Tehoa kuvaava muuttuja | TAGRISSO | Solunsalpaajahoito (pemetreksedi/sisplatiini tai pemetreksedi/karboplatiini) |
Keskushermostoon liittyvä objektiivinen hoitovastea | ||
Keskushermostoon liittyvä hoitovaste % (n/N) (95 %:n luottamusväli) | 70 % (21/30) (51, 85) | 31 % (5/16) (11 %, 59 %) |
Ristitulosuhde, OR, (95 %:n luottamusväli); p-arvo | 5,1 (1,4, 21); p = 0,015 | |
Keskushermostoon liittyvän vasteen kestob | ||
Keskushermostoon liittyvän vasteen keston mediaani, kuukautta (95 %:n luottamusväli) | 8,9 (4,3, NC) | 5,7 (NC, NC) |
Keskushermostoon liittyvän sairauden hallinnan aste | ||
Potilaat, joilla keskushermostoon metastasoitunut tauti oli hallinnassa (DCR), % (n/N) (95 %:n luottamusväli) | 87 % (65/75) (77, 93) | 68 % (28/41) (52, 82) |
Ristitulosuhde, OR, (95 %:n luottamusväli); p-arvo | 3 (1,2, 7,9); p = 0,021 | |
Keskushermostoon liittyvä etenemisvapaa elossaoloaikac | N = 75 | N = 41 |
Tapahtumien lukumäärä (maturiteetti, %) | 19 (25) | 16 (39) |
Keskushermostoon liittyvän etenemisvapaan elossaoloajan mediaani, kuukautta (95 %:n luottamusväli) | 11,7 (10, NC) | 5,6 (4,2, 9,7) |
Riskisuhde (95 %:n luottamusväli); p-arvo | 0,32 (0,15, 0,69); p = 0,004 | |
a Keskushermostoon liittyvä objektiivinen hoitovaste ja vasteen kesto keskushermoston BICR-arvion mukaan RECIST v1.1 ‑kriteereillä määritettynä populaatiossa, jossa vaste oli arvioitavissa (lähtötilanteessa mitattavissa olevia keskushermostoleesioita BICR:n mukaan), n = 30 TAGRISSO-valmisteella ja n = 16 solunsalpaajahoidolla.
b Perustuu ainoastaan potilaisiin, jotka saivat vasteen; DoR määriteltiin ajaksi ensimmäisestä dokumentoidusta vasteesta (täydellisestä vasteesta tai osittaisesta vasteesta) taudin etenemistapahtumaan tai kuolemaan; DCR määriteltiin niiden potilaiden osuudeksi, jotka saivat vasteen (täydellisen vasteen tai osittaisen vasteen) tai joiden tauti oli vakaa vähintään kuuden viikon ajan.
c Keskushermostoon liittyvä etenemisvapaa elossaoloaika keskushermoston BICR-arvion mukaan RECIST v1.1 ‑kriteereillä määritettynä koko analyysisarjanpopulaatiossa (lähtötilanteessa mitattavissa ja ei-mitattavissa olevia keskushermostoleesioita BICR:n mukaan), n = 75 TAGRISSO-valmisteella ja n = 41 solunsalpaajahoidolla.
Riskisuhde, joka on alle 1, suosii TAGRISSO-valmistetta.
AURA3-tutkimuksessa arvioitiin ennalta määritetty etenemisvapaan elossaoloajan alaryhmä, joka perustui keskushermostometastaasistatukseen tutkimuksessa aloittamisen hetkellä, ja arvioinnin tulokset on esitetty kuvassa 12.
Kuva 12. Etenemisvapaa elossaoloaika (tutkijan arvio) keskushermostometastaasistatuksen mukaan tutkimuksessa aloittamisen hetkellä, Kaplan–Meier-käyrä (koko analyysisarja) AURA3-tutkimuksessa
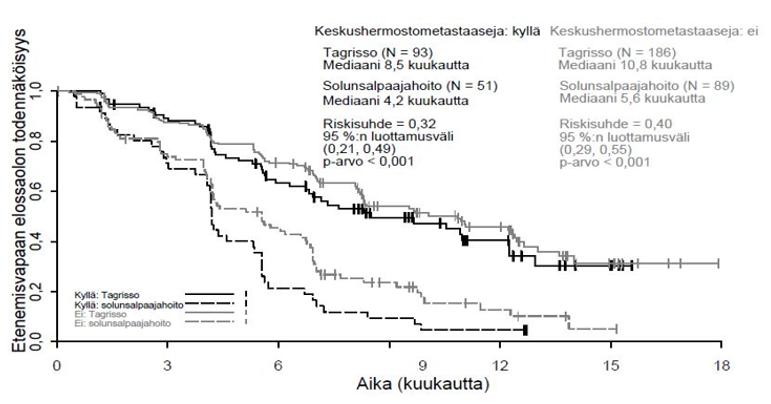
AURA3-tutkimuksessa osoitettiin tilastollisesti merkitsevä etenemisvapaan elossaoloajan piteneminen TAGRISSO-valmistetta saaneilla potilailla verrattuna solunsalpaajahoitoa saaneisiin potilaisiin riippumatta keskushermostometastaasistatuksesta tutkimuksen sisäänottovaiheessa.
