
KYPROLIS infuusiokuiva-aine, liuosta varten 10 mg, 30 mg, 60 mg
Vaikuttavat aineet ja niiden määrät
Kyprolis 10 mg infuusiokuiva‑aine, liuosta varten
Yksi injektiopullo sisältää 10 mg karfiltsomibia.
Apuaine, jonka vaikutus tunnetaan
Yksi injektiopullo sisältää 37 mg natriumia.
Yksi injektiopullo sisältää 500 mg syklodekstriiniä (beetadeksisulfobutyylieetterinatriumia).
Kyprolis 30 mg infuusiokuiva‑aine, liuosta varten
Yksi injektiopullo sisältää 30 mg karfiltsomibia.
Apuaine, jonka vaikutus tunnetaan
Yksi injektiopullo sisältää 109 mg natriumia.
Yksi injektiopullo sisältää 1 500 mg syklodekstriiniä (beetadeksisulfobutyylieetterinatriumia).
Kyprolis 60 mg infuusiokuiva‑aine, liuosta varten
Yksi injektiopullo sisältää 60 mg karfiltsomibia.
Apuaine, jonka vaikutus tunnetaan
Yksi injektiopullo sisältää 216 mg natriumia.
Yksi injektiopullo sisältää 3 000 mg syklodekstriiniä (beetadeksisulfobutyylieetterinatriumia).
Käyttökuntoon saattamisen jälkeen 1 ml liuosta sisältää 2 mg karfiltsomibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Infuusiokuiva-aine, liuosta varten.
Kliiniset tiedot
Käyttöaiheet
Kyprolis yhdessä daratumumabin ja deksametasonin, lenalidomidin ja deksametasonin tai pelkän deksametasonin kanssa on tarkoitettu multippelin myelooman hoitoon aikuispotilaille, jotka ovat saaneet aikaisemmin vähintään yhtä hoitoa (ks. kohta Farmakodynamiikka).
Ehto
Valmiste annetaan syövän lääkehoitoon perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Kyprolis‑hoito on toteutettava syövän hoitoon perehtyneen lääkärin valvonnassa.
Annostus
Annoksen laskemiseen käytetään potilaan kehon pinta‑alaa (BSA) lähtötilanteessa. Jos potilaan kehon pinta‑ala on yli 2,2 m2, annos lasketaan 2,2 m2:n pinta‑alan mukaan. Annosta ei tarvitse muuttaa, jos potilaan paino muuttuu 20 % tai vähemmän.
Kyprolis yhdessä lenalidomidin ja deksametasonin kanssa
Kun Kyprolista annetaan yhdessä lenalidomidin ja deksametasonin kanssa, Kyprolis annetaan 10 minuutin infuusiona laskimoon kahtena peräkkäisenä päivänä joka viikko kolmen viikon ajan (päivinä 1, 2, 8, 9, 15 ja 16), jonka jälkeen on 12 vuorokauden tauko (päivät 17–28), kuten taulukossa 1 on kuvattu. Jokainen 28 vuorokauden jakso on yksi hoitojakso.
Kyprolis‑hoito aloitetaan annoksella 20 mg/m2 (enimmäisannos 44 mg), joka annetaan ensimmäisen hoitojakson 1. ja 2. päivänä. Jos potilas sietää tämän, annos nostetaan ensimmäisen hoitojakson 8. päivänä tasolle 27 mg/m2 (enimmäisannos 60 mg). Hoitojaksosta 13 lähtien 8. ja 9. päivän Kyprolis‑annokset jätetään pois.
Hoitoa voidaan jatkaa taudin etenemiseen tai kestämättömien haittavaikutusten ilmaantumiseen asti.
Jos Kyprolis‑hoitoa yhdessä lenalidomidin ja deksametasonin kanssa halutaan jatkaa kauemmin kuin 18 hoitojakson ajan, se voidaan tehdä vain yksilöllisen hyöty‑riskiarvion perusteella, koska karfiltsomibin siedettävyydestä ja toksisuudesta 18 hoitojaksoa pidemmässä hoidossa on vähän tietoja (ks. kohta Farmakodynamiikka).
Yhdessä Kyprolis‑hoidon kanssa annetaan lenalidomidia 25 mg suun kautta 28 päivän hoitojakson 1.−21. päivänä ja deksametasonia 40 mg suun kautta tai laskimoon 28 päivän hoitojakson 1., 8., 15. ja 22. päivänä. Lenalidomidin aloitusannoksen pienentämistä on harkittava lenalidomidin voimassa olevan valmisteyhteenvedon suositusten mukaisesti, esimerkiksi jos potilaalla on munuaisten vajaatoimintaa lähtötilanteessa. Deksametasoni tulee antaa vähintään 30 minuuttia ja enintään 4 tuntia ennen Kyprolis‑annosta.
Taulukko 1. Kyprolis yhdessä lenalidomidin ja deksametasonin kanssa
Hoitojakso 1 | |||||||||||
1. viikko | 2. viikko | 3. viikko | 4. viikko | ||||||||
1. päivä | 2. päivä | 3.–7. päivä | 8. päivä | 9. päivä | 10.–14. päivä | 15. päivä | 16. päivä | 17.–21. päivä | 22. päivä | 23.–28. päivä | |
Kyprolis (mg/m2)a | 20 | 20 | - | 27 | 27 | - | 27 | 27 | - | - | - |
Deksametasoni (mg) | 40 | - | - | 40 | - | - | 40 | - | - | 40 | - |
Lenalidomidi | 25 mg/vrk | - | - | ||||||||
Hoitojaksot 2–12 | |||||||||||
1. viikko | 2. viikko | 3. viikko | 4. viikko | ||||||||
1. päivä | 2. päivä | 3.–7. päivä | 8. päivä | 9. päivä | 10.–14. päivä | 15. päivä | 16. päivä | 17.–21. päivä | 22. päivä | 23.–28. päivä | |
Kyprolis (mg/m2)a | 27 | 27 | - | 27 | 27 | - | 27 | 27 | - | - | - |
Deksametasoni (mg) | 40 | - | - | 40 | - | - | 40 | - | - | 40 | - |
Lenalidomidi | 25 mg/vrk | - | - | ||||||||
Hoitojaksosta 13 alkaen | |||||||||||
1. viikko | 2. viikko | 3. viikko | 4. viikko | ||||||||
1. päivä | 2. päivä | 3.–7. päivä | 8. päivä | 9. päivä | 10.–14. päivä | 15. päivä | 16. päivä | 17.–21. päivä | 22. päivä | 23.–28. päivä | |
Kyprolis (mg/m2)a | 27 | 27 | - | - | - | - | 27 | 27 | - | - | - |
Deksametasoni (mg) | 40 | - | - | 40 | - | - | 40 | - | - | 40 | - |
Lenalidomidi | 25 mg/vrk | - | - | ||||||||
| a. Infuusioaika on 10 minuuttia ja pysyy samana koko hoito‑ohjelman ajan | |||||||||||
Kyprolis yhdessä deksametasonin kanssa
Kun Kyprolista annetaan yhdessä deksametasonin kanssa, Kyprolis annetaan 30 minuutin infuusiona laskimoon kahtena peräkkäisenä päivänä joka viikko kolmen viikon ajan (päivinä 1, 2, 8, 9, 15 ja 16), jonka jälkeen on 12 vuorokauden tauko (päivät 17–28), kuten taulukossa 2 on kuvattu. Jokainen 28 vuorokauden jakso on yksi hoitojakso.
Kyprolis‑hoito aloitetaan annoksella 20 mg/m2 (enimmäisannos 44 mg), joka annetaan ensimmäisen hoitojakson 1. ja 2. päivänä. Jos potilas sietää tämän, annos nostetaan ensimmäisen hoitojakson 8. päivänä tasolle 56 mg/m2 (enimmäisannos 123 mg).
Hoitoa voidaan jatkaa taudin etenemiseen tai kestämättömien haittavaikutusten ilmaantumiseen asti.
Kun Kyprolis‑hoitoon yhdistetään pelkkä deksametasoni, deksametasonia annetaan 20 mg suun kautta tai laskimoon 28 päivän hoitojakson 1., 2., 8., 9., 15., 16., 22. ja 23. päivänä. Deksametasoni tulee antaa vähintään 30 minuuttia ja enintään 4 tuntia ennen Kyprolis‑annosta.
Taulukko 2. Kyprolis yhdessä pelkän deksametasonin kanssa
1. hoitojakso | ||||||||||||||
1. viikko | 2. viikko | 3. viikko | 4. viikko | |||||||||||
1. päivä | 2. päivä | 3.–7. päivä | 8. päivä | 9. päivä | 10.–14. päivä | 15. päivä | 16. päivä | 17.–21. päivä | 22. päivä | 23. päivä | 24.–28. päivä | |||
Kyprolis (mg/m2)a | 20 | 20 | - | 56 | 56 | - | 56 | 56 | - | - | - | - | ||
Deksametasoni (mg) | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | ||
2. hoitojakso ja kaikki myöhemmät hoitojaksot | ||||||||||||||
1. viikko | 2. viikko | 3. viikko | 4. viikko | |||||||||||
1. päivä | 2. päivä | 3.–7. päivä | 8. päivä | 9. päivä | 10.–14. päivä | 15. päivä | 16. päivä | 17.–21. päivä | 22. päivä | 23. päivä | 24.–28. päivä | |||
Kyprolis (mg/m2)a | 56 | 56 | - | 56 | 56 | - | 56 | 56 | - | - | - | - | ||
Deksametasoni (mg) | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | ||
| a. Infuusioaika on 30 minuuttia ja pysyy samana koko hoito‑ohjelman ajan | ||||||||||||||
Kyprolis yhdessä daratumumabin ja deksametasonin kanssa
Kun Kyprolista annetaan yhdessä daratumumabin ja deksametasonin kanssa, Kyprolis annetaan 30 minuutin infuusiona laskimoon kahtena peräkkäisenä päivänä joka viikko kolmen viikon ajan (päivinä 1, 2, 8, 9, 15 ja 16), jonka jälkeen on 12 vuorokauden tauko (päivät 17–28), kuten taulukossa 3 on kuvattu. Jokainen 28 vuorokauden jakso on yksi hoitojakso.
Kyprolis‑hoito aloitetaan annoksella 20 mg/m2 (enimmäisannos 44 mg), joka annetaan ensimmäisen hoitojakson 1. ja 2. päivänä. Jos potilas sietää tämän, annos nostetaan ensimmäisen hoitojakson 8. päivänä tasolle 56 mg/m2 (enimmäisannos 123 mg).
Hoitoa voidaan jatkaa taudin etenemiseen tai kestämättömien haittavaikutusten ilmaantumiseen asti.
Deksametasonia annetaan jokaisen 28 päivän hoitojakson 1., 2., 8., 9., 15. ja 16. päivänä 20 mg suun kautta tai laskimoon ja 22. päivänä 40 mg suun kautta tai laskimoon. Yli 75‑vuotiaille potilaille deksametasonia annetaan ensimmäisen viikon jälkeen 20 mg kerran viikossa suun kautta tai laskimoon. Deksametasoni tulee antaa vähintään 30 minuuttia ja enintään 4 tuntia ennen Kyprolis‑annosta.
Daratumumabi voidaan antaa laskimoon tai ihon alle.
Jos daratumumabi annetaan laskimoon, annos on 16 mg/kg (todellinen kehonpaino), mutta ensimmäisen hoitojakson 1. ja 2. päivänä annetaan jaettu annos 8 mg/kg. Tämän jälkeen daratumumabia annetaan ensin 16 mg/kg kerran viikossa 1. hoitojakson 8., 15. ja 22. päivänä ja 2. hoitojakson 1., 8., 15. ja 22. päivänä, sitten joka toinen viikko neljän hoitojakson ajan (3.–6. hoitojakso) ja lopuksi neljän viikon välein kaikkien seuraavien hoitojaksojen ajan tai taudin etenemiseen saakka.
Vaihtoehtoisesti daratumumabia voidaan antaa ihon alle 1 800 mg:n annos 1. hoitojakson 1., 8., 15. ja 22. päivänä ja 2. hoitojakson 1., 8., 15. ja 22. päivänä, sitten joka toinen viikko neljän hoitojakson ajan (3.–6. hoitojakso) ja lopuksi neljän viikon välein kaikkien seuraavien hoitojaksojen ajan tai taudin etenemiseen saakka.
Katso daratumumabin valmisteyhteenvedosta lisätietoja ihon alle annettavan lääkemuodon käyttämisestä.
Kun enemmän kuin yksi mainituista lääkkeistä annetaan samana päivänä, ne suositellaan antamaan seuraavassa järjestyksessä: ensin deksametasoni, sitten daratumumabi-infuusion esilääkitys (katso kohta Muut samanaikaiset lääkevalmisteet), karfiltsomibi, daratumumabi ja lopuksi daratumumabi-infuusion jälkeen annettava lääkitys (katso kohta Muut samanaikaiset lääkevalmisteet).
Lisätietoja annostelusta on daratumumabin ja deksametasonin valmisteyhteenvedoissa.
Taulukko 3. Kyprolis yhdessä deksametasonin ja daratumumabin kanssa
| 1. hoitojakso | |||||||||||
1. viikko | 2. viikko | 3. viikko | 4. viikko | |||||||||
1. päivä | 2. päivä | 3.–7. päivä | 8. päivä | 9. päivä | 10.–14. päivä | 15. päivä | 16. päivä | 17.–21. päivä | 22. päivä | 23. päivä | 24.–28. päivä | |
Kyprolis (mg/m2)a | 20 | 20 | - | 56 | 56 | - | 56 | 56 | - | - | - | - |
Deksametasoni (mg)b | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 40 | - | - |
Daratumumabi (laskimoon TAI ihon alle) | ||||||||||||
Laskimoon annettuna (mg/kg) | 8 | 8 | - | 16 | - | - | 16 | - | - | 16 | - | - |
Ihon alle annettuna (mg) | 1 800 | - | - | 1 800 | - | - | 1 800 | - | - | 1 800 | - | - |
| 2. hoitojakso | |||||||||||
1. viikko | 2. viikko | 3. viikko | 4. viikko | |||||||||
1. päivä | 2. päivä | 3.–7. päivä | 8. päivä | 9. päivä | 10.–14. päivä | 15. päivä | 16. päivä | 17.–21. päivä | 22. päivä | 23. päivä | 24.–28. päivä | |
Kyprolis (mg/m2)a | 56 | 56 | - | 56 | 56 | - | 56 | 56 | - | - | - | - |
Deksametasoni (mg)b | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 40 | - | - |
Daratumumabi (laskimoon TAI ihon alle) | ||||||||||||
Laskimoon annettuna (mg/kg) | 16 | - | - | 16 | - | - | 16 | - | - | 16 | - | - |
Ihon alle annettuna (mg) | 1 800 | - | - | 1 800 | - | - | 1 800 | - | - | 1 800 | - | - |
| 3.–6. hoitojakso | |||||||||||
1. viikko | 2. viikko | 3. viikko | 4. viikko | |||||||||
1. päivä | 2. päivä | 3.–7. päivä | 8. päivä | 9. päivä | 10.–14. päivä | 15. päivä | 16. päivä | 17.–21. päivä | 22. päivä | 23. päivä | 24.–28. päivä | |
Kyprolis (mg/m2)a | 56 | 56 | - | 56 | 56 | - | 56 | 56 | - | - | - | - |
Deksametasoni (mg)b | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 40 | - | - |
Daratumumabi (laskimoon TAI ihon alle) | ||||||||||||
Laskimoon annettuna (mg/kg) | 16 | - | - | - | - | - | 16 | - | - | - | - | - |
Ihon alle annettuna (mg) | 1 800 | - | - | - | - | - | 1 800 | - | - | - | - | - |
| 7. hoitojakso ja kaikki myöhemmät hoitojaksot | |||||||||||
1. viikko | 2. viikko | 3. viikko | 4. viikko | |||||||||
1. päivä | 2. päivä | 3.–7. päivä | 8. päivä | 9. päivä | 10.–14. päivä | 15. päivä | 16. päivä | 17.–21. päivä | 22. päivä | 23. päivä | 24.–28. päivä | |
Kyprolis (mg/m2)a | 56 | 56 | - | 56 | 56 | - | 56 | 56 | - | - | - | - |
Deksametasoni (mg)b | 20 | 20 | - | 20 | 20 | - | 20 | 20 | - | 40 | - | - |
Daratumumabi (laskimoon TAI ihon alle) | ||||||||||||
Laskimoon annettuna (mg/kg) | 16 | - | - | - | - | - | - | - | - | - | - | - |
Ihon alle annettuna (mg) | 1 800 | - | - | - | - | - | - | - | - | - | - | - |
a. Infuusioaika on 30 minuuttia ja pysyy samana koko hoito‑ohjelman ajan b. Yli 75‑vuotiaille potilaille deksametasonia annetaan ensimmäisen viikon jälkeen 20 mg kerran viikossa suun kautta tai laskimoon. | ||||||||||||
Muut samanaikaiset lääkevalmisteet
Ehkäisevän viruslääkityksen aloittamista on harkittava Kyprolis‑hoitoa saaville potilaille herpes zoster ‑viruksen uudelleenaktivoitumisen välttämiseksi (ks. kohta Haittavaikutukset).
