
OPDIVO infuusiokonsentraatti, liuosta varten 10 mg/ml
Vaikuttavat aineet ja niiden määrät
Yksi ml infuusiokonsentraattia liuosta varten sisältää 10 mg nivolumabia.
Yksi 4 ml:n injektiopullo sisältää 40 mg nivolumabia.
Yksi 10 ml:n injektiopullo sisältää 100 mg nivolumabia.
Yksi 12 ml:n injektiopullo sisältää 120 mg nivolumabia.
Yksi 24 ml:n injektiopullo sisältää 240 mg nivolumabia.
Nivolumabi on tuotettu yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasoluissa.
Apuaine(et), joiden vaikutus tunnetaan
Yksi ml konsentraattia sisältää 0,1 mmol (eli 2,5 mg) natriumia.
Yksi 4 ml:n injektiopullo konsentraattia sisältää 0,94 mg polysorbaatti 80:tä.
Yksi 10 ml:n injektiopullo konsentraattia sisältää 2,14 mg polysorbaatti 80:tä.
Yksi 12 ml:n injektiopullo konsentraattia sisältää 2,6 mg polysorbaatti 80:tä.
Yksi 24 ml:n injektiopullo konsentraattia sisältää 5,0 mg polysorbaatti 80:tä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Yksi ml infuusiokonsentraattia liuosta varten sisältää 10 mg nivolumabia.
Lääkemuoto
Infuusiokonsentraatti, liuosta varten (steriili konsentraatti).
Kliiniset tiedot
Käyttöaiheet
Melanooma
OPDIVO monoterapiana on tarkoitettu aikuisten sekä 12‑vuotiaiden ja sitä vanhempien nuorten asteen IIB tai IIC melanooman liitännäishoitoon, tai kun melanooma on levinnyt imusolmukkeisiin tai kyseessä on etäpesäkkeinen sairaus ja kun potilaalle on tehty täydellinen poistoleikkaus (ks. kohta Farmakodynamiikka).
OPDIVO monoterapiana tai yhdistelmähoitona ipilimumabin kanssa on tarkoitettu aikuisten sekä 12‑vuotiaiden ja sitä vanhempien nuorten edenneen melanooman hoitoon (jota ei voida kirurgisesti poistaa tai joka on metastasoinut).
Nivolumabi–ipilimumabi-yhdistelmähoito on osoittanut nivolumabi-monoterapiaan verrattuna etenemisvapaan elinajan ja kokonaiselinajan kasvua ainoastaan potilailla, joilla on vähäinen kasvaimen PD-L1-ilmentymä (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Ei-pienisoluinen keuhkosyöpä (NSCLC)
OPDIVO yhdistelmähoitona platinapohjaisen kemoterapian kanssa on tarkoitettu kirurgisesti poistettavissa olevan ei‑pienisoluisen keuhkosyövän esiliitännäishoitoon aikuisille, joilla on suuri riski taudin uusiutumiseen ja joiden kasvainten PD‑L1:n ilmentymistaso on ≥ 1 % (valintakriteerit, ks. kohta Farmakodynamiikka).
OPDIVO yhdistelmähoitona platinapohjaisen kemoterapian kanssa on tarkoitettu kirurgisesti poistettavissa olevan ei‑pienisoluisen keuhkosyövän esiliitännäishoitoon ja sen jälkeen Opdivo-monoterapiana liitännäishoitoon aikuisille, joilla on suuri riski taudin uusiutumiseen ja joiden kasvainten PD‑L1:n ilmentymistaso on ≥ 1 % (valintakriteerit, ks. kohta Farmakodynamiikka).
OPDIVO yhdistelmähoitona ipilimumabin ja kahden platinapohjaisen kemoterapiajakson kanssa on tarkoitettu etäpesäkkeisen ei-pienisoluisen keuhkosyövän ensilinjan hoitoon aikuisille, joiden kasvaimissa ei ole herkistävää EGFR-mutaatiota tai ALK-translokaatiota.
OPDIVO monoterapiana on tarkoitettu paikallisesti edenneen tai etäpesäkkeisen ei-pienisoluisen keuhkosyövän hoitoon kemoterapian jälkeen aikuisille.
Keuhkopussin pahanlaatuinen mesoteliooma (MPM)
OPDIVO yhdistelmähoitona ipilimumabin kanssa on tarkoitettu ensilinjan hoitoon aikuisille, joilla on leikkaukseen soveltumaton keuhkopussin pahanlaatuinen mesoteliooma.
Munuaiskarsinooma (RCC)
OPDIVO yhdistelmähoitona ipilimumabin kanssa on tarkoitettu ensilinjan hoidoksi aikuisille, joilla on kohtalaisen/huonon ennusteen edennyt munuaiskarsinooma (ks. kohta Farmakodynamiikka).
OPDIVO yhdistelmähoitona kabotsantinibin kanssa on tarkoitettu ensilinjan hoidoksi aikuisille, joilla on edennyt munuaiskarsinooma (ks. kohta Farmakodynamiikka).
OPDIVO monoterapiana on tarkoitettu aikuisten edenneen munuaiskarsinooman hoitoon aiemman hoidon jälkeen.
Klassinen Hodgkinin lymfooma (cHL)
OPDIVO yhdistelmähoitona doksorubisiinin, vinblastiinin ja dakarbatsiinin (AVD) kanssa on tarkoitettu aikuisten ja 12‑vuotiaiden ja sitä vanhempien nuorten aiemmin hoitamattoman asteen III tai IV klassisen Hodgkinin lymfooman hoitoon.
OPDIVO yhdistelmähoitona brentuksimabivedotiinin kanssa on tarkoitettu 5‑vuotiaiden ja sitä vanhempien lasten, nuorten ja enintään 30‑vuotiaiden aikuisten uusiutuneen tai refraktaarisen klassisen Hodgkinin lymfooman hoitoon yhden aiemman hoitolinjan jälkeen (ks. kohta Farmakodynamiikka).
OPDIVO monoterapiana on tarkoitettu aikuisten uusiutuneen tai refraktaarisen klassisen Hodgkinin lymfooman hoitoon autologisen kantasolujen siirron (ASCT) sekä brentuksimabivedotiinihoidon jälkeen.
Pään ja kaulan alueen levyepiteelisyöpä (SCCHN)
OPDIVO monoterapiana on tarkoitettu aikuisten pään ja kaulan alueen uusiutuneen tai etäpesäkkeisen levyepiteelisyövän hoitoon, kun syöpä etenee platinapohjaisen hoidon aikana tai sen jälkeen (ks. kohta Farmakodynamiikka).
Uroteelikarsinooma (UC)
OPDIVO monoterapiana on tarkoitettu lihakseen tunkeutuvan uroteelikarsinooman (MIUC) liitännäishoitoon aikuisille, joiden kasvainsolujen PD‑L1-ilmentymä on ≥ 1 % ja joilla on suuri riski taudin uusiutumiseen lihakseen tunkeutuvan uroteelikarsinooman radikaaliresektion jälkeen (ks. kohta Farmakodynamiikka).
OPDIVO yhdistelmähoitona sisplatiinin ja gemsitabiinin kanssa on tarkoitettu ensilinjan hoitoon aikuisille, joilla on leikkaukseen soveltumaton tai etäpesäkkeinen uroteelikarsinooma.
OPDIVO monoterapiana on tarkoitettu aikuisten paikallisesti edenneen leikkaukseen soveltumattoman tai etäpesäkkeisen uroteelikarsinooman hoitoon silloin, kun aikaisempi platinaa sisältänyt hoito ei ole tehonnut.
Kolorektaalisyöpä (CRC), johon liittyy DNA:n kopiovirheenkorjaus- eli MMR-järjestelmän geenien puutteellinen toiminta (dMMR) tai mikrosatelliitti-instabiliteetti (MSI-H)
OPDIVO yhdistelmähoitona ipilimumabin kanssa on tarkoitettu sellaisen aikuisten kolorektaalisyövän hoitoon, johon liittyy DNA:n kopiovirheenkorjaus- eli MMR-järjestelmän geenien puutteellinen toiminta tai mikrosatelliitti-instabiliteetti seuraavissa tilanteissa:
-
leikkaukseen soveltumattoman tai etäpesäkkeisen kolorektaalisyövän ensilinjan hoito
-
etäpesäkkeisen kolorektaalisyövän hoito aiemman fluoropyrimidiinipohjaisen yhdistelmäkemoterapian jälkeen (ks. kohta Farmakodynamiikka).
Ruokatorven levyepiteelikarsinooma (OSCC)
OPDIVO yhdistelmähoitona ipilimumabin kanssa on tarkoitettu ensilinjan hoitoon aikuisille, joilla on leikkaukseen soveltumaton, edennyt, uusiutunut tai etäpesäkkeinen ruokatorven levyepiteelikarsinooma, jonka kasvainsolujen PD-L1‑ilmentymä on ≥ 1 %.
OPDIVO yhdistelmähoitona fluoropyrimidiini‑ ja platinapohjaisen yhdistelmäkemoterapian kanssa on tarkoitettu ensilinjan hoitoon aikuisille, joilla on leikkaukseen soveltumaton, edennyt, uusiutunut tai etäpesäkkeinen ruokatorven levyepiteelikarsinooma, jonka kasvainsolujen PD‑L1‑ilmentymä on ≥ 1 %.
OPDIVO monoterapiana on tarkoitettu aikuisten leikkaukseen soveltumattoman, edenneen, uusiutuneen tai etäpesäkkeisen ruokatorven levyepiteelikarsinooman hoitoon aiemman fluoropyrimidiini- ja platinapohjaisen yhdistelmäkemoterapian jälkeen.
Ruokatorvisyövän (OC) tai ruokatorvi-mahalaukkurajan syövän (GEJC) liitännäishoito
OPDIVO monoterapiana on tarkoitettu aikuisten ruokatorvisyövän tai ruokatorvi-mahalaukkurajan syövän liitännäishoitoon, kun potilaalla on patologista jäännöstautia aiemman esiliitännäishoitona annetun kemosädehoidon jälkeen (ks. kohta Farmakodynamiikka).
Mahalaukun, ruokatorvi-mahalaukkurajan (GEJ) tai ruokatorven adenokarsinooma
OPDIVO yhdistelmähoitona fluoropyrimidiini‑ ja platinapohjaisen yhdistelmäkemoterapian kanssa on tarkoitettu ensilinjan hoidoksi aikuisille, joilla on HER2‑negatiivinen edennyt tai etäpesäkkeinen mahalaukun, ruokatorvi-mahalaukkurajan tai ruokatorven adenokarsinooma ja joilla kasvaimen PD-L1:n ilmentymisen CPS-pistemäärä (Combined Positive Score) on ≥ 5.
Hepatosellulaarinen karsinooma (HCC)
OPDIVO yhdistelmähoitona ipilimumabin kanssa on tarkoitettu ensilinjan hoitoon aikuisille, joilla on leikkaukseen soveltumaton tai edennyt hepatosellulaarinen karsinooma.
Ehto
Hoito tulee aloittaa syövän hoitoon tarkoitettujen lääkevalmisteiden antamiseen perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Nivolumabihoidon aloittavan ja sitä valvovan lääkärin on oltava perehtynyt syövän hoitoon.
PD-L1‑määritys
Jos kyseisessä käyttöaiheessa näin mainitaan, potilaat valitaan saamaan OPDIVO‑valmistetta sen perusteella, todetaanko heillä CE-merkityllä lääkinnällisellä in vitro (IVD) -laitteella arvioitu PD‑L1-ligandin ilmentyminen. Jos CE-merkittyä IVD-laitetta ei ole käytettävissä, on käytettävä vaihtoehtoista, lainsäädännön vaatimukset täyttävää lääkinnällistä laitetta (ks. kohdat Käyttöaiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
MSI-/MMR-testaus
Jos käyttöaiheessa mainitaan, potilaan lääkehoidon OPDIVO-valmisteella on perustuttava potilaan kasvaimen MSI‑H-/dMMR‑statukseen. Kasvaimen statuksen määritys on tehtävä CE-merkityllä (diagnostiikan ja lääkehoidon yhdistävällä), tähän käyttötarkoitukseen tarkoitetulla, lääkinnällisellä laitteella. Mikäli tällaista ei ole saatavilla, on käytettävä vaihtoehtoista, lainsäädännön vaatimukset täyttävää lääkinnällistä laitetta (ks. kohdat Käyttöaiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakodynamiikka).
Annostus
OPDIVO monoterapiana
Suositeltu OPDIVO-annos on joko 240 mg nivolumabia 2 viikon välein tai 480 mg 4 viikon välein käyttöaiheen ja potilasryhmän mukaan (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka) taulukossa 1 esitetyllä tavalla.
Taulukko 1: Laskimoon monoterapiana annettavan nivolumabin suositeltu annos ja infuusioaika
Käyttöaihe* | Suositeltu annos ja infuusioaika |
|---|---|
Melanooma (edennyt melanooma tai melanooman liitännäishoitona**) | Aikuiset ja nuoret (12‑vuotiaat ja sitä vanhemmat, jotka painavat vähintään 50 kg): |
Nuoret (12‑vuotiaat ja sitä vanhemmat, jotka painavat alle 50 kg): | |
Munuaiskarsinooma | 240 mg 2 viikon välein 30 minuutin kuluessa tai |
Ruokatorvisyöpä tai ruokatorvi-mahalaukkurajan syöpä (liitännäishoito**) | 240 mg 2 viikon välein 30 minuutin kuluessa tai |
Ei-pienisoluinen keuhkosyöpä | 240 mg 2 viikon välein 30 minuutin kuluessa |
* Koskee kohdan Käyttöaiheet monoterapiakäyttöaiheita.
** Liitännäishoidossa OPDIVO-hoito voi kestää enintään 12 kuukautta.
Jos melanoomaa, munuaiskarsinoomaa, ruokatorvisyöpää, ruokatorvi-mahalaukkurajan syöpää tai lihakseen tunkeutuvaa uroteelikarsinoomaa (liitännäishoito) sairastavan potilaan pitää vaihtaa annostusohjelmasta 240 mg 2 viikon välein annostusohjelmaan 480 mg 4 viikon välein, ensimmäinen 480 mg:n annos pitää antaa kaksi viikkoa viimeisen 240 mg:n annoksen jälkeen. Jos taas potilaan pitää vaihtaa annostusohjelmasta 480 mg 4 viikon välein annostusohjelmaan 240 mg 2 viikon välein, ensimmäinen 240 mg:n annos pitää antaa neljä viikkoa viimeisen 480 mg:n annoksen jälkeen.
OPDIVO yhdistelmähoitona ipilimumabin kanssa
Melanooma
Taulukko 2: Laskimoon yhdistelmähoitona annettavien nivolumabin ja ipilimumabin ja niitä seuraavan nivolumabimonoterapian suositellut annokset ja infuusioajat melanooman hoidossa (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka)
| Yhdistelmähoitovaihe, 4 annosta | Monoterapiavaihe |
|---|---|---|
Nivolumabi | Aikuiset sekä 12‑vuotiaat ja sitä vanhemmat nuoret: (painosta riippumatta) | Aikuiset ja nuoret (12‑vuotiaat ja sitä vanhemmat, jotka painavat vähintään 50 kg): Ensimmäinen annos tulee antaa:
Nuoret (12‑vuotiaat ja sitä vanhemmat, jotka painavat alle 50 kg): Ensimmäinen annos tulee antaa:
|
Ipilimumabi | Aikuiset sekä 12‑vuotiaat ja sitä vanhemmat nuoret (painosta riippumatta): | - |
Keuhkopussin pahanlaatuinen mesoteliooma (MPM)
Taulukko 3: Laskimoon yhdistelmähoitona annettavien nivolumabin ja ipilimumabin suositellut annokset ja infuusioajat keuhkopussin pahanlaatuisen mesoteliooman hoidossa
| Yhdistelmähoito* |
|---|---|
Nivolumabi | 360 mg 3 viikon välein 30 minuutin kuluessa |
Ipilimumabi | 1 mg/kg 6 viikon välein 30 minuutin kuluessa |
* Hoitoa jatketaan enintään 24 kuukauden ajan potilailla, joiden tauti ei etene.
Munuaiskarsinooma (RCC)
Taulukko 4: Laskimoon yhdistelmähoitona annettavien nivolumabin ja ipilimumabin ja niitä seuraavan nivolumabimonoterapian suositellut annokset ja infuusioajat munuaiskarsinooman hoidossa
| Yhdistelmähoitovaihe, 4 annosta | Monoterapiavaihe |
Nivolumabi | 3 mg/kg 3 viikon välein 30 minuutin kuluessa | 240 mg 2 viikon välein 30 minuutin kuluessa tai Ensimmäinen annos tulee antaa:
|
Ipilimumabi | 1 mg/kg 3 viikon välein 30 minuutin kuluessa | - |
dMMR‑ tai MSI‑H-kolorektaalisyöpä (CRC)
Taulukko 5: Laskimoon yhdistelmähoitona annettavien nivolumabin ja ipilimumabin ja niitä seuraavan nivolumabimonoterapian suositellut annokset ja infuusioajat dMMR- tai MSI‑H‑tyyppisen kolorektaalisyövän hoidossa
| Yhdistelmähoitovaihe, enintään 4 annosta | Monoterapiavaihe | |
|---|---|---|---|
Nivolumabi | Ensilinjan hoito | 240 mg 3 viikon välein 30 minuutin kuluessa | 240 mg 2 viikon välein 30 minuutin kuluessa tai Ensimmäinen annos annetaan 3 viikon kuluttua viimeisestä nivolumabi- ja ipilimumabiyhdistelmän annoksesta. Nivolumabihoitoa suositellaan, kunnes tauti etenee, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan potilaille, joiden tauti ei etene. |
Aiemman fluoropyrimidiinipohjaisen yhdistelmäkemoterapian jälkeinen hoito | 3 mg/kg 3 viikon välein 30 minuutin kuluessa | 240 mg 2 viikon välein 30 minuutin kuluessa Ensimmäinen nivolumabiannos annetaan 3 viikon kuluttua viimeisestä nivolumabi- ja ipilimumabiyhdistelmän annoksesta. | |
Ipilimumabi | 1 mg/kg 3 viikon välein 30 minuutin kuluessa | - | |
Ruokatorven levyepiteelikarsinooma (OSCC)
Taulukko 6: Laskimoon yhdistelmähoitona annettavien nivolumabin ja ipilimumabin suositellut annokset ja infuusioajat ruokatorven levyepiteelikarsinooman hoidossa
| Yhdistelmähoito* |
|---|---|
Nivolumabi | 3 mg/kg 2 viikon välein 30 minuutin kuluessa tai |
Ipilimumabi | 1 mg/kg 6 viikon välein 30 minuutin kuluessa |
* Hoitoa suositellaan, kunnes tauti etenee, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan potilaille, joiden tauti ei etene.
Hepatosellulaarinen karsinooma (HCC)
Taulukko 7: Laskimoon yhdistelmähoitona annettavien nivolumabin ja ipilimumabin ja niitä seuraavan nivolumabimonoterapian suositellut annokset ja infuusioajat hepatosellulaarisen karsinooman hoidossa (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka)
| Yhdistelmähoitovaihe, enintään 4 annosta | Monoterapiavaihe∗ |
|---|---|---|
Nivolumabi | 1 mg/kg 3 viikon välein 30 minuutin kuluessa | 240 mg 2 viikon välein 30 minuutin kuluessa tai Ensimmäinen annos annetaan 3 viikon kuluttua viimeisestä nivolumabi- ja ipilimumabiyhdistelmän annoksesta. |
Ipilimumabi | 3 mg/kg 3 viikon välein 30 minuutin kuluessa | - |
* Nivolumabihoitoa on suositeltavaa jatkaa, kunnes tauti etenee tai kunnes ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan.
OPDIVO yhdistelmähoitona kabotsantinibin kanssa
Munuaiskarsinooma (RCC)
Taulukko 8: Laskimoon annettavan nivolumabin suositellut annokset ja infuusioajat ja yhdistelmähoitona suun kautta annettavan kabotsantinibin suositellut annokset munuaiskarsinooman hoidossa
| Yhdistelmähoito* |
Nivolumabi | 240 mg 2 viikon välein 30 minuutin kuluessa tai |
Kabotsantinibi | 40 mg kerran päivässä suun kautta |
*Kun OPDIVO-hoitoa annetaan yhdistelmähoitona kabotsantinibin kanssa, OPDIVO-hoitoa jatketaan, kunnes tauti etenee, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan potilaille, joiden tauti ei etene. Kabotsantinibihoitoa jatketaan, kunnes tauti etenee tai ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä. Lue lisätietoa kabotsantinibin valmisteyhteenvedosta.
OPDIVO yhdistelmähoitona ipilimumabin ja kemoterapian kanssa
Ei-pienisoluinen keuhkosyöpä (NSCLC)
Taulukko 9: Laskimoon yhdistelmähoitona annettavien nivolumabin ja ipilimumabin ja platinapohjaisen kemoterapian sekä niitä seuraavan nivolumabin ja ipilimumabin yhdistelmähoidon suositellut annokset ja infuusioajat ei‑pienisoluisen keuhkosyövän hoidossa
| Yhdistelmävaihe platinapohjaisen kemoterapian 2 hoitojakson ajan | Nivolumabin ja ipilimumabin yhdistelmävaihe* |
|---|---|---|
Nivolumabi | 360 mg 3 viikon välein 30 minuutin kuluessa | 360 mg 3 viikon välein 30 minuutin kuluessa |
Ipilimumabi | 1 mg/kg 6 viikon välein 30 minuutin kuluessa | 1 mg/kg 6 viikon välein 30 minuutin kuluessa |
Platinapohjainen kemoterapia | 3 viikon välein | – |
* Hoitoa suositellaan, kunnes tauti etenee, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan potilaille, joiden tauti ei etene.
OPDIVO yhdistelmähoitona kemoterapian kanssa
Ei‑pienisoluisen keuhkosyövän esiliitännäishoito
Taulukko 10: Laskimoon esiliitännäishoitona annettavan nivolumabin ja platinapohjaisen kemoterapian yhdistelmän suositellut annokset ja infuusioajat ei-pienisoluisen keuhkosyövän hoidossa (ks. kohta Farmakodynamiikka)
| Yhdistelmähoito 3 hoitojakson ajan |
|---|---|
Nivolumabi | 360 mg 3 viikon välein 30 minuutin kuluessa |
Platinapohjainen kemoterapia | 3 viikon välein |
Ei‑pienisoluisen keuhkosyövän esiliitännäishoito ja liitännäishoito
Taulukko 11: Laskimoon esiliitännäishoitona annettavan nivolumabin ja platinapohjaisen kemoterapian yhdistelmän sekä niitä seuraavan liitännäishoitona annettavan nivolumabimonoterapian suositellut annokset ja infuusioajat ei‑pienisoluisen keuhkosyövän hoidossa
| Yhdistelmähoitovaihe Esiliitännäishoitovaihe 4 jakson ajan | Monoterapiavaihe* Liitännäishoitovaihe |
|---|---|---|
Nivolumabi | 360 mg 3 viikon välein 30 minuutin kuluessa | 480 mg 4 viikon välein 30 minuutin kuluessa |
Platinapohjainen kemoterapia | 3 viikon välein | – |
* Hoitoa on suositeltavaa jatkaa, kunnes tauti etenee tai uusiutuu, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 13 jakson ajan (ks. kohta Farmakodynamiikka).
Ruokatorven levyepiteelikarsinooma (OSCC)
Taulukko 12: Laskimoon yhdistelmähoitona annettavien nivolumabin ja fluoropyrimidiini‑ ja platinapohjaisen kemoterapian suositellut annokset ja infuusioajat ruokatorven levyepiteelikarsinooman hoidossa (ks. kohta Farmakodynamiikka)*
| Yhdistelmähoito |
|---|---|
Nivolumabi | 240 mg 2 viikon välein 30 minuutin kuluessa tai |
Fluoropyrimidiini‑ ja platinapohjainen kemoterapia | 4 viikon välein |
* Nivolumabihoitoa suositellaan, kunnes tauti etenee, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan potilaille, joiden tauti ei etene.
Mahalaukun, ruokatorvi-mahalaukkurajan (GEJ) tai ruokatorven adenokarsinooma
Taulukko 13: Laskimoon yhdistelmähoitona annettavien nivolumabin ja fluoropyrimidiini‑ ja platinapohjaisen kemoterapian suositellut annokset ja infuusioajat mahalaukun, ruokatorvi-mahalaukkurajan tai ruokatorven adenokarsinooman hoidossa (ks. kohta Farmakodynamiikka)*
| Yhdistelmähoito |
|---|---|
Nivolumabi | 360 mg 3 viikon välein 30 minuutin kuluessa tai |
Fluoropyrimidiini‑ ja platinapohjainen kemoterapia | 2 viikon välein tai 3 viikon välein hoito-ohjelman mukaisesti |
* Nivolumabihoitoa suositellaan, kunnes tauti etenee, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan potilaille, joiden tauti ei etene.
Uroteelikarsinooma (UC)
Taulukko 14: Laskimoon yhdistelmähoitona annettavien nivolumabin sekä sisplatiinin ja gemsitabiinin ja niitä seuraavan nivolumabimonoterapian suositellut annokset ja infuusioajat uroteelikarsinooman hoidossa (ks. kohta Farmakodynamiikka)
| Yhdistelmävaihe, enintään 6 jakson ajan | Monoterapiavaihe* |
|---|---|---|
Nivolumabi | 360 mg 3 viikon välein 30 minuutin kuluessa | 240 mg 2 viikon välein 30 minuutin kuluessa tai |
Sisplatiini ja gemsitabiini | 3 viikon välein | – |
* Nivolumabihoitoa suositellaan jatkettavan, kunnes tauti etenee, ilmaantuu toksisia vaikutuksia, joita ei voida hyväksyä, tai enintään 24 kuukauden ajan ensimmäisestä annoksesta sen mukaan, mikä näistä tapahtuu ensin.
Aiemmin hoitamaton asteen III tai IV klassinen Hodgkinin lymfooma (cHL)
Taulukko 15: Laskimoon yhdistelmähoitona annettavien nivolumabin ja AVD-kemoterapian suositellut annokset ja infuusioaika aiemmin hoitamattoman cHL:n hoidossa (ks. kohta Farmakodynamiikka)
Yhdistelmähoitovaihe 6 hoitojakson ajan* | ||
Nivolumabi | Aikuiset ja nuoret (12‑vuotiaat ja sitä vanhemmat, jotka painavat vähintään 80 kg) | 240 mg 2 viikon välein 30 minuutin kuluessa |
Nivolumabi | Nuoret (12‑vuotiaat ja sitä vanhemmat, jotka painavat alle 80 kg) | 3 mg/kg 2 viikon välein 30 minuutin kuluessa |
Doksorubisiini, vinblastiini ja dakarbatsiini (AVD) | 2 viikon välein. |
*Yksi nivolumabin ja AVD:n yhdistelmähoitojakso on 28 vuorokautta.
OPDIVO yhdistelmähoitona brentuksimabivedotiinin kanssa
Uusiutunut tai refraktaarinen cHL
Taulukko 16: Laskimoon yhdistelmähoitona annettavien nivolumabin ja brentuksimabivedotiinin suositellut annokset ja infuusioajat uusiutuneen tai refraktaarisen klassisen Hodgkinin lymfooman hoidossa
| Yhdistelmähoito* |
|---|---|
Nivolumabi | 3 mg/kg 3 viikon välein 30 minuutin kuluessa |
Brentuksimabivedotiini | 1,8 mg/kg, enintään 180 mg, 3 viikon välein 30 minuutin kuluessa |
* Hoito on riskiperusteista ja sitä mukautetaan hoitovasteen mukaan. Tietoa riskiluokituksesta on kohdassa Farmakodynamiikka. Potilaiden kasvaimia on arvioitava 2 hoitojakson välein ja hoito on lopetettava, jos missä tahansa arviointivaiheessa todetaan taudin eteneminen:
-
pieni uusiutumisriski: enintään 6 yhdistelmähoitojaksoa, minkä jälkeen edetään konsolidaatiohoitoon, jos täydellinen vaste saavutetaan
-
tavanomainen uusiutumisriski: enintään 4 yhdistelmähoitojaksoa, minkä jälkeen edetään konsolidaatiohoitoon, jos täydellinen vaste saavutetaan.
Jos vaste 4 yhdistelmähoitojakson jälkeen ei ole optimaalinen, potilaille on annettava tehostettua hoitoa ennen etenemistä konsolidaatiohoitoon täydellisen vasteen saavuttamisen jälkeen (ks. kohta Farmakodynamiikka).
Hoidon kesto
OPDIVO‑hoitoa jatketaan monoterapiana tai ipilimumabin tai muiden lääkeaineiden kanssa yhdistelmähoitona niin kauan kuin siitä todetaan olevan kliinistä hyötyä tai kunnes potilas ei enää siedä sitä (ja enintään hoidon enimmäiskeston ajan, jos indikaatiolle on sellainen määritetty).
Epätyypillisiä vasteita (esim. kasvain kasvaa aluksi tilapäisesti tai pieniä uusia leesioita kehittyy ensimmäisten kuukausien aikana kasvaimen pienenemisen jälkeen) on havaittu. Kliinisesti vakaiden potilaiden nivolumabihoitoa tai nivolumabi–ipilimumabi-yhdistelmähoitoa suositellaan jatkamaan taudin etenemisestä kertovien ensimmäisten merkkien ilmettyä, kunnes taudin eteneminen on vahvistettu.
Annoksen suurentamista tai pienentämistä ei suositella annettaessa OPDIVO-hoitoa monoterapiana tai yhdessä muiden lääkeaineiden kanssa. Annosten siirtäminen myöhemmäksi tai hoidon keskeytys voi olla tarpeen yksilöllisen turvallisuuden ja siedettävyyden vuoksi. Taulukossa 17 on ohjeet hoidon lopettamiseen ja keskeyttämiseen. Kohdassa Varoitukset ja käyttöön liittyvät varotoimet on yksityiskohtaiset ohjeet immuunivälitteisten haittavaikutusten hoitoon. Kun nivolumabia annetaan yhdistelmähoitona muiden lääkeaineiden kanssa, lue lisätietoa näiden muiden yhdistelmänä käytettävien lääkkeiden annostuksesta niiden valmisteyhteenvedoista.
Taulukko 17: OPDIVO-hoidon tai OPDIVO–yhdistelmähoidon suositellut muutokset
Immuunivälitteinen haittavaikutus | Vaikeusaste | Hoidon muutos |
Immuunivälitteinen keuhkotulehdus | Asteen 2 keuhkotulehdus | Keskeytä hoito, kunnes oireet häviävät, radiologisesti havaittavat poikkeavuudet paranevat ja kortikosteroidihoito on toteutettu kokonaan. |
Asteen 3 tai 4 keuhkotulehdus | Lopeta hoito pysyvästi | |
Immuunivälitteinen koliitti | Asteen 2 ripuli tai koliitti | Keskeytä hoito, kunnes oireet häviävät ja mahdollisesti tarvittava kortikosteroidihoito on toteutettu kokonaan. |
Asteen 3 ripuli tai koliitti | ||
| Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito on toteutettu kokonaan | |
| Lopeta hoito pysyvästi | |
Asteen 4 ripuli tai koliitti | Lopeta hoito pysyvästi | |
Immuunivälitteinen maksatulehdus ilman hepatosellulaarista karsinoomaa
HUOMIO: Hoidon muutokset niille OPDIVO–kabotsantinibi-yhdistelmähoitoa saaville munuaiskarsinoomapotilaille, joiden maksa-arvot ovat kohonneet, on esitetty tämän taulukon jälkeen. | Asteen 2 nousu aspartaattiaminotransferaasi- (ASAT), alaniiniaminotransferaasi- (ALAT) tai kokonaisbilirubiiniarvossa | Keskeytä hoito, kunnes laboratorioarvot palaavat ennalleen ja mahdollisesti tarvittava kortikosteroidihoito on toteutettu kokonaan. |
Asteen 3 tai 4 nousu ASAT-, ALAT- tai kokonaisbilirubiiniarvossa | Lopeta hoito pysyvästi | |
Immuunivälitteinen maksatulehdus ja hepatosellulaarinen karsinooma | Jos ASAT/ALAT on lähtötilanteessa normaalin rajoissa ja nousee tasolle > 3 ja ≤ 10 kertaa normaalin yläraja (ULN) | Keskeytä hoito, kunnes laboratorioarvot palaavat ennalleen ja mahdollisesti tarvittava kortikosteroidihoito on toteutettu kokonaan. |
ASAT/ALAT nousee tasolle > 10 kertaa ULN | Lopeta hoito pysyvästi | |
Immuunivälitteinen munuaistulehdus ja munuaisten toimintahäiriö
| Asteen 2 tai 3 nousu kreatiniiniarvossa. | Keskeytä hoito, kunnes kreatiniiniarvo palaa ennalleen ja kortikosteroidihoito on toteutettu kokonaan. |
Asteen 4 nousu kreatiniiniarvossa | Lopeta hoito pysyvästi. | |
Immuunivälitteiset umpierityshäiriöt | Oireiset asteen 2 tai 3 kilpirauhasen vajaatoiminta, kilpirauhasen liikatoiminta, hypofysiitti | Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito (jos sitä tarvitaan akuutin tulehduksen oireisiin) on toteutettu kokonaan. Hoitoa pitää jatkaa yhdessä hormonikorvaushoidonb kanssa niin kauan kuin oireita on. |
Asteen 4 kilpirauhasen vajaatoiminta | Lopeta hoito pysyvästi | |
Immuunivälitteiset ihohaitat
| Asteen 3 ihottuma | Annos/annokset on jätettävä antamatta, kunnes oireet häviävät ja kortikosteroidihoito on toteutettu kokonaan. |
Asteen 4 ihottuma Stevens–Johnsonin oireyhtymä (SJS) tai toksinen epidermaalinen nekrolyysi (TEN) | Lopeta hoito pysyvästi. Lopeta hoito pysyvästi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). | |
Immuunivälitteinen myokardiitti | Asteen 2 myokardiitti | Annos/annokset on jätettävä antamatta, kunnes oireet häviävät ja kortikosteroidihoito on toteutettu kokonaanc. |
Asteen 3 tai 4 myokardiitti | Lopeta hoito pysyvästi. | |
Muut immuunivälitteiset haittavaikutukset | Asteen 3 (ensimmäistä kertaa) | Keskeytä hoito. |
Asteen 4 tai uusiutunut asteen 3; jatkuva asteen 2 tai 3 huolimatta hoidon muutoksista; kortikosteroidin vuorokausiannosta ei pystytä vähentämään 10 mg:aan prednisonia tai vastaavaa | Lopeta hoito pysyvästi. | |
Myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymä d | Asteen 2 myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymä | Keskeytä hoito, kunnes oireet häviävät ja kortikosteroidihoito on toteutettu kokonaan. |
Asteen 3 tai 4 myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymä | Lopeta hoito pysyvästi. | |
Huom.: Toksisuus on luokiteltu NCI-CTCAE-haittavaikutusluokituksen (National Cancer Institute Common Terminology Criteria for Adverse Events) version 4.0 mukaan.
a Lopeta hoito pysyvästi, jos yhdistelmähoitoa seuraavan toisen vaiheen (nivolumabi-monoterapia) aikana ilmenee asteen 3 ripulia tai koliittia.
b Suositukset hormonikorvaushoidon käytöstä on annettu kohdassa Varoitukset ja käyttöön liittyvät varotoimet.
c Nivolumabihoidon tai nivolumabi–ipilimumabi-yhdistelmähoidon uudelleen aloittamisen turvallisuutta potilaille, joilla on aikaisemmin ollut immuunivälitteinen myokardiitti, ei tunneta.
d Ilmenee näistä joko kahden tai kaikkien kolmen sairauden limittymisenä. Hoitoon suositeltavan muutoksen arvioinnissa on huomioitava yksittäisten tapahtumien vaikein CTCAE-aste.
OPDIVO monoterapiana tai OPDIVO–yhdistelmähoitona muiden lääkeaineiden kanssa on lopetettava pysyvästi, jos potilaalla on:
-
asteen 4 tai uusiutunut asteen 3 haittavaikutus
-
jatkuva asteen 2 tai 3 haittavaikutus sen hoidosta huolimatta.
Potilaille, joita hoidetaan OPDIVO-valmisteella on annettava potilaskortti ja kerrottava OPDIVO-hoidon riskeistä (ks. myös pakkausseloste).
Jos yhden lääkeaineen antaminen keskeytetään annettaessa yhdistelmähoitona OPDIVO-valmistetta ipilimumabin kanssa, tulee myös toisen lääkeaineen antaminen keskeyttää. Jos hoitoa jatketaan, sitä voidaan potilaan arvioinnin pohjalta jatkaa joko yhdistelmähoitona tai OPDIVO-monoterapiana.
Kun OPDIVO‑valmistetta annetaan yhdistelmähoitona kemoterapian kanssa, lue lisätietoa näiden muiden yhdistelmänä käytettävien lääkkeiden annostuksesta niiden valmisteyhteenvedoista. Jos jonkin lääkeaineen antaminen keskeytetään, voidaan muiden lääkeaineiden antamista jatkaa. Jos hoitoa jatketaan, sitä voidaan potilaan tilanteen arvioinnin pohjalta jatkaa joko yhdistelmähoitona, OPDIVO‑monoterapiana tai pelkkänä kemoterapiana.
OPDIVO yhdistelmähoitona kabotsantinibin kanssa munuaiskarsinooman hoidossa
Kun OPDIVO-valmistetta annetaan yhdistelmähoitona kabotsantinibin kanssa, yllä olevat taulukossa 17 kuvatut hoidon muutokset koskevat myös OPDIVO-valmistetta. Lisäksi seuraava koskee OPDIVO–kabotsantinibi-yhdistelmähoitoa saavia munuaiskarsinoomapotilaita, joiden maksa-arvot ovat kohonneet:
-
Jos ALAT tai ASAT on > 3 kertaa normaalin yläraja (ULN) mutta ≤ 10 kertaa ULN ilman, että kokonaisbilirubiini on samanaikaisesti ≥ 2 kertaa ULN, sekä OPDIVO- että kabotsantinibihoito on keskeytettävä, kunnes haittavaikutukset ovat lievittyneet asteeseen 0–1. Kortikosteroidihoitoa voidaan harkita. Hoidon aloittamista uudelleen yhdellä lääkkeellä tai molemmilla lääkkeillä voidaan harkita haittavaikutusten lievityttyä. Jos hoito aloitetaan uudelleen kabotsantinibilla, katso lisätiedot kabotsantinibin valmisteyhteenvedosta.
-
Jos ALAT tai ASAT > 10 kertaa ULN tai > 3 kertaa ULN siten, että kokonaisbilirubiini on samanaikaisesti ≥ 2 kertaa ULN, sekä OPDIVO- että kabotsantinibihoito on lopetettava pysyvästi ja kortikosteroidihoitoa voidaan harkita.
Erityisryhmät
Iäkkäät potilaat
Annoksen muuttaminen ei ole tarpeen hoidettaessa iäkkäitä potilaita (≥ 65 vuotta) (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Populaatiofarmakokineettisten tulosten perusteella annoksen muuttaminen ei ole tarpeen hoidettaessa lievää tai kohtalaista munuaisten vajaatoimintaa sairastavia (ks. kohta Farmakokinetiikka). Vaikeaa munuaisten vajaatoimintaa sairastavista on niin vähän tietoa, ettei ryhmää koskevia johtopäätöksiä voida tehdä.
Maksan vajaatoiminta
Populaatiofarmakokineettisten tulosten perusteella annoksen muuttaminen ei ole tarpeen hoidettaessa lievää tai kohtalaista maksan vajaatoimintaa sairastavia (ks. kohta Farmakokinetiikka). Vaikeaa maksan vajaatoimintaa sairastavista on niin vähän tietoa, ettei tätä ryhmää koskevia johtopäätöksiä voida tehdä. OPDIVO-valmistetta on annettava varoen potilaille, jotka sairastavat vaikeaa (kokonaisbilirubiini > 3 × ULN ja mikä ASAT-arvo tahansa) maksan vajaatoimintaa.
Pediatriset potilaat
OPDIVO‑valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu, lukuun ottamatta melanoomaa tai aiemmin hoitamatonta klassista Hodgkinin lymfoomaa sairastavien 12‑vuotiaiden ja sitä vanhempien nuorten hoidossa sekä uusiutunutta tai refraktaarista klassista Hodgkinin lymfoomaa sairastavien 5‑vuotiaiden tai sitä vanhempien pediatristen potilaiden hoidossa. Tämänhetkiset tiedot OPDIVO-monoterapiasta, OPDIVO‑valmisteesta yhdistelmähoitona ipilimumabin kanssa, yhdistelmähoitona brentuksimabivedotiinin kanssa tai yhdistelmähoitona AVD:n kanssa kuvataan kohdissa Annostus ja antotapa, Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka.
Antotapa
OPDIVO on tarkoitettu annosteltavaksi laskimoon. Se annetaan infuusiona laskimoon 30 tai 60 minuutin kuluessa annoksesta riippuen (ks. taulukot 1 - 16). Infuusio annetaan steriilin, ei-pyrogeenisen, vähän proteiineja sitovan infuusiosuodattimen kautta, jonka huokoskoko on 0,2–1,2 mikrom.
OPDIVO-valmistetta ei saa antaa laskimoon nopeana injektiona eikä boluksena.
Koko tarvittavan OPDIVO-annoksen voi infusoida suoraan 10 mg/ml:n vahvuisena liuoksena tai sen voi laimentaa joko 9 mg/ml:n vahvuisella (0,9 %) natriumkloridi-injektioliuoksella tai 50 mg/ml:n vahvuisella (5 %) glukoosi-injektioliuoksella (ks. kohta Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet).
Kun OPDIVO‑valmistetta annetaan yhdistelmähoitona ipilimumabin ja/tai kemoterapian kanssa, annetaan ensin OPDIVO‑valmistetta, sen jälkeen samana päivänä ipilimumabi (jos annetaan) ja sitten kemoterapia, paitsi yhdistelmähoitona AVD:n kanssa potilaille, joilla on aiemmin hoitamaton klassinen Hodgkinin lymfooma. Kun OPDIVO‑valmistetta annetaan yhdistelmähoitona AVD:n kanssa, annetaan ensin doksorubisiini, vinblastiini ja dakarbatsiini ja sen jälkeen samana päivänä OPDIVO‑valmiste. Kun OPDIVO‑valmistetta annetaan yhdistelmähoitona brentuksimabivedotiinin kanssa, hoitojaksolla 1 brentuksimabivedotiini annetaan päivänä 1 ja OPDIVO päivänä 8. Hoitojaksoilla 2–6 OPDIVO annetaan samana päivänä aikaisintaan 30 minuutin kuluttua brentuksimabivedotiini-infuusion päättymisestä. Käytä kullekin infuusiolle erillistä infuusiopussia ja suodatinta.
Ks. kohdasta Käyttö- ja käsittelyohjeet (Erityiset varotoimet hävittämiselle ja muut käsittelyohjeet) ohjeet lääkevalmisteen valmistamiseen ja käsittelyyn ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
PD-L1-statuksen arviointi
Kasvaimen PD-L1-statuksen määritykseen on tärkeää valita hyvin validoitu ja luotettava menetelmä.
MSI-/MMR-statuksen määritys
Kasvaimen MSI‑H- ja dMMR-statuksen määritykseen on tärkeää valita hyvin validoitu ja luotettava menetelmä.
Immuunivälitteiset haittavaikutukset
Kun nivolumabia annetaan yhdistelmähoitona, lue yhdistelmähoidon muiden lääkeaineiden valmisteyhteenvedot ennen hoidon aloittamista. Immuunivälitteisiä haittavaikutuksia on havaittu enemmän nivolumabi–ipilimumabi-yhdistelmähoidon kuin nivolumabi-monoterapian yhteydessä. Immuunivälitteisiä haittavaikutuksia on havaittu yhtä paljon OPDIVO-hoidon ja kabotsantinibin yhdistelmähoidon, brentuksimabivedotiinin yhdistelmähoidon tai AVD-yhdistelmähoidon kuin nivolumabimonoterapian yhteydessä. Tästä syystä seuraavat immuunivälitteisiä haittavaikutuksia koskevat ohjeet koskevat yhdistelmähoidon OPDIVO-osaa, ellei toisin mainita. Useimmat immuunivälitteiset haittavaikutukset paranivat tai hävisivät asianmukaisella hoidolla, johon kuului kortikosteroidihoito ja muutos hoidossa (ks. kohta Annostus ja antotapa).
Useampaan kuin yhteen elinjärjestelmään vaikuttavia immuunijärjestelmään liittyviä haittavaikutuksia voi ilmetä samanaikaisesti.
Yhdistelmähoidossa on raportoitu myös sydämeen ja keuhkoihin liittyviä haittavaikutuksia mukaan lukien keuhkoemboliaa. Potilaita on seurattava sydämeen ja keuhkoihin liittyvien haittavaikutusten varalta jatkuvasti sekä elektrolyyttihäiriön ja kuivumisen kliinisten merkkien ja oireiden sekä laboratorioarvojen huononemisen varalta ennen hoidon aloittamista ja säännöllisesti sen aikana. Nivolumabi–ipilimumabi-yhdistelmähoito tulee keskeyttää, jos hengenvaarallisia tai uusiutuvia sydämeen ja keuhkoihin liittyviä haittavaikutuksia ilmenee (ks. kohta Annostus ja antotapa).
Potilaita on seurattava jatkuvasti (vähintään viisi kuukautta viimeisen annoksen jälkeen), sillä haittavaikutus voi tulla missä vaiheessa nivolumabihoitoa tai nivolumabi–ipilimumabi-yhdistelmähoitoa hyvänsä tai vasta sen päätyttyä.
Epäiltyjen immuunivälitteisten haittavaikutusten riittävä arviointi on tehtävä syiden vahvistamiseksi tai muiden aiheuttajien poissulkemiseksi. Haittavaikutuksen vakavuuden perusteella nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja annettava kortikosteroideja. Jos haittavaikutuksen hoitoon käytetään kortikosteroideilla aikaansaatua immunosuppressiota, kortikosteroidiannoksen vähentäminen on aloitettava tilan alkaessa korjaantua, ja siihen on käytettävä vähintään kuukausi. Kortikosteroidihoidon liian nopea lopetus voi pahentaa haittavaikutusta tai aiheuttaa haittavaikutuksen uusiutumisen. Muu immunosuppressiivinen lääke on syytä lisätä hoito-ohjelmaan, jos haittavaikutus pahenee tai ei parane kortikosteroidien käytöstä huolimatta.
Havainnoivista tutkimuksista saadut tiedot viittaavat siihen, että immuunivälitteisten haittavaikutusten riski immuunivasteen tarkistuspisteen estäjällä annetun hoidon jälkeen voi olla suurempi potilailla, joilla on jo ennestään autoimmuunisairaus, kuin potilailla, joilla ei ole ennestään autoimmuunisairautta. Lisäksi taustalla olevan autoimmuunisairauden pahenemisvaiheita ilmaantui usein, mutta suurin osa tapauksista oli lieviä ja hallittavissa.
Nivolumabin käyttöä tai nivolumabi–ipilimumabi-yhdistelmähoitoa ei saa aloittaa uudelleen potilaan saadessa immunosuppressiivisia annoksia kortikosteroideja tai muuta immunosuppressiivista lääkettä. Immunosuppressiivista hoitoa saaville potilaille on annettava profylaktista antibioottihoitoa opportunististen infektioiden ehkäisemiseksi.
Nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi, jos yksikään vakava immunivälitteinen haittavaikutus uusiutuu tai on hengenvaarallinen.
Immuunivälitteinen keuhkotulehdus
Nivolumabihoidon tai nivolumabi–ipilimumabi-yhdistelmähoidon yhteydessä on todettu vaikeaa keuhkotulehdusta tai interstitiaalista keuhkosairautta, joka on joissain tapauksissa johtanut kuolemaan (ks. kohta Haittavaikutukset). Potilaita on seurattava keuhkotulehduksen löydösten ja oireiden varalta. Näitä ovat esimerkiksi radiologiset muutokset (kuten fokaaliset mattalasimuutokset, läiskikkäiset infiltraatit), hengenahdistus ja hypoksia. Infektioihin ja sairauksiin liittyvät syyt on suljettava pois.
Jos potilaalla on asteen 3 tai 4 keuhkotulehdus, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 2–4 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 2 (oireinen) keuhkotulehdus, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua nivolumabin käyttöä tai nivolumabi–ipilimumabi-yhdistelmähoitoa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannos on suurennettava tasolle, joka vastaa 2–4 mg/kg/vrk metyyliprednisolonia ja nivolumabin tai nivolumabi–ipilimumabi-yhdistelmähoidon käyttö on lopetettava pysyvästi.
Immuunivälitteinen koliitti
Nivolumabihoidon tai nivolumabi–ipilimumabi-yhdistelmähoidon yhteydessä on todettu vaikeaa ripulia ja koliittia (ks. kohta Haittavaikutukset). Potilaita on seurattava ripulin ja koliitin oireiden varalta, joita ovat esimerkiksi vatsakipu ja veriset tai limaiset ulosteet. Sytomegalovirus (CMV) ‑infektioita tai niiden uudelleenaktivoitumista on ilmoitettu potilailla, joilla on kortikosteroidihoitoon reagoimaton immuunivälitteinen koliitti. Infektiot ja ripulin muut syyt on suljettava pois, joten asianmukaiset laboratoriotestit ja muut tutkimukset on tehtävä. Jos kortikosteroidihoitoon reagoimattoman immuunivälitteisen koliitin diagnoosi varmistuu, vaihtoehtoisen immunosuppressantin lisäämistä kortikosteroidihoitoon tai kortikosteroidihoidon korvaamista on harkittava.
Jos potilaalla on asteen 4 ripuli tai koliitti, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 3 ripuli tai koliitti, nivolumabihoito on keskeytettävä ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua nivolumabi-monoterapiaa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, nivolumabi-monoterapia on lopetettava pysyvästi. Jos nivolumabi–ipilimumabi-yhdistelmähoitoa saavalla potilaalla on asteen 3 ripuli tai koliitti, hoito on lopetettava pysyvästi ja kortikosteroidien käyttö on aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 2 ripuli tai koliitti, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä. Jos ripuli tai koliitti jatkuu, se on hoidettava antamalla kortikosteroideja annoksina, jotka vastaavat 0,5–1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua nivolumabin käyttöä tai nivolumabi–ipilimumabi-yhdistelmähoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on suurennettava tasolle, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia, ja nivolumabin käyttö tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi.
Immuunivälitteinen maksatulehdus
Nivolumabi-monoterapian tai nivolumabi–ipilimumabi-yhdistelmähoidon yhteydessä on todettu vaikeaa maksatulehdusta (ks. kohta Haittavaikutukset). Potilaita on seurattava maksatulehduksen merkkien ja oireiden, kuten transaminaasi- ja kokonaisbilirubiiniarvon nousun, varalta. Infektiot ja sairauksiin liittyvät syyt on suljettava pois.
Jos potilaalla on asteen 3 tai 4 transaminaasi- tai kokonaisbilirubiiniarvon nousu, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 2 transaminaasi- tai kokonaisbilirubiiniarvon nousu, nivolumabin käyttö tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä. Jos nämä laboratorioarvot ovat jatkuvasti koholla, potilaalle on annettava kortikosteroideja annoksina, jotka vastaavat 0,5–1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua nivolumabin käyttöä tai nivolumabi–ipilimumabi-yhdistelmähoitoa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannosta on suurennettava tasolle, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia, ja nivolumabin käyttö tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi.
Immuunivälitteinen munuaistulehdus ja munuaisten toimintahäiriö
Nivolumabi-monoterapian tai nivolumabi–ipilimumabi-yhdistelmähoidon yhteydessä on todettu vaikeaa munuaistulehdusta ja munuaisten toimintahäiriöitä (ks. kohta Haittavaikutukset). Potilaita on seurattava munuaistulehduksen tai munuaisten toimintahäiriön löydösten ja oireiden varalta. Useimpien potilaiden seerumin kreatiniiniarvo suurenee ilman oireita. Sairauksiin liittyvät syyt on suljettava pois.
Jos potilaalla on asteen 4 seerumin kreatiniinipitoisuuden nousu, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 1–2 mg/kg/vrk metyyliprednisolonia.
Jos potilaalla on asteen 2 tai 3 seerumin kreatiniinipitoisuuden nousu, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja kortikosteroidien käyttö aloitettava annoksina, jotka vastaavat 0,5–1 mg/kg/vrk metyyliprednisolonia. Tilan parannuttua nivolumabin käyttöä tai nivolumabi–ipilimumabi-yhdistelmähoitoa voi jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidihoidon aloittamisesta huolimatta, kortikosteroidiannos on suurennettava tasolle, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia, ja nivolumabin käyttö tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi.
Immuunivälitteiset umpierityshäiriöt
Nivolumabihoidon tai nivolumabi–ipilimumabi-yhdistelmähoidon yhteydessä on todettu vaikeita umpierityshäiriöitä, kuten kilpirauhasen vajaa- ja liikatoimintaa, lisämunuaisten vajaatoimintaa (mukaan lukien lisämunuaiskuoren sekundaarista vajaatoimintaa), hypofysiittiä (mukaan lukien aivolisäkkeen etulohkon vajaatoimintaa), diabetesta ja diabeettista ketoasidoosia (ks. kohta Haittavaikutukset).
Potilaita on tarkkailtava umpierityshäiriöiden kliinisten merkkien ja oireiden ja hyperglykemian sekä kilpirauhasen toiminnan muutosten varalta (hoidon alussa, jaksoittain hoidon aikana ja kliinisen arvioinnin pohjalta). Potilailla saattaa esiintyä väsymystä, päänsärkyä, mielialamuutoksia, vatsakipuja, epätavallista vatsantoimintaa ja matalaa verenpainetta tai epäspesifisiä oireita, jotka voivat muistuttaa muita syitä kuten aivojen etäpesäke tai taustasairaus. Jos vaihtoehtoisia syitä ei ole tunnistettu, umpierityshäiriöiden oireet ja löydökset on katsottava immuunivälitteiseksi.
Jos potilaalla on kilpirauhasen vajaatoiminnan oireita, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja korvaushoito kilpirauhashormonilla aloitettava tarpeen mukaan. Jos potilaalla on kilpirauhasen liikatoiminnan oireita, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja kilpirauhasen toimintaa estävä lääkitys on aloitettava tarpeen mukaan. Kortikosteroidien käyttöä 1–2 mg/kg/vrk metyyliprednisolonia vastaavina annoksina on harkittava myös, jos epäillään akuuttia kilpirauhastulehdusta. Tilan parannuttua nivolumabin käyttöä tai nivolumabi–ipilimumabi-yhdistelmähoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Kilpirauhasen toimintaa on seurattava edelleen, jotta pystytään varmistamaan, että hormonikorvaushoito on asianmukaisella tasolla. Nivolumabi tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi, jos potilaalla on henkeäuhkaava kilpirauhasen liika- tai vajaatoiminta.
Jos potilaalla on asteen 2 lisämunuaisen vajaatoiminnan oireita, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja fysiologinen kortikosteroidikorvaushoito aloitettava tarpeen mukaan. Nivolumabi tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi, jos potilaalla on vakava (asteen 3) tai henkeäuhkaava (asteen 4) lisämunuaisen vajaatoiminta. Lisämunuaisten toimintaa ja hormonipitoisuuksia on seurattava edelleen, jotta pystytään varmistamaan, että kortikosteroidikorvaushoito on asianmukaisella tasolla.
Jos potilaalla on asteen 2 tai 3 hypofysiitin oireita, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja hormonikorvaushoito aloitettava tarpeen mukaan. Kortikosteroidien käyttöä 1–2 mg/kg/vrk metyyliprednisolonia vastaavina annoksina on harkittava myös, jos epäillään akuuttia aivolisäkkeen tulehdusta. Tilan parannuttua nivolumabin käyttöä tai nivolumabi–ipilimumabi-yhdistelmähoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Nivolumabi tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi, jos potilaalla on henkeäuhkaava (asteen 4) hypofysiitti. Aivolisäkkeen toimintaa ja hormonipitoisuuksia on seurattava edelleen, jotta pystytään varmistamaan, että hormonikorvaushoito on asianmukaisella tasolla.
Jos potilaalla on diabeteksen oireita, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja insuliinikorvaushoito aloitettava tarpeen mukaan. Verensokeripitoisuuksia on seurattava edelleen, jotta pystytään varmistamaan, että insuliinikorvaushoito on asianmukaisella tasolla. Nivolumabi tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi, jos potilaalla on henkeäuhkaava diabetes.
Immuunivälitteiset ihohaitat
Nivolumabi–ipilimumabi-yhdistelmähoidon yhteydessä on todettu vaikeaa ihottumaa, nivolumabi-monoterapiahoidon yhteydessä vaikeaa ihottumaa on todettu harvemmin. (ks. kohta Haittavaikutukset). Nivolumabi tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä asteen 3 ihottumassa ja lopetettava asteen 4 ihottumassa. Vaikeaa ihottumaa on hoidettava suurella kortikosteroidiannoksella, joka vastaa 1–2 mg/kg/vrk metyyliprednisolonia.
SJS:aa ja TEN:iä on havaittu harvoin, ja joissain tapauksissa tila on johtanut kuolemaan. Jos SJS:n tai TEN:n oireita tai merkkejä ilmenee, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja potilas on ohjattava niihin erikoistuneeseen yksikköön arviointia ja hoitoa varten. Jos potilaalle kehittyy SJS tai TEN nivolumabihoidon tai nivolumabi–ipilimumabi-yhdistelmähoidon aikana, suositellaan hoidon lopettamista pysyvästi (ks. kohta Annostus ja antotapa).
Varovaisuutta on noudatettava, kun harkitaan nivolumabin käyttöä potilaalle, jolla on aikaisemmin ollut vaikea tai henkeäuhkaava ihohaitta aiemman toisen immuunijärjestelmää stimuloivan syöpälääkehoidon aikana.
Muut immuunivälitteiset haittavaikutukset
Seuraavia immuunivälitteisiä haittavaikutuksia on raportoitu alle 1 %:lla nivolumabi-monoterapiaa tai nivolumabi–ipilimumabi-yhdistelmähoitoa saaneista potilaista kliinisissä tutkimuksissa eri annoksilla ja kasvaintyypeillä: haimatulehdus, suonikalvoston tulehdus, demyelinaatio, autoimmuunineuropatia (mukaan lukien kasvo- ja loitontajahermon halvaus), Guillain–Barrén oireyhtymä, myasthenia gravis, myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymä, myasteeninen oireyhtymä, aseptinen aivokalvotulehdus, aivotulehdus, gastriitti, sarkoidoosi, duodeniitti, myosiitti, myokardiitti, rabdomyolyysi ja myeliitti. Vogt–Koyanagi–Haradan oireyhtymän (VKH), lisäkilpirauhasten vajaatoiminnan ja ei-infektiivisen virtsarakkotulehduksen tapauksista on ilmoitettu markkinoille tulon jälkeen (katso kohdat Annostus ja antotapa ja Haittavaikutukset).
Immuunivälitteistä haittavaikutusta epäiltäessä potilaan tila on arvioitava riittävän tarkasti, jotta pystytään vahvistamaan haittavaikutuksen etiologia tai sulkemaan pois muut syyt. Jos haittavaikutus on vaikea, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja on annettava kortikosteroideja. Tilan parannuttua nivolumabin käyttöä tai nivolumabi–ipilimumabi-yhdistelmähoitoa voi tarpeen mukaan jatkaa kortikosteroidiannoksen vähentämisen jälkeen. Nivolumabin tai nivolumabi–ipilimumabi-yhdistelmähoidon käyttö on lopetettava pysyvästi, jos tulee toistuva, vaikea immuunivälitteinen haittavaikutus tai hengenvaarallinen immuunivälitteinen haittavaikutus.
Myotoksisuutta (myosiitti, myokardiitti ja rabdomyolyysi) on raportoitu nivolumabihoidon tai nivolumabi–ipilimumabi‑yhdistelmähoidon yhteydessä, ja joissain tapauksissa tila on johtanut kuolemaan. Jos potilaalle kehittyy myotoksisuuden oireita tai merkkejä, on potilasta seurattava huolellisesti ja hänet on välittömästi ohjattava asiantuntijalle arviointia ja hoitoa varten. Myotoksisuuden vakavuuden perusteella nivolumabihoito tai nivolumabi–ipilimumabi‑yhdistelmähoito on keskeytettävä tai lopetettava (ks. kohta Annostus ja antotapa) ja asianmukainen hoito on aloitettava.
Myokardiitin diagnosointi vaatii vahvan epäilyn. Potilaat, joilla on sydän- tai sydän- ja keuhko-oireita, on syytä arvioida mahdollisen myokardiitin varalta. Myokardiittia epäiltäessä on aloitettava ripeästi hoito suurella steroidiannoksella (prednisoni 1–2 mg/kg/päivä tai metyyliprednisoloni 1–2 mg/kg/päivä) ja konsultoitava viipymättä kardiologian asiantuntijaa diagnoosin varmistamista varten nykyisten hoitosuositusten mukaisesti. Myokardiittidiagnoosin varmistuttua nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä tai lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymän (ilmenee näistä joko kahden tai kaikkien kolmen sairauden limittymisenä) tapauksia, jotka ovat joskus johtaneet kuolemaan, on raportoitu nivolumabihoidossa ja nivolumabin ja muiden lääkeaineiden yhdistelmähoidossa. Oireyhtymän varhainen tunnistaminen ja aggressiivinen hoito ovat oleellisia siihen liittyvän sairastuvuuden ja kuolleisuusriskin vuoksi.
Jos potilaalla todetaan asteen 3 tai 4 myokardiitti-myosiitti-myasthenia gravis overlap -oireyhtymä, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa). Kortikosteroidien käyttö on aloitettava kliinisen tarpeen mukaan.
Jos potilaalla todetaan asteen 2 myokardiitti-myosiitti-myasthenia gravis overlap -oireyhtymä, nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on keskeytettävä ja kortikosteroidien käyttö aloitettava kliinisen tarpeen mukaan (ks. kohta Annostus ja antotapa). Tilan parannuttua nivolumabihoitoa voidaan harkita kortikosteroidiannoksen asteittaisen pienentämisen jälkeen. Jos potilaan tila pahenee tai ei parane kortikosteroidien käytön aloittamisesta huolimatta, kortikosteroidiannosta on muutettava kliinisen tarpeen mukaan ja nivolumabihoito tai nivolumabi–ipilimumabi-yhdistelmähoito on lopetettava pysyvästi.
PD-1:n estäjillä hoidetuilta potilailta on ilmoitettu hyljintäreaktioita kiinteän elimen siirron jälkeen valmisteen markkinoilletulon jälkeen. Nivolumabihoito saattaa suurentaa hyljintäreaktion riskiä kiinteän elimen saaneilla potilailla. Näillä potilailla nivolumabihoidon hyötyjä mahdollisen hyljintäreaktion riskiin nähden on arvioitava huolellisesti.
Hemofagosyyttistä lymfohistiosytoosia (HLH) on havaittu monoterapiana annetun nivolumabin sekä nivolumabin ja ipilimumabin yhdistelmähoidon yhteydessä. On noudatettava varovaisuutta, kun nivolumabia annetaan yksilääkehoitona tai yhdistelmähoitona ipilimumabin kanssa. Jos potilaalla on vahvistettu HLH, on lopetettava nivolumabin tai nivolumabi–ipilimumabi-yhdistelmähoidon käyttö ja aloitettava HLH:n hoito.
Infuusioreaktiot
Kliinisissä tutkimuksissa on raportoitu infuusioreaktioita, myös vaikeita infuusioreaktioita, nivolumabi-monoterapian tai nivolumabi–ipilimumabin tai nivolumabin ja muiden lääkeaineiden yhdistelmähoidon yhteydessä. (ks. kohta Haittavaikutukset). Vaikeissa tai henkeäuhkaavissa infuusioreaktiotapauksissa nivolumabi-infuusio tai nivolumabi–ipilimumabin tai nivolumabin ja muiden lääkeaineiden infuusio täytyy keskeyttää ja antaa asianmukaista lääketieteellistä hoitoa. Potilaat, jotka saavat lievän tai keskivaikean infuusioreaktion, voivat saada nivolumabia tai nivolumabi–ipilimumabin tai nivolumabin ja muiden lääkeaineiden yhdistelmähoitoa huolellisessa seurannassa ja käyttämällä infuusioreaktioita estävää esilääkitystä paikallisten hoitosuositusten mukaisesti.
Sairauskohtaiset varotoimet
Edennyt melanooma
Potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, joilla oli aktiivisia leptomeningeaalisia tai aivometastaaseja tai autoimmuunisairaus tai jotka olivat saaneet systeemistä immunosuppressiivista hoitoa ennen tutkimukseen ottamista, suljettiin pois kliinisistä pivotaalitutkimuksista, jotka koskivat nivolumabia tai nivolumabi–ipilimumabi-yhdistelmähoitoa (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Pivotaalisista melanoomatutkimuksista suljettiin pois potilaat, joilla oli silmän/uvean melanooma. CA209037-tutkimukseen ei otettu myöskään potilaita, joilla oli ollut anti-CTLA-4-hoitoon liittyvä asteen 4 haittavaikutus (ks. kohta Farmakodynamiikka). Potilaat, joiden lähtötason toimintakykyluokka oli 2, joiden leptomeningeaalisia metastaaseja, silmän/uvean melanoomaa, autoimmuunisairautta oli hoidettu ja potilaat, joilla oli ollut aiempaan anti‑CTLA‑4‑hoitoon liittyvä asteen 3–4 haittavaikutus, otettiin mukaan CA209172-tutkimukseen (ks. kohta Farmakodynamiikka). Tutkimustiedon puuttuessa koskien potilaita, jotka olivat saaneet systeemistä immunosuppressiivista hoitoa ennen tutkimukseen ottamista, sekä potilaita, joilla oli aktiivisia leptomeningeaalisia tai aivometastaaseja, nivolumabia on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Nivolumabi–ipilimumabi-yhdistelmähoito on osoittanut nivolumabi-monoterapiaan verrattuna etenemisvapaan elinajan kasvavan ainoastaan potilailla, joilla on vähäinen kasvaimen PD-L1-ilmentymä. Kokonaiselinajan parantuminen oli samaa luokkaa nivolumabi–ipilimumabi-yhdistelmähoitoa ja nivolumabi-monoterapiaa saaneilla potilailla, joilla oli suuri kasvaimen PD-L1-ilmentymä (PD-L1 ≥ 1 %). Ennen yhdistelmähoidon aloittamista lääkärin tulee arvioida tarkkaan potilas ja kasvaimen ominaisuudet sekä huomioida yhdistelmähoidon havaitut edut ja toksisuus nivolumabi-monoterapiaan verrattuna (ks. kohdat Haittavaikutukset ja Farmakodynamiikka).
Nivolumabin käyttö potilaille, joilla on nopeasti etenevä melanooma
Lääkärien tulee huomioida nivolumabin viivästynyt vaikutuksen alkaminen ennen hoidon aloittamista potilaille, joilla on nopeasti etenevä sairaus (ks. kohta Farmakodynamiikka).
Melanooman liitännäishoito
Melanooman liitännäishoidosta ei ole tietoa niiden potilaiden osalta, joilla on seuraavat riskitekijät (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka):
-
potilaat, joilla oli ollut autoimmuunisairaus ja mikä tahansa systeemistä hoitoa joko kortikosteroideilla (≥ 10 mg prednisonia tai vastaavaa vuorokaudessa) tai muulla immunosuppressiivisella lääkkeellä vaativa sairaus
-
potilaat, joiden melanoomaa oli hoidettu aiemmin (pois lukien potilaat, jotka oli leikattu, jotka olivat saaneet sädehoitoa liitännäishoitona keskushermostojärjestelmän leesioihin neurokirurgisen poistoleikkauksen jälkeen ja jotka olivat saaneet aiemmin interferonia liitännäishoitona, joka oli päättynyt ≥ 6 kuukautta ennen satunnaistamista)
-
potilaat, jotka olivat saaneet aiemmin anti-PD-1‑, anti-PD-L1‑, anti-PD-L2‑, anti-CD137‑ tai anti-CTLA-4‑hoitoa (mukaan lukien ipilimumabi tai mikä tahansa muu vasta-aine tai lääkeaine, joka kohdistuu erityisesti T-solujen kostimulointiin tai tarkistuspisteiden signalointireitteihin)
-
alle 18‑vuotiaat potilaat.
Tutkimustiedon puuttuessa nivolumabia on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Ei-pienisoluinen keuhkosyöpä
Ei-pienisoluisen keuhkosyövän ensilinjan hoito
Ei-pienisoluisen keuhkosyövän ensilinjan hoitoa koskevasta kliinisestä avaintutkimuksesta suljettiin pois ne potilaat, joilla oli aktiivinen autoimmuunisairaus, oireinen interstitiaalinen keuhkosairaus, systeemistä immunosuppressiota vaativa tila, aktiivinen (hoitamaton) aivojen etäpesäke, tai jotka olivat saaneet aiempaa systeemistä hoitoa edenneeseen tautiin tai joilla oli herkistäviä EGFR-mutaatioita tai ALK:n translokaatioita (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tietoja käytöstä iäkkäille potilaille (≥ 75‑vuotiaille) on vain vähän (ks. kohta Farmakodynamiikka). Näiden potilaiden osalta nivolumabin annossa yhdessä ipilimumabin ja kemoterapian kanssa on noudatettava varovaisuutta; tällöin hoidon mahdollisia hyötyjä ja riskejä on arvioitava huolellisesti potilaskohtaisesti.
Ei-pienisoluisen keuhkosyövän hoito aiemman kemoterapian jälkeen
Potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, joilla oli aktiivinen aivometastaasi tai autoimmuunisairaus, oireinen interstitiaalinen keuhkosairaus, sekä potilaat jotka olivat saaneet systeemistä immunosuppressiivista hoitoa ennen tutkimukseen ottamista, suljettiin pois kliinisistä ei‑pienisoluista keuhkosyöpää koskevista pivotaalitutkimuksista (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Potilaat, joiden lähtötason toimintakykyluokka oli 2, otettiin mukaan CA209171-tutkimukseen (ks. kohta Farmakodynamiikka). Tutkimustiedon puuttuessa koskien potilaita, joilla oli autoimmuunisairaus, oireinen interstitiaalinen keuhkosairaus, aktiivisia aivometastaaseja, sekä potilaita, jotka olivat saaneet systeemistä immunosuppressiivista hoitoa ennen tutkimukseen ottamista, nivolumabia on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Lääkärien tulee huomioida nivolumabin viivästynyt vaikutuksen alkaminen ennen hoidon aloittamista potilaille, joilla on huonompi ennuste ja/tai aggressiivinen sairaus. Ei-levyepiteeliperäistä ei‑pienisoluista keuhkosyöpää sairastavista ja nivolumabia saaneista potilaista kuoli kolmen kuukauden aikana useampi kuin dosetakselia saaneista. Varhaiseen kuolemaan liittyvät tekijät olivat huonompi ennuste ja/tai aggressiivisempi sairaus yhdistettynä kasvaimen matalaan PD-L1:n ilmentymiseen tai sen puuttumiseen (ks. kohta Farmakodynamiikka).
Ei‑pienisoluisen keuhkosyövän esiliitännäishoito
Kirurgisesti poistettavissa olevan ei‑pienisoluisen keuhkosyövän esiliitännäishoitoa koskevasta kliinisestä avaintutkimuksesta suljettiin pois ne potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, joilla oli aktiivinen autoimmuunisairaus, oireinen interstitiaalinen keuhkosairaus, systeemistä immunosuppressiota vaativa tila, leikkaukseen soveltumaton tai metastaattinen sairaus, kirurgisesti poistettavissa olevaan tautiin aiempaa syöpähoitoa saaneet tai joilla oli tunnistettu EGFR-mutaatioita tai ALK:n translokaatioita (ks. kohta Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabin ja kemoterapian yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Ei‑pienisoluisen keuhkosyövän esiliitännäishoito ja liitännäishoito
Ei‑pienisoluisen keuhkosyövän esiliitännäishoitoa ja liitännäishoitoa koskevasta avaintutkimuksesta suljettiin pois ne potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2 tai joilla oli vähintään asteen 2 perifeerinen neuropatia, aktiivinen autoimmuunisairaus, oireinen interstitiaalinen keuhkosairaus, systeemistä immunosuppressiota vaativa tila tai leikkaukseen soveltumaton tai metastaattinen sairaus, kirurgisesti poistettavissa olevaan tautiin aiempaa syöpähoitoa saaneet potilaat sekä potilaat, joilla oli EGFR-mutaatioita tai tunnistettuja ALK:n translokaatioita tai aivometastaaseja (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabin ja kemoterapian yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Keuhkopussin pahanlaatuinen mesoteliooma
Keuhkopussin pahanlaatuisen mesoteliooman ensilinjan hoitoa koskevista pivotaalitutkimuksista suljettiin pois potilaat, joilla oli primitiivinen vatsakalvon, sydänpussin, kiveksen tai tuppikalvon mesoteliooma, interstitiaalinen keuhkosairaus, aktiivinen autoimmuunisairaus, systeemistä immunosuppressiota vaativa tila tai aivojen etäpesäke (ellei etäpesäkettä ollut poistettu kirurgisesti tai hoidettu stereotaktisella sädehoidolla siten, ettei etäpesäke ollut kasvanut kolmeen kuukauteen ennen tutkimukseen ottamista) (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabi–ipilimumabi yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Munuaiskarsinooma
Nivolumabi ja nivolumabi–ipilimumabi‑yhdistelmähoito
Nivolumabia ja nivolumabi–ipilimumabi-yhdistelmähoitoa koskevista kliinisistä tutkimuksista suljettiin pois ne potilaat joilla oli, tai joilla oli ollut, aivometastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabia ja nivolumabi–ipilimumabi-yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Nivolumabi–kabotsantinibi-yhdistelmähoito
Nivolumabi–kabotsantinibi-yhdistelmähoitoa koskevista kliinisistä tutkimuksista suljettiin pois ne potilaat, joilla oli aktiivisia aivometastaaseja, autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabi–kabotsantinibi-yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Kun nivolumabia on annettu kabotsantinibin kanssa, edennyttä munuaiskarsinoomaa sairastavilla potilailla on ilmoitettu enemmän asteen 3 ja 4 ALAT- ja ASAT-arvojen kohoamista kuin nivolumabi-monoterapiaa saaneilla edennyttä munuaiskarsinoomaa sairastavilla potilailla (ks. kohta Haittavaikutukset). Maksaentsyymiarvoja on seurattava ennen hoidon aloittamista ja säännöllisesti koko hoidon ajan. Molempien lääkkeiden käyttöä koskevia ohjeita on noudatettava (ks. kohta Annostus ja antotapa ja kabotsantinibin valmisteyhteenveto).