Potilaan ilmoittamat vaikutukset (Patient Reported Outcomes, PRO)
Potilaiden ilmoittamat oireet ja terveyteen liittyvää elämänlaatua koskevat tiedot kerättiin sähköisesti EORTC QLQ-C30:llä ja sen keuhkosyöpämoduulilla (EORTC QLQ-LC13). LC13 täytettiin aluksi kerran viikossa ensimmäisten 6 viikon ajan ja sen jälkeen 3 viikon välein ennen taudin etenemistä ja sen jälkeen. C30 arvioitiin 6 viikon välein ennen taudin etenemistä ja sen jälkeen.
Keskeisten keuhkosyövän oireiden analyysi
TAGRISSO paransi tilastollisesti merkitsevästi potilaiden ilmoittamia keuhkosyövän oireita solunsalpaajahoitoon verrattuna viiden ennalta määritellyn keskeisimmän potilaan ilmoittaman oireen (ruokahaluttomuus, yskä, rintakipu, hengenahdistus ja väsymys) osalta, kun mitattiin muutosta lähtötilanteesta kuuteen kuukauteen saakka, kuten on esitetty taulukossa 10.
Taulukko 10. Toistettujen mittausten sekamalli – keskeiset keuhkosyövän oireet – keskimääräinen muutos lähtötilanteesta TAGRISSO-valmistetta saaneilla potilailla solunsalpaajahoitoon verrattuna
| Ruokahaluttomuus | Yskä | Rintakipu | Hengenahdistus | Väsymys | |||||
Hoitohaarat | TAGRISSO (279) | Solunsalpaajahoito (140) | TAGRISSO (279) | Solunsalpaajahoito (140) | TAGRISSO (279) | Solunsalpaajahoito (140) | TAGRISSO (279) | Solunsalpaajahoito (140) | TAGRISSO (279) | Solunsalpaajahoito (140) |
N | 239 | 97 | 228 | 113 | 228 | 113 | 228 | 113 | 239 | 97 |
Korjattu keskiarvo | -5,51 | 2,73 | -12,22 | -6.69 | -5,15 | 0,22 | -5,61 | 1,48 | -5,68 | 4,71 |
Arvioitu ero (95 %:n luottamusväli) | -8,24 (-12,88, 3,60) | -5,53 (-8,89, -2,17) | -5,36 (-8,20, -2,53) | -7,09 (-9,86, -4,33) | -10,39 (-14,55, -6,23) | |||||
p-arvo | p < 0,001 | p = 0,001 | p < 0,001 | p < 0,001 | p < 0,001 | |||||
Korjattu keskiarvo ja arvioidut erot saatiin toistettujen mittausten sekamallin (Mixed Model Repeated Measures, MMRM) analyysistä. Mallissa oli mukana potilas, hoito, lääkärissä käynti, käyntikohtainen hoidon vuorovaikutus, lähtötilanteen oirepistemäärä ja lähtötilanteen oireiden käyntikohtaiset vuorovaikutuksen pistemäärät, ja siinä käytettiin strukturoimatonta kovarianssimatriisia.
Terveyteen liittyvän elämänlaadun ja fyysisen toimintakyvyn paranemisen analyysi
TAGRISSO-hoitoa saaneilla potilailla oli solunsalpaajahoitoon verrattuna merkittävästi suuremmat mahdollisuudet saavuttaa kliinisesti merkittävä paraneminen eli vähintään 10 pisteen parannus yleistä terveydentilaa ja fyysistä toimintakykyä mittaavassa EORTC-C30-kyselyssä tutkimusjakson aikana: ristitulosuhde (OR) yleiselle terveydentilalle: 2,11 (95 %:n luottamusväli 1,24, 3,67, p = 0,007); OR fyysiselle toimintakyvylle 2,79 (95 %:n luottamusväli: 1,50, 5,46, p = 0,002).