Tromboosiprofylaksia suositellaan potilaille, jotka saavat Kyprolista yhdessä daratumumabin ja deksametasonin, lenalidomidin ja deksametasonin tai pelkän deksametasonin kanssa, ja sen tulee perustua potilaan yksilöllisten riskien ja kliinisen tilan arviointiin. Muita mahdollisesti tarvittavia samanaikaisia lääkkeitä, kuten ehkäisevää antasidilääkitystä, koskevat tiedot on tarkistettava lenalidomidin ja deksametasonin valmisteyhteenvedoista.
Potilaille, jotka saavat Kyprolista yhdessä daratumumabin ja deksametasonin kanssa, tulee antaa infuusion esilääkitys daratumumabi-infuusioon liittyvien reaktioiden riskin vähentämiseksi.
Daratumumabin valmisteyhteenvedossa on lisätietoja samanaikaisesti annettavista lääkevalmisteista, mukaan lukien infuusion esilääkitys ja infuusion jälkeen annettava lääkitys.
Nesteytyksen ja neste- ja elektrolyyttitasapainon seuranta
Riittävä nesteytys on tarpeen ennen annosten antamista ensimmäisen hoitojakson aikana, varsinkin jos tuumorilyysioireyhtymän tai munuaisiin kohdistuvien haittavaikutusten riski on suuri. Kaikkien potilaiden tilaa on tarkkailtava nesteylimäärään viittaavien merkkien havaitsemiseksi, ja nesteiden antaminen on mitoitettava potilaan yksilöllisten tarpeiden mukaan. Kokonaisnestemäärää voidaan sovittaa kliinisen tarpeen mukaan, jos potilaalla on lähtötilanteessa sydämen vajaatoiminta tai vajaatoiminnan riski (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Suositeltava nestehoito koostuu sekä suun kautta annettavista nesteistä (30 ml/kg/vrk 48 tunnin ajan ennen ensimmäisen hoitojakson ensimmäistä päivää) että laskimoon annettavista nesteistä (250–500 ml sopivaa laskimoon annettavaa nestettä ennen jokaista annosta ensimmäisen hoitojakson aikana). Tarvittaessa annetaan 250–500 ml nesteitä laskimoon myös Kyprolis‑annoksen jälkeen ensimmäisen hoitojakson aikana. Suun kautta ja/tai laskimoon annettavaa nesteytystä jatketaan tarpeen mukaan seuraavien hoitojaksojen aikana.
Kun valmistetta annetaan yhdessä laskimoon annettavan daratumumabin kanssa, suun kautta ja/tai laskimoon annettava nesteytys ei ole tarpeen niinä päivinä, joina daratumumabia annetaan laskimoon.
Seerumin kaliumarvoja on seurattava Kyprolis‑hoidon aikana kuukausittain tai useammin kliinisen tarpeen mukaan ja riippuen ennen hoidon aloittamista mitatuista kaliumarvoista, muusta samanaikaisesta lääkityksestä (esim. lääkkeet, joiden tiedetään lisäävän hypokalemian riskiä) ja muista samanaikaisista sairauksista.
Suositellut annosmuutokset
Annostusta muutetaan Kyproliksen haittavaikutusten perusteella. Taulukossa 4 on kuvattu suositeltavat toimenpiteet ja annosmuutokset. Annostason laskua koskevat ohjeet ovat taulukossa 5.
Taulukko 4. Annosmuutokset Kyprolis‑hoidon aikana
Hematologinen toksisuus | Suositeltava toimenpide |
|
|
|
|
|
|
Muu kuin hematologinen toksisuus (munuaistoksisuus) | Suositeltava toimenpide |
|
|
Muu (ei hematologinen) toksisuus | Suositeltava toimenpide |
|
|
| a. Ks. annostason laskua koskevat ohjeet taulukosta 5 | |
Taulukko 5. Annostason lasku Kyprolis‑hoidossa
Hoito‑ohjelma | Kyprolis‑annos | Ensimmäinen Kyprolis-annoksen lasku | Toinen Kyprolis-annoksen lasku | Kolmas Kyprolis-annoksen lasku |
Kyprolis, lenalidomidi ja deksametasoni | 27 mg/m2 | 20 mg/m2 | 15 mg/m2 a | — |
Kyprolis ja deksametasoni | 56 mg/m2 | 45 mg/m2 | 36 mg/m2 | 27 mg/m2 a |
Kyprolis, daratumumabi ja deksametasoni | 56 mg/m2 | 45 mg/m2 | 36 mg/m2 | 27 mg/m2 a |
Huom: Kyproliksen infuusioajat pysyvät muuttumattomina annosta pienennettäessä a. Elleivät oireet lievity, Kyprolis‑hoito lopetetaan | ||||
Erityisryhmät
Munuaisten vajaatoiminta
Potilaat, joilla on kohtalainen tai vaikea munuaisten vajaatoiminta, otettiin mukaan Kyproliksen ja deksametasonin yhdistelmällä tehtyihin tutkimuksiin mutta suljettiin pois Kyproliksen ja lenalidomidin yhdistelmällä tehdyistä tutkimuksista. Siksi Kyproliksen, lenalidomidin ja deksametasonin yhdistelmästä on vain vähän tutkimustietoa niiden potilaiden hoidossa, joiden kreatiniinipuhdistuma (CrCl) on < 50 ml/min. Lenalidomidin aloitusannoksen pienentämistä on harkittava lenalidomidin valmisteyhteenvedon suositusten mukaisesti, jos potilaalla on munuaisten vajaatoimintaa lähtötilanteessa.
Käytettävissä olevien farmakokineettisten tietojen perusteella Kyproliksen aloitusannoksen muuttamista ei suositella hoidettaessa potilaita, joilla on lähtötilanteessa lievä, kohtalainen tai vaikea munuaisten vajaatoiminta tai jotka ovat dialyysihoidossa (ks. kohta Farmakokinetiikka). Kolmannen vaiheen kliinisissä tutkimuksissa haittatapahtumaksi luokitellun akuutin munuaisten vajaatoiminnan ilmaantuvuus oli kuitenkin suurempi potilailla, joiden kreatiniinipuhdistuma oli pienempi lähtötilanteessa, kuin potilailla, joilla lähtötilanteen kreatiniinipuhdistuma oli suurempi.
Munuaisten toiminta on arvioitava hoidon alkaessa ja sitä on seurattava vähintään kuukausittain tai hyväksyttyjen hoitosuositusten mukaisesti, varsinkin, jos lähtötilanteen kreatiniinipuhdistuma on alentunut (CrCl < 30 ml/min). Annosta on muutettava tarvittaessa haittavaikutusten perusteella (ks. taulukko 4). Hoidon tehosta ja turvallisuudesta on vain vähän tutkimustietoa niiden potilaiden hoidossa, joilla kreatiniinipuhdistuman lähtöarvo on < 30 ml/min.
Kyproliksen poistumista dialyysissä ei ole tutkittu, joten se on annettava dialyysin jälkeen.
Maksan vajaatoiminta
Kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavat potilaat suljettiin pois tutkimuksista, joissa Kyprolista annettiin yhdessä lenalidomidin ja deksametasonin tai pelkän deksametasonin kanssa.
Kyproliksen farmakokinetiikkaa ei ole arvioitu vaikeaa maksan vajaatoimintaa sairastavien potilaiden hoidossa. Käytettävissä olevien farmakokineettisten tietojen perusteella aloitusannoksen muuttamista ei suositella hoidettaessa potilaita, joilla on lievä tai kohtalainen maksan vajaatoiminta. Maksan toiminnan poikkeavuuksien, ≥ 3. asteen haittatapahtumien ja vakavien haittatapahtumien potilaskohtainen ilmaantuvuus oli kuitenkin suurempi potilailla, joilla oli lievä tai kohtalainen maksan vajaatoiminta lähtötilanteessa, kuin potilailla, joiden maksan toiminta oli normaali (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). Maksaentsyymi- ja bilirubiiniarvot on määritettävä karfiltsomibihoitoa aloitettaessa ja niitä on seurattava kuukausittain hoidon aikana lähtöarvoista riippumatta, ja annosta on muutettava tarvittaessa haittavaikutusten perusteella (ks. taulukko 4). Kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavien potilaiden tilaa on seurattava erityisen huolellisesti, koska hoidon tehosta ja turvallisuudesta on vain vähän tutkimustietoa tässä potilasryhmässä.
Iäkkäät potilaat
Tiettyjen haittatapahtumien (myös sydämen vajaatoiminnan) potilaskohtainen ilmaantuvuus oli kliinisissä tutkimuksissa yleisesti suurempi ≥ 75‑vuotiailla kuin alle 75‑vuotiailla potilailla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Kyprolis‑valmisteen turvallisuutta ja tehoa lapsipotilaiden hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Kyprolis annetaan infuusiona laskimoon. Annos 20/27 mg/m2 annetaan 10 minuutin infuusiona. Annos 20/56 mg/m2 on annettava 30 minuutin infuusiona.
Kyprolis‑annosta ei saa antaa nopeana injektiona eikä boluksena laskimoon.
Laskimoletku on huuhdeltava fysiologisella natriumkloridiliuoksella tai 5‑prosenttisella glukoosi‑injektionesteellä juuri ennen Kyprolis‑annoksen antamista ja heti sen jälkeen.
Kyprolista ei saa sekoittaa eikä antaa infuusiona muiden lääkevalmisteiden kanssa.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ennen lääkkeen antoa.
Vasta-aiheet
- Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
- Imettäminen (ks. kohta Raskaus ja imetys).
Koska Kyprolista annetaan yhdessä muiden lääkevalmisteiden kanssa, muut vasta-aiheet on tarkistettava näiden valmisteiden valmisteyhteenvedoista.
Varoitukset ja käyttöön liittyvät varotoimet
Koska Kyprolista annetaan yhdessä muiden lääkevalmisteiden kanssa, näiden muiden valmisteiden valmisteyhteenvetoihin on tutustuttava ennen Kyprolis‑hoidon aloittamista. Koska lenalidomidia voidaan käyttää yhdessä Kyproliksen kanssa, on kiinnitettävä erityistä huomiota lenalidomidin vaatimiin raskaustesteihin ja raskaudenehkäisyyn (ks. kohta Raskaus ja imetys).
Sydän
Sydämen vajaatoimintaa tai aikaisemman vajaatoiminnan vaikeutumista (esim. sydämen kongestiivinen vajaatoiminta, keuhkoedeema, pienentynyt ejektiofraktio), sydänlihasiskemiaa ja sydäninfarktitapauksia on esiintynyt Kyproliksen antamisen jälkeen. Kuolemaan johtanut sydänpysähdys on todettu vuorokauden kuluessa Kyprolis‑annoksen antamisesta, ja sydämen vajaatoimintaan ja sydäninfarktiin liittyviä kuolemantapauksia on raportoitu. Mahdolliset annokseen liittyvät vaikutukset, ks. kohta Haittavaikutukset.
Vaikka riittävä nesteytys on välttämätöntä ennen annosten antamista ensimmäisen hoitojakson aikana, kaikkia potilaita on tarkkailtava nesteylimäärään viittaavien merkkien havaitsemiseksi, varsinkin jos potilaalla on sydämen vajaatoiminnan riski. Kokonaisnestemäärää voidaan sovittaa kliinisen tarpeen mukaan, jos potilaalla on lähtötilanteessa sydämen vajaatoiminta tai vajaatoiminnan riski (ks. kohta Annostus ja antotapa).
Jos potilaalla todetaan 3. tai 4. asteen sydäntapahtuma, Kyprolis‑hoito keskeytetään, kunnes tila on parantunut. Tämän jälkeen tehdään hyöty‑riskiarvio, jonka perusteella harkitaan, voidaanko Kyprolis aloittaa uudelleen yhtä annostasoa pienemmällä annoksella (ks. kohta Annostus ja antotapa).
Sydämen vajaatoiminnan riski on suurentunut iäkkäillä (≥ 75‑vuotiailla) potilailla. Sydämen vajaatoiminnan riski on suurentunut myös aasialaisilla potilailla.
Ennen hoidon aloittamista suositellaan perusteellista kardiovaskulaaristen riskitekijöiden arviointia.
Kliinisiin tutkimuksiin ei otettu potilaita, joilla oli NYHA (New York Heart Association) ‑luokan III tai IV sydämen vajaatoiminta, hiljattain sairastettu sydäninfarkti tai sydämen johtumishäiriöitä, joita ei saatu hallintaan lääkehoidolla. Näillä potilailla saattaa olla suurentunut sydänkomplikaatioiden riski. Jos potilaalla on NYHA‑luokan III tai IV sydämen vajaatoimintaan viittaavia löydöksiä tai oireita, hiljattain (4 edellisen kuukauden aikana) sairastettu sydäninfarkti tai huonossa hoitotasapainossa oleva angina pectoris tai hallitsemattomia rytmihäiriöitä, perusteellinen kardiologinen arviointi on tehtävä ennen Kyprolis‑hoidon aloittamista. Arvioinnin yhteydessä potilaan tila pyritään samaan hyvään tasapainoon kiinnittäen erityistä huomiota verenpaineen ja nestetasapainon hallintaan. Tämän jälkeen potilaan hoidossa on noudatettava varovaisuutta ja hänen tilaansa on seurattava tarkoin.
EKG‑muutokset
Kliinisissä tutkimuksissa ja markkinoille tulon jälkeisessä seurannassa on raportoitu QT‑ajan pitenemistä. Kyprolis‑hoitoa saavilla potilailla on raportoitu kammiotakykardiaa.
Keuhkoihin kohdistuvat haittavaikutukset
Kyprolis‑hoitoa saaneilla potilailla on esiintynyt äkillistä hengitysvajausoireyhtymää (ARDS), äkillistä hengitysvajausta ja akuutteja diffuuseja infiltratiivisia keuhkosairauksia, kuten pneumoniittia ja interstitiaalista keuhkosairautta. Jotkin näistä tapahtumista ovat johtaneet kuolemaan. Potilas tutkitaan ja Kyprolis‑hoito keskeytetään, kunnes tila on parantunut, minkä jälkeen harkitaan hyöty‑riskiarvion perusteella, voidaanko Kyprolis aloittaa uudelleen (ks. kohta Annostus ja antotapa).
Kohonnut keuhkoverenpaine
Kyprolis‑hoitoa saaneilla potilailla on raportoitu kohonnutta keuhkoverenpainetta. Jotkin näistä tapahtumista ovat johtaneet kuolemaan. Tila arvioidaan tarpeen mukaan. Jos keuhkoverenpaine on kohonnut, Kyprolis‑hoito keskeytetään, kunnes tila paranee tai palautuu lähtötasolle, minkä jälkeen harkitaan hyöty‑riskiarvion perusteella, voidaanko Kyprolis aloittaa uudelleen (ks. kohta Annostus ja antotapa).
Hengenahdistus
Hengenahdistusta raportoitiin yleisesti Kyprolis‑hoitoa saaneilla potilailla. Hengenahdistus tutkitaan, jotta voidaan sulkea pois sydämestä tai keuhkoista johtuvat syyt, kuten sydämen vajaatoiminta tai keuhkoperäiset oireyhtymät. Jos potilaalla on 3. tai 4. asteen hengenahdistusta, Kyprolis‑hoito on keskeytettävä, kunnes tila paranee tai palautuu lähtötasolle, minkä jälkeen harkitaan hyöty‑riskiarvion perusteella, voidaanko Kyprolis aloittaa uudelleen (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Hypertensio
Kyprolis‑hoitoa saaneilla potilailla on todettu hypertensiota, myös hypertensiivisiä kriisejä ja hypertensiivisiä hätätilanteita. Jotkin näistä tapahtumista ovat johtaneet kuolemaan. Tutkimuksessa 20160275 hypertensiota raportoitiin useammin potilailla, jotka saivat Kyprolista yhdessä daratumumabin kanssa. Verenpaineen tulisi olla hallinnassa ennen hoidon aloittamista ja sen aikana. Kaikki potilaat on tutkittava rutiininomaisesti Kyprolis‑hoidon aikana hypertension havaitsemiseksi ja hoidettava tarvittaessa. Ellei hypertensiota saada hallintaan, Kyprolis‑annosta on pienennettävä. Jos potilaalla todetaan hypertensiivinen kriisi, Kyprolis‑hoito on keskeytettävä, kunnes tila paranee tai palautuu lähtötasolle, minkä jälkeen harkitaan hyöty‑riskiarvion perusteella, voidaanko Kyprolis aloittaa uudelleen (ks. kohta Annostus ja antotapa).