Klassinen Hodgkinin lymfooma
Nivolumabi-monoterapia
Kliinisistä klassista Hodgkinin lymfoomaa koskevista tutkimuksista suljettiin pois ne potilaat, joilla oli aktiivinen autoimmuunisairaus ja oireinen interstitiaalinen keuhkosairaus (ks. kohta Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabia on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Nivolumabi yhdistelmähoitona AVD‑kemoterapian kanssa
Nivolumabin ja AVD‑kemoterapian yhdistelmähoitoa koskevasta kliinisestä tutkimuksesta suljettiin pois ne potilaat, joilla oli edellisten 2 vuoden aikana systeemistä hoitoa vaatinut aktiivinen autoimmuunisairaus, vaikeusasteen > 2 perifeerinen neuropatia, keskushermostolymfooma, aktiivinen interstitiaalinen pneumoniitti tai interstitiaalinen keuhkosairaus. Tutkimustiedon puuttuessa nivolumabin ja AVD‑kemoterapian yhdistelmähoitoa on käytettävä varoen näille ryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Kun nivolumabia on annettu yhdistelmähoitona AVD:n kanssa, klassista Hodgkinin lymfoomaa sairastavilla potilailla on ilmoitettu enemmän vaikeusasteen 3 ja 4 neutropeniaa. Täydellinen verenkuva on tarkistettava ennen jokaista hoitojaksoa ja kliinisen tarpeen mukaan. Potilaita, joilla on suurentunut kuumeisen neutropenian riski, kuten iäkkäämpiä aikuisia, on seurattava tiiviisti, ja ennaltaehkäisevää hoitoa granulosyyttikasvutekijällä (G‑CSF) on harkittava paikallisten hoitosuositusten mukaisesti.
Nivolumabi yhdistelmähoitona brentuksimabivedotiinin kanssa
Tutkimukseen, jossa selvitettiin nivolumabin yhdistelmähoitoa brentuksimabivedotiinin kanssa cHL:n hoidossa, ei otettu potilaita, joilla oli aktiivinen autoimmuunisairaus, nodulaarinen runsaslymfosyyttinen HL tai jotka olivat saaneet aiemmin anti‑PD‑1‑, anti‑PD‑L1‑, anti‑PD‑L2‑, anti‑CD137‑ tai anti‑CTLA‑4-hoitoa tai jotakin muuta vasta-ainetta tai lääkeainetta, joka kohdistuu erityisesti T‑solujen kostimulointiin tai tarkistuspisteiden signalointireitteihin (ks. kohta Farmakodynamiikka).
Allogeenisen hematopoieettisten kantasolujen siirron (HSCT) komplikaatiot klassista Hodgkinin lymfoomaa sairastavilla potilailla
Klassista Hodgkinin lymfoomaa sairastavien potilaiden, jotka olivat saaneet allogeenisen hematopoieettisten kantasolujen siirron ja joita oli tätä ennen hoidettu nivolumabilla, seurannassa on todettu akuuttia käänteishyljintää (GVHD) ja siirtoon liittyvää kuolleisuutta (TRM). Hematopoieettisten kantasolujen siirron mahdolliset hyödyt ja siirtoon liittyvät mahdolliset lisääntyneet riskit on arvioitava tarkasti kunkin potilaan kohdalla (ks. kohta Haittavaikutukset).
Potilailla, joita hoidettiin nivolumabilla allogeenisen hematopoieettisten kantasolujen siirron jälkeen, on ilmoitettu nopeasti alkavaa ja vaikea-asteista käänteishyljintää valmisteen markkinoilletulon jälkeen. Joissakin tapauksissa käänteishyljintä johti kuolemaan. Nivolumabihoito saattaa lisätä vaikea-asteisen käänteishyljinnän ja kuoleman riskiä potilailla, jotka ovat saaneet allogeenisen hematopoieettisten kantasolujen siirron; erityisesti potilailla, joilla on aiemminkin ollut käänteishyljintää. Näillä potilailla nivolumabihoidon hyödyt on arvioitava mahdolliseen riskiin nähden (ks. kohta Haittavaikutukset).
Pään ja kaulan alueen syöpä
Potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, joilla oli aktiivisia leptomeningeaalisia tai aivometastaaseja, aktiivinen autoimmuunisairaus, systeemistä immunosuppressiota vaativa tila tai joilla alkuperäinen kasvain sijaitsi nenänielussa tai sylkirauhasessa, suljettiin pois kliinisestä SCCHN-tutkimuksesta (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabia on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Lääkärien tulee huomioida nivolumabin viivästynyt vaikutuksen alkaminen ennen hoidon aloittamista potilaille, joilla on huonompi ennuste ja/tai aggressiivinen sairaus. Pään tai kaulan alueen syöpää sairastavista ja nivolumabia saaneista potilaista kuoli kolmen kuukauden aikana useampi kuin dosetakselia saaneista. Varhaiseen kuolemaan liittyvät tekijät olivat heikko ECOG-toimintakyky, nopeasti etenevä sairaus aiemman platinapohjaisen hoidon jälkeen ja suuri kasvaintaakka.
Uroteelikarsinooma
Edenneen uroteelikarsinooman hoito
Potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, joilla oli aktiivisia leptomeningeaalisia tai aivometastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila, suljettiin pois uroteelikarsinoomaa koskevista kliinisistä tutkimuksista (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabia on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Uroteelikarsinooman liitännäishoito
Potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2 (pois lukien potilaat, joiden lähtötason toimintakykyluokka oli 2, jotka eivät olleet saaneet esiliitännäishoitona annettua sisplatiinipohjaista kemoterapiaa ja joiden ei katsottu soveltuvan saamaan liitännäishoitona annettavaa sisplatiinikemoterapiaa), joilta löydettiin näyttöä taudista leikkauksen jälkeen, joilla oli aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila, suljettiin pois uroteelikarsinooman liitännäishoitoa koskevasta kliinisestä tutkimuksesta (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabia on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
dMMR- tai MSI-H-tyyppinen kolorektaalisyöpä
Potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, joilla oli aktiivisia aivometastaaseja tai leptomeningeaalisia metastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila, suljettiin pois dMMR- ja MSI-H-tyyppistä metastaattista kolorektaalisyöpää koskevista kliinisistä tutkimuksista (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabi–ipilimumabi-yhdistelmähoitoa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Ruokatorven levyepiteelikarsinooma
Ruokatorven levyepiteelikarsinooman ensilinjan hoito
Potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, joilla oli tai oli ollut aivometastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila tai joilla oli suuri riski verenvuodolle tai fistelille kasvaimen havaittavan invaasion ruokatorven kasvainta lähellä oleviin elimiin aiheuttamana, suljettiin pois ruokatorven levyepiteelikarsinoomaa koskevasta kliinisestä tutkimuksesta (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabia yhdistelmähoitona ipilimumabin tai kemoterapian kanssa on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Ruokatorven levyepiteelikarsinooman ensilinjan hoidon tutkimuksessa nivolumabi–ipilimumabi‑yhdistelmähoitoa saaneista potilaista kuoli 4 kuukauden aikana useampi kuin kemoterapiaa saaneista. Lääkärien tulee huomioida nivolumabi– ipilimumabi‑yhdistelmähoidon viivästynyt vaikutuksen alkaminen ennen hoidon aloittamista potilaille, joilla on huonompi ennuste ja/tai aggressiivinen sairaus (ks. kohta Farmakodynamiikka).
Ruokatorven levyepiteelikarsinooman hoito aiemman ensilinjan kemoterapian jälkeen
Suurin osa ruokatorven levyepiteelikarsinoomaa koskevista kliinisistä tiedoista perustuu aasialaista syntyperää oleviin potilaisiin (ks. kohta Farmakodynamiikka).
Ruokatorven levyepiteelikarsinoomaa koskevasta kliinisestä tutkimuksesta suljettiin pois potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, joilla oli oireisia tai hoitoa vaativia aivometastaaseja, kasvaimen havaittavaa invaasiota ruokatorven lähellä oleviin elimiin (esim. aorttaan tai hengitysteihin), aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa, nivolumabia on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Lääkärien tulee huomioida nivolumabin viivästynyt vaikutuksen alkaminen ennen hoidon aloittamista potilailla, joilla on ruokatorven levyepiteelikarsinooma. Nivolumabia saaneista potilaista kuoli satunnaistamisen jälkeen 2,5 kuukauden aikana useampi kuin kemoterapiaa saaneista. Varhaiseen kuolemaan liittyviä tekijöitä ei voitu tunnistaa (ks. kohta Farmakodynamiikka).
Ruokatorvisyövän tai ruokatorvi-mahalaukkurajan syövän liitännäishoito
Ruokatorvisyöpää ja ruokatorvi-mahalaukkurajan syöpää koskevasta kliinisestä tutkimuksesta suljettiin pois potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, jotka eivät saaneet samanaikaista kemosädehoitoa (CRT) ennen leikkausta, joilla oli kirurgisesti poistettavissa oleva IV asteen syöpä, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabia on käytettävä varoen näille erityisryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Mahalaukun, ruokatorvi-mahalaukkurajan tai ruokatorven adenokarsinooma
Mahalaukun, ruokatorvi-mahalaukkurajan tai ruokatorven adenokarsinoomaa koskevasta kliinisestä tutkimuksesta suljettiin pois potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, joilla oli hoitamattomia keskushermoston metastaaseja, aktiivinen, tiedossa oleva tai epäilty autoimuunisairaus tai systeemistä immunosuppressiivista hoitoa vaativa tila (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Tutkimustiedon puuttuessa nivolumabi–kemoterapia‑yhdistelmähoitoa on käytettävä varoen näille ryhmille ja mahdollinen hyöty‑riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
CA209649‑tutkimuksesta suljettiin pois potilaat, joiden HER2‑statuksen tiedettiin olevan positiivinen. Potilaat, joiden statusta ei tiedetty, saivat osallistua tutkimukseen, ja 40,3 % potilaista oli tällaisia potilaita (ks. kohta Farmakodynamiikka).
Hepatosellulaarinen karsinooma
Hepatosellulaarista karsinoomaa koskevasta kliinisestä tutkimuksesta suljettiin pois potilaat, joiden lähtötason ECOG-toimintakykyluokka oli ≥ 2, joille oli tehty aiemmin maksansiirto tai joilla oli Child‑Pugh-luokan C maksasairaus, aiemmin todettuja samanaikaisia aivometastaaseja, aiempi maksaperäinen enkefalopatia (12 kuukauden sisällä satunnaistamisesta), kliinisesti merkittävä askites, HIV-infektio tai aktiivinen samanaikainen hepatiitti B -virusinfektio (HBV) ja hepatiitti C -virusinfektio (HCV) tai HBV ja hepatiitti D -virusinfektio (HDV), aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiivista hoitoa vaativa tila (ks. kohdat Yhteisvaikutukset ja Farmakodynamiikka). Hepatosellulaarista karsinoomaa sairastavista potilaista, joilla on Child‑Pugh-luokan B maksasairaus, on saatavilla vain vähän tietoa. Tutkimustiedon puuttuessa nivolumabi–ipilimumabi-yhdistelmähoitoa ja sen jälkeen annettavaa nivolumabihoitoa on käytettävä varoen näille ryhmille ja mahdollinen hyöty-riskisuhde on arvioitava tarkasti kunkin potilaan kohdalla.
Hepatosellulaarista karsinoomaa sairastavilla potilailla nivolumabi–ipilimumabi-yhdistelmähoitoa saaneista potilaista kuoli 6 kuukauden aikana useampi kuin lenvatinibia tai sorafenibia saaneista. Suurempi kuolemanriski voi olla yhteydessä huonoon ennusteeseen. Lääkäreiden tulee huomioida tämä riski ennen nivolumabi–ipilimumabi-yhdistelmähoidon aloittamista potilaille, joiden ennuste on huono.
Vähänatriumista ruokavaliota noudattavat potilaat
Yksi ml tätä lääkevalmistetta sisältää 0,1 mmol (eli 2,5 mg) natriumia. Tämä lääkevalmiste sisältää 10 mg natriumia per 4 ml:n injektiopullo, 25 mg natriumia per 10 ml:n injektiopullo, 30 mg natriumia per 12 ml:n injektiopullo tai 60 mg natriumia per 24 ml:n injektiopullo, joka vastaa 0,5 %:a (4 ml:n injektiopullo), 1,25 %:a (10 ml:n injektiopullo), 1,5 %:a (12 ml:n injektiopullo) tai 3 %:a (24 ml:n injektiopullo) WHO:n suosittelemasta natriumin 2 g:n päivittäisestä enimmäissaannista aikuisille. Natriumin saanti saattaa vaihdella, mikäli laimennusvaiheissa käytetään natriumkloridia.
OPDIVO sisältää polysorbaatti 80:tä (E433)
Tämä lääkevalmiste sisältää 0,94 mg polysorbaatti 80:tä per 4 ml:n injektiopullo; 2,14 mg polysorbaatti 80:tä per 10 ml:n injektiopullo; 2,6 mg polysorbaatti 80:tä per 12 ml:n injektiopullo; ja 5,0 mg polysorbaatti 80:tä per 24 ml:n injektiopullo. Polysorbaatit saattavat aiheuttaa allergisia reaktioita.
Potilaskortti
Kaikkien OPDIVO-valmistetta määräävien lääkäreiden on perehdyttävä lääkärin tiedotus- ja hoito-ohjeisiin. Lääkettä määräävän lääkärin on keskusteltava OPDIVO–hoidon riskeistä potilaan kanssa. Potilaalle toimitetaan potilaskortti jokaisen lääkemääräyksen yhteydessä.
Yhteisvaikutukset
Nivolumabi on humaani monoklonaalinen vasta-aine eikä sen farmakokineettisiä yhteisvaikutuksia ole tutkittu. Koska monoklonaaliset vasta-aineet eivät metaboloidu sytokromi P450 (CYP) entsyymien eivätkä muiden lääkeaineita metaboloivien entsyymien vaikutuksesta, muiden samaan aikaan käytettyjen lääkevalmisteiden näitä entsyymejä estävien tai indusoivien vaikutusten ei odoteta vaikuttavan nivolumabin farmakokinetiikkaan.
Muut yhteisvaikutukset
Systeeminen immunosuppressio
Systeemisten kortikosteroidien ja muiden immunosuppressiivisten lääkkeiden käyttöä ennen nivolumabihoidon aloitusta on vältettävä, koska ne saattaisivat vaikuttaa nivolumabin farmakodynamiikkaan. Systeemisiä kortikosteroideja ja muita immunosuppressiivisia lääkkeitä voi kuitenkin käyttää nivolumabihoidon alettua immuunivälitteisten haittavaikutusten hoitoon. Alustavat tulokset osoittavat, että systeemisten immunosuppressiivisten lääkkeiden käyttö nivolumabihoidon alettua ei näytä estävän nivolumabin vastetta
Raskaus ja imetys
Raskaus
Nivolumabin käytöstä raskaana oleville ei ole tietoa. Eläinkokeissa on todettu alkio- ja sikiötoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Ihmisen IgG4:n tiedetään läpäisevän veri-istukkaesteen, ja nivolumabi on IgG4-luokan vasta-aine. Siksi nivolumabi voi potentiaalisesti siirtyä äidistä kehittyvään sikiöön. Nivolumabia ei suositella käytettäväksi raskauden aikana eikä sellaisten naisten hoitoon, jotka saattaisivat tulla raskaaksi mutta eivät käytä tehokasta ehkäisyä, paitsi siinä tapauksessa, että kliininen hyöty on merkittävämpi kuin mahdollinen riski. Tehokasta ehkäisymenetelmää on käytettävä vähintään 5 kuukauden ajan viimeisen nivolumabiannoksen jälkeen.
Imetys
Ei tiedetä, erittyykö nivolumabi ihmisen rintamaitoon. Koska monet lääkevalmisteet, kuten antibiootit, voivat erittyä ihmisen rintamaitoon, vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida sulkea pois. On päätettävä, lopetetaanko rintaruokinta vai lopetetaanko nivolumabihoito, ottaen huomioon rintaruokinnasta lapselle ja hoidosta äidille koituva hyöty.
Hedelmällisyys
Nivolumabin vaikutusta hedelmällisyyteen ei ole tutkittu. Siksi sen vaikutusta miehen ja naisen hedelmällisyyteen ei tiedetä.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Nivolumabilla tai nivolumabi-ipilimumabilla tai nivolumabin ja muiden lääkeaineiden yhdistelmähoidolla saattaa olla vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Koska haittavaikutukset, kuten uupumus, ovat mahdollisia (ks. kohta Haittavaikutukset), potilaita on kehotettava varovaisuuteen ajamisessa ja koneiden käytössä, kunnes he ovat varmoja, ettei nivolumabilla ole heihin haitallista vaikutusta.
Haittavaikutukset
Nivolumabi-monoterapia (ks. kohta Annostus ja antotapa)
Tiivistelmä turvallisuusprofiilista
Yhdistetyissä tutkimustiedoissa eri kasvaintyyppeihin käytetyn nivolumabi-monoterapian (n = 4646) yleisimmät haittavaikutukset (≥ 10 %), kun seuranta-aika oli vähintään 2,3–28 kuukautta, olivat uupumus (44 %), muskuloskeletaalinen kipu (28 %), ripuli (26 %), ihottuma (24 %), yskä (22 %), pahoinvointi (22 %), kutina (19 %), ruokahalun heikkeneminen (17 %), artralgia (17 %), ummetus (16 %), dyspnea (16 %), vatsakipu (15 %), ylähengitystieinfektio (15 %), kuume (13 %), päänsärky (13 %), anemia (13 %) ja oksentelu (12 %). Suurin osa haittavaikutuksista oli lieviä tai kohtalaisia (asteen 1 tai 2). Asteen 3–5 haittavaikutusten ilmaantuvuus oli 44 %, ja tutkimuslääkkeestä johtuvien, kuolemaan johtaneiden haittavaikutusten ilmaantuvuus oli 0,3 %. Ei‑pienisoluisen keuhkosyövän vähintään 63 kuukauden seurannassa ei havaittu uusia turvallisuussignaaleja.
Tiivistelmä haittavaikutuksista taulukkona
Taulukossa 18 on esitetty yhdistetyistä tutkimustiedoista kootut nivolumabia monoterapiana saaneiden potilaiden (n = 4646) raportoidut haittavaikutukset. Haittavaikutukset on esitetty elinjärjestelmäluokan ja yleisyyden mukaan. Yleisyydet määritellään seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000); tuntematon (koska myyntiluvan myöntämisen jälkeiset tutkimustiedot ovat vielä riittämättömät). Haittavaikutukset on lueteltu kussakin yleisyysryhmässä vakavimmasta lähtien.
Taulukko 18: Nivolumabi‑monoterapiaan liittyvät haittavaikutukset
| Nivolumabi‑monoterapia |
Infektiot | |
Hyvin yleinen | ylähengitystieinfektio |
Yleinen | keuhkokuumea, keuhkoputkentulehdus |
Harvinainen | aseptinen aivokalvotulehdus |
Hyvän‑ ja pahanlaatuiset kasvaimet (mukaan lukien kystat ja polyypit) | |
Harvinainen | histiosyyttinen nekrotisoiva lymfadeniitti (Kikuchin lymfadeniitti) |
Veri ja imukudos | |
Hyvin yleinen | lymfopeniab, anemiab,i, leukopeniab, neutropeniaa,b, trombosytopeniab |
Melko harvinainen | eosinofilia |
Tuntematon | hemofagosyyttinen lymfohistiosytoosi |
Immuunijärjestelmä | |
Yleinen | infuusioreaktiot (mukaan lukien sytokiinioireyhtymä), yliherkkyys (mukaan lukien anafylaktinen reaktio) |
Melko harvinainen | sarkoidoosi |
Tuntematon | hyljintäreaktio kiinteän elimen siirron jälkeenf |
Umpieritys | |
Yleinen | kilpirauhasen vajaatoiminta, kilpirauhasen liikatoiminta, kilpirauhastulehdus |
Melko harvinainen | lisämunuaisten vajaatoimintaj, hypopituitarismi, hypofysiitti, diabetes mellitus |
Harvinainen | diabeettinen ketoasidoosi, lisäkilpirauhasten vajaatoiminta |
Aineenvaihdunta ja ravitsemus | |
Hyvin yleinen | ruokahalun väheneminen, hyperglykemiab |
Yleinen | kuivumistila, painon lasku, hypoglykemiab |
Melko harvinainen | metabolinen asidoosi |
Tuntematon | tuumorilyysioireyhtymäg |
Hermosto | |
Hyvin yleinen | päänsärky |
Yleinen | perifeerinen neuropatia, heitehuimaus |
Melko harvinainen | polyneuropatia, autoimmuunineuropatia (mukaan lukien kasvo- ja loitontajahermon halvaus), myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymäm |
Harvinainen | Guillain–Barrén oireyhtymä, demyelinaatio, myasteeninen oireyhtymä, aivotulehdusa,k, näköhermotulehdus |
Tuntematon | myeliitti (mukaan lukien transversaalinen myeliitti) |
Silmät | |
Yleinen | sumentunut näkö, kuivat silmät |
Melko harvinainen | uveiitti |
Tuntematon | Vogt–Koyanagi–Haradan oireyhtymäf |
Sydän | |
Yleinen | sydämen tiheälyöntisyys, eteisvärinä |
Melko harvinainen | myokardiittia, perikardiaaliset häiriöth, rytmihäiriö (myös kammioperäinen rytmihäiriö) |
Verisuonisto | |
Yleinen | kohonnut verenpaine |
Harvinainen | verisuonitulehdus |
Hengityselimet, rintakehä ja välikarsina | |
Hyvin yleinen | hengenahdistusa, yskä |
Yleinen | keuhkotulehdusa, keuhkopussin nestekertymä |
Melko harvinainen | keuhkoinfiltraatio |
Ruoansulatuselimistö | |
Hyvin yleinen | ripuli, oksentelu, pahoinvointi, vatsakipu, ummetus |
Yleinen | koliittia, suutulehdus, suun kuivuminen |
Melko harvinainen | haimatulehdus, gastriitti |
Harvinainen | pohjukaissuolen haavauma, haiman eksokriininen vajaatoiminta, keliakia |
Maksa ja sappi | |
Melko harvinainen | hepatiitti, kolestaasi |
Iho ja ihonalainen kudos | |
Hyvin yleinen | ihottumac, kutina |
Yleinen | vitiligo, kuiva iho, eryteema, alopesia |
Melko harvinainen | psoriaasi, ruusufinni, erythema multiforme, nokkosihottuma |
Harvinainen | toksinen epidermaalinen nekrolyysia,d, Stevens–Johnsonin oireyhtymäa |
Tuntematon | valkojäkäläg, muut jäkälätaudit |
Luusto, lihakset ja sidekudos | |
Hyvin yleinen | muskuloskeletaalinen kipue, artralgia |
Yleinen | niveltulehdus |
Melko harvinainen | polymyalgia rheumatica (monilihaskipu) |
Harvinainen | Sjögrenin oireyhtymä, myopatia, myosiitti (myös monilihastulehdus)a, rabdomyolyysia,d |
Munuaiset ja virtsatiet | |
Yleinen | munuaisten vajaatoiminta (myös akuutti munuaisvaurio)a |
Harvinainen | tubulointerstitiaalinen nefriitti, ei-infektiivinen virtsarakkotulehdus |
Yleisoireet ja antopaikassa todettavat haitat | |
Hyvin yleinen | uupumus, kuume |
Yleinen | kipu, rintakipu, turvotusl |
Tutkimuksetb | |
Hyvin yleinen | lisääntynyt ASAT, hyponatremia, hypoalbuminemia, lisääntynyt alkalinen fosfataasi, lisääntynyt kreatiniini, lisääntynyt ALAT, lisääntynyt lipaasi, hyperkalemia, lisääntynyt amylaasi, hypokalsemia, hypomagnesemia, hypokalemia, hyperkalsemia |
Yleinen | lisääntynyt kokonaisbilirubiinin määrä, hypernatremia, hypermagnesemia |
Taulukossa 18 esitetyt yleisyysluokat eivät välttämättä liity täysin pelkästään nivolumabiin, vaan niihin voi vaikuttaa myös perussairaus.
a Kuolemaan johtaneita tapauksia on ilmoitettu päättyneissä ja jatkuvissa kliinisissä tutkimuksissa.
b Yleisyys laskettiin sen mukaan, kuinka suurella osalla potilaista laboratorioarvo huononi lähtötasolta. Katso jäljempää ”Valikoitujen haittavaikutusten kuvaus, Poikkeavat laboratorioarvot”.
c Ihottuma on yhdistelmätermi, joka sisältää makulopapulaarisen ihottuman, punoittavan ihottuman, kutiavan ihottuman, follikulaarisen ihottuman, makulaarisen ihottuman, tuhkarokkomaisen ihottuman, näppyläisen ihottuman, märkärakkulaihottuman, vesirakkulaisen ihottuman, hilseilevän ihottuman, ihotulehduksen, aknea muistuttavan ihottuman, allergisen ihottuman, atooppisen ihottuman, rakkulaisen ihottuman, hilseilevän dermatiitin, psoriaasia muistuttavan ihottuman, lääkeihottuman ja pemfigoidin.
d Ilmoitettu myös yhdistetyn aineiston ulkopuolisissa tutkimuksissa. Yleisyys perustuu kehitysohjelman laajuiseen altistukseen.
e Muskuloskeletaalinen kipu on termi, joka kattaa selkäkivun, luukivun, muskuloskeletaalisen rintakivun, muskuloskeletaaliset vaivat, lihaskivun, kylkivälilihasten kivun, niskakivun, raajakivun ja selkärankakivun.
f Markkinoilletulon jälkeen (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
g Ilmoitettu kliinisissä tutkimuksissa ja valmisteen markkinoilletulon jälkeen.
h Perikardiaaliset häiriöt on termi, joka kattaa perikardiitin, perikardiaalisen effuusion, perikardiumin tamponaation ja Dresslerin oireyhtymän.
i Anemia on yhdistelmätermi, joka sisältää muiden syiden lisäksi hemolyyttisen anemian ja autoimmuunianemian, alhaisen hemoglobiinin, raudanpuuteanemian ja pienentyneen punasolumäärän.
j Kattaa lisämunuaisten vajaatoiminnan, akuutin lisämunuaiskuoren vajaatoiminnan ja lisämunuaiskuoren sekundaarisen vajaatoiminnan.
k Kattaa aivotulehduksen ja limbisen aivotulehduksen.
l Turvotus on yhdistelmätermi, joka sisältää yleistyneen edeeman, periferaalisen edeeman, periferaalisen turvotuksen ja turvotuksen.
m Myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymän (ilmenee näistä joko kahden tai kaikkien kolmen sairauden limittymisenä) tapauksia, jotka ovat joskus johtaneet kuolemaan, on raportoitu nivolumabihoidossa ja nivolumabin ja muiden lääkeaineiden yhdistelmähoidossa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Nivolumabi yhdistelmähoitona muiden lääkeaineiden kanssa (ks. kohta Annostus ja antotapa)
Tiivistelmä turvallisuusprofiilista
Kun nivolumabia annetaan yhdistelmähoitona muiden lääkeaineiden kanssa, lue muiden lääkeaineiden valmisteyhteenvedosta lisätietoa turvallisuusprofiilista ennen hoidon aloittamista.
Nivolumabi–ipilimumabi-yhdistelmähoito (kemoterapian kanssa tai ilman)
Yhdistetyissä tutkimustiedoissa (n = 2626) eri kasvaintyyppien hoidossa käytetyn nivolumabi–ipilimumabi-yhdistelmähoidon (kemoterapian kanssa tai ilman) yleisimmät haittavaikutukset (≥ 10 %) olivat uupumus (47 %), ripuli (35 %), ihottuma (37 %), pahoinvointi (27 %), kutina (29 %), muskuloskeletaalinen kipu (26 %), kuume (23 %), ruokahalun väheneminen (22 %), yskä (21 %), vatsakipu (18 %), oksentelu (18 %), ummetus (18 %), artralgia (18 %), hengenahdistus (17 %), kilpirauhasen vajaatoiminta (16 %), päänsärky (15 %), ylähengitystieinfektio (13 %), turvotus (13 %) ja heitehuimaus (10 %), kun seuranta-aika oli vähintään 6–47 kuukautta. Asteen 3–5 haittavaikutusten ilmaantuvuus oli 66 %, kun nivolumabia käytettiin yhdistelmähoitona ipilimumabin kanssa (kemoterapian kanssa tai ilman). Tutkimuslääkkeestä johtuvien, kuolemaan johtaneiden haittavaikutusten ilmaantuvuus oli 1,0 %. Seuraavien haittavaikutusten ilmaantuvuus oli ≥ 10 % suurempi potilailla, jotka saivat yhdistelmähoitona nivolumabia 1 mg/kg ja ipilimumabia 3 mg/kg melanooman hoitoon, kuin ilmaantuvuus yhdistetyissä tutkimustiedoissa, jotka koskivat nivolumabi–ipilimumabi-yhdistelmähoitoa (kemoterapian kanssa tai ilman): uupumus (62 %), ihottuma (57 %), ripuli (52 %), pahoinvointi (42 %), kutina (40 %), kuume (36 %) ja päänsärky (26 %). Seuraavien haittavaikutusten ilmaantuvuus oli ≥ 10 % suurempi potilailla, jotka saivat yhdistelmähoitona ei-pienisoluisen keuhkosyövän (NSCLC) hoidoon nivolumabia 360 mg ja ipilimumabia 1 mg/kg sekä kemoterapiaa, kuin ilmaantuvuus yhdistetyissä tutkimustiedoissa, jotka koskivat nivolumabi–ipilimumabi-yhdistelmähoitoa (kemoterapian kanssa tai ilman): anemia (32 %) ja neutropenia (15 %).
Nivolumabin ja kemoterapian yhdistelmähoito
Yhdistetyissä tutkimustiedoissa eri kasvaintyyppien hoidossa käytetyn nivolumabin (240 mg joka toinen viikko tai 360 mg joka kolmas viikko) ja kemoterapian yhdistelmähoidon (n = 1800) yleisimmät haittavaikutukset (≥ 10 %), kun seuranta-aika oli vähintään 7,4–23,6 kuukautta, olivat kirurgisesti poistettavissa olevan ei‑pienisoluisen keuhkosyövän (NSCLC) kolmen hoitojakson hoidossa pahoinvointi (48 %), väsymys (40 %), perifeerinen neuropatia (33 %), ruokahalun väheneminen (31 %), ummetus (31 %), ripuli (28 %), oksentelu (24 %), ihottuma (19 %), vatsakipu (18 %), stomatiitti (18 %), muskuloskeletaalinen kipu (18 %), kuume (16 %), yskä (13 %), turvotus (mukaan lukien perifeerinen ödeema) (12 %) ja kutina (11 %). Vaikeusasteen 3–5 haittavaikutuksia oli 69 %:lla nivolumabin ja kemoterapian yhdistelmähoitoa saaneista, ja nivolumabin ja kemoterapian yhdistelmähoidon aiheuttamista haittavaikutuksista 1,2 % johti kuolemaan. Nivolumabin ja kemoterapian yhdistelmähoidon keston mediaani oli 6,14 kuukautta (95 %:n luottamusväli: 5,78–6,60). Kirurgisesti poistettavissa olevan ei‑pienisoluisen keuhkosyövän hoidossa potilaista 93 % sai kolme jaksoa nivolumabin ja kemoterapian yhdistelmähoitoa.
Nivolumabi yhdistelmähoitona AVD‑kemoterapian kanssa
Tutkimustiedoissa cHL:n ensilinjan hoidossa käytetyn nivolumabin (240 mg tai 3 mg/kg joka toinen viikko) ja AVD‑kemoterapian yhdistelmähoidon (n = 490) yleisimmät haittavaikutukset (≥ 10 %), kun seuranta-aika oli vähintään 30,4 kuukautta, olivat pahoinvointi (70,4 %), neutropenia (61 %), väsymys (59,0 %), anemia (52 %), ummetus (49,2 %), leukopenia (45,1 %), muskuloskeletaalinen kipu (42,7 %), suurentuneet transaminaasiarvot (41,2 %), perifeerinen neuropatia (41 %), oksentelu (33,5 %), stomatiitti (30,6 %), hyperglykemia (29,8 %), ripuli (27,6 %), lymfopenia (25,9 %), yskä (25,7 %), ihottuma (24,7 %), päänsärky (23,9 %), alopesia (22,7 %), vatsakipu (21,8 %), kuume (21,6 %), dyspepsia (20,4 %), hypoalbuminemia (18 %), lisääntynyt veren alkalinen fosfataasi (17,6 %), kutina (17,6 %), kohonnut verenpaine (16,3 %), ruokahalun väheneminen (16,3 %), kipu (15,7 %), hypokalsemia (13,3 %), trombosytopenia (12 %), hyponatremia (12 %), heitehuimaus (12 %), hypokalemia (11 %), sydämen tiheälyöntisyys (10,8 %), rintakipu (10,8 %) ja dysgeusia (10 %). Vaikeusasteen 3–5 haittavaikutusten ilmaantuvuus oli 71,2 %, ja nivolumabin ja AVD:n yhdistelmähoidosta johtuvien, kuolemaan johtaneiden haittavaikutusten ilmaantuvuus oli 0,6 %.
Nivolumabi yhdistelmähoitona kabotsantinibin kanssa
Tutkimustiedoissa munuaiskarsinooman hoidossa (n = 320) käytetyn nivolumabin (240 mg joka toinen viikko) ja kabotsantinibin kanssa (40 mg kerran päivässä) yhdistelmähoidon yleisimmät haittavaikutukset (≥ 10 %), kun seuranta-aika oli vähintään 16,0 kuukautta, olivat ripuli (64,7 %), väsymys (51,3 %), käsi-jalkaoireyhtymä (40,0 %), stomatiitti (38,8 %), muskuloskeletaalinen kipu (37,5 %), hypertensio (37,2 %), ihottuma (36,3 %), kilpirauhasen vajaatoiminta (35,6 %), ruokahalun väheneminen (30,3 %), pahoinvointi (28,8 %), vatsakipu (25,0 %), makuhäiriö (23,8 %), ylähengitystietulehdus (20,6 %), yskä (20,6 %), kutina (20,6 %), artralgia (19,4 %), oksentelu (18,4 %), dysfonia (17,8 %), päänsärky (16,3 %), dyspepsia (15,9 %), huimaus (14,1 %), ummetus (14,1 %), kuume (14,1 %), turvotus (13,4 %), lihaskouristukset (12,2 %), dyspnea (11,6 %), proteiinivirtsaisuus (10,9 %) ja kilpirauhasen liikatoiminta (10,0 %). Asteen 3–5 haittavaikutusten ilmaantuvuus oli 78 %, ja tutkimuslääkkeestä johtuvien, kuolemaan johtaneiden haittavaikutusten ilmaantuvuus oli 0,3 %.
Nivolumabi yhdistelmähoitona brentuksimabivedotiinin kanssa
Tutkimustiedoissa uusiutuneen tai refraktaarisen cHL:n hoidossa (kaikki hoitoa saaneet potilaat, n = 72) käytetyn nivolumabin (3 mg/kg 3 viikon välein) ja brentuksimabivedotiinin (1,8 mg/kg 3 viikon välein) yhdistelmähoidon yleisimmät haittavaikutukset (≥ 10 %), kun seuranta-aika oli vähintään 40,7 kuukautta, olivat pahoinvointi (50,0 %), ihottuma (29,2 %), kuume (26,4 %), päänsärky (25,0 %), ripuli (25,0 %), muskuloskeletaalinen kipu (25,0 %), vatsakipu (22,2 %), yliherkkyys (20,8 %), infuusioreaktio (18,1 %), oksentelu (18,1 %), väsymys (18,1 %), suurentuneet transaminaasiarvot (13,9 %) ja ummetus (11,1 %). Asteen 3–5 haittavaikutusten ilmaantuvuus oli 36,1 %. Lääkkeeseen liittyneitä kuolemaan johtaneita haittavaikutuksia ei ilmoitettu.
Haittavaikutustaulukko
Taulukossa 19 esitetään yhdistetyistä tutkimustiedoista kootut haittavaikutukset potilailla, jotka saivat nivolumabin ja ipilimumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) (n = 2626), nivolumabin ja kemoterapian yhdistelmähoitoa (n = 1800) ja nivolumabin ja kabotsantinibin yhdistelmähoitoa (n = 320). Haittavaikutukset on esitetty elinjärjestelmäluokan ja yleisyyden mukaan. Yleisyysluokkien määritelmät ovat: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa vakavimmasta lievimpään.
Taulukko 19: Nivolumabin ja muiden lääkeaineiden yhdistelmähoitoon liittyvät haittavaikutukset
| Yhdistelmähoitona ipilimumabin kanssa (kemoterapian kanssa tai ilman) | Yhdistelmähoitona kemoterapian kanssa | Yhdistelmähoitona kabotsantinibin kanssa |
|---|---|---|---|
Infektiot | |||
Hyvin yleinen | ylähengitystieinfektio |
| ylähengitystieinfektio |
Yleinen | keuhkokuume, keuhkoputkentulehdus, konjunktiviitti | ylähengitystieinfektio, keuhkokuumea | keuhkokuume |
Harvinainen | aseptinen aivokalvotulehdus |
|
|
Veri ja imukudos | |||
Hyvin yleinen | anemiab,j, trombosytopeniab, leukopeniab, lymfopeniab, neutropeniab | neutropeniab, anemiab,j, leukopeniab, lymfopeniab, trombosytopeniab | anemiab, trombosytopeniab, leukopeniab, lymfopeniab, neutropeniab |
Yleinen | eosinofilia | kuumeinen neutropeniaa | eosinofilia |
Melko harvinainen | kuumeinen neutropenia | eosinofilia |
|
Tuntematon | hemofagosyyttinen lymfohistiosytoosi |
|
|
Immuunijärjestelmä | |||
Yleinen | infuusioreaktio (mukaan lukien sytokiinioireyhtymä), yliherkkyys | yliherkkyys, infuusioreaktio (mukaan lukien sytokiinioireyhtymä) | yliherkkyys (mukaan lukien anafylaktinen reaktio) |
Melko harvinainen |
|
| infuusioon liittyvä yliherkkyysreaktio |
Harvinainen | sarkoidoosi |
|
|
Tuntematon | hyljintäreaktio kiinteän elimen siirron jälkeeng |
|
|
Umpieritys | |||
Hyvin yleinen | kilpirauhasen vajaatoiminta |
| kilpirauhasen vajaatoiminta, kilpirauhasen liikatoiminta |
Yleinen | kilpirauhasen liikatoiminta, kilpirauhastulehdus, lisämunuaisten vajaatoiminta, hypofysiitti, hypopituitarismi, diabetes mellitus | kilpirauhasen vajaatoiminta, kilpirauhasen liikatoiminta, diabetes mellitus | lisämunuaisten vajaatoiminta |
Melko harvinainen | diabeettinen ketoasidoosi | lisämunuaisten vajaatoiminta, kilpirauhastulehdus, hypopituitarismi, hypofysiitti | hypofysiitti, kilpirauhastulehdus |
Harvinainen | lisäkilpirauhasten vajaatoiminta |
|
|
Aineenvaihdunta ja ravitsemus | |||
Hyvin yleinen | ruokahalun väheneminen, hyperglykemiab, hypoglykemiab | ruokahalun väheneminen, hyperglykemiab, hypoglykemiab | ruokahalun väheneminen, hypoglykemiab, hyperglykemiab, painon lasku |
Yleinen | kuivumistila, hypoalbuminemia, hypofosfatemia, painon lasku | hypoalbuminemia, hypofosfatemia | kuivumistila |
Melko harvinainen | metabolinen asidoosi |
|
|
Harvinainen |
| tuumorilyysioireyhtymä |
|
Tuntematon | tuumorilyysioireyhtymäh |
|
|
Hermosto | |||
Hyvin yleinen | päänsärky | perifeerinen neuropatia | dysgeusia, heitehuimaus, päänsärky |
Yleinen | heitehuimaus, perifeerinen neuropatia | parestesia, heitehuimaus, päänsärky | perifeerinen neuropatia |
Melko harvinainen | polyneuropatia, peroneuspareesi, autoimmuunineuropatia (ml. kasvohalvaus ja loitontajahermon halvaus), aivotulehdus, myasthenia gravis, myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymäl | Guillain–Barrén oireyhtymä | autoimmuunienkefaliitti, Guillain–Barrén oireyhtymä, myasteeninen oireyhtymä |
Harvinainen | Guillain–Barrén oireyhtymä, neuriitti, myeliitti (mukaan lukien transversaalinen myeliitti), näköhermotulehdus | aivotulehdus, myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymäl |
|
Tuntematon |
| myeliitti (mukaan lukien transversaalinen myeliitti), näköhermotulehdus | myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymäl |
Kuulo ja tasapainoelin | |||
Yleinen |
|
| tinnitus |
Silmät | |||
Yleinen | sumentunut näkö, kuivat silmät | kuivat silmät, sumentunut näkö | kuivat silmät, sumentunut näkö |
Melko harvinainen | uveiitti, episkleriitti | uveiitti | uveiitti |
Harvinainen | Vogt–Koyanagi–Haradan oireyhtymä |
|
|
Sydän | |||
Yleinen | sydämen tiheälyöntisyys, eteisvärinä | sydämen tiheälyöntisyys, eteisvärinä | eteisvärinä, sydämen tiheälyöntisyys |
Melko harvinainen | myokardiittia, rytmihäiriö (myös kammioperäinen rytmihäiriö)a, sydämen harvalyöntisyys | myokardiitti | myokardiitti |
Tuntematon | perikardiaaliset häiriöti |
|
|
Verisuonisto | |||
Hyvin yleinen |
|
| kohonnut verenpaine |
Yleinen | kohonnut verenpaine | tromboosia,k, kohonnut verenpaine, verisuonitulehdus | tromboosik |
Hengityselimet, rintakehä ja välikarsina | |||
Hyvin yleinen | yskä, hengenahdistus | yskä | dysfonia, hengenahdistus, yskä |
Yleinen | keuhkotulehdusa, keuhkoemboliaa, keuhkopussin nestekertymä | keuhkotulehdusa, hengenahdistus | keuhkotulehdus, keuhkoembolia, keuhkopussin nestekertymä, nenäverenvuoto |
Ruoansulatuselimistö | |||
Hyvin yleinen | ripuli, oksentelu, pahoinvointi, vatsakipu, ummetus | ripulia, suutulehdus, oksentelu, pahoinvointi, vatsakipu, ummetus | ripuli, oksentelu, pahoinvointi, ummetus, suutulehdus, vatsakipu, dyspepsia |
Yleinen | koliittia, haimatulehdus, suutulehdus, gastriitti, suun kuivuminen | koliitti, suun kuivuminen | koliitti, gastriitti, suukipu, suun kuivuminen, peräpukamat |
Melko harvinainen | duodeniitti | haimatulehdus | haimatulehdus, ohutsuolen puhkeaminena, kielikipu |
Harvinainen | suolen puhkeaminena, haiman eksokriininen vajaatoiminta, keliakia |
|
|
Tuntematon |
| haiman eksokriininen vajaatoiminta, keliakia | haiman eksokriininen vajaatoiminta, keliakia |
Maksa ja sappi | |||
Yleinen | maksatulehdus |
| maksatulehdus |
Melko harvinainen |
| maksatulehdus |
|
Iho ja ihonalainen kudos | |||
Hyvin yleinen | ihottumac, kutina | ihottumac,kutina | käsi-jalkaoireyhtymä, ihottumac, kutina |
Yleinen | alopesia, vitiligo, nokkosihottuma, kuiva iho, eryteema | käsi-jalkaoireyhtymä, ihon hyperpigmentaatio, alopesia, kuiva iho, eryteema | alopesia, kuiva iho, eryteema, hiusten värin muuttuminen |
Melko harvinainen | Stevens–Johnsonin oireyhtymä, erythema multiforme, psoriaasi, muut jäkälätauditd |
| psoriaasi, nokkosihottuma |
Harvinainen | toksinen epidermaalinen nekrolyysia,e, valkojäkälä |
|
|
Tuntematon |
|
| valkojäkälä, muut jäkälätaudit |
Luusto, lihakset ja sidekudos | |||
Hyvin yleinen | muskuloskeletaalinen kipuf, artralgia | muskuloskeletaalinen kipuf | muskuloskeletaalinen kipuf, artralgia, lihaskouristukset |
Yleinen | lihaskouristukset, lihasheikkous, niveltulehdus | artralgia, lihasheikkous | niveltulehdus |
Melko harvinainen | polymyalgia rheumatica (monilihaskipu), myopatia, myosiitti (myös monilihastulehdus)a |
| myopatia, leuan osteonekroosi, fisteli |
Harvinainen | spondylartropatia, Sjögrenin oireyhtymä, rabdomyolyysia |
|
|
Munuaiset ja virtsatiet | |||
Hyvin yleinen |
|
| proteiinivirtsaisuus |
Yleinen | munuaisten vajaatoiminta (myös akuutti munuaisvaurio)a | munuaisten vajaatoimintaa | munuaisten vajaatoiminta, akuutti munuaisvaurio |
Melko harvinainen | tubulointerstitiaalinen nefriitti, nefriitti | ei-infektiivinen virtsarakkotulehdus,nefriitti | nefriitti |
Harvinainen | ei-infektiivinen virtsarakkotulehdus |
| ei-infektiivinen virtsarakkotulehdush |
Yleisoireet ja antopaikassa todettavat haitat | |||
Hyvin yleinen | uupumus, kuume, turvotus (mukaan lukien perifeerinen edeema) | uupumus, kuume, turvotus (mukaan lukien perifeerinen edeema) | uupumus, kuume, turvotus |
Yleinen | rintakipu, kipu, vilunväristykset | huonovointisuus | kipu, rintakipu |
Tutkimukset | |||
Hyvin yleinen | lisääntynyt alkalinen fosfataasib, lisääntynyt ASATb, lisääntynyt ALATb, lisääntynyt kokonaisbilirubiinib, lisääntynyt kreatiniinib, lisääntynyt amylaasib, lisääntynyt lipaasib, hyponatremiab, hyperkalemiab, hypokalemiab, hyperkalsemiab, hypokalsemiab,hypomagnesemiab | hypokalsemiab, lisääntynyt ASATb, lisääntynyt ALATb, hyponatremiab, lisääntynyt amylaasib, hypomagnesemiab, lisääntynyt alkalinen fosfataasib, hypokalemiab, lisääntynyt kreatiniinib, lisääntynyt lipaasib, hyperkalemiab, lisääntynyt kokonaisbilirubiinib | lisääntynyt alkalinen fosfataasib, lisääntynyt ALATb, lisääntynyt ASATb, lisääntynyt kokonaisbilirubiinib, lisääntynyt kreatiniinib, lisääntynyt amylaasib, lisääntynyt lipaasib, hypokalemiab, hypomagnesemiab, hyponatremiab, hypokalsemiab, hyperkalsemiab, hypofosfatemiab, hyperkalemiab, hypermagnesemiab, hypernatremiab |
Yleinen | hypernatremiab, hypermagnesemiab, kilpirauhasta stimuloivan hormonin lisääntyminen, lisääntynyt gammaglutamyylitransferaasi | hypernatremiab, hyperkalsemiab, hypermagnesemiab | kohonnut veren kolesterolipitoisuus, hypertriglyseridemia |
Taulukossa 19 esitetyt yleisyysluokat eivät välttämättä liity täysin pelkästään nivolumabiin tai nivolumabin ja muiden lääkeaineiden yhdistelmähoitoihin, vaan niihin voivat vaikuttaa myös perussairaudet tai muut yhdistelmänä käytetyt lääkevalmisteet.
a Kuolemaan johtaneita tapauksia on ilmoitettu päättyneissä ja jatkuvissa kliinisissä tutkimuksissa.
b Yleisyys laskettiin sen mukaan, kuinka suurella osalla potilaista laboratorioarvo huononi lähtötasolta. Katso jäljempää ”Valikoitujen haittavaikutusten kuvaus, Poikkeavat laboratorioarvot”.
c Ihottuma on yhdistelmätermi, joka sisältää makulopapulaarisen ihottuman, punoittavan ihottuman, kutiavan ihottuman, follikulaarisen ihottuman, makulaarisen ihottuman, tuhkarokkomaisen ihottuman, näppyläisen ihottuman, märkärakkulaihottuman, näppyläisen ja suomuilevan ihottuman, vesirakkulaisen ihottuman, laajalle levinneen ihottuman, hilseilevän ihottuman, ihotulehduksen, aknea muistuttavan ihottuman, allergisen ihottuman, atooppisen ihottuman, rakkulaisen ihottuman, hilseilevän dermatiitin, psoriaasia muistuttavan ihottuman, lääkeihottuman, nodulaarisen ihottuman ja pemfigoidin.
d Jäkälätaudit on termi, joka kattaa jäkälän aiheuttaman keratoosin ja punajäkälän.
e Ilmoitettu myös yhdistetyn aineiston ulkopuolisissa tutkimuksissa. Yleisyys perustuu kehitysohjelman laajuiseen altistukseen.
f Muskuloskeletaalinen kipu on termi, joka kattaa selkäkivun, luukivun, muskuloskeletaalisen rintakivun, muskuloskeletaaliset vaivat, lihaskivun, kylkivälilihasten kivun, niskakivun, raajakivun ja selkärankakivun.
g Markkinoilletulon jälkeen (ks. myös kohta Varoitukset ja käyttöön liittyvät varotoimet).
h Ilmoitettu kliinisissä tutkimuksissa ja valmisteen markkinoilletulon jälkeen.
i Perikardiaaliset häiriöt on termi, joka kattaa perikardiitin, perikardiaalisen effuusion, perikardiumin tamponaation ja Dresslerin oireyhtymän.
j Anemia on yhdistelmätermi, joka sisältää muiden syiden lisäksi hemolyyttisen anemian ja autoimmuunianemian, alhaisen hemoglobiinin, raudanpuuteanemian ja pienentyneen punasolumäärän.
k Tromboosi on yhdistelmätermi, joka kattaa porttilaskimotromboosin, keuhkoverisuonen tromboosin, keuhkotromboosin, aortan tromboosin, valtimotromboosin, syvän laskimotromboosin, lantion laskimotromboosin, vena cavan tromboosin, laskimotromboosin ja raajan laskimotromboosin.
l Myokardiitti-myosiitti-myasthenia gravis overlap ‑oireyhtymän (ilmenee näistä joko kahden tai kaikkien kolmen sairauden limittymisenä) tapauksia, jotka ovat joskus johtaneet kuolemaan, on raportoitu nivolumabihoidossa ja nivolumabin ja muiden lääkeaineiden yhdistelmähoidossa (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Taulukossa 20 on esitetty nivolumabin ja brentuksimabivedotiinin yhdistelmähoitoa saaneiden potilaiden (n = 72) ja nivolumabin ja AVD:n yhdistelmähoitoa saaneiden potilaiden (n = 490) raportoidut haittavaikutukset. Haittavaikutukset on esitetty elinjärjestelmäluokan ja yleisyyden mukaan. Yleisyydet määritellään seuraavasti: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100).
Taulukko 20: Nivolumabin ja muiden lääkeaineiden yhdistelmähoitoon liittyvät haittavaikutukset
| Yhdistelmähoito brentuksimabivedotiinin kanssa | Yhdistelmähoito AVD:n kanssa |
Infektiot | ||
|---|---|---|
Hyvin yleinen | ylähengitystieinfektio | |
Yleinen |
| ylähengitystieinfektio, keuhkokuume |
Veri ja imukudos | ||
Hyvin yleinen |
| neutropeniaa, anemia, leukopeniab, lymfopeniac, trombosytopenia |
Yleinen | neutropenia, anemia, lymfopenia, kuumeinen neutropenia, leukopenia, trombosytopenia | kuumeinen neutropenia, eosinofilia |
Immuunijärjestelmä | ||
Hyvin yleinen | yliherkkyysda, infuusioreaktio |
|
Yleinen |
| infuusioreaktio, yliherkkyysd |
Melko harvinainen |
| sarkoidoosi |
Umpieritys | ||
Yleinen | kilpirauhasen liikatoiminta, kilpirauhasen vajaatoiminta | kilpirauhasen vajaatoiminta, kilpirauhasen liikatoiminta |
Melko harvinainen |
| kilpirauhastulehdus, lisämunuaisten vajaatoiminta, lisäkilpirauhasten vajaatoiminta |
Aineenvaihdunta ja ravitsemus | ||
Hyvin yleinen | ruokahalun väheneminen, hyperglykemia, hypokalemia, hypernatremia | hyperglykemia, hypoalbuminemia, ruokahalun väheneminen, hypokalsemia, hyponatremia, hypokalemia |
Yleinen |
| hypoglykemia, kuivumistila, hypofosfatemia, hyperkalemia, hyperkalsemia, hypomagnesemia, hypernatremia, hypermagnesemia |
Melko harvinainen |
| tuumorilyysioireyhtymä, diabetes mellitus |
Hermosto | ||
Hyvin yleinen | päänsärky | perifeerinen neuropatia, päänsärky, heitehuimaus, dysgeusia |
Yleinen | perifeerinen neuropatia, heitehuimaus |
|
Melko harvinainen |
| aivotulehdus |
Sydän | ||
Hyvin yleinen |
| sydämen tiheälyöntisyys |
Yleinen |
| perikardiaalinen effuusio |
Melko harvinainen |
| eteisvärinä, perikardiitti |
Silmät | ||
Yleinen |
| sumentunut näkö, kuivat silmät |
Melko harvinainen |
| uveiitti |
Verisuonisto | ||
Hyvin yleinen |
| kohonnut verenpaine |
Yleinen | tromboosi | tromboosi |
Hengityselimet, rintakehä ja välikarsina | ||
Hyvin yleinen | yskä | yskä, hengenahdistus |
Yleinen | keuhkotulehdus, hengenahdistus | keuhkotulehdus, keuhkopussin nestekertymä |
Ruoansulatuselimistö | ||
Hyvin yleinen | ripuli, oksentelu, pahoinvointi, vatsakipu, ummetus | pahoinvointi, ummetus, oksentelu, suutulehdus, ripuli, vatsakipu, dyspepsia |
Yleinen | suutulehdus | suun kuivuminen, gastriitti, koliitti |
Melko harvinainen |
| haimatulehdus |
Maksa ja sappi | ||
Melko harvinainen |
| immuunivälitteinen maksatulehdus |
Iho ja ihonalainen kudos | ||
Hyvin yleinen | ihottumae | ihottumae, alopesia, kutina |
Yleinen | alopesia, kutina, kuiva iho, eryteema, nokkosihottuma | kuiva iho, ihon hyperpigmentaatio |
Melko harvinainen |
| käsi-jalkaoireyhtymä |
Luusto, lihakset ja sidekudos | ||
Hyvin yleinen | muskuloskeletaalinen kipuf | muskuloskeletaalinen kipuf |
Yleinen |
| lihaskouristukset, lihasheikkous |
Melko harvinainen |
| niveltulehdus, myosiitti |
Munuaiset ja virtsatiet | ||
Yleinen | munuaisten vajaatoiminta | |
Melko harvinainen |
| munuaisten vajaatoiminta |
Yleisoireet ja antopaikassa todettavat haitat | ||
Hyvin yleinen | kuume, väsymys | väsymys, kuume, kipu, rintakipu |
Yleinen | turvotus | turvotus, vilunväristykset |
Tutkimukset | ||
Hyvin yleinen | suurentuneet transaminaasiarvotg | suurentuneet transaminaasiarvotg, lisääntynyt veren alkalinen fosfataasi |
Yleinen | lisääntynyt kokonaisbilirubiini, lisääntynyt amylaasi, lisääntynyt lipaasi | lisääntynyt veren kreatiniini, painon lasku, lisääntynyt veren bilirubiini |
Melko harvinainen |
| lisääntynyt gammaglutamyylitransferaasi, lisääntynyt lipaasi, lisääntynyt amylaasi |
Taulukossa 20 esitetyt yleisyysluokat eivät välttämättä liity täysin pelkästään nivolumabiin tai nivolumabin yhdistelmähoitoon muiden lääkeaineiden kanssa, vaan niihin voivat vaikuttaa myös perussairaudet tai muut yhdistelmänä käytetyt lääkevalmisteet.
a Neutropenia sisältää neutropenian ja neutrofiilien määrän vähenemisen.
b Leukopenia sisältää leukopenian ja valkosolujen määrän vähenemisen.
c Lymfopenia sisältää lymfopenian ja lymfosyyttien määrän vähenemisen.
d Yliherkkyys on yhdistelmätermi, joka sisältää anafylaktisen reaktion.
e Ihottuma on yhdistelmätermi, joka sisältää makulopapulaarisen ihottuman, punoittavan ihottuman, kutiavan ihottuman, näppyläisen ihottuman, aknea muistuttavan ihottuman ja allergisen ihottuman.
f Muskuloskeletaalinen kipu on yhdistelmätermi, joka kattaa selkäkivun, luukivun, muskuloskeletaalisen rintakivun, lihaskivun, niskakivun, raajakivun, selkärankakivun, nivelsäryn ja leukakivun.
g Suurentuneet transaminaasiarvot on yhdistelmätermi, joka sisältää suurentuneet alaniiniaminotransferaasiarvot ja suurentuneet aspartaattiaminotransferaasiarvot.
Valikoitujen haittavaikutusten kuvaus
Nivolumabihoitoon ja nivolumabi-yhdistelmähoitoon muiden lääkeaineiden kanssa liittyy immuunivälitteisiä haittavaikutuksia. Immuunivälitteiset haittavaikutukset hävisivät useimmissa tapauksissa oikeanlaisella lääkehoidolla. Hoito piti yleensä lopettaa pysyvästi nivolumabin ja muiden lääkeaineiden yhdistelmähoitoa saaneiden kohdalla useammin kuin pelkkää nivolumabi-monoterapiaa saaneiden kohdalla. Taulukossa 21 esitetään niiden potilaiden prosenttiosuus, joilla oli immuunivälitteisiä haittavaikutuksia ja joiden yhdistelmähoito lopetettiin pysyvästi. Lisäksi sellaisten potilaiden osalta, joilla oli haittatapahtuma, taulukossa 21 esitetään niiden potilaiden prosenttiosuus hoito-ohjelman mukaan jaoteltuna, jotka tarvitsivat suuriannoksista kortikosteroidihoitoa (joka vastasi vähintään 40 mg:aa prednisonia vuorokaudessa). Kohdassa Varoitukset ja käyttöön liittyvät varotoimet on haittavaikutusten hoito-ohjeet.
Taulukko 21: Immuunivälitteiset haittavaikutukset, joiden takia hoito lopetettiin pysyvästi tai jotka vaativat suuriannoksista kortikosteroidihoitoa (nivolumabi-monoterapiaa, nivolumabia yhdistelmähoitona ipilimumabin kanssa [kemoterapian kanssa tai ilman], nivolumabia yhdistelmähoitona kemoterapian kanssa tai nivolumabia yhdistelmähoitona kabotsantinibin kanssa)
| Nivolumabi-monoterapia % | Nivolumabi–ipilimumabi-yhdistelmähoito (kemoterapian kanssa tai ilman) % | Nivolumabi–kemoterapia-yhdistelmähoito % | Nivolumabi–kabotsantinibi-yhdistelmähoito % |
|---|---|---|---|---|
Immuunivälitteiset haittavaikutukset, joiden takia hoito lopetettiin pysyvästi | ||||
Keuhkotulehdus | 1,4 | 2,1 | 2,0 | 2,5 |
Koliitti | 1,2 | 6 | 1,8 | 2,5 |
Hepatiitti | 1,1 | 5 | 0,7 | 4,1 |
Munuaistulehdus ja munuaisten toimintahäiriö | 0,3 | 1,1 | 3,1 | 0,6 |
Umpierityshäiriöt | 0,5 | 2,2 | 0,6 | 1,3 |
Iho | 0,8 | 1,0 | 0,9 | 2,2 |
Yliherkkyys-/ infuusioreaktio | 0,1 | 0,3 | 1,7 | 0 |
Immuunivälitteiset haittavaikutukset, joiden takia potilas tarvitsi suuriannoksista kortikosteroidihoitoaa,b | ||||
Keuhkotulehdus | 65 | 59 | 59 | 56 |
Koliitti | 14 | 32 | 9 | 8 |
Hepatiitti | 21 | 39 | 7 | 23 |
Munuaistulehdus ja munuaisten toimintahäiriö | 22 | 27 | 9 | 9 |
Umpierityshäiriöt | 5 | 18 | 4,3 | 4,2 |
Iho | 3,3 | 8 | 6 | 8 |
Yliherkkyys-/ infuusioreaktio | 18 | 18 | 22 | 0 |
a Hoito vastasi vähintään 40 mg:aa prednisonia vuorokaudessa.
b Yleisyys perustuu niiden potilaiden lukumäärään, jotka saivat immuunivälitteisen haittavaikutuksen.
Immuunivälitteinen keuhkotulehdus
Keuhkotulehduksen ilmaantuvuus (interstitiaalinen keuhkosairaus ja keuhkoinfiltraatio mukaan luettuna) potilailla, jotka saivat nivolumabia monoterapiana, oli 3,3 % (155/4646). Suurin osa oli vaikeusastetta 1, jota ilmoitettiin 0,9 prosentilla (42/4646) potilaista, ja vaikeusastetta 2, jota ilmoitettiin 1,7 prosentilla (77/4646) potilaista. Asteen 3 keuhkotulehduksia oli 0,7 prosentilla (33/4646) potilaista, ja asteen 4 keuhkotulehduksia oli < 0,1 prosentilla (1/4646) potilaista. Kuuden potilaan (0,1 %) tapaus johti kuolemaan. Oireiden ilmenemisajan mediaani oli 15,1 viikkoa (vaihteluväli 0,7–85,1). Haittavaikutus korjaantui 107 potilaalla (69,0 %). Korjaantumisajan mediaani oli 6,7 viikkoa (vaihteluväli 0,1+–109,1+); + tarkoittaa sensuroitunutta havaintoa.
Nivolumabin ja ipilimumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla keuhkotulehduksen, mukaan lukien interstitiaalinen keuhkosairaus, ilmaantuvuus oli 6,0 % (157/2626). Potilaista 3,0 prosentilla (78/2626) ilmoitettiin vaikeusasteen 2 tapauksia, 1,0 prosentilla (27/2626) vaikeusasteen 3 tapauksia ja 0,3 prosentilla (8/2626) vaikeusasteen 4 tapauksia. Neljän potilaan (0,2 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 2,7 kuukautta (vaihteluväli: 0,1–56,8). Haittavaikutus korjaantui 129 potilaalla (82,2 %). Korjaantumisajan mediaani oli 6,1 viikkoa (vaihteluväli: 0,1+–149,3+).
Nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla potilailla pneumoniitin, mukaan lukien interstitiaalinen keuhkosairaus, ilmaantuvuus oli 4,4 % (80/1800). Potilaista 2,2 prosentilla (40/1800) ilmoitettiin vaikeusasteen 2 tapauksia, 0,9 prosentilla (17/1800) vaikeusasteen 3 tapauksia ja 0,2 prosentilla (3/1800) vaikeusasteen 4 tapauksia. Kolmen potilaan (0,2 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 24,6 viikkoa (vaihteluväli: 0,6–96,9). Haittavaikutus korjaantui 58 potilaalla (72,5 %). Korjaantumisajan mediaani oli 10,4 viikkoa (vaihteluväli: 0,3+–171,4+).
Nivolumabia ja kabotsantinibia yhdistelmähoitona saaneilla potilailla pneumoniitin, mukaan lukien interstitiaalinen keuhkosairaus, ilmaantuvuus oli 5,6 % (18/320). Potilaista 1,9 prosentilla (6/320) ilmoitettiin asteen 2 tapauksia ja 1,6 prosentilla (5/320) asteen 3 tapauksia. Haittavaikutusten ilmenemisajan mediaani oli 26,9 viikkoa (vaihteluväli 12,3–74,3 viikkoa). Haittavaikutus korjaantui 14 potilaalla (77,8 %). Korjaantumisajan mediaani oli 7,5 viikkoa (vaihteluväli 2,1–60,7+ viikkoa).
Immuunivälitteinen koliitti
Nivolumabia monoterapiana saaneilla potilailla ripulin, koliitin tai ulostamistiheyden lisääntymisen ilmaantuvuus oli 15,4 % (716/4646). Suurin osa tapauksista oli vaikeusastetta 1, joita ilmoitettiin 9,9 prosentilla potilaista (462/4646), ja vaikeusastetta 2, joita ilmoitettiin 4,0 prosentilla potilaista (186/4646). Asteen 3 tapauksia oli 1,4 prosentilla potilaista (67/4646) ja asteen 4 tapauksia < 0,1 prosentilla potilaista (1/4646). Haittavaikutusten ilmenemisajan mediaani oli 8,3 viikkoa (vaihteluväli 0,1–115,6). Haittavaikutus parani 639 potilaalla (90,3 %). Korjaantumisajan mediaani oli 2,9 viikkoa (vaihteluväli 0,1–124,4+).
Nivolumabia ja ipilimumabia yhdistelmähoitona (kemoterapian kanssa tai ilman) saaneilla potilailla ripulin tai koliitin ilmaantuvuus oli 26,0 % (682/2626). Potilaista 8,1 prosentilla (212/2626) ilmoitettiin vaikeusasteen 2 tapauksia, 6,4 prosentilla (167/2626) vaikeusasteen 3 tapauksia ja 0,2 prosentilla (4/2626) vaikeusasteen 4 tapauksia. Kahden potilaan (< 0,1 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 1,4 kuukautta (vaihteluväli: 0,0–48,9). Haittavaikutus korjaantui 618 potilaalla (91 %). Korjaantumisajan mediaani oli 2,9 viikkoa (vaihteluväli: 0,1–170,0+). Nivolumabin (1 mg/kg) ja ipilimumabin (3 mg/kg) yhdistelmähoitoa melanooman hoitoon saaneilla potilailla ripulin tai koliitin ilmaantuvuus oli 46,7 %, johon sisältyi asteen 2 tapauksia (13,6 %), asteen 3 tapauksia (15,8 %) ja asteen 4 tapauksia (0,4 %).
Nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla potilailla ripulin tai koliitin ilmaantuvuus oli 22,5 % (405/1800). Potilaista 7,2 prosentilla (130/1800) ilmoitettiin vaikeusasteen 2 tapauksia, 3,1 prosentilla (56/1800) vaikeusasteen 3 tapauksia ja 0,3 prosentilla (6/1800) vaikeusasteen 4 tapauksia. Yhden potilaan (< 0,1 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 4,4 viikkoa (vaihteluväli: 0,1–93,6). Haittavaikutus korjaantui 357 potilaalla (88,6 %). Korjaantumisajan mediaani oli 1,6 viikkoa (vaihteluväli: 0,1–212,3+).
Nivolumabia ja kabotsantinibia yhdistelmähoitona saaneilla potilailla ripulin, koliitin, ulostustiheyden lisääntymisen tai enteriitin ilmaantuvuus oli 59,1 prosenttia (189/320). Potilaista 25,6 prosentilla ilmoitettiin vaikeusasteen 2 tapauksia (82/320) ja 6,3 prosentilla (20/320) vaikeusasteen 3 tapauksia. Vaikeusasteen 4 tapauksia ilmoitettiin 0,6 prosentilla (2/320) potilaista. Haittavaikutusten ilmenemisajan mediaani oli 12,9 viikkoa (vaihteluväli: 0,3–110,9 viikkoa). Haittavaikutus korjaantui 143 potilaalla (76,1 %). Korjaantumisajan mediaani oli 12,9 viikkoa (vaihteluväli: 0,1–139,7+ viikkoa).
Immuunivälitteinen maksatulehdus
Nivolumabia monoterapiana saaneista potilaista maksan toimintakokeista saatiin poikkeavia tuloksia 8,0 prosentilta (371/4646). Suurin osa tapauksista oli vaikeusastetta 1, joita ilmoitettiin 4,3 prosentilla potilaista (200/4646), ja vaikeusastetta 2, joita ilmoitettiin 1,8 prosentilla potilaista (82/4646). Asteen 3 muutoksia oli 1,6 prosentilla potilaista (74/4646), ja asteen 4 muutoksia oli 0,3 prosentilla (15/4646) potilaista. Muutoksen ilmenemisajan mediaani oli 10,6 viikkoa (vaihteluväli 0,1–132,0). Haittavaikutus parani 298 potilaalla (81,4 %). Korjaantumisajan mediaani oli 6,1 viikkoa (vaihteluväli 0,1–126,4+).
Nivolumabia ja ipilimumabia yhdistelmähoitona (kemoterapian kanssa tai ilman) saaneilla potilailla maksan toimintakokeiden poikkeavien tulosten ilmaantuvuus oli 21,2 % (556/2626). Potilaista 5,0 prosentilla (132/2626) ilmoitettiin vaikeusasteen 2 tapauksia, 8,3 prosentilla (218/2626) vaikeusasteen 3 tapauksia ja 1,3 prosentilla (34/2626) vaikeusasteen 4 tapauksia. Seitsemän potilaan (0,3 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 1,5 kuukautta (vaihteluväli: 0,0–36,6). Haittavaikutus korjaantui 482 potilaalla (87,0 %). Korjaantumisajan mediaani oli 5,9 viikkoa (vaihteluväli: 0,1–175,9+). Nivolumabin (1 mg/kg) ja ipilimumabin (3 mg/kg) yhdistelmähoitoa melanooman hoitoon saaneilla potilailla maksan toimintakokeiden poikkeavien tulosten ilmaantuvuus oli 30,1 %. Näistä asteen 2 tapauksia oli 6,9 %, asteen 3 tapauksia 15,8 % ja asteen 4 tapauksia 1,8 %. Nivolumabin (1 mg/kg) ja ipilimumabin (3 mg/kg) yhdistelmähoitoa hepatosellulaarisen karsinooman hoitoon saaneilla potilailla maksan toimintakokeiden poikkeavien tulosten ilmaantuvuus oli 34,3 %. Näistä asteen 2 tapauksia oli 8,4 %, asteen 3 tapauksia 14,2 % ja asteen 4 tapauksia 2,7 %.
Nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla potilailla maksan toimintakokeiden poikkeavien tulosten ilmaantuvuus oli 18 % (322/1800). Potilaista 5,1 prosentilla (92/1800) ilmoitettiin vaikeusasteen 2 tapauksia, 2,6 prosentilla (47/1800) vaikeusasteen 3 tapauksia ja < 0,1 prosentilla (1/1800) vaikeusasteen 4 tapauksia. Haittavaikutusten ilmenemisajan mediaani oli 7,0 viikkoa (vaihteluväli: 0,1–99,0). Haittavaikutus korjaantui 258 potilaalla (81,1 %). Korjaantumisajan mediaani oli 7,4 viikkoa (vaihteluväli: 0,4–240,0+).
Nivolumabin ja AVD:n yhdistelmähoitoa saaneilla potilailla maksan toimintakokeiden poikkeavien tulosten ilmaantuvuus oli 41,2 % (202/490). Potilaista 6,1 prosentilla (30/490) ilmoitettiin vaikeusasteen 2 tapauksia, 4,7 prosentilla (23/490) vaikeusasteen 3 tapauksia ja 0,6 prosentilla (3/490) vaikeusasteen 4 tapauksia.
Nivolumabia ja kabotsantinibia yhdistelmähoitona saaneilla potilailla maksan toimintakokeiden poikkeavien tulosten ilmaantuvuus oli 41,6 % (133/320). Potilaista 14,7 prosentilla (47/320) ilmoitettiin asteen 2 tapauksia, 10,3 prosentilla (33/320) asteen 3 tapauksia ja 0,6 prosentilla (2/320) asteen 4 tapauksia. Haittavaikutusten ilmenemisajan mediaani oli 8,3 viikkoa (vaihteluväli: 0,1–107,9 viikkoa). Haittavaikutus korjaantui 101 potilaalla (75,9 %). Korjaantumisajan mediaani oli 9,6 viikkoa (vaihteluväli: 0,1–89,3+ viikkoa).