Aiemmin hoitoa saaneet T790M-positiiviset NSCLC-potilaat – AURAex ja AURA2
Kahdessa yksihaaraisessa avoimessa kliinisessä tutkimuksessa, AURAex (faasin 2 kohortin laajennustutkimus [n = 201]) ja AURA2 (n = 210), tutkittiin potilaita, jotka sairastivat EGFR T790M-mutaatiopositiivista keuhkosyöpää, joka oli edennyt yhden tai useamman systeemisen hoidon aikana, mukaan lukien EGFR-TKI-hoidot. Kaikilla potilailla edellytettiin ennen hoidon aloittamista olevan keskuslaboratoriossa Cobas-EGFR‑mutaatiotestillä määritetty EGFR T790M-mutaatiopositiivinen NSCLC. T790M-mutaatiostatus arvioitiin myös retrospektiivisesti käyttämällä tutkimuksen sisäänottohetkellä otetusta plasmanäytteestä eristettyä ctDNA:ta. Kaikki potilaat saivat TAGRISSO‑valmistetta 80 mg kerran vuorokaudessa. Näiden kahden tutkimuksen ensisijainen tehoa kuvaava päätetapahtuma oli RECIST v.1.1 ‑kriteerien mukainen objektiivinen hoitovaste (objective response rate, ORR), jonka arvioi sokkoutettu riippumaton keskitetty arvioijataho (BICR). Toissijaiset tehoa kuvaavat päätetapahtumat olivat vasteen kesto (DoR) ja etenemisvapaa elossaoloaika (PFS).
Kokonaistutkimuspopulaation tiedot (AURAex‑ ja AURA2‑tutkimuksissa) lähtötilanteessa olivat: mediaani‑ikä oli 63 vuotta, 13 % potilaista oli vähintään 75‑vuotiaita, naisia oli 68 %, valkoihoisia 36 % ja aasialaisia 60 %. Kaikki potilaat olivat saaneet vähintään yhtä aiemman linjan hoitoa. 31 % (N = 129) potilaista oli saanut yhtä aiemman linjan hoitoa (ainoastaan EGFR‑TKI‑hoitoa), 69 % (N = 282) oli saanut kahta tai useampaa aiemman linjan hoitoa. 72 % potilaista ei ollut koskaan tupakoinut, 100 %:lla potilaista WHO:n mukainen suorituskykyluokitus oli 0 tai 1. 59 %:lla potilaista oli rintakehän ulkopuolisia metastaaseja sisäelimissä, mukaan lukien 39 % potilaita, joilla oli keskushermostometastaaseja (todettu keskushermostossa sijaitseva leesio lähtötilanteessa, todettu aiemmin sairaushistoriassa ja/tai aiemman leikkauksen ja/tai keskushermostometastaaseihin aiemmin annetun sädehoidon perusteella), ja 29 % potilaita, joilla oli maksametastaaseja. 47 %:lla potilaista oli metastaattinen luusairaus. Etenemisvapaan elossaoloajan seurannan mediaanikesto oli 12,6 kuukautta.
411 potilaalla, jotka olivat saaneet aiempaa hoitoa ja jotka olivat positiivisia EGFR:n T790M-mutaation suhteen, kokonais-ORR oli sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) mukaan 66 % (95 %:n luottamusväli: 61, 71). Potilailla, jotka saivat BICR:n vahvistaman vasteen, mediaani-DoR oli 12,5 kuukautta (95 %:n luottamusväli: 11, 1, ei arvioitu). ORR oli BICR:n mukaan AURAex-tutkimuksessa 62 % (95 %:n luottamusväli: 55, 68) ja AURA2-tutkimuksessa 70 % (95 %:n luottamusväli: 63, 77). Etenemisvapaan elossaoloajan (PFS) mediaani oli 11,0 kuukautta (95 %:n luottamusväli: 9,6, 12,4).
Sokkoutetun riippumattoman keskitetyn arvioijatahon (BICR) arvioima yli 50 %:n objektiivinen hoitovaste havaittiin kaikissa analysoiduissa ennalta määritellyissä alaryhmissä, mukaan lukien hoitolinja, etninen tausta, ikä ja maantieteellinen alue.
Vasteen suhteen arvioitavissa olevasta populaatiosta 85 %:lla (223 tutkittavalla 262:sta) dokumentoitiin vaste ensimmäisen kuvantamisen ajankohtana (6 viikon kohdalla); 94 %:lla (247 tutkittavalla 262:sta) dokumentoitiin vaste toisen kuvantamisen ajankohtana (12 viikon kohdalla).