Akuutti munuaisten vajaatoiminta
Kyprolis‑hoitoa saaneilla potilailla on raportoitu akuuttia munuaisten vajaatoimintaa. Jotkin näistä tapahtumista ovat johtaneet kuolemaan. Akuuttia munuaisten vajaatoimintaa esiintyi useammin potilailla, joilla oli pitkälle edennyt uusiutunut ja refraktaarinen multippeli myelooma ja jotka saivat Kyprolista ainoana lääkkeenä. Kolmannen vaiheen kliinisissä tutkimuksissa haittatapahtumaksi luokitellun akuutin munuaisten vajaatoiminnan ilmaantuvuus oli suurempi potilailla, joiden kreatiniinipuhdistuma oli pienempi lähtötilanteessa, kuin potilailla, joilla lähtötilanteen kreatiniinipuhdistuma oli suurempi. Suurimmalla osalla potilaista kreatiniinipuhdistuma pysyi muuttumattomana ajan myötä. Munuaisten toimintaa on seurattava vähintään kuukausittain tai hyväksyttyjen hoitosuositusten mukaisesti, varsinkin, jos lähtötilanteen kreatiniinipuhdistuma on alentunut. Annosta pienennetään tai hoito keskeytetään tarpeen mukaan (ks. kohta Annostus ja antotapa).
Tuumorilyysioireyhtymä
Kyprolis‑hoitoa saaneilla potilailla on raportoitu tuumorilyysioireyhtymää, joka on joissakin tapauksissa johtanut kuolemaan. Tuumorilyysioireyhtymän riskin katsotaan olevan tavanomaista suurempi potilailla, joilla on suuri kasvaintaakka. Potilaiden hyvä nesteytys on varmistettava ennen Kyproliksen antamista ensimmäisen hoitojakson aikana ja tarvittaessa myös myöhempien hoitojaksojen yhteydessä (ks. kohta Annostus ja antotapa). Virtsahappopitoisuutta pienentävää lääkitystä on harkittava, jos potilaalla on suuri tuumorilyysioireyhtymän riski. Tuumorilyysioireyhtymään viittaavia merkkejä on seurattava hoidon aikana, myös säännöllisten seerumin elektrolyyttipitoisuuksien mittausten avulla, ja häiriöt on hoidettava heti. Kyprolis‑hoito on keskeytettävä, kunnes tuumorilyysioireyhtymä on parantunut (ks. kohta Annostus ja antotapa).
Infuusioreaktiot
Kyprolis‑hoitoa saaneilla potilailla on raportoitu infuusioreaktioita, jotka ovat joissakin tapauksissa olleet hengenvaarallisia. Oireita voivat olla kuume, vilunväristykset, nivelkipu, lihaskipu, kasvojen punoitus, kasvojen turvotus, oksentelu, heikkous, hengenahdistus, verenpaineen lasku, pyörtyminen, bradykardia, puristava tunne rinnassa tai iskeeminen rintakipu. Näitä reaktioita voi ilmaantua heti Kyprolis‑annoksen jälkeen tai viimeistään 24 tunnin kuluttua annoksesta. Deksametasonia on annettava ennen Kyprolis‑annosta reaktioiden vähentämiseksi ja lievittämiseksi (ks. kohta Annostus ja antotapa).
Verenvuoto ja trombosytopenia
Kyprolis‑hoitoa saavilla potilailla on raportoitu verenvuotoja (esim. ruoansulatuskanavan verenvuotoa, keuhkoverenvuotoa ja kallonsisäistä verenvuotoa), joihin on usein liittynyt trombosytopeniaa. Jotkin näistä tapahtumista ovat johtaneet kuolemaan (ks. kohta Haittavaikutukset).
Kyprolis aiheuttaa trombosytopeniaa. Pienimmät trombosyyttipitoisuudet havaittiin jokaisen 28 päivän hoitojakson 8. tai 15. päivänä, ja trombosyyttipitoisuus palautui lähtötasolle seuraavan hoitojakson alkuun mennessä (ks. kohta Haittavaikutukset). Trombosyyttiarvoa on seurattava usein Kyprolis‑hoidon aikana. Annosta pienennetään tai hoito keskeytetään tarpeen mukaan (ks. kohta Annostus ja antotapa).
Laskimoiden tromboemboliset tapahtumat
Kyprolis‑hoitoa saavilla potilailla on raportoitu laskimoiden tromboembolisia tapahtumia, esimerkiksi syviä laskimotrombooseja ja kuolemaan johtaneita keuhkoembolioita.
Jos potilaalla on tunnettuja tromboembolian riskitekijöitä – aikaisempi tromboosi mukaan luettuna – hänen tilaansa on seurattava huolellisesti. Kaikkia muutettavissa olevia riskitekijöitä (esim. tupakointia, hypertensiota ja hyperlipidemiaa) on pyrittävä vähentämään tehokkaasti. Varovaisuutta on noudatettava, jos annetaan samanaikaisesti muita lääkkeitä, jotka voivat lisätä tromboosiriskiä (esim. erytropoieesia stimuloivia lääkeaineita tai hormonikorvaushoitoa). Potilaiden ja lääkäreiden tulisi tarkkailla mahdollisia tromboemboliaan viittaavia muutoksia ja oireita. Potilaita on neuvottava hakeutumaan lääkärin hoitoon, jos heille ilmaantuu oireita, kuten hengenahdistusta, rintakipua, veriysköksiä, käsivarsien tai jalkojen turvotusta tai kipua.
Tromboosiprofylaksia on harkittava yksilöllisen hyöty‑riskiarvion perusteella.
Maksaan kohdistuvat haittavaikutukset
Maksan vajaatoimintaa, myös kuolemaan johtaneita tapauksia, on esiintynyt. Kyprolis voi aiheuttaa seerumin aminotransferaasiarvojen nousua (ks. kohta Haittavaikutukset). Annosta pienennetään tai hoito keskeytetään tarvittaessa (ks. kohta Annostus ja antotapa). Maksaentsyymi- ja bilirubiiniarvoja on seurattava karfiltsomibihoitoa aloitettaessa ja kuukausittain hoidon aikana, lähtöarvoista riippumatta.
Tromboottinen mikroangiopatia
Kyprolis‑hoitoa saaneilla potilailla on raportoitu tromboottista mikroangiopatiaa, kuten tromboottista trombosytopeenista purppuraa ja hemolyyttis‑ureemista oireyhtymää (TTP/HUS). Jotkin näistä tapahtumista ovat johtaneet kuolemaan. Tromboottiseen trombosytopeeniseen purppuraan / hemolyyttis‑ureemiseen oireyhtymään viittaavia merkkejä ja oireita on tarkkailtava. Jos TTP/HUS‑diagnoosia epäillään, Kyprolis‑hoito on lopetettava ja potilas on tutkittava. Jos TTP/HUS‑diagnoosi suljetaan pois, Kyprolis voidaan aloittaa uudelleen. Ei tiedetä, onko Kyprolis‑hoidon uudelleenaloittaminen turvallista potilaille, joilla on aikaisemmin todettu TTP/HUS.
Hypertensiivinen enkefalopatia
Kyprolis‑hoitoa saaneilla potilailla on raportoitu hypertensiivistä enkefalopatiaa. Hypertensiivinen enkefalopatia, josta on aiemmin käytetty nimeä posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES), on harvinainen neurologinen sairaus, jonka oireita voivat olla kouristuskohtaus, päänsärky, horros, sekavuus, sokeutuminen, tajunnan häiriöt sekä muut näköhäiriöt ja neurologiset häiriöt ja hypertensio. Diagnoosi varmistetaan neuroradiologisten kuvantamistutkimusten avulla. Jos hypertensiivistä enkefalopatiaa epäillään, Kyprolis‑hoito on lopetettava. Ei tiedetä, onko Kyprolis‑hoidon uudelleenaloittaminen turvallista potilaille, joilla on aikaisemmin todettu hypertensiivinen enkefalopatia.
Hepatiitti B -viruksen (HBV) uudelleenaktivoituminen
Karfiltsomibilla hoidetuilla potilailla on raportoitu hepatiitti B -viruksen (HBV) uudelleenaktivoitumista.
Kaikki potilaat on seulottava HBV-tartunnan varalta ennen karfiltsomibihoidon aloittamista. Potilaille, joilla todetaan HBV-vasta-aineita, on harkittava ennaltaehkäisevää viruslääkitystä. Tällaisia potilaita on tarkkailtava hepatiitti B -viruksen uudelleenaktivoitumisen kliinisten ja laboratoriotuloksissa ilmenevien merkkien varalta sekä hoidon aikana että hoidon päättymisen jälkeen. Tarvittaessa on konsultoitava hepatiitti B:n hoitoon perehtyneitä asiantuntijoita. Ei ole olemassa tietoa siitä, onko karfiltsomibihoitoa turvallista jatkaa uudelleen aktivoituneen hepatiitti B -infektion hallintaan saamisen jälkeen. Tämän vuoksi hoidon jatkamisesta on keskusteltava hepatiitti B:n hoitoon perehtyneiden asiantuntijoiden kanssa.
Etenevä multifokaalinen leukoenkefalopatia
Etenevää multifokaalista leukoenkefalopatiaa (PML) on raportoitu niillä karfiltsomibilla hoidetuilla potilailla, jotka ovat saaneet aiemmin tai samanaikaisesti immunosuppressiivista hoitoa.
Karfiltsomibilla hoidettavia potilaita on seurattava siltä varalta, että heillä ilmenee uusia tai pahenevia neurologisia, kognitiivisia tai käyttäytymiseen liittyviä merkkejä tai oireita, jotka saattavat viitata etenevään multifokaaliseen leukoenkefalopatiaan, kun niitä tarkastellaan osana keskushermoston sairauksien erotusdiagnostiikkaa.
Etenevää multifokaalista leukoenkefalopatiaa epäiltäessä lääkkeen antaminen on keskeytettävä, kunnes erikoislääkäri on sulkenut tämän mahdollisuuden pois asianmukaisilla diagnostisilla testeillä. Jos potilaalla todetaan etenevä multifokaalinen leukoenkefalopatia, hoito karfiltsomibilla on lopettava.
Raskauden ehkäisy
Naispotilaan, joka voi tulla raskaaksi, (ja/tai hänen kumppaninsa) on käytettävä tehokasta ehkäisyä hoidon aikana ja kuukauden ajan hoidon päättymisen jälkeen. Miespotilaan on käytettävä tehokasta ehkäisyä hoidon aikana ja 3 kuukautta hoidon päättymisen jälkeen, jos hänen kumppaninsa on raskaana tai jos kumppani voi tulla raskaaksi eikä käytä tehokasta ehkäisyä (ks. kohta Raskaus ja imetys). Karfiltsomibi saattaa heikentää ehkäisytablettien tehoa (ks. kohta Yhteisvaikutukset).
Natriumsisältö
Kyprolis 10 mg infuusiokuiva‑aine, liuosta varten
Tämä lääkevalmiste sisältää 37 mg natriumia per 10 mg:n injektiopullo, joka vastaa 1,9 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Kyprolis 30 mg infuusiokuiva‑aine, liuosta varten
Tämä lääkevalmiste sisältää 109 mg natriumia per 30 mg:n injektiopullo, joka vastaa 5,5 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Kyprolis 60 mg infuusiokuiva‑aine, liuosta varten
Tämä lääkevalmiste sisältää 216 mg natriumia per 60 mg:n injektiopullo, joka vastaa 11 % WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille.
Syklodekstriinisisältö
Kyprolis 10 mg infuusiokuiva‑aine, liuosta varten
Tämä lääkevalmiste sisältää 500 mg syklodekstriiniä (beetadeksisulfobutyylieetterinatriumia) per 10 mg:n injektiopullo, joka vastaa 88 mg/kg aikuiselle, joka painaa 70 kg.
Kyprolis 30 mg infuusiokuiva‑aine, liuosta varten
Tämä lääkevalmiste sisältää 1 500 mg syklodekstriiniä (beetadeksisulfobutyylieetterinatriumia) per 30 mg:n injektiopullo, joka vastaa 88 mg/kg aikuiselle, joka painaa 70 kg.
Kyprolis 60 mg infuusiokuiva‑aine, liuosta varten
Tämä lääkevalmiste sisältää 3 000 mg syklodekstriiniä (beetadeksisulfobutyylieetterinatriumia) per 60 mg:n injektiopullo, joka vastaa 88 mg/kg aikuiselle, joka painaa 70 kg.
Yhteisvaikutukset
Karfiltsomibi metaboloituu pääasiassa peptidaasi- ja epoksidihydrolaasientsyymien välityksellä, joten sytokromi P450 ‑entsyymien estäjien tai induktoreiden samanaikainen käyttö ei todennäköisesti vaikuta karfiltsomibin farmakokinetiikkaan.
In vitro ‑tutkimukset osoittivat, ettei karfiltsomibi indusoinut ihmisen CYP3A4-entsyymiä viljellyissä ihmisen maksasoluissa. Kliininen tutkimus, jossa suun kautta annettua midatsolaamia käytettiin CYP3A:n koetinsubstraattina ja karfiltsomibia annettiin 27 mg/m2 (2–10 minuutin infuusiona), osoitti, ettei samanaikaisesti annettu karfiltsomibi vaikuttanut midatsolaamin farmakokinetiikkaan. Tämä viittaa siihen, ettei karfiltsomibi todennäköisesti estä CYP3A4/5:n substraattien metaboliaa eikä ole CYP3A4:n induktori ihmiselimistössä. Annoksella 56 mg/m2 ei ole tehty kliinisiä tutkimuksia. Ei kuitenkaan tiedetä, onko karfiltsomibi CYP1A2-, 2C8-, 2C9-, 2C19- ja 2B6-entsyymien induktori hoitopitoisuuksina. Varovaisuutta on noudatettava, jos karfiltsomibia annetaan sellaisten lääkevalmisteiden kanssa, jotka ovat näiden entsyymien substraatteja. Tällaisia ovat esimerkiksi suun kautta otettavat ehkäisyvalmisteet. Raskauden ehkäisyyn on käytettävä tehokkaita ehkäisymenetelmiä (ks. kohta Raskaus ja imetys, lisäksi on tutustuttava lenalidomidin voimassa olevaan valmisteyhteenvetoon). Ehkäisytabletteja käyttävien potilaiden on siirryttävä käyttämään jotakin muuta tehokasta ehkäisymenetelmää.
Karfiltsomibi ei estä CYP1A2-, 2B6-, 2C8-, 2C9-, 2C19- eikä 2D6-entsyymin toimintaa in vitro, joten ei ole todennäköistä, että se muuttaisi estovaikutuksen seurauksena sellaisten lääkeaineiden pitoisuuksia, jotka ovat näiden entsyymien substraatteja.
Karfiltsomibi on P-glykoproteiinin (P‑gp:n) mutta ei BCRP:n substraatti. Koska Kyprolis annetaan laskimoon ja se metaboloituu tehokkaasti, P‑gp:n tai BCRP:n estäjät tai induktorit eivät todennäköisesti vaikuta karfiltsomibin farmakokinetiikkaan. In vitro, pitoisuuksina (3 µM), jotka ovat pienempiä kuin hoitoannosten aikaansaamat pitoisuudet, karfiltsomibi vähentää P‑gp:n substraatin, digoksiinin, ulospumppausta 25 %. Varovaisuutta on noudatettava, jos karfiltsomibia annetaan yhdessä P‑gp:n substraattien (esim. digoksiinin, kolkisiinin) kanssa.
Karfiltsomibi estää in vitro OATP1B1:n toimintaa (IC50 = 2,01 µM), mutta ei tiedetä, estääkö karfiltsomibi muiden kuljetusmekanismien, OATP1B3:n, OAT1:n, OAT3:n, OCT2:n ja BSEP:n, toimintaa systeemisellä tasolla. Karfiltsomibi ei estä ihmisen UGT2B7:n mutta estää ihmisen UGT1A1:n toimintaa (IC50 = 5,5 µM). Kuitenkin, kun otetaan huomioon karfiltsomibin nopea eliminoituminen, etenkin systeemisen pitoisuuden nopea pieneneminen 5 minuutin kuluttua infuusion päättymisestä, kliinisesti merkittävien yhteisvaikutusten riski OATP1B1:n ja UGT1A1:n substraattien kanssa on todennäköisesti pieni.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Ehkäisyohjeet miehille ja naisille
Kyprolis-hoitoa saavan naispotilaan, joka voi tulla raskaaksi, (ja/tai hänen kumppaninsa) on käytettävä tehokasta ehkäisyä hoidon aikana ja kuukauden ajan hoidon päättymisen jälkeen.
On mahdollista, että ehkäisytablettien teho saattaa heikentyä karfiltsomibihoidon aikana (ks. kohta Yhteisvaikutukset). Koska karfiltsomibihoitoon liittyy myös suurentunut laskimoiden tromboembolisten tapahtumien vaara, naisten on karfiltsomibihoidon aikana vältettävä hormonaalisia ehkäisyvalmisteita, joiden käyttöön liittyy tromboosiriski (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Jos potilas käyttää ehkäisytabletteja tai hormonaalista ehkäisymenetelmää, jonka käyttöön liittyy tromboosiriski, hänen on siirryttävä käyttämään jotakin muuta tehokasta ehkäisymenetelmää.
Miespotilaan on käytettävä tehokasta ehkäisyä hoidon aikana ja 3 kuukautta hoidon päättymisen jälkeen, jos hänen kumppaninsa on raskaana tai jos kumppani voi tulla raskaaksi eikä käytä tehokasta ehkäisyä.
Raskaus
Ei ole olemassa tietoja karfiltsomibin käytöstä raskaana oleville naisille.
Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutusmekanisminsa vuoksi ja eläinkokeissa saatujen löydösten perusteella Kyprolis voi vahingoittaa sikiötä, jos sitä annetaan raskauden aikana. Kyprolista ei pidä käyttää raskauden aikana, paitsi jos hoidon mahdollinen hyöty on suurempi kuin sikiölle mahdollisesti aiheutuva vaara. Potilaalle on kerrottava mahdollisesta sikiöön kohdistuvasta vaarasta, jos Kyprolista annetaan raskauden aikana tai jos potilas tulee raskaaksi tämän lääkkeen käytön aikana.
Lenalidomidi on rakenteellisesti sukua talidomidille. Talidomidin tiedetään olevan ihmiselle teratogeeninen lääkeaine, joka aiheuttaa vaikeita hengenvaarallisia synnynnäisiä epämuodostumia. Lenalidomidi voi aiheuttaa sikiöepämuodostumia, jos sitä käytetään raskauden aikana. Kaikkien potilaiden on täytettävä lenalidomidin raskaudenehkäisyohjelman ehdot, paitsi jos on luotettavia todisteita siitä, että potilas ei voi saada lapsia. Lisätietoja on lenalidomidin voimassa olevassa valmisteyhteenvedossa.
Imetys
Ei tiedetä, erittyykö karfiltsomibi tai erittyvätkö sen metaboliitit ihmisen rintamaitoon. Sen farmakologisten ominaisuuksien perusteella imeväiseen kohdistuvaa riskiä ei voida poissulkea. Siksi on varmuuden vuoksi vältettävä imettämistä Kyprolis-hoidon aikana ja vähintään 2 vuorokautta hoidon jälkeen.
Hedelmällisyys
Hedelmällisyystutkimuksia ei ole tehty eläimillä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Kyprolis‑valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn.
Kliinisissä tutkimuksissa on havaittu väsymystä, huimausta, pyörtymisiä, näön hämärtymistä, uneliaisuutta ja/tai verenpaineen laskua. Kyprolis‑hoitoa saavia potilaita on kehotettava välttämään ajamista ja koneiden käyttöä, jos heillä esiintyy tällaisia oireita.
Haittavaikutukset
Tiivistelmä turvallisuustiedoista
Vakavia haittavaikutuksia, joita voi esiintyä Kyprolis‑hoidon aikana, ovat: sydämen vajaatoiminta, sydäninfarkti, sydänpysähdys, sydänlihasiskemia, interstitiaalinen keuhkosairaus, pneumoniitti, akuutti hengitysvaikeusoireyhtymä, akuutti hengitysvajaus, kohonnut keuhkoverenpaine, hengenahdistus, hypertensio, myös hypertensiiviset kriisit, akuutti munuaisvaurio, tuumorilyysioireyhtymä, infuusioon liittyvä reaktio, ruoansulatuskanavan verenvuoto, kallonsisäinen verenvuoto, keuhkoverenvuoto, trombosytopenia, maksan vajaatoiminta, hepatiitti B -viruksen uudelleenaktivoituminen, hypertensiivinen enkefalopatia, tromboottinen mikroangiopatia ja tromboottinen trombosytopeeninen purppura / hemolyyttis‑ureeminen oireyhtymä (TTP/HUS). Kyproliksen kliinisissä tutkimuksissa sydämeen kohdistuneita haittavaikutuksia ja hengenahdistusta esiintyi yleensä Kyprolis‑hoidon varhaisvaiheessa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Yleisimmät haittavaikutukset (joita esiintyi yli 20 %:lla tutkittavista) olivat: anemia, väsymys, trombosytopenia, pahoinvointi, ripuli, kuume, hengenahdistus, hengitystieinfektio, yskä ja neutropenia.
Karfiltsomibin aloitusannos oli 20 mg/m2, ja se nostettiin tasolle 27 mg/m2 tutkimuksessa PX‑171‑009 ja tasolle 56 mg/m2 tutkimuksessa 2011‑003 (ks. kohta Farmakodynamiikka). Haittavaikutusten esiintymistä verrattiin tutkimuksen 2011‑003 Kyprolista ja deksametasonia (Kd) saaneen hoitohaaran ja tutkimuksen PX‑171‑009 Kyprolista, lenalidomidia ja deksametasonia (KRd) saaneen hoitohaaran välillä, ja tulokset viittaavat siihen, että seuraavat haittavaikutukset saattavat olla yhteydessä annokseen: sydämen vajaatoiminta (Kd 8,2 %, KRd 6,4 %), hengenahdistus (Kd 30,9 %, KRd 22,7 %), hypertensio (Kd 25,9 %, KRd 15,8 %) ja kohonnut keuhkoverenpaine (Kd 1,3 %, KRd 0,8 %).
Tutkimuksessa 20160275 (ks. kohta Farmakodynamiikka) Kyproliksen, daratumumabin ja deksametasonin yhdistelmähoitoa (KdD) verrattiin Kyproliksen ja deksametasonin yhdistelmähoitoon (Kd). 30 päivän kuluessa viimeisestä minkä tahansa tutkimuslääkkeen annoksesta kuoli haittapahtuman vuoksi 10 % KdD‑hoitohaaran potilaista ja 5 % Kd‑hoitohaaran potilaista. Yleisimpänä kuolemansyynä kummassakin hoitohaarassa olivat infektiot (KdD 5 %, Kd 3 %). Hoidon yhteydessä esiintyvien kuolemaan johtavien haittatapahtumien riski oli korkeampi ≥ 65‑vuotiailla tutkittavilla. Vakavia haittatapahtumia raportoitiin KdD‑hoitohaarassa 56 %:lla ja Kd‑hoitohaarassa 46 %:lla potilaista. Yleisimpiä raportoituja vakavia haittatapahtumia olivat anemia (KdD‑hoitohaarassa 2 % ja Kd‑hoitohaarassa 1 %), ripuli (KdD 2 %, Kd 0 %), kuume (KdD 4 %, Kd 2 %), keuhkokuume (KdD 12 %, Kd 9 %), influenssa (KdD 4 %, Kd 1 %), sepsis (KdD 4 %, Kd 1 %) ja bronkiitti (KdD 2 %, Kd 0 %).
Haittavaikutustaulukko
Haittavaikutukset luetellaan alla elinjärjestelmäluokan ja yleisyyden mukaan (ks. taulukko 6). Yleisyysluokat määritettiin kunkin haittavaikutuksen vakioimattomien ilmaantuvuuslukujen perusteella kliinisten tutkimusten yhdistetystä aineistosta (n = 3 878). Haittavaikutukset on esitetty kussakin elinjärjestelmä- ja yleisyysluokassa vakavuuden mukaan alenevassa järjestyksessä.
Taulukko 6. Haittavaikutustaulukko
| Elinjärjestelmä (MedDRA) | Hyvin yleinen (≥ 1/10) | Yleinen (≥ 1/100, < 1/10) | Melko harvinainen (≥ 1/1000, < 1/100) | Harvinainen (≥ 1/10 000, < 1/1000) |
| Infektiot | Keuhkokuume Hengitystieinfektio | Sepsis Keuhkoinfektio Influenssa Vyöruusu* Virtsatieinfektio Bronkiitti Maha‑suolitulehdus Virusinfektio Nenänielun tulehdus Nuha | Clostridium difficile ‑koliitti Sytomegalovirusinfektio Hepatiitti B -viruksen uudelleenaktivoituminen | |
| Immuunijärjestelmä | Lääkeyliherkkyys | |||
| Veri ja imukudos | Trombosytopenia Neutropenia Anemia Lymfopenia Leukopenia | Kuumeinen neutropenia | Hemolyyttis-ureeminen oireyhtymä Tromboottinen trombosytopeeninen purppura | Tromboottinen mikroangiopatia |
| Aineenvaihdunta ja ravitsemus | Hypokalemia Heikentynyt ruokahalu | Kuivuminen Hyperkalemia Hypomagnesemia Hyponatremia Hyperkalsemia Hypokalsemia Hypofosfatemia Hyperurikemia Hypoalbuminemia Hyperglykemia | Tuumorilyysioireyhtymä | |
| Psyykkiset häiriöt | Unettomuus | Ahdistuneisuus Sekavuustila | ||
| Hermosto | Huimaus Perifeerinen neuropatia Päänsärky | Parestesiat Heikentynyt tuntoaisti | Kallonsisäinen verenvuoto Äkillinen aivoverenkiertohäiriö Hypertensiivinen enkefalopatia | |
| Silmät | Kaihi Näön hämärtyminen | |||
| Kuulo ja tasapainoelin | Korvien soiminen | |||
| Sydän | Sydämen vajaatoiminta Sydäninfarkti Eteisvärinä Takykardia Pienentynyt ejektiofraktio Sydämentykytys | Sydänpysähdys Kardiomyopatia Sydänlihasiskemia Sydänpussitulehdus Sydänpussin nestekertymä Kammiotakykardia | ||
| Verisuonisto | Hypertensio | Syvä laskimotromboosi Hypotensio Kasvojen ja kaulan punoitus | Hypertensiivinen kriisi Verenvuoto | Hypertensiivinen hätätila |
| Hengityselimet, rintakehä ja välikarsina | Hengenahdistus Yskä | Keuhkoembolia Keuhkoedeema Nenäverenvuoto Suunielun kipu Ääntöhäiriö Hengityksen vinkuminen Kohonnut keuhkoverenpaine | ARDS Äkillinen hengitysvajaus Keuhkoverenvuoto Interstitiaalinen keuhkosairaus Pneumoniitti | |
| Ruoansulatuselimistö | Oksentelu Ripuli Ummetus Vatsakipu Pahoinvointi | Ruoansulatuskanavan verenvuoto Ruoansulatusvaivat Hammassärky | Ruoansulatuskanavan perforaatio Äkillinen haimatulehdus | |
| Maksa ja sappi | Kohonnut alaniiniaminotrans-feraasiarvo Kohonnut aspartaatti-aminotransferaasi-arvo Kohonnut gammaglutamyyli-transferaasiarvo Hyperbilirubinemia | Maksan vajaatoiminta Kolestaasi | ||
| Iho ja ihonalainen kudos | Ihottuma Kutina Punoitus Runsas hikoilu | Angioedeema | ||
| Luusto, lihakset ja sidekudos | Selkäkipu Nivelsärky Raajakipu Lihaskouristukset | Lihas- ja luustokipu Lihas- ja luustoperäinen rintakipu Luukipu Lihaskipu Lihasheikkous | ||
| Munuaiset ja virtsatiet | Kohonnut veren kreatiniiniarvo | Akuutti munuaisvaurio Munuaisten vajaatoiminta Heikentynyt munuaistoiminta Heikentynyt kreatiniinin munuaispuhdistuma | ||
| Yleisoireet ja antopaikassa todettavat haitat | Kuume Perifeerinen edeema Voimattomuus Väsymys Vilunväristykset | Rintakipu Kipu Infuusiokohdan reaktiot Influenssan kaltainen sairaus Yleinen huonovointisuus | Monielinvaurio-oireyhtymä | |
| Tutkimukset | Kohonnut C‑reaktiivinen proteiini (CRP) Kohonnut veren virtsahappoarvo | |||
| Vammat ja myrkytykset | Infuusioon liittyvä reaktio | |||
| * Esiintymistiheys on laskettu sellaisten kliinisten tutkimusten tiedoista, joissa suurin osa potilaista sai ehkäisevää hoitoa | ||||
Tärkeimpien haittavaikutusten kuvaus
Sydämen vajaatoiminta, sydäninfarkti ja sydänlihasiskemia
Kyproliksen kliinisissä tutkimuksissa sydämen vajaatoiminta raportoitiin noin 5 %:lla tutkittavista (noin 3 %:lla tutkittavista oli ≥ 3. asteen tapahtumia), sydäninfarkti raportoitiin noin 1 %:lla tutkittavista (noin 1 %:lla tutkittavista oli ≥ 3. asteen tapahtumia) ja sydänlihasiskemia raportoitiin < 1 %:lla tutkittavista (< 1 %:lla tutkittavista oli ≥ 3. asteen tapahtumia). Nämä tapahtumat ilmaantuivat yleensä Kyprolis‑hoidon alkuvaiheessa (5 ensimmäisen hoitojakson aikana).
Tutkimuksessa 20160275 sydänoireiden kokonaisilmaantuvuus (kaikki ja kaiken asteiset tapahtumat) potilailla, joilla oli lähtötilanteessa verisuonisairauksia, oli 29,9 % KdD‑hoitohaarassa ja 19,8 % Kd‑hoitohaarassa. Potilailla, joilla oli lähtötilanteessa hypertensiota, kokonaisilmaantuvuus oli 30,6 % KdD‑hoitohaarassa ja 18,1 % Kd‑hoitohaarassa. Kuolemaan johtaneiden sydäntapahtumien ilmaantuvuus oli ensin mainitussa alaryhmässä 1,9 % KdD‑hoitohaarassa ja 0,0 % Kd‑hoitohaarassa, ja jälkimmäisessä alaryhmässä 1,5 % KdD‑hoitohaarassa ja 0,0 % Kd‑hoitohaarassa. Mikään yksittäinen sydäntapahtumatyyppi ei selittänyt KdD- ja Kd‑hoitohaarojen välistä eroa alaryhmissä, joiden potilailla oli lähtötilanteessa verisuonisairauksia tai hypertensiota.
Kyprolis‑hoidon aikana ilmaantuvien sydänoireiden kliinistä hoitoa kuvataan kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Hengenahdistus
Hengenahdistusta raportoitiin noin 24 %:lla tutkittavista Kyproliksen kliinisissä tutkimuksissa. Suurin osa haittavaikutuksena ilmaantuneista hengenahdistustapauksista ei ollut vakavia (< 5 %:lla tutkittavista oli ≥ 3. asteen tapahtumia), hengenahdistus korjaantui, johti harvoin hoidon keskeyttämiseen ja alkoi tutkimuksen varhaisessa vaiheessa (3 ensimmäisen hoitojakson aikana). Kyprolis‑hoidon aikana ilmaantuvan hengenahdistuksen kliinistä hoitoa kuvataan kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Hypertensio ja hypertensiiviset kriisit
Hypertensiivisiä kriisejä (kiireellistä hoitoa vaativia tiloja tai hypertensiivisiä hätätilanteita) on esiintynyt Kyprolis‑annosten antamisen jälkeen. Jotkin näistä tapahtumista ovat johtaneet kuolemaan. Kliinisissä tutkimuksissa hypertensiota esiintyi haittatapahtumana noin 21 %:lla tutkittavista ja 8 %:lla tutkittavista oli ≥ 3. asteen hypertensiotapahtumia mutta hypertensiivisiä kriisejä esiintyi < 0,5 %:lla tutkittavista. Hypertension ilmaantuvuus haittatapahtumana oli samanlainen potilailla, joilla oli aikaisemmin todettu hypertensiota, kuin niillä, joilla ei ollut esiintynyt hypertensiota. Kyprolis‑hoidon aikana ilmaantuvan hypertension kliinistä hoitoa kuvataan kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Trombosytopenia
Trombosytopeniaa raportoitiin Kyproliksen kliinisissä tutkimuksissa noin 33 %:lla tutkittavista, ja noin 20 %:lla tutkittavista oli ≥ 3. asteen tapahtumia. Tutkimuksessa 20160275 ≥ 3. asteen trombosytopenian ilmaantuvuus oli KdD‑hoitohaarassa 24,4 % ja Kd‑hoitohaarassa 16,3 %. Kyprolis aiheuttaa trombosytopeniaa estämällä trombosyyttien kuroutumisen megakaryosyyteistä, mikä johtaa klassiseen sykliseen trombosytopeniaan. Pienimmät trombosyyttipitoisuudet havaittiin jokaisen 28 päivän hoitojakson 8. tai 15. päivänä, ja trombosyyttipitoisuus palautui yleensä lähtötasolle seuraavan hoitojakson alkuun mennessä. Kyprolis‑hoidon aikana ilmaantuvan trombosytopenian kliinistä hoitoa kuvataan kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Laskimoiden tromboemboliset tapahtumat
Kyprolis‑hoitoa saavilla potilailla on raportoitu laskimoiden tromboembolisia tapahtumia, esimerkiksi syviä laskimotrombooseja ja kuolemaan johtaneita keuhkoembolioita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Kolmessa kolmannen vaiheen tutkimuksessa laskimoiden tromboembolisten tapahtumien kokonaisilmaantuvuus oli suurempi Kyprolista saaneissa hoitohaaroissa. Tutkimuksessa PX‑171‑009 laskimoiden tromboembolisten tapahtumien ilmaantuvuus oli KRd‑haarassa 15,6 % ja Rd‑haarassa 9,0 %. Vaikeusasteluokan ≥ 3 laskimoiden tromboembolisia tapahtumia raportoitiin KRd‑hoitohaarassa 5,6 %:lla ja Rd‑hoitohaarassa 3,9 %:lla potilaista. Tutkimuksessa 2011‑003 laskimoiden tromboembolisten tapahtumien ilmaantuvuus oli Kd‑hoitohaarassa 12,5 % ja bortetsomibia ja deksametasonia (Vd) saaneessa haarassa 3,3 %. Vaikeusasteluokan ≥ 3 laskimoiden tromboembolisia tapahtumia raportoitiin Kd‑hoitohaarassa 3,5 %:lla ja Vd‑hoitohaarassa 1,8 %:lla potilaista. Tutkimuksessa 20160275 laskimoiden tromboembolisten tapahtumien ilmaantuvuus oli KdD‑hoitohaarassa 6,2 % ja Kd‑hoitohaarassa 11,1 %. Vaikeusasteluokan ≥ 3 laskimoiden tromboembolisia tapahtumia raportoitiin KdD‑hoitohaarassa 1,9 %:lla ja Kd‑hoitohaarassa 6,5 %:lla potilaista.