Immuunivälitteinen munuaistulehdus ja munuaisten toimintahäiriö
Nivolumabia monoterapiana saaneilla potilailla munuaistulehduksen tai munuaisten toimintahäiriön ilmaantuvuus oli 2,6 % (121/4646). Suurin osa tapauksista oli vaikeusastetta 1, joita ilmoitettiin 1,5 prosentilla potilaista (69/4646), ja vaikeusastetta 2, joita ilmoitettiin 0,7 prosentilla potilaista (32/4646). Asteen 3 munuaistulehdus tai toimintahäiriö oli 0,4 prosentilla potilaista (18/4646), ja asteen 4 munuaistulehdus tai toimintahäiriö oli < 0,1 prosentilla (2/4646) potilaista. Haittavaikutusten ilmenemisajan mediaani oli 12,1 viikkoa (vaihteluväli 0,1–79,1). Haittavaikutus korjaantui 80 potilaalla (69,0 %). Korjaantumisajan mediaani oli 8,0 viikkoa (vaihteluväli 0,3–79,1+).
Nivolumabin ja ipilimumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla munuaistulehduksen tai munuaisten toimintahäiriön ilmaantuvuus oli 5,4 % (141/2626). Potilaista 2,0 prosentilla (52/2626) ilmoitettiin vaikeusasteen 2 tapauksia, 0,8 prosentilla (21/2626) vaikeusasteen 3 tapauksia ja 0,4 prosentilla (11/2626) vaikeusasteen 4 tapauksia. Kahden potilaan (< 0,1 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 2,6 kuukautta (vaihteluväli: 0,0–34,8). Haittavaikutus korjaantui 110 potilaalla (78,0 %). Korjaantumisajan mediaani oli 5,9 viikkoa (vaihteluväli: 0,1–172,1+).
Nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla potilailla munuaistulehduksen ja munuaisten toimintahäiriön ilmaantuvuus oli 10,9 % (196/1800). Potilaista 3,7 prosentilla (66/1800) ilmoitettiin vaikeusasteen 2 tapauksia, 1,4 prosentilla (25/1800) vaikeusasteen 3 tapauksia ja 0,2 prosentilla (3/1800) vaikeusasteen 4 tapauksia. Kahden potilaan (0,1 %) tapaus johti kuolemaan. Haittavaikutusten ilmenemisajan mediaani oli 6,7 viikkoa (vaihteluväli: 0,1–60,7). Haittavaikutus korjaantui 133 potilaalla (67,9 %). Korjaantumisajan mediaani oli 9,1 viikkoa (vaihteluväli 0,1–226,0+).
Nivolumabia ja kabotsantinibia yhdistelmähoitona saaneilla potilailla nefriitin, immuunivälitteisen nefriitin, munuaisten vajaatoiminnan, akuutin munuaisvaurion, kohonneen veren kreatiniiniarvon tai kohonneen veren ureapitoisuuden ilmaantuvuus oli 10,0 prosenttia (32/320). Potilaista 3,4 prosentilla (11/320) ilmoitettiin asteen 2 tapauksia, ja potilaista 1,3 % prosentilla (4/320) asteen 3 tapauksia. Haittavaikutusten ilmenemisajan mediaani oli 14,2 viikkoa (vaihteluväli: 2,1–87,1 viikkoa). Haittavaikutus korjaantui 18 potilaalla (58,1 %). Korjaantumisajan mediaani oli 10,1 viikkoa (vaihteluväli: 0,6–90,9+ viikkoa).
Immuunivälitteiset umpierityshäiriöt
Nivolumabia monoterapiana saaneilla potilailla kilpirauhasen häiriöiden, kuten vajaa- ja liikatoiminnan, ilmaantuvuus oli 13,0 % (603/4646). Suurin osa tapauksista oli vaikeusastetta 1, joita ilmoitettiin 6,6 prosentilla potilaista (305/4646), ja vaikeusastetta 2, joita ilmoitettiin 6,2 prosentilla potilaista (290/4646). Asteen 3 kilpirauhasen häiriö oli 0,2 prosentilla (8/4646) potilaista. Hypofysiittiä (3 tapausta astetta 1, 7 tapausta astetta 2, 9 tapausta astetta 3 ja 1 tapaus astetta 4), aivolisäkkeen etulohkon vajaatoimintaa (6 tapausta astetta 2 ja 1 tapaus astetta 3), lisämunuaisten vajaatoimintaa (mukaan lukien lisämunuaiskuoren sekundaarista vajaatoimintaa, akuuttia lisämunuaiskuoren vajaatoimintaa ja veren kortikotropiinin vähenemistä) (2 tapausta astetta 1, 23 tapausta astetta 2, 11 tapausta astetta 3), diabetesta (mukaan lukien tyypin 1 diabetesta ja diabeettista ketoasidoosia) (1 tapaus astetta 1, 3 tapausta astetta 2, 8 tapausta astetta 3 ja 2 tapausta astetta 4) ilmoitettiin. Näiden umpierityshäiriöiden ilmenemisajan mediaani oli 11,1 viikkoa (vaihteluväli 0,1–126,7). Haittavaikutus korjaantui 323 potilaalla (48,7 %). Korjaantumisajan mediaani oli 48,6 viikkoa (vaihteluväli: 0,4–204,4+).
Nivolumabin ja ipilimumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla kilpirauhasen toimintahäiriöiden ilmaantuvuus oli 23,2 % (608/2626). Potilaista 12,7 prosentilla (333/2626) ilmoitettiin vaikeusasteen 2 kilpirauhasen toimintahäiriöitä ja 1,0 prosentilla (27/2626) vaikeusasteen 3 kilpirauhasen toimintahäiriöitä. Potilaista 1,9 prosentilla (49/2626) ilmoitettiin vaikeusasteen 2 hypofysiittitapauksia (mukaan lukien lymfosyyttista hypofysiittia) ja 1,5 prosentilla (40/2626) vaikeusasteen 3 hypofysiittitapauksia. Vaikeusasteen 2 hypopituitarismia ilmeni 0,6 prosentilla potilaista (16/2626) ja vaikeusasteen 3 hypopituitarismia 0,5 prosentilla (13/2626) potilaista. Vaikeusasteen 2 lisämunuaisten vajaatoimintaa (mukaan lukien lisämunuaiskuoren sekundaarista vajaatoimintaa, akuuttia lisämunuaiskuoren vajaatoimintaa, pienentynyttä veren kortikotropiinipitoisuutta ja immuunivälitteistä lisämunuaisen vajaatoimintaa) ilmoitettiin 2,7 prosentilla (72/2626) potilaista, vaikeusasteen 3 vajaatoimintaa ilmoitettiin 1,6 prosentilla (43/2626) ja vaikeusasteen 4 vajaatoimintaa ilmoitettiin 0,2 prosentilla (4/2626) potilaista. Asteen 1 diabetes mellitusta (mukaan lukien tyypin 1 diabetesta ja diabeettista ketoasidoosia) esiintyi < 0,1 prosentilla (1/2626), asteen 2 diabetes mellitusta 0,3 prosentilla (8/2626), asteen 3 diabetes mellitusta 0,3 prosentilla (7/2626) ja asteen 4 diabetes mellitusta 0,2 prosentilla (6/2626) potilaista. Näiden umpierityshäiriöiden ilmenemisajan mediaani oli 2,1 kuukautta (vaihteluväli: 0,0–28,1). Haittavaikutus korjaantui 297 potilaalla (40,0 %). Korjaantumisajan vaihteluväli oli 0,3–257,1+ viikkoa.
Nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla potilailla kilpirauhasen toimintahäiriöiden ilmaantuvuus oli 12,8 % (230/1800). Potilaista 6,3 prosentilla (114/1800) ilmoitettiin vaikeusasteen 2 ja 0,1 prosentilla (2/1800) vaikeusasteen 3 kilpirauhasen toimintahäiriöitä. Potilaista 0,1 prosentilla (2/1800) ilmoitettiin vaikeusasteen 3 hypofysiittiä. Potilaista 0,2 prosentilla (4/1800) ilmoitettiin vaikeusasteen 2 hypopituitarismia ja vaikeusasteen 3 hypopituitarismia. Potilaista 0,6 prosentilla (11/1800) ilmoitettiin vaikeusasteen 2 lisämunuaisten vajaatoimintaa, 0,2 prosentilla (3/1800) vaikeusasteen 3 tapauksia ja < 0,1 prosentilla (1/1800) vaikeusasteen 4 tapauksia. Lisämunuaisten vajaatoiminta johti yhden potilaan (< 0,1 %) kuolemaan. Diabetes mellitusta, mukaan lukien tyypin 1 diabetes mellitusta ja fulminanttia tyypin 1 diabetes mellitusta (4 tapausta astetta 2, 2 tapausta astetta 3 ja 1 tapaus astetta 4) sekä diabeettista ketoasidoosia (1 tapaus astetta 2 ja 1 tapaus astetta 4) ilmoitettiin. Näiden umpierityshäiriöiden ilmenemisajan mediaani oli 15,3 viikkoa (vaihteluväli: 1,1–124,3). Haittavaikutus korjaantui 101 potilaalla (40,1 %). Korjaantumisaika vaihteli 0,3+ viikosta 233,6+ viikkoon.
Nivolumabia ja kabotsantinibia yhdistelmähoitona saaneilla potilailla kilpirauhasen toimintahäiriöiden ilmaantuvuus oli 43,1 prosenttia (138/320). Potilaista 23,1 prosentilla (74/320) ilmoitettiin vaikeusasteen 2 kilpirauhasen toimintahäiriöitä ja 0,9 prosentilla (3/320) vaikeusasteen 3 kilpirauhasen toimintahäiriöitä. Potilaista 0,6 prosentilla (2/320) ilmoitettiin hypofysiittiä; kaikki tapaukset olivat vaikeusasteen 2 tapauksia. Potilaista 4,7 prosentilla (15/320) ilmoitettiin lisämunuaisten vajaatoimintaa (mukaan lukien lisämunuaiskuoren sekundaarista vajaatoimintaa). Potilaista 2,2 prosentilla (7/320) ilmoitettiin vaikeusasteen 2 lisämunuaisten vajaatoimintaa ja 1,9 prosentilla (6/320) vaikeusasteen 3 lisämunuaisten vajaatoimintaa. Umpierityshäiriöiden ilmenemisajan mediaani oli 12,3 viikkoa (vaihteluväli: 2,0–89,7 viikkoa). Haittavaikutus korjaantui 50 potilaalla (35,2 %). Korjaantumisajan vaihteluväli oli 0,9–132,0+ viikkoa.
Immuunivälitteiset ihohaitat
Nivolumabia monoterapiana saaneilla potilailla ihottuman ilmaantuvuus oli 30,0 % (1396/4646). Suurin osa tapauksista oli vaikeusastetta 1, joita ilmoitettiin 22,8 prosentilla potilaista (1060/4646). Asteen 2 ihottumaa oli 5,9 prosentilla (274/4646) potilaista ja asteen 3 ihottumaa oli 1,3 prosentilla (62/4646) potilaista. Haittavaikutusten ilmenemisajan mediaani oli 6,7 viikkoa (vaihteluväli 0,1–121,1). Haittavaikutus korjaantui 896 potilaalla (64,6 %). Korjaantumisajan mediaani oli 20,1 viikkoa (vaihteluväli 0,1–192,7+).
Nivolumabin ja ipilimumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla ihottuman ilmaantuvuus oli 46,1 % (1210/2626). Potilaista 14,3 prosentilla (375/2626) ilmoitettiin vaikeusasteen 2 tapauksia, 4,6 prosentilla (120/2626) vaikeusasteen 3 tapauksia ja 0,1 prosentilla (3/2626) vaikeusasteen 4 tapauksia. Haittavaikutusten ilmenemisajan mediaani oli 0,7 kuukautta (vaihteluväli: 0,0–33,8). Haittavaikutus korjaantui 843 potilaalla (70 %). Korjaantumisajan mediaani oli 12,1 viikkoa (vaihteluväli: 0,1–268,7+). Nivolumabin (1 mg/kg) ja ipilimumabin (3 mg/kg) yhdistelmähoitoa melanooman hoitoon saaneilla potilailla ihottuman ilmaantuvuus oli 65,2 %, mukaan lukien vaikeusasteen 2 tapaukset (20,3 %) ja vaikeusasteen 3 tapaukset (7,8 %).
Nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla potilailla ihottuman ilmaantuvuus oli 25,4 % (457/1800). Potilaista 6,2 prosentilla (111/1800) ilmoitettiin vaikeusasteen 2 tapauksia ja 2,3 prosentilla (42/1800) vaikeusasteen 3 tapauksia. Haittavaikutusten ilmenemisajan mediaani oli 6,4 viikkoa (vaihteluväli: 0,1–97,4). Haittavaikutus korjaantui 320 potilaalla (70,2 %). Korjaantumisajan mediaani oli 12,1 viikkoa (vaihteluväli 0,1–258,7+).
Nivolumabia ja kabotsantinibia yhdistelmähoitona saaneilla potilailla ihottuman ilmaantuvuus oli 62,8 prosenttia (201/320). Potilaista 23,1 prosentilla (74/320) ilmoitettiin vaikeusasteen 2 tapauksia ja 10,6 prosentilla (34/320) vaikeusasteen 3 tapauksia. Haittavaikutusten ilmenemisajan mediaani oli 6,14 viikkoa (vaihteluväli: 0,1–104,4 viikkoa). Haittavaikutus korjaantui 137 potilaalla (68,2 %). Korjaantumisajan mediaani oli 18,1 viikkoa (vaihteluväli: 0,1–130,6+ viikkoa).
SJS:aa ja TEN:iä on havaittu harvoin, ja joissain tapauksissa tila on johtanut kuolemaan (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Infuusioreaktiot
Nivolumabia monoterapiana saaneilla potilailla yliherkkyys-/infuusioreaktioiden ilmaantuvuus oli 4,0 % (188/4646), joista yhdeksällä se oli astetta 3 ja kolmella astetta 4.
Nivolumabin ja ipilimumabin yhdistelmähoitoa (kemoterapian kanssa tai ilman) saaneilla potilailla yliherkkyys-/infuusioreaktioiden ilmaantuvuus oli 4,5 % (118/2626). Potilaista 1,9 prosentilla (49/2626) ilmoitettiin vaikeusasteen 1 tapauksia; 2,4 prosentilla (62/2626) vaikeusasteen 2 tapauksia, 0,2 prosentilla (6/2626) vaikeusasteen 3 tapauksia ja < 0,1 prosentilla (1/2626) vaikeusasteen 4 tapauksia. Nivolumabia (3 mg/kg) ja ipilimumabia (1 mg/kg) yhdistelmähoitona keuhkopussin pahanlaatuisen mesoteliooman (MPM) hoitoon saaneilla potilailla yliherkkyys-/infuusioreaktioiden ilmaantuvuus oli 12 %.
Nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla potilailla yliherkkyys-/infuusioreaktioiden ilmaantuvuus oli 8,2 % (148/1800). Potilaista 4,6 prosentilla (83/1800) ilmoitettiin vaikeusasteen 2 tapauksia, 1,1 prosentilla (20/1800) vaikeusasteen 3 tapauksia ja 0,2 prosentilla (3/1800) vaikeusasteen 4 tapauksia.
Nivolumabia ja kabotsantinibia yhdistelmähoitona saaneilla potilailla yliherkkyys-/infuusioreaktioiden ilmaantuvuus oli 2,5 prosenttia (8/320) Kaikki tapaukset olivat vaikeusastetta 1 tai 2. Vaikeusasteen 2 tapauksia ilmoitettiin 0,3 prosentilla (1/320) potilaista.
Allogeenisen hematopoieettisten kantasolujen siirron komplikaatiot klassista Hodgkinin lymfoomaa sairastavilla potilailla
Nopeasti alkavaa käänteishyljintää on ilmoitettu esiintyneen potilailla, joita hoidettiin nivolumabilla ennen allogeenista hematopoieettisten kantasolujen siirtoa sekä potilailla, joita hoidettiin nivolumabilla kantasolusiirron jälkeen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Kahdessa klassisen Hodgkinin lymfooman tutkimuksessa arvioitiin hoitoa autologisen kantasolusiirron jälkeen aikuispotilailla, joilla oli uusiutunut tai refraktaarinen cHL ja jotka saivat allogeenisen hematopoieettisten kantasolujen siirron nivolumabi-monoterapian lopettamisen jälkeen. Arvioiduista 62 potilaasta seitsemällätoista (27,4 %:lla) raportoitiin asteen 3 tai 4 akuuttia käänteishyljintää. Neljällä potilaalla (6 %:lla) raportoitiin ilmenneen hyperakuuttia käänteishyljintää (akuuttia käänteishyljintää, joka ilmeni 14 päivän kuluessa kantasoluinfuusion jälkeen). Kuudella potilaalla (12 %:lla) raportoitiin steroideja vaatinut kuumeinen oireyhtymä, jolle ei ollut tunnistettavaa infektioosia syytä, kuuden ensimmäisen viikon aikana siirron jälkeen. Steroideja annettiin neljälle potilaalle, ja kolme näistä potilaista sai vasteen steroideille. Maksan veno‑okklusiivista tautia ilmeni kahdella potilaalla, joista toinen kuoli käänteishyljintään ja monielinvaurioon. Yhdeksäntoista potilasta 62:sta (30,6 %) kuoli nivolumabihoidon jälkeisen allogeenisen hematopoieettisten kantasolujen siirron komplikaatioihin. 62 potilaan seurannan mediaani oli 38,5 kuukautta (vaihteluväli 0–68 kuukautta) nivolumabihoitoa seuranneen allogeenisen hematopoieettisten kantasolujen siirron jälkeen.
Maksaentsyymiarvojen kohoaminen, kun nivolumabia käytetään yhdessä kabotsantinibin kanssa munuaiskarsinooman hoidossa
Kliinisessä tutkimuksessa, jossa nivolumabia annettiin yhdistelmänä kabotsantinibin kanssa munuaiskarsinoomaa sairastaville potilaille, jotka eivät olleet aiemmin saaneet hoitoa, havaittiin enemmän vaikeusasteiden 3 ja 4 ALAT-arvon kohoamista (10,1 %) ja ASAT-arvon kohoamista (8,2 %) kuin nivolumabi-monoterapiaa saaneilla, edennyttä munuaiskarsinoomaa sairastavilla potilalla. Potilailla, joilla ALAT- tai ASAT-arvot kohosivat vaikeusastelle ≥ 2 (n = 85), haittavaikutusten ilmenemisajan mediaani oli 10,1 viikkoa (vaihteluväli: 2,0–106,6 viikkoa). 26 prosenttia sai kortikosteroidihoitoa, jonka mediaanikesto oli 1,4 viikkoa (vaihteluväli: 0,9–75,3 viikkoa). Haittavaikutus korjaantui 91 prosentilla asteelle 0–1. Korjaantumisajan mediaani oli 2,3 viikkoa (vaihteluväli: 0,4–108,1+ viikkoa). Niillä 45 potilaalla, joiden ALAT- tai ASAT-arvo oli kohonnut asteseen ≥ 2 ja joiden hoito aloitettiin uudelleen joko nivolumabilla (n = 10), kabotsantinibilla (n = 10) tai näiden yhdistelmähoidolla (n = 25), ALAT- tai ASAT-arvo palasi asteelle ≥ 2 kolmella nivolumabia saaneella potilaalla, neljällä kabotsantinibia saaneella potilalla ja kahdeksalla nivolumabin ja kabotsantinibin yhdistelmähoitoa saaneella potilaalla.
Poikkeavat laboratorioarvot
Nivolumabia monoterapiana saaneista potilaista niiden potilaiden osuudet, joilla laboratorioarvo muuttui lähtötasolta asteen 3 tai 4 poikkeavuudeksi, olivat seuraavat: anemia (kaikki astetta 3) 3,4 %, trombosytopenia 0,7 %, leukopenia 0,7 %, lymfopenia 8,7 %, neutropenia 0,9 %, lisääntynyt alkalinen fosfataasi 1,7 %, lisääntynyt ASAT 2,6 %, lisääntynyt ALAT 2,3 %, lisääntynyt kokonaisbilirubiini 0,8 % ja lisääntynyt kreatiniini 0,7 %, hyperglykemia 2,0 %, hypoglykemia 0,7 %, lisääntynyt amylaasi 3,8 %, lisääntynyt lipaasi 6,9 %, hyponatremia 4,7 %, hyperkalemia 1,6 %, hypokalemia 1,3 %, hyperkalsemia 1,1 %, hypermagnesemia 0,6 %, hypomagnesemia 0,4 %, hypokalsemia 0,6 %, hypoalbuminemia 0,6 % ja hypernatremia < 0,1 %.
Nivolumabia ja ipilimumabia yhdistelmähoitona (kemoterapian kanssa tai ilman) saaneista potilaista niiden potilaiden osuudet, joilla laboratorioarvo muuttui lähtötasolta asteen 3 tai 4 poikkeavuudeksi, olivat seuraavat: anemia 4,8 %, trombosytopenia 1,8 %, leukopenia 2,2 %, lymfopenia 6,9 %, neutropenia 3,3 %, lisääntynyt alkalinen fosfataasi 2,7 %, lisääntynyt ASAT 9,8 %, lisääntynyt ALAT 9,3 %, lisääntynyt kokonaisbilirubiini 2,3 %, lisääntynyt kreatiniini 1,8 %, hypoalbuminemia 1,4 %, hyperglykemia 7,1 %, hypoglykemia 0,7 %, lisääntynyt amylaasi 7,8 %, lisääntynyt lipaasi 16,3 %, hypokalsemia 0,8 %, hypernatremia 0,2 %, hyperkalsemia 0,8 %, hyperkalemia 2,0 %, hypermagnesemia 0,8 %, hypomagnesemia 0,4 %, hypokalemia 3,0 % ja hyponatremia 8,7 %.
Nivolumabin (1 mg/kg) ja ipilimumabin (3 mg/kg) yhdistelmähoitoa melanooman hoitoon saaneilla potilailla lisääntynyt ALAT-arvo muuttui suuremmalla osalla lähtötasosta asteeseen 3 tai 4 (15,3 %).
Nivolumabin ja kemoterapian yhdistelmähoitoa saaneista potilaista niiden potilaiden osuudet, joilla laboratorioarvo muuttui lähtötasolta asteen 3 tai 4 poikkeavuudeksi, olivat seuraavat: anemia 14,7 %, trombosytopenia 6,2 %, leukopenia 11,7 %, lymfopenia 13,6 %, neutropenia 26,3 %, lisääntynyt alkalinen fosfataasi 2,0 %, lisääntynyt ASAT 3,3 %, lisääntynyt ALAT 2,6 %, lisääntynyt bilirubiini 1,9 %, lisääntynyt kreatiniini 1,3 %, lisääntynyt amylaasi 4,5 %, lisääntynyt lipaasi 5,2 %, hypernatremia 0,4 %, hyponatremia 8,1 %, hyperkalemia 1,8 %, hypokalemia 5,1 %, hyperkalsemia 0,7 %, hypokalsemia 1,8 %, hypermagnesemia 1,5 %, hypomagnesemia 2,9 %, hyperglykemia 3,7 % ja hypoglykemia 0,6 %.
Nivolumabia ja kabotsantinibia yhdistelmähoitona saaneilla potilailla niiden potilaiden osuudet, joilla laboratorioarvo muuttui lähtötasolta asteen 3 tai 4 poikkeavuudeksi, olivat seuraavat: anemia (kaikki astetta 3) 3,5 %, trombosytopenia 0,3 %, leukopenia 0,3 %, lymfopenia 7,5 %, neutropenia 3,5 %, lisääntynyt alkalinen fosfataasi 3,2 %, lisääntynyt ASAT 8,2 %, lisääntynyt ALAT 10,1 %, lisääntynyt kokonaisbilirubiini 1,3 %, lisääntynyt kreatiniini 1,3 %, lisääntynyt amylaasi 11,9 %, lisääntynyt lipaasi 15,6 %, hyperglykemia 3,5 %, hypoglykemia 0,8 %, hypokalsemia 2,2 %, hyperkalsemia 0,3 %, hyperkalemia 5,4 %, hypermagnesemia 4,2%, hypomagnesemia 1,9 %, hypokalemia 3,2 %, hyponatremia 12,3 % ja hypofosfatemia 21,2 %.
Immunogeenisuus
Niistä 3529 potilaasta, joille annettiin 3 mg/kg tai 240 mg nivolumabia monoterapiana joka toinen viikko ja joilla lääkevasta‑aineet pystyttiin arvioimaan, 328 potilaan tulokset (9,3 %) olivat positiiviset hoidosta johtuvien lääkevasta‑aineiden suhteen. 21 potilaaĺla (0,6 %) testitulos oli positiivinen neutraloivien vasta‑aineiden osalta.
Samanaikainen anto kemoterapian kanssa ei vaikuttanut nivolumabin immunogeenisuuteen. Niistä 1407 potilaasta, joille annettiin nivolumabia 240 mg joka toinen viikko tai 360 mg joka kolmas viikko yhdistelmähoitona kemoterapian kanssa ja joilla lääkevasta-aineet pystyttiin arvioimaan, 7,2 prosentilla tulokset olivat positiiviset hoidosta johtuvien lääkevasta-aineiden suhteen. 0,5 prosentilla testitulos oli positiivinen neutraloivien vasta-aineiden osalta.
Potilaista, jotka saivat nivolumabi–ipilimumabi‑yhdistelmähoitoa ja joilta voitiin arvioida nivolumabin vasta‑aineiden esiintyvyys, nivolumabi 3 mg/kg‑ ja ipilimumabi 1 mg/kg ‑yhdistelmähoitoa joka kolmas viikko saaneista 26,0 prosentille kehittyi nivolumabin vasta‑aineita, nivolumabia 3 mg/kg joka toinen viikko ja ipilimumabia 1 mg/kg joka kuudes viikko saaneista 24,9 prosentille kehittyi nivolumabin vasta‑aineita ja nivolumabi 1 mg/kg‑ ja ipilimumabi 3 mg/kg ‑yhdistelmähoitoa joka kolmas viikko saaneista 37,8 prosentille kehittyi nivolumabin vasta‑aineita. Nivolumabi 3 mg/kg‑ ja ipilimumabi 1 mg/kg ‑yhdistelmähoitoa joka kolmas viikko saaneista 0,8 prosentille kehittyi nivolumabia neutraloivia vasta‑aineita. Nivolumabia 3 mg/kg joka toinen viikko ja ipilimumabia 1 mg/kg joka kuudes viikko saaneista 1,5 prosentille kehittyi nivolumabia neutraloivia vasta‑aineita. Nivolumabi 1 mg/kg‑ ja ipilimumabi 3 mg/kg ‑yhdistelmähoitoa joka kolmas viikko saaneista 4,6 prosentille kehittyi nivolumabia neutraloivia vasta‑aineita. Potilailla, joilta voitiin arvioida ipilimumabin vasta‑aineiden esiintyvyys, 6,3–13,7 prosentille kehittyi ipilimumabin vasta‑aineita ja 0–0,4 prosentille kehittyi ipilimumabia neutraloivia vasta‑aineita.
Niistä potilaista, jotka saivat nivolumabin, ipilimumabin ja kemoterapian yhdistelmähoitoa ja joilta voitiin arvioida nivolumabin vasta-aineiden tai nivolumabia neutraloivien vasta-aineiden esiintyvyys, 33,8 %:lle kehittyi nivolumabin vasta-aineita ja 2,6 %:lle kehittyi nivolumabia neutraloivia vasta-aineita. Niistä potilaista, jotka saivat nivolumabin, ipilimumabin ja kemoterapian yhdistelmähoitoa ja joilta voitiin arvioida ipilimumabin vasta-aineiden tai ipilimumabia neutraloivien vasta-aineiden esiintyvyys, 7,5 %:lle kehittyi ipilimumabin vasta-aineita ja 1,6 %:lle kehittyi ipilimumabia neutraloivia vasta-aineita.
Niistä potilaista, jotka saivat nivolumabin ja brentuksimabivedotiinin yhdistelmähoitoa ja joilta voitiin arvioida nivolumabin vasta-aineiden esiintyvyys, nivolumabin vasta-aineita kehittyi 36 %:lle potilaista, joilla uusiutumisriski oli pieni, ja 12,5 %:lle potilaista, joilla uusiutumisriski oli tavanomainen. Yhdelläkään potilaalla ei todettu nivolumabia neutraloivia vasta-aineita.
Niistä potilaista, jotka saivat nivolumabin ja brentuksimabivedotiinin yhdistelmähoitoa ja joilta voitiin arvioida brentuksimabivedotiinin vasta-aineiden esiintyvyys, brentuksimabivedotiinin vasta-aineita kehittyi 95,8 %:lle potilaista, joilla uusiutumisriski oli pieni, ja 58,5 %:lle potilaista, joilla uusiutumisriski oli tavanomainen. Kaikilla potilailla, joille kehittyi brentuksimabivedotiinin vasta-aineita, todettiin myös brentuksimabivedotiinin neutraloivia vasta-aineita.
Vaikka nivolumabin puhdistuma kasvoi 20 prosentilla nivolumabin vasta-aineiden läsnäollessa, farmakokinetiikka- ja altistus-vasteanalyyseihin perustuen vasta-aineiden läsnäololla ei ollut yhteyttä tehon menetykseen tai toksisuusprofiilin muutoksiin monoterapiassa tai yhdistelmähoidossa.
Pediatriset potilaat
Nivolumabi-monoterapia tai nivolumabi–ipilimumabi-yhdistelmähoito
Nivolumabin turvallisuutta monoterapiana (3 mg/kg 2 viikon välein) ja yhdistelmähoitona ipilimumabin kanssa (nivolumabia 1 mg/kg tai 3 mg/kg yhdessä ipilimumabin 1 mg/kg kanssa 3 viikon välein 4 ensimmäistä annosta, minkä jälkeen nivolumabia 3 mg/kg monoterapiana 2 viikon välein) arvioitiin kliinisessä CA209070-tutkimuksessa 97 pediatrisella potilaalla, jotka olivat ≥ 1–< 18 vuoden ikäisiä (mukaan lukien 53 potilasta, jotka olivat 12–< 18 vuoden ikäisiä) ja joilla oli uusiutunut tai refraktaarinen kiinteä tai hematologinen kasvain, edennyt melanooma mukaan lukien. Pediatristen potilaiden turvallisuusprofiili oli yleisesti ottaen samankaltainen kuin nivolumabia monoterapiana tai yhdistelmähoitona ipilimumabin kanssa saaneilla aikuisilla. Uusia turvallisuussignaaleja ei tunnistettu. Pitkän aikavälin turvallisuustietoja nivolumabin käytöstä 12‑vuotiailla ja sitä vanhemmilla nuorilla ei ole saatavilla.
Nivolumabi-monoterapian yleisimmin ilmoitetut haittavaikutukset (ilmoitettiin vähintään 20 %:lla pediatrisista potilaista) olivat uupumus (35,9 %) ja ruokahalun heikkeneminen (21,9 %). Suurin osa nivolumabi-monoterapian haittavaikutuksista oli vaikeusastetta 1 tai 2. Vaikeusasteen 3 tai 4 haittavaikutuksia ilmoitettiin yksi tai useampi kahdellakymmenelläyhdellä potilaalla (33 %).
Nivolumabi–ipilimumabi-yhdistelmähoidon yleisimmin ilmoitetut haittavaikutukset (ilmoitettiin vähintään 20 %:lla pediatrisista potilaista) olivat uupumus (33,3 %) ja makulopapulaarinen ihottuma (21,2 %). Suurin osa nivolumabi–ipilimumabi-yhdistelmähoidon haittavaikutuksista oli vaikeusastetta 1 tai 2. Vaikeusasteen 3 tai 4 haittavaikutuksia ilmoitettiin yksi tai useampi kymmenellä potilaalla (30 %).
Nivolumabin turvallisuutta monoterapiana (3 mg/kg 2 viikon välein) ja yhdistelmähoitona ipilimumabin kanssa (nivolumabia 3 mg/kg ja sen jälkeen ipilimumabia 1 mg/kg 3 viikon välein 4 annosta, minkä jälkeen nivolumabia monoterapiana 3 mg/kg 2 viikon välein) arvioitiin 151 pediatrisella potilaalla, jotka olivat ≥ 6 kuukauden – < 18 vuoden ikäisiä ja joilla oli korkean asteen primaarinen keskushermoston syöpä. Uusia turvallisuussignaaleja ei tunnistettu (ks. kohta Farmakodynamiikka) verrattuna aikuispotilaista saatuihin tietoihin kaikista indikaatioista.
Nivolumabi yhdistelmähoitona brentuksimabivedotiinin kanssa
Kliinisessä CA209744‑tutkimuksessa arvioitiin nivolumabin (3 mg/kg 3 viikon välein) turvallisuutta yhdistelmähoitona brentuksimabivedotiinin (1,8 mg/kg 3 viikon välein) kanssa 49 pediatrisella potilaalla, jotka olivat ≥ 5 – < 18 vuoden ikäisiä ja joilla oli uusiutunut tai refraktaarinen cHL. Uusia turvallisuussignaaleja ei tunnistettu.
Nivolumabin ja brentuksimabivedotiinin yhdistelmähoidon yleisimmin ilmoitetut haittavaikutukset vähintään 20 %:lla pediatrisista potilaista olivat pahoinvointi (57,1 %), kuume (30,6 %), vatsakipu (26,5 %), ihottuma (24,5 %), päänsärky (22,4 %), muskuloskeletaalinen kipu (22,4 %), ripuli (20,4 %), oksentelu (20,4 %), väsymys (20,4 %), infuusioreaktio (20,4 %) ja yliherkkyys (20,4 %). Suurin osa nivolumabi–brentuksimabivedotiini-yhdistelmähoidon haittavaikutuksista oli vaikeusastetta 1 tai 2. Vaikeusasteen 3 tai 4 haittavaikutuksia ilmoitettiin seitsemällätoista potilaalla (34,7 %).
Nivolumabi yhdistelmähoitona AVD:n kanssa
Kliinisessä CA2098UT‑tutkimuksessa arvioitiin nivolumabin turvallisuutta yhdistelmähoitona AVD:n kanssa 120 pediatrisella potilaalla, jotka olivat 12‑vuotiaita tai sitä vanhempia ja joilla oli aiemmin hoitamaton asteen III tai IV cHL (ks. kohta Farmakodynamiikka). Pediatristen potilaiden turvallisuusprofiili oli yleisesti ottaen samankaltainen kuin nivolumabia yhdistelmähoitona AVD‑kemoterapian kanssa saaneilla aikuisilla.
Iäkkäät potilaat
Kaikkiaan iäkkäiden (≥ 65 vuotta) ja nuorempien (< 65 vuotta) potilaiden välillä ei raportoitu olleen eroja lääkkeen turvallisuudessa. Yli 75‑vuotiaista ja sitä vanhemmista pään ja kaulan alueen levyepiteelisyöpää sairastavista potilaista, potilaista, jotka ovat saaneet nivolumabia melanooman liitännäishoitona, sekä potilaista, jotka ovat saaneet nivolumabia ruokatorvisyövän tai ruokatorvi-mahalaukkurajan syövän liitännäishoitona, on niin vähän tietoa, ettei ryhmää koskevia johtopäätöksiä voida tehdä (ks. kohta Farmakodynamiikka). 75-vuotiaista ja sitä vanhemmista dMMR- tai MSI-H-tyyppistä kolorektaalisyöpää sairastavista potilaista on vain vähän tietoa (ks. kohta Farmakodynamiikka). 65‑vuotiaista ja sitä vanhemmista cHL‑potilaista on niin vähän tietoa, ettei ryhmää koskevia johtopäätöksiä voida tehdä (ks. kohta Farmakodynamiikka).
Keuhkopussin pahanlaatuista mesotelioomaa sairastavista potilaista 75-vuotiailla ja sitä vanhemmilla oli enemmän vakavia haittavaikutuksia (68 %) ja lääkitys lopetettiin useammin haittavaikutusten vuoksi (35 %) kuin yhteensä kaikilla nivolumabi–ipilimumabi-yhdistelmähoitoa saaneilla potilailla, joista 54 %:lla oli vakavia haittavaikutuksia ja lääkitys lopetettiin haittavaikutusten vuoksi 28 %:lla. Hepatosellulaarista karsinoomaa sairastavista potilaista 75‑vuotiailla ja sitä vanhemmilla oli enemmän vakavia haittavaikutuksia (67 %) ja lääkitys lopetettiin useammin haittavaikutusten vuoksi (35 %) kuin yhteensä kaikilla nivolumabi–ipilimumabi-yhdistelmähoitoa saaneilla potilailla, joista 53 %:lla oli vakavia haittavaikutuksia ja lääkitys lopetettiin haittavaikutusten vuoksi 27 %:lla.
Yli 75-vuotiaista ja sitä vanhemmista munuaiskarsinoomaa sairastavista potilaista, jotka ovat saaneet nivolumabia yhdistelmähoitona kabotsantinibin kanssa, on niin vähän tutkimustietoa, ettei ryhmää koskevia johtopäätöksiä voida tehdä (ks. kohta Farmakodynamiikka).
Niistä kolmestakymmenestä ≥ 65‑vuotiaasta potilaasta, jotka saivat nivolumabia yhdistelmähoitona AVD:n kanssa, 70 % (21/30) sai G‑CSF-hoitoa verrattuna 56,1 %:iin (258/460) < 65‑vuotiaista potilaista. Neutropenian ilmoitettu ilmaantuvuus oli numeerisesti pienempi G‑CSF-hoitoa saaneilla potilailla (52,4 %) kuin potilailla, jotka eivät saaneet sitä (66,7 %). Kuumeisen neutropenian ilmaantuvuus oli suurempi ≥ 65‑vuotiailla potilailla (16,7 %) kuin < 65‑vuotiailla potilailla (5,9 %).
Maksan tai munuaisten vajaatoiminta
Ei-levyepiteeliperäisessä NSCLC-tutkimuksessa (CA209057) munuaisten- tai maksan vajaatoimintaa lähtötasolla sairastavilla potilailla nivolumabin turvallisuusprofiili oli verrattavissa koko populaatioon. Näitä tuloksia pitää tulkita varoen alaryhmän pienen otoskoon vuoksi.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa ei ole raportoitu yliannostustapauksia. Yliannostuksen sattuessa potilaita on seurattava tarkasti haittavaikutusten oireiden ja löydösten varalta, ja heille on aloitettava välittömästi tarvittava oireenmukainen hoito.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset lääkeaineet, monoklonaaliset vasta‑aineet ja vasta-aine-konjugoidut solunsalpaajat, PD-1/PDL-1 (ohjelmoituneen solukuoleman proteiinin 1 / ligandin 1) estäjät. ATC‑koodi: L01FF01.
Vaikutusmekanismi
Nivolumabi on humaani monoklonaalinen immunoglobuliini G4 (IgG4) ‑vasta-aine (HuMAb), joka sitoutuu PD-1-reseptoriin (PD-1 = programmed death-1) ja estää sen interaktion PD-L1:n ja PD-L2:n kanssa. PD-1-reseptori on T-solujen aktiivisuuden negatiivinen säätelijä, jonka on todettu olevan osallisena T-soluvälitteisessä immuunivasteessa. PD-1:n sitoutuminen ligandeihin PD-L1 ja PD-L2, jotka ilmentyvät antigeenejä esittelevissä soluissa ja joita kasvaimet tai muut niiden mikroympäristön solut voivat ilmentää, johtaa T-solujen proliferaation ja sytokiinierityksen estymiseen. Nivolumabi tehostaa T-soluvasteita, myös kasvaimia vastaan, estämällä PD-1:n sitoutumisen PD-L1- ja PD-L2-ligandeihin. Syngeenisissä hiirimalleissa PD-1-aktiivisuuden salpaaminen vähensi kasvainten kasvua.
Nivolumabihoito (anti-PD-1) ja ipilimumabihoito (anti-CTLA-4) yhdessä johtivat metastaattista melanoomaa sairastavilla parempiin vasteisiin kasvaimia vastaan. Syngeenisissä hiirimalleissa sekä PD-1:n että CTLA-4:n sitoutumisen estämisellä oli synerginen vaikutus kasvaimia vastaan.
Kliininen teho ja turvallisuus
Annoksen/altistuksen tehon ja turvallisuuden suhteiden mallintamisen perusteella nivolumabiannosten 240 mg 2 viikon välein tai 3 mg/kg 2 viikon välein välillä ei ole kliinisesti merkittäviä eroja tehossa ja turvallisuudessa. Lisäksi näiden suhteiden perusteella nivolumabiannosten 480 mg 4 viikon välein tai 3 mg/kg 2 viikon välein välillä ei ole kliinisesti merkittäviä eroja melanooman liitännäishoidossa tai hoidettaessa edennyttä melanoomaa tai edennyttä munuaiskarsinoomaa.
Melanooma
Melanooman liitännäishoito
Satunnaistettu vaiheen 3 tutkimus, nivolumabi vs. lumelääke (CA20976K)
Nivolumabin (480 mg) tehoa ja turvallisuutta ainoana hoitona niiden potilaiden hoidossa, joiden melanooma oli täydellisesti poistettavissa kirurgisesti, arvioitiin vaiheen 3 satunnaistetussa kaksoissokkotutkimuksessa (CA20976K).Tutkimuksessa oli mukana potilaita, joiden ECOG-toimintakykyluokka oli 0 tai 1 ja joilla oli American Joint Committee on Cancer (AJCC) ‑luokittelun 8. painoksen mukaisesti asteen IIB tai IIC histologisesti vahvistettu melanooma, joka oli täydellisesti kirurgisesti poistettu. Tutkimukseen ottaminen edellytti satunnaistamista edeltävien 12 viikon aikana tehtyä primaarimelanooman täydellistä poistoleikkausta, joka sisälsi tervekudosmarginaalit ja negatiivisen vartijaimusolmukebiopsian. Tutkimukseen otettiin potilaita riippumatta kasvaimen PD‑L1-statuksesta. Tutkimuksesta suljettiin pois ne potilaat, joilla oli silmän/uvean melanooma tai limakalvomelanooma, aktiivinen autoimmuunisairaus, mikä tahansa systeemistä hoitoa joko kortikosteroideilla (≥ 10 mg prednisonia tai vastaavaa vuorokaudessa) tai muulla immunosuppressiivisella lääkkeellä vaativa sairaus, sekä potilaat, joiden melanoomaa oli hoidettu aiemmin (pois lukien potilaat, jotka oli leikattu).
Kaikkiaan 790 potilasta satunnaistettiin (2:1) saamaan joko 480 mg nivolumabia (n = 526) annosteltuna 30 minuutin kuluessa laskimoon joka neljäs viikko tai lumelääkettä (n = 264) enintään 1 vuoden ajan tai kunnes tauti uusiutui tai ilmaantui ei-hyväksyttävää toksisuutta. Satunnaistaminen ositettiin AJCC-luokittelun 8. painoksen T‑luokan mukaisesti (T3b vs. T4a vs. T4b). Kasvain arvioitiin 26 viikon välein 1–3 vuoden ajan ja 52 viikon välein vuodesta 3 vuoteen 5. Ensisijainen tehokkuusmuuttuja oli relapsivapaa elinaika (recurrence-free survival, RFS). Relapsivapaa elinaika, tutkijan arvioimana, määriteltiin ajaksi satunnaistamispäivämäärän ja jonkin seuraavan päivämäärän välillä sen mukaan, mikä seuraavista tapahtui ensimmäisenä: ensimmäinen uusiutuminen (paikallinen, alueellinen tai etäinen etäpesäke), uusi primaarimelanooma tai kuolema mistä tahansa syystä. Toissijainen tulosmuuttuja sisälsi kokonaiselinajan ja etäpesäkevapaan elinajan (distant metastasis-free survival, DMFS).
Näiden kahden ryhmän lähtötason ominaisuudet olivat pääosin tasapainossa. Iän mediaani oli 62 vuotta (vaihteluväli: 19–92 vuotta), 61 % oli miehiä ja 98 % valkoihoisia. Lähtötason ECOG-toimintakykyluokka oli 0 (94 %) tai 1 (6 %). Taudin aste oli 60 %:lla IIB ja 40 %:lla IIC.
Relapsivapaan elinajan osoitettiin primaarisessa ennalta määritetyssä välianalyysissä (seuranta-aika vähintään 7,8 kuukautta) olevan tilastollisesti merkitsevästi parempi nivolumabia saaneilla potilailla verrattuna lumelääkettä saaneisiin potilaisiin; riskitiheyssuhde oli 0,42 (95 %:n luottamusväli: 0,30–0,59; p‑arvo < 0,0001). Päivitetyssä deskriptiivisessä relapsivapaan elinajan analyysissä (seuranta-aika vähintään 15,6 kuukautta) nivolumabiryhmän relapsivapaan elinajan osoitettiin edelleen parantuneen: riskitiheyssuhde oli 0,53 (95 %:n luottamusväli: 0,40–0,71). Kokonaiselinaikatulokset eivät olleet kypsiä analysoitaviksi. Ylimääräisessä deskriptiivisessä relapsivapaan elinajan analyysissä (seuranta-aika vähintään 26,9 kuukautta) nivolumabiryhmän relapsivapaan elinajan osoitettiin edelleen parantuneen: riskitiheyssuhde oli 0,62 (95 %:n luottamusväli: 0,47–0,80). Seuranta-ajan mediaanikesto oli nivolumabiryhmässä 34,25 kuukautta ja lumelääkeryhmässä 33,92 kuukautta. Tulokset olivat yhdenmukaisia virallisen välianalyysin kanssa. Taulukossa 22 ja kuvassa 1 esitetään analyysien raportoidut tulokset, joiden seuranta-aika on vähintään 15,6 kuukautta.
Taulukko 22: Tulokset lääkeaineen tehosta (CA20976K)
| nivolumabi (n = 526) | lumelääke (n = 264) |
|---|---|---|
Relapsivapaa elinaika, kun seuranta-aika oli vähintään 15,6 kuukautta | ||
Relapsivapaa elinaika | ||
Tapahtumia | 102 (19,4 %) | 84 (31,8 %) |
Riskitiheyssuhdea | 0,53 | |
95 %:n luottamusväli | (0,40–0,71) | |
| ||
Mediaani (95 %:n luottamusväli) kuukausina | NR | 36,14 (24,77–NR) |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdallab | 88,8 (85,6–91,2) | 81,1 (75,7–85,4) |
Osuus (95 %:n luottamusväli) 18 kuukauden kohdallab | 83,9 (80,3–86,9) | 70,7 (64,5–76,1) |
| ||
a Perustuu ositettuun Coxin suhteellisen vaaran malliin.
b Perustuu Kaplan–Meier-arvioihin.
Hyöty relapsivapaassa elinajassa oli johdonmukainen kaikissa tärkeimmissä alaryhmissä, kuten taudin asteeseen, T‑luokkaan ja ikään perustuvissa ryhmissä.
Kuva 1: Relapsivapaa elinaika (CA20976K)
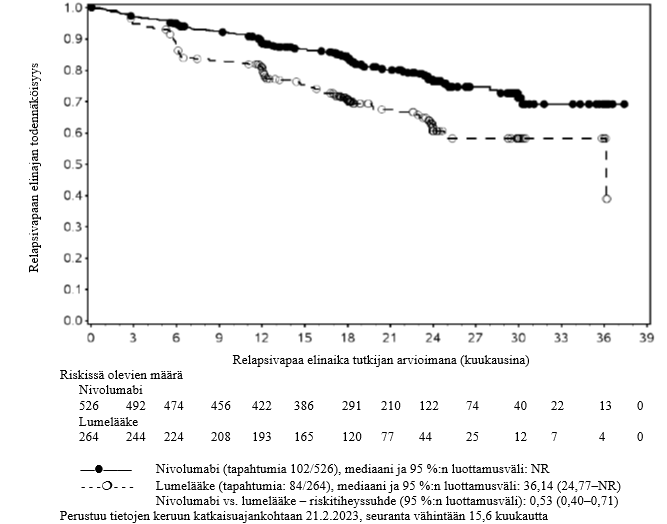
Kasvaimen PD‑L1-ilmentymän tiedot olivat saatavilla 302:sta/790 (38,2 %) satunnaistetusta potilaasta (36,3 % nivolumabi- ja 42,0 % lumelääkeryhmässä), sillä PD‑L1-ilmentymä ei ollut satunnaistamisen ositustekijä. PD‑L1-ilmentymien eksploratiiviset RFS-analyysit osoittivat, että nivolumabin riskitiheyssuhde oli lumelääkkeeseen verrattuna 0,43 (95 %:n luottamusväli: 0,22–0,84) potilailla (N = 167), joiden PD‑L1-ilmentymä oli ≥ 1 %, 0,82 (95 %:n luottamusväli: 0,44–1,54) potilailla (N = 135), joiden PD‑L1-ilmentymä oli < 1 %, ja 0,50 (95 %:n luottamusväli: 0,34–0,73) potilailla (N = 488), joiden PD‑L1-ilmentymä oli määrittämätön / ei ilmoitettu / ei arvioitavissa.
Satunnaistettu vaiheen 3 tutkimus, nivolumabi verrattuna ipilimumabiin 10 mg/kg (CA209238)
Nivolumabin (3 mg/kg) tehoa ja turvallisuutta ainoana hoitona niiden potilaiden hoidossa, joiden melanooma oli täydellisesti resekoitu, arvioitiin vaiheen 3 satunnaistetussa kaksoissokkotutkimuksessa (CA209238). Tutkimuksessa oli mukana aikuispotilaita, joiden ECOG-toimintakykyluokka oli 0 tai 1 ja joilla oli AJCC ‑luokittelun 7. painoksen mukaisesti IIIB/C tai IV asteen histologisesti vahvistettu melanooma, joka oli täydellisesti kirurgisesti resekoitu. AJCC-luokittelun 8. painoksen mukaan kyseessä ovat potilaat, joiden sairaus on levinnyt imusolmukkeisiin tai sairauteen liittyy etäpesäkkeitä. Tutkimukseen otettiin potilaita riippumatta kasvaimen PD‑L1-statuksesta. Tutkimuksesta suljettiin pois ne potilaat, joilla oli ollut autoimmuunisairaus ja mikä tahansa systeemistä hoitoa joko kortikosteroideilla (≥ 10 mg prednisonia tai vastaavaa vuorokaudessa) tai muulla immunosuppressiivisella lääkkeellä vaativa sairaus sekä potilaat, joiden melanoomaa oli hoidettu aiemmin (pois lukien potilaat, jotka oli leikattu, jotka olivat saaneet sädehoitoa liitännäishoitona keskushermostojärjestelmän leesioihin neurokirurgisen poistoleikkauksen jälkeen ja jotka olivat saaneet aiemmin interferonia liitännäishoitona, joka oli päättynyt ≥ 6 kuukautta ennen satunnaistamista), jotka olivat saaneet aiemmin anti‑PD‑1‑, anti‑PD‑L1‑, anti‑PD‑L2‑, anti‑CD137‑ tai anti‑CTLA‑4-hoitoa (mukaan lukien ipilimumabi tai mikä tahansa muu vasta-aine tai lääkeaine, joka kohdistuu erityisesti T‑solujen kostimulointiin tai tarkistuspisteiden signalointireitteihin).
Yhteensä 906 potilasta satunnaistettiin saamaan joko nivolumabia 3 mg/kg (n = 453) 2 viikon välein tai ipilimumabia 10 mg/kg (n = 453) 3 viikon välein 4 annoksen ajan, minkä jälkeen 12 viikon välein alkaen viikosta 24 enintään 1 vuoden ajan. Satunnaistaminen ositettiin kasvaimen PD‑L1-ilmentymän (≥ 5 % vs. < 5 %/määrittämätön) ja sairauden AJCC-luokittelun määrittämän asteen mukaan. Kasvaimet arvioitiin 12 viikon välein ensimmäisten 2 vuoden ajan ja sen jälkeen 6 kuukauden välein. Ensisijainen päätetapahtuma oli relapsivapaa elinaika (recurrence-free survival, RFS). Relapsivapaa elinaika, tutkijan arvioimana, arvioitiin ajaksi satunnaistamispäivämäärän ja jonkin seuraavan päivämäärän välillä sen mukaan, mikä seuraavista tapahtui ensimmäisenä: ensimmäinen uusiutuminen (paikallinen, alueellinen tai etäinen etäpesäke), uusi primaarimelanooma tai kuolema mistä tahansa syystä.
Ryhmien lähtötason ominaisuudet olivat tasapainossa. Iän mediaani oli 55 vuotta (vaihteluväli: 18–86 vuotta), 58 % oli miehiä ja 95 % valkoihoisia. Lähtötason ECOG-toimintakykyluokka oli 0 (90 %) tai 1 (10 %). Useimmilla potilailla oli AJCC-luokittelun mukaisesti III asteen sairaus (81 %), ja 19 %:lla potilaista oli IV asteen sairaus. 48 %:lla potilaista oli makroskooppisia imusolmukkeita ja 32 %:lla kasvaimen ulseraatiota. 42 % potilaista oli BRAF V600 ‑mutaatiopositiivisia, 45 % oli BRAF‑villityypin potilaita ja 13 %:lla BRAF-status oli tuntematon. 34 %:lla potilaista kasvaimen PD‑L1:n ilmentyminen oli ≥ 5 % ja 62 %:lla < 5 % kliinisen tutkimuksen testauksen mukaan. Potilaat, joilla oli määritettävissä oleva kasvaimen PD‑L1-ilmentyminen, olivat jakaantuneet tasaisesti hoitoryhmiin. Kasvaimen PD‑L1-ilmentyminen määriteltiin PD‑L1 IHC 28–8 pharmDx ‑testausmenetelmän avulla.
Relapsivapaan elinajan osoitettiin primaarisessa ennalta määritetyssä välianalyysissä (seuranta-aika vähintään 18 kuukautta) olevan tilastollisesti merkitsevästi parempi nivolumabia saaneilla potilailla verrattuna ipilimumabia saaneisiin potilaisiin; riskitiheyssuhde oli 0,65 (97,56 %:n luottamusväli: 0,51–0,83; ositettu log‑rank-testin p‑arvo < 0,0001). Relapsivapaan elinajan paraneminen vahvistettiin päivitetyssä deskriptiivisessä relapsivapaan elinajan analyysissä, jossa seuranta-aika oli vähintään 24 kuukautta ja riskitiheyssuhde 0,66 (95 %:n luottamusväli: 0,54–0,81; p < 0,0001), ja jossa kokonaiselinaikatiedot eivät olleet kypsiä analysoitaviksi. Tulokset tehosta, kun seuranta-aika oli vähintään 36 kuukautta (relapsivapaan elinajan ennalta määritetty lopullinen analyysi) ja 48 kuukautta (kokonaiselinajan ennalta määritetty lopullinen analyysi) esitetään taulukossa 23 sekä kuvissa 2 ja 3 (koko satunnaistettu populaatio).
Taulukko 23: Tulokset lääkeaineen tehosta (CA209238)
| nivolumabi | ipilimumabi 10 mg/kg |
|---|---|---|
Lopullinen ennalta määritetty analyysi Relapsivapaa elinaika, kun seuranta-aika oli vähintään 36 kuukautta | ||
Tapahtumia | 188 (41,5 %) | 239 (52,8 %) |
Riskitiheyssuhdea | 0,68 | |
95 %:n luottamusväli | (0,56–0,82) | |
p‑arvo | p < 0,0001 | |
| ||
Mediaani (95 %:n luottamusväli) kuukausina | NR (38,67–NR) | 24,87 (16,62–35,12) |
Relapsivapaa elinaika, kun seuranta-aika oli vähintään 48 kuukautta | ||
Tapahtumia | 212 (46,8 %) | 253 (55,8 %) |
Riskitiheyssuhdea | 0,71 | |
95 %:n luottamusväli | (0,60–0,86) | |
Mediaani (95 %:n luottamusväli) kuukausina | 52,37 (42,51–NR) | 24,08 (16,56–35,09) |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 70,4 (65,9–74,4) | 60,0 (55,2–64,5) |
Osuus (95 %:n luottamusväli) 18 kuukauden kohdalla | 65,8 (61,2–70,0) | 53,0 (48,1–57,6) |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 62,6 (57,9–67,0) | 50,2 (45,3–54,8) |
Osuus (95 %:n luottamusväli) 36 kuukauden kohdalla | 57,6 (52,8–62,1) | 44,4 (39,6–49,1) |
Osuus (95 %:n luottamusväli) 48 kuukauden kohdalla | 51,7 (46,8–56,3) | 41,2 (36,4–45,9) |
Lopullinen ennalta määritetty analyysi Kokonaiselinaika, kun seuranta-aika oli vähintään 48 kuukautta | ||
Tapahtumia | 100 (22,1 %) | 111 (24,5 %) |
Riskitiheyssuhdea | 0,87 | |
95,03 %:n luottamusväli | (0,66–1,14) | |
p‑arvo | 0,3148 | |
Mediaani (95 %:n luottamusväli) kuukausina | Ei saavutettu | Ei saavutettu |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 96,2 (93,9–97,6) | 95,3 (92,8–96,9) |
Osuus (95 %:n luottamusväli) 18 kuukauden kohdalla | 91,9 (88,9–94,1) | 91,8 (88,8–94,0) |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 88,0 (84,6–90,7) | 87,8 (84,4–90,6) |
Osuus (95 %:n luottamusväli) 36 kuukauden kohdalla | 81,7 (77,8–85,1) | 81,6 (77,6–85,0) |
Osuus (95 %:n luottamusväli) 48 kuukauden kohdalla | 77,9 (73,7–81,5) | 76,6 (72,2–80,3) |
a Johdettu ositetusta verrannollisten riskitiheyksien mallista.
Kun seuranta-aika oli vähintään 36 kuukautta, tutkimuksessa osoitettiin relapsivapaan elinajan olevan tilastollisesti merkitsevästi parempi potilailla, jotka oli satunnaistettu saamaan nivolumabia, verrattuna ipilimumabi 10 mg/kg ‑hoitoryhmään. Hyöty relapsivapaassa elinajassa osoitettiin johdonmukaisesti kaikissa alaryhmissä, mukaan lukien kasvaimen PD‑L1-ilmentyminen, BRAF-status ja taudin vaihe. Kun seuranta-aika oli vähintään 48 kuukautta (ks. kuva 2), tutkimuksessa todettiin edelleen paranemista relapsivapaassa elinajassa nivolumabiryhmässä ipilimumabiryhmään verrattuna. Hyöty relapsivapaassa elinajassa säilyi kaikissa alaryhmissä.
Kuva 2: Relapsivapaa elinaika (CA209238)
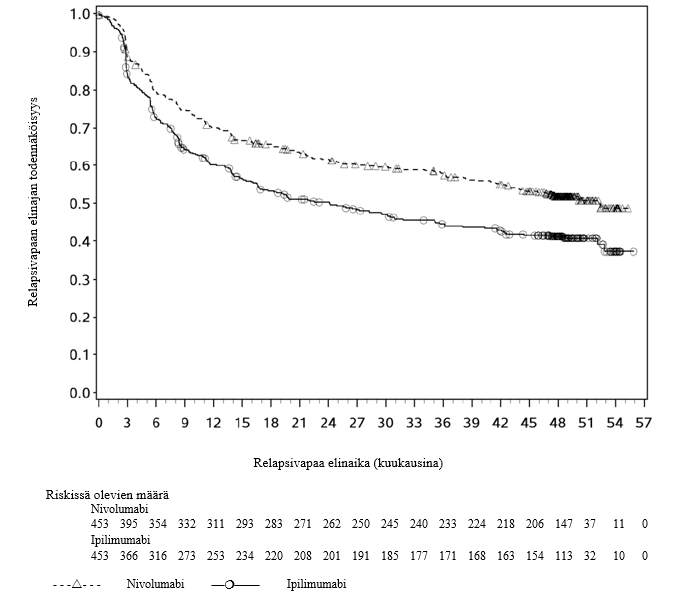
Kuva 3: Kokonaiselinaika (CA209238)
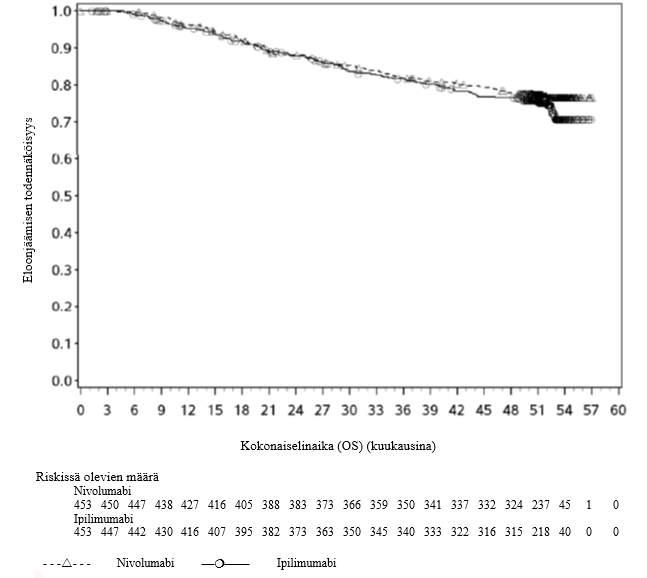
Kokonaiselinajan mediaania ei saavutettu kummassakaan ryhmässä (HR = 0,87, 95,03 %:n luottamusväli: 0,66–1,14; p‑arvo: 0,3148), kun seuranta-aika oli vähintään 48 kuukautta (ks. kuva 3). Tutkimuslääkehoidon jälkeiset tehokkaat syöpähoidot vaikuttavat sekoittavasti kokonaiselinaikatietoihin. Myöhempää systeemistä hoitoa sai 33 prosenttia nivolumabia saaneista potilaista ja 42 prosenttia ipilimumabia saaneista potilaista. Myöhempää immuunihoitoa (mukaan lukien anti-PD‑1-hoitoa, anti-CTLA‑4-hoitoa tai muuta immuunihoitoa) sai 23 prosenttia nivolumabia saaneista potilaista ja 34 prosenttia ipilimumabia saaneista potilaista.
Elämänlaatu (Quality of life, QoL) pysyi vakaana ja lähellä lähtötason arvoja hoidon aikana nivolumabilla hoidetuilla potilailla validien ja luotettavien asteikkojen mukaan arvioituna. Näitä asteikkoja olivat esimerkiksi EORTC:n QLQ‑C30 ja EQ‑5D-hyötyindeksi sekä visuaalis-analoginen asteikko (VAS).
Edenneen melanooman hoito
Satunnaistettu vaiheen 3 tutkimus, vertailu dakarbatsiiniin (CA209066)
Nivolumabin (3 mg/kg) tehoa ja turvallisuutta edenneen melanooman (jota ei voi kirurgisesti poistaa tai joka on metastasoinut) hoidossa arvioitiin vaiheen 3 satunnaistetussa kaksoissokkotutkimuksessa (CA209066). Tutkimukseen otettiin mukaan diagnosoituja, aiemmin hoitamattomia aikuisia 18 vuotta täyttäneitä potilaita, joilla oli III tai IV asteen BRAF-villityypin melanooma ja joiden ECOG-toimintakykyluokka oli 0 tai 1. Niitä, joilla oli aktiivinen autoimmuunisairaus, silmän melanooma tai aktiivisia leptomeningeaalisia tai aivometastaaseja, ei otettu mukaan tutkimukseen.
Kaikkiaan 418 potilasta satunnaistettiin saamaan joko 3 mg/kg nivolumabia (n = 210) annosteltuna 60 minuutin kuluessa laskimoon joka toinen viikko tai 1000 mg/m2 dakarbatsiinia (n = 208) joka kolmas viikko. Satunnaistaminen ositettiin kasvaimen PD-L1-statuksen ja M-luokan mukaan (M0/M1a/M1b vs M1c). Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt sitä. Hoitoa voitiin jatkaa sairauden edettyä, jos potilas tutkijan arvion mukaan sai tutkimuslääkkeestä kliinistä hyötyä eikä se aiheuttanut huomattavia haittavaikutuksia. Kasvaimet arvioitiin kiinteiden kasvainten vasteenarviointikriteerien (Response Evaluation Criteria in Solid Tumours, RECIST) version 1.1 mukaan 9 viikon kuluttua satunnaistamisesta ja sitten ensimmäisen vuoden ajan joka 6. viikko ja sen jälkeen joka 12. viikko. Ensisijainen tehokkuusmuuttuja oli kokonaiselinaika. Tärkeimmät toissijaiset tehokkuusmuuttujat olivat tutkijan arvioima etenemisvapaa elinaika (PFS) ja objektiivisen vasteen saavuttaneiden osuus (objective response rate, ORR).
Ryhmien lähtötason ominaisuudet olivat tasapainossa. Iän mediaani oli 65 vuotta (vaihteluväli 18–87), 59 % oli miehiä ja 99,5 % valkoihoisia. Useimpien potilaiden ECOG-toimintakykyluokka oli 0 (64 %) tai 1 (34 %). 61 prosentilla potilaista sairaus oli tutkimukseen otettaessa luokkaa M1c. 74 prosentilla potilaista oli ihomelanooma ja 11 prosentilla limakalvomelanooma. 35 prosentilla potilaista melanooma oli PD-L1-positiivinen (≥ 5 %:ssa kasvainsolun membraanissa ilmentyvä). 16 prosenttia potilaista oli saanut aiemmin liitännäishoitoa; yleisin liitännäishoito oli interferoni (9 %). 4 prosentilla potilaista oli ollut aivometastaaseja ja 37 prosentilla LDH-arvo oli tutkimukseen otettaessa normaalia suurempi.
Kuvassa 4 on esitetty kokonaiselinajan Kaplan-Meier-kuvaajat.
Kuva 4: Kokonaiselinajan Kaplan–Meier-kuvaajat (CA209066)
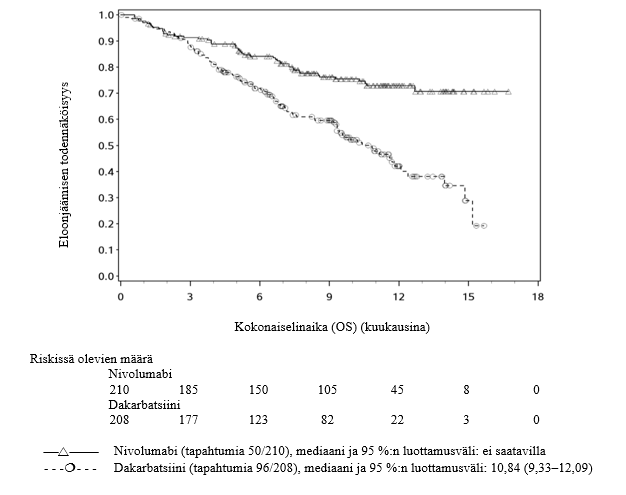
Kokonaiselinaikaa pidentävä vaikutus todettiin johdonmukaisesti kaikissa alaryhmissä, jotka oli jaettu mm. lähtötason ECOG-toimintakyvyn, M-luokan, aivometastaasianamneesin ja lähtötason LDH-arvon mukaan. Elinaikaa pidentävä vaikutus havaittiin riippumatta siitä oliko potilaan kasvain PD-L1-negatiivinen vai PD-L1-positiivinen (kasvainmembraanissa ilmentymien raja-arvo 5 % tai 10 %).
Saatavilla olevat tiedot osoittavat, että nivolumabin vaikutuksen alkaminen viivästyy siten, että nivolumabin hyödyt kemoterapian jälkeen ilmenevät 2–3 kuukaudessa.
Tulokset tehosta näkyvät taulukossa 24.
Taulukko 24:Tulokset tehosta(CA209066)
| nivolumabi | dakarbatsiini | ||
Kokonaiselinaika (OS) | ||||
Tapahtumat | 50 (23,8 %) | 96 (46,2 %) | ||
Riskitiheyssuhde | 0,42 | |||
99,79 %:n luottamusväli | (0,25–0,73) | |||
95 %:n luottamusväli | (0,30–0,60) | |||
p‑arvo | < 0,0001 | |||
Mediaani (95 %:n luottamusväli) | Ei saavutettu | 10,8 (9,33–12,09) | ||
Osuus (95 %:n luottamusväli) |
|
| ||
6 kuukauden kohdalla | 84,1 (78,3–88,5) | 71,8 (64,9–77,6) | ||
12 kuukauden kohdalla | 72,9 (65,5–78,9) | 42,1 (33,0–50,9) | ||
Etenemisvapaa elinaika | ||||
Tapahtumat | 108 (51,4 %) | 163 (78.4 %) | ||
Riskitiheyssuhde | 0,43 | |||
95 %:n luottamusväli | (0,34–0,56) | |||
p‑arvo | < 0,0001 | |||
Mediaani (95 %:n luottamusväli) | 5,1 (3,48–10,81) | 2,2 (2,10–2,40) | ||
Osuus (95 %:n luottamusväli) |
|
| ||
6 kuukauden kohdalla | 48,0 (40,8–54,9) | 18,5 (13,1–24,6) | ||
12 kuukauden kohdalla | 41,8 (34,0–49,3) | Ei sovellettavissa | ||
Objektiivinen vaste | 84 (40,0 %) | 29 (13,9 %) | ||
(95%:n luottamusväli) | (33,3–47,0) | (9,5–19,4) | ||
Ristitulosuhde (95 %:n luottamusväli) | 4,06 (2,52–6,54) | |||
p‑arvo | < 0,0001 | |||
Täydellinen vaste (CR) | 16 (7,6 %) | 2 (1,0 %) | ||
Osittainen vaste (PR) | 68 (32,4 %) | 27 (13,0 %) | ||
Stabiili tauti (SD) | 35 (16,7 %) | 46 (22,1 %) | ||
Vasteen keston mediaani | ||||
Kuukausina (vaihteluväli) | Ei saavutettu | (0+–12,5+) | 6,0 | (1,1–10,0+) |
Vasteen saavuttamisajan mediaani | ||||
Kuukausina (vaihteluväli) | 2,1 (1,2–7,6) | 2,1 (1,8–3,6) | ||
”+” tarkoittaa sensuroitunutta havaintoa.
Satunnaistettu vaiheen 3 tutkimus, vertailu kemoterapiaan (CA209037)
Nivolumabin (3 mg/kg) tehoa ja turvallisuutta edenneen melanooman (jota ei voi kirurgisesti poistaa tai joka on metastasoinut) hoidossa arvioitiin vaiheen 3 satunnaistetussa, avoimessa tutkimuksessa (CA209037). Siinä tutkittiin aikuisia, joiden sairaus oli edennyt ipilimumabihoidon aikana tai jälkeen ja, jos heillä oli BRAF V600 ‑mutaatio, myös BRAF-kinaasinestäjää käytettäessä tai sen jälkeen. Tutkimukseen ei otettu potilaita, joilla oli aktiivinen autoimmuunisairaus, silmän melanooma, aktiivisia leptomeningeaalisia tai aivometastaaseja tai anamneesissa ipilimumabiin liittyvä korkean asteen (CTCAE v4.0:n mukaan asteen 4) haittavaikutus, paitsi korjaantunut pahoinvointi, uupumus, infuusioreaktio tai umpierityshäiriö.
Kaikkiaan 405 potilasta satunnaistettiin saamaan joko 3 mg/kg nivolumabia (n = 272) laskimoon 60 minuutin kuluessa 2 viikon välein tai kemoterapiaa (n = 133). Kemoterapiana käytettiin tutkijan valinnan mukaan joko dakarbatsiinia (1000 mg/m2 3 viikon välein) tai karboplatiinia (AUC 6 kolmen viikon välein) ja paklitakselia (175 mg/m2 3 viikon välein). Satunnaistaminen ositettiin BRAF- ja kasvaimen PD-L1-statuksen ja aiemmasta ipilimumabihoidosta saadun parhaan vasteen mukaan.
Primaariset tehokkuusmuuttujat olivat vahvistettu objektiivisen vasteen saavuttaneiden osuus (ORR), jonka riippumaton radiologinen arviointiryhmä (Independent Radiology Review Committee, IRRC) mittasi RECIST 1.1-kriteerien perusteella ensimmäisellä 120 nivolumabilla hoidetulla potilaalla, sekä nivolumabia ja kemoterapiaa saaneiden kokonaiselinaika (OS). Muita tulosmuuttujia olivat vasteen kesto ja aika.
Iän mediaani oli 60 vuotta (vaihteluväli 23–88). 64 % potilaista oli miehiä ja 98 % valkoihoisia. ECOG-toimintakykyluokka oli 61 prosentilla potilaista 0 ja 39 prosentilla 1. Potilaista suurimman osan (75 %) sairaus oli tutkimukseen otettaessa luokkaa M1c. 73 prosentilla potilaista oli ihomelanooma ja 10 prosentilla limakalvomelanooma. 27 % potilaista oli aiemmin saanut systeemistä hoitoa kerran, 51 % kaksi kertaa ja 21 % > 2 kertaa. 22 prosentilla potilaista oli kasvaimia, joissa todettin BRAF-mutaatio, ja 50 %:lla potilaista oli kasvaimia, jotka katsottiin PD-L1-positiivisiksi. 64 % potilaista ei ollut aiemmin hyötynyt kliinisesti ipilimumabista (täydellinen/osittainen hoitovaste (complete/partial response, CR/PR) tai stabiili tauti (stable disease, SD)). Lähtötason ominaisuudet olivat tasapainossa ryhmien välillä lukuun ottamatta niiden potilaiden osuutta, joilla oli anamneesissa aivometastaasi (nivolumabiryhmässä 19 % ja kemoterapiaryhmässä 13 %), ja niiden osuutta, joiden LDH-arvo oli lähtötasolla normaalia suurempi (nivolumabiryhmässä 51 %, kemoterapiaryhmässä 35 %).
