
MABTHERA infuusiokonsentraatti, liuosta varten 100 mg, 500 mg
Vaikuttavat aineet ja niiden määrät
MabThera 100 mg infuusiokonsentraatti, liuosta varten
Jokainen millilitra sisältää 10 mg rituksimabia.
Jokainen 10 ml:n injektiopullo sisältää 100 mg rituksimabia.
MabThera 500 mg infuusiokonsentraatti, liuosta varten
Jokainen millilitra sisältää 10 mg rituksimabia.
Jokainen 50 ml:n injektiopullo sisältää 500 mg rituksimabia.
Rituksimabi on geeniteknologisesti tuotettu, kimeerinen hiiren/ihmisen monoklonaalinen vasta-aine. Se on glykosyloitu immunoglobuliini, joka muodostuu ihmisen IgG1:n vakioalueiden ja hiiren kevyt- ja raskasketjujen vaihtelevien osien sekvensseistä. Vasta-aine on tuotettu nisäkkäältä (kiinalaisen hamsterin munasarja- eli CHO-soluista) peräisin olevassa solususpensioviljelmässä ja puhdistettu affiniteettikromatografialla ja ioninvaihtotekniikalla. Valmistusprosessiin kuuluu erityisiä virusten inaktivaatio- ja poistotoimenpiteitä.
Apuaine, jonka vaikutus tunnetaan
Jokainen 10 ml:n injektiopullo sisältää natriumia 2,3 mmol (52,6 mg).
Jokainen 50 ml:n injektiopullo sisältää natriumia 11,5 mmol (263,2 mg).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Non-Hodgkin-lymfooma (NHL)
MabThera on tarkoitettu yhdessä solunsalpaajahoitojen kanssa III–IV levinneisyysasteen follikulaarisen lymfooman hoitoon aikuisille potilaille, jotka eivät ole saaneet aikaisempaa hoitoa.
MabThera-ylläpitohoito on tarkoitettu follikulaarista lymfoomaa sairastaville aikuisille potilaille, jotka ovat saaneet vasteen induktiohoitoon.
MabThera-monoterapia on tarkoitettu III–IV levinneisyysasteen follikulaarisen lymfooman hoitoon aikuisille potilaille, kun tauti on solunsalpaajille resistentti tai kun tauti on uusiutunut kahdesti tai useammin solunsalpaajahoidon jälkeen.
MabThera on tarkoitettu yhdessä CHOP-solunsalpaajahoidon (CHOP = syklofosfamidi, doksorubisiini, vinkristiini, prednisoloni) kanssa aikuisille potilaille, joilla on CD20-positiivinen diffuusi suurisoluinen non-Hodgkin-B-solulymfooma.
MabThera on tarkoitettu yhdessä solunsalpaajahoitojen kanssa pediatrisille potilaille (iältään 6 kuukaudesta alle 18-vuotiaisiin), joilla on aiemmin hoitamaton pitkälle edennyt CD20-positiivinen diffuusi suurisoluinen B-solulymfooma (DLBCL), Burkittin lymfooma (BL) / Burkittin leukemia (kypsien B‑solujen akuutti leukemia, BAL) tai Burkitt-tyyppinen lymfooma (BLL).
Krooninen lymfaattinen leukemia (KLL)
MabThera on tarkoitettu yhdessä solunsalpaajahoitojen kanssa kroonisen lymfaattisen leukemian hoitoon aikuisille potilaille, jotka eivät ole saaneet aiempaa hoitoa ja joilla on uusiutunut tai refraktorinen tauti. Tietoa tehosta ja turvallisuudesta on rajallisesti saatavilla potilailla, joita on aikaisemmin hoidettu monoklonaalisilla vasta-aineilla mukaan lukien MabThera, tai potilaista, joiden tauti ei ole reagoinut aiempaan MabThera- ja solunsalpaajahoitoon.
Ks. lisätietoja kohdasta Farmakodynamiikka.
Nivelreuma
MabThera on tarkoitettu yhdessä metotreksaatin kanssa aikuispotilaiden vaikean aktiivisen nivelreuman hoitoon, kun muilla tautiprosessia hidastavilla reumalääkkeillä (DMARD), joihin on sisältynyt yksi tai useampi tuumorinekroositekijän (TNF) estäjä, ei ole saatu riittävää hoitovastetta, tai potilas ei siedä niitä.
MabTheran on osoitettu yhdessä metotreksaatin kanssa vähentävän nivelvaurion etenemistä röntgenkuvista mitattuna ja parantavan fyysistä toimintakykyä.
Granulomatoottinen polyangiitti ja mikroskooppinen polyangiitti
MabThera on tarkoitettu yhdessä glukokortikoidien kanssa aikuispotilaiden vaikean aktiivisen granulomatoottisen polyangiitin (GPA) (aiemmin Wegenerin granulomatoosi) ja mikroskooppisen polyangiitin (MPA) hoitoon.
MabThera on tarkoitettu yhdessä glukokortikoidien kanssa pediatristen potilaiden (2 vuoden – alle 18 vuoden ikäisten) vaikean aktiivisen granulomatoottisen polyangiitin (GPA) (aiemmin Wegenerin granulomatoosi) ja mikroskooppisen polyangiitin (MPA) remission induktioon.
Tavallinen pemfigus (pemphigus vulgaris)
MabThera on tarkoitettu aikuisille potilaille keskivaikean tai vaikean tavallisen pemfiguksen hoitoon.
Ehto
Valmiste on annettava kokeneen terveydenhuollon ammattilaisen valvonnassa sellaisessa hoitopaikassa, jossa kaikki potilaan elvyttämiseen tarvittavat välineet ovat välittömästi saatavilla.
Annostus ja antotapa
MabThera on annettava kokeneen terveydenhuollon ammattilaisen valvonnassa sellaisessa hoitopaikassa, jossa kaikki potilaan elvyttämiseen tarvittavat välineet ovat välittömästi saatavilla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Esilääkitys ja estohoito
Kaikki käyttöaiheet
Esilääkitys antipyreettisellä lääkkeellä ja antihistamiinilla, esim. parasetamolilla ja difenhydramiinilla, pitää antaa ennen jokaista MabThera-infuusiota.
Non-Hodgkin-lymfooma ja krooninen lymfaattinen leukemia
Non-Hodgkin-lymfoomaa ja kroonista lymfaattista leukemiaa sairastaville aikuispotilaille pitää harkita esilääkitystä glukokortikoideilla, ellei MabTheraa anneta yhdessä glukokortikoideja sisältävien solunsalpaajahoitoyhdistelmien kanssa.
Non‑Hodgkin-lymfoomaa sairastaville pediatrisille potilaille pitää antaa esilääkityksenä parasetamolia ja H1-antihistamiinia (= difenhydramiinia tai vastaavaa) 30–60 minuuttia ennen MabThera-infuusion aloittamista. Lisäksi annetaan prednisonia taulukon 1 mukaisesti.
Tuumorilyysisyndrooman riskin vähentämiseksi on suositeltavaa, että kroonista lymfaattista leukemiaa sairastaville potilaille annetaan 48 tuntia ennen hoidon aloittamista estolääkitystä, johon kuuluu riittävä hydraatio ja veren virtsahappopitoisuutta tasapainottava lääkitys. Kroonista lymfaattista leukemiaa sairastaville potilaille, joiden lymfosyyttien kokonaismäärä on > 25 x 109/l, on suositeltavaa antaa 100 mg prednisonia/prednisolonia laskimoon juuri ennen MabThera-infuusiota akuuttien infuusioreaktioiden ja/tai sytokiinioireyhtymän vähentämiseksi ja lievittämiseksi.
Nivelreuma, granulomatoottinen polyangiitti ja mikroskooppinen polyangiitti sekä tavallinen pemfigus (pemphigus vulgaris)
Nivelreumapotilaille, granulomatoottista polyangiittia tai mikroskooppista polyangiittia tai tavallista pemfigusta sairastaville potilaille pitää antaa esilääkityksenä 100 mg metyyliprednisolonia laskimoon 30 minuuttia ennen kunkin MabThera‑infuusion aloittamista vähentämään infuusioon liittyvien reaktioiden määrää ja vaikeusastetta.
Granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastaville aikuispotilaille suositellaan annettavaksi metyyliprednisolonia 1000 mg:n vuorokausiannoksina 1–3 vuorokauden ajan laskimoon (viimeinen metyyliprednisoloniannos voidaan antaa samana päivänä, jona ensimmäinen MabThera-infuusio annetaan) ennen ensimmäistä MabThera-infuusiota. Tämän jälkeen MabThera-hoidon aikana ja sen 4 viikon induktiojakson jälkeen annetaan suun kautta prednisonia 1 mg/kg/vrk (enintään 80 mg/vrk, jota pienennetään mahdollisimman nopeasti kliinisen tarpeen mukaan).
Granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastaville aikuisille ja pediatrisille potilaille ja tavallista pemfigusta sairastaville aikuisille potilaille suositellaan MabThera-hoidon aikana ja sen jälkeen annettavaksi Pneumocystis jirovecii ‑pneumonian (PJP) estohoitoa tarpeen mukaan paikallisen kliinisen hoitokäytännön mukaisesti.
Granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastaville pediatrisille potilaille pitää ennen ensimmäistä MabThera-infuusiota antaa vaikea-asteisen vaskuliitin oireiden hoitoon metyyliprednisolonia laskimonsisäisesti kolme vuorokausiannosta 30 mg/kg/vrk (enintään 1 g/vrk). Metyyliprednisolonin laskimonsisäisiä lisäannoksia voidaan antaa ennen ensimmäistä MabThera-infuusiota enintään kolme vuorokausiannosta 30 mg/kg.
Laskimonsisäisesti annetun metyyliprednisolonin jälkeen pediatrisille potilaille pitää lisäksi antaa suun kautta prednisonia annoksella 1 mg/kg/vrk (enintään 60 mg/vrk). Annosta pienennetään mahdollisimman nopeasti kliinisen tarpeen mukaan (ks. kohta Farmakodynamiikka).
Annostus
On tärkeää tarkistaa valmisteen etiketistä, että potilaalle annetaan lääkemääräyksen mukaista (laskimoon tai ihon alle annettavaa) lääkemuotoa.
Annoksen muuttaminen hoidon aikana
MabTheran annosta ei suositella pienennettäväksi. Kun MabTheraa annetaan yhdistelmähoitona solunsalpaajien kanssa, solunsalpaajalääkkeiden yleisiä annosten pienentämistä koskevia ohjeita on noudatettava.
Non-Hodgkin-lymfooma
Follikulaarinen non-Hodgkin-lymfooma
Yhdistelmähoito
MabTheran suositeltu aloitusannos solunsalpaajahoidon kanssa follikulaarista lymfoomaa sairastaville aikaisemmin hoitamattomille potilaille tai potilaille, joilla on uusiutunut tai refraktorinen tauti, on 375 mg/m2 jokaisessa syklissä, enintään 8 syklin ajan.
MabThera tulisi antaa jokaisen solunsalpaajasyklin ensimmäisenä päivänä solunsalpaajahoitoon mahdollisesti kuuluvan glukokortikoidin i.v.-annon jälkeen.
Ylläpitohoito
- Aiemmin hoitamaton follikulaarinen lymfooma
Ylläpitohoidossa MabTheran suositeltu annos potilaille, joilla on aiemmin hoitamaton follikulaarinen lymfooma ja jotka ovat saaneet vasteen induktiohoitoon, on 375 mg/m2 joka toinen kuukausi (alkaen kaksi kuukautta induktiohoidon viimeisen annoksen jälkeen), kunnes tauti etenee tai pisimmillään 2 vuoden ajan (yhteensä 12 infuusiota).
- Uusiutunut/refraktorinen follikulaarinen lymfooma
Ylläpitohoidossa MabTheran suositeltu annos potilaille, joilla on uusiutunut tai refraktorinen tauti ja jotka ovat saaneet vasteen induktiohoitoon, on 375 mg/m2 joka 3. kuukausi (alkaen kolme kuukautta induktiohoidon viimeisen annoksen jälkeen), kunnes tauti etenee tai pisimmillään 2 vuoden ajan (yhteensä 8 infuusiota).
Monoterapia
Uusiutunut/refraktorinen follikulaarinen lymfooma
Monoterapiassa suositeltu MabThera-annos aikuisille potilaille, joilla on III–IV levinneisyysasteen follikulaarinen lymfooma ja joiden tauti on solunsalpaajille resistentti tai uusiutunut kahdesti tai useammin solunsalpaajahoidon jälkeen, on 375 mg/m2 annosteltuna laskimonsisäisenä infuusiona kerran viikossa 4 viikon ajan.
Kun hoito uusitaan monoterapiassa potilaille, joilla on uusiutunut tai refraktorinen follikulaarinen lymfooma ja jotka ovat vastanneet aikaisemmin annetulle MabThera-monoterapiahoidolle, suositusannos on 375 mg/m2 annettuna laskimonsisäisenä infuusiona kerran viikossa 4 viikon ajan (ks. kohta Farmakodynamiikka).
Aikuisten diffuusi suurisoluinen non-Hodgkin-B-solulymfooma
MabTheraa tulisi käyttää yhdessä CHOP-solunsalpaajahoidon kanssa. Suositeltu annostus on 375 mg/m2 jokaisen kemoterapiasyklin ensimmäisenä päivänä 8 syklin ajan CHOP-hoitoon kuuluvan glukokortikoidin i.v.-infuusion jälkeen. Suurisoluisen non-Hodgkin-B-solulymfooman hoidossa MabTheran turvallisuutta ja tehoa ei ole tutkittu yhdistelmähoidossa muiden solunsalpaajahoitojen kanssa.
Krooninen lymfaattinen leukemia
MabTheran suositeltu annos solunsalpaajahoidon kanssa aikaisemmin hoitamattomille potilaille ja potilaille, joilla on uusiutunut tai refraktorinen tauti, on 375 mg/m2 päivänä 0 ensimmäisessä hoitosyklissä. Sen jälkeen suositeltu annos on 500 mg/m2 jokaisen seuraavan hoitosyklin ensimmäisenä päivänä, yhteensä 6 syklin ajan. Solunsalpaajahoito annetaan MabThera-infuusion jälkeen.
Nivelreuma
MabThera-hoitoa saaville potilaille on annettava MabThera-potilaskortti jokaisen infuusion yhteydessä.
MabThera-hoitojakso koostuu kahdesta 1000 mg:n laskimonsisäisestä infuusiosta. Suositeltu MabThera-annostus on 1000 mg infuusiona laskimoon ja sen jälkeen toinen 1000 mg:n laskimonsisäinen infuusio kahden viikon kuluttua.
Toistohoidon tarve tulisi arvioida 24 viikon kuluttua edellisestä hoitojaksosta. Toistohoito on annettava, jos tällöin todetaan taudin aktiivisuutta, muussa tapauksessa toistohoito annetaan, kun taudin aktiivisuus palaa.
Saatavilla olevat tutkimustiedot viittaavat siihen, että kliininen vaste saavutetaan yleensä 16–24 viikon kuluessa ensimmäisestä hoitojaksosta. Hoidon jatkamista on harkittava huolellisesti uudelleen, jos potilaalla ei havaita näyttöä hoitovasteesta tämän ajan kuluessa.
Granulomatoottinen polyangiitti (GPA) ja mikroskooppinen polyangiitti (MPA)
MabThera-hoitoa saaville potilaille on annettava MabThera-potilaskortti jokaisen infuusion yhteydessä.
Remission induktio aikuisilla
Suositeltu MabThera-annostus granulomatoottisen polyangiitin ja mikroskooppisen polyangiitin hoitoon remission induktiossa aikuispotilaille on 375 mg/m2 kehon pinta-alan perusteella, joka annetaan infuusiona laskimoon kerran viikossa 4 viikon ajan (yhteensä neljä infuusiota).
Aikuisten ylläpitohoito
MabThera-valmisteella indusoidun remission ylläpitohoito aikuisille GPA- ja MPA-potilaille aloitetaan aikaisintaan 16 viikon kuluttua viimeisen MabThera-infuusion jälkeen.
Muilla hoitokäytännön mukaisilla immunosuppressiivisilla valmisteilla indusoidun remission MabThera-ylläpitohoito aloitetaan remission saavuttamisen jälkeisten 4 viikon aikana.
MabThera pitää antaa kahtena 500 mg:n annoksena infuusiona laskimoon kahden viikon välein. Tämän jälkeen annetaan 500 mg:n annos infuusiona laskimoon 6 kuukauden välein. MabTheraa pitää antaa vähintään 24 kuukauden ajan remission (ei sairauden oireita tai löydöksiä) saavuttamisen jälkeen. Jos potilaalla voi olla tavanomaista suurempi relapsin riski, lääkärin pitää harkita pidempää MabThera-ylläpitohoitoa enimmillään 5 vuoteen saakka.
Tavallinen pemfigus (pemphigus vulgaris)
MabThera-hoitoa saaville potilaille on annettava jokaisen infuusion yhteydessä potilaskortti.
Suositeltu MabThera-annostus tavallisen pemfiguksen hoitoon on 1000 mg infuusiona laskimoon ja kahden viikon kuluttua toinen 1000 mg:n infuusio laskimoon yhdistelmänä glukokortikoidien kanssa, joiden annosta pienennetään vähitellen.
Ylläpitohoito
500 mg:n ylläpitoannos annetaan infuusiona laskimoon hoitokuukautena 12 ja 18, ja sen jälkeen 6 kuukauden välein tarvittaessa kliinisen arvion perusteella.
Relapsin hoito
Relapsin yhteydessä potilaalle voidaan antaa 1000 mg laskimoon. Terveydenhoitohenkilöstön pitää myös harkita glukokortikoidihoidon aloittamista uudelleen tai annoksen suurentamista kliinisen arvion perusteella.
Seuraavat infuusiot voidaan antaa aikaisintaan 16 viikkoa edellisen infuusion jälkeen.
Erityisryhmät
Pediatriset potilaat
Non-Hodgkin-lymfooma
Pediatrisille potilaille (iältään 6 kuukaudesta alle 18-vuotiaisiin), jotka sairastavat aiemmin hoitamatonta pitkälle edennyttä CD20-positiivista diffuusia suurisoluista B-solulymfoomaa, Burkittin lymfoomaa, Burkittin leukemiaa tai Burkitt-tyyppistä lymfoomaa, MabThera-valmistetta pitää antaa yhdistelmänä systeemisen LMB (Lymphome Malin B) ‑solunsalpaajahoito-ohjelman kanssa (ks. taulukot 1 ja 2). Suositeltu MabThera-annostus on 375 mg/m2 kehon pinta-alan perusteella infuusiona laskimoon. MabThera-annosta ei tarvitse muuttaa muutoin kuin kehon pinta-alan perusteella.
MabTheran turvallisuutta ja tehoa 6 kuukauden – alle 18 vuoden ikäisten pediatristen potilaiden hoidossa ei ole varmistettu muissa käyttöaiheissa kuin aiemmin hoitamattoman pitkälle edenneen CD20-positiivisen diffuusin suurisoluisen B-solulymfooman, Burkittin lymfooman, Burkittin leukemian ja Burkitt-tyyppisen lymfooman hoidossa. Tietoja on vain rajallisesti saatavilla alle 3-vuotiailla lapsipotilailla. Ks. lisätietoja kohdasta Farmakodynamiikka.
MabTheraa ei saa käyttää vastasyntyneiden ja < 6 kuukauden ikäisten pediatristen potilaiden CD20-positiivisen diffuusin suurisoluisen B-solulymfooman hoitoon (ks. kohta Farmakodynamiikka).
Taulukko 1 MabThera-annostus pediatristen potilaiden non-Hodgkin-lymfooman hoidossa
| Hoitosykli | Hoitopäivä | Valmisteen antoa koskevat tiedot |
| Esivaihe (COP) | MabTheraa ei anneta | - |
Induktiojakso 1 (COPADM1) | Päivä -2 (vastaa esivaiheen päivää 6) 1. MabThera-infuusio | 1. induktiojakson aikana annetaan prednisonia osana solunsalpaajahoitoa, ja se pitää antaa ennen MabThera-infuusiota. |
Päivä 1 2. MabThera-infuusio | MabThera annetaan 48 tuntia ensimmäisen MabThera-infuusion jälkeen. | |
Induktiojakso 2 (COPADM2) | Päivä -2 3. MabThera-infuusio | 2. induktiojaksossa prednisonia ei anneta MabThera-infuusion annon aikana. |
Päivä 1 4. MabThera-infuusio | MabThera annetaan 48 tuntia kolmannen MabThera-infuusion jälkeen. | |
Konsolidaatiojakso 1 (CYM/CYVE) | Päivä 1 5. MabThera-infuusio | Prednisonia ei anneta MabThera-infuusion annon aikana. |
Konsolidaatiojakso 2 (CYM/CYVE) | Päivä 1 6. MabThera-infuusio | Prednisonia ei anneta MabThera-infuusion annon yhteydessä. |
| Ylläpitojakso 1 (M1) | 2. konsolidaatiojakson päivät 25–28 (CYVE) MabTheraa ei anneta | Alkaa kun 2. konsolidaatiojakson (CYVE) jälkeen perifeerisiksi määriksi on korjautunut ANC > 1,0 x 109/l ja trombosyytit > 100 x 109/l |
| Ylläpitojakso 2 (M2) | 1. ylläpitojakson päivä 28 (M1) MabTheraa ei anneta | - |
| ANC = absoluuttinen neutrofiilimäärä (Absolute Neutrophil Count); COP = syklofosfamidi, vinkristiini, prednisoni; COPADM = syklofosfamidi, vinkristiini, prednisoloni, doksorubisiini, metotreksaatti; CYM = sytarabiini (Aracytine, Ara-C), metotreksaatti; CYVE = sytarabiini (Aracytine, Ara-C), veposidi (VP16) | ||
Taulukko 2 Hoitosuunnitelma non-Hodgkin-lymfoomaa sairastaville pediatrisille potilaille: solusalpaajahoitojen ja MabTheran samanaikainen käyttö
| Hoitosuunnitelma | Potilaan taudin levinneisyysaste | Antoa koskevat tiedot |
| Ryhmä B | III levinneisyysaste sekä suuri LDH-pitoisuus (> N x 2), IV levinneisyysaste, ei levinnyt keskushermostoon | Esivaiheen jälkeen 4 jaksoa: 2 induktiojaksoa (COPADM) sekä HDMTX 3 g/m2 ja 2 konsolidaatiojaksoa (CYM) |
| Ryhmä C | Ryhmä C1: Burkittin leukemia ei levinnyt keskushermostoon, IV levinneisyysaste + Burkittin leukemia levinnyt keskushermostoon, mutta ei levinnyt aivo-selkäydinnesteeseen | Esivaiheen jälkeen 6 jaksoa: 2 induktiojaksoa (COPADM) sekä HDMTX 8 g/m², 2 konsolidaatiojaksoa (CYVE) ja 2 ylläpitojaksoa (M1 ja M2) |
Ryhmä C3: Burkittin leukemia levinnyt aivo-selkäydinnesteeseen, IV levinneisyysaste levinnyt aivo-selkäydinnesteeseen | ||
| Seuraava jakso pitää antaa heti, kun verisolumäärä on korjautunut ja potilaan tila sen sallii, lukuun ottamatta ylläpitojaksoja, jotka annetaan 28 päivän välein | ||
| Burkittin leukemia = kypsien B-solujen akuutti leukemia; HDMTX = metotreksaatti suurina annoksina (High-dose Methotrexate); LDH = laktaattidehydrogenaasi (Lactic Acid Dehydrogenase) | ||
Granulomatoottinen polyangiitti (GPA) ja mikroskooppinen polyangiitti (MPA)
Remission induktio
Suositeltu MabThera-annostus pediatristen potilaiden vaikean, aktiivisen granulomatoottisen polyangiitin tai mikroskooppisen polyangiitin remission induktioon on 375 mg/m2 kehon pinta-alan perusteella infuusiona laskimoon kerran viikossa 4 viikon ajan.
MabTheran turvallisuutta ja tehoa 2 vuoden – alle 18 vuoden ikäisten pediatristen potilaiden hoidossa ei ole varmistettu muissa käyttöaiheissa kuin vaikean, aktiivisen granulomatoottisen polyangiitin tai mikroskooppisen polyangiitin hoidossa.
MabTheraa ei pidä käyttää alle 2 vuoden ikäisten pediatristen potilaiden vaikean, aktiivisen granulomatoottisen polyangiitin tai mikroskooppisen polyangiitin hoitoon, sillä näillä potilailla immuunivaste yleisiä rokotuksella estettävissä olevia lapsuusiän sairauksia (esim. tuhkarokko, sikotauti, vihurirokko ja polio) vastaan annettaviin rokotuksiin voi olla riittämätön (ks. kohta Farmakodynamiikka).
Iäkkäät
Annoksen muuttaminen ei ole tarpeen vähintään 65-vuotiaita potilaita hoidettaessa.
Antotapa
Kaikki käyttöaiheet
Laimennettu MabThera-infuusioneste on annettava erillisen laskimonsisäisen infuusiolinjan kautta. Laimennettuja infuusioliuoksia ei saa antaa laskimoon nopeina infuusioina eikä boluksina.
Potilaiden tilaa on seurattava huolellisesti mahdollisen sytokiinioireyhtymän ilmaantumisen varalta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Infuusio on heti keskeytettävä potilailla, joilla ilmenee oireita vakavasta reaktiosta, etenkin jos esiintyy vakavaa hengenahdistusta, bronkospasmia tai hypoksiaa. Non-Hodgkin-lymfoomaa sairastavia potilaita on tämän jälkeen tutkittava mahdollisen tuumorilyysisyndrooman varalta mm. laboratoriokokein sekä keuhkoinfiltraattien varalta keuhkojen röntgenkuvauksella. Seuraava koskee kaikkia potilaita: infuusiota ei saa jatkaa, ennen kuin kaikki oireet ovat hävinneet ja laboratoriokokeiden tulokset sekä keuhkoröntgenkuvat ovat normalisoituneet. Tällöin infuusiota voidaan jatkaa, mutta infuusionopeus saa olla enintään puolet edellisen infuusion nopeudesta. Jos samoja vakavia haittavaikutuksia taas ilmaantuu, on vakavasti ja tapauskohtaisesti harkittava lääkityksen lopettamista kokonaan.
Lievät tai keskivaikeat infuusioon liittyvät oireet (kohta Haittavaikutukset) yleensä vähenevät, kun infuusionopeutta hidastetaan. Kun oireet vähenevät, infuusionopeutta voidaan taas nostaa.
Aikuisten potilaiden non-Hodgkin-lymfooma, krooninen lymfaattinen leukemia, nivelreuma, tavallinen pemfigus (pemphigus vulgaris), aikuisten ja pediatristen potilaiden granulomatoottinen polyangiitti ja mikroskooppinen polyangiitti
Ensimmäinen infuusio
Suositeltu infuusion aloitusnopeus on 50 mg/tunti. Ensimmäisten 30 minuutin jälkeen infuusionopeutta voidaan asteittain lisätä 50 mg/tunti joka 30. minuutti, korkeintaan nopeuteen 400 mg/tunti.
Seuraavat infuusiot
Seuraavat MabThera-annokset voidaan aloittaa nopeudella 100 mg/tunti. Infuusionopeutta voidaan asteittain lisätä 100 mg/tunti joka 30. minuutti, korkeintaan nopeuteen 400 mg/tunti.
Non-Hodgkin-lymfooma – pediatriset potilaat
Ensimmäinen infuusio
Suositeltu infuusion aloitusnopeus on 0,5 mg/kg/tunti (korkeintaan 50 mg/tunti). Jos yliherkkyyttä tai infuusioon liittyviä reaktioita ei ilmene, infuusionopeutta voidaan asteittain lisätä 0,5 mg/kg/tunti joka 30. minuutti, korkeintaan nopeuteen 400 mg/tunti.
Seuraavat infuusiot
Seuraavat MabThera-annokset voidaan aloittaa nopeudella 1 mg/kg/tunti (korkeintaan 50 mg/tunti). Infuusionopeutta voidaan asteittain lisätä 1 mg/kg/tunti joka 30. minuutti, korkeintaan nopeuteen 400 mg/tunti.
Nivelreuma
Seuraavien infuusioiden vaihtoehtoinen ja nopeampi antotapa
Jos potilaalle ei ole ilmaantunut vakavia infuusioon liittyviä reaktioita ensimmäisen tai sen jälkeen tavanomaisella antonopeudella annettujen 1000 mg:n MabThera-infuusioiden yhteydessä, toinen ja sitä seuraavat infuusiot voidaan antaa nopeammin käyttämällä samaa pitoisuutta kuin aikaisemmissa infuusioissa (4 mg/ml 250 ml:n tilavuudessa). Infuusio aloitetaan nopeudella 250 mg/tunti ensimmäisen 30 minuutin ajan. Tämän jälkeen infuusionopeus nostetaan 600 mg:aan/tunti seuraavan 90 minuutin ajaksi. Jos potilas sietää nopeamman infuusion, myös seuraavat infuusiot voidaan antaa tällä nopeammalla antotavalla.
Potilaille, joilla on kliinisesti merkittävä sydän- ja verisuonitauti, kuten sydämen rytmihäiriöitä, tai joilla on esiintynyt vakavia infuusioreaktioita jonkin aiemmin annetun biologisen lääkehoidon tai rituksimabin yhteydessä, ei saa antaa nopeampaa infuusiota.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai hiiren proteiineille tai kohdassa Apuaineet mainituille apuaineille.
Aktiivinen vaikea infektio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Vaikeasti immuunipuutteiset potilaat.
Vaikea sydämen vajaatoiminta (NYHA-luokka IV, NYHA = New York Heart Association) tai vaikea, kontrolloimaton sydänsairaus vain nivelreuman, granulomatoottisen polyangiitin, mikroskooppisen polyangiitin ja tavallisen pemfiguksen hoidossa (muut kardiovaskulaariset taudit, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Progressiivinen multifokaalinen leukoenkefalopatia
Kaikille MabThera-hoitoa saaville nivelreumapotilaille, granulomatoottista polyangiittia, mikroskooppista polyangiittia tai tavallista pemfigusta sairastaville potilaille on annettava MabThera-potilaskortti jokaisen infuusion yhteydessä (ks. Liite IIIA, myyntipäällysmerkinnät). Potilaskortti sisältää potilaille tärkeitä turvallisuustietoja, jotka liittyvät infektioiden, mukaan lukien progressiivisen multifokaalisen leukoenkefalopatian (PML), mahdollisesti kohonneeseen riskiin.