Maksan vajaatoiminta
Maksan vajaatoimintaa, myös kuolemaan johtaneita tapauksia, on raportoitu alle yhdellä prosentilla tutkittavista Kyproliksen kliinisissä tutkimuksissa. Kyprolis‑hoidon aikana ilmaantuvien maksavaikutusten kliinistä hoitoa kuvataan kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
Perifeerinen neuropatia
Avoimessa satunnaistetussa monikeskustutkimuksessa, jossa potilaat saivat Kyprolista 20/56 mg/m2 30 minuutin infuusiona yhdessä deksametasonin kanssa (Kd, n = 464) tai bortetsomibia ja deksametasonia (Vd, n = 465), uusiutunutta multippelia myeloomaa sairastaneista Kd‑hoitohaaran potilaista 7 %:lla ja Vd‑hoitohaaran potilaista 35 %:lla raportoitiin vähintään vaikeusasteluokan 2 perifeeristä neuropatiaa etukäteen suunnitellun elinaika‑analyysin ajankohtana. Tutkimuksessa 20160275 uusiutunutta multippelia myeloomaa sairastaneista KdD‑hoitohaaran potilaista 10,1 %:lla ja Kd‑hoitohaaran potilaista 3,9 %:lla raportoitiin vähintään vaikeusasteluokan 2 perifeeristä neuropatiaa.
Infuusioreaktio
Tutkimuksessa 20160275 infuusioreaktion riski osoittautui suuremmaksi, kun karfiltsomibia annetaan yhdessä daratumumabin kanssa.
Hengitystieinfektiot
Tutkimuksessa 20160275 vakavina haittavaikutuksina raportoituja hengitystieinfektioita ilmeni kummassakin hoitoryhmässä (KdD 27,6 %, Kd 15,0 %). Tutkimuksessa 20160275 vakavana haittavaikutuksena raportoitua keuhkokuumetta ilmeni kummassakin hoitoryhmässä (KdD 15,3 %, Kd 9,8 %). Tapahtumista kuolemaan johtavia oli 1,3 % KdD‑hoitohaarassa ja 0 % Kd‑hoitohaarassa.
Uudet sekundaariset primaarisyövät
Tutkimuksessa 20160275 uusia sekundaarisia primaarisyöpiä on raportoitu kummassakin hoitoryhmässä (KdD 1,9 %, Kd 1,3 %).
Opportunisti‑infektiot
Tutkimuksessa 20160275 opportunisti‑infektioita on raportoitu kummassakin hoitoryhmässä (KdD 9,4 %, Kd 3,9 %). KdD‑hoitohaarassa ≥ 1 %:lla tutkittavista ilmenneitä opportunisti‑infektioita olivat vyöruusu, sammas, oraaliherpes ja herpes simplex.
Hepatiitti B ‑viruksen uudelleenaktivoituminen
Tutkimuksessa 20160275 hepatiitti B -viruksen uudelleenaktivoitumista ilmeni 0,6 %:lla KdD‑hoitohaaran potilaista ja 0 %:lla Kd‑hoitohaaran potilaista.
Muut erityisryhmät
Iäkkäät potilaat
Kyproliksen kliinisissä tutkimuksissa tiettyjen haittatapahtumien (myös sydämen rytmihäiriöiden, sydämen vajaatoiminnan (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet), hengenahdistuksen, leukopenian ja trombosytopenian) potilaskohtainen ilmaantuvuus oli yleisesti suurempi ≥ 75‑vuotiailla kuin alle 75‑vuotiailla potilailla.
Tutkimuksessa 20160275 KdD‑hoitoa sai annoksella 20/56 mg/m2 kahdesti viikossa 308 potilasta, joista 47 % oli ≥ 65‑vuotiaita. Tutkimuksen KdD‑hoitohaarassa hoidon yhteydessä esiintyviä kuolemaan johtavia haittatapahtumia ilmeni 6 %:lla alle 65‑vuotiaista potilaista ja 14 %:lla ≥ 65‑vuotiaista potilaista. Kd‑hoitohaarassa tällaisia tapahtumia ilmeni 8 %:lla alle 65‑vuotiaista potilaista ja 3 %:lla ≥ 65‑vuotiaista potilaista.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‑haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta: www.fimea.fi.
Yliannostus
Toistaiseksi ei ole riittävästi tietoa, jotta voitaisiin tehdä johtopäätöksiä kliinisissä tutkimuksissa käytettyjä annoksia suurempien annosten turvallisuudesta. Äkillisesti alkaneita vilunväristyksiä, verenpaineen laskua, munuaisten vajaatoimintaa, trombosytopeniaa ja lymfopeniaa raportoitiin, kun oli annettu erehdyksessä 200 mg:n Kyprolis-annos.
Karfiltsomibin yliannokseen ei ole tunnettua spesifistä vastalääkettä. Yliannostapauksessa potilasta on tarkkailtava, erityisesti kohdassa Haittavaikutukset mainittujen Kyproliksen haittavaikutusten varalta.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Solunsalpaajat, muut syöpälääkkeet, ATC-koodi: L01XG02
Vaikutusmekanismi
Karfiltsomibi on epoksiketonipohjainen tetrapeptidirakenteinen proteasomin estäjä, joka sitoutuu selektiivisesti ja palautumattomasti N-terminaaliseen treoniiniin, jossa ovat 26S-proteasomikompleksin sisällä olevan proteolyyttisen ydinpartikkelin, 20S-proteasomin, aktiiviset kohdat, ja se vaikuttaa muihin proteaasiluokkiin vain vähän tai ei lainkaan. Karfiltsomibilla oli proliferaatiota estäviä ja apoptoosia edistäviä vaikutuksia hematologisten kasvainten prekliinisissä malleissa. Eläinkokeissa karfiltsomibi esti proteasomien toimintaa veressä ja kudoksessa ja hidasti kasvaimen kasvua multippelin myelooman malleissa. In vitro karfiltsomibin neurotoksinen vaikutus oli hyvin vähäinen, ja se reagoi hyvin heikosti muihin proteaaseihin kuin proteasomeihin.
Farmakodynaamiset vaikutukset
Laskimoon annettu karfiltsomibi esti proteasomin kymotrypsiinin kaltaista (CT‑L) vaikutusta mitattuna verestä tunnin kuluttua ensimmäisestä annoksesta. Annokset ≥ 15 mg/m2 estivät johdonmukaisesti proteasomin kymotrypsiinin kaltaista vaikutusta (≥ 80-prosenttisesti). Lisäksi karfiltsomibi esti immunoproteasomin LMP2-alayksikön (latent membrane protein 2) toimintaa 26−32-prosenttisesti ja MECL1-alayksikön (multicatalytic endopeptidase complex‑like 1) toimintaa 41–49-prosenttisesti annostasolla 20 mg/m2. Proteasomin toimintaa estävä vaikutus säilyi ≥ 48 tuntia kunkin hoitoviikon ensimmäisen karfiltsomibiannoksen jälkeen. Yhteiskäyttö lenalidomidin ja deksametasonin kanssa ei vaikuttanut proteasomia estävään tehoon.
Suurempi annos (56 mg/m2) esti tehokkaammin CT-L-alayksiköiden (≥ 90-prosenttisesti) ja myös proteasomin muiden alayksiköiden (LMP7, MECL1 ja LMP2) toimintaa kuin pienemmät annokset (15–20 mg/m2). Estovaikutus oli LMP7-alayksikön osalta noin 8 %, MECL1:n osalta noin 23 % ja LMP2:n osalta noin 34 % tehokkaampi annostasolla 56 mg/m2 kuin annostasolla 15–20 mg/m2. Karfiltsomibin proteasomin toimintaa estävä vaikutus oli samanlainen 2–10 minuutin ja 30 minuutin infuusioaikaa käytettäessä kahdella testatulla annostasolla (20 ja 36 mg/m2).
Kliininen teho ja turvallisuus
Kyprolis yhdessä lenalidomidin ja deksametasonin kanssa uusiutuneen multippelin myelooman hoidossa – tutkimus PX‑171‑009 (ASPIRE)
Kyprolis-hoidon turvallisuutta ja tehoa arvioitiin satunnaistetussa avoimessa monikeskustutkimuksessa 792 potilaalla, joilla oli uusiutunut multippeli myelooma. Kyproliksen, lenalidomidin ja deksametasonin yhdistelmähoitoa verrattiin pelkkään lenalidomidin ja deksametasonin yhdistelmään, ja potilaat satunnaistettiin suhteessa 1:1.
Tässä tutkimuksessa Kyproliksen aloitusannos oli 20 mg/m2, joka nostettiin tasolle 27 mg/m2 ensimmäisen hoitojakson 8. päivänä. Kyprolis annettiin 10 minuutin infuusiona kaksi kertaa viikossa kolmena viikkona neljästä. Kyprolis-hoitoa annettiin enintään 18 hoitojakson ajan, ellei sitä lopetettu ennenaikaisesti taudin etenemisen tai kestämättömien haittavaikutusten vuoksi. Lenalidomidi- ja deksametasonihoitoa voitiin jatkaa taudin etenemiseen tai kestämättömien haittavaikutusten ilmaantumiseen saakka.
Tutkimuksen poissulkukriteereitä olivat: kreatiniinipuhdistuma < 50 ml/min, NYHA-luokan III tai IV sydämen kongestiivinen vajaatoiminta tai sydäninfarkti 4 edellisen kuukauden aikana, taudin eteneminen bortetsomibia sisältäneen hoito-ohjelman aikana tai taudin eteneminen 3 ensimmäisen kuukauden aikana lenalidomidi- ja deksametasonihoidon aloittamisen jälkeen tai taudin eteneminen milloin tahansa lenalidomidi- ja deksametasonihoidon aikana, jos tämä oli potilaan viimeisin hoitolinja.Tutkimuksen valintakriteerit sallivat bortetsomibille (n = 118) tai lenalidomidille (n = 57) resistenttien myeloomapotilaiden pienen alaryhmän mukaanoton. Tutkimukseen otetut potilaat määriteltiin hoitoresistenteiksi, jos he täyttivät jonkin seuraavista kolmesta kriteeristä: vasteen puuttuminen (< hyvin heikko vaste) mihin tahansa hoito-ohjelmaan, taudin eteneminen minkä tahansa hoito-ohjelman aikana tai taudin eteneminen 60 vuorokauden kuluessa minkä tahansa hoito-ohjelman päättymisestä. Tässä tutkimuksessa ei arvioitu hyöty‑riskisuhdetta tässä laajemmassa refraktaarisessa potilasjoukossa.
Potilaiden tautitila ja muut ominaisuudet lähtötilanteessa olivat molemmissa hoitohaaroissa hyvin samankaltaiset. Näitä olivat ikä (64 vuotta, vaihteluväli 31–91 vuotta), sukupuoli (56 % miehiä), ECOG-toimintakykyluokka (48 %:lla toimintakykyluokka 1), suuren riskin geneettisiä mutaatioita, joita olivat geneettiset alatyypit t(4;14), t(14;16) tai 17p deleetio ≥ 60 prosentissa plasmasoluista (13 %), geneettisen mutaation riskiluokitus ei tiedossa, koska potilaiden tuloksia ei kerätty tai niitä ei analysoitu (47 %), ja lähtötilanteessa ISS-vaikeusasteluokan III tauti (20 %). Potilaat olivat saaneet 1–3 aikaisempaa hoitolinjaa (mediaani 2), joihin kuuluivat aikaisemmat hoidot bortetsomibilla (66 %), talidomidilla (44 %) ja lenalidomidilla (20 %).
Taulukossa 7 ja kuvissa 1 ja 2 on tiivistelmä tutkimuksen PX‑171‑009 tuloksista.
Taulukko 7. Tiivistelmä tehon analyysistä uusiutuneen multippelin myelooman hoidossa tutkimuksessa PX‑171‑009
| KRd-yhdistelmähoito | ||
KRd-hoitohaaraa (N = 396) | Rd-hoitohaaraa (N = 396) | |
| PFS-mediaani, kk (95 % CI) | 26,3 (23,3–30,5) | 17,6 (15,0–20,6) |
| HR (95 % CI); 1‑suuntainen p‑arvob | 0,69 (0,57–0,83); < 0,0001 | |
| OS-mediaani, kk (95 % CI) | 48,3 (42,4–52,8) | 40,4 (33,6–44,4) |
| HR (95 % CI); 1‑suuntainen p‑arvob | 0,79 (0,67–0,95); 0,0045 | |
| ORR, n (%) | 345 (87,1) | 264 (66,7) |
| sCR | 56 (14,1) | 17 (4,3) |
| CR | 70 (17,7) | 20 (5,1) |
| VGPR | 151 (38,1) | 123 (31,1) |
| PR | 68 (17,2) | 104 (26,3) |
| ORR, 95 % CI | 83,4–90,3 | 61,8–71,3 |
| 1‑suuntainen p‑arvo | < 0,0001 | |
KRd = Kyprolis, lenalidomidi ja deksametasoni; Rd = lenalidomidi ja deksametasoni; PFS = elinaika ilman taudin etenemistä; HR = vaarasuhde; CI = luottamusväli; OS = kokonaiselinaika; ORR = kokonaisvasteosuus; sCR (stringent complete response) = täydellinen vaste lisäehdoin; CR = täydellinen vaste; VGPR = erittäin hyvä osittainen vaste; PR = osittainen vaste; IMWG = kansainvälinen myeloomatyöryhmä; EBMT = Euroopan kantasolusiirtojärjestö a. Riippumattoman arviointikomitean määrittelemänä standardeja objektiivisia IMWG/EBMT-vastekriteerejä käyttäen b. Tilastollisesti merkitsevä | ||
Kyprolis-, lenalidomidi- ja deksametasonihoitohaaran (KRd) potilailla elinaika ilman taudin etenemistä (PFS) oli parempi kuin lenalidomidia ja deksametasonia (Rd) saaneen haaran potilailla (HR = 0,69, yksisuuntainen p‑arvo < 0,0001). Tämä tarkoittaa PFS:n paranemista 45 %:lla tai tapahtumariskin pienenemistä 31 %:lla riippumattoman arviointikomitean määrittelemänä kansainvälisen myeloomatyöryhmän ja Euroopan kantasolusiirtojärjestön (IMWG/EBMT) standardeja objektiivisia vastekriteerejä käyttäen.
KRd-hoidon aikaansaama PFS-hyöty tuli esiin johdonmukaisesti kaikissa alaryhmissä, joita olivat ≥ 75-vuotiaat potilaat (n = 96), potilaat, joilla oli suuren riskin geneettisiä mutaatioita (n = 100) tai geneettisen mutaation riskiluokitus ei ollut tiedossa (n = 375), ja potilaat, joilla kreatiniinipuhdistuman lähtöarvo oli 30 – < 50 ml/min (n = 56).
Kuva 1. Kaplan–Meier-kuvaaja uusiutunutta multippelia myeloomaa sairastavien potilaiden elinajasta ilman taudin etenemistä (PFS)a
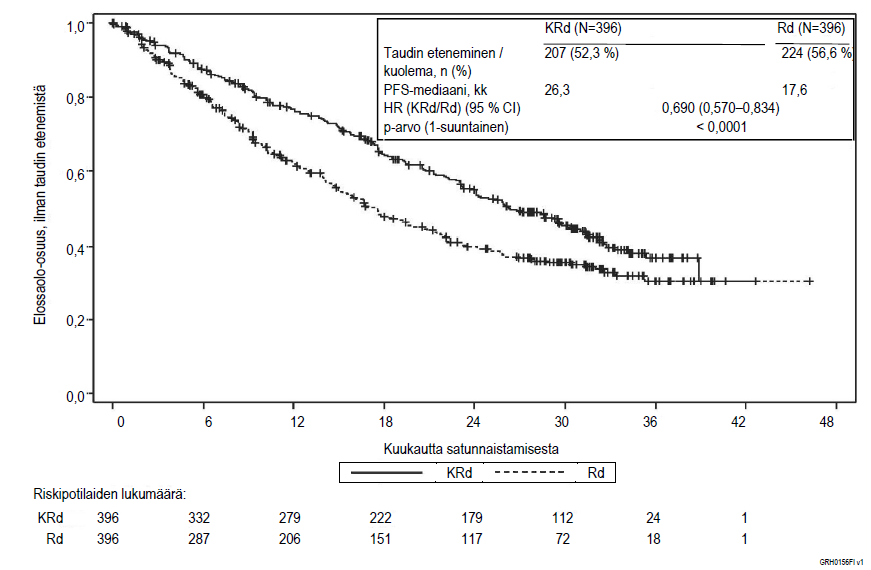
KRd = Kyprolis, lenalidomidi ja deksametasoni; Rd = lenalidomidi ja deksametasoni; PFS = elinaika ilman taudin etenemistä; HR = vaarasuhde; CI = luottamusväli; IMWG = kansainvälinen myeloomatyöryhmä; EBMT = Euroopan kantasolusiirtojärjestö; kk = kuukautta
Huom: Hoitovaste ja taudin eteneminen määritettiin standardeja objektiivisia IMWG/EBMT-vastekriteerejä käyttäen.
a. Tutkimus PX‑171‑009
Etukäteen suunniteltu kokonaiselinajan (OS) analyysi tehtiin, kun KRd-hoitohaaran potilaista 246 ja Rd-hoitohaaran potilaista 267 oli kuollut. Seuranta-ajan mediaani oli noin 67 kuukautta. KRd-hoitohaaran potilailla havaittiin tilastollisesti merkitsevä elinaikaetu Rd-hoitohaaran potilaisiin verrattuna. KRd-hoitohaaran potilaiden kuolemanvaara pieneni 21 % verrattuna Rd-hoitohaaran potilaisiin (HR = 0,79; 95 %:n luottamusväli 0,67–0,95; p‑arvo = 0,0045). Elinajan mediaani oli 7,9 kuukautta pidempi KRd-hoitohaaran potilailla kuin Rd-hoitohaaran potilailla (ks. taulukko 7 ja kuva 2).