Tässä lopullisessa ORR-analyysissa oli mukana tulokset 120:ltä nivolumabia ja 47:ltä kemoterapiaa saaneelta potilaalta, joiden seuranta oli kestänyt vähintään 6 kuukautta. Tehokkuustulokset on esitetty taulukossa 25.
Taulukko 25: Paras kokonaisvaste, vasteen saavuttamisaika ja kesto (CA209037)
| nivolumabi | kemoterapia | ||
Vahvistettu objektiivinen vaste (IRRC) | 38 (31,7 %) | 5 (10,6 %) | ||
(95 %:n luottamusväli) | (23,5–40,8) | (3,5–23,1) | ||
Täydellinen vaste (CR) | 4 (3,3 %) | 0 | ||
Osittainen vaste (PR) | 34 (28,3 %) | 5 (10,6 %) | ||
Stabiili tauti (SD) | 28 (23,3 %) | 16 (34,0 %) | ||
Vasteen keston mediaani | ||||
Kuukausina (vaihteluväli) | Ei saavutettu | 3,6 (Ei saatavilla) | ||
Vasteen saavuttamisajan mediaani | ||||
Kuukausina (vaihteluväli) | 2,1 (1,6–7,4) | 3,5 (2,1–6,1) | ||
Saatavilla olevat tiedot osoittavat, että nivolumabin vaikutuksen alkaminen viivästyy siten, että nivolumabilla todettavat hyödyt kemoterapiaan verrattuna voivat kestää 2–3 kuukautta.
Päivitetty analyysi (24 kuukauden seuranta)
Kaikista satunnaistetuista potilaista ORR oli nivolumabia saaneessa ryhmässä 27,2 % (95 %:n luottamusväli 22,0–32,9) ja kemoterapiaa saaneessa ryhmässä 9,8 % (95 %:n luottamusväli 5,3–16,1). Vasteen keston mediaani oli nivolumabilla 31,9 kuukautta (vaihteluväli 1,4+–31,9) ja kemoterapialla 12,8 kuukautta (vaihteluväli 1,3+–13,6+). Etenemisvapaan elinajan riskitiheyssuhde oli nivolumabilla kemoterapiaan verrattuna 1,03 (95 %:n luottamusväli 0,78–1,36). IRRC arvioi ORR:n ja PFS:n RECIST 1.1-kriteerien perusteella.
Lopullisissa kokonaiselinaika-analyyseissa nivolumabin ja kemoterapian välillä ei ollut tilastollisesti merkitsevää eroa. Alustavassa kokonaiselinaika-analyysissa ei huomioitu tutkimuslääkkeen jälkeistä lääkehoitoa, jossa 54 kemoterapiaryhmän potilasta (40,6 %) sai myöhempää anti‑PD1 ‑hoitoa. Kokonaiselinaikaan voivat vaikuttaa tutkimushoidon keskeyttämiset, myöhempien hoitojen epätasapaino tutkimushaaroissa sekä erot tutkittavien lähtötason tekijöissä. Nivolumabiryhmässä oli kemoterapiaryhmään verrattuna enemmän potilaita, joilla oli huonompi ennuste (kohonnut lähtötason LDH-arvo ja aivometastaaseja).
Teho BRAF-statuksen mukaan: Nivolumabilla havaittiin objektiivisia vasteita (ensisijaisen yhdistetyn päätetapahtuman määritelmän mukaan) potilaan BRAF-mutaatiostatuksesta riippumatta. ORR oli BRAF‑mutaatiopositiivisessa alaryhmässä nivolumabia saaneilla 17 % (95 %:n luottamusväli 8,4–29,0) ja kemoterapiaa saaneilla 11 % (95 %:n luottamusväli 2,4–29,2) ja BRAF-villityypin alaryhmässä nivolumabia saaneilla 30 % (95 %:n luottamusväli 24,0–36,7) ja kemoterapiaa saaneilla 9 % (95 %:n luottamusväli 4,6–16,7).
Verrattaessa nivolumabia kemoterapiaan etenemisvapaan elinajan riskitiheyssuhteet olivat BRAF‑mutaatiopositiivisille potilailla 1,58 (95 %:n luottamusväli 0,87–2,87) ja BRAF-villityypin potilailla 0,82 (95 %:n luottamusväli 0,60–1,12). Kokonaiselinajan riskitiheyssuhteet olivat verrattaessa nivolumabia kemoterapiaan BRAF-mutaatiopositiivisille potilailla 1,32 (95 %:n luottamusväli 0,75–2,32) ja BRAF-villityypin potilailla 0,83 (95 %:n luottamusväli 0,62–1,11).
Teho kasvaimen PD-L1:n ilmentymän mukaan: Nivolumabilla havaittiin objektiivisia vasteita kasvaimen PD‑L1-ilmentymästä riippumatta. Tämän biomarkkerin (kasvaimen PD-L1:n ilmentymä) roolia ei ole kuitenkaan täysin selvitetty.
Potilailla, joilla PD-L1:n ilmentymistaso oli ≥ 1 %, ORR oli nivolumabihoidossa 33,5 % (n = 179; 95%:n luottamusväli 26,7–40,9) ja kemoterapiassa 13,5 % (n = 74; 95 %:n luottamusväli 6,7–23,5. Potilailla, joilla PD-L1:n ilmentymistaso oli < 1 %, ORR oli IRRC:n mukaan nivolumabihoidossa 13,0 % (n = 69; 95 %:n luottamusväli 6,1–23,3 ja kemoterapiassa 12,0 %(n = 25; 95 %:n luottamusväli 2,5–31,2).
Verrattaessa nivolumabia kemoterapiaan etenemisvapaan elinajan riskitiheyssuhde oli 0,76 (95 %:n luottamusväli 0,54–1,07) potilailla, joiden PD-L1:n ilmentymistaso oli ≥ 1 % ja 1,92 (95 %:n luottamusväli 1,05–3,5) potilailla, joiden PD‑L1:n ilmentymistaso oli < 1 %.
Verrattaessa nivolumabia kemoterapiaan kokonaiselinajan riskitiheyssuhteet olivat 0,69 (95 %:n luottamusväli 0,49–0,96) potilailla, joiden PD‑L1:n ilmentymistaso oli ≥ 1 % ja 1,52 (95 %:n luottamusväli 0,89–2,57) potilailla, joilla oli PD‑L1:n ilmentymistaso oli < 1 %.
Näiden alaryhmien analyysejä tulee tulkita varauksella alaryhmien pienen koon perusteella ja koska kokonaispopulaatiossa ei osoitettu tilastollisesti merkitsevää eroa kokonaiselinajassa.
Avoin, suurenevin annoksin tehty vaiheen 1 tutkimus (MDX1106‑03)
Nivolumabin turvallisuutta ja siedettävyyttä selvitettiin avoimessa, suurenevin annoksin tehdyssä vaiheen 1 tutkimuksessa, jossa tutkittiin montaa kasvaintyyppiä ja myös edennyttä melanoomaa. Tutkimukseen otetuista aiemmin hoidetuista 306 potilaasta 107:llä oli melanooma, ja he saivat nivolumabia joko 0,1 mg/kg, 0,3 mg/kg, 1 mg/kg, 3 mg/kg tai 10 mg/kg korkeintaan 2 vuoden ajan. Tässä potilasryhmässä objektiivinen vaste saavutettiin 33 potilaalla (31 %), ja vasteen keston mediaani oli 22,9 kuukautta (95 %:n luottamusväli 17,0–NR). PFS-mediaani (progression-free survival, etenemisvapaa elinaika) oli 3,7 kuukautta (95 %:n luottamusväli 1,9–9,3). Kokonaiselinajan mediaani oli 17,3 kuukautta (95 %:n luottamusväli 12,5–37,8), ja arvioidut elossaolo-osuudet olivat 3 vuoden kuluttua 42 % (95 %:n luottamusväli 32–51), 4 vuoden kuluttua 35 % (95 %:n luottamusväli 26–44) ja 5 vuoden kuluttua 34 % (95 %:n luottamusväli 25–43) (seuranta vähintään 45 kuukautta).
Yhden hoitohaaran vaiheen 2 tutkimus (CA209172)
CA209172-tutkimus oli yksihaarainen avoin nivolumabi-monoterapiaa koskeva tutkimus potilailla, joilla oli III asteen melanooma (jota ei voida kirurgisesti poistaa) tai IV asteen metastasoinut melanooma ja joiden aiempi hoito sisälsi monoklonaalisen anti-CTLA-4-vasta-aineen. Turvallisuus oli ensisijainen päätetapahtuma ja teho toissijainen päätetapahtuma. Hoidetusta 1008 potilaasta 103:lla (10 %:lla) oli silmän/uvean melanooma, 66:lla (7 %:lla) ECOG‑toimintakykyluokka oli 2, 165:llä (16 %:lla) oli oireettomia hoidettuja ja hoitamattomia keskushermoston metastaaseja, 13:lla (1,3 %:lla) oli hoidettuja leptomeningeaalisia metastaaseja, 25:llä (2 %:lla) oli autoimmuunisairaus ja 84:llä (8 %:lla) potilaalla, jotka olivat aiemmin saaneet anti‑CTLA‑4‑hoitoa, oli asteen 3–4 immuunivälitteisiä haittatapahtumia. Uusia turvallisuussignaaleja ei havaittu hoidetuissa potilaissa, ja nivolumabin yleinen turvallisuusprofiili oli samanlainen kaikissa alaryhmissä. Taulukossa 26 alla on esitetty tutkijan arvioimiin vasteosuuksiin perustuvat tulokset tehosta viikon 12 kohdalla.
Taulukko 26: Vasteosuus viikon 12 kohdalla – kaikki potilaat, joilta vaste oli arvioitavissa, ja alaryhmän mukaan jaoteltuna (CA209172)
| Yhteensä | Silmän/ uvean melanooma | ECOG-toiminta-kykypiste-määrä 2
| Keskus-hermoston metastaasi | Auto-immuuni-sairaus | Aiempi anti-CTLA-4-hoito ja asteen 3–4 immuuni-välitteisiä haitta-tapahtumia | |
N (%)a | 161/588 (27,4) | 4/61 (6,6) | 4/20 (20,0) | 20/73 (27,4) | 3/16 (18,8) | 13/46 (28,3) | |
a Vasteet arvioitiin RECIST 1.1 ‑kriteerien perusteella niiden 588:n potilaan osalta 1008:sta (58,3 %), jotka jatkoivat hoitoa viikon 12 loppuun ja joille tehtiin seurantakuvaus viikolla 12.
Satunnaistettu vaiheen 3 tutkimus, nivolumabi–ipilimumabi-yhdistelmähoito tai nivolumabi-monoterapia vs. ipilimumabi-monoterapia (CA209067)
Nivolumabi 1 mg/kg ja ipilimumabi 3 mg/kg -yhdistelmähoidon tai nivolumabi 3 mg/kg ‑monoterapian vs. ipilimumabi 3 mg/kg -monoterapian turvallisuutta ja tehoa edenneen melanooman (jota ei voida kirurgisesti poistaa tai joka on metastasoinut) hoidossa arvioitiin satunnaistetussa vaiheen 3 kaksoissokkoutetussa tutkimuksessa (CA209067). Kahden nivolumabia saaneen ryhmän eroja arvioitiin deskriptiivisesti. Tutkimukseen otettiin mukaan diagnosoituja, aiemmin hoitamattomia aikuisia potilaita, joilla oli III tai IV asteen melanooma. Potilaiden ECOG-toimintakykystatuksen pisteiden tuli olla 0 tai 1. Tutkimukseen otettiin mukaan potilaita, jotka eivät olleet aikaisemmin saaneet systemaattista syöpähoitoa melanoomaan, jota ei voida kirurgisesti poistaa tai joka on metastasoinut. Aikaisempi liitännäishoito tai esiliitännäishoito sallittiin, jos se oli päättynyt vähintään kuusi viikkoa ennen satunnaistamista. Potilaita, joilla oli aktiivinen autoimmuunisairaus, silmän/uvean melanooma tai aktiivisia leptomeningeaalisia tai aivometastaaseja, ei otettu mukaan tutkimukseen.
Kaikkiaan 945 potilaista satunnaistettiin saamaan nivolumabi–ipilimumabi-yhdistelmähoitoa (n = 314), nivolumabi-monoterapiaa (n = 316) tai ipilimumabi-monoterapiaa (n = 315). Yhdistelmähoitohaarassa potilaat saivat ensimmäiset neljä annosta nivolumabia 1 mg/kg 60 minuutin kuluessa ja ipilimumabia 3 mg/kg 90 minuutin kuluessa laskimonsisäisesti kolmen viikon välein, jonka jälkeen heille annettiin nivolumabia 3 mg/kg monoterapiana joka toinen viikko. Monoterapiaryhmässä potilaille annettiin nivolumabia 3 mg/kg joka toinen viikko. Vertailuhaaran potilaat saivat neljä ensimmäistä annosta ipilimumabia 3 mg/kg ja nivolumabin lumelääkettä laskimonsisäisesti joka kolmas viikko, jonka jälkeen heille annettiin lumelääkettä joka toinen viikko. Satunnaistaminen ositettiin PD-L1-statuksen (≥ 5 % vs. < 5 %:ssa kasvainsolun membraanissa ilmentyvä), BRAF-statuksen ja American Joint Committee on Cancer (AJCC) -luokittelun määrittelemän M-vaiheen mukaan. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt hoitoa. Kasvaimet arvioitiin 12 viikkoa satunnaistamisen jälkeen, sen jälkeen ensimmäisen vuoden ajan joka kuudes viikko ja sen jälkeen joka kahdestoista viikko. Primaariset muuttujat olivat etenemisvapaa elinaika ja kokonaiselinaika (OS). Myös objektiivisen vasteen saavuttaneiden osuus (ORR) ja vasteen kesto arvioitiin.
Ryhmien lähtötason ominaisuudet olivat kussakin ryhmässä tasapainossa. Iän mediaani oli 61 vuotta (vaihteluväli 18–90 vuotta), 65 % oli miehiä ja 97 % valkoihoisia. ECOG-toimintakykyluokka oli 0 (73 %) tai 1 (27 %). Useimmilla potilailla oli AJCC-luokittelun mukaisesti IV asteen sairaus (93 %), 58 prosentilla potilaista sairaus oli tutkimukseen otettaessa luokkaa M1c. 22 % potilaista oli saanut aiemmin liitännäishoitoa. 32:lla prosentilla potilaista oli BRAF-mutaatiopositiivinen melanooma; 26,5:lla prosentilla oli PD-L1 ≥5 %:ssa kasvainsolun membraanissa ilmentyvänä. 4:llä prosentilla potilaista oli aiemmin ollut aivometastaasi ja 36:lla prosentilla LDH-arvo oli tutkimukseen otettaessa normaalia suurempi. Potilaat, joilla oli määritettävissä oleva PD-L1:n ilmentyminen, olivat jakaantuneet tasaisesti kolmeen hoitoryhmään. Kasvaimen PD-L1-ilmentyminen määriteltiin PD-L1 IHC 28–8 pharmDx -testausmenetelmän avulla.
Primaarianalyysissa (seuranta‑aika vähintään 9 kuukautta) etenemisvapaan elinajan mediaani oli 6,9 kuukautta nivolumabiryhmässä, kun taas ipilimumabiryhmässä se oli 2,9 kuukautta (riskitiheyssuhde = 0,57, 99,5 %:n luottamusväli: 0,43–0,76; p < 0,0001). Etenemisvapaan elinajan mediaani oli 11,5 kuukautta nivolumabin ja ipilimumabin yhdistelmää saaneiden ryhmässä, kun taas ipilimumabiryhmässä se oli 2,9 kuukautta (riskitiheyssuhde = 0,42, 99,5 %:n luottamusväli: 0,31–0,57; p < 0,0001).
Deskriptiivisen analyysin mukaiset etenemisvapaan elinajan tulokset (seuranta-ajan ollessa vähintään 90 kuukautta) esitetään kuvassa 5 (kaikki satunnaistetut populaatiot), kuvassa 6 (PD-L1‑ilmentymän raja-arvolla 5 %) ja kuvassa 7 (PD-L1‑ilmentymän raja-arvolla 1 %).
Kuva 5: Etenemisvapaa elinaika (CA209067)
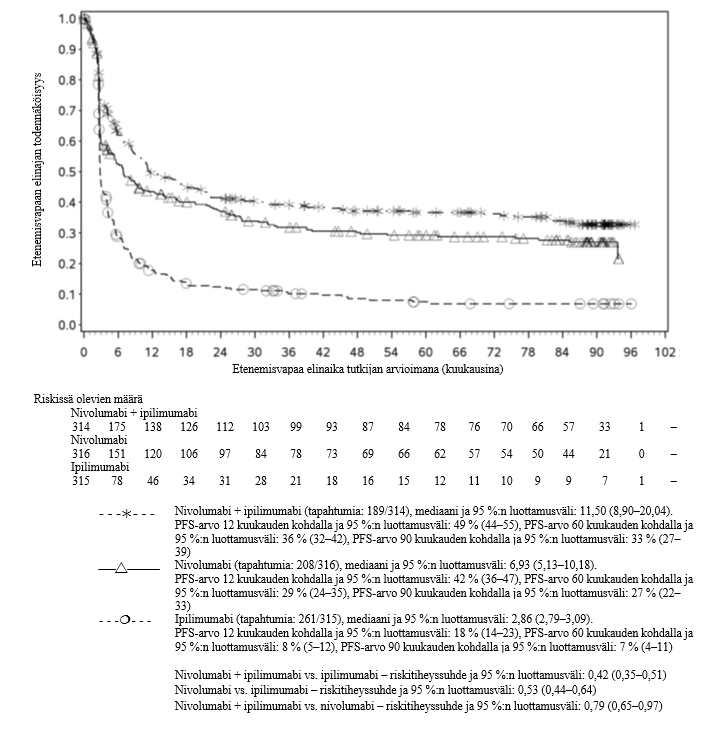
Kuva 6: Etenemisvapaa elinaika PD‑L1-ilmentymittäin: 5 %:n raja arvo (CA209067)
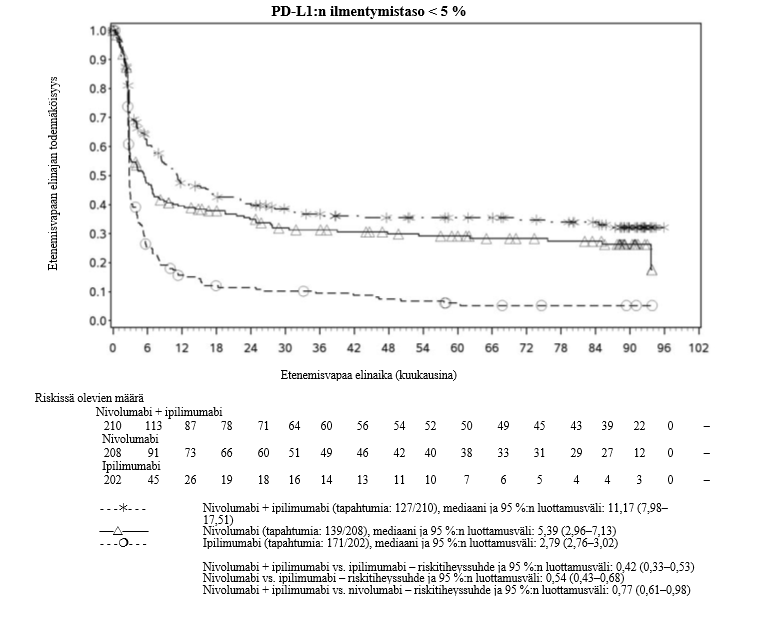
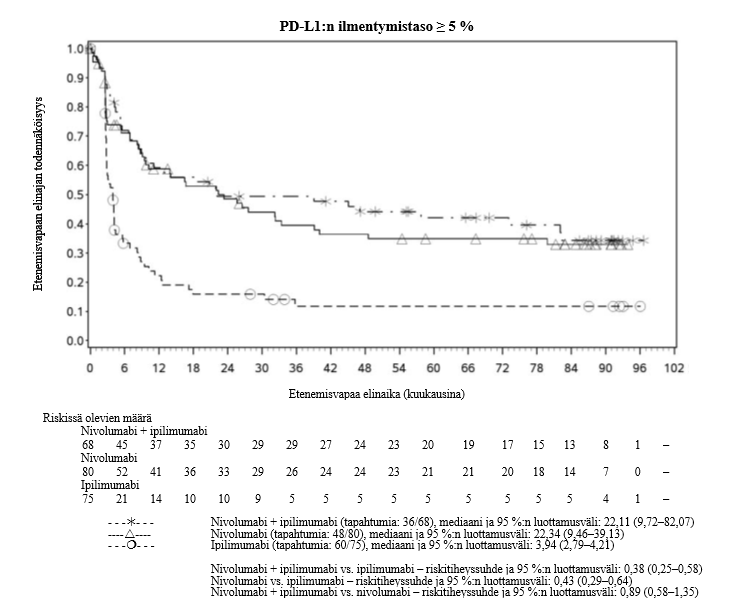
Kuva 7: Etenemisvapaa elinaika PD‑L1-ilmentymittäin: 1 %:n raja-arvo (CA209067)
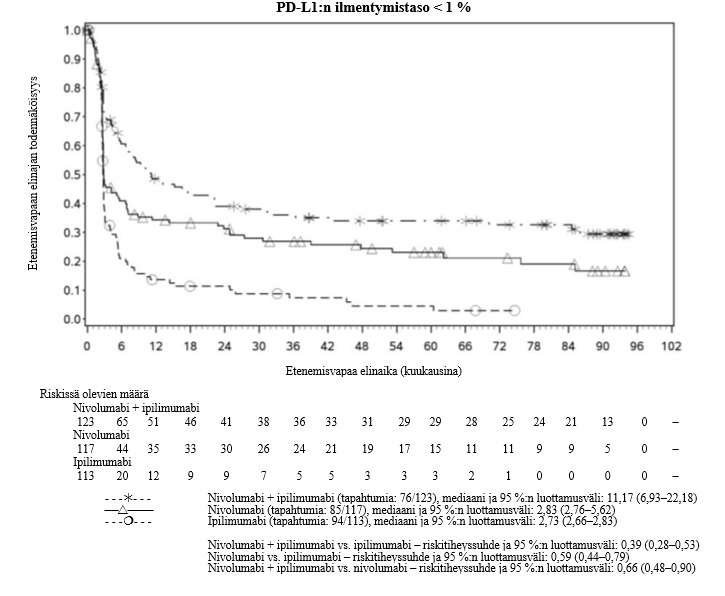
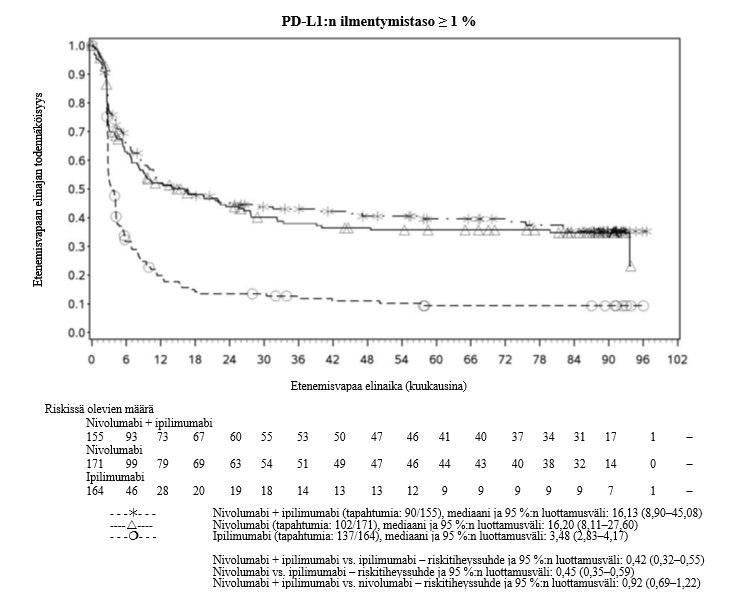
Lopullinen kokonaiselinajan (primaari)analyysi tehtiin, kun kaikkia potilaita oli seurattu vähintään 28 kuukauden ajan. Kokonaiselinajan mediaania ei saavutettu 28 kuukauden kohdalla nivolumabia saaneiden ryhmässä, kun taas ipilimumabia saaneiden ryhmässä kokonaiselinajan mediaani oli 19,98 kuukautta (riskitiheyssuhde = 0,63, 98 %:n luottamusväli: 0,48–0,81; p‑arvo: < 0,0001). Kokonaiselinajan mediaania ei saavutettu nivolumabin ja ipilimumabin yhdistelmähoitoa saaneiden ryhmässä ipilimumabia saaneiden ryhmään verrattuna (riskitiheyssuhde = 0,55, 98 %:n luottamusväli 0,42–0,72; p‑arvo: < 0,0001).
Vähintään 90 kuukauden seuranta‑ajan jälkeen tehdyn ylimääräisen deskriptiivisen analyysin mukaiset kokonaiselinajan tulokset olivat yhteneviä alkuperäisen primaarianalyysin kanssa. Tämän seuranta‑analyysin mukaiset kokonaiselinajan tulokset esitetään kuvassa 8 (kaikki satunnaistetut), kuvissa 9 ja 10 (kasvaimen PD‑L1:n ilmentymän raja‑arvoilla 5 % ja 1 %).
Kokonaiselinaika-analyysissa ei huomioitu tutkimuslääkkeen jälkeistä lääkehoitoa. Potilaat saivat myöhempää systeemistä hoitoa seuraavasti: 36,0 % yhdistelmähoitoa saaneista, 49,1 % nivolumabi-monoterapiaa saaneista ja 66,3 % ipilimumabia saaneista. Potilaat saivat myöhempää immuunihoitoa (mukaan lukien anti‑PD1-hoitoa, anti‑CTLA‑4-hoitoa tai muuta immuunihoitoa) seuraavasti: 19,1 % yhdistelmähoitoa saaneista, 34,2 % nivolumabi-monoterapiaa saaneista ja 48,3 % ipilimumabia saaneista
Kuva 8: Kokonaiselinaika (OS) (CA209067) – vähintään 90 kuukauden seuranta-aika
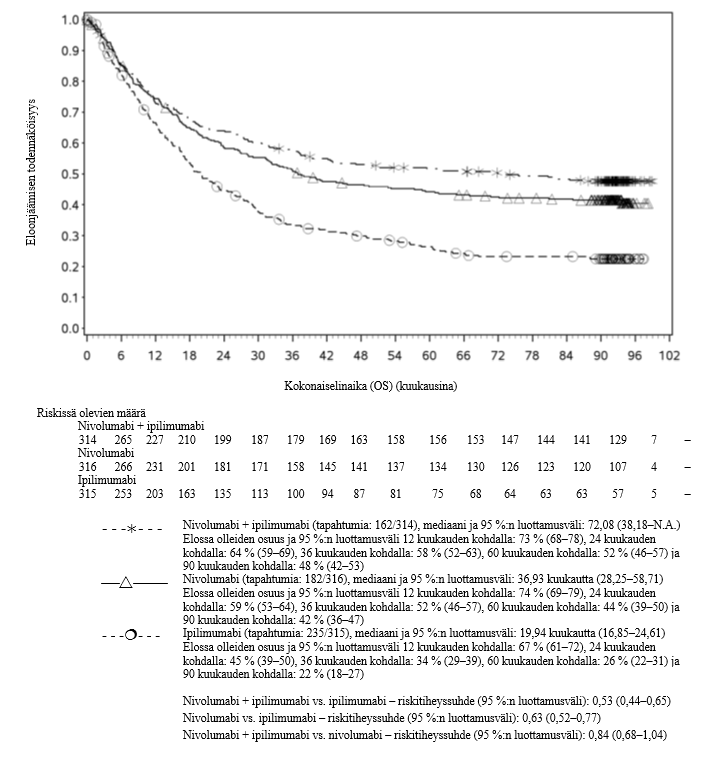
Kuva 9: Kokonaiselinaika PD‑L1-ilmentymittäin: 5 %:n raja-arvo (CA209067) – vähintään 90 kuukauden seuranta-aika
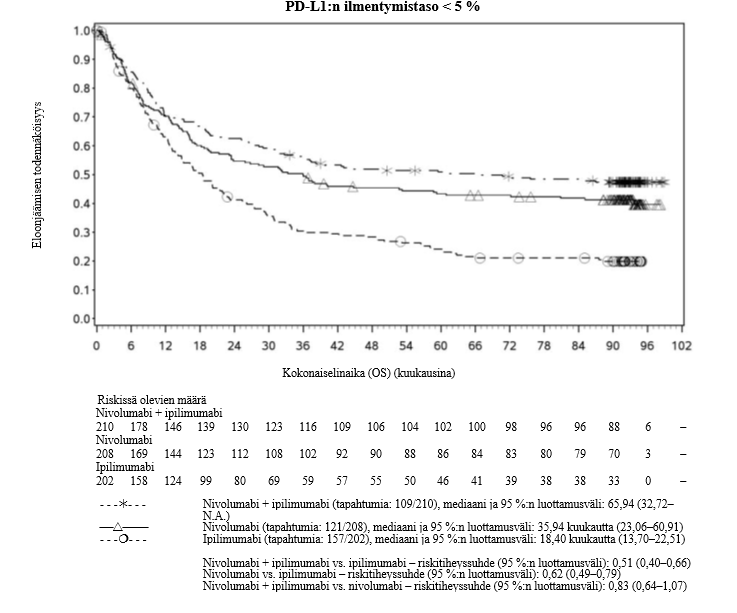
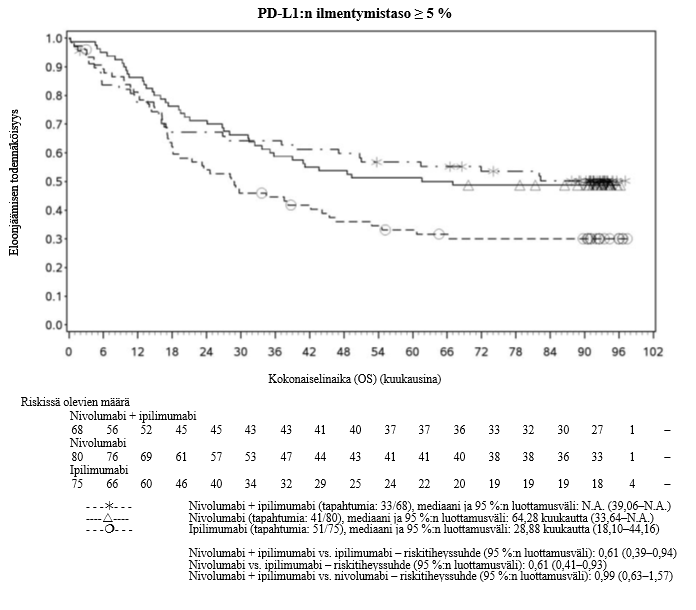
Kuva 10: Kokonaiselinaika (OS) PD‑L1-ilmentymittäin: 1 %:n raja-arvo (CA209067) – vähintään 90 kuukauden seuranta-aika
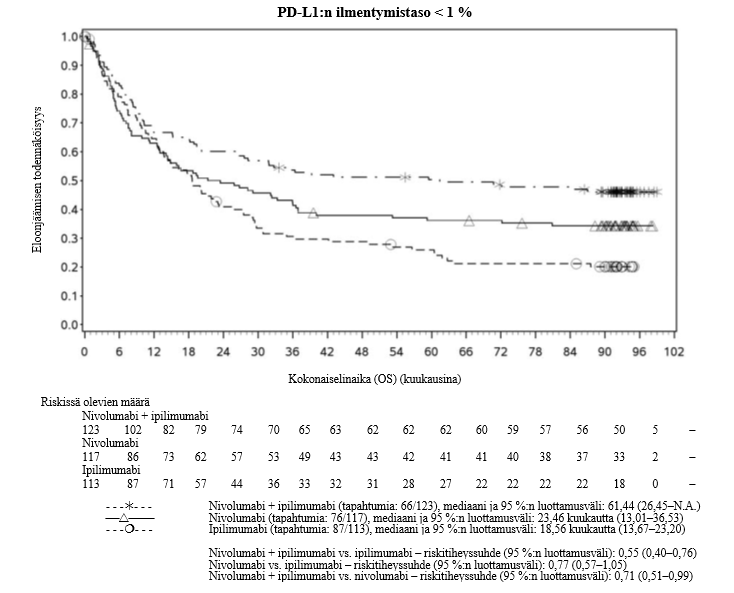
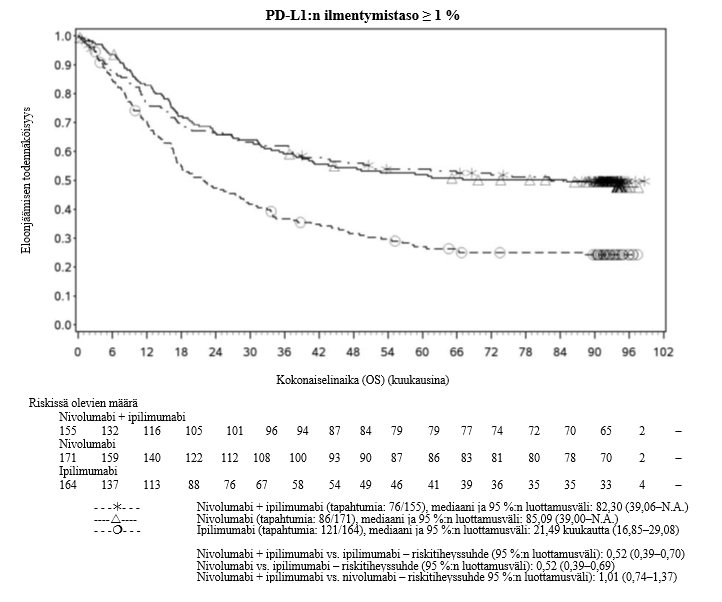
Objektiivisen vasteen saavuttaneiden osuuden analyysin seuranta-aika oli vähintään 90 kuukautta. Vasteiden yhteenveto on taulukossa 27.
Taulukko 27: Objektiivinen vaste (CA209067)
| nivolumabi + |
nivolumabi |
ipilimumabi |
|---|---|---|---|
Objektiivinen vaste | 183 (58 %) | 142 (45 %) | 60 (19 %) |
(95 %:n luottamusväli) | (52,6–63,8) | (39,4–50,6) | (14,9–23,8) |
Ristitulosuhde (vs. ipilimumabi) | 6,35 | 3,5 |
|
(95 %:n luottamusväli) | (4,38–9,22) | (2,49–5,16) |
|
Täydellinen vaste (CR) | 71 (23 %) | 59 (19 %) | 19 (6 %) |
Osittainen vaste (PR) | 112 (36 %) | 83 (26 %) | 41 (13 %) |
Stabiili tauti (SD) | 38 (12 %) | 29 (10 %) | 69 (22 %) |
Vasteen kesto |
|
|
|
Mediaani (vaihteluväli), kuukausina | N.A. (69,1–N.A.) | 90,8 (45,7–N.A.) | 19,3 (8,8–47,4) |
Osuus ≥ 12 kuukauden keston kohdalla | 68 % | 73 % | 44 % |
Osuus ≥ 24 kuukauden keston kohdalla | 58 % | 63 % | 30 % |
Objektiivisen vasteen saavuttaneiden osuus (95 %:n luottamusväli) kasvaimen PD‑L1‑ilmentymittäin |
|
| |
< 5 % | 56 % (48,7–62,5) | 43 % (36–49,8) | 18 % (12,8–23,8) |
≥ 5 % | 72 % (59,9–82,3) | 59 % (47,2–69,6) | 21 % (12,7–32,3) |
< 1 % | 54 % (44,4–62,7) | 36 % (27,2–45,3) | 18 % (11,2–26,0) |
≥ 1 % | 65 % (56,4–72) | 55 % (47,2–62,6) | 20 % (13,7–26,4) |
Kummatkin nivolumabihoitoa sisältäneet tutkimushaarat osoittivat pelkkään ipilimumabihoitoon verrattuna merkitsevää etua etenemisvapaan elinajan ja kokonaiselinajan sekä objektiivisen vasteen saavuttaneiden osuuden suhteen. Havaitut etenemisvapaan elinajan (PFS) tulokset osoitettiin 18 kuukauden kohdalla ja objektiivisen vasteen saavuttaneiden osuuden sekä kokonaiselinajan tulokset 28 kuukauden kohdalla tehdyssä seurannassa johdonmukaisesti kaikissa alaryhmissä, joihin sisältyi ECOG-toimintakykystatus, BRAF-status, M‑vaihe, ikä, aiempi aivometastaasi ja LDH lähtötilanteessa. Tämä havainto pysyi muuttumattomana kokonaiselinajan tulosten osalta seuranta-ajan ollessa vähintään 90 kuukautta.
Haittavaikutuksen vuoksi yhdistelmähoidon 28 kuukauden seuranta-ajan jälkeen keskeyttäneiden 131 potilaan objektiivisen vasteen saavuttaneiden osuus oli 71 % (93/131), josta 20 % (26/131) saavutti täydellisen vasteen, eikä kokonaiselinajan mediaania saavutettu.
Kummallakin nivolumabihoitoa sisältäneellä tutkimushaaralla oli pelkkään ipilimumabihoitoon verrattuna parempi objektiivisen vasteen saavuttaneiden osuus PD‑L1-ilmentymistasosta riippumatta. Objektiivisen vasteen saavuttaneiden osuudet olivat kaikilla PD‑L1-ilmentymistasoilla korkeammat nivolumabi–ipilimumabi-yhdistelmähoitoa saaneessa ryhmässä kuin nivolumabi-monoterapiaa saaneessa ryhmässä (taulukko 27) 90 kuukauden seuranta-ajan jälkeen parhaan saavutetun kokonaisvasteen, täydellisen vasteen, korreloidessa paremman eloonjäämisasteen kanssa.
Potilailla, joilla oli PD‑L1 ≥ 5 % -ilmentymistason kasvain, vasteen mediaanikesto oli 78,19 kuukautta 90 kuukauden seuranta-ajan jälkeen yhdistelmähoitoa saaneessa ryhmässä (vaihteluväli 18,07–N.A.), 77,21 kuukautta nivolumabi-monoterapiaa saaneessa ryhmässä (vaihteluväli 26,25–N.A.) ja 31,28 kuukautta (vaihteluväli 6,08–N.A.) ipilimumabia saaneessa ryhmässä. Potilailla, joilla oli PD‑L1 < 5 % ‑ilmentymistason kasvain, vasteen mediaanikestoa ei saavutettu yhdistelmähoitoa saaneessa ryhmässä (vaihteluväli 61,93–N.A.). Vasteen mediaanikesto oli 90,84 kuukautta nivolumabi-monoterapiaa saaneessa ryhmässä (vaihteluväli 50,43–N.A.) ja 19,25 kuukautta (vaihteluväli 5,32–47,44) ipilimumabi-monoterapiaa saaneessa ryhmässä.
Kasvaimen vasteita ja etenemisvapaata elinaikaa sekä kokonaiselinaikaa koskevat merkitykselliset päätetapahtumat huomioon ottaen selkeää raja-arvoa PD‑L1-ilmentymiselle ei voida luotettavasti määritellä. Tämän havainnollistavan usean satunnaismuuttujan analyysin tulosten perusteella voitiin tunnistaa potilaan ja kasvaimen erityispiirteitä (ECOG-toimintakykystatus, M‑vaihe, LDH lähtötilanteessa, BRAF-mutaatiostatus, PD‑L1-status ja sukupuoli), joilla voi olla merkitystä eloonjäämisen kannalta.
Teho BRAF-statuksen mukaan:
Nivolumabi–ipilimumabi-yhdistelmähoidolle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla PFS‑mediaani oli 90 kuukauden seurannan jälkeen 16,76 kuukautta (95 %:n luottamusväli 8,28–32,0) ja potilailla, joilla oli BRAF‑villityypin kasvain, PFS‑mediaani oli 11,7 kuukautta (95 %:n luottamusväli 7,0–19,32). Nivolumabi-monoterapialle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla PFS‑mediaani oli 5,62 kuukautta (95 %:n luottamusväli 2,79–9,46) ja potilailla, joilla oli BRAF‑villityypin kasvain, PFS‑mediaani oli 8,18 kuukautta (95 %:n luottamusväli 5,13–19,55. Ipilimumabi-monoterapialle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla etenemisvapaan elinajan mediaani oli 3,09 kuukautta (95 %:n luottamusväli 2,79–5,19) ja potilailla, joilla oli BRAF-villityypin kasvain, etenemisvapaan elinajan mediaani oli 2,83 kuukautta (95 %:n luottamusväli: 2,76–3,06).
Nivolumabi–ipilimumabi-yhdistelmähoidolle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla objektiivisen vasteen saavuttaneiden osuus oli 90 kuukauden seurannan jälkeen 67,0 % (95 %:n luottamusväli 57,0–75,9; n = 103) ja potilailla, joilla oli BRAF‑villityypin kasvain, objektiivisen vasteen saavuttaneiden osuus oli 54,0 % (95 %:n kokonaisvaste 47,1–60,9; n = 211). Nivolumabi-monoterapialle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla objektiivisen vasteen saavuttaneiden osuus oli 37,87 % (95 %:n luottamusväli 28,2–48,1; n = 98) ja potilailla, joilla oli BRAF‑villityypin kasvain, objektiivisen vasteen saavuttaneiden osuus oli 48,2 % (95 %:n luottamusväli 41,4–55,0; n = 218). Ipilimumabi-monoterapialle satunnaistetuilla BRAF V600 ‑mutaatiopositiivisilla potilailla objektiivisen vasteen saavuttaneiden osuus oli 23,0 % (95 %:n luottamusväli 15,2–32,5; n = 100) ja potilailla, joilla oli BRAF‑villityypin kasvain, objektiivisen vasteen saavuttaneiden osuus oli 17,2 % (95 %:n luottamusväli 12,4–22,9; n = 215).
Kokonaiselinajan mediaania ei saavutettu BRAF V600 ‑mutaatiopositiivisilla potilailla 90 kuukauden seuranta-ajan jälkeen yhdistelmähoitoa saaneessa ryhmässä, ja nivolumabi-monoterapiaryhmässä kokonaiselinajan mediaani oli 45,5 kuukautta. Kokonaiselinajan mediaani oli BRAF V600 ‑mutaatiopositiivisilla potilailla ipilimumabi-monoterapiaryhmässä 24,6 kuukautta. Kokonaiselinajan mediaani potilailla, joilla oli BRAF‑villityypin kasvain, oli yhdistelmähoitoa saaneessa ryhmässä 39,06 kuukautta, nivolumabi-monoterapiaryhmässä 34,37 kuukautta ja ipilimumabi-monoterapiaryhmässä 18,5 kuukautta. Kokonaiselinajan riskitiheyssuhteet olivat verrattaessa nivolumabi–ipilimumabi-yhdistelmähoitoa nivolumabi-monoterapiaan BRAF V600 ‑mutaatiopositiivisilla potilailla 0,66 (95 %:n luottamusväli 0,44–0,98) ja BRAF‑villityypin potilailla 0,95 (95 %:n luottamusväli 0,74–1,22).
Satunnaistettu vaiheen 2 tutkimus, nivolumabi–ipilimumabi vs. ipilimumabi (CA209069)
CA209069 oli satunnaistettu, vaiheen 2 kaksoissokkoutettu tutkimus, jossa verrattiin nivolumabi–ipilimumabi-yhdistelmähoitoa ipilimumabihoitoon. Tutkimukseen osallistui 142 potilasta, joilla oli edennyt melanooma, jota ei voida kirurgisesti poistaa tai joka on metastasoinut. Kelpoisuusvaatimukset olivat samanlaiset kuin tutkimuksessa CA209067 ja primaarianalyysi potilaiden BRAF‑villityypin melanoomasta oli samanlainen (77 prosentilla potilaista). Tutkijan arvioima objektiivisen vasteen saavuttaneiden osuus (ORR) oli 61 % (95 %:n luottamusväli 48,9–72,4) ja yhdistelmähoitoryhmässä (n = 72) kun se ipilimumabi-hoitohaarassa (n = 37) oli 11 % (95 %:n luottamusväli 3,0–25,4). Arvioidut 2 ja 3 vuoden kokonaiselinaikaluvut olivat yhdistelmähoitoryhmässä (n = 73) 68 % (95 %:n luottamusväli 56–78) ja 61 % (95 %:n luottamusväli 49–71) ja ipilimumabiryhmässä (n = 37) 53 % (95 %:n luottamusväli 36–68) ja 44 % (95 %:n luottamusväli 28–60).
Ei-pienisoluinen keuhkosyöpä
Ei‑pienisoluisen keuhkosyövän esiliitännäishoito
Satunnaistettu, avoin vaiheen 3 tutkimus nivolumabista yhdistelmähoitona platinapohjaisen kemoterapian kanssa vs. platinapohjainen kemoterapia (CA209816)
Nivolumabin ja kolmen platinapohjaisen kemoterapiajakson yhdistelmähoidon turvallisuutta ja tehoa arvioitiin vaiheen 3 satunnaistetussa avoimessa tutkimuksessa (CA209816). Tutkimuksessa oli mukana potilaita, joiden ECOG-toimintakykyluokka oli 0 tai 1, joiden tauti oli mitattavissa (RECIST 1.1 ‑kriteerien perusteella) ja joilla oli kirurgisesti poistettavissa oleva, histologisesti vahvistettu IB asteen (≥ 4 cm), II asteen tai IIIA asteen ei‑pienisoluinen keuhkosyöpä (AJCC / Union for International Cancer Control [UICC]) -luokittelun 7. painoksen mukaisesti).
Seuraavien valintakriteereiden perusteella voidaan määritellä käyttöaiheen mukaiset potilaat, joilla on suuri riski taudin uusiutumiseen ja jotka edustavat potilasryhmää, jolla on AJCC/UICC:n luokittelun 7. painoksen mukaisesti II–IIIA asteen tauti: potilaan kasvaimen koko on ≥ 5 cm; potilaalla on N1‑ tai N2-tauti (primaarisen kasvaimen koosta riippumatta); potilaalla on useita kasvainkyhmyjä joko yhdessä lohkossa tai useammassa ipsilateraalisessa lohkossa; potilaan kasvaimet ovat tunkeutuneet rintakehän rakenteisiin (kasvain tunkeutunut suoraan viskeraaliseen pleuraan, parietaaliseen pleuraan, rintakehän seinämään, palleaan, palleahermoon, mediastinaaliseen pleuraan, parietaaliseen perikardiumiin, välikarsinaan, sydämeen, suuriin verisuoniin, henkitorveen, palaavahermoon, ruokatorveen, nikamansolmuun tai henkitorven harjuun); tai potilaalla on pääkeuhkoputkeen levinneitä kasvaimia; tai potilaan kasvaimiin liittyy atelektaasi tai hiluksen seutuun ulottuva tai koko keuhkon laajuinen obstruktiivinen pneumoniitti.
Tutkimukseen ei osallistunut potilaita, joiden taudin levinneisyys oli N2 ja joilla kasvaimet olivat lisäksi tunkeutuneet välikarsinaan, sydämeen, suuriin verisuoniin, henkitorveen, palaavahermoon, ruokatorveen, nikamansolmuun tai henkitorven harjuun, tai joilla oli yksi tai useampia erillisiä kasvainkyhmyjä eri ipsilateraalisessa lohkossa.
Tutkimuksesta suljettiin pois ne potilaat, joilla oli leikkaukseen soveltumaton tai metastaattinen NSCLC, joilla oli tunnistettu EGFR-mutaatioita tai ALK-translokaatioita (EGFR-mutaatioiden tai ALK-translokaatioiden testaus ei ollut pakollista tutkimukseen otettaessa) ja joilla oli vähintään asteen 2 perifeerinen neuropatia, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila. Satunnaistaminen ositettiin kasvaimen PD‑L1-ilmentymistason (≥ 1 % vs. < 1 % tai ei määritettävissä), taudin asteen (IB/II vs. IIIA) ja sukupuolen (mies vs. nainen) mukaan. Potilaat otettiin mukaan tutkimukseen kasvaimen PD‑L1:n ilmentymisestä riippumatta. Kasvaimen PD‑L1:n ilmentyminen määritettiin käyttämällä PD‑L1 IHC 28–8 pharmDx ‑testausmenetelmää.
Kaikkiaan 358 potilasta satunnaistettiin saamaan joko nivolumabin ja platinapohjaisen kemoterapian yhdistelmähoitoa (n = 179) tai platinapohjaista kemoterapiaa (n = 179). Nivolumabin ja kemoterapian yhdistelmähoitoa saaneen ryhmän potilaat saivat nivolumabia 360 mg laskimoon 30 minuutin kuluessa yhdessä platinapohjaisen kemoterapian kanssa joka kolmas viikko enintään kolmen hoitojakson ajan. Kemoterapiaryhmän potilaat saivat platinapohjaista kemoterapiaa kolmen viikon välein enintään kolmen hoitojakson ajan. Platinapohjainen kemoterapia koostui tutkijan valinnan mukaan paklitakselista (175 mg/m2 tai 200 mg/m2) ja karboplatiinista (AUC 5 tai AUC 6) (kaikki histologiat); pemetreksedistä (500 mg/m2) ja sisplatiinista (75 mg/m2) (ei‑levyepiteeliperäinen histologia) tai gemsitabiinista (1000 mg/m2 tai 1250 mg/m2) ja sisplatiinista (75 mg/m2) (levyepiteeliperäinen histologia). Kemoterapiaryhmässä oli lisäksi kaksi muuta hoitovaihtoehtoa: vinorelbiini (25 mg/m2 tai 30 mg/m2) ja sisplatiini (75 mg/m2) tai dosetakseli (60 mg/m2 tai 75 mg/m2) ja sisplatiini (75 mg/m2) (kaikki histologiat).
Kasvaimet arvioitiin lähtötasolla, 14 vuorokauden kuluessa leikkauksesta, leikkauksen jälkeen 12 viikon välein kahden vuoden ajan, sen jälkeen kuuden kuukauden välein kolmen vuoden ajan ja sen jälkeen kerran vuodessa viiden vuoden ajan taudin uusiutumiseen tai etenemiseen saakka. Ensisijaiset tehokkuusmuuttujat olivat päätetapahtumaton elinaika (event-free survival, EFS) riippumattoman keskitetyn arvioijatahon (Blinded Independent Central Review, BICR) arvioimana ja täydellisen patologisen vasteen (pCR) saavuttaneet riippumattoman sokkoutetun patologisen arvioinnin (BIPR) perusteella. Kokonaiselinaika oli tärkein toissijainen tehokkuusmuuttuja, ja havainnollistaviin päätetapahtumiin kuului leikkauksen toteutettavuus.
Hoitoryhmien lähtötason ominaisuudet lähtöryhmien mukaisessa populaatiossa olivat pääosin tasapainossa. Iän mediaani oli 65 vuotta (vaihteluväli 34–84). Potilaista 51 % oli ≥ 65‑vuotiaita ja 7 % potilaista oli ≥ 75‑vuotiaita. Potilaista 50 % oli aasialaisia, 47 % valkoihoisia ja 71 % miehiä. Lähtötason ECOG-toimintakykyluokka oli 0 (67 %) tai 1 (33 %). Potilaista 50 prosentilla PD‑L1 oli ≥ 1 % ja 43 prosentilla PD‑L1 oli < 1 %. Potilaista 5 prosentilla taudin aste oli IB, 17 prosentilla IIA, 13 prosentilla IIB ja 64 prosentilla IIIA. Histologia oli 51 prosentilla levyepiteeliperäinen ja 49 prosentilla ei‑levyepiteeliperäinen. Potilaista 89 % oli entisiä/nykyisiä tupakoitsijoita. Definitiivinen leikkaus tehtiin 83 prosentille potilaista nivolumabin ja kemoterapian yhdistelmähoitoa saaneessa ryhmässä ja 75 prosentille kemoterapiaryhmässä. Systeemistä liitännäishoitoa sai 14,8 % potilaista nivolumabin ja kemoterapian yhdistelmähoitoa saaneessa ryhmässä ja 25 % kemoterapiaryhmässä.
Lopullisessa pCR-analyysissä ja ennalta määritetyssä päätetapahtumattoman elinajan (EFS) välianalyysissä (seuranta-aika vähintään 21 kuukautta) todettiin tilastollisesti merkitsevää pCR:n ja EFS:n paranemista potilailla, jotka oli satunnaistettu saamaan nivolumabin ja kemoterapian yhdistelmähoitoa, verrattuna potilaisiin, jotka saivat vain kemoterapiaa. Täydellinen patologinen vaste (pCR) saavutettiin 24 %:lla nivolumabin ja kemoterapian yhdistelmähoitoa saaneessa ryhmässä ja 2,2 %:lla pelkkää kemoterapiaa saaneessa ryhmässä (pCR:n ero 21,6, 99 %:n luottamusväli: 13,0–30,3; pCR:n ristitulosuhde 13,9, 99 %:n luottamusväli: 3,49–55,75; ositettu p‑arvo < 0,0001). Päätetapahtumattoman elinajan (EFS) mediaani oli 31,6 kuukautta nivolumabin ja kemoterapian yhdistelmähoitoa saaneessa ryhmässä ja 20,8 kuukautta pelkkää kemoterapiaa saaneessa ryhmässä (HR = 0,63, 97,38 %:n luottamusväli: 0,43–0,91; ositetun log‑rank-testin p‑arvo 0,0052). Ennalta määritetyssä välianalyysissä kokonaiselinajan riskitiheyssuhde (seuranta-aika vähintään 21 kuukautta) oli nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla 0,57 (99,67 %:n luottamusväli: 0,30–1,07) verrattuna kemoterapiaa saaneisiin.
Lopullisessa kokonaiselinajan analyysissä, jonka seuranta-aika oli vähintään 59,9 kuukautta, kokonaiselinajan riskitiheyssuhde oli 0,72 (95,18 %:n luottamusväli: 0,52–1,00), ja tulos oli tilastollisesti merkitsevä p‑arvolla 0,0479 (ositettu log‑rank-testi) verrattaessa nivolumabin ja kemoterapian yhdistelmähoitoa pelkkään kemoterapiaan.
Eksploratiivinen alaryhmäanalyysi kasvaimen PD‑L1-ilmentymistason ja taudin asteen mukaan
Taulukossa 28 on yhteenveto eksploratiivisen analyysin tärkeimmistä tehoon liittyvistä tuloksista potilaiden alaryhmässä, jossa kasvaimen PD‑L1-ilmentymistaso oli ≥ 1 %, taudin aste II–IIIA ja seuranta-aika vähintään 32,9 kuukautta.
Taulukko 28: Tulokset tehosta potilailla, joiden kasvaimen PD‑L1 oli ≥ 1 % ja taudin aste II–IIIA* (CA209816)
| nivolumabi + kemoterapia | kemoterapia |
|---|---|---|
Päätetapahtumaton elinaika (EFS) BICR:n arvioimana | ||
Tapahtumia | 22 (27,2 %) | 39 (45,3 %) |
Riskitiheyssuhdea (95 %:n luottamusväli) | 0,49 (0,29–0,83) | |
Mediaani (kuukautta)b (95 %:n luottamusväli) | NR (44,42–NR) | 26,71 (13,40–NR) |
Täydellinen patologinen vaste (pCR) BIPR:n perusteella | ||
Vasteet | 26 (32,1 %) | 2 (2,3 %) |
(95 %:n luottamusväli)c | (22,2–43,4) | (0,3–8,1) |
pCR, ero (95 %:n luottamusväli)d | 29,8 % (19,0–40,7) | |
a Perustuu osittamattomaan Coxin suhteellisen vaaran malliin.
b Kaplan–Meier-arvio.
c Perustuu Clopper–Pearsonin menetelmään.
d Kaksisuuntainen 95 %:n luottamusväli painottamattomalle erolle laskettiin Newcomben menetelmällä.
* AJCC-/UICC-luokittelun 7. painos.
Päätetapahtumattoman elinajan (EFS) seuranta vähintään 32,9 kuukautta, tietojen keruun katkaisuajankohta: 6.9.2022
pCR-tietojen keruun katkaisuajankohta: 28.7.2020
Kuvassa 11 on Kaplan–Meier-kuvaajat päätetapahtumattomasta elinajasta (EFS) potilaiden alaryhmässä, jossa kasvaimen PD‑L1-ilmentymistaso oli ≥ 1 %, taudin aste II–IIIA ja seuranta-aika vähintään 32,9 kuukautta.
Kuva 11: Kaplan-Meier-kuvaajat päätetapahtumattomasta elinajasta potilailla, joiden kasvaimen PD‑L1 oli ≥ 1 % ja taudin aste II–IIIA (CA209816)
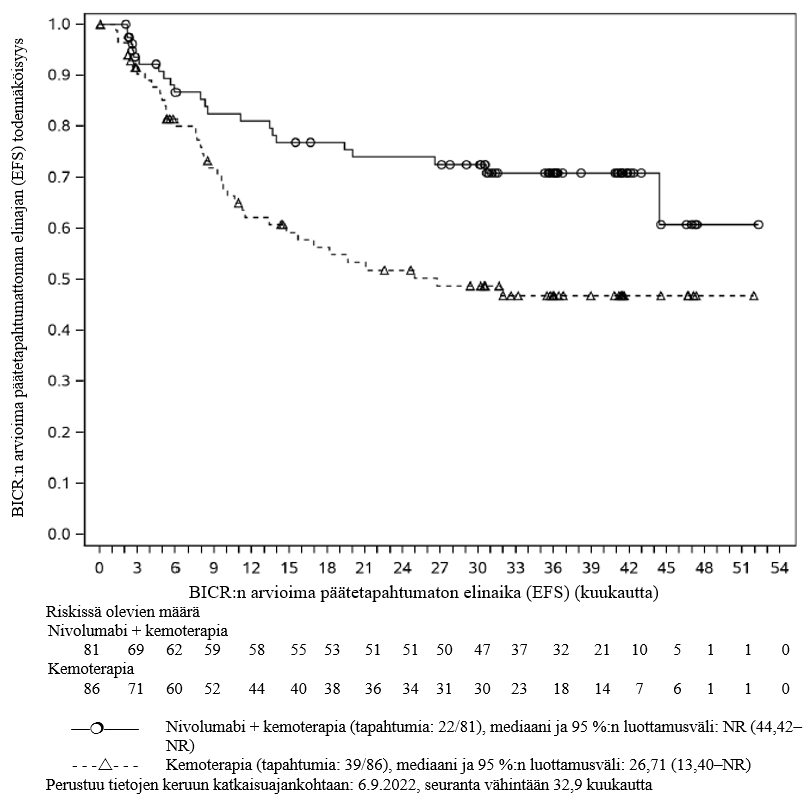
Samaan aikaan päivitetyn EFS-analyysin kanssa tehtiin ennalta määritetty kokonaiselinajan välianalyysi (seuranta-aika vähintään 32,9 kuukautta). Kokonaiselinajan eksploratiivinen, deskriptiivinen riskitiheyssuhde oli 0,43 (95 %:n luottamusväli: 0,22–0,83) potilailla, joilla kasvaimen PD‑L1-ilmentymistaso oli ≥ 1 % ja taudin aste II–IIIA nivolumabin ja kemoterapian yhdistelmähoitoa saaneessa ryhmässä verrattuna pelkkää kemoterapiaa saaneeseen ryhmään.
Lopullisessa analyysissä (seuranta-aika vähintään 59,9 kuukautta) kokonaiselinajan riskitiheyssuhde oli 0,59 (95 %:n luottamusväli: 0,35–0,98) potilailla, joilla kasvaimen PD‑L1-ilmentymistaso oli ≥ 1 % ja taudin aste II–IIIA, nivolumabin ja kemoterapian yhdistelmähoitoa saaneessa ryhmässä verrattuna pelkkää kemoterapiaa saaneeseen ryhmään. Kuvassa 12 on esitetty vastaavat kokonaiselinajan Kaplan–Meier-kuvaajat.
Kuva 12: Kaplan-Meier-kuvaajat kokonaiselinajasta potilailla, joiden kasvaimen PD‑L1 oli ≥ 1 % ja taudin aste II–IIIA (CA209816)
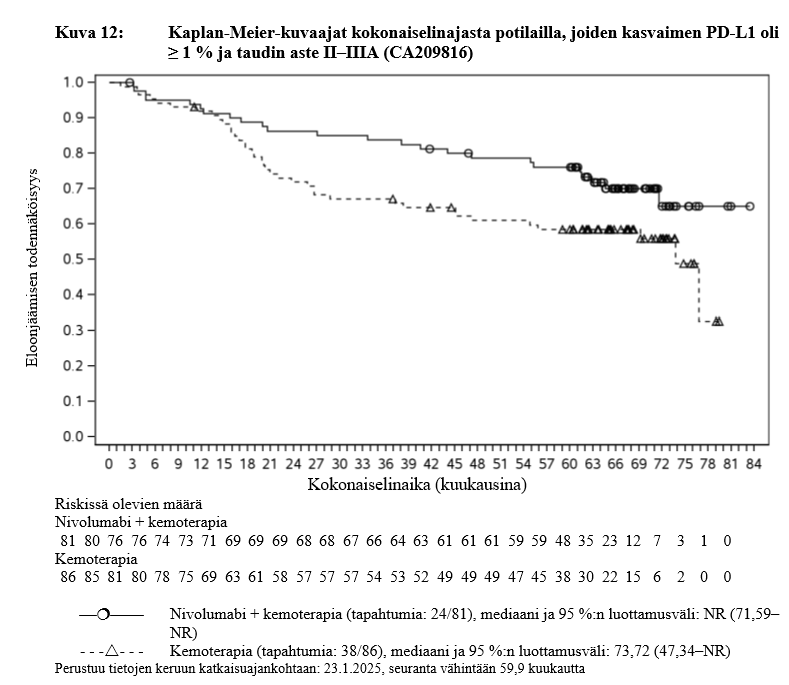
Ei‑pienisoluisen keuhkosyövän esiliitännäishoito ja liitännäishoito
Satunnaistettu, kaksoissokkoutettu vaiheen 3 tutkimus esiliitännäishoitona annetusta nivolumabista yhdistelmähoitona platinapohjaisen kemoterapian kanssa vs. platinapohjainen kemoterapia sekä liitännäishoitona annetusta nivolumabimonoterapiasta vs. lumehoito (CA20977T)
Nivolumabin ja neljän platinapohjaisen kemoterapiajakson yhdistelmähoidon ja sitä seuraavan nivolumabimonoterapian turvallisuutta ja tehoa arvioitiin satunnaistetussa kaksoissokkoutetussa tutkimuksessa (CA20977T). Tutkimuksessa oli mukana potilaita, joiden ECOG-toimintakykyluokka oli 0 tai 1 ja joiden kasvaimet olivat kirurgisesti poistettavissa ja joilla epäiltiin olevan tai oli histologisesti vahvistettu IIA asteen (> 4 cm) – IIIB asteen (T3–T4 N2) ei‑pienisoluinen keuhkosyöpä (American Joint Committee on Cancer [AJCC] ‑luokittelun 8. painoksen mukaisesti). Potilaat otettiin mukaan tutkimukseen kasvaimen PD‑L1:n ilmentymisestä riippumatta.
Seuraavien valintakriteereiden perusteella voidaan määritellä käyttöaiheen mukaiset potilaat, joilla on suuri riski taudin uusiutumiseen ja jotka edustavat potilasryhmää, jolla on AJCC/UICC:n luokittelun 8. painoksen mukaisesti IIA–IIIB asteen tauti: potilaan kasvaimen koko on > 4 cm; potilaalla on N1‑ tai N2‑tauti (primaarisen kasvaimen koosta riippumatta); potilaalla on useita kasvainkyhmyjä joko yhdessä lohkossa tai useammassa ipsilateraalisessa lohkossa; potilaan kasvaimet ovat tunkeutuneet rintakehän rakenteisiin (kasvain tunkeutunut suoraan viskeraaliseen pleuraan, parietaaliseen pleuraan, rintakehän seinämään, palleaan, palleahermoon, mediastinaaliseen pleuraan, parietaaliseen perikardiumiin, välikarsinaan, sydämeen, suuriin verisuoniin, henkitorveen, palaavahermoon, ruokatorveen, nikamansolmuun tai henkitorven harjuun); potilaalla on pääkeuhkoputkeen levinneitä kasvaimia; tai potilaan kasvaimiin liittyy atelektaasi tai hiluksen seutuun ulottuva tai koko keuhkon laajuinen obstruktiivinen pneumoniitti.
Tutkimuksesta suljettiin pois ne potilaat, joilla oli leikkaukseen soveltumaton tai metastaattinen ei‑pienisoluinen keuhkosyöpä, EGFR-mutaatioita tai tunnistettuja ALK-translokaatioita (ALK-translokaatioiden testaus ei ollut pakollista tutkimukseen otettaessa), aivometastaaseja, vähintään asteen 2 perifeerinen neuropatia, interstitiaalinen keuhkosairaus tai aktiivinen ei‑infektiivinen pneumoniitti (oireinen ja/tai hoitoa vaativa), aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila.
Kaikkiaan 461 potilasta satunnaistettiin saamaan joko nivolumabin ja platinapohjaisen kemoterapian yhdistelmähoitoa ja sen jälkeen nivolumabimonoterapiaa (n = 229) tai platinapohjaista kemoterapiaa ja sen jälkeen lumelääkettä (n = 232). Esiliitännäishoitovaiheessa potilaat saivat enintään neljän jakson ajan joko 360 mg nivolumabia laskimoon 30 minuutin kuluessa ja platinapohjaista kahden solunsalpaajan yhdistelmää joka kolmas viikko tai lumelääkettä ja platinapohjaista kahden solunsalpaajan yhdistelmää joka kolmas viikko, kunnes sairaus eteni tai ilmaantui ei‑hyväksyttävää toksisuutta. Molempien hoitoryhmien potilaille voitiin antaa leikkauksenjälkeistä sädehoitoa (PORT) tavanomaisena hoitona. Liitännäishoitovaiheessa, 90 vuorokauden sisällä leikkauksesta, potilaat saivat enintään 13 jakson ajan joko 480 mg nivolumabia laskimoon 30 minuutin kuluessa joka neljäs viikko tai lumelääkettä joka neljäs viikko, kunnes sairaus eteni tai uusiutui tai ilmaantui ei‑hyväksyttävää toksisuutta. Platinapohjainen kemoterapia koostui paklitakselista (175 mg/m2 tai 200 mg/m2) ja karboplatiinista (AUC 5 tai AUC 6) (kaikki histologiat); pemetreksedistä (500 mg/m2) ja sisplatiinista (75 mg/m2) tai karboplatiinista (AUC 5 tai AUC 6) (ei‑levyepiteeliperäinen histologia) tai sisplatiinista (75 mg/m2) ja dosetakselista (75 mg/m2) (levyepiteeliperäinen histologia).
Satunnaistamisen ositustekijöitä olivat kasvaimen PD‑L1:n ilmentymistaso (≥ 1 % vs. < 1 % vs. määrittämätön / ei arvioitavissa), taudin aste (II aste vs. III aste) ja kasvaimen histologia (levyepiteeliperäinen vs. ei‑levyepiteeliperäinen). Kasvaimen PD‑L1:n ilmentymistasot arvioitiin PD‑L1 IHC 28-8 pharmDx -testillä. Kasvaimet arvioitiin lähtötasolla, 14 vuorokauden kuluessa esiliitännäishoidon viimeisestä annoksesta ja ennen leikkausta, leikkauksenjälkeisen liitännäishoidon aloittamista edeltävien 7 vuorokauden aikana, liitännäishoidon ensimmäisen annoksen jälkeen 12 viikon välein kahden vuoden ajan ja sitten 24 viikon välein enintään viiden vuoden ajan tai kunnes BICR vahvisti sairauden uusiutuneen tai edenneen.
CA20977T‑tutkimuksen 442 potilaasta 256:lla (58 %:lla) kasvaimen PD‑L1:n ilmentymä oli ≥ 1 % PD‑L1 IHC 28‑8 pharmDx -testin perusteella. Iän mediaani oli 66 vuotta (vaihteluväli: 35–86). Potilaista 55 % oli ≥ 65‑vuotiaita ja 7 % oli ≥ 75‑vuotiaita. Potilaista 69 % oli valkoihoisia, 28 % aasialaisia, 2 % tummaihoisia ja 75 % miehiä. Lähtötason ECOG-toimintakykyluokka oli 0 (59 %) tai 1 (41 %). Potilaista 36 prosentilla taudin aste oli II, 63 prosentilla III, 24 prosentilla N1 ja 39 prosentilla N2. Potilaista 25 prosentilla oli yhden alueen ja 14 prosentilla usean alueen tauti. Kasvainten histologia oli 61 prosentilla levyepiteeliperäinen ja 39 prosentilla ei-levyepiteeliperäinen. Potilaista 91 % oli entisiä/nykyisiä tupakoitsijoita.
Definitiivinen leikkaus tehtiin 78 prosentille potilaista, jotka saivat esiliitännäishoitona nivolumabin ja platinapohjaista kahden solunsalpaajan yhdistelmää ja liitännäishoitona nivolumabia, ja 77 prosentille potilaista, jotka saivat esiliitännäishoitona lumelääkettä ja platinapohjaista kahden solunsalpaajan yhdistelmää ja sen jälkeen lumelääkettä. Leikkauksenjälkeistä sädehoitoa sai noin 5 prosenttia kummankin hoitoryhmän potilaista.
Ensisijainen tehokkuusmuuttuja oli BICR:n arvioima päätetapahtumaton elinaika (EFS). Muita tehokkuusmuuttujia olivat kokonaiselinaika (OS), täydellinen patologinen vaste (pCR) ja merkittävä patologinen vaste riippumattoman sokkoutetun patologisen arvioinnin (BIPR) perusteella.
Ennalta määritetyssä välianalyysissä, jossa olivat mukana kaikki satunnaistetut potilaat ja seuranta-ajan mediaani oli 25,4 kuukautta (vaihteluväli: 15,7–44,2 kuukautta), päätetapahtumattoman elinajan osoitettiin parantuneen tilastollisesti merkitsevästi. Päätetapahtumattoman elinajan mediaania ei saavutettu (95 %:n luottamusväli: 28,94–NE) nivolumabin ja kemoterapian yhdistelmää / nivolumabia saaneessa ryhmässä, ja lumelääkkeen ja kemoterapian yhdistelmää / lumelääkettä saaneessa ryhmässä se oli 18,43 kuukautta (95 %:n luottamusväli: 13,63–28,06; HR = 0,58, 97,36 %:n luottamusväli: 0,42–0,81; ositetun log‑rank-testin p‑arvo 0,00025). Ennalta määritetyssä välianalyysissä, jossa olivat mukana kaikki satunnaistetut potilaat ja seuranta-ajan mediaani oli 41 kuukautta (vaihteluväli: 31,3–59,8 kuukautta), kokonaiselinajan mediaania ei saavutettu nivolumabin ja kemoterapian yhdistelmää / nivolumabia saaneessa ryhmässä eikä lumelääkkeen ja kemoterapian yhdistelmää / lumelääkettä saaneessa ryhmässä (HR = 0,85, 97,63 %:n luottamusväli: 0,58–1,25).
Eksploratiivinen alaryhmäanalyysi kasvaimen PD‑L1-ilmentymistason mukaan
Taulukossa 29 ja kuvassa 13 esitetään päätetapahtumaton elinaika potilaiden alaryhmässä, jossa kasvaimen PD‑L1:n ilmentymistaso oli ≥ 1 % ja seuranta-ajan mediaani 41 kuukautta (vaihteluväli: 31,3–59,8 kuukautta).
Taulukko 29: Tulokset tehosta potilailla, joiden kasvaimen PD‑L1 oli ≥ 1 % (CA20977T)
nivolumabi ja kemoterapia / nivolumabi | lumelääke ja kemoterapia / lumelääke | |
|---|---|---|
Päätetapahtumaton elinaika (EFS) BICR:n arvioimana | ||
Tapahtumia (%) | 47 (37 %) | 70 (55 %) |
Mediaani (kuukautta)a (95 %:n luottamusväli) | 46,55 (35,81–NE) | 15,08 (9,33–31,41) |
Riskitiheyssuhdeb (95 %:n luottamusväli) | 0,53 (0,36–0,76) | |
NE = ei arvioitavissa
Päätetapahtumattoman elinajan (EFS) seuranta vähintään 31,3 kuukautta; tietojen keruun katkaisuajankohta: 11.11.2024.
a Kaplan–Meier-arvio.
b Perustuu osittamattomaan Coxin suhteellisen vaaran malliin.
