
PRAXBIND injektio/infuusioneste, liuos 2,5 g/50 ml
Vaikuttavat aineet ja niiden määrät
Yksi millilitra injektio-/infuusionestettä sisältää 50 mg idarusitsumabia.
Yksi injektiopullo sisältää 2,5 g idarusitsumabia 50 ml:ssa.
Idarusitsumabi on valmistettu yhdistelmä-DNA‑tekniikalla kiinanhamsterin munasarjasoluissa.
Apuaineet, joiden vaikutus tunnetaan
Yksi injektiopullo sisältää 2 g sorbitolia ja 25 mg natriumia 50 ml:ssa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektio-/infuusioneste, liuos
Kliiniset tiedot
Käyttöaiheet
Praxbind on dabigatraanin spesifinen vastalääke ja se on tarkoitettu dabigatraanieteksilaattihoitoa saaneille aikuispotilaille, kun sen antikoagulaatiovaikutukset on kumottava nopeasti:
- hätäleikkauksia / kiireellisiä toimenpiteitä varten
- henkeä uhkaavan tai hallitsemattoman verenvuodon yhteydessä.
Ehto
Vain sairaalakäyttöön.
Annostus ja antotapa
Vain sairaalakäyttöön.
Annostus
Suositeltu annos on 5 g idarusitsumabia (kaksi 2,5 g:n / 50 ml:n injektiopulloa).
Sitoutumattoman dabigatraanin esiintymistä uudelleen plasmassa ja siitä johtuvaa hyytymisaikojen pidentymistä on esiintynyt eräässä potilasalaryhmässä enintään 24 tuntia idarusitsumabin annostelun jälkeen (ks. kohta Farmakodynamiikka).
Toisen 5 g:n idarusitsumabi‑annoksen annostelua voidaan harkita seuraavissa tilanteissa:
- kliinisesti merkittävää verenvuotoa esiintyy uudelleen, ja siihen liittyy hyytymisaikojen pitenemistä, tai
- jos mahdollisesti uusiutuva verenvuoto olisi henkeä uhkaavaa ja havaitaan pidentyneitä hyytymisaikoja, tai
- potilaat tarvitsevat toisen hätäleikkauksen / kiireellisen toimenpiteen, ja hyytymisajat ovat pidentyneet.
Asiaankuuluvia hyytymisparametreja ovat aktivoitu partiaalinen tromboplastiiniaika (APTT), laimennettu trombiiniaika (dTT) tai ekariiniaktivoitu hyytymisaika (ECT) (ks. kohta Farmakodynamiikka).
Suurinta vuorokausiannosta ei ole tutkittu.
Antitromboottisen hoidon aloittaminen uudelleen
Dabigatraanieteksilaattihoito voidaan aloittaa uudelleen 24 tuntia idarusitsumabin annostelun jälkeen, jos potilaan kliininen tila on vakaa ja riittävä hemostaasi on saavutettu.
Idarusitsumabin annostelun jälkeen muu antitromboottinen hoito (esim. pienimolekyylinen hepariini) voidaan aloittaa milloin tahansa, jos potilaan kliininen tila on vakaa ja riittävä hemostaasi on saavutettu.
Antitromboottisen hoidon puuttuminen altistaa potilaan perussairaudesta tai -tilasta johtuvalle tromboosiriskille.
Erityisryhmät
Iäkkäät
Annoksen muuttaminen iäkkäillä, 65 vuotta täyttäneillä ja sitä vanhemmilla potilailla ei ole tarpeen (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoimintapotilaat
Annoksen muuttaminen munuaisten vajaatoimintapotilailla ei ole tarpeen. Munuaisten vajaatoiminta ei vaikuttanut idarusitsumabin kumoavaan vaikutukseen (ks. kohta Farmakokinetiikka).
Maksan vajaatoimintapotilaat
Annoksen muuttaminen potilailla, joilla on maksavaurio, ei ole tarpeen (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Praxbind‑valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Saatavissa oleva tieto on kuvattu kohdassa Farmakodynamiikka.
Antotapa
Laskimoon.
Praxbind (kaksi 2,5 g:n / 50 ml:n injektiopulloa) annostellaan laskimoon kahtena peräkkäisenä infuusiona, kumpikin 5‑10 minuutin aikana, tai bolusinjektiona.
Muut käyttö- ja käsittelyohjeet, ks. kohta Käyttö- ja käsittelyohjeet.
Vasta-aiheet
Ei ole.
Varoitukset ja käyttöön liittyvät varotoimet
Idarusitsumabi sitoutuu spesifisesti dabigatraaniin ja kumoaa sen antikoagulaatiovaikutuksen. Se ei kumoa muiden antikoagulanttien vaikutuksia (ks. kohta Farmakodynamiikka).
Praxbind‑hoitoa voidaan käyttää yhdessä tavanomaisten tukitoimien kanssa, joita on harkittava, jos tämä on lääketieteellisesti asianmukaista.
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Yliherkkyys
Praxbind‑valmisteen käyttöön liittyvää riskiä potilailla, joiden tiedetään olevan yliherkkiä (esim. anafylaktoidinen reaktio) idarusitsumabille tai jollekin valmisteen apuaineelle, on punnittava tarkasti suhteessa tällaisen hätätoimenpiteen mahdolliseen hyötyyn. Jos potilas saa anafylaktisen reaktion tai muun vakavan allergisen reaktion, Praxbind‑valmisteen annostelu on keskeytettävä välittömästi ja asianmukainen hoito aloitettava.
Perinnöllinen fruktoosi‑intoleranssi
Suositeltu Praxbind‑annos sisältää apuaineena 4 g sorbitolia. Perinnöllistä fruktoosi‑intoleranssia sairastavilla potilailla sorbitolin parenteraaliseen annosteluun on raportoitu liittyneen hypoglykemiaa, hypofosfatemiaa, metabolista asidoosia, virtsahappopitoisuuden nousua, akuuttia maksan vajaatoimintaa, jossa sekä eritys- että synteesitoiminta lakkaa, sekä kuolemantapauksia. Siksi Praxbind‑annosteluun perinnöllistä fruktoosi‑intoleranssia sairastaville potilaille liittyvää riskiä on punnittava tarkasti suhteessa tällaisen hätätoimenpiteen mahdolliseen hyötyyn. Jos näille potilaille annetaan Praxbind‑valmistetta, tarvitaan tehostettua hoitoa Praxbind‑altistuksen aikana ja 24 tuntia sen jälkeen.