Kuva 2. Kaplan–Meier-kuvaaja uusiutunutta multippelia myeloomaa sairastavien potilaiden kokonaiselinajastaa
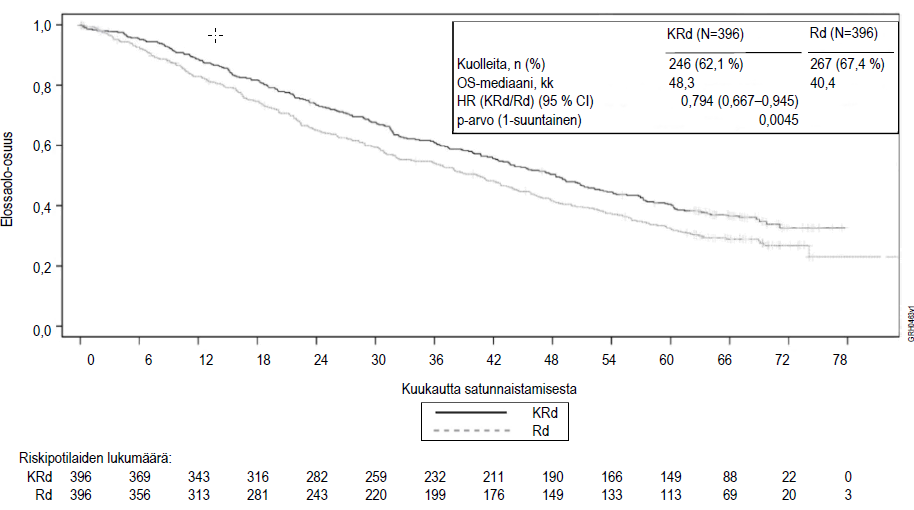
KRd = Kyprolis, lenalidomidi ja deksametasoni; Rd = lenalidomidi ja deksametasoni; OS = kokonaiselinaika; HR = vaarasuhde; CI = luottamusväli; kk = kuukautta
a. Tutkimus PX‑171‑009
KRd-hoitoa saaneiden potilaiden raportoima yleinen terveydentila ja yleistä terveydentilaa / elämänlaatua mittaavat pistearvot olivat parempia Rd-hoitohaaraan verrattuina 18 hoitojakson jälkeen (1‑suuntainen p‑arvo = 0,0001 ilman monivertailukorjausta) mitattuna EORTC QLQ‑C30 ‑asteikolla, joka on validoitu multippelin myelooman arviointiasteikko.
Kyprolis yhdessä deksametasonin kanssa uusiutuneen multippelin myelooman hoidossa – tutkimus 2011‑003 (ENDEAVOR)
Kyprolis-hoidon turvallisuutta ja tehoa arvioitiin kolmannen vaiheen satunnaistetussa avoimessa monikeskustutkimuksessa, jossa Kyproliksen ja deksametasonin yhdistelmää (Kd) verrattiin bortetsomibin ja deksametasonin yhdistelmään (Vd). Yhteensä 929 potilasta, joilla oli uusiutunut tai refraktaarinen multippeli myelooma ja jotka olivat saaneet 1–3 aikaisempaa hoitolinjaa, otettiin mukaan tutkimukseen ja satunnaistettiin (464 Kd-hoitohaaraan ja 465 Vd-hoitohaaraan).
Tässä tutkimuksessa Kyproliksen aloitusannos oli 20 mg/m2, joka nostettiin tasolle 56 mg/m2 ensimmäisen hoitojakson 8. päivänä. Kyprolis annettiin 30 minuutin infuusiona kaksi kertaa viikossa kolmena viikkona neljästä taudin etenemiseen tai kestämättömien haittavaikutusten ilmaantumiseen asti.
Vd-hoitohaaraan satunnaistetuille potilaille voitiin antaa bortetsomibia joko laskimoon (n = 108) tai ihon alle (n = 357). Tutkimuksen poissulkukriteereitä olivat: kreatiniinipuhdistuma < 15 ml/min, NYHA-luokan III-IV sydämen kongestiivinen vajaatoiminta, sydäninfarkti neljän edellisen kuukauden aikana tai vasemman kammion ejektiofraktio (LVEF) < 40 %. Tutkimuksen valintakriteerit sallivat aikaisemman karfiltsomibi- (n = 3) tai bortetsomibihoidon (n = 502), mikäli aikaisemmalla proteasominestäjähoidolla oli saavutettu vähintään osittainen hoitovaste (PR) eikä proteasominestäjähoitoa jouduttu lopettamaan haittavaikutusten vuoksi ja proteasominestäjähoidon viimeisestä annoksesta oli kulunut vähintään 6 kuukautta.
Potilaiden demografiset ja muut ominaisuudet lähtötilanteessa olivat hyvin samankaltaiset tutkimuksen 2011‑003 molemmissa hoitohaaroissa. Niitä olivat aikaisempi bortetsomibihoito (54 %), aikaisempi lenalidomidihoito (38 %), lenalidomidiresistenssi (25 %), ikä (65 vuotta, vaihteluväli 30–89 vuotta), sukupuoli (51 % miehiä), ECOG-toimintakykyluokka (45 %:lla toimintakykyluokka 1), suuren riskin geneettisiä mutaatioita, joita olivat geneettiset alatyypit t(4;14) tai t(14;16) vähintään 10 prosentissa seulotuista plasmasoluista tai 17p deleetio ≥ 20 prosentissa plasmasoluista (23 %), geneettisen mutaation riskiluokitus ei tiedossa, koska potilaiden tuloksia ei kerätty tai niitä ei analysoitu (9 %), ja lähtötilanteessa ISS-vaikeusasteluokan III tauti (24 %).
Taulukossa 8 on tiivistelmä tutkimuksen 2011‑003 tuloksista.
Taulukko 8. Tiivistelmä tehon analyysistä uusiutuneen multippelin myelooman hoidossa tutkimuksessa 2011‑003
Kd-hoitohaara (N = 464) | Vd-hoitohaara (N = 465) | |
| PFS-mediaani, kk (95 % CI)a | 18,7 (15,6–NE) | 9,4 (8,4–10,4) |
| HR (95 % CI); 1‑suuntainen p‑arvob | 0,533 (0,44–0,65); < 0,0001 | |
| Kokonaiselinaika, kk, mediaani (95 % CI) | 47,6 (42,5–NE) | 40,0 (32,6–42,3) |
| HR (95 % CI); 1-suuntainen p-arvob | 0,791 (0,65–0,96); 0,010 | |
| ORR n (%)a, c | 357 (76,9) | 291 (62,6) |
| ≥ CRd | 58 (12,5) | 29 (6,2) |
| ≥ VGPRe | 252 (54,3) | 133 (28,6) |
| ORR, 95 % CI | 72,8–80,7 | 58,0–67,0 |
| 1‑suuntainen p‑arvob | < 0,0001 | |
Kd = Kyprolis + deksametasoni; Vd = bortetsomibi ja deksametasoni; CI = luottamusväli; NE = ei arvioitavissa; HR = vaarasuhde; ORR = kokonaisvasteosuus; CR = täydellinen vaste; VGPR = erittäin hyvä osittainen vaste a. Nämä päätetapahtumat olivat riippumattoman arviointikomitean (IRC) määrittelemiä b. Tilastollisesti merkitsevä c. Kokonaisvasteeksi katsotaan paras saavutettu kokonaisvaste, joka voi olla osittainen vaste (PR), erittäin hyvä osittainen vaste (VGPR), täydellinen vaste (CR) tai täydellinen vaste lisäehdoin (sCR) d. Tilastollisesti merkitsevä, 1‑suuntainen p‑arvo = 0,0005 e. Tilastollisesti merkitsevä, 1‑suuntainen p‑arvo = 0,0001 | ||
Tutkimus osoitti, että elinaika ilman taudin etenemistä (PFS) oli Kd-hoitohaaran potilailla merkitsevästi parempi kuin Vd-hoitohaaran potilailla (HR: 0,53, 95 % CI: 0,44–0,65 [p‑arvo < 0,0001]) (ks. kuva 3).
PFS-tulokset olivat samanlaiset potilailla, jotka olivat aikaisemmin saaneet bortetsomibihoitoa (HR 0,56, 95 % CI: 0,44–0,73), ja niillä, jotka eivät olleet aikaisemmin saaneet bortetsomibia (HR 0,48, 95 % CI: 0,36–0,66).
Kd-hoidon aikaansaama PFS-hyöty tuli esiin johdonmukaisesti kaikissa alaryhmissä, joita olivat ≥ 75‑vuotiaat potilaat (n = 143), potilaat, joilla oli suuren riskin geneettisiä mutaatioita (n = 210), ja potilaat, joilla kreatiniinipuhdistuman lähtöarvo oli 30 – < 50 ml/min (n = 128).
Potilailla, jotka olivat aikaisemmin saaneet bortetsomibia (54 %), PFS-mediaani oli 15,6 kuukautta Kd-hoitohaarassa ja 8,1 kuukautta Vd-haarassa (HR = 0,56, 95 % CI: 0,44–0,73), kokonaisvasteosuus (ORR) oli Kd-haarassa 71,2 % ja Vd-haarassa 60,3 %.
Potilailla, jotka olivat aikaisemmin saaneet lenalidomidia (38 %), PFS-mediaani oli 12,9 kuukautta Kd-hoitohaarassa ja 7,3 kuukautta Vd-haarassa (HR = 0,69, 95 % CI: 0,52–0,92), kokonaisvasteosuus (ORR) oli Kd-haarassa 70,1 % ja Vd-haarassa 59,3 %. Potilailla, jotka olivat resistenttejä lenalidomidille (25 %), PFS-mediaani oli 8,6 kuukautta Kd-hoitohaarassa ja 6,6 kuukautta Vd-haarassa (HR = 0,80, 95 % CI: 0,57–1,11), kokonaisvasteosuus (ORR) oli Kd-haarassa 61,9 % ja Vd-haarassa 54,9 %.
Kuva 3. Kaplan–Meier-kuvaaja elinajasta ilman taudin etenemistä (PFS) riippumattoman arviointikomitean (IRC) määrittelemänä (hoitoaikeen [ITT] mukainen potilasjoukko) tutkimuksessa 2011‑003
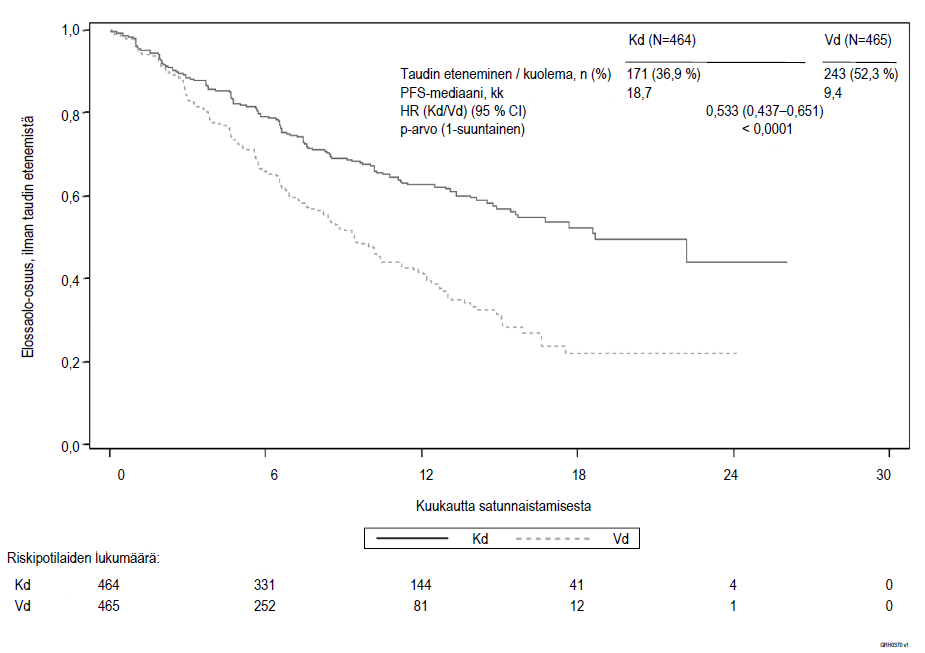
Kd = Kyprolis + deksametasoni; Vd = bortetsomibi + deksametasoni; PFS = elinaika ilman taudin etenemistä; kk = kuukautta; HR = vaarasuhde; CI = luottamusväli
Etukäteen suunniteltu toinen elinajan välianalyysi tehtiin, kun Kd-hoitohaaran potilaista 189 ja Vd-hoitohaaran potilaista 209 oli kuollut. Analyysin ajankohtana oli rekisteröity 80 % kohteena olleista tapahtumista. Seuranta-ajan mediaani oli noin 37 kuukautta. Kd-hoitohaaran potilailla havaittiin tilastollisesti merkitsevä elinaikaetu Vd-hoitohaaran potilaisiin verrattuna (HR = 0,791; 95 % CI: 0,65–0,96; p-arvo = 0,010) (ks. kuva 4).
Kuva 4. Kaplan–Meier-kuvaaja uusiutunutta multippelia myeloomaa sairastavien potilaiden kokonaiselinajasta tutkimuksessa 2011‑003
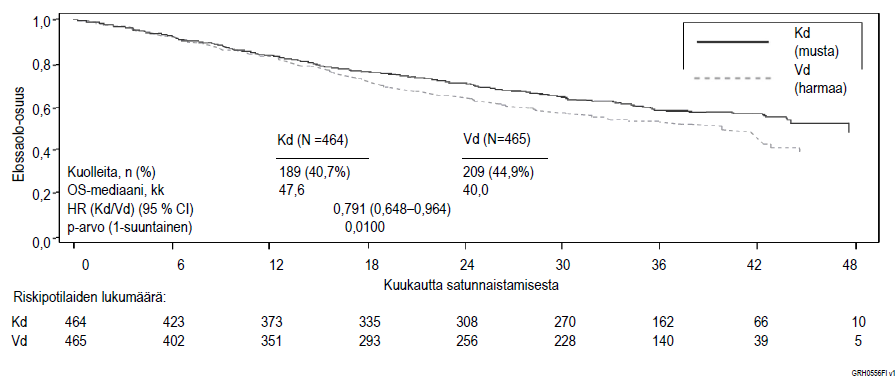
Kd = Kyprolis + deksametasoni; Vd = bortetsomibi + deksametasoni; OS = kokonaiselinaika; kk = kuukautta; HR = vaarasuhde; CI = luottamusväli
Kyprolis yhdessä daratumumabin ja deksametasonin kanssa uusiutuneen tai refraktaarisen multippelin myelooman hoidossa – tutkimus 20160275 (CANDOR)
Kyprolis‑hoidon turvallisuutta ja tehoa arvioitiin kolmannen vaiheen satunnaistetussa avoimessa monikeskustutkimuksessa (paremmuustutkimus), jossa Kyproliksen, daratumumabin ja deksametasonin yhdistelmää (KdD) verrattiin Kyproliksen ja deksametasonin yhdistelmään (Kd). Yhteensä 466 potilasta, joilla oli uusiutunut tai refraktaarinen multippeli myelooma ja jotka olivat saaneet 1–3 aikaisempaa hoitolinjaa, otettiin mukaan tutkimukseen ja satunnaistettiin suhteessa 2:1 (312 KdD‑hoitohaaraan ja 154 Kd‑hoitohaaraan).
Sekä KdD- että Kd‑hoitohaarassa Kyproliksen aloitusannos oli 20 mg/m2, joka nostettiin tasolle 56 mg/m2 ensimmäisen hoitojakson 8. päivänä. Kyprolis annettiin 30 minuutin infuusiona kaksi kertaa viikossa kolmena viikkona neljästä.
