
PRALUENT injektioneste, liuos, esitäytetty kynä 75 mg, 150 mg, 300 mg
Vaikuttavat aineet ja niiden määrät
Praluent 75 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kerta-annoskynä sisältää 75 mg alirokumabia 1 ml:ssa liuosta.
Praluent 150 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kerta-annoskynä sisältää 150 mg alirokumabia 1 ml:ssa liuosta.
Praluent 300 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kerta-annoskynä sisältää 300 mg alirokumabia 2 ml:ssa liuosta.
Alirokumabi on ihmisen monoklonaalinen IgG1-vasta-aine, joka on tuotettu kiinanhamsterin munasarjasoluissa yhdistelmä-DNA-tekniikalla.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Primaarinen hyperkolesterolemia ja sekamuotoinen dyslipidemia
Praluent on tarkoitettu käytettäväksi ruokavalion lisänä aikuisille, joilla on primaarinen hyperkolesterolemia (heterotsygoottinen familiaalinen tai ei-familiaalinen) tai sekamuotoinen dyslipidemia, ja vähintään 8-vuotiaille pediatrisille potilaille, joilla on heterotsygoottinen familiaalinen hyperkolesterolemia:
- yhdistettynä statiinihoitoon, mahdollisesti muiden rasva-arvoja alentavien hoitojen kanssa, potilaille, jotka eivät saavuta LDL-kolesterolitavoitteita suurimmalla siedetyllä statiiniannoksella tai
- yksin tai yhdessä muiden rasva-arvoja alentavien hoitojen kanssa potilaille, jotka eivät siedä statiineja tai joille statiinin käyttö on vasta-aiheista.
Todettu ateroskleroottinen sydän- ja verisuonitauti
Praluent on tarkoitettu käytettäväksi muita riskitekijöitä korjaavan hoidon lisänä aikuisille, joilla on todettu ateroskleroottinen sydän- ja verisuonitauti, sydän- ja verisuonitautiriskin pienentämiseen alentamalla LDL-kolesteroliarvoja:
- yhdistettynä suurimpaan siedettyyn statiiniannokseen muiden rasva-arvoja alentavien hoitojen kanssa tai ilman niitä tai
- yksin tai yhdessä muiden rasva-arvoja alentavien hoitojen kanssa potilaille, jotka eivät siedä statiineja tai joille statiinin käyttö on vasta-aiheista.
Katso LDL-kolesteroliin, sydän- ja verisuonitapahtumiin ja tutkittuihin populaatioihin kohdistuvia vaikutuksia koskevat tutkimustulokset kohdasta Farmakodynamiikka.
Annostus ja antotapa
Annostus
Aikuiset
Ennen alirokumabihoidon aloittamista on suljettava pois hyperlipidemian tai sekamuotoisen dyslipidemian sekundaariset aiheuttajat (kuten nefroottinen oireyhtymä ja hypotyreoosi).
Suositellut alirokumabiannokset ovat 75 mg kahden viikon välein, 150 mg kahden viikon välein ja 300 mg neljän viikon välein (kuukausittain) ihon alle annettuna. Kaikkia annoksia voidaan käyttää hoidon aloittamiseen.
Alirokumabiannos voidaan säätää yksilöllisesti potilaan ominaisuuksien, kuten lähtötason LDL-kolesteroliarvon, hoitotavoitteen ja hoitovasteen, mukaan. Lipidipitoisuus voidaan arvioida 4–8 viikon kuluttua hoidon aloittamisesta tai annoksen titrauksesta, ja annosta voidaan muuttaa sen mukaan (annoksen suurentaminen tai pienentäminen). LDL-kolesteroliarvon odotetaan pienenevän voimakkaasti, jos alirokumabiannos on 150 mg kahden viikon välein tai 300 mg neljän viikon välein (kuukausittain), jolloin enimmäisannos on 150 mg kahden viikon välein (ks. kohta Farmakodynamiikka).
Vähintään 8-vuotiaat pediatriset potilaat, joilla on heterotsygoottinen familiaalinen hyperkolesterolemia
| Potilaan paino | Suositeltu annos | Suositeltu annos, jos LDL-kolesteroliarvoa on tarpeen pienentää edelleen* |
| Alle 50 kg | 150 mg 4 viikon välein | 75 mg 2 viikon välein |
| Vähintään 50 kg | 300 mg 4 viikon välein | 150 mg 2 viikon välein |
* Lipidiarvot voidaan arvioida 8 viikon kuluttua hoidon aloittamisesta tai annoksen titrauksesta, ja annosta voidaan muuttaa sen mukaan.
Unohtunut annos
Jos potilaan annos jää väliin, annos on annettava mahdollisimman pian ja lääkkeenantoa on sen jälkeen jatkettava normaalin annosaikataulun mukaisesti.
Erityisryhmät
Iäkkäät potilaat
Annosta ei tarvitse muuttaa iäkkäille potilaille.
Maksan vajaatoiminta
Annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä tai keskivaikea maksan vajaatoiminta. Tietoa ei ole potilaista, joilla on vaikea maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annoksen muuttaminen ei ole tarpeen, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta. Tietoa on saatavilla vähän potilaista, joilla on vaikea munuaisten vajaatoiminta (ks. kohta Farmakokinetiikka).
Kehonpaino
Annosta ei tarvitse muuttaa potilaan painon perusteella.
Pediatriset potilaat
Praluent-valmisteen turvallisuutta ja tehoa alle 8-vuotiaiden lasten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Ihon alle.
Alirokumabi annetaan injektiona ihon alle reiteen, vatsaan tai olkavarteen.
Esitäytetty kynä on tarkoitettu vain yhtä käyttökertaa varten.
300 mg:n annoksen antamiseksi joko yksi 300 mg:n annos tai kaksi 150 mg:n annosta on pistettävä peräjälkeen kahteen eri pistoskohtaan.
Pistoskohdan vaihtamista suositellaan jokaisella pistoskerralla.
Alirokumabia ei pidä injektoida kohtaan, jossa on aktiivinen ihosairaus tai ‑vaurio, kuten auringonpolttama, ihottuma, tulehdus tai ihoinfektio.
Alirokumabia ei saa antaa yhdessä muiden injektoitavien lääkevalmisteiden kanssa samaan pistoskohtaan.
Ennen lääkkeen käsittelyä tai antoa huomioon otettavat varotoimet
Liuoksen on annettava lämmetä huoneenlämpöiseksi ennen käyttöä (ks. kohta Käyttö- ja käsittelyohjeet).
Vähintään 8-vuotiaat pediatriset potilaat
Jos potilas on vähintään 12-vuotias nuori, suositellaan että Praluent-valmisteen antaa tai sen antamista valvoo aikuinen.
Jos potilas on alle 12-vuotias lapsi, huoltajan on annettava Praluent.
Aikuiset
Aikuinen potilas voi joko pistää alirokumabin itse tai toinen henkilö voi antaa alirokumabipistoksen, kun terveydenhuollon ammattilainen on neuvonut oikean ihon alle pistämisen tekniikan.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Allergiset reaktiot
Kliinisissä tutkimuksissa on ilmoitettu yleisiä allergisia reaktioita, kuten kutinaa, sekä harvinaisia, joskus vakavia allergisia reaktioita, kuten yliherkkyyttä, läiskäekseemaa, nokkosihottumaa ja yliherkkyysvaskuliittia. Myyntiluvan myöntämisen jälkeen on ilmoitettu angioedeemaa (ks. kohta Haittavaikutukset). Jos vakavan allergisen reaktion oireita tai merkkejä ilmaantuu, alirokumabihoito täytyy keskeyttää ja potilaalle on aloitettava sopiva oireenmukainen hoito (ks. kohta Vasta-aiheet).
Munuaisten vajaatoiminta
Vaikeaa munuaisten vajaatoimintaa (eGFR < 30 ml/min/1,73 m2) sairastavia potilaita osallistui vähän kliinisiin tutkimuksiin (ks. kohta Farmakokinetiikka). Alirokumabia on käytettävä varoen potilaille, joilla on vaikea munuaisten vajaatoiminta.
Maksan vajaatoiminta
Potilaita, joilla on vaikea maksan vajaatoiminta (Child-Pugh luokitus C), ei ole tutkittu (ks. kohta Farmakokinetiikka). Alirokumabia on käytettävä varoen potilaille, joilla on vaikea maksan vajaatoiminta.
Yhteisvaikutukset
Alirokumabin vaikutukset muihin lääkevalmisteisiin
Alirokumabi on biologinen lääkevalmiste, joten ei ole odotettavissa, että alirokumabilla olisi farmakokineettisiä vaikutuksia muihin lääkevalmisteisiin tai vaikutuksia sytokromi P450 ‑entsyymeihin.
Muiden lääkevalmisteiden vaikutukset alirokumabiin
Statiinien ja lipideihin vaikuttavien muiden hoitojen tiedetään lisäävän alirokumabin kohdeproteiinin PCSK9:n tuotantoa. Tämä suurentaa kohdeproteiinin välittämää alirokumabin puhdistumaa ja pienentää systeemistä alirokumabialtistusta. Alirokumabimonoterapiaan verrattuna altistus alirokumabille on statiinien kanssa samanaikaisesti käytettynä noin 40 % pienempi, etsetimibin kanssa samanaikaisesti käytettynä noin 15 % pienempi ja fenofibraatin kanssa samanaikaisesti käytettynä noin 35 % pienempi. LDL-kolesterolitason lasku kuitenkin säilyy annosvälien aikana, kun alirokumabia annetaan kahden viikon välein.
Raskaus ja imetys
Raskaus
Ei ole olemassa tietoja Praluent-valmisteen käytöstä raskaana oleville naisille. Alirokumabi on yhdistelmä-DNA-tekniikalla valmistettu IgG1-vasta-aine, joten sen odotetaan läpäisevän veri-istukkaesteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Eläinkokeissa ei ole havaittu suoria tai epäsuoria haitallisia vaikutuksia raskauden jatkumiseen tai alkion/sikiön kehitykseen. Ihmisten annoksia suuremmilla annoksilla todettiin emoon kohdistuvaa toksisuutta rotilla, mutta ei apinoilla, ja apinoiden jälkeläisillä havaittiin heikompi sekundaarinen immuunivaste antigeenialtistukselle (ks. kohta Prekliiniset tiedot turvallisuudesta).
Praluent-valmisteen käyttöä ei suositella raskauden aikana, ellei raskaana olevan potilaan kliininen tilanne edellytä hoitoa alirokumabilla.
Imetys
Ei tiedetä, erittyykö alirokumabi ihmisen rintamaitoon. Ihmisen immunoglobuliini G (IgG) erittyy ihmisen rintamaitoon, erityisesti ternimaitoon; Praluent-valmisteen käyttöä ei suositella rintaruokinnan alkuvaiheessa. Imetyksen alkuvaiheen jälkeen tapahtuvan altistuksen uskotaan olevan vähäinen.
Koska alirokumabin vaikutuksia imeväisiin ei tiedetä, on päätettävä, lopetetaanko rintaruokinta vai lopetetaanko Praluent-hoito rintaruokinnan ajaksi.
Hedelmällisyys
Eläinkokeissa ei todettu haitallisia vaikutuksia hedelmällisyyden sijaismarkkereihin (ks. kohta Prekliiniset tiedot turvallisuudesta). Ei ole olemassa tietoja haitallisista vaikutuksista ihmisten hedelmällisyyteen.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Praluent-valmisteella ei ole haitallista vaikutusta ajokykyyn tai koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimpiä haittavaikutuksia suositelluilla annoksilla ovat paikalliset pistoskohdan reaktiot (6,1 %), ylähengitysteiden oireet (2,0 %) ja kutina (1,1 %). Alirokumabia saaneilla potilailla yleisimpiä hoidon keskeyttämiseen johtaneita haittavaikutuksia olivat paikalliset pistoskohdan reaktiot.
ODYSSEY OUTCOMES ‑tutkimuksen turvallisuusprofiili vastasi vaiheen 3 kontrolloiduissa tutkimuksissa kuvattua kokonaisturvallisuusprofiilia.
Vaiheen 3 tutkimusohjelmassa käytettyjen kahden annoksen (75 mg ja 150 mg) turvallisuusprofiilien välillä ei havaittu eroja.
Haittavaikutustaulukko
Seuraavia haittavaikutuksia ilmoitettiin alirokumabilla hoidetuilla potilailla yhdistetyissä kontrolloiduissa tutkimuksissa ja/tai kauppaan tuonnin jälkeisessä seurannassa (ks. taulukko 1).