Tromboemboliset tapahtumat
Dabigatraanihoitoa saavilla potilailla on perussairauksia, jotka altistavat heidät tromboembolisille tapahtumille. Dabigatraanihoidon kumoaminen altistaa potilaan hänen perussairaudestaan johtuvalle tromboosiriskille. Tämän riskin vähentämiseksi antikoagulaatiohoidon jatkamista on harkittava niin pian kuin se on lääketieteellisesti mahdollista (ks. kohta Annostus ja antotapa).
Virtsan valkuaisaineiden testaus
Praxbind aiheuttaa ohimenevää proteinuriaa. Se on fysiologinen reaktio suuresta proteiinimäärästä munuaisissa sen jälkeen, kun on annettu 5 g idarusitsumabia laskimoon boluksena tai lyhytkestoisena infuusiona (ks. kohta Farmakokinetiikka). Virtsakokeita otettaessa on huomioitava, että ohimenevä proteinuria ei merkitse munuaisvauriota.
Natriumpitoisuus
Tämä lääkevalmiste sisältää 50 mg natriumia per annos, joka vastaa 2,5 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Yhteisvaikutukset
Muodollisia yhteisvaikutustutkimuksia Praxbind‑valmisteella ja muilla lääkevalmisteilla ei ole tehty. Farmakokineettisten ominaisuuksien ja hyvin spesifisen dabigatraaniin sitoutumisen perusteella kliinisesti merkittäviä yhteisvaikutuksia muiden lääkevalmisteiden kanssa pidetään epätodennäköisinä.
Prekliinisissä tutkimuksissa idarusitsumabilla ei ole havaittu yhteisvaikutuksia
- plasman laajentajien kanssa.
- hyytymistekijöitä sisältävien konsentraattien, kuten protrombiinikompleksikonsentraattien (PCC-valmisteet, esim. faktori 3 ja faktori 4), aktivoitujen protrombiinikompleksikonsentraattien (aPCC-valmisteet) ja rekombinantin tekijän VIIa kanssa.
- muiden antikoagulanttien (esim. muiden trombiinin estäjien kuin dabigatraanin, hyytymistekijä Xa:n estäjien, mukaan lukien pienimolekyylinen hepariini, K‑vitamiiniantagonistit, hepariini) kanssa. Idarusitsumabi ei siis kumoa muiden antikoagulanttien vaikutuksia.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja idarusitsumabin käytöstä raskaana oleville naisille. Lääkevalmisteen luonteesta ja kliinisistä käyttöaiheista johtuen lisääntymis- ja kehitystoksisuustutkimuksia ei ole tehty. Praxbind‑valmistetta voi käyttää raskauden aikana, jos odotettavissa oleva kliininen hyöty on suurempi kuin mahdolliset riskit.
Imetys
Ei tiedetä, erittyvätkö idarusitsumabi/metaboliitit ihmisen rintamaitoon.
Hedelmällisyys
Idarusitsumabin vaikutuksesta hedelmällisyyteen ei ole olemassa tietoja (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ei merkityksellinen.
Haittavaikutukset
Praxbind‑valmisteen turvallisuutta on arvioitu vaiheen III tutkimuksessa 503 potilaalla, joilla on ollut hallitsematon verenvuoto tai jotka ovat tarvinneet hätäleikkausta tai -toimenpiteitä Pradaxa (dabigatraanieteksilaatti) -hoidon aikana, sekä myös vaiheen I tutkimuksissa 224 vapaaehtoisella. Lisäksi tosielämän käyttökokemuksia kerättiin maailmanlaajuisessa idarusitsumabin annostelun seurantaohjelmassa 359 potilaasta. Yksi pediatrinen potilas sai hoitoa pediatrisen turvallisuustutkimuksen yhteydessä.
Haittavaikutuksia ei ole todettu.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Idarusitsumabin yliannostuksesta ei ole kliinistä kokemusta.
Korkein terveillä henkilöillä tutkittu idarusitsumabin kerta-annos oli 8 g. Ryhmässä ei ole havaittu turvallisuuteen liittyviä huolenaiheita.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: muut lääkevalmisteet, myrkytysten hoitoon käytettävät lääkeaineet, ATC‑koodi: V03AB37
Vaikutusmekanismi
Idarusitsumabi on dabigatraanin spesifinen vastalääke. Se on humanisoitu monoklonaalinen vasta‑ainefragmentti (Fab), joka sitoutuu dabigatraaniin hyvin voimakkaalla affiniteetilla. Affiniteetti on noin 300 kertaa voimakkaampi kuin dabigatraanin sitoutumisaffiniteetti trombiiniin. Idarusitsumabi‑dabigatraanikompleksille tunnusomaista on sen hyvin nopea muodostuminen ja hyvin hidas hajoaminen, mikä tekee kompleksista erittäin vakaan. Idarusitsumabi sitoutuu voimakkaasti ja spesifisesti dabigatraaniin ja sen metaboliitteihin ja neutraloi niiden antikoagulaatiovaikutuksen.
Farmakodynaamiset vaikutukset
Idarusitsumabin farmakodynamiikkaa dabigatraanieteksilaatin annostelun jälkeen tutkittiin 141:llä tutkimushenkilöllä vaiheen I tutkimuksissa. Tiedot kuuden 45–64-vuotiaan terveen henkilön edustavasta alaryhmästä, jotka saivat 5 g:n annoksen infuusiona laskimoon, esitetään tässä. Dabigatraanialtistuman huippuarvon mediaani oli tutkituilla terveillä henkilöillä samalla alueella kuin 150 mg dabigatraanieteksilaattia kahdesti vuorokaudessa saavilla potilailla.