Tutkimuksen poissulkukriteereitä olivat: todettu keskivaikea tai vaikea yhtämittainen astma edellisen 2 vuoden aikana, todettu keuhkoahtaumatauti (COPD), jossa uloshengityksen sekuntikapasiteetti FEV1 on < 50 % ennustetusta normaaliarvosta, sekä aktiivinen sydämen kongestiivinen vajaatoiminta.
Potilaiden demografiset ja muut ominaisuudet lähtötilanteessa olivat molemmissa hoitohaaroissa samankaltaiset. Niitä olivat sukupuoli (57,5 % miehiä), etninen tausta (78,5 % valkoihoisia), ikä (64 vuotta, vaihteluväli 29–84 vuotta), aiempi bortetsomibihoito (90 %), bortetsomibiresistenssi (29 %), suuren riskin geneettiset mutaatiot, eli geneettiset alatyypit t(4;14), t(14;16) tai 17p‑deleetio (16 %) sekä geneettiset mutaatiot, joiden riskiluokitus ei ole tiedossa, koska potilaiden näytteiden analyysia ei tehty, se epäonnistui, tai näytteen määrä ei ollut riittävä (51 %). KdD‑hoitoryhmässä ≥ 75‑vuotiaiden osuus (9,0 %) oli pienempi kuin Kd‑ryhmässä (14,3 %). Tutkittaville oli annettu 1–4 aikaisempaa hoitolinjaa (mediaani 2,0). KdD‑hoitoryhmässä aiemman kantasolusiirron saaneiden osuus (62,5 %) oli suurempi kuin Kd‑ryhmässä (48,7 %). Vain yksi KdD‑hoitoryhmän potilas oli aiemmin saanut monoklonaalista anti‑CD38‑vasta‑ainehoitoa.
Taulukossa 9 ja kuvissa 5 ja 6 on tiivistelmä tutkimuksen 20160275 primaarisen analyysin tuloksista.
Taulukko 9. Tiivistelmä tehosta tutkimuksen 20160275 primaarisessa analyysissa
KdD‑hoitohaara (N = 312) | Kd‑hoitohaara (N = 154) | |
| PFS‑mediaani, kk (95 % CI)a | NE (NE–NE) | 15,8 (12,1–NE) |
| HR (95 % CI); 1‑suuntainen p‑arvob | 0,630 (0,464–0,854); 0,0014 | |
| ORR (%) (95 % CI)a, c | 84,3 (79,8–88,1) | 74,7 (67,0–81,3) |
| Vasteen luokka, n (%) | ||
| N (vasteen saaneet) | 263 | 115 |
| CR | 89 (28,5) | 16 (10,4) |
| MRD [-] CR | 43 (13,8) | 5 (3,2) |
| VGPR | 127 (40,7) | 59 (38,3) |
| PR | 47 (15,1) | 40 (26,0) |
| Kerroinsuhde | 1,925 (1,184–3,129) | |
| 1‑suuntainen p‑arvob | 0,0040 | |
| MRD[-]CR 12 kk kohdalla | 12,5 (9,0–16,7) | 1,3 (0,2–4,6) |
| Kerroinsuhde | 11,329 (2,703–47,476) | |
| 1‑suuntainen p‑arvob | < 0,0001 | |
KdD = Kyprolis + deksametasoni ja daratumumabi; Kd = Kyprolis + deksametasoni; CI = luottamusväli; NE = ei arvioitavissa; HR = vaarasuhde; ORR = kokonaisvasteosuus; CR = täydellinen vaste; VGPR = erittäin hyvä osittainen vaste; MRD[-]CR = täydellinen vaste, ei minimaalista jäännöstautia a. Nämä päätetapahtumat olivat riippumattoman arviointikomitean (IRC) määrittelemiä IMWG:n vastekriteerejä käyttäen. b. Tilastollisesti merkitsevä c. Kokonaisvasteeksi katsotaan paras saavutettu kokonaisvaste, joka voi olla osittainen vaste (PR), erittäin hyvä osittainen vaste (VGPR), täydellinen vaste (CR) tai parempi. | ||
Tutkimuksessa KdD‑hoitohaaran potilailla elinaika ilman taudin etenemistä (PFS) oli primaarisen PFS‑analyysin ajankohtana parempi kuin Kd‑hoitohaaran potilailla (vaarasuhde [HR] = 0,630 ja 95 % CI: 0,464–0,854; p = 0,0014) edustaen 37 %:n vähenemää riskiin taudin etenemisen tai kuoleman suhteen potilailla, jotka saivat KdD-hoitoa. PFS‑mediaani ei ollut arvioitavissa KdD‑hoitohaarassa, ja Kd‑hoitohaarassa se oli 15,8 kuukautta.
Potilailla, jotka olivat aikaisemmin saaneet lenalidomidia (42,3 %), PFS‑mediaani ei ollut arvioitavissa KdD‑hoitohaarassa, ja Kd‑hoitohaarassa se oli 12,1 kuukautta (HR = 0,52, 95 % CI: 0,34–0,80), kokonaisvasteosuus (ORR) oli KdD‑haarassa 78,9 % ja Kd‑haarassa 74,3 % (OR = 1,29, 95 % CI: 0,65–2,54), ja MRD[-]CR 12 kuukauden kohdalla oli KdD‑haarassa 11,4 % ja Kd‑haarassa 0,0 % (OR = NE, 95 % CI: NE–NE). Potilailla, jotka olivat resistenttejä lenalidomidille (33 %), PFS‑mediaani ei ollut arvioitavissa KdD‑hoitohaarassa, ja Kd‑hoitohaarassa se oli 11,1 kuukautta (HR = 0,45, 95 % CI: 0,28–0,74), kokonaisvasteosuus (ORR) oli KdD‑haarassa 79,8 % ja Kd‑haarassa 72,7 % (OR = 1,48, 95 % CI: 0,69–3,20), ja MRD[-]CR 12 kuukauden kohdalla oli KdD‑haarassa 13,1 % ja Kd‑haarassa 0,0 % (OR = NE, 95 % CI: NE–NE).
Iäkkäistä (≥ 75‑vuotiaista) potilaista on saatavilla vain vähän tutkimustietoa. Tutkimukseen 20160275 osallistui yhteensä 43 yli 75‑vuotiasta potilasta: 25 potilasta KdD‑hoitoryhmässä ja 18 potilasta Kd‑hoitoryhmässä. PFS‑analyysissa HR oli 1,459 (95 % CI: 0,504–4,223). Hoidon yhteydessä esiintyvien kuolemaan johtavien haittatapahtumien riski oli korkeampi ≥ 65‑vuotiailla tutkittavilla (ks. kohta Haittavaikutukset). KdD‑hoidossa on noudatettava varovaisuutta ≥ 75‑vuotiailla potilailla, ja mahdolliset hyödyt ja haitat on punnittava huolellisesti kussakin tapauksessa.
Kuva 5. Kaplan–Meier‑kuvaaja elinajasta ilman taudin etenemistä (PFS) riippumattoman arviointikomitean (IRC) määrittelemänä (hoitoaikeen [ITT] mukainen potilasjoukko) tutkimuksessa 20160275
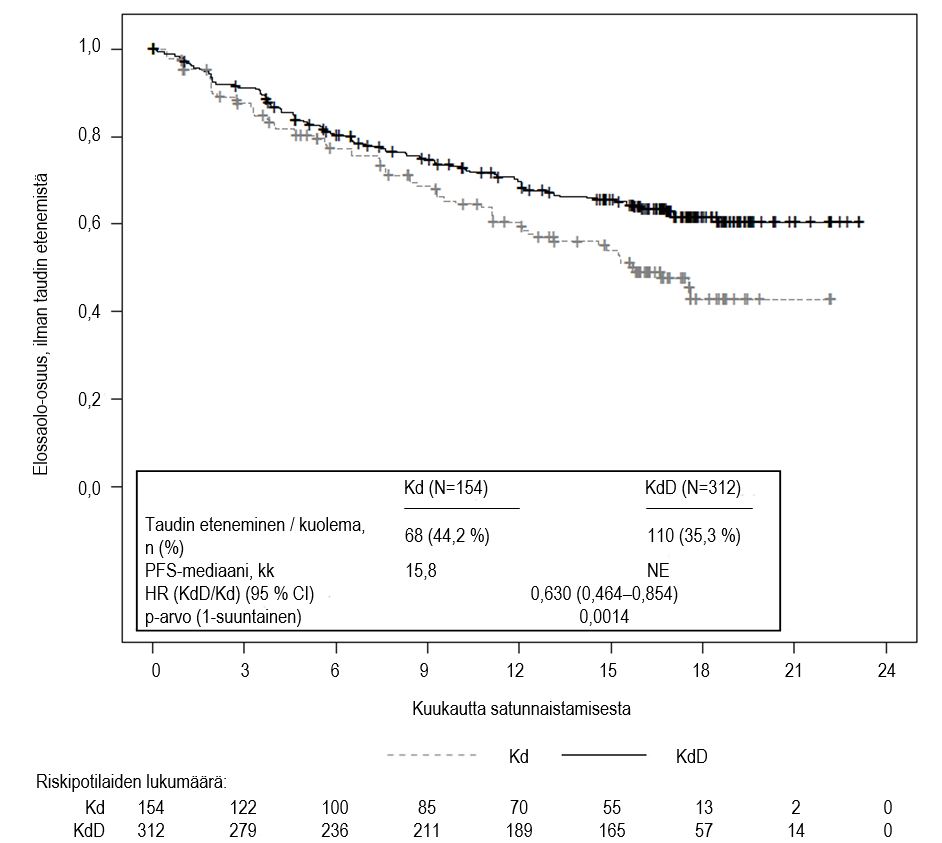
Kokonaisvasteosuus (ORR) oli KdD‑hoitohaarassa 84,3 % ja Kd‑hoitohaarassa 74,7 % (ks. taulukko 9). Vasteen keston mediaani ei ollut arvioitavissa KdD‑hoitohaarassa, ja Kd‑hoitohaarassa se oli 16,6 kuukautta (13,9–NE). Mediaaniaika vasteen saavuttamiseen oli KdD‑hoitohaarassa 1,0 (1, 14) kuukautta ja Kd‑hoitohaarassa 1,0 (1, 10) kuukautta.
Loppuanalyysin ajankohtana 148 (47,4 %) KdD‑hoitoryhmän potilasta ja 80 (51,9 %) Kd‑hoitoryhmän potilasta oli kuollut. Kokonaiselinajan mediaani (95 % CI) oli 50,8 (44,7–NE) kuukautta KdD‑hoitoryhmässä ja 43,6 (35,3–NE) kuukautta Kd‑hoitoryhmässä (HR [KdD/Kd] = 0,784; 95 % CI: 0,595–1,033; 1‑suuntainen p = 0,0417). Tämä yksisuuntainen p‑arvo ei saavuttanut loppuanalyysissa tilastollisesti merkitsevää tasoa 0,021. Seuranta‑ajan mediaani oli 50,6 kuukautta KdD‑hoitoryhmässä ja 50,1 kuukautta Kd‑hoitoryhmässä.
Kuva 6. Kaplan–Meier‑kuvaaja kokonaiselinajasta tutkimuksessa 20160275
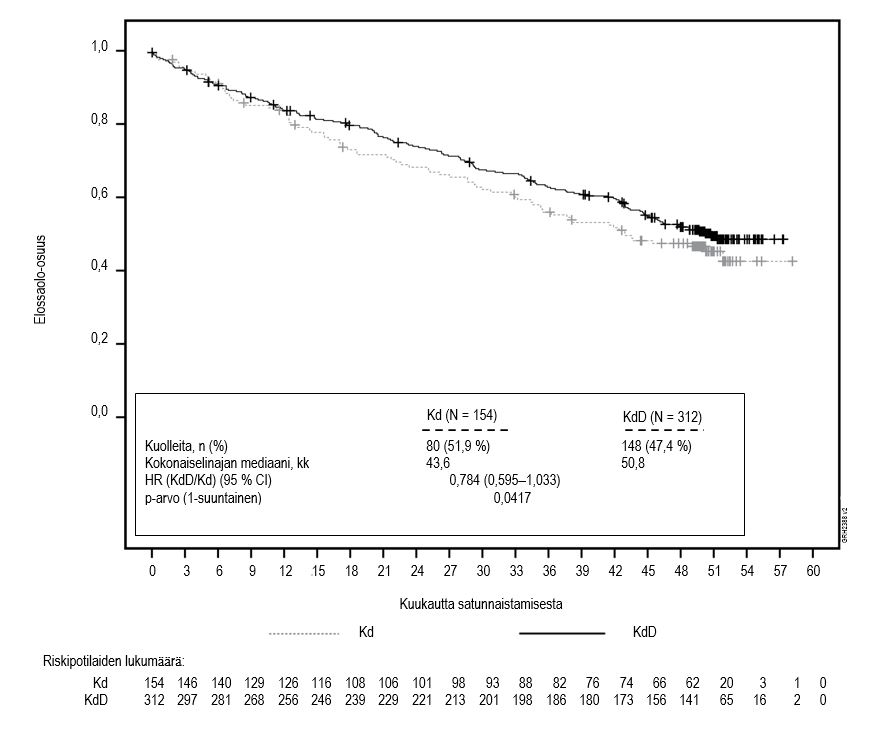
Kyprolis ainoana lääkkeenä uusiutuneen ja refraktaarisen multippelin myelooman hoidossa
Kliinisiä lisäkokemuksia on saatu, kun Kyprolista on annettu ainoana lääkkeenä uusiutuneen ja refraktaarisen multippelin myelooman hoitoon. Tutkimus PX‑171‑011 oli avoin satunnaistettu vaiheen 3 tutkimus (N = 315; valintakriteerinä oli ≥ 3 aikaisempaa hoitoa). Tutkimukseen PX‑171‑011 otetut potilaat olivat saaneet aikaisemmin raskaampia hoitoja ja elintoiminnot ja luuytimen toiminta olivat heikommat kuin tutkimukseen PX‑171‑009 otetuilla potilailla. Tutkimuksessa PX‑171‑011 verrattiin Kyprolista ainoana lääkkeenä vertailuhoitoon (kortikosteroideihin ja syklofosfamidiin). Tutkimus ei saavuttanut sille asetettua ensisijaista tehoa mittaavaa päätetapahtumaa eli se ei pystynyt osoittamaan ainoana lääkkeenä käytetyn Kyproliksen paremmuutta vaikuttavaan vertailuhoitoon verrattuna kokonaiselinajalla mitattuna (HR = 0,975 [95 % CI: 0,760–1,249]). PX‑171‑003A1 oli yhden hoitohaaran, vaiheen 2 tutkimus (N = 266; valintakriteerinä oli ≥ 2 aikaisempaa hoitoa), joka saavutti sille asetetun ensisijaisen tehoa mittaavan päätetapahtuman eli riippumattoman arviointikomitean arvioiman kokonaisvasteosuuden (22,9 %).
Sydämen sähköinen toiminta
Karfiltsomibin mahdollisia vaikutuksia sydämen toimintaan arvioitiin analysoimalla, keskitettyä sokkoutettua tulkintamenettelyä käyttäen, EKG:n kolmoisrekisteröinnin tulokset 154 potilaalta, joilla oli pitkälle edennyt pahanlaatuinen sairaus, kuten multippeli myelooma. Karfiltsomibin vaikutus sydämen repolarisaatioon Friderician kaavalla korjatun QT-ajan (QTcF-ajan) perusteella ja pitoisuus-QTc-suhteiden analyysi eivät anna selvää signaalia minkäänlaisesta annokseen liittyvästä vaikutuksesta. Ennustettu QTcF-aikaan kohdistuva vaikutus maksimipitoisuuden (Cmax) aikana oli 4,8 millisekuntia (yksisuuntaisen 95 %:n luottamusvälin (CI) yläraja). Bazettin kaavalla korjattu (QTcB-aika) ennustettu vaikutus QTcB-aikaan oli maksimipitoisuuden aikana 5,9 millisekuntia (yksisuuntaisen 95 %:n luottamusvälin (CI) yläraja).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Kyprolis-valmisteen käytöstä multippelin myelooman hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Laskimoon 2–10 minuutin infuusiona annetun 27 mg/m2 annoksen jälkeen Cmax-arvo oli 4232 ng/ml ja AUC-arvo 379 ng•hr/ml. Toistuvien 15 ja 20 mg/m2 Kyprolis-annosten jälkeen systeeminen altistus (AUC) ja puoliintumisaika olivat samanlaiset ensimmäisen hoitojakson 1. päivänä ja 15. tai 16. päivänä, mikä viittaa siihen, ettei karfiltsomibi kumuloidu systeemisesti. Annostasolla 20−56 mg/m2 altistus suureni annoksesta riippuvasti.
Kun sama annos annettiin 30 minuutin ja 2–10 minuutin infuusiona, puoliintumisaika ja AUC-arvo olivat molempien infuusioiden jälkeen samanlaiset mutta Cmax-arvo oli 2–3 kertaa pienempi 30 minuutin infuusion jälkeen. Kun annos 56 mg/m2 annettiin 30 minuutin infuusiona, AUC (948 ng•hr/ml) oli noin 2,5-kertainen verrattuna annoksen 27 mg/m2 jälkeen mitattuun AUC-arvoon ja Cmax (2079 ng/ml) oli pienempi kuin 2–10 minuutin infuusiona annetun 27 mg/m2 annoksen jälkeen.