Kaikkien kliinisissä tutkimuksissa todettujen haittavaikutusten esiintymistiheydet on laskettu niiden ilmaantuvuuksista yhdistetyissä vaiheen 3 kliinisissä tutkimuksissa. Haittavaikutukset on esitetty elinjärjestelmäluokituksen mukaisesti. Esiintymistiheydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Kauppaan tuonnin jälkeisessä seurannassa ilmoitettujen haittavaikutusten yleisyyksiä ei voi määrittää, sillä ne on saatu spontaaneista raporteista. Siten näiden haittavaikutusten yleisyydeksi on määritetty ”tuntematon”.
Taulukko 1 – Haittavaikutukset
| Elinjärjestelmä | Yleinen | Harvinainen | Tuntematon |
| Immuunijärjestelmä | Yliherkkyys, yliherkkyysvaskuliitti | ||
| Hengityselimet, rintakehä ja välikarsina | Ylähengitysteiden oireet* | ||
| Iho ja ihonalainen kudos | Kutina | Nokkosihottuma, läiskäekseema | Angioedeema |
| Yleisoireet ja antopaikassa todettavat haitat | Pistoskohdan reaktiot** | Flunssan kaltainen sairaus |
* Näitä voivat olla pääasiassa kipu nielussa, nuha ja aivastelu.
** Näitä voivat olla punoitus, kutina, turvotus, kipu/arkuus
Valikoitujen haittavaikutusten kuvaus
Paikalliset pistoskohdan reaktiot
Paikallisia pistoskohdan reaktioita, kuten punoitusta, kutinaa, turvotusta ja kipua/arkuutta, ilmoitettiin 6,1 %:lla alirokumabia saaneista potilaista ja 4,1 %:lla vertailuryhmästä (jossa annettiin lumeinjektioita). Useimmat pistoskohdan reaktiot olivat tilapäisiä ja vaikeusasteeltaan lieviä. Paikallisista pistoskohdan reaktioista johtuneiden keskeyttämisten määrä oli verrattavissa toisiinsa näissä kahdessa ryhmässä (0,2 % alirokumabiryhmässä ja 0,3 % vertailuryhmässä). Sydän- ja verisuonivaikutuksia arvioineessa tutkimuksessa (ODYSSEY OUTCOMES) pistoskohdan reaktioita ilmeni myös useammin alirokumabia kuin lumelääkettä saaneilla potilailla (alirokumabi: 3,8 %, lumelääke: 2,1 %).
Yleiset allergiset reaktiot
Yleisiä allergisia reaktioita ilmoitettiin useammin alirokumabiryhmässä (8,1 %:lla potilaista) kuin vertailuryhmässä (7,0 %:lla potilaista), lähinnä siksi, että kutinan ilmaantuvuudessa oli eroa. Todetut kutinatapaukset olivat tyypillisesti lieviä ja tilapäisiä. Lisäksi kontrolloiduissa kliinisissä tutkimuksissa on ilmoitettu harvinaisia ja joskus vakavia allergisia reaktioita, kuten yliherkkyyttä, läiskäekseemaa, nokkosihottumaa ja yliherkkyysvaskuliittia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Sydän- ja verisuonivaikutuksia arvioineessa tutkimuksessa (ODYSSEY OUTCOMES) yleisiä allergisia reaktioita todettiin alirokumabia saaneilla potilailla saman verran kuin lumelääkettä saaneilla potilailla (alirokumabi: 7,9 %, lumelääke: 7,8 %). Kutinan ilmaantuvuudessa ei havaittu eroa.
Erityisryhmät
Iäkkäät potilaat
Vaikka yli 75-vuotiailla potilailla ei ole todettu turvallisuuteen liittyviä seikkoja, tietoja tästä ikäryhmästä on vähän. Primaarista hyperkolesterolemiaa ja sekamuotoista dyslipidemiaa koskeneissa vaiheen 3 kontrolloiduissa tutkimuksissa alirokumabia käyttäneistä potilaista 1 158 (34,7 %) oli vähintään 65-vuotiaita ja 241 (7,2 %) oli vähintään 75-vuotiaita. Sydän‑ ja verisuonivaikutuksia arvioineessa kontrolloidussa tutkimuksessa alirokumabia käyttäneistä potilaista 2 505 (26,5 %) oli vähintään 65‑vuotiaita ja 493 (5,2 %) oli vähintään 75‑vuotiaita. Ikääntymisen ei todettu muuttavan turvallisuutta ja tehoa merkittävästi.
Pediatriset potilaat
Praluent-valmisteen turvallisuus ja teho heterotsygoottista familiaalista hyperkolesterolemiaa sairastavien lasten ja nuorten hoidossa on varmistettu. Kliininen tutkimus Praluent-valmisteen vaikutuksista toteutettiin 153 potilaalla, jotka olivat 8–17-vuotiaita ja joilla oli heterotsygoottinen familiaalinen hyperkolesterolemia. Uusia turvallisuuslöydöksiä ei havaittu, ja tässä populaatiossa havaitut turvallisuustiedot vastasivat valmisteen tunnettua turvallisuusprofiilia heterotsygoottista familiaalista hyperkolesterolemiaa sairastavilla aikuisilla.
Kokemukset alirokumabin käytöstä pediatrisille potilaille, joilla on homotsygoottinen familiaalinen hyperkolesterolemia, rajoittuvat 18 potilaaseen, jotka olivat 8–17-vuotiaita. Tunnettuun aikuisilla todettuun turvallisuusprofiiliin verrattuna ei tehty yhtään uutta turvallisuuteen liittyvää löydöstä.
Tutkimus koskien annostelua 4 viikon välein
Turvallisuusprofiili potilailla, joita oli hoidettu 300 mg:n annoksella 4 viikon välein (kuukausittain), oli samanlainen kuin kliinisen tutkimuksen ohjelmassa annettaessa lääkettä 2 viikon välein, lukuun ottamatta suurempaa määrää paikallisia pistoskohdan reaktioita. Ilmoitettujen paikallisten pistospaikan reaktioiden esiintymistiheys oli kaiken kaikkiaan 16,6 % hoitoryhmässä, jossa lääkettä annettiin 300 mg 4 viikon välein, ja 7,9 % lumeryhmässä. Alirokumabiryhmän (300 mg 4 viikon välein) potilaat saivat vuoroittain lumepistoksia sokkouttamisen säilyttämiseksi pistostiheyden suhteen. Lumepistosten jälkeen ilmaantuneita pistospaikan reaktioita lukuun ottamatta pistospaikan reaktioiden esiintymistiheys oli 11,8 %. Pistospaikan reaktioista johtuen 0,7 % alirokumabiryhmän (300 mg 4 viikon välein) potilaista ja 0 % lumeryhmän potilaista keskeytti hoidon.
LDL-kolesteroliarvot < 0,65 mmol/l (< 25 mg/dl)
Kaikissa kliinisissä tutkimuksissa tutkimusasetelma ei sallinut rasva-arvoja alentavan taustalääkityksen muuttamista. Alle 0,65 mmol/l:n (< 25 mg/dl) LDL-kolesteroliarvon saavuttaneiden potilaiden prosentuaaliseen osuuteen vaikutti sekä lähtötilanteen LDL-kolesteroliarvo että alirokumabiannos.
Yhdistetyissä kontrolloiduissa tutkimuksissa, joissa aloitusannos oli 75 mg 2 viikon välein ja joissa annos suurennettiin 150 mg:aan 2 viikon välein, jos potilaan LDL-kolesteroliarvo ei ollut alle 1,81 mmol/l tai alle 2,59 mmol/l (< 70 mg/dl tai < 100 mg/dl), 29,3 %:lla alirokumabia saaneista potilaista, joilla lähtötilanteen LDL-kolesteroliarvo oli alle 2,59 mmol/l (< 100 mg/dl), ja 5,0 %:lla alirokumabia saaneista potilaista, joilla lähtötilanteen kolesteroliarvo oli vähintään 2,59 mmol/l (≥ 100 mg/dl), kaksi peräkkäistä LDL-kolesteroliarvoa oli alle 0,65 mmol/l (< 25 mg/dl). ODYSSEY OUTCOMES ‑tutkimuksessa, jossa alirokumabin aloitusannos oli 75 mg 2 viikon välein ja jossa annos suurennettiin 150 mg:aan 2 viikon välein, jos potilaan LDL-kolesteroliarvo ei ollut alle 1,29 mmol/l (< 50 mg/dl), 54,8 %:lla alirokumabia saaneista potilaista, joilla lähtötilanteen LDL-kolesteroliarvo oli alle 2,59 mmol/l (< 100 mg/dl), ja 24,2 %:lla alirokumabia saaneista potilaista, joilla lähtötilanteen LDL-kolesteroliarvo oli vähintään 2,59 mmol (≥ 100 mg/dl), kaksi peräkkäistä LDL-kolesteroliarvoa oli alle 0,65 mmol/l (< 25 mg/dl).
Vaikka alirokumabilla tehdyissä tutkimuksissa ei havaittu hyvin pieneen LDL-kolesteroliarvoon liittyviä haitallisia vaikutuksia, pitkäkestoisten hyvin pienten LDL-kolesteroliarvojen pitkäaikaisvaikutuksia ei tunneta.
Immunogeenisuus/lääkevasta-aineet (anti-drug-antibodies, ADA)
ODYSSEY OUTCOMES -tutkimuksessa todettiin lääkevasta-aineita (ADA) hoidon aloittamisen jälkeen 5,5 %:lla alirokumabia 75 mg ja/tai 150 mg 2 viikon välein saaneista potilaista ja 1,6 %:lla lumelääkettä saaneista potilaista. Suurin osa näistä vasteista oli ohimeneviä. Pitkäkestoisia ADA-vasteita havaittiin 0,7 %:lla alirokumabia saaneista potilaista ja 0,4 %:lla lumelääkettä saaneista potilaista. Neutraloivien vasta-aineiden vasteita havaittiin 0,5 %:lla alirokumabia saaneista potilaista ja alle 0,1 %:lla lumelääkettä saaneista potilaista.
Lääkevasta-ainevasteiden, neutraloivat vasta-aineet mukaan lukien, titteri oli pieni, eikä vasteilla vaikuttanut olevan kliinisesti merkittävää vaikutusta alirokumabin tehoon tai turvallisuuteen lukuun ottamatta pistoskohdan reaktioiden suurempaa esiintyvyyttä potilailla, joilla ilmeni hoidon aikana ADA-vaste (7,5 %), verrattuna ADA-negatiivisiin potilaisiin (3,6 %). Alirokumabihoidon jatkamisen pitkäaikaisvaikutuksia potilailla, joilla on todettu lääkevasta-aineita, ei tunneta.
Kymmenessä yhdistetyssä lume- ja aktiivikontrolloidussa tutkimuksessa, joissa potilaille annettiin 75 mg ja/tai 150 mg alirokumabia 2 viikon välein, sekä erillisessä kliinisessä tutkimuksessa, jossa potilaille annettiin alirokumabia 75 mg 2 viikon välein tai 300 mg 4 viikon välein (joillakin potilailla annos muutettiin 150 mg:aan 2 viikon välein), todettujen lääkevasta-aineiden ja neutraloivien vasta-aineiden ilmaantuvuus oli samankaltainen kuin edellä kuvatuissa ODYSSEY OUTCOMES -tutkimuksen tuloksissa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Alirokumabin yliannostukseen ei ole erityistä hoitoa. Yliannostustapauksessa potilasta on hoidettava oireenmukaisesti ja tarvittaessa on aloitettava peruselintoimintoja tukeva hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Lipidejä muuntavat lääkeaneet, muut lipidejä muuntavat lääkeaineet, ATC-koodi: C10AX14.
Vaikutusmekanismi
Alirokumabi on täysin humaani IgG1 monoklonaalinen vasta-aine, joka sitoutuu suurella affiniteetilla ja spesifisyydellä proproteiini-konvertaasi-subtilisiini-keksiini tyyppiin 9 (PCSK9). PCSK9 sitoutuu pienitiheyksisen lipoproteiinin (LDL) reseptoreihin maksasolujen pinnalla ja edistää LDL-reseptorin hajoamista maksassa. LDL-reseptori on ensisijainen reseptori, joka poistaa LDL:ää verestä, joten LDL-reseptorien määrän väheneminen PCSK9-entsyymin vaikutuksesta suurentaa veren LDL-kolesterolipitoisuutta. Alirokumabi estää PCSK9-entsyymin sitoutumista LDL-reseptoriin ja siten lisää LDL:n poistamiseen käytettävissä olevien LDL-reseptorien määrää, jolloin LDL-kolesterolipitoisuus pienenee.