Idarusitsumabin vaikutus dabigatraanialtistukseen ja dabigatraanin antikoagulaatiovaikutukseen
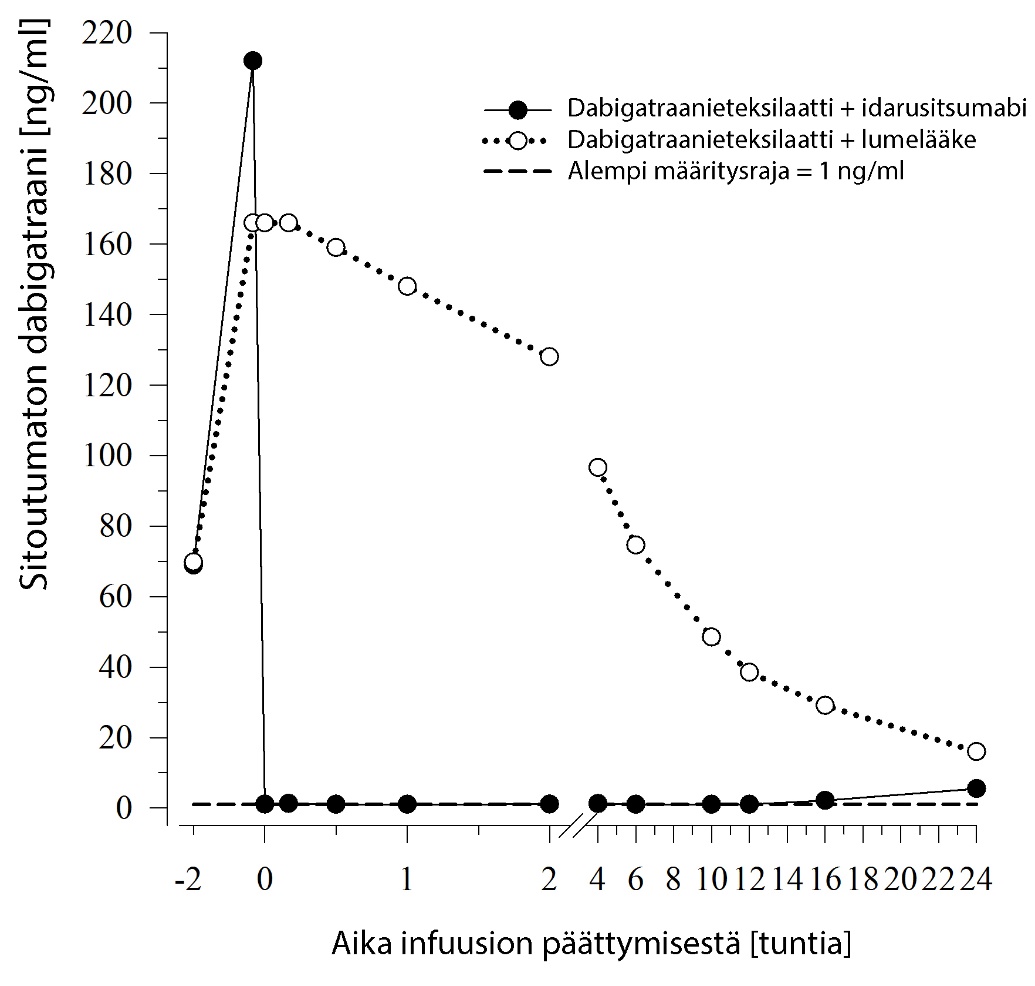
Sitoutumattoman dabigatraanin pitoisuudet plasmassa pienenivät välittömästi idarusitsumabiannostelun jälkeen yli 99 % ja saavuttivat tason, jolla ei enää ole antikoagulaatiovaikutusta.
Suurimmalla osalla potilaista dabigatraanin pitoisuus plasmassa kumoutui 12 tuntiin asti (≥ 90 %). Sitoutumattoman dabigatraanin esiintymistä uudelleen plasmassa ja siitä johtuvaa hyytymisaikojen pidentymistä havaittiin eräässä potilasalaryhmässä, mikä mahdollisesti johtui dabigatraanin uudelleenjakautumisesta perifeerisestä tilasta plasmaan. Tämä tapahtui 1‑24 tuntia idarusitsumabin annostelun jälkeen, pääasiassa aikapisteissä ≥ 12 tuntia.