Hyvin harvinaisia fataaleja PML-tapauksia on raportoitu käytettäessä MabThera-valmistetta nivelreuman ja autoimmuunisairauksien (mukaan lukien SLE-taudin ja vaskuliitiin) hoitoon sekä MabThera-valmisteen markkinoille tulon jälkeen käytettäessä sitä non-Hodgkin-lymfooman ja kroonisen lymfaattisen leukemian hoitoon (jolloin valtaosa potilaista oli saanut MabThera-valmistetta yhdistelmänä solunsalpaajahoidon kanssa tai osana hematopoieettista kantasolusiirtoa). Potilaita on säännöllisin väliajoin seurattava kaikkien uusien tai pahenevien, mahdollisesti PML:ään viittaavien neurologisten oireiden ja merkkien varalta. Jos PML:ää epäillään, annostelu on keskeytettävä, kunnes PML on poissuljettu. Lääkärin tulee arvioida potilaan oireita selvittääkseen viittaavatko ne neurologiseen toimintahäiriöön, ja jos näin on, ovatko oireet mahdollisesti PML:ään viittaavia. Neurologin konsultaatiota on harkittava kliinisen tarpeen mukaan.
Jos PML:ää epäillään, lisätutkimuksia on harkittava. Tällaisia lisätutkimuksia ovat mm. MRI-kuvaus (mieluummin varjoaineella tehtynä), JC-viruksen DNA:n testaus aivo-selkäydinnesteestä (CSF) ja toistuvat neurologiset arvioinnit.
Lääkärin on oltava erityisen valpas PML:ään viittaavissa oireissa, joita potilas ei ehkä itse havaitse (esim. kognitiiviset, neurologiset ja psykiatriset oireet). Potilaita tulee myös neuvoa kertomaan hoidostaan lähiomaisilleen, koska on mahdollista, että he huomaavat sellaisia oireita, joista potilas itse ei ole tietoinen.
Jos potilaalle kehittyy PML, MabThera-hoito on lopetettava pysyvästi. Immuunivajausta ja PML:ää sairastavilla potilailla on havaittu immuunijärjestelmän toipumisen jälkeen tilanteen vakaantumista tai kohentumista. On edelleen epäselvää, voivatko PML:n varhainen havaitseminen ja MabThera-hoidon keskeyttäminen johtaa tilanteen samankaltaiseen vakaantumiseen tai kohentumiseen.
Sydänvaivat
Rasitusrintakivun oireita, sydämen rytmihäiriöitä, kuten eteislepatusta ja eteisvärinää, sydämen vajaatoimintaa ja/tai sydäninfarkteja on esiintynyt MabTheralla hoidetuilla potilailla. Potilaita, joilla aikaisemmin on ollut jokin sydänsairaus ja/tai joille on annettu kardiotoksista kemoterapiaa, on siksi seurattava huolellisesti (ks. jäljempänä infuusioon liittyvät reaktiot).
Infektiot
MabThera-hoitoa saavilla potilailla saattaa olla kohonnut riski sairastua infektioihin, mikä johtuu sekä MabTheran vaikutusmekanismista että B-solujen tärkeästä roolista normaalin immuunivasteen ylläpitämisessä (ks. kohta Farmakodynamiikka). Vakavia infektioita, myös kuolemantapauksia, saattaa esiintyä MabThera-hoidon aikana (ks. kohta Haittavaikutukset). MabTheraa ei pidä antaa potilaille, joilla on aktiivinen vaikea-asteinen infektio (esim. tuberkuloosi, sepsis ja opportunistisia infektioita, ks. kohta Vasta-aiheet), eikä potilaille, joilla on vaikea immuunivajavuus (esim. jos CD4- tai CD8-pitoisuus on hyvin alhainen). MabThera-hoitoa harkittaessa on noudatettava varovaisuutta, jos potilaalla on anamneesissa uusiutuvia tai kroonisia infektioita tai perussairaus, esim. hypogammaglobulinemia, joka saattaa lisätä alttiutta vakavalle infektiolle (ks. kohta Haittavaikutukset). Immunoglobuliinitasojen määrittämistä suositellaan ennen MabThera-hoidon aloittamista.
Jos MabThera-hoidon jälkeen ilmaantuu infektioon viittaavia merkkejä tai oireita, potilas on tutkittava viipymättä ja aloitettava asianmukainen hoito. Ennen seuraavan MabThera-hoitojakson antamista potilaan tila on arvioitava uudelleen mahdollisen infektioriskin suhteen.
Progressiivista multifokaalista enkefalopatiaa (PML) koskevat tiedot, ks. edellä kohta Progressiivinen multifokaalinen leukoenkefalopatia.
Enteroviraalista meningoenkefaliittia, myös fataalia, on raportoitu rituksimabin annostelun jälkeen.
Hepatiitti B ‑infektiot
Hepatiitti B ‑viruksen uudelleen aktivoitumista, myös kuolemaan johtaneita tapauksia, on raportoitu MabTheraa saaneilla potilailla. Valtaosa näistä potilaista oli altistunut myös sytotoksiselle solunsalpaajahoidolle. Suppeat tiedot yhdestä tutkimuksesta uusiutunutta tai hoitoon reagoimatonta kroonista lymfaattista leukemiaa sairastavilla potilailla viittaavat siihen, että MabThera-hoito voi myös heikentää primaarien hepatiitti B ‑infektioiden hoitotulosta.
Kaikille potilaille pitää tehdä hepatiitti B -viruksen (HBV) seulonta ennen MabThera-hoidon aloittamista. Minimissään tähän on sisällytettävä HBsAg- ja HBcAb-status. Seulontaa voidaan täydentää muilla sopivilla markkereilla paikallisten ohjeistojen mukaan. Jos potilaalla on aktiivinen hepatiitti B -infektio, hänelle ei saa antaa MabThera-hoitoa. Jos potilaan serologia todetaan HBV-positiiviseksi (joko HBsAg tai HBcAb), maksasairauksiin erikoistunutta lääkäriä pitää konsultoida ennen hoidon aloittamista, ja potilasta pitää seurata ja hoitaa paikallisten hoitokäytäntöjen mukaisesti hepatiitti B -viruksen uudelleen aktivoitumisen estämiseksi.
Infektioita koskeva virheellinen negatiivinen serologinen testitulos
Koska infektioita koskevat serologiset testit voivat antaa virheellisiä negatiivisia tuloksia, vaihtoehtoisia diagnosointityökaluja on harkittava potilaille, joilla on harvinaiseen infektiotautiin viittaavia oireita, kuten Länsi-Niilin virusinfektio tai neuroborrelioosi.
Ihoreaktiot
Vaikea-asteisia ihoreaktioita, kuten toksista epidermaalista nekrolyysiä (Lyellin oireyhtymä) ja Stevens–Johnsonin oireyhtymää, jotka toisinaan johtivat potilaan kuolemaan, on raportoitu (ks. kohta Haittavaikutukset). Tällaisen tapahtuman ilmaantuessa, johon liittyy epäilty syy-yhteys MabTheraan, hoito on lopetettava pysyvästi.
Non-Hodgkin-lymfooma ja krooninen lymfaattinen leukemia
Infuusioon liittyvät reaktiot
MabThera-hoidon yhteydessä on esiintynyt infuusioon liittyneitä reaktioita, jotka saattavat liittyä sytokiinien ja/tai muiden kemiallisten välittäjäaineiden vapautumiseen. Sytokiinioireyhtymä ei välttämättä ole kliinisesti erotettavissa akuuteista yliherkkyysreaktioista.
Tällaiset reaktiot, kuten sytokiinioireyhtymä, tuumorilyysisyndrooma sekä anafylaktiset ja yliherkkyysreaktiot, on kuvattu jäljempänä.
MabTheran laskimoon annettavan lääkemuodon käytössä on valmisteen markkinoille tulon jälkeen raportoitu vaikea-asteisia, kuolemaan johtaneita infuusioon liittyneitä reaktioita. Ne ovat ilmaantuneet 30 minuutista 2 tuntiin ensimmäisen laskimoon annettavan MabThera-infuusion aloittamisen jälkeen. Näille reaktioille tyypillistä olivat keuhkotapahtumat ja joissakin tapauksissa nopea tuumorilyysi ja tuumorilyysisyndrooman piirteet sekä lisäksi kuume, vilunväristykset, kankeus, hypotensio, urtikaria, angioödeema ja muut oireet (ks. kohta Haittavaikutukset).
Vakavaan sytokiinioireyhtymään kuuluu vaikea hengenahdistus, johon usein liittyy bronkospasmia ja hypoksiaa. Lisäksi esiintyy kuumetta, vilunväristyksiä, jäykkyyttä, urtikariaa ja angioödeemaa. Tähän reaktioon voi myös liittyä tuumorilyysisyndrooman oireita, kuten hyperurikemia, hyperkalemia, hypokalsemia, hyperfosfatemia, akuutti munuaisten vajaatoiminta, kohonnut LDH-arvo (laktaattidehydrogenaasi) ja lisäksi akuutti hengitystoiminnan pettäminen ja kuolema. Akuuttiin hengitystoiminnan pettämiseen voi liittyä esim. interstitielli keuhkoinfiltraatti tai ödeema, jotka näkyvät keuhkoröntgenkuvissa. Oireyhtymä ilmaantuu useimmiten yhden tai kahden tunnin kuluessa ensimmäisen infuusion aloittamisesta. Huonon lopputuloksen riski saattaa olla suurempi potilailla, joilla on/on ollut keuhkojen vajaatoimintaa, sekä niillä, joilla on tuumori-infiltraatteja keuhkoissa. Näiden potilaiden hoidossa on siksi noudatettava erityistä varovaisuutta.
Vakavan sytokiinioireyhtymän ilmaantuessa infuusio on heti lopetettava (ks. kohta Annostus ja antotapa) ja potilaalle on annettava aggressiivista oireidenmukaista hoitoa. Kliinisten oireiden vähennyttyä saattaa jälleen seurata niiden paheneminen, joten potilaiden tilaa on seurattava tarkoin, kunnes tuumorilyysisyndrooma ja keuhkoinfiltraatit ovat hävinneet tai on todettu, että niitä ei ole. Vakava sytokiinioireyhtymä on uusiutunut vain harvoissa tapauksissa, kun hoitoa on jatkettu kaikkien oireiden häviämisen jälkeen.
Jos potilaan kasvaintaakka on suuri tai verenkierrossa on paljon (≥ 25 x 109/l) syöpäsoluja, kuten kroonista lymfaattista leukemiaa sairastavilla potilailla, vakavan sytokiinioireyhtymän riski saattaa olla suurempi ja potilaan hoidossa on oltava äärimmäisen varovainen. Näitä potilaita hoidettaessa on noudatettava äärimmäistä varovaisuutta ja heitä on seurattava erityisen huolellisesti koko ensimmäisen infuusion ajan. Lääkärin on harkittava tavallista hitaamman infuusionopeuden käyttöä ensimmäisen infuusion ajan tai annoksen jakamista kahdelle päivälle ensimmäisen syklin aikana ja myös seuraavien syklien aikana, jos veren lymfosyyttimäärä yhä ylittää 25 x 109/l.
Erilaisia infuusioon liittyviä haittavaikutuksia on havaittu 77 %:lla MabTheralla hoidetuista potilaista (muun muassa sytokiinioireyhtymä, johon liittyy hypotensiota ja bronkospasmia, ilmeni 10 %:lla potilaista) ks. kohta Haittavaikutukset. Nämä oireet menevät yleensä ohi, kun MabThera-infuusio keskeytetään ja potilaalle annetaan antipyreettistä lääkettä, antihistamiinia sekä joskus tarvittaessa happea, fysiologista keittosuolaliuosta laskimonsisäisesti tai bronkodilataattoreita ja glukokortikoideja. Vakavien reaktioiden varalta, ks. edellä mainittu sytokiinioireyhtymä.
Anafylaktisia ja muita yliherkkyysreaktioita on raportoitu proteiinien laskimonsisäisen annon yhteydessä. Toisin kuin sytokiinireaktion kohdalla, todelliset yliherkkyysreaktiot useimmiten ilmaantuvat muutamien minuuttien kuluessa infuusion aloittamisesta. Yliherkkyysreaktion hoitamiseksi tarvittavia lääkkeitä eli adrenaliinia, antihistamiineja ja glukokortikoideja on oltava välittömästi saatavilla siltä varalta, että MabThera-hoidon aikana ilmaantuu allerginen reaktio. Anafylaktisen reaktion kliiniset oireet voivat vaikuttaa sytokiinireaktion kliinisiltä oireilta (kuvattu edellä). Yliherkkyyteen liittyviä oireita on raportoitu harvemmin kuin sytokiinireaktion oireita.
Muita yksittäisissä tapauksissa raportoituja infuusion liittyviä reaktioita ovat sydäninfarkti, eteisvärinä, pulmonaarinen ödeema ja akuutti ohimenevä trombosytopenia.
Koska MabThera-infuusion aikana saattaa esiintyä hypotensiota, on harkittava verenpainelääkityksestä pidättäytymistä 12 tuntia ennen infuusion antamista.
Hematologinen toksisuus
Vaikka MabThera ei ole myelosuppressiivinen monoterapiassa, varovaisuutta on noudatettava harkittaessa MabThera-hoitoa potilaille, joilla neutrofiilien määrä on < 1,5 x 109/l ja/tai trombosyyttien määrä < 75 x 109/l, sillä tästä potilasryhmästä kliinistä kokemusta on vain rajoitetusti. MabTheraa on annettu 21:lle autologisen luuydinsiirron saaneelle potilaalle ja muille sellaisiin riskiryhmiin kuuluville potilaille, joiden luuytimen toiminnan voitiin olettaa olevan heikentynyt. Kummassakaan edellä mainitussa potilasryhmässä ei esiintynyt myelotoksisuutta.
MabThera-hoidon aikana on otettava täydellinen verenkuva mukaan lukien neutrofiili- ja trombosyyttimäärät.
Rokotukset
Eläviä viruksia sisältävien rokotteiden turvallisuutta ei ole tutkittu MabThera-hoidon jälkeen non-Hodgkin-lymfoomaa eikä kroonista lymfaattista leukemiaa sairastavilla potilailla. Rokottamista elävillä virusrokotteilla ei suositella. MabThera-hoitoa saaville potilaille voidaan antaa inaktivoituja mikrobeja sisältäviä rokotteita. Hoitovaste inaktivoiduille rokotteille saattaa kuitenkin olla alentunut. Ei-satunnaistetussa tutkimuksessa rokotteiden vasteita verrattiin aikuisilla potilailla, jotka saivat MabThera-monoterapiaa uusiutuneeseen matala-asteiseen non-Hodgkin-lymfoomaan, ja terveillä vapaaehtoisilla henkilöillä. Vaste tetanus tehosterokotukselle oli heikompi MabThera-ryhmässä (16 %) kuin verrokkiryhmässä (81 %). Myös vaste Keyhole Limpet haemocyan (KLH) ‑neoantigeenille oli heikompi MabThera-ryhmässä (4 %) verrattuna verrokkiryhmään (76 %). Molemmissa tapauksissa vasteeksi arvioitiin yli 2-kertainen lisäys vasta-ainetitterissä. Kroonista lymfaattista leukemiaa sairastavilla potilailla samantapaisia tuloksia on odotettavissa näiden kahden taudin samankaltaisuuden vuoksi, mutta asiaa ei ole varmistettu kliinisillä tutkimuksilla.
Keskimääräiset hoitoa edeltäneet vasta-ainetitterit lukuisia antigeeneja vastaan (pneumokokki, influenssa A, sikotauti, vihurirokko, vesirokko) säilyivät vähintään 6 kuukautta MabThera-hoidon jälkeen.
Pediatriset potilaat
Tietoja on vain rajallisesti saatavilla alle 3-vuotiailla lapsipotilailla. Ks. lisätietoja kohdasta Farmakodynamiikka.
Nivelreuma, granulomatoottinen polyangiitti (GPA), mikroskooppinen polyangiitti (MPA) ja tavallinen pemfigus
Nivelreumapotilaat, joita ei ole aiemmin hoidettu metotreksaatilla
MabTheraa ei suositella potilaille, joita ei ole aiemmin hoidettu metotreksaatilla (MTX), koska suotuisaa hyöty-riski-suhdetta ei ole osoitettu.
Infuusioon liittyvät reaktiot
MabThera voi aiheuttaa infuusioon liittyviä reaktioita, jotka voivat liittyä sytokiinien ja/tai muiden kemiallisten välittäjäaineiden vapautumiseen.
Kuolemaan johtaneita vaikeita infuusioon liittyviä reaktioita on raportoitu nivelreumapotilailla myyntiluvan myöntämisen jälkeisessä seurannassa. Suurin osa kliinisissä tutkimuksissa nivelreumapotilailla raportoiduista infuusioon liittyvistä tapahtumista oli vaikeusasteeltaan lieviä tai kohtalaisia. Yleisimpiin oireisiin kuuluivat allergiset reaktiot, kuten päänsärky, kutina, kurkun ärsytys, punoitus, ihottuma, urtikaria, hypertensio ja kuume. Infuusioon liittyviä reaktioita saavien potilaiden osuus oli yleensä suurempi ensimmäisen infuusion jälkeen kuin minkä tahansa hoitojakson toisen infuusion jälkeen. Infuusioon liittyvien reaktioiden ilmaantuvuus laski myöhempien infuusioiden yhteydessä (ks. kohta Haittavaikutukset). Raportoidut reaktiot korjautuivat yleensä, kun MabTheran infuusionopeutta pienennettiin tai infuusio keskeytettiin ja potilaalle annettiin kuumelääkettä, antihistamiinia sekä joissakin tapauksissa tarvittaessa happea, fysiologista keittosuolaliuosta laskimonsisäisesti tai bronkodilataattoreita ja glukokortikoideja. Sydänpotilaita ja kardiopulmonaalisia haittavaikutuksia aiemmin saaneita potilaita on tarkkailtava huolellisesti. Infuusioon liittyvän reaktion vaikeusasteesta ja tarvittavista interventioista riippuen MabThera-infuusio keskeytetään tai lopetetaan kokonaan. Infuusio voidaan useimmiten aloittaa uudelleen 50 % pienemmällä nopeudella (esim. nopeus lasketaan 100 mg:sta 50 mg:aan tunnissa), kun oireet ovat kokonaan hävinneet.
Yliherkkyysreaktion hoitamiseksi tarvittavia lääkkeitä, esim. epinefriinia (adrenaliinia), antihistamiineja ja glukokortikoideja, on oltava välittömästi saatavilla siltä varalta, että allerginen reaktio ilmaantuu MabThera-hoidon aikana.
MabThera-hoidon turvallisuudesta ei ole tutkimustietoa kohtalaista sydämen vajaatoimintaa (NYHA-luokka III) tai vaikeaa, kontrolloimatonta sydänsairautta sairastavien potilaiden hoidossa. MabThera-hoitoa saaneilla potilailla on todettu olemassa olevien iskeemisten sydänsairauksien, kuten angina pectoriksen, oireita sekä eteisvärinää ja eteislepatusta. Sydänpotilailla ja kardiopulmonaalisia haittavaikutuksia aiemmin saaneilla potilailla infuusioreaktioista johtuvien kardiovaskulaaristen komplikaatioiden riski on otettava huomioon ennen MabThera-hoidon aloittamista, ja näiden potilaiden tilaa on seurattava tarkoin lääkkeen annon aikana. Koska MabThera-infuusion aikana saattaa esiintyä hypotensiota, on harkittava verenpainelääkityksestä pidättäytymistä 12 tuntia ennen MabThera-infuusion antamista.
Kliinisissä tutkimuksissa ja valmisteen markkinoille tulon jälkeen havaitut infuusioon liittyneet reaktiot olivat yhdenmukaisia granulomatoottista polyangiittia, mikroskooppista polyangiittia tai tavallista pemfigusta sairastavilla potilailla ja nivelreumapotilailla (ks. kohta Haittavaikutukset).
Viivästynyt neutropenia
Veren neutrofiilitasot tulee määrittää ennen jokaista MabThera-infuusiota ja säännöllisin välein kuuden kuukauden ajan hoidon päättymisestä sekä tapauksissa, joissa infektioon viittaavia oireita ilmenee (ks. kohta Haittavaikutukset).
Rokotukset
Ennen MabThera-hoidon aloittamista lääkärin pitää tarkistaa potilaan rokotusstatus, ja potilaan pitää saattaa kaikki rokotuksensa ajan tasalle voimassa olevan rokotusohjeistuksen mukaisesti, jos mahdollista. Viimeiset rokotukset on annettava vähintään 4 viikkoa ennen ensimmäistä MabThera-infuusiota.
Eläviä viruksia sisältävien rokotteiden turvallisuutta ei ole tutkittu MabThera-hoidon jälkeen. Siksi rokottamista elävillä virusrokotteilla ei suositella potilaille, joita hoidetaan MabTheralla tai joilla on matala perifeerinen B-solumäärä.
MabThera-hoitoa saavat potilaat voidaan rokottaa inaktivoiduilla rokotteilla. Hoitovaste inaktivoiduille rokotteille saattaa kuitenkin olla alentunut. Satunnaistetussa tutkimuksessa nivelreumapotilailla havaittiin, että vaste tetanus-tehosterokotukselle oli vastaava MabTheralla ja metotreksaatilla hoidetuilla potilailla (39 %) kuin pelkkää metotreksaattia saavilla potilailla (42 %). Kuusi kuukautta MabThera-hoidon jälkeen annetuille pneumokokki-polysakkaridirokotteelle ja KLH neoantigeenille vasteet olivat alentuneet MabTheraa ja metotreksaattia saavien ryhmässä verrattuna pelkkää metotreksaattia saavien ryhmään. MabTheraa ja metotreksaattia saavilla vaste polysakkaridirokotteelle oli 43 % (ainakin kahdelle pneumokokki vasta-aineen serotyypille) ja pelkkää metotreksaattia saavilla 82 %. Vasteet KLH neoantigeenille olivat 47 % ja 93 %. Jos inaktivoidut rokotteet ovat tarpeellisia MabThera-hoidon aikana, rokotukset on annettava vähintään 4 viikkoa ennen seuraavaa MabThera-infuusiota.
Kokemukset yhden vuoden aikana nivelreumapotilaille annetuista MabTheran toistohoidoista osoittivat, että potilaita, joilla oli positiivisia vasta-ainetittereitä pneumokokkia, influenssaa, sikotautia, vihurirokkoa, vesirokkoa ja tetanustoksoidia vastaan, oli yleisesti saman verran kuin hoidon alussa.
Nivelreumapotilaille samanaikaisesti/myöhemmin annettu DMARD-lääkitys
MabTheran ja muiden antireumaattisten lääkkeiden samanaikaista käyttöä ei suositella (lukuun ottamatta kohdissa ”Käyttöaiheet” ja ”Annostus” mainittuja nivelreumalääkkeitä).
Kliinisistä tutkimuksista ei ole riittävästi tietoa, jotta voitaisiin täysin arvioida MabThera-hoidon jälkeen annettujen muiden tautiprosessia hidastavien reumalääkkeiden (DMARD), mukaan lukien TNF-estäjien ja muiden biologisten valmisteiden turvallisuutta (ks. kohta Yhteisvaikutukset). Saatavilla oleva tieto viittaa siihen, että kliinisesti merkittävien infektioiden määrä pyysyy muuttumattomana annettaessa tällaista hoitoa aikaisemmin MabTheraa saaneille potilaille. Potilaita on kuitenkin seurattava tarkasti infektioon viittaavien merkkien varalta, jos biologisia ja/tai tautiprosessia hidastavia reumalääkkeitä (DMARD) käytetään MabThera-hoidon jälkeen.
Maligniteetit
Immuunivasteen muuntajat voivat lisätä maligniteettien riskiä. Saatavilla olevat tiedot eivät kuitenkaan viittaa siihen, että rituksimabin käyttö autoimmuunisairauksien hoidossa lisäisi maligniteettien riskiä verrattuna jo olemassa olevaan autoimmuuniperussairauksiin liittyvään maligniteettien riskiin.
Apuaineet
Tämä lääkevalmiste sisältää 2,3 mmol (eli 52,6 mg) natriumia per 10 ml:n injektiopullo ja 11,5 mmol (eli 263,2 mg) natriumia per 50 ml:n injektiopullo, mikä vastaa 2,6 %:a (10 ml:n injektiopullo) ja 13,2 %:a (50 ml:n injektiopullo) WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Tämä lääkevalmiste sisältää 7,0 mg polysorbaattia 80 per 10 ml:n injektiopullo, mikä vastaa 0,7 mg:aa/ml, ja 35,0 mg polysorbaattia 80 per 50 ml:n injektiopullo, mikä vastaa 0,7 mg:aa/ml. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Yhteisvaikutukset
Tällä hetkellä MabTheran ja muiden lääkeaineiden mahdollisista interaktioista on rajallisesti tietoa.
KLL-potilailla samanaikainen MabThera-hoito ei näyttänyt vaikuttavan fludarabiinin tai syklofosfamidin farmakokinetiikkaan. Hoito fludarabiinilla ja syklofosfamidilla ei myöskään aiheuttanut selvää vaikutusta MabTheran farmakokinetiikkaan.
Samanaikainen metotreksaattihoito ei vaikuttanut MabTheran farmakokinetiikkaan nivelreumapotilailla.
Potilaat, joilla on humaani vasta-aineita hiiren proteiineille (HAMA) tai lääkevasta-aineita (ADA), saattavat saada allergisia reaktioita tai yliherkkyysreaktioita, kun heille annetaan diagnostista tai terapeuttista monoklonaalista vasta-ainetta.
283 nivelreumapotilasta sai jatkohoitoa biologisella DMARDilla MabThera-hoidon jälkeen. Näillä potilailla kliinisesti merkittävien infektioiden määrä oli 6,01 tapausta 100 potilasvuotta kohden, kun potilaat saivat pelkkää MabTheraa ja 4,97 tapausta 100 potilasvuotta kohden, kun potilaat saivat jatkohoitoa biologisella DMARDilla.
Raskaus ja imetys
Ehkäisy miehille ja naisille
Rituksimabin retentioajan tiedetään olevan pitkä potilailla, joilla on vähän B-soluja. Hedelmällisessä iässä olevien naisten on siksi huolehdittava raskauden tehokkaasta ehkäisystä MabThera-hoidon aikana ja 12 kuukautta MabThera-hoidon päättymisen jälkeen.
Raskaus
IgG-immunoglobuliinien tiedetään läpäisevän istukan.
Vastasyntyneiden lasten B-solupitoisuuksia raskaudenaikaisen MabThera-altistuksen jälkeen ei ole tutkittu kliinisissä tutkimuksissa. MabTheran käytöstä raskauden aikana ei ole saatavilla riittävää, kontrolloitua kliinistä tutkimustietoa. B-solumäärän ohimenevää vähenemistä ja lymfosytopeniaa on kuitenkin jonkin verran raportoitu raskauden aikana MabTheralle altistuneiden naisten vastasyntyneillä vauvoilla. Eläinkokeissa on havaittu samankaltaisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Siksi MabTheraa ei saa antaa raskaana oleville naisille, ellei hoidon mahdollista hyötyä arvioida suuremmaksi kuin mahdollista riskiä.
Imetys
Vähäiset tiedot rituksimabin erittymisestä rintamaitoon viittaavat siihen, että rituksimabin pitoisuudet rintamaidossa ovat hyvin pienet (lapsen saama suhteellinen annos alle 0,4 %). Muutamien rintaruokittujen vauvojen jatkoseurannassa huomattiin normaalia kasvua ja kehitystä 2 ikävuoteen asti. Koska nämä tiedot ovat rajalliset ja pitkäaikaisen käytön vaikutukset rintaruokittuihin vauvoihin ovat edelleen tuntemattomia, imettämistä ei suositella rituksimabihoidon aikana eikä mieluiten 6 kuukauteen rituksimabihoidon päättymisen jälkeen.
Hedelmällisyys
Eläinkokeissa ei ole havaittu haitallisia vaikutuksia lisääntymiselimiin.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia MabThera-valmisteen vaikutuksesta ajokykyyn tai koneidenkäyttökykyyn ei ole tehty, vaikka farmakologinen vaikutus ja tähän mennessä raportoidut haittavaikutukset viittaavat siihen, että MabTheralla ei ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Kokemukset aikuisten non-Hodgkin-lymfooman ja kroonisen lymfaattisen leukemian hoidosta
Turvallisuusprofiilin yhteenveto
MabTheran yleinen turvallisuusprofiili non-Hodgkin-lymfooman ja kroonisen lymfaattisen leukemian hoidossa perustuu kliinisistä tutkimuksista ja markkinoille tulon jälkeen saatuihin kokemuksiin. Näitä potilaita oli hoidettu joko MabThera-monoterapialla (induktiohoitona tai ylläpitohoitona induktiohoidon jälkeen) tai yhdistelmähoidolla solunsalpaajien kanssa.
MabTheralla hoidetuilla potilailla yleisimmin havaitut haittavaikutukset olivat infuusioon liittyvät reaktiot, joita esiintyi suurimmalla osalla potilaista ensimmäisen rituksimabi-infuusion aikana. Infuusioreaktioiden esiintyvyys laskee huomattavasti seuraavien infuusioiden yhteydessä ja on vähäisempi kuin 1 % kahdeksannen MabThera-annoksen jälkeen.
Infektiotapahtumia (lähinnä bakteeri- ja virusinfektioita) esiintyi kliinisissä tutkimuksissa noin 30–55 %:lla non-Hodgkin-lymfoomaa sairastavista potilaista ja 30–50 %:lla kroonista lymfaattista leukemiaa sairastavista potilaista.
Yleisimmin raportoidut tai havaitut vakavat haittavaikutukset olivat:
- Infuusioon liittyvät reaktiot (mukaan lukien sytokiini- ja tuumorilyysioireyhtymät), ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
- Infektiot, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
- Kardiovaskulaariset tapahtumat, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Muihin raportoituihin vakaviin haittavaikutuksiin kuuluvat B-hepatiitin uudelleen aktivoituminen ja progressiivinen multifokaalinen enkefalopatia (PML) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Haittavaikutustaulukko
Haittavaikutusten yleisyydet MabThera-monoterapiassa tai yhdistelmähoidossa solunsalpaajien kanssa on koottu alla olevaan taulukkoon (taulukko 3). Esiintyvyys määritetään seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Jokaisessa yleisyysluokassa haittavaikutukset on esitetty haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Valmisteen markkinoille tulon jälkeen havaitut haittavaikutukset on listattu sarakkeessa ”Tuntematon”. Näiden haittavaikutusten esiintyvyyttä ei voida arvioida, ks. alaviitteet.