Keskushermostometastaaseihin liittyvät tehoa koskevat tiedot faasin 2 tutkimuksissa (AURAex ja AURA2)
Keskushermostoon liittyvän tehon RECIST v1.1 ‑kriteereihin perustuva BICR-arvio tehtiin 50 potilaan alaryhmässä (411 potilaasta), jossa potilailla oli todettu lähtötilanteen aivokuvauksessa mitattavissa olevia keskushermostometastaaseja. 54 %:lla potilaista (27 potilaalla 50 potilaasta; 95 %:n luottamusväli: 39,3, 68,2) todettiin keskushermostoon liittyvä objektiivinen vaste (ORR). 12 % näistä vasteista oli täydellisiä vasteita.
Kliinisiä tutkimuksia ei ole tehty potilailla, joilla on de novo EGFR T790M‑mutaatiopositiivinen NSCLC.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset TAGRISSO-valmisteen käytöstä NSCLC:n hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Osimertinibin farmakokineettiset parametrit määritettiin terveillä tutkittavilla ja NSCLC:tä sairastavilla potilailla. Perustuen populaatiofarmakokineettiseen analyysiin osimertinibin näennäinen plasmapuhdistuma on 14,3 l/h, näennäinen jakautumistilavuus 918 l ja terminaalinen puoliintumisaika noin 44 tuntia. Farmakokinetiikka potilailla, jotka ovat saaneet osimertinibia yhdistelmänä pemetreksedin ja platinapohjaisen solunsalpaajahoidon kanssa, on samankaltainen kuin potilailla, jotka ovat saaneet osimertinibia monoterapiana. AUC ja Cmax suurenivat suhteessa annokseen annosvälillä 20−240 mg. Osimertinibin annostelu kerran vuorokaudessa johtaa noin kolminkertaiseen akkumuloitumiseen ja vakaan tilan altistus saavutetaan 15 vuorokauden hoidolla. Vakaassa tilassa kiertävät pitoisuudet plasmassa säilyvät tavallisesti 1,6‑kertaisen vaihteluvälin sisällä annosvälin ollessa 24 tuntia.
Imeytyminen
TAGRISSO‑valmisteen suun kautta annostelun jälkeen mediaani‑tmax osimertinibin huippupitoisuuden saavuttamiseen plasmassa oli 6 (3–24) tuntia. Joillakin potilailla havaittiin useita huippupitoisuuksia ensimmäisten 24 tunnin aikana. TAGRISSO-valmisteen absoluuttinen hyötyosuus on 70 % (90 %:n luottamusväli 67, 73). Kliiniseen farmakokineettiseen tutkimukseen perustuen 80 mg:n annosta saaneilla potilailla ruoka ei muuttanut osimertinibin hyötyosuutta kliinisesti merkittävästi (AUC suureni 6 % [90 %:n luottamusväli ‑5, 19] ja Cmax pieneni 7 % [90 %:n luottamusväli ‑19, 6]). Altistus osimertinibille ei muuttunut (AUC suureni 7 % ja Cmax 2 %) ja altistussuhteen 90 %:n luottamusväli oli 80–125 %:n rajoissa annettaessa terveillä vapaaehtoisilla 80 mg:n tabletti ja omepratsolia 5 vuorokauden ajan mahalaukun pH-arvon suurentamiseksi.
Jakautuminen
Populaation arvioitu osimertinibin keskimääräinen vakaan tilan jakautumistilaisuus (Vss/F) on 918 l, mikä viittaa laajaan jakautumiseen kudoksissa. In vitro osimertinibin sitoutumisaste plasman proteiineihin on 94,7 % (5,3 % vapaana). Osimertinibin on myös osoitettu sitoutuvan kovalenttisesti rotan ja ihmisen plasman proteiineihin, ihmisen seerumialbumiiniin sekä rotan ja ihmisen maksasoluihin.
Biotransformaatio
In vitro -tutkimuksissa on osoitettu, että osimertinibi metaboloituu pääasiassa CYP3A4:n ja CYP3A5:n välityksellä. Tällä hetkellä saatavissa olevan tiedon mukaan vaihtoehtoisia metaboliareittejä ei voida kuitenkaan täysin sulkea pois. In vitro ‑tutkimuksissa havaittua kahta farmakologisesti aktiivista metaboliittia (AZ7550 ja AZ5104) on havaittu myös prekliinisten lajien plasmassa ja ihmisillä osimertinibin oraalisen annostelun jälkeen. AZ7550:lla on osoitettu olevan samankaltainen farmakologinen profiili kuin TAGRISSO-valmisteella, kun taas AZ5104:n on osoitettu olevan tehokkaampi sekä mutatoituneeseen että villityypin EGFR:ään. Molemmat metaboliitit olivat havaittavissa plasmassa hitaasti TAGRISSO-valmisteen antamisen jälkeen; niiden mediaani-tmax-arvot (vähimmäis- ja enimmäisajat) olivat 24 (4–72) ja 24 (6–72) tuntia. Ihmisen plasmassa muuttumattoman osimertinibin osuus on 0,8 % ja näiden kahden metaboliitin osuudet ovat 0,08 % ja 0,07 % kokonaisradioaktiivisuudesta, suurimman osan radioaktiivisuudesta ollessa sitoutunut kovalenttisesti plasman proteiineihin. Sekä AZ5104- että AZ7550-metaboliitin AUC-arvoon perustuvan altistuksen geometrinen keskiarvo oli noin 10 % osimertinibialtistuksesta vakaassa tilassa.