Kuva 1 – Sitoutumattoman dabigatraanin pitoisuudet plasmassa edustavassa terveiden henkilöiden ryhmässä (idarusitsumabin tai lumelääkkeen annostelu 0 tunnin kohdalla)
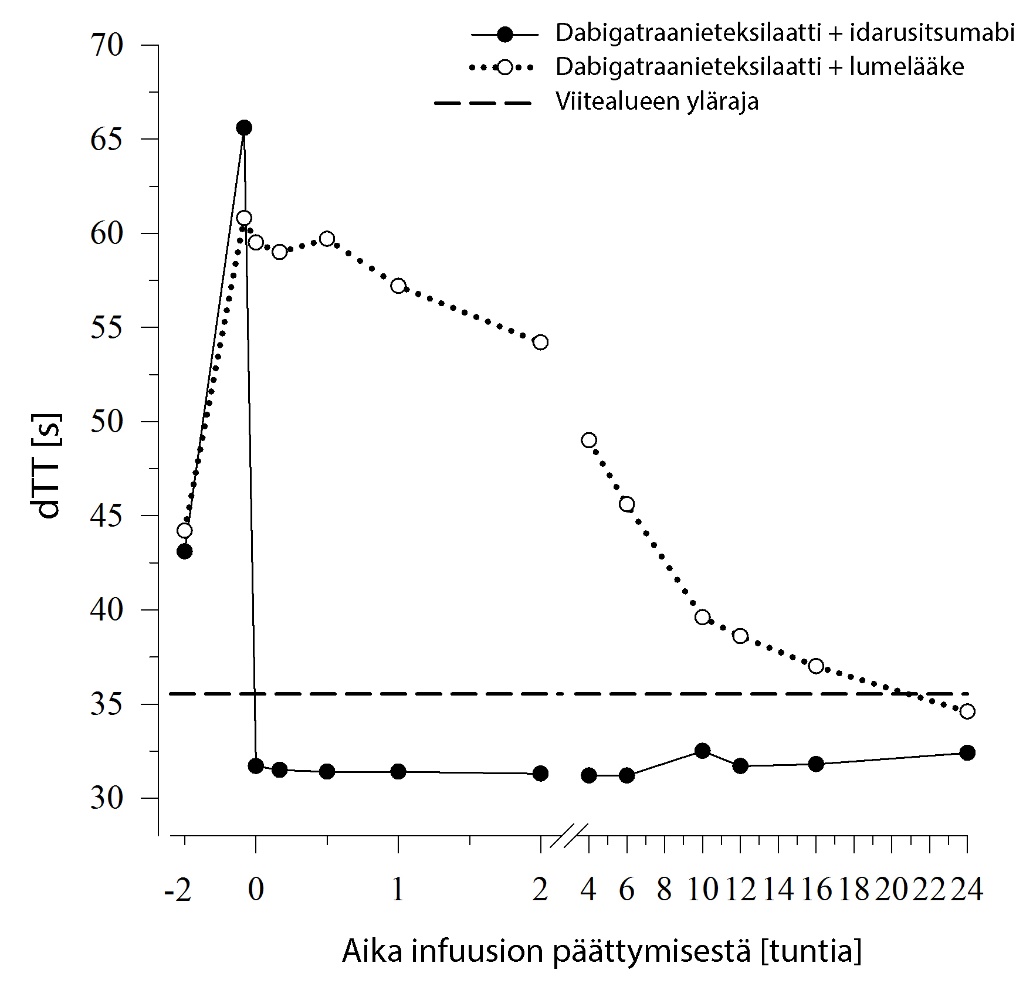
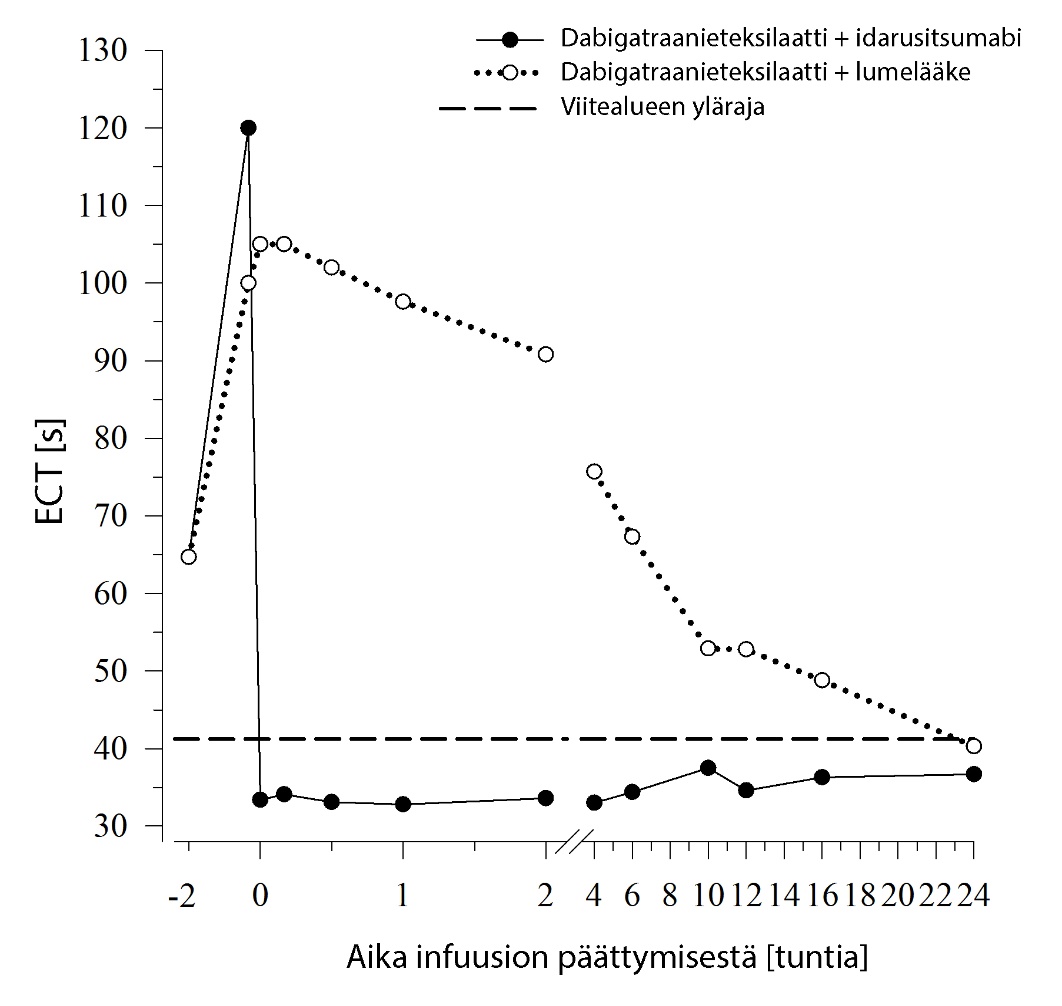
Dabigatraani pidentää koagulaatiota kuvaavien merkkiaineiden, kuten dTT, TT, APTT ja ECT, hyytymisaikaa, mikä osoittaa suunnilleen antikoagulaation voimakkuuden. Idarusitsumabiannostelun jälkeen viitealueelle sijoittuva arvo viittaa siihen, että potilaalla ei ole enää antikoagulaatiovaikutusta. Viitealueen yläpuolelle sijoittuva arvo saattaa olla merkki jäljellä olevasta aktiivisesta dabigatraanista tai muusta kliinisestä tilanteesta, esim. muista vaikuttavista aineista tai verensiirtoon liittyvästä koagulopatiasta. Näitä testejä käytettiin dabigatraanin antikoagulaatiovaikutuksen määrittämiseen. Dabigatraanin aiheuttaman hyytymisajan pitenemisen havaittiin kumoutuvan täysin ja pitkäkestoisesti välittömästi idarusitsumabi‑infuusion jälkeen, ja vaikutus kesti koko havainnointijakson (vähintään 24 tunnin) ajan.
