
SYNJARDY tabletti, kalvopäällysteinen 5/850 mg, 5/1000 mg, 12,5/850 mg, 12,5/1000 mg
Vaikuttavat aineet ja niiden määrät
Synjardy 5 mg/850 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää 5 mg empagliflotsiinia ja 850 mg metformiinihydrokloridia.
Synjardy 5 mg/1 000 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää 5 mg empagliflotsiinia ja 1 000 mg metformiinihydrokloridia.
Synjardy 12,5 mg/850 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää 12,5 mg empagliflotsiinia ja 850 mg metformiinihydrokloridia.
Synjardy 12,5 mg/1 000 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää 12,5 mg empagliflotsiinia ja 1 000 mg metformiinihydrokloridia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti)
Kliiniset tiedot
Käyttöaiheet
Synjardy on tarkoitettu ruokavalion ja liikunnan ohella tyypin 2 diabetesta sairastavien aikuisten ja vähintään 10-vuotiaiden lasten hoitoon
- kun potilaan sairaus ei pysy riittävän hyvin hallinnassa metformiinin suurimmalla siedetyllä annoksella yksinään
- yhdessä muiden diabeteksen hoitoon tarkoitettujen lääkkeiden kanssa potilailla, joiden sairaus ei pysy riittävän hyvin hallinnassa metformiinilla ja näillä lääkkeillä
- kun potilas käyttää jo empagliflotsiinin ja metformiinin yhdistelmää erillisinä tabletteina.
Tutkimustulokset eri yhdistelmähoidoista, vaikutuksista glukoositasapainoon ja sydän- ja verisuonitapahtumiin sekä tutkimuspopulaatioista on luettavissa kohdissa Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakodynamiikka.
Annostus ja antotapa
Annostus
Aikuiset, joiden munuaistoiminta on normaali (eGFR ≥ 90 ml/min/1,73 m2)
Suositeltu annos on yksi tabletti kahdesti vuorokaudessa. Annostus on yksilöllistettävä potilaan nykyisen hoidon, tehon ja siedettävyyden perusteella käyttäen suositeltua 10 mg tai 25 mg empagliflotsiinivuorokausiannosta, ylittämättä kuitenkaan metformiinin suositeltua enimmäisvuorokausiannosta.
Potilaat, joiden hoitotasapaino on riittämätön, kun käytössä on pelkkä metformiini tai metformiini yhdessä muiden diabeteksen hoitoon tarkoitettujen lääkkeiden kanssa
Potilaille, joiden hoitotasapaino on riittämätön, kun käytössä on pelkkä metformiini tai metformiini yhdessä muiden diabeteksen hoitoon tarkoitettujen lääkkeiden kanssa, suositeltu Synjardy-aloitusannos on 5 mg empagliflotsiinia kahdesti vuorokaudessa (10 mg vuorokausiannos) ja metformiiniannos sama kuin entuudestaan käytössä oleva annos. Potilailla, jotka sietävät empagliflotsiinin 10 mg kokonaisvuorokausiannoksen ja jotka tarvitsevat parempaa glukoositasapainoa, empagliflotsiinin kokonaisvuorokausiannos voidaan nostaa 25 mg:aan.
Kun Synjardy-valmistetta käytetään yhdessä sulfonyyliurean ja/tai insuliinin kanssa, pienempi sulfonyyliurea- ja/tai insuliiniannos voi olla tarpeen hypoglykemiariskin pienentämiseksi (ks. kohdat Yhteisvaikutukset ja Haittavaikutukset).
Potilaat, jotka käyttävät empagliflotsiinin ja metformiinin yhdistelmää erillisinä tabletteina
Potilaiden, jotka vaihtavat erillisistä empagliflotsiinitableteista (10 mg tai 25 mg kokonaisvuorokausiannos) ja metformiinitableteista Synjardy-valmisteeseen, on käytettävä samaa empagliflotsiini- ja metformiinivuorokausiannosta kuin aiemmin tai lähintä terapeuttisesti sopivaa metformiiniannosta (saatavilla olevat vahvuudet, ks. kohta Vaikuttavat aineet ja niiden määrät).
Unohtunut annos
Jos annos jää väliin, se on otettava heti, kun potilas muistaa asian; kaksinkertaista annosta ei kuitenkaan saa ottaa kerralla. Tässä tapauksessa unohtunut annos on jätettävä väliin.
Erityisryhmät
Munuaisten vajaatoiminta
Empagliflotsiinin glukoositasapainoa parantava teho riippuu munuaisten toiminnasta. Kardiovaskulaarisen riskin vähentämiseksi tavanomaisen hoidon lisänä on käytettävä empagliflotsiinin 10 mg:n vuorokausiannosta potilaille, joiden eGFR on alle 60 ml/min/1,73 m2 (ks. taulukko 1). Empagliflotsiinin glukoosipitoisuutta alentava teho on heikentynyt potilailla, joilla on kohtalainen munuaisten vajaatoiminta, ja todennäköisesti puuttuu kokonaan potilailta, joilla on vaikea munuaisten vajaatoiminta. Jos glykeeminen lisähallinta on tarpeen, näiden potilaiden hoitoon on harkittava muiden veren glukoosipitoisuutta alentavien lääkeaineiden lisäämistä.
Suositukset annoksen muuttamisesta eGFR- tai CrCl-arvon perusteella on esitetty taulukossa 1.
eGFR-arvo on arvioitava ennen metformiinia sisältävien valmisteiden käytön aloittamista ja vähintään kerran vuodessa sen jälkeen. Jos munuaisten vajaatoiminnan etenemisriski on suurentunut tai kyseessä on iäkäs potilas, munuaistoiminta on arvioitava tiheämmin, esim. 3–6 kuukauden välein.
Mikäli Synjardy-valmisteesta ei ole saatavilla asianmukaista vahvuutta, vaikuttavia aineita on käytettävä erillisinä valmisteina kiinteän yhdistelmävalmisteen sijasta.
Annossuositukset pediatrisille potilaille, ks. pediatrisia potilaita koskeva kohta alla.
Taulukko 1: Annostus munuaisten vajaatoimintaa sairastavilla aikuispotilaillaa
eGFR [ml/min/1,73 m²] tai CrCl [ml/min] | Metformiini | Empagliflotsiini |
≥ 60 | Enimmäisvuorokausiannos on 3 000 mg. Voidaan harkita annoksen pienentämistä suhteessa munuaistoiminnan heikkenemiseen. | Aloitusannos on 10 mg. |
45 – < 60 | Enimmäisvuorokausiannos on 2 000 mg. Aloitusannos on enintään puolet enimmäisannoksesta. | Aloitusannos on 10 mg.b |
30 – < 45 | Enimmäisvuorokausiannos on 1 000 mg Aloitusannos on enintään puolet enimmäisannoksesta. | Aloitusannos on 10 mg.b |
< 30 | Metformiini on vasta-aiheinen. | Empagliflotsiinia ei suositella. |
a Ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka
b Potilaat, joilla on tyypin 2 diabetes ja todettu sydän- ja verisuonitauti
Maksan vajaatoiminta
Tätä lääkevalmistetta ei saa käyttää, jos potilaalla on maksan vajaatoiminta (ks. kohdat Vasta-aiheet, Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Iäkkäät
Vaikutusmekanismista johtuen heikentynyt munuaistoiminta johtaa empagliflotsiinin glykeemisen tehon heikentymiseen. Metformiini erittyy munuaisten kautta ja iäkkäillä potilailla munuaisten vajaatoiminta on todennäköisempää, joten Synjardy-valmistetta on käytettävä varoen näille potilaille. Munuaistoimintaa on seurattava metformiiniin liittyvän maitohappoasidoosin riskin vuoksi etenkin iäkkäillä potilailla (ks. kohdat Vasta-aiheet ja Varoitukset ja käyttöön liittyvät varotoimet). 75‑vuotiailla ja sitä vanhemmilla potilailla on otettava huomioon suurempi nestehukan riski (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Pediatriset potilaat
Annostus määritetään yksilöllisesti potilaan senhetkisen hoidon, tehokkuuden ja siedettävyyden perusteella.
Jos metformiinia saavan potilaan hoitoon lisätään empagliflotsiini, metformiiniannos on pidettävä samana kuin aiemmin. Suositeltu aloitusannos on 5 mg empagliflotsiinia kahdesti vuorokaudessa (kokonaisvuorokausiannos 10 mg). Potilailla, jotka sietävät empagliflotsiiniannosta 5 mg kahdesti vuorokaudessa ja jotka tarvitsevat parempaa glukoositasapainoa, annos voidaan nostaa 12,5 mg:aan kahdesti vuorokaudessa (kokonaisvuorokausiannos 25 mg).
Potilaiden, jotka vaihtavat erillisistä empagliflotsiini- ja metformiinitableteista Synjardy-valmisteeseen, on käytettävä samaa empagliflotsiini- ja metformiinivuorokausiannosta kuin aiemmin tai lähintä terapeuttisesti sopivaa metformiiniannosta.
Synjardy-valmisteen suurin suositeltu vuorokausiannos on 25 mg empagliflotsiinia ja 2 000 mg metformiinia (ks. yleiset tiedot yllä kohdassa Annostus ja antotapa). Tietoja ei ole saatavilla lapsista, joiden eGFR‑arvo on < 60 ml/min/1,73 m2, eikä alle 10‑vuotiaista lapsista.
Antotapa
Synjardy otetaan kahdesti vuorokaudessa aterioiden yhteydessä metformiinin käyttöön liittyvien ruuansulatuskanavan haittojen vähentämiseksi. Niele tabletit kokonaisina veden kanssa. Kaikkien potilaiden on jatkettava ruokavalionsa noudattamista siten, että hiilihydraattien saanti jakautuu tasaisesti päivän aikana. Ylipainoisten potilaiden on jatkettava vähäenergisen ruokavalion noudattamista.
Vasta-aiheet
-
Yliherkkyys vaikuttavalle aineelle (vaikuttaville aineille) tai kohdassa Apuaineet mainituille apuaineille.
-
Akuutti metabolinen asidoosi tyypistä riippumatta (esim. maitohappoasidoosi, diabeettinen ketoasidoosi) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
-
Diabeettinen prekooma.
-
Vaikea munuaisten vajaatoiminta (eGFR < 30 ml/min/1,73 m2) (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
-
Akuutit tilat, jotka saattavat vaikuttaa munuaistoimintaan, esim. nestehukka, vaikea infektio tai sokki (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
-
Sairaus, joka voi aiheuttaa kudosten hypoksiaa (erityisesti akuutti sairaus tai paheneva krooninen sairaus), esim. dekompensoitu sydämen vajaatoiminta, hengitysvajaus, äskettäin sairastettu sydäninfarkti, sokki (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
-
Maksan vajaatoiminta, akuutti alkoholi-intoksikaatio, alkoholismi (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Varoitukset ja käyttöön liittyvät varotoimet
Yleistä tietoa
Empagliflotsiinia ei pidä käyttää tyypin 1 diabetesta sairastaville potilaille (ks. ”Diabeettinen ketoasidoosi” kohdassa Varoitukset ja käyttöön liittyvät varotoimet).
Maitohappoasidoosi
Maitohappoasidoosi on hyvin harvinainen, mutta vakava metabolinen komplikaatio, jota ilmenee useimmiten munuaistoiminnan akuutin heikkenemisen yhteydessä tai kardiorespiratorisen sairauden tai sepsiksen yhteydessä. Munuaistoiminnan akuutin heikkenemisen yhteydessä metformiinia kertyy elimistöön, mikä suurentaa maitohappoasidoosin riskiä.
Nestehukan (vaikea ripuli tai oksentelu, kuume tai vähentynyt nesteen saanti) yhteydessä metformiinin käyttö on tauotettava, ja on suositeltavaa ottaa yhteys terveydenhuoltohenkilöstöön.
Jos potilas saa metformiinihoitoa, munuaistoimintaa potentiaalisesti heikentävien lääkevalmisteiden (kuten verenpainelääkkeiden, diureettien ja tulehduskipulääkkeiden) käyttö on aloitettava varoen. Muita maitohappoasidoosin riskitekijöitä ovat liiallinen alkoholinkäyttö, maksan vajaatoiminta, huonossa hoitotasapainossa oleva diabetes, ketoosi, pitkittynyt paasto ja kaikki tilat, joihin liittyy hypoksiaa, sekä maitohappoasidoosia potentiaalisesti aiheuttavien lääkevalmisteiden samanaikainen käyttö (ks. kohdat Vasta-aiheet ja Yhteisvaikutukset).
Potilaille ja/tai hoitajille on kerrottava maitohappoasidoosin riskistä. Maitohappoasidoosin tyyppioireita ovat asidoottinen hengenahdistus, vatsakipu, lihaskrampit, voimattomuus ja hypotermia. Tilan edetessä kehittyy kooma. Jos potilaalla epäillään näitä oireita, hänen on lopetettava metformiinin käyttö ja hakeuduttava välittömästi lääkärin hoitoon. Diagnostisia laboratoriolöydöksiä ovat veren matala pH (< 7,35), suurentunut plasman laktaattipitoisuus (> 5 mmol/l), suurentunut anionivaje ja suurentunut laktaatti-pyruvaattisuhde.
Potilaat, joilla on tai joilla epäillään olevan mitokondriotauteja
Potilaille, joilla tiedetään olevan mitokondriotauteja kuten MELAS (Mitochondrial Encephalopathy with Lactic Acidosis, and Stroke-like episodes) -oireyhtymä ja MIDD (Maternal inherited diabetes and deafness) -diabetes, ei suositella metformiinia maitohappoasidoosin pahenemisen ja mahdollisesti taudin pahenemiseen johtavien neurologisten komplikaatioiden vaaran vuoksi.
Jos metformiinin ottamisen jälkeen ilmenee MELAS-oireyhtymään tai MIDD-diabetekseen viittaavia merkkejä tai oireita, metformiinihoito on keskeytettävä välittömästi ja diagnostinen arviointi tehtävä.
Diabeettinen ketoasidoosi
Harvinaisia, toisinaan henkeä uhanneita tai kuolemaan johtaneita diabeettisen ketoasidoosin tapauksia on raportoitu potilailla, joita on hoidettu SGLT2:n estäjillä, mukaan lukien empagliflotsiini. Diabeettinen ketoasidoosi oli monissa näistä tapauksista epätyypillistä siten, että verensokeripitoisuuksien havaittiin olevan vain kohtalaisesti suurentuneet, alle 14 mmol/l (250 mg/dl). Ei tiedetä, onko diabeettisen ketoasidoosin ilmeneminen todennäköisempää suuremmilla empagliflotsiiniannoksilla.
Jos potilaalla on epäspesifisiä oireita, kuten pahoinvointia, oksentelua, ruokahaluttomutta, vatsakipua, voimakasta janoa, hengitysvaikeuksia, sekavuutta, epätavallista uupumusta tai uneliaisuutta, diabeettisen ketoasidoosin riski on otettava huomioon. Potilaat tulee tutkia ketoasidoosin varalta välittömästi, jos näitä oireita ilmenee, veren glukoosipitoisuudesta huolimatta.
Jos potilaalla epäillään tai todetaan diabeettinen ketoasidoosi, hoito empagliflotsiinilla pitää heti lopettaa.
Hoito on keskeytettävä potilailla, jotka joutuvat sairaalahoitoon suuren kirurgisen toimenpiteen tai äkillisen vakavan sairauden takia. Näillä potilailla suositellaan ketonien seurantaa. Ketonipitoisuus kannattaa mitata verestä eikä virtsasta. Hoito empagliflotsiinilla voidaan aloittaa uudelleen, kun ketonipitoisuus on normaali ja potilaan tila on jälleen vakaa.
Potilaan ketoasidoosille altistavat aiemmat sairaudet pitää ottaa huomioon ennen kuin hoito empagliflotsiinilla aloitetaan.
Empagliflotsiinin käytön yhteydessä on havaittu pitkittynyttä diabeettista ketoasidoosia ja pitkittynyttä glukosuriaa. Diabeettinen ketoasidoosi voi jatkua empagliflotsiinihoidon keskeyttämisen jälkeen pidempään kuin puoliintumisaika plasmassa antaisi odottaa (ks. kohta Farmakokinetiikka). Empagliflotsiinista riippumattomat tekijät, kuten insuliinin puute, voivat vaikuttaa diabeettisen ketoasidoosin pitkittymiseen.
Diabeettisen ketoasidoosin riski saattaa olla suurempi potilailla, joilla on pieni beetasolujen toimintareservi (esim. tyypin 2 diabetesta sairastavat potilaat, joilla on pieni C‑peptidipitoisuus, aikuisen piilevä autoimmuunidiabetes (LADA) tai haimatulehduksen aiemmin sairastaneet potilaat); potilailla, joilla on ruoan saantia rajoittava sairaus tai vaikea elimistön nestevajaus; potilailla, joiden insuliiniannosta on pienennetty sekä potilailla, joilla on akuutin sairauden, leikkaustoimenpiteen tai alkoholin väärinkäytön vuoksi lisääntynyt insuliinintarve. SGLT2:n estäjien käytössä näiden potilasryhmien hoitoon pitää olla varovainen.
SGLT2:n estäjähoidon aloittamista uudelleen ei suositella, jos potilaalla on aiemmin ollut diabeettinen ketoasidoosi SGLT2:n estäjähoidon aikana, paitsi jos tunnistetaan jokin toinen diabeettista ketoasidoosia edistävä tekijä ja se on hävinnyt.