Taulukko 3 Kliinisissä tutkimuksissa tai markkinoille tulon jälkeen raportoidut haittavaikutukset non-Hodgkin-lymfoomaa ja kroonista lymfaattista leukemiaa sairastavilla potilailla, jotka saivat MabTheraa monoterapiassa/ylläpitohoitona tai yhdistettynä solunsalpaajiin
| MedDRA-elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Hyvin harvinainen | Tuntematon |
| Infektiot | Bakteeri-infektio, virusinfektio, +keuhkoputken-tulehdus | Verenmyrkytys, +keuhkokuume, +kuumeinen infektio, +herpes zoster, +hengitystie-infektio, sieni-infektiot, infektiot, joiden aiheuttaja tuntematon, +akuutti keuhkoputken tulehdus, +sivuontelo-tulehdus, B-hepatiitti1 | Vakava virusinfektio2, pneumocystis jirovecii -pneumonia | progressiivinen multifokaalinen leukoenkefalopatia | Enteroviraalinen meningo-enkefaliitti2, 3 | |
| Veri ja imukudos | Neutropenia, leukopenia, +kuumeinen neutropenia, +trombosyto-penia | Anemia, +pansytopenia, +granulosyto-penia | Koagulaatiohäiriö, aplastinen anemia, hemolyyttinen anemia, lymfadenopatia | Ohimenevä nousu seerumin IgM-tasoissa4 | Viivästynyt neutropenia4 | |
| Immuunijärjestelmä | Infuusioon liittyvät reaktiot5, angioödeema | Yliherkkyys | Anafylaksia | Tuumorilyysisyndrooma, sytokiinioireyhtymä5, seerumitauti | Infuusioon liittyvä akuutti palautuva trombosytopenia5 | |
| Aineenvaihdunta ja ravitsemus | Hyperglykemia, painonlasku, perifeerinen ödeema, kasvoödeema, kohonnut LDH, hypokalsemia | |||||
| Psyykkiset häiriöt | Depressio, hermostuneisuus | |||||
| Hermosto | Parestesia, hypoestesia, agitaatio, unettomuus, vasodilataatio, huimaus, ahdistuneisuus | Dysgeusia | Perifeerinen neuropatia, kasvohermohalvaus6 | Kraniaalinen neuropatia, muiden aistien menetys6 | ||
| Silmät | Kyynelnesteen erityshäiriö, konjunktiviitti | Vaikea näönmenetys6 | ||||
| Kuulo ja tasapainoelin | Tinnitus, korvakipu | Kuulonmenetys6 | ||||
| Sydän | +Sydäninfarkti5,7, arytmia, +eteisvärinä, takykardia, +sydänhäiriöt | +Vasemman kammion toimintahäiriö, +supraventrikulaarinen takykardia, +ventrikulaarinen takykardia, +angina, +sydänlihas-iskemia, bradykardia | Vaikeat sydämen toimintahäiriöt5, 7 | Sydämen vajaatoiminta5,7 | ||
| Verisuonisto | Hypertensio, ortostaattinen hypotensio, hypotensio | Vaskuliitti (pääasiassa ihoon liittyvä), leukosytoklastinen vaskuliitti | ||||
| Hengityselimet, rintakehä ja välikarsina | Bronkospasmi5, hengityselinsairaus, rintakipu, hengenahdistus, lisääntynyt yskiminen, nuha | Astma, ahtauttava bronkioliitti, keuhkojen toimintahäiriö, hypoksia | Interstitiaalinen keuhkosairaus8 | Hengitysvaje5 | Keuhkoinfiltraatit | |
| Ruoansulatuselimistö | Pahoinvointi | Oksentamien, ripuli, mahakipu, nielemishäiriö, suutulehdus, ummetus, dyspepsia, anoreksia, nieluärsytys | Suurentunut vatsa | Ruoansulatuskanavan perforaatio8 | ||
| Iho ja ihonalainen kudos | Kutina, ihottuma, +alopesia | Urtikaria, hikoilu, yöhikoilu, +iho-oireet | Vaikea rakkulainen ihoreaktio, Stevens–Johnsonin oireyhtymä, toksinen epidermaalinen nekrolyysi (Lyellin oireyhtymä)8 | |||
| Luusto, lihakset ja sidekudos | Hypertonia, lihaskipu, nivelsärky, selkäkipu, niskakipu, kipu | |||||
| Munuaiset ja virtsatiet | Munuaisten vajaatoiminta5 | |||||
| Yleisoireet ja antopaikassa todettavat haitat | Kuume, vilunväristykset, voimattomuus, päänsärky | Tuumorikipu, ihon punoitus, huonovointisuus, vilustumisen kaltaiset oireet, +uupumus, +vilunväristykset, +usean elimen toimintavajaus5 | Infuusiokohdan kipu | |||
| Tutkimukset | Alentuneet IgG-pitoisuudet | |||||
| Esiintymistiheydet perustuvat haittavaikutusten kokonaismäärään (kaikki vakavuusasteet mukaan luettuina), lukuun ottamatta niitä haittavaikutuksia, jotka on merkitty "+". Niiden esiintymistiheys perustuu vain vakaviin tapauksiin (≥ 3 aste NCI:n (National Cancer Institute) Common Toxicity Criteria -luokituksen mukaisesti). Tutkimuksissa vain suurin havaittu esiintymistiheys on raportoitu. 1 Myös taudin uusiutumista ja primääri-infektioita, frekvenssi perustuu R-FC-hoitoon KLL-potilailla, joilla on uusiutunut tai refraktorinen tauti 2 Katso myös kohta Infektiot edempänä. 3 Havaittu markkinoille tulon jälkeisessä seurannassa 4 Katso myös kohta Hematologiset haittavaikutukset edempänä. 5 Katso myös kohta Infuusioon liittyvät reaktiot edempänä. Kuolemaan johtaneita tapauksia raportoitiin harvoin. 6 Kraniaaliseen neuropatiaan viittaavat signaalit ja oireet, joita esiintyi useita kertoja jopa monta kuukautta MabThera-hoidon päättymisen jälkeen. 7 Havaittu lähinnä potilailla, joilla on ollut aikaisempia sydänongelmia ja/tai jotka ovat saaneet kardiotoksista solunsalpaajahoitoa. Useimmiten yhteydessä infuusioon liittyviin reaktioihin. 8 Myös kuolemaan johtaneita tapauksia. | ||||||
Seuraavia kliinisissä tutkimuksissa haittavaikutuksina raportoituja oireita esiintyi vastaavasti tai vähemmän MabThera-haarassa verrattuna kontrollihaaraan: hematotoksisuus, neutropeeninen infektio, virtsatietulehdus, aistihäiriöt, kuume.
Infuusioon liittyvän reaktion tunnusmerkkejä ja oireita raportoitiin kliinisissä tutkimuksissa yli 50 %:lla potilaista, ja niitä havaittiin lähinnä ensimmäisen infuusion yhteydessä, tavallisesti sen ensimmäisen kahden tunnin kuluessa. Näihin oireisiin kuuluivat pääasiallisesti kuume, vilunväristykset ja jäykkyys. Muita oireita olivat ihon punoitus, angioödeema, bronkospasmi, oksentelu, pahoinvointi, urtikaria/ihottuma, väsymys, päänsärky, nieluärsytys, nuha, kutina, kipu, takykardia, hypertensio, hypotensio, hengenahdistus, ruoansulatushäiriö, astenia ja tuumorilyysisyndrooman piirteitä. Vakavia infuusioon liittyviä reaktioita (kuten bronkospasmia ja hypotensiota) esiintyi korkeintaan 12 %:ssa tapauksista. Lisäksi muutamissa tapauksissa raportoitiin sydäninfarktia, eteisvärinää, keuhkoödeemaa ja akuuttia palautuvaa trombosytopeniaa. Seuraavien raportoitujen haittavaikutuksien yleisyys on vähäinen tai tuntematon: olemassa olevien sydänsairauksien paheneminen (esim. angina pectoris tai sydämen vajaatoiminta), sydämen toimintahäiriöt (sydämen vajaatoiminta, sydäninfarkti, eteisvärinä), keuhkoödeema, usean elimen toiminnanvajaus, tuumorilyysisyndrooma, sytokiinioireyhtymä, munuaisten vajaatoiminta ja hengitysvaje. Seuraavilla infuusiokerroilla infuusioon liittyvien oireiden esiintyvyys väheni selvästi ja niitä havaittiin < 1 %:lla potilaista kahdeksanteen MabThera-hoitosykliin mennessä.
Valittujen haittavaikutusten kuvaus
Infektiot
MabThera aiheuttaa B-solujen vähäisyyttä noin 70–80 %:lla potilaista, mutta vain pienellä osalla seerumin immunoglobuliinien määrä laskee.
Satunnaistetuissa tutkimuksissa raportoitiin MabTheraa saavien haarassa enemmän paikallisia Candida-infektioita ja herpes zoster -infektioita. Vaikeita infektioita raportoitiin noin 4 %:lla potilaista, joita hoidettiin MabThera-monoterapialla. Seurantaryhmään verrattuna infektioiden kokonaisesiintyvyyden lisääntymistä (mukaan lukien asteet 3 ja 4) havaittiin MabThera-ylläpitohoitoa (pisimmillään 2 vuotta) saavassa ryhmässä. Kahden vuoden ylläpitojakson aikana ei ilmaantunut kumulatiivista infektioiden aiheuttamaa toksisuutta. Lisäksi muita vakavia virusinfektioita raportoitiin MabThera-hoidon yhteydessä. Kyseessä oli joko uusi infektio tai olemassa olevan infektion uudelleen aktivoituminen tai paheneminen. Osa tapauksista oli kuolemaan johtavia. Enemmistö potilaista oli saanut MabTheraa yhdessä solunsalpaajahoidon kanssa tai hematopoieettisen kantasolusiirteen yhteydessä. Näitä vakavia virusinfektioita ovat esimerkiksi herpesvirusten (sytomegalovirus, vesirokkovirus ja herpes simplex -virus), JC-viruksen (progressiivinen multifokaalinen leukoenkefalopatia, PML), enteroviruksen (meningoenkefaliitti) ja C-hepatiittiviruksen aiheuttamat infektiot (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Taudin etenemisen ja toistohoidon jälkeen ilmenneitä kuolemaan johtaneita PML-tapauksia on myös raportoitu kliinisissä tutkimuksissa. B-hepatiitin uudelleen aktivoitumista on raportoitu potilailla, joista suurin osa sai MabTheraa yhdessä sytotoksisen solunsalpaajahoidon kanssa. Uusiutunutta KLL:aa tai refraktorista tautia sairastavilla potilailla asteiden 3 ja 4 B-hepatiitti-infektion (reaktivoitunut ja primääri-infektio) esiintymistiheys oli 2 % R-FC-ryhmässä ja 0 % FC-ryhmässä. MabTheralle altistuneilla, Kaposin sarkoomaa sairastavilla potilailla on havaittu kyseisen taudin etenemistä. Nämä tapaukset ilmenivät hyväksyttyjen käyttöaiheiden ulkopuolella ja suurin osa potilaista oli HIV-positiivisia.
Hematologiset haittavaikutukset
Kliinisissä tutkimuksissa, joissa MabTheraa annettiin monoterapiana 4 viikon ajan, pienellä osalla potilaista esiintyi hematologisia poikkeavuuksia, jotka olivat yleensä lieviä ja ohimeneviä. Potilailla raportoitiin vaikeaa asteiden 3 ja 4 neutropeniaa 4,2 %, anemiaa 1,1 % ja trombosytopeniaa 1,7 %. MabTheran ylläpitohoidossa (pisimmillään 2 vuotta) asteiden 3 ja 4 leukopeniaa ja neutropeniaa raportointiin useammin MabThera-ryhmässä kuin seurantaryhmässä. Leukopenia: MabThera-ryhmässä 5 % ja seurantaryhmässä 2 %. Neutropenia: MabThera-ryhmässä 10 % ja seurantaryhmässä 4 %. Trombosytopenian esiintyvyys oli matala (< 1 %, asteet 3 ja 4), eikä tutkimushaarojen välillä havaittu eroa. Tutkimuksissa, joissa MabTheraa käytettiin yhdessä solunsalpaajien kanssa, seuraavien haittavaikutusten esiintyvyys hoitojakson aikana oli yleensä korkeampi verrattuna pelkkään solunsalpaajahoitoon: asteiden 3 ja 4 leukopenia (88 % R-CHOP-ryhmässä vs. 79 % CHOP-ryhmässä, 23 % R-FC-ryhmässä vs. 12 % FC-ryhmässä), neutropenia (24 % R-CVP-ryhmässä vs. 14% CVP-ryhmässä; 97 % R-CHOP-ryhmässä vs. 88 % CHOP-ryhmässä, 30 % R-FC-ryhmässä vs. 19 % FC-ryhmässä aikaisemmin hoitamattomassa KLL:ssa) ja pansytopenia (3 % R-FC-ryhmässä vs. 1 % FC-ryhmässä aikaisemmin hoitamattomassa KLL:ssa). Pelkkää solunsalpaajahoitoa saaviin verrattuna neutropeniaa esiintyi enemmän MabTheralla ja solunsalpaajilla hoidetuilla potilailla, mutta tähän ei kuitenkaan liittynyt infektioiden eikä loistartuntojen määrän lisääntymistä. Tutkimuksissa aikaisemmin hoitamattomassa ja uusiutuneessa/refraktorisessa KLL:ssa on osoitettu, että jopa 25 %:lla R-FC-hoitoa saaneilla potilailla neutropenia oli pitkäkestoinen (neutrofiilien määrä jäi alle 1 x 109/l päivinä 24–42 viimeisestä annoksesta), tai kehittyi myöhemmin MabThera-FC-hoidon jälkeen (neutrofiilien määrä alle 1 x 109/l 42. päivän jälkeen viimeisestä annoksesta potilailla, joilla ei aikaisemmin ollut pitkittynyt neutropenia tai jotka olivat toipuneet neutropeniasta ennen päivää 42). Anemian esiintyvyydessä ei havaittu eroa. Muutamia viivästyneen neutropenian tapauksia havaittiin yli neljä viikkoa viimeisen MabThera-infuusion jälkeen. Ensilinjan KLL-tutkimuksessa Binet C -vaiheen tautia sairastavat potilaat kokivat enemmän haittavaikutuksia R-FC-haarassa kuin FC-haarassa (83 % R-FC-haarassa ja 71 % FC-haarassa). Uusiutuneessa/refraktorisessa KLL-tutkimuksessa asteiden 3 ja 4 trombosytopeniaa raportoitiin 11 %:lla R-FC-ryhmän potilaista ja 9 %:lla FC-ryhmän potilaista.
MabTheralla tehdyissä tutkimuksissa Waldenströmin makroglobulinemiaa sairastavilla potilailla on havaittu hoidon aloituksen jälkeen ohimenevää nousua seerumin IgM-tasoissa, mihin saattaa liittyä veren viskositeetin nousua ja siihen yhteydessä olevia oireita. Ohimenevä IgM-tason nousu palautui tavallisesti ainakin perustasolle neljän kuukauden sisällä.
Kardiovaskulaariset haittavaikutukset
Kliinisissä tutkimuksissa, joissa MabTheraa annettiin monoterapiana, kardiovaskulaarisia haittavaikutuksia raportoitiin 18,8 %:lla potilaista. Useimmin ilmoitetut haitat olivat hypotensio ja hypertensio. Infuusion aikaista asteen 3 tai 4 arytmiaa (mukaan lukien ventrikulaarinen ja supraventrikulaarinen takykardia) ja angina pectorista raportoitiin. Ylläpitohoidossa asteiden 3 ja 4 sydänhäiriöiden esiintyvyys oli samankaltainen MabTheralla hoidetuilla potilailla ja seurantaryhmissä. Sydäntapahtumia raportoitiin vakavina haittavaikutuksina (mukaan lukien eteisvärinä, sydäninfarkti, vasemman kammion vajaatoiminta, sydänlihaksen hapenpuute) 3 %:lla MabThera-ryhmän potilaista ja < 1 %:lla seurantaryhmän potilaista. Kliinisissä tutkimuksissa, joissa arvioitiin rituksimabin käyttöä yhdessä solunsalpaajien kanssa, sydämen rytmihäiriöiden asteiden 3 ja 4, pääasiassa supraventrikulaaristen häiriöiden, kuten takykardian ja eteislepatuksen/eteisvärinän, esiintyvyys oli suurempi R-CHOP-ryhmässä (14 potilasta, 6,9 %) verrattuna CHOP-ryhmään (3 potilasta, 1,5 %). Kaikki edellä mainitut sydämen rytmihäiriöt tapahtuivat joko MabThera-infuusion yhteydessä tai niihin liittyi altistavia tekijöitä, kuten kuume, infektio, akuutti sydäninfarkti tai jo olemassa oleva hengityselimiin liittyvä tai kardiovaskulaarinen sairaus. R-CHOP- ja CHOP-ryhmien välillä ei havaittu eroa muiden, sydämeen kohdistuvien asteiden 3 ja 4 haittatapahtumien, kuten sydämen vajaatoiminnan, sydänlihassairauden tai sepelvaltimotaudin oireiden suhteen. KLL:ssa asteiden 3 ja 4 sydänhäiriöiden esiintyvyys oli vähäistä sekä ensilinjan tutkimuksessa (4 % R-FC-haarassa, 3 % FC-haarassa) että uusiutuneen/refraktorisen taudin tutkimuksessa (4 % R-FC-haarassa, 4 % FC-haarassa).
Hengityselimet
Interstitiaalisia keuhkotautitapauksia (joskus kuolemaan johtavia) on raportoitu.
Neurologiset häiriöt
Hoitojakson aikana (hoidon induktiovaihe, joka oli enintään kahdeksan hoitosyklin R-CHOP-hoito) R-CHOP-ryhmästä neljä potilasta (2 %), joilla kaikilla oli kardiovaskulaarisia riskitekijöitä, sai tromboembolisen aivoverisuonitapahtuman ensimmäisen hoitosyklin aikana. Muiden tromboembolisten haittatapahtumien esiintyvyydessä ei havaittu eroa hoitoryhmien välillä. Sitä vastoin kolme potilasta (1,5 %) CHOP-ryhmästä koki aivoverisuonitapahtumia, jotka kaikki sattuivat seurantajakson aikana. KLL:ssa asteiden 3 ja 4 hermostojärjestelmän häiriöiden esiintyvyys oli vähäistä sekä ensilinjan tutkimuksessa (4 % R-FC-haarassa, 4 % FC-haarassa) että uusiutuneen/refraktorisen taudin tutkimuksessa (3 % R-FC-haarassa, 3 % FC-haarassa).
Tapauksia posteriorisesta palautuvasta enkefalopatiaoireyhtymästä (PRES)/palautuvasta posteriorisesta leukoenkefaliittioireyhtymästä (RPLS) on raportoitu. Tunnusmerkkeihin ja oireisiin kuuluvat muun muassa näköhäiriöt, pääsärky, kouristuskohtaukset ja muutokset mielialassa, ja johon voi liittyä hypertensiota. PRES/RPLS-diagnoosi pitää varmentaa aivojen kuvantamisella. Raportoiduissa tapauksissa potilailla oli PRES/RPLS:n tunnettuja riskitekijöitä kuten muu perussairaus, hypertensio, immunosuppressiivinen lääkitys ja/tai solusalpaajahoito.
Ruoansulatuselimistö
Ruoansulatuskanavan perforaatioita on havaittu potilailla, jotka ovat saaneet MabTheraa non-Hodgkin-lymfooman hoitoon, ja joissakin tapauksissa ne ovat olleet kuolemaan johtavia. Suurimmassa osassa tapauksista MabTheraa annettiin yhdessä solunsalpaajahoidon kanssa.
IgG-pitoisuus
Kliinisessä tutkimuksessa, joissa arvioitiin MabTheran käyttöä ylläpitohoidossa uusiutunutta/refraktorista follikulaarista lymfoomaa sairastavilla potilailla, seerumin IgG-pitoisuuksien mediaanit olivat induktiohoidon jälkeen alle viitealueen alarajan (eli < 7 g/l) sekä seuranta- että MabThera-ryhmissä. Tämän jälkeen seurantaryhmässä IgG-pitoisuuden mediaani suureni viitealueen alarajan yläpuolelle, mutta pysyi muuttumattomana MabThera-ryhmässä. Kahden vuoden hoitojakson aikana potilaita, joiden IgG-pitoisuudet olivat alle viitealueen alarajan, oli noin 60 % MabThera-ryhmässä, kun taas näiden potilaiden osuus pieneni seurantaryhmässä (36 % kahden vuoden kuluttua).
Spontaaniraportoinnissa ja kirjallisuudessa on havaittu pieni määrä hypogammaglobulinemiatapauksia MabTheralla hoidetuilla pediatrisilla potilailla. Joissakin tapauksissa oire on ollut vakava ja pitkäkestoista immunoglobuliinikorvaushoitoa vaativaa. Pitkäaikaisen B-soluvajeen seuraukset pediatrisille potilaille eivät ole tiedossa.
Iho- ja ihonalainen kudos
Toksista epidermaalista nekrolyysiä (Lyellin oireyhtymä), ja Stevens–Johnsonin oireyhtymää, jotka ovat toisinaan johtaneet potilaan kuolemaan, on raportoitu hyvin harvoin.
Erityisryhmien potilaat - MabThera-monoterapia
Iäkkäät (vähintään 65-vuotiaat):
Haittavaikutusten kaikkien asteiden, myös asteiden 3 ja 4, esiintyvyys oli samanlainen iäkkäillä potilailla ja nuoremmilla (alle 65-vuotiaita) potilailla.
Sairaus, johon liittyy suuri tautimassa
Asteiden 3 ja 4 haittavaikutusten esiintyvyys oli korkeampi potilailla, joilla oli suuri tautimassa, kuin potilailla ilman suurta tautimassaa (25,6 % vs. 15,4 %). Haittavaikutusten kaikkien asteiden esiintyvyys oli samanlainen näissä kahdessa ryhmässä.
Uusintahoito
Uudelleen hoidetuilla potilailla, jotka saivat toistuvia MabThera-hoitojaksoja, raportoitiin saman verran haittavaikutuksia kuin ensimmäisen hoitojakson saaneilla potilailla (kaikki asteet sekä asteiden 3 ja 4 haittavaikutukset).
Erityisryhmien potilaat – MabThera-yhdistelmähoito
Iäkkäät (vähintään 65-vuotiaat):
Aikaisemmin hoitamattomassa KLL:ssa tai uusiutuneessa tai refraktorisessa taudissa hematologisten ja lymfaattisen järjestelmän asteiden 3 ja 4 haittavaikutuksia esiintyi enemmän iäkkäillä potilailla kuin alle 65-vuotiailla.
Kokemukset pediatristen potilaiden diffuusista suurisoluisesta B-solulymfoomasta, Burkittin lymfoomasta, Burkittin leukemiasta ja Burkitt-tyyppisestä lymfoomasta
Turvallisuusprofiilin yhteenveto
Avoimessa, satunnaistetussa monikeskustutkimuksessa tutkittiin LMB-solunsalpaajahoitoa (Lymphome Malin B) yhdessä MabTheran kanssa ja ilman MabTheraa pediatrisilla potilailla (ikä 6 kuukaudesta alle 18 vuoteen), jotka sairastivat aiemmin hoitamatonta pitkälle edennyttä CD20-positiivista diffuusia suurisoluista B-solulymfoomaa, Burkittin lymfoomaa, Burkittin leukemiaa tai Burkitt-tyyppistä lymfoomaa.
Yhteensä 309 pediatrista potilasta sai MabThera-hoitoa ja otettiin mukaan turvallisuusanalyysin potilasjoukkoon. Pediatriset potilaat satunnaistettiin LMB-solunsalpaajahaaraan, jossa potilaat saivat myös MabTheraa, tai otettiin mukaan tutkimuksen yksihaaraiseen osaan, jossa potilaille annettiin MabThera-annoksia 375 mg/m2 kehon pinta-alan mukaan, ja he saivat yhteensä kuusi MabThera-infuusiota laskimoon (kaksi infuusiota kummassakin kahdesta induktiojaksosta ja yksi infuusio LMB-hoito-ohjelman kummassakin kahdesta konsolidaatiojaksosta).
Aiemmin hoitamatonta pitkälle edennyttä CD20-positiivista diffuusia suurisoluista B-solulymfoomaa, Burkittin lymfoomaa, Burkittin leukemiaa tai Burkitt-tyyppistä lymfoomaa sairastavilla pediatrisilla potilailla (ikä 6 kuukaudesta alle 18 vuoteen) MabTheran turvallisuusprofiili oli tyypiltään, luonteeltaan ja vaikeusasteeltaan yleisesti yhdenmukainen non-Hodgkin-leukemiaa ja kroonista lymfaattista leukemiaa sairastavien aikuisten potilaiden tunnetun turvallisuusprofiilin kanssa. MabTheran lisääminen solunsalpaajahoitoon johti joidenkin tapahtumien, kuten infektioiden (mukaan lukien sepsis), lisääntyneeseen riskiin verrattuna pelkkään solunsalpaajahoitoon.
Kokemukset nivelreumasta
Turvallisuusprofiilin yhteenveto
MabTheran turvallisuusprofiili nivelreuman hoidossa perustuu kliinisiin tutkimuksiin ja markkinoille tulon jälkeen saatuihin kokemuksiin.
Yhteenveto MabTheran turvallisuusprofiilista vaikeaa nivelreumaa sairastavilla potilailla on esitetty seuraavissa kappaleissa. Kliinisissä tutkimuksissa yli 3100 potilasta sai vähintään yhden hoitojakson, ja seuranta-ajan pituus vaihteli 6 kuukaudesta yli 5 vuoteen. Noin 2400 potilasta sai kaksi tai useampia hoitojaksoja ja yli 1000 potilasta sai viisi tai useampia hoitojaksoja. Myyntiluvan myöntämisen jälkeen kerätyt turvallisuustiedot vastasivat odotettua, MabTheran kliinisissä tutkimuksissa havaittua haittavaikutusprofiilia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Potilaat saivat 2 x 1000 mg MabTheraa kahden viikon välein metotreksaattihoidon (10–25 mg viikossa) lisäksi. MabThera-infuusiot annettiin 100 mg:n laskimonsisäisen metyyliprednisoloni-infuusion jälkeen. Lisäksi potilaille annettiin prednisonia suun kautta 15 vuorokauden ajan.
Haittavaikutustaulukko
Haittavaikutukset on listattu taulukossa 4. Yleisyys määritetään seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Jokaisessa yleisyysluokassa haittavaikutukset on esitetty haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä.
Valmisteen markkinoille tulon jälkeen havaitut haittavaikutukset on listattu sarakkeessa ”Tuntematon”. Näiden haittavaikutusten esiintyvyyttä ei voida arvioida, ks. alaviitteet.
Infuusioon liittyvät reaktiot olivat yleisimpiä haittavaikutuksia, joiden katsottiin johtuneen MabTherasta. Infuusioon liittyvien reaktioiden ilmaantuvuus kliinisissä tutkimuksissa oli 23 % ensimmäisen infuusion yhteydessä ja väheni seuraavien infuusioiden yhteydessä. Vakavat infuusioon liittyvät reaktiot olivat melko harvinaisia (0,5 %:lla potilaista) ja niitä havaittiin lähinnä ensimmäisen hoitojakson yhteydessä. Kliinisissä tutkimuksissa MabTheran käytöstä reumatoidiartriitin hoidossa ilmenneiden haittavaikutusten lisäksi, markkinoille tulon jälkeen on raportoitu progressiivisesta multifokaalisesta leukoenkefalopatiasta (PML) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) ja seerumitaudin kaltaisista reaktioista.
Taulukko 4 Yhteenveto haittavaikutuksista nivelreumapotilailla, jotka saivat MabTheraa kliinisissä tutkimuksissa tai markkinoille tulon jälkeen
| MedDRA-elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Hyvin harvinainen | Tuntematon |
| Infektiot | Ylempien hengitysteiden tulehdus, virtsatietulehdukset | Keuhkoputkentulehdus, sivuontelotulehdus, mahasuolitulehdus, jalkasilsa | Progressiivinen multifokaalinen leukoenkefalopatia, B-hepatiitin uudelleen aktivoitumista | Vakava virusinfektio1, enteroviraalinen meningoenkefaliitti2 | ||
| Veri ja imukudos | Neutropenia3 | Viivästynyt neutropenia4 | Seerumitaudin kaltaiset reaktiot | |||
| Immuunijärjestelmä | 5Infuusioon liittyvät reaktiot (hypertensio, pahoinvointi, ihottuma, kuume, kutina, urtikaria, kurkun ärsytys, kuumat aallot, hypotensio, nuha, vilunväristykset, takykardia, uupumus, suun ja nielun kipu, perifeerinen ödeema, eryteema) | 5Infuusioon liittyvät reaktiot (yleistynyt ödeema, bronkospasmi, hengityksen vinkuminen, kurkunpään turvotus, angioödeema, yleistynyt kutina, anafylaksi, anafylaksin kaltainen reaktio) | ||||
| Yleisoireet ja antopaikassa todettavat haitat | ||||||
| Aineenvaihdunta ja ravitsemus | Hyperkolesterolemia | |||||
| Psyykkiset häiriöt | Masennus, ahdistuneisuus | |||||
| Hermosto | Päänsärky | Parestesia, migreeni, huimaus, iskias | ||||
| Sydän | Angina pectoris, eteisvärinä, sydämen vajaatoiminta, sydäninfarkti | Eteislepatus | ||||
| Ruoansulatuselimistö | Dyspepsia, ripuli, gastroesofageaalinen refluksi, suun haavauma, ylävatsakipu | |||||
| Iho ja ihonalainen kudos | Alopesia | Toksinen epidermaalinen nekrolyysi (Lyellin oireyhtymä, Stevens–Johnsonin oireyhtymä7 | ||||
| Luusto, lihakset ja sidekudos | Nivelsärky/lihaskipu, nivelrikko, bursiitti | |||||
| Tutkimukset | IgM-tasojen lasku6 | IgG-tasojen lasku6 | ||||
| 1 Katso myös kohta infektiot alempana. 2. Havaittu markkinoille tulon jälkeisessä seurannassa 3 Esiintymistiheys perustuu laboratorioarvoihin, jotka on kerätty kliinisten tutkimusten rutiininomaisen laboratorioseurannan yhteydessä. 4 Esiintymistiheys perustuu myyntiluvan myöntämisen jälkeen saatuun tietoon. 5 Reaktiot, jotka esiintyivät infuusion aikana tai 24 tunnin sisällä infuusion antamisesta. Katso myös infuusioon liittyvät reaktiot alempana. Infuusioon liittyvät reaktiot voivat johtua yliherkkyydestä ja/tai lääkkeen vaikutusmekanismista. 6 Mukaan luettu havainnot, jotka on kerätty rutiininomaisen laboratorioseurannan yhteydessä. 7 Mukaan luettu fataalit tapaukset. | ||||||
Toistuvat hoitojaksot
Toistuvien hoitojaksojen haittavaikutusprofiili on samankaltainen kuin ensimmäisessä altistuksessa. Ensimmäisen MabThera-altistuksen jälkeen kaikkien haittavaikutusten esiintyvyys oli korkeimmillaan ensimmäisten kuuden kuukauden aikana ja väheni sen jälkeen. Nämä haitat olivat lähinnä infuusioon liittyviä reaktioita (ilmenevät yleisesti ensimmäisen hoitojakson yhteydessä), nivelreuman pahenemista ja infektioita, jotka kaikki olivat yleisempiä hoidon kuuden ensimmäisen kuukauden aikana.