Kuva 2 – Dabigatraanin aiheuttaman hyytymisajan pitenemisen kumoutuminen dTT‑arvolla määritettynä edustavassa terveiden henkilöiden ryhmässä (idarusitsumabin tai lumelääkkeen annostelu 0 tunnin kohdalla)
Kuva 3 – Dabigatraanin aiheuttaman hyytymisajan pitenemisen kumoutuminen ECT‑arvolla määritettynä edustavassa terveiden henkilöiden ryhmässä (idarusitsumabin tai lumelääkkeen annostelu 0 tunnin kohdalla)
Trombiinin muodostumista kuvaavat parametrit
Dabigatraani vaikuttaa voimakkaasti endogeenisen trombiinin potentiaalia (ETP) kuvaaviin parametreihin. Idarusitsumabihoito normalisoi sekä trombiinin muodostumista edeltäneen viiveen että huippuarvon saavuttamiseen kuluneen ajan takaisin lähtötasolle, kun määritys tehtiin 0,5‑12 tuntia idarusitsumabi‑infuusion päättymisestä. Idarusitsumabi yksin ei endogeenisen trombiinin potentiaalin (ETP) perusteella vaikuta hyytymistä edistävästi. Tämä viittaa siihen, että idarusitsumabilla ei ole protromboottista vaikutusta.
Dabigatraanieteksilaatin uudelleenannostelu
Dabigatraanieteksilaatin uudelleenannostelu 24 tuntia idarusitsumabi‑infuusion jälkeen sai aikaan odotetun antikoagulaatiovaikutuksen.
Prekliininen farmakodynamiikka
Vammamallissa sioille aiheutettiin tylppä maksavaurio dabigatraanin annostelun jälkeen, käyttäen annosta, jolla saavutettiin ihmisen plasman pitoisuuksiin nähden noin 10 kertaa terapeuttisia pitoisuuksia suuremmat dabigatraanipitoisuudet. Idarusitsumabi kumosi henkeä uhkaavan verenvuodon tehokkaasti ja nopeasti 15 minuutin kuluessa injektiosta. Yksikään sioista ei kuollut käytettäessä noin 2,5‑5 g:n idarusitsumabiannoksia. Ilman idarusitsumabia kuolleisuus antikoagulanttia saaneessa ryhmässä oli 100 %.
Kliininen teho ja turvallisuus
Idarusitsumabin turvallisuutta, tehoa, siedettävyyttä, farmakokinetiikkaa ja farmakodynamiikkaa tutkittiin kolmessa vaiheen I satunnaistetussa, lumelääkekontrolloidussa kaksoissokkotutkimuksessa, joissa oli mukana 283 henkilöä (224 henkilöä sai idarusitsumabia). Idarusitsumabia annettiin joko yksinään tai dabigatraanieteksilaatin annostelun jälkeen. Tutkimuspopulaatio koostui terveistä henkilöistä ja henkilöistä, jotka täyttivät tietyt populaatiokriteerit, joita olivat ikä, paino, etninen tausta, sukupuoli ja munuaisten vajaatoiminta. Näissä tutkimuksissa idarusitsumabiannos vaihteli 20 mg:sta 8 g:aan ja infuusioaika 5 minuutista 1 tuntiin.
Farmakokinetiikan ja farmakodynamiikan parametreja kuvaavat arvot määritettiin 45–64‑vuotiaiden, 5 g idarusitsumabia saavien terveiden henkilöiden perusteella (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Yhdessä prospektiivisessa, avoimessa, satunnaistamattomassa, kontrolloimattomassa tutkimuksessa (RE‑VERSE AD) hoitoa tutkittiin aikuispotilailla, joilla oli dabigatraaniin liittyvää henkeä uhkaavaa tai hallitsematonta verenvuotoa (ryhmä A) tai jotka tarvitsivat hätäleikkauksen tai kiireellisiä toimenpiteitä (ryhmä B). Ensisijainen päätetapahtuma oli dabigatraanin antikoagulaatiovaikutuksen kumoamisen maksimaalinen prosenttiarvo dTT:n tai ECT:n keskuslaboratoriomäärityksen perusteella 4 tuntia idarusitsumabin annostelun jälkeen. Tärkeä toissijainen päätetapahtuma oli hemostaasin palautuminen.
RE‑VERSE AD‑tutkimuksessa oli mukana 503 potilaan tiedot: 301 vakavan verenvuodon saanutta potilasta (ryhmä A) ja 202 potilasta, jotka tarvitsivat kiireellisen toimenpiteen/leikkauksen (ryhmä B). Noin puolet kummankin ryhmän potilaista oli miehiä. Mediaani‑ikä oli 78 vuotta ja kreatiniinipuhdistuman mediaani 52,6 ml/min. Noin 61,5 % ryhmän A potilaista ja 62,4 % ryhmän B potilaista sai 110 mg dabigatraania kahdesti vuorokaudessa.