Osimertinibin pääasiallinen metaboliareitti oli hapettuminen ja dealkylaatio. Yhdistetyissä ihmisiltä kerätyistä virtsa- ja ulostenäytteissä havaittiin vähintään 12:ta komponenttia, joista 5 komponentin osuus oli yli 1 % annoksesta. Muuttumattoman osimertinibin osuus oli 1,9 %, AZ5104:n osuus 6,6 % ja AZ7550:n osuus 2,7 % annoksesta, kun taas kysteinyyliadduktin (M21) osuus oli 1,5 % ja tuntemattoman metaboliitin (M25) osuus 1,9 % annoksesta.
In vitro ‑tutkimusten perusteella osimertinibi on kliinisesti merkittävinä pitoisuuksina kompetitiivinen CYP3A4/5:n, mutta ei CYP1A2:n, ‑2A6:n, ‑2B6:n, ‑2C8:n, ‑2C9:n, ‑2C19:n, ‑2D6:n tai ‑2E1:n inhibiittori. In vitro ‑tutkimusten perusteella osimertinibi ei ole kliinisesti merkittävinä pitoisuuksina UGT1A1:n tai UGT2B7:n inhibiittori maksassa. UGT1A1:n inhibointi suolistossa on mahdollista, mutta sen kliinistä vaikutusta ei tunneta.
Eliminaatio
Suun kautta annostellun 20 mg:n kerta-annoksen jälkeen 67,8 % annoksesta erittyi ulosteeseen (1,2 % muuttumattomana lääkeaineena) ja 14,2 % annoksesta todettiin virtsasta (0,8 % muuttumattomana lääkeaineena) 84 päivän näytteiden keräämisen aikana. Muuttumattoman osimertinibin osuus oli noin 2 % eliminaatiosta, ja siitä poistui virtsaan 0,8 % ja ulosteeseen 1,2 %.
Yhteisvaikutukset kuljettajaproteiinien kanssa
In vitro ‑tutkimukset ovat osoittaneet, että osimertinibi ei ole OATP1B1:n eikä OATP1B3:n substraatti. Osimertinibi ei inhiboi OAT1:tä, OAT3:a, OATP1B1:tä, OATP1B3:a, MATE1:tä, OCT2:ta tai MATE2K:ta in vitro kliinisesti merkittävinä pitoisuuksina.
In vitro ‑tutkimusten perusteella osimertinibi on P-gp:n ja BCRP:n substraatti, mutta kliinisinä annoksina sillä ei todennäköisesti ole kliinisesti merkittäviä yhteisvaikutuksia. In vitro ‑tutkimusten perusteella osimertinibi on BCRP:n ja P-gp:n inhibiittori (ks. kohta Yhteisvaikutukset).
Erityisryhmät
Populaatioon perustuvassa farmakokineettisessä analyysissä (n = 1 367) ei todettu kliinisesti merkittäviä yhteyksiä ennustetun vakaan tilan altistuksen (AUCss) ja potilaan iän (vaihteluväli: 25−91 vuotta), sukupuolen (naisia 65 %), etnisen taustan (mukaan lukien valkoihoiset, aasialaiset, japanilaiset, kiinalaiset ja ei-aasialaiset, ei-valkoihoiset potilaat), hoitolinjan tai tupakoinnin (n = 34 tupakoiville potilaille, n = 419 aiemmin tupakoineille potilaille) välillä. Populaatiofarmakokineettisen analyysin mukaan paino oli merkittävä kovariaatti, sillä osimertinibin odotettu AUCss-arvon muutos oli vähemmän kuin 20 % vastaavasti painovälillä 88 kg – 43 kg (kvantiilit 95 % – 5 %) verrattuna mediaanipainon (61 kg) AUCss-arvoon. Huomioiden potilaiden painojen vaihtelun rajat, < 43 kg – > 88 kg, AZ5104-metaboliitin suhteellinen osuus oli välillä 11,8−9,6 %, kun taas AZ7550-metaboliitin suhteellinen osuus oli välillä 12,8–8,1 %. Populaatiofarmakokineettisen analyysin perusteella seerumin albumiinin todettiin olevan merkittävä kovariaatti, sillä osimertinibin odotettu AUCss-arvon muutos oli < 30 %, vastaavasti albumiiniarvojen muutos välillä 29–46 g/l (kvantiilit 95 % – 5 %), verrattuna lähtötason albumiinin mediaani (39 g/l) AUCss-arvoon. Näiden painoeroista tai lähtötason albumiinin eroista johtuvien altistusmuutosten ei katsota olevan kliinisesti merkittäviä.