Valittujen haittavaikutusten kuvaus
Infuusioon liittyvät reaktiot
Yleisin haittavaikutus, joka kliinisissä tutkimuksissa ilmeni MabThera-infuusion yhteydessä, oli infuusioon liittyvät reaktiot (ks. taulukko 4). MabThera-hoitoa saaneista 3189 potilaasta 36 % (1135) koki vähintään yhden infuusioon liittyvän reaktion ja 23 % (733/3189) koki infuusioon liittyvän reaktion ensimmäisen MabThera-altistuksen ensimmäisen infuusion jälkeen. Myöhempien infuusioiden yhteydessä infuusioon liittyvien reaktioiden ilmaantuvuus vähenee. Kliinisissä tutkimuksissa alle 1 % (17/3189) potilaista sai vakavan infuusioon liittyvän reaktion. CTC-luokituksen mukaisia asteen 4 infuusioon liittyviä reaktioita eikä kuolemaan johtaneita tapauksia ole raportoitu kliinisissä tutkimuksissa. CTC-luokituksen mukaiset asteen 3 tapahtumat ja hoidon keskeyttämiseen johtaneet infuusioon liittyvät reaktiot vähenivät joka hoitojaksolla, ja olivat harvinaisia kolmannesta hoitojaksosta eteenpäin. Esilääkityksenä annettu laskimonsisäinen glukokortikoidi vähensi merkittävästi infuusioon liittyvien reaktioiden ilmaantuvuutta ja vaikeusastetta (ks. kohta Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet). Vaikeita kuolemaan johtaneita infuusioon liittyviä reaktioita on raportoitu markkinoille tulon jälkeen.
Tutkimuksessa, jonka tarkoituksena oli arvioida nopeamman MabThera-infuusion turvallisuutta nivelreumapotilailla, MabThera voitiin antaa keskivaikeaa tai vaikeaa aktiivista nivelreumaa sairastaville potilaille laskimoon 2 tunnin kestoisena infuusiona, jos heillä ei esiintynyt vakavaa infuusioreaktiota ensimmäisen tutkitun infuusion aikana tai 24 tunnin kuluessa sen jälkeen. Potilaat, jotka olivat saaneet aikaisemmin vakavan infuusioreaktion jonkin nivelreuman hoidossa käytetyn biologisen lääkehoidon yhteydessä, suljettiin pois tutkimuksesta. Infuusioreaktioiden ilmaantuvuus, luonne ja vaikeusaste olivat yhdenmukaiset aikaisempien havaintojen kanssa. Vakavia infuusioreaktioita ei havaittu.
Infektiot
Infektioiden kokonaisesiintyvyys kliinisissä tutkimuksissa oli MabThera-hoitoa saaneilla potilailla noin 94 tapausta 100 potilasvuotta kohti. Infektiot olivat lähinnä lieviä tai kohtalaisia ja ilmenivät useimmiten ylempien hengitysteiden infektioina ja virtsatieinfektioina. Vakavia tai laskimonsisäisiä antibiootteja vaativia infektioita oli noin 4 tapausta 100 potilasvuotta kohti. Vakavien infektioiden määrä ei noussut merkittävästi MabTheran toistohoitojen jälkeen. Alempien hengitysteiden tulehduksia (myös keuhkokuume) on raportoitu kliinisissä tutkimuksissa MabThera-haaroissa saman verran kuin vertailuhaaroissa.
Markkinoille tulon jälkeen on raportoitu vakavia virusinfektioita rituksimabilla hoidetuilla nivelreumapotilailla.
Kuolemaan johtavia progressiivisia multifokaalisia leukoenkefalopatiatapauksia on raportoitu potilailla, joiden autoimmuunisairautta on hoidettu MabTheralla. Näihin sairauksiin kuuluvat nivelreuma ja mm. systeeminen lupus erythematosus (SLE) ja vaskuliitti (hyväksyttyjen käyttöaiheiden ulkopuolinen käyttö).
B-hepatiitin uudelleen aktivoitumista on raportoitu non-Hodgkin-lymfoomaa sairastavilla potilailla, jotka saivat MabTheraa yhdessä sytotoksisen solunsalpaajahoidon kanssa (ks. non-Hodgkin-lymfooma). B-hepatiitin uudelleenaktivoitumista on hyvin harvoin raportoitu MabThera-hoitoa saaneilla nivelreumapotilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kardiovaskulaariset haittavaikutukset
Vakavia sydämeen kohdistuvia haittavaikutuksia raportoitiin 1,3 tapausta 100 potilasvuotta kohti MabThera-hoitoa saavilla potilailla. Vertailuryhmässä plasebolla hoidetuilla potilailla raportoituja tapauksia oli saman verran (1,3 tapausta 100 potilasvuotta kohti). Sydämeen kohdistuvia haittavaikutuksia (kaikki asteet, myös vakavat) saaneiden potilaiden osuus ei lisääntynyt toistohoitojen yhteydessä.
Neurologiset tapahtumat
Posteriorista reversiibeliä enkefalopatiaoireyhtymää (PRES) / reversiibeliä posteriorista leukoenkefalopatiaoireyhtymää (RPLS) on raportoitu. Oireita olivat näköhäiriöt, päänsärky, kouristuskohtaukset ja mielentilan muutokset, joihin saattoi liittyä hypertensiota. PRES/RPLS-diagnoosi on varmistettava aivojen kuvantamisella. Raportoituihin tapauksiin liittyi PRES:n/RPLS:n tunnistettuja riskitekijöitä, kuten potilaan perussairaus, hypertensio, immunosuppressiivinen hoito ja/tai solunsalpaajahoito.
Neutropenia
MabThera-hoidon yhteydessä on havaittu neutropeniatapahtumia. Suurin osa näistä tapahtumista oli ohimeneviä ja vaikeusasteeltaan lieviä tai kohtalaisia. Neutropenia voi ilmetä useita kuukausia MabThera-annon jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kliinisten tutkimusten plasebokontrolloidussa vaiheessa vaikea neutropenia kehittyi 0,94 %:lle (13/1382) MabTheralla hoidetuille potilaille ja 0,27 %:lle (2/731) plaseboryhmän potilaille.
Myyntiluvan myöntämisen jälkeisessä seurannassa on raportoitu harvoin neutropeenisiä tapahtumia, mukaan lukien viivästynyt ja pitkään jatkuva neutropenia. Joissakin tapauksissa tapahtumiin liittyi kuolemaan johtaneita infektioita.
Iho- ja ihonalainen kudos
Toksista epidermaalista nekrolyysiä (Lyellin oireyhtymä), ja Stevens–Johnsonin oireyhtymää, jotka ovat toisinaan johtaneet potilaan kuolemaan, on raportoitu hyvin harvoin.
Poikkeavat laboratorioarvot
Hypogammaglobulinemiaa (IgG tai IgM alle normaaliarvon alarajan) on havaittu MabTheralla hoidetuilla nivelreumapotilailla. Infektioiden kokonaisesiintyvyys tai vakavien infektioiden esiintyvyys ei lisääntynyt alentuneiden IgG- tai IgM-arvojen kehittymisen jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Spontaaniraportoinnissa ja kirjallisuudessa on havaittu pieni määrä hypogammaglobulinemiatapauksia MabTheralla hoidetuilla pediatrisilla potilailla. Joissakin tapauksissa oire on ollut vakava ja pitkäkestoista immunoglobuliinikorvaushoitoa vaativaa. Pitkäaikaisen B-soluvajeen seuraukset pediatrisille potilaille eivät ole tiedossa.
Kokemukset granulomatoottisesta polyangiitista (GPA) ja mikroskooppisesta polyangiitista (MPA)
MabThera-valmisteen kokonaisturvallisuusprofiili granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavilla aikuisilla ja pediatrisilla potilailla perustuu kolmesta kliinisestä tutkimuksesta ja valmisteen markkinoille tulon jälkeisestä seurannasta saatuihin tietoihin.
Remission induktio aikuisilla (GPA/MPA-tutkimus 1)
99 aikuispotilasta sai GPA/MPA-tutkimuksessa 1 MabThera-hoitoa (375 mg/m2 kerran viikossa 4 viikon ajan) ja glukokortikoideja granulomatoottisen polyangiitin (GPA) ja mikroskooppisen polyangiitin (MPA) remission induktioon (ks. kohta Farmakodynamiikka).
Kaikkien taulukossa 5 lueteltujen granulomatoottista polyangiittia tai mikroskooppista polyangiittia koskeneessa Study 1 ‑tutkimuksessa havaittujen haittavaikutusten, joiden esiintyvyys on yleinen tai hyvin yleinen, ilmaantuvuus MabThera-ryhmässä oli ≥ 5 %. Esiintymistiheys oli suurempi kuin vertailuryhmässä.
Valmisteen markkinoille tulon jälkeen havaitut haittavaikutukset on listattu sarakkeessa ”Tuntematon”. Näiden haittavaikutusten esiintyvyyttä ei voida arvioida, ks. alaviitteet.
Taulukko 5 GPA/MPA-tutkimuksessa 1 kuuden kuukauden hoidon jälkeen esiintyneet haittavaikutukset, joita esiintyi ≥ 5 %:lla MabThera-hoitoa saaneista aikuispotilaista (rituksimabi n=99, suurempi esiintymistiheys kuin vertailuryhmässä) tai markkinoille tulon jälkeisessä seurannassa.
| MedDRA-elinjärjestelmä | Hyvin yleinen | Yleinen | Tuntematon |
| Infektiot | virtsatietulehdus, keuhkoputkentulehdus, herpes zoster ‑infektio, nasofaryngiitti | vakava virusinfektio1,2, enteroviraalinen meningoenkefaiitti1 | |
| Veri ja imukudos | trombosytopenia | ||
| Immuunijärjestelmä | sytokiinioireyhtymä | ||
| Aineenvaihdunta ja ravitsemus | hyperkalemia | ||
| Psyykkiset häiriöt | unettomuus | ||
| Hermosto | huimaus, vapina | ||
| Verisuonisto | hypertensio | punastelu | |
| Hengityselimet, rintakehä ja välikarsina | yskä, hengenahdistus, nenäverenvuoto | nenän tukkoisuus | |
| Ruoansulatuselimistö | ripuli | dyspepsia, ummetus | |
| Iho ja ihonalainen kudos | akne | ||
| Luusto, lihakset ja sidekudos | lihasspasmit, nivelsärky, selkäkipu | lihasheikkous, tuki- ja liikuntaelimistön kipu, raajakipu | |
| Yleisoireet ja antopaikassa todettavat haitat | perifeerinen ödeema | ||
| Tutkimukset | alentunut hemoglobiinipitoisuus | ||
| 1 Havaittu markkinoille tulon jälkeisessä seurannassa. 2 Katso myös kohta infektiot alempana. | |||
Ylläpitohoito (GPA/MPA-tutkimus 2)
GPA/MPA-tutkimuksessa 2 yhteensä 57 aikuispotilasta, joilla oli vaikea-asteinen, aktiivinen granulomatoottinen polyangiitti tai mikroskooppinen polyangiitti, sai MabThera-hoitoa remission ylläpitoon (ks. kohta Farmakodynamiikka).
Taulukko 6 GPA/MPA-tutkimuksessa 2 havaitut haittavaikutukset, joita esiintyi ≥ 5 %:lla MabTheraa saaneista aikuispotilaista (rituksimabi n=57) ja suuremmalla esiintymistiheydellä kuin vertailuryhmässä tai markkinoille tulon jälkeisessä seurannassa
| MedDRA-elinjärjestelmä | Hyvin yleinen | Yleinen | Tuntematon |
| Infektiot | keuhkoputkentulehdus | nuha | vakava virusinfektio1,2, enteroviraalinen meningoenkefaliitti1 |
| Hengityselimet, rintakehä ja välikarsina | hengenahdistus | ||
| Ruoansulatuselimistö | ripuli | ||
| Yleisoireet ja antopaikassa todettavat haitat | kuume, influenssan kaltainen sairaus, perifeerinen ödeema | ||
| Vammat, myrkytykset ja hoitokomplikaatiot | infuusioon liittyvät reaktiot3 | ||
| 1 Havaittu markkinoille tulon jälkeisessä seurannassa. 2 Katso myös kohta infektiot alempana. 3 Tiedot infuusioon liittyvistä reaktioista esitetään kohdassa Valittujen haittavaikutusten kuvaus. | |||
Kokonaisturvallisuusprofiili oli yhdenmukainen MabTheran autoimmuunisairauksien, mukaan lukien granulomatoottisen polyangiitin ja mikroskooppisen polyangiitin, hoidossa havaitun vakiintuneen turvallisuusprofiilin kanssa. Kaikkiaan 4 %:lle MabThera-hoitohaaran potilaista ilmaantui hoidon lopettamiseen johtaneita haittavaikutuksia. Suurin osa MabThera-hoitohaarassa esiintyneistä haittavaikutuksista oli vaikeusasteeltaan lieviä tai keskivaikeita. MabThera-hoitohaaran potilaat eivät kokeneet kuolemaan johtaneita haittatapahtumia.
Yleisimmin raportoituja haittavaikutuksiksi katsottuja tapahtumia olivat infuusioon liittyneet reaktiot ja infektiot.
Pitkäaikaisseuranta (GPA/MPA-tutkimus 3)
Pitkäkestoisessa havainnoivassa turvallisuustutkimuksessa 97 potilasta, jotka sairastivat granulomatoottista polyangiittia ja mikroskooppista polyangiittia, sai MabThera-hoitoa (keskimäärin 8 infuusiota [vaihteluväli 1–28]) enimmillään 4 vuoden ajan lääkärinsä hoitokäytännön ja harkinnan mukaan. Kokonaisturvallisuusprofiili oli yhdenmukainen nivelreuman sekä granulomatoottisen polyangiitin ja mikroskooppisen polyangiitin MabThera-hoidossa vakiintuneen turvallisuusprofiilin kanssa, eikä uusia haittavaikutuksia raportoitu.
Pediatriset potilaat
Avoin yhden haaran tutkimus tehtiin 25 pediatrisella potilaalla, joilla oli vaikea, aktiivinen granulomatoottinen polyangiitti tai mikroskooppinen polyangiitti. Koko tutkimusjakso koostui 6 kuukauden remission induktiovaiheesta ja vähintään 18 kuukauden pituisesta seurantavaiheesta enintään 4,5 vuoteen saakka. MabThera-valmistetta annettiin seurantavaiheen aikana tutkijan harkinnan mukaan (25 potilaasta 17 potilasta sai MabThera-lisähoitoa). Samanaikainen hoito muilla immunosuppressiivisilla valmisteilla oli sallittua (ks. kohta Farmakodynamiikka).
Haittavaikutuksiksi katsottiin ne haittatapaukset, joiden ilmaantuvuus oli ≥ 10 %. Näitä olivat infektiot (17 potilaalla [68 %] remission induktiovaiheessa, 23 potilaalla [92 %] koko tutkimusjakson aikana), infuusioon liittyvät reaktiot (15 potilaalla [60 %] remission induktiovaiheessa, 17 potilaalla [68 %] koko tutkimusjakson aikana) ja pahoinvointi (4 potilaalla [16 %] remission induktiovaiheessa, 5 potilaalla [20 %] koko tutkimusjakson aikana).
MabTheran turvallisuusprofiili oli koko tutkimusjakson aikana yhdenmukainen remission induktiovaiheen aikana raportoidun turvallisuusprofiilin kanssa.
MabTheran turvallisuusprofiili granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavilla pediatrisilla potilailla oli tyypiltään, luonteeltaan ja vaikeusasteeltaan yhdenmukainen aikuispotilaiden autoimmuunitauteihin liittyvien hyväksyttyjen käyttöaiheiden tunnetun turvallisuusprofiilin kanssa (mukaan lukien aikuisten granulomatoottinen polyangiitti ja mikroskooppinen polyangiitti).
Valittujen haittavaikutusten kuvaus
Infuusioon liittyvät reaktiot
GPA/MPA-tutkimuksessa 1 (remission induktiota koskeva tutkimus aikuisilla) infuusioon liittyviksi reaktioiksi määriteltiin mikä tahansa haittatapahtuma, joka esiintyi 24 tunnin kuluessa infuusiosta ja joka tutkijalääkärin arvion mukaan liittyi infuusioon potilasjoukossa, jossa selvitettiin valmisteen turvallisuutta. Niistä 99 potilaasta, jotka saivat MabThera-hoitoa, 12:lla (12 %) esiintyi vähintään yksi infuusioon liittyvä reaktio. Kaikki infuusioon liittyvät reaktiot olivat CTC-luokituksen mukaan asteen 1 tai 2 tapahtumia. Yleisimmät infuusioon liittyvät reaktiot olivat sytokiinioireyhtymä, punastelu, kurkun ärsytys ja vapina. MabThera annettiin yhdistelmänä laskimoon annettujen glukokortikoidien kanssa, mikä saattaa vähentää tällaisten tapahtumien ilmaantuvuutta ja vaikeusastetta.
GPA/MPA-tutkimuksessa 2 (ylläpitohoitotutkimus aikuisilla) 57 MabThera-hoitohaaran potilaasta 7 potilaalle (12 %) ilmaantui vähintään yksi infuusioon liittynyt reaktio. Infuusioon liittyneiden reaktioiden oireiden ilmaantuvuus oli suurin ensimmäisen infuusion aikana tai jälkeen (9 %) ja väheni seuraavien infuusioiden yhteydessä (< 4 %). Kaikki infuusioon liittyneet reaktiot olivat lieviä tai keskivaikeita, ja valtaosan niistä raportoitiin koskeneen elinjärjestelmiä Hengityselimet, rintakehä ja välikarsina sekä Iho ja ihonalainen kudos.
Kliinisessä tutkimuksessa, joka koski granulomatoottista polyangiittia ja mikroskooppista polyangiittia sairastavia pediatrisia potilaita, raportoidut infuusioon liittyneet reaktiot todettiin pääasiassa ensimmäisen infuusion yhteydessä (8 potilaalla [32 %]), ja ne vähenivät ajan mittaan MabThera-infuusioiden lukumäärän myötä (20 %:lla toisen infuusion yhteydessä, 12 %:lla kolmannen infuusion yhteydessä ja 8 %:lla neljännen infuusion yhteydessä). Remission induktiovaiheen aikana infuusioon liittyneen reaktion yleisimmät raportoidut oireet olivat päänsärky, ihottuma, nuha ja kuume (kutakin oiretta 8 %:lla potilaista). Infuusioon liittyvien reaktioiden havaitut oireet olivat samankaltaisia kuin MabThera-hoitoa saaneilla granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavilla aikuispotilailla. Valtaosa infuusioon liittyvistä reaktioista oli asteen 1 ja asteen 2 tapahtumia, kaksi oli ei-vakavia asteen 3 infuusioon liittyviä reaktioita. Asteen 4 tai 5 infuusioon liittyviä reaktioita ei raportoitu. Yhdellä potilaalla raportoitiin yksi vakava asteen 2 infuusioon liittyvä reaktio (yleistynyt edeema, joka hävisi hoidon avulla) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Infektiot
Infektioiden kokonaisesiintyvyys GPA/MPA-tutkimuksessa 1, jossa oli mukana 99 vaikeaa granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavaa aikuispotilasta, oli noin 237 tapausta 100 potilasvuotta kohti (95 %:n luottamusväli 197–285) 6 kuukauden ensisijaisen päätetapahtuman ajankohtana. Infektiot olivat pääasiassa lieviä tai keskivaikeita ja ne olivat useimmiten ylempien hengitysteiden infektioita, herpes zoster ‑infektioita ja virtsatietulehduksia. Vakavien infektioiden esiintyvyys oli noin 25 tapausta 100 potilasvuotta kohti. Yleisimmin raportoitu vakava infektio MabThera-ryhmässä oli keuhkokuume, jonka esiintyvyys oli 4 %.
GPA/MPA-tutkimuksessa 2 infektioita ilmaantui 30 potilaalle MabThera-hoitohaaran 57 potilaasta (53 %). Kaikkien vaikeusasteiden infektioiden ilmaantuvuus oli hoitohaarojen välillä samankaltainen. Infektiot olivat pääasiassa lieviä tai keskivaikeita. Yleisimpiä infektioita MabThera-hoitohaarassa olivat ylempien hengitysteiden infektiot, gastroenteriitti, virtsatieinfektiot ja vyöruusu (herpes zoster). Vakavien infektioiden ilmaantuvuus oli kummassakin hoitohaarassa samankaltainen (noin 12 %). MabThera-ryhmässä yleisimmin raportoitu vakava infektio oli lievä tai keskivaikea keuhkoputkentulehdus.
Kliinisessä tutkimuksessa, jossa oli mukana vaikeaa, aktiivista granulomatoottista polyangiittia ja mikroskooppista polyangiittia sairastavia pediatrisia potilaita, 91 % raportoiduista infektioista ei ollut vakavia, ja 90 % niistä oli lieviä tai keskivaikeita.
Yleisimmät infektiot koko jakson aikana olivat ylähengitystieinfektiot (48 %), influenssa (24 %), konjunktiviitti (20 %), nasofaryngiitti (20 %), alahengitystieinfektiot (16 %), sinuiitti (16 %), virusperäiset ylähengitystieinfektiot (16 %), korvainfektio (12 %), gastroenteriitti (12 %), faryngiitti (12 %), virtsatieinfektio (12 %). Vakavia infektioita raportoitiin 7 potilaalla (28 %), jolloin yleisimmin raportoituja tapahtumia olivat influenssa (2 potilaalla [8 %]) ja alahengitystieinfektiot (2 potilaalla [8 %]).
Markkinoille tulon jälkeen on raportoitu vakavia virusinfektioita rituksimabilla hoidetuilla GPA/MPA-potilailla.
Maligniteetit
Maligniteettien ilmaantuvuus GPA/MPA-tutkimuksessa 1 MabThera-hoitoa saaneilla potilailla granulomatoottista polyangiittia ja mikroskooppista polyangiittia koskevassa kliinisessä tutkimuksessa oli 2,00 tapausta 100 potilasvuotta kohti tutkimuksen yleisenä päättymispäivänä (kun viimeisen potilaan seurantajakso päättyi). Syöpien ilmaantuvuus vaikuttaa vakioitujen ilmaantuvuussuhteiden perusteella olevan samankaltainen kuin aiemmin on raportoitu ANC-vasta-aineisiin liittyvää vaskuliittia sairastavilla potilailla.
Pediatrisilla potilailla tehdyssä kliinisessä tutkimuksessa ei raportoitu maligniteetteja, kun seurannan kesto oli enintään 54 kuukautta.
Kardiovaskulaariset haittavaikutukset
GPA/MPA-tutkimuksessa 1 sydäntapahtumia esiintyi noin 273 tapausta 100 potilasvuotta kohti (95 %:n luottamusväli 149–470) 6 kuukauden ensisijaisen päätetapahtuman ajankohtana. Vakavien sydäntapahtumien esiintyvyys oli 2,1 tapausta 100 potilasvuotta kohti (95 %:n luottamusväli 3–15). Yleisimmin raportoituja tapahtumia olivat takykardia (4 %) ja eteisvärinä (3 %) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Neurologiset tapahtumat
Autoimmuunisairauksien yhteydessä on raportoitu posteriorista reversiibeliä enkefalopatiaoireyhtymää (PRES) / reversiibeliä posteriorista leukoenkefalopatiaoireyhtymää (RPLS). Oireita olivat näköhäiriöt, päänsärky, kouristuskohtaukset ja mielentilan muutokset, joihin saattoi liittyä hypertensiota. PRES/RPLS-diagnoosi on varmistettava aivojen kuvantamisella. Raportoituihin tapauksiin liittyi PRES:n/RPLS:n tunnistettuja riskitekijöitä, kuten potilaan perussairaus, hypertensio, immunosuppressiivinen hoito ja/tai solunsalpaajahoito.
B-hepatiitin uudelleen aktivoituminen
Granulomatoottista polyangiittia ja mikroskooppista polyangiittia sairastavilla MabThera-hoitoa saaneilla potilailla on raportoitu valmisteen markkinoille tulon jälkeen pieni määrä B-hepatiitin uudelleenaktivoitumistapauksia, joista osa johti potilaan kuolemaan.
Hypogammaglobulinemia
MabThera-hoitoa saaneilla granulomatoottista polyangiittia ja mikroskooppista polyangiittia sairastavilla pediatrisilla ja aikuispotilailla on raportoitu hypogammaglobulinemiaa (IgA-, IgG- tai IgM-pitoisuus normaalin viitealueen alapuolella).
MabThera-hoitoa GPA/MPA-tutkimuksessa 1 saaneista potilaista, joiden immunoglobuliinipitoisuus oli lähtötilanteessa normaali, 27 %:lla oli pieni IgA-pitoisuus, 58 %:lla oli pieni IgG-pitoisuus ja 51 %:lla oli pieni IgM-pitoisuus hoitokuukautena 6. Syklofosfamidiryhmässä vastaavat osuudet olivat 25 % (pieni IgA-pitoisuus), 50 % (pieni IgG-pitoisuus) ja 46 % (pieni IgM-pitoisuus). Kaikkien infektioiden ja vakavien infektioiden esiintyvyys ei suurentunut IgA-, IgG- tai IgM-pitoisuuden pienenemisen jälkeen.
GPA/MPA-tutkimuksessa 2 näiden kahden hoitohaaran välillä ei ollut kliinisesti merkittäviä eroja. Tutkimuksen aikana ei myöskään havaittu kokonaisimmunoglobuliinin, IgG:n, IgM:n tai IgA:n pitoisuuksien pienenemistä.
Pediatrisilla potilailla tehdyssä kliinisessä tutkimuksessa koko tutkimusjakson aikana kaikkiaan 25 potilaasta 3 potilasta (12 %) raportoi hypogammaglobulinemiatapahtuman, 18 potilaalla (72 %) oli pitkään (määritelty viitearvojen alarajaa pienemmäksi Ig-pitoisuudeksi vähintään 4 kuukauden ajan) pieni IgG-pitoisuus (näistä 15 potilaalla oli pitkään myös pieni IgM-pitoisuus). Kolme potilasta sai hoitona laskimoon annettavaa immunoglobuliinia (IV-IG). Rajallisen tiedon perusteella ei voida tehdä varmoja johtopäätöksiä siitä, johtaako pitkäaikaiset pienet IgG- ja IgM-pitoisuudet vakavien infektioiden suurentuneeseen riskiin näillä potilailla. Pitkäaikaisen B-soluvajeen seuraukset pediatrisille potilaille eivät ole tiedossa.
Neutropenia
MabThera-hoitoa GPA/MPA-tutkimuksessa 1 saaneista potilaista 24 %:lle MabThera-ryhmän potilaista (yksi hoitojakso) ja 23 %:lle syklofosfamidiryhmän potilaista kehittyi CTC-luokituksen mukainen asteen 3 tai vaikeampiasteinen neutropenia. Neutropeniaan ei havaittu MabThera-hoitoa saaneilla potilailla liittyneen vakavien infektioiden lisääntymistä.
GPA/MPA-tutkimuksessa 2 kaikkien vaikeusasteiden neutropenian ilmaantuvuus oli MabThera-hoitoa saaneilla potilailla 0 % verrattuna 5 %:iin atsatiopriinihoitoa saaneilla potilailla.
Iho- ja ihonalainen kudos
Toksista epidermaalista nekrolyysiä (Lyellin oireyhtymä), ja Stevens–Johnsonin oireyhtymää, jotka ovat toisinaan johtaneet potilaan kuolemaan, on raportoitu hyvin harvoin.
Kokemukset tavallisesta pemfiguksesta
MabThera-valmisteen kokonaisturvallisuusprofiili tavallista pemfigusta sairastavilla potilailla perustuu kahdesta kliinisestä tutkimuksesta ja valmisteen markkinoille tulon jälkeisestä seurannasta saatuihin tietoihin.
Turvallisuusprofiilin yhteenveto PV-tutkimuksesta 1 (tutkimus ML22196) ja PV-tutkimuksesta 2 (tutkimus WA29330)
MabTheran ja lyhytaikaisesti pieninä annoksina käytetyn glukokortikoidihoidon yhdistelmän turvallisuusprofiilia tavallista pemfigusta sairastavien potilaiden hoidossa tutkittiin faasin 3, satunnaistetussa, kontrolloidussa, avoimessa monikeskustutkimuksessa pemfigusta sairastavilla potilailla, joista 38 tavallista pemfigusta (PV) sairastavaa potilasta satunnaistettiin MabThera-ryhmään (PV-tutkimus 1). MabThera-ryhmään satunnaistetut potilaat saivat aloitusannoksena 1000 mg laskimoon 1. tutkimuspäivänä ja toisen 1000 mg:n annoksen laskimoon 15. tutkimuspäivänä. 500 mg:n ylläpitoannoksia annettiin laskimoon kuukausina 12 ja 18. Relapsin yhteydessä potilaille voitiin antaa 1000 mg laskimoon (ks. kohta Farmakodynamiikka).
PV-tutkimus 2 oli satunnaistettu, kaksoissokkoutettu, kaksoislumevalmisteella toteutettu ja vaikuttavaan aineeseen vertaileva monikeskustutkimus, jossa MabTheran tehoa ja turvallisuutta verrattiin mykofenolaattimofetiiliin (MMF) keskivaikeaa tai vaikeaa tavallista pemfigusta sairastavilla potilailla, jotka tarvitsivat suun kautta otettavia kortikosteroideja. 67 tavallista pemfigusta sairastavaa potilasta sai MabThera-hoitoa (aluksi 1000 mg laskimoon tutkimuspäivänä 1 ja toinen 1000 mg:n annos laskimoon tutkimuspäivänä 15 viikkoina 24 ja 26 toistettuina) enintään 52 viikon ajan (ks. kohta Farmakodynamiikka).
MabThera-valmisteen turvallisuusprofiili tavallisen pemfiguksen hoidossa oli yhdenmukainen muiden autoimmuunitauteja koskevien hyväksyttyjen käyttöaiheiden varmistetun turvallisuusprofiilin kanssa.