LDL-reseptoriin sitoutuu myös runsaasti triglyseridejä sisältäviä hyvin pienitiheyksisiä lipoproteiineja (VLDL jäännöspartikkelit) ja keskitiheyksisiä lipoproteiineja (IDL). Siksi alirokumabihoito voi vähentää näitä jäännöslipoproteiineja, minkä osoittaa sen apolipoproteiinia B (apo B), non-HDL-kolesterolia (kokonaiskolesterolin ja HDL-kolesterolin erotusta) ja triglyseridejä (TG) vähentävä vaikutus. Alirokumabi vähentää myös lipoproteiinia(a) [Lp(a)], joka on apolipoproteiiniin(a) sitoutunut LDL:n muoto. LDL-reseptorilla on kuitenkin osoitettu olevan heikko affiniteetti Lp(a):ta kohtaan, joten tarkkaa mekanismia, jolla alirokumabi vähentää Lp(a):ta, ei täysin ymmärretä.
Ihmisillä tehdyissä geneettisissä tutkimuksissa on todettu PCSK9-variantteja, joissa on joko toiminnanvähennys- (loss-of-function) tai toiminnanlisäysmutaatioita (gain-of-function). Ihmisillä, joilla on PCSK9-geenin yhdessä alleelissa toiminnanvähennysmutaatio, on pienemmät LDL-kolesteroliarvot, mikä korreloi sepelvaltimotaudin huomattavasti pienemmän ilmaantuvuuden kanssa. On raportoitu muutamia potilaita, joilla on PCSK9-toiminnanvähennysmutaatiot kahdessa alleellissa. Heidän LDL-kolesteroliarvonsa ovat todella pienet ja HDL-kolesteroli- ja triglyseridiarvot ovat normaalilla tasolla. PCSK9-geenin toiminnanlisäysmutaatioita on puolestaan todettu potilailla, joiden LDL-kolesteroliarvot ovat kohonneet ja joilla on kliinisesti diagnosoitu familiaalinen hyperkolesterolemia.
Kaksoissokkoutetussa, lumekontrolloidussa, 14 viikkoa kestäneessä monikeskustutkimuksessa satunnaistettiin 13 potilasta, joilla oli PCSK9-geenin toiminnanlisäysmutaatioista johtuva heterotsygoottinen familiaalinen hyperkolesterolemia (heFH), saamaan joko alirokumabia 150 mg kahden viikon välein tai lumelääkettä. LDL-kolesterolin keskimääräinen lähtöarvo oli 3,90 mmol/l (151,5 mg/dl). Viikon 2 kohdalla LDL-kolesterolipitoisuus oli pienentynyt lähtötilanteesta keskimäärin 62,5 % alirokumabia saaneilla ja 8,8 % lumelääkettä saaneilla potilailla. Viikon 8 kohdalla LDL-kolesterolipitoisuus oli kaikilla alirokumabia saaneilla potilailla pienentynyt lähtötilanteesta keskimäärin 72,4 %.
Farmakodynaamiset vaikutukset
In vitro ‑analyyseissä alirokumabi ei indusoinut Fc-välitteisen efektorin aktiivisuutta (vasta-aineesta riippuvainen soluvälitteinen toksisuus ja komplementista riippuvainen sytotoksisuus) PCSK9-entsyymin läsnä ollessa eikä ilman sitä, eikä PCSK9-entsyymiin sitoutuneelle alirokumabille todettu liukoisia immuunikomplekseja, jotka pystyvät sitomaan komplementtiproteiineja.
Kliininen teho ja turvallisuus primaarisen hyperkolesterolemian ja sekamuotoisen dyslipidemian hoidossa
Vaiheen 3 kliinisen tutkimusohjelman yhteenveto – annostelu 75 mg ja/tai 150 mg 2 viikon välein
Alirokumabin tehoa tutkittiin kymmenessä vaiheen 3 tutkimuksessa (viisi lumekontrolloitua ja viisi etsetimibikontrolloitua tutkimusta), joihin osallistui 5 296 satunnaistettua potilasta, joilla oli hyperkolesterolemia (heterotsygoottinen familiaalinen tai ei-familiaalinen) tai sekamuotoinen dyslipidemia. 3 188 potilasta satunnaistettiin saamaan alirokumabia. Vaiheen 3 tutkimuksissa 31 %:lla potilaista oli tyypin 2 diabetes ja 64 %:lla potilaista oli aiemmin todettu sepelvaltimotauti. Kymmenestä tutkimuksesta kolme toteutettiin pelkästään potilailla, joilla oli heterotsygoottinen familiaalinen hyperkolesterolemia (heFH). Suurimmalla osalla vaiheen 3 ohjelmaan osallistuneista potilaista oli käytössä lipideihin vaikuttava hoito, jonka muodosti suurin siedetty annos statiinia muiden lipideihin vaikuttavien hoitojen kanssa tai ilman niitä, ja heillä oli suuri tai hyvin suuri sydän- ja verisuonitautiriski. Kaksi tutkimusta tehtiin potilailla, jotka eivät saaneet samanaikaisesti statiinia, ja näistä toinen tutkimus tehtiin potilailla, joilla oli dokumentoitu statiini-intoleranssi.
Kaksi tutkimusta (LONG TERM ja HIGH FH), joihin osallistui yhteensä 2 416 potilasta, tehtiin ainoastaan 150 mg:n annoksella kahden viikon välein. Kahdeksan tutkimusta tehtiin 75 mg:n annoksella kahden viikon välein ja annos suurennettiin kriteerien perusteella 150 mg:aan kahden viikon välein viikolla 12 potilailla, jotka eivät olleet saavuttaneet heidän viikolla 8 arvioituun sydän- ja verisuonitautiriskiinsä perustuvaa ennalta määrättyä tavoitettaan LDL-kolesterolin suhteen.
Kaikkien vaiheen 3 tutkimusten ensisijainen tehoa mittaava päätetapahtuma oli viikolla 24 todettu LDL-kolesterolipitoisuuden keskimääräinen prosentuaalinen pieneneminen lähtötilanteesta verrattuna lumelääkkeeseen tai etsetimibiin. Kaikissa tutkimuksissa saavutettiin niiden ensisijainen päätetapahtuma. Yleensä alirokumabin antaminen vähensi kokonaiskolesterolia (TotalC), non-HDL-kolesterolia (kokonaiskolesterolin ja HDL-kolesterolin erotusta, non-HDL-C), apolipoproteiinia B (apo B) ja lipoproteiinia(a) [Lp(a)] myös prosentuaalisesti tilastollisesti merkitsevästi enemmän kuin lumelääke tai etsetimibi riippumatta siitä, saivatko potilaat samanaikaisesti statiinia. Alirokumabi myös vähensi triglyseridejä (TG) ja lisäsi suuritiheyksiseen lipoproteiiniin sitoutunutta kolesterolia (HDL-kolesterolia) ja apolipoproteiinia A-I (apo A-I) lumelääkkeeseen verrattuna. Katso yksityiskohtaiset tulokset jäljempänä esitetystä taulukosta 2. LDL-kolesterolin väheneminen havaittiin iästä, sukupuolesta, painoindeksistä, rodusta ja lähtötilanteen LDL-kolesteroliarvoista riippumatta familiaalista ja ei-familiaalista hyperkolesterolemiaa, sekamuotoista dyslipidemiaa ja diabetesta sairastavilla potilailla. Vaikka yli 75-vuotiailla potilailla todettiin yhtä suuri teho, tietoja tästä ikäryhmästä on vähän. LDL-kolesteroli väheni johdonmukaisesti riippumatta samanaikaisesti käytetyistä statiineista ja annoksista. Alirokumabiryhmän potilaista huomattavasti suurempi osa saavutti < 1,81 mmol/l:n (< 70 mg/dl:n) LDL-kolesterolipitoisuuden kuin lume- ja etsetimibiryhmissä viikolla 12 ja viikolla 24. Tutkimuksissa, joissa käytettiin kriteereihin perustuvaa annoksen suurentamisohjelmaa, suurin osa potilaista saavutti ennalta (sydän- ja verisuonitautiriskin perusteella) määrätyn LDL-kolesterolin tavoitearvon 75 mg:n annoksella kahden viikon välein, ja suurin osa potilaista jatkoi hoitoa 75 mg:n annoksella kahden viikon välein. Alirokumabin lipidejä vähentävä vaikutus todettiin 15 päivän kuluessa ensimmäisen annoksen jälkeen ja maksimaalinen vaikutus saavutettiin suunnilleen 4 viikossa. Pitkäaikaishoidossa teho säilyi tutkimusten keston ajan (2 vuoteen asti). Alirokumabihoidon lopettamisen jälkeen LDL-kolesteroliarvoissa ei todettu rebound-ilmiötä ja LDL-kolesteroliarvot palautuivat vähitellen lähtötasolle.
Kahdeksassa tutkimuksessa, joissa potilaat aloittivat kahden viikon välein annetulla 75 mg:n annoksen ohjelmalla, ennalta määritellyissä analyyseissä saavutettiin 44,5–49,2 %:n keskimääräinen LDL-kolesteroliarvojen pieneneminen, ennen mahdollista annoksen suurentamista viikolla 12. Kahdessa tutkimuksessa, joissa potilaat aloittivat ja jatkoivat kahden viikon välein annetulla 150 mg:n annoksella, saavutettiin keskimäärin 62,6 %:n LDL-kolesteroliarvojen pieneneminen viikolla 12. Sellaisten yhdistettyjen vaiheen 3 tutkimusten analyyseissä, joissa annoksen suurentaminen oli mahdollista, alaryhmässä, jossa annosta suurennettiin, kahden viikon välein annostellusta 75 mg:sta 150 mg:aan viikolla 12 edelleen pienensi statiinihoitoa saaneiden potilaiden LDL-kolesterolipitoisuutta keskimäärin 14 %. Potilailla, jotka eivät saaneet statiinihoitoa, alirokumabiannoksen suurentaminen pienensi LDL-kolesterolipitoisuutta edelleen keskimäärin 3 % ja suurin osa tästä pienenemästä todettiin noin 25 %:lla potilaista, joilla LDL-kolesterolipitoisuus pieneni edelleen vähintään 10 % annoksen suurentamisen jälkeen. Potilailla, joiden annos suurennettiin 150 mg:aan kahden viikon välein, oli lähtötilanteessa keskimäärin korkeampi LDL-kolesteroliarvo.
Sydän- ja verisuonitapahtumien arviointi
Yhdistettyjen vaiheen 3 tutkimusten ennalta määritellyissä analyyseissä ilmoitettiin hoidon aikana ilmaantuneet varmistetut sydän- ja verisuonitapahtumat, joita olivat sepelvaltimotaudista (CHD) johtunut kuolema, sydäninfarkti, iskeeminen aivohalvaus, sairaalahoitoa vaatinut epävakaa sepelvaltimotauti, sairaalahoito kongestiivisen sydämen vajaatoiminnan vuoksi ja revaskularisaatio. Näitä oli 110 potilaalla (3,5 %) alirokumabiryhmässä ja 53 potilaalla (3,0 %) vertailuryhmässä (lumelääke tai aktiivinen verrokki), jolloin riskisuhde oli 1,08 (95 %:n luottamusväli 0,78–1,50). Varmistettuja merkittäviä sydän- ja verisuonitapahtumia (MACE plus, eli sepelvaltimotaudista johtunut kuolema, sydäninfarkti, iskeeminen aivohalvaus tai sairaalahoitoa vaativa epävakaa sepelvaltimotauti) ilmoitettiin alirokumabiryhmässä 52 potilaalla 3 182:sta (1,6 %) ja vertailuryhmässä (lumelääke tai aktiivinen verrokki) 33 potilaalla 1 792:sta (1,8 %); riskisuhde = 0,81 (95 %:n luottamusväli 0,52–1,25).
LONG TERM ‑tutkimuksen ennalta määritellyissä lopullisissa analyyseissä ilmeni hoidon aikana ilmaantuneita sydän- ja verisuonitapahtumia alirokumabiryhmässä 72 potilaalla 1 550:stä (4,6 %) ja lumeryhmässä 40 potilaalla 788:sta (5,1 %); varmistettuja merkittäviä sydän- ja verisuonitapahtumia (MACE plus) ilmoitettiin alirokumabiryhmässä 27 potilaalla 1 550:stä (1,7 %) ja lumeryhmässä 26 potilaalla 788:sta (3,3 %). Riskisuhteet laskettiin post-hoc; kaikkien sydän- ja verisuonitapahtumien riskisuhde oli 0,91 (95 %:n luottamusväli 0,62–1,34); merkittävien sydän- ja verisuonitapahtumien (MACE plus) riskisuhde oli 0,52 (95 %:n luottamusväli 0,31–0,90).