Maksan vajaatoiminta
Osimertinibi eliminoituu pääasiassa maksan kautta. Potilailla, joilla oli erityyppisiä pitkälle edenneitä kiinteitä kasvaimia ja lievä maksan vajaatoiminta (Child–Pugh-luokka A, keskimääräinen pistemäärä 5,3, n = 7) tai kohtalainen maksan vajaatoiminta (Child–Pugh-luokka B, keskimääräinen pistemäärä 8,2, n = 5), ei kliinisessä tutkimuksessa todettu TAGRISSO-valmisteen 80 mg:n kerta-annoksen antamisen jälkeen suurentunutta altistusta verrattuna potilaisiin, joiden maksan toiminta oli normaali (n = 10). Verrattuna altistukseen potilailla, joilla maksan toiminta oli normaali, geometristen keskiarvojen suhteet (90 %:n luottamusväli) olivat osimertinibin AUC-arvolle 63,3 % (47,3, 84,5) ja Cmax-arvolle 51,4 % (36,6, 72,3) lievää maksan vajaatoimintaa sairastavilla potilailla sekä AUC-arvolle 68,4 % (49,6, 94,2) ja Cmax-arvolle 60,7 % (41,6, 88,6) kohtalaista maksan vajaatoimintaa sairastavilla potilailla. Vastaavat geometristen keskiarvojen suhteet olivat AZ5104-metaboliitin AUC-arvolle 66,5 % (43,4, 101,9) ja Cmax-arvolle 66,3 % (45,3, 96,9) lievää maksan vajaatoimintaa sairastavilla potilailla sekä AUC-arvolle 50,9 % (31,7, 81,6) ja Cmax-arvolle 44,0 % (28,9, 67,1) kohtalaista maksan vajaatoimintaa sairastavilla potilailla. Populaatiofarmakokineettisen analyysin perusteella maksan toimintaa kuvaavien arvojen (ALAT, ASAT, bilirubiini) ja osimertinibialtistuksen välillä ei ole yhteyttä. Maksan vajaatoimintaa kuvaavan arvon, seerumialbumiinin, osoitettiin vaikuttavan osimertinibin farmakokinetiikkaan. Kliinisistä tutkimuksista suljettiin pois potilaat, joiden ASAT tai ALAT oli yli 2,5-kertainen viitealueen ylärajaan (ULN) verrattuna tai taustalla olevan pahanlaatuisen sairauden vuoksi yli viisinkertainen viitealueen ylärajaan verrattuna tai joiden kokonaisbilirubiini oli yli 1,5-kertainen viitealueen ylärajaan verrattuna. Farmakokineettisen analyysin perusteella osimertinibialtistus oli samanlainen 134:llä lievää maksan vajaatoimintaa sairastavalla potilaalla ja kahdeksalla kohtalaista maksan vajaatoimintaa sairastavalla potilaalla kuin 1 216 potilaalla, joiden maksan toiminta oli normaali. Käytöstä vaikeaa maksan vajaatoimintaa sairastaville potilaille ei ole saatavilla tietoa (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Potilailla, joilla oli vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma vähintään 15 ja alle 30 ml/min; n = 7), havaittiin kliinisessä tutkimuksessa TAGRISSO-valmisteen 80 mg:n oraalisen kerta-annoksen antamisen jälkeen 1,85-kertainen AUC-arvon suureneminen (90 %:n luottamusväli 0,94, 3,64) ja 1,19-kertainen Cmax-arvon suureneminen (90 %:n luottamusväli 0,69, 2,07) verrattuna potilaisiin, joilla oli normaali munuaisten toiminta (kreatiniinipuhdistuma vähintään 90 ml/min; n = 8). Populaatiofarmakokineettisen analyysin perusteella osimertinibialtistukset olivat lisäksi samankaltaiset 593 potilaalla, joilla oli lievä munuaisten vajaatoiminta (kreatiniinipuhdistuma vähintään 60 ja alle 90 ml/min), 254 potilaalla, joilla oli kohtalainen munuaisten vajaatoiminta (kreatiniinipuhdistuma vähintään 30 ja alle 60 ml/min), 5 potilaalla, joilla oli vaikea munuaisten vajaatoiminta (kreatiniinipuhdistuma vähintään 15 ja alle 30 ml/min), ja 502 potilaalla, joilla oli normaali munuaisten toiminta (vähintään 90 ml/min). Potilaita, joiden kreatiniinipuhdistuma oli enintään 10 ml/min, ei otettu mukaan kliinisiin tutkimuksiin.