PV-tutkimuksia 1 ja 2 sekä valmisteen markkinoille tulon jälkeistä seurantaa koskeva haittavaikutustaulukko
Taulukossa 7 esitetään haittavaikutukset, joiden esiintyvyys PV-tutkimuksissa 1 ja 2 oli yleinen tai hyvin yleinen. PV-tutkimuksessa 1 haittavaikutuksiksi määriteltiin haittatapahtumat, joita esiintyi ≥ 5 %:lla MabTheraa tavallisen pemfiguksen hoitoon saaneista potilaista ja joiden ilmaantuvuudessa oli ≥ 2 %:n absoluuttinen ero MabThera-hoitoa saaneen ryhmän ja tavanomaisia prednisoniannoksia saaneen ryhmän välillä kuukauteen 24 saakka. Yksikään PV-tutkimuksen 1 potilas ei lopettanut hoitoa haittavaikutusten vuoksi. PV-tutkimuksessa 2 haittavaikutuksiksi määriteltiin haittatapahtumat, joita esiintyi ≥ 5 %:lla potilaista MabThera-haarassa ja joiden arvioitiin liittyneen lääkitykseen.
Valmisteen markkinoille tulon jälkeen havaitut haittavaikutukset on listattu sarakkeessa ”Tuntematon”. Näiden haittavaikutusten esiintyvyyttä ei voida arvioida, ks. alaviitteet.
Taulukko 7 Haittavaikutukset PV-tutkimuksessa 1 (kuukauteen 24 saakka) ja PV-tutkimuksessa 2 (viikkoon 52 saakka) MabThera-hoitoa saaneilla tavallista pemfigusta sairastavilla potilailla tai markkinoille tulon jälkeisessä seurannassa
| MedDRA-elinjärjestelmä | Hyvin yleinen | Yleinen | Tuntematon |
| Infektiot | ylähengitystieinfektio | herpesvirusinfektio, vyöruusu, suun herpesinfektio, konjunktiviitti, nasofaryngiitti, sammas, virtsatieinfektio | vakava virusinfektio1, 2, enteroviraalinen meningoenkefaliitti1 |
| Hyvän- ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | ihon papillooma | ||
| Psyykkiset häiriöt | pitkittyvä masennus | vakava masennus, ärtyisyys | |
| Hermosto | päänsärky | huimaus | |
| Sydän | takykardia | ||
| Ruoansulatuselimistö | ylävatsakipu | ||
| Iho ja ihonalainen kudos | hiustenlähtö | kutina, urtikaria, iho-oireet | |
| Luusto, lihakset ja sidekudos | tuki- ja liikuntaelimistön kipu nivelsärky, selkäkipu | ||
| Yleisoireet ja antopaikassa todettavat haitat | väsymys, voimattomuus, kuume | ||
| Vammat, myrkytykset ja hoitokomplikaatiot | infuusioon liittyvät reaktiot3 | ||
1 Havaittu markkinoille tulon jälkeisessä seurannassa. PV-tutkimuksessa 2 yleisimmät infuusioon liittyvien reaktioiden oireet / Preferred Term ‑termit olivat hengenahdistus, eryteema, liikahikoilu, ihon punoitus / kuumat aallot, hypotension / matala verenpaine ja ihottuma / kutiseva ihottuma. | |||
Valittujen haittavaikutusten kuvaus
Infuusioon liittyvät reaktiot
PV-tutkimuksessa 1 infuusioon liittyvät reaktiot olivat yleisiä (58 %). Lähes kaikki infuusioon liittyneet reaktiot olivat lieviä tai keskivaikeita. Niiden potilaiden osuus, joilla oli infuusioon liittynyt reaktio, oli ensimmäisen infuusion yhteydessä 29 % (11 potilasta), toisen infuusion yhteydessä 40 % (15 potilasta), kolmannen infuusion yhteydessä 13 % (5 potilasta) ja neljännen infuusion yhteydessä 10 % (4 potilasta). Yksikään potilas ei lopettanut hoitoa infuusioon liittyneiden reaktioiden vuoksi. Infuusioon liittyneiden reaktioiden oireet olivat tyypiltään ja vaikeusasteeltaan samankaltaisia kuin nivelreumaa, granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavilla potilailla on todettu.
PV-tutkimuksessa 2 infuusioon liittyviä reaktioita esiintyi pääasiassa ensimmäisen infuusion yhteydessä ja niiden esiintyvyys väheni seuraavien infuusioiden yhteydessä: infuusioon liittyviä reaktiota esiintyi ensimmäisen infuusion yhteydessä 17,9 %:lla potilaista, toisen infuusion yhteydessä 4,5 %:lla potilaista, kolmannen infuusion yhteydessä 3 %:lla potilaista ja neljännen infuusion yhteydessä 3 %:lla potilaista. Niistä 15 potilaasta, joilla esiintyi vähintään yksi infuusioon liittynyt reaktio, 11 potilaalla oli asteen 1 tai 2 infuusioon liittynyt reaktio. 15 potilaasta 4 potilaalla raportoitiin asteen ≥ 3 infuusioon liittynyt reaktio, joka johti MabThera-hoidon keskeyttämiseen, ja näistä neljästä potilaasta kolmella oli vakava (hengenvaarallinen) infuusioon liittynyt reaktio. Vakavia infuusioon liittyneitä reaktioita esiintyi ensimmäisen (2 potilaalla) tai toisen (1 potilaalla) infuusion yhteydessä, ja ne hävisivät oireenmukaisella hoidolla.
Infektiot
PV-tutkimuksessa 1 hoitoon liittyneitä infektioita todettiin 14 potilaalla (37 %) MabThera-ryhmässä verrattuna 15 potilaaseen (42 %) tavanomaisia prednisoniannoksia saaneessa ryhmässä. MabThera-ryhmässä yleisimpiä infektioita olivat yskänrokko (herpes simplex) ja vyöruusu (herpes zoster), keuhkoputkentulehdus, virtsatieinfektio, sieni-infektio ja konjunktiviitti. Kolmella MabThera-ryhmän potilaalla (8 %) oli yhteensä 5 vakavaa infektiota (Pneumocystis jirovecii ‑pneumonia, tulehdusperäinen tromboosi, nikamavälilevyn tulehdus, keuhkoinfektio, stafylokokkisepsis) ja yhdellä tavanomaisia prednisoniannoksia saaneen ryhmän potilaalla (3 %) oli vakava infektio (Pneumocystis jirovecii ‑pneumonia).
PV-tutkimuksessa 2 infektioita esiintyi MabThera-haarassa 42 potilaalla (62,7 %). MabThera-ryhmässä yleisimpiä infektioita olivat ylähengitystieinfektiot, nasofaryngiitti, sammas ja virtsatieinfektio. Kuudella MabThera-ryhmän potilaalla (9 %) esiintyi vakavia infektioita.
Markkinoille tulon jälkeen on raportoitu vakavia virusinfektioita rituksimabilla hoidetuilla tavallista pemfigusta sairastavilla potilailla.
Laboratorioarvojen poikkeavuudet
PV-tutkimuksen 2 MabThera-haarassa havaittiin infuusion jälkeen hyvin yleisesti ohimenevää lymfosyyttimäärän vähenemistä, joka johtui perifeeristen T‑solupopulaatioiden vähenemisestä, sekä fosforipitoisuuden ohimenevää pienenemistä. Näiden katsottiin olleen esilääkityksenä laskimoon annetun metyyliprednisoloni-infuusion indusoimia.
PV-tutkimuksessa 2 havaittiin yleisesti pieniä IgG-pitoisuuksia ja hyvin yleisesti pieniä IgM‑pitoisuuksia, mutta IgG- tai IgM-pitoisuuksien pienenemisen jälkeen ei havaittu lisääntynyttä vakavien infektioiden riskiä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Tehdyistä kliinisissä tutkimuksissa ihmisille on rajoitetusti tietoa saatavilla suuremmista kuin hyväksytyistä laskimoon annettavista MabTheran annoksista. Suurin ihmisellä tutkittu laskimoon annettu MabThera-annos on tähän mennessä ollut 5000 mg (2250 mg/m2), jota tutkittiin annoseskalaatiotutkimuksessa kroonista lymfaattista leukemiaa sairastavilla potilailla. Uusia turvallisuuteen liittyviä signaaleja ei ilmennyt.
Jos potilas saa yliannoksen, infuusion anto on välittömästi keskeytettävä ja potilasta on tarkkailtava huolellisesti.
Myyntiluvan myöntämisen jälkeisessä seurannassa on raportoitu viisi MabTheran yliannostustapausta. Kolmessa tapauksessa ei raportoitu haittatapahtumia. Kaksi haittatapahtumaa raportoitiin, toinen oli influenssan tapaisia oireita rituksimabiannoksella 1,8 g ja toinen oli fataali hengityslama rituksimabiannoksella 2 g.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: antineoplastiset lääkeaineet, monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, ATC-koodi: L01FA01
Vaikutusmekanismi
Rituksimabi sitoutuu spesifisesti transmembraaniseen CD20-antigeeniin, joka on non-glykosyloitu fosfoproteiini. Tätä antigeenia esiintyy pre-B-soluissa ja kypsissä B-lymfosyyteissä. Yli 95 % B-solu non-Hodgkinin lymfoomista ilmaisee tätä antigeenia.
CD20:tä esiintyy normaaleissa ja maligneissa B-soluissa, mutta ei hematopoieettisissa kantasoluissa, pro-B-soluissa, normaaleissa plasmasoluissa eikä muussa terveessä kudoksessa. CD20-antigeeni ei internalisoidu vasta-aineen sitoutuessa eikä sitä irtoa solun pinnalta. CD20-antigeeni ei kierrä vapaana plasmassa, joten kilpailua vasta-ainesitoutumisesta ei esiinny.
Rituksimabin Fab-alayksikkö sitoutuu B-lymfosyytin CD20-antigeeniin ja Fc-alayksikkö voi aktivoida immuunijärjestelmän välittämänä B-solun tuhoutumisen. Mahdollisia efektorin välittämiä solun tuhoutumisen mekanismeja ovat C1q-sitoutumisen laukaisema, komplementista riippuva sytotoksisuus (CDC) ja vasta-aineista riippuvainen soluvälitteinen sytotoksisuus (ADCC), jota välittävät granulosyyttien, makrofagien ja NK-solujen pinnassa olevat Fcγ-reseptorit. Rituksimabin sitoutumisen B-lymfosyytin CD20-antigeeniin on myös osoitettu aiheuttavan ohjelmoitua solujen kuolemaa (apoptoosia).
Farmakodynaamiset vaikutukset
Perifeeristen B-solujen määrät laskivat alle normaaliarvojen ensimmäisen MabThera-annoksen jälkeen. Pahanlaatuisten hematologisten tautien vuoksi hoidetuilla potilailla B-solujen palautuminen alkoi 6 kuukauden sisällä hoidon päättymisestä ja yleensä arvot palasivat normaaleiksi 12 kuukauden kuluttua hoidon päättymisestä, vaikka joillakin potilailla palautuminen voi kestää kauemmin (palautumisajan mediaani jopa 23 kuukautta hoidon aloituksesta). Nivelreumapotilailla perifeerisen veren B-solumäärä pieneni heti, kun potilaille oli annettu kaksi 1000 mg:n MabThera-infuusiota 14 vuorokauden välein. Perifeerisen veren B-solumäärä alkoi lisääntyä viikolla 24, ja merkkejä solupopulaation uusiutumisesta havaittiin suurimmalla osalla potilaista viikkoon 40 mennessä riippumatta siitä, annettiinko MabTheraa yksinään vai yhdessä metotreksaatin kanssa. Pienellä osalla potilaista oli pitkittynyt perifeerinen B-soluvaje, joka viimeisen MabThera-annoksen jälkeen kesti kaksi vuotta tai pitempään. Granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavilla potilailla, perifeeristen B-solujen määrä laski tasolle < 10 solua/μl kahden kerran viikossa annetun rituksimabi-infuusion jälkeen (375 mg/m2), ja useimmilla potilailla määrä pysyi tällä tasolla hoidosta seuraavat 6 kuukautta. Suurimalla osalla potilaista (81 %) havaittiin viitteitä B-solujen palautumisesta ja kun hoidon päättymisestä oli kulunut 12 kuukautta, heiltä mitattiin arvoja, jotka olivat > 10 solua/μl. 18 kuukautta hoidon päättymisestä osuus oli kasvanut 87 %:iin.
Kliininen teho ja turvallisuus
Kliininen teho ja turvallisuus non-Hodgkin-lymfooman ja kroonisen lymfaattisen leukemian hoidossa
Follikulaarinen lymfooma
Monoterapia
Hoito kerran viikossa, yhteensä neljä annosta
Avaintutkimuksessa MabTheraa annettiin 375 mg/m2 laskimonsisäisenä infuusiona kerran viikossa neljän viikon ajan 166 potilaalle, joilla oli uusiutunut tai solunsalpaajaresistentti, matala-asteinen tai follikulaarinen B‑solu-NHL. Kokonaishoitovaste oli 48 % intent-to-treat (ITT) -ryhmässä (95 %:n luottamusväli 41–56 %), 6 %:lla potilaista saavutettiin täydellinen hoitovaste ja 42 %:lla osittainen hoitovaste. Hoitoon reagoivilla potilailla arvioitu mediaaniaika taudin etenemiseen (TTP) oli 13 kuukautta. Alaryhmäanalyysissä havaittiin, että kokonaishoitovasteluku oli suurempi potilailla, joilla oli IWF-luokituksen mukainen histologinen alatyyppi B, C tai D verrattuna alatyyppiin A (58 % vs. 12 %). Kokonaishoitovasteluku oli myös suurempi potilailla, joiden leesion suurin halkaisija oli alle 5 cm verrattuna potilaisiin, joilla halkaisija oli suurempi kuin 7 cm (53 % vs. 38 %) tai jos potilailla oli kemoterapiaan reagoiva relapsi verrattuna solunsalpaajaresistenttiin (= hoitovasteen kesto alle kolme kuukautta) relapsiin (50 % vs. 22 %). Kokonaishoitovaste potilailla, joita oli aikaisemmin hoidettu autologisella luuydinsiirrolla (ABMT), oli 78 %, kun taas potilailla, jotka eivät olleet saaneet luuydinsiirtoa, hoitovaste oli 43 %. Seuraavilla tekijöillä ei ollut tilastollisesti merkitsevää vaikutusta (Fisherin testillä analysoituna) hoitovasteeseen MabTheralle: ikä, sukupuoli, lymfooman pahanlaatuisuusaste, alkudiagnoosi, suuri tautimassa / ei suurta tautimassaa, normaali tai korkea LDH tai ekstranodaalinen tauti. Hoitovastelukujen ja luuytimeen levinneen taudin välillä havaittiin tilastollisesti merkitsevä korrelaatio: hoitovasteen sai 40 % potilaista, joilla oli luuytimeen levinnyt tauti. Potilaista, joilla ei ollut luuytimeen levinnyttä tautia, hoitovasteen sai 59 % (p = 0,0186). Tätä tulosta ei tukenut asteittainen logistinen regressioanalyysi, jossa seuraavat tekijät havaittiin hoitovastetta ennustaviksi: histologinen tyyppi, lähtötilanteen bcl-2-positiivisuus, resistenssi viimeiselle edeltävälle solunsalpaajahoidolle ja suuri tautimassa.
Hoito kerran viikossa, yhteensä kahdeksan annosta
Monikeskustutkimuksessa, jossa oli vain yksi hoitohaara, 37:lle uusiutunutta tai solunsalpaajaresistenttiä, matala-asteista tai follikulaarista non-Hodgkin-B-solulymfoomaa sairastavalle potilaalle annettiin 375 mg/m2 MabTheraa laskimonsisäisenä infuusiona kerran viikossa yhteensä kahdeksan annosta. Kokonaishoitovaste oli 57 % (95 %:n luottamusväli 41–73 %, täydellinen hoitovaste 14 %, osittainen hoitovaste 43 %) ja arvioitu mediaaniaika taudin etenemiseen hoitoon reagoivilla potilailla 19,4 kuukautta (vaihteluväli 5,3–38,9 kk).
Hoito kerran viikossa, yhteensä neljä annosta, suuri tautimassa
Kolmessa tutkimuksessa 39 potilaalle annettiin 375 mg/m2 MabTheraa laskimonsisäisenä infuusiona kerran viikossa yhteensä neljä annosta. Potilaat sairastivat uusiutunutta tai solunsalpaajaresistenttiä, suuren tautimassan (yksittäisen leesion halkaisija ≥ 10 cm) matala-asteista tai follikulaarista non-Hodgkin-B-solulymfoomaa. Tässä yhdistetyssä aineistossa kokonaishoitovaste oli 36 % (95 %:n luottamusväli 21–51 %, täydellinen hoitovaste 3 %, osittainen hoitovaste 33 %) ja mediaaniaika taudin etenemiseen hoitoon reagoivilla potilailla 9,6 kuukautta (vaihteluväli 4,5–26,8 kk).
Uusintahoito, annostus kerran viikossa, yhteensä neljä annosta
Monikeskustutkimuksessa, jossa oli vain yksi hoitohaara, 58:lle uusiutunutta tai solunsalpaajaresistenttiä, matala-asteista tai follikulaarista non-Hodgkin-B-solulymfoomaa sairastavalle potilaalle, jotka olivat saaneet objektiivisen kliinisen hoitovasteen ensimmäiselle MabThera-hoitojaksolle, annettiin uudelleen 375 mg/m2 MabTheraa laskimonsisäisenä infuusiona kerran viikossa yhteensä neljä annosta. Kolme potilasta oli saanut jo kaksi MabThera-hoitojaksoa ennen tutkimukseen mukaantuloa, joten he saivat kolmannen hoitojakson tutkimuksen aikana. Kahta potilasta hoidettiin kaksi kertaa uudelleen tutkimuksen aikana. Tutkimuksen 60 uusintahoidon kokonaishoitovaste oli 38 % (95 %:n luottamusväli 26–51 %, täydellinen hoitovaste 10 %, osittainen hoitovaste 28 %) ja arvioitu mediaaniaika taudin etenemiseen hoitoon reagoivilla potilailla 17,8 kuukautta (vaihteluväli 5,4–26,6 kk). Tämä tulos kestää hyvin vertailun ensimmäiseen MabThera-hoitojaksoon, jolloin aika taudin etenemiseen oli 12,4 kuukautta.
Yhdistelmähoito solunsalpaajien kanssa
Satunnaistetussa, avoimessa kliinisessä tutkimuksessa 322 aikaisemmin hoitamatonta, follikulaarista lymfoomaa sairastavaa potilasta satunnaistettiin kahteen ryhmään. Toisen ryhmän potilaille annettiin CVP-solunsalpaajahoitoa (syklofosfamidi 750 mg/m2, vinkristiini 1,4 mg/m2 aina 2 mg:n annokseen saakka päivänä 1 ja prednisolonia 40 mg/m2/vrk päivinä 1–5) joka kolmas viikko kahdeksan syklin ajan ja toisen ryhmän potilaille MabTheraa 375 mg/m2 yhdistettynä CVP-hoitoon (R-CVP). MabThera annettiin jokaisen hoitosyklin ensimmäisenä päivänä. Hoitoa sai kaikkiaan 321 potilasta, joilla teho voitiin arvioida (162 R-CVP-ryhmässä ja 159 CVP-ryhmässä). Potilaiden seuranta-ajan mediaani oli 53 kuukautta. Tutkimuksen primaarisella päätepisteellä (aika hoidon tehon menetykseen) mitattuna R-CVP-hoidolla saavutettiin merkittävä hyöty verrattuna CVP-hoitoon (27 kuukautta vs. 6,6 kuukautta, p < 0,0001, log-rank-testi). Kasvaimelle hoitovasteen (CR, CRu, PR) saaneiden potilaiden osuus oli merkitsevästi suurempi (p < 0,0001, Chi-Square-testi) R-CVP-ryhmässä (80,9 %) verrattuna CVP-ryhmään (57,2 %). Verrattuna CVP-hoitoon R-CVP-hoito pidensi merkitsevästi aikaa taudin etenemiseen (33,6 kuukautta) tai kuolemaan (14,7 kuukautta), p < 0,0001, log-rank-testi. Hoitovasteen keston mediaani oli R-CVP-ryhmässä 37,7 kuukautta ja CVP‑ryhmässä 13,5 kuukautta (p < 0,0001, log-rank-testi).
Hoitoryhmien välinen ero kokonaiselinajassa osoitti merkitsevän kliinisen eron (p = 0,029, keskuksen mukaan stratifioitu log-rank-testi): 53 kuukauden kohdalla eloonjäämisprosentti oli R-CVP-ryhmän potilailla 80,9 % ja CVP-ryhmän potilailla 71,1 %.
Tulokset kolmesta muusta satunnaistetusta tutkimuksesta, joissa MabTheraa käytettiin yhdistelmähoidossa muiden solunsalpaajien kanssa (CHOP, MCP, CHVP/interferoni-α), ovat myös osoittaneet vasteiden, aikaan sidonnaisten parametrien ja kokonaiselinajan merkittävää paranemista. Keskeiset tulokset kaikista neljästä tutkimuksesta on esitetty taulukossa 8.
Taulukko 8 Yhteenveto neljän faasin III satunnaistetun tutkimuksen keskeisistä tuloksista. Tutkimuksissa selvitettiin MabTherasta saatavaa hyötyä erilaisten solunsalpaajahoitojen kanssa follikulaarisessa lymfoomassa.
| Tutkimus | Hoito, potilaiden määrä | Seuranta-ajan mediaani (kk) | Kokonais-hoitovaste (%) | Täydellinen hoitovaste (%) | Mediaani TTF/PFS/ EFS (kuukautta) | Kokonaiseloon-jääminen (%) |
| M39021 | CVP: 159 R-CVP: 162 | 53 | 57 81 | 10 41 | Mediaani TTP: 14,7 33,6 p < 0,0001 | 53 kk kohdalla 71,1 80,9 p = 0,029 |
| GLSG’00 | CHOP: 205 R-CHOP: 223 | 18 | 90 96 | 17 20 | Mediaani TTF: 2,6 v Ei saavutettu p < 0,001 | 18 kk kohdalla 90 95 p = 0,016 |
| OSHO-39 | MCP: 96 R-MCP: 105 | 47 | 75 92 | 25 50 | Mediaani PFS: 28,8 Ei saavutettu p < 0,0001 | 48 kk kohdalla 74 87 p = 0,0096 |
| FL2000 | CHVP-IFN: 183 R-CHVP-IFN: 175 | 42 | 85 94 | 49 76 | Mediaani EFS: 36 Ei saavutettu p < 0,0001 | 42 kk kohdalla 84 91 p = 0,029 |
EFS – Tapahtumavapaa elinaika
TTP – Aika taudin etenemiseen tai kuolemaan
PFS – Taudin etenemisestä vapaa elinaika
TTF – Aika hoidon tehon menetykseen
Kokonaiseloonjääminen – eloonjääminen analysointihetkellä
Ylläpitohoito
Aiemmin hoitamaton follikulaarinen lymfooma
Prospektiivisessa, avoimessa, kansainvälisessä, faasin III monikeskustutkimuksessa 1193 potilaalle, joilla oli aiemmin hoitamaton pitkälle edennyt follikulaarinen lymfooma, annettiin tutkijan valinnan mukaan induktiohoitona R-CHOP (n = 881), R-CVP (n = 268) tai R-FCM (n = 44). Hoitovasteen sai yhteensä 1078 potilasta, joista 1018 oli satunnaistettu MabThera-ylläpitohoitoryhmään (n = 505) tai seurantaryhmään (n = 513). Nämä kaksi hoitoryhmää olivat hyvin tasapainossa potilaiden perusominaisuuksien ja tautistatuksen suhteen. MabThera-ylläpitohoidossa annettiin kertainfuusio MabTheraa annoksella 375 mg/m2 joka toinen kuukausi, kunnes tauti eteni tai pisimmillään 2 vuoden ajan.
Ennalta määritelty primaarianalyysi tehtiin, kun seuranta-aika (mediaani) oli 25 kuukautta satunnaistamisesta. Siinä havaittiin, että ylläpitohoito MabTheralla johti primaarisen päätemuuttujan (PFS) kliinisesti merkittävään ja tilastollisesti merkitsevään paranemiseen verrattuna seurantaryhmän potilaisiin, joilla oli aiemmin hoitamaton follikulaarinen lymfooma (taulukko 9).
Primaarianalyysissa osoitettiin MabTheran ylläpitohoidosta merkittävää hyötyä myös seuraavissa toissijaisissa päätetapahtumissa: tapahtumavapaa elinaika (EFS), aika seuraavaan lymfoomahoitoon (TNLT), aika seuraavaan solunsalpaajahoitoon (TNCT) ja kokonaishoitovaste (ORR) (taulukko 9).
Tiedot tutkimuksessa mukana olleiden potilaiden pidennetystä seurannasta (seurannan keston mediaani 9 vuotta) vahvistivat MabThera-ylläpitohoidon pitkäaikaishyödyt seuraavien päätetapahtumien suhteen: taudin etenemisestä vapaa elinaika, tapahtumavapaa elinaika, aika seuraavaan lymfoomahoitoon ja aika seuraavaan solunsalpaajahoitoon (taulukko 9).
Taulukko 9 Yhteenveto MabThera-ylläpitohoidon vs. seurantaryhmän tehoa koskevista tuloksista tutkimussuunnitelmassa määritellyssä primaarianalyysissa ja 9 vuoden (mediaani) seurannan jälkeen (loppuanalyysi)
| Primaarianalyysi (seuranta-ajan mediaani 25 kuukautta) | Loppuanalyysi (seuranta-ajan mediaani 9,0 vuotta) | |||
| Seuranta N = 513 | MabThera N = 505 | Seuranta N = 513 | MabThera N = 505 | |
| Primaarinen teho | ||||
| PFS (mediaani) | NR | NR | 4,06 vuotta | 10,49 vuotta |
| log-rank-testin p-arvo | < 0,0001 | < 0,0001 | ||
| riskisuhde (95 %:n vaihteluväli) | 0,50 (0,39, 0,64) | 0,61 (0,52, 0,73) | ||
| riskin vähenemä | 50 % | 39 % | ||
| Sekundaarinen teho | ||||
| OS (mediaani) | NR | NR | NR | NR |
| log-rank-testin p-arvo | 0,7246 | 0,7948 | ||
| riskisuhde (95 %:n vaihteluväli) | 0,89 (0,45; 1,74) | 1,04 (0,77; 1,40) | ||
| riskin vähenemä | 11 % | -6 % | ||
| EFS (mediaani) | 38 kuukautta | NR | 4,04 vuotta | 9,25 vuotta |
| log-rank-testin p-arvo | < 0,0001 | < 0,0001 | ||
| riskisuhde (95 %:n vaihteluväli) | 0,54 (0,43; 0,69) | 0,64 (0,54; 0,76) | ||
| riskin vähenemä | 46 % | 36 % | ||
| TNLT (mediaani) | NR | NR | 6,11 vuotta | NR |
| log-rank-testin p-arvo | 0,0003 | < 0,0001 | ||
| riskisuhde (95 %:n vaihteluväli) | 0,61 (0,46; 0,80) | 0,66 (0,55; 0,78) | ||
| riskin vähenemä | 39 % | 34 % | ||
| TNCT (mediaani) | NR | NR | 9,32 vuotta | NR |
| log-rank-testin p-arvo | 0,0011 | 0,0004 | ||
| riskisuhde (95 %:n vaihteluväli) | 0,60 (0,44; 0,82) | 0,71 (0,59; 0,86) | ||
| riskin vähenemä | 40 % | 39 % | ||
| ORR* | 55 % | 74 % | 61 % | 79 % |
| khiin neliö ‑testillä todettu p-arvo | < 0,0001 | < 0,0001 | ||
| kerroinsuhde (95 %:n vaihteluväli) | 2,33 (1,73; 3,15) | 2,43 (1,84; 3,22) | ||
| Täydellisen vasteen osuus (CR/CRu)* | 48 % | 67 % | 53 % | 72 % |
| khiin neliö ‑testillä todettu p-arvo | < 0,0001 | < 0,0001 | ||
| kerroinsuhde (95 %:n vaihteluväli) | 2,21 (1,65; 2,94) | 2,34 (1,80; 3,03) | ||
* ylläpitohoidon/seurannan päättyessä; loppuanalyysin tulokset perustuvat 73 kuukauden seuranta-aikaan (mediaani).
NR: ei saavutettu kliinisen tiedonkeruun katkaisuajankohtana (not reached at time of clinical cut off), TNCT: aika seuraavaan solunsalpaajahoitoon (time to next chemotherapy treatment); TNLT: aika seuraavaan lymfoomahoitoon (time to next anti lymphoma treatment).
MabThera-ylläpitohoidon hyöty osoitettiin kaikissa tutkituissa ennalta määritellyissä alaryhmissä: sukupuoli (mies, nainen), ikä (< 60 vuotta, >= 60 vuotta), FLIPI-pisteet (<= 1, 2 tai >= 3), induktiohoito (R-CHOP, R-CVP tai R-FCM) riippumatta induktiohoidon hoitovasteen laadusta (CR, CRu tai PR). Ylläpitohoidon hyötyä selvittävissä eksploratiivisissä analyyseissä havaittiin, että iäkkäillä potilailla (> 70-vuotiaat) hyöty ei ollut yhtä selkeä, otanta oli kuitenkin pieni.
Uusiutunut/refraktorinen follikulaarinen lymfooma
Prospektiivisessa, avoimessa, kansainvälisessä, faasin III monikeskustutkimuksessa satunnaistettiin 465 uusiutunutta/refraktorista follikulaarista lymfoomaa sairastavaa potilasta saamaan ensimmäisessä vaiheessa induktiohoitoa joko CHOP-yhdistelmällä (syklofosfamidi, doksorubisiini, vinkristiini, prednisoloni; n = 231) tai MabTheran ja CHOP-hoidon yhdistelmällä (R-CHOP, n = 234). Nämä kaksi hoitoryhmää olivat hyvin tasapainossa potilaiden perusominaisuuksien ja tautistatuksen suhteen. Kaikkiaan 334 potilasta, jotka saivat täydellisen tai osittaisen remission induktiohoidon jälkeen, satunnaistettiin toisessa vaiheessa saamaan joko MabThera-ylläpitohoitoa (n = 167) tai seurantaan (n = 167). MabThera-ylläpitohoidossa annettiin kertainfuusio MabTheraa annoksella 375 mg/m2 joka 3. kuukausi, kunnes tauti eteni tai pisimmillään 2 vuoden ajan.