Kaikki kuolinsyyt kattava kuolleisuus
Vaiheen 3 tutkimuksissa kaikki kuolinsyyt kattava kuolleisuus oli 0,6 % (20 potilasta 3 182:sta) alirokumabiryhmässä ja 0,9 % (17 potilasta 1 792:sta) vertailuryhmässä. Ensisijaisena kuolinsyynä suurimmalla osalla näistä potilaista olivat sydän- ja verisuonitapahtumat.
Yhdistelmähoito statiinin kanssa
Lumekontrolloidut vaiheen 3 tutkimukset (statiinihoito taustalla) primaarista hyperkolesterolemiaa tai sekamuotoista dyslipidemiaa sairastavilla potilailla
LONG TERM ‑tutkimus
Tähän kaksoissokkoutettuun, lumekontrolloituun, 18 kuukautta kestäneeseen monikeskustutkimukseen osallistui 2 310 potilasta, joilla oli primaarinen hyperkolesterolemia, johon liittyi suuri tai hyvin suuri sydän- ja verisuonitautiriski, ja jotka saivat statiinia suurimmalla siedetyllä annoksella, sekä jotka saivat tai eivät saaneet muuta lipideihin vaikuttavaa hoitoa. Potilaat saivat joko alirokumabia 150 mg:n annoksella kahden viikon välein tai lumelääkettä lipideihin vaikuttavan hoitonsa lisäksi. LONG TERM ‑tutkimuksen potilaista 17,7 %:lla oli heterotsygoottinen familiaalinen hyperkolesterolemia, 34,6 %:lla oli tyypin 2 diabetes ja 68,6 %:lla oli todettu aiemmin sepelvaltimotauti. Viikolla 24 LDL-kolesterolipitoisuuden prosentuaalisen muutoksen keskimääräinen ero alirokumabi- ja lumeryhmien välillä oli -61,9 % (95 %:n luottamusväli: ‑64,3 %, ‑59,4 %; p-arvo: < 0,0001). Katso yksityiskohtaiset tulokset taulukosta 2. Viikolla 12 LDL-kolesterolipitoisuuden < 1,81 mmol/l (< 70 mg/dl) oli saavuttanut alirokumabiryhmässä 82,1 % potilaista ja lumeryhmässä 7,2 % potilaista. Ero lumelääkkeeseen nähden oli tilastollisesti merkitsevä viikolla 24 kaikkien lipidien/lipoproteiinien osalta.
COMBO I ‑tutkimus
Kaksoissokkoutettuun, lumekontrolloituun, 52 viikkoa kestäneeseen monikeskustutkimukseen osallistui 311 potilasta, jotka luokiteltiin hyvin suuren sydän- ja verisuonitautiriskin ryhmään ja jotka eivät olleet saavuttaneet ennalta määriteltyä LDL-kolesterolin tavoitearvoa suurimmalla siedetyllä statiiniannoksella ja jotka saivat tai eivät saaneet muuta lipideihin vaikuttavaa hoitoa. Potilaat saivat joko 75 mg alirokumabia kahden viikon välein tai lumelääkettä lipideihin vaikuttavan hoitonsa lisäksi. Viikolla 12 alirokumabiannos suurennettiin 150 mg:aan kahden viikon välein potilailla, joiden LDL-kolesteroliarvo oli vähintään 1,81 mmol/l (≥ 70 mg/dl). Viikolla 24 LDL-kolesterolipitoisuuden prosentuaalisen muutoksen keskimääräinen ero alirokumabi- ja lumeryhmien välillä oli -45,9 % (95 %:n luottamusväli: ‑52,5 %, ‑39,3 %; p-arvo: < 0,0001). Katso yksityiskohtaiset tulokset taulukosta 2. Viikolla 12 (ennen annoksen suurentamista) LDL-kolesterolipitoisuuden < 1,81 mmol/l (< 70 mg/dl) oli saavuttanut alirokumabiryhmässä 76,0 % potilaista ja lumeryhmässä 11,3 % potilaista. Annos suurennettiin 150 mg:aan kahden viikon välein 32 potilaalla (16,8 %), jotka saivat hoitoa yli 12 viikon ajan. Viikolla 24 niiden potilaiden alaryhmässä, joiden annosta suurennettiin viikolla 12, LDL-kolesterolipitoisuus oli edelleen pienentynyt keskimäärin 22,8 %. Ero lumelääkkeeseen oli tilastollisesti merkitsevä viikolla 24 kaikkien lipidien/lipoproteiinien osalta triglyseridejä ja apo A-I:tä lukuun ottamatta.
Lumekontrolloidut vaiheen 3 tutkimukset (statiinihoito taustalla) heterotsygoottista familiaalista hyperkolesterolemiaa (heFH) sairastavilla potilailla
FH I ja FH II ‑tutkimukset
Kahteen lumekontrolloituun, kaksoissokkoutettuun, 18 kuukautta kestäneeseen monikeskustutkimukseen osallistui 732 potilasta, joilla oli heterotsygoottinen familiaalinen hyperkolesterolemia ja jotka saivat statiinia suurimmalla siedetyllä annoksella ja jotka saivat tai eivät saaneet muuta lipideihin vaikuttavaa hoitoa. Potilaat saivat joko 75 mg alirokumabia kahden viikon välein tai lumelääkettä lipideihin vaikuttavan hoitonsa lisäksi. Viikolla 12 alirokumabiannos suurennettiin 150 mg:aan kahden viikon välein potilaille, joiden LDL-kolesteroliarvo oli vähintään 1,81 mmol/l (≥ 70 mg/dl). Viikolla 24 LDL-kolesterolipitoisuuden prosentuaalisen muutoksen ero alirokumabi- ja lumeryhmien välillä oli -55,8 % (95 %:n luottamusväli: ‑60,0 %, ‑51,6 %; p-arvo: < 0,0001). Katso yksityiskohtaiset tulokset taulukosta 2. Viikolla 12 (ennen annoksen suurentamista) LDL-kolesterolipitoisuuden < 1,81 mmol/l (< 70 mg/dl) oli saavuttanut alirokumabiryhmässä 50,2 % potilaista ja lumeryhmässä 0,6 % potilaista. Viikolla 24 niiden potilaiden alaryhmässä, joiden annosta suurennettiin viikolla 12, LDL-kolesterolipitoisuus oli edelleen pienentynyt keskimäärin 15,7 %. Ero lumelääkkeeseen nähden oli tilastollisesti merkitsevä viikolla 24 kaikkien lipidien/lipoproteiinien osalta.
HIGH FH ‑tutkimus
Kolmanteen kaksoissokkoutettuun, lumekontrolloituun, 18 kuukautta kestäneeseen monikeskustutkimukseen osallistui 106 heterotsygoottista familiaalista hyperkolesterolemiaa sairastavaa potilasta, jotka saivat statiinia suurimmalla siedetyllä annoksella ja jotka saivat tai eivät saaneet muuta lipideihin vaikuttavaa hoitoa ja joiden lähtötilanteen LDL-kolesteroliarvo oli vähintään 4,14 mmol/l (≥ 160 mg/dl). Potilaat saivat lipideihin vaikuttavan hoitonsa lisäksi joko alirokumabia 150 mg:n annoksella kahden viikon välein tai lumelääkettä. Viikolla 24 LDL-kolesterolipitoisuuden prosentuaalisen muutoksen ero alirokumabi- ja lumeryhmien välillä oli keskimäärin -39,1 % (95 %:n luottamusväli: ‑51,1 %, ‑27,1 %; p-arvo: < 0,0001). Katso yksityiskohtaiset tulokset taulukosta 2. Keskimääräiset muutokset kaikkien muiden lipidien/lipoproteiinien osalta olivat samankaltaiset kuin FH I- ja FH II ‑tutkimuksissa, mutta triglyseridien, HDL-kolesterolin ja apo A-I:n suhteen ei saavutettu tilastollista merkitsevyyttä.
Etsetimibikontrolloitu vaiheen 3 tutkimus (statiinihoito taustalla) primaarista hyperkolesterolemiaa tai sekamuotoista dyslipidemiaa sairastavilla potilailla
COMBO II ‑tutkimus
Kaksoissokkoutettuun, etsetimibikontrolloituun, kaksi vuotta kestäneeseen monikeskustutkimukseen osallistui 707 potilasta, jotka luokiteltiin hyvin suuren sydän- ja verisuonitautiriskin ryhmään ja jotka eivät olleet saavuttaneet ennalta määriteltyä LDL-kolesterolin tavoitearvoa suurimmalla siedetyllä statiiniannoksella. Potilaat saivat joko 75 mg alirokumabia kahden viikon välein tai 10 mg etsetimibia statiinihoitonsa lisäksi. Viikolla 12 alirokumabiannos suurennettiin 150 mg:aan kahden viikon välein potilailla, joiden LDL-kolesteroliarvo oli vähintään 1,81 mmol/l (≥ 70 mg/dl). Viikolla 24 LDL-kolesterolipitoisuuden prosentuaalisen muutoksen keskimääräinen ero alirokumabi- ja etsetimibiryhmien välillä oli -29,8 % (95 %:n luottamusväli: ‑34,4 %, ‑25,3 %; p-arvo: < 0,0001). Katso yksityiskohtaiset tulokset taulukosta 2. Viikolla 12 (ennen annoksen suurentamista) LDL-kolesterolipitoisuuden < 1,81 mmol/l (< 70 mg/dl) oli saavuttanut alirokumabiryhmässä 77,2 % potilaista ja etsetimibiryhmässä 46,2 % potilaista. Viikolla 24 niiden potilaiden alaryhmässä, joiden annosta suurennettiin viikolla 12, LDL-kolesterolipitoisuus oli edelleen pienentynyt keskimäärin 10,5 %. Ero etsetimibiin nähden oli tilastollisesti merkitsevä viikolla 24 kaikkien lipidien/lipoproteiinien osalta triglyseridejä ja apo A-I:tä lukuun ottamatta.
Monoterapiana tai muun kuin statiinipohjaisen lipideihin vaikuttavan hoidon lisälääkkeenä
Etsetimibikontrolloidut vaiheen 3 tutkimukset primaarista hyperkolesterolemiaa sairastavilla potilailla (ilman statiinihoitoa taustalla)
ALTERNATIVE-tutkimus
Kaksoissokkoutettuun, etsetimibikontrolloituun, 24 viikkoa kestäneeseen monikeskustutkimukseen osallistui 248 potilasta, joilla oli dokumentoitu statiini-intoleranssi luurankolihaksiin liittyvien oireiden vuoksi. Potilaat saivat joko alirokumabia 75 mg kahden viikon välein, etsetimibiä 10 mg kerran vuorokaudessa tai atorvastatiinia 20 mg kerran vuorokaudessa (tutkimushaara, jossa hoito lopetettiin ja aloitettiin uudelleen). Viikolla 12 alirokumabiannos suurennettiin 150 mg:aan kahden viikon välein potilaille, joiden LDL-kolesteroliarvo oli vähintään 1,81 mmol/l (≥ 70 mg/dl) tai vähintään 2,59 mmol/l (≥ 100 mg/dl) sydän- ja verisuonitautiriskin mukaan. Viikolla 24 LDL-kolesterolipitoisuuden prosentuaalisen muutoksen keskimääräinen ero alirokumabi- ja etsetimibiryhmien välillä oli -30,4 % (95 %:n luottamusväli: ‑36,6 %, ‑24,2 %; p-arvo: < 0,0001). Katso yksityiskohtaiset tulokset taulukosta 2. Viikolla 12 (ennen annoksen suurentamista) LDL-kolesterolipitoisuuden < 1,81 mmol/l (< 70 mg/dl) oli saavuttanut alirokumabiryhmässä 34,9 % potilaista ja etsetimibiryhmässä 0 % potilaista. Viikolla 24 niiden potilaiden alaryhmässä, joiden annosta suurennettiin viikolla 12, LDL-kolesterolipitoisuus oli edelleen pienentynyt keskimäärin 3,6 %. Ero etsetimibiin nähden oli tilastollisesti merkitsevä viikolla 24 LDL-kolesterolin, kokonaiskolesterolin, non-HDL-kolesterolin, apo B:n ja Lp(a):n osalta.