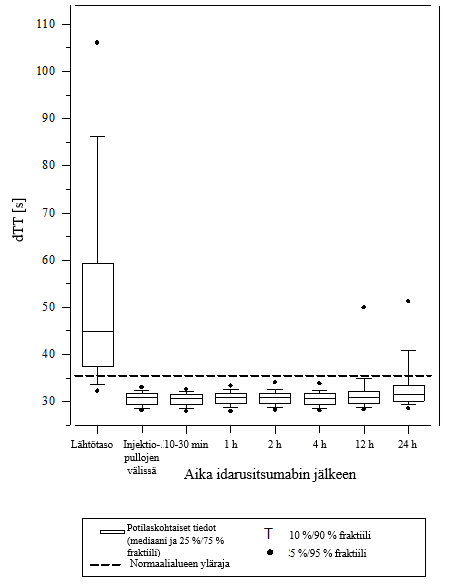
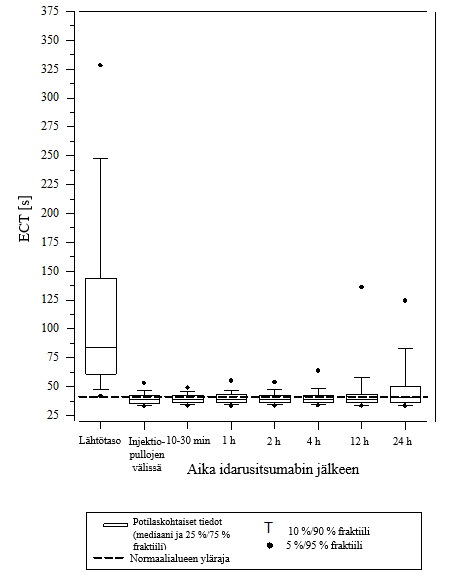
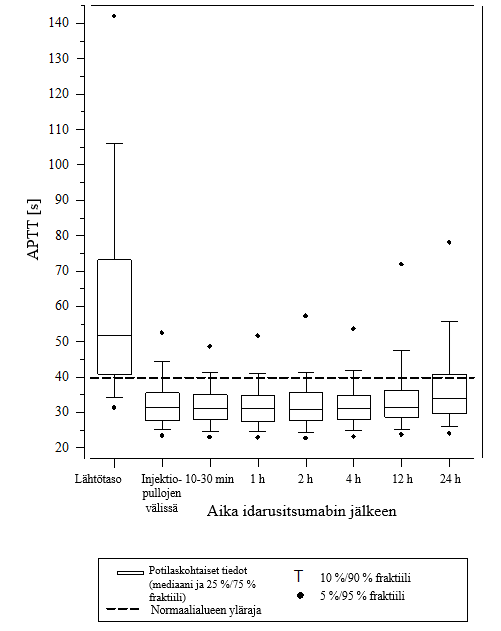
Vaikutuksen kumoamista voitiin arvioida vain potilailla, joiden hyytymisajat olivat pidentyneet ennen idarusitsumabihoitoa. Useimmilla potilailla molemmissa ryhmissä A ja B dabigatraanin antikoagulaatiovaikutus kumoutui (dTT: 98,7 %; ECT: 82,2 %; APTT: 92,5 % arviointikelpoisista potilaista) kokonaan ensimmäisten 4 tunnin aikana 5 g idarusitsumabin annostelun jälkeen. Kumoavat vaikutukset olivat ilmeisiä heti annostelun jälkeen.
Kuva 4 – Dabigatraanin aiheuttaman hyytymisajan pitenemisen kumoutuminen dTT‑arvolla määritettynä RE‑VERSE AD‑tutkimuksessa mukana olleilla potilailla (N = 487)
Kuva 5 – Dabigatraanin aiheuttaman hyytymisajan pitenemisen kumoutuminen ECT‑arvolla määritettynä RE‑VERSE AD‑tutkimuksessa mukana olleilla potilailla (N = 487)
Kuva 6 – Dabigatraanin aiheuttaman hyytymisajan pitenemisen kumoutuminen APTT‑arvolla määritettynä RE‑VERSE AD‑tutkimuksessa mukana olleilla potilailla (N = 486)
Hemostaasin palautuminen saavutettiin 80,3 %:lla arvioitavissa olleista potilaista, joilla oli esiintynyt vakavaa verenvuotoa. Hemostaasi todettiin normaaliksi 93,4 %:lla potilaista, jotka tarvitsivat kiireellisen toimenpiteen.
Koko 503 potilaan ryhmästä 101 potilasta kuoli; jokaisen näistä kuolemantapauksista voitiin katsoa olevan indeksitapahtuman komplikaatio tai liittyvän toiseen samanaikaiseen sairauteen. Tromboottisia tapahtumia raportoitiin 34 potilaalla (23 potilasta 34:stä ei saanut tapahtumahetkellä antitromboottista hoitoa). Näistä jokaisessa tapauksessa tromboottinen tapahtuma voitiin liittää potilaan lääketieteelliseen tilaan. Lieviä oireita mahdollisesta yliherkkyydestä (kuume, bronkospasmi, hyperventilaatio, ihottuma tai kutina) raportoitiin. Syy‑yhteyttä idarusitsumabihoitoon ei voitu osoittaa.
Pediatriset potilaat
Yksi pediatrinen potilas osallistui avoimeen turvallisuutta koskevaan kerta-annostutkimukseen, jossa idarusitsumabia annettiin laskimoon. Tutkimukseen otettiin mukaan pediatrisia potilaita dabigatraanieteksilaatin käyttöä laskimotromboembolian hoitoon ja sekundaariseen ennaltaehkäisyyn käsittelevistä tutkimuksista. Tutkimukseen hyväksyttiin mukaan potilaat, joilla dabigatraanin antikoagulaatiovaikutus piti saada kumottua nopeasti. Kyseistä potilasta (16-<18-vuotias) hoidettiin dabigatraanieteksilaatilla laskimotromboembolian ennaltaehkäisemiseksi kliinisen riskitekijän vuoksi. Verenvuototapahtuma vaati kirurgisen intervention ja riittävän hemostaasin. Hoito 5 g:n idarusitsumabiannoksella johti dabigatraanin antikoagulaatiovaikutuksen nopeaan ja täydelliseen kumoutumiseen. Idarusitsumabin farmakokinetiikka ja vaikutukset farmakodynamiikkaan olivat yhdenmukaiset aikuisten hoidosta saatujen tietojen kanssa.
Immunogeenisuus
Idarusitsumabivasta‑aineet tutkittiin vaiheen I tutkimuksissa 283 tutkittavan (224 vapaaehtoista sai idarusitsumabia) ja 501 potilaan seeruminäytteistä ennen hoitoa ja sen jälkeen. Noin 12 %:lla (33/283) vaiheen I tutkimuksiin osallistuneista henkilöistä ja 3,8 %:lla (19/501) potilaista havaittiin ennestään olevia vasta‑aineita, joilla oli ristireaktiivisuutta idarusitsumabin kanssa. Yliherkkyysreaktioita tai vaikutusta idarusitsumabin farmakokinetiikkaan tai kumoavaan vaikutukseen ei havaittu.