Synjardy-valmistetta ei pidä käyttää potilaille, joilla on tyypin 1 diabetes. Tyypin 1 diabetesta sairastavilla potilailla tehdystä kliinisestä tutkimusohjelmasta saatujen tietojen mukaan diabeettisen ketoasidoosin esiintyvyys suureni lumelääkkeeseen verrattuna yleiseksi potilailla, jotka saivat 10 mg ja 25 mg empagliflotsiinia insuliinin lisänä.
Jodivarjoaineiden anto
Jodivarjoaineiden intravaskulaarinen anto voi johtaa varjoainenefropatiaan, joka johtaa metformiinin kumuloitumiseen ja maitohappoasidoosin riskin suurenemiseen. Metformiinihoito on lopetettava ennen kuvantamistutkimusta tai sen yhteydessä ja aloitettava uudelleen vasta vähintään 48 tunnin kuluttua, kun munuaistoiminta on ensin arvioitu uudelleen ja todettu stabiiliksi (ks. kohdat Annostus ja antotapa ja Yhteisvaikutukset).
Munuaisten vajaatoiminta
Vaikutusmekanismista johtuen heikentynyt munuaistoiminta johtaa empagliflotsiinin glykeemisen tehon heikentymiseen. Empagliflotsiini/metformiini on vasta-aiheinen potilailla, joiden eGFR-arvo on < 30 ml/min/1,73 m2, ja se on tauotettava, jos potilaalla on jokin munuaistoimintaan vaikuttava tila (ks. kohta Vasta-aiheet).
Munuaistoiminnan seuranta
Munuaisten toimintaa on suositeltavaa arvioida seuraavasti:
-
ennen empagliflotsiini-/metformiinihoidon aloittamista ja säännöllisin väliajoin hoidon aikana ts. ainakin vuosittain (ks. kohta Annostus ja antotapa)
-
ennen minkään sellaisen samanaikaisesti käytettävän lääkevalmisteen käytön aloittamista, jolla voi olla haitallinen vaikutus munuaisten toimintaan.
Sydämen toiminta
Sydämen vajaatoiminnasta kärsivillä potilailla hypoksian ja munuaisten vajaatoiminnan riski on suurempi. Potilailla, joilla on vakaa krooninen sydämen vajaatoiminta, Synjardy-valmistetta voidaan käyttää, jos sydämen ja munuaisten toimintaa seurataan säännöllisesti. Potilailla, jotka kärsivät akuutista ja epävakaasta sydämen vajaatoiminnasta, Synjardy-valmisteen käyttö on metformiinista johtuen vasta-aiheista (ks. kohta Vasta-aiheet).
Leikkaus
Metformiinihoito on tauotettava yleisanestesiassa tai spinaali- tai epiduraalianestesiassa tehtävän leikkauksen yhteydessä. Hoito voidaan aloittaa uudelleen aikaisintaan 48 tunnin kuluttua leikkauksesta tai peroraalisen ravitsemuksen aloittamisesta, mikäli munuaistoiminta on arvioitu uudelleen ja todettu stabiiliksi.
Nestehukan riski
Perustuen SGLT2‑estäjien vaikutusmekanismiin, hoidolliseen glukosuriaan liittyvä osmoottinen diureesi voi johtaa verenpaineen lievään laskuun (ks. kohta Farmakodynamiikka). Varovaisuutta on siksi noudatettava potilailla, joilla empagliflotsiinin aiheuttama verenpaineen aleneminen voi aiheuttaa riskin kuten potilailla, joilla tiedetään olevan sydän- ja verisuonitauti; potilailla, joilla on verenpainelääkitys ja anamneesissa hypotensio, tai 75‑vuotiailla ja sitä vanhemmilla potilailla.
Jos Synjardya käyttävillä potilailla on sairauksia, jotka voivat johtaa nesteen menetykseen (esim. ruoansulatuskanavan sairaus), suositellaan nestetasapainon (esim. lääkärintarkastus, verenpainemittaukset, laboratoriotutkimukset mukaan lukien hematokriitti) ja elektrolyyttitasapainon huolellista seurantaa. Synjardy-hoidon tilapäistä keskeyttämistä on harkittava, kunnes nesteen menetys on korjattu.
Iäkkäät
Empagliflotsiinin vaikutus glukoosin erittymiseen virtsaan liitetään osmoottiseen diureesiin, joka saattaa vaikuttaa nesteytykseen. 75‑vuotiailla ja sitä vanhemmilla potilailla saattaa olla lisääntynyt riski nestehukkaan. Siksi näiden potilaiden kohdalla on kiinnitettävä erityistä huomiota nesteen nauttimiseen, jos he käyttävät samanaikaisesti lääkkeitä, jotka voivat aiheuttaa nestehukkaa (esim. diureetteja, ACE:n estäjiä).
Virtsatieinfektiot
Komplisoituneita virtsatieinfektioita, mukaan lukien pyelonefriitti ja urosepsis, on raportoitu valmisteen markkinoilletulon jälkeen empagliflotsiinia saaneilla potilailla (ks. kohta Haittavaikutukset). Hoidon tilapäistä keskeyttämistä on harkittava potilailla, joilla on komplisoituneita virtsatieinfektioita.
Välilihan nekrotisoiva faskiitti (Fournier’n gangreeni)
Välilihan nekrotisoivan faskiitin (tämä tunnetaan myös nimellä Fournier’n gangreeni) tapauksista on ilmoitettu nais- ja miespotilailla, jotka käyttävät SGLT2:n estäjiä, kuten empagliflotsiinia. Tämä on harvinainen, mutta vakava ja mahdollisesti hengenvaarallinen tapahtuma, joka edellyttää kiireellistä leikkausta ja antibioottihoitoa.
Potilaita on kehotettava kääntymään lääkärin puoleen, jos heillä on kipua, aristusta, punoitusta tai turvotusta genitaali- tai perineaalialueella ja tähän liittyy kuumetta tai huonovointisuutta. On syytä huomioida, että nekrotisoivaa faskiittia voi edeltää urogenitaali-infektio tai perineaaliabsessi. Jos Fournier’n gangreenia epäillään, Synjardy-valmisteen käyttö on keskeytettävä ja hoito (mukaan lukien antibioottihoito ja puhdistusleikkaus) on aloitettava.
Alaraajan amputaatiot
Pitkän aikavälin kliinisissä tutkimuksissa toisella SGLT2:n estäjällä on havaittu alaraajan (pääasiallisesti varpaan) amputaatioiden määrän lisääntymistä. Ei tiedetä, onko kyseessä lääkeluokkaan liittyvä vaikutus. Kuten kaikkien diabetespotilaiden kohdalla, on tärkeää antaa potilaille neuvoja säännöllisestä ehkäisevästä jalkahoidosta.
Maksavaurio
Kliinisissä tutkimuksissa on raportoitu maksavauriotapauksia empagliflotsiinia käytettäessä. Syy-yhteytttä empagliflotsiinin ja maksavaurion välillä ei ole osoitettu.
Kohonnut hematokriitti
Hematokriitin kohoamista havaittiin empagliflotsiinihoidon yhteydessä (ks. kohta Haittavaikutukset). Potilaita, joiden hematokriitti on kohonnut huomattavasti, on seurattava, ja heille on tehtävä tutkimuksia taustalla olevan hematologisen sairauden varalta.
Krooninen munuaistauti
Empagliflotsiinin käytöstä on saatu kokemusta diabeteksen hoidossa potilailla, joilla on krooninen munuaistauti (eGFR ≥ 30 ml/min/1,73 m2) ja jolla on tai ei ole albuminuriaa. Albuminuriapotilailla empagliflotsiinihoidosta voi olla enemmän hyötyä.
Virtsan laboratoriotutkimukset
Synjardy-valmisteen vaikutusmekanismin vuoksi potilaiden virtsan glukoosimääritys on positiivinen.
Häiriöt 1,5‑anhydroglusitolin (1,5‑AG) määrityksessä
Glukoositasapainon seurantaa 1,5‑AG‑määrityksellä ei suositella, sillä SGLT2:n estäjiä käyttäviltä potilailta 1,5‑AG‑määrityksellä mitatut arvot eivät luotettavasti kuvaa glukoositasapainoa. Vaihtoehtoisten menetelmien käyttöä glukoositasapainon seurantaan suositellaan.
B12‑vitamiini
Metformiini saattaa pienentää B12‑vitamiiniarvoa. B12‑vitamiiniarvon alenemisen riski kasvaa metformiiniannoksen suurentuessa, hoidon pidentyessä ja/tai potilailla, joilla on B12‑vitamiinin puutokselle altistavia riskitekijöitä. Jos on syytä epäillä B12‑vitamiinin puutosta (kuten anemia tai neuropatia), tulee seerumin B12‑vitamiiniarvoa seurata. Ajoittainen B12‑vitamiiniarvon seuranta voi olla tarpeen potilailla, joilla on B12‑vitamiinin puutoksen riskitekijöitä. Metformiinihoitoa tulisi jatkaa niin kauan, kuin potilas sietää sitä, eikä vasta-aiheita ole. Asianmukaista korjaavaa hoitoa B12‑vitamiinin puutokseen on annettava voimassa olevien hoitosuositusten mukaisesti.
Pediatriset potilaat
DINAMO-tutkimuksessa (ks. kohta Farmakodynamiikka), turvallisuusprofiili lapsilla ja nuorilla oli yleisesti ottaen samanlainen kuin tiedossa oleva turvallisuusprofiili aikuispotilailla, eikä kasvun arvioinneissa tai sukupuolisessa kypsymisessä todettu merkityksellisiä eroja lumelääkkeen ja empagliflotsiinin välillä 26 viikkoa kestäneen hoidon jälkeen.
Metformiinin ei ole havaittu vaikuttavan kasvuun eikä murrosikään vuoden kestäneissä kontrolloiduissa kliinisissä tutkimuksissa, mutta näistä nimenomaisista seikoista ei ole saatavilla pitkäaikaistietoja. Siksi metformiinin vaikutusta näihin parametreihin on suositeltavaa seurata huolellisesti metformiinihoitoa saavilla lapsilla ja erityisesti esimurrosikäisillä lapsilla.
10–12-vuotiaat lapset
Lapsilla ja nuorilla tehtyihin kontrolloituihin kliinisiin metformiinitutkimuksiin osallistui vain 15 potilasta, joiden ikä oli 10–12 vuotta.
DINAMO-tutkimukseen osallistui 157 potilasta, joista 91 % sai metformiinia taustahoitona. Näistä potilaista 25 oli 10–12-vuotiaita.
Vaikka metformiinin teho ja turvallisuus näillä lapsilla ei eronnut tehosta ja turvallisuudesta muilla lapsilla ja nuorilla, on suositeltavaa noudattaa erityistä varovaisuutta, jos sitä määrätään 10–12-vuotiaille lapsille.
Yhteisvaikutukset
Toistuvien empagliflotsiini- ja metformiiniannosten samanaikainen anto ei muuta merkittävästi empagliflotsiinin eikä metformiinin farmakokinetiikkaa terveillä tutkittavilla.
Synjardy-valmisteen yhteisvaikutustutkimuksia ei ole tehty. Saatavilla olevat tiedot molemmista vaikuttavista aineista kerrotaan alla.
Empagliflotsiini
Farmakodynaamiset yhteisvaikutukset
Diureetit
Empagliflotsiini saattaa voimistaa tiatsidi- ja loop-diureettien diureettista vaikutusta ja lisätä elimistön kuivumisen ja hypotension riskiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Insuliini ja insuliinin eritystä lisäävät lääkeaineet
Insuliini ja insuliinin eritystä lisäävät lääkeaineet, kuten sulfonyyliureat, saattavat lisätä hypoglykemian riskiä. Tästä syystä insuliinin ja insuliinin eritystä lisäävien lääkeaineiden pienempi annos saattaa olla tarpeen hypoglykemiariskin pienentämiseksi, kun niitä käytetään yhdessä empagliflotsiinin kanssa (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Farmakokineettiset yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset empagliflotsiiniin
In vitro -tiedot viittaavat siihen, että empagliflotsiini metaboloituu ihmisillä ensisijaisesti glukuronidaatiolla uridiini‑5’‑difosfo-glukuronosyylitransferaasien UGT1A3:n, UGT1A8:n, UGT1A9:n ja UGT2B7:n vaikutuksesta. Empagliflotsiini on ihmisen sisäänotto-kuljettajaproteiinien OAT3:n, OATP1B1:n ja OATP1B3:n substraatti mutta ei OAT1:n eikä OCT2:n substraatti. Empagliflotsiini on P‑glykoproteiinin (P‑gp) ja rintasyöpäresistenssiproteiinin (BCRP) substraatti.
Kun empagliflotsiinia annettiin UGT‑entsyymien ja OAT3:n estäjä probenesidin kanssa, empagliflotsiinin huippupitoisuudet plasmassa (Cmax) nousivat 26 % ja pitoisuus–aika-käyrän alle jäävä pinta-ala (AUC) suureni 53 %. Muutoksia ei pidetty kliinisesti merkittävinä.
UGT:n induktion (esim. rifampisiinin tai fenytoiinin aiheuttaman induktion) vaikutusta empagliflotsiiniin ei ole tutkittu. Samanaikaista hoitoa tunnettujen UGT‑entsyymien indusorien kanssa ei suositella mahdollisen tehoa alentavan vaikutuksen vuoksi. Jos UGT‑entsyymien indusoreja joudutaan antamaan samanaikaisesti, glukoositasapainoa on syytä seurata Synjardy-valmisteen vasteen arvioimiseksi.
Yhteisvaikutustutkimus gemfibrotsiilin kanssa, joka on OAT3- ja OATP1B1/1B3‑kuljettajaproteiinien in vitro -estäjä, osoitti että empagliflotsiinin Cmax nousi 15 % ja AUC suureni 59 % yhtäaikaisen käytön jälkeen. Näitä muutoksia ei pidetty kliinisesti merkittävinä.
OATP1B1/1B3‑kuljettajaproteiinien estäminen samanaikaisesti annetun rifampisiinin avulla suurensi empagliflotsiinin Cmax‑arvoa 75 % ja AUC‑arvoa 35 %. Näitä muutoksia ei pidetty kliinisesti merkittävinä.
Empagliflotsiinialtistus oli samankaltaista annettaessa yhdessä P‑gp:n estäjän verapamiilin kanssa ja ilman verapamiilia. Tämä viittaa siihen, että P‑gp:n estolla ei ole kliinisesti merkittävää vaikutusta empagliflotsiiniin.
Yhteisvaikutustutkimusten tiedot viittaavat siihen, että samanaikaisesti annetut metformiini, glimepiridi, pioglitatsoni, sitagliptiini, linagliptiini, varfariini, verapamiili, ramipriili, simvastatiini, torasemidi tai hydroklooritiatsidi eivät vaikuttaneet empagliflotsiinin farmakokinetiikkaan.
Empagliflotsiinin vaikutukset muihin lääkevalmisteisiin
Empagliflotsiini saattaa lisätä litiumin erittymistä munuaisten kautta, ja veren litiumpitoisuudet saattavat pienentyä. Seerumin litiumpitoisuutta on seurattava useammin empagliflotsiinihoidon aloittamisen ja annosmuutosten jälkeen. Potilas tulee ohjata litiumia määränneen lääkärin vastaanotolle seerumin litiumpitoisuuden seurantaa varten.
In vitro -tutkimusten perusteella empagliflotsiini ei estä, inaktivoi eikä indusoi CYP450‑isoformeja. Empagliflotsiini ei estä UGT1A1:tä, UGT1A3:a, UGT1A8:aa, UGT1A9:ää eikä UGT2B7:ää. Siksi yhteisvaikutukset merkittävimpien CYP450‑ ja UGT‑isoformien kanssa ovat epätodennäköisiä, kun empagliflotsiinia annetaan samanaikaisesti näiden entsyymien substraattien kanssa.
Empagliflotsiini ei estä P‑gp:tä terapeuttisilla annoksilla. In vitro ‑tutkimusten perusteella on epätodennäköistä, että empagliflotsiinilla on yhteisvaikutuksia sellaisten vaikuttavien aineiden kanssa, jotka ovat P‑gp:n substraatteja. Kun digoksiinia, P‑gp:n substraattia, annettiin samanaikaisesti empagliflotsiinin kanssa, digoksiinin AUC‑arvo suureni 6 % ja Cmax‑arvo 14 %. Näitä muutoksia ei pidetty kliinisesti merkittävinä.
Empagliflotsiini ei estä ihmisen sisäänotto-kuljettajaproteiineja kuten OAT3:a, OATP1B1:tä ja OATP1B3:a in vitro kliinisesti merkittävillä pitoisuuksilla plasmassa, ja näin ollen lääkeaineiden väliset yhteisvaikutukset näiden sisäänotto-kuljettajaproteiinien substraattien kanssa ovat epätodennäköisiä.
Terveillä vapaaehtoisilla tehtyjen yhteisvaikutustutkimusten tiedot viittaavat siihen, että empagliflotsiinilla ei ollut kliinisesti merkittävää vaikutusta metformiinin, glimepiridin, pioglitatsonin, sitagliptiinin, linagliptiinin, simvastatiinin, varfariinin, ramipriilin, digoksiinin, diureettien ja suun kautta otettavien ehkäisyvalmisteiden farmakokinetiikkaan.
Metformiini
Samanaikaista käyttöä ei suositella
Alkoholi
Alkoholi-intoksikaatioon liittyy suurentunut maitohappoasidoosin riski etenkin paaston, vajaaravitsemuksen tai maksan vajaatoiminnan yhteydessä.