Kuva 13: Kaplan–Meier-kuvaajat päätetapahtumattomasta elinajasta potilailla, joiden kasvaimen PD‑L1 oli ≥ 1 % (CA20977T)
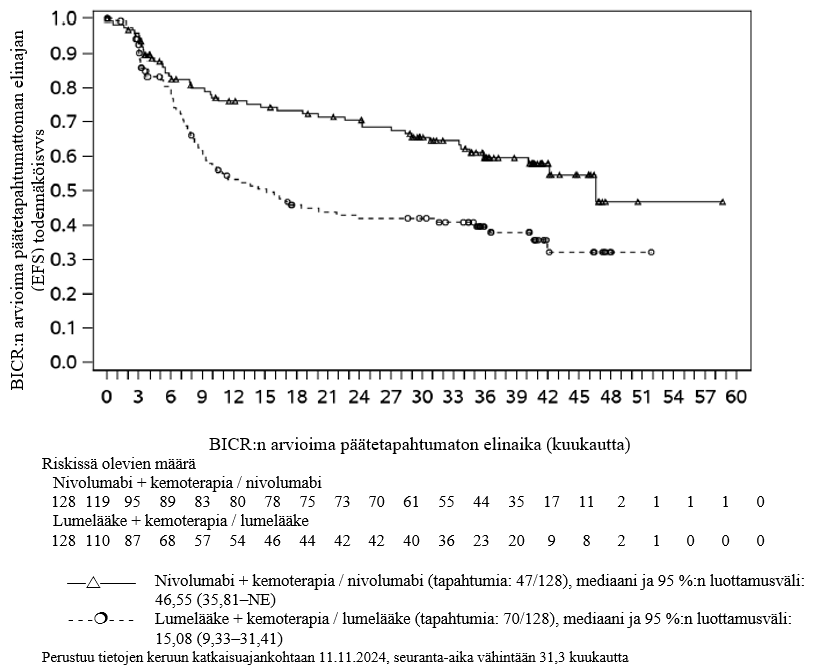
Samaan aikaan päivitetyn EFS-analyysin kanssa tehtiin ennalta määritetty kokonaiselinajan välianalyysi (seuranta-aika vähintään 31,3 kuukautta). Kokonaiselinajan eksploratiivinen, deskriptiivinen riskitiheyssuhde oli 0,61 (95 %:n luottamusväli: 0,39–0,97) potilailla, joilla kasvaimen PD‑L1-ilmentymistaso oli ≥ 1 % nivolumabin ja kemoterapian yhdistelmää / nivolumabia saaneessa ryhmässä verrattuna lumelääkkeen ja kemoterapian yhdistelmää / lumelääkettä saaneeseen ryhmään. Kuvassa 14 on Kaplan–Meier-kuvaajat kokonaiselinajasta (OS) potilaiden alaryhmässä, jossa kasvaimen PD‑L1-ilmentymistaso oli ≥ 1 %.
Kuva 14: Kaplan–Meier-kuvaajat kokonaiselinajasta potilailla, joiden kasvaimen PD‑L1 oli ≥ 1 % (CA20977T)
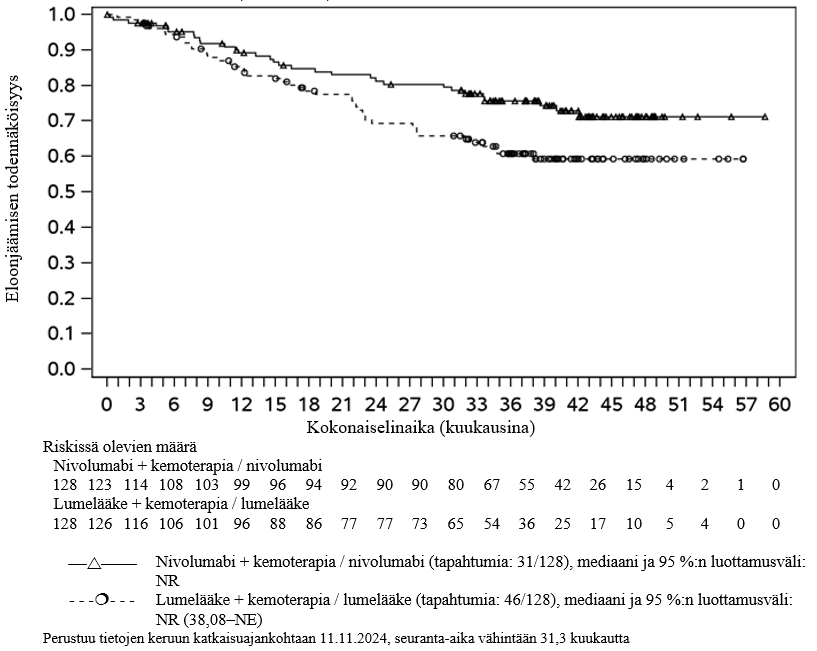
Ei-pienisoluisen keuhkosyövän ensilinjan hoito
Satunnaistettu faasin 3 tutkimus nivolumabista yhdistelmähoitona ipilimumabin ja kahden platinapohjaisen kemoterapiajakson kanssa vs. neljä platinapohjaista kemoterapiajaksoa (CA2099LA)
Kolmen viikon välein annetun nivolumabin (360 mg) ja kuuden viikon välein annetun ipilimumabin (1 mg/kg) ja sekä kahden platinapohjaisen kemoterapiajakson yhdistelmähoidon turvallisuutta ja tehoa arvioitiin satunnaistetussa faasin 3 avoimessa tutkimuksessa (CA2099LA). Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli histologisesti vahvistettu ei-levyepiteeliperäinen tai levyepiteeliperäinen IV asteen tai uusiutunut ei-pienisoluinen keuhkosyöpä (International Association for the Study of Lung Cancer ‑yhdistyksen 7:nnen luokittelun mukaisesti), joiden ECOG‑toimintakykyluokka oli 0 tai 1 ja jotka eivät olleen saaneet aiempaa syöpähoitoa (mukaan lukien EGFR:n ja ALK:n estäjät). Potilaan kasvaimen PD-L1-status ei vaikuttanut tutkimukseen mukaan ottamiseen.
Tutkimuksesta suljettiin pois ne potilaat, joilla oli herkistäviä EGFR-mutaatioita tai ALK:n translokaatioita, aktiivinen (hoitamaton) aivojen etäpesäke, karsinomatoottinen aivokalvotulehdus, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila. Ne potilaat, jotka olivat saaneet aiempaa hoitoa aivojen etäpesäkkeisiin, voitiin ottaa mukaan tutkimukseen, jos heidän neurologinen tilansa palautui lähtötasolle vähintään 2 viikkoa ennen tutkimukseen ottamista ja jos he eivät joko saaneet lainkaan kortikosteroideja tai saivat vakaata tai pienentyvää annosta, joka vastasi < 10 mg:aa prednisonia vuorokaudessa. Satunnaistaminen ositettiin histologian (levyepiteeliperäinen vs. ei-levyepiteeliperäinen), kasvaimen PD-L1‑ilmentymistason (≥ 1 % vs. < 1 %) ja sukupuolen (mies vs. nainen) mukaan.
Kaikkiaan 719 potilaista satunnaistettiin saamaan joko nivolumabin, ipilimumabin ja platinapohjaisen kemoterapian yhdistelmähoitoa (n = 361) tai platinapohjaista kemoterapiaa (n = 358). Nivolumabin, ipilimumabin ja platinapohjaisen kemoterapian yhdistelmähoitoa saaneen ryhmän potilaat saivat nivolumabia 360 mg laskimoon annosteltuna 30 minuutin kuluessa joka kolmas viikko yhdessä ipilimumabin kanssa, jota he saivat 1 mg/kg laskimoon annosteltuna 30 minuutin kuluessa joka kuudes viikko, sekä platinapohjaisen kemoterapian kanssa, jota annettiin joka kolmas viikko kahden hoitojakson ajan. Kemoterapiaryhmän potilaat saivat platinapohjaista kemoterapiaa, jota annettiin kolmen viikon välein neljän hoitojakson ajan; ne potilaat, joiden syöpä oli ei-levyepiteeliperäinen, saattoivat saada valinnaista pemetreksedi-ylläpitohoitoa.
Platinapohjainen kemoterapia koostui karboplatiinista (AUC 5 tai 6) ja pemetreksedistä (500 mg/m2); tai sisplatiinista (75 mg/m2) ja pemetreksedistä (500 mg/m2) ei-levyepiteeliperäisen ei-pienisoluisen keuhkosyövän hoitoon; tai karboplatiinista (AUC 6) ja paklitakselista (200 mg/m2) levyepiteeliperäisen ei-pienisoluisen keuhkosyövän hoitoon.
Hoitoa jatkettiin, kunnes tauti eteni, ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä, tai enintään 24 kuukauden ajan. Hoitoa voitiin jatkaa taudin etenemisen jälkeenkin, jos potilas oli kliinisesti stabiili ja jos tutkija arvioi, että potilas sai hoidosta kliinistä hyötyä. Potilaat, jotka eivät jatkaneet yhdistelmähoitoa ipilimumabin aiheuttaman haittavaikutuksen takia, saivat jatkaa nivolumabi-monoterapiaa. Kasvaimet arvioitiin kuuden viikon välein ensimmäisen tutkimushoidon saamisen jälkeen ensimmäisten 12 kuukauden ajan, minkä jälkeen 12 viikon välein, kunnes tauti eteni tai tutkimushoito lopetettiin.
CA2099LA‑tutkimuksen kaikkien ryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Iän mediaani oli 65 vuotta (vaihteluväli 26–86). 51 % potilaista oli ≥ 65‑vuotiaita ja 10 % oli ≥ 75‑vuotiaita. Suurin osa potilasta oli valkoihoisia (89 %) ja miehiä (70 %). Lähtötason ECOG‑toimintakykyluokka oli 0 (31 %) tai 1 (68 %). 57 %:lla potilaista PD-L1 oli ≥ 1 % ja 37 %:lla PD-L1 oli < 1 %. 31 %:lla histologia oli levyepiteeliperäinen ja 69 %:lla ei-levyepiteeliperäinen. 17 %:lla oli aivojen etäpesäkkeitä, ja 86 % oli entisiä/nykyisiä tupakoitsijoita. Kukaan potilaista ei ollut saanut aiempaa immunoterapiaa.
CA2099LA‑tutkimuksen ensisijainen tehokkuusmuuttuja oli kokonaiselinaika. Tehon muita päätetapahtumia olivat etenemisvapaa elinaika (PFS), objektiivisen vasteen saavuttaneiden osuus (ORR) ja vasteen kesto BICR:n arvioimana.
Tutkimuksen ennalta määritetty välianalyysi, joka tehtiin, kun 351 tapahtumaa oli todettu (87 % lopullisen analyysin tapahtumien suunnitellusta määrästä), osoitti tilastollisesti merkitsevää etua kokonaiselinajassa, etenemisvapaassa elinajassa ja objektiivisen vasteen saavuttaneiden osuudessa potilailla, jotka saivat nivolumabin, ipilimumabin ja platinapohjaisen kemoterapian yhdistelmähoitoa, verrattuna potilaisiin, jotka saivat vain platinapohjaista kemoterapiaa. Kokonaiselinajan seuranta-aika oli vähintään 8,1 kuukautta.
Tehon tulokset esitetään kuvassa 15 (päivitetty kokonaiselinajan analyysi, kun seuranta-aika oli vähintään 12,7 kuukautta) ja taulukossa 30 (primaarianalyysi, kun seuranta-aika oli vähintään 8,1 kuukautta).
Tehon päivitetty analyysi tehtiin, kun kaikki potilaat olivat olleet seurannassa vähintään 12,7 kuukautta (ks. kuva 15). Analyysihetkellä kokonaiselinajan riskitiheyssuhde oli 0,66 (95 %:n luottamusväli: 0,55–0,80) ja etenemisvapaan elinajan riskitiheyssuhde oli 0,68 (95 %:n luottamusväli: 0,57–0,82).
Kuva 15: Kokonaiselinajan Kaplan–Meier-kuvaaja (CA2099LA)
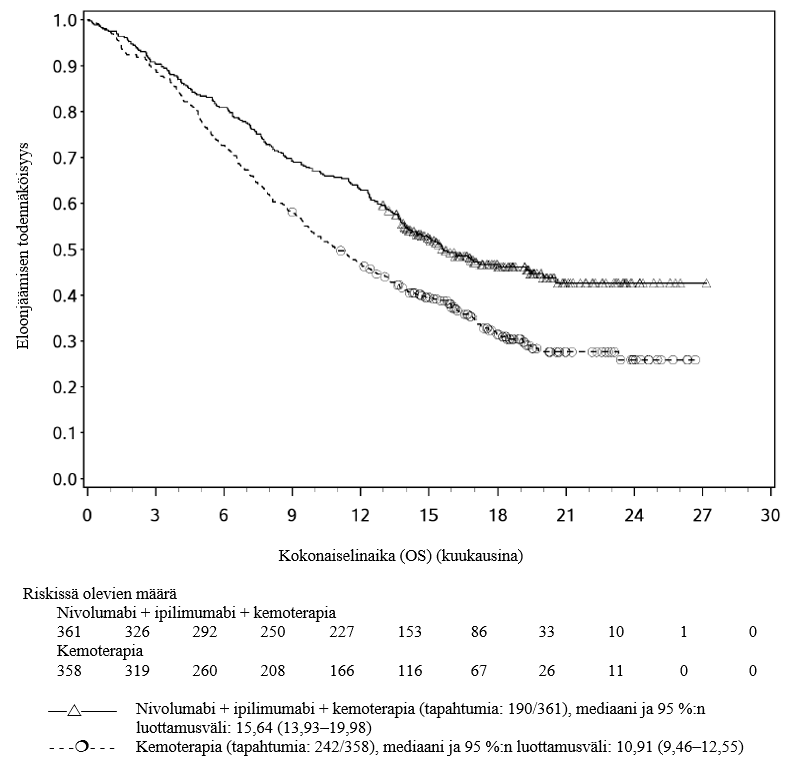
Taulukko 30: Tehoon liittyvät tulokset (CA2099LA)
| nivolumabi + ipilimumabi + kemoterapia | kemoterapia |
|---|---|---|
Kokonaiselinaika | ||
Tapahtumat | 156 (43,2 %) | 195 (54,5 %) |
Riskitiheyssuhde (96,71 %:n luottamusväli)a | 0,69 (0,55–0,87) | |
Ositettu log-rank-testin p‑arvob | 0,0006 | |
Mediaani (kuukautta) (95 %:n luottamusväli) | 14,1 | 10,7 |
Osuus (95 %:n luottamusväli) 6 kuukauden kohdalla | 80,9 (76,4–84,6) | 72,3 (67,4–76,7) |
|
|
|
Etenemisvapaa elinaika | ||
Tapahtumat | 232 (64,3 %) | 249 (69,6 %) |
Riskitiheyssuhde (97,48 %:n luottamusväli)a | 0,70 (0,57–0,86) | |
Ositettu log-rank-testin p‑arvoc | 0,0001 | |
Mediaani (kuukautta)d | 6,83 | 4,96 |
Osuus (95 %:n luottamusväli) 6 kuukauden kohdalla | 51,7 (46,2–56,8) | 35,9 (30,5–41,3) |
|
|
|
Kokonaishoitovastee | 136 (37,7 %) | 90 (25,1 %) |
(95 %:n luottamusväli) | (32,7–42,9) | (20,7–30,0) |
Ositettu CMH-testin p-arvof | 0,0003 | |
Täydellinen vaste (CR) | 7 (1,9 %) | 3 (0,8 %) |
Osittainen vaste (PR) | 129 (35,7 %) | 87 (24,3 %) |
|
|
|
Vasteen kesto | ||
Mediaani (kuukautta) | 10,02 | 5,09 |
%, kun kesto oli ≥ 6 kuukauttag | 74 | 41 |
a Perustuu ositettuun Coxin suhteellisen vaaran malliin.
b p‑arvoa on verrattu allokoituun α 0,0329:een tätä välianalyysia varten.
c p‑arvoa on verrattu allokoituun α 0,0252:een tätä välianalyysia varten.
d Kaplan–Meier-arvio.
e Osuus täydellisellä tai osittaisella vasteella; luottamusväli perustuu Clopper–Pearsonin menetelmään.
f p‑arvoa on verrattu allokoituun α 0,0025:een tätä välianalyysia varten.
g Perustuu Kaplan–Meierin arvioihin vasteen kestosta.
CMH = Cochran–Mantel–Haenszel
Potilaat saivat myöhempää systeemistä hoitoa seuraavasti: 28,8 % yhdistelmähoitoa saaneista ja 41,1 % kemoterapiaa saaneista. Potilaat saivat myöhempää immuunihoitoa (mukaan lukien anti-PD-1-hoitoa, anti-PD-L1-hoitoa ja anti-CTLA-4-hoitoa) seuraavasti: 3,9 % yhdistelmähoitoa saaneista ja 27,9 % kemoterapiaa saaneista.
CA2099LA‑tutkimuksessa kemoterapiaan liittyvä alaryhmien deskriptiivinen analyysi osoitti kokonaiselinaikahyötyä potilailla, joita hoidettiin nivolumabin, ipilimumabin ja kemoterapian yhdistelmähoidolla ja joilla histologia oli levyepiteeliperäinen (riskitiheyssuhde [95 %:n luottamusväli] 0,65 [0,46–0,93], n = 227) ja joilla histologia oli ei-levyepiteeliperäinen (riskitiheyssuhde [95 %:n luottamusväli] 0,72 [0,55–0,93], n = 492).
Taulukossa 31 on yhteenveto tehoon liittyvistä ennalta määritettyjen alaryhmien analyysien tuloksista mitattuna kokonaiselinajalla, etenemisvapaalla elinajalla ja objektiivisen vasteen saavuttaneiden osuudella kasvaimen PD-L1-ilmentymistason mukaan.
Taulukko 31: Tehoon liittyvät tulokset kasvaimen PD-L1-ilmentymistason mukaan (CA2099LA)
| nivolumabi + ipilimumabi + kemoterapia | kemo-terapia | nivolumabi + ipilimumabi + kemoterapia | kemo-terapia | nivolumabi + ipilimumabi + kemoterapia | kemo-terapia | nivolumabi + ipilimumabi + kemoterapia | kemo-terapia |
| PD-L1:n ilmentymistaso < 1 % (n = 264) | PD-L1:n ilmentymistaso ≥ 1 % (n = 406) | PD-L1:n ilmentymistaso ≥ 1 % – 49 % (n = 233) | PD-L1:n ilmentymistaso ≥ 50 % (n = 173) | ||||
Kokonais-elinajan riskitiheys-suhde (95 %:n luottamus-väli)a | 0,65 | 0,67 | 0,69 | 0,64 | ||||
Etenemis-vapaan elinajan riskitiheys-suhde (95 %:n luottamus-väli)a | 0,77 | 0,67 | 0,71 | 0,59 | ||||
Objektiivisen vasteen saavuttaneiden osuus, % | 31,1 | 20,9 | 41,9 | 27,6 | 37,8 | 24,5 | 48,7 | 30,9 |
a Riskitiheyssuhde perustuu ei-ositettuun Coxin suhteellisen vaaran malliin.
Yhteensä 70 ei-pienisoluista keuhkosyöpää sairastavaa ≥ 75‑vuotiasta potilasta otettiin mukaan CA2099LA‑tutkimukseen (37 potilasta nivolumabin, ipilimumabin ja kemoterapian yhdistelmähoitoa saaneiden ryhmässä ja 33 potilasta kemoterapiaa saaneiden ryhmässä). Tutkimuksen tässä alaryhmässä todettiin nivolumabin, ipilimumabin ja kemoterapian yhdistelmähoitoa saaneilla kokonaiselinajan riskitiheyssuhde 1,36 (95 %:n luottamisväli: 0,74–2,52) ja etenemisvapaan elinajan riskitiheyssuhde 1,12 (95 %:n luottamusväli: 0,64–1,96) verrattuna kemoterapiaa saaneisiin. Objektiivisen vasteen saavuttaneiden osuus oli 27,0 % nivolumabin, ipilimumabin ja kemoterapian yhdistelmähoitoa saaneiden ryhmässä ja 15,2 % kemoterapiaa saaneiden ryhmässä. 43 %:lla potilaista, jotka olivat ≥ 75‑vuoden ikäisiä, nivolumabin. ipilimumabin ja kemoterapian yhdistelmähoito lopetettiin. Tietoa nivolumabin, ipilimumabin ja kemoterapian yhdistelmähoidon tehosta ja turvallisuudesta tässä potilasjoukossa on vain vähän.
Alaryhmäanalyysissa todettiin pienentynyt elinaikahyöty nivolumabin, ipilimumabin ja kemoterapian yhdistelmähoitoa saaneilla verrattuna kemoterapiaa saaneisiin potilailla, jotka eivät olleet koskaan tupakoineet. Pienen potilasmäärän vuoksi näistä tiedoista ei voida kuitenkaan tehdä luotettavia johtopäätöksiä.
Ei-pienisoluisen keuhkosyövän hoito aiemman kemoterapian jälkeen
Levyepiteeliperäinen NSCLC
Satunnaistettu vaiheen 3 tutkimus, vertailu dosetakseliin (CA209017)
Nivolumabin (3 mg/kg) tehoa ja turvallisuutta ainoana hoitona edenneen tai etäpesäkkeisen ei-pienisoluisen levyepiteeliperäisen keuhkosyövän hoidossa arvioitiin vaiheen 3 satunnaistetussa avoimessa tutkimuksessa (CA209017). Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita joiden sairaus oli edennyt yhden aiemman platinapohjaisen kemoterapian aikana tai sen jälkeen ja ECOG–toimintakykystatuksen pisteet olivat 0 tai 1. Potilaat otettiin mukaan tutkimukseen kasvaimen PD-L1:n ilmentymisestä riippumatta. Niitä potilaita, joilla oli aktiivinen autoimmuunisairaus, oireinen interstitiaalinen keuhkosairaus tai aktiivisia aivometastaaseja ei otettu mukaan tutkimukseen. Potilaat, joiden aivometastaaseja oli hoidettu, voitiin ottaa mukaan tutkimukseen jos neurologinen tila oli palautunut lähtötasolle vähintään 2 viikkoa ennen tutkimukseen mukaan tuloa, ja jos kortikosteroidihoito oli joko lopetettu, tai he saivat muuttumattomia tai pieneneviä annoksia, jotka vastasivat alle 10 mg:aa prednisonia päivässä.
Kaikkiaan 272 potilasta satunnaistettiin saamaan joko nivolumabia, 3 mg/kg (n = 135) annosteltuna 60 minuutin kuluessa laskimoon joka toinen viikko, tai dosetakselia (n = 137), 75 mg/m2 joka kolmas viikko. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt hoitoa. Kasvaimet arvioitiin kiinteiden kasvainten vasteenarviointikriteerien (RECIST) version 1.1 mukaan 9 viikon kuluttua satunnaistamisesta, ja sen jälkeen joka 6. viikko. Ensisijainen tehokkuusmuuttuja oli OS. Tärkeimmät toissijaiset tehokkuusmuuttujat olivat tutkijan arvioima ORR ja PFS. Lisäksi arvioitiin oireiden paranemista keuhkosyövän oireasteikon (Lung Cancer Symptom Score, LCSS) keskimääräisen oiretaakkaindeksin avulla sekä yleistä terveydentilaa EQ-5D visuaalisen analogisen asteikon (EQ-VAS) avulla.
Lähtötason ominaisuudet näissä kahdessa ryhmässä oli tasapainotettu. Iän mediaani oli 63 vuotta (vaihteluväli 39–85); 44 % oli iältään ≥ 65 vuotta ja 11 % ≥ 75 vuotta. Suurin osa potilaista oli valkoihoisia (93 %) ja miehiä (76 %). 31 prosentilla oli etenevä sairaus tutkimushoitoa edeltäneen viimeisimmän hoidon parhaan vasteen mukaan raportoituna, ja 45 % sai nivolumabia 3 kuukauden kuluessa tutkimusta edeltäneen viimeisimmän hoidon päättymisestä. Lähtötason ECOG (Eastern Cooperative Oncology Group) –toimintakykyluokka oli 0 (24 %) tai 1 (76 %).
Kokonaiselinajan Kaplan-Meier-kuvaajat esitetään kuvassa 16.
Kuva 16: Kokonaiselinajan Kaplan–Meier-kuvaajat (CA209017)
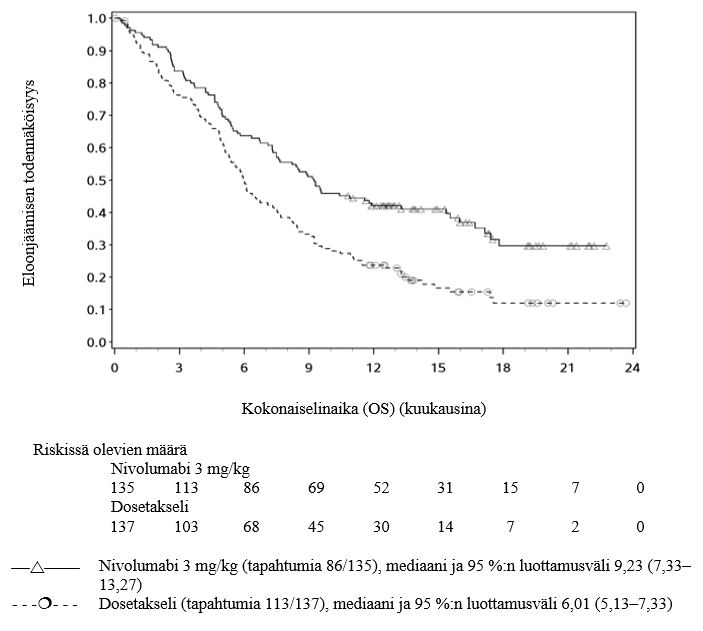
Havaitut kokonaiselinajassa saavutetut hyödyt osoitettiin johdonmukaisesti kaikissa potilasalaryhmissä. Elinajan hyödyt havaittiin riippumatta siitä oliko potilailla kasvaimia, jotka oli määritelty PD-L1‑negatiivisiksi tai PD-L1‑positiivisiksi (tuumorikalvoilmentyminen cut off –tasoilla 1 %, 5 % ja 10 %) Kuitenkaan, tämän biomarkkerin (kasvaimen PD-L1-ilmentymä) roolia ei ole täysin selvennetty. Kokonaiselinajassa saavutetut hyödyt osoitettiin johdonmukaisesti kaikissa potilasalaryhmissä myös vähintään 62,6 kuukauden seurannassa.
CA209017-tutkimuksessa oli rajallinen määrä ≥ 75-vuotiaita potilaita (11 nivolumabiryhmässä ja 18 dosetakseliryhmässä). Nivolumabi vaikutti lukumääräisesti vähemmän kokonaiselinaikaan (riskitihyessuhde 1,85; 95 %:n luottamusväli 0,76–4,51), etenemisvapaaseen elinaikaan (riskitiheyssuhde 1,76; 95 %:n luottamusväli 0,77–4,05) ja vahvistettuun objektiivisen vasteen saavuttaneiden osuuteen (9,1 % vs. 16,7 %). Pienen otoskoon vuoksi näistä tiedoista ei voida tehdä lopullista johtopäätöstä.
Tulokset tehosta näkyvät taulukossa 32.
Taulukko 32: Tulokset lääkeaineen tehosta (CA209017)
| nivolumabi | dosetakseli |
Primaarianalyysi Seuranta-aika vähintään: 10,6 kuukautta | ||
Kokonaiselinaika (OS) |
|
|
Tapahtumia | 86 (63,7 %) | 113 (82,5 %) |
Riskitiheyssuhde | 0,59 | |
96,85 %:n luottamusväli | (0,43–0,81) | |
p‑arvo | 0,0002 | |
|
|
|
Mediaani (95 %:n luottamusväli) kuukausina | 9,23 (7,33–13,27) | 6,01 (5,13–7,33) |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 42,1 (33,7–50,3) | 23,7 (16,9–31,1) |
|
|
|
Vahvistettu objektiivinen vaste | 27 (20,0 %) | 12 (8,8 %) |
(95 %:n luottamusväli) | (13,6–27,7) | (4,6–14,8) |
Ristitulosuhde (95 %:n luottamusväli) | 2,64 (1,27–5,49) | |
p‑arvo | 0,0083 | |
|
|
|
Täydellinen vaste (CR) | 1 (0,7 %) | 0 |
Osittainen vaste (PR) | 26 (19,3 %) | 12 (8,8 %) |
Stabiili tauti (SD) | 39 (28,9 %) | 47 (34,3 %) |
|
|
|
Vasteen keston mediaani |
|
|
Kuukausina (vaihteluväli) | Ei saavutettu (2,9–20,5+) | 8,4 (1,4+–15,2+) |
|
|
|
Vasteen saavuttamisajan mediaani |
|
|
Kuukausina (vaihteluväli) | 2,2 (1,6–11,8) | 2,1 (1,8–9,5) |
|
|
|
Etenemisvapaa elinaika |
|
|
Tapahtumia | 105 (77,8 %) | 122 (89,1 %) |
Riskitiheyssuhde | 0,62 | |
95 %:n luottamusväli | (0,47–0,81) | |
p‑arvo | < 0,0004 | |
|
|
|
Mediaani (95 %:n luottamusväli) kuukausina | 3,48 (2,14–4,86) | 2,83 (2,10–3,52) |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 20,8 (14,0–28,4) | 6,4 (2,9–11,8) |
Päivitetty analyysi Seuranta-aika vähintään: 24,2 kuukautta | ||
Kokonaiselinaika (OS)a |
|
|
Tapahtumia | 110 (81,4 %) | 128 (93,4 %) |
Riskitiheyssuhde | 0,62 | |
95 %:n luottamusväli | (0,47–0,80) | |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 22,9 (16,2–30,3) | 8 (4,3–13,3) |
|
|
|
Vahvistettu objektiivinen vaste | 20,0 % | 8,8 % |
(95 %:n luottamusväli) | (13,6–27,7) | (4,6–14,8) |
|
|
|
Vasteen keston mediaani |
|
|
Kuukausina (vaihteluväli) | 25,2 (2,9–30,4) | 8,4 (1,4+–18,0+) |
|
|
|
Etenemisvapaa elinaika |
|
|
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 15,6 (9,7–22,7) | Kaikkien potilaiden sairaus joko eteni, heidät sensuroitiin tutkimuksesta tai menetettiin seurannassa |
Päivitetty analyysi Seuranta-aika vähintään: 62,6 kuukautta | ||
Kokonaiselinaika (OS)a |
|
|
Tapahtumia | 118 (87,4 %) | 133 (97,1 %) |
Riskitiheyssuhde | 0,62 (0,48–0,79) | |
95 %:n luottamusväli | ||
Osuus (95 %:n luottamusväli) 60 kuukauden kohdalla | 12,3 (7,4–18,5) | 3,6 (1,4–7,8) |
|
|
|
Vahvistettu objektiivinen vaste | 20,0 % | 8,8 % |
(95 %:n luottamusväli) | (13,6–27,7) | (4,6–14,8) |
|
|
|
Vasteen keston mediaani |
|
|
Kuukausina (vaihteluväli) | 25,2 (2,9–70,6+) | 7,5 (0,0+–18,0+) |
|
|
|
Etenemisvapaa elinaika |
|
|
Osuus (95 %:n luottamusväli) 60 kuukauden kohdalla | 9,4 (4,8–15,8) | Kaikkien potilaiden sairaus joko eteni, heidät sensuroitiin tutkimuksesta tai menetettiin seurannassa |
a Kuusi dosetakselille satunnaistettua potilasta (4 %) siirrettiin missä tahansa vaiheessa saamaan nivolumabihoitoa.
”+” tarkoittaa sensuroitunutta havaintoa.
LCSS-asteikolla mitattu sairauteen liittyvien oireiden paraneminen oli samaa luokkaa nivolumabiryhmässä (18,5 %) ja dosetakseliryhmässä (21,2 %). Keskimääräinen EQ-VAS nousi ajan myötä molemmissa ryhmissä, mikä osoittaa, että hoitoa jatkaneiden potilaiden kokonaisterveydentila oli parempi.
Yhden hoitohaaran vaiheen 2 tutkimus (CA209063)
CA209063-tutkimus oli yksihaarainen avoin tutkimus 117:llä potilaalla joilla oli paikallisesti edennyt tai metastasoinut ei-pienisoluinen levyepiteelikeuhkosyöpä kahden tai useamman hoitolinjan jälkeen; muutoin kelpoisuusehdot olivat samat kuin tutkimuksessa CA209017. Nivolumabi 3 mg/kg –annoksen objektiivisen vasteen saavuttaneiden osuus oli 14,5 % (95 %:n luottamusväli 8,7–22,2), kokonaiselinajan mediaani 8,21 kuukautta (95 %:n luottamusväli: 6,05–10,9), ja etenemisvapaan elinajan mediaani 1,87 kuukautta (95 %:n luottamusväli 1,77–3,15). Etenemisvapaa elinaika mitattiin RECIST 1.1 –kriteerien perusteella. Arvioitu yhden vuoden eloonjäämisaste oli 41 %.
Yhden hoitohaaran vaiheen 2 tutkimus (CA209171)
CA209171-tutkimus oli yksihaarainen avoin nivolumabi-monoterapiaa koskeva tutkimus potilailla, joilla oli aiemmin hoidettu edennyt tai etäpesäkkeinen ei-pienisoluinen levyepiteeliperäinen keuhkosyöpä. Turvallisuus oli ensisijainen päätetapahtuma ja teho toissijainen päätetapahtuma. Hoidetusta 811 potilaasta 103:lla (13 %:lla) ECOG‑toimintakykyluokka oli 2, 686 (85 %) oli < 75‑vuotiaita ja 125 (15 %) oli ≥ 75‑vuotiaita. Uusia turvallisuussignaaleja ei havaittu hoidetuissa potilaissa, ja nivolumabin yleinen turvallisuusprofiili oli samanlainen kaikissa alaryhmissä. Taulukossa 33 alla on esitetty tutkijan arvioimaan objektiivisen vasteen saavuttaneiden osuuteen perustuvat tulokset tehosta.
Taulukko 33: objektiivisen vasteen saavuttaneiden osuuteen niiden potilaiden perusteella, joilta vaste oli arvioitavissa – yhteensä ja alaryhmän mukaan jaoteltuna (CA209171)
Tulokset | Yhteensä | ECOG-toimintakykyluokka 2 | < 75‑vuotiaat | ≥ 75‑vuotiaat |
N vasteen saavuttaneet / N vaste arvioitavissaa (%)
95 %:n luottamusvälib | 66/671 (9,8)
(7,7–12,3) | 1/64 (6,1)
(0,0–8,4) | 55/568 (9,7)
(7,4–12,4) | 11/103 (10,7)
(5,5–18,3) |
a Sisältää vahvistetut ja vahvistamattomat vasteet, kuvaukset olivat pakolliset vain viikolla 8/9 ja viikolla 52.
b Täydellinen vaste + osittainen vaste, luottamusväli perustuu Clopper–Pearsonin menetelmään.
Ei-levyepiteeliperäinen NSCLC
Satunnaistettu vaiheen 3 tutkimus, vertailu dosetakseliin (CA209057)
Nivolumabin (3 mg/kg) tehoa ja turvallisuutta ainoana hoitona edenneen tai etäpesäkkeisen ei-levyepiteeliperäisen NSCLC:n hoidossa arvioitiin vaiheen 3 satunnaistetussa avoimessa tutkimuksessa (CA209057). Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joiden sairaus oli edennyt yhden aiemman platinapohjaisen solunsalpaajayhdistelmähoidon, joka on voinut sisältää ylläpitohoitojakson, aikana tai sen jälkeen ja joiden ECOG–toimintakykystatuksen pisteet olivat 0 tai 1. Ylimääräinen TKI-hoitolinja sallittiin potilailla, joilla oli tunnistettu EGFR-mutaatio tai ALK-translokaatio. Potilaat otettiin mukaan tutkimukseen kasvaimen PD-L1:n ilmentymisestä riippumatta. Niitä potilaita, joilla oli aktiivinen autoimmuunisairaus, oireinen interstitiaalinen keuhkosairaus tai aktiivisia aivometastaaseja ei otettu mukaan tutkimukseen. Potilaat, joiden aivometastaaseja oli hoidettu, voitiin ottaa mukaan tutkimukseen jos neurologinen tila oli palautunut lähtötasolle vähintään 2 viikkoa ennen tutkimukseen mukaan tuloa, ja jos kortikosteroidihoito oli joko lopetettu, tai he saivat muuttumattomia tai pieneneviä annoksia, jotka vastasivat alle 10 mg:aa prednisonia päivässä.
Kaikkiaan 582 potilasta satunnaistettiin saamaan joko nivolumabia, 3 mg/kg (n = 292) annosteltuna 60 minuutin kuluessa laskimoon joka toinen viikko, tai dosetakselia (n = 290), 75 mg/m2 joka kolmas viikko. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt hoitoa. Kasvaimet arvioitiin RECIST:n version 1.1 mukaan. Ensisijainen tehokkuusmuuttuja oli OS. Tärkeimmät toissijaiset tehokkuusmuuttujat olivat tutkijan arvioima ORR ja PFS. Ylimääräiset alaryhmäanalyysit tehokkuuden arvioimiseksi tehtiin etukäteen määritellyillä kasvaimen PD-L1 ilmentymistasoilla 1 %, 5 % ja 10 %. Arviointi erillisten PD-L1:n ilmentymisen intervallien mukaan ei ollut mukana etukäteen määritellyissä analyyseissä intervallien pienen otoskoon vuoksi.
Prekliiniset kasvainkudosnäytteet kerättiin systemaattisesti ennen satunnaistamista, jotta voitiin tehdä ennaltasuunnitellut tehokkuusanalyysit kasvaimen PD-L1:n ilmentymisten mukaisesti. Kasvaimen PD-L1:n ilmentyminen määritettiin käyttämällä PD-L1 IHC 28–8 pharmDx-testausmenetelmää.
Iän mediaani oli 62 vuotta (vaihteluväli 21–85); 34 % oli iältään > 65 vuotta ja 7 % > 75 vuotta. Suurin osa potilaista oli valkoihoisia (92 %) ja miehiä (55 %). Lähtötason ECOG-toimintakykyluokka oli 0 (31 %) tai 1 (69 %). 79 % potilaista oli aiempia/nykyisiä tupakoitsijoita.
Kokonaiselinajan Kaplan-Meier-kuvaajat esitetään kuvassa 17.
Kuva 17:Kokonaiselinajan Kaplan–Meier-kuvaaja (CA209057)
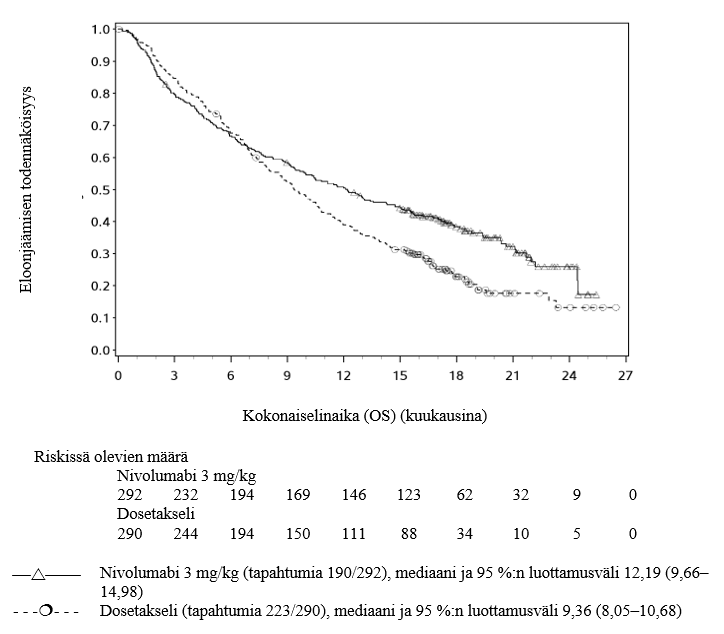
Dosetakseliin verrattuna tutkimuksessa todettiin tilastollisesti merkitsevä paraneminen kokonaiselinajassa potilailla, jotka oli satunnaistettu saamaan nivolumabia. Tulos perustui ennaltamääriteltyyn välianalyysiin, jossa arvioitiin 413 tapausta (93 % suunnitellusta loppuanalyysin tapahtumien määrästä). Tulokset tehosta esitetään taulukossa 34.
Taulukko 34: Tulokset tehosta (CA209057)
| nivolumabi | dosetakseli |
Ennalta määritetty välianalyysi Seuranta-aika vähintään: 13,2 kuukautta | ||
Kokonaiselinaika (OS) |
|
|
Tapahtumia | 190 (65,1 %) | 223 (76,9 %) |
Riskitiheyssuhdea | 0,73 | |
(95,92 %:n luottamusväli) | (0,59–0,89) | |
p‑arvob | 0,0015 | |
|
|
|
Mediaani (95 %:n luottamusvälillä) kuukausina | 12,19 (9,66–14,98) | 9,36 (8,05–10,68) |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 50,5 (44,6–56,1) | 39,0 (33,3–44,6) |
|
|
|
Vahvistettu objektiivinen vaste | 56 (19,2 %) | 36 (12,4 %) |
(95 %:n luottamusväli) | (14,8–24,2) | (8,8–16,8) |
Ristitulosuhde (95 %:n luottamusväli) | 1,68 (1,07–2,64) | |
p‑arvo | 0,0246 | |
|
|
|
Täydellinen vaste (CR) | 4 (1,4 %) | 1 (0,3 %) |
Osittainen vaste (PR) | 52 (17,8 %) | 35 (12,1 %) |
Stabiili tauti (SD) | 74 (25,3 %) | 122 (42,1 %) |
|
|
|
Vasteen keston mediaani |
|
|
Kuukausina (vaihteluväli) | 17,15 (1,8–22,6+) | 5,55 (1,2+–15,2+) |
|
|
|
Vasteen saavuttamisajan mediaani |
|
|
Kuukausina (vaihteluväli) | 2,10 (1,2–8,6) | 2,61 (1,4–6,3) |
|
|
|
Etenemisvapaa elinaika |
|
|
Tapahtumia | 234 (80,1 %) | 245 (84,5 %) |
Riskitiheyssuhde | 0,92 | |
95 %:n luottamusväli | (0,77–1,11) | |
p‑arvo | 0,3932 | |
|
|
|
Mediaani (95 %:n luottamusväli) kuukausina | 2,33 (2,17–3,32) | 4,21 (3,45–4,86) |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 18,5 (14,1–23,4) | 8,1 (5,1–12,0) |
Päivitetty analyysi Seuranta-aika vähintään: 24,2 kuukautta | ||
Kokonaiselinaika (OS)c |
|
|
Tapahtumia | 228 (78,1 %) | 247 (85,1 %) |
Riskitiheyssuhdea | 0,75 | |
(95 %:n luottamusväli) | (0,63–0,91) | |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 28,7 (23,6–34,0) | 15,8 (11,9–20,3) |
|
|
|
Vahvistettu objektiivinen vaste | 19,2 % | 12,4 % |
(95 %:n luottamusväli) | (14,8–24,2) | (8,8–16,8) |
|
|
|
Vasteen keston mediaani |
|
|
Kuukausina (vaihteluväli) | 17,2 (1,8–33,7+) | 5,6 (1.2+–16,8) |
|
|
|
Etenemisvapaa elinaika |
|
|
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 11,9 (8,3–16,2) | 1,0 (0,2–3,3) |
Päivitetty analyysi Seuranta-aika vähintään: 62,7 kuukautta | ||
Kokonaiselinaika (OS)d |
|
|
Tapahtumia | 250 (85,6 %) | 279 (96,2 %) |
Riskitiheyssuhdea | 0,70 | |
(95 %:n luottamusväli) | (0,58–0,83) | |
Osuus (95 %:n luottamusväli) 60 kuukauden kohdalla | 14,0 (10,2–18,3) | 2,1 (0,9–4,4) |
|
|
|
Vahvistettu objektiivinen vaste | 19,5 % | 12,4 % |
(95 %:n luottamusväli) | (15,1–24,5) | (8,8–16,8) |
|
|
|
Vasteen keston mediaani |
|
|
Kuukausina (vaihteluväli) | 17,2 (1,8–70,4+) | 5,6 (0,0+–33,4) |
|
|
|
Etenemisvapaa elinaika |
|
|
Osuus (95 %:n luottamusväli) 60 kuukauden kohdalla | 7,5 (4,5–11,4) | Kaikkien potilaiden sairaus joko eteni, heidät sensuroitiin tutkimuksesta tai menetettiin seurannassa |
a Johdettu ositetusta verrannollisten riskitiheyksien mallista.
b P‑arvo perustuu logrank merkitsevyystestiin ositettuna aiemman ylläpitohoidon ja hoitolinjan mukaan; vastaava O’Brien‑Fleming ‑keskeytysrajan merkitsevyystaso on 0,0408.
c Kuusitoista dosetakselille satunnaistettua potilasta (6 %) siirrettiin missä tahansa vaiheessa saamaan nivolumabihoitoa.
d Seitsemäntoista dosetakselille satunnaistettua potilasta (6 %) siirrettiin missä tahansa vaiheessa saamaan nivolumabihoitoa.
”+” tarkoittaa sensuroitunutta havaintoa.
Laskennallinen kasvaimen PD-L1:n ilmentyminen mitattiin 79 %:lta nivolumabiryhmän potilaista ja 77 %:lta dosetakseliryhmän potilaista. Kasvaimen PD-L1:n ilmentymistasojen raja-arvot olivat tasapainossa näiden kahden ryhmän välillä (nivolumabi vs. dosetakseli), joissa kummassakin ennaltamääritetyt kasvaimen PD-L1:n ilmentymistasojen raja-arvot olivat ≥ 1 % (53 % vs. 55 %), ≥ 5 % (41 % vs. 38 %) tai ≥ 10 % (37 % vs. 35 %).
Nivolumabiryhmän potilailla, joiden kasvaimet ilmensivät PD‑L1:ää kaikilla ennaltamääritetyillä raja-arvoilla oli suurempi mahdollisuus pidempään kokonaiselinaikaan verrattuna dosetakseliryhmän potilaisiin, kun taas elinaika oli samanlainen dosetakseliryhmään verrattuna, jos kasvaimen PD‑L1:n ilmentymistaso oli matala tai sitä ei ollut lainkaan. Objektiivisen vasteen saavuttaneiden osuuden arvioinnin kannalta lisääntynyt PD‑L1:n ilmentyminen liittyi suurempaan objektiivisen vasteen saavuttaneiden osuuteen. Koko populaatioon liittyvässä vertailussa vasteen keston mediaani oli suurempi nivolumabiryhmässä (18,3 kuukautta) verrattuna dosetakseliryhmään (5,6 kuukautta) potilailla, joilla ei ollut PD‑L1:n ilmentymistä ja nivolumabiryhmässä (16,0 kuukautta) verrattuna dosetakseliryhmään (5,6 kuukautta) potilailla, joilla oli PD‑L1:n ilmentymistä.
Taulukossa 35 esitetään tulosten yhteenveto kasvaimen PD‑L1:n ilmentymisen mukaisesta objektiivisen vasteen saavuttaneiden osuudesta ja kokonaiselinajasta.
Taulukko 35: PD‑L1:n ilmentymisen mukaiset objektiivisen vasteen saavuttaneiden osuudet ja kokonaiselinajat (CA209057)
PD-L1:n ilmentyminen | nivolumabi | dosetakseli |
|
|---|---|---|---|
Kasvaimen PD-L1:n ilmentymisen mukainen objektiivisen vasteen saavuttaneiden osuus | |||
|
|
| Ristitulosuhde (95 %:n luottamusvälillä) |
< 1 % | 10/108 (9,3 %) 95 %:n luottamusväli 4,5–16,4 | 15/101 (14,9 %) 95 %:n luottamusväli 8,6–23,3 | 0,59 (0,22–1,48) |
≥ 1 % | 38/123 (30,9 %) 95 %:n luottamusväli 22,9–39,9 | 15/123 (12,2 %) 95 %:n luottamusväli 7,0–19,3 | 3,22 (1,60–6,71) |
≥ 1 % – < 10 %a | 6/37 (16,2 %) 95 %:n luottamusväli 6,2–32,0 | 5/44 (11,4 %) 95 %:n luottamusväli 3,8–24,6 | 1,51 (0,35–6,85) |
≥ 10 % – < 50 %a | 5/20 (25,0 %) 95 %:n luottamusväli 8,7–49,1 | 7/33 (21,2 %) 95 %:n luottamusväli 9,0–38,9 | 1,24 (0,26–5,48) |
≥ 50 %a | 27/66 (40,9 %) 95 %:n luottamusväli 29,0–53,7 | 3/46 (6,5 %) 95 %:n luottamusväli 1,4,–17,9 | 9,92 (2,68–54,09) |
Kasvaimen PD-L1:n ilmentyminen kokonaiselinajan mukaan | |||
| Tapahtumien määrä (potilasmäärät) | Osittamaton riskitiheyssuhde (95 %:n luottamusväli) | |
< 1 % | 77 (108) | 75 (101) | 0,90 (0,66–1,24) |
≥ 1 % | 68 (123) | 93 (123) | 0,59 (0,43–0,82) |
≥ 1 % – < 10 %a | 27 (37) | 30 (44) | 1,33 (0,79–2,24) |
≥ 10 % – < 50 %a | 11 (20) | 26 (33) | 0,61 (0,30–1,23) |
≥ 50 %a | 30 (66) | 37 (46) | 0,32 (0,20–0,53) |
| |||
Päivitetty analyysi | |||
< 1 % | 91 (108) | 86 (101) | 0,91 (0,67–1,22) |
≥ 1 % | 87 (123) | 103 (123) | 0,62 (0,47–0,83) |
|
|
|
|
Päivitetty analyysi | |||
< 1 % | 100 (109) | 96 (101) | 0,87 (0,66–1,16) |
≥ 1 % | 96 (122) | 119 (123) | 0,55 (0,42–0,73) |
a Post hoc ‑analyysi: Näitä tuloksia pitää tulkita varoen, koska alaryhmän otoskoko on pieni ja analyysin aikana PD‑L1 IHC 28–8 pharmDx‑testausmenetelmää ei oltu analyyttisesti validoitu 10 % tai 50 % ilmentymistasoille.
Nivolumabiryhmässä potilaita kuoli enemmän ensimmäisen kolmen kuukauden aikana (59/292, 20,2 %) kuin dosetakseliryhmässä (44/290, 15,2 %). Tämän jälkikäteen tehdyn havainnollistavan usean satunnaismuuttujan analyysin tulokset näyttävät, että nivolumabilla hoidetuilla potilailla joilla on huonompi ennuste ja/tai aggressiivinen tauti yhdistettynä matalaan (esim. < 50 %) kasvaimen PD-L1:n ilmentymiseen tai sen puuttumiseen, voi olla suurempi kuolemanriski hoidon ensimmäisen kolmen kuukauden aikana.
Alaryhmäanalyyseissä, elinajan hyötyä dosetakseliin verrattuna ei osoittautunut potilaille jotka eivät olleet koskaan tupakoineet ja joiden kasvaimissa oli aktivoiva EGFR-mutaatio. Kuitenkin pienen potilasmäärän vuoksi näistä tiedoista ei voida tehdä lopullista johtopäätöstä.
Keuhkopussin pahanlaatuinen mesoteliooma
Satunnaistettu vaiheen 3 tutkimus: nivolumabi–ipilimumabi-yhdistelmähoito vs. kemoterapia (CA209743)
Nivolumabi–ipilimumabi-yhdistelmähoidon (nivolumabia 3 mg/kg kahden viikon välein ja ipilimumabia 1 mg/kg kuuden viikon välein) turvallisuutta ja tehoa arvioitiin vaiheen 3 satunnaistetussa avoimessa tutkimuksessa (CA209743). Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli histologisesti vahvistettu ja aiemmin hoitamaton keuhkopussin pahanlaatuinen mesoteliooma, jonka histologia oli epiteloidinen tai ei-epiteloidinen; joiden ECOG-toimintakykyluokka oli 0 tai 1 ja jotka eivät olleet saaneet palliatiivista sädehoitoa 14 vuorokauden sisällä ensimmäisen tutkimushoidon saannista. Tutkimukseen otettiin potilaita riippumatta kasvaimen PD-L1-statuksesta.
Tutkimuksen ulkopuolelle suljettiin potilaat, joilla oli primitiivinen vatsakalvon, sydänpussin, kiveksen tai tuppikalvon mesoteliooma, interstitiaalinen keuhkosairaus, aktiivinen autoimmuunisairaus, systeemistä immunosuppressiota vaativa tila tai aivojen etäpesäke (ellei etäpesäkettä ollut poistettu kirurgisesti tai hoidettu stereotaktisella sädehoidolla siten, ettei etäpesäke ollut kasvanut kolmeen kuukauteen ennen tutkimukseen ottamista). Satunnaistaminen ositettiin histologian (epiteloidinen vs. sarkomatoidinen tai sekamuotoinen histologinen alatyyppi) ja sukupuolen (miehet vs. naiset) mukaan.
Yhteensä 605 potilasta satunnaistettiin saamaan joko nivolumabi–ipilimumabi-yhdistemähoitoa (n = 303) tai kemoterapiaa (n = 302).Nivolumabi–ipilimumabi-yhdistelmähoitoryhmän potilaat saivat 3 mg/kg nivolumabia laskimoon annosteltuna 30 minuutin kuluessa joka toinen viikko ja 1 mg/kg ipilimumabia laskimoon annosteltuna 30 minuutien kuluessa joka kuudes viikko enintään kahden vuoden ajan. Kemoterapiaryhmän potilaat saivat enintään 6 hoitojaksoa kemoterapiaa (yksi hoitojakso oli 21 päivää). Kemoterapia koostui sisplatiinista (75 mg/m2) ja pemetreksedistä (500 mg/m2) tai karboplatiinista (5 AUC) ja pemetreksedistä (500 mg/m2).
Hoitoa jatkettiin, kunnes tauti eteni tai kunnes ilmeni ei-hyväksyttävää toksisuutta tai enintään 24 kuukauden ajan. Hoitoa voitiin jatkaa taudin etenemisen jälkeenkin, jos potilas oli kliinisesti stabiili ja jos tutkija arvioi, että potilas sai hoidosta kliinistä hyötyä. Potilaat, jotka eivät jatkaneet yhdistelmähoitoa ipilimumabin aiheuttaman haittavaikutuksen takia, saivat jatkaa nivolumabi-monoterapiaa. Kasvaimet arvioitiin kuuden viikon välein ensimmäisen tutkimushoidon saamisen jälkeen ensimmäisten 12 kuukauden ajan, minkä jälkeen 12 viikon välein, kunnes tauti eteni tai tutkimushoito lopetettiin.
CA209743-tutkimuksen kaikkien ryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Iän mediaani oli 69 vuotta (vaihteluväli: 25–89). 72 prosenttia potilaista oli ≥ 65-vuotiaita ja 26 prosenttia ≥ 75-vuotiaita. Suurin osa potilaista oli valkoihoisia (85 %) ja miehiä (77 %). Lähtötason ECOG-toimintakykyluokka oli 0 (40 %) tai 1 (60 %). 80 prosentilla potilaista PD-L1 oli ≥ 1 % ja 20 prosentilla PD-L1 oli < 1 %. 75 prosentilla histologia oli epiteloidinen ja 25 prosentilla ei-epiteloidinen.
CA209743-tutkimuksen ensisijainen tehokkuusmuuttuja oli kokonaiselinaika. Keskeisiä toissijaisia päätetapahtumia olivat etenemisvapaa elinaika (PFS), objektiivisen vasteen saavuttaneiden osuus (ORR) ja riippumattoman keskitetyn arvioijatahon (BICR) arvioima vasteen kesto, jonka arvioinnissa hyödynnettiin keuhkopussin mesotelioomaa varten muokattuja kiinteiden kasvainten vasteenarviointikriteerejä (RECIST). Toissijaisten päätetapahtumien deskriptiiviset analyysit esitetään taulukossa 36.
Kemoterapiaan verrattuna tutkimuksessa todettiin tilastollisesti merkitsevää hyötyä kokonaiselinajassa potilailla, jotka oli satunnaistettu saamaan nivolumabi–ipilimumabi-yhdistelmähoitoa. Tulos perustui ennaltamääriteltyyn välianalyysiin, jossa arvioitiin 419 tapahtumaa (89 % suunnitellusta loppuanalyysin tapahtumien määrästä). Kokonaiselinajan seuranta-aika oli vähintään 22 kuukautta.
Tulokset tehosta esitetään kuvassa 18 ja taulukossa 36.
Kuva 18: Kokonaiselinajan Kaplan–Meier-kuvaaja (CA209743)
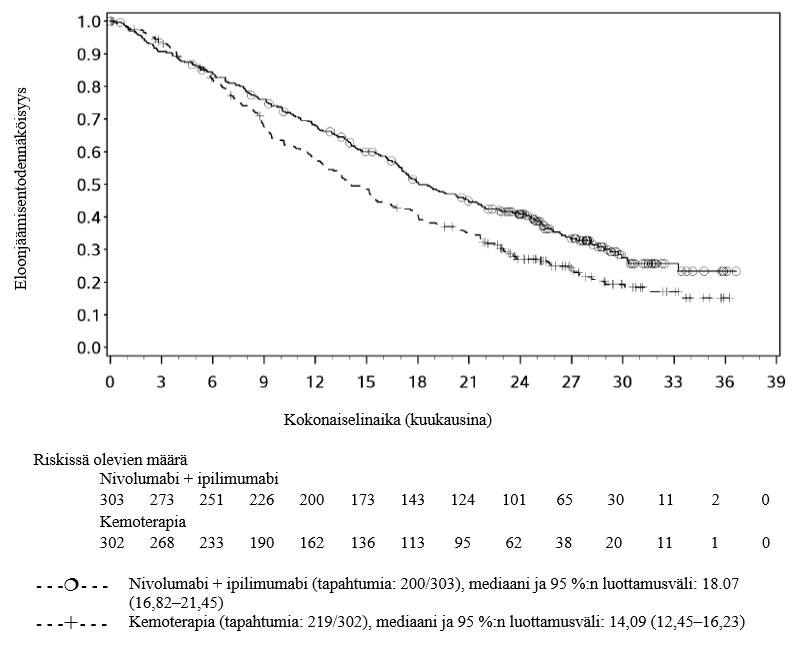
Taulukko 36: Tulokset tehosta (CA209743)
| nivolumabi + ipilimumabi | kemoterapia |
|---|---|---|
Kokonaiselinaika (OS) |
|
|
Tapahtumia | 200 (66 %) | 219 (73 %) |
Riskitiheyssuhde (96,6 %:n luottamusväli)a | 0,74 (0,60–0,91) | |
Ositettu log-rank-testin p-arvob | 0,002 | |
Mediaani (kuukautta)c (95 %:n luottamusväli) | 18,1 | 14,1 |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdallac | 41 % (35,1–46,5) | 27 % (21,9–32,4) |
Etenemisvapaa elinaika |
|
|
Tapahtumia | 218 (72 %) | 209 (69 %) |
Riskitiheyssuhde (95 %:n luottamusväli)a | 1,0 (0,82–1,21) | |
Mediaani (kuukautta)c | 6,8 | 7,2 |
|
|
|
Kokonaishoitovaste | 40 % | 43 % |
(95 %:n luottamusväli) | (34,1–45,4) | (37,1–48,5) |
Täydellinen vaste (CR) | 1,7% | 0 |
Osittainen vaste (PR) | 38 % | 43 % |
|
|
|
Vasteen kesto |
|
|
Mediaani (kuukautta)c | 11,0 | 6,7 |
a Ositettu Coxin suhteellisen vaaran malli.
b p‑arvoa on verrattu allokoituun α 0,0345:een tätä välianalyysiä varten.
c Kaplan–Meier-arvio.
Potilaat saivat myöhempää systeemistä hoitoa seuraavasti: 44,2 prosenttia yhdistelmähoitoa saaneista ja 40,7 prosenttia kemoterapiaa saaneista. Potilaat saivat myöhempää immuunihoitoa (mukaan lukien anti-PD-1-hoitoa, anti-PD-L1-hoitoa ja anti-CTLA-4-hoitoa) seuraavasti: 3,3 prosenttia yhdistelmähoitoa saaneista ja 20,2 prosenttia kemoterapiaa saaneista.
Taulukossa 37 on yhteenveto tehoon liittyvistä ennalta määritettyjen alaryhmien analyysien tuloksista histologian mukaan ja mitattuna kokonaiselinajalla, etenemisvapaalla elinajalla ja objektiivisen vasteen saavuttaneiden osuudella.
Taulukko 37: Tulokset tehosta histologian mukaan (CA209743)
| Epiteloidinen (n = 471) | Ei-epiteloidinen (n = 134) | ||
| nivolumabi + ipilimumabi (n = 236) | kemoterapia (n = 235) | nivolumabi + ipilimumabi (n = 67) | kemoterapia (n = 67) |
Kokonaiselinaika (OS) | ||||
Tapahtumia | 157 | 164 | 43 | 55 |
Riskitiheyssuhde (95 %:n luottamusväli)a | 0,85 | 0,46 | ||
Mediaani (kuukautta) (95 %:n luottamusväli) | 18,73 (17,05–21,72) | 16,23 (14,09–19,15) | 16,89 (11,83–25,20) | 8,80 (7,62–11,76) |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 41,2 (34,7–47,6) | 31,8 (25,7–38,1) | 39,5 (27,5–51,2) | 9,7 (3,8–18,9) |
| ||||
Etenemisvapaa elinaika | ||||
Riskitiheyssuhde (95 %:nluottamusväli)a | 1,14 (0,92–1,41) | 0,58 (0,38–0,90) | ||
Mediaani (kuukautta) (95 %:n luottamusväli) | 6,18 (5,49–7,03) | 7,66 (7,03–8,31) | 8,31 (3,84–11,01) | 5,59 (5,13–7,16) |
|
|
|
|
|
Kokonaisvaste | 38,6 % | 47,2 % | 43,3 % | 26,9 % |
(95 %:n luottamusväli)b | (32,3–45,1) | (40,7–53,8) | (31,2–56,0) | (16,8–39,1) |
| ||||
Vasteen kesto | 8,44 | 6,83 | 24,02 | 4,21 |
Mediaani (kuukautta) (95 %:n luottamusväli)c | (7,16–14,59) | (5,59–7,13) | (8,31–N.A.) | (2,79–7,03) |
a Riskitiheyssuhde perustuu ei-ositettuun Coxin suhteellisen vaaran malliin.
b Luottamusväli perustuu Clopper–Pearsonin menetelmään.
c Mediaani laskettu Kaplan–Meierin menetelmän avulla.
Taulukossa 38 on yhteenveto tehoon liittyvistä ennalta määritettyjen alaryhmien analyysien tuloksista mitattuna kokonaiselinajalla, etenemisvapaalla elinajalla ja objektiivisen vasteen saavuttaneiden osuudella kasvaimen lähtötason PD‑L1-ilmentymistason mukaan.
Taulukko 38: Tehoon liittyvät tulokset kasvaimen PD-L1-ilmentymistason mukaan (CA209743)
| PD-L1 < 1 % (n = 135) | PD-L1 ≥ 1 % (n = 451) | ||
| nivolumabi + ipilimumabi (n = 57) | kemoterapia (n = 78) | nivolumabi + ipilimumabi (n = 232) | kemoterapia (n = 219) |
Kokonaiselinaika (OS) | ||||
Tapahtumia | 40 | 58 | 150 | 157 |
Riskithieyssuhde (95 %:n luottamusväli)a | 0,94 | 0,69 | ||
Mediaani (kuukautta) (95 %:n luottamusväli)b | 17,3 (10,1–24,3) | 16,5 (13,4–20,5) | 18,0 (16,8–21,5) | 13,3 (11,6–15,4) |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 38,7 (25,9–51,3) | 24,6 (15,5–35,0) | 40,8 (34,3–47,2) | 28,3 (22,1–34,7) |
| ||||
Etenemisvapaa elinaika | ||||
Riskitiheyssuhde (95 %:n luottamusväli)a | 1,79 (1,21–2,64) | 0,81 (0,64–1,01) | ||
Mediaani (kuukautta) (95 %:n luottamusväli)b | 4,1 (2,7–5,6) | 8,3 (7,0–11,1) | 7,0 (5,8–8,5) | 7,1 (6,2–7,6) |
|
|
|
|
|
Kokonaisvaste | 21,1 % | 38,5 % | 43,5 % | 44.3 % |
(95 %:n luottamusväli)c | (11,4–33,9) | (27,7–50,2) | (37,1–50,2) | (37,6–51,1) |
a Riskitiheyssuhde perustuu ei-ositettuun Coxin suhteellisen vaaran malliin.
b Mediaani laskettu Kaplan–Meierin menetelmän avulla.
c Luottamusväli perustuu Clopper–Pearsonin menetelmään.
CA209743-tutkimukseen osallistui yhteensä 157 keuhkopussin pahanlaatuista mesotelioomaa sairastavaa ≥ 75-vuotiasta potilasta (78 nivolumabi–ipilimumabi-yhdistelmähoitoa saaneessa ryhmässä ja 79 kemoterapiaryhmässä). Tässä alaryhmässä kokonaiselinajan riskitiheyssuhde nivolumabi–ipilimumabi-yhdistelmähoitoryhmässä oli 1,02 (95 %:n luottamusväli: 0,70–1,48) kemoterapiaryhmään verrattuna. Tutkimuksessa todettiin, että 75-vuotiailla ja tätä vanhemmilla ilmeni enemmän vakavia haittavaikutuksia kuin kaikilla nivolumabi–ipilimumabi-yhdistelmähoitoa saaneilla ja että 75-vuotiaat ja tätä vanhemmat keskeyttivät hoidon haittavaikusten takia useammin kuin kaikki nivolumabi–ipilimumabi-yhdistelmähoitoa saaneet potilaat (ks. kohta Haittavaikutukset). Alaryhmäanalyysin havainnollistavan luonteen vuoksi luotettavia johtopäätöksiä ei kuitenkaan voida tehdä.
Munuaiskarsinooma (RCC)
Satunnaistettu vaiheen 3 tutkimus, ipilimumabi–nivolumabi-yhdistelmähoito vs. sunitinibi (CA209214)
Nivolumabi 3 mg/kg- ja ipilimumabi 1 mg/kg ‑yhdistelmähoidon turvallisuutta ja tehoa edenneen/metastasoituneen munuaiskarsinooman hoidossa arvioitiin satunnaistetussa vaiheen 3 avoimessa tutkimuksessa (CA209214). Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli aiemmin hoitamaton, edennyt tai metastasoitunut kirkassoluinen munuaiskarsinooma. Tehon arviointiin ensisijaisesti käytettävässä potilasjoukossa oli kohtalaisen/huonon ennusteen potilaita, joilla oli vähintään 1 tai useampi International Metastatic RCC Database Consortiumin (IMDC) kriteerien mukaisista kuudesta prognostisesta riskitekijästä (alle vuosi ensimmäisestä munuaiskarsinoomadiagnoosista satunnaistamiseen, Karnofskyn asteikolla pisteitä < 80 %, viitearvon alarajaa matalampi hemoglobiini, korjattu kalsiumpitoisuus yli 10 mg/dl, viitearvon ylärajaa korkeampi trombosyyttien määrä ja viitearvon ylärajaa korkeampi neutrofiilien määrä). Tutkimukseen otettiin potilaita riippumatta kasvaimen PD‑L1‑statuksesta. Tutkimuksesta suljettiin pois ne potilaat, joilla oli Karnofskyn asteikolla pisteitä < 70 %, sekä potilaat joilla oli tai joilla oli ollut aivometastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila. Potilaat ositettiin IMDC:n prognostisen pistemäärän ja esiintymispaikan mukaan.
Tutkimukseen satunnaistettiin yhteensä 1 096 potilasta, joista 847 potilaalla oli kohtalaisen/huonon ennusteen munuaiskarsinooma. Potilaille annettiin joko 3 mg/kg nivolumabia (n = 425) laskimoon 60 minuutin kuluessa sekä 1 mg/kg ipilimumabia laskimoon 30 minuutin kuluessa joka kolmas viikko yhteensä 4 annosta, minkä jälkeen annettiin nivolumabi-monoterapiaa 3 mg/kg:n annoksena joka toinen viikko tai 50 mg/vrk sunitinibia (n = 422) suun kautta neljän viikon ajan, minkä jälkeen pidettiin aina kahden viikon tauko. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt hoitoa. Kasvaimet arvioitiin ensimmäisen kerran 12 viikkoa satunnaistamisen jälkeen, sen jälkeen ensimmäisen vuoden ajan joka kuudes viikko ja sen jälkeen joka kahdestoista viikko taudin etenemiseen tai hoidon lopettamiseen saakka, kumpi näistä tapahtui jälkimmäisenä. Hoito taudin etenemisen jälkeen sallittiin tutkijan suorittaman ensimmäisen kiinteiden kasvainten vasteenarviointikriteerien (RECIST) version 1.1 mukaisesti tekemän arvioinnin jälkeen, jos tutkija määritteli, että potilaalle on kliinistä hyötyä tutkimuslääkkeestä ja hän sietää hoitoa. Ensisijaiset tehokkuusmuuttujat olivat OS, ORR ja PFS riippumattoman keskitetyn arvioijatahon (BICR) arvioimana kohtalaisen/huonon ennusteen potilailla.
Ryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Iän mediaani oli 61 vuotta (vaihteluväli 21–85). 38 % potilaista oli ≥ 65 vuotta ja 8 % oli ≥ 75 vuotta. Suurin osa potilaista oli miehiä (73 %) ja valkoihoisia (87 %). 31 %:lla potilaista lähtötason Karnofskyn toimintakykypisteet (KPS) olivat 70–80 % ja 69 %:lla potilaista 90–100 %. Ajan mediaani ensimmäisestä diagnoosista satunnaistamiseen oli 0,4 vuotta sekä ryhmässä, joka sai 3 mg/kg nivolumabia ja 1 mg/kg ipilimumabia, että ryhmässä, joka sai sunitinibia. Ipilimumabia ja nivolumabia saaneiden potilaiden hoidon keston mediaani oli 7,9 kuukautta (vaihteluväli 1 päivä – 21,4+ kuukautta) ja sunitinibia saaneiden 7,8 kuukautta (vaihteluväli 1 päivä – 20,2+ kuukautta). Nivolumabi- ja ipilimumabihoitoa jatkettiin taudin etenemisen jälkeen 29 %:lla potilaista.
Kohtalaisen/huonon ennusteen potilaiden tehoon liittyvät tulokset esitetään taulukossa 39 (primaarianalyysi, kun seuranta-aika oli vähintään 17,5 kuukautta ja vähintään 60 kuukautta) sekä kuvassa 19 (seuranta-aika vähintään 60 kuukautta).
Vähintään 60 kuukauden seuranta‑ajan jälkeen tehdyn ylimääräisen deskriptiivisen analyysin mukaiset kokonaiselinajan tulokset olivat yhteneviä alkuperäisen primaarianalyysin kanssa.
Taulukko 39: Kohtalaisen/huonon ennusteen potilaiden tehon tulokset (CA209214)
| nivolumabi + ipilimumabi | sunitinibi | ||
| Primaarianalyysi Seuranta‑aika vähintään: 17,5 kuukautta | |||
Kokonaiselinaika (OS) |
|
| ||
Tapahtumat | 140 (33 %) | 188 (45 %) | ||
Riskitiheyssuhdea | 0,63 | |||
99,8 %:n luottamusväli | (0,44–0,89) | |||
p‑arvob, c | < 0,0001 | |||
| ||||
Mediaani (95 %:n luottamusväli) | NE (28,2–NE) | 25,9 (22,1–NE) | ||
Osuus (95 %:n luottamusväli) |
|
| ||
6 kuukauden kohdalla | 89,5 (86,1–92,1) | 86,2 (82,4–89,1) | ||
12 kuukauden kohdalla | 80,1 (75,9–83,6) | 72,1 (67,4–76,2) | ||
Etenemisvapaa elinaika |
|
| ||
Tapahtumat | 228 (53,6 %) | 228 (54,0 %) | ||
Riskitiheyssuhdea | 0,82 | |||
99,1 %:n luottamusväli | (0,64–1,05) | |||
p‑arvob,h | 0,0331 | |||
Mediaani (95 %:n luottamusväli)
| 11,6 (8,71–15,51) | 8,4 (7,03–10,81) | ||
Vahvistettu objektiivinen vaste (BICR) | 177 (41,6 %) | 112 (26,5 %) | ||
(95 %:n luottamusväli) | (36,9–46,5) | (22,4–31,0) | ||
Ero objektiivisen vasteen saavuttaneiden osuudessa (95 %:n luottamusväli)d | 16,0 (9,8–22,2) | |||
p‑arvoe,f | < 0,0001 | |||
| ||||
Täydellinen vaste (CR) | 40 (9,4 %) | 5 (1,2 %) | ||
Osittainen vaste (PR) | 137 (32,2 %) | 107 (25,4 %) | ||
Stabiili tauti (SD) | 133 (31,3 %) | 188 (44,5 %) | ||
| ||||
Vasteen keston mediaanig |
|
|
| |
Kuukausina (vaihteluväli) | NE (1,4+–25,5+) | 18,17 (1,3+–23,6+) | ||
| ||||
Vasteen saavuttamisajan mediaani |
|
| ||
Kuukausina (vaihteluväli) | 2,8 (0,9–11,3) | 3,0 (0,6–15,0) | ||
| Päivitetty analyysi* Seuranta-aika vähintään: 60 kuukautta | |||
Kokonaiselinaika (OS) |
|
| ||
Tapahtumat | 242 (57 %) | 282 (67 %) | ||
Riskitiheyssuhdea | 0,68 | |||
95 %:n luottamusväli | (0,58–0,81) | |||
| ||||
Mediaani (95 %:n luottamusväli) | 46,95 (35,35–57,43) | 26,64 (22,08–33,54) | ||
Osuus (95 %:n luottamusväli) |
|
| ||
24 kuukauden kohdalla | 66,3 (61,5–70,6) | 52,4 (47,4–57,1) | ||
36 kuukauden kohdalla | 54,6 (49,7–59,3) | 43,7 (38,7–48,5) | ||
48 kuukauden kohdalla | 49,9 (44,9–54,6) | 35,8 (31,1–40,5) | ||
60 kuukauden kohdalla | 43,0 (38,1–47,7) | 31,3 (26,8–35,9) | ||
Etenemisvapaa elinaika |
|
| ||
Tapahtumat | 245 (57,6 %) | 253 (60,0 %) | ||
Riskitiheyssuhdea | 0,73 | |||
95 %:n luottamusväli | (0,61–0,87) | |||
Mediaani (95 %:n luottamusväli) | 11,6 (8,44–16,63) | 8,3 (7,03–10,41) | ||
Vahvistettu objektiivinen vaste (BICR) | 179 (42,1 %) | 113 (26,8 %) | ||
(95 %:n luottamusväli) | (37,4–47,0) | (22,6–31,3) | ||
Ero objektiivisen vasteen saavuttaneiden osuudessa (95 %:n luottamusväli)d,e | 16,2 (10,0–22,5) | |||
| ||||
Täydellinen vaste (CR) | 48 (11,3 %) | 9 (2,1 %) | ||
Osittainen vaste (PR) | 131 (30,8 %) | 104 (24,6 %) | ||
Stabiili tauti (SD) | 131 (30,8 %) | 187 (44,3 %) | ||
| ||||
Vasteen keston mediaanig |
|
| ||
Kuukausina (vaihteluväli) | NE (50,89–NE) | 19,38 (15,38–25,10) | ||
| ||||
Vasteen saavuttamisajan mediaani |
|
| ||
Kuukausina (vaihteluväli) | 2,8 (0,9–35,0) | 3,1 (0,6–23,6) | ||
a Perustuen ositettuun verrannollisten riskitiheyksien malliin.
b Perustuen ositettuun log‑rank‑merkitsevyystestiin.
c Tilastollisen merkitsevyyden saavuttamiseksi p‑arvoa on verrattu α 0,002:een.
d Osajoukkojen suhteen korjattu ero.
e Perustuen ositettuun DerSimonian‑Lairdin testiin.
f Tilastollisen merkitsevyyden saavuttamiseksi p‑arvoa on verrattu α 0,001:een.
g Laskettu Kaplan‑Meierin menetelmällä.
h Tilastollisen merkitsevyyden saavuttamiseksi p‑arvoa on verrattu α 0,009:een.