Tässä tutkimuksessa arvioitiin potilaita, jotka eivät olleet sietäneet vähintään kahta statiinia (joista ainakin toisen annos oli pienin hyväksytty annos). Näillä potilailla ilmeni luurankolihaksiin liittyviä haittatapahtumia vähemmän alirokumabiryhmässä (32,5 %) kuin atorvastatiiniryhmässä (46,0 %) (riskisuhde = 0,61 [95 %:n luottamusväli 0,38–0,99]), ja atorvastatiiniryhmään (22,2 %) verrattuna pienempi prosentuaalinen osuus alirokumabiryhmän potilaista (15,9 %) keskeytti tutkimushoidon luurankolihaksiin liittyvien haittavaikutusten vuoksi. Viidessä lumekontrolloidussa tutkimuksessa suurinta siedettyä statiiniannosta käyttäneillä potilailla (n = 3 752) luurankolihaksiin liittyvistä haittatapahtumista johtunut keskeyttämisten määrä oli 0,4 % alirokumabiryhmässä ja 0,5 % lumeryhmässä.
MONO-tutkimus
Kaksoissokkoutettuun, etsetimibikontrolloituun, 24 viikkoa kestäneeseen monikeskustutkimukseen osallistui 103 potilasta, joilla oli kohtalainen sydän- ja verisuonitautiriski ja jotka eivät saaneet statiineja tai muita lipideihin vaikuttavia hoitoja ja joiden lähtötilanteen LDL-kolesteroliarvot olivat 2,59–4,91 mmol/l (100–190 mg/dl). Potilaat saivat joko 75 mg alirokumabia kahden viikon välein tai 10 mg etsetimibia kerran vuorokaudessa. Viikolla 12 alirokumabiannos suurennettiin 150 mg:aan kahden viikon välein potilailla, joiden LDL-kolesteroliarvo oli vähintään 1,81 mmol/l (≥ 70 mg/dl). Viikolla 24 LDL-kolesterolipitoisuuden prosentuaalisen muutoksen keskimääräinen ero alirokumabi- ja etsetimibiryhmien välillä oli -31,6 % (95 %:n luottamusväli: ‑40,2 %, ‑23,0 %; p-arvo: < 0,0001). Katso yksityiskohtaiset tulokset taulukosta 2. Viikolla 12 (ennen annoksen suurentamista) LDL-kolesterolipitoisuuden < 1,81 mmol/l (< 70 mg/dl) oli saavuttanut alirokumabiryhmässä 57,7 % potilaista ja etsetimibiryhmässä 0 % potilaista. Annos suurennettiin 150 mg:aan kahden viikon välein 14 potilaalla (30,4 %), jotka saivat hoitoa yli 12 viikon ajan. Viikolla 24 niiden potilaiden alaryhmässä, joiden annosta suurennettiin viikolla 12, LDL-kolesterolipitoisuus oli edelleen pienentynyt keskimäärin 1,4 %. Ero etsetimibiin nähden oli tilastollisesti merkitsevä viikolla 24 LDL-kolesterolin, kokonaiskolesterolin, non-HDL-kolesterolin ja apo B:n osalta.
Taulukko 2: LDL-kolesterolin ja muiden lipidien/lipoproteiinien keskimääräinen prosentuaalinen muutos lähtötilanteesta lume- ja etsetimibikontrolloiduissa tutkimuksissa – annostelu 75 mg ja/tai 150 mg 2 viikon välein
| Keskimääräinen prosentuaalinen muutos lähtötilanteesta lumekontrolloiduissa tutkimuksissa statiinihoito taustalla | ||||||||
| LONG TERM (N = 2 310) | FH I ja FH II (N = 732) | High FH (N = 106) | COMBO I (N = 311) | |||||
| Lumelääke | Alirokumabi | Lumelääke | Alirokumabi | Lumelääke | Alirokumabi | Lumelääke | Alirokumabi | |
| Potilaiden määrä | 780 | 1 530 | 244 | 488 | 35 | 71 | 106 | 205 |
| Keskimääräinen lähtötason LDL-kolesteroli mmol/l (mg/dl) | 3,16 (122,0)
| 3,18 (122,8)
| 3,65 (140,9)
| 3,66 (141,3)
| 5,21 (201,0)
| 5,10 (196,3)
| 2,71 (104,6)
| 2,60 (100,3)
|
| Viikko 12 | ||||||||
| LDL-kolesteroli (ITT)a | 1,5 | -63,3 | 5,4 | -43,6 | -6,6 | -46,9 | 1,1 | -46,3 |
| LDL-kolesteroli (hoidon aikana)b | 1,4 | -64,2 | 5,3 | -44,0 | -6,6 | -46,9 | 1,7 | -47,6 |
| Viikko 24 | ||||||||
| LDL-kolesteroli (ITT)a | 0,8 | -61,0c | 7,1 | -48,8d | -6,6 | -45,7e | -2,3 | -48,2f |
| LDL-kolesteroli (hoidon aikana)b | 0,7 | -62,8 | 6,8 | -49,3 | -6,6 | -45,5 | -0,8 | -50,7 |
| Non-HDL-kolesteroli | 0,7 | -51,6 | 7,4 | -42,8 | -6,2 | -41,9 | -1,6 | -39,1 |
| Apo B | 1,2 | -52,8 | 1,9 | -41,7 | -8,7 | -39,0 | -0,9 | -36,7 |
| Total-C | -0,3 | -37,8 | 5,5 | -31,2 | -4,8 | -33,2 | -2,9 | -27,9 |
| Lp(a) | -3,7 | -29,3 | -8,5 | -26,9 | -8,7 | -23,5 | -5,9 | -20,5 |
| TG | 1,8 | -15,6 | 4,3 | -9,8 | -1,9 | -10,5 | -5,4 | -6,0 |
| HDL-kolesteroli | -0,6 | 4,0 | 0,2 | 7,8 | 3,9 | 7,5 | -3,8 | 3,5 |
| Apo A-I | 1,2 | 4,0 | -0,4 | 4,2 | 2,0 | 5,6 | -2,5 | 3,3 |
| Keskimääräinen prosentuaalinen muutos lähtötilanteesta etsetimibikontrolloiduissa tutkimuksissa | ||||||
| Statiinihoito taustalla | Ei statiinihoitoa taustalla | |||||
| COMBO II (N = 707) | ALTERNATIVE (N = 248) | MONO (N = 103) | ||||
| Etsetimibi | Alirokumabi | Etsetimibi | Alirokumabi | Etsetimibi | Alirokumabi | |
| Potilaiden määrä | 240 | 467 | 122 | 126 | 51 | 52 |
| Lähtötilanteen keskimääräinen LDL-kolesteroli mmol/l (mg/dl) | 2,71 (104,5)
| 2,81 (108,3)
| 5,03 (194,2)
| 5,0 (191,1)
| 3,58 (138,3)
| 3,65 (141,1)
|
| Viikko 12 | ||||||
| LDL-kolesteroli (ITT)a | -21,8 | -51,2 | -15,6 | -47,0 | -19,6 | -48,1 |
| LDL-kolesteroli (hoidon aikana)b | -22,7 | -52,4 | -18,0 | -51,2 | -20,4 | -53,2 |
| Viikko 24 | ||||||
| LDL-kolesteroli (ITT)a | -20,7 | -50,6g | -14,6 | -45,0h | -15,6 | -47,2i |
| LDL-kolesteroli (hoidon aikana)b | -21,8 | -52,4 | -17,1 | -52,2 | -17,2 | -54,1 |
| Non-HDL-kolesteroli | -19,2 | -42,1 | -14,6 | -40,2 | -15,1 | -40,6 |
| Apo B | -18,3 | -40,7 | -11,2 | -36,3 | -11,0 | -36,7 |
| Total-C | -14,6 | -29,3 | -10,9 | -31,8 | -10,9 | -29,6 |
| Lp(a) | -6,1 | -27,8 | -7,3 | -25,9 | -12,3 | -16,7 |
| TG | -12,8 | -13,0 | -3,6 | -9,3 | -10,8 | -11,9 |
| HDL-kolesteroli | 0,5 | 8,6 | 6,8 | 7,7 | 1,6 | 6,0 |
| Apo A-I | -1,3 | 5,0 | 2,9 | 4,8 | -0,6 | 4,7 |
a ITT-analyysi (lähtöryhmien mukainen potilasjoukko, intent-to-treat), sisältää kaikki lipiditulokset tutkimuksen koko ajalta tutkimushoitoon sitoutumisesta riippumatta.
b Hoitoa saaneiden analyysi – analyysi, joka rajoittuu ajanjaksoon, jolloin potilaat todella saavat hoitoa.
LDL-kolesterolipitoisuuden prosentuaalinen pieneneminen viikolla 24 vastaa seuraavaa keskimääräistä absoluuttista muutosta:
c ‑1,92 mmol/l (‑74,2 mg/dl); d ‑1,84 mmol/l (‑71,1 mg/dl); e ‑2,35 mmol/l (‑90,8 mg/dl); f ‑1,30 mmol/l (‑50,3 mg/dl); g ‑1,44 mmol/l (‑55,4 mg/dl); h ‑2,18 mmol/l (‑84,2 mg/dl); i ‑1,73 mmol/l (‑66,9 mg/dl)
Annostelu 4 viikon välein
CHOICE I -tutkimus
Kaksoissokkoutetussa, lumekontrolloidussa, 48 viikkoa kestäneessä monikeskustutkimuksessa oli mukana 540 potilasta, jotka saivat suurimman siedetyn annoksen statiinia ja mahdollisesti muuta lipidejä alentavaa hoitoa (308 potilasta ryhmässä, jossa alirokumabia annettiin 300 mg:n annoksena 4 viikon välein, 76 potilasta ryhmässä, jossa alirokumabia annettiin 75 mg:n annoksena 2 viikon välein, ja 156 potilasta lumeryhmässä), ja 252 potilasta, joita ei hoidettu statiinilla (144 potilasta ryhmässä, jossa alirokumabia annettiin 300 mg:n annoksena 4 viikon välein, 37 potilasta ryhmässä, jossa alirokumabia annettiin 75 mg:n annoksena 2 viikon välein, ja 71 potilasta lumeryhmässä). Potilaat saivat lipidejä alentavan lääkityksensä (statiinin, muun kuin statiinihoidon tai pelkän ruokavaliohoidon) lisäksi alirokumabia 300 mg 4 viikon välein, alirokumabia 75 mg 2 viikon välein tai lumelääkettä. Alirokumabiryhmän (300 mg 4 viikon välein) potilaat saivat vuoroittain lumepistoksia sokkouttamisen säilyttämiseksi pistostiheyden suhteen. Kaiken kaikkiaan 71,6 % potilaista luokiteltiin korkean tai hyvin korkean kardiovaskulaaririskin potilaiksi ja he eivät olleet saavuttaneet LDL-kolesterolitavoitettaan. Viikolla 12 annos muutettiin alirokumabiryhmissä 150 mg:aan 2 viikon välein, jos potilaan LDL-kolesteroliarvo oli ≥ 1,81 mmol/l (≥ 70 mg/dl) tai ≥ 2,59 mmol/l (≥ 100 mg/dl) kardiovaskulaaririskin tasosta riippuen, tai jos potilaan LDL-kolesteroli ei ollut laskenut vähintään 30 % lähtötilanteesta.
Statiinia taustalääkityksenä käyttäneiden kohortissa keskimääräinen LDL-kolesteroli lähtötilanteessa oli 2,91 mmol/l (112,7 mg/dl). Alirokumabiryhmässä (300 mg 4 viikon välein) viikolla 12 keskimääräinen prosentuaalinen muutos lähtötilanteen LDL-kolesterolissa (ITT-analyysi) oli -55,3 % ja lumeryhmässä +1,1 %. Viikolla 12 (ennen annoksen muuttamista) 77,3 % alirokumabiryhmän (300 mg 4 viikon välein) potilaista ja 9,3 % lumeryhmän potilaista saavutti LDL-kolesteroliarvon < 1,81 mmol/l (˂ 70 mg/dl). Alirokumabiryhmässä (300 mg 4 viikon välein/150 mg 2 viikon välein) viikolla 24 keskimääräinen prosentuaalinen muutos lähtötilanteen LDL-kolesterolissa (ITT-analyysi) oli -58,8 % ja lumeryhmässä -0,1 %. Viikolla 24 alirokumabin (300 mg 4 viikon välein/150 mg 2 viikon välein) keskimääräinen hoitoero lumeeseen verrattuna LDL-kolesterolin prosentuaalisessa muutoksessa lähtötilanteesta oli -58,7 % (97,5 % CI: -65,0 %, -52,4 %; p-arvo: ˂ 0,0001). 12 hoitoviikon jälkeen annos muutettiin 150 mg:aan 2 viikon välein 56 potilaalla alirokumabilla (300 mg 4 viikon välein) hoidetusta 290 potilaasta (19,3 %). Niiden potilaiden alaryhmässä, joiden annos muutettiin 150 mg:aan 2 viikon välein viikolla 12, LDL-kolesteroli laski edelleen 25,4 % viikolle 24 tultaessa. Potilailla, jotka aloittivat hoidon alirokumabiannoksella 300 mg neljän viikon välein ja joiden annosta ei muutettu 150 mg:aan kahden viikon välein (80,7 % potilaista), laskennallisen LDL-kolesteroliarvon keskimääräinen prosentuaalinen muutos lähtötilanteesta oli ‑63,4 % viikolla 12 ja ‑62,0 % viikolla 24.