Hoidon myötä syntyneitä ja mahdollisesti pitkäkestoisia idarusitsumabivasta‑aineita havaittiin pieninä pitoisuuksina 4 %:lla vaiheen I tutkimuksiin osallistuneista henkilöistä (10/224) ja 1,6 %:lla potilaista (8/501), mikä viittaa idarusitsumabin vähäiseen immunogeenisuuteen. Kuuden vaiheen I tutkimuksiin osallistuneen henkilön alaryhmässä idarusitsumabia annosteltiin toistamiseen kaksi kuukautta ensimmäisen annostelun jälkeen. Näillä henkilöillä ei havaittu anti‑idarusitsumabivasta‑aineita ennen toista annostelua. Yhdellä henkilöllä havaittiin hoidon myötä syntyneitä anti‑idarusitsumabivasta‑aineita toisen annostelun jälkeen. Yhdeksälle potilaalle annettiin uudestaan idarusitsumabia. Uusi annos annettiin kaikille yhdeksälle potilaalle 6 päivän sisällä ensimmäisestä idarusitsumabiannoksesta. Anti-idarusitsumabivasta-aineita ei todettu yhdelläkään niistä potilaista, jotka olivat saaneet uudestaan idarusitsumabia.
Farmakokinetiikka
Idarusitsumabin farmakokinetiikkaa tutkittiin 224:llä tutkimushenkilöllä vaiheen I tutkimuksissa. Tiedot kuuden 45–64-vuotiaan terveen henkilön edustavasta alaryhmästä, jotka saivat 5 g:n annoksen infuusiona laskimoon, esitetään tässä.
Jakautuminen
Idarusitsumabi noudatti monivaiheista jakautumiskinetiikkaa ja jakautui ekstravaskulaariseen tilaan vain vähän. Kun laskimoon annettiin infuusiona 5 g:n annos vakaan tilan jakautumistilavuuden (Vdss) geometrinen keskiarvo oli 8,9 l (geometrinen variaatiokerroin 24,8 %).
Biotransformaatio
Vasta‑aineiden metaboliaan mahdollisesti liittyviä reittejä on kuvattu useita. Kaikissa reiteissä vasta‑aine hajoaa biologisesti pienemmiksi molekyyleiksi eli pieniksi peptideiksi tai aminohapoiksi, jotka sitten imeytyvät uudestaan ja osallistuvat yleiseen proteiinisynteesiin.
Eliminaatio
Idarusitsumabi eliminoitui nopeasti. Kokonaispuhdistuma oli 47,0 ml/min (geometrinen variaatiokerroin 18,4 %), initiaalinen puoliintumisaika (t1/2) 47 minuuttia (geometrinen variaatiokerroin 11,4 %) ja terminaalinen t½ 10,3 h (geometrinen variaatiokerroin 18,9 %). Kun idarusitsumabia annosteltiin 5 g laskimoon, 32,1 % (geometrinen variaatiokerroin 60,0 %) annoksesta erittyi virtsaan 6 tunnin keräysjakson aikana ja alle 1 % seuraavien 18 tunnin aikana. Loppuannoksen arvellaan eliminoituvan proteiinikatabolian kautta, pääasiassa munuaisessa.
Idarusitsumabihoidon jälkeen on havaittu proteinuriaa. Ohimenevä proteinuria on fysiologinen reaktio suuresta proteiinimäärästä munuaisissa sen jälkeen, kun on annettu 5 g idarusitsumabia laskimoon boluksena tai lyhytkestoisena infuusiona. Ohimenevä proteinuria oli yleensä voimakkaimmillaan neljä tuntia idarusitsumabin annostelun jälkeen ja normalisoitui 12‑24 tunnin kuluessa. Yksittäisissä tapauksissa ohimenevä proteinuria kesti yli 24 tuntia.
Munuaisten vajaatoimintapotilaat
Vaiheen I tutkimuksissa Praxbind‑valmistetta on tutkittu henkilöillä, joiden kreatiniinipuhdistuma on välillä 44–213 ml/min. Henkilöitä, joiden kreatiniinipuhdistuma on alle 44 ml/min, ei ole tutkittu vaiheessa I. Munuaisten vajaatoiminnan vaikeusasteesta riippuen kokonaispuhdistuma pieneni terveisiin henkilöihin verrattuna, mikä johti idarusitsumabialtistuksen suurenemiseen.
Niiden farmakokineettisten tietojen perusteella, jotka kerättiin 347 potilaalta, joiden munuaistoiminta oli eriasteista (kreatiniinipuhdistuman mediaani 21–99 ml/min), arvioidaan, että idarusitsumabialtistuksen keskiarvo (pitoisuus-aikakäyrän alle jäävä pinta-ala (AUC0‑24h)) nousee 38 % lievää (kreatiniinipuhdistuma 50 – < 80 ml/min), 90 % keskivaikeaa (30 – < 50 ml/min) ja 146 % vaikeaa (0 – < 30 ml/min) munuaisten vajaatoimintaa sairastavilla potilailla. Koska dabigatraani erittyy myös pääasiassa munuaisten kautta, myös dabigatraanialtistus suurenee munuaistoiminnan heikentyessä.
Näiden tietojen perusteella sekä dabigatraanin antikoagulaatiovaikutuksen voimakkaan kumoutumisen perusteella munuaisten vajaatoiminta ei muuta idarusitsumabin kumoavaa vaikutusta.
Maksan vajaatoimintapotilaat
Maksan toimintakoearvojen nousun perusteella määritettyyn maksavaurioon perustuvan maksan vajaatoiminnan ei ole todettu vaikuttavan idarusitsumabin farmakokinetiikkaan.