Orgaaniset kationiset kuljetusaineet (OCT)
Metformiini on substraatti molemmille kuljetusaineille OCT1 ja OCT2. Metformiinin samanaikainen ottaminen seuraavien aineiden kanssa:
-
OCT1-estäjät (kuten verapamiili) saattavat vähentää metformiinin vaikutusta
-
OCT1:tä indusoivat aineet (kuten rifampisiini) saattavat lisätä metformiinin imeytymistä ruoansulatuskanavasta ja lisätä sen vaikutusta
-
OCT2-estäjät (kuten simetidiini, dolutegraviiri, ranolatsiini, trimetopriimi, vandetanibi, isavukonatsoli) saattavat vähentää metformiinin eliminaatiota munuaisten kautta, ja näin lisätä metformiinin pitoisuutta plasmassa
-
aineet, jotka estävät sekä OCT1:tä että OCT2:ta (kuten kritsotinibi ja olaparibi) saattavat vaikuttaa metformiinin tehoon ja sen eliminaatioon munuaisten kautta.
Varovaisuutta tulisi näin ollen noudattaa otettaessa näitä lääkkeitä samanaikaisesti metformiinin kanssa, etenkin potilailla joilla on munuaisten vajaatoimintaa, koska tämä voi nostaa metformiinin pitoisuutta plasmassa. Tarvittaessa voidaan harkita annoksen muuttamista, koska OCT:tä estävät/indusoivat aineet voivat vaikuttaa metformiinin tehoon (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Jodivarjoaineet
Metformiinihoito on lopetettava ennen kuvantamistutkimusta tai sen yhteydessä ja aloitettava uudelleen vasta vähintään 48 tunnin kuluttua, kun munuaistoiminta on ensin arvioitu uudelleen ja todettu stabiiliksi (ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet).
Varotoimia vaativat yhdistelmät
Jotkin lääkevalmisteet voivat huonontaa munuaistoimintaa, mikä voi suurentaa maitohappoasidoosin riskiä. Tällaisia ovat esimerkiksi tulehduskipulääkkeet, myös selektiiviset syklo-oksigenaasi (COX) II:n estäjät, ACE:n estäjät, angiotensiini II ‑reseptorin salpaajat ja diureetit, etenkin loop-diureetit. Munuaistoiminnan tarkka seuranta on tarpeen, jos tällaisia valmisteita otetaan käyttöön tai käytetään yhdessä metformiinin kanssa.
Glukokortikoideilla (systeemiset ja paikalliset), beeta‑2‑agonisteilla ja diureeteilla on luontainen veren glukoosipitoisuutta suurentava vaikutus. Tästä on kerrottava potilaille, ja glukoosiarvoja on seurattava tavanomaista tiheämmin, erityisesti kun hoito näillä lääkkeillä aloitetaan. Diabeteslääkkeen annosta on muutettava tarvittaessa näiden lääkkeiden käytön aikana sekä käytön lopettamisen yhteydessä.
Insuliini ja insuliinin eritystä lisäävät lääkeaineet
Insuliini ja insuliinin eritystä lisäävät lääkeaineet, kuten sulfonyyliureat, saattavat suurentaa hypoglykemian riskiä. Tästä syystä insuliinin ja insuliinin eritystä lisäävien lääkeaineiden pienempi annos saattaa olla tarpeen hypoglykemiariskin pienentämiseksi, kun valmistetta käytetään yhdessä metformiinin kanssa (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Tämän lääkevalmisteen tai empagliflotsiinin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehtyjen tutkimusten mukaan empagliflotsiini läpäisee istukan raskauden myöhäisvaiheessa hyvin rajallisessa määrin, mutta sillä ei vaikuta olevan suoria eikä epäsuoria haitallisia vaikutuksia varhaiseen alkion kehitykseen. Eläimillä tehdyissä tutkimuksissa on kuitenkin havaittu haittavaikutuksia postnataaliseen yksilönkehitykseen. Vähäiset tiedot metformiinin käytöstä raskauden aikana eivät viittaa synnynnäisten epämuodostumien suurentuneeseen riskiin. Eläimillä tehdyissä tutkimuksissa empagliflotsiinin ja metformiinin yhdistelmällä ja pelkällä metformiinilla on havaittu lisääntymistoksisuutta suurempien metformiinimonoterapia-annosten yhteydessä (ks. kohta Prekliiniset tiedot turvallisuudesta).
Raskautta suunniteltaessa ja raskauden aikana on suositeltavaa, että diabetesta ei hoideta tällä lääkevalmisteella. Sen sijaan suositellaan insuliinin käyttöä pitämään veren glukoosipitoisuus mahdollisimman normaalina, jotta poikkeaviin veren glukoosiarvoihin liittyvä sikiön epämuodostumariski pienenisi.
Imetys
Metformiini erittyy ihmisillä äidinmaitoon. Vaikutuksia ei ole havaittu hoitoa saaneiden naisten imettämissä vauvoissa. Ei ole tietoa empagliflotsiinin erittymisestä äidinmaitoon ihmisillä. Olemassa olevat tiedot koe-eläimistä ovat osoittaneet empagliflotsiinin ja metformiinin erittyvän maitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida poissulkea.
Tätä lääkevalmistetta ei pidä käyttää imetyksen aikana.
Hedelmällisyys
Tutkimuksia tämän lääkevalmisteen tai empagliflotsiinin vaikutuksista ihmisen hedelmällisyyteen ei ole tehty. Eläimillä tehdyissä tutkimuksissa empagliflotsiinilla ja metformiinilla ei ole havaittu suoria eikä epäsuoria haitallisia vaikutuksia hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Synjardy-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaita on neuvottava noudattamaan varotoimenpiteitä hypoglykemian välttämiseksi ajamisen ja koneiden käytön aikana, etenkin kun Synjardy-valmistetta käytetään yhdessä sulfonyyliurean ja/tai insuliinin kanssa.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kliinisissä tutkimuksissa yleisimmin ilmoitettuja haittavaikutuksia olivat hypoglykemia, kun käytössä oli myös insuliini ja/tai sulfonyyliurea, sekä ruuansulatuskanavan oireet kuten pahoinvointi, oksentelu, ripuli, vatsakipu ja ruokahaluttomuus. Kliinisissä tutkimuksissa, joissa empagliflotsiini oli yhdistetty metformiinihoitoon, empagliflotsiinin ei havaittu aiheuttavan sellaisia haittavaikutuksia, jotka eroaisivat empagliflotsiinin tai metformiinin monoterapian haittavaikutuksista.
Haittavaikutustaulukko
Haittavaikutukset on luokiteltu absoluuttisen esiintyvyyden mukaan. Esiintyvyydet määritellään seuraavasti: hyvin yleiset (≥ 1/10), yleiset (≥ 1/100, < 1/10), melko harvinaiset (≥ 1/1 000, < 1/100), harvinaiset (≥ 1/10 000, < 1/1 000), hyvin harvinaiset (< 1/10 000) ja yleisyys tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 2: Taulukoitu luettelo haittavaikutuksista (MedDRA) lumekontrolloiduissa tutkimuksissa ja myyntiluvan myöntämisen jälkeisessä käytössä
Elinjärjestelmä | Hyvin yleiset | Yleiset | Melko harvinaiset | Harvinaiset | Hyvin harvinaiset |
Infektiot |
| Emättimen kandidiaasi, vulvovaginiitti, balaniitti ja muu sukuelinten infektio1, 2 Virtsatieinfektio (mukaan lukien pyelonefriitti ja urosepsis)1, 2 |
| Välilihan nekrotisoiva faskiitti (Fournier’n gangreeni)a |
|
Aineenvaihdunta ja ravitsemus | Hypoglykemia (käytettäessä yhdessä sulfonyyliurean tai insuliinin kanssa)1 | Jano2 B12-vitamiiniarvon lasku / B12-vitamiinin puutos3,a |
| Diabeettinen ketoasidoosia | Maitohappoasidoosi3 |
Hermosto |
| Makuaistin häiriö3 |
|
|
|
Verisuonisto |
|
| Nestehukka1, 2, d |
|
|
Ruoansulatus-elimistö | Ruuansulatuskanavan oireet3, 4 | Ummetus |
|
|
|
Maksa ja sappi |
|
|
|
| Poikkeavuudet maksan toiminta-kokeissa3 |
Iho ja ihonalainen kudos |
| Kutina (yleistynyt)2,3 | Urtikaria |
| Punoitus3 |
Munuaiset ja virtsatiet |
| Lisääntynyt virtsaaminen1, 2 | Dysuria2 |
| Tubulo-interstitiaalinen nefriitti |
Tutkimukset |
| Kohonneet seerumin lipidiarvot2,b | Kohonnut veren kreatiniinipitoisuus / Alentunut glomerulusten suodatusnopeus1 |
|
|
1 Lisätiedot, ks. alakohdat jäljempänä
2 Empagliflotsiinimonoterapian tunnistetut haittavaikutukset
3 Metformiinimonoterapian tunnistetut haittavaikutukset
4 Ruuansulatuskanavan oireita kuten pahoinvointia, oksentelua, ripulia, vatsakipua ja ruokahaluttomuutta esiintyy yleisimmin hoidon alussa, ja ne menevät useimmiten ohi itsestään.
a Ks. kohta Varoitukset ja käyttöön liittyvät varotoimet
b Keskimääräinen prosentuaalinen nousu lähtötilanteeseen nähden oli empagliflotsiini 10 mg- ja 25 mg ‑hoidossa lumehoitoon verrattuna kokonaiskolestrolin kohdalla 5,0 % ja 5,2 % vs. 3,7 %, HDL-kolesterolin kohdalla 4,6 % ja 2,7 % vs. ‑0,5 %, LDL-kolesterolin kohdalla 9,1 % ja 8,7 % vs. 7,8 % ja triglyseridien kohdalla 5,4 % ja 10,8 % vs. 12,1 %.
c Keskimääräiset muutokset hematokriitissä olivat lähtötilanteeseen nähden 3,6 % empagliflotsiini 10 mg ‑hoidossa, 4,0 % empagliflotsiini 25 mg ‑hoidossa ja 0 % lumehoidossa. EMPA-REG OUTCOME -tutkimuksessa hematokriittiarvot palasivat kohti lähtöarvoja hoidon päättymistä seuranneen 30 päivän seurantajakson jälkeen.
d Yhdistetyt tiedot empagliflotsiinilla tehdyistä tutkimuksista, joihin osallistuneilla potilailla oli sydämen vajaatoiminta (ja puolella potilaista tyypin 2 diabetes), osoittivat, että nestehukkaa esiintyi yleisemmin (”hyvin yleinen”: 11,4 % empagliflotsiinilla vs. 9,7 % lumelääkkeellä).
Valikoitujen haittavaikutusten kuvaus
Hypoglykemia
Hypoglykemian esiintyvyys tutkimuksissa riippui potilaalla käytössä olleesta taustahoidosta ja se oli samaa luokkaa, kun empagliflotsiinia tai lumetta käytettiin metformiinin kanssa, linagliptiinin ja metformiinin kanssa, empagliflotsiinin ja metformiinin yhdistelmähoidossa potilailla, jotka eivät aiemmin olleet saaneet lääkehoitoa, verrattuna potilaisiin, joita hoidettiin empagliflotsiinilla ja metformiinilla yksittäisinä ainesosina ja yhdistettynä tavanomaiseen hoitoon. Hypoglykemiaa esiintyi useammin, kun empagliflotsiinia annettiin metformiinin ja sulfonyyliurean lisänä (empagliflotsiini 10 mg: 16,1 %, empagliflotsiini 25 mg: 11,5 % ja lume: 8,4 %) tai metformiinin ja insuliinin lisänä (empagliflotsiini 10 mg: 31,3 %, empagliflotsiini 25 mg: 36,2 % ja lume: 34,7 %).
Merkittävä hypoglykemia (hoitoa vaativat tapahtumat)
Merkittävää hypoglykemiaa esiintyi kaikkiaan vähän (< 1 %). Esiintyvyys oli samaa luokkaa, kun empagliflotsiinia tai lumetta käytettiin metformiinin kanssa, ja empagliflotsiinin ja metformiinin yhdistelmähoidossa potilailla, jotka eivät aiemmin olleet saaneet lääkehoitoa, verrattuna potilaisiin, joita hoidettiin empagliflotsiinilla ja metformiinilla yksittäisinä ainesosina ja yhdistettynä tavanomaiseen hoitoon. Merkittävää hypoglykemiaa esiintyi 0,5 %:lla 10 mg empagliflotsiinia metformiinin ja insuliinin kanssa saaneista potilaista, 0 %:lla 25 mg empagliflotsiinia metformiinin ja insuliinin kanssa saaneista potilaista ja 0,5 %:lla lumetta metformiinin ja insuliinin kanssa saaneista potilaista. Yhdelläkään metformiinin ja sulfonyyliurean yhdistelmähoitoa ja linagliptiinia metformiinin kanssa saaneista potilaista ei esiintynyt merkittävää hypoglykemiaa.
Virtsatieinfektio
Haittatapahtumana ilmoitettujen virtsatieinfektioiden kokonaisesiintyvyys oli metformiinia ja 10 mg empagliflotsiinia saaneilla potilailla suurempi (8,8 %) kuin potilailla, jotka saivat metformiinin lisänä 25 mg empagliflotsiinia (6,6 %) tai lumetta (7,8 %). Samoin kuin lumelääkettä saaneilla, virtsatieinfektioita ilmoitettiin empagliflotsiinihoidon yhteydessä useammin potilailla, joilla oli ollut aikaisemmin kroonisia tai toistuvia virtsatieinfektioita. Virtsatieinfektioiden vaikeusaste (lievä/keskivaikea/vaikea) oli samankaltainen empagliflotsiinia ja lumelääkettä saaneilla potilailla. Virtsatieinfektiotapahtumia ilmoitettiin 10 mg empagliflotsiinia saaneilla useammin kuin lumetta saaneilla naispotilailla, mutta ei 25 mg empagliflotsiinia saaneilla. Virtsatieinfektioita esiintyi miespotilailla vähän, ja esiintyvyys oli samankaltainen eri hoitoryhmissä.
Emättimen kandidiaasi, vulvovaginiitti, balaniitti ja muu sukuelinten infektio
Emättimen kandidiaasia, vulvovaginiittia, balaniittia ja muita sukuelinten infektioita raportoitiin yleisemmin potilailla, jotka saivat metformiinin lisäksi 10 mg empagliflotsiinia (4,0 %) tai 25 mg empagliflotsiinia (3,9 %) verrattuna potilaisiin, jotka saivat metformiinin lisäksi lumelääkettä (1,3 %), ja niitä raportoitiin yleisemmin empagliflotsiinia saaneilla kuin lumelääkettä saaneilla naispotilailla. Esiintyvyyden ero ei ollut yhtä selkeä miespotilailla. Sukuelinten infektiot olivat vaikeusasteeltaan lieviä ja keskivaikeita, yksikään ei ollut vaikea.
Fimoosia / hankinnaista fimoosia on raportoitu esiintyvän samanaikaisesti sukupuolielinten infektioiden kanssa, ja joissakin tapauksissa tarvittiin ympärileikkaus.
Lisääntynyt virtsaaminen
Kuten vaikutusmekanismin perusteella oli odotettavissa, lisääntynyttä virtsaamista (mukaan lukien ennalta määritellyt termit pollakisuria, polyuria, nokturia) havaittiin useammin metformiinia saaneilla potilailla, jotka saivat 10 mg empagliflotsiinia (3,0 %) tai 25 mg empagliflotsiinia (2,9 %) verrattuna lumetta metformiinihoidon lisänä saaneisiin (1,4 %). Lisääntynyt virtsaaminen oli useimmiten voimakkuudeltaan lievää tai kohtalaista. Raportoidun nokturian esiintyvyys oli samanlainen lumelääke- ja empagliflotsiiniryhmässä (< 1 %).
Nestehukka
Metformiinia ja empagliflotsiinia saaneilla potilailla nestehukan (mukaan lukien etukäteen määritellyt termit verenpaineen (ambulatorinen) aleneminen, systolisen verenpaineen aleneminen, dehydraatio, hypotensio, hypovolemia, ortostaattinen hypotensio ja pyörtyminen) kokonaisesiintyvyys oli pieni: 0,6 % 10 mg empagliflotsiinia saaneilla, 0,3 % 25 mg empagliflotsiinia saaneilla ja 0,1 % lumetta saaneilla. Empagliflotsiini vaikuttaa glukoosin erittymiseen virtsaan, ja tähän vaikutukseen liittyy osmoottinen diureesi, joka voi vaikuttaa 75-vuotiaiden ja sitä vanhempien potilaiden nestetasapainoon. 75‑vuotiailla ja tätä vanhemmilla potilailla nestehukkaa raportoitiin yhdellä potilaalla, joka sai 25 mg empagliflotsiinia metformiinihoidon lisänä.
Kohonnut veren kreatiniinipitoisuus / Alentunut glomerulusten suodatusnopeus
Niiden potilaiden, joilla veren kreatiniinipitoisuus oli kohonnut ja glomerulusten suodatusnopeus alentunut, kokonaisesiintyvyys oli yhtä suuri empagliflotsiini- ja lumelääkeryhmissä metformiinin kanssa (veren kreatiniinipitoisuus kohonnut: empagliflotsiini 10 mg 0,5 %, empagliflotsiini 25 mg 0,1 %, lumelääke 0,4 %; glomerulusten suodatusnopeus alentunut: empagliflotsiini 10 mg 0,1 %, empagliflotsiini 25 mg 0 %, lumelääke 0,2 %).
Empagliflotsiinihoitoa metformiinin lisälääkkeenä saaneilla potilailla hoidon alussa havaittu kreatiniinipitoisuuden nousu ja hoidon alussa arvioidussa glomerulusten suodatusnopeudessa havaittu alenema oli hoidon jatkuessa yleensä ohimenevää tai lääkehoidon keskeytyessä palautuvaa.
Vastaavasti EMPA-REG OUTCOME -tutkimuksessa empagliflotsiinia saaneiden potilaiden laskennallinen glomerulusten suodatusnopeus (eGFR) pieneni aluksi (keskiarvo: 3 ml/min/1,73 m2). Sen jälkeen eGFR pysyi samalla tasolla, kun hoitoa jatkettiin. Keskimääräinen eGFR palasi lähtöarvoihin hoidon lopettamisen jälkeen, mikä viittaa siihen, että äkillisillä hemodynaamisilla muutoksilla voi olla osuutensa näissä munuaistoiminnan muutoksissa.
Pediatriset potilaat
DINAMO‑tutkimuksessa hoidettiin 157 lasta, jotka olivat iältään vähintään 10 vuotta ja joilla oli tyypin 2 diabetes. Tutkimuksessa 52 potilasta sai empagliflotsiinia, 52 linagliptiinia ja 53 lumelääkettä (ks. kohta Farmakodynamiikka). Lumelääkekontrolloidun vaiheen aikana yleisin haittavaikutus oli hypoglykemia (empagliflotsiini 10 mg ja 25 mg, yhdistettynä: 23,1 %, lumelääke: 9,4 %). Mikään haittatapahtumista ei ollut vaikea tai edellyttänyt hoitoa.
Yleisesti ottaen turvallisuusprofiili lapsilla oli samanlainen kuin turvallisuusprofiili aikuisilla, joilla on tyypin 2 diabetes.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‑haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet
Empagliflotsiini
Kontrolloiduissa kliinisissä tutkimuksissa, joissa terveille vapaaehtoisille annettiin enintään 800 mg:n kerta-annoksia empagliflotsiinia (vastaa 32‑kertaista korkeinta suositeltua vuorokausiannosta) ja tyypin 2 diabetesta sairastaville potilaille enintään 100 mg toistuvia vuorokausiannoksia empagliflotsiinia (vastaa 4‑kertaista korkeinta suositeltua vuorokausiannosta), ei ilmennyt minkäänlaista toksisuutta. Empagliflotsiini lisäsi glukoosin erittymistä virtsaan, mikä johti virtsamäärän suurenemiseen. Havaittu virtsamäärän suureneminen ei ollut annosriippuvaista eikä kliinisesti merkittävää. Yli 800 mg:n suuruisista annoksista ihmisillä ei ole kokemusta.
Metformiini
Alle 85 g:n metformiiniannokset eivät ole aiheuttaneet hypoglykemiaa, mutta tällaiset annokset ovat aiheuttaneet maitohappoasidoosia. Suuri metformiinin yliannostus tai samanaikaiset muut riskitekijät voivat johtaa maitohappoasidoosiin. Maitohappoasidoosi on lääketieteellinen hätätila ja vaatii sairaalahoitoa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Yhteisvaikutukset).
Hoito
Yliannostustapauksessa on aloitettava hoito potilaan kliinisen tilan mukaisesti. Laktaatin ja metformiinin eliminaatio onnistuu tehokkaimmin hemodialyysin avulla. Empagliflotsiinin poistoa hemodialyysillä ei ole tutkittu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: diabeteslääkkeet, oraalisten veren glukoosipitoisuutta pienentävien lääkkeiden yhdistelmävalmisteet, ATC‑koodi: A10BD20
Vaikutusmekanismi
Synjardy on yhdistelmävalmiste, joka sisältää kahta diabeteslääkettä. Niiden vaikutusmekanismit täydentävät toisiaan, mikä parantaa tyypin 2 diabeetikon glukoositasapainoa. Nämä lääkkeet ovat empagliflotsiini, joka on natriumin- ja glukoosinkuljettajaproteiini 2:n (SGLT2) estäjä, ja metformiinihydrokloridi, joka kuuluu biguanidien lääkeryhmään.
Empagliflotsiini
Empagliflotsiini on reversiibeli, erittäin voimakas (IC50 1,3 nmol) ja selektiivinen kilpaileva SGLT2:n estäjä. Empagliflotsiini ei estä muita glukoosinkuljettajaproteiineja, jotka ovat tärkeitä glukoosin kuljetuksessa ääreiskudoksiin, ja se on 5 000 kertaa selektiivisempi SGLT2:n kuin SGLT1:n suhteen. SGLT1 on kuljettajaproteiini, joka pääasiallisesti vastaa glukoosin imeytymisestä suolessa. SGLT2:ta esiintyy pääosin munuaisissa, kun taas muissa kudoksissa sitä ei ole lainkaan tai sen määrä on hyvin alhainen. Se vastaa pääasiallisesti glukoosin takaisinimeytymisestä glomerulussuodoksesta takaisin verenkiertoon. Tyypin 2 diabetesta ja hyperglykemiaa sairastavilla potilailla suurempi määrä glukoosia suodattuu ja imeytyy takaisin.
Empagliflotsiini parantaa tyypin 2 diabetesta sairastavien potilaiden glukoositasapainoa vähentämällä glukoosin takaisinimeytymistä munuaisissa. Munuaisten poistaman glukoosin määrä tällä glukosuurisella mekanismilla riippuu veren glukoosipitoisuudesta ja GFR-arvosta. SGLT2:n esto tyypin 2 diabetesta ja hyperglykemiaa sairastavilla potilailla johtaa liialliseen glukoosin erittymiseen virtsaan. Lisäksi empagliflotsiinihoidon aloittaminen lisää natriumin eritystä, mikä johtaa osmoottiseen diureesiin ja suontensisäisen tilavuuden laskuun.
Tyypin 2 diabetesta sairastavilla potilailla glukoosin erittyminen virtsaan lisääntyi välittömästi empagliflotsiinin ensimmäisen annoksen jälkeen ja jatkuu 24 tunnin annosvälin ajan. Glukoosin lisääntynyt erittyminen virtsaan jatkui 4 viikon hoitojakson loppuun asti, ja sitä erittyi keskimäärin 78 g/vrk käytettäessä 25 mg:aa empagliflotsiinia. Glukoosin lisääntynyt erittyminen virtsaan alensi välittömästi plasman glukoosiarvoja tyypin 2 diabetesta sairastavilla potilailla.
Empagliflotsiini parantaa plasman glukoosiarvoja sekä paaston että aterian jälkeen. Empagliflotsiinin vaikutusmekanismi on riippumaton beetasolujen toiminnasta ja insuliinierityksestä ja tämä vaikuttaa osaltaan hypoglykemian vähäiseen riskiin. Beetasolujen toimintaa kuvaavissa markkereissa, mukaan lukien homeostaasimallimääritys (HOMA‑β), havaittiin paranemista. Lisäksi glukoosin erittyminen virtsaan aiheuttaa kalorien hävikkiä, mihin liittyy kehon rasvakudoksen vähenemistä ja painon laskua. Empagliflotsiinista aiheutuvaan glukosuriaan liittyy lievä diureesi, joka voi osaltaan johtaa pitkäaikaiseen kohtalaiseen verenpaineen laskuun. Empagliflotsiinista johtuva glukosuria, natriureesi ja osmoottinen diureesi voivat olla osasyy sydän- ja verisuonisairauksia koskevien tulosten paranemiseen.
Metformiini
Metformiini on biguanidien ryhmään kuuluva lääke, joka alentaa kohonneita verenglukoosiarvoja. Se alentaa plasman glukoosipitoisuutta sekä perustilanteessa että aterioiden jälkeen. Se ei stimuloi insuliinieritystä eikä siten aiheuta hypoglykemiaa.
Metformiinilla on 3 mahdollista vaikutusmekanismia:
-
maksan glukoosituotanto vähenee, kun glukoneogeneesi ja glykogenolyysi estyvät
-
lihasten insuliiniherkkyys paranee, mikä tehostaa glukoosin soluunottoa ja käyttöä perifeerisissä kudoksissa
-
glukoosin imeytyminen suolesta hidastuu.
Metformiini stimuloi solunsisäistä glykogeenisynteesiä vaikuttamalla glykogeenisyntaasientsyymiin. Metformiini parantaa solukalvon kaikkien tunnettujen glukoosinkuljettajaproteiinien (GLUT) kuljetuskapasiteettia.
Metformiinilla on suotuisa vaikutus ihmisen lipidiaineenvaihduntaan. Vaikutus ei riipu sen glukoosiarvoihin kohdistuvasta vaikutuksesta. Tämä on todettu terapeuttisilla annoksilla kontrolloiduissa keskipitkissä ja pitkissä kliinisissä tutkimuksissa, joissa metformiini on alentanut kokonaiskolesterolia, LDL‑arvoja ja triglyseridiarvoja.
Kliininen teho ja turvallisuus
Sekä glukoositasapainon parantaminen että sydän- ja verisuonisairauksien ja -kuolemien vähentäminen ovat olennainen osa tyypin 2 diabeteksen hoitoa.
Glukoositasapainoa parantavaa tehoa ja sydän- ja verisuonisairauksia koskevia tuloksia arvoitiin yhteensä 10 366:lla tyypin 2 diabetesta sairastavalla potilaalla, joita hoidettiin yhdeksässä kaksoissokkoutetussa, lume- tai aktiivikontrolloidussa kliinisessä tutkimuksessa, jotka kestivät vähintään 24 viikkoa. Potilaista 2 950 sai 10 mg empagliflotsiinia ja 3 701 sai 25 mg empagliflotsiinia metformiinihoidon lisänä. Näistä 266 potilasta sai 10 mg empagliflotsiinia ja 264 sai 25 mg empagliflotsiinia metformiinin ja insuliinin lisänä.
Empagliflotsiinin ja metformiinin yhdistelmähoito yhdessä muiden diabeteslääkkeiden (pioglitatsoni, sulfonyyliurea, DPP‑4:n estäjät ja insuliini) kanssa tai ilman paransi kliinisesti merkittävästi HbA1c‑arvoa, paastoglukoosiarvoa, painoa sekä systolista ja diastolista verenpainetta. Suurempi osa 25 mg:n empagliflotsiiniannoksella hoidetuista potilaista saavutti tavoitteen HbA1c alle 53 mmol/mol (7 %) ja pienempi osa tarvitsi lisälääkitystä glukoositasapainon korjaamiseksi kuin 10 mg:n empagliflotsiiniannoksella tai lumelääkkeellä hoidetuista potilaista. HbA1c‑arvot alenivat numeerisesti vähemmän empagliflotsiinihoitoa saaneilla 75‑vuotiailla ja sitä vanhemmilla potilailla. Potilailla, joilla HbA1c‑arvo tutkimuksen alussa oli korkeampi, havaittiin myös suurempi HbA1c‑arvon aleneminen. Lisäksi empagliflotsiini yhdistettynä tavanomaiseen hoitoon vähensi sydän- ja verisuonikuolemia tyypin 2 diabetespotilailla, joilla oli sydän- ja verisuonisairaus.
Empagliflotsiini yhdistettynä metformiiniin, sulfonyyliureaan tai pioglitatsoniin
Empagliflotsiini metformiinin, metformiinin ja sulfonyyliurean tai pioglitatsonin ja metformiinin lisänä alensi tilastollisesti merkitsevästi (p < 0,0001) HbA1c‑arvoa ja painoa lumelääkkeeseen verrattuna (taulukko 3). Lisäksi se alensi kliinisesti merkittävästi paastoglukoosiarvoa sekä systolista ja diastolista verenpainetta lumelääkkeeseen verrattuna.
Näiden tutkimusten kaksoissokkoutetussa, lumekontrolloidussa jatkovaiheessa HbA1c‑arvon, painon ja verenpaineen aleneminen säilyi viikolle 76 asti.
Taulukko 3: 24 viikon empagliflotsiinin lumekontrolloitujen tutkimusten tehon tulokset
Yhdistettynä metformiiniina | |||
Lumehoito | Empagliflotsiini | ||
10 mg | 25 mg | ||
N | 207 | 217 | 213 |
HbA1c mmol/mol [%] | |||
Lähtötilanne (keskiarvo) | 62,87 [7,90] | 63,28 [7,94] | 62,35 [7,86] |
Muutos lähtötilanteesta1 | ‑1,45 [-0,13] | ‑7,64 [‑0,70] | ‑8,40 [‑0,77] |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑6,20* (‑7,85, ‑4,54) [-0,57* (-0,72, -0,42)] | ‑6,95* (‑8,62, ‑5,29) [-0,64* (-0,79, -0,48)] | |
N | 184 | 199 | 191 |
HbA1c‑arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c‑arvon ollessa ≥ 53 mmol/mol (7 %)2 | 12,5 | 37,7 | 38,7 |
N | 207 | 217 | 213 |
Paino (kg) | |||
Lähtötilanne (keskiarvo) | 79,73 | 81,59 | 82,21 |
Muutos lähtötilanteesta1 | ‑0,45 | ‑2,08 | ‑2,46 |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑1,63* (‑2,17, ‑1,08) | ‑2,01* (‑2,56, ‑1,46) | |
N | 207 | 217 | 213 |
SVP (mmHg)2 | |||
Lähtötilanne (keskiarvo) | 128,6 | 129,6 | 130,0 |
Muutos lähtötilanteesta1 | ‑0,4 | ‑4,5 | ‑5,2 |
Ero lumelääkkeeseen1 (95 % lv) | ‑4,1* (‑6,2, ‑2,1) | ‑4,8* (‑6,9, ‑2,7) | |
Yhdistettynä metformiiniin ja sulfonyyliureaana | |||
Lumehoito | Empagliflotsiini | ||
10 mg | 25 mg | ||
N | 225 | 225 | 216 |
HbA1c mmol/mol [%] | |||
Lähtötilanne (keskiarvo) | 65,57 [8,15] | 64,66 [8,07] | 64,98 [8,10] |
Muutos lähtötilanteesta1 | ‑1,90 [-0,17] | ‑8,92 [-0,82] | ‑8,38 [-0,77] |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑7,02* (‑8,67, ‑5,38) [-0,64* (-0,79, -0,49)] | ‑6,48* (‑8,14, ‑4,81) [-0,59* (-0,74, -0,44)] | |
N | 216 | 209 | 202 |
HbA1c‑arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c‑arvon ollessa ≥ 53 mmol/mol (7 %)2 | 9,3 | 26,3 | 32,2 |
N | 225 | 225 | 216 |
Paino (kg) | |||
Lähtötilanne (keskiarvo) | 76,23 | 77,08 | 77,50 |
Muutos lähtötilanteesta1 | ‑0,39 | ‑2,16 | ‑2,39 |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑1,76* (-2,25, ‑1,28) | ‑1,99* (‑2,48, ‑1,50) | |
N | 225 | 225 | 216 |
Systolinen verenpaine (SVP, mmHg)2 | |||
Lähtötilanne (keskiarvo) | 128,8 | 128,7 | 129,3 |
Muutos lähtötilanteesta1 | ‑1,4 | ‑4,1 | ‑3,5 |
Ero lumelääkkeeseen1 (95 % lv) | ‑2,7 (‑4,6, ‑0,8) | ‑2,1 (‑4,0, ‑0,2) | |
Yhdistettynä pioglitatsoni + metformiini -hoitoonb | |||
Lumehoito | Empagliflotsiini | ||
10 mg | 25 mg | ||
N | 124 | 125 | 127 |
HbA1c mmol/mol [%] | |||
Lähtötilanne (keskiarvo) | 65,60 [8,15] | 64,72 [8,07] | 65,05 [8,10] |
Muutos lähtötilanteesta1 | ‑1,18 [‑0,11] | ‑6,05 [‑0,55] | ‑7,69 [‑0,70] |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑4,87* (‑7,50, ‑2,24) [‑0,45* (‑0,69, ‑0,21)] | ‑6,51* (‑9,13, ‑3,89) [‑0,60* (‑0,83, ‑0,36)] | |
N | 118 | 116 | 123 |
HbA1c‑arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c‑arvon ollessa ≥ 53 mmol/mol (7 %)2 | 8,5 | 22,4 | 28,5 |
N | 124 | 125 | 127 |
Paino (kg) | |||
Lähtötilanne (keskiarvo) | 79,45 | 79,44 | 80,98 |
Muutos lähtötilanteesta1 | 0,40 | ‑1,74 | ‑1,59 |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑2,14* (‑2,93, ‑1,35) | ‑2,00* (‑2,78, ‑1,21) | |
N | 124 | 125 | 127 |
SVP (mmHg)2, 3 | |||
Lähtötilanne (keskiarvo) | 125,5 | 126,3 | 126,3 |
Muutos lähtötilanteesta1 | 0,8 | ‑3,5 | ‑3,3 |
Ero lumelääkkeeseen1 (95 % lv) | ‑4,2** (‑6,94, ‑1,53) | ‑4,1** (‑6,76, ‑1,37) | |
a Koko analyysijoukko (FAS) käyttäen viimeisimmästä havainnosta laskettua arviota (last observation carried forward, LOCF), ennen lisälääkityksen ottamista glukoositasapainon korjaamiseksi
b Alaryhmäanalyysi potilaista, jotka saivat lisäksi metformiinihoitoa (FAS, LOCF)
1 Korjattu keskiarvo lähtötilanteeseen nähden
2 Tilastollista merkitsevyyttä ei arvioitu perättäisen vahvistavan testimenetelmän vuoksi
3 LOCF, verenpainetta alentavan varalääkkeen jälkeiset arvot on poistettu
* p‑arvo < 0,0001
** p‑arvo < 0,01
Empagliflotsiini ja metformiini yhdistelmähoitona potilailla, jotka eivät aiemmin olleet saaneet lääkehoitoa
Empagliflotsiinin tehoa ja turvallisuutta arvioitiin 24 viikon pituisessa faktorikoeasetelmatutkimuksessa potilailla, jotka eivät aiemmin olleet saaneet lääkehoitoa. Empagliflotsiinin ja metformiinin yhdistelmähoito (5 mg ja 500 mg; 5 mg ja 1 000 mg; 12,5 mg ja 500 mg ja 12,5 mg ja 1 000 mg kaksi kertaa vuorokaudessa annettuna) paransi HbA1c‑arvoja tilastollisesti merkitsevästi (taulukko 4) ja johti suurempiin paastoglukoosiarvojen (yksittäisiin ainesosiin verrattuna) ja painon alenemiseen (metformiiniin verrattuna).
Taulukko 4: Tehon tulokset 24 viikon kohdalla, kun empagliflotsiinin ja metformiinin yhdistelmähoitoa verrattiin yksittäisiin ainesosiina
| Empagliflotsiini 10 mgb | Empagliflotsiini 25 mgb | Metformiinic | |||||
| + Met 1 000 mgc | + Met 2 000 mgc | Ei met-formii-nia | + Met 1 000 mgc | + Met 2 000 mgc | Ei met-formii-nia | 1 000 mg | 2 000 mg |
N | 161 | 167 | 169 | 165 | 169 | 163 | 167 | 162 |
HbA1c mmol/mol [%] | ||||||||
Lähtö-tilanne (keski-arvo) | 71,40 [8,68] | 71,04 [8,65] | 70,71 [8,62] | 73,07 [8,84] | 71,18 [8,66] | 73,27 [8,86] | 71,46 [8,69] | 69,97 [8,55] |
Muutos lähtötilan-teeseen nähden1 | ‑21,60 [‑1,98] | ‑22,66 [‑2,07] | ‑14,75 [‑1,35] | ‑21,11 [‑1,93] | ‑22,69 [‑2,08] | ‑14,85 [‑1,36] | ‑12,93 [‑1,18] | ‑19,09 [‑1,75] |
Vertailu empagli-flotsiiniin nähden (95 % lv)1 | ‑6,85* (‑9,38, ‑4,32) [-0,63* (-0,86, -0,40)] | ‑7,92* (‑10,43, ‑5,40) [-0,72* (-0,96, -0,49)] |
| ‑6,26* (‑8,82, ‑3,70) [-0,57* (-0,81, -0,34)] | ‑7,84* (‑10,38, ‑5,30) [-0,72* (-0,95, -0,48)] |
|
|
|
Vertailu metformii-niin nähden (95 % lv)1 | ‑8,67* (‑11,23, ‑6,11) [-0,79* (-1,03, -0,56)] | ‑3,57* (‑6,13, ‑1,01) [-0,33* (-0,56, -0,09)] |
| ‑8,18* (‑10,74, ‑5,63) [-0,75* (-0,98, -0,51)] | ‑3,60* (‑6,14, ‑1,05) [-0,33* (-0,56, -0,10)] |
|
|
|
Met = metformiini
1 Korjattu keskiarvo lähtötilanteeseen nähden
a Analyysit suoritettiin koko analyysiaineiston (FAS) perusteella havaittujen tapausten (OC) lähestymistapaa noudattaen
b Annettiin jaettuna kahteen yhtä suureen annokseen vuorokaudessa annettaessa yhdessä metformiinin kanssa
c Annettiin jaettuna kahteen yhtä suureen annokseen vuorokaudessa
* HbA1c‑arvon osalta p ≤ 0,0062
Empagliflotsiini potilailla, joiden hoitotasapaino on rittämätön, kun käytössä on metformiini ja linagliptiini
Potilailla, joiden hoitotasapaino oli riittämätön metformiinilla ja 5 mg linagliptiinilla, empagliflotsiinihoito 10 mg:n tai 25 mg:n annoksella alensi tilastollisesti merkitsevästi (p < 0,0001) HbA1c‑arvoa ja painoa lumelääkkeeseen verrattuna (taulukko 5). Lisäksi se alensi kliinisesti merkittävästi paastoglukoosiarvoa sekä systolista ja diastolista verenpainetta lumelääkkeeseen verrattuna.
Taulukko 5: Tehon tulokset 24 viikon lumekontrolloidussa tutkimuksessa potilailla, joiden hoitotasapaino oli riittämätön metformiinilla ja 5 mg linagliptiinilla
Metformiinin ja 5 mg linagliptiinin kanssa | |||
Lumehoito5 | Empagliflotsiini6 | ||
10 mg | 25 mg | ||
N | 106 | 109 | 110 |
HbA1c mmol/mol [%]3 | |||
Lähtötilanne (keskiarvo) | 63,45 [7,96] | 63,63 [7,97] | 63,64 [7,97] |
Muutos lähtötilanteesta1 | 1,49 [0,14] | ‑7,14 [-0,65] | ‑6,12 [-0,56] |
Ero lumelääkkeeseen(95 % lv) | ‑8,63* (‑11,19, ‑6,06) [-0,79* (-1,02, -0,55)] | ‑7,61* (‑10,18, ‑5,04) [-0,70* (-0,93, -0,46)] | |
N | 100 | 100 | 107 |
HbA1c‑arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c‑arvon ollessa ≥ 53 mmol/mol (7 %)2 | 17,0 | 37,0 | 32,7 |
N | 106 | 109 | 110 |
Paino (kg)3 | |||
Lähtötilanne (keskiarvo) | 82,3 | 88,4 | 84,4 |
Muutos lähtötilanteesta1 | ‑0,3 | ‑3,1 | ‑2,5 |
Ero lumelääkkeeseen(95 % lv) | ‑2,8* (‑3,5, ‑2,1) | ‑2,2* (‑2,9, ‑1,5) | |
N | 106 | 109 | 110 |
SVP (mmHg)4 | |||
Lähtötilanne (keskiarvo) | 130,1 | 130,4 | 131,0 |
Muutos lähtötilanteesta1 | ‑1,7 | ‑3,0 | ‑4,3 |
Ero lumelääkkeeseen(95 % CI) | ‑1,3 (‑4,2, 1,7) | ‑2,6 (‑5,5, 0,4) | |
1 Korjattu keskiarvo lähtötilanteeseen nähden
2 Tilastollista merkitsevyyttä ei arvioitu; ei mukana toissijaisten päätetapahtumien sekventiaalisessa testimenetelmässä.
3 FAS:n (OC) MMRM‑malli; mukana lähtötilanteen HbA1c‑arvo, lähtötilanteen eGFR‑arvo (MDRD), maantieteellinen alue, käynti ja hoito sekä käynnin ja hoidon vuorovaikutus. Painon osalta huomioitiin lähtötilanteen paino.
4 MMRM‑malli; mukana lineaarisina kovariaatteina lähtötilanteen SVP ja lähtötilanteen HbA1c‑arvo ja kiinteinä vaikutuksina lähtötilanteen eGFR-arvo, maantieteellinen alue, hoito, käynti sekä käynnin ja hoidon vuorovaikutus.
5 Lumeryhmään satunnaistetut potilaat saivat lumelääkettä ja 5 mg linagliptiinia sekä lisäksi metformiinia
6 Empagliflotsiini 10 mg tai empagliflotsiini 25 mg -ryhmään satunnaistetut potilaat saivat 10 mg tai 25 mg empagliflotsiinia ja 5 mg linagliptiinia sekä lisäksi metformiinia
* p‑arvo < 0,0001
Ennalta määrätyssä alaryhmässä, jossa potilaiden lähtötilanteen HbA1c‑arvo oli suurempi tai yhtä suuri kuin 69 mmol/mol (8,5 %), HbA1c‑arvon alenema oli lähtötilanteeseen nähden ‑13,6 mmol/mol (‑1,3 %) empagliflotsiini 10 mg tai empagliflotsiini 25 mg -hoidossa viikolla 24 (p < 0,0001) lumelääkkeeseen verrattuna.
Empagliflotsiini-hoidon 24 kuukauden tulokset yhdistettynä metformiiniin ja verrattuna glimepiridiin
Tutkimuksessa, jossa verrattiin 25 mg empagliflotsiinin tehoa ja turvallisuutta glimepiridiin (korkeintaan 4 mg/vrk) potilailla, joiden glukoositasapaino oli riittämätön pelkkää metformiinia käytettäessä, päivittäinen empagliflotsiinihoito alensi HbA1c‑arvoa enemmän kuin glimepiridi (taulukko 6). Lisäksi päivittäinen empagliflotsiinihoito alensi paastoglukoosiarvoa kliinisesti merkittävästi glimepiridiin verrattuna. Päivittäin käytetty empagliflotsiini alensi glimepiridiin verrattuna tilastollisesti merkitsevästi painoa, systolista ja diastolista verenpainetta ja pienensi tilastollisesti merkitsevästi niiden potilaiden osuutta, joilla oli hypoglykeemisiä tapahtumia (2,5 % empagliflotsiinihoitoa ja 24,2 % glimepiridihoitoa saaneista, p < 0,0001).
Taulukko 6: Tehon tulokset viikolla 104 aktiivikontrolloidussa tutkimuksessa, jossa verrattiin empagliflotsiinia glimepiridiin metformiinin lisähoitonaa
Empagliflotsiini 25 mg | Glimepiridib | |
N | 765 | 780 |
HbA1c mmol/mol [%] | ||
Lähtötilanne (keskiarvo) | 63,02 [7,92] | 63,01 [7,92] |
Muutos lähtötilanteesta1 | ‑7,18 [-0,66] | ‑6,02 [-0,55] |
Ero glimepiridiin1 (97,5 % lv) | ‑1,16* (‑2,24, ‑0,09) [-0,11* (-0,20, -0,01)] | |
N | 690 | 715 |
HbA1c‑arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c‑arvon ollessa ≥ 53 mmol/mol (7 %)2 | 33,6 | 30,9 |
N | 765 | 780 |
Paino (kg) | ||
Lähtötilanne (keskiarvo) | 82,52 | 83,03 |
Muutos lähtötilanteesta1 | ‑3,12 | 1,34 |
Ero glimepiridiin1 (97,5 % lv) | ‑4,46** (‑4,87, ‑4,05) | |
N | 765 | 780 |
SVP (mmHg)3 | ||
Lähtötilanne (keskiarvo) | 133,4 | 133,5 |
Muutos lähtötilanteesta1 | ‑3,1 | 2,5 |
Ero glimepiridiin1 (97,5 % lv) | ‑5,6** (‑7,0, ‑4,2) | |
a Koko analyysijoukko (FAS) käyttäen viimeisimmästä havainnosta laskettua arviota (last observation carried forward, LOCF), ennen lisälääkityksen ottamista glukoositasapainon korjaamiseksi
b Korkeintaan 4 mg glimepiridiä
1 Korjattu keskiarvo lähtötilanteeseen nähden
2 Tilastollista merkitsevyyttä ei arvioitu perättäisen vahvistavan testimenetelmän vuoksi
3 LOCF, verenpainetta alentavan varalääkkeen jälkeiset arvot on poistettu
* p‑arvo < 0,0001 samanveroisuudelle ja p‑arvo = 0,0153 paremmuudelle
** p‑arvo < 0,0001
Yhdistettynä insuliinihoitoon
Empagliflotsiini yhdistettynä päivittäiseen moniannoksiseen insuliiniin
Empagliflotsiinin tehoa ja turvallisuutta yhdistettynä päivittäiseen moniannoksiseen insuliiniin samanaikaisesti käytetyn metformiinihoidon kanssa arvioitiin kaksoissokkoutetussa, lumekontrolloidussa, 52 viikon pituisessa tutkimuksessa. Ensimmäisten 18 viikon ja viimeisten 12 viikon aikana insuliiniannos pidettiin vakaana, mutta viikkojen 19 ja 40 välillä sitä säädettiin, jotta saavutettiin ateriaa edeltävä glukoosiarvo < 5,5 mmol/l (< 100 mg/dl) ja aterianjälkeinen glukoosiarvo < 7,8 mmol/l (< 140 mg/dl).
Viikolla 18 empagliflotsiini paransi tilastollisesti merkitsevästi HbA1c‑arvoa lumelääkkeeseen verrattuna (taulukko 7).
Viikolla 52 empagliflotsiinihoito alensi tilastollisesti merkitsevästi HbA1c‑arvoa ja vähensi insuliinin tarvetta lumelääkkeeseen verrattuna sekä alensi painoa.
Taulukko 7: Tehon tulokset viikoilla 18 ja 52 lumekontrolloidussa tutkimuksessa, jossa empagliflotsiini yhdistettiin päivittäiseen moniannoksiseen insuliiniin metformiinihoidon kanssa
Lumehoito | Empagliflotsiini | |||
10 mg | 25 mg | |||
N | 135 | 128 | 137 | |
HbA1c mmol/mol [%] viikolla 18a | ||||
Lähtötilanne (keskiarvo) | 67,05 [8,29] | 68,55 [8,42] | 67,07 [8,29] | |
Muutos lähtötilanteesta1 | ‑6,22 [‑0,58] | ‑10,84 [‑0,99] | ‑11,27 [‑1,03] | |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑4,62* (‑6,83, ‑2,42) [‑0,41* (‑0,61, ‑0,21)] | ‑5,06* (‑7,22, ‑2,89) [‑0,45* (‑0,65, ‑0,25)] | ||
N | 86 | 84 | 87 | |
HbA1c mmol/mol (%) viikolla 52b | ||||
Lähtötilanne (keskiarvo) | 66,77 [8,26] | 68,62 [8,43] | 68,08 [8,38] | |
Muutos lähtötilanteesta1 | ‑9,30 [‑0,86] | ‑13,50 [‑1,23] | ‑14,31 [‑1,31] | |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑4,20** (‑7,32, ‑1,08) [‑0,37** (‑0,67, ‑0,08)] | ‑5,00* (‑8,08, ‑1,92) [‑0,45* (‑0,74, ‑0,16)] | ||
N | 84 | 84 | 87 | |
HbA1c‑arvon < 53 mmol/mol (7 %) viikolla 52 saavuttaneet potilaat (%) lähtötilanteen HbA1c‑arvon ollessa ≥ 53 mmol/mol (7 %)b, 2 | 27,4 | 41,7 | 48,3 | |
N | 86 | 83 | 86 | |
Insuliiniannos (ky/vrk) viikolla 52b, 3 | ||||
Lähtötilanne (keskiarvo) | 91,01 | 91,77 | 90,22 | |
Muutos lähtötilanteesta1 | 12,84 | 0,22 | ‑2,25 | |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑12,61** (‑21,43, ‑3,80) | ‑15,09** (‑23,79, ‑6,40) | ||
N | 86 | 84 | 87 | |
Paino (kg) viikolla 52b | ||||
Lähtötilanne (keskiarvo) | 97,78 | 98,86 | 94,93 | |
Muutos lähtötilanteesta1 | 0,42 | ‑2,47 | ‑1,94 | |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑2,89* (‑4,29, ‑1,49) | ‑2,37* (‑3,75, ‑0,98) | ||
a Alaryhmäanalyysi potilaista, jotka saivat lisäksi metformiinihoitoa (koko analyysijoukko, LOCF)
b Alaryhmäanalyysi potilaista, jotka saivat lisäksi metformiinihoitoa (tutkimussuunnitelman mukaisen hoidon suorittanut populaatio, LOCF)
1 Korjattu keskiarvo lähtötilanteeseen nähden
2 Tilastollista merkitsevyyttä ei arvioitu perättäisen vahvistavan testimenetelmän vuoksi
3 Viikot 19‑40: Etukäteen määriteltyjen tavoiteglukoosiarvojen saavuttamiseen pyrkivä hoito (ateriaa edeltävä < 5,5 mmol/l [< 100 mg/dl], aterianjälkeinen < 7,8 mmol/l [< 140 mg/dl]) insuliiniannoksia muuttamalla
* p‑arvo ≤ 0,0005
** p‑arvo < 0,005
Empagliflotsiini yhdistettynä perusinsuliiniin
Empagliflotsiinin tehoa ja turvallisuutta yhdistettynä perusinsuliiniin metformiinin kanssa arvioitiin kaksoissokkoutetussa, lumekontrolloidussa, 78 viikon pituisessa tutkimuksessa. Ensimmäisten 18 viikon aikana insuliiniannos pidettiin vakaana, mutta seuraavien 60 viikon aikana sitä muutettiin, jotta saavutettiin paastoglukoosiarvo < 6,1 mmol/l.
Viikolla 18 empagliflotsiini paransi HbA1c‑arvoa tilastollisesti merkitsevästi. Suurempi osa empagliflotsiinia saaneista potilaista, joilla HbA1c‑arvo oli lähtötilanteessa ≥ 53 mmol/mol (7,0 %), saavutti HbA1c‑tavoitearvon < 53 mmol/mol (7 %) verrattuna lumelääkkeeseen (taulukko 8).
Viikolla 78 empagliflotsiinista johtuva alentunut HbA1c‑arvo ja vähentynyt insuliinin tarve säilyivät ennallaan. Empagliflotsiini alensi myös paastoglukoosiarvoja, painoa ja verenpainetta.
Taulukko 8: Tehon tulokset viikoilla 18 ja 78 lumekontrolloidussa tutkimuksessa, jossa empagliflotsiini yhdistettiin perusinsuliiniin metformiinin kanssa a
Lumehoito | Empagliflotsiini 10 mg | Empagliflotsiini 25 mg | |
N | 96 | 107 | 99 |
HbA1c mmol/mol [%] viikolla 18 | |||
Lähtötilanne (keskiarvo) | 64,14 [8,02] | 66,18 [8,21] | 67,71 [8,35] |
Muutos lähtötilanteesta1 | ‑0,93 [‑0,09] | ‑6,79 [‑0,62] | ‑7,83 [‑0,72] |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑5,86* (‑8,46, ‑3,25) [‑0,54* (‑0,77, ‑0,30)] | ‑6,90* (‑9,58, ‑4,22) [‑0,63* (‑0,88, ‑0,39)] | |
N | 89 | 105 | 94 |
HbA1c mmol/mol [%] viikolla 78 | |||
Lähtötilanne (keskiarvo) | 64,21 [8,03] | 66,52 [8,24] | 67,04 [8,29] |
Muutos lähtötilanteesta1 | ‑0,88 [‑0,08] | ‑4,63 [‑0,42] | ‑7,73 [‑0,71] |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑3,76** (‑6,96, ‑0,56) [‑0,34** (‑0,64, ‑0,05)] | ‑6,85* (‑10,14, ‑3,56) [‑0,63* (‑0,93, ‑0,33)] | |
N | 89 | 105 | 94 |
Perusinsuliiniannos (ky/vrk) viikolla 78 | |||
Lähtötilanne (keskiarvo) | 49,61 | 47,25 | 49,37 |
Muutos lähtötilanteesta1 | 4,14 | ‑2,07 | ‑0,28 |
Ero lumelääkkeeseen1 (97,5 % lv) | ‑6,21** (‑11,81, ‑0,61) | ‑4,42 (‑10,18, 1,34) |
a Alaryhmäanalyysi koko analyysijoukosta (FAS), joka sai lisäksi metformiinihoitoa (käyttäen viimeisimmästä havainnosta laskettua arviota [LOCF] ennen lisälääkityksen ottamista glukoositasapainon korjaamiseksi)
1 Korjattu keskiarvo lähtötilanteeseen nähden
* p‑arvo < 0,0001
** p‑arvo ≤ 0,025
Empagliflotsiini ja linagliptiini yhdistettynä metformiiniin
Kaksoissokkoutetussa tutkimuksessa potilailla, joiden glukoositasapaino oli riittämätön, 24 viikon hoito molemmilla empagliflotsiiniannoksilla ja linagliptiinilla metformiinin lisänä alensi tilastollisesti merkitsevästi (p < 0,0001) HbA1c‑arvoja (muutos lähtötilanteesta ‑11,82 mmol/mol (‑1,08 %) empagliflotsiini 10 mg + linagliptiini 5 mg ‑ryhmässä, ‑13,05 mmol/mol (‑1,19 %) empagliflotsiini 25 mg + linagliptiini 5 mg ‑ryhmässä ja ‑7,61 mmol/mol (‑0,70 %) linagliptiini 5 mg ‑ryhmässä). Monoterapiana käytettyyn linagliptiiniin (5 mg) verrattuna molemmat empagliflotsiiniannokset yhdessä linagliptiini 5 mg:n kanssa alensivat paastoglukoosiarvoja ja verenpainetta tilastollisesti merkitsevästi. Molemmat annokset alensivat samankaltaisesti ja tilastollisesti merkitsevästi painoa, ilmaistuna kilogrammoina ja prosentuaalisena muutoksena. Suurempi osa empagliflotsiinia yhdessä linagliptiinin kanssa saaneista potilaista, joilla HbA1c‑arvo oli lähtötilanteessa ≥ 53 mmol/mol (7,0 %), saavutti HbA1c‑tavoitearvon < 53 mmol/mol (7 %) verrattuna monoterapiana linagliptiinia 5 mg saaneiden ryhmään. Kliinisesti merkittävästi alentuneet HbA1c‑arvot säilyivät 52 viikon ajan.
Empagliflotsiini kahdesti vuorokaudessa vs. kerran vuorokaudessa yhdistettynä metformiiniin
Kahdesti vuorokaudessa ja kerran vuorokaudessa annetun empagliflotsiinin (10 mg ja 25 mg vuorokausiannos) tehoa ja turvallisuutta metformiinin lisänä arvioitiin kaksoissokkoutetussa, lumekontrolloidussa, 16 viikon pituisessa tutkimuksessa metformiinihoitoa saaneilla potilailla, joiden glukoositasapaino oli riittämätön. Kaikki empagliflotsiinihoidot alensivat HbA1c‑arvoja merkitsevästi lähtötilanteesta [kokonaiskeskiarvo 61,7 mmol/mol (7,8 %)] 16 viikon jälkeen lumelääkkeeseen verrattuna. Kun empagliflotsiiniannos annettiin kahdesti vuorokaudessa metformiinitaustahoidon lisänä, HbA1c‑arvot alenivat vertailukelpoisesti verrattuna kerran vuorokaudessa tapahtuvaan lääkkeen annosteluun. Hoitojen väliset erot HbA1c‑arvojen alenemisessa lähtötilanteesta viikolle 16 olivat ‑0,22 mmol/mol (‑0,02 %) [95 % lv ‑1,80, 1,37 (‑0,16; 0,13)] empagliflotsiini 5 mg kahdesti vuorokaudessa vs. 10 mg kerran vuorokaudessa ja ‑1,26 mmol/mol (‑0,11 %) [95 % lv ‑2,84, 0,33 (‑0,26; 0,03)] empagliflotsiini 12,5 mg kahdesti vuorokaudessa vs. 25 mg kerran vuorokaudessa.
Sydän- ja verisuonisairauksiin liittyvät tulokset
Lumekontrolloitu EMPA-REG OUTCOME -kaksoissokkotutkimus vertasi yhdistetyssä analyysissä 10 mg:n ja 25 mg:n empagliflotsiiniannoksia lumelääkkeeseen; lisättynä tavanomaisen hoidon rinnalle tyypin 2 diabetespotilailla, joilla oli sydän- ja verisuonisairaus. Kaikkiaan 7 020 potilasta sai hoitoa (empagliflotsiini 10 mg: 2 345, empagliflotsiini 25 mg: 2 342, lumelääke: 2 333), ja seurantajakson mediaanipituus oli 3,1 vuotta. Keski-ikä oli 63 vuotta, keskimääräinen HbA1c‑arvo oli 64,7 mmol/mol (8,1 %) ja 71,5 % potilaista oli miehiä. Lähtötilanteessa 74 % potilaista sai metformiini‑, 48 % insuliini- ja 43 % sulfonyyliureahoitoa. Noin puolella potilaista (52,2 %) eGFR oli 60‑90 ml/min/1,73 m2, 17,8 %:lla 4560 ml/min/1,73 m2 ja 7,7 %:lla 30‑45 ml/min/1,73 m2.
Viikolla 12 korjattu keskimääräinen parannus (keskivirhe) HbA1c‑arvossa lähtötilanteeseen nähden oli lumeryhmässä 1,20 mmol/mol (0,16) [0,11 % (0,02)], empagliflotsiini 10 mg ‑ryhmässä ‑7,06 mmol/mol (0,16) [0,65 % (0,02)] ja empagliflotsiini 25 mg ‑ryhmässä ‑7,71 mmol/mol (0,16) [0,71 % (0,02)]. Ensimmäisten 12 viikon jälkeen glukoositasapainoa optimoitiin tutkimushoidosta riippumatta. Siksi vaikutus oli viikolla 94 heikentynyt; korjattu keskimääräinen parannus (keskivirhe) HbA1c‑arvossa oli lumeryhmässä ‑0,89 mmol/mol (0,25) [0,08 % (0,02)], empagliflotsiini 10 mg ‑ryhmässä ‑5,49 mmol/mol (0,25) [0,50 % (0,02)] ja empagliflotsiini 25 mg ‑ryhmässä ‑6,05 mmol/mol (0,25) [0,55 % (0,02)].
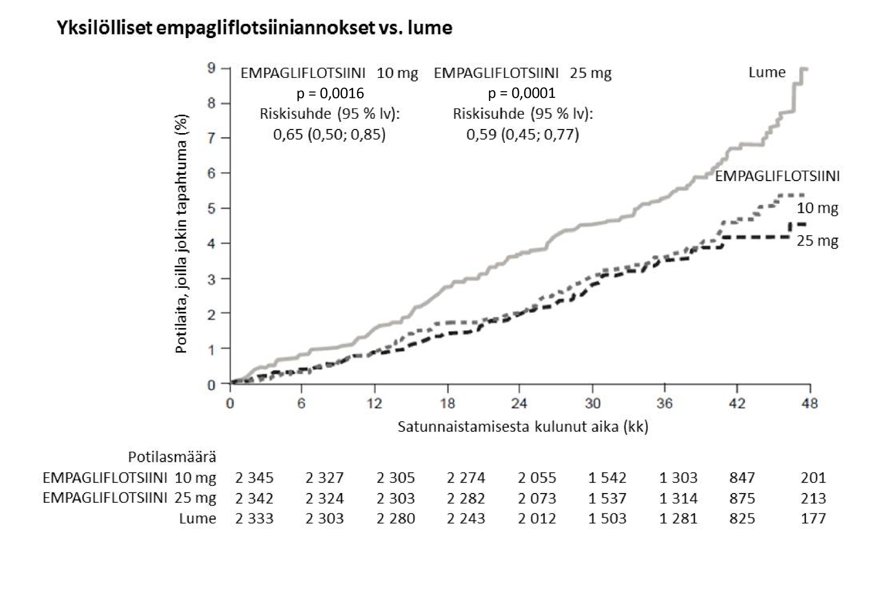
Empagliflotsiini oli parempi ehkäisemään ensisijaista yhdistelmäpäätetapahtumaa eli sydän- ja verisuonikuolemia, ei-kuolemaan johtaneita sydäninfarkteja tai ei-kuolemaan johtaneita aivoinfarkteja lumehoitoon verrattuna. Hoidon vaikutus johtui pääasiassa sydän‑ ja verisuonikuolemien merkitsevästä vähenemisestä. Ei-kuolemaan johtaneissa sydäninfarkteissa tai ei-kuolemaan johtaneissa aivoinfarkteissa ei ollut merkitsevää muutosta. Sydän- ja verisuonikuolemat vähenivät empagliflotsiini 10 mg- ja 25 mg ‑hoidossa yhtä paljon (kuva 1), ja se näkyi myös kokonaiseloonjäännin paranemisena (taulukko 9). EMPA-REG OUTCOME ‑tutkimuksessa empagliflotsiinin vaikutus ensisijaiseen yhdistelmäpäätetapahtumaan eli sydän- ja verisuonikuolemiin, ei-kuolemaan johtaneisiin sydäninfarkteihin tai ei-kuolemaan johtaneisiin aivoinfarkteihin oli yleisesti riippumaton glukoositasapainosta tai munuaistoiminnasta (eGFR). Empagliflotsiinin vaikutus oli yleisesti yhtäläinen eri eGFR‑luokissa aina eGFR‑tasoon 30 ml/min/1,73 m2 saakka.
EMPA-REG OUTCOME -tutkimuksessa DPP‑4:n estäjien käyttäjät ja tummaihoiset potilaat olivat rajallisesti edustettuina. Tehoa sydän‑ ja verisuonikuolemien ehkäisemisessä ei ole vakuuttavasti osoitettu tällaisilla potilailla, jotka käyttävät samanaikaisesti empagliflotsiinia.
Taulukko 9: Hoidon vaikutus ensisijaiseen yhdistelmäpäätetapahtumaan, sen osatekijöihin ja kuolleisuuteena
Lumelääke | Empagliflotsiinib | |
N | 2 333 | 4 687 |
Ensimmäiseen sydän- ja verisuonikuolemaan, ei-kuolemaan johtaneeseen sydäninfarktiin tai ei-kuolemaan johtaneeseen aivoinfarktiin kulunut aika N (%) | 282 (12,1) | 490 (10,5) |
Riskisuhde vs. lume (95,02 % lv)* |
| 0,86 (0,74; 0,99) |
Paremmuuden p‑arvo |
| 0,0382 |
Sydän- ja verisuonikuolemat N (%) | 137 (5,9) | 172 (3,7) |
Riskisuhde vs. lume (95 % lv) |
| 0,62 (0,49; 0,77) |
p‑arvo |
| < 0,0001 |
Ei-kuolemaan johtanut sydäninfarkti N (%) | 121 (5,2) | 213 (4,5) |
Riskisuhde vs. lume (95 % lv) |
| 0,87 (0,70; 1,09) |
p‑arvo |
| 0,2189 |
Ei-kuolemaan johtanut aivoinfarkti N (%) | 60 (2,6) | 150 (3,2) |
Riskisuhde vs. lume (95 % lv) |
| 1,24 (0,92; 1,67) |
p‑arvo |
| 0,1638 |
Kokonaiskuolleisuus N (%) | 194 (8,3) | 269 (5,7) |
Riskisuhde vs. lume (95 % lv) |
| 0,68 (0,57; 0,82) |
p‑arvo |
| < 0,0001 |
Muu kuin sydän- ja verisuonikuolleisuus N (%) | 57 (2,4) | 97 (2,1) |
Riskisuhde vs. lume (95 % lv) |
| 0,84 (0,60; 1,16) |
a Hoidetut potilaat eli potilaat, jotka olivat saaneet vähintään yhden annoksen tutkittavaa lääkettä
b 10 mg:n ja 25 mg:n empagliflotsiiniannosten yhdistetty analyysi
* Koska tutkimustulokset otettiin mukaan välianalyysiin, sovellettiin kaksipuolista 95,02 %:n luottamusväliä, joka vastaa merkitsevyyttä kuvaavaa p‑arvoa < 0,0498.
Kuva 1. Sydän- ja verisuonikuolemaan kulunut aika EMPA-REG OUTCOME -tutkimuksessa
Sairaalahoitoa vaativa sydämen vajaatoiminta
EMPA-REG OUTCOME -tutkimuksessa empagliflotsiini vähensi sydämen vajaatoiminnasta johtuvien sairaalahoitojen riskiä lumelääkkeeseen verrattuna (empagliflotsiini 2,7 %; lumelääke 4,1 %; riskisuhde 0,65; 95 %:n luottamusväli 0,50; 0,85).
Nefropatia
EMPA-REG OUTCOME -tutkimuksessa ensimmäiseen nefropatiatapahtumaan kuluneen ajan riskisuhde oli 0,61 (95 %:n luottamusväli 0,53; 0,70) empagliflotsiinille (12,7 %) lumelääkkeeseen verrattuna (18,8 %).
Lisäksi empagliflotsiinihoitoa saaneilla potilailla, joilla oli lähtötilanteessa makroalbuminuria, esiintyi enemmän (riskisuhde 1,82; 95 %:n luottamusväli 1,40; 2,37) pitkäkestoista normo- tai mikroalbuminuriaa (49,7 %) lumelääkkeeseen verrattuna (28,8 %).
Glukoosipitoisuus 2 tuntia aterian jälkeen
Empagliflotsiinihoito metformiinin tai metformiinin ja sulfonyyliurean lisänä alensi kliinisesti merkittävästi 2 tuntia aterian jälkeen mitattua glukoosipitoisuutta (ateriarasituskoe) viikolla 24 (metformiinin lisänä: lume: + 0,3 mmol/l, empagliflotsiini 10 mg: ‑2,6 mmol/l, empagliflotsiini 25 mg: ‑2,5 mmol/l; metformiinin + sulfonyyliurean lisänä: lume: ‑0,1 mmol/l, empagliflotsiini 10 mg: ‑2,0 mmol/l, empagliflotsiini 25 mg: ‑2,0 mmol/l).
Potilaat, joilla lähtötilanteen HbA1c‑arvo ≥ 75 mmol/mol (9 %)
Kun ennalta määritelty analyysi tehtiin tutkittavista, joilla HbA1c‑arvo oli lähtötilanteessa ≥ 75 mmol/mol (9,0 %), empagliflotsiini 10 mg tai 25 mg metformiinin lisänä alensi HbA1c‑arvoja tilastollisesti merkitsevästi viikolla 24 [korjattu keskimuutos lähtötilanteesta ‑16,29 mmol/mol (‑1,49 %) empagliflotsiini 25 mg ‑ryhmässä, ‑15,27 mmol/mol (‑1,40 %) empagliflotsiini 10 mg ‑ryhmässä ja ‑4,81 mmol/mol (‑0,44 %) lumeryhmässä].
Paino
Neljän lumekontrolloidun tutkimuksen ennalta määritellyssä yhdistetyssä analyysissä empagliflotsiinihoito (68 % kaikista potilaista sai metformiinitaustahoitoa) alensi painoa lumelääkkeeseen verrattuna (‑2,04 kg 10 mg empagliflotsiinia saaneilla, ‑2,26 kg 25 mg empagliflotsiinia saaneilla ja ‑0,24 kg lumelääkettä saaneilla) viikolla 24. Painon lasku säilyi viikolle 52 asti (‑1,96 kg 10 mg empagliflotsiinia saaneilla, ‑2,25 kg 25 mg empagliflotsiinia saaneilla ja ‑0,16 kg lumelääkettä saaneilla).
Verenpaine
Empagliflotsiinin tehoa ja turvallisuutta arvioitiin kaksoissokkoutetussa, lumekontrolloidussa, 12 viikon pituisessa tutkimuksessa, johon osallistui tyypin 2 diabetesta sairastavia potilaita, joilla oli kohonnut verenpaine ja käytössään eri diabeteslääkkeitä ja korkeintaan kaksi verenpainetta alentavaa lääkitystä. Empagliflotsiinihoito kerran vuorokaudessa paransi tilastollisesti merkitsevästi HbA1c‑arvoa sekä 24 tunnin keskimääräistä systolista ja diastolista verenpainetta ambulatorisessa verenpaineseurannassa (taulukko 10). Empagliflotsiinihoito alensi istuma-asennossa mitattua systolista ja diastolista verenpainetta.
Taulukko 10: Tehon tulokset viikolla 12 lumekontrolloidussa empagliflotsiinitutkimuksessa, johon osallistui tyypin 2 diabetesta ja kontrolloimatonta verenpainetta sairastavia potilaitaa
Lumehoito | Empagliflotsiini | ||
10 mg | 25 mg | ||
N | 271 | 276 | 276 |
HbA1c mmol/mol [%] viikolla 121 | |||
Lähtötilanne (keskiarvo) | 62,88 [7,90] | 62,55 [7,87] | 63,03 [7,92] |
Muutos lähtötilanteesta2 | 0,31 [0,03] | ‑6,49 [-0,59] | ‑6,82 [-0,62] |
Ero lumelääkkeeseen1 (95 % lv)2 | ‑6,80* (‑7,88, ‑5,73) [-0,62* (-0,72, -0,52)] | ‑7,12* (‑8,19, ‑6,06) [-0,65* (-0,75, -0,55)] | |
Systolinen verenpaine 24 tunnin kohdalla viikolla 123 | |||
Lähtötilanne (keskiarvo) | 131,72 | 131,34 | 131,18 |
Muutos lähtötilanteesta4 | 0,48 | ‑2,95 | ‑3,68 |
Ero lumelääkkeeseen4 (95 % lv) | ‑3,44* (‑4,78, ‑2,09) | ‑4,16* (‑5,50, ‑2,83) | |
Diastolinen verenpaine 24 tunnin kohdalla viikolla 123 | |||
Lähtötilanne (keskiarvo) | 75,16 | 75,13 | 74,64 |
Muutos lähtötilanteesta5 | 0,32 | ‑1,04 | ‑1,40 |
Ero lumelääkkeeseen5 (95 % lv) | ‑1,36** (‑2,15, ‑0,56) | ‑1,72* (‑2,51, ‑0,93) | |
a Koko analyysijoukko
1 LOCF, diabetestasapainoa korjaavan lisälääkityksen ottamisen jälkeiset arvot on poistettu
2 Korjattu keskiarvo lähtötilanteen HbA1c‑arvoon, lähtötilanteen eGFR‑arvoon, maantieteelliseen alueeseen ja verenpainelääkevalmisteiden lukumäärään nähden
3 LOCF, diabetestasapainoa korjaavan lisälääkityksen ottamisen jälkeiset arvot tai verenpainetta alentavan lisälääkkeen vaihtamisen jälkeiset arvot on poistettu
4 Korjattu keskiarvo lähtötilanteen systoliseen verenpaineeseen, lähtötilanteen HbA1c‑arvoon, lähtötilanteen eGFR‑arvoon, maantieteelliseen alueeseen ja verenpainetta alentavien lääkevalmisteiden lukumäärään nähden
5 Korjattu keskiarvo lähtötilanteen diastoliseen verenpaineeseen, lähtötilanteen HbA1c‑arvoon, lähtötilanteen eGFR‑arvoon, maantieteelliseen alueeseen ja verenpainetta alentavien lääkevalmisteiden lukumäärään nähden
* p‑arvo < 0,0001
** p‑arvo < 0,001
Neljän lumekontrolloidun tutkimuksen ennalta määritellyssä yhteisanalyysissä empagliflotsiinihoito (68 % kaikista potilaista sai metformiinitaustahoitoa) alensi systolista verenpainetta (10 mg empagliflotsiinia saaneilla ‑3,9 mmHg, 25 mg empagliflotsiinia saaneilla ‑4,3 mmHg) verrattuna lumelääkkeeseen (‑0,5 mmHg) ja diastolista verenpainetta (10 mg empagliflotsiinia saaneilla ‑1,8 mmHg, 25 mg empagliflotsiinia saaneilla ‑2,0 mmHg) verrattuna lumelääkkeeseen (‑0,5 mmHg). Verenpaineen aleneminen havaittiin viikolla 24, ja se säilyi viikolle 52 asti.
Metformiini
Tehostetun glukoositasapainon hoidon pitkäaikaiset edut tyypin 2 diabeteksen hoidossa on vahvistettu prospektiivisessa, satunnaistetussa UKPDS‑tutkimuksessa. Kun ylipainoisia potilaita hoidettiin metformiinilla pelkän ruokavaliohoidon epäonnistuttua, tulosten analyysissä todettiin seuraavaa:
-
minkä tahansa diabeteskomplikaation absoluuttinen riski oli metformiiniryhmässä merkitsevästi pienempi (29,8 tapahtumaa/1 000 potilasvuotta) kuin pelkkää ruokavaliohoitoa käytettäessä (43,3 tapahtumaa/1 000 potilasvuotta), p = 0,0023, ja merkitsevästi pienempi kuin pelkkää sulfonyyliureaa tai pelkkää insuliinia käyttäneissä ryhmissä yhteensä (40,1 tapahtumaa/1 000 potilasvuotta), p = 0,0034
-
diabetekseen liittyvän kuolleisuuden absoluuttinen riski pieneni merkitsevästi (metformiini: 7,5 tapahtumaa/1 000 potilasvuotta, pelkkä ruokavalio: 12,7 tapahtumaa/1 000 potilasvuotta, p = 0,017)
-
kokonaiskuolleisuuden absoluuttinen riski oli metformiiniryhmässä merkitsevästi pienempi (13,5 tapahtumaa/1 000 potilasvuotta) kuin pelkkää ruokavaliohoitoa käytettäessä (20,6 tapahtumaa/1 000 potilasvuotta), p = 0,011, ja merkitsevästi pienempi kuin pelkkää sulfonyyliureaa tai pelkkää insuliinia käyttäneissä ryhmissä yhteensä (18,9 tapahtumaa/1 000 potilasvuotta), p = 0,021
-
sydäninfarktin absoluuttinen riski pieneni merkitsevästi (metformiini: 11 tapahtumaa/1 000 potilasvuotta, pelkkä ruokavalio: 18 tapahtumaa/1 000 potilasvuotta, p = 0,01).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Synjardy-valmisteen käytöstä tyypin 2 diabeteksen hoidossa kaikissa pediatrisissa potilasryhmissä syntymästä alle 10 vuoden ikään (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Kerran vuorokaudessa otettavan empagliflotsiinin (10 mg ja mahdollinen annoksen suurennus 25 mg:aan) ja linagliptiinin (5 mg) kliinistä tehoa ja turvallisuutta on tutkittu 10–17‑vuotiailla lapsilla ja nuorilla, joilla oli tyypin 2 diabetes, lumelääkekontrolloidussa tutkimuksessa (DINAMO) 26 viikon ajan ja turvallisuusjatkovaiheessa enintään 52 viikon ajan. Taustahoidot ruokavalion ja liikunnan lisänä sisälsivät metformiinia (51 %), metformiinin ja insuliinin yhdistelmää (40,1 %), insuliinia (3,2 %) tai ei mitään (5,7 %).
HbA1c-arvon mukautettu keskimääräinen muutos viikolla 26 empagliflotsiinin (N = 52) ja lumelääkkeen (N = 53) välillä oli ‑9,23 mmol/mol (‑0,84 %), ja se katsottiin kliinisesti merkittäväksi ja tilastollisesti merkitseväksi [95 % lv ‑16,39, ‑2,07 (‑1,50; ‑0,19); p = 0,0116]. Lisäksi empagliflotsiinihoidolla saavutettiin lumelääkkeeseen verrattuna kliinisesti merkittävä plasman paastoglukoosin mukautettu keskimääräinen muutos, ‑1,95 mmol/l (95 %:n luottamusväli: ‑3,25 – ‑0,65) (‑35,2 mg/dl [95 % lv ‑58,6; ‑11,7]). Arvot olivat ‑0,76 % (95 %:n luottamusväli: ‑1,45 %; ‑0,08 %) HbA1c:n osalta ja ‑2,12 mmol/l (95 %:n luottamusväli: ‑3,36 – ‑0,89) (‑38,28 mg/dl [95 %:n luottamusväli: ‑60,47 – ‑16,10]) paastoglukoosin osalta metformiinia saaneessa alaryhmässä (N = 48 empagliflotsiini, N = 47 lumelääke).
Farmakokinetiikka
Synjardy
Bioekvivalenssitutkimusten tulokset terveillä tutkittavilla osoittivat, että Synjardy-yhdistelmätablettihoito (empagliflotsiini/metformiinihydrokloridi, 5 mg/850 mg, 5 mg/1 000 mg, 12,5 mg/850 mg ja 12,5 mg/1 000 mg) on bioekvivalentti verrattuna erillisten empagliflotsiini- ja metformiinihydrokloriditablettien vastaaviin annoksiin.
Kun empagliflotsiini/metformiini 12,5 mg/1 000 mg -valmistetta annettiin ruokailun jälkeen empagliflotsiinin AUC‑arvo aleni 9 % ja Cmax‑arvo 28 % verrattuna paasto-olosuhteisiin. Metformiinin AUC‑arvo pieneni 12 % ja Cmax‑arvo 26 % verrattuna paasto-olosuhteisiin. Ruoan vaikutusta empagliflotsiiniin ja metformiiniin ei pidetä kliinisesti merkittävänä. Metformiini on kuitenkin suositeltavaa ottaa aterian yhteydessä, joten myös Synjardy on suositeltavaa ottaa ruokailun yhteydessä.
Synjardy-valmisteen kummankin vaikuttavan aineen farmakokineettisistä ominaisuuksista kerrotaan alla.
Empagliflotsiini
Imeytyminen
Empagliflotsiinin farmakokinetiikkaa on kuvattu laajasti terveillä vapaaehtoisilla ja tyypin 2 diabetesta sairastavilla potilailla. Suun kautta annettu empagliflotsiini imeytyi nopeasti, ja huippupitoisuus plasmassa (tmax‑arvon mediaani) saavutettiin keskimäärin 1,5 tuntia annoksen jälkeen. Tämän jälkeen pitoisuudet plasmassa alenivat kaksivaiheisesti (nopea jakautumisvaihe ja suhteellisen hidas terminaalinen vaihe). Vakaan tilan keskimääräinen plasman AUC oli 1 870 nmol.h/l ja Cmax 259 nmol/l, kun empagliflotsiinia otettiin 10 mg kerran vuorokaudessa, ja vastaavasti 4 740 nmol.h/l ja 687 nmol/l, kun empagliflotsiinia otettiin 25 mg kerran vuorokaudessa. Systeeminen empagliflotsiinialtistus suureni suhteessa annokseen. Farmakokineettiset parametrit olivat samaa luokkaa empagliflotsiinin kerta-annoksen jälkeen ja vakaassa tilassa, mikä viittaa farmakokinetiikan olevan lineaarinen ajan suhteen. Empagliflotsiinin farmakokinetiikassa ei ollut kliinisesti oleellisia eroja terveiden vapaaehtoisten ja tyypin 2 diabetesta sairastavien potilaiden välillä.
Kahdesti vuorokaudessa annettavan empagliflotsiinin (5 mg) ja kerran vuorokaudessa annettavan empagliflotsiinin (10 mg) farmakokinetiikkaa verrattiin terveillä tutkittavilla. Kun empagliflotsiinia annettiin 5 mg kahdesti vuorokaudessa, empagliflotsiinin kokonaisaltistus (AUCss) 24 tunnin aikana oli samaa luokkaa kuin silloin, kun empagliflotsiinia annettiin 10 mg kerran vuorokaudessa. Kuten oli odotettavissa, 5 mg empagliflotsiinia kahdesti vuorokaudessa pienensi Cmax‑arvoa enemmän ja suurensi empagliflotsiinin minimipitoisuutta plasmassa (Cmin) enemmän kuin 10 mg empagliflotsiinia kerran vuorokaudessa.
Kun empagliflotsiinia annettiin 25 mg runsasrasvaisen ja kaloripitoisen aterian jälkeen, altistus oli hieman vähäisempää; AUC pieneni noin 16 % ja Cmax noin 37 % paastototilanteeseen verrattuna. Havaittua ruoan vaikutusta empagliflotsiinin farmakokinetiikkaan ei pidetty kliinisesti merkittävänä ja empagliflotsiinia voidaan antaa ruoan kanssa tai ilman. Tulokset olivat samankaltaisia, kun Synjardy-yhdistelmätabletteja (empagliflotsiini/metformiini) annettiin runsaasti rasvaa ja kaloreita sisältävän aterian yhteydessä.
Jakautuminen
Näennäisen vakaan tilan jakautumistilavuuden arvioitiin olevan 73,8 litraa perustuen populaatiofarmakokineettisen analyysin. Kun terveille vapaaehtoisille annettiin suun kautta [14C]‑empagliflotsiiniliuosta, punasoluihin jakautui noin 37 % ja plasman proteiineihin sitoutui 86 %.
Biotransformaatio
Empagliflotsiinin merkittäviä metaboliitteja ei havaittu ihmisen plasmassa, kun määritelmänä oli vähintään 10 % lääkeaineen kokonaismäärästä, ja yleisimmät metaboliitit olivat kolme glukuronidikonjugaattia (2‑, 3‑ ja 6‑O‑glukuronidi). In vitro ‑tutkimukset viittaavat siihen, että empagliflotsiinin ensisijainen metaboliareitti ihmisillä on glukuronidaatio uridiini‑5’‑difosfo-glukuronosyylitransferaasien UGT2B7, UGT1A3, UGT1A8 ja UGT1A9 vaikutuksesta.
Eliminaatio
Populaatiofarmakokineettisen analyysin perusteella empagliflotsiinin näennäisen terminaalisen eliminaation puoliintumisajan arvioitiin olevan 12,4 tuntia ja näennäinen oraalinen puhdistuma oli 10,6 litraa/tunti. Suun kautta otetun empagliflotsiinin puhdistuma vaihteli tutkittavien välillä 39,1 % ja jäännöspitoisuus 35,8 %. Kun empagliflotsiinia otettiin kerran vuorokaudessa, vakaan tilan pitoisuus plasmassa saavutettiin viidennellä annoksella. Vakaassa tilassa havaittiin korkeintaan 22 % kertyminenplasman AUC:n suhteen, mikä oli yhdenmukaista puoliintumisajan kanssa. Kun terveille vapaaehtoisille annettiin suun kautta [14C]‑empagliflotsiiniliuosta, noin 96 % lääkeaineeseen liittyvästä radioaktiivisuudesta poistui ulosteen mukana (41 %) ja virtsan mukana (54 %). Ulosteessa suurin osa lääkeaineeseen liittyvästä radioaktiivisuudesta oli muuttumatonta kantalääkettä. Virtsaan erittyneestä lääkeaineeseen liittyvästä radioaktiivisuudesta noin puolet oli muuttumatonta kantalääkettä.
Erityisryhmät
Munuaisten vajaatoiminta
Lievää, kohtalaista tai vaikeaa munuaisten vajaatoimintaa sairastavilla (kreatiniinipuhdistuma < 30‑< 90 ml/min) ja loppuvaiheen munuaisten vajaatoimintaa (ESRD) sairastavilla empagliflotsiinin AUC suureni vastaavasti noin 18 %, 20 %, 66 % ja 48 % verrattuna tutkittaviin, joilla oli normaali munuaistoiminta. Empagliflotsiinin huippupitoisuudet plasmassa olivat kohtalaista munuaisten vajaatoimintaa ja loppuvaiheen munuaisten vajaatoimintaa sairastavilla samaa luokkaa kuin potilailla, joiden munuaistoiminta oli normaali. Empagliflotsiinin huippupitoisuudet plasmassa lievää ja vaikeaa munuaisten vajaatoimintaa sairastavilla olivat noin 20 % suuremmat kuin tutkittavilla, joiden munuaistoiminta oli normaali. Populaatiofarmakokineettinen analyysi osoitti, että suun kautta otetun empagliflotsiinin näennäinen puhdistuma pieneni kreatiniinipuhdistuman alenemisen myötä, mikä lisäsi lääkeainealtistusta.
Maksan vajaatoiminta
Child‑Pugh‑luokituksen mukaisesti lievää, kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavilla tutkittavilla empagliflotsiinin AUC suureni vastaavasti noin 23 %, 47 % ja 75 % ja Cmax vastaavasti noin 4 %, 23 % ja 48 % verrattuna tutkittaviin, joiden maksantoiminta oli normaali.
Painoindeksi
Painoindeksillä ei ollut kliinisesti merkittävää vaikutusta empagliflotsiinin farmakokinetiikkaan populaatiofarmakokineettisen analyysin perusteella. Tässä analyysissä AUC‑arvon arvioitiin olevan 5,82 %, 10,4 % ja 17,3 % alempi tutkittavilla, joiden painoindeksi oli vastaavasti 30, 35 ja 45 kg/m2, verrattuna tutkittaviin, joiden painoindeksi oli 25 kg/m2.