Jakautuminen
Karfiltsomibiannoksen 20 mg/m2 vakaan tilan jakautumistilavuus oli 28 litraa (keskiarvo). In vitro ‑testeissä karfiltsomibi sitoutui plasman proteiineihin keskimäärin 97-prosenttisesti 0,4–4 mikromolaarisella pitoisuusalueella.
Biotransformaatio
Karfiltsomibi metaboloitui nopeasti ja laajasti. Pääasialliset metaboliitit, joita on mitattu ihmisen plasmasta ja virtsasta ja joita ihmisen maksasolut muodostavat in vitro, olivat peptidifragmentit ja karfiltsomibin dioli, mikä viittaa siihen, että tärkeimmät metaboloitumistiet olivat peptidaasien aikaansaama pilkkoutuminen ja epoksidin hydrolyysi. Sytokromi P450 ‑välitteisillä mekanismeilla oli vain vähäinen merkitys karfiltsomibin metaboliassa. Metaboliiteilla ei ole tunnettuja biologisia vaikutuksia.
Eliminaatio
Laskimoon annettujen ≥ 15 mg/m2 annosten jälkeen karfiltsomibi poistui nopeasti systeemisestä verenkierrosta, ja sen puoliintumisaika oli ≤ 1 tunti ensimmäisen hoitojakson ensimmäisenä päivänä. Systeeminen puhdistuma oli 151–263 litraa tunnissa ja ylitti maksan verenvirtauksen, mikä viittaa siihen, että karfiltsomibin poistuminen elimistöstä tapahtui suureksi osaksi maksan ulkopuolella. Karfiltsomibi eliminoituu pääasiassa metaboloitumalla, minkä jälkeen sen metaboliitit erittyvät virtsaan.
Erityisryhmät
Populaatiofarmakokineettiset analyysit osoittavat, ettei ikä, sukupuoli eikä etninen tausta vaikuta karfiltsomibin farmakokinetiikkaan.
Maksan vajaatoiminta
Farmakokineettisessä tutkimuksessa arvioitiin 33 potilasta, joilla oli uusiutuneita tai eteneviä levinneitä pahanlaatuisia sairauksia (kiinteitä kasvaimia, n = 31, tai pahanlaatuisia verisairauksia, n = 2) ja normaali maksan toiminta (bilirubiini ≤ normaalialueen yläraja; aspartaattiaminotransferaasi [ASAT] ≤ normaalialueen yläraja, n = 10), lievä maksan vajaatoiminta (bilirubiini > 1−1,5 × normaalialueen yläraja tai ASAT > normaalialueen yläraja mutta bilirubiini ≤ normaalialueen yläraja, n = 14) tai kohtalainen maksan vajaatoiminta (bilirubiini > 1,5−3 × normaalialueen yläraja, ASAT-arvo mikä tahansa, n = 9). Karfiltsomibin farmakokinetiikkaa ei ole tutkittu potilailla, joilla on vaikea maksan vajaatoiminta (bilirubiini > 3 × normaalialueen yläraja, ASAT-arvo mikä tahansa). Kyprolista annettiin ainoana lääkeaineena 30 minuutin infuusiona laskimoon ensimmäisen hoitojakson 1. ja 2. päivänä 20 mg/m2 ja 8., 9., 15. ja 16. päivänä 27 mg/m2. Jos potilas sieti tämän, annos nostettiin toisesta hoitojaksosta alkaen tasolle 56 mg/m2. Maksan toiminta lähtötilanteessa ei vaikuttanut merkittävästi karfiltsomibin systeemiseen kokonaisaltistukseen (AUClast) kerta-annoksen eikä toistuvien annosten jälkeen (AUClast-arvojen geometristen keskiarvojen suhde oli annostasolla 27 mg/m2 ensimmäisen hoitojakson 16. päivänä lievää maksan vajaatoimintaa sairastavilla potilailla 144,4 % ja kohtalaista maksan vajaatoimintaa sairastavilla 126,1 % verrattuna potilaisiin, joiden maksan toiminta oli normaali, ja annostasolla 56 mg/m2 toisen hoitojakson 1. päivänä vastaavat luvut olivat 144,7 % ja 121,1 %). Potilailla, joilla oli lievä tai kohtalainen maksan vajaatoiminta lähtötilanteessa ja joilla kaikilla oli kiinteitä kasvaimia, maksan toimintahäiriöiden, ≥ 3. asteen haittatapahtumien ja vakavien haittatapahtumien potilaskohtainen ilmaantuvuus oli kuitenkin suurempi kuin potilailla, joiden maksan toiminta oli normaali (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Karfiltsomibin farmakokinetiikkaa tutkittiin kahdessa erityisesti munuaisten vajaatoimintaan keskittyneessä tutkimuksessa.
Ensimmäisessä tutkimuksessa oli mukana 50 multippelia myeloomaa sairastavaa potilasta, joiden munuaisten toiminta oli normaali (CrCl > 80 ml/min, n = 12) tai joilla oli lievä (CrCl 50–80 ml/min, n = 12), kohtalainen (CrCl 30–49 ml/min, n = 10) tai vaikea (CrCl < 30 ml/min, n = 8) munuaisten vajaatoiminta tai jotka olivat dialyysihoidossa (n = 8). Kyprolista annettiin ainoana lääkeaineena 2−10 minuutin infuusiona laskimoon enintään annoksina 20 mg/m2. Farmakokineettiset tiedot kerättiin ensimmäisellä hoitojaksolla 15 mg/m2 annoksen jälkeen ja toisella hoitojaksolla 20 mg/m2 annoksen jälkeen. Toisessa tutkimuksessa oli mukana 23 potilasta, joilla oli uusiutunut multippeli myelooma ja joiden kreatiniinipuhdistuma oli ≥ 75 ml/min (n = 13) tai joilla oli dialyysihoitoa vaativa loppuvaiheen munuaistauti (n = 10). Farmakokineettiset tiedot kerättiin 30 minuutin infuusiona annetun 27 mg/m2 annoksen jälkeen ensimmäisen hoitojakson 16. päivänä ja 56 mg/m2 annoksen jälkeen toisen hoitojakson 1. päivänä.
Molempien tutkimusten tulokset osoittavat, ettei munuaisten toiminnalla ollut merkittävää vaikutusta karfiltsomibialtistukseen kerta-annoksen eikä toistuvien annosten jälkeen. AUClast-arvojen geometristen keskiarvojen suhde oli annostasolla 15 mg/m2 ensimmäisen hoitojakson 1. päivänä lievää munuaisten vajaatoimintaa sairastavilla potilailla 124,36 %, kohtalaista vajaatoimintaa sairastavilla 111,07 %, vaikeaa vajaatoimintaa sairastavilla 84,73 % ja dialyysipotilailla 121,72 % verrattuna potilaisiin, joiden munuaisten toiminta oli normaali. AUClast-arvojen geometristen keskiarvojen suhde oli loppuvaiheen munuaistautia sairastavilla potilailla annostasolla 27 mg/m2 ensimmäisen hoitojakson 16. päivänä 139,72 % ja annostasolla 56 mg/m2 toisen hoitojakson 1. päivänä 132,75 % verrattuna potilaisiin, joiden munuaisten toiminta oli normaali. Ensimmäisessä tutkimuksessa yleisimmän verenkierrossa esiintyvän metaboliitin, M14-metaboliitin (peptidifragmentti) määrä kaksinkertaistui kohtalaista munuaisten vajaatoimintaa sairastavilla potilailla, kolminkertaistui vaikeaa munuaisten vajaatoimintaa sairastavilla ja seitsenkertaistui dialyysipotilailla (AUClast-arvon perusteella). Toisessa tutkimuksessa M14-altistus oli suurempi (noin nelinkertainen) loppuvaiheen munuaistautia sairastavilla potilailla kuin potilailla, joiden munuaisten toiminta oli normaali. Tällä metaboliitilla ei ole tunnettuja biologisia vaikutuksia. Heikkenevään munuaisten toimintaan liittyvät vakavat haittatapahtumat olivat yleisempiä potilailla, joilla esiintyi munuaisten vajaatoimintaa lähtötilanteessa (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Karfiltsomibi oli klastogeeninen in vitro perifeerisen veren lymfosyyteissä tehdyssä kromosomipoikkeavuustestissä. Karfiltsomibi ei ollut mutageeninen bakteereilla tehdyissä takaisinmutaatiokokeessa (Ames) in vitro eikä klastogeeninen hiiren luuytimen mikrotumatestissä in vivo.
Kun apinoille annettiin karfiltsomibia 3 mg/kg kertaboluksena laskimoon (mikä vastaa annosta 36 mg/m2 ja on samansuuruinen kuin ihmisille suositeltu kehon pinta-alan mukainen annos 27 mg/m2), havaittiin verenpaineen laskua, sydämen sykkeen nopeutumista ja seerumin troponiini T ‑pitoisuuden suurenemista. Kun karfiltsomibia annettiin toistuvina boluksina laskimoon rotille ≥ 2 mg/kg/annos ja apinoille 2 mg/kg/annos noudattaen samaa annostusohjelmaa kuin kliinisessä käytössä, kuolemaan johtaneita haittavaikutuksia todettiin verenkiertoelimistössä (sydämen vajaatoiminta, sydänfibroosi, sydänpussin nestekertymä, sydämen verenvuoto/rappeutuminen), ruoansulatuskanavassa (nekroosi/verenvuoto), munuaisissa (glomerulonefropatia, munuaistiehyiden nekroosi, toimintahäiriö) ja keuhkoissa (verenvuoto/tulehdus). Rotille annettu 2 mg/kg/annos on noin puolet ihmisille suositellusta kehon pinta-alaan perustuvasta 27 mg/m2 annoksesta. Apinoilla suurin annos, johon ei liittynyt vaikeita haittavaikutuksia, 0,5 mg/kg, aiheutti interstitiaalisen munuaistulehduksen sekä lievän glomerulopatian ja lievän sydäntulehduksen. Nämä löydökset raportoitiin annostasolla 6 mg/m2, joka on pienempi kuin ihmisille suositeltu annos 27 mg/m2.
Karfiltsomibilla ei ole tehty hedelmällisyystutkimuksia. Lisääntymiskudoksiin kohdistuvia vaikutuksia ei havaittu rotilla ja apinoilla tehdyissä 28 vuorokauden toistuvien annosten toksisuustutkimuksissa eikä pitkäaikaisissa 6 kuukauden toksisuustutkimuksessa rotilla ja 9 kuukauden toksisuustutkimuksessa apinoilla. Tiineille kaniineille annettu karfiltsomibi aiheutti alkio- ja sikiötoksisuutta annoksina, jotka olivat pienempiä kuin potilaille suositeltu annos. Karfiltsomibi ei ollut teratogeeninen, kun sitä annettiin tiineille rotille organogeneesivaiheen aikana, rotille enintään 2 mg/kg/vrk, mikä on noin puolet ihmisille suositellusta kehon pinta-alaan perustuvasta 27 mg/m2 annoksesta.
Farmaseuttiset tiedot
Apuaineet
Beetadeksisulfobutyylieetterinatrium, vedetön sitruunahappo (E330), natriumhydroksidi (pH:n säätämiseen)
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kyprolis infuusiokuiva‑ainetta, liuosta varten, ei saa sekoittaa natriumkloridi 9 mg/ml (0,9 %) ‑injektionesteen kanssa.
Kestoaika
Kuiva-ainetta sisältävä injektiopullo (avaamaton)
3 vuotta.
Käyttövalmis liuos
Injektiopullossa, ruiskussa tai infuusiopussissa säilytettävien käyttövalmiiden liuosten kemiallisen ja fysikaalisen käytönaikaisen säilyvyyden on osoitettu olevan 24 tuntia 2–8 °C:ssa tai 4 tuntia 25 °C:ssa. Valmisteen käyttökuntoon saattamisen ja annoksen antamisen välinen aika ei saa olla yli 24 tuntia.
Mikrobiologisista syistä valmiste on käytettävä heti. Ellei sitä käytetä heti, käytönaikaiset säilytysajat ja olosuhteet ovat käyttäjän vastuulla, eikä säilytysaika saa ylittää 24 tuntia 2–8 °C:ssa.
Säilytys
Säilytä jääkaapissa (2°C - 8°C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Käyttökuntoon saatetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
KYPROLIS infuusiokuiva-aine, liuosta varten
10 mg (L:ei) 1 kpl (305,58 €)
30 mg (L:ei) 1 kpl (874,62 €)
60 mg (L:ei) 1 kpl (1704,85 €)
PF-selosteen tieto
Kyprolis 10 mg infuusiokuiva-aine, liuosta varten
10 ml:n injektiopullo (kirkasta tyypin I lasia), jossa on fluoropolymeerilla pinnoitettu elastomeeritulppa, alumiinisuljin ja vaaleansininen muovinen suojakansi.
Kyprolis 30 mg infuusiokuiva-aine, liuosta varten
30 ml:n injektiopullo (kirkasta tyypin I lasia), jossa on fluoropolymeerilla pinnoitettu elastomeeritulppa, alumiinisuljin ja oranssi muovinen suojakansi.
Kyprolis 60 mg infuusiokuiva-aine, liuosta varten
50 ml:n injektiopullo (kirkasta tyypin I lasia), jossa on fluoropolymeerilla pinnoitettu elastomeeritulppa, alumiinisuljin ja violetti muovinen suojakansi.
Pakkauskoko: 1 injektiopullo.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen kylmäkuivattu jauhe.
Käyttö- ja käsittelyohjeet
Yleiset varotoimet
Karfiltsomibi on sytotoksinen aine. Siksi Kyproliksen käytössä ja valmistelussa on noudatettava varovaisuutta. Käsineiden ja muiden suojavarusteiden käyttöä suositellaan.
Käyttökuntoon saattaminen ja valmistaminen laskimoon antoa varten
Kyprolis‑injektiopulloissa ei ole antimikrobisia säilytysaineita, ja ne on tarkoitettu vain yhtä käyttökertaa varten. Asianmukaista aseptista menettelytapaa on noudatettava.
Käyttökuntoon saatettu liuos sisältää karfiltsomibia 2 mg/ml. Lue valmistusohjeet kokonaan ennen käyttökuntoon saattamista:
- Laske annos (mg/m2) ja tarvittavien Kyprolis‑injektiopullojen lukumäärä käyttäen laskuperusteena potilaan kehon pinta‑alaa lähtötilanteessa. Jos potilaan kehon pinta‑ala on yli 2,2 m2, annettava annos lasketaan 2,2 m2:n pinta‑alan mukaan. Annosta ei tarvitse muuttaa, jos paino muuttuu 20 % tai vähemmän.
- Ota injektiopullo jääkaapista juuri ennen käyttöä.
- Käytä ainoastaan 21 G:n tai pienempää (ulkoläpimitta enintään 0,8 mm) neulaa. Saata jokainen injektiopullo käyttökuntoon aseptisesti ruiskuttamalla hitaasti 5 ml (10 mg:n injektiopulloon), 15 ml (30 mg:n injektiopulloon) tai 29 ml (60 mg:n injektiopulloon) steriiliä injektionesteisiin käytettävää vettä tulpan läpi. Suuntaa vesi INJEKTIOPULLON SISÄSEINÄMÄÄN niin, että muodostuu mahdollisimman vähän vaahtoa.
- Pyörittele injektiopulloa varovasti ja/tai kääntele sitä hitaasti ylösalaisin noin 1 minuutin ajan tai kunnes kuiva‑aine on kokonaan liuennut. ÄLÄ RAVISTA. Jos vaahtoa muodostuu, anna liuoksen asettua injektiopullossa, kunnes vaahto häviää (noin 5 minuuttia) ja liuos on kirkasta.
- Tarkista ennen antamista silmämääräisesti, ettei liuoksessa ole hiukkasia eikä sen väri ole muuttunut. Käyttövalmiin liuoksen on oltava kirkasta ja väritöntä tai kellertävää. Sitä ei saa antaa, jos siinä havaitaan värimuutoksia tai hiukkasia.
- Hävitä injektiopulloon jäänyt käyttämätön lääke.
- Kyprolis voidaan antaa suoraan infuusiona laskimoon tai vaihtoehtoisesti infuusiopussin kautta. Ei saa antaa nopeana injektiona eikä boluksena laskimoon.
- Jos liuos annetaan infuusiopussin kautta, vedä laskettu annos injektiopullosta käyttäen ainoastaan 21 G:n tai pienempää (ulkoläpimitta enintään 0,8 mm) neulaa ja ruiskuta se 50 tai 100 ml:n infuusiopussiin, jossa on 5‑prosenttista glukoosi‑infuusionestettä.
Hävittäminen
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
KYPROLIS infuusiokuiva-aine, liuosta varten
10 mg 1 kpl
30 mg 1 kpl
60 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01XG02
Valmisteyhteenvedon muuttamispäivämäärä
14.12.2023
Yhteystiedot
Keilaranta 10, PL 86
02101 Espoo
09 5490 0500