Statiinia käyttämättömien potilaiden kohortissa keskimääräinen LDL-kolesteroli lähtötilanteessa oli 3,67 mmol/l (142,1 mg/dl). Alirokumabiryhmässä (300 mg 4 viikon välein) viikolla 12 keskimääräinen prosentuaalinen muutos lähtötilanteen LDL-kolesterolissa (ITT-analyysi) oli -58,4 % ja lumeryhmässä +0,3 %. Viikolla 12 (ennen annoksen muuttamista) 65,2 % alirokumabiryhmän (300 mg 4 viikon välein) potilaista ja 2,8 % lumeryhmän potilaista saavutti LDL-kolesteroliarvon < 1,81 mmol/l (˂ 70 mg/dl). Alirokumabiryhmässä (300 mg 4 viikon välein/150 mg 2 viikon välein) viikolla 24 keskimääräinen prosentuaalinen muutos lähtötilanteen LDL-kolesterolissa (ITT-analyysi) oli -52,7 % ja lumeryhmässä -0,3 %. Viikolla 24 alirokumabin (300 mg 4 viikon välein/150 mg 2 viikon välein) keskimääräinen hoitoero lumeeseen verrattuna LDL-kolesterolin prosentuaalisessa muutoksessa lähtötilanteesta oli -52,4 % (97,5 % CI: -59,8 %, -45,0 %; p-arvo: ˂ 0,0001). 12 hoitoviikon jälkeen annos muutettiin 150 mg:aan 2 viikon välein 19 potilaalla alirokumabilla (300 mg 4 viikon välein) hoidetusta 129 potilaasta (14,7 %). Niiden potilaiden alaryhmässä, joiden annos muutettiin 150 mg:aan 2 viikon välein viikolla 12, LDL-kolesteroli laski edelleen keskimäärin 7,3 % viikolle 24 tultaessa.
Molemmissa kohorteissa ero lumeeseen verrattuna oli tilastollisesti merkitsevä viikolla 24 kaikkien lipidiparametrien suhteen lukuun ottamatta Apo A-1:ä statiinia taustalääkityksenä saaneiden potilaiden alaryhmässä.
Toinen tutkimuksen rinnakkaisista ensisijaisista päätetapahtumista oli LDL-kolesterolipitoisuuden keskimääräinen pieneneminen lähtötilanteesta viikkoon 21–24 mennessä. Potilailla, jotka saivat alirokumabia 300 mg neljän viikon välein tai 150 mg kahden viikon välein, LDL-kolesterolipitoisuus pieneni lähtötilanteesta viikkoon 21–24 mennessä keskimäärin merkitsevästi enemmän kuin lumelääkettä saaneilla potilailla. Tämä koski sekä potilaita, jotka eivät saaneet statiineja (alirokumabiryhmä: ‑56,9 %, lumeryhmä: ‑1,6 %), että potilaita, jotka saivat statiineja (alirokumabiryhmä: ‑65,8 %, lumeryhmä: ‑0,8 %).
Useimmat potilaat (344/419, 82,1 %), jotka aloittivat annostuksella 300 mg neljän viikon välein, olivat saavuttaneet tutkimuksessa määritellyt LDL-kolesterolitavoitteet viikolla 8, eikä heidän annostaan tarvinnut muuttaa viikolla 12. LDL-kolesterolipitoisuudet olivat pienentyneet 60,5–71,9 % viikoilla 20–24 potilailla, joilla annostus oli 300 mg neljän viikon välein, ja 57,2–63,0 % potilailla, joilla annostus muutettiin 300 mg:sta neljän viikon välein 150 mg:aan kahden viikon välein.
SYDNEY-tutkimus
Satunnaistetussa, avoimessa, 16 viikkoa kestäneessä monikeskustutkimuksessa arvioitiin esitäytetyn alirokumabikynän (2 ml) käytettävyyttä. Tutkimuksessa oli kaksi rinnakkaista hoitohaaraa (anto kerran kuukaudessa: yksi injektio 2 ml:n esitäytetyllä kynällä [1 x 300 mg] tai kaksi erillistä 150 mg:n injektiota 1 ml:n esitäytetyllä kynällä [2 x 150 mg]). Tutkimukseen osallistui 69 potilasta, joilla oli suuri tai hyvin suuri sydän- ja verisuonitautiriski sekä hyperkolesterolemia, jota ei ollut saatu riittävästi hallintaan lipidejä muuntavalla lääkityksellä (lähtötilanteen LDL-kolesteroliarvo ≥ 1,81 mmol/l [≥ 70 mg/dl]). LDL-kolesteroliarvot olivat pienentyneet lähtötilanteesta viikkoon 4 mennessä keskimäärin 66,2 %, kun antoon kerran kuukaudessa käytettiin 2 ml:n esitäytettyä kynää, ja 51,2 % käytettäessä 1 ml:n esitäytettyä kynää. Kun ryhmien välillä oli tehty korjaukset sukupuolten välisten erojen suhteen, LDL-kolesteroliarvot olivat pienentyneet keskimäärin 63,5 % (2 ml:n esitäytetty kynä) ja 53,9 % (1 ml:n esitäytetty kynä). LDL-kolesteroliarvoa pienentävä vaikutus säilyi 16 viikon ajan.
Kliininen teho ja turvallisuus sydän- ja verisuonitapahtumien ehkäisyssä
ODYSSEY OUTCOMES -tutkimus
Kaksoissokkoutetussa, lumekontrolloidussa monikeskustutkimuksessa seurattiin 18 924:ää aikuispotilasta (9 462 sai alirokumabia ja 9 462 lumelääkettä) enintään 5 vuoden ajan. Potilailla oli ilmennyt äkilliseen sepelvaltimo-oireyhtymään (ACS) liittyvä tapahtuma 4–52 viikkoa ennen satunnaistamista, ja he saivat lipideihin vaikuttavaa hoitoa, johon sisältyi intensiivinen statiinilääkitys (määritelmän mukaan 40 tai 80 mg atorvastatiinia tai 20 tai 40 mg rosuvastatiinia), tai näitä statiineja suurimmalla siedetyllä annoksella joko toisen lipideihin vaikuttavan hoidon kanssa tai ilman sitä. Potilaat satunnaistettiin suhteessa 1:1 saamaan joko 75 mg alirokumabia 2 viikon välein tai lumelääkettä 2 viikon välein. Jos LDL‑kolesterolia oli ennalta määriteltyjen LDL‑kolesterolia koskevien kriteerien perusteella kuukauden 2 kohdalla tarpeen alentaa edelleen (LDL kolesteroli ≥ 1,29 mmol/l tai ≥ 50 mg/dl), alirokumabiannos suurennettiin 150 mg:aan 2 viikon välein. Potilailla, joiden annos oli suurennettu 150 mg:aan 2 viikon välein ja joilla kaksi peräkkäistä LDL-kolesteroliarvoa oli alle 0,65 mmol/l (< 25 mg/dl), annosta pienennettiin 150 mg:sta 2 viikon välein 75 mg:aan 2 viikon välein. Potilaat, jotka saivat 75 mg 2 viikon välein ja joilla kaksi peräkkäistä LDL-kolesteroliarvoa oli alle 0,39 mmol/l (< 15 mg/dl), siirrettiin sokkoutetusti saamaan lumelääkettä. Alirokumabia saaneista 9 451 potilaasta noin 2 615 potilaan (27,7 %) annos oli suurennettava 150 mg:aan 2 viikon välein. Näistä 2 615 potilaasta 805 potilaalla (30,8 %) annos pienennettiin 75 mg:aan 2 viikon välein. Kaikkiaan 730 (7,7 %) 9 451 potilaasta siirrettiin saamaan lumelääkettä. Potilaista 99,5 %:n elossaoloa seurattiin tutkimuksen loppuun asti. Seuranta-ajan keston mediaani oli 33 kuukautta.
Äkilliseen sepelvaltimo-oireyhtymään liittyvä indeksitapahtuma oli 83,2 %:lla potilaista sydäninfarkti (34,6 %:lla ST-nousuinfarkti ja 48,6 %:lla ST-nousuton infarkti) ja 16,8 %:lla potilaista epävakaaseen sepelvaltimotautiin liittyvä kohtaus. Suurin osa (88,8 %) potilaista sai satunnaistamishetkellä intensiivistä statiinihoitoa joko toisen lipideihin vaikuttavan hoidon kanssa tai ilman sitä. Keskimääräinen LDL-kolesteroliarvo lähtötilanteessa oli 2,39 mmol/l (92,4 mg/dl).
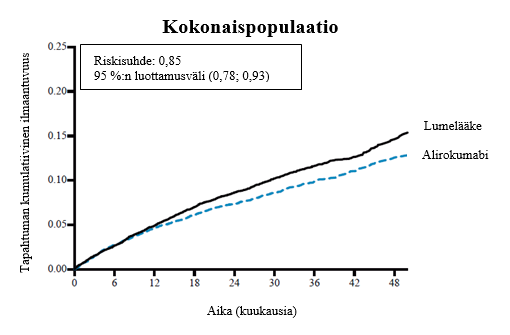
Alirokumabi pienensi merkitsevästi ensisijaisen yhdistelmäpäätetapahtuman riskiä. Ensisijainen yhdistelmäpäätetapahtuma oli aika ensimmäisen merkittävän sydän- ja verisuonitapahtuman (MACE plus) eli sepelvaltimotaudista johtuneen kuoleman, ei‑fataalin sydäninfarktin, fataalin tai ei‑fataalin iskeemisen aivohalvauksen tai sairaalahoitoa vaatineen epävakaan sepelvaltimotaudin ilmaantumiseen (riskisuhde 0,85, 95 %:n luottamusväli 0,78–0,93; p‑arvo = 0,0003). Alirokumabi vähensi myös merkitsevästi seuraavia yhdistelmäpäätetapahtumia: sepelvaltimotautitapahtuman riski; merkittävä sepelvaltimotautitapahtuma; sydän- ja verisuonitapahtuma; sekä kaikki kuolinsyyt kattavan kuolleisuuden, ei-fataalin sydäninfarktin ja ei-fataalin iskeemisen aivohalvauksen yhdistelmä. Kaikki kuolinsyyt kattavan kuolleisuuden havaittiin myös vähentyneen, ja vähentyminen oli hierarkkisen testauksen perusteella vain nimellisesti tilastollisesti merkitsevä (riskisuhde 0,85, 95 %:n luottamusväli 0,78–0,98). Tulokset on esitetty taulukossa 3.