Lopullisessa tehoa koskevassa analyysissa olivat mukana kaikki potilaat, jotka oli satunnaistettu tutkimuksen molemmissa vaiheissa. Kun seuranta-ajan mediaani oli 31 kuukautta, havaittiin, että ennuste parani merkitsevästi uusiutunutta/refraktorista follikulaarista lymfoomaa sairastavilla potilailla, jotka oli induktiovaiheessa satunnaistettu saamaan R-CHOP-hoitoa verrattuna CHOP-hoitoa saaneisiin potilaisiin (ks. taulukko 10).
Taulukko 10 Induktiovaihe: yhteenveto CHOP- ja R-CHOP-hoitojen tehoa koskevista tuloksista (seuranta-ajan mediaani 31 kuukautta)
| CHOP | R-CHOP | p-arvo | Riskin väheneminen1) | |
| Primaariset tehomuuttujat | ||||
| ORR2) | 74 % | 87 % | 0,0003 | NA |
| CR2) | 16 % | 29 % | 0,0005 | NA |
| PR2) | 58 % | 58 % | 0,9449 | NA |
1) Estimaatit laskettiin riskisuhteina (hazard ratio)
2) Viimeinen tutkijan arvioima hoitovaste kasvaimeen. ”Primaarinen” tilastotesti ”hoitovasteen” mittaamiseen oli trenditesti (CR vs. PR vs. ei hoitovastetta, p < 0.0001).
Lyhenteet: NA, tietoa ei käytettävissä (not available); ORR, kokonaishoitovaste (overall response rate); CR, täydellinen hoitovaste (complete response); PR, osittainen hoitovaste (partial response)
Tutkimuksen ylläpitovaiheeseen satunnaistettujen potilaiden seuranta-ajan mediaani oli 28 kuukautta ajankohdasta, jolloin ylläpitohoitoon satunnaistaminen tapahtui. Ylläpitohoito MabTheralla johti primaarisen päätemuuttujan (PFS) kliinisesti merkittävään ja tilastollisesti merkitsevään paranemiseen verrattuna pelkkään seurantaan (p < 0,0001, log-rank-testi). Taudin etenemisestä vapaa elinaika (PFS) tarkoittaa aikaa ylläpitohoitoon satunnaistamisesta taudin uusiutumiseen, taudin etenemiseen tai kuolemaan. Taudin etenemisestä vapaan elinajan mediaani oli 42,2 kuukautta MabThera-ylläpitohoitoa saaneilla potilailla ja 14,3 kuukautta pelkässä seurannassa olleilla. Coxin regressioanalyysi osoitti, että etenevän taudin tai kuoleman riski pieneni 61 % MabThera-ylläpitohoitoa saaneilla potilailla verrattuna seurantaryhmään (95 %:n luottamusväli 45–72 %). Kaplan-Meier-menetelmällä arvioituna niiden potilaiden osuus, jolla tauti ei edennyt, oli 78 % MabThera-ylläpitohoitoryhmässä ja 57 % seurantaryhmässä 12 kuukauden kohdalla. Kokonaiseloonjäämisaikaa koskeva analyysi vahvisti, että MabThera-ylläpitohoidolla saavutettiin merkitsevä hyöty verrattuna potilaisiin, jotka olivat pelkässä seurannassa (p = 0,0039, log-rank-testi). MabThera-ylläpitohoito pienensi kuoleman riskiä 56 % (95 %:n luottamusväli 22–75 %).
Taulukko 11 Ylläpitovaihe: Yhteenveto MabThera-hoidon ja pelkän seurannan tehoa koskevista tuloksista (seuranta-ajan mediaani 28 kuukautta)
| Tehomuuttuja | Kaplan-Meier-estimaatti (kk): mediaaniaika tapahtumaan | Riskin väheneminen | ||
Seuranta (N = 167) | MabThera (N = 167) | Log-rank-testin p-arvo | ||
| Taudin etenemisestä vapaa elinaika (PFS) | 14,3 | 42,2 | < 0,0001 | 61 % |
| Kokonaiseloonjäämisaika (OS) | NR | NR | 0,0039 | 56 % |
| Aika ennen uuden lymfoomahoidon aloittamista | 20,1 | 38,8 | < 0,0001 | 50 % |
| Taudista vapaa elinaika (DFSa) | 16,5 | 53,7 | 0,0003 | 67 % |
Alaryhmäanalyysi Taudin etenemisestä vapaa elinaika (PFS) | ||||
CHOP R-CHOP Täydellinen hoitovaste Osittainen hoitovaste | 11,6 22,1 14,3 14,3 | 37,5 51,9 52,8 37,8 | < 0,0001 0,0071 0,0008 < 0,0001 | 71 % 46 % 64 % 54 % |
| Kokonaiseloonjäämisaika (OS) | ||||
CHOP R-CHOP | NR NR | NR NR | 0,0348 0,0482 | 55 % 56 % |
NR: ei saavutettu; a: koskee vain täydellisen hoitovasteen saaneita potilaita
MabThera-ylläpitohoidon suotuisa vaikutus varmistui kaikissa analysoiduissa alaryhmissä riippumatta induktiohoidosta (CHOP tai R-CHOP) tai induktiohoidon hoitovasteen laadusta (täydellinen tai osittainen hoitovaste) (taulukko 11). MabThera-ylläpitohoito pidensi merkitsevästi taudin etenemisestä vapaan elinajan mediaania potilailla, jotka vastasivat CHOP-induktiohoitoon (mediaani PFS 37,5 kk vs. 11,6 kk, p < 0,0001) sekä potilailla, jotka vastasivat R-CHOP-induktiohoitoon (mediaani PFS 51,9 kk vs. 22,1 kk, p = 0,0071). Vaikka alaryhmät olivat pieniä, MabThera-ylläpitohoito toi merkitsevää hyötyä kokonaiseloonjäämisajassa mitattuna sekä CHOP-hoitoon että R-CHOP-hoitoon vastaaville potilaille. Tämän havainnon varmistamiseksi tarvitaan pidempi seuranta-aika.
Aikuisten diffuusi suurisoluinen non-Hodgkin-B-solulymfooma
Satunnaistetussa, avoimessa kliinisessä tutkimuksessa 399:lle aikaisemmin hoitamattomalle, iäkkäälle potilaalle (ikä 60–80 vuotta), joilla oli diffuusi suurisoluinen B-solulymfooma, annettiin standardi-CHOP-hoitoa (syklofosfamidi 750 mg/m2, doksorubisiini 50 mg/m2, vinkristiini 1,4 mg/m2 aina 2 mg:n annokseen saakka päivänä 1 ja prednisoloni 40 mg/m2/vrk päivinä 1–5) joka kolmas viikko kahdeksan hoitosyklin ajan tai MabTheraa 375 mg/m2 ja CHOP-hoitoa (R-CHOP). MabThera annettiin hoitosyklin ensimmäisenä päivänä.
Tehoa koskevassa loppuanalyysissä olivat mukana kaikki satunnaistetusti eri ryhmiin jaetut potilaat (197 CHOP- ja 202 R-CHOP-ryhmässä). Seuranta-ajan keston mediaani oli noin 31 kuukautta. Potilaat oli jaettu eri hoitoa saaviin ryhmiin tasapuolisesti ottaen huomioon lähtötilanteen sairauden ominaisuudet ja aste. Loppuanalyysi vahvisti, että R-CHOP-hoito pidensi kliinisesti merkittävästi ja tilastollisesti merkitsevästi tapahtumista vapaata aikaa (primaarinen tehoa osoittava parametri; tapahtumia olivat kuolema, taudin uusiutuminen, lymfooman eteneminen tai uuden lymfoomahoidon aloittaminen) (p = 0,0001). Tapahtumista vapaan ajan Kaplan-Meier-estimaatti (mediaani) oli 35 kuukautta R-CHOP-ryhmässä ja 13 kuukautta CHOP-haarassa vastaten riskin vähenemistä 41 %:lla. Kokonaiseloonjäämisajan 24 kuukauden estimaatti oli 68,2 % R-CHOP-ryhmässä ja 57,4 % CHOP-haarassa. Kun seuranta-ajan mediaani oli 60 kuukautta, suoritettiin seuraava kokonaiseloonjäämisaikaa koskeva analyysi, joka vahvisti R-CHOP-hoidon paremmuuden verrattuna CHOP-hoitoon (p = 0,0071): riski väheni 32 %.
Sekundaariset parametrit (hoitovaste, taudin etenemisestä vapaa aika, taudista vapaa aika, hoitovasteen kesto) vahvistivat R-CHOP-hoidon vaikutuksen verrattuna CHOP-hoitoon. Täydellinen hoitovaste kahdeksan syklin jälkeen oli 76,2 % R-CHOP-haarassa ja 62,4 % CHOP-haarassa (p = 0,0028). Taudin etenemisen riski väheni 46 % ja taudin uusiutumisen riski 51 %.
Kaikissa potilaiden alaryhmissä (sukupuoli, ikä, iän huomioon ottava IPI eli International NHL Prognostic Index, Ann Arborin luokitus, ECOG, β2-mikroglobuliini, LDH, albumiini, B-oireet, suuri tautimassa, ekstranodaalinen tauti, luuytimeen levinnyt tauti) tapahtumista vapaan ajan ja kokonaiseloonjäämisajan riskisuhteet (R-CHOP verrattuna CHOP:iin) olivat vastaavasti alle 0,83 ja 0,95. R-CHOP-hoito hyödytti sekä korkean että matalan riskin potilaita käytettäessä iän huomioon ottavaa IPI-systeemiä.
Kliiniset laboratoriolöydökset
Humaani anti-hiiri (HAMA) -vastetta ei löytynyt 67 potilaalta, joilta HAMA tutkittiin. Lääkevasta-aine (ADA) -vaste oli positiivinen neljällä potilaalla (1,1 %) 356 tutkitusta.
Krooninen lymfaattinen leukemia
Kahdessa avoimessa, satunnaistetussa kliinisessä tutkimuksessa yhteensä 817 aikaisemmin hoitamatonta KLL-potilasta ja 552 uusiutunutta tai refraktorista tautia sairastavaa potilasta satunnaistettiin saamaan joko FC-solunsalpaajahoitoa (fludarabiinia 25 mg/m2, syklofosfamidia 250 mg/m2, päivinä 1–3) joka neljäs viikko kuuden syklin ajan tai MabTheraa yhdistettynä FC-solunsalpaajahoitoon (R-FC). MabTheraa annettiin annostuksella 375 mg/m2 ensimmäisen syklin ensimmäisenä päivänä ennen solunsalpaajahoitoa ja annostuksella 500 mg/m2 seuraavien hoitosyklien ensimmäisenä päivänä. Potilaat suljettiin pois tutkimuksesta, jos heitä oli aikaisemmin hoidettu monoklonaalisella vasta-aineella tai jos heidän sairautensa ei ollut reagoinut (määritellään osittaisen vasteen puuttumisena vähintään 6 kk:n ajan) fludarabiinille tai jollekin muulle nukleosidianalogille. Tehon selvittämiseksi analysoitiin ensilinjan tutkimuksesta yhteensä 810 potilaan (403 R-FC, 407 FC) tiedot (taulukot 12a ja 12b) ja uusiutuneen/refraktorisen taudin tutkimuksesta 552 potilaan (276 R-FC, 276 FC) tiedot (taulukko 13).
Ensilinjan tutkimuksessa, jossa mediaaniseuranta-aika oli 48,1 kuukautta, mediaani PFS oli 55 kuukautta R-FC-ryhmässä ja 33 kuukautta FC-ryhmässä (p < 0,0001, log-rank-testi). Tulokset kokonaiselinajan analyysistä osoittivat R-FC-hoidon merkitsevää etua verrattuna pelkkään FC-hoitoon (p = 0,0319, log-rank-testi) (taulukko 12a). Etenemisvapaaseen elinaikaan liittyvää etua havaittiin tasaisesti useimmissa potilasalaryhmissä, jotka analysoitiin lähtötason sairausriskin mukaan (Binet luokat A-C) (taulukko 12b).
Taulukko 12a Kroonisen lymfaattisen leukemian ensilinjan hoito
Yhteenveto tehoa koskevista tuloksista: MabThera + FC vs. pelkkä FC - seuranta-ajan mediaani 48,1 kuukautta
| Tehomuuttuja | Kaplan-Meier-estimaatti (kk): mediaaniaika tapahtumaan | Riskin väheneminen | ||
FC (N = 409) | R-FC (N = 408) | Log-rank-testin p-arvo | ||
| Taudin etenemisestä vapaa elinaika (PFS) | 32,8 | 55,3 | < 0,0001 | 45 % |
| Kokonaiseloonjäämisaika (OS) | NR | NR | 0,0319 | 27 % |
| Tapahtumavapaa elinaika | 31,3 | 51,8 | < 0,0001 | 44 % |
| Hoitovaste (CR, nPR, tai PR) | 72,6 % | 85,8 % | < 0,0001 | n.a. |
| CR-vaste | 16,9 % | 36,0 % | < 0,0001 | n.a. |
| Vasteen kesto* | 36,2 | 57,3 | < 0,0001 | 44 % |
| Taudista vapaa elinaika (DFS)** | 48,9 | 60,3 | 0,0520 | 31 % |
| Aika ennen uuden hoidon aloittamista | 47,2 | 69,7 | < 0,0001 | 42 % |
Hoitovaste ja CR-vasteet analysoitu Chi-Square-testin avulla. NR (not reached): ei saavutettu; n.a. ei käytettävissä
* Koskee vain täydellisen tai osittaisen hoitovasteen saaneita potilaita (CR, nPR, PR).
** Koskee vain täydellisen hoitovasteen saaneita.
Taulukko 12b Kroonisen lymfaattisen leukemian ensilinjan hoito
Riskisuhde taudin etenemisestä vapaalle elinajalle (PFS) Binet-vaiheessa (ITT) - seuranta-ajan mediaani 48,1 kuukautta
| Taudin etenemisestä vapaa elinaika (PFS) | Potilasmäärä | Riskisuhde (hazard ratio, (95 %:n luottamusväli) | P-arvo (ei säädetty Wald testi) | |
| FC | R-FC | |||
| Binet-vaihe A | 22 | 18 | 0,39 (0,15; 0,98) | 0,0442 |
| Binet-vaihe B | 259 | 263 | 0,52 (0,41; 0,66) | < 0,0001 |
| Binet-vaihe C | 126 | 126 | 0,68 (0,49; 0,95) | 0,0224 |
Uusiutuneen/refraktorisen taudin tutkimuksessa taudin etenemisestä vapaan elinajan (ensisijainen päätetapahtuma) mediaani oli 30,6 kuukautta R-FC-ryhmässä ja 20,6 kuukautta FC-ryhmässä (p = 0,0002, log-rank-testi). Etenemisvapaaseen elinaikaan liittyvää etua havaittiin lähes kaikissa potilasalaryhmissä, jotka analysoitiin lähtötason sairausriskin mukaan. R-FC-haarasta raportoitiin vähäistä, mutta ei merkittävää parannusta kokonaiselinajassa FC-haaraan verrattuna.
Taulukko 13 Uusiutuneen/refraktorisen kroonisen lymfaattisen leukemian hoito - yhteenveto tehoa koskevista tuloksista: MabThera + FC vs. pelkkä FC (seuranta-ajan mediaani 25,3 kuukautta)
| Tehomuuttuja | Kaplan-Meier-estimaatti (kk): mediaaniaika tapahtumaan | Riskin väheneminen | ||
FC (N = 276) | R-FC (N = 276) | Log-rank-testin p-arvo | ||
| Taudin etenemisestä vapaa elinaika (PFS) | 20,6 | 30,6 | 0,0002 | 35 % |
| Kokonaiseloonjäämisaika (OS) | 51,9 | NR | 0,2874 | 17 % |
| Tapahtumavapaa elinaika | 19,3 | 28,7 | 0,0002 | 36 % |
| Hoitovaste (CR, nPR, tai PR) | 58,0 % | 69,9 % | 0,0034 | n.a. |
| CR-vaste | 13,0 % | 24,3 % | 0,0007 | n.a. |
| Vasteen kesto* | 27,6 | 39,6 | 0,0252 | 31 % |
| Taudista vapaa elinaika (DFS)** | 42,2 | 39,6 | 0,8842 | -6 % |
| Aika ennen uuden KLL-hoidon aloittamista | 34,2 | NR | 0,0024 | 35 % |
Hoitovaste ja CR-vasteet analysoitu Chi-Square-testin avulla.
* Koskee vain täydellisen tai osittaisen hoitovasteen saaneita potilaita (CR, nPR, PR). NR (not reached): ei saavutettu n.a. ei käytettävissä
** Koskee vain täydellisen hoitovasteen saaneita.
Tulokset lisätutkimuksista, joissa MabTheraa käytettiin yhdessä muiden solunsalpaajien kanssa (mukaan lukien CHOP, FCM, PC, PCM, bendamustiini ja kladribiini) aikaisemmin hoitamattomilla KLL-potilailla ja potilailla, joilla on uusiutunut tai refraktorinen tauti, ovat myös osoittaneet korkeita kokonaisvasteita ja etenemisvapaaseen elinaikaan liittyvää etua, vaikkakin toksisuus hieman lisääntyi (erityisesti myelotoksisuus). Nämä tutkimukset tukevat MabTheran käyttöä yhdessä minkä tahansa solunsalpaajan kanssa. Tutkimustulokset 180 potilaasta osoittavat, että aikaisemmasta MabThera-hoidosta on kliinistä hyötyä (mukaan lukien CR) ja ne tukevat MabTheran uusintahoitoa.
Pediatriset potilaat
Avoimessa, satunnaistetussa monikeskustutkimuksessa tutkittiin LMB-solunsalpaajahoitoa (Lymphome Malin B, eli kortikosteroidit, vinkristiini, syklofosfamidi, metotreksaatti suurina annoksina, sytarabiini, doksorubisiini, etoposidi ja selkäydinnesteeseen annettava kolmoislääkehoito [metotreksaatti/sytarabiini/kortikosteroidi]) yksinään ja yhdistelmänä MabTheran kanssa pediatrisilla potilailla, jotka sairastivat aiemmin hoitamatonta pitkälle edennyttä CD20-positiivista diffuusia suurisoluista B‑solulymfoomaa, Burkittin lymfoomaa, Burkittin leukemiaa tai Burkitt-tyyppistä lymfoomaa. Pitkälle edenneeksi levinneisyysasteeksi määriteltiin III levinneisyysaste sekä kohonnut laktaattidehydrogenaasipitoisuus (LDH) (”B-high”) (LDH > kaksinkertainen laitoskohtaiseen aikuisten normaaliarvojen ylärajaan nähden [> N x 2]) tai mikä tahansa IV levinneisyysaste tai Burkittin leukemia. Potilaat satunnaistettiin joko LMB-solunsalpaajahoito-ohjelmaan tai saamaan kuusi MabThera-infuusiota laskimoon annoksina 375 mg/m2 kehon pinta-alan perusteella yhdistelmänä LMB-solunsalpaajahoito-ohjelman kanssa (kaksi infuusiota kummassakin kahdesta induktiojaksosta ja yksi infuusio kummassakin kahdesta konsolidaatiojaksosta) LMB‑hoito-ohjelman mukaisesti. Tehoa koskeviin analyyseihin otettiin mukaan yhteensä 328 satunnaistettua potilasta, joista yksi alle 3-vuotias potilas sai MabTheraa yhdessä LMB-solunsalpaajahoidon kanssa.
Lähtötilanteen ominaisuudet olivat näissä kahdessa hoitohaarassa eli LMB-hoidossa (LMB-solunsalpaajahoito-ohjelma) ja R-LMB-hoidossa (LMB-solunsalpaajahoito-ohjelma ja MabThera) hyvin tasapainossa. Potilaiden iän mediaani oli LMB-haarassa 7 vuotta ja R-LMB-haarassa 8 vuotta. Noin puolet potilaista oli ryhmässä B (LMB-haarassa 50,6 % ja R-LMB-haarassa 49,4 %), kummassakin hoitohaarassa 39,6 % oli ryhmässä C1, ja LMB-haarassa 9,8 % ja R-LMB-haarassa 11,0 % oli ryhmässä C3. Murphyn levinneisyysasteiden perusteella useimpien potilaiden taudin levinneisyysaste oli joko Burkittin lymfooman III levinneisyysaste (LMB-haarassa 45,7 % ja R-LMB-haarassa 43,3 %) tai ei keskushermostoon levinnyt Burkittin leukemia (LMB-haarassa 21,3 % ja R‑LMB-haarassa 24,4 %). Alle puolella potilaista (kummassakin hoitohaarassa 45,1 %) tauti oli levinnyt luuytimeen, ja valtaosalla potilaista (LMB-haarassa 72,6 % ja R-LMB-haarassa 73,2 %) tauti ei ollut levinnyt keskushermostoon. Tehon ensisijainen päätetapahtuma oli tapahtumavapaa elinaika, jossa tapahtumaksi määriteltiin jonkin seuraavista ilmeneminen: taudin eteneminen, relapsi, uusi syöpä, kuolema mistä tahansa syystä tai vasteen puuttuminen, joka osoitetaan toteamalla jäännöstautina elinkykyisiä soluja toisen CYVE-hoitojakson jälkeen. Toissijaisia tehon päätetapahtumia olivat kokonaiseloonjäämisaika ja täydellinen remissio.
Noin 1 vuoden (mediaani) seurannan jälkeen tehdyssä ennalta määritellyssä välianalyysissa havaittiin ensisijaisen päätetapahtuman eli tapahtumavapaan elinajan pidentyneen oleellisesti, ja 1 vuoden estimaatit olivat R-LMB-haarassa 94,2 % (95 %:n luottamusväli 88,5–97,2 %) vs. LMB-haarassa 81,5 % (95 %:n luottamusväli 73,0–87,8 %), ja korjattu Coxin riskisuhde 0,33 (95 %:n luottamusväli 0,14–0,79). Riippumattoman seurantaryhmän (independent data monitoring committee) tämän tuloksen perusteella antaman suosituksen mukaisesti satunnaistaminen keskeytettiin, ja LMB-haaran potilaille sallittiin MabThera-hoito.
Ensisijaiset tehon analyysit tehtiin 328 satunnaistetusta potilaasta, joiden seuranta-ajan mediaani oli 3,1 vuotta. Tulokset kuvataan taulukossa 14.
Taulukko 14 Tiivistelmä ensisijaisista tehon tuloksista (ITT-potilasjoukko)
| Analyysi | LMB (N = 164) | R-LMB (N = 164) |
| Tapahtumavapaa elinaika | 28 tapahtumaa | 10 tapahtumaa |
| Yksitahoisen log-rank-testin p-arvo 0,0006 | ||
| Korjattu Coxin riskisuhde (HR) 0,32 (90 %:n luottamusväli: 0,17, 0,58) | ||
| 3 vuoden tapahtumavapaata elinaikaa kuvaavat luvut | 82,3 % (95 %:n luottamusväli: 75,7 %, 87,5 %) | 93,9 % (95 %:n luottamusväli: 89,1 %, 96,7 %) |
| Kokonaiseloonjäämisaika | 20 kuolemaa | 8 kuolemaa |
| Yksitahoisen log-rank-testin p-arvo 0,0061 | ||
| Korjattu Coxin mallin riskisuhde (HR) 0,36 (95 %:n luottamusväli: 0,16; 0,81) | ||
| 3 vuoden kokonaiseloonjäämisaikaa kuvaavat luvut | 87,3 % (95 %:n luottamusväli: 81,2 %, 91,6 %) | 95,1 % (95 %:n luottamusväli: 90,5 %, 97,5 %) |
| CR-vaste | 93,6 % (95 %:n luottamusväli: 88,2 %; 97,0 %) | 94,0 % (95 %:n luottamusväli: 88,8 %, 97,2 %) |
Ensisijainen tehon analyysi osoitti, että MabThera-valmisteen lisäämisellä LMB-solunsalpaajahoito-ohjelmaan saatiin tapahtumavapaan elinajan suhteen hyöty pelkkään LMB-solunsalpaajahoito-ohjelmaan verrattuna, sillä kansallisen ryhmän, histologian ja hoitoryhmän perusteella korjatusta Coxin regressioanalyysista saatu tapahtumavapaan elinajan riskisuhde (HR) oli 0,32 (90 %:n luottamusväli 0,17–0,58). Sen sijaan kahden hoitohaaran välillä ei havaittu merkittävää eroa kliinisen vasteen (CR) saavuttaneiden potilaiden lukumäärässä. MabThera-hoidon lisäämisestä LMB-solunsalpaajahoito-ohjelmaan osoitettiin hyötyä myös toissijaisen päätetapahtuman eli kokonaiseloonjäämisajan suhteen, ja kokonaiseloonjäämisajan riskisuhde (HR) oli 0,36 (95 %:n luottamusväli 0,16–0,81).
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset MabTheran käytöstä follikulaarisen lymfooman ja kroonisen lymfaattisen leukemian hoidossa kaikissa pediatrisissa potilasryhmissä sekä vastasyntyneiden ja < 6 kuukauden ikäisten pediatristen potilaiden CD20-positiivisen diffuusin suurisoluisen B-solulymfooman hoidossa. Ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Kliininen teho ja turvallisuus nivelreuman hoidossa
MabTheran teho ja turvallisuus potilailla, joilla on riittämätön hoitovaste TNF-estäjille, osoitettiin nivelreuman tunnusmerkkien ja oireiden lievittämisessä yhdessä keskeisessä satunnaistetussa kontrolloidussa kaksoissokkoutetussa monikeskustutkimuksessa (tutkimus 1).
Tutkimuksessa 1 evaluoitiin 517 potilasta, joiden aikaisempi hoito yhdellä tai useammalla TNF-estäjällä ei ollut tuonut riittävää hoitovastetta, tai potilaat eivät olleet sietäneet hoitoa. Tutkimukseen hyväksytyillä potilailla oli ACR-kriteerien (American College of Rheumatology) mukaan diagnosoitu aktiivinen nivelreuma. MabTheraa annettiin kahtena 1000 mg:n infuusiona laskimoon 15 vuorokauden välein. Potilaat saivat metotreksaattiin yhdistettynä laskimonsisäisesti kaksi 1000 mg:n MabThera-infuusiota tai plaseboa. Kaikki potilaat saivat samanaikaisesti prednisonia suun kautta 60 mg 2.−7. päivänä ja 30 mg 8.–14. päivänä ensimmäisen infuusion jälkeen. Ensisijainen päätetapahtuma oli niiden potilaiden osuus, joilla saavutettiin ACR20-vaste viikkoon 24 mennessä. Potilaiden seuranta jatkui viikon 24 jälkeen pitkäaikaisten hoitovasteiden arvioimiseksi, muun muassa radiologinen arviointi tehtiin viikoilla 56 ja 104. Tänä aikana 81 % plaseboryhmän potilaista sai MabTheraa viikoilla 24–56 avoimessa jatkotutkimuksessa.
Ensisijaiset päätetapahtumat saavutettiin MabThera-tutkimuksissa tuoretta nivelreumaa sairastavilla potilailla (metotreksaatilla aiemmin hoitamattomat tai metotreksaatille riittämättömän vasteen saaneet potilaat, jotka eivät vielä olleet saaneet TNF-alfa estäjiä). MabTheralla ei ole indikaatiota näille potilaille, koska turvallisuustiedot MabTheran pitkäaikaishoidosta ovat riittämättömät, erityisesti kiinteiden kasvainten ja PML:n muodostumista koskevaa riskiä ei ole selvitetty.
Taudin aktiivisuuteen perustuvat tulokset
MabThera yhdessä metotreksaatin kanssa nosti merkitsevästi niiden potilaiden osuutta, joiden ACR-pistearvo parani vähintään 20 % pelkkää metotreksaattia saaneisiin potilaisiin verrattuna (taulukko 15). Kaikissa kehittämistutkimuksissa hoidon hyöty oli sama riippumatta potilaiden iästä, sukupuolesta, ihon pinta-alasta, etnisestä taustasta, aikaisempien hoitojen lukumäärästä tai taudin tilasta.
Kliinisesti ja tilastollisesti merkitsevää paranemista todettiin myös kaikissa yksittäisissä ACR-vasteen osatekijöissä [(aristavien ja turvonneiden nivelten määrä, potilaan ja lääkärin yleisarvio, toimintakykyindeksi (HAQ), kipuarvio ja CRP-arvo (mg/dl)].
Taulukko 15 Kliininen vaste ensisijaisen päätetapahtuman kohdalla tutkimuksessa 1 (ITT-populaatio)
| Tulos† | Plasebo + MTX | MabThera + MTX (2 x 1000 mg) | |
| Tutkimus 1 | N = 201 | N = 298 | |
| ACR20 | 36 (18 %) | 153 (51 %)*** | |
| ACR50 | 11 (5 %) | 80 (27 %)*** | |
| ACR70 | 3 (1 %) | 37 (12 %)***1 | |
| EULAR-vaste (hyvä/kohtalainen) | 44 (22 %) | 193 (65 %)*** | |
| DAS-arvon keskimääräinen muutos | -0,34 | -1,83*** |
† Tulos viikolla 24
Ensisijaisessa päätepisteessä merkitsevä muutos verrattuna yhdistelmään plasebo + metotreksaatti:***p ≤ 0,0001
Taudin aktiivisuutta mittaava indeksi (DAS28) laski merkitsevästi enemmän MabThera- ja metotreksaattihoitoa saaneilla potilailla kuin pelkkää metotreksaattia saaneilla potilailla (taulukko 15). Vastaavasti kaikissa tutkimuksissa hyvä tai kohtalainen European League Against Rheumatism (EULAR) -vaste saavutettiin merkitsevästi useammalla MabThera- ja metotreksaattihoitoa saaneella potilaalla kuin pelkkää metotreksaattia saaneella potilaalla (taulukko 15).
Radiologinen vaste
Rakenteellinen nivelvaurio arvioitiin radiologisesti, ja muutos ilmaistiin käyttämällä modifioitua Sharpin kokonaispistearvoa, eroosiota ja nivelraon kaventumista.
Tutkimukseen 1 osallistui potilaita, joiden aikaisempi hoito yhdellä tai useammalla TNF-estäjällä ei ollut tuonut riittävää hoitovastetta. MabTheran ja metotreksaatin yhdistelmähoitoa saaneilla potilailla havaittiin radiologisesti viikolla 56 merkitsevästi vähemmän etenemistä kuin pelkkää metotreksaattia saaneilla potilailla. Alun perin pelkkää metotreksaattia saaneista potilaista 81 % sai myös MabTheraa joko rescue-hoitona viikoilla 16–24 tai jatkotutkimuksessa, kuitenkin ennen viikkoa 56. Alun perin MabTheraa ja metotreksaattia saaneessa ryhmässä oli suurempi osuus potilaita, joilla ei todettu erosiivista etenemistä 56 viikon aikana (taulukko 16).