Potilaat, joilla on aivometastaaseja
EGFR-mutaatiopositiivisille NSCLC-potilaille (n = 4), joilla oli aivometastaaseja, ja terveille vapaaehtoisille (n = 7) tehtiin PET-kuvaus sen jälkeen, kun heille oli annettu mikroannoksia [11C]osimertinibia. Kuvat osoittivat, että aivokudoksen ja plasman [11C]osimertinibipitoisuuksien suhde (Kp) oli samankaltainen potilailla ja terveillä vapaaehtoisilla ja että [11C]osimertinibi läpäisi nopeasti veri-aivoesteen ja jakautui homogeenisesti kaikille aivojen alueille molemmissa ryhmissä.
Prekliiniset tiedot turvallisuudesta
Tärkeimmät löydökset toistuvan altistuksen aiheuttamaa toksisuutta koskevissa tutkimuksissa rotilla ja koirilla olivat atrofisia, inflammatorisia ja/tai degeneratiivisia muutoksia silmän sarveiskalvon epiteeleissä (joihin liittyi koirien oftalmologisissa tutkimuksissa sarveiskalvon kirkkautta ja sameutta), ruuansulatuskanavassa (mukaan lukien kieli), ihossa, urosten ja naaraiden lisääntymiselimissä sekä sekundäärisiä muutoksia pernassa. Nämä löydökset todettiin sellaisilla pitoisuuksilla plasmassa, jotka olivat pienempiä kuin 80 mg:n terapeuttista annosta saaneilla potilailla havaitut pitoisuudet. Löydökset, jotka ilmaantuivat yhden kuukauden hoidon jälkeen, korjaantuivat pääosin yhden kuukauden sisällä hoidon lopettamisesta, poikkeuksena osa sarveiskalvon muutoksista, joissa havaittiin osittaista korjaantumista.
Mykiön syiden degeneraatiota todettiin 104 viikkoa kestäneessä rotilla tehdyssä karsinogeenisuutta koskeneessa tutkimuksessa altistuksilla, jotka olivat 0,2-kertaisia verrattuna ihmisellä todettavaan AUC-arvoon käytettäessä suositeltua 80 mg:n vuorokausiannosta. Mykiön sameutta todettiin ensimmäisen kerran tämän tutkimuksen viikosta 52 lähtien, ja löydöksen ilmaantuvuus ja vaikeusaste suurenivat asteittain valmisteen käytön keston pidentyessä. Tämän löydöksen kliinistä merkitystä ei voida poissulkea.
Osimertinibi läpäisi makakiapinoiden (annostelu laskimoon), rottien ja hiirten (oraalinen annostelu) vahingoittumattoman veri-aivoesteen.
Ei-kliinisten tietojen perusteella osimertinibi ja sen metaboliitit (AZ5104) inhiboivat h-ERG-kanavaa, eikä QTc-aikaa pidentävää vaikutusta voida poissulkea.
Osimertinibi ei aiheuttanut geneettisiä vaurioita in vitro- tai in vivo ‑tutkimuksissa. Osimertinibilla ei todettu olevan karsinogeenista potentiaalia, kun sitä annettiin siirtogeenisille Tg rasH2-hiirille suun kautta 26 viikon ajan. Proliferatiivisten verisuonileesioiden (angiomatoosinen hyperplasia ja hemangiooma) ilmaantuvuuden suurenemista mesenteerisessä imusolmukkeessa todettiin 104 viikkoa kestäneessä rotilla tehdyssä karsinogeenisuutta koskeneessa tutkimuksessa altistuksilla, jotka olivat 0,2-kertaisia verrattuna AUC-arvoon suositellulla 80 mg:n vuorokausiannoksella. Tällä ei todennäköisesti ole merkitystä ihmisille.
Lisääntymistoksisuus
Rotilla ja koirilla, joita oli altistettu osimertinibille vähintään yhden kuukauden ajan, ilmeni degeneratiivisia muutoksia kiveksissä. Tutkimukset osoittivat urosten hedelmällisyyden heikentyneen rotilla kolmen kuukauden osimertinibialtistuksen jälkeen. Nämä löydökset olivat havaittavissa kliinisesti merkittävillä pitoisuuksilla plasmassa. Yhden kuukauden altistuksen jälkeen kiveksissä havaitut patologiset löydökset korjaantuivat rotilla. Näiden vaurioiden korjaantumisesta koirilla ei kuitenkaan ole varmuutta.