Taulukko 3: Alirokumabin teho ODYSSEY OUTCOMES -tutkimuksessa (kokonaispopulaatio)
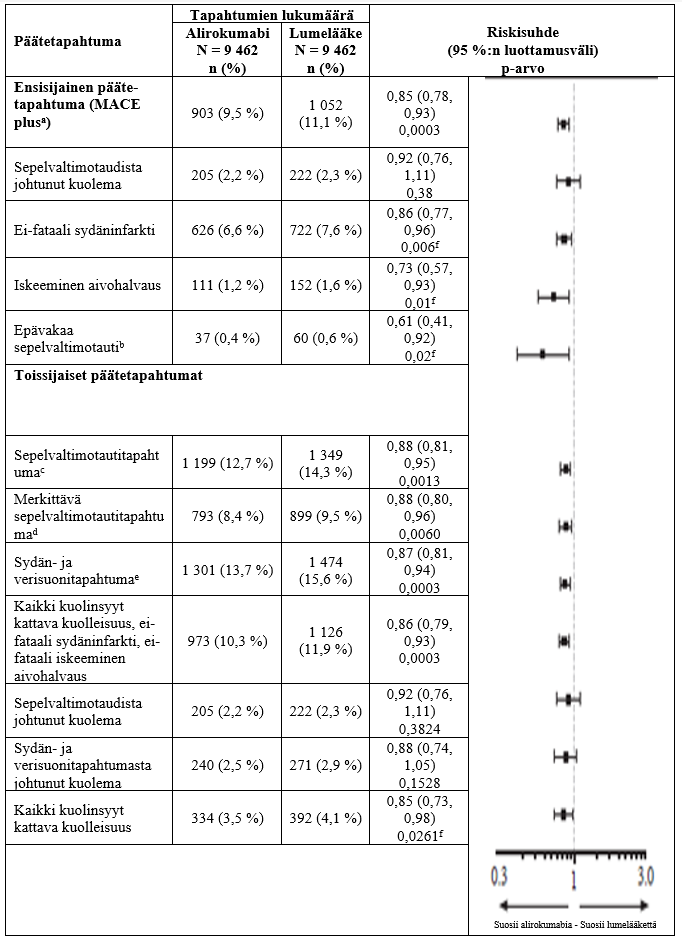
a Mace plus, merkittävä sydän- ja verisuonitapahtuma, eli sepelvaltimotaudista johtunut kuolema, ei‑fataali sydäninfarkti, fataali tai ei‑fataali iskeeminen aivohalvaus tai sairaalahoitoa vaativa epävakaa sepelvaltimotauti
b Sairaalahoitoa vaatinut epävakaa sepelvaltimotauti
c Sepelvaltimotautitapahtuma, määritelmän mukaan jokin seuraavista: merkittävä sepelvaltimotautitapahtumad, sairaalahoitoa vaativa epävakaa sepelvaltimotauti tai iskemian vuoksi tehty sepelvaltimon revaskularisaatiotoimenpide
d Merkittävä sepelvaltimotautitapahtuma, määritelmän mukaan jompikumpi seuraavista: sepelvaltimotaudista johtuva kuolema tai ei-fataali sydäninfarkti
e Sydän- ja verisuonitapahtuma, määritelmän mukaan jokin seuraavista: sydän- ja verisuonitapahtumasta johtuva kuolema, mikä tahansa ei-fataali sepelvaltimotautitapahtuma tai ei-fataali iskeeminen aivohalvaus
f Nimellinen merkitsevyys
Kuvassa 1 on esitetty ensisijaisen päätetapahtuman kumulatiivisen ilmaantuvuuden Kaplan-Meier-estimaatit koko potilaspopulaatiolle ajan funktiona.
Kuva 1 Ensisijaisen yhdistelmäpäätetapahtuman kumulatiivinen ilmaantuvuus 4 vuoden aikana ODYSSEY OUTCOMES -tutkimuksessa
Neurokognitiivinen toiminta
96 viikon pituisessa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa arvioitiin alirokumabin vaikutusta neurokognitiiviseen toimintaan 96 hoitoviikon (noin 2 vuoden) jälkeen potilailla, joilla oli heterotsygoottinen familiaalinen hyperkolesterolemia tai ei-familiaalinen hyperkolesterolemia ja suuri tai hyvin suuri sydän- ja verisuonitautiriski.
Neurokognitiivista toimintaa arvioitiin CANTAB-testisarjalla (Cambridge Neuropsychological Test Automated Battery). Yhteensä 2 171 potilasta satunnaistettiin; 1 087 potilasta sai alirokumabia 75 mg ja/tai 150 mg kahden viikon välein ja 1 084 potilasta sai lumelääkettä. Suurin osa (> 80 %) kummankin ryhmän potilaista jatkoi 96 viikon pituisen kaksoissokkoutetun hoitojakson loppuun asti.
96 hoitoviikon aikana alirokumabilla ei havaittu olevan vaikutusta neurokognitiiviseen toimintaan. Niiden potilaiden osuus, joilla ilmeni neurokognitiivisia häiriöitä, oli alirokumabia saaneiden ryhmissä pieni (1,3 %) ja verrattavissa lumelääkettä saaneisiin (1,7 %). Turvallisuuteen liittyviä huolenaiheita neurokognitiivisen toiminnan suhteen ei havaittu alirokumabia saaneilla potilailla, joilla kaksi peräkkäistä LDL-kolesteroliarvoa oli hoitojakson aikana joko alle 0,65 mmol/l (< 25 mg/dl) tai alle 0,39 mmol/l (< 15 mg/dl).
Pediatriset potilaat
Homotsygoottista familiaalista hyperkolesterolemiaa sairastavien pediatristen potilaiden hoito
48 viikon pituisessa avoimessa tutkimuksessa arvioitiin alirokumabin tehoa ja turvallisuutta taustahoitojen lisänä kahden viikon välein annetulla 75 mg:n annoksella (jos kehonpaino oli < 50 kg) tai kahden viikon välein annetulla 150 mg:n annoksella (jos kehonpaino oli ≥ 50 kg) 18 pediatrisella potilaalla (8–17-vuotiaita), joilla oli homotsygoottinen familiaalinen hyperkolesterolemia. Potilaat saivat alirokumabia 75 tai 150 mg kahden viikon välein ilman annosmuutoksia viikkoon 12 asti.
LDL-kolesterolin keskimääräinen lähtöarvo oli 9,6 mmol/l (373 mg/dl). LDL-kolesterolin keskimääräinen prosentuaalinen muutos lähtötilanteesta viikkoon 12 oli lähtöryhmien mukaisessa potilasjoukossa (ITT) (N = 18) ‑4,1 % (95 %:n luottamusväli: ‑23,1 %, 14,9 %), ja siihen liittyi vasteen suuri vaihtelu LDL-kolesterolin vähenemisen osalta. Vasteen saaneita, joilla saavutettiin ≥ 15 %:n väheneminen lähtötilanteesta, oli viikon 12 kohdalla 50 %, viikon 24 kohdalla 50 % ja viikon 48 kohdalla 39 %.
Heterotsygoottista familiaalista hyperkolesterolemiaa sairastavien pediatristen potilaiden hoito
Alirokumabin tehoa ja turvallisuutta arvioitiin vaiheen 3 monikeskustutkimuksessa 153 potilaalla (8–17-vuotiaita), joilla oli heterotsygoottinen familiaalinen hyperkolesterolemia. Tämä tutkimus koostui 24 viikon pituisesta satunnaistetusta, kaksoissokkoutetusta hoidosta, jossa potilaat saivat lumelääkettä tai alirokumabia. Tämän jälkeen potilaille annettiin alirokumabia 80 viikon pituisessa avoimessa vaiheessa. Potilaiden piti noudattaa vähärasvaista ruokavaliota ja saada rasva-arvoja alentavaa taustahoitoa. Tutkimukseen mukaan otetut potilaat satunnaistettiin suhteessa 2:1 saamaan alirokumabia 2 viikon tai 4 viikon välein tai lumelääkettä. Annostusohjelmassa, jossa valmistetta annettiin 4 viikon välein, 79 potilasta sai hoitoa 150 mg:n annoksella, jos potilaan paino oli < 50 kg, tai 300 mg:n annoksella, jos potilaan paino oli ≥ 50 kg. Viikon 12 kohdalla alirokumabiannos muutettiin 75 mg:aan 2 viikon välein (potilaan paino < 50 kg) tai 150 mg:aan 2 viikon välein (potilaan paino ≥ 50 kg) potilailla, joiden LDL-kolesteroliarvo oli vähintään 2,84 mmol/l (≥ 110 mg/dl).
Kaksoissokkoutettu hoitojakso:
Tässä tutkimuksessa ensisijainen tehoa mittaava päätetapahtuma oli LDL-kolesterolipitoisuuden prosentuaalinen muutos lähtötilanteesta viikkoon 24 mennessä. Tiedot on esitetty tarkemmin taulukossa 4. Absoluuttisten LDL-kolesteroliarvojen keskiarvo viikon 24 kohdalla oli 2,847 mmol/l (110,09 mg/dl) alirokumabiryhmässä ja 4,177 mmol/l (161,52 mg/dl) lumeryhmässä kohortissa, jossa hoitoa annettiin 4 viikon välein. LDL-kolesterolipitoisuuden pienenemistä todettiin ensimmäisessä lähtötilanteen jälkeisessä arvioinnissa viikon 8 kohdalla, ja tulokset säilyivät koko 24 viikon pituisen kaksoissokkoutetun hoitojakson ajan.
Taulukko 4: Alirokumabin ja lumelääkkeen hoitovaikutukset heterotsygoottista familiaalista hyperkolesterolemiaa sairastavilla pediatrisilla potilailla
| Prosentuaalisen muutoksen keskiarvo lähtötilanteesta viikon 24 kohdalla (prosentteina) | ||
| Annostelu 4 viikon välein | ||
| Lumelääke | Alirokumabi | |
| Potilaiden määrä | N = 27 | N = 52 |
| LDL-kolesteroli | -4,4 | -38,2 |
| Non-HDL-kolesteroli | -3,7 | -35,6 |
| Kokonaiskolesteroli | -3,6 | -34,6 |
| Apo B | -3,6 | -34,3 |
LDL = pienitiheyksinen lipoproteiini; HDL = suuritiheyksinen lipoproteiini;
Apo B = apolipoproteiini B. Kaikki korjatut p-arvot < 0,0001.
Avoin hoitojakso:
Yhteensä 74 potilasta kohortista, jossa hoitoa annettiin 4 viikon välein, osallistui 80 viikkoa kestäneeseen avoimeen yksihaaraiseen tutkimukseen. Aloitusannoksena käytettiin kaksoissokkoutettua hoitojaksoa varten valittua alirokumabiannosta, joka perustui painoon ja annostusohjelmaan. Tutkijoiden oli mahdollista suurentaa tai pienentää annosta lääketieteellisen arvioinnin perusteella. LDL-kolesterolin prosentuaalinen keskimuutos (keskivirhe) lähtötilanteesta (satunnaistaminen kaksoissokkoutetun hoitojakson aikana) oli -23,4 % (4,7) viikon 104 kohdalla. Muiden lipidipäätemuuttujien prosentuaaliset keskimuutokset (keskivirhe) lähtötilanteesta viikolle 104 olivat: non-HDL-kolesteroli -21,5 % (26,2), Apo B -17,8 % (21,7), kokonaiskolesteroli -17,4 % (19,9).
Farmakokinetiikka
Imeytyminen
Kun alirokumabia annettiin ihon alle 50–300 mg, mediaaniajat huippupitoisuuden saavuttamiseen seerumissa (tmax) olivat 3–7 vuorokautta.
Farmakokinetiikka oli samanlainen, kun 75 mg alirokumabia annettiin kerta-annoksena ihon alle vatsaan, olkavarteen tai reiteen.
Populaatiofarmakokineettisellä analyysillä määritettynä alirokumabin absoluuttinen hyötyosuus ihon alle annon jälkeen oli noin 85 %. Kuukausittainen altistus oli sama käytettäessä hoitoannosta 300 mg 4 viikon välein tai 150 mg 2 viikon välein. Cmax- ja Ctrough-arvojen vaihtelut olivat suurempia, kun hoitoväli oli 4 viikkoa.
Vakaa tila saavutettiin 2–3 annoksen jälkeen ja kumulaatiosuhde oli enintään suunnilleen kaksinkertainen.
Jakautuminen
Laskimoon annon jälkeen jakautumistilavuus oli noin 0,04–0,05 l/kg, mikä viittaa siihen, että alirokumabi jakautuu lähinnä verenkiertoon.
Biotransformaatio
Erityisiä aineenvaihduntatutkimuksia ei ole tehty, koska alirokumabi on proteiini. Alirokumabin odotetaan hajoavan pieniksi peptideiksi ja yksittäisiksi aminohapoiksi.
Eliminaatio
Alirokumabilla on todettu kaksi eliminaatiovaihetta. Pienillä pitoisuuksilla eliminaatio tapahtuu lähinnä saturoituvalla sitoutumisella kohteeseen (PCSK9), kun taas suuremmilla pitoisuuksilla alirokumabi eliminoituu pääasiassa ei-saturoituvan proteolyyttisen reitin kautta.
Populaatiofarmakokineettisen analyysin perusteella alirokumabin vakaan tilan näennäisen puoliintumisajan mediaani oli 17–20 vuorokautta potilailla, jotka saivat alirokumabia monoterapiana joko 75 mg:n tai 150 mg:n annoksina ihon alle kahden viikon välein. Statiinin kanssa samanaikaisesti annettuna alirokumabin näennäisen puoliintumisajan mediaani oli 12 vuorokautta.
Lineaarisuus/ei-lineaarisuus
Pitoisuuden todettiin suurenevan suhteellisesti hiukan annosta enemmän: kun alirokumabin annos kaksinkertaistettiin 75 mg:sta 150 mg:aan kahden viikon välein, alirokumabin kokonaispitoisuudet suurenivat 2,1–2,7-kertaisiksi.
Erityisryhmät
Iäkkäät potilaat
Populaatiofarmakokineettisen analyysin perusteella ikä aiheutti pienen eron alirokumabialtistuksessa vakaassa tilassa mutta sillä ei ollut vaikutusta tehoon tai turvallisuuteen.