Taulukko 16 Yhden vuoden radiologiset tulokset (mITT-populaatio)
| Plasebo+MTX | MabThera +MTX 2 × 1000 mg | |
| Tutkimus 1 | (n = 184) | (n = 273) |
Keskimääräinen muutos lähtötasosta: Modifioitu Sharpin kokonaispistearvo Nivelten eroosio Nivelraon kaventuminen Potilaat, joilla ei todettu radiologista muutosta Potilaat, joilla ei todettu erosiivista muutosta | 2,30 1,32 0,98 46 % 52 % | 1,01* 0,60* 0,41** 53 %, NS 60 %*, NS |
150 potilasta, jotka alun perin randomoitiin saamaan plaseboa + MTX tutkimuksessa 1, sai vuoden aikana vähintään yhden hoitojakson MabThera + MTX
* p < 0,05, ** p < 0,001. NS (non significant) = ei-merkitsevä
Nivelvaurioiden etenemisen estämistä on havaittu myös pitkäaikaisesti. Radiologinen analyysi kahden vuoden kohdalla tutkimuksessa 1 osoitti merkitsevää vähenemistä nivelvaurioiden etenemisessä potilailla, jotka olivat saaneet MabTheraa yhdistettynä metotreksaattiin verrattuna pelkkää metotreksaattia saaneisiin potilaisiin. Merkitsevästi suuremmalla osalla yhdistelmähoitoa saaneista potilaista nivelvauriot eivät edenneet kahden vuoden jakson aikana.
Tulokset fyysisessä toimintakyvyssä ja elämänlaadussa
MabTheraa saaneilla potilailla todettiin merkittävää parantumista toimintakykyä mittaavassa indeksissä (HAQ-DI) ja väsymystä mittaavissa FACIT-F-pisteissä pelkkää metotreksaattia saaneisiin potilaisiin verrattuna. Myös niiden MabTheraa saaneiden potilaiden osuus, joilla havaittiin pienin kliinisesti merkittävä muutos (MCID = minimal clinically important difference) HAQ-DI-indeksissä (yksittäinen kokonaispistearvo pieneni > 0,22), oli suurempi kuin pelkkää metotreksaattia saaneiden ryhmässä (taulukko 17).
Terveyteen liittyvässä elämänlaadussa osoitettiin myös merkittävää parannusta käyttäen SF-36-asteikon sekä fyysistä (PHS) että psyykkistä (MHS) terveydentilaa mittaavia osa-alueita. Lisäksi merkitsevästi suurempi osuus potilaista saavutti MCID-arvoja näissä mittauksissa (taulukko 17).
Taulukko 17 Fyysistä toimintakykyä ja elämänlaatua mittaavat tulokset viikolla 24 tutkimuksessa 1
| Tulos† | Plasebo+MTX | MabThera+MTX (2 x 1000 mg) |
| n = 201 | n = 298 | |
| Keskimääräinen muutos HAQ-DI-indeksissä | 0,1 | -0,4*** |
| % HAQ-DI MCID | 20 % | 51 % |
| Keskimääräinen muutos FACIT-F-pisteissä | -0,5 | -9,1*** |
| Keskimääräinen muutos SF-36 PHS-asteikossa | n = 197 0,9 | n = 294 5,8*** |
| % SF-36 PHS MCID | 13 % | 48 %*** |
| Keskimääräinen muutos SF-36 MHS-asteikossa | 1,3 | 4,7** |
| % SF-36 PHS MCID | 20 % | 38 %* |
† Tulos viikolla 24
Ensisijaisessa päätepisteessä merkitsevä muutos verrattuna plaseboon:* p < 0,05, **p < 0,001, ***p ≤ 0,0001
MCID HAQ-DI ≥0,22, MCID SF-36 PHS > 5,42, MCID SF-36 MHS > 6,33
Teho autovasta-aine (RF ja/tai anti-CCP) -seropositiivisilla potilailla
Autovasta-aineiden RF (reumafaktori) ja/tai anti-CCP:tä (syklisten sitrullinoituneiden peptidien vasta-aine) suhteen seropositiiviset potilaat saivat paremman vasteen MabTheralla yhdessä metotreksaatin kanssa kuin seronegatiiviset potilaat.
MabThera-hoidon tehoa analysoitiin potilaiden lähtötilanteen autovasta-ainestatuksen perusteella. Viikon 24 kohdalla havaittiin, että hoidon alussa reumafaktori- ja/tai anti-CCP-seropositiivisilla potilailla oli huomattavasti suuremmat mahdollisuudet saavuttaa ACR20- ja ACR50-vasteita verrattuna seronegatiivisiin potilaisiin (p = 0,0312 ja p = 0,0096) (taulukko 18). Nämä löydökset toistuivat viikolla 48, jolloin autovasta-aineille positiivisilla potilailla todettiin olevan huomattavasti suurempi todennäköisyys saavuttaa myös ACR70-vaste. Viikolla 48, seropositiivisilla potilailla oli 2−3 kertaa suurempi todennäköisyys saavuttaa ACR-vasteita verrattuna seronegatiivisiin potilaisiin. Myös DAS28-ESR-arvo laski merkitsevästi enemmän seropositiivisilla potilailla verrattuna seronegatiivisiin potilaisiin (kuva 1).
Taulukko 18 Yhteenveto tehosta lähtötilanteen autovasta-ainestatuksen perusteella
| Viikko 24 | Viikko 48 | |||
Seropositii-vinen (n = 514) | Seronegatii-vinen (n = 106) | Seropositii-vinen (n = 506) | Seronegatii-vinen (n = 101) | |
| ACR20 (%) | 62,3* | 50,9 | 71,1* | 51,5 |
| ACR50 (%) | 32,7* | 19,8 | 44,9** | 22,8 |
| ACR70 (%) | 12,1 | 5,7 | 20,9* | 6,9 |
| EULAR-vaste (%) | 74,8* | 62,9 | 84,3* | 723 |
| Keskimääräinen muutos DAS28-ESR | -1,97** | -1,50 | -2,48*** | -1,72 |
Merkitsevät tasot määriteltiin seuraavasti * p < 0,05, **p < 0,001, ***p < 0,0001.
Kuva 1: DAS28-ESR-arvon muutos lähtötilanteesta autovasta-ainestatuksen perusteella
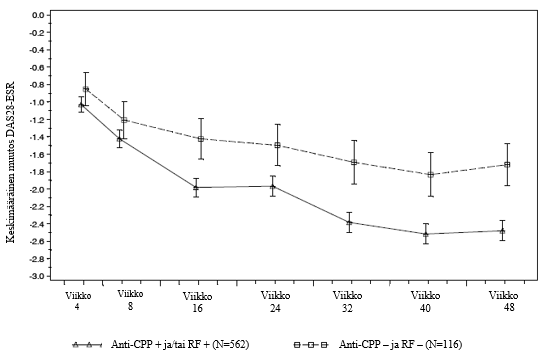
Toistuvien hoitojen pitkäaikaisteho
Annettaessa toistuvia hoitoja MabTheralla metotreksaattiin yhdistettynä saavutettiin pysyvää parannusta nivelreuman kliinisissä oireissa, kun tulokset määriteltiin ACR-, DAS28-ESR- ja EULAR-vasteina. Kaikissa tutkituissa potilaspopulaatioissa tulokset olivat ilmeisiä (kuva 2). Pysyvää parannusta fyysisessä toimintakyvyssä havaittiin arvioitaessa tuloksia sekä HAQ-DI-indeksin että HAQ-DI MCID-arvojen avulla.
Kuva 2: Neljän hoitojakson ACR-vasteet (24 viikkoa jokaisen hoitokerran jälkeen/potilas/käynti) potilailla, joilla riittämätön vaste TNF-estäjille (N = 146)
Kliiniset laboratoriolöydökset
Lääkevasta-aineita (ADA), todettiin MabThera-hoidon jälkeen yhteensä 392 potilaalla (12,7 %) 3095 nivelreumapotilaasta. ADA-vasta-aineiden ilmenemiseen ei liittynyt kliinisen tilan heikkenemistä, eikä myöhempien infuusioiden aiheuttamien reaktioiden havaittu lisääntyneen suurimmalla osalla näistä potilaista. ADA-vasta-aineisiin saattaa liittyä infuusio- tai allergiareaktioiden pahenemista myöhempien hoitojaksojen toisen infuusion jälkeen.
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset MabTheran käytöstä kaikkien pediatristen potilasryhmien hoidossa nivelreumassa. Ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Kliininen teho ja turvallisuus granulomatoottisen polyangiitin ja mikroskooppisen polyangiitin yhteydessä
Remission induktio aikuisilla
GPA/MPA-tutkimuksessa 1 yhteensä 197 vähintään 15-vuotiasta potilasta, jotka sairastivat vaikea-asteista, aktiivista granulomatoottista polyangiittia (75 %) ja mikroskooppista polyangiittia (24 %), otettiin mukaan ja sai hoitoa satunnaistetussa, kaksoissokkoutetussa lumekontrolloidussa monikeskustutkimuksessa, jossa käytettiin vaikuttavaa vertailuainetta ja jossa selvitettiin valmisteen vertailukelpoisuutta (non-inferiority).
Potilaat satunnaistettiin suhteessa 1:1 saamaan joko suun kautta päivittäin syklofosfamidia (2 mg/kg/vrk) 3–6 kuukauden ajan tai MabThera-valmistetta (375 mg/m2) kerran viikossa 4 viikon ajan. Kaikki syklofosfamidiryhmän potilaat saivat seurannan aikana atsatiopriinia ylläpitohoitona. Kummankin hoitoryhmän potilaat saivat pulssihoitona laskimoon (i.v.) 1000 mg metyyliprednisolonia (tai vastaavan annoksen muuta glukokortikoidia) vuorokaudessa 1–3 päivän ajan, minkä jälkeen he saivat suun kautta prednisonia (1 mg/kg/vrk, enintään 80 mg/vrk). Prednisonihoidon vähitellen toteutettavan lopettamisen tuli päättyä 6. kuukauteen mennessä hoidon aloittamisesta.
Ensisijainen hoitotuloksen mittari oli täydellisen remission saavuttaminen hoitokuukautena 6, mikä määriteltiin sairauden aktiivisuuspisteinä (Birmingham Vasculitis Activity Score for Wegener’s Granulomatosis, BVAS/WG) 0 ja sillä, ettei potilas käyttänyt glukokortikoidihoitoa. Hoitojen välisen eron ennalta määritelty vertailukelpoisuuden marginaali (non-inferiority margin) oli 20 %. Tutkimuksessa osoitettiin, että MabThera oli täydellisen remission suhteen vertailukelpoinen syklofosfamidin kanssa hoitokuukautena 6 (taulukko 19).
Teho havaittiin sekä äskettäin diagnosoiduilla potilailla että potilailla, joiden tauti oli relapsoitunut (taulukko 20).
Taulukko 19 Täydellisen remission saavuttaneiden aikuispotilaiden prosenttiosuus hoitokuukautena 6 (Intent‑to‑Treat-potilaat*)
| MabThera (n = 99) | Syklofosfamidi (n = 98) | Hoitojen ero (MabThera- syklofosfamidi) | |
| Prosenttiosuus | 63,6 % | 53,1 % | 10,6 % 95,1 %b CI (‑3,2 %, 24,3 %) a |
a Vertailukelpoisuus osoitettiin, koska alempi tulos (‑3,2 %) oli suurempi kuin ennalta määritetty vertailukelpoisuuden marginaali (‑20 %). | |||
Taulukko 20 Täydellinen remissio hoitokuukautena 6 taudin tilanteen mukaan
| MabThera | Syklofosfamidi | Ero (CI 95 %) | |
| Kaikki potilaat | n = 99 | n = 98 | |
| Äskettäin diagnosoidut | n = 48 | n = 48 | |
| Relapsoituneet | n = 51 | n = 50 | |
| Täydellinen remissio | |||
| Kaikki potilaat | 63,6 % | 53,1 % | 10,6 % (-3,2, 24,3) |
| Äskettäin diagnosoidut | 60,4 % | 64,6 % | ‑4,2 % (‑23,6, 15,3) |
| Relapsoituneet | 66,7 % | 42,0 % | 24,7 % (5,8, 43,6) |
Huonoimman tapauksen paikkaus koskee potilaita, joilta tiedot puuttuvat
Täydellinen remissio hoitokuukausina 12 ja 18
MabThera-ryhmässä 48 % potilaista oli saavuttanut täydellisen remission hoitokuukautena 12 ja 39 % potilasta oli saavuttanut täydellisen remission hoitokuukautena 18. Syklofosfamidihoitoa (ja tämän jälkeen atsatiopriinia täydellisen remission ylläpitämiseen) saaneista potilaista täydellisen remission oli saavuttanut 39 % potilaista hoitokuukautena 12 ja 33 % potilaista oli saavuttanut täydellisen remission hoitokuukautena 18. Hoitokuukaudesta 12 hoitokuukauteen 18 MabThera-ryhmässä oli havaittu 8 relapsia verrattuna neljään relapsiin syklofosfamidiryhmässä.
Laboratoriotutkimukset
MabThera-hoitoa remission induktiota koskeneessa tutkimuksessa saaneista potilaista yhteensä 23/99 (23 %) todettiin testissä lääkevasta-aineille (ADA) positiivisiksi hoitokuukauteen 18 mennessä. Yksikään 99 MabThera-hoitoa saaneesta potilaasta ei ollut seulonnassa lääkevasta-aineille positiivinen. Lääkevasta-aineilla ei ollut remission induktiota koskeneessa tutkimuksessa selkeää trendiä tai haitallista vaikutusta turvallisuuteen tai tehoon.
Aikuisten ylläpitohoito remission yhteydessä
Prospektiivisessa, kontrolloidussa, avoimessa monikeskustutkimuksessa yhteensä 117 potilasta (88 sairasti granulomatoottista polyangiittia, 24 sairasti mikroskooppista polyangiittia ja 5 sairasti munuaisiin rajoittunutta ANCA-vaskuliittia), joiden sairaus oli remissiossa, satunnaistettiin saamaan atsatiopriinia (59 potilasta) tai MabThera-valmistetta (58 potilasta). Mukana olleet potilaat olivat iältään 21–75-vuotiaita ja sairastivat äskettäin diagnosoitua tai uusiutunutta sairautta, joka oli täydellisessä remissiossa glukokortikoideista ja syklofosfamidipulssihoidosta koostuneen yhdistelmähoidon jälkeen. Valtaosa potilaista oli diagnoosin saadessaan tai sairauden aikana ANCA-positiivisia; potilailla oli histologisesti varmistettu nekrotisoiva pienten suonten vaskuliitti sekä granulomatoottisen polyangiitin tai mikroskooppisen polyangiitin kliininen fenotyyppi tai munuaisiin rajoittunut ANCA-vaskuliitti tai molemmat.
Remission induktioon annettuun hoitoon kuului tutkijan harkinnan mukaan laskimoon annettava prednisoni, jota ennen osa potilaista sai metyyliprednisolonia pulssihoitona, ja syklofosfamidi pulssihoitona, kunnes saavutettiin remissio 4–6 kuukauden kuluttua. Potilaat satunnaistettiin tänä ajankohtana, kuitenkin enintään 1 kuukauden kuluessa viimeisen syklofosfamidipulssihoidon jälkeen, saamaan joko MabThera-valmistetta (kaksi 500 mg:n infuusiota laskimoon kahden viikon välein [päivänä 1 ja päivänä 15], jonka jälkeen he saivat 500 mg laskimoon 6 kuukauden välein 18 kuukauden ajan) tai atsatiopriinia (suun kautta annoksina 2 mg/kg/vrk 12 kuukauden ajan, minkä jälkeen 1,5 mg/kg/vrk 6 kuukauden ajan, ja lopuksi 1 mg/kg/vrk 4 kuukauden ajan [näiden 22 kuukauden jälkeen hoito päättyi]). Prednisoniannosta pienennettiin vähitellen, ja sen jälkeen hoitoa jatkettiin pieninä annoksina (noin 5 mg vuorokaudessa) vähintään 18 kuukautta satunnaistamisen jälkeen. Prednisoniannoksen pienentäminen vähitellen ja päätös prednisonihoidon lopettamisesta 18 kuukauden jälkeen jätettiin tutkijan harkintaan.
Kaikkia potilaita seurattiin kuukauteen 28 saakka (10 kuukautta viimeisen MabThera-infuusion jälkeen tai 6 kuukautta viimeisen atsatiopriiniannoksen jälkeen). Pneumocystis jirovecii ‑pneumoniaestohoito vaadittiin kaikille potilaille, joiden CD4+-reseptoria ilmentävien T-solujen määrä oli alle 250/kuutiomillimetri.
Hoitotuloksen ensisijainen mittari oli vakavien relapsien lukumäärä kuukautena 28.
Tulokset
Vakava relapsi (määriteltiin vaskuliitin aktiivisuuden kliinisten oireiden ja/tai laboratoriolöydösten uusiutumiseksi [BVAS > 0], josta saattoi aiheutua elimen vajaatoimintaa tai elinvaurio tai joka saattoi olla hengenvaarallinen) todettiin kuukautena 28 MabThera-ryhmässä 3 potilaalla (5 %) ja atsatiopriiniryhmässä 17 potilaalla (29 %) (p = 0,0007). Lieviä relapseja (eivät hengenvaarallisia eikä niihin liittynyt vakavia elinvaurioita) todettiin MabThera-ryhmässä seitsemällä potilaalla (12 %) ja atsatiopriiniryhmässä kahdeksalla potilaalla (14 %).
Kumulatiivista ilmaantuvuutta esittävät käyrät osoittavat, että MabThera-hoitoa saaneilla potilailla aika ensimmäiseen vakavaan relapsiin oli pidempi alkaen kuukaudesta 2. Ero säilyy aina 28 kuukauteen saakka (kuva 3).
Kuva 3: Ensimmäisen vakavan relapsin kumulatiivinen ilmaantuvuus ajan mittaan
Huom.: Potilaat sensoroitiin kuukautena 28, jos heillä ei ollut tapahtumia.
Laboratoriotutkimukset
Kliinisessä tutkimuksessa yhteensä 34 MabThera-ylläpitohoitoa saaneesta potilaasta 6 potilaalle (18 %) kehittyi lääkevasta-aineita. Lääkevasta-aineista ei aiheutunut ylläpitohoidon kliinisessä tutkimuksessa selkeää trendiä tai haitallista vaikutusta turvallisuuteen tai tehoon.
Pediatriset potilaat
Tutkimus WA25615 (PePRS) oli avoin, yhden hoitohaaran kontrolloimaton monikeskustutkimus 25 pediatrisella potilaalla (≥ 2-vuotiaista < 18-vuotiaisiin), joilla oli vaikea, aktiivinen granulomatoottinen polyangiitti tai mikroskooppinen polyangiitti. Tutkimuspotilaiden iän mediaani oli 14 vuotta (vaihteluväli 6–17 vuotta), ja valtaosa potilaista (20/25 [80 %]) oli tyttöjä. Lähtötilanteessa yhteensä 19 potilaalla (76 %) oli granulomatoottinen polyangiitti ja 6 potilaalla (24 %) oli mikroskooppinen polyangiitti. Tutkimukseen tullessa kahdeksallatoista potilaalla (72 %) tauti oli diagnosoitu äskettäin (13 potilaalla granulomatoottinen polyangiitti ja 5 potilaalla mikroskooppinen polyangiitti), ja 7 potilaalla oli relapsoituva tauti (6 potilaalla granulomatoottinen polyangiitti ja 1 potilaalla mikroskooppinen polyangiitti).
Tutkimusasetelma käsitti alkuvaiheen 6 kuukauden pituisen remission induktiovaiheen ja vähintään 18 kuukauden seurantavaiheen, joka kaiken kaikkiaan kesti enimmillään 54 kuukautta (4,5 vuotta). Potilaille oli tarkoitus antaa vähintään 3 metyyliprednisoloniannosta laskimoon (30 mg/kg/vrk, enintään 1 g/vrk) ennen ensimmäistä laskimoon annettavaa MabThera-infuusiota. Metyyliprednisolonia voitiin antaa ylimääräisinä vuorokausiannoksina laskimoon (enintään kolme annosta), jos se oli kliinisesti aiheellista. Remission induktiohoito koostui neljästä kerran viikossa laskimoon annetusta MabThera-infuusiosta, jonka annos oli 375 mg/m2 kehon pinta-alan perusteella, tutkimuspäivinä 1, 8, 15 ja 22 yhdistelmänä suun kautta otettavan prednisolonin tai prednisonin kanssa, joita otettiin annoksina 1 mg/kg/vrk (enintään 60 mg/vrk), ja annos pienennettiin kuukauteen 6 mennessä pienimpään annokseen 0,2 mg/kg/vrk (enintään 10 mg/vrk). Remission induktiovaiheen jälkeen potilaille voitiin tutkijan harkinnan perusteella antaa seuraavat MabThera-infuusiot kuukautena 6 tai sen jälkeen, jotta PVAS-remissio säilyi ja sairauden aktiivisuus (mukaan lukien etenevä tauti tai pahenemisvaihe) pysyi hallinnassa tai saavutettiin ensimmäinen remissio.
Kaikki 25 potilasta saivat kaikki neljä kerran viikossa annettavaa infuusiota laskimoon 6 kuukautta kestäneen remission induktiovaiheen aikana. Kaikkiaan 25 potilaasta 24 potilasta oli mukana vähintään 18 kuukauden seurannan ajan.
Tämän tutkimuksen tarkoituksena oli arvioida MabThera-hoidon turvallisuutta, farmakokineettisiä parametreja ja tehoa granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavilla pediatrisilla potilailla (≥ 2-vuotiaista < 18-vuotiaisiin). Tutkimuksen tehoa koskevat tavoitteet olivat eksploratiivisia, ja ne arvioitiin pääasiassa PVAS-pisteytyksen (Pediatric Vasculitis Activity Score) avulla (taulukko 21).
Kumulatiivinen glukokortikoidiannos (laskimoon ja suun kautta) kuukauteen 6 mennessä:
WA25615-tutkimuksessa mukana olleista 25 potilaasta 24 potilaan (96 %) suun kautta otettava glukokortikoidiannos pienennettiin kuukautena 6 tai kuukauteen 6 mennessä tasolle 0,2 mg/kg/vrk (tai enintään 10 mg/vrk, kumpi näistä oli pienempi) suun kautta otettavan steroidiannoksen tutkimussuunnitelmassa määritellyn pienentämisjakson aikana.
Suun kautta otettavien glukokortikoidien kokonaiskäytössä havaittiin väheneminen viikosta 1 (mediaani = 45 mg:aa prednisonia vastaava annos [kvartiilivälin pituus: 35–60]) kuukauteen 6 (mediaani = 7,5 mg [kvartiilivälin pituus: 4–10]), minkä jälkeen se oli säilynyt ennallaan kuukautena 12 (mediaani = 5 mg [kvartiilivälin pituus: 2–10]) ja kuukautena 18 (mediaani = 5 mg [kvartiilivälin pituus: 1–5]).
Jatkohoito
Potilaat saivat koko tutkimusjakson aikana 4–28 MabThera-infuusiota (enintään 4,5 vuotta [53,8 kuukautta]). Potilaat saivat neljä kertaa enintään 375 mg/m2 MabThera-annoksen noin 6 kuukauden välein tutkijan harkinnan mukaan. Yhteensä 25 potilaasta 17 potilasta (68 %) sai lisäksi rituksimabia kuukautena 6 tai sen jälkeen tutkimuksen yleiseen päättymispäivämäärään (Common Close Out) saakka, ja näistä 17 potilaasta 14 sai lisäksi rituksimabihoitoa kuukausina 6–18.
Taulukko 21 Tutkimus WA25615 (PePRS) – PVAS-remissio kuukauteen 1, 2, 4, 6, 12 ja 18 mennessä
| Tutkimuskäynti | Vasteena PVAS-remission* saaneiden lukumäärä (vasteluku [%]) n = 25 | 95 %:n luottamusväliα |
| Kuukausi 1 | 0 | 0,0%, 13,7% |
| Kuukausi 2 | 1 (4,0%) | 0,1%, 20,4% |
| Kuukausi 4 | 5 (20,0%) | 6,8%, 40,7% |
| Kuukausi 6 | 13 (52,0%) | 31,3%, 72,2% |
| Kuukausi 12 | 18 (72,0%) | 50,6%, 87,9% |
| Kuukausi 18 | 18 (72,0%) | 50,6%, 87,9% |
| *PVAS-pisteet 0 ja glukokortikoidihoito vähennetty annokseen 0,2 mg/kg/vrk (tai 10 mg/vrk) kumpi on pienempi arviointihetkellä αTehon tulokset ovat eksploratiiviset eikä näistä päätetapahtumista ole tehty varsinaista tilastollista testausta MabThera-hoito (375 mg/m2 x 4 infuusiota) kuukauteen 6 asti oli kaikille potilaille samanlainen. Jatkohoito kuukauden 6 jälkeen tutkijan päätöksen mukaan. | ||
Laboratoriotutkimukset
Koko tutkimusjakson aikana yhteensä 25 potilaasta 4 potilaalle (16 %) kehittyi lääkevasta-aineita. Suppeat tiedot osoittavat, että potilailla, joilla oli todettu lääkevasta-aineita, ei havaittu raportoitujen haittavaikutusten osalta mitään kehityskulkua.
Granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavilla pediatrisilla potilailla tehdyissä kliinisissä tutkimuksissa lääkevasta-aineilla ei ollut selkeää kehityskulkua tai haitallista vaikutusta turvallisuuteen tai tehoon.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset MabThera-valmisteen käytöstä vaikeaa, aktiivista granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavien < 2-vuotiaiden pediatristen potilasryhmien hoidossa. Ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa.
Kliininen teho ja turvallisuus tavallisen pemfiguksen hoidossa
PV-tutkimus 1 (tutkimus ML22196)
Tässä satunnaistetussa, avoimessa, kontrolloidussa monikeskustutkimuksessa arvioitiin MabTheran ja lyhytaikaisesti pieninä annoksina käytetyn glukokortikoidihoidon (prednisoni) yhdistelmän tehoa ja turvallisuutta potilailla, joilla oli äskettäin diagnosoitu keskivaikea tai vaikea pemfigus (74 potilaalla tavallinen pemfigus ja 16 potilaalla kesivä pemfigus). Potilaiden ikä oli 19–79 vuotta, eivätkä he olleet saaneet pemfigukseen aiempaa hoitoa. Tavallista pemfigusta sairastavassa potilasjoukossa 5 potilaalla (13 %) MabThera-ryhmässä ja 3 potilaalla (8 %) tavanomaista prednisonihoitoa saaneessa ryhmässä oli keskivaikea tauti, ja 33 potilaalla (87 %) MabThera-ryhmässä ja 33 potilaalla (92 %) tavanomaisia prednisoniannoksia saaneessa ryhmässä oli vaikea-asteinen tauti. Taudin vaikeusaste määriteltiin Harmanin kriteerien mukaisesti.
Potilaat ositettiin taudin lähtötilanteen vaikeusasteen (keskivaikea tai vaikea) mukaan ja satunnaistettiin suhteessa 1:1 saamaan joko MabTheraa ja pieniä prednisoniannoksia tai tavanomaisia prednisoniannoksia. MabThera-ryhmään satunnaistetut potilaat saivat aluksi 1. tutkimuspäivänä 1000 mg infuusiona laskimoon yhdistelmänä suun kautta otettavan prednisoniannoksen kanssa. Keskivaikeaa tautia sairastavien potilaiden prednisonihoito annostuksella 0,5 mg/kg/vrk lopetettiin vähitellen 3 kuukauden kuluessa, ja vaikea-asteista tautia sairastavien potilaiden suun kautta otettava prednisonihoito annostuksella 1 mg/kg/vrk lopetettiin vähitellen 6 kuukauden aikana. Toinen 1000 mg:n infuusio laskimoon annettiin 15. tutkimuspäivänä. MabThera-ylläpitohoidon 500 mg:n infuusiot annettiin kuukausina 12 ja 18. Tavanomaisia prednisoniannoksia saamaan satunnaistetun ryhmän keskivaikeaa tautia sairastavat potilaat saivat aluksi suun kautta otettavaa prednisonia annoksina 1 mg/kg/vrk, ja hoito lopetettiin vähitellen 12 kuukauden aikana, ja vaikea-asteista tautia sairastavat potilaat saivat suun kautta otettavaa prednisonia annoksina 1,5 mg/kg/vrk, ja hoito lopetettiin vähitellen 18 kuukauden aikana. MabThera-ryhmässä potilaille, joille ilmeni relapsi, voitiin antaa 1000 mg:n MabThera-lisäinfuusio yhdistelmänä uudelleen aloitetun prednisonihoidon tai suurennettujen prednisoniannosten kanssa. Ylläpitohoitoon tai relapsin yhteydessä annetut infuusiot annettiin aikaisintaan 16 viikon kuluttua edellisestä infuusiosta.
Tutkimuksen ensisijainen tavoite oli täydellinen remissio (täydellinen epiteelin muodostuminen eikä uusia ja/tai vakiintuneita leesioita) kuukautena 24 ilman prednisonihoitoa vähintään kahteen kuukauteen (CRoff ≥ 2 kuukautta).
PV-tutkimuksen 1 tulokset
Tutkimuksessa todettiin tilastollisesti merkitseviä tuloksia MabTheran ja pienten prednisoniannosten käytössä verrattuna tavanomaisiin prednisoniannoksiin, kun tavallista pemfigusta sairastavien potilaiden täydellistä remissiota ilman prednisonihoitoa vähintään kahteen kuukauteen (CRoff ≥ 2 kuukautta) tarkasteltiin kuukautena 24 (ks. taulukko 22).
Taulukko 22 Niiden tavallista pemfigusta sairastavien potilaiden prosenttiosuus kuukautena 24, jotka saavuttivat täydellisen remission ilman kortikosteroidihoitoa vähintään kahteen kuukauteen (hoitoaikeen mukainen potilasjoukko, ITT – tavallinen pemfigus)
Rituksimabi + prednisoni N = 38 | Prednisoni N = 36 | p-arvo a | CI 95 %b | |
Vasteen saaneiden lukumäärä (vasteosuus [%]) | 34 (89,5 %) | 10 (27,8 %) | < 0,0001 | 61,7 % (38,4, 76,5) |
| ap-arvo saatu Fisherin eksaktilla testillä, jossa mid-p-arvo on korjattu b 95 %:n luottamusväli on korjattu Newcomben menetelmällä laskettu luottamusväli | ||||
Niiden potilaiden lukumäärä, jotka saivat rituksimabin ja pienten prednisoniannosten yhdistelmää ja olivat ilman prednisonihoitoa tai saivat vain minimaalista hoitoa (prednisoniannos 10 mg tai vähemmän vuorokaudessa) verrattuna tavanomaisia prednisoniannoksia saaneisiin potilaisiin 24 kuukauden hoitojakson aikana, osoittaa MabTheran steroideja säästävän tehon (kuva 4).