Idarusitsumabia on tutkittu 58 potilaalla, joilla oli eriasteista maksan vajaatoimintaa. Verrattuna 272 potilaaseen, joilla ei ollut maksan vajaatoimintaa, idarusitsumabin AUC-arvon mediaani muuttui 6 % potilailla, joiden ASAT-/ALAT-arvo oli 1 – < 2-kertainen normaaliarvon ylärajaan nähden (N = 34), ja vastaavasti 37 % ja 10 % potilailla, joiden ASAT-/ALAT-arvo oli 2 – < 3-kertainen normaaliarvon ylärajaan nähden (N = 3) ja > 3-kertainen normaaliarvon ylärajaan nähden (N = 21). Kahdeltatoista maksasairautta sairastavalta potilaalta saatujen farmakokineettisten tietojen perusteella idarusitsumabin AUC-arvo nousi 10 % verrattuna potilaisiin, joilla ei ollut maksasairautta.
Iäkkäät potilaat/sukupuoli/etninen tausta
Iällä, sukupuolella tai etnisellä taustalla ei populaatiofarmakokineettisten analyysien perusteella ollut kliinisesti merkittävää vaikutusta idarusitsumabin farmakokinetiikkaan.
Prekliiniset tiedot turvallisuudesta
Toistuvan altistuksen aiheuttamaa toksisuutta koskevien ei-kliinisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Tutkimukset kestivät rotilla enintään neljä viikkoa ja apinoilla kaksi viikkoa. Farmakologista turvallisuutta koskevissa tutkimuksissa ei todettu hengityselimistöön, keskushermostoon eikä sydämeen tai verisuoniin kohdistuvia vaikutuksia.
Idarusitsumabin mutageenisuutta ja karsinogeenisuutta ei ole tutkittu. Sen vaikutusmekanismin ja proteiinien ominaisuuksien perusteella karsinogeeniset tai genotoksiset vaikutukset eivät ole todennäköisiä.
Idarusitsumabin mahdollisia lisääntymiseen kohdistuvia vaikutuksia ei ole tutkittu. Hoitoon liittyviä vaikutuksia ei ole todettu lisääntymiskudoksissa kummallakaan sukupuolella laskimoon annettujen toistuvien annosten yhteydessä toksisuustutkimusten aikana (enintään neljä viikkoa rotilla ja kaksi viikkoa apinoilla). Kudosten ristireaktiivisuustutkimuksessa idarusitsumabin ei havaittu myöskään sitoutuvan ihmisen lisääntymiskudoksiin. Prekliinisten tulosten perusteella ei siis ole viitteitä hedelmällisyyttä eikä alkio‑sikiökehitystä koskevasta riskistä.
Paikallista verisuoniärsytystä ei havaittu, kun idarusitsumabia oli annettu laskimoon tai laskimon viereen. Idarusitsumabivalmiste ei aiheuttanut hemolyysiä ihmisen kokoveressä in vitro.
Farmaseuttiset tiedot
Apuaineet
natriumasetaattitrihydraatti (E262), etikkahappo (E260 pH:n säätämiseen), sorbitoli (E420), polysorbaatti 20 (E432), injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
4 vuotta.
Injektiopullon avaamisen jälkeen idarusitsumabin on osoitettu säilyvän käytön aikana kemiallisesti ja fysikaalisesti stabiilina 6 tunnin ajan huoneenlämmössä (enintään 30 °C).
Mikrobiologiselta kannalta lääkevalmiste on käytettävä välittömästi avaamisen jälkeen, ellei avaustapa estä mikrobikontaminaation riskiä. Jos valmistetta ei käytetä välittömästi, käytönaikaiset säilytysajat ja käyttöä edeltävät säilytysolosuhteet ovat käyttäjän vastuulla.
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Avaamatonta injektiopulloa voidaan ennen käyttöä pitää huoneenlämpötilassa (enintään 30 °C) korkeintaan 48 tuntia, jos sitä säilytetään alkuperäispakkauksessa suojassa valolta. Liuosta ei saa altistaa valolle 6 tuntia pidempään (ei avaamattomana eikä avattuna).
Avatun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
PRAXBIND injektio/infuusioneste, liuos
2,5 g/50 ml (L:ei) 2 x 50 ml (3867,08 €)
PF-selosteen tieto
50 ml liuosta lasisessa injektiopullossa (tyypin 1 lasia), jossa on butyylikumitulppa, alumiinikorkki ja etiketti, jossa on integroitu ripustin.
Pakkauksessa on kaksi injektiopulloa.
Valmisteen kuvaus:
Kirkas tai hieman opaalinhohtoinen, väritön tai hieman kellertävä liuos.
Käyttö- ja käsittelyohjeet
Parenteraaliset lääkevalmisteet, kuten Praxbind, tulee tarkastaa silmämääräisesti hiukkasten ja värimuutosten varalta ennen annostelua.
Praxbind‑valmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa. Praxbind‑valmisteen annostelussa voidaan käyttää potilaalla entuudestaan olevaa infuusioletkua. Letku on huuhdeltava ennen infuusiota ja infuusion jälkeen injektionesteisiin käytettävällä 9 mg/ml (0,9 %) natriumkloridiliuoksella. Muita infuusioita ei saa antaa yhtä aikaa saman laskimoyhteyden kautta.
Praxbind on tarkoitettu ainoastaan yhtä käyttökertaa varten, eikä se sisällä säilöntäaineita (ks. kohta Kestoaika).
Praxbind‑valmisteen ja polyvinyylikloridi-, polyeteeni- ja polyuretaani‑infuusiolaitteistojen ja polypropeeniruiskujen välillä ei ole havaittu yhteensopimattomuuksia.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
PRAXBIND injektio/infuusioneste, liuos
2,5 g/50 ml 2 x 50 ml
- Ei korvausta.
ATC-koodi
V03AB37
Valmisteyhteenvedon muuttamispäivämäärä
20.11.2024
Yhteystiedot
 BOEHRINGER INGELHEIM FINLAND KY
BOEHRINGER INGELHEIM FINLAND KY Tammasaarenkatu 5
00180 Helsinki
010 310 2800
www.boehringer-ingelheim.fi
medinfo.finland@boehringer-ingelheim.com