Sukupuoli
Populaatiofarmakokineettisen analyysin perusteella sukupuolella ei ollut vaikutusta alirokumabin farmakokinetiikkaan.
Rotu
Populaatiofarmakokineettisen analyysin perusteella rodulla ei ollut vaikutusta alirokumabin farmakokinetiikkaan. Japanilaisten ja valkoihoisten terveiden tutkittavien välillä ei todettu merkitsevää eroa altistuksessa ihon alle annetun alirokumabin 100–300 mg:n kerta-annoksen jälkeen.
Kehonpaino
Kehonpaino todettiin yhdeksi merkittäväksi alirokumabin farmakokinetiikkaan vaikuttavaksi lopullisen populaatiofarmakokineettisen mallin kovariaatiksi. Vakaan tilan alirokumabialtistus (AUC0-14vrk) pieneni kahden viikon välein annetulla 75 mg:n annoksella 29 % ja kahden viikon välein annetulla 150 mg:n annoksella 36 % yli 100 kg painavilla potilailla verrattuna potilaisiin, jotka painoivat 50–100 kg. Tämä ei johtanut kliinisesti merkitykselliseen eroon LDL-kolesterolipitoisuuden pienenemisessä.
Maksan vajaatoiminta
Vaiheen 1 tutkimuksessa alirokumabin farmakokineettiset profiilit olivat ihon alle annetun 75 mg:n kerta-annoksen jälkeen lievää tai keskivaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla samanlaiset kuin tutkittavilla, joiden maksan toiminta oli normaali. Tietoa ei ole potilaista, joilla on vaikea maksan vajaatoiminta.
Munuaisten vajaatoiminta
Monoklonaalisten vasta-aineiden ei tiedetä eliminoituvan munuaisten kautta, joten munuaisten toiminnan ei odoteta vaikuttavan alirokumabin farmakokinetiikkaan. Populaatiofarmakokineettiset analyysit osoittivat, että vakaan tilan alirokumabialtistus (AUC0-14vrk) suureni kahden viikon välein annetulla 75 mg:n annoksella 22–35 % ja kahden viikon välein annetulla 150 mg:n annoksella 49–50 % lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla verrattuna potilaisiin, joiden munuaisten toiminta oli normaali. Kehonpainon ja iän, kahden alirokumabialtistukseen vaikuttavan kovariaatin, jakaumat olivat erilaisia munuaisten eri toimintakykyluokissa, mikä mitä todennäköisimmin selittää havaitut farmakokineettiset erot. Tietoa on vähän saatavilla potilaista, joilla on vaikea munuaisten vajaatoiminta; näillä potilailla alirokumabialtistus oli noin kaksi kertaa suurempi verrattuna koehenkilöihin, joiden munuaisten toiminta oli normaali.
Pediatriset potilaat
Praluent-valmisteen farmakokinetiikkaa arvioitiin 140:llä 8–17-vuotiaalla pediatrisella potilaalla, joilla oli heterotsygoottinen familiaalinen hyperkolesterolemia. Vakaan tilan keskimääräiset Ctrough-pitoisuudet saavutettiin viimeistään viikolla 8 (ensimmäinen farmakokineettinen näytteenotto toistuvan annostelun aikana) suositellulla annostusohjelmalla (ks. kohta Annostus ja antotapa).
Farmakokineettisiä tietoja on saatavilla vain vähän: 18 pediatrisesta potilaasta (8–17‑vuotiaita), joilla oli homotsygoottinen familiaalinen hyperkolesterolemia (HoFH). Alirokumabin vakaan tilan keskimääräiset Ctrough-pitoisuudet saavutettiin viimeistään viikolla 12 molemmissa ryhmissä, joissa tutkittavat saivat alirokumabia 75 mg kahden viikon välein tai 150 mg kahden viikon välein. Alirokumabia koskevia tutkimuksia ei ole tehty alle 8‑vuotiailla pediatrisilla potilailla (ks. kohta Farmakodynamiikka).
Farmakokineettiset/farmakodynaamiset suhteet
Alirokumabilla on epäsuora farmakodynaaminen vaikutus LDL-kolesterolipitoisuuden pienenemiseen, ja vaikutus välittyy PCSK9-entsyymiin sitoutumisen kautta. Vapaan PCSK9-entsyymin ja LDL-kolesterolin on todettu vähenevän pitoisuudesta riippuvaisella tavalla, kunnes saavutetaan alirokumabin kohteen saturaatio. Kun PCSK9-sitoutumisen saturaatio on saavutettu, alirokumabipitoisuuden suurentaminen ei enää vähennä LDL-kolesterolia, mutta on havaittu, että LDL-kolesterolia vähentävä vaikutus säilyy pidempään.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta ja toistuvan altistuksen aiheuttamaa toksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Lisääntymistoksisuutta koskevat tulokset rotilla ja apinoilla ovat osoittaneet, että kuten muut IgG-vasta-aineet, alirokumabi läpäisee veri-istukkaesteen.
Apinoilla ei todettu haitallisia vaikutuksia hedelmällisyyden sijaismarkkereihin (kuten kiimakiertoon, kivesten tilavuuteen, ejakulaatin tilavuuteen, siittiöiden liikkuvuuteen tai siittiöiden kokonaismäärään ejakulaatissa), eikä missään rotilla tai apinoilla tehdyssä toksikologisessa tutkimuksessa todettu alirokumabiin liittyviä anatomisesti patologisia tai histopatologisia löydöksiä lisääntymiskudoksissa.
Rotilla tai apinoilla ei todettu haitallisia vaikutuksia sikiön kasvuun tai kehitykseen. Tiineillä apinoilla ei todettu selvää emoon kohdistuvaa toksisuutta systeemisillä altistuksilla, jotka olivat 81-kertaisia ihmisen altistukseen nähden, kun käytetään 150 mg:n annosta kahden viikon välein. Tiineillä rotilla havaittiin kuitenkin emoon kohdistuvaa toksisuutta systeemisillä altistuksilla, joiden arvioitiin olevan suunnilleen 5,3-kertaisia ihmisen altistukseen nähden, kun käytetään 150 mg:n annosta kahden viikon välein (laskettuna altistuksesta, joka mitattiin ei-tiineillä rotilla viisi viikkoa kestäneen toksikologisen tutkimuksen aikana).
Kerran viikossa koko tiineyden ajan suuria alirokumabiannoksia saaneiden apinoiden jälkeläisillä oli heikompi sekundaarinen immuunivaste antigeenialtistukselle kuin vertailueläinten jälkeläisillä. Jälkeläisillä ei todettu muita viitteitä alirokumabiin liittyvästä immuunijärjestelmän toimintahäiriöstä.
Farmaseuttiset tiedot
Apuaineet
Histidiini
Sakkaroosi
Polysorbaatti 20
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
Praluent 75 mg injektioneste, liuos, esitäytetty kynä
3 vuotta
Praluent 150 mg injektioneste, liuos, esitäytetty kynä
2 vuotta
Praluent 300 mg injektioneste, liuos, esitäytetty kynä
2 vuotta
Säilytys
Säilytä jääkaapissa (2 °C ‑ 8 °C). Ei saa jäätyä.
Praluent voidaan säilyttää poissa jääkaapista (alle 25 °C) valolta suojattuna yhden, korkeintaan 30 vuorokautta kestävän, ajanjakson ajan. Kun lääkevalmiste on otettu jääkaapista, se on käytettävä 30 vrk:n kuluessa tai hävitettävä.
Pidä kynä ulkopakkauksessa. Herkkä valolle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
PRALUENT injektioneste, liuos, esitäytetty kynä
75 mg (L:ei) 6 x 1 ml (ilman aktivointipainiketta) (1339,95 €), 6 x 1 ml (1339,95 €)
150 mg (L:ei) 6 x 1 ml (1339,95 €), 6 x 1 ml (ilman aktivointipainiketta) (1339,95 €)
300 mg (L:ei) 3 x 2 ml (1339,95 €), 3 x 2 ml (ilman aktivointipainiketta, uritettu korkki) (1339,95 €)
PF-selosteen tieto
1 ml tai 2 ml liuosta silikonoidussa ruiskussa (tyypin 1 kirkasta lasia), jossa on ruostumattomasta teräksestä valmistettu kiinteä neula, styreenibutadieenikumista valmistettu neulansuojus ja eteenitetrafluorieteenipinnoitettu bromibutyylikumista valmistettu männän pysäytin.
75 mg injektioneste, liuos, esitäytetty kynä
Ruiskun osat on koottu esitäytetyksi kerta-annoskynäksi, jossa on sininen korkki ja vaaleanvihreä aktivointipainike.
Pakkauskoko:
1, 2 tai 6 esitäytettyä kynää.
Tai
Ruiskun osat on koottu esitäytetyksi kerta-annoskynäksi, jossa on sininen korkki ja jossa ei ole aktivointipainiketta.
Pakkauskoko:
1, 2, tai 3 esitäytettyä kynää, joissa ei ole aktivointipainiketta, tai monipakkaus, joka sisältää 6 (2 kpl 3 kynän pakkauksia) esitäytettyä kynää, joissa ei ole aktivointipainiketta.
150 mg injektioneste, liuos, esitäytetty kynä
Ruiskun osat on koottu esitäytetyksi kerta-annoskynäksi, jossa on sininen korkki ja tummanharmaa aktivointipainike.
Pakkauskoko:
1, 2 tai 6 esitäytettyä kynää.
Tai
Ruiskun osat on koottu esitäytetyksi kerta-annoskynäksi, jossa on sininen korkki ja jossa ei ole aktivointipainiketta.
Pakkauskoko:
1, 2, tai 3 esitäytettyä kynää, joissa ei ole aktivointipainiketta, tai monipakkaus, joka sisältää 6 (2 kpl 3 kynän pakkauksia) esitäytettyä kynää, joissa ei ole aktivointipainiketta.
300 mg injektioneste, liuos, esitäytetty kynä
Ruiskun osat on koottu esitäytetyksi kerta-annoskynäksi, jossa on sininen korkki ja jossa ei ole aktivointipainiketta.
Pakkauskoko:
1 tai 3 esitäytettyä kynää, joissa ei ole aktivointipainiketta.
Kaikkia lääkemuotoja ja pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Kirkas, väritön tai kellertävä liuos.
pH: 5,7–6,3
Osmolaalisuus:
Praluent 75 mg injektioneste, liuos
293–439 mOsm/kg
Praluent 150 mg injektioneste, liuos
383–434 mOsm/kg
Praluent 300 mg injektioneste, liuos
383–434 mOsm/kg
Käyttö- ja käsittelyohjeet
Esitäytetty kynä on laitettava käytön jälkeen terävälle jätteelle tarkoitettuun astiaan. Astiaa ei pidä toimittaa kierrätykseen.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
PRALUENT injektioneste, liuos, esitäytetty kynä
75 mg 6 x 1 ml, 6 x 1 ml
150 mg 6 x 1 ml, 6 x 1 ml
300 mg 3 x 2 ml, 3 x 2 ml
- Alempi erityiskorvaus (65 %). Alirokumabi ja evolokumabi familiaalisen hyperkolesterolemian hoidossa (lapset ja nuoret): Familiaalisen hyperkolesterolemian hoito lapsille ja nuorille erityisin edellytyksin (254), Alirokumabi ja evolokumabi familiaalisen hyperkolesterolemian hoidossa (aikuiset): Familiaalisen hyperkolesterolemian hoito erityisin edellytyksin (292), Alirokumabi ja evolokumabi: Krooniseen sepelvaltimotautiin liittyvän hyperkolesterolemian ja sekamuotoisen dyslipidemian hoito erityisin edellytyksin (294).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Alirokumabi, evolokumabi ja inklisiraani familiaalisen hyperkolesterolemian hoidossa (aikuiset): Familiaalisen hyperkolesterolemian hoito erityisin edellytyksin (388), Alirokumabi, evolokumabi ja inklisiraani: Hyperkolesterolemian ja sekamuotoisen dyslipidemian hoito erityisin edellytyksin (3015), Alirokumabi ja evolokumabi familiaalisen hyperkolesterolemian hoidossa (lapset ja nuoret): Familiaalisen hyperkolesterolemian hoito lapsille ja nuorille erityisin edellytyksin (3074).
ATC-koodi
C10AX14
Valmisteyhteenvedon muuttamispäivämäärä
17.10.2025
Yhteystiedot
 SANOFI OY
SANOFI OY Revontulenkuja 1
02100 Espoo
0201 200 300
www.sanofi.fi