Kuva 4: Niiden potilaiden lukumäärä ajan mittaan, jotka eivät käyttäneet kortikosteroideja tai käyttivät kortikosteroideja minimaalisesti (≤ 10 mg/vrk)
Laboratorioarvojen retrospektiivinen post-hoc-arvio
Yhteensä 34 tavallista pemfigusta sairastavasta potilaasta 19 potilaan (56 %) lääkevasta-ainetesti oli positiivinen 18 kuukauteen mennessä. Tavallista pemfigusta sairastaville MabThera-hoitoa saaville potilaille muodostuvien lääkevasta-aineiden kliininen merkitys on epäselvä.
PV-tutkimus 2 (tutkimus WA29330)
Satunnaistetussa, kaksoissokkoutetussa, kaksoislumevalmisteella toteutetussa ja vaikuttavaan aineeseen vertailevassa monikeskustutkimuksessa MabTheran tehoa ja turvallisuutta mykofenolaattimofetiiliin (MMF) verrattuna arvioitiin potilailla, jotka sairastivat kohtalaista tai vaikeaa tavallista pemfigusta. Tutkimuksen alussa potilaat saivat suun kautta 60–120 mg:n suuruisen vuorokausiannoksen prednisonia tai vastaavaa (1.0–1,5 mg/kg/vrk), joka vähennettiin annostasolle 60 tai 80 mg/vrk:ssa päivään 1 mennessä. Potilailla oli diagnostisoitu tavallinen pemfigus edeltävien 24 kuukauden aikana ja heidän tautinsa oli havaittu olevan kohtalainen tai vaikea (määritelty taudin aktiivisuutta osoittavaksi PDAI-kokonaispistemääräksi ≥ 15 [PDAI = Pemphigus Disease Area Index]).
Satakolmekymmentäviisi potilasta satunnaistettiin MabThera 1000 mg ‑hoitoon, jota annettiin päivänä 1, päivänä 15, viikolla 24 ja viikolla 26, tai suun kautta annettavaan hoitoon mykofenolaattimofetiiliannoksilla 2 g/vrk 52 viikon ajan yhdistelmänä suun kautta otettavien 60 mg:n tai 80 mg:n prednisoniannosten kanssa; prednisonihoidossa oli tavoitteena pienentää annos tasolle 0 mg/vrk viikkoon 24 mennessä.
Tässä tutkimuksessa ensisijainen tehoa koskeva tavoite oli arvioida viikolla 52 MabTheran tehoa mykofenolaattimofetiiliin verrattuna pitkäkestoisen täydellisen remission saavuttamisessa. Pitkäkestoiseksi täydelliseksi remissioksi määriteltiin leesioiden paraneminen ilman uusia aktiivisia leesioita (eli taudin aktiivisuutta osoittava PDAI-pistemäärä 0) prednisoniannosten ollessa 0 mg/vrk tai vastaavalla hoidolla ja tämän vasteen säilyminen 52 viikon hoitojakson aikana vähintään 16 peräkkäisen viikon ajan.
PV-tutkimuksen 2 tulokset
Tutkimus osoitti MabThera-hoidon paremmuuden mykofenolaattimofetiiliin ja suun kautta pienenevinä annoksina otettavien kortikosteroidien yhdistelmään verrattuna. Mittarina käytettiin tavallista pemfigusta sairastavien potilaiden viikolla 52 saavutettu pitkäkestoinen täydellinen remissio ilman kortikosteroidihoitoa ≥ 16 viikon ajaksi (taulukko 23). Valtaosa potilaista modifioidussa hoitoaikeen mukaisessa potilasjoukossa oli saanut diagnoosin äskettäin (74 %), ja 26 %:lla potilaista tauti oli vakiintunut (sairauden kesto ≥ 6 kuukautta, ja aiemmin hoidettu tavallinen pemfigus).
Taulukko 23 Niiden tavallista pemfigusta sairastavien potilaiden prosenttiosuus, jotka olivat viikolla 52 saavuttaneet pitkäkestoisen täydellisen remission ilman kortikosteroidihoitoa vähintään 16 viikon ajaksi (modifioitu hoitoaikeen mukainen potilasjoukko)
| MabThera (N = 62) | MMF (N = 63) | Ero (95 %:n luottamusväli) | p-arvo | |
| Vasteen saaneiden lukumäärä (vasteosuus [%]) | 25 (40,3 %) | 6 (9,5 %) | 30,80 % (14,70 %; 45,15 %) | < 0,0001 |
| Äskettäin diagnoosin saaneet potilaat | 19 (39,6 %) | 4 (9,1 %) | ||
| Potilaat, joilla vakiintunut tauti | 6 (42,9 %) | 2 (10,5 %) | ||
| MMF = mykofenolaattimofetiili. Äskettäin diagnoosin saaneet potilaat = sairauden kesto < 6 kuukautta, ei aiemmin hoidettu tavallinen pemfigus. Potilaat, joilla vakiintunut tauti = sairauden kesto ≥ 6 kuukautta, ja aiemmin hoidettu tavallinen pemfigus. P-arvo perustuu Cochran-Mantel-Haenszelin testiin. | ||||
Kaikkien toissijaisten parametrien (mukaan lukien kumulatiivinen kortikosteroidiannos suun kautta, sairauden pahenemisvaiheiden kokonaislukumäärä ja muutos terveyteen liittyvässä elämänlaadussa DLQI-indeksillä [Dermatology Life Quality Index] mitattuna) analyysi vahvisti MabTheran tilastollisesti merkitsevät tulokset mykofenolaattimofetiiliin verrattuna. Toissijaisia päätetapahtumia testattiin ja tarkastettiin monimuotoisuuden varalta.
Altistus glukokortikoideille
Suun kautta otettu kortikosteroidiannos oli huomattavasti pienempi MabThera-hoitoa saaneilla potilailla. Kumulatiivisen prednisoniannoksen mediaani (min, max) viikolla 52 oli MabThera-ryhmässä 2775 mg (450; 22180) ja mykofenolaattimofetiiliryhmässä 4005 mg (900; 19920) (p = 0,0005).
Taudin pahenemisvaihe
Taudin pahenemisvaiheiden kokonaislukumäärä oli huomattavasti pienempi MabThera-hoitoa saaneilla kuin mykofenolaattimofetiilia saaneilla (6 vs. 44, p < 0,0001), ja potilaita, joilla oli vähintään yksi pahenemisvaihe, oli vähemmän (8,1 % vs. 41,3 %).
Laboratoriotulosten arviot
Yhteensä 63:sta MabTheraa tavallisen pemfiguksen hoitoon saaneesta potilaasta 20 potilaalla (31,7 %) todettiin viikkoon 52 mennessä lääkevasta-aineita (ADA-positiivinen) (19:llä aiheutunut hoidosta ja 1:llä pahentunut hoidosta). Lääkevasta-aineista ei todettu PV-tutkimuksessa 2 selkeää haitallista vaikutusta turvallisuuteen tai tehoon.
Farmakokinetiikka
Aikuisten non-Hodgkin-lymfooma
Populaatiofarmakokineettinen analyysi tehtiin 298:lla non-Hodgkin-lymfoomaa sairastavalla potilaalla, jotka olivat saaneet MabTheraa kerta- tai toistoinfuusiona joko yksinään tai CHOP-hoitoon yhdistettynä (käytetyt MabThera-annokset vaihtelivat 100–500 mg/m2 välillä). Analyysin perusteella populaation tyypillinen arvioitu ei-spesifinen puhdistuma (CL1) oli 0,14 l/vuorokausi. Arvioitu spesifinen puhdistuma (CL2), johon B-solut ja tuumorin koko todennäköisesti vaikuttavat, oli 0,59 l/vuorokausi, kun taas arvioitu sentraalinen jakautumistilavuus (V1) oli 2,7 l. MabTheran eliminaation terminaalinen puoliintumisaika oli 22 vuorokautta (arvioitu mediaani), vaihteluväli oli 6,1–52 vuorokautta. CD19-positiivisten solujen määrä lähtötasolla ja mitattavissa olevien tuumorileesioiden koko vaikuttivat osittain MabTheran CL2-arvon vaihtelevuuteen aineistossa, jossa 161 potilaalle annettiin MabTheraa 375 mg/m2 laskimonsisäisenä infuusiona kerran viikossa neljän viikon ajan. Potilailla, joilla oli isompi määrä CD19-positiivisia soluja tai leesioita (tuumoreita), oli korkeampi CL2-arvo. Arvon yksilölliset vaihtelut jäivät kuitenkin suuriksi, vaikka huomioitiin CD19-positiivisten solujen määrä ja leesioiden koko. V1-arvo vaihteli ihon pinta-alan ja CHOP-hoidon mukaan. Tämä V1-arvon vaihtelu (27,1 % ja 19,0 %) oli melko pientä, ja siihen vaikuttivat sekä ihon pinta-ala (1,53–2,32 m2) että samanaikainen CHOP-hoito. Ikä, sukupuoli, rotu tai WHO:n suorituskykyluokka eivät vaikuttaneet MabTheran farmakokinetiikkaan. Tämän analyysin perusteella voidaan olettaa, että MabThera-annoksen muuttaminen mitattujen kovariaattien mukaan ei vähennä merkitsevästi vaihtelua MabTheran farmakokineettisissä parametreissa.
MabTheraa annettiin laskimonsisäisenä infuusiona annoksella 375 mg/m2 kerran viikossa neljän viikon ajan 203:lle non-Hodgkin-lymfoomaa sairastavalle potilaalle, jotka eivät aikaisemmin olleet saanet MabThera-hoitoa. Keskimääräinen Cmax neljännen infuusion jälkeen oli 486 µg/ml (vaihteluväli 77,5–996,6 µg/ml). Rituksimabia oli osoitettavissa potilaiden seerumissa noin 3–6 kuukauden ajan viimeisen hoidon jälkeen.
MabTheraa annettiin annoksella 375 mg/m2 laskimonsisäisenä infuusiona kerran viikossa kahdeksan viikon ajan 37:lle non-Hodgkin-lymfoomaa sairastavalle potilaalle. Keskimääräinen Cmax nousi jokaisen peräkkäisen infuusion jälkeen. Ensimmäisen infuusion jälkeen Cmax-arvo oli keskimäärin 243 µg/ml (vaihteluväli 16–582 µg/ml) ja kahdeksannen infuusion jälkeen 550 µg/ml (vaihteluväli 171–1177 µg/ml).
MabTheran farmakokineettinen profiili yhdessä CHOP-solunsalpaajahoidon kanssa käytettynä (kuusi MabThera-infuusiota annoksella 375 mg/m2 kuuden CHOP-hoitosyklin ajan) oli samankaltainen kuin pelkällä MabTheralla havaittu farmakokineettinen profiili.
Pediatristen potilaiden diffuusi suurisoluinen B-solulymfooma, Burkittin lymfooma, Burkittin leukemia ja Burkitt-tyyppinen lymfooma
Pediatristen potilaiden diffuusia suurisoluista B-solulymfoomaa, Burkittin lymfoomaa, Burkittin leukemiaa ja Burkitt-tyyppistä lymfoomaa koskeneessa kliinisessä tutkimuksessa tutkittiin farmakokinetiikkaa 35:n vähintään 3-vuotiaan potilaan alaryhmässä. Farmakokinetiikka oli verrannollinen kahdessa ikäryhmässä (≥ 3 – < 12-vuotiaat vs. ≥ 12 – < 18-vuotiaat). Kummassakin kahdesta induktiosyklistä (hoitosyklit 1 ja 2) annettiin kaksi MabThera-infuusiota annoksina 375 mg/m2 laskimoon, minkä jälkeen kummassakin konsolidaatiosyklissä (hoitosyklit 3 ja 4) annettiin yksi MabThera-infuusio annoksena 375 mg/m2 laskimoon; tämän jälkeen maksimipitoisuus oli suurimmillaan neljännen infuusion (hoitosykli 2) jälkeen, jolloin geometrinen keskiarvo oli 347 µg/ml. Sen jälkeen maksimipitoisuuden geometrinen keskiarvo oli pienempi (hoitosykli 4: 247 μg/ml). Pienimmät pitoisuudet säilyivät tällä annostuksella (geometriset keskiarvot: 41,8 µg/ml [ennen hoitosyklin 2 annosta; yhden hoitosyklin jälkeen], 67,7 µg/ml [ennen hoitosyklin 3 annosta, kahden hoitosyklin jälkeen] ja 58,5 µg/ml [ennen hoitosyklin 4 annosta, kolmen hoitosyklin jälkeen]). Eliminaation puoliintumisajan mediaani iältään vähintään 3-vuotiailla pediatrisilla potilailla oli 26 päivää.
MabTheran farmakokineettiset ominaisuudet diffuusia suurisoluista B-solulymfoomaa, Burkittin lymfoomaa, Burkittin leukemiaa tai Burkitt-tyyppistä lymfoomaa sairastavilla pediatrisilla potilailla olivat samankaltaiset kuin aikuisilla non-Hodgkin-lymfoomaa sairastavilla potilailla on havaittu.
Farmakokineettisiä tietoja ei ole käytettävissä ikäryhmässä ≥ 6 kuukautta – < 3-vuotiaat, mutta farmakokineettinen ennuste tukee sitä, että altistus (AUC, Ctrough) tässä ikäryhmässä on verrattavissa ≥ 3-vuotiaiden ryhmään (taulukko 24). Kasvaimen pienempi koko lähtötilanteessa yhdistetään suurempaan altistukseen pienemmän aikariippuvaisen puhdistuman takia. Kasvaimen koon vaikutus systeemiseen altistukseen pysyy kuitenkin alueella, jossa altistus oli tehokas ja turvallisuusprofiili hyväksyttävä.
Taulukko 24: Farmakokineettisen ennusteen muuttujat rituksimabi-hoito-ohjelman jälkeen diffuusia suurisoluista B-solulymfoomaa, Burkittin lymfoomaa, Burkittin leukemiaa tai Burkitt-tyyppistä lymfoomaa sairastavilla pediatrisilla potilailla
Ikäryhmä | ≥ 6 kuukautta – < 3-vuotiaat | ≥ 3 – < 12-vuotiaat | ≥ 12 – < 18-vuotiaat |
Ctrough (µg/ml) | 47,5 (0,01-179) | 51,4 (0,00-182) | 44,1 (0,00-149) |
AUC1-4 syklit (µg*vrk/ml) | 13501 (278-31070) | 11609 (135-31157) | 11467 (110-27066) |
Tulokset esitetään mediaanina (min – max); Ctrough ennen 4. syklin annostusta.
Krooninen lymfaattinen leukemia
KLL-potilaille annettiin MabTheraa yhdistettynä fludarabiiniin ja syklofosfamidiin laskimonsisäisenä infuusiona annoksella 375 mg/m2 ensimmäisessä hoitosyklissä ja nostettiin annokseen 500 mg/m2 jokaisessa seuraavassa viidessä hoitosyklissä. Viidennen 500 mg/m2 -annoksen jälkeen keskimääräinen Cmax (n = 15) oli 408 µg/ml (vaihteluväli 97–764 µg/ml) ja keskimääräinen loppuvaiheen puoliintumisaika 32 vuorokautta (vaihteluväli 14–62 vrk).
Nivelreuma
Kun MabTheraa annettiin kahtena 1000 mg:n infuusiona laskimoon kahden viikon välein, terminaalisen puoliintumisajan keskiarvo oli 20,8 vuorokautta (vaihteluväli 8,58–35,9 vuorokautta), systeemisen puhdistuman keskiarvo oli 0,23 l/vrk (vaihteluväli 0,091–0,67 l/vrk) ja vakaan tilan aikaisen jakautumistilavuuden keskiarvo oli 4,6 l (vaihteluväli 1,7–7,51 l). Samojen tulosten populaatiofarmakokineettisessä analyysissä saatiin vastaavat keskiarvot systeemiselle puhdistumalle (0,26 l/vrk) ja puoliintumisajalle (20,4 vuorokautta). Populaatiofarmakokineettinen analyysi osoitti, että ihon pinta-ala ja sukupuoli olivat merkittävimmät farmakokineettisten parametrien yksilöllisiä eroja selittävät kovariaatit. Kun tulokset korjattiin ihon pinta-alan suhteen, miespuolisilla tutkimushenkilöillä jakautumistilavuus oli suurempi ja puhdistuma oli nopeampi kuin naispuolisilla tutkimushenkilöillä. Sukupuoleen liittyvien farmakokineettisten erojen ei katsota olevan kliinisesti merkittäviä, eivätkä ne vaadi annostuksen muuttamista. Farmakokineettisiä tietoja ei ole käytettävissä maksan tai munuaisten vajaatoimintaa sairastavista potilaista.
Rituksimabin farmakokinetiikkaa arvioitiin neljässä tutkimuksessa päivinä 1 ja 15 kahden laskimonsisäisesti (i.v.) annetun annoksen jälkeen (2 x 500 mg ja 2 x 1000 mg). Kaikissa näissä tutkimuksissa rituksimabin farmakokinetiikka oli annosriippuvainen tutkitulla rajoitetulla annosalueella. Ensimmäisen 2 x 500 mg:n infuusion jälkeen rituksimabin keskimääräinen Cmax-arvo plasmassa vaihteli arvosta 157 µg/ml arvoon 171 µg/ml. Annoksella 2 x 1000 mg vastaava vaihteluväli oli 298–341 µg/ml. Toisen infuusion jälkeen keskimääräinen Cmax-arvo oli 183–198 µg/ml annoksella 2 x 500 mg ja 355–404 µg/ml annoksella 2 x 1000 mg. Keskimääräinen loppuvaiheen puoliintumisaika annoksella 2 x 500 mg oli 15–16 vuorokautta ja annoksella 2 x 1000 mg 17−21 vuorokautta. Keskimääräinen Cmax-arvo oli molemmilla annoksilla 16–19 % korkeampi toisen infuusion jälkeen verrattuna ensimmäiseen infuusioon.
Rituksimabin farmakokinetiikkaa arvioitiin toistohoidon toisen hoitojakson päätyttyä kahden laskimonsisäisesti annetun annoksen jälkeen (2 x 500 mg ja 2 x 1000 mg). Ensimmäisen 2 x 500 mg:n infuusion jälkeen rituksimabin keskimääräinen Cmax-arvo plasmassa vaihteli arvosta 170 µg/ml arvoon 175 µg/ml. Annoksella 2 x 1000 mg vastaava vaihteluväli oli 317–370 µg/ml. Toisen infuusion jälkeen keskimääräinen Cmax-arvo oli 207 µg/ml annoksella 2 x 500 mg ja vaihteli välillä 377−386 µg/ml annoksella 2 x 1000 mg. Keskimääräinen loppuvaiheen eliminaation puoliintumisaika toisen hoitojakson toisen infuusion jälkeen annoksella 2 x 500 mg oli 19 vuorokautta ja annoksella 2 x 1000 mg 21–22 vuorokautta. Rituksimabin farmakokineettiset parametrit olivat vastaavanlaisia kahden hoitojakson aikana.
Populaatiossa, jossa TNF-estäjät eivät olleet tuoneet riittävää hoitovastetta, farmakokineettiset parametrit olivat saman hoito-ohjelman jälkeen (2 x 1000 mg i.v. 2 viikon välein) samanlaiset, ja maksimipitoisuus seerumissa oli 369 μg/ml ja terminaalisen puoliintumisajan keskiarvo oli 19,2 vuorokautta.
Granulomatoottinen polyangiitti (GPA) ja mikroskooppinen polyangiitti (MPA)
Aikuiset potilaat
Analyysi 97 granulomatoottista polyangiittia ja mikroskooppista polyangiittia sairastavan potilaan populaatiofarmakokineettisistä tiedoista, kun potilaat saivat MabTheraa 375 mg/m2 kerran viikossa neljän viikon ajan, osoitti, että arvioitu terminaalisen eliminaation puoliintumisajan mediaani oli 23 vuorokautta (vaihteluväli 9–49 vuorokautta). Rituksimabin keskimääräinen puhdistuma oli 0,313 l/vrk (vaihteluväli 0,116–0,726 l/vrk) ja jakautumistilavuus oli 4,50 l (vaihteluväli 2,25–7,39 l). Ensimmäisten 180 päivän aikana maksimipitoisuus (Cmax) oli 372,6 (252,3–533,5) mikrog/ml, päivänä 180 minimipitoisuus (C180) oli 2,1 (0–29,3) mikrog/ml, ja 180 päivän kumulatiivinen aika-pitoisuuskuvaajan alle jäävä pinta-ala (AUC180) oli 10302 (3653–21874) mikrog/ml*vrk (mediaani [vaihteluväli]). Rituksimabin farmakokineettiset parametrit vaikuttavat olevan granulomatoottista polyangiittia ja mikroskooppista polyangiittia sairastavilla aikuisilla samankaltaiset kuin nivelreumapotilailla on havaittu.
Pediatriset potilaat
Kahdestakymmenestäviidestä granulomatoottista polyangiittia ja mikroskooppista polyangiittia sairastavasta lapsesta (ikä 6–17 vuotta), jotka saivat MabTheraa 375 mg/m2 kerran viikossa neljä annosta, tehtiin populaatiofarmakokineettinen analyysi, ja tämän analyysin perusteella laskennallisen terminaalisen eliminaation puoliintumisajan mediaani oli 22 vuorokautta (vaihteluväli 11–42 vuorokautta). Rituksimabin keskimääräinen puhdistuma oli 0,221 l/vrk (vaihteluväli 0,0996–0,381 l/vrk), ja jakautumistilavuus oli 2,27 l (vaihteluväli 1,43–3,17 l). Ensimmäisten 180 päivän aikana maksimipitoisuus (Cmax) oli 382,8 (270,6–513,6) mikrog/ml, päivänä 180 minimipitoisuus (C180) oli 0,9 (0–17,7) mikrog/ml, ja 180 päivän kumulatiivinen aika-pitoisuuskuvaajan alle jäävä pinta-ala (AUC180) oli 9787 (4838–20446) mikrog/ml*vrk (mediaani [vaihteluväli]). Rituksimabin farmakokineettiset parametrit olivat granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavilla pediatrisilla potilailla samankaltaiset kuin granulomatoottista polyangiittia tai mikroskooppista polyangiittia sairastavilla aikuisilla, kun otetaan huomioon kehon pinta-alan vaikutus puhdistuman ja jakautumistilavuuden parametreihin.
Tavallinen pemfigus
Yhteenveto MabThera-annoksia 1000 mg päivinä 1, 15, 168 ja 182 saavien tavallista pemfigusta sairastavien aikuisten potilaiden farmakokineettisistä parametreista on taulukossa 25.
Taulukko 25Tavallista pemfigusta sairastavien aikuisten potilaiden populaatiofarmakokinetiikka PV-tutkimuksessa 2
Parametri | Infuusiosykli | |
1. hoitosykli (1000 mg) päivänä 1 ja päivänä 15 N = 67 | 2. hoitosykli (1000 mg) päivänä 168 ja päivänä 182 N = 67 | |
Terminaalinen puoliintumisaika (vrk) Mediaani (vaihteluväli) | 21,0 (9,3–36,2) | 26,5 (16,4–42,8) |
Puhdistuma (l/vrk) Keskiarvo (vaihteluväli) | 391 (159–1510) | 247 (128–454) |
Keskusjakautumistilavuus (l) Keskiarvo (vaihteluväli) | 3,52 (2,48–5,22) | 3,52 (2,48–5,22) |
Rituksimabin kahden ensimmäisen antokerran (päivinä 1 ja 15, vastaa 1. hoitosykliä) jälkeen rituksimabin farmakokineettiset parametrit olivat tavallista pemfigusta sairastavilla potilailla samankaltaiset kuin granulomatoottista polyangiittia ja mikroskooppista polyangiittia sairastavilla potilailla ja nivelreumapotilailla. Kahden viimeisen antokerran (päivinä 168 ja 182, vastaa 2. hoitosykliä) jälkeen rituksimabin puhdistuma väheni, kun taas keskusjakautumistilavuus pysyi muuttumattomana.
Prekliiniset tiedot turvallisuudesta
Rituksimabi on osoittautunut erittäin spesifiseksi B-solujen CD20-antigeeniä kohtaan. Ainoa Cynomolgus-apinoilla suoritetuissa toksisuustutkimuksissa ilmennyt vaikutus oli odotettu farmakologinen vaikutus, joka näkyi perifeerisen veren ja lymfoidisen kudoksen B-solujen määrän vähenemisenä.
Alkion-/sikiönkehitykseen kohdistuvaa toksisista vaikutusta on tutkittu Cynomolgus-apinoilla, joille annettiin enintään 100 mg/kg (20.–50. tiineyspäivänä). Tutkimuksissa ei havaittu viitteitä rituksimabin sikiötoksisuudesta. Sikiöiden lymfoidisissa elimissä havaittiin kuitenkin annoksesta riippuvaa farmakologista B-solujen vähenemistä, mikä jatkui vielä syntymän jälkeen, ja siihen liittyi myös vastasyntyneiden eläinten IgG-pitoisuuden pieneneminen. Eläinten B-soluarvot palautuivat normaalitasolle 6 kuukauden kuluessa syntymän jälkeen, eikä tämä heikentänyt rokotusvastetta.
Mutageenisuuden selvittämiseksi ei ole suoritettu standarditutkimuksia, sillä ne eivät ole tälle molekyylille merkityksellisiä. Rituksimabin karsinogeenisen potentiaalin selvittämiseksi ei ole suoritettu pitkäaikaisia tutkimuksia eläimillä.
Rituksimabin hedelmällisyyteen kohdistuvien vaikutusten selvittämiseksi ei ole tehty spesifisiä tutkimuksia. Yleistä toksisuutta selvittäneissä tutkimuksissa cynomolgus-apinoilla ei havaittu haitallisia vaikutuksia urosten tai naaraiden lisääntymiselimiin.
Farmaseuttiset tiedot
Apuaineet
Natriumsitraatti (E331)
Polysorbaatti 80 (E433)
Natriumkloridi
Natriumhydroksidi (pH:n säätöön) (E524)
Kloorivetyhappo (pH:n säätöön) (E507)
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
MabTheran ja polyvinyylikloridi- tai polyetyleenimuovipussien tai infuusiovälineiden välillä ei ole havaittu yhteensopimattomuutta.
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kestoaika
Avaamaton injektiopullo
3 vuotta
Laimennettu lääkevalmiste
-
Natriumkloridiliuoksella tehdyn aseptisen laimentamisen jälkeen
0,9-prosenttisella natriumkloridiliuoksella laimennettu MabThera-infuusioneste on fysikaalisesti ja kemiallisesti stabiili 30 vuorokautta +2 °C−+8 °C:ssa ja sen jälkeen vielä 24 tuntia ≤ 30 °C:ssa. -
D-glukoosiliuoksella tehdyn aseptisen laimentamisen jälkeen
5-prosenttisella D-glukoosiliuoksella laimennettu MabThera-infuuusioneste on fysikaalisesti ja kemiallisesti stabiili 24 tuntia +2 °C−+8 °C:ssa ja sen jälkeen vielä 12 tuntia huoneenlämmössä.
Mikrobiologiselta kannalta katsottuna laimennettu infuusioneste tulisi käyttää heti. Mikäli infuusionestettä ei käytetä heti, säilytysajat ja -olosuhteet ennen käyttöä ovat käyttäjän vastuulla eikä niiden normaalisti tulisi ylittää 24 tuntia +2 °C–+8 °C:ssa, ellei laimentamista ole tehty kontrolloiduissa ja validoiduissa, aseptisissa olosuhteissa.
Säilytys
Säilytä jääkaapissa (2 °C–8 °C). Ei saa jäätyä. Pidä injektiopullo ulkopakkauksessa. Herkkä valolle.
Laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
MABTHERA infuusiokonsentraatti, liuosta varten
100 mg (L:ei) 2 x 10 ml (824,86 €)
500 mg (L:ei) 50 ml (1987,21 €)
PF-selosteen tieto
MabThera 100 mg infuusiokonsentraatti, liuosta varten
10 ml:n injektiopullo (väritöntä tyypin I borosilikaattilasia, fluoriresiinilaminaattikumitulppa ja alumiinisinetti sekä punainen muovinen irti napsautettava [flip-off] korkki) sisältää rituksimabia 100 mg. Pakkauksessa on 2 injektiopulloa.
MabThera 500 mg infuusiokonsentraatti, liuosta varten
50 ml:n injektiopullo (väritöntä tyypin I borosilikaattilasia, fluoriresiinilaminaattikumitulppa ja alumiinisinetti sekä harmaa muovinen irti napsautettava [flip-off] korkki) sisältää rituksimabia 500 mg. Pakkauksessa on 1 injektiopullo.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas tai opaalinhohtoinen, väritön tai vaaleankeltainen neste, jonka pH on 6,2–6,8 ja osmolaliteetti on 324–396 mOsmol/kg.
Käyttö- ja käsittelyohjeet
MabThera on pakattu steriileihin infuusiokonsentraattipulloihin, jotka on tarkoitettu kerta-annoskäyttöön. MabThera on pyrogeeniton eikä siinä ole säilytysaineita.
Käytä steriiliä neulaa ja ruiskua MabTheran käyttökuntoon saattamista varten. Tarvittava MabThera-annos vedetään aseptisesti infuusiokonsentraattipullosta ja laimennetaan laskettuun rituksimabikonsentraatioon 1–4 mg/ml lisäämällä annos infuusiopussiin, jossa on steriiliä, pyrogeenitonta 0,9-prosenttista natriumkloridi- tai 5-prosenttista D-glukoosi-infuusionestettä. Sekoitetaan varovaisesti kääntämällä pussia ylösalaisin. Näin menetellen vaahtoaminen voidaan välttää. Laimennettujen infuusionesteiden steriiliys on varmistettava. Aseptinen työskentely on välttämätöntä, sillä MabThera ei sisällä mitään antimikrobista säilytysainetta eikä bakteriostaattista ainetta. Parenteraalisia lääkevalmisteita on tarkastettava visuaalisesti ennen käyttöönottoa mahdollisten hiukkasten esiintymisen tai liuosten värjääntymisen varalta.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
MABTHERA infuusiokonsentraatti, liuosta varten
100 mg 2 x 10 ml
500 mg 50 ml
- Ei korvausta.
- Oikeus käyttää samaa biologista lääkettä (sama kauppanimi) 6 kk ajan.
ATC-koodi
L01FA01
Valmisteyhteenvedon muuttamispäivämäärä
12.02.2026
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com