Eläimillä tehtyjen tutkimusten mukaan hoito osimertinibillä voi heikentää naisten hedelmällisyyttä. Toistuvan altistuksen aiheuttamaa toksisuutta koskeneissa tutkimuksissa todettiin kiimattomuutta, keltarauhasen rappeutumista munasarjoissa sekä kohdun ja emättimen epiteelin ohentumista rotilla, jotka oli altistettu osimertinibille vähintään yhden kuukauden ajan kliinisesti merkittävinä pitoisuuksina plasmassa. Yhden kuukauden altistuksen jälkeen todetut löydökset munasarjoissa olivat korjaantuvia. Naarasrotilla tehdyssä hedelmällisyystutkimuksessa päivittäinen annostus osimertinibia annoksella 20 mg/kg (vastaten suurin piirtein suositeltua päivittäistä kliinistä 80 mg annosta) ei vaikuttanut kiimajaksoihin tai kantaviksi tulleiden naaraiden määrään, mutta johti aikaisiin alkiokuolemiin. On todisteita, että kuukausi annostuksen lopettamisen jälkeen nämä löydökset alkavat korjaantua.
Alkion/sikiön kehitystä koskevassa modifioidussa tutkimuksessa osimertinibi aiheutti rotilla alkiokuolleisuutta, kun sitä annettiin tiineille rotille ennen alkion istutusta. Tällaisia vaikutuksia havaittiin emojen sietämällä annoksella 20 mg/kg, jolloin altistus vastasi ihmisen altistusta suositellulla 80 mg:n vuorokausiannoksella (kokonais-AUC-arvon perusteella). Altistus vähintään 20 mg/kg:n annoksella organogeneesin aikana pienensi sikiön painoa, mutta sillä ei ollut haitallisia vaikutuksia sikiön ulkoiseen tai viskeraaliseen morfologiaan. Kun osimertinibia annettiin tiineille naarasrotille koko tiineyden ajan ja sen jälkeen varhaisen laktaation ajan, vieroittamattomilla poikasilla voitiin osoittaa altistus osimertinibille ja sen metaboliiteille sekä eloonjääneiden poikasten määrän väheneminen ja huono poikasten kasvu (vähintään annoksella 20 mg/kg).
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
Mannitoli
Mikrokiteinen selluloosa
Matalasubstituutioasteinen hydroksipropyyliselluloosa
Natriumstearyylifumaraatti
Tabletin päällyste
Polyvinyylialkoholi
Titaanidioksidi (E171)
Makrogoli 3350
Talkki
Keltainen rautaoksidi (E172)
Punainen rautaoksidi (E172)
Musta rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
TAGRISSO tabletti, kalvopäällysteinen
40 mg (L:ei) 30 x 1 fol (5587,01 €)
80 mg (L:ei) 30 x 1 fol (5587,01 €)
PF-selosteen tieto
Al/Al repäisyviivallinen yksittäispakattu läpipainopakkaus. 30 x 1 tabletin pakkaus (3 läpipainopakkausta).
Al/Al repäisyviivallinen yksittäispakattu läpipainopakkaus. 28 x 1 tabletin pakkaus (4 läpipainopakkausta).
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
TAGRISSO 40 mg tabletti
Beigenvärinen, 9 mm, pyöreä, kaksoiskupera tabletti, jonka toiselle puolelle on kaiverrettu "AZ" ja "40" ja toinen puoli on ilman kaiverrusta.
TAGRISSO 80 mg tabletti
Beigenvärinen, 7,25 x 14,5 mm, soikea, kaksoiskupera tabletti, jonka toiselle puolelle on kaiverrettu "AZ" ja "80" ja toinen puoli on ilman kaiverrusta.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
TAGRISSO tabletti, kalvopäällysteinen
40 mg 30 x 1 fol
80 mg 30 x 1 fol
- Ylempi erityiskorvaus (100 %). Osimertinibi: Ei-pienisoluisen keuhkosyövän hoito aikuispotilailla erityisin edellytyksin (1519).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Osimertinibi: Ei-pienisoluisen keuhkosyövän hoito aikuispotilailla erityisin edellytyksin (3004).
ATC-koodi
L01EB04
Valmisteyhteenvedon muuttamispäivämäärä
21.05.2026
Yhteystiedot
Keilaranta 18
02150 Espoo
010 23 010
www.astrazeneca.fi