“+” tarkoittaa sensuroitunutta havaintoa.
NE = non‑estimable, ei arvioitavissa
* Deskriptiivinen analyysi perustuu tietojen keruun katkaisuajankohtaan 26.2.2021.
Kuva 19: Kohtalaisen/huonon ennusteen potilaiden kokonaiselinajan Kaplan–Meier‑kuvaaja (CA209214) – seuranta-aika vähintään 60 kuukautta
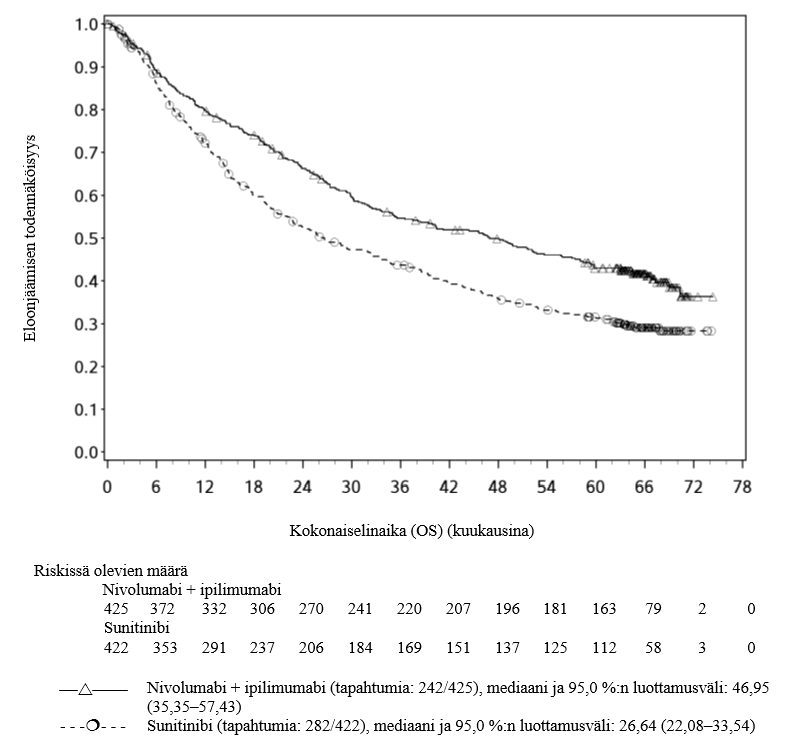
Kokonaiselinajan päivitetty deskriptiivinen analyysi tehtiin, kun kaikki potilaat olivat olleet seurannassa vähintään 24 kuukautta. Analyysihetkellä riskitihyessuhde oli 0,66 (99,8 %:n luottamusväli: 0,48–0,91), ja tapahtumia oli yhdistelmähoitohaarassa 166/425 ja sunitinibihaarassa 209/422. Nivolumabi–ipilimumabi‑yhdistelmähoitoa saaneilla kohtalaisen/huonon ennusteen potilailla kokonaiselinajan hyöty oli sunitinibia saaneita parempi riippumatta kasvainten PD‑L1:n ilmentymisasteesta. PD‑L1:n ilmentymistason ollessa ≥ 1 % nivolumabi–ipilimumabi‑yhdistelmähoitoa saaneiden ryhmässä kokonaiselinajan mediaania ei saavutettu, ja sunitinibiryhmässä se oli 19,61 kuukautta (riskitiheyssuhde = 0,52; 95 %:n luottamusväli: 0,34–0,78). PD‑L1:n ilmentymistason ollessa < 1 % nivolumabi–ipilimumabi‑yhdistelmähoitoa saaneiden ryhmässä kokonaiselinajan mediaani oli 34,7 kuukautta ja sunitinibiryhmässä 32,2 kuukautta (riskitiheyssuhde = 0,70; 95 %:n luottamusväli: 0,54–0,92).
CA209214‑tutkimuksessa satunnaistettiin IMDC:n kriteerien mukaisesti lisäksi 249 hyvän ennusteen potilasta saamaan joko nivolumabia ja ipilimumabia (n = 125) tai sunitinibia (n = 124). Näitä potilaita ei arvioitu osana tehon arviointiin ensisijaisesti käytettyä potilasjoukkoa. Kun seuranta-aika oli vähintään 24 kuukautta, kokonaiselinajan riskitiheyssuhde oli nivolumabia ja ipilimumabia saaneilla hyvän ennusteen potilailla sunitinibiin verrattuna 1,13 (95 %:n luottamusväli: 0,64–1,99; p = 0,6710). Kun seuranta-aika oli vähintään 60 kuukautta, kokonaiselinajan riskitiheyssuhde oli 0,94 (95 %:n luottamusväli: 0,65–1,37).
Nivolumabin yhteiskäytöstä ipilimumabin kanssa ei ole tietoa munuaiskarsinooman ensilinjan hoidossa potilailla, joilla on ainoastaan ei-kirkassoluinen histologinen kasvaintyyppi.
CA209214‑tutkimuksessa 8 % kaikista kohtalaisen/huonon ennusteen potilaista oli ≥ 75‑vuotiaita, ja koko potilasjoukkoon verrattuna tässä alaryhmässä nivolumabi–ipilimumabi‑hoito vaikutti lukumääräisesti vähemmän kokonaiselinaikaan (riskitiheyssuhde 0,97, 95 %:n luottamusväli: 0,48–1,95), kun seuranta-aika oli vähintään 17,5 kuukautta. Koska alaryhmä oli pieni, sen tiedoista ei voida tehdä luotettavia johtopäätöksiä.
Satunnaistettu vaiheen 3 tutkimus, nivolumabi–kabotsantinibi-yhdistelmähoito vs. sunitinibi (CA2099ER)
Satunnaistetussa vaiheen 3 avoimessa tutkimuksessa (CA2099ER) arvioitiin nivolumabin (240 mg laskimoon kahden viikon välein) ja kabotsantinibin (40 mg suun kautta kerran vuorokaudessa) yhdistelmähoidon turvallisuutta ja tehoa edenneen/metastasoituneen munuaiskarsinooman ensilinjan hoidossa. Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli edennyt tai metastasoitunut kirkassoluinen munuaiskarsinooma; tutkimukseen otettiin potilaita kasvaimen PD-L1-statuksesta tai potilaiden IMDC-riskiryhmästä riippumatta. Tutkimuksessa mukana olleiden potilaiden Karnofskyn toimintakykypisteet (Karnofsky Performance Status, KPS) olivat ≥ 70 %, ja tauti oli mitattavissa RECIST 1.1 -kriteerien perusteella. Tutkimuksesta suljettiin pois potilaat, joilla oli autoimmuunisairaus tai jokin muu systeemistä immunosuppressiota vaativa tila, jotka olivat aiemmin saaneet hoitoa PD-1-, PD-L1-, PD-L2-, CD137- tai CTLA-4-vasta-aineilla tai joilla oli huonossa hoitotasapainossa oleva hypertensio verenpainelääkityksestä huolimatta, aktiivisia aivometastaaseja tai huonossa hoitotasapainossa oleva lisämunuaisten vajaatoiminta. Potilaat ositettiin IMDC:n prognostisen pistemäärän, kasvaimen PD-L1:n ilmentymisen ja maantieteellisen alueen mukaan.
Yhteensä 651 potilasta satunnaistettiin saamaan joko nivolumabia 240 mg laskimoon kahden viikon välein sekä kabotsantinibia 40 mg suun kautta kerran vuorokaudessa (n = 323) tai sunitinibia (n = 328) 50 mg suun kautta kerran vuorokaudessa neljän viikon ajan, minkä jälkeen pidettiin aina kahden viikon tauko. Hoitoa jatkettiin, kunnes sairaus eteni tai kunnes enintään 24 kuukauden pituisen nivolumabihoidon yhteydessä ilmaantui ei-hyväksyttävää toksisuutta. Hoito taudin etenemisen jälkeen sallittiin tutkijan suorittaman ensimmäisen kiinteiden kasvainten vasteenarviointikriteerien (RECIST) version 1.1 mukaisesti tekemän arvioinnin jälkeen, jos tutkija arvioi, että potilaalle on kliinistä hyötyä tutkimuslääkkeestä ja hän sietää hoitoa. Kasvaimet arvioitiin ensimmäisen kerran lähtötilanteen jälkeen 12 viikon (± 7 päivän) kuluttua satunnaistamisesta. Sen jälkeen kasvaimet arvioitiin 6 viikon (± 7 päivän) välein viikkoon 60 saakka, ja tämän jälkeen 12 viikon (± 14 päivän) välein sairauden radiologiseen etenemiseen saakka, minkä riippumaton keskitetty arvioijataho (Blinded Independent Central Review, BICR) varmisti. Ensisijainen tehokkuusmuuttuja oli sokkoutetun riippumattoman keskitetyn arvioijatahon määrittelemä etenemisvapaa elinaika (PFS). Muita tehokkuusmuuttujia olivat keskeiset toissijaiset päätetapahtumat kokonaiselinaika (OS) ja objektiivisen vasteen saavuttaneiden osuus (ORR).
Ryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Iän mediaani oli 61 vuotta (vaihteluväli: 28–90). Potilaista 38,4 % oli ≥ 65-vuotiaita ja 9,5 % ≥ 75-vuotiaita. Suurin osa potilaista oli miehiä (73,9 %) ja valkoihoisia (81,9 %). Kahdeksan prosenttia potilaista oli aasialaisia, ja 23,2 %:lla potilaista lähtötason Karnofskyn toimintakykypisteet (KPS) olivat 79–80 % ja 76,5 %:lla potilaista 90–100 %. Potilaat jakautuivat IMDC-ennusteriskiluokituksen osalta siten, että 22,6 %:lla ennuste oli hyvä, 57,6 %:lla kohtalainen ja 19,7 %:lla huono. Kasvaimen PD-L1:n ilmentymisen osalta 72,5 %:lla potilaista PD-L1:n ilmentymistaso oli < 1 % tai määrittämätön ja 24,9 %:lla potilaista PD-L1:n ilmentymistaso oli ≥ 1 %. Potilaista 11,5 %:lla oli kasvaimessa sarkomatoidisia piirteitä. Nivolumabin ja kabotsantinibin yhdistelmähoitoa saaneiden potilaiden hoidon keston mediaani oli 14,26 kuukautta (vaihteluväli: 0,2–27,3 kuukautta) ja sunitinibihoitoa saaneiden 9,23 kuukautta (vaihteluväli: 0,8–27,6 kuukautta).
Tutkimus osoitti tilastollisesti merkitsevää hyötyä etenemisvapassa elinajassa, kokonaiselinajassa ja objektiivisen vasteen saavuttaneiden osuudessa potilailla, jotka oli satunnaistettu saamaan nivolumabin ja kabotsantinibin yhdistelmähoitoa, verrattuna potilaisiin, jotka oli satunnaistettu saamaan sunitinibihoitoa. Primaarianalyysin tehoa koskevat tulokset (seuranta-aika vähintään 10,6 kuukautta; seuranta-ajan mediaani 18,1 kuukautta) esitetään taulukossa 40.
Taulukko 40: Tulokset tehosta (CA2099ER)
| nivolumabi + kabotsantinibi | sunitinibi |
Etenemisvapaa elinaika |
|
|
Tapahtumat | 144 (44,6 %) | 191 (58,2 %) |
Riskitiheyssuhdea | 0,51 | |
95 %:n luottamusväli | (0,41–0,64) | |
p‑arvob, c | < 0,0001 | |
| ||
Mediaani (95 %:n luottamusväli)d | 16,59 (12,45–24,94) | 8,31 (6,97–9,69) |
Kokonaiselinaika (OS) |
|
|
Tapahtumat | 67 (20,7 %) | 99 (30,2 %) |
Riskitiheyssuhdea | 0,60 | |
98,89 %:n luottamusväli | (0,40–0,89) | |
p‑arvob,c,e | 0,0010 | |
Mediaani (95 %:n luottamusväli)
| NE | NE (22,6–NE) |
Osuus (95 %:n luottamusväli) |
|
|
6 kuukauden kohdalla | 93,1 (89,7–95,4) | 86,2 (81,9–89,5) |
Vahvistettu objectiivinen vaste (BICR) |
180 (55,7 %) |
89 (27,1 %) |
(95 %:n luottamusväli)f | (50,1–61,2) | (22,4–32,3) |
Ero objektiivisen vasteen saavuttaneiden osuudessa (95 %:n luottamusväli g | 28,6 (21,7–35,6) | |
p‑arvoh | < 0,0001 | |
| ||
Täydellinen vaste (CR) | 26 (8,0%) | 15 (4,6%) |
Osittainen vaste (PR) | 154 (47,7%) | 74 (22,6%) |
Stabiili tauti (SD) | 104 (32,2%) | 138 (42,1%) |
| ||
Vasteen keston mediaanid |
|
|
Kuukausina (vaihteluväli) | 20,17 (17,31–NE) | 11.47 (8,31–18,43) |
| ||
Vasteen saavuttamisajan mediaani |
|
|
Kuukausina (vaihteluväli) | 2,83 (1,0–19,4) | 4,17 (1.7–12,3) |
a Ositettu Coxin suhteellisen vaaran malli. Riskitiheyssuhde on nivolumabin ja kabotsantinibin osalta parempi kuin sunitinibin.
b Log‑rank‑testi ositettu IMDC-ennusteriskipisteiden (0, 1–2, 3–6), kasvaimen PD‑L1:n ilmentymän (≥ 1 % versus < 1 % tai määrittämätön) ja maantieteellisen alueen (Yhdysvallat/Kanada/Länsi-Eurooppa/Pohjois-Eurooppa, muu maailma) mukaan siten kuin ne on syötetty IRT-järjestelmään (interaktiivinen vastausjärjestelmä)
c 2‑tahoiset p‑arvot saatu ositetusta tavallisesta log‑rank‑testistä.
d Perustuu Kaplan–Meier-arvioihin.
e Tilastollisen merkitsevyyden p‑arvon raja < 0,0111.
f Luottamusväli perustuu Clopperin ja Pearsonin menetelmään.
g Osajoukkojen suhteen korjattu ero objektiivisen vasteen saavuttaneiden osuudessa (nivolumabi + kabotsantinibi – sunitinibi) perustuu DerSimonianin ja Lairdin menetelmään.
h CMH-testin 2‑tahoinen p‑arvo.
NE = non estimable, ei arvioitavissa
Etenemisvapaan elinajan primaarianalyysiin sisältyi uuden syöpähoidon sensurointi (taulukko 40). Etenemisvapaata elinaikaa koskevat sensuroidut ja sensuroimattomat tulokset olivat uuden syöpähoidon osalta yhdenmukaiset.
Nivolumabin–kabotsantinibi-yhdistelmähoitoa saaneen ryhmän etenemisvapaassa elinajassa havaittiin hyötyä sunitinibiryhmään verrattuna riskiluokasta (IMDC) riippumatta. Hyvän ennusteen ryhmässä nivolumabi–kabotsantinibi-yhdistelmähoitoa saaneet potilaat eivät saavuttaneet etenemisvapaan elinajan mediaania, ja sunitinibiryhmässä se oli 12,81 kuukautta (riskitiheyssuhde = 0,60; 95 %:n luottamusväli: 0,37–0,98). Kohtalaisen ennusteen ryhmässä etenemisvapaan elinajan mediaani oli nivolumabi–kabotsantinibi-yhdistelmähoitoa saaneiden ryhmässä 17,71 kuukautta ja sunitinibiryhmässä 8,38 kuukautta (riskitiheyssuhde = 0,54; 95 %:n luottamusväli: 0,41–0,73). Huonon ennusteen ryhmässä etenemisvapaan elinajan mediaani oli nivolumabi–kabotsantinibi-yhdistelmähoitoa saaneiden ryhmässä 12,29 kuukautta ja sunitinibiryhmässä 4,21 kuukautta (riskitiheyssuhde = 0,36; 95 %:n luottamusväli: 0,23–0,58).
Nivolumabin–kabotsantinibi-yhdistelmähoitoa saaneen ryhmän etenemisvapaassa elinajassa havaittiin hyötyä sunitinibihoitoa saaneeseen ryhmään verrattuna riippumatta PD-L1:n ilmentymisestä kasvaimessa. Kasvaimen PD-L1:n ilmentymistason ollessa ≥ 1 prosenttia etenemisvapaan elinajan mediaani oli nivolumabi–kabotsantinibi-yhdistelmähoitoa saaneiden ryhmässä 13,08 kuukautta ja sunitinibiryhmässä 4,67 kuukautta (riskitiheyssuhde = 0,45; 95 %:n luottamusväli: 0,29–0,68). Kasvaimen PD-L1:n ilmentymistason ollessa < 1 prosenttia etenemisvapaan elinajan mediaani oli nivolumabi–kabotsantinibi-yhdistelmähoitoa saaneiden ryhmässä 19,84 kuukautta ja sunitinibiryhmässä 9,26 kuukautta (riskitiheyssuhde = 0,50; 95 %:n luottamusväli: 0,38–0,65).
Etenemisvapaan elinajan ja kokonaiselinajan päivitetty analyysi tehtiin, kun kaikkia potilaita oli seurattu vähintään 16,0 kuukauden ajan ja seuranta-ajan mediaani oli 23,5 kuukautta (ks. kuvat 20 ja 21). Etenemisvapaan elinajan riskitiheyssuhde oli 0,52 (95 %:n luottamusväli: 0,43–0,64). Kokonaiselinajan riskitiheyssuhde oli 0,66 (95 %:n luottamusväli: 0,50–0,87). IMDC-riskiluokitusten alaryhmien tehoa koskevat päivitetyt tiedot (etenemisvapaa elinaika ja kokonaiselinaika) ja PD-L1:n ilmentymistasot vahvistivat alkuperäiset tulokset. Hyvän ennusteen ryhmä saavutti päivitetyssä analyysissä etenemisvapaan elinajan mediaanin.
Kuva 20: Etenemisvapaan elinajan Kaplan–Meier-kuvaajat (CA2099ER)
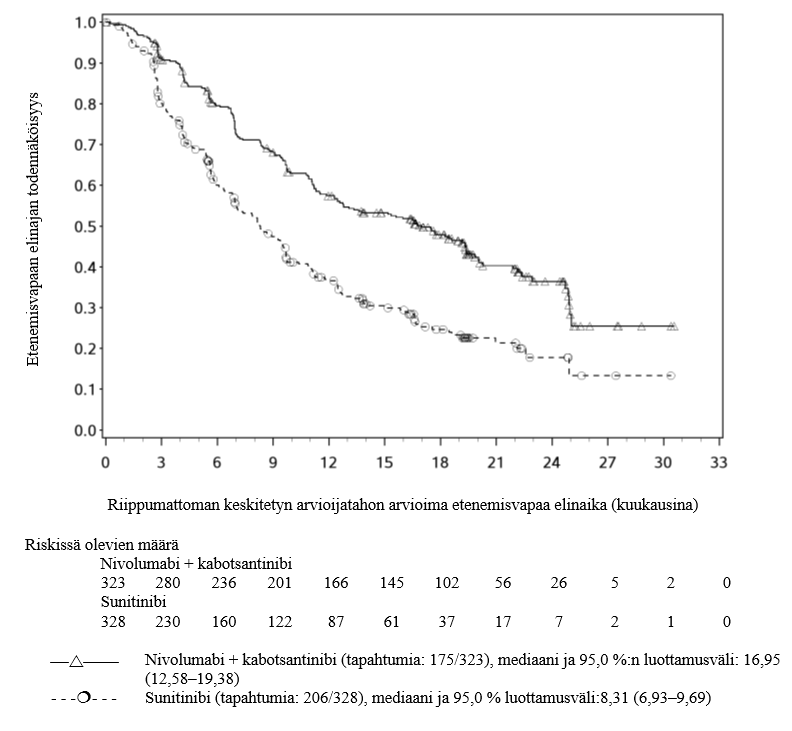
Kuva 21: Kokonaiselinajan Kaplan–Meier-kuvaajat (CA2099ER)
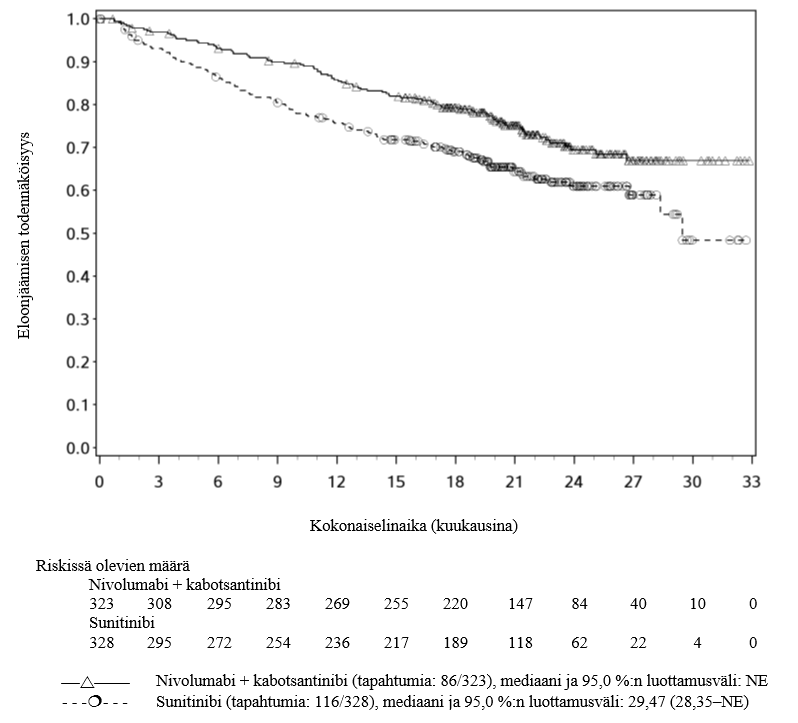
Satunnaistettu vaiheen 3 tutkimus, nivolumabi-monoterapia vs. everolimuusi (CA209025)
Nivolumabin (3 mg/kg) tehoa ja turvallisuutta monoterapiana edenneen kirkassoluisen munuaiskarsinooman hoidossa arvioitiin vaiheen 3 satunnaistetussa avoimessa tutkimuksessa (CA209025). Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joiden sairaus oli edennyt yhden tai kahden aiemman angiogeneesinestäjähoidon aikana tai sen jälkeen ja joilla oli korkeintaan 3 aiempaa systeemistä hoitoa. Potilailla piti olla Karnofskyn asteikolla pisteitä ≥ 70 %. Tutkimukseen otettiin potilaita riippumatta kasvaimen PD‑L1-statuksesta. Tutkimuksesta suljettiin pois potilaat, joilla oli tai oli ollut aivomestastaaseja, joita oli aiemmin hoidettu nisäkkään rapamysiinin kohteen (mammalian target of rapamycin, mTOR) estäjällä ja joilla oli aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila.
Yhteensä 821 potilasta satunnaistettiin saamaan joko nivolumabia 3 mg/kg (n = 410) suonensisäisesti 60 minuutin ajan joka toinen viikko tai everolimuusia (n = 411) 10 mg päivittäin suun kautta. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt sitä. Ensimmäinen arvio kasvaimista tehtiin 8 viikkoa satunnaistamisen jälkeen ja tämän jälkeen joka 8. viikko ensimmäisen vuoden ajan ja sitten joka 12. viikko taudin etenemiseen tai hoidon lopettamiseen saakka, kumpi näistä tapahtui jälkimmäisenä. Kasvainten arvioimista jatkettiin hoidon lopettamisen jälkeen niillä potilailla, jotka lopettivat hoidon muista kuin taudin etenemisestä johtuvista syistä. Nivolumabihoito taudin etenemisen jälkeen sallittiin tutkijan suorittaman ensimmäisen kiinteiden kasvainten vasteenarviointikriteerien (RECIST) version 1.1 mukaisesti tekemän arvioinnin jälkeen, jos tutkija totesi, että potilaalle on kliinistä hyötyä tutkimuslääkkeestä ja hän sietää hoitoa. Ensisijainen tehokkuusmuuttuja oli OS. Toissijaiset tehokkuusmuuttujat olivat tutkijan arvioima ORR ja PFS.
Ryhmien lähtötason ominaisuudet olivat tasapainossa. Iän mediaani oli 62 vuotta (vaihteluväli 18–88), 40 % oli 65‑vuotiaita tai sitä vanhempia ja 9 % oli 75‑vuotiaita tai sitä vanhempia. Suurin osa tutkittavista oli miehiä (75 %) ja valkoihoisia (88 %), kaikki MSKCC:n (Memorial Sloan Kettering Cancer Center) mukaiset riskiryhmät olivat edustettuina ja 34 %:lla potilaista lähtötason Karnofskyn toimintakykypisteet (KPS) olivat 70–80 % ja 66 %:lla potilaista KPS oli 90–100 %. Suurin osa potilaista (72 %) oli saanut yhtä aiempaa angiogeneesiestäjähoitoa. Ajan mediaani ensimmäisestä diagnoosista satunnaistamiseen oli 2,6 vuotta sekä nivolumabi‑, että everolimuusiryhmissä. Hoidon keston mediaani oli 5,5 kuukautta (vaihteluväli 0–29,6+ kuukautta) nivolumabihoitoa saaneilla potilailla ja 3,7 kuukautta (vaihteluväli 6 päivästä 25,7 +kuukauteen) everolimuusihoitoa saaneilla potilailla.
Nivolumabihoitoa jatkettiin taudin etenemisen jälkeen 44 %:lla potilaista.
Kuvassa 22 on esitetty kokonaiselinajan Kaplan–Meier-kuvaajat.
Kuva 22: Kokonaiselinajan Kaplan–Meier-kuvaajat (CA209025)
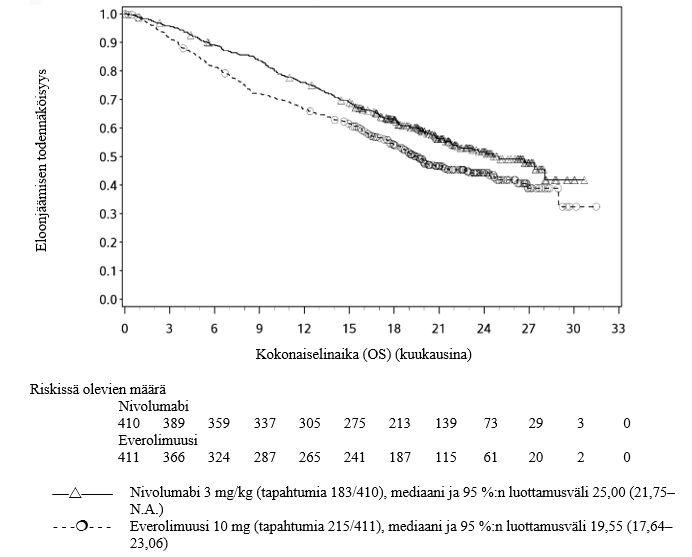
Everolimuusiin verrattuna tutkimuksessa todettiin tilastollisesti merkitsevä paraneminen kokonaiselinajassa potilailla, jotka oli satunnaistettu saamaan nivolumabia. Tulos perustui ennaltamääriteltyyn välianalyysiin, jossa arvioitiin 398 tapausta (70 % suunnitellusta loppuanalyysin tapahtumien määrästä) (taulukko 41 ja kuva 22). Kokonaiselinajan hyöty oli riippumaton kasvainten PD‑L1:n ilmentymisasteesta.
Tulokset tehosta näkyvät taulukossa 41.
Taulukko 41: Tulokset lääkeaineen tehosta (CA209025)
| nivolumabi | everolimuusi |
|---|---|---|
Kokonaiselinaika (OS) | ||
Tapahtumia | 183 (45 %) | 215 (52 %) |
Riskitiheyssuhde | 0,73 | |
98,52 %:n luottamusväli | (0,57–0,93) | |
p‑arvo | 0,0018 | |
| ||
Mediaani (95 %:n luottamusväli) | 25,0 (21,7–NE) | 19,6 (17,6–23,1) |
Osuus (95 %:n luottamusväli) | ||
6 kuukauden kohdalla | 89,2 (85,7–91,8) | 81,2 (77,0–84,7) |
12 kuukauden kohdalla | 76,0 (71,5–79,9) | 66,7 (61,8–71,0) |
| ||
Objektiivinen vaste | 103 (25,1 %) | 22 (5,4 %) |
(95 %:n luottamusväli) | (21,0–29,6) | (3,4, 8,0) |
Ristitulosuhde (95 %:n luottamusväli) | 5,98 (3,68–9,72) | |
p‑arvo | < 0,0001 | |
| ||
Täydellinen vaste (CR) | 4 (1,0 %) | 2 (0,5 %) |
Osittainen vaste (PR) | 99 (24,1 %) | 20 (4,9 %) |
Stabiili tauti (SD) | 141 (34,4 %) | 227 (55,2 %) |
| ||
Vasteen keston mediaani | ||
Kuukausina (vaihteluväli) | 11,99 (0,0–27,6+) | 11,99 (0,0+–22,2+) |
| ||
Vasteen saavuttamisajan mediaani | ||
Kuukausina (vaihteluväli) | 3,5 (1,4–24,8) | 3,7 (1,5–11,2) |
Etenemisvapaa elinaika | ||
Tapahtumia | 318 (77,6 %) | 322 (78,3 %) |
Riskitiheyssuhde | 0,88 | |
95 %:n luottamusväli | (0,75–1,03) | |
p‑arvo | 0,1135 | |
Mediaani (95 %:n luottamusväli) | 4,6 (3,71–5,39) | 4,4 (3,71–5,52) |
”+” tarkoittaa sensuroitunutta havaintoa.
NE = ei arvioitavissa
Objektiivisen vasteen saavuttamisajan mediaani oli 3,5 kuukautta (vaihteluväli 1,4–24,8 kuukautta) nivolumabihoidon aloituksesta. 49:llä vasteensaajalla (47,6 %) vasteen kesto oli 0,0–27,6+ kuukautta.
Kokonaiselinaikaan voi ajan kuluessa liittyä sairauteen liittyvien oireiden helpottuminen ja yleisen elämänlaadun kohentuminen (QoL). Näitä arvioitiin käyttäen valideja ja luotettavia asteikkoja FKSI‑DRS (Functional Assessment of Cancer Therapy‑Kidney Symptom Index‑Disease Related Symptoms) ja EuroQoL EQ‑5D. Oireiden merkittävä helpottuminen (MID = 2 pisteen muutos FKSI‑DRS ‑asteikolla, p < 0,001) ja tähän kulunut aika (riskitiheyssuhde = 1,66 (1,33–2,08), p < 0,001) olivat merkitsevästi parempia potilailla, jotka saivat nivolumabia. Koska kumpikin tutkimushaara sai aktiivista hoitoa, elämänlaatuaineistoa tulee tulkita harkiten avoimen tutkimusasetelman puitteissa.
Vaiheen 3b/4 turvallisuustutkimus (CA209374)
Lisää turvallisuustietoja ja deskriptiivisiä tietoja tehosta saatiin CA209374-tutkimuksessa, joka oli avoin, vaiheen 3b/4 turvallisuustutkimus. Tutkimuksessa nivolumabi-monoterapiaa (240 mg joka toinen viikko) annettiin potilaille, joilla oli edennyt tai metastasoinut munuaiskarsinooma (n = 142). Mukana tutkimuksessa oli 44 potilasta, joilla oli ei‑kirkassoluinen histologinen kasvaintyyppi.
Potilailla, joilla oli ei‑kirkassoluinen histologinen kasvaintyyppi, objektiivisen vasteen saavuttaneiden osuus oli vähintään noin 16,7 kuukautta kestäneessä seurannassa 13,6 % ja vasteen keston mediaani 10,2 kuukautta. Kliinistä aktiivisuutta havaittiin kasvaimen PD‑L1:n ilmentymisestä riippumatta.
Klassinen Hodgkinin lymfooma
Satunnaistettu vaiheen 3 tutkimus, nivolumabi–AVD‑yhdistelmähoito vs. brentuksimabivedotiini–AVD‑yhdistelmähoito (CA2098UT)
Nivolumabin tehoa ja turvallisuutta yhdistelmähoitona doksorubisiinin, vinblastiinin ja dakarbatsiinin (AVD) kanssa aikuisilla ja 12‑vuotiailla ja sitä vanhemmilla nuorilla aiemmin hoitamattoman asteen III tai IV cHL:n hoidossa arvioitiin vaiheen 3 satunnaistetussa avoimessa monikeskustutkimuksessa.
Yhteensä 994 potilasta satunnaistettiin suhteessa 1:1 saamaan joko nivolumabin ja AVD‑kemoterapian yhdistelmähoitoa (n = 496) tai brentuksimabivedotiinin ja AVD‑kemoterapian yhdistelmähoitoa (BV + AVD) (n = 498). Satunnaistamisen ositustekijöitä olivat ikä (12–17 vs. 18–60 vs. > 60 vuotta), kansainvälinen ennustepisteytys (0–3 vs. 4–7) ja ennalta määritetty jäännöstaudin PET‑sädehoito (kyllä vs. ei). Kasvaimet arvioitiin 4–8 viikon kuluttua hoitojaksosta 6 tai kun tutkimushoito lopetettiin. Ensisijainen tehokkuusmuuttuja oli etenemisvapaa elinaika (PFS) tutkijan arvioimana. Muita tehokkuusmuuttujia olivat kokonaiselinaika (OS), päätetapahtumaton elinaika (EFS) ja täydellisen metabolisen vasteen saavuttaneiden osuus hoidon päättyessä.
Tutkimuspopulaatio koostui tutkittavista, joilla oli histologisesti vahvistettu, äskettäin diagnosoitu, aiemmin hoitamaton asteen III tai IV cHL (sidekudoskyhmyinen, sekasoluinen, runsaslymfosyyttinen tai vähälymfosyyttinen tai määrittelemätön) ja kaksiulotteisesti mitattavissa oleva tauti (vähintään yksi leesio, jonka pisin halkaisija oli > 1,5 cm). Henkilöt, joilla oli nodulaarinen runsaslymfosyyttinen Hodgkinin lymfooma, eivät soveltuneet tutkimukseen. Tutkittaville tehtiin koko kehon PET‑TT‑kuvaus tai rajattu koko kehon PET‑TT‑kuvaus 42 vuorokauden sisällä ennen satunnaistamista. Tutkimuksesta suljettiin pois potilaat, joilla oli aktiivinen autoimmuunisairaus, systeemistä immunosuppressiota vaativia sairauksia, vaikeusasteen > 2 perifeerinen neuropatia, aktiivinen interstitiaalinen pneumoniitti tai interstitiaalinen keuhkosairaus tai keskushermostolymfooma.
Molempien hoitoryhmien potilaat saivat laskimonsisäisesti jotakin seuraavista kunkin 28 vuorokauden hoitojakson päivinä 1 ja 15 kuuden hoitojakson ajan:
-
nivolumabi 240 mg 30 minuutin kuluessa sekä doksorubisiini (25 mg/m2), vinblastiini (6 mg/m2) ja dakarbatsiini (375 mg/m2) joka toinen viikko aikuisille ja nuorille (12‑vuotiaille ja sitä vanhemmille, jotka painoivat vähintään 80 kg) tai
-
nivolumabi 3 mg/kg 30 minuutin kuluessa sekä doksorubisiini (25 mg/m2), vinblastiini (6 mg/m2) ja dakarbatsiini (375 mg/m2) joka toinen viikko 12‑vuotiaille ja sitä vanhemmille nuorille, jotka painoivat alle 80 kg tai
-
brentuksimabivedotiini 1,2 mg/kg sekä doksorubisiini (25 mg/m2), vinblastiini (6 mg/m2) ja dakarbatsiini (375 mg/m2) joka toinen viikko aikuisille ja nuorille.
Kaikkien satunnaistettujen tutkittavien lähtötason ominaisuudet olivat tasapainossa molemmissa hoitoryhmissä. Tutkimuspopulaation mediaani-ikä oli 27 vuotta (vaihteluväli: 12–83) ja 31 (6 %) oli ≥ 65‑vuotiaita. Suurin osa potilaista oli valkoihoisia (75 %) ja 56 % oli miehiä. Lähtötason ECOG-toimintakykyluokka oli 0 (56 %), 1 (38 %) tai 2 (5 %). Potilaista 36 %:lla oli diagnoosivaiheessa asteen III cHL ja 64 %:lla asteen IV cHL. Nivolumabi–AVD-hoitoryhmässä 338 potilaan (68,1 %) IPS oli 0–3 ja 158 potilaan (31,9 %) IPS oli 4–7 diagnoosivaiheessa.
Etukäteen määritellyn välianalyysin ajankohtana (seuranta vähintään 2,4 kuukautta) etenemisvapaan elinajan osoitettiin parantuneen tilastollisesti merkitsevästi nivolumabin ja AVD‑kemoterapian yhdistelmähoitoa saaneilla verrattuna BV + AVD -yhdistelmähoitoa saaneisiin, ja HR oli 0,42 (95 %:n luottamusväli: 0,27–0,67). Päivitetyssä kuvailevassa analyysissa, jossa seuranta oli vähintään 30,4 kuukautta, kokonaiselinaikatulokset eivät olleet kypsiä analysoitaviksi. Tulokset tehosta esitetään taulukossa 42 ja kuvassa 23.
Taulukko 42 Tehoon liittyvät tulokset (CA2098UT)
| nivolumabi + AVD | BV + AVD |
| Seuranta-aika vähintään: 2,4 kuukautta | |
Etenemisvapaa elinaika | ||
Tapahtumia (%) | 28/496 (5,6) | 62/498 (12,4) |
Riskitiheyssuhde
|
| |
(95 %:n luottamusväli)a
| 0,42 (0,27–0,67) | |
p‑arvob | < 0,0001 | |
Mediaani (kuukautta)c (95 %:n luottamusväli) | Ei sovellettavissa | Ei sovellettavissa |
a Riskitiheyssuhde on nivolumabi + AVD -hoito BV + AVD -hoitoon nähden Coxin suhteellisen vaaran mallissa, jossa ositustekijöitä ovat ikä (12–17 vs. 18–60 vs. > 60 vuotta), IPS (0–3 vs. 4–7) ja etukäteen määritetty suunnitelma jäännöstaudin PET‑sädehoidosta (kyllä vs. ei) Oncology Patient Enrollment Network (OPEN) -ryhmän suorittamana.
b Yksipuolinen paremmuuden log‑rank‑testi, jossa käytettiin samaa ositustekijää kuin Coxin suhteellisen vaaran mallissa.
c Perustuu Kaplan–Meierin arvioihin
Kuva 23: Etenemisvapaan elinajan Kaplan–Meier-kuvaaja (CA2098UT)
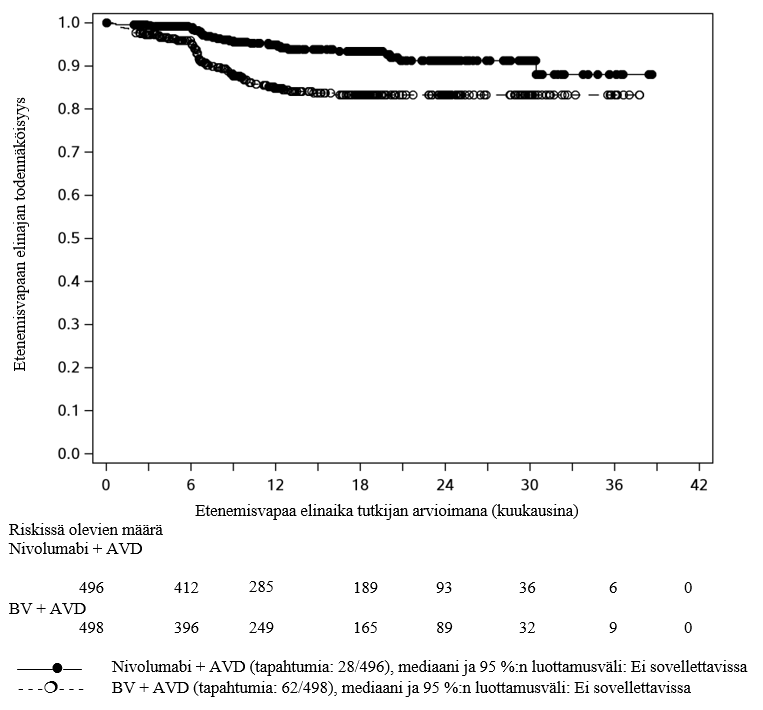
Perustuu kliinisten tietojen keruun katkaisuajankohtaan 15.12.2022, seuranta-aika vähintään 2,4 kuukautta
Avoin vaiheen 2 tutkimus nivolumabista yhdistelmähoitona brentuksimabivedotiinin kanssa (CA209744)
Yhdistelmähoitona brentuksimabivedotiinin (1,8 mg/kg) kanssa annetun nivolumabin (3 mg/kg) turvallisuutta ja tehoa arvioitiin yhden aiemman hoitolinjan jälkeen vaiheen 2 riskiperusteisessa, hoitovasteeseen mukautetussa avoimessa tutkimuksessa potilailla, joilla oli uusiutunut tai refraktaarinen cHL. Kohortissa R1 oli 28 potilasta, joilla uusiutumisriski oli pieni, ja kohortissa R2 oli 44 potilasta, joilla uusiutumisriski oli tavanomainen. Riskin ositus perustui seuraaviin: sairauden aste ensimmäisen diagnoosin tekohetkellä; aika uusiutumiseen (hoidon päättymisestä); B‑oireet tai ekstranodaalinen sairaus uusiutumishetkellä; laaja sairaus, jossa sädehoito oli vasta-aiheista uusiutumishetkellä; tai uusiutuma aiemmassa säteilykentässä, kuten taulukossa 43 on esitetty. CD30:n ilmentymistä ei mitattu ennen tutkimukseen osallistumista.
Taulukko 43: Riskin ositusalgoritmi CA209744‑tutkimuksessa
Aste ensimmäisen diagnoosin tekohetkellä | Aika uusiutumiseen | B‑oireet tai ekstranodaalinen sairaus uusiutumishetkellä; laaja sairaus, jossa sädehoito oli vasta-aiheista uusiutumishetkellä; tai uusiutuma aiemmassa säteilykentässä | Uusiutumisriskin luokka |
IA, IIA | ≥ 12 kuukautta | Ei | Kohortti R1 Pieni riski |
3–12 kuukautta (≤ 3 jaksoa, ei sädehoitoa) | Ei | ||
IB, IIB, IIIA | > 12 kuukautta | Ei | |
Kaikki muut | Kohortti R2 Tavanomainen riski | ||
Ensimmäinen arvio kasvaimista tehtiin 6 viikkoa hoidon alkamisen jälkeen, ja tämän jälkeen arviointeja tehtiin taudin etenemiseen tai hoidon päättymiseen tai lopettamiseen saakka. Ensisijaiset tehokkuusmuuttujat olivat kohortissa R1 täydellisen metabolisen vasteen (complete metabolic response, CMR) saavuttaneiden osuus koska tahansa ennen sädehoitoa BICR:n arvioimana ja päätetapahtumattoman elinajan (EFS) saavuttaneiden osuus 3 vuoden kohdalla riippumattoman arvioijatahon arvioimana. Kohortin R2 ensisijainen tehokkuusmuuttuja oli täydellisen metabolisen vasteen saavuttaneiden osuus koska tahansa ennen suuriannoksista solunsalpaajahoitoa (HDCT) / autologista kantasolujen siirtoa (ASCT), riippumattoman arvioijatahon arvioimana. Muita tehokkuusmuuttujia olivat BICR:n arvioima objektiivisen vasteen saavuttaneiden osuus (ORR) nivolumabin ja brentuksimabivedotiinin 4 yhdistelmähoitojakson jälkeen ja BICR:n arvioima etenemisvapaan elinajan (PFS) saavuttaneiden osuus 3 vuoden kohdalla. Tutkimuksesta poissuljettiin potilaat, jotka olivat altistuneet aiemmin anti-PD(L)‑1:lle/2:lle tai brentuksimabivedotiinille.
Kaikki potilaat siirtyivät induktiovaiheeseen ja saivat nivolumabin ja brentuksimabivedotiinin yhdistelmähoitoa. Hoito lopetettiin potilailta, joilla kasvain oli tutkijan arvion mukaan edennyt kahden jakson jälkeen. Potilaat, jotka eivät saavuttaneet täydellistä metabolista vastetta nivolumabin ja brentuksimabivedotiinin 4 yhdistelmähoitojakson jälkeen, siirtyivät tehostusvaiheeseen ja saivat joko 2 jaksoa (kohortti R1) tai 2–4 jaksoa (kohortti R2) brentuksimabivedotiinia yhdessä bendamustiinin (90 mg/m2/vrk) kanssa täydellisen metabolisen vasteen saavuttamisen edistämiseksi. Kohortin R1 potilaat, jotka saavuttivat täydellisen metabolisen vasteen nivolumabin ja brentuksimabivedotiinin 4 yhdistelmähoitojakson jälkeen, saivat 2 lisäjaksoa nivolumabin ja brentuksimabivedotiinin yhdistelmähoitoa ennen konsolidaatiovaihetta. Potilaat, jotka saavuttivat täydellisen metabolisen vasteen induktion tai tehostuksen jälkeen siirtyivät konsolidaatiovaiheeseen, jossa he saivat sädehoitoa (kohortti R1) tai suuriannoksista solunsalpaajahoitoa (HDCT) / autologisen kantasolujen siirron (kohortti R2).
Hoitoryhmien lähtötason ominaisuudet olivat tasapainossa. Useimmat potilaat kuuluivat ikäryhmään ≥ 5 – < 18 vuotta sekä kohortissa R1 (64,3 %) että kohortissa R2 (70,5 %). Kohortissa R1 hoitoa sai 3 lasta (5–11 vuotta), 15 nuorta (12–17 vuotta) ja 10 aikuista (18–30 vuotta), ja kohortissa R2 hoitoa sai 4 lasta (5–11 vuotta), 27 nuorta (12–17 vuotta) ja 13 aikuista (18–30 vuotta). Kohortissa R1 64,3 % oli naisia, ja kohortissa R2 65,9 % oli miehiä. Kohortissa R1 64,3 %:lla oli asteen IIA sairaus tutkimukseen otettaessa, ja kohortissa R2 sairaus oli tutkimukseen otettaessa 25 %:lla astetta IIA, 22,7 %:lla astetta IIB ja 25 %:lla astetta IVA. Karnofskyn ja Lanskyn keskimääräiset toimintakykypisteet olivat > 95 % molemmissa kohorteissa. Kohortissa R1 aika uusiutumiseen aiemman hoidon lopusta oli 82,1 %:lla ≥ 12 kuukautta. Kohortissa R2 aika uusiutumiseen aiemman hoidon lopusta oli 36,4 %:lla < 3 kuukautta ja 40,9 %:lla ≥ 3 – < 12 kuukautta. Kohortin R1 tutkittavista 96,4 %:lla tauti oli uusiutunut aiemman hoitolinjan jälkeen, kun taas kohortissa R2 jakauma oli tasainen niiden potilaiden välillä, joilla tauti oli uusiutunut (45,5 %) tai joilla vaste aiempaan hoitolinjaan oli refraktaarinen (54,5 %).
Kaikki kohorttien R1 ja R2 potilaat saivat aiempaa syöpähoitoa. Yleisin aiempi syöpähoito oli OEPA/COPDAC sekä kohortissa R1 (42,9 %) että kohortissa R2 (45,5 %).
Hoidettujen pediatristen potilaiden lähtötason ominaisuudet ja aiemmat syöpähoidot olivat yhdenmukaiset kaikkien CA209744‑tutkimuksessa hoidettujen potilaiden kanssa.
Tulokset tehosta on esitetty taulukossa 44.
Taulukko 44: Tehoon liittyvät tulokset (CA209744)
| Kohortti R1 Pieni uusiutumisriski | Kohortti R2 Tavanomainen uusiutumisriski | ||||
| Kaikki hoidetut potilaat (n = 28) | Hoidetut pediatriset potilaat (n = 18) | Kaikki hoidetut potilaat (n = 44) | Hoidetut pediatriset potilaat (n = 31) | ||
Täydellinen metabolinen vaste ennen konsolidaatiotaa,b | ||||||
n (%) | 26 (92,9) | 16 (88,9) | 39 (88,6) | 28 (90,3) | ||
95 %:n luottamusväli | (76,5–99,1) | (65,3–98,6) | (75,4–96,2) | (74,2–98,0) | ||
Kokonaisvasteen 4 induktiojakson jälkeen saavuttaneetc | ||||||
n (%) 95 %:n luottamusväli | 27 (96,4) (81,7–99,9) | 17 (94,4) (72,7–99,9) | 41 (93,2) (81,3–98,6) | 29 (93,5) (78,6–99,2) | ||
Täydellinen metabolinen vaste, n (%) | ||||||
| 23 (82,1) | 13 (72,2) | 33 (75,0) | 23 (74,2) | ||
Osittainen metabolinen vaste, n (%) | ||||||
| 4 (14,3) | 4 (22,2) | 8 (18,2) | 6 (19,4) | ||
a Nimittäjä on niiden potilaiden lukumäärä, joilla vaste on arvioitavissa. Vaste oli arvioitavissa kaikilta hoidetuilta potilailta / hoidetuilta pediatrisilta potilailta.
Kohortin R1 potilaista 6 ja kohortin R2 potilaista 11 sai brentuksimabivedotiinia yhdessä bendamustiinin kanssa tehostavana kemoterapiana ennen konsolidaatiota, koska kyseiset potilaat eivät saavuttaneet täydellistä metabolista vastetta nivolumabin ja brentuksimabivedotiinin 4 induktiojakson jälkeen.
b Kohortin R1 potilaista 22 eteni sädehoidolla toteutettuun vakautukseen tutkimusaikana. Kohortin R2 potilaista 41 sai konsolidaatiota suuriannoksisen solunsalpaajahoidon (HDCT) ja autologisen kantasolujen siirron yhdistelmällä (32 tutkimuksen aikana ja 9 tutkimuksen ulkopuolella lääkärin harkinnan mukaan).
c Nimittäjä on niiden potilaiden lukumäärä, jotka olivat arvioitavissa nivolumabin ja brentuksimabivedotiinin 4 yhdistelmähoitojakson jälkeen.
Avoin vaiheen 2 (CA209205) ja avoin vaiheen 1b (CA209039) nivolumabimonoterapiatutkimus
Nivolumabin (3 mg/kg) tehoa ja turvallisuutta monoterapiana uusiutuneen tai refraktaarisen klassisen Hodgkinin lymfooman hoidossa autologisen kantasolujen siirron jälkeen arvioitiin kahdessa avoimessa yhden hoitohaaran monikeskustutkimuksessa (CA209205 ja CA209039).
CA209205 on vaiheen 2 yhden hoitohaaran avoin monikohorttitutkimus nivolumabista klassisen Hodgkinin lymfooman hoidossa. Tutkimuksessa on 243 potilasta, jotka saivat autologisen kantasolujen siirron; kohortissa A oli 63 potilasta (26 %), joita ei ollut aiemmin hoidettu brentuksimabivedotiinilla; kohortissa B oli 80 potilasta (33 %), joita oli hoidettu brentuksimabivedotiinilla sen jälkeen kun autologinen kantasolujen siirto ei ollut aikaansaanut vastetta; ja kohortissa C oli 100 potilasta (41 %), joita oli hoidettu brentuksimabivedotiinilla ennen autologista kantasolujen siirtoa ja/tai sen jälkeen ja joista 33:a (14 %:a) oli hoidettu brentuksimabivedotiinilla ainoastaan ennen autologista kantasolujen siirtoa. Kaikki potilaat saivat nivolumabia 3 mg/kg monoterapiana laskimonsisäisesti 60 minuutin ajan joka toinen viikko. Ensimmäiset arviot kasvaimista tehtiin 9 viikkoa hoidon alkamisen jälkeen ja tämän jälkeen taudin etenemiseen tai hoidon lopettamiseen saakka. Ensisijainen tehokkuusmuuttuja oli riippumattoman radiologisen arviointiryhmän määrittämä objektiivisen vasteen saavuttaneiden osuus. Vasteen kesto, etenemisvapaa elinaika ja kokonaiselinaika olivat muita tehon mittareita.
CA209039 on vaiheen 1b avoin suurenevin annoksin tehty moniannos- ja monikeskustutkimus nivolumabista uusiutuneiden / refraktaaristen hematologisten syöpien hoidossa. Tutkimukseen osallistui 23 klassista Hodgkinin lymfoomaa sairastavaa potilasta, joita hoidettiin nivolumabimonoterapialla 3 mg/kg. Näistä potilasta 15 oli saanut brentuksimabivedotiinihoitoa autologisen kantasolujen siirron jälkeen salvage-hoitona, kuten kohortissa B tutkimuksessa CA209205. Ensimmäiset arviot kasvaimista tehtiin 4 viikkoa hoidon alkamisen jälkeen ja tämän jälkeen taudin etenemiseen tai hoidon lopettamiseen saakka. Tehokkuusmuuttujia olivat tutkijan arvioima objektiivisen vasteen saavuttaneiden osuus, jonka riippumaton radiologinen arviointiryhmä arvioi jälkikäteen, sekä vasteen kesto.
Tiedot CA209205‑tutkimuksen kohortti B:n 80 potilaasta sekä CA209039‑tutkimuksen 15 stä brentuksimabivedotiinihoitoa autologisen kantasolujen siirron jälkeen saaneesta potilaasta yhdistettiin. Lisäksi CA209205‑tutkimuksen kohortti C:n 100 potilaasta, joita oli hoidettu brentuksimabilla ennen autologista kantasolujen siirtoa ja/tai sen jälkeen, on esitetty lisätietoa. Lähtötason ominaisuudet olivat samanlaiset näissä kahdessa tutkimuksessa sekä kohorteissa (ks. taulukko 45 alla).
Taulukko 45: Potilaiden lähtötason ominaisuudet CA209205‑tutkimuksen kohortti B:ssä, kohortti C:ssä ja CA209039‑tutkimuksessa
| CA209205, kohortti Bja CA209039 | CA209205, kohortti Ba (n = 80) | CA209039 (n = 15) | CA209205, (n = 100) |
Iän mediaani, vuosina (vaihteluväli) | 37,0 (18–72) | 37,0 (18–72) | 40,0 (24–54) | 32,0 (19–69) |
Sukupuoli | 61 (64 %) M 34 (36 %) N | 51 (64 %) M 29 (36 %) N | 10 (67 %) M 5 (33 %) N | 56 (56 %) M 44 (44 %) N
|
ECOG‑toimintakykyluokka |
|
|
|
|
0 | 49 (52 %) | 42 (52,5 %) | 7 (47 %) | 50 (50 %) |
1 | 46 (48 %) | 38 (47,5 %) | 8 (53 %) | 50 (50 %) |
≥ 5 aiempaa systeemistä hoitolinjaa | 49 (52 %) | 39 (49 %) | 10 (67 %) | 30 (30 %) |
Aiempi sädehoito | 72 (76%) | 59 (74%) | 13 (87%) | 69 (69 %) |
Aiempi autologinen kantasolujen siirto |
|
|
|
|
1 | 87 (92 %) | 74 (92,5 %) | 13 (87 %) | 100 (100 %) |
≥ 2 | 8 (8 %) | 6 (7,5 %) | 2 (13 %) | 0 (0 %) |
Vuodet viimeisimmästä siirrosta ensimmäiseen tutkimushoitoannokseen, mediaani (min–max) | 3,5 (0,2–19,0) | 3,4 (0,2–19,0) | 5,6 (0,5–15,0) | 1,7 (0,2–17,0) |
a 18:lla potilaalla 80:stä (22,5 %:lla) CA209205-tutkimuksen kohortti B:ssä oli B‑oireita lähtötasolla.
b 25:llä potilaalla 100:sta (25 %:lla) CA209205-tutkimuksen kohortti C:ssä oli B‑oireita lähtötasolla.
Sama riippumaton radiologinen arviointiryhmä arvioi tehoa molemmissa tutkimuksissa. Tulokset on esitetty taulukossa 46.
Taulukko 46:Uusiutunutta tai refraktaarista klassista Hodgkinin lymfoomaa sairastaneiden potilaiden tehokkuustulokset
Lukumäärä (n) / seuranta-aika vähintään (kuukausina) | CA209205, kohortti Ba ja CA209039 | CA209205, kohortti Ba
| CA209039
|
Objektiivinen vaste, n (%); (95 %:n luottamusväli) | 63 (66 %); (56–76) | 54 (68 %); (56–78) | 9 (60 %); (32–84) |
Täydellinen remissio (CR), n (%); (95 %:n luottamusväli) | 6 (6 %); (2–13) | 6 (8 %); (3–16) | 0 (0 %); (0–22) |
Osittainen remissio (PR), n (%); (95 %:n luottamusväli) | 57 (60 %); (49–70) | 48 (60 %); (48–71) | 9 (60 %); (32–84) |
Stabiili tauti, n (%) | 22 (23) | 17 (21) | 5 (33) |
Vasteen kesto (kuukausina)b Mediaani (95 %:n luottamusväli) Vaihteluväli |
13,1 (9,5–NE) 0,0+–23,1+ | 13,1 (8,7–NE) 0,0+–14,2+ | 12,0 (1,8–NE) 1,8–23,1+ |
Vasteen saavuttamisajan mediaani Kuukausina (vaihteluväli) |
2,0 (0,7–11,1) | 2,1 (1,6–11,1) | 0,8 (0,7–4,1) |
Seurannan keston mediaani Kuukausina (vaihteluväli) |
15,8 (1,9–27,6) | 15,4 (1,9–18,5) | 21,9 (11,2–27,6) |
Etenemisvapaa elinaika Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla |
57 (45–68) | 55 (41–66) | 69 (37–88) |
“+” tarkoittaa sensuroitunutta havaintoa.
a Seuranta jatkui vielä tietojen toimittamisen aikaan.
b Tiedot ovat epävarmoja sensuroinnin seurauksena rajallisen vasteen keston suhteen kohortti B:ssä.
NE = ei arvioitavissa.
Alla olevassa taulukossa 47 on esitetty päivitetyt tiedot tehosta CA209205‑tutkimuksen kohortti B:n pidemmältä seuranta‑ajalta (vähintään 68,7 kuukautta) sekä kohortti C:n pidemmältä seuranta‑ajalta (vähintään 61,9 kuukautta).
Taulukko 47Uusiutunutta tai refraktaarista klassista Hodgkinin lymfoomaa sairastaneiden potilaiden päivitetyt tehokkuustulokset pidemmältä seuranta-ajalta CA209205‑tutkimuksessa
| CA209205, kohortti B | CA209205, kohortti C |
Lukumäärä (n) / seuranta‑aika vähintään (kuukausina) | (n = 80/68,7) | (n = 100/61,9)a |
Objektiivinen vaste, n (%);(95 %:n luottamusväli) | 57 (71 %); (60–81) | 75 (75 %); (65–83) |
Täydellinen remissio (CR), n (%); (95 %:n luottamusväli) | 11 (14 %); (7–23) | 21 (21 %); (14–30) |
Osittainen remissio (PR), n (%); (95 %:n luottamusväli) | 46 (58 %); (46–69) | 54 (54 %); (44–64) |
Stabiili tauti, n (%) | 14 (18 %) | 12 (12 %) |
Vasteen kesto kaikilla, jotka saivat vasteen (kuukausina)b |
|
|
Mediaani (95 %:n luottamusväli) | 16,6 (9,3–25,7) | 18,2 (11,6–30,9) |
Vaihteluväli | 0,0+–71,0+ | 0,0+–59,8+ |
Vasteen kesto täydellisessä remissiossa (kuukausina) |
|
|
Mediaani (95 %:n luottamusväli) | 30,3 (2,4–NE) | 26,4 (7,1–NE) |
Vaihteluväli | 0,7+–50,0+ | 0,0+–55,7+ |
Vasteen kesto osittaisessa remissiossa (kuukausina) |
|
|
Mediaani (95 %:n luottamusväli) | 10,6 (7,5–25,3) | 14,7 (9,4–30,4) |
Vaihteluväli | 0,0+–67,9+ | 0,0+–55,9+ |
Vasteen saavuttamisajan mediaani |
|
|
Kuukausina (vaihteluväli) | 2,2 (1,6–11,1) | 2,1 (0,8–17,9) |
Seuranta‑ajan mediaani |
|
|
Kuukausina (vaihteluväli) | 58,5 (1,9–74,3) | 53,5 (1,4–70,4) |
Etenemisvapaa elinaika |
|
|
Mediaani (95 %:n luottamusväli) | 14,8 (11,0–19,8) | 15,1 (11,1–19,1) |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 52 (39–63) | 53 (42–64) |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 36 (24–48) | 37 (25–48) |
Osuus (95 %:n luottamusväli) 60 kuukauden kohdalla | 16 (6–29) | 15 (6–28) |
Kokonaiselinaika (OS) |
|
|
Mediaani | Ei saavutettu | Ei saavutettu |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 95 (87–98) | 90 (82–94) |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 87 (77–93) | 86 (77–91) |
Osuus (95 %:n luottamusväli) 60 kuukauden kohdalla | 72 (60–81) | 67 (56–75) |
”+” tarkoittaa sensuroitunutta havaintoa.
a Kohortin C potilaiden (n = 33), joita on hoidettu brentuksimabivedotiinilla ainoastaan ennen autologista kantasolujen siirtoa, objektiivisen vasteen saavuttaneiden osuus oli 73 % (95 %:n luottamusväli: 55–87), täydellinen remissio 21 % (95 %:n luottamusväli: 9–39), osittainen remissio 52 % (95 %:n luottamusväli: 34–69). Vasteen keston mediaani oli 13,5 kuukautta (95 %:n luottamusväli: 9,4–30,9).
b Määritetty niiden potilaiden osalta, jotka saavuttivat täydellisen tai osittaisen remission.
NE = ei arvioitavissa.
22 %:lla (53/243) potilaista CA209205‑tutkimuksessa oli B‑oireita lähtötasolla. Nivolumabihoito johti B‑oireiden nopeaan korjaantumiseen 88,7 %:lla (47/53) potilaista, ja korjaantumisajan mediaani oli 1,9 kuukautta.
Jälkikäteen tehdyssä analyysissa CA209205‑tutkimuksen kohortti B:n 80:stä potilaasta 37 ei saanut vastetta aiempaan brentuksimabivedotiinihoitoon. Näillä 37 potilaalla nivolumabihoito johti 62,2 %:n (23/37) objektiivisen vasteen saavuttaneiden osuuteen. Niiden 23 potilaan, jotka saivat vasteen nivolumabille, mutta joille aiempi brentuksimabivedotiinihoito ei ollut aikaansaanut vastetta, vasteen keston mediaani oli 25,6 kuukautta (10,6–56,5).
Pään ja kaulan alueen levyepiteelisyöpä
Nivolumabin (3 mg/kg) turvallisuutta ja tehoa ainoana hoitona etäpesäkkeisen tai uusiutuneen SCCHN:n hoidossa arvioitiin satunnaistetussa vaiheen 3 avoimessa tutkimuksessa (CA209141). Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli histologisesti vahvistettu uusiutunut tai etäpesäkkeinen III/IV asteen pään ja kaulan alueen levyepiteelisyöpä (suuontelo, nielu, kurkunpää), jota ei voitu hoitaa kuratiivisella paikallishoidolla (leikkaus tai sädehoito joko kemoterapian kanssa tai ilman), ja joiden sairaus oli edennyt platinapohjaisen hoidon aikana tai 6 kuukauden kuluessa sen jälkeen ja joiden ECOG-toimintakykyluokka oli 0 tai 1. Edeltävä platinapohjainen hoito annettiin jossakin seuraavista tilanteista: liitännäishoito, esiliitännäishoito, primaarinen syöpä, uusiutuminen tai etäpesäkkeiden muodostuminen. Tutkimukseen otettiin potilaita riippumatta kasvaimen PD-L1-statuksesta tai ihmisen papilloomavirusinfektion (HPV) statuksesta. Potilaat, joilla oli aktiivinen autoimmuunisairaus, immunosuppressiota vaativa tila, nenänielun uusiutunut tai etäpesäkkeinen syöpä, primaariselta histologialtaan tuntematon levyepiteelisyöpä tai primaarisen kasvaimen sijainti oli sylkirauhasessa tai ei-levyepiteeliperäinen (esim. limakalvomelanooma), tai aktiivisia leptomeningeaalisia tai aivometastaaseja, suljettiin pois tutkimuksesta. Potilaat, joiden aivometastaaseja oli hoidettu, voitiin ottaa mukaan tutkimukseen, jos neurologinen tila oli palautunut lähtötasolle vähintään 2 viikkoa ennen tutkimukseen mukaan tuloa ja jos kortikosteroidihoito oli joko lopetettu tai he saivat muuttumattomia tai pieneneviä annoksia, jotka vastasivat alle 10 mg:aa prednisonia päivässä.
Kaikkiaan 361 potilasta satunnaistettiin saamaan joko nivolumabia, 3 mg/kg (n = 240) annosteltuna 60 minuutin kuluessa laskimoon joka toinen viikko, tai tutkijan valinnan mukaan joko setuksimabia (n = 15) latausannoksella 400 mg/m2, minkä jälkeen viikoittain 250 mg/m2, tai metotreksaattia (n = 52) viikoittain 40–60 mg/ m2 tai dosetakselia (n = 54) viikoittain 30–40 mg/ m2. Satunnaistaminen ositettiin aiemman setuksimabihoidon mukaan. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt hoitoa. Kasvaimet arvioitiin kiinteiden kasvainten vasteenarviointikriteerien (RECIST) version 1.1 mukaan 9 viikon kuluttua satunnaistamisesta, ja sen jälkeen joka 6. viikko. Hoito taudin etenemisen jälkeen sallittiin tutkijan suorittaman ensimmäisen kiinteiden kasvainten vasteenarviointikriteerien (RECIST) version 1.1 mukaisesti tekemän arvioinnin jälkeen, jos tutkija totesi, että nivolumabia saavalle potilaalle on kliinistä hyötyä tutkimuslääkkeestä ja hän sietää hoitoa. Ensisijainen tehokkuusmuuttuja oli OS. Tärkeimmät toissijaiset tehokkuusmuuttujat olivat tutkijan arvioima PFS ja ORR. Ylimääräiset ennalta määritellyt alaryhmäanalyysit tehon arvioimiseksi tehtiin etukäteen määritellyillä kasvaimen PD-L1-ilmentymistasoilla 1 %, 5 % ja 10 %.
Tutkimusta edeltävät kasvainkudosnäytteet kerättiin systemaattisesti ennen satunnaistamista, jotta voitiin tehdä ennalta suunnitellut tehokkuusanalyysit kasvaimen PD-L1:n ilmentymisten mukaisesti. Kasvaimen PD-L1:n ilmentyminen määritettiin käyttämällä PD-L1 IHC 28–8 pharmDx ‑testausmenetelmää.
Ryhmien lähtötason ominaisuudet olivat tasapainossa. Iän mediaani oli 60 vuotta (vaihteluväli: 28–83); 31 % potilaista oli 65‑vuotiaita tai sitä vanhempia ja 5 % oli 75‑vuotiaita tai sitä vanhempia; 83 % oli miehiä ja 83 % valkoihoisia. Lähtötason ECOG (Eastern Cooperative Oncology Group) ‑toimintakykyluokka oli 0 (20 %) tai 1 (78 %); 77 % potilaista oli aiempia/nykyisiä tupakoitsijoita, 90 %:lla oli IV asteen sairaus, 66 %:lla oli yksi tai useampi leesio, 45 %:a oli aiemmin hoidettu yhdellä, 34 %:a kahdella ja 20 %:a kolmella tai useammalla systeemisellä hoitolinjalla ja 25 %:lla HPV-16-status oli positiivinen.
Minimiseuranta-ajan ollessa 11,4 kuukautta tutkimuksessa todettiin tilastollisesti merkitsevä paraneminen kokonaiselinajassa potilailla, jotka oli satunnaistettu saamaan nivolumabia, verrattuna tutkijan valintaan. Kokonaiselinajan Kaplan–Meier-kuvaajat esitetään kuvassa 24. Tulokset tehosta esitetään taulukossa 48.
Kuva 24: Kokonaiselinajan Kaplan–Meier-kuvaajat (CA209141)
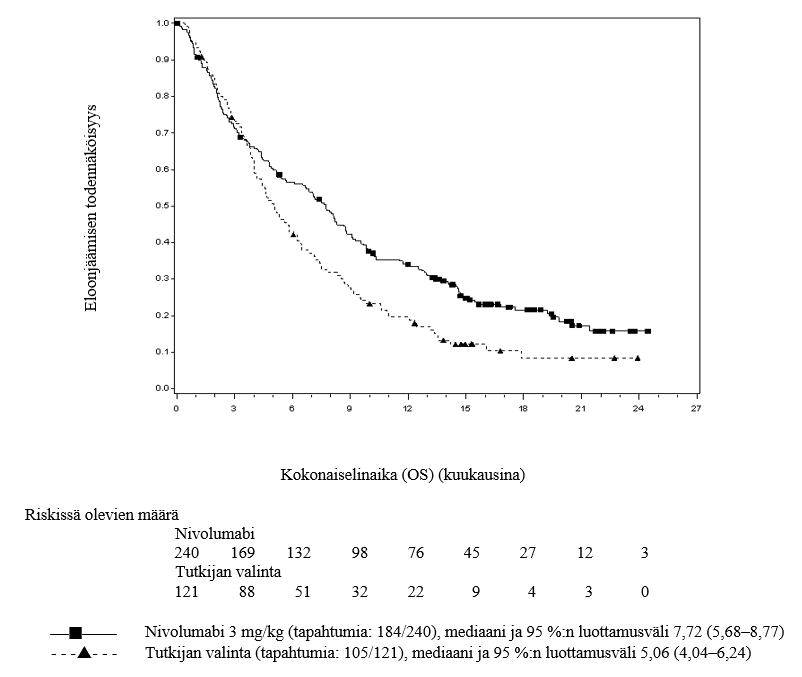
Taulukko48:Tulokset tehosta (CA209141)
| nivolumabi | tutkijan valinta | |
Kokonaiselinaika (OS) |
|
| |
Tapahtumat | 184 (76,7 %) | 105 (86,8 %) | |
Riskitiheyssuhdea | 0,71 | ||
(95 %:n luottamusväli) | (0,55–0,90) | ||
p-arvob | 0,0048 | ||
Mediaani (95 %:n luottamusväli) (kuukausina) | 7,72 (5,68–8,77) | 5,06 (4,04–6,24) | |
Osuus (95 %:n luottamusväli) 6 kuukauden kohdalla | 56,5 (49,9–62,5) | 43,0 (34,0–51,7) | |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 34,0 (28,0–40,1) | 19,7 (13,0–27,3) | |
Osuus (95 %:n luottamusväli) 18 kuukauden kohdalla | 21,5 (16,2–27,4) | 8.3 (3,6–15,7) | |
Etenemisvapaa elinaika |
|
| |
Tapahtumat | 204 (85,0 %) | 104 (86,0 %) | |
Riskitiheyssuhde | 0,87 | ||
95 %:n luottamusväli | (0,69–1,11) | ||
p-arvo | 0,2597 | ||
Mediaani (95 %:n luottamusväli) (kuukausina) | 2,04 (1,91–2,14) | 2,33 (1,97–3,12) | |
Osuus (95 %:n luottamusväli) 6 kuukauden kohdalla | 21,0 (15,9–26,6) | 11,1 (5,9–18,3) | |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 9,5 (6,0–13,9) | 2,5 (0,5–7,8) | |
Vahvistettu objektiivinen vastec | 32 (13,3 %) | 7 (5,8 %) | |
(95 %:n luottamusväli) | (9,3–18,3) | (2,4–11,6) | |
Ristitulosuhde (95 %:n luottamusväli) | 2,49 (1,07–5,82) | ||
Täydellinen vaste (CR) | 6 (2,5 %) | 1 (0,8 %) | |
Osittainen vaste (PR) | 26 (10,8 %) | 6 (5,0 %) | |
Stabiili tauti (SD) | 55 (22,9 %) | 43 (35,5 %) | |
Vasteen saavuttamisajan mediaani |
|
| |
Kuukausina (vaihteluväli) | 2,1 (1,8–7,4) | 2,0 (1,9–4,6) | |
Vasteen keston mediaani |
|
| |
Kuukausina (vaihteluväli) | 9,7 (2,8–20,3+) | 4,0 (1,5+–8,5+) | |
a Johdettu ositetusta verrannollisten riskitiheyksien mallista.
b P‑arvo perustuu logrank‑merkitsevyystestiin ositettuna aiemman setuksimabihoidon mukaan; vastaava O’Brien–Fleming-keskeytysrajan merkitsevyystaso on 0,0227.
c Nivolumabiryhmässä oli kaksi potilasta, jotka saavuttivat täydellisen vasteen, ja seitsemän, jotka saavuttivat osittaisen vasteen ja joilla kasvaimen PD‑L1:n ilmentyminen oli < 1 %.
Laskennallinen kasvaimen PD-L1:n ilmentyminen mitattiin 67 %:lta nivolumabiryhmän potilaista ja 82 %:lta tutkijan valitseman hoidon ryhmän potilaista. Kasvaimen PD-L1:n ilmentymistasojen raja-arvot olivat tasapainossa näiden kahden ryhmän välillä (nivolumabi vs. tutkijan valinta), joissa kummassakin ennalta määritetyt kasvaimen PD-L1:n ilmentymistasojen raja-arvot olivat ≥ 1 % (55 % vs. 62 %), ≥ 5 % (34 % vs. 43 %) tai ≥ 10 % (27 % vs. 34 %).
Nivolumabiryhmän potilailla, joiden kasvaimet ilmensivät PD-L1:ää kaikilla ennalta määritetyillä raja-arvoilla, oli suurempi mahdollisuus pidempään kokonaiselinaikaan verrattuna tutkijan valitseman hoidon ryhmän potilaisiin. Kokonaiselinajassa saavutettujen hyötyjen laajuus osoitettiin johdonmukaisesti kasvaimen PD-L1:n ilmentymistasoilla ≥ 1 %, ≥ 5 % tai ≥ 10 % (ks. taulukko 49).
Taulukko 49: Kasvaimen PD‑L1:n ilmentymisen mukainen kokonaiselinaika (CA209141)
PD-L1:n ilmentyminen | nivolumabi | tutkijan valinta |
|
|---|---|---|---|
Kasvaimen PD‑L1:n ilmentymisen mukainen kokonaiselinaika | |||
| Tapahtumien määrä (potilaiden määrä) | Osittamaton riskitiheyssuhde (95 %:n luottamusväli) | |
< 1 % | 56 (73) | 32 (38) | 0,83 (0,54–1,29) |
≥ 1 % | 66 (88) | 55 (61) | 0,53 (0,37–0,77) |
≥ 5 % | 39 (54) | 40 (43) | 0,51 (0,32–0,80) |
≥ 10 % | 30 (43) | 31 (34) | 0,57 (0,34–0,95) |
Jälkikäteen tehdyssä havainnollistavassa analyysissa, jossa käytettiin ei‑validoitua määritystapaa, sekä kasvainsolujen PD‑L1:n ilmentyminen että kasvaimeen liittyvien immuunisolujen (TAIC) PD‑L1:n ilmentyminen analysoitiin suhteessa nivolumabihoidon tehon merkittävyyteen verrattuna tutkijan valitsemaan hoitoon. Analyysi osoitti, että kasvainsolujen PD‑L1:n ilmentymisen lisäksi myös kasvaimeen liittyvien immuunisolujen PD‑L1:n ilmentyminen vaikutti liittyvän nivolumabihoidosta saatavaan hyötyyn verrattuna tutkijan valitsemasta hoidosta saatavaan hyötyyn (ks. taulukko 50). Koska potilaita oli alaryhmissä vain vähän ja analyysi luonteeltaan havainnollistava, näiden tietojen perusteella ei voi tehdä lopullisia johtopäätöksiä.
Taulukko 50: Kasvainsolujen ja kasvaimeen liittyvien immuunisolujen (TAIC) PD‑L1:n ilmentymisen mukainen teho (CA209141)
| Kokonaiselinajan mediaania (kuukausina) | Etenemisvapaan elinajan mediaania (kuukausina) | Objektiivisen vasteen saavuttaneiden osuus (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Riskitiheyssuhdeb (95 %:n luottamusväli) | Riskitiheyssuhdeb (95 %:n luottamusväli) | (95 %:n luottamusväli)c | ||||||
| nivolumabi | tutkijan valinta | nivolumabi | tutkijan valinta | nivolumabi | tutkijan valinta | |||
PD‑L1 ≥ 1 %, | 9,10 | 4,60 | 3,19 | 1,97 | 19,7 | 0 | |||
0,43 (0,28–0,67) | 0,48 (0,31–0,75) | (10,6–31,8) | (0–7,5) | ||||||
PD‑L1 ≥ 1 %, | 6,67 | 4,93 | 1,99 | 2,04 | 11,1 | 7,1 | |||
0,89 (0,44–1,80) | 0,93 (0,46–1,88) | (2,4–29,2) | (0,2–33,9) | ||||||
PD‑L1 < 1 %, | 11,73 | 6,51 | 2,10 | 2,73 | 18,6 | 12,0 | |||
0,67 (0,38–1,18) | 0,96 (0,55–1,67) | (8,4–33,4) | (2,5–31,2) | ||||||
PD-L1 < 1 %, | 3,71 | 4,85 | 1,84 | 2,12 | 3,7 | 10,0 | |||
1,09 (0,50–2,36) | 1,91 (0,84–4,36) | (< 0,1–19,0) | (0,3–44,5) | ||||||
a Kokonaiselinaika ja etenemisvapaa elinaika arvioitiin Kaplan–Meierin menetelmällä.
b Kunkin alaryhmän riskitiheyssuhde saatiin Coxin suhteellisen vaaran regressiomallilla hoidon ollessa ainoa muuttuja.
c Objektiivisen vasteen saavuttaneiden osuuden luottamusväli laskettiin Clopper–Pearsonin menetelmällä.
d PD‑L1+ TAIC kasvaimen mikroympäristössä arvioitiin laadullisesti ja luonnehdittiin ”lukuisiksi”, ”keskitasoisiksi” ja ”vähäisiksi” patologien arvioiden mukaan. Ryhmät ”lukuisat” ja ”keskitason” yhdistettiin, ja niistä muodostettiin ryhmä ”runsaat”.
Potilailta, joilla ensisijainen kasvain sijaitsi tutkijan arvion mukaan suunielussa, testattiin HPV (määritettiin p16-immunohistokemialla [IHC]). Todettu kokonaiselinajan hyöty oli riippumaton HPV‑statuksesta (HPV‑positiivinen: riskitiheyssuhde = 0,63; 95 %:n luottamusväli 0,38–1,04, HPV‑negatiivinen: riskitiheyssuhde = 0,64; 95 %:n luottamusväli 0,40–1,03 ja HPV tuntematon: riskitiheyssuhde = 0,78; 95 %:n luottamusväli 0,55–1,10).
Potilaiden ilmoittamat tulokset (PRO:t) analysoitiin seuraavilla mittareilla EORTC QLQ-C30, EORTC QLQ-H&N35 ja EQ-5D-3L. Yli 15 viikon seurannan aikana nivolumabihoitoa saaneet potilaat raportoivat vointinsa pysyvän vakaana, kun taas tutkijan valitseman mukaista hoitoa saaneet potilaat raportoivat merkittävää toimintakyvyn alentumista (esim. fyysisen tai sosiaalisen toimintakyvyn tai roolitoimintakyvyn) ja terveydentilan huonontumista sekä oireiden lisääntymistä (esim. uupumuksen, hengenahdistuksen, ruokahalun vähenemisen, kipujen, sensoristen ongelmien, sosiaalisten kontaktien ongelmien). Potilaiden ilmoittamien tietojen tulkinnassa on huomioitava avoin tutkimusasetelma, ja siksi niihin on suhtauduttava varovaisuudella.
Edennyt uroteelikarsinooma
Uroteelikarsinooman liitännäishoito
Satunnaistettu vaiheen 3 tutkimus nivolumabista liitännäishoitona vs. lumelääke (CA209274)
Nivolumabi-monoterapian turvallisuutta ja tehoa uroteelikarsinooman liitännäishoitona arvioitiin vaiheen 3 satunnaistetussa, lumekontrolloidussa, kaksoissokkoutetussa monikeskustutkimuksessa (CA209274). Tutkimukseen otettiin potilaita (18‑vuotiaita tai tätä vanhempia), joille oli tehty rakosta tai ylemmistä virtsateistä (munuaisaltaasta tai virtsanjohtimesta) peräisin olevan lihakseen tunkeutuvan uroteelikarsinooman radikaaliresektio ja joilla oli suuri riski taudin uusiutumiseen. Lihakseen tunkeutuvan uroteelikarsinooman patologian luokittelukriteerit, joiden avulla määritettiin korkean riskin potilaat, olivat ypT2‑ypT4a tai ypN+ aikuispotilaille, jotka saivat sisplatiinikemoterapiaa esiliitännäishoitona, ja pT3-pT4a tai pN+ aikuispotilaille, jotka eivät saaneet sisplatiinikemoterapiaa esiliitännäishoitona ja joille ei voitu antaa sisplatiinikemoterapiaa liitännäishoitona tai jotka kieltäytyivät siitä. Tutkimukseen otettiin potilaita riippumatta kasvaimen PD‑L1-statuksesta. Potilaiden ECOG-toimintakykyluokka oli 0 tai 1 (ECOG-toimintakykyluokka 2 sallittiin potilaille, jotka eivät soveltuneet saamaan sisplatiinikemoterapiaa esiliitännäishoitona). Kasvainsolujen PD‑L1-ilmentyminen määriteltiin PD‑L1 IHC 28–8 pharmDx ‑testausmenetelmän avulla. Tutkimuksesta suljettiin pois potilaat, joilla oli aktiivinen, tiedossa oleva tai epäilty autoimuunisairaus, potilaat, joita oli hoidettu millä tahansa kemoterapialla, sädehoidolla, biologisella syöpälääkkeellä, intravesikaalihoidolla tai tutkimushoidolla 28 vuorokauden aikana ennen tutkimuslääkkeen ensimmäistä annosta.
Yhteensä 709 potilasta satunnaistettiin saamaan joko nivolumabia 240 mg (n = 353) kahden viikon välein tai lumelääkettä (n = 356) kahden viikon välein, kunnes tauti uusiutui tai ilmaantui ei‑hyväksyttävää toksisuutta, enintään 1 vuoden ajan. Näistä potilaista 282 potilaan kasvainsolujen PD‑L1-ilmentymä oli ≥ 1 %; 140 potilaalla nivolumabiryhmässä ja 142 potilaalla lumelääkeryhmässä. Satunnaistaminen ositettiin patologisen imusolmukestatuksen mukaan (N+ vs. N0/x, kun < 10 imusolmuketta poistettu vs. N0, kun ≥ 10 imusolmuketta poistettu), kasvainsolujen PD‑L1-ilmentymän (≥ 1 % vs. < 1 % / määrittämätön) mukaan sekä esiliitännäishoitona annettavan sisplatiinikemoterapian saamisen mukaan. Kasvaimet oli määrä arvioida kuvantamalla 12 viikon välein ensimmäisen annoksen antopäivästä viikkoon 96 asti, minkä jälkeen 16 viikon välein viikosta 96 viikkoon 160 asti, minkä jälkeen 24 viikon välein, kunnes tauti uusiutui muualla kuin uroteelissa tai hoito lopetettiin (sen mukaan, kumpi näistä tapahtui jälkimmäisenä) enintään 5 vuoden ajan. Ensisijaiset tehokkuusmuuttujat olivat tautivapaa elossaoloaika (DFS) kaikilla satunnaistetuilla potilailla ja tautivapaa elossaoloaika satunnaistetuilla potilailla, joiden kasvainsolujen PD‑L1-ilmentymä oli ≥ 1 %. Tautivapaa elossaoloaika arvioitiin ajaksi satunnaistamispäivämäärän ja jonkin seuraavan päivämäärän välillä sen mukaan, mikä seuraavista tapahtui ensimmäisenä: ensimmäinen dokumentoitu uusiutuminen tutkijan arvioimana (paikallinen uroteelin, paikallinen muun kuin uroteelin tai etäinen) tai kuolema (mistä tahansa syystä). Toissijaisiin tehokkuusmuuttujiin kuului muun muassa kokonaiselinaika.
Hoitoryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Potilasryhmässä, jossa kasvainsolujen PD‑L1-ilmentymä oli ≥ 1 %, iän mediaani oli 66 vuotta (vaihteluväli: 34–92 vuotta), 76 % oli miehiä ja 76 % oli valkoihoisia. 82 prosentilla oli lihakseen tunkeutuva virtsarakon syöpä, 18 %:lla oli ylävirtsateiden uroteelikarsinooma (munuaisaltaassa tai virtsanjohtimessa). 42 % potilaista oli saanut aiemmin sisplatiinia esiliitännäishoitona. 45 % potilaista oli N+ radikaaliresektiossa. Potilaiden ECOG-toimintakykyluokka oli 0 (61 %), 1 (37 %) tai 2 (2 %), ja 7 %:lla potilaista hemoglobiini oli < 10 g/dl.
Tautivapaan elossaoloajan osoitettiin primaarisessa ennalta määritetyssä välianalyysissä potilaista, joiden kasvainsolujen PD‑L1-ilmentymä oli ≥ 1 % (seuranta aika vähintään 6,3 kuukautta ja seuranta-ajan mediaani 22,1 kuukautta nivolumabiryhmässä), olevan tilastollisesti merkitsevästi parempi potilailla, jotka oli satunnaistettu saamaan nivolumabia, verrattuna potilaisiin, jotka oli satunnaistettu saamaan lumelääkettä. Tautivapaan elossaoloajan mediaania tutkijan määrittämänä (95 %:n luottamusväli: 21,19–ei saavutettu) ei saavutettu nivolumabia saaneiden ryhmässä, ja lumelääkettä saaneiden ryhmässä se oli 8,41 kuukautta (95 %:n luottamusväli: 5,59–21,19), riskitiheyssuhde 0,55 (98,72 %:n luottamusväli: 0,35–0,85), p‑arvo = 0,0005. Tautivapaan elossaoloajan primaarianalyysiin sisältyi uuden syöpähoidon sensurointi. Tautivapaan elossaoloajan sensuroidut ja sensuroimattomat tulokset olivat uuden syöpähoidon osalta yhdenmukaiset.
Tautivapaan elossaoloajan paraneminen vahvistettiin päivitetyssä deskriptiivisessä tautivapaan elossaoloajan analyysissä potilaista, joiden kasvainsolujen PD‑L1-ilmentymä oli ≥ 1 % (seuranta-aika vähintään 11,4 kuukautta ja seuranta-ajan mediaani 25,5 kuukautta nivolumabiryhmässä).
Tämän deskriptiivisen päivitetyn analyysin tehoa koskevat tulokset esitetään taulukossa 51 ja kuvassa 25.
Taulukko 51: Tulokset tehosta potilailla, joiden kasvainsolujen PD‑L1 oli ≥ 1 % (CA209274)
nivolumabi (n = 140) | lumelääke (n = 142) | ||
|---|---|---|---|
Tautivapaa elossaoloaika | Seuranta-aika vähintään 11,4 kuukautta |
| |
Tapahtumia (%) | 56 (40,0) | 85 (59,9) | |
Riskitiheyssuhde (95 %:n luottamusväli)a | 0,53 (0,38–0,75) |
| |
Mediaani (95 %:n luottamusväli) (kuukautta)b | NR (22,11–NE) | 8,41 (5,59–20,04) | |
Osuus (95 %:n luottamusväli) 6 kuukauden kohdalla | 74,5 (66,2–81,1) | 55,7 (46,8–63,6) | |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 67,6 (59,0–74,9) | 46,3 (37,6–54,5) | |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 58,6 (49,3–66,9) | 37,4 (29,0–45,8) | |
NR: ei saavutettu, NE: ei arvioitavissa.
a Ositettu Coxin suhteellisen vaaran malli. Riskitiheyssuhde on nivolumabin osalta parempi kuin lumelääkkeen.
b Perustuu Kaplan–Meier-arvioihin.
Kuva 25: Tautivapaan elossaoloajan Kaplan–Meier-kuvaajat potilailla, joiden kasvainsolujen PD‑L1-ilmentymä oli ≥ 1 % (CA209274)
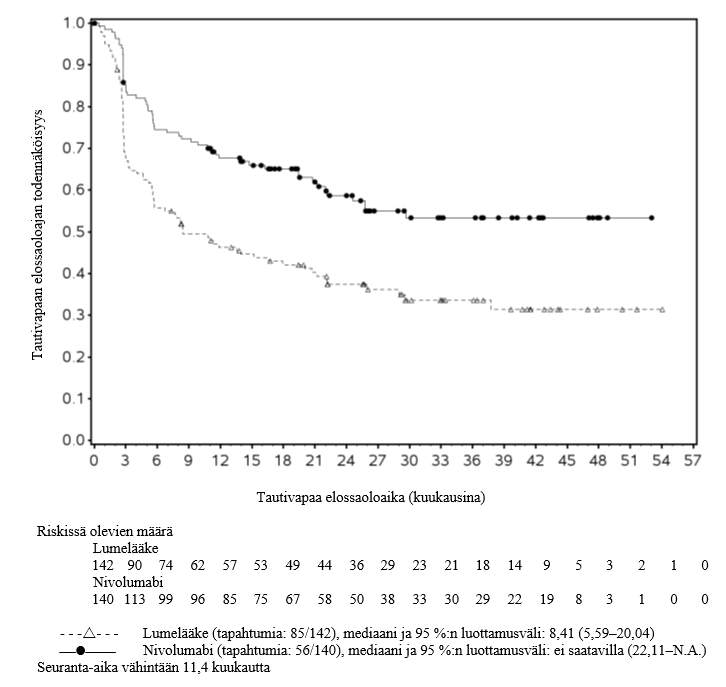
Aiempaa sisplatiinihoitoa esiliitännäishoitona saaneista potilaista tehtiin havainnollistavia, ennalta määritettyjä alaryhmien deskriptiivisiä analyysejä.
Niiden potilaiden alaryhmässä, joiden kasvainsolujen PD‑L1-ilmentymä oli ≥ 1 % ja jotka saivat aiempaa sisplatiinihoitoa esiliitännäishoitona (n = 118), tautivapaan elossaoloajan riskitiheyssuhde oli 0,37 (95 %:n luottamusväli: 0,22–0,64). Tautivapaan elossaoloajan mediaania ei saavutettu nivolumabiryhmässä, ja lumelääkeryhmässä se oli 8,41 kuukautta. Niiden potilaiden alaryhmässä, joiden kasvainsolujen PD‑L1-ilmentymä oli ≥ 1 % ja jotka eivät saaneet aiempaa sisplatiinihoitoa esiliitännäishoitona (n = 164), tautivapaan elossaoloajan riskitiheyssuhde oli 0,69 (95 %:n luottamusväli: 0,44–1,08). Tautivapaan elossaoloajan mediaani oli 29,67 kuukautta nivolumabiryhmässä ja 11,37 kuukautta lumelääkeryhmässä.
Satunnaistettu, avoin vaiheen 3 tutkimus nivolumabista yhdistelmähoitona kemoterapian kanssa vs. kemoterapia (CA209901)
Yhdistelmähoitona sisplatiinin ja gemsitabiinin kanssa annetun nivolumabin ja sitä seuraavan nivolumabimonoterapian turvallisuutta ja tehoa arvioitiin satunnaistetussa, avoimessa tutkimuksessa (CA209901) potilailla, joille voitiin antaa sisplatiinia ja joilla oli leikkaukseen soveltumaton tai etäpesäkkeinen uroteelikarsinooma. Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli histologisesti tai sytologisesti vahvistettu etäpesäkkeinen tai leikkaukseen soveltumaton munuaisaltaan, virtsanjohtimen, virtsarakon tai virtsaputken välimuotoisen epiteelin uroteelikarsinooma (TCC) ja joille voitiin antaa sisplatiinia ja gemsitabiinia. Vähäiset histologiset variantit (yhteensä < 50 %) sallittiin (TCC kuitenkin histologiassa dominoiva). Kaikkien tutkittavien taudin täytyi olla mitattavissa joko tietokonetomografialla (TT) tai magneettikuvauksella (MRI) RECIST 1.1 ‑kriteerien perusteella. Aiempaa systeemistä syöpähoitoa etäpesäkkeisen tai leikkaukseen soveltumattoman uroteelikarsinooman hoidossa ei sallittu. Radikaalin kystektomian jälkeinen aikaisempi esiliitännäishoitona annettu kemoterapia tai aikaisempi liitännäishoitona annettu platinapohjainen kemoterapia sallittiin, jos tauti uusiutui ≥ 12 kuukautta hoidon päättymien jälkeen. Aikaisempi intravesikaalihoito sallittiin, jos hoito oli päättynyt vähintään 4 viikkoa ennen tutkimushoidon aloittamista. Parantavassa tarkoituksessa annettu sädehoito (kemoterapian kanssa tai ilman) sallittiin, jos hoito oli päättynyt ≥ 12 kuukautta ennen tutkimukseen osallistumista. Palliatiivinen sädehoito sallittiin, jos se oli päättynyt vähintään 2 viikkoa ennen hoidon aloittamista.
Yhteensä 608 potilasta satunnaistettiin saamaan joko nivolumabia yhdistelmähoitona sisplatiinin ja gemsitabiinin kanssa (n = 304) tai sisplatiinia ja gemsitabiinia (n = 304). Satunnaistaminen ositettiin kasvaimen PD‑L1‑statuksen (≥ 1 % vs. < 1 % tai määrittämätön) ja maksan etäpesäkkeiden (kyllä/ei) perusteella. Iän mediaani oli 65 vuotta (vaihteluväli: 32–86), ja potilaista 51 % oli iältään ≥ 65 vuotta ja 12 % oli iältään ≥ 75 vuotta; 23 % oli aasialaisia, 72 % valkoihoisia ja 0,3 % oli tummaihoisia; 77 % oli miehiä ja 23 % oli naisia. Lähtötason ECOG-toimintakykyluokka oli 0 (53 %) tai 1 (46 %). Nivolumabia yhdistelmähoitona sisplatiinin ja gemsitabiinin kanssa saaneet potilaat saivat nivolumabia 360 mg kolmen viikon välein yhdessä sisplatiinin ja gemsitabiinin kanssa enintään kuuden jakson ajan, minkä jälkeen potilaat saivat nivolumabia monoterapiana 480 mg neljän viikon välein yhteensä enintään 24 kuukauden ajan. Potilaat saivat gemsitabiinia 1000 mg/m2 laskimoon 30 minuutin kuluessa 3‑viikkoisen hoitojakson päivinä 1 ja 8 ja sisplatiinia 70 mg/m2 laskimoon 30–120 minuutin kuluessa 3‑viikkoisen hoitojakson päivänä 1. Yhteensä 92 potilasta (49 potilasta nivolumabia yhdistelmähoitona sisplatiinin ja gemsitabiinin kanssa saavien ryhmästä ja 43 potilasta sisplatiinia ja gemsitabiinia saavien ryhmästä) siirtyi sisplatiinista karboplatiiniin vähintään yhden sisplatiinijakson jälkeen.
Tutkimus osoitti tilastollisesti merkitsevää hyötyä kokonaiselinajassa ja etenemisvapaassa elinajassa potilailla, jotka oli satunnaistettu saamaan nivolumabia yhdistelmähoitona sisplatiinin ja gemsitabiinin kanssa, verrattuna potilaisiin, jotka oli satunnaistettu saamaan vain sisplatiinia ja gemsitabiinia. Tulokset tehosta esitetään taulukossa 52 sekä kuvissa 26 ja 27.
Taulukko 52: Tulokset tehosta (CA209901)
| nivolumabi ja sisplatiini-gemsitabiinikemoterapia | sisplatiini-gemsitabiinikemoterapia |
|---|---|---|
Kokonaiselinaika (OS)a |
|
|
Tapahtumia | 172 (56,6) | 193 (63,5) |
Mediaani (kuukautta) (95 %:n luottamusväli) | 21,7 (18,6–26,4) | 18,9 (14,7–22,4) |
Riskitiheyssuhde (95 %:n luottamusväli)b | 0,78 (0,63–0,96) | |
p‑arvoc | 0,0171 | |
Etenemisvapaa elinaikaa |
|
|
Tapahtumia | 211 (69,4) | 191 (62,8) |
Mediaani (kuukautta) (95 %:n luottamusväli) | 7,92 (7,62–9,49) | 7,56 (6,05–7,75) |
Riskitiheyssuhde (95 %:n luottamusväli)b | 0,72 (0,59–0,88) | |
p‑arvoc | 0,0012 | |
Objektiivisen vasteen saavuttaneiden osuus |
| |
Vasteen saavuttaneet | 175 (57,6) | 131 (43,1) |
(95 %:n luottamusväli) | (51,8–63,2) | (37,5–48,9) |
a Perustuu Kaplan–Meier‑arvioihin.
b Ositettu Coxin suhteellisen vaaran malli.
c 2‑tahoinen p‑arvo ositetusta log‑rank‑testistä.
Kuva 26: Etenemisvapaan elinajan Kaplan–Meier-kuvaajat (CA209901)
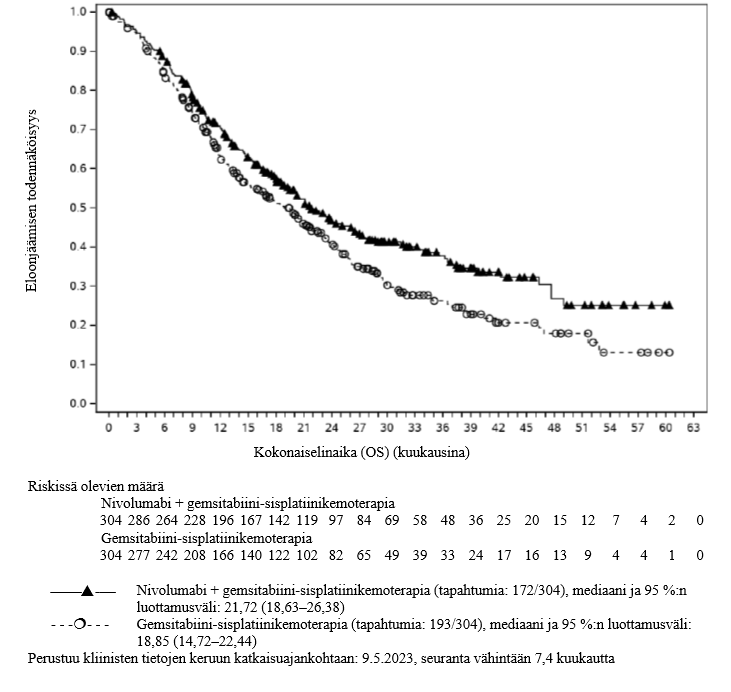
Kuva 27: Etenemisvapaan elinajan Kaplan–Meier-kuvaajat (CA209901)
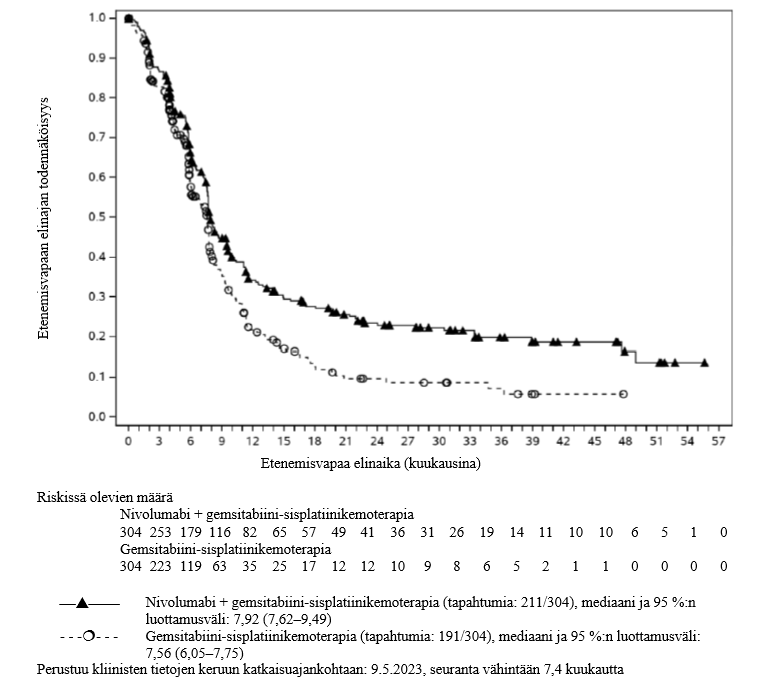
Etenemisvapaan elinajan primaarianalyysiin sisältyi uuden syöpähoidon sensurointi ennen taudin etenemistä (taulukko 52). Etenemisvapaata elinaikaa koskevat sensuroidut ja sensuroimattomat tulokset olivat uuden syöpähoidon osalta yhdenmukaiset ennen taudin etenemistä.
Avoin vaiheen 2 tutkimus (CA209275)
Ainoana lääkkeenä 3 mg/kg annoksena annetun nivolumabin turvallisuutta ja tehoa arvioitiin yksihaaraisessa avoimessa vaiheen 2 monikeskustutkimuksessa (CA209275) potilailla, joilla oli paikallisesti edennyt tai metastasoinut uroteelikarsinooma.
Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joiden edennyt tai metastaattinen sairaus eteni platinaa sisältävän kemoterapian aikana tai sen jälkeen tai joiden sairaus eteni 12 kuukauden kuluessa platinapohjaisen esiliitännäishoidon tai liitännäishoidon antamisesta. Potilaiden ECOG-toimintakykyluokka oli 0 tai 1, ja heidät otettiin mukaan tutkimukseen PD-L1-statuksesta riippumatta. Potilaat, joilla oli aktiivisia aivometastaaseja, leptomeningeaalisia metastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila, suljettiin pois tutkimuksesta. Potilaat, joilla oli maksametastaaseja ja jotka olivat saaneet aikaisemmin useampaa kuin kahta kemoterapian hoitolinjaa, suljettiin pois tutkimuksesta.
Lääkkeen tehon arviointiin sisällytettiin yhteensä 270 potilasta, joille annettiin joka toinen viikko laskimonsisäisesti 3 mg/kg ‑annos nivolumabia 60 minuutin aikana ja joita seurattiin vähintään 8,3 kuukauden ajan. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt sitä. Ensimmäinen arvio kasvaimista tehtiin 8 viikkoa hoidon alkamisen jälkeen ja tämän jälkeen joka 8. viikko 48:n viikon ajan ja sitten joka 12. viikko taudin etenemiseen tai hoidon lopettamiseen saakka, kumpi näistä tapahtui jälkimmäisenä. Kasvainten arvioimista jatkettiin hoidon lopettamisen jälkeen niillä potilailla, jotka lopettivat hoidon muista kuin taudin etenemisestä johtuvista syistä. Hoito taudin etenemisen jälkeen sallittiin tutkijan suorittaman ensimmäisen kiinteiden kasvainten vasteenarviointikriteerien (RECIST) version 1.1 mukaisesti tekemän arvioinnin jälkeen, jos tutkija totesi potilaan saavan kliinistä hyötyä tutkimuslääkkeestä, sairaus ei edennyt nopeasti ja potilas sieti hoitoa. Ensisijainen tehokkuusmuuttuja oli ORR BICR:n arvioimana. Toissijaiset tehokkuusmuuttujat olivat vasteen kesto, PFS ja OS.
Iän mediaani oli 66 vuotta (vaihteluväli 38–90); 55 % oli iältään ≥ 65 vuotta ja 14 % oli iältään ≥ 75 vuotta. Suurin osa potilaista oli valkoihoisia (86 %) ja miehiä (78 %). Lähtötason ECOG-toimintakykystatus oli 0 (54 %) tai 1 (46 %).
Taulukko 53: Tulokset tehosta (CA209275)a
| nivolumabi | ||
Vahvistettu objektiivinen vaste | 54 (20,0 %) | ||
(95 %:n luottamusväli) | (15,4–25,3) | ||
Täydellinen vaste (CR) | 8 (3,0 %) | ||
Osittainen vaste (PR) | 46 (17,0 %) | ||
Stabiili tauti (SD) | 60 (22,2 %) | ||
Vasteen keston mediaanib |
| ||
Kuukausina (vaihteluväli) | 10,4 (1,9+–12,0+) | ||
Vasteen saavuttamisajan mediaani |
| ||
Kuukausina (vaihteluväli) | 1,9 (1,6–7,2) | ||
Etenemisavapaa elinaika |
| ||
Tapahtumia (%) | 216 (80) | ||
Mediaani (95 %:n luottamusväli) kuukausina | 2,0 (1,9–2,6) | ||
Osuus (95 %:n luottamusväli) 6 kuukauden kohdalla | 26,1 (20,9–31,5) | ||
Kokonaiselinaika (OS)c |
| ||
Tapahtumia (%) | 154 (57) | ||
Mediaani (95 %:n luottamusväli) kuukausina | 8,6 (6,05–11,27) | ||
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 41,0 (34,8–47,1) | ||
Kasvaimen PD-L1-ilmentymistaso | |||
| < 1 % | ≥ 1 % | |
Vahvistettu objektiivinen svaste (95 %:n luottamusväli) | |||
| 16 % (10,3–22,7) n = 146 | 25 % (17,7–33,6) n = 124 | |
Vasteen keston mediaani Kuukausina (vaihteluväli)) | |||
| 10,4 (3,7–12,0+) | Ei saavutettu (1,9+–12,0+) | |
Etenemisvapaa elinaika | |||
Mediaani (95 %:n luottamusväli) kuukausina | 1,9 (1,8–2,0) | 3,6 (1,9–3,7) | |
Osuus (95 %:n luottamusväli) 6 kuukauden kohdalla | 22,0 (15,6–29,2) | 30,8 (22,7–39,3) | |
Kokonaiselinaika (OS) |
|
| |
Mediaani (95 %:n luottamusväli) kuukausina | 5,9 (4,37–8,08) | 11,6 (9,10–NE) | |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 34,0 (26,1–42,1) | 49,2 (39,6–58,1) | |
”+” tarkoittaa sensuroitunutta havaintoa.
a seurannan mediaani 11,5 kuukautta.
b Tiedot ovat epävarmoja rajallisen vasteen keston vuoksi.
c mukaan lukien 4 lääkkeestä aiheutunutta kuolemantapausta: 1 pneumoniitti, 1 äkillinen hengitysvajaus, 1 hengitysvajaus ja 1 verenkiertoelimistön toiminnan pettäminen.
NE: ei arvioitavissa
Jälkikäteen tehtyjen havainnollistavien analyysien tulokset osoittavat, että potilailla, joilla kasvaimen PD-L1:n ilmentymistaso oli matala (esim. < 1 %) tai sitä ei ollut lainkaan, muut potilaan ominaisuudet (esim. maksan metastaasit, viskeraaliset metastaasit, lähtötason hemoglobiini < 10 g/dl ja ECOG‑toimintakykystatus = 1) saattavat vaikuttaa kliiniseen lopputulokseen.
Avoin vaiheen 1/2 tutkimus (CA209032)
CA209032 oli avoin vaiheen 1/2 monikeskustutkimus, jossa oli mukana 78 potilaan kohortti (mukaan lukien 18 tutkittavaa, jotka saivat suunnitellusti tutkimushoidon jälkeistä yhdistelmähoitoa 3 mg/kg ‑nivolumabiannoksella ja 1 mg/kg ‑ipilimumabiannoksella), jonka kelpoisuusvaatimukset olivat samanlaiset kuin tutkimuksessa CA209275 ja jossa uroteelikarsinoomaa hoidettiin nivolumabi‑monoterapialla 3 mg/kg ‑annoksella. Vähintään 9 kuukauden seuranta-ajan jälkeen tutkijan arvioima vahvistettu ORR oli 24,4 % (95 %:n luottamusväli 15,3–35,4). Vasteen keston mediaania ei saavutettu (vaihteluväli 4,4–16.6+ kuukautta). Kokonaiselinajan mediaani oli 9,7 kuukautta (95 %:n luottamusväli 7,26–16,16) ja arvioidut kokonaiselinajan osuudet olivat 6 kuukauden kohdalla 69,2 % (luottamusväli 57,7–78,2) ja 12 kuukauden kohdalla 45,6 % (luottamusväli 34,2–56,3).
dMMR- tai MSI-H-tyyppinen kolorektaalisyöpä
Avoin tutkimus nivolumabin ja ipilimumabin yhdistelmähoidosta verrattuna kemoterapiaan dMMR- tai MSI‑H-tyyppisen kolorektaalisyövän hoidossa potilailla, jotka eivät olleet saaneet aiempaa hoitoa etäpesäkkeiseen tautiin
Nivolumabin (240 mg) ja ipilimumabin (1 mg/kg) yhdistelmän, jota annettiin 3 viikon välein enintään 4 annosta ja jonka jälkeen nivolumabia annettiin monoterapiana 480 mg 4 viikon välein, turvallisuutta ja tehoa leikkaukseen soveltumattoman tai etäpesäkkeisen kolorektaalisyövän (kasvaimen MSI‑H- tai dMMR-status oli tiedossa) ensilinjan hoidossa arvioitiin satunnaistetussa, useilla hoitoryhmillä tehdyssä, vaiheen 3 avoimessa tutkimuksessa (CA2098HW). Tutkimuksen hoitoryhmiä olivat nivolumabi monoterapiana, nivolumabin ja ipilimumabin yhdistelmä tai tutkijan valitsema kemoterapia. Kasvaimen MSI‑H- tai dMMR-status määritettiin paikallisen käytännön mukaan PCR-, NGS- tai IHC-menetelmillä. MSI‑H-status määritettiin keskitetysti PCR-testillä (Idylla MSI) ja dMMR-status IHC-testillä (Omnis MMR) retrospektiivisesti potilaiden kasvainnäytteistä, joita käytettiin MSI‑H-/dMMR-statuksen määrittämiseen paikallisesti. Ensisijainen tehopopulaatio koostui potilaista, joiden MSI‑H-/dMMR-status vahvistettiin kummalla tahansa keskitetyllä testillä. Tutkimuksesta suljettiin pois potilaat, joilla oli oireita aiheuttavia aivometastaaseja tai aktiivinen autoimmuunisairaus tai jotka käyttivät systeemisiä kortikosteroideja tai immunosuppressantteja tai joita oli hoidettu tarkistuspisteen estäjillä. Satunnaistaminen ositettiin kasvaimen sijainnin (oikea vs. vasen) perusteella. Solunsalpaajaryhmään satunnaistetuille potilaille voitiin antaa nivolumabin ja ipilimumabin yhdistelmää BICR:n arvioiman taudin etenemisen jälkeen.
Tutkimukseen satunnaistettiin yhteensä 303 aiemmin hoitamatonta potilasta, joilla oli etäpesäkkeinen tauti. 202 potilasta satunnaistettiin saamaan nivolumabin ja ipilimumabin yhdistelmää ja 101 satunnaistettiin saamaan kemoterapiaa. Näistä potilaista 255:n MSI‑H-/dMMR-status oli vahvistettu keskitetysti. Heistä 171 oli nivolumabin ja ipilimumabin yhdistelmää saavassa ryhmässä ja 84 kemoterapiaa saavassa ryhmässä. Nivolumabin ja ipilimumabin yhdistelmää saavassa ryhmässä potilaat saivat nivolumabia 240 mg ja ipilimumabia 1 mg/kg 3 viikon välein enintään 4 annosta. Tämän jälkeen nivolumabia annettiin monoterapiana 480 mg 4 viikon välein. Solunsalpaajaryhmän potilaat saivat hoitoa seuraavasti: mFOLFOX6-hoito (oksaliplatiinia, leukovoriinia ja fluorourasiilia) joko bevasitsumabin tai setuksimabin kanssa tai ilman niitä: oksaliplatiinia 85 mg/m2, leukovoriinia 400 mg/m2 ja fluorourasiilia 400 mg/m2 boluksena, minkä jälkeen fluorourasiilia 2400 mg/m2 46 tunnin aikana 2 viikon välein. Bevasitsumabia 5 mg/kg tai setuksimabia 500 mg/m2 annettiin ennen mFOLFOX6-hoitoa 2 viikon välein; tai FOLFIRI-hoito (irinotekaania, leukovoriinia ja fluorourasiilia) joko bevasitsumabin tai setuksimabin kanssa tai ilman niitä: irinotekaania 180 mg/m2, leukovoriinia 400 mg/m2 ja fluorourasiilia 400 mg/m2 boluksena, minkä jälkeen fluorourasiilia 2400 mg/m2 46 tunnin aikana 2 viikon välein. Bevasitsumabia 5 mg/kg tai setuksimabia 500 mg/m2 annettiin ennen FOLFIRI-hoitoa 2 viikon välein. Hoitoa jatkettiin, kunnes tauti eteni tai kunnes ilmeni ei-hyväksyttävää toksisuutta tai nivolumabin ja ipilimumabin yhdistelmän kohdalla enintään 24 kuukauden ajan. Potilaat, jotka eivät jatkaneet yhdistelmähoitoa ipilimumabin aiheuttaman haittavaikutuksen takia, saivat jatkaa pelkän nivolumabin käyttöä. RECIST v1.1:n mukaiset kasvainten arvioinnit tehtiin 6 viikon välein ensimmäisten 24 viikon ajan, sen jälkeen 8 viikon välein viikolle 96 asti, sen jälkeen 16 viikon välein viikolle 146 asti ja sen jälkeen 24 viikon välein.
Lähtötason ominaisuudet kaikilla satunnaistetuilla potilailla, jotka eivät olleet saaneet aiemmin hoitoa etäpesäkkeiseen tautiin: iän mediaani oli 63 vuotta (vaihteluväli: 21–87) ja 46 % oli ≥ 65‑vuotiaita ja 18 % ≥ 75‑vuotiaita; 46 % oli miehiä ja 86 % oli valkoihoisia. Lähtötason ECOG-toimintakykyluokka oli 0 (54 %) ja ≥ 1 (46 %), kasvain sijaitsi oikealla puolella 68 %:lla potilaista ja vasemmalla puolella 32 %:lla potilaista, ja 39 potilaalla niistä 223 potilaista, joiden status tunnettiin, oli vahvistettu Lynchin oireyhtymä. Lähtötason ominaisuudet potilailla, jotka eivät olleet saaneet aiemmin hoitoa etäpesäkkeiseen tautiin ja joiden MSI‑H-/dMMR-status oli keskitetysti vahvistettu, olivat yhdenmukaiset kaikkien niiden satunnaistettujen potilaiden kanssa, jotka eivät olleet saaneet aiemmin hoitoa. Kemoterapiaan satunnaistetuista 101 potilaasta 88 sai tutkimussuunnitelman mukaista kemoterapiaa, mukaan lukien oksaliplatiinia sisältäviä hoitoja (58 %) ja irinotekaania sisältäviä hoitoja (42 %). Tämän lisäksi 66 potilasta sai täsmälääkkeenä joko bevasitsumabia (64 %) tai setuksimabia (11 %).
Tutkimuksen ensisijainen tehokkuusmuuttuja oli BICR:n arvioima RECIST 1.1 -kriteerien mukainen etenemisvapaa elinaika (PFS). Muita tehokkuusmuuttujia olivat BICR:n arvioima objektiivisen vasteen saavuttaneiden osuus (ORR), kokonaiselinaika (OS) ja vasteen kesto.
Tutkimuksen ensisijainen päätetapahtuma saavutettiin suunnitellussa välianalyysissa, jossa BICR:n arvioiman etenemisvapaan elinajan osoitettiin parantuneen tilastollisesti merkitsevästi nivolumabin ja ipilimumabin yhdistelmää saaneilla potilailla, joiden MSI‑H-/dMMR-status oli keskitetysti vahvistettu, kemoterapiaa saaneeseen ryhmään verrattuna. BICR:n arvioiman etenemisvapaan elinajan tulokset on esitetty taulukossa 54 ja kuvassa 28. Välianalyysin ajankohtana muita päätetapahtumia (mukaan lukien tiedot pelkkää nivolumabia saaneesta ryhmästä) ei testattu testaushierarkian takia.
Taulukko 54: Tulokset tehosta keskitetysti vahvistetun MSI‑H-/dMMR-tyyppisen kolorektaalisyövän ensilinjan hoidossa (CA2098HW)a
nivolumabi + ipilimumabi | kemoterapia | |
|---|---|---|
Etenemisvapaa elinaika |
|
|
Tapahtumia | 48 (28 %) | 52 (62 %) |
Riskitiheyssuhde | 0,21 | |
95 %:n luottamusväli | (0,14–0,32) | |
p‑arvob | < 0,0001 | |
Mediaani (95 %:n luottamusväli) (kuukautta) | NR (38,4–NR) | 5,9 (4,4–7,8) |
a Seurannan mediaani 31,5 kuukautta (vaihteluväli: 6,1–48,4 kuukautta).
b Perustuu ositettuun kaksipuoliseen log‑rank‑merkitsevyystestiin.
Kuva 28: Etenemisvapaan elinajan Kaplan‑Meier-kuvaaja keskitetysti vahvistetun MSI‑H-/dMMR-tyyppisen kolorektaalisyövän ensilinjan hoidossa (CA2098HW)
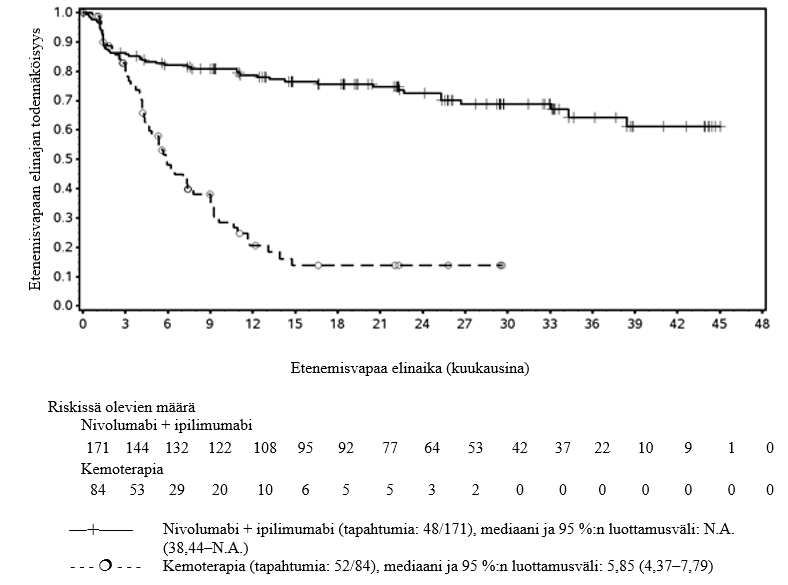
Avoin tutkimus nivolumabin ja ipilimumabin yhdistelmähoidosta dMMR- tai MSI‑H-tyyppisen kolorektaalisyövän hoidossa potilailla, jotka olivat saaneet aiempaa fluoropyrimidiinipohjaista yhdistelmäkemoterapiaa
Nivolumabi 3 mg/kg- ja ipilimumabi 1 mg/kg -yhdistelmähoidon turvallisuutta ja tehoa dMMR- ja MSI-H-tyyppisen metastaattisen kolorektaalisyövän hoidossa arvioitiin yksihaaraisessa avoimessa vaiheen 2 monikeskustutkimuksessa (CA209142).
Tutkimuksessa oli mukana 18 vuotta täyttäneitä potilaita, joilla oli paikallisesti määritetty dMMR- tai MSI-H-status ja joiden tauti oli edennyt aiemman fluoropyrimidiini- ja oksaliplatiinihoidon tai irinotekaanihoidon aikana tai sen jälkeen tai jotka eivät olleet sietäneet fluoropyrimidiini- ja oksaliplatiinihoitoa tai irinotekaanihoitoa. Niillä potilailla, joiden viimeisin aiempi hoito oli liitännäishoito, taudin oli pitänyt edetä hoidon aikana tai kuuden kuukauden sisällä liitännäishoitona annetusta kemoterapiasta. Potilaiden ECOG-toimintakykyluokka oli 0 tai 1, ja heidät otettiin mukaan tutkimukseen kasvaimen PD-L1-ilmentymistasosta riippumatta. Potilaat, joilla oli aktiivisia aivometastaaseja, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila, suljettiin pois tutkimuksesta.
Yhteensä 119 potilasta sai joka kolmas viikko neljän annoksen ajan yhdistelmähoitoa, jossa nivolumabia 3 mg/kg annosteltiin laskimoon 60 minuutin ajan ja ipilimumabia 1 mg/kg annosteltiin laskimoon 90 minuutin ajan. Tämän jälkeen potilaille annettiin nivolumabia monoterapiana 3 mg/kg joka toinen viikko. Hoitoa jatkettiin niin kauan kuin siitä todettiin olevan kliinistä hyötyä tai kunnes potilas ei enää sietänyt hoitoa. Kasvaimet arvioitiin kiinteiden kasvainten vasteenarviointikriteerien (RECIST) version 1.1 mukaisesti ensimmäisen 24 viikon ajan joka 6. viikko ja tämän jälkeen joka 12. viikko. Ensisijainen muuttuja oli tutkijan arvioima objektiivisen vasteen saavuttaneiden osuus (ORR). Toissijaisia muuttujia olivat riippumattoman keskitetyn arvioijatahon (Blinded Independent Central Review, BICR) arvioima objektiivisen vasteen saavuttaneiden osuus ja niiden potilaiden määrä, joilla sairaus saatiin hallintaan. Vasteen kesto ja vasteen saavuttamisaika sisältyivät objektiivisen vasteen saavuttaneiden osuuden analyysiin. Havainnollistavia (eksploratiivisia) muuttujia olivat etenemisvapaa elinaika (PFS) ja kokonaiselinaika (OS).
Potilaiden mediaani-ikä oli 58 vuotta (vaihteluväli 21–88). Potilaista 32 % oli iältään ≥ 65-vuotiaita ja 9 % oli iältään ≥ 75-vuotiaita. Potilaista 59 % oli miehiä ja 92 % valkoihoisia. Lähtötason ECOG-toimintakykyluokka oli 0 (45 %) tai 1 (55 %). Potilaista 25 %:lla oli BRAF-mutaatioita, 37 %:lla oli KRAS-mutaatioita ja 12 %:lla mutaatiostatus oli tuntematon. 119:stä hoidetusta potilaasta 109 oli saanut aiempaa fluoropyrimidiinipohjaista kemoterapiaa metastaattiseen tautiin ja 9 liitännäishoitona. Ennen tutkimukseen osallistumista kaikista 119:sta hoitoa saaneesta potilaasta 118 potilasta (99 %) oli saanut fluorourasiilia, 111 potilasta (93 %) oli saanut oksaliplatiinia, 87 potilasta (73 %) oli saanut irinotekaania osana aiempia hoitoja ja 82 potilasta (69 %) oli saanut aiempaa fluoropyrimidiini-, oksaliplatiini- ja irinotekaanihoitoa. 23 % potilaista oli saanut aiemmin yhtä hoitoa, 36 % kahta hoitoa, 24 % kolmea hoitoa ja 16 % neljää tai useampaa hoitoa. 29 % potilaista oli saanut EGFR:n estäjää.
Tulokset tehosta (seuranta-aika vähintään 46,9 kuukautta; seuranta-ajan mediaani 51,1 kuukautta) esitetään taulukossa 55.
Taulukko 55: Tulokset tehosta (CA209142)*
| nivolumabi + ipilimumabi (n = 119) |
Vahvistettu objektiivinen svaste, n (%) | 77 (64,7) |
(95 %:n luottamusväli) | (55,4–73,2) |
Täydellinen vaste (CR), n (%) | 15 (12,6) |
Osittainen vaste (PR), n (%) | 62 (52,1) |
Stabiili tauti (SD), n (%) | 25 (21,0) |
Vasteen kesto |
|
Mediaani kuukausina (vaihteluväli) | NR (1,4– 58,0+) |
Vasteen saavuttamisajan mediaani |
|
Kuukausina (vaihteluväli) | 2,8 (1,1–37,1) |
* tutkijan arvion mukaan
”+” tarkoittaa sensuroitunutta havaintoa.
NR = ei saavutettu
BICR:n arvioima objektiivisen vasteen saavuttaneiden osuus oli 61,3 % (95 %:n luottamusväli: 52,0–70,1), mukaan lukien täydellisen vasteen osuus 20,2 % (95 %:n luottamusväli: 13,4–28,5), osittaisen vasteen osuus 41,2 % (95 %:n luottamusväli: 32,2–50,6) ja stabiilin taudin raportoitu osuus 22,7 %. BICR:n arviot olivat yleisesti ottaen yhteneviä tutkijan arvion kanssa. Vahvistettuja vasteita havaittiin kasvaimen BRAF- tai KRAS-mutaatiostatuksesta ja PD‑L1-ilmentymistasosta riippumatta.
119 potilaasta 11 (9,2 %) oli ≥ 75-vuotiaita. Tutkijan arvioima objektiivisen vasteen saavuttaneiden osuus ≥ 75-vuotiailla potilailla oli 45,5 % (95 %:n luottamusväli: 16,7–76,6).
Ruokatorven levyepiteelikarsinooma
Satunnaistettu vaiheen 3 tutkimus nivolumabista yhdistelmähoitona ipilimumabin kanssa vs. kemoterapia ja nivolumabi yhdistelmähoitona kemoterapian kanssa vs. kemoterapia ensilinjan hoitona (CA209648)
Nivolumabin ja ipilimumabin yhdistelmähoidon sekä nivolumabin ja kemoterapian yhdistelmähoidon turvallisuutta ja tehoa arvioitiin satunnaistetussa, aktiivisella vertailuvalmisteella kontrolloidussa avoimessa tutkimuksessa (CA209648). Tutkimukseen osallistui aikuisia potilaita (18‑vuotiaita tai tätä vanhempia), joilla oli aiemmin hoitamaton, leikkaukseen soveltumaton, edennyt, uusiutunut tai etäpesäkkeinen ruokatorven levyepiteelikarsinooma (OSCC). Tutkimukseen otettiin potilaita riippumatta kasvaimen PD-L1‑statuksesta, ja kasvainsolujen PD-L1‑ilmentyminen määritettiin PD‑L1 IHC 28‑8 pharmDx ‑ testausmenetelmän avulla. Potilailla piti olla ruokatorven levyepiteelikarsinooma tai adenoskvamoosi karsinooma, jota ei voitu hoitaa kemosädehoidolla ja/tai leikkauksella. Aikaisempi liitännäishoitona, esiliitännäishoitona tai definitiivisenä hoitona annettu kemoterapia, sädehoito tai kemosädehoito sallittiin, jos se oli annettu osana parantavassa tarkoituksessa annettua hoitoa ennen tutkimukseen osallistumista. Potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, joilla oli oireisia aivometastaaseja tai aktiivinen autoimmuunisairaus, jotka käyttivät systeemisiä kortikosteroideja tai immunosuppressiivisia lääkkeitä tai joilla oli suuri riski verenvuodolle tai fistelille kasvaimen havaittavan invaasion ruokatorven kasvainta lähellä oleviin elimiin aiheuttamana, suljettiin pois tutkimuksesta. Satunnaistaminen ositettiin kasvainsolujen PD‑L1‑statuksen (≥ 1 % vs. < 1 % tai määrittämätön), alueen (Itä-Aasia vs. muu Aasia vs. muu maailma), ECOG‑toimintakykyluokan (0 vs. 1) ja metastaaseja sisältävien elinten lukumäärän (≤ 1 vs. ≥ 2) mukaan.
Yhteensä 970 potilasta satunnaistettiin saamaan joko nivolumabin ja ipilimumabin yhdistelmähoitoa (n = 325), nivolumabin ja kemoterapian yhdistelmähoitoa (n = 321) tai kemoterapiaa (n = 324). Näistä potilaista 473 potilaalla kasvainsolujen PD-L1‑ilmentymä oli ≥ 1 %; 158 potilaalla nivolumabi–ipilimumabi‑yhdistelmähoidon ryhmässä, 158 potilaalla nivolumabi–kemoterapia‑yhdistelmähoidon ryhmässä ja 157 potilaalla kemoterapiaryhmässä. Nivolumabi–ipilimumabi‑yhdistelmähoitoryhmässä olleet potilaat saivat nivolumabia 3 mg/kg kahden viikon välein ja ipilimumabia 1 mg/kg kuuden viikon välein, ja nivolumabi–kemoterapia‑yhdistelmähoitoryhmässä olleet potilaat saivat nivolumabia 240 mg kahden viikon välein päivinä 1 ja 15, fluorourasiilia 800 mg/m2/vrk laskimoon päivästä 1 päivään 5 (5 päivän ajan) ja sisplatiinia 80 mg/m2 laskimoon päivänä 1 (4 viikon jaksosta). Kemoterapiaryhmässä olleet potilaat saivat fluorourasiilia 800 mg/m2/vrk laskimoon päivästä 1 päivään 5 (5 päivän ajan) ja sisplatiinia 80 mg/m2 laskimoon päivänä 1 (4 viikon jaksosta). Hoitoa jatkettiin, kunnes tauti eteni tai kunnes ilmeni ei-hyväksyttävää toksisuutta tai enintään 24 kuukauden ajan. Nivolumabi–ipilimumabi‑yhdistelmähoitoryhmän potilaat, jotka eivät jatkaneet yhdistelmähoitoa ipilimumabin aiheuttaman haittavaikutuksen takia, saivat jatkaa nivolumabi-monoterapiaa. Nivolumabi–kemoterapia‑yhdistelmähoitoryhmän potilaat, joille joko fluorourasiilin ja/tai sisplatiinin anto keskeytettiin, saivat jatkaa muiden hoito-ohjelman lääkkeiden käyttöä.
Hoitoryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Potilasryhmässä, jossa kasvainsolujen PD-L1‑ilmentymä oli ≥ 1 %, iän mediaani oli 63 vuotta (vaihteluväli: 26–85), 8,2 % oli ≥ 75‑vuotiaita, 81,8 % oli miehiä, 73,1 % oli aasialaisia ja 23,3 % oli valkoihoisia. Potilaiden ruokatorven levyepiteelikarsinooma (98,9 %) tai adenoskvamoosi karsinooma (1,1 %) oli histologisesti vahvistettu. Lähtötason ECOG‑toimintakykyluokka oli 0 (45,2 %) tai 1 (54,8 %).
Nivolumabi yhdistelmähoitona ipilimumabin kanssa vs. kemoterapia
Ensisijaiset tehokkuusmuuttujat olivat etenemisvapaa elinaika (BICR:n arvioimana) ja kokonaiselinaika, jotka arvioitiin potilailla, joiden kasvainsolujen PD-L1‑ilmentymä oli ≥ 1 %. Toissijaiset päätetapahtumat ennalta määritellyn hierarkkisen testausjärjestyksen mukaan olivat muun muassa kokonaiselinaika, etenemisvapaa elinaika (BICR:n arvioimana) ja objektiivisen vasteen saavuttaneiden osuus (BICR:n arvioimana) kaikilla satunnaistetuilla potilailla. Kasvaimet arvioitiin RECIST v1.1 ‑kriteerien mukaan joka 6. viikko viikkoon 48 asti, viikko 48 mukaan lukien, ja sen jälkeen joka 12. viikko.
Kokonaiselinajan osoitettiin primaarisessa ennalta määritetyssä analyysissä, kun seuranta‑aika oli vähintään 13,1 kuukautta, olevan tilastollisesti merkitsevästi parempi potilailla, joiden kasvainsolujen PD-L1‑ilmentymä oli ≥ 1 %. Tulokset tehosta esitetään taulukossa 56.
Taulukko 56: Tulokset tehosta potilailla, joiden kasvainsolujen PD-L1 oli ≥ 1 % (CA209648)
| nivolumabi + ipilimumabi | kemoterapiaa | |
Kokonaiselinaika |
|
| |
Tapahtumat | 106 (67,1 %) | 121 (77,1 %) | |
Riskitiheyssuhde (98,6 %:n luottamusväli)b | 0,64 (0,46–0,90) | ||
p-arvoc | 0,0010 | ||
Mediaani (95 %:n luottamusväli) (kuukausina)d | 13,70 (11,24–17,02) | 9,07 (7,69–9,95) | |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdallad | 57,1 (49,0–64,4) | 37,1 (29,2–44,9) | |
Etenemisvapaa elinaikae |
|
| |
Tapahtumat | 123 (77,8 %) | 100 (63,7 %) | |
Riskitiheyssuhde (98,5 %:n luottamusväli)b | 1,02 (0,73–1,43) | ||
p-arvoc | 0,8958 | ||
Mediaani (95 %:n luottamusväli) (kuukausina)d | 4,04 (2,40–4,93) | 4,44 (2,89–5,82) | |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdallad | 26,4 (19,5–33,9) | 10,5 (4,7–18,8) | |
Objektiivisen vasteen saavuttaneiden osuus, n (%)e | 56 (35,4) | 31 (19,7) | |
(95 %:n luottamusväli) | (28,0–43,4) | (13,8–26,8) | |
Täydellinen vaste | 28 (17,7) | 8 (5,1) | |
Osittainen vaste | 28 (17,7) | 23 (14,6) | |
Vasteen kestoe |
|
| |
Mediaani (95 %:n luottamusväli) (kuukausina)d | 11,83 (7,10–27,43) | 5,68 (4,40–8,67) | |
Vaihteluväli | 1,4+––34,5+ | 1,4+–31,8+ | |
a Fluorourasiili ja sisplatiini.
b Perustuu ositettuun Coxin suhteellisen vaaran malliin.
c Perustuu ositettuun kaksipuoliseen log‑rank‑merkitsevyystestiin.
d Perustuu Kaplan–Meierin arvioihin.
e BICR:n arvioimana.
Vähintään 20 kuukauden seuranta‑ajan jälkeen tehdyn päivitetyn deskriptiivisen analyysin mukaiset kokonaiselinajan tulokset olivat yhteneviä primaarianalyysin kanssa. Kokonaiselinajan mediaani oli 13,70 kuukautta (95 %:n luottamusväli: 11,24–17,41) nivolumabin ja ipilimumabin yhdistelmähoitoa saaneilla vs. 9,07 kuukautta (95 %:n luottamusväli: 7,69–10,02) kemoterapiaa saaneilla (riskitiheyssuhde = 0,63; 95 %:n luottamusväli: 0,49–0,82). Etenemisvapaan elinajan mediaani oli 4,04 kuukautta (95 %:n luottamusväli: 2,40–4,93) nivolumabin ja ipilimumabin yhdistelmähoitoa saaneilla vs. 4,44 kuukautta (95 %:n luottamusväli: 2,89–5,82) kemoterapiaa saaneilla (riskitiheyssuhde = 1,02; 95 %:n luottamusväli: 0,77–1,34). Objektiivisen vasteen saavuttaneiden osuus oli 35,4 % (95 %:n luottamusväli: 28,0,–43,4) nivolumabin ja ipilimumabin yhdistelmähoitoa saaneilla vs. 19,7 % (95 %:n luottamusväli: 13,8–26,8) kemoterapiaa saaneilla.
Kuvassa 29 esitetään Kaplan–Meier-kuvaajat kokonaiselinajasta vähintään 20 kuukauden seuranta-ajalla.
Kuva 29: Kaplan–Meier-kuvaajat kokonaiselinajasta potilailla, joiden kasvainsolujen PD‑L1 oli ≥ 1 % (CA209648)
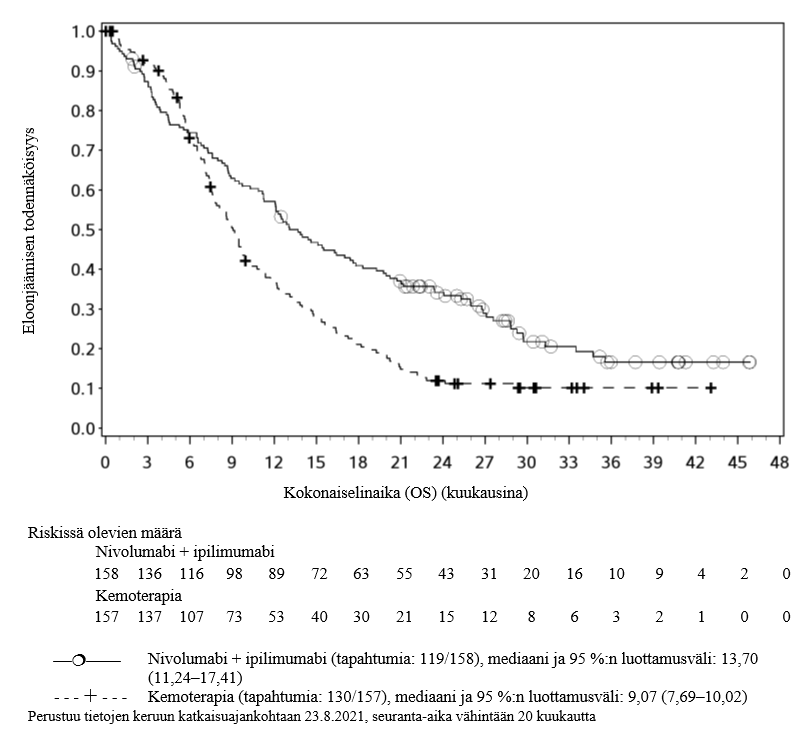
Nivolumabi yhdistelmähoitona kemoterapian kanssa vs. kemoterapia
Ensisijaiset tehokkuusmuuttujat olivat etenemisvapaa elinaika (BICR:n arvioimana) ja kokonaiselinaika potilailla, joiden kasvainsolujen PD-L1‑ilmentymä oli ≥ 1 %. Toissijaiset päätetapahtumat ennalta määritellyn hierarkkisen testausjärjestyksen mukaan olivat muun muassa kokonaiselinaika, etenemisvapaa elinaika (BICR:n arvioimana) ja objektiivisen vasteen saavuttaneiden osuus (BICR:n arvioimana) kaikilla satunnaistetuilla potilailla. Kasvaimet arvioitiin RECIST v1.1 ‑kriteerien mukaan joka 6. viikko viikkoon 48 asti, viikko 48 mukaan lukien, ja sen jälkeen joka 12. viikko.
Kokonaiselinajan ja etenemisvapaan elinajan osoitettiin primaarisessa ennalta määritetyssä analyysissä, kun seuranta aika oli vähintään 12,9 kuukautta, olevan tilastollisesti merkitsevästi parempia potilailla, joiden kasvainsolujen PD-L1‑ilmentymä oli ≥ 1 %. Tulokset tehosta esitetään taulukossa 57.
Taulukko 57: Tulokset tehosta potilailla, joiden kasvainsolujen PD-L1 oli ≥ 1 % (CA209648)
| nivolumabi + kemoterapia | kemoterapiaa | |
Kokonaiselinaika |
|
| |
Tapahtumat | 98 (62,0 %) | 121 (77,1 %) | |
Riskitiheyssuhde (99,5 %:n luottamusväli)b | 0,54 (0,37– 0,80) | ||
p-arvoc | < 0,0001 | ||
Mediaani (95 %:n luottamusväli) (kuukausina)d | 15,44 (11,93–19,52) | 9,07 (7,69–9,95) | |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdallad | 58,0 (49,8–65,3) | 37,1 (29,2–44,9) | |
Etenemisvapaa elinaikae |
|
| |
Tapahtumat | 117 (74,1 %) | 100 (63,7 %) | |
Riskitiheyssuhde (98,5 %:n luottamusväli)b | 0,65 (0,46, 0,92) | ||
p-arvoc | 0,0023 | ||
Mediaani (95 %:n luottamusväli) (kuukausina)d | 6,93 (5,68–8,34) | 4,44 (2,89–5,82) | |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdallad | 25,4 (18,2–33,2) | 10,5 (4,7–18,8) | |
Kokonaisvasta, n (%)e | 84 (53,2) | 31 (19,7) | |
(95 %:n luottamusväli) | (45,1–61,1) | (13,8–26,8) | |
Täydellinen vaste | 26 (16,5) | 8 (5,1) | |
Osittainen vaste | 58 (36,7) | 23 (14,6) | |
Vasteen kestoe |
|
| |
Mediaani (95 %:n luottamusväli) (kuukausina)d | 8,38 (6,90–12,35) | 5,68 (4,40–8,67) | |
Vaihteluväli | 1,4+–34,6 | 1,4+–31,8+ | |
a Fluorourasiili ja sisplatiini.
b Perustuu ositettuun Coxin suhteellisen vaaran malliin.
c Perustuu ositettuun kaksipuoliseen log‑rank‑merkitsevyystestiin.
d Perustuu Kaplan–Meierin arvioihin.
e BICR:n arvioimana.
Vähintään 20 kuukauden seuranta‑ajan jälkeen tehdyn päivitetyn deskriptiivisen analyysin mukaiset kokonaiselinajan tulokset olivat yhteneviä primaarianalyysin kanssa. Kokonaiselinajan mediaani oli 15,05 kuukautta (95 %:n luottamusväli: 11,93–18,63) nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla vs. 9,07 kuukautta (95 %:n luottamusväli: 7,69, 10,02) kemoterapiaa saaneilla (riskitiheyssuhde = 0,59; 95 %:n luottamusväli: 0,46, 0,76). Etenemisvapaan elinajan mediaani oli 6,93 kuukautta (95 %:n luottamusväli: 5,68–8,35) nivolumabin ja kemoterapia yhdistelmähoitoa saaneilla vs. 4,44 kuukautta (95 %:n luottamusväli: 2,89–5,82) kemoterapiaa saaneilla (riskitiheyssuhde = 0,66; 95 %:n luottamusväli: 0,50–0,87). Objektiivisen vasteen saavuttaneiden osuus oli 53,2 % (95 %:n luottamusväli: 45,1–61,1) nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla vs. 19,7 % (95 %:n luottamusväli: 13,8–26,8) kemoterapiaa saaneilla.
Kuvissa 30 ja 31 esitetään Kaplan–Meier-kuvaajat kokonaiselinajasta ja etenemisvapaasta elinajasta vähintään 20 kuukauden seuranta-ajalla.
Kuva 30: Kaplan–Meier-kuvaajat kokonaiselinajasta potilailla, joiden kasvainsolujen PD‑L1 oli ≥ 1 % (CA209648)
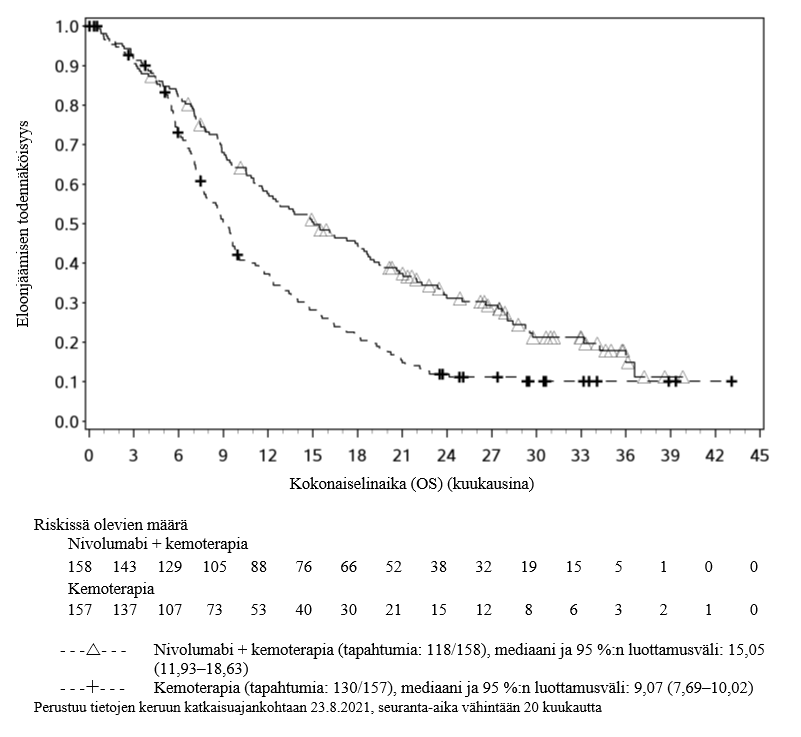
Kuva 31: Kaplan–Meier-kuvaajat etenemisvapaasta elinajasta potilailla, joiden kasvainsolujen PD‑L1 oli ≥ 1 % (CA209648)
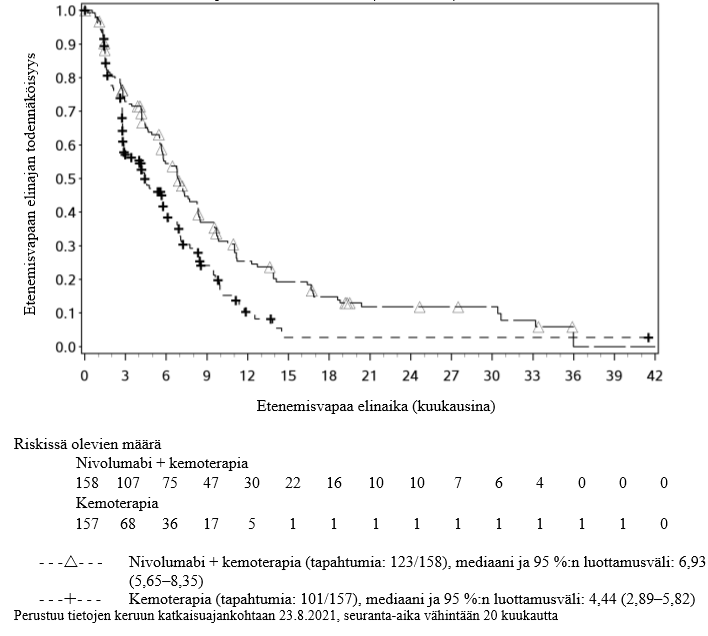
Satunnaistettu vaiheen 3 tutkimus nivolumabista monoterapiana potilailla, jotka olivat saaneet aiempaa hoitoa (ONO‑4538‑24/ CA209473)
Nivolumabi-monoterapian tehoa ja turvallisuutta 240 mg:n annoksella leikkaukseen sopimattoman, edenneen, uusiutuneen tai etäpesäkkeisen ruokatorven levyepiteelikarsinooman (OSCC) hoidossa arvioitiin vaiheen 3 satunnaistetussa, aktiivisella vertailuvalmisteella kontrolloidussa avoimessa tutkimuksessa (ONO‑4538‑24/CA209473). Tutkimukseen osallistui aikuisia (20‑vuotiaita tai vanhempia) potilaita, joiden sairaus oli refraktaari vähintään yhdelle fluoropyrimidiini- ja platinapohjaiselle yhdistelmähoidolle tai jotka eivät sietäneet vähintään yhtä fluoropyrimidiini- ja platinapohjaista yhdistelmähoitoa. Potilaat otettiin mukaan tutkimukseen kasvaimen PD‑L1:n ilmentymistasosta riippumatta. Tutkimuksesta suljettiin pois potilaat, joiden sairaus oli taksaanihoidolle refraktaari tai jotka eivät sietäneet taksaanihoitoa, joilla oli oireisia tai hoitoa vaativia aivometastaaseja, aktiivinen autoimmuunisairaus, systeemistä immuunisuppressiota vaativa tila tai joilla oli kasvaimen havaittavaa invaasiota ruokatorven lähellä oleviin elimiin (esim. aorttaan tai hengitysteihin).
Kaikkiaan 419 potilasta satunnaistettiin suhteessa 1:1 saamaan joko nivolumabia 240 mg laskimoon 30 minuutin ajan joka toinen viikko (n = 210) tai tutkijan valitsemaa taksaanikemoterapiaa: joko dosetakselia (n = 65) 75 mg/m2 laskimoon joka kolmas viikko tai paklitakselia (n = 144) 100 mg/m2 laskimoon kerran viikossa kuuden viikon ajan, mitä seurasi yhden viikon tauko. Satunnaistaminen ositettiin sijainnin (Japani vs. muu maailma), metastaaseja sisältävien elinten lukumäärän (≤ 1 vs. ≥ 2) ja kasvaimen PD‑L1:n ilmentymisen (≥ 1 % vs. < 1 % tai määrittämätön) mukaan. Hoitoa jatkettiin sairauden etenemiseen saakka tutkijan RECIST 1.1 -kriteerien perusteella tekemän arvion mukaan tai kunnes ilmeni ei-hyväksyttävää toksisuutta. Kasvaimet arvioitiin 6 viikon välein 1 vuoden ajan ja tämän jälkeen 12 viikon välein. Hoito tutkijan arvioiman taudin etenemisen jälkeen sallittiin nivolumabia saaville potilaille, jos sairaus ei edennyt nopeasti, jos hoidosta oli tutkijan arvion mukaan hyötyä potilaalle, jos potilas sieti hoitoa, jos potilaan toimintakyky oli vakaa ja jos hoito taudin etenemisen jälkeen ei viivyttäisi mahdollisia toimenpiteitä, jotka liittyvät taudin etenemiseen liittyviin vakaviin komplikaatioihin (esim. aivometastaaseihin). Ensisijainen tehokkuusmuuttuja oli OS. Tärkeimmät toissijaiset tehokkuusmuuttujat olivat tutkijan arvioima ORR ja PFS. Ylimääräiset ennalta määritellyt alaryhmäanalyysit tehokkuuden arvioimiseksi tehtiin etukäteen määritellyllä kasvaimen PD‑L1-ilmentymistasolla 1 %. Kasvaimen PD‑L1:n ilmentyminen määritettiin käyttämällä PD‑L1 IHC 28–8 pharmDx ‑testausmenetelmää.
Kahden ryhmän lähtötason ominaisuudet olivat tasapainossa. Iän mediaani oli 65 vuotta (vaihteluväli: 33–87), 53 % oli iältään ≥ 65 vuotta ja 10 % oli iältään ≥ 75 vuotta; 87 % oli miehiä, 96 % oli aasialaisia ja 4 % valkoihoisia. Lähtötason ECOG-toimintakykyluokka oli 0 (50 %) tai 1 (50 %).
Kun minimiseuranta-aika oli 17,6 kuukautta, tutkimus osoitti tilastollisesti merkitsevää hyötyä kokonaiselinajassa potilailla, jotka oli satunnaistettu saamaan nivolumabia, verrattuna potilaisiin, jotka oli satunnaistettu saamaan tutkijan valitsemaa taksaanikemoterapiaa. Tulokset tehosta esitetään taulukossa 58 ja kuvassa 32.
Nivolumabiryhmässä potilaita kuoli enemmän ensimmäisen 2,5 kuukauden aikana (32/210, 15,2 %) kuin kemoterapiaryhmässä (15/209, 7,2 %). Mitään tiettyjä varhaiseen kuolemaan liittyviä tekijöitä ei voitu tunnistaa.
Taulukko 58: Tulokset tehosta (ONO‑4538‑24/CA209473)
nivolumabi | tutkijan valinta | |
|---|---|---|
Kokonaiselinaika (OS)a | ||
Tapahtumia | 160 (76 %) | 173 (83 %) |
Riskitiheyssuhde (95 %:n luottamusväli)b | 0,77 (0,62–0,96) | |
p‑arvoc | 0,0189 | |
Mediaani (95 %:n luottamusväli) (kuukausina) | 10,9 (9,2–13,3) | 8,4 (7,2–9,9) |
Objektiivisen vasteen saavuttaneiden osuusd,e | 33 (19,3 %) | 34 (21,5 %) |
(95 %:n luottamusväli) | (13,7–26,0) | (15,4–28,8) |
Täydellinen vaste | 1 (0,6 %) | 2 (1,3 %) |
Osittainen vaste (PR) | 32 (18,7 %) | 32 (20,3 %) |
Stabiili tauti (SD) | 31 (18,1 %) | 65 (41,1 %) |
Vasteen keston mediaani (95 %:n luottamusväli) (kuukausina) | 6,9 (5,4–11,1) | 3,9 (2,8–4,2) |
Etenemisvapaa elinaikaa | ||
Tapahtumat | 187 (89 %) | 176 (84 %) |
Mediaani (95 %:n luottamusväli) (kuukausina) | 1,7 (1,5–2,7) | 3,4 (3,0–4,2) |
Riskitiheyssuhde (95 %:n luottamusväli)b | 1,1 (0,9–1,3) | |
a Perustuen lähtöryhmien mukaiseen analyysiin (ITT).
b Perustuen ositettuun verrannollisten riskitiheyksien malliin.
c Perustuen ositettuun log‑rank‑merkitsevyystestiin.
d Perustuen RES-analyysiin (Response Evaluable Set); nivolumabiryhmässä n = 171 ja tutkijan valitseman hoidon ryhmässä n = 158.
e Ei merkitsevä, p‑arvo 0,6323.
Kuva 32: Kokonaiselinajan Kaplan–Meier-kuvaajat (ONO‑4538‑24/CA209473)
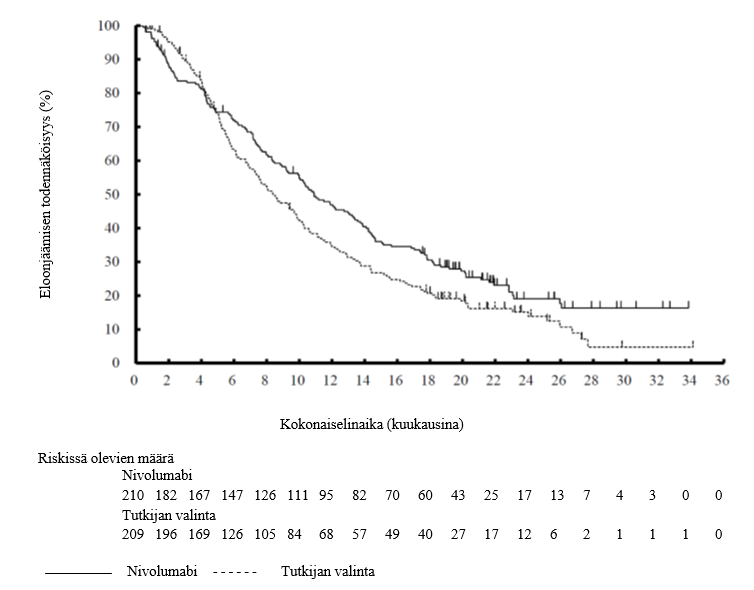
419 potilaasta 48 %:lla kasvaimen PD‑L1-ilmentymä oli ≥ 1 %. Lopuilla 52 %:lla potilaista kasvaimen PD‑L1-ilmentymä oli < 1 %. PD‑L1-positiivisen kasvaimen alaryhmässä kokonaiselinajan riskitiheyssuhde oli 0,69 (95 %:n luottamusväli: 0,51–0,94) ja elinajan mediaani 10,9 kuukautta nivolumabiryhmässä ja 8,1 kuukautta tutkijan valitseman taksaanikemoterapian ryhmässä. PD‑L1-negatiivisen kasvaimen ruokatorven levyepiteelikarsinoomaa sairastavien alaryhmässä riskitiheyssuhde oli 0,84 (95 %:n luottamusväli: 0,62–1,14) ja elinajan mediaani 10,9 kuukautta nivolumabiryhmässä ja 9,3 kuukautta kemoterapiaryhmässä.
Ruokatorvisyövän tai ruokatorvi-mahalaukkurajan syövän liitännäishoito
Nivolumabi-monoterapian turvallisuutta ja tehoa ruokatorvisyövän tai ruokatorvi-mahalaukkurajan syövän liitännäishoidossa arvioitiin vaiheen 3 satunnaistetussa, lumekontrolloidussa, kaksoissokkoutetussa monikeskustutkimuksessa (CA209577). Tutkimukseen osallistui aikuispotilaita, jotka olivat saaneet kemosädehoitoa (CRT), minkä jälkeen syöpä oli täydellisesti kirurgisesti resekoitu satunnaistamista edeltävien 16 viikon aikana, ja joilla oli patologista jäännöstautia tutkijan vahvistamana, vähintään ypN1 tai ypT1. Tutkimuksesta suljettiin pois potilaat, joiden lähtötason toimintakykyluokka oli ≥ 2, jotka eivät saaneet samanaikaista kemosädehoitoa ennen leikkausta, joilla oli kirurgisesti poistettavissa oleva IV asteen syöpä, aktiivinen autoimmuunisairaus tai systeemistä immunosuppressiota vaativa tila. Potilaat otettiin mukaan tutkimukseen kasvaimen PD‑L1:n ilmentymistasosta riippumatta.
Yhteensä 794 potilasta satunnaistettiin suhteessa 2:1 saamaan joko nivolumabia 240 mg (n = 532) tai lumelääkettä (n = 262). Potilaille annettiin nivolumabia laskimoon 30 minuutin kuluessa 2 viikon välein 16 viikon ajan, minkä jälkeen 480 mg infusoituna 30 minuutin kuluessa 4 viikon välein viikosta 17 alkaen. Potilaille annettiin lumelääkettä 30 minuutin kuluessa samalla antoaikataululla kuin nivolumabia. Satunnaistaminen ositettiin kasvaimen PD‑L1-statuksen (≥ 1 % vs. < 1 % tai määrittämätön tai ei arvioitavissa), patologisen imusolmukestatuksen (positiivinen ≥ypN1 vs. negatiivinen ypN0) ja histologian (levyepiteeliperäinen vs. adenokarsinooma) mukaan. Hoitoa jatkettiin, kunnes sairaus uusiutui, ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä, tai yhteensä enintään 1 vuoden ajan. Ensisijainen tehokkuusmuuttuja oli tautivapaa elossaoloaika (disease-free survival (DFS)) tutkijan arvioimana. Se määriteltiin ajaksi satunnaistamispäivämäärän ja jommankumman seuraavan päivämäärän välillä sen mukaan, kumpi tapahtui ensimmäisenä: ensimmäinen uusiutuminen (paikallinen, alueellinen tai etäinen uusiutuminen alkuperäisen resektiokohdan ulkopuolella) tai kuolema mistä tahansa syystä. Hoidon aikana potilaat kuvannettiin kasvainten uusiutumisen varalta 12 viikon välein 2 vuoden ajan, ja vuosien 3–5 ajan tehtiin vähintään yksi kuvaus 6–12 kuukauden välein.
Ryhmien lähtötason ominaisuudet olivat tasapainossa. Iän mediaani oli 62 vuotta (vaihteluväli: 26–86), ja 36 % oli ≥ 65‑vuotiaita ja 5 % ≥ 75‑vuotiaita. Suurin osa potilaista oli valkoihoisia (82 %) ja miehiä (85 %). Lähtötason ECOG‑toimintakykyluokka oli 0 (58 %) tai 1 (42 %).
Tautivapaan elossaoloajan osoitettiin primaarisessa ennalta määritetyssä välianalyysissä (seuranta‑aika vähintään 6,2 kuukautta ja seuranta-ajan mediaani 24,4 kuukautta) olevan tilastollisesti merkitsevästi parempi potilailla, jotka oli satunnaistettu saamaan nivolumabia, verrattuna potilaisiin, jotka oli satunnaistettu saamaan lumelääkettä. Tautivapaan elossaoloajan mediaani tutkijan määrittämänä oli 22,41 kuukautta (95 %:n luottamusväli: 16,62–34,00) nivolumabia saaneilla ja 11,04 kuukautta (95 %:n luottamusväli: 8,34–14,32) lumelääkettä saaneilla; riskitihyessuhde 0,69 (96,4 %:n luottamusväli: 0,56–0,86); p-arvo < 0,0003. Tautivapaan elossaoloajan primaarianalyysiin sisältyi uuden syöpähoidon sensurointi. Tautivapaan elossaoloajan sensuroidut ja sensuroimattomat tulokset olivat uuden syöpähoidon osalta yhdenmukaiset. Tautivapaan elossaoloajan paraneminen vahvistettiin päivitetyssä deskriptiivisessä tautivapaan elossaoloajan analyysissä, jossa seuranta‑aika oli vähintään 14 kuukautta ja seuranta‑ajan mediaani 32,2 kuukautta. Tämän deskriptiivisen sekundaarianalyysin tehoa koskevat tulokset esitetään taulukossa 59 ja kuvassa 33.
Taulukko 59: Tulokset tehosta (CA209577)
| nivolumabi | lumelääke |
|---|---|---|
Tautivapaa elossaoloaikaaseuranta-ajan ollessa vähintään 14 kuukauttac | ||
|
|
|
Tapahtumia (%) | 268 (50) | 171 (65) |
Riskitiheyssuhde (95 %:n luottamusväli)b | 0,67 (0,55–0,81) | |
Mediaani (95 %:n luottamusväli) (kuukautta) | 22,4 (17,0–33,6) | 10,4 (8,3–13,9) |
|
|
|
Osuus (95 %:n luottamusväli) 6 kuukauden kohdalla | 72,6 (68,5–76,3) | 61,5 (55,3–67,1) |
Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 61,8 (57,4–65,8) | 45,5 (39,3–51,4) |
Osuus (95 %:n luottamusväli) 24 kuukauden kohdalla | 48,3 (43,7–52,8) | 36,0 (29,9–42,0) |
a Perustuu kaikkiin satunnaistettuihin potilaisiin.
b Perustuu ositettuun Coxin suhteellisen vaaran malliin.
c Deskriptiivinen analyysi perustuu tietojen keruun katkaisuajankohtaan 18.2.2021.
Kuva 33: Tautivapaan elossaoloajan Kaplan–Meier-kuvaajat (CA209577)
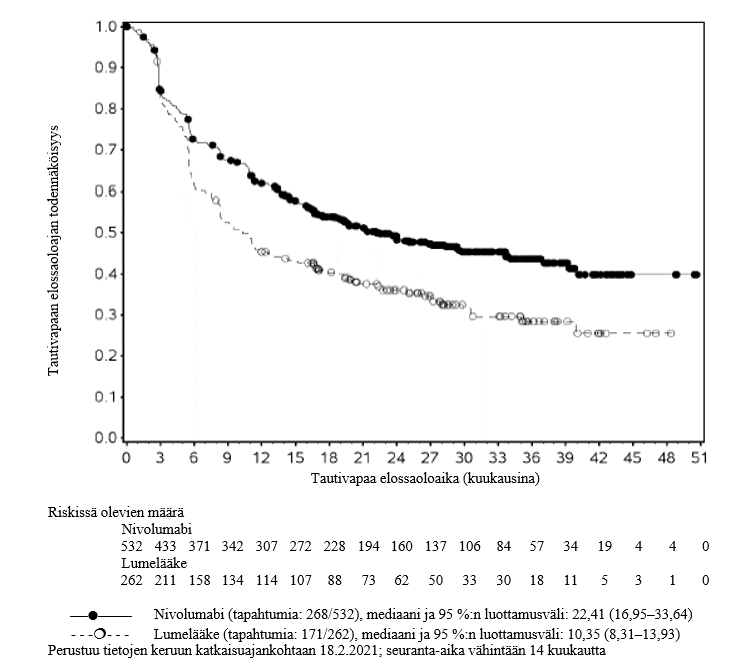
Lopullisessa kokonaiselinajan analyysissä, jonka seuranta-aika oli vähintään 60 kuukautta, kokonaiselinajan riskitiheyssuhde oli 0,85 (95,87 %:n luottamusväli: 0,70–1,04), p‑arvo = 0,1064. Kokonaiselinajan mediaani oli 51,71 kuukautta (95 %:n luottamusväli: 41,03–61,63) nivolumabiryhmässä, kun taas lumelääkeryhmässä se oli 35,25 kuukautta (95 %:n luottamusväli: 30,72–48,76). Kuvassa 34 esitetään Kaplan–Meier-kuvaajat kokonaiselinajasta vähintään 60 kuukauden seuranta-ajalla.
Kuva 34: Kokonaiselinajan Kaplan–Meier-kuvaajat (CA209577)
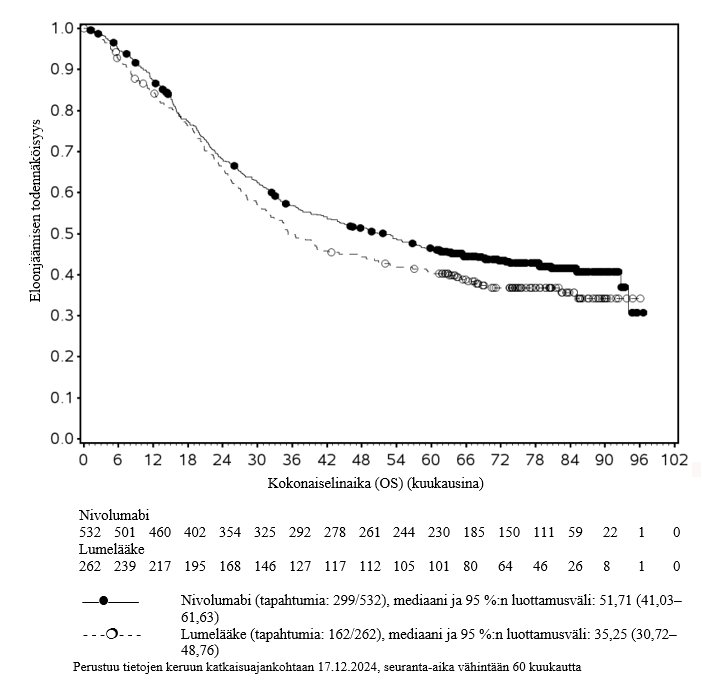
Mahalaukun, ruokatorvi-mahalaukkurajan tai ruokatorven adenokarsinooma
Nivolumabin (240 mg joka toinen viikko tai 360 mg joka kolmas viikko) ja kemoterapian yhdistelmähoidon (nivolumabin annos ja annosaikataulu valittiin käytetyn kemoterapiahoidon mukaan, ks. alla) turvallisuutta ja tehoa arvioitiin vaiheen 3 satunnaistetussa, avoimessa tutkimuksessa (CA209649). Tutkimukseen osallistui aikuispotilaita (18‑vuotiaita tai tätä vanhempia), joilla oli aiemmin hoitamaton edennyt tai etäpesäkkeinen mahalaukun, ruokatorvi-mahalaukkurajan (GEJ) tai ruokatorven adenokarsinooma, jotka eivät olleet aiemmin saaneet systeemistä hoitoa (mukaan lukien HER2:n estäjiä) ja joiden ECOG‑toimintakykyluokan pisteet olivat 0 tai 1. Tutkimukseen otettiin potilaita riippumatta kasvainsolujen PD‑L1‑statuksesta, ja kasvainsolujen PD-L1‑ilmentyminen määritettiin PD-L1 IHC 28-8 pharmDx ‑testausmenetelmän avulla. Potilaan kasvaimen PD‑L1‑status pisteytettiin retrospektiivisesti uudestaan CPS‑pistemäärällä (Combined Positive Score) käyttäen PD‑L1‑värjättyjä satunnaistamiseen käytettyjä kasvainnäytteitä. Tutkimuksesta suljettiin pois potilaat, joiden kasvainten tiedettiin olevan HER2‑positiivisia, joiden lähtötason ECOG‑toimintakykyluokka oli ≥ 2, joilla oli hoitamattomia keskushermoston metastaaseja, aktiivinen, tiedossa oleva tai epäilty autoimuunisairaus tai systeemistä immunosuppressiivista hoitoa vaativa tila. Yhteensä 643 potilasta, joiden HER2‑statusta ei tiedetty (40,3 % tutkimuspopulaatiosta), otettiin mukaan tutkimukseen. Satunnaistaminen ositettiin kasvainsolujen PD‑L1‑statuksen (≥ 1 % vs. < 1 % tai määrittämätön), alueen (Aasia vs. USA vs. muu maailma), ECOG‑toimintakykyluokan (0 vs. 1) ja kemoterapiahoidon mukaan. Kemoterapia oli FOLFOX‑hoitoa (fluorourasiili, leukovoriini ja oksaaliplatiini) tai CAPOX‑hoitoa (kapesitabiini ja oksaaliplatiini).
Yhteensä 1581 potilasta satunnaistettiin saamaan joko nivolumabia yhdessä kemoterapian kanssa tai kemoterapiaa. Näistä potilaista 955 potilaalla PD-L1:n CPS‑pistemäärä oli ≥ 5; 473 potilaalla nivolumabin ja kemoterapian yhdistelmähoidon ryhmässä ja 482 potilaalla kemoterapiaryhmässä. Nivolumabin ja kemoterapian yhdistelmähoidon ryhmän potilaat saivat jompaakumpaa hoitoa: nivolumabi 240 mg laskimoon annosteltuna infuusiona 30 minuutin kuluessa yhdessä FOLFOX‑hoidon kanssa (oksaaliplatiini 85 mg/m2, leukovoriini 400 mg/m2 ja fluorourasiili 400 mg/m2 laskimoon annosteltuna päivänä 1 ja fluorourasiili 1200 mg/m2 laskimoon annosteltuna jatkuvana infuusiona 24 tunnin kuluessa päivittäin tai paikallisten ohjeiden mukaisesti päivinä 1 ja 2) joka toinen viikko; tai nivolumabi 360 mg laskimoon annosteltuna infuusiona 30 minuutin kuluessa yhdessä CAPOX‑hoidon kanssa (oksaaliplatiini 130 mg/m2 laskimoon annosteltuna päivänä 1 ja kapesitabiini 1000 mg/m2 suun kautta annosteltuna kahdesti päivässä päivinä 1–14) joka kolmas viikko. Hoitoa jatkettiin, kunnes tauti eteni, ilmaantui toksisia vaikutuksia, joita ei voitu hyväksyä, tai enintään 24 kuukauden ajan ainoastaan nivolumabilla. Nivolumabia ja kemoterapiaa saaneille potilaille, joiden kemoterapiaa ei jatkettu, voitiin antaa nivolumabi‑monoterapiaa 240 mg joka toinen viikko, 360 mg joka kolmas viikko tai 480 mg joka neljäs viikko enintään 24 kuukauden ajan hoidon aloittamisen jälkeen. Kasvaimet arvioitiin joka kuudes viikko enintään viikkoon 48 asti, viikko 48 mukaan lukien, ja tämän jälkeen joka 12. viikko.
Hoitoryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Potilasryhmässä, jossa PD‑L1:n CPS‑pistemäärä oli ≥ 5, iän mediaani oli 62 vuotta (vaihteluväli: 18–90), 11 % oli ≥ 75‑vuotiaita, 71 % oli miehiä, 25 % oli aasialaisia ja 69 % oli valkoihoisia. Lähtötason ECOG‑toimintakykyluokka oli 0 (42 %) tai 1 (58 %). Kasvaimet sijaitsivat mahalaukussa (70 %), ruokatorvi‑mahalaukkurajalla (18 %) ja ruokatorvessa (12 %).
Ensisijaiset tehokkuusmuuttujat olivat etenemisvapaa elinaika (BICR:n arvioimana) ja kokonaiselinaika, jotka arvioidaan potilailla, joiden PD-L1:n CPS‑pistemäärä oli ≥ 5 PD-L1 IHC 28-8 pharmDX ‑testausmenetelmän mukaan. Toissijaiset päätetapahtumat ennalta määritellyn hierarkkisen testausjärjestyksen mukaan olivat kokonaiselinaika potilailla, joiden PD-L1:n CPS‑pistemäärä oli ≥ 1, ja kaikilla satunnaistetuilla potilailla; muita päätetapahtumia olivat muun muassa objektiivisen vasteen saavuttaneiden osuus (BICR:n arvioimana) potilailla, joiden PD-L1:n CPS‑pistemäärä oli ≥ 5, ja kaikilla satunnaistetuilla potilailla. Kokonaiselinajan ja etenemisvapaan elinajan osoitettiin primaarisessa ennalta määritetyssä analyysissä, kun seuranta aika oli vähintään 12,1 kuukautta, olevan tilastollisesti merkitsevästi parempi potilailla, joiden PD-L1:n CPS‑pistemäärä oli ≥ 5. Kokonaiselinajan mediaani oli 14,4 kuukautta (95 %:n luottamusväli: 13,1–16,2) nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla ja 11,1 kuukautta (95 %:n luottamusväli: 10,0–12,1) kemoterapiaa saaneilla (riskitiheyssuhde = 0,71; 98,4 %:n luottamusväli: 0,59–0,86; p-arvo < 0,0001). Etenemisvapaan elinajan mediaani oli 7,69 kuukautta (95 %:n luottamusväli: 7,03–9,17) nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla ja 6,05 kuukautta (95 %:n luottamusväli: 5,55–6,90) kemoterapiaa saaneilla (riskitiheyssuhde = 0,68; 98 %:n luottamusväli: 0,56–0,81; p-arvo < 0,0001). Objektiivisen vasteen saavuttaneiden osuus oli 60 % (95 %:n luottamusväli: 55–65) nivolumabin ja kemoterapian yhdistelmähoitoa saaneilla ja 45 % (95 %:n luottamusväli: 40–50) kemoterapiaa saaneilla.
Vähintään 19,4 kuukauden seuranta‑ajan jälkeen tehdyn päivitetyn deskriptiivisen analyysin mukaiset kokonaiselinajan tulokset olivat yhteneviä primaarianalyysin kanssa. Tulokset tehosta esitetään taulukossa 60 sekä kuvissa 35 ja 36.
Taulukko 60: Tulokset tehosta potilailla, joiden PD-L1:n CPS‑pistemäärä oli ≥ 5 (CA209649)
| nivolumabi + kemoterapia | kemoterapia | ||
|---|---|---|---|---|
Seuranta-aika vähintään 19,4 kuukauttaa | ||||
Kokonaiselinaika |
|
| ||
Tapahtumat | 344 (73 %) | 397 (82 %) | ||
Riskitiheyssuhde (95 %:n luottamusväli)b | 0,69 (0,60–0,81) | |||
Mediaani (95 %:n luottamusväli) (kuukausina)c Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 14,4 (13,1–16,3)
57,3 (52,6–61,6) | 11,1 (10,0–12,1)
46,4 (41,8–50,8) | ||
Etenemisvapaa elinaikad |
|
|
|
|
Tapahtumat | 342 (72,3 %) | 366 (75,9 %) | ||
Riskitiheyssuhde (95 %:n luottamusväli)b | 0,68 (0,59–0,79) | |||
Mediaani (95 %:n luottamusväli) (kuukausina)c Osuus (95 %:n luottamusväli) 12 kuukauden kohdalla | 8,31 (7,03–9,26)
36,3 (31,7–41,0) | 6,05 (5,55–6,90)
21,9 (17,8–26,1) | ||
Objektiivisen vasteen saavuttaneiden osuus, nd,e | 227/378 (60 %) | 176/390 (45 %) | ||
(95 %:n luottamusväli) | (54,9–65,0) | (40,1–50,2) | ||
Täydellinen vaste | 12,2 % | 6,7 % | ||
Osittainen vaste | 47,9 % | 38,5 % | ||
Vasteen kestod,e |
|
| ||
Mediaani (95 %:n luottamusväli) (kuukausina)c | 9,69 (8,25–12,22) | 6,97 (5,62–7,85) | ||
a Deskriptiivinen analyysi perustuu tietojen keruun katkaisuajankohtaan 4.1.2021.
b Perustuu ositettuun Coxin suhteellisen vaaran log-malliin.
c Kaplan–Meierarvio.
d BICR:n vahvistama.
e Perustuu potilaisiin, joiden tauti oli mitattavissa lähtötasolla.
Kuva 35: Kokonaiselinajan Kaplan–Meier kuvaaja (CA209649); potilaat, joiden PD‑L1:n CPS‑pistemäärä oli ≥ 5
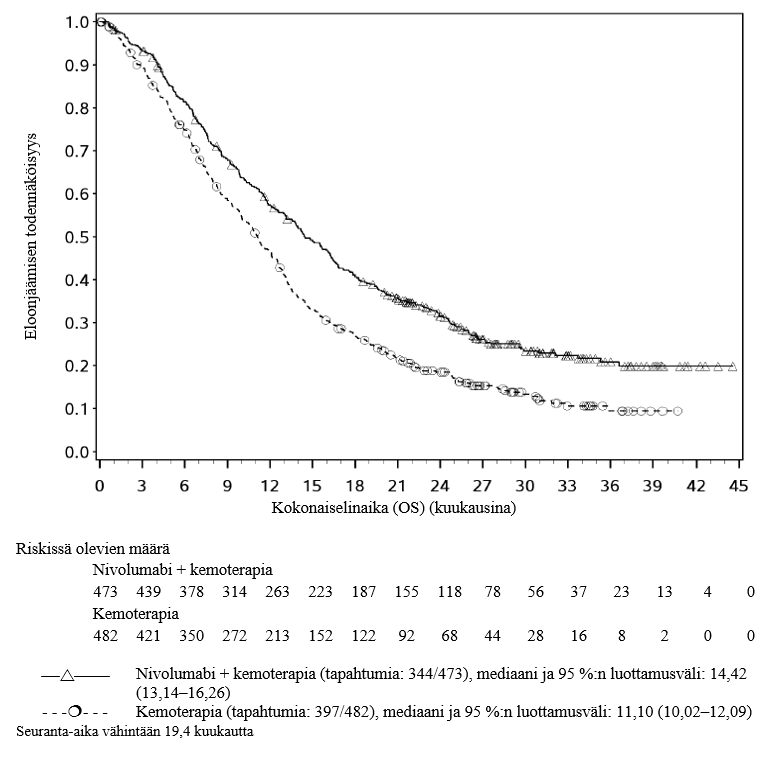
Kuva 36: Etenemisvapaan elinajan Kaplan–Meier kuvaaja (CA209649); potilaat, joiden PD‑L1:n CPS‑pistemäärä oli ≥ 5
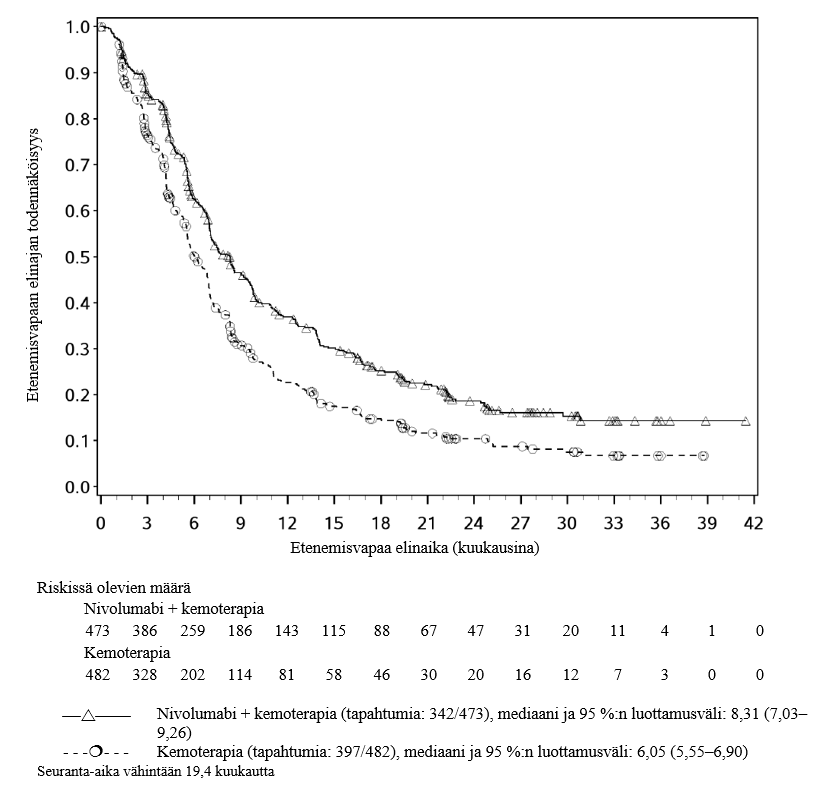
Hepatosellulaarinen karsinooma
Nivolumabin (1 mg/kg) ja ipilimumabin (3 mg/kg) yhdistelmän, jota annettiin 3 viikon välein enintään 4 annosta ja jonka jälkeen nivolumabia annettiin monoterapiana 480 mg 4 viikon välein, turvallisuutta ja tehoa leikkaukseen soveltumattoman tai edenneen hepatosellulaarisen karsinooman (HCC) ensilinjan hoidossa arvioitiin satunnaistetussa, aktiivikontrolloidussa, vaiheen 3 avoimessa tutkimuksessa (CA2099DW). Tutkimuksessa oli mukana aikuispotilaita (vähintään 18‑vuotiaita), joilla oli histologisesti vahvistettu hepatosellulaarinen karsinooma, Child‑Pugh-luokan A maksasairaus ja ECOG-toimintakykyluokka 0 tai 1 ja jotka eivät olleet saaneet aiemmin systeemistä hoitoa edenneeseen tautiin. Esofagogastroduodenoskopia ei ollut pakollinen ennen tutkimukseen ottamista. Tutkimukseen otettiin aikuisia, joiden tauti ei soveltunut hoidettavaksi leikkauksella ja/tai lokoregionaalisella hoidolla tai oli edennyt näiden hoitojen jälkeen. Aiemmat systeemiset esiliitännäis- tai adjuvanttihoidot olivat sallittuja. Tutkimuksesta suljettiin pois potilaat, joille oli tehty aiemmin maksansiirto tai joilla oli aktiivinen autoimmuunisairaus, aivometastaaseja tai leptomeningeaalisia metastaaseja, aiempi maksaperäinen enkefalopatia (12 kuukauden sisällä satunnaistamisesta), kliinisesti merkittävä askites, systeemistä immunosuppressiivista hoitoa vaativa tila, HIV-infektio tai aktiivinen samanaikainen hepatiitti B -virusinfektio (HBV) ja hepatiitti C -virusinfektio (HCV) tai HBV ja hepatiitti D -virusinfektio (HDV). Satunnaistaminen ositettiin etiologian (HBV vs. HCV vs. ei-virusperäinen), makrovaskulaarisen invaasion ja/tai maksan ulkopuolelle leviämisen (kyllä tai ei) sekä alfafetoproteiiniarvojen (≥ 400 tai < 400 ng/ml) perusteella.
Yhteensä 668 potilasta satunnaistettiin saamaan joko nivolumabi–ipilimumabi-yhdistemähoitoa (n = 335) tai tutkijan valitsemaa (n = 333) lenvatinibi- tai sorafenibihoitoa. Tutkijan valitsemaa hoitoa saaneessa ryhmässä 85 % potilaista sai lenvatinibia ja 15 % sorafenibia. Nivolumabin ja ipilimumabin yhdistelmää saavassa ryhmässä potilaat saivat nivolumabia 1 mg/kg ja ipilimumabia 3 mg/kg 3 viikon välein enintään 4 annosta. Tämän jälkeen nivolumabia annettiin monoterapiana 480 mg 4 viikon välein. Tutkijan valitsemaa hoitoa saaneessa ryhmässä potilaat saivat joko lenvatinibia 8 mg vuorokaudessa suun kautta (jos paino oli < 60 kg) tai 12 mg vuorokaudessa suun kautta (jos paino oli ≥ 60 kg) tai sorafenibia 400 mg kahdesti vuorokaudessa suun kautta. Hoitoa jatkettiin, kunnes tauti eteni tai kunnes ilmeni ei-hyväksyttävää toksisuutta tai enintään 24 kuukauden ajan. Potilaat, jotka eivät jatkaneet yhdistelmähoitoa ipilimumabin aiheuttaman haittavaikutuksen takia, saivat jatkaa pelkän nivolumabin käyttöä. Kasvaimet arvioitiin lähtötasolla, satunnaistamisen jälkeen viikolla 9 ja viikolla 16, sen jälkeen 8 viikon välein viikolle 48 saakka ja sen jälkeen 12 viikon välein taudin etenemiseen, hoidon lopettamiseen tai seuraavan hoidon aloittamiseen saakka.
Hoitoryhmien lähtötason ominaisuudet olivat pääosin tasapainossa. Iän mediaani oli 66 vuotta (vaihteluväli: 20–89). Potilaista 53 % oli ≥ 65‑vuotiaita ja 16 % oli ≥ 75‑vuotiaita. Potilaista 53 % oli valkoihoisia, 44 % aasialaisia, 2,2 % tummaihoisia ja 82 % miehiä. Lähtötason ECOG-toimintakykyluokka oli 0 (71 %) tai 1 (29 %). Potilaista 34 %:lla oli HBV-infektio, 28 %:lla oli HCV-infektio ja 36 %:lla ei ollut näyttöä HBV- tai HCV-infektiosta. Potilaista 19 %:lla oli alkoholin aiheuttama maksasairaus ja 11 %:lla alkoholiin liittymätön rasvamaksa. Suurimmalla osalla potilaista oli lähtötilanteessa BCLC-luokituksen mukainen asteen C tauti (73 %), 19 %:lla oli asteen B tauti ja 6 %:lla asteen A tauti. Child‑Pugh-pistearvo oli 77 %:lla potilaista 5, 20 %:lla potilaista 6 ja 3 %:lla potilaista ≥ 7. Kaikkiaan 54 %:lla potilaista tauti oli levinnyt maksan ulkopuolelle, 25 %:lla esiintyi makrovaskulaarista invaasiota ja 33 %:lla AFP-arvo oli ≥ 400 µg/l.
Tutkimus osoitti tilastollisesti merkitsevää hyötyä kokonaiselinajassa ja kokonaisvasteessa potilailla, jotka oli satunnaistettu saamaan nivolumabia yhdistelmähoitona ipilimumabin kanssa, verrattuna potilaisiin, jotka oli satunnaistettu saamaan tutkijan valitsemaa lenvatinibi- tai sorafenibihoitoa. Tulokset tehosta esitetään taulukossa 61 ja kuvassa 37.
Taulukko 61: Tulokset tehosta hepatosellulaarisen karsinooman ensilinjan hoidossa (CA2099DW)a
nivolumabi + ipilimumabi | lenvatinibi tai sorafenibi | |
|---|---|---|
Kokonaiselinaika | ||
Tapahtumia | 194 (58 %) | 228 (68 %) |
Mediaani (kuukautta) (95 %:n luottamusväli) | 23,7 (18,8–29,4) | 20,6 (17,5–22,5) |
Riskitiheyssuhde (95 %:n luottamusväli)b | 0,79 (0,65–0,96) | |
p‑arvoc | 0,0180 | |
Kokonaisvaste, n (%)d | 121 (36,1) | 44 (13,2) |
(95 %:n luottamusväli) | (31,0–41,5) | (9,8–17,3) |
p‑arvoe | < 0,0001 | |
Täydellinen vaste (%) | 23 (6,9) | 6 (1,8) |
Osittainen vaste (%) | 98 (29,3) | 38 (11,4) |
Vasteen kesto (kuukausina)d |
|
|
Mediaani (95 %:n luottamusväli) | 30,4 (21,2–N.A.) | 12,9 (10,2–31,2) |
a Seuranta-aika vähintään 26,8 kuukautta. Seurannan mediaani 35,2 kuukautta.
b Perustuu ositettuun Coxin suhteellisen vaaran malliin.
c Perustuu kaksipuoliseen ositettuun log‑rank‑merkitsevyystestiin. Tilastollisen merkitsevyyden p‑arvon raja ≤ 0,0257.
d BICR:n arvioima ja RECIST 1.1 -kriteerien mukainen.
e Perustuu kaksipuoliseen ositettuun Cochran-Mantel-Haenszelin testiin. Tilastollisen merkitsevyyden p‑arvon raja ≤ 0,025.
Kuva 37: Kokonaiselinajan Kaplan‑Meier-kuvaaja hepatosellulaarisen karsinooman ensilinjan hoidossa (CA2099DW)
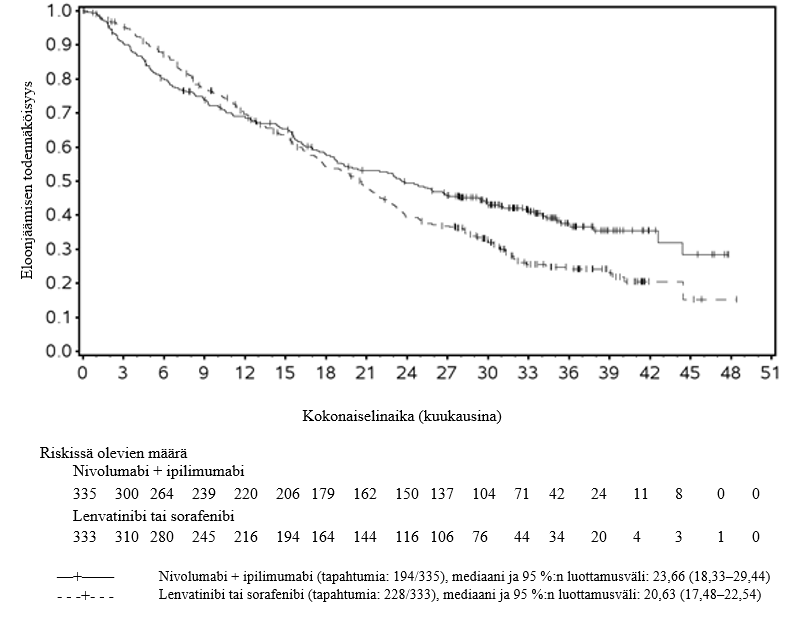
Pediatriset potilaat
Avoin vaiheen 1/2 tutkimus (CA209070)
CA209070-tutkimus oli avoin vaiheen 1/2 yksihaarainen annoksen varmistus- ja laajennustutkimus, jossa nivolumabia tutkittiin ainoana lääkkeenä ja yhdistelmähoitona ipilimumabin kanssa pediatrisilla potilailla ja nuorilla aikuispotilailla, joilla oli uusiutunut tai refraktaarinen kiinteä tai hematologinen kasvain, mukaan lukien neuroblastooma, osteosarkooma, rabdomyosarkooma, Ewingin sarkooma, edennyt melanooma, klassinen Hodgkinin lymfooma ja non-Hodgkin-lymfooma (NHL). Hoidetuista 126 potilaasta 97 oli pediatrisia potilaita (12 kuukauden ikäisistä < 18‑vuotiaisiin). Näistä 97 pediatrisesta potilaasta 64 sai nivolumabi-monoterapiaa (3 mg/kg annosteltuna 60 minuutin kuluessa laskimoon joka toinen viikko) ja 33 sai nivolumabi–ipilimumabi-yhdistelmähoitoa (nivolumabia 1 mg/kg tai 3 mg/kg annosteltuna 60 minuutin kuluessa laskimoon yhdessä ipilimumabiannoksen 1 mg/kg kanssa, joka annosteltiin 90 minuutin kuluessa laskimoon, joka kolmas viikko ensimmäiset neljä annosta, minkä jälkeen nivolumabi-monoterapiaa 3 mg/kg joka toinen viikko). Potilaat saivat joko nivolumabi-monoterapiaa kaksi annosta (mediaani, vaihteluväli: 1–89) tai nivolumabi–ipilimumabi-yhdistelmähoitoa kaksi annosta (mediaani, vaihteluväli: 1–24). Tärkeimmät ensisijaiset muuttujat olivat turvallisuus, siedettävyys ja kasvaimeen kohdistuva estovaikutus arvioituna deskriptiivisillä objektiivisen vasteen saavuttaneiden osuudella ja kokonaiselinajalla.
Nivolumabi-monoterapiaa saaneista 64 pediatrisesta potilaasta 60 potilaan vaste oli arvioitavissa (melanooma n = 1, kiinteitä kasvaimia n = 47 ja hematologisia kasvaimia n = 12). Niillä 48 pediatrisella potilaalla, joilla oli melanooma tai kiinteitä kasvaimia ja joiden vaste oli arvioitavissa, ei havaittu objektiivisia vasteita. Niillä 12 pediatrisella potilaalla, joilla oli hematologisia kasvaimia ja joiden vaste oli arvioitavissa, objektiivisen vasteen saavuttaneiden osuus oli 25,0 % (95 %:n luottamusväli: 5,5–57,2), mukaan lukien yksi täydellinen vaste klassista Hodgkinin lymfoomaa sairastaneella ja kaksi osittaista vastetta, joista toinen klassista Hodgkinin lymfoomaa sairastaneella ja toinen non-Hodgkin-lymfoomaa sairastaneella. Näiden nivolumabi-monoterapiaa saaneiden 64 pediatrisen potilaan deskriptiivisissä analyyseissä kokonaiselinajan mediaani oli 6,67 kuukautta (95 %:n luottamusväli: 5,98–NA), 6,14 kuukautta (95 %:n luottamusväli: 5,39–24,67) potilailla, joilla oli melanooma tai kiinteitä kasvaimia, eikä sitä saavutettu potilailla, joilla oli hematologisia kasvaimia.
Niillä 30 pediatrisella potilaalla, jotka saivat nivolumabi–ipilimumabi-yhdistelmähoitoa (vain muita kiinteitä kasvaimia kuin melanooma) ja joiden vaste oli arvioitavissa, ei havaittu objektiivisia vasteita. Nivolumabi–ipilimumabi-yhdistelmähoitoa saaneilla 33 pediatrisella potilaalla kokonaiselinajan mediaani oli 8,25 kuukautta (95 %:n luottamusväli: 5,45–16,95) deskriptiivisessä analyysissä.
Avoin vaiheen 1b/2 tutkimus (CA209908)
CA209908‑tutkimus oli avoin, sekventiaalisten hoitoryhminen, vaiheen 1b/2 kliininen tutkimus nivolumabi-monoterapiasta ja nivolumabin ja ipilimumabin yhdistelmähoidosta pediatrisilla potilailla ja nuorilla aikuispotilailla, joilla oli korkean asteen primaarinen keskushermoston syöpä, mukaan lukien diffuusi sisäinen aivosillan gliooma (DIPG), korkean asteen gliooma, medulloblastooma, ependymooma ja muut korkean asteen keskushermoston syövän uusiutuneet alatyypit (esim. pineoblastooma, epätyypillinen teratoidi-rhabdoidikasvain ja keskushermoston embryonaaliset kasvaimet). Tutkimukseen otetuista 151 pediatrisesta potilaasta (≥ 6 kuukauden ikäisistä < 18‑vuotiaisiin) 77:ää hoidettiin nivolumabi-monoterapialla (3 mg/kg joka toinen viikko) ja 74:ää hoidettiin nivolumabin ja ipilimumabin yhdistelmähoidolla (3 mg/kg nivolumabia, minkä jälkeen 1 mg/kg ipilimumabia joka kolmas viikko 4 annoksen ajan, minkä jälkeen nivolumabi‑monoterapiaa 3 mg/kg joka toinen viikko). Ensisijaiset tehokkuusmuuttujat olivat kokonaiselinaika DIPG‑kohortissa ja etenemisvapaa elinaika tutkijan arvioimana RANO‑kriteerien mukaan kaikissa muissa kasvaintyypeissä. Kokonaiselinajan mediaani DIPG‑kohortissa oli 10,97 kuukautta (80 %:n luottamusväli: 9,92–12,16) nivolumabi‑monoterapialla hoidetuilla potilailla ja 10,50 kuukautta (80 %:n luottamusväli: 9,10–12,32) nivolumabin ja ipilimumabin yhdistelmähoidolla hoidetuilla potilailla. Kaikkien muiden tutkimuksessa tutkittujen pediatristen keskushermoston kasvaintyyppien osalta etenemisvapaan elinajan mediaani vaihteli 1,23 kuukaudesta 2,35 kuukauteen nivolumabi‑monoterapialla hoidetuilla potilailla ja 1,45 kuukaudesta 3,09 kuukauteen nivolumabin ja ipilimumabin yhdistelmähoidolla hoidetuilla potilailla. Tutkimuksessa ei havaittu objektiivisia vasteita, paitsi yhdellä ependymoomaa sairastavalla, nivolumabi-monoterapialla hoidetulla potilaalla, joka sai osittaisen vasteen. CA209908‑tutkimuksessa havaitut tulokset kokonaiselinajassa, etenemisvapaassa elinajassa ja objektiivisen vasteen saavuttaneiden osuudessa eivät näytä antavan viitteitä kliinisesti merkittävästä edusta verrattuna siihen, mitä tällaisilta potilaspopulaatioilta voidaan muuten odottaa.
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset nivolumabin käytöstä pahanlaatuisten imukudoskasvainten hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Turvallisuus ja tehokkuus iäkkäillä potilailla
Kaikkiaan iäkkäiden (≥ 65 vuotta) ja nuorempien (< 65 vuotta) potilaiden välillä ei raportoitu olleen turvallisuus‑ eikä tehokkuuseroja. Yli 75‑vuotiaista ja sitä vanhemmista pään ja kaulan alueen levyepiteelisyöpää sairastavista potilaista, potilaista, jotka ovat saaneet nivolumabia liitännäishoitona melanoomaan, sekä potilaista, jotka ovat saaneet nivolumabia liitännäishoitona ruokatorvisyöpään tai ruokatorvi-mahalaukkurajan syöpään, on niin vähän tietoa, ettei ryhmää koskevia johtopäätöksiä voida tehdä. 65‑vuotiaista ja sitä vanhemmista klassista Hodgkinin lymfoomaa sairastavista potilaista on niin vähän tietoa, ettei ryhmää koskevia johtopäätöksiä voida tehdä. Tiedot keuhkopussin pahanlaatuista mesotelioomaa sairastavista potilaista osoittavat, että 75-vuotiailla ja sitä vanhemmilla on enemmän vakavia haittavaikutuksia (68 %) kuin kaikilla nivolumabi–ipilimumabi-yhdistelmähoitoa saaneilla potilailla (54 %) ja että tässä potilasryhmässä lääkitys keskeytään haittavaikutuksen takia useammin (35 %) kuin nivolumabi–ipilimumabi-yhdistelmähoitoa saaneiden ryhmässä yleisesti (28 %).
Farmakokinetiikka
Nivolumabi-monoterapia
Nivolumabin farmakokinetiikka on lineaarinen annosvälillä 0,1–10 mg/kg. Populaatiofarmakokineettisen analyysin perusteella nivolumabin puhdistuman geometrinen keskiarvo oli 7,9 ml/h, terminaalinen puoliintumisaika 25,0 vuorokautta ja vakaan tilan altistuksen keskiarvo, kun nivolumabia annosteltiin 3 mg/kg joka toinen viikko, 86,6 mikrog/ml.
Nivolumabin puhdistuma klassista Hodgkinin lymfoomaa sairastavilla potilailla oli noin 32 % vähäisempi kuin ei-pienisoluista keuhkosyöpää sairastavilla. Nivolumabin lähtötason puhdistuma oli sitä melanooman liitännäishoitona saavilla potilailla noin 40 % vähäisempi ja vakaan tilan puhdistuma noin 20 % vähäisempi kuin potilailla, jotka saivat nivolumabia edenneen melanooman hoitoon. Saatavilla olevien turvallisuustietojen valossa puhdistumien vähentyminen ei ollut kliinisesti merkittävää.
Nivolumabin aineenvaihduntareittiä ei ole selvitetty. Nivolumabin odotetaan hajoavan kataboliareittien kautta pieniksi peptideiksi ja aminohapoiksi samaan tapaan kuin endogeeninen IgG.
Nivolumabi yhdistelmähoitona ipilimumabin kanssa:
Kun nivolumabia annettiin 1 mg/kg yhdistelmähoitona ipilimumabin 3 mg/kg kanssa, nivolumabin puhdistuma kasvoi 29:llä prosentilla ja ipilimumabin 9:llä prosentilla, mitä ei pidetä kliinisesti merkityksellisenä. Kun nivolumabia annettiin 3 mg/kg yhdistelmähoitona ipilimumabin 1 mg/kg kanssa, nivolumabin puhdistuma kasvoi yhdellä prosentilla ja ipilimumabin puhdistuma väheni 1,5:llä prosentilla. Näitä ei pidetä kliinisesti merkityksellisinä.
Yhdistelmähoidossa ipilimumabin kanssa nivolumabin puhdistuma kasvoi 20:lla prosentilla nivolumabin vasta-aineiden läsnäollessa ja ipilimumabin puhdistuma kasvoi 5,7:llä prosentilla ipilimumabin vasta-aineiden läsnäollessa. Näitä muutoksia ei pidetä kliinisesti merkityksellisinä.
Nivolumabi yhdistelmähoitona ipilimumabin ja kemoterapian kanssa
Kun nivolumabia (360 mg joka kolmas viikko) annettiin yhdistelmähoitona ipilimumabin (1 mg/kg joka kuudes viikko) ja kahden kemoterapiajakson kanssa, nivolumabin puhdistuma vähentyi noin 10 %:lla ja ipilimumabin puhdistuma kasvoi noin 22 %:lla. Näitä muutoksia ei pidetty kliinisesti merkityksellisinä.
Erityisryhmät
Populaatiofarmakokineettisen analyysin perusteella ikä, sukupuoli, rotu, kiinteä kasvaintyyppi, kasvaimen koko tai maksan vajaatoiminta eivät näytä vaikuttavan nivolumabin puhdistumaan. Vaikka ECOG-luokka, lähtötason glomerulusten suodattumisnopeus (GFR), albumiini, paino ja lievä maksan vajaatoiminta vaikuttivat nivolumabin puhdistumaan, vaikutus ei ollut kliinisesti merkittävä.
Pediatriset potilaat
Nivolumabi-monoterapiassa 12‑vuotiaiden ja sitä vanhempien vähintään 50 kg painavien nuorten nivolumabialtistusten oletetaan vastaavan aikuispotilaiden vastaavia suositellulla annoksella. Painoon perustuvaa annostusta suositellaan 12‑vuotiaille ja sitä vanhemmille nuorille, jotka painavat alle 50 kg.
Nivolumabi–ipilimumabi-yhdistelmähoidossa 12‑vuotiaiden ja sitä vanhempien nuorten nivolumabi- ja ipilimumabialtistusten oletetaan vastaavan aikuispotilaiden vastaavia suositellulla annoksella.
Samoin kuin aikuispotilaiden ryhmässä nivolumabin puhdistuma oli pienempi pediatrisilla cHL‑potilailla kuin potilailla, joilla oli kiinteitä kasvaimia. Puhdistuman pienenemä oli suuruudeltaan samaa luokkaa pediatrisilla ja aikuisilla cHL‑potilailla. Nivolumabialtistukset olivat samaa luokkaa (geometristen keskiarvojen ero < 20 %) pediatrisilla cHL‑potilailla, jotka saivat nivolumabia 3 mg/kg i.v. 3 viikon välein, ja aikuisilla cHL‑potilailla, jotka saivat nivolumabia 3 mg/kg i.v. 3 viikon välein, tai aikuispotilailla, joilla oli kiinteitä kasvaimia ja jotka saivat nivolumabia 3 mg/kg i.v. 2 viikon välein.
Munuaisten vajaatoiminta
Populaatiofarmakokineettisissä analyyseissä arvioitiin munuaisten vajaatoiminnan vaikutusta nivolumabin puhdistumaan potilailla, joilla oli lievä (GFR < 90 ja ≥ 60 ml/min/1,73 m2; n = 379), kohtalainen (GFR < 60 ja ≥ 30 ml/min/1,73 m2; n = 179) tai vaikea (GFR < 30 ja ≥ 15 ml/min/1,73 m2; n = 2) munuaisten vajaatoiminta, verrattuna potilaisiin, joiden munuaisten toiminta oli normaali (GFR ≥ 90 ml/min/1,73 m2; n = 342). Nivolumabin puhdistumassa ei todettu kliinisesti merkittäviä eroja lievää tai kohtalaista munuaisten vajaatoimintaa sairastavien ja niiden potilaiden välillä, joiden munuaiset toimivat normaalisti. Tiedot munuaisten vajaatoimintaa sairastavista potilaista ovat liian rajallisia, jotta voitaisiin tehdä johtopäätöksiä tästä potilasryhmästä (ks. kohta Annostus ja antotapa).
Maksan vajaatoiminta
Populaatiofarmakokineettisessä analyysissä arvioitiin maksan vajaatoiminnan vaikutusta nivolumabin puhdistumaan potilailla, joilla oli eri tyyppisiä kasvaimia (NSCLC, SCLC, melanooma, RCC, SCCHN, UC, GC ja cHL) ja lievä maksan vajaatoiminta (National Cancer Instituten maksan vajaatoiminnan kriteerien mukaisesti kokonaisbilirubiini 1,0–1,5 × ULN tai ASAT > ULN; n = 351), sekä potilailla, joilla oli kohtalainen maksan vajaatoiminta (kokonaisbilirubiini > 1,5–3 × ULN ja mikä tahansa ASAT-arvo; n = 10), potilaisiin, joiden maksan toiminta oli normaali (kokonaisbilirubiini ja ASAT ≤ ULN; n = 3 096). Nivolumabin puhdistumassa ei todettu kliinisesti merkittäviä eroja lievää tai kohtalaista maksan vajaatoimintaa sairastavien ja niiden potilaiden välillä, joiden maksa toimi normaalisti. Samankaltaisia tuloksia todettiin potilailla, joilla oli hepatosellulaarinen karsinooma (lievä maksan vajaatoiminta: n = 152; kohtalainen maksan vajaatoiminta: n = 13). Nivolumabin käyttöä vaikeaa maksan vajaatoimintaa (kokonaisbilirubiini > 3 × ULN ja mikä tahansa ASAT-arvo) hoidossa ei ole tutkittu (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
PD-L1-signaloinnin eston on hiirimalleissa todettu huonontavan sikiön sietämistä ja lisäävän sikiökuolemia. Nivolumabin vaikutusta pre- ja postnataaliseen kehitykseen arvioitiin apinoilla, joille annettiin nivolumabia 2 kertaa viikossa organogeneesin alusta ensimmäisellä raskauskolmanneksella synnytykseen asti. Altistus oli 8 tai 35 kertaa suurempi kuin on todettu käytettäessä kliinisesti 3 mg/kg nivolumabia (AUC-arvon perusteella). Kolmannesta raskauskolmanneksesta lähtien todettiin annoksen suuruudesta riippuvaa sikiökuolemien lisääntymistä sekä vastasyntyneiden kuolleisuuden lisääntymistä.
Muut nivolumabihoitoa saaneiden naarasapinoiden jälkeläiset säilyivät hengissä suunniteltuun keskeytykseen saakka ilman hoitoon liittyviä kliinisiä merkkejä, normaalin kehityksen muutoksia, vaikutuksia elinten painoon, makro- tai mikroskooppisia patologisia muutoksia. Tulokset olivat kasvuindeksien sekä teratogeenisten, neurobehavioraalisten, immunologisten ja kliinispatologisten parametrien perusteella koko 6 kuukauden postnataalivaiheen ajan vastaavat kuin verrokkiryhmässä. Vaikutusmekanismin perusteella sikiön altistuminen nivolumabille voi kuitenkin lisätä immuunivälitteisten sairauksien tai normaalin immuunivasteen muutosten riskiä, ja PD-1-poistogeenisillä hiirillä on raportoitu esiintyneen immuunivälitteisiä sairauksia.
Nivolumabin vaikutusta hedelmällisyyteen ei ole tutkittu.
Farmaseuttiset tiedot
Apuaineet
Natriumsitraattidihydraatti, natriumkloridi, mannitoli (E421), pentetiinihappo (dietyleenitriamiinipentaetikkahappo), polysorbaatti 80 (E433), natriumhydroksidi (pH:n säätämiseen), kloorivetyhappo (pH:n säätämiseen), injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa. OPDIVO-valmistetta ei saa infusoida samaan aikaan saman laskimoletkun kautta kuin muita lääkevalmisteita.
Kestoaika
Avaamaton injektiopullo
3 vuotta
Infuusion valmistamisen jälkeen
Kemiallinen ja fysikaalinen käytönaikainen säilyvyys valmistamisajankohdan jälkeen on osoitettu seuraavasti (aikoihin on sisällytetty valmisteen antoaika):
Infuusion valmistaminen | Kemiallinen ja fysikaalinen käytönaikainen säilyvyys | |
Säilytys 2–8ºC:ssa valolta suojattuna | Säilytys huoneenlämmössä (≤ 25 °C) ja huoneenvalossa | |
Laimentamaton tai laimennettu 9 mg/ml:n vahvuisella (0,9 %) natriumkloridi‑injektioliuoksella | 30 vrk | 24 tuntia |
Laimennettu 50 mg/ml:n (5 %) vahvuisella glukoosi‑injektioliuoksella | 7 päivää | 8 tuntia |
Valmistettu infuusioliuos on mikrobiologiselta kannalta käytettävä välittömästi riippumatta käytetystä laimentimesta. Ellei liuosta käytetä heti, säilytysajat ja ‑olosuhteet ennen käyttöä ovat käyttäjän vastuulla, eikä niiden normaalisti tulisi ylittää 7:ää päivää 2–8 °C:ssa tai 8:aa tuntia (yhteensä 7 päivän säilytyksestä) huoneenlämmössä (≤ 25 °C). Infuusion valmistuksessa on toimittava aseptisesti (ks. kohta Käyttö- ja käsittelyohjeet).
Säilytys
Säilytä jääkaapissa (2 °C–8 °C).
Ei saa jäätyä.
Säilytä alkuperäispakkauksessa. Herkkä valolle.
Avaamattoman injektiopullon voi säilyttää kontrolloidussa huoneenlämmössä enintään 25 °C:ssa huoneenvalossa enintään 48 tunnin ajan.
Valmistetun infuusion säilytys, ks. kohta Kestoaika
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
OPDIVO infuusiokonsentraatti, liuosta varten
10 mg/ml (L:ei) 4 ml (769,10 €), 10 ml (1856,13 €), 12 ml (2193,64 €), 24 ml (4206,67 €)
PF-selosteen tieto
4 ml konsentraattia 10 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (päällystettyä butyylikumia) ja tummansininen irti napsautettava (flip-off) korkki (alumiinia). Pakkauksessa on 1 injektiopullo.
10 ml konsentraattia 10 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (päällystettyä butyylikumia) ja harmaa irti napsautettava (flip-off) korkki (alumiinia). Pakkauksessa on 1 injektiopullo.
12 ml konsentraattia 25 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (päällystettyä butyylikumia) ja sininen irti napsautettava (flip‑off) korkki (alumiinia). Pakkauksessa on 1 injektiopullo.
24 ml konsentraattia 25 ml:n injektiopullossa (tyypin I lasia), jossa on tulppa (päällystettyä butyylikumia) ja matanpunainen irti napsautettava (flip-off) korkki (alumiinia). Pakkauksessa on 1 injektiopullo.
Valmisteen kuvaus:
Kirkas tai opalisoiva, väritön tai vaaleankeltainen neste , joka saattaa sisältää joitakin vaaleita hiukkasia. Liuoksen pH-arvo on noin 6,0 ja osmolaalisuus noin 340 mOsm/kg.
Käyttö- ja käsittelyohjeet
Koulutetun henkilökunnan on valmistettava lääke käyttökuntoon noudattaen erityisesti aseptiikkaan liittyviä hyviä toimintatapoja.
Valmistaminen ja annostelu
Annoksen laskeminen
Potilaan tarvitsemaan kokonaisannokseen voidaan tarvita enemmän kuin yksi injektiopullollinen OPDIVO‑konsentraattia.
Nivolumabi monoterapiana
Lääkäri määrää aikuispotilaalle annoksen 240 mg tai 480 mg painosta riippumatta käyttöaiheen mukaan (ks. kohta Annostus ja antotapa).
Melanooma (edennyt melanooma tai melanooman liitännäishoitona) nuorilla. Lääkäri määrää 12‑vuotiaille ja sitä vanhemmille vähintään 50 kg painaville nuorille annoksen 240 mg tai 480 mg. Lääkäri määrää 12‑vuotiaille ja sitä vanhemmille nuorille, jotka painavat alle 50 kg, annoksen milligrammoina painokiloa kohti. Annosteltava kokonaisannos lasketaan määrätyn annoksen perusteella.
-
Nivolumabin kokonaisannos milligrammoina = potilaan paino kiloina × lääkärin määräämä annos, mg/kg.
-
Annoksen (ml) valmistamiseen tarvittava määrä OPDIVO-konsentraattia = nivolumabin kokonaisannos milligrammoina jaettuna kymmenellä (OPDIVO-konsentraatin vahvuus on 10 mg/ml).
Nivolumabi yhdistelmähoitona ipilimumabin kanssa
Lääkäri määrää potilaalle annoksen milligrammoina painokiloa kohti. Annosteltava kokonaisannos lasketaan määrätyn annoksen perusteella (ks. edellä).
Nivolumabi yhdistelmähoitona ipilimumabin kanssa keuhkopussin pahanlaatuisen mesoteliooman hoidossa
Potilaalle määrätty annos on 360 mg kehonpainosta riippumatta.
Nivolumabi yhdistelmähoitona ipilimumabin kanssa edenneen kolorektaalisyövän hoidossa
Potilaalle määrätty annos voi perustua kehonpainoon (3 mg/kg) tai olla 240 mg kehonpainosta riippumatta.
Nivolumabi yhdistelmähoitona ipilimumabin kanssa ruokatorven levyepiteelikarsinooman hoidossa
Potilaalle määrätty annos voi perustua kehonpainoon (3 mg/kg) tai olla 360 mg kehonpainosta riippumatta.
Nivolumabi yhdistelmähoitona kemoterapian kanssa kirurgisesti poistettavissa olevan ei‑pienisoluisen keuhkosyövän hoidossa
Potilaalle määrätty annos on 360 mg kehonpainosta riippumatta.
Nivolumabi yhdistelmähoitona kemoterapian kanssa ruokatorven levyepiteelikarsinooman hoidossa
Potilaalle määrätty annos on 240 mg tai 480 mg kehonpainosta riippumatta.
Nivolumabi yhdistelmähoitona kemoterapian kanssa mahalaukun, ruokatorvi-mahalaukkurajan tai ruokatorven adenokarsinooman hoidossa
Potilaalle määrätty annos on 360 mg tai 240 mg kehonpainosta riippumatta.
Nivolumabi yhdistelmähoitona ipilimumabin ja kemoterapian kanssa
Potilaalle määrätty annos on 360 mg kehonpainosta riippumatta.
Nivolumabi yhdistelmähoitona kabotsantinibin kanssa
Potilaalle määrätty annos on 240 mg tai 480 mg nivolumabia kehonpainosta riippumatta.
Nivolumabi yhdistelmähoitona brentuksimabivedotiinin kanssa
Lääkäri määrää potilaalle annoksen milligrammoina painokiloa kohti. Annosteltava kokonaisannos lasketaan määrätyn annoksen perusteella (ks. edellä).
Infuusion valmistaminen
Muista toimia aseptisesti valmistaessasi infuusion.
OPDIVO-valmisteen voi annostella laskimoon joko:
-
laimentamatta, kun se on siirretty sopivalla steriilillä ruiskulla infuusiosäiliöön tai
-
laimennettuna seuraavien ohjeiden mukaan:
Lopullisen infuusion pitoisuuden tulee olla 1–10 mg/ml.
Infuusion kokonaismäärä ei saa ylittää 160 ml:aa. Jos potilas painaa alle 40 kg, infuusion kokonaismäärä ei saa ylittää 4 ml:aa per potilaan yksi painokilo.
-
OPDIVO-konsentraatin laimentamiseen voi käyttää joko
9 mg/ml:n (0,9 %) vahvuista natriumkloridi-injektioliuosta tai
50 mg/ml:n (5 %) vahvuista glukoosi-injektioliuosta.
VAIHE 1
-
Tarkista onko OPDIVO-konsentraatissa hiukkasia tai värjäymiä. Älä ravista injektiopulloa. OPDIVO-konsentraatti on kirkasta tai opalisoivaa, väritöntä tai vaaleankeltaista nestettä. Hylkää injektiopullo, jos liuos on sameaa, värjäytynyttä, tai sisältää muunlaisia hiukkasia kuin muutaman väriltään läpinäkyvän tai vaalean hiukkasen.
-
Ota sopivaan steriiliin ruiskuun tarvittava määrä OPDIVO-konsentraattia.
VAIHE 2
-
Siirrä konsentraatti steriiliin, lasiseen tyhjiöpulloon tai liuospussiin(PVC:tä tai polyolefiinia).
-
Laimenna tarvittaessa tarpeellisella määrällä 9 mg/ml:n (0,9 %) vahvuista natriumkloridi-injektioliuosta tai 50 mg/ml:n (5 %) vahvuista glukoosi-injektioliuosta. Valmistamisen helpottamiseksi konsentraatin voi myös siirtää suoraan esitäytettyyn pussiin, jossa on oikea määrä 9 mg/ml:n (0,9 %) vahvuista natriumkloridi-injektioliuosta tai 50 mg/ml:n (5 %) vahvuista glukoosi-injektioliuosta.
-
Sekoita infuusio varovasti käsin pyörittelemällä. Älä ravista.
Annostelu
OPDIVO-infuusiota ei saa antaa laskimoon nopeana injektiona eikä boluksena.
Annostele OPDIVO-infuusio laskimoon 30 tai 60 minuutin kuluessa annoksesta riippuen.
OPDIVO-infuusiota ei saa infusoida samaan aikaan saman laskimoletkun kautta kuin muita aineita. Käytä infuusiolle erillistä infuusioletkua.
Käytä infuusiolaitetta ja linjassa steriiliä, ei-pyrogeenistä, vähän proteiineja sitovaa suodatinta (huokoskoko 0,2–1,2 mikrom).
OPDIVO-infuusio on yhteensopiva seuraavien kanssa: PVC-säiliöt ja polyolefiinisäiliöt, lasipullot, PVC-infuusiolaitteet ja linjasuodattimet, joiden polyeetterisulfonikalvon huokoskoko on 0,2–1,2 mikrom.
Nivolumabin annostelun jälkeen huuhdo linja 9 mg/ml:n (0,9 %) vahvuisella natriumkloridi-injektioliuoksella tai 50 mg/ml:n (5 %) vahvuisella glukoosi-injektioliuoksella.
Hävittäminen
Älä säilytä käyttämätöntä osaa infuusioliuoksesta uutta käyttöä varten. Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
OPDIVO infuusiokonsentraatti, liuosta varten
10 mg/ml 4 ml, 10 ml, 12 ml, 24 ml
- Ei korvausta.
ATC-koodi
L01FF01
Valmisteyhteenvedon muuttamispäivämäärä
28.05.2026
Yhteystiedot
09 2512 1244
www.bms.com/fi
medinfo.finland@bms.com