Sukupuoli
Sukupuolella ei ollut kliinisesti merkittävää vaikutusta empagliflotsiinin farmakokinetiikkaan populaatiofarmakokineettisen analyysin perusteella.
Etninen tausta
Populaatiofarmakokineettisessä analyysissä AUC‑arvon arvioitiin olevan 13,5 % suurempi aasialaisilla, joiden painoindeksi oli 25 kg/m2, verrattuna ei-aasialaisiin, joiden painoindeksi oli 25 kg/m2.
Iäkkäät
Iällä ei ollut kliinisesti merkittävää vaikutusta empagliflotsiinin farmakokinetiikkaan populaatiofarmakokineettisen analyysin perusteella.
Pediatriset potilaat
Pediatrisessa vaiheen 1 tutkimuksessa tutkittiin empagliflotsiinin (5 mg, 10 mg ja 25 mg) farmakokinetiikkaa ja farmakodynamiikkaa ≥ 10 ‑ < 18 vuoden ikäisillä tyypin 2 diabetesta sairastavilla lapsilla ja nuorilla. Havaitut farmakokineettiset ja farmakodynaamiset vasteet vastasivat aikuispotilaiden vasteita.
Pediatrisessa vaiheen 3 tutkimuksessa tutkittiin empagliflotsiinin 10 mg:n annoksen sekä mahdollisen 25 mg:aan suurennetun annoksen farmakokinetiikkaa ja farmakodynamiikkaa (HbA1c‑arvon muutosta lähtötilanteesta) 10–17 vuoden ikäisillä tyypin 2 diabetesta sairastavilla lapsilla ja nuorilla. Havaittu altistus-vastesuhde oli yleisesti ottaen verrattavissa aikuisten, lasten ja nuorten kesken. Empagliflotsiinin oraalinen annostelu tuotti altistuksen, joka oli aikuispotilailla havaitulla alueella.
Havaitut geometriset keskimääräiset pohjapitoisuudet ja geometriset keskimääräiset pitoisuudet 1,5 tuntia lääkkeen annon jälkeen vakaassa tilassa olivat 26,6 nmol/l ja 308 nmol/l empagliflotsiinin 10 mg:n vuorokausiannoksella ja 67,0 nmol/l ja 525 nmol/l empagliflotsiinin 25 mg:n vuorokausiannoksella.
Metformiini
Imeytyminen
Kun metformiini otetaan suun kautta, tmax on 2,5 tuntia. 500 mg tai 850 mg metformiinihydrokloriditablettien absoluuttinen biologinen hyötyosuus on terveillä henkilöillä noin 50‑60 %. 20‑30 % suun kautta otetusta annoksesta erittyi imeytymättömänä ulosteeseen. Suun kautta otetun metformiinin imeytyminen on saturoituvaa ja epätäydellistä. Metformiinin imeytymisen farmakokinetiikka on todennäköisesti ei-lineaarinen. Kun metformiinia käytetään suositusannoksina ja -ajankohtina, plasman vakaan tilan pitoisuudet saavutetaan 24‑48 tunnissa ja ne ovat yleensä alle 1 mikrog/ml. Kontrolloiduissa kliinisissä tutkimuksissa metformiinin maksimipitoisuudet plasmassa (Cmax) olivat maksimiannoksillakin enintään 5 mikrog/ml.
Ruoka vähentää ja hieman hidastaa metformiinin imeytymistä. Kun metformiinihydrokloridia otettiin 850 mg annos, plasman huippupitoisuus pieneni 40 %, AUC‑arvo pieneni 25 % ja plasman huippupitoisuuden saavuttamiseen kulunut aika piteni 35 minuuttia. Näiden havaintojen kliinistä merkitystä ei tunneta.
Jakautuminen
Sitoutuminen plasman proteiineihin on merkityksettömän vähäistä. Metformiini jakautuu punasoluihin. Veren huippupitoisuudet ovat pienemmät kuin plasman huippupitoisuudet, ja ne saavutetaan suurin piirtein samaan aikaan. Punasolut ovat todennäköisesti toissijainen jakautumisaitio. Keskimääräinen jakautumistilavuus (Vd) oli 63‑276 l.
Biotransformaatio
Metformiini erittyy muuttumattomana virtsaan. Ihmisellä ei ole havaittu metaboliitteja.
Eliminaatio
Metformiinin munuaispuhdistuma on > 400 ml/min, mikä viittaa siihen, että metformiini poistuu elimistöstä glomerulaarisen filtraation ja tubulaarisen sekreetion kautta. Suun kautta otetun annoksen näennäinen terminaalinen eliminaation puoliintumisaika on noin 6,5 h.
Jos munuaistoiminta heikkenee, munuaispuhdistuma pienenee suhteessa kreatiniinipuhdistumaan ja eliminaation puoliintumisaika pitenee, jolloin plasman metformiinipitoisuudet suurenevat.
Erityisryhmät
Pediatriset potilaat
Kerta-annostutkimus: 500 mg kerta-annosten jälkeen metformiinihydrokloridin farmakokinetiikka on ollut lapsipotilailla samankaltainen kuin terveillä aikuisilla.
Toistuvaisannostutkimus: Kun toistuvia 500 mg × 2 annoksia annettiin lapsipotilaille 7 vrk ajan, huippupitoisuus plasmassa (Cmax) pieneni noin 33 % ja systeeminen altistus (AUC0‑t) noin 40 % verrattuna aikuisiin diabeetikkoihin, jotka saivat toistuvia 500 mg × 2 annoksia 14 vrk ajan. Annos titrataan yksilöllisesti glukoositasapainon mukaan, joten ilmiön kliininen merkitys on vähäinen.
Prekliiniset tiedot turvallisuudesta
Empagliflotsiini ja metformiini
Empagliflotsiinin ja metformiinin yhdistelmää arvioitiin yleisissä toksisuustutkimuksissa rotilla enintään 13 viikon ajan. Tutkimuksissa ei havaittu toksisuutta muihin kohde-elimiin verrattuna empagliflotsiinin tai metformiinin käyttöön yksinään. Yhdistelmähoito lisäsi joitain vasteita, kuten vaikutuksia munuaisten fysiologiaan, elektrolyyttitasapainoon ja happo‑emästasapainoon. Kuitenkin ainoastaan hypokloremiaa pidettiin haittavaikutuksena altistuksen ollessa noin 9-kertainen verrattuna empagliflotsiinin suositellun enimmäisannoksen kliiniseen AUC-altistukseen ja 3-kertainen verrattuna metformiinin suositellun enimmäisannoksen kliiniseen AUC-altistukseen.
Alkion- ja sikiönkehitystä koskenut tutkimus tiineillä rotilla ei osoittanut empagliflotsiinin ja metformiinin samanaikaisen käytön aiheuttavan teratogeenistä vaikutusta, kun altistukset olivat noin 14-kertaiset verrattuna empagliflotsiinin suurimpaan annokseen liittyvään kliiniseen AUC-altistukseen ja 4-kertaiset verrattuna 2 000 mg metformiiniannokseen liittyvään kliiniseen AUC-altistukseen.
Empagliflotsiini
Farmakologista turvallisuutta, genotoksisuutta, hedelmällisyyttä ja varhaista alkionkehitystä koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Jyrsijöillä ja koirilla tehdyissä pitkäaikaisissa toksisuustutkimuksissa toksisuuden merkkejä havaittiin, kun altistukset olivat vähintään kymmenkertaisia empagliflotsiinin kliiniseen annokseen verrattuna. Suurin osa toksisuudesta oli yhdenmukaista glukoosin virtsaan erittymiseen ja elektrolyyttien epätasapainoon liittyvän sekundaarisen farmakologian kanssa, kuten painon ja kehon rasvakudoksen määrän aleneminen, lisääntynyt ravinnon nauttiminen, ripuli, dehydraatio, seerumin glukoosin aleneminen ja muiden lisääntyneeseen proteiinimetaboliaan ja glukoneogeneesiin viittaavien seerumin parametrien lisääntyminen, muutokset virtsassa kuten polyuria ja glukosuria, ja mikroskooppiset muutokset, mukaan lukien mineralisaatio munuaisissa ja joissain pehmyt- ja verisuonikudoksissa. Joillain lajeilla havaittiin mikroskooppisesti liiallisia farmakologisia vaikutuksia munuaisissa kuten munuaistiehyiden laajenemista ja munuaistiehyiden ja -altaan mineralisaatiota. Näitä havaittiin, kun altistus oli noin nelinkertainen verrattuna kliiniseen AUC‑altistukseen 25 mg empagliflotsiiniannoksella.
Empagliflotsiini ei ole genotoksinen.
Kahden vuoden mittaisessa karsinogeenisuustutkimuksessa empagliflotsiini ei lisännyt kasvainten esiintyvyyttä naarasrotilla, jotka saivat korkeintaan 700 mg/kg:n vuorokausiannoksia vastaten keskimäärin 72‑kertaista maksimaalista empagliflotsiinin kliinistä AUC‑altistusta. Urosrotilla hoitoon liittyviä hyvänlaatuisia vaskulaarisia proliferatiivisia leesioita (hemangioomia) havaittiin mesenteerisessä imusolmukkeessa suurimmilla annoksilla, mutta ei annoksella 300 mg/kg/vrk, joka vastaa keskimäärin 26‑kertaista maksimaalista kliinistä empagliflotsiinialtistusta. Kiveksisssä olevien interstitiaalisten kasvainten esiintyvyys rotilla oli suurempi 300 mg/kg/vrk ja tätä suuremmilla annoksilla, mutta ei 100 mg/kg/vrk annoksella, joka vastaa keskimäärin 18‑kertaista maksimaalista kliinistä empagliflotsiinialtistusta. Molemmat kasvaimet ovat yleisiä rotilla eivätkä ne todennäköisesti ole merkityksellisiä ihmisille.
Empagliflotsiini ei lisännyt kasvainten ilmaantuvuutta naarashiirillä, jotka saivat korkeintaan 1 000 mg/kg:n vuorokausiannoksia, joka vastaa keskimäärin 62‑kertaista maksimaalista kliinistä empagliflotsiinialtistusta. Empagliflotsiini aiheutti munuaisten kasvaimia uroshiirillä 1 000 mg/kg:n vuorokausiannoksilla, mutta ei 300 mg/kg:n vuorokausiannoksilla, joka vastaa keskimäärin 11‑kertaista maksimaalista kliinistä empagliflotsiinialtistusta. Näiden kasvainten kehittyminen on riippuvainen uroshiirten luontaisesta munuaispatologiasta ja metaboliareitistä, jotka eivät vastaa munuaispatologiaa ja metaboliareittiä ihmisessä. Uroshiirten munuaiskasvaimia ei pidetä merkityksellisinä ihmisille.
Ihmisen terapeuttista annosta riittävästi suuremmilla altistustasoilla empagliflotsiinilla ei ollut haitallisia vaikutuksia hedelmällisyyteen tai varhaiseen alkionkehitykseen. Organogeneesivaiheen aikana annettu empagliflotsiini ei ollut teratogeeninen. Ainoastaan emolle toksisina annoksina empagliflotsiini aiheutti myös taipuneita raajan luita rotalla ja lisäsi alkio- ja sikiökuolemia kanilla.
Rotilla tehdyissä pre- ja postnataalitoksisuustutkimuksissa havaittiin poikasten painonnousun vähenevän, kun emo altistettiin annoksille, jotka vastasivat keskimäärin nelinkertaista maksimaalista kliinistä empagliflotsiinialtistusta. Vastaavaa vaikutusta ei havaittu systeemisellä altistuksella, joka vastasi maksimaalista kliinistä empagliflotsiinialtistusta. Tämän löydöksen merkitys ihmisille on epäselvä.
Nuorilla rotilla tehdyssä toksisuustutkimuksessa, jossa empagliflotsiinia annosteltiin 21 päivän iästä 90 päivän ikään, nuorilla rotilla havaittiin hyvin vähäistä tai lievää, ei haittavaikutukseksi luokiteltavaa munuaistiehyiden ja munuaisaltaan laajentumaa ainoastaan annoksella 100 mg/kg/vrk, joka on suunnilleen 11‑kertainen annos kliiniseen 25 mg:n enimmäisannokseen nähden. Näitä löydöksiä ei enää 13 viikon lääkkeettömän toipumisjakson jälkeen havaittu.
Metformiini
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta, genotoksisuutta ja karsinogeenisuutta sekä lisääntymistoksisuutta koskevien konventionaalisten metformiinitutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille. Kun Wistar Hannover ‑rotille annettu annos oli 500 mg/kg/vrk eli 7-kertainen verrattuna metformiinin ihmisille suositeltuun enimmäisannokseen, metformiinin havaittiin aiheuttavan teratogeenisuutta. Tämä näkyi selvimmin luuston epämuodostumien lukumäärän lisääntymisenä.
Farmaseuttiset tiedot
Apuaineet
Synjardy 5 mg/850 mg kalvopäällysteiset tabletit ja Synjardy 5 mg/1 000 mg kalvopäällysteiset tabletit
Tabletin ydin:
Maissitärkkelys, kopovidoni (nimellinen K-arvo 28), vedetön kolloidinen piidioksidi, magnesiumstearaatti
Kalvopäällyste:
Hypromelloosi, makrogoli 400, titaanidioksidi (E171), talkki, keltainen rautaoksidi (E172)
Synjardy 12,5 mg/850 mg kalvopäällysteiset tabletit ja Synjardy 12,5 mg/1 000 mg kalvopäällysteiset tabletit
Tabletin ydin:
Maissitärkkelys, kopovidoni (nimellinen K-arvo 28), vedetön kolloidinen piidioksidi, magnesiumstearaatti
Kalvopäällyste:
Hypromelloosi, makrogoli 400, titaanidioksidi (E171), talkki, musta rautaoksidi (E172), punainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
SYNJARDY tabletti, kalvopäällysteinen
5/850 mg (L:ei) 60 x 1 fol (55,60 €), 180 x 1 fol (2x(90x1 fol)) (141,74 €)
5/1000 mg (L:ei) 60 x 1 fol (55,60 €), 180 x 1 fol (2x(90x1 fol)) (141,74 €)
12,5/850 mg (L:ei) 60 x 1 fol (55,60 €), 180 x 1 fol (2x(90x1 fol)) (141,74 €)
12,5/1000 mg (L:ei) 60 x 1 fol (55,60 €), 180 x 1 fol (2x(90x1 fol)) (141,74 €)
PF-selosteen tieto
Perforoidut yksittäispakatut PVC/PVDC/alumiiniläpipainopakkaukset.
Pakkauskoot: 10 × 1, 14 × 1, 30 × 1, 56 × 1, 60 × 1, 90 × 1 ja 100 × 1 kalvopäällysteistä tablettia ja monipakkaukset, jotka sisältävät 120 (2 pakkausta, joissa 60 × 1 tablettia), 180 (2 pakkausta, joissa 90 × 1 tablettia) ja 200 (2 pakkausta, joissa 100 × 1 tablettia) kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Synjardy 5 mg/850 mg kalvopäällysteiset tabletit
Kellertävän valkoinen, soikea, kaksoiskupera kalvopäällysteinen tabletti, jossa toisella puolella merkintä ”S5” ja Boehringer Ingelheimin logo ja toisella puolella ”850” (tabletin pituus: 19,2 mm, tabletin leveys: 9,4 mm).
Synjardy 5 mg/1 000 mg kalvopäällysteiset tabletit
Ruskehtavan keltainen, soikea, kaksoiskupera kalvopäällysteinen tabletti, jossa toisella puolella merkintä ”S5” ja Boehringer Ingelheimin logo ja toisella puolella ”1 000” (tabletin pituus: 21,1 mm, tabletin leveys: 9,7 mm).
Synjardy 12,5 mg/850 mg kalvopäällysteiset tabletit
Vaaleanpunertavan valkoinen, soikea, kaksoiskupera kalvopäällysteinen tabletti, jossa toisella puolella merkintä ”S12” ja Boehringer Ingelheimin logo ja toisella puolella ”850” (tabletin pituus: 19,2 mm, tabletin leveys: 9,4 mm).
Synjardy 12,5 mg/1 000 mg kalvopäällysteiset tabletit
Tummanruskehtavan violetti, soikea, kaksoiskupera kalvopäällysteinen tabletti, jossa toisella puolella merkintä ”S12” ja Boehringer Ingelheimin logo ja toisella puolella ”1 000” (tabletin pituus: 21,1 mm, tabletin leveys: 9,7 mm).
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
SYNJARDY tabletti, kalvopäällysteinen
5/850 mg 60 x 1 fol, 180 x 1 fol
5/1000 mg 60 x 1 fol, 180 x 1 fol
12,5/850 mg 60 x 1 fol, 180 x 1 fol
12,5/1000 mg 60 x 1 fol, 180 x 1 fol
- Alempi erityiskorvaus (65 %). Diabetes, muu kuin insuliinihoito (215).
- Peruskorvaus (40 %).
ATC-koodi
A10BD20
Valmisteyhteenvedon muuttamispäivämäärä
21.05.2026
Yhteystiedot
 BOEHRINGER INGELHEIM FINLAND KY
BOEHRINGER INGELHEIM FINLAND KY Tammasaarenkatu 5
00180 Helsinki
010 310 2800
www.boehringer-ingelheim.fi
medinfo.finland@boehringer-ingelheim.com