
LENVIMA kapseli, kova 4 mg, 10 mg
Vaikuttavat aineet ja niiden määrät
Yksi kova kapseli sisältää lenvatinibimesilaattia vastaten 4 mg lenvatinibia.
Yksi kova kapseli sisältää lenvatinibimesilaattia vastaten 10 mg lenvatinibia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, kova.
Kliiniset tiedot
Käyttöaiheet
Erilaistunut kilpirauhaskarsinooma (DTC)
LENVIMA monoterapiana on tarkoitettu etenevän, paikallisesti pitkälle edenneen tai metastaattisen, erilaistuneen (papillaarinen/follikulaarinen/hürthle-solu) radiojodihoidolle resistentin kilpirauhaskarsinooman hoitoon aikuisilla potilailla.
Hepatosellulaarinen karsinooma (HCC)
LENVIMA monoterapiana on tarkoitettu edenneen tai ei-resektoitavissa olevan hepatosellulaarisen karsinooman (HCC) hoitoon aikuisilla potilailla, jotka eivät ole aiemmin saaneet systeemistä hoitoa (ks. kohta Farmakodynamiikka).
Endometriumkarsinooma (EC)
LENVIMA yhdistelmähoitona pembrolitsumabin kanssa on tarkoitettu edenneen tai uusiutuneen endometriumkarsinooman (EC) hoitoon aikuisilla potilailla, joiden tauti on edennyt aiemmin missä tahansa tilanteessa annetun platinapohjaisen hoidon aikana tai sen jälkeen ja jotka eivät sovellu saamaan kuratiivista leikkaus- tai sädehoitoa.
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
LENVIMA-hoidon tulee aloittaa ja sitä tulee valvoa terveydenhuollon ammattilaisen, jolla on kokemusta syöpähoidoista.
Pahoinvointiin, oksenteluun ja ripuliin tulee antaa optimaalista lääketieteellistä hoitoa ennen kuin lenvatinibihoito keskeytetään tai annostusta pienennetään. Maha-suolikanavan toksisuutta on hoidettava aktiivisesti munuaisten toiminnan heikkenemisen tai vajaatoiminnan riskin vähentämiseksi (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Annostus
Jos potilas unohtaa annoksen eikä ota sitä 12 tunnin sisällä, annos jätetään väliin ja seuraava annos otetaan tavalliseen ottamisaikaan.
Hoitoa on jatkettava niin kauan kuin kliinistä hyötyä havaitaan tai kunnes esiintyy toksisuutta, joka ei ole hyväksyttävissä.
Erilaistunut kilpirauhassyöpä (DTC)
Lenvatinibin suositeltu vuorokausiannos on 24 mg (kaksi 10 mg:n kapselia ja yksi 4 mg:n kapseli) kerran päivässä. Vuorokausiannosta säädetään tarvittaessa annosta ja toksisuutta koskevan hallintasuunnitelman mukaisesti.
Annoksen säätäminen ja hoidon lopettaminen (DTC)
Haittavaikutusten hallinta saattaa vaatia lenvatinibihoidon keskeyttämistä, annoksen muuttamista tai lenvatinibihoidon lopettamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Lievät tai keskivaikeat (asteen 1 tai 2) haittavaikutukset eivät yleensä edellytä lenvatinibihoidon keskeyttämistä, paitsi jos ne ovat optimaalisesta hoidosta huolimatta sietämättömiä potilaalle. Vaikea-asteiset (asteen 3) tai sietämättömät haittavaikutukset edellyttävät lenvatinibihoidon keskeyttämistä siihen saakka, kunnes vaikutukset lieventyvät asteeseen 0 tai 1 tai lähtötilanteen tasolle.
Lenvatinibiin liittyvät toksisuudet (ks. taulukko 4): Haittavaikutuksen hävittyä tai lievennyttyä asteeseen 0 tai 1 tai lähtötilanteen tasolle hoitoa tulee jatkaa pienennetyllä lenvatinibiannoksella taulukon 1 suosituksen mukaisesti.
| Taulukko 1 Levantinibin suositellun vuorokausiannoksen muuttaminen DTC-potilaillaa | |||
| Annostaso | Vuorokausiannos | Kapselien määrä | |
| Suositeltu vuorokausiannos | 24 mg suun kautta kerran päivässä | Kaksi 10 mg:n kapselia + yksi 4 mg:n kapseli | |
| Ensimmäinen annoksen pienennys | 20 mg suun kautta kerran päivässä | Kaksi 10 mg:n kapselia | |
| Toinen annoksen pienennys | 14 mg suun kautta kerran päivässä | Yksi 10 mg:n kapseli + yksi 4 mg:n kapseli | |
| Kolmas annoksen pienennys | 10 mg suun kautta kerran päivässäa | Yksi 10 mg:n kapseli | |
| a: Tämän jälkeiset annosten pienennykset on harkittava potilaskohtaisesti, sillä alle 10 mg:n annoksia koskevat tiedot ovat rajallisia. | |||
Hoito on lopetettava, jos hengenvaarallisia (asteen 4) haittavaikutuksia esiintyy, lukuun ottamatta tapauksia, joissa poikkeavien laboratorioarvojen ei katsota aiheuttavan hengenvaaraa, jolloin ne täytyy hoitaa vaikeina (esim. asteen 3) haittavaikutuksina.
Hepatosellulaarinen karsinooma
Lenvatinibin suositeltu vuorokausiannos on 8 mg (kaksi 4 mg:n kapselia) kerran vuorokaudessa potilailla, jotka painavat alle 60 kg, ja 12 mg (kolme 4 mg:n kapselia) kerran vuorokaudessa potilailla, jotka painavat vähintään 60 kg. Annoksen muutokset perustuvat vain havaittuihin toksisuuksiin eivätkä painon muutoksiin hoidon aikana. Vuorokausiannosta on muutettava tarpeen mukaan annosta ja toksisuutta koskevan suunnitelman mukaisesti.
Annoksen säätäminen ja hoidon lopettaminen (HCC)
Joidenkin haittavaikutusten hoito voi edellyttää hoidon keskeytystä, annoksen muuttamista tai lenvatinibihoidon lopettamista. Lievät tai keskivaikeat haittavaikutukset (esim. aste 1 tai 2) eivät yleensä edellytä lenvatinibihoidon keskeyttämistä, ellei hoito ole huonosti siedettyä optimaalisesta hallinnasta huolimatta. Ks. lenvatinibiin liittyvät toksisuudet taulukosta 4. Tarkempia tietoja seurannasta, annoksen säätämisestä ja hoidon lopettamisesta on annettu taulukossa 2.
Taulukko 2 HCC-potilaiden annoksen säätäminen suositellusta lenvatinibin vuorokausiannoksesta
| Aloitusannos | Paino ≥ 60 kg 12 mg (kolme 4 mg:n kapselia suun kautta kerran vuorokaudessa) | Paino < 60 kg 8 mg (kaksi 4 mg:n kapselia suun kautta kerran vuorokaudessa) | ||
| Jatkuvat ja ei-siedettävät asteen 2 tai 3 toksisuudeta | ||||
| Haittavaikutus | Muutos | Säädetty annosb (paino ≥ 60 kg) | Säädetty annosb (paino < 60 kg) | |
| Ilmaantuu ensimmäisen kerran c | Keskeytä hoito, kunnes lieventynyt asteeseen 0–1 tai lähtötasoond | 8 mg (kaksi 4 mg:n kapselia suun kautta kerran vuorokaudessa) | 4 mg (yksi 4 mg:n kapseli) suun kautta kerran vuorokaudessa | |
| Ilmaantuu toisen kerran (sama reaktio tai uusi reaktio) | Keskeytä hoito, kunnes lieventynyt asteeseen 0–1 tai lähtötasoond | 4 mg (yksi 4 mg:n kapseli) suun kautta kerran vuorokaudessa | 4 mg (yksi 4 mg:n kapseli) suun kautta joka toinen päivä | |
Ilmaantuu kolmannen kerran (sama reaktio tai uusi reaktio) | Keskeytä hoito, kunnes lieventynyt asteeseen 0–1 tai lähtötasoond | 4 mg (yksi 4 mg:n kapseli) suun kautta joka toinen päivä | Lopeta hoito | |
| Hengenvaaralliset toksisuudet (aste 4): lopeta hoitoe | ||||
| ||||
Haittavaikutusten asteet perustuvat NCI:n (National Cancer Institute) CTCAE-luokitukseen (Common Terminology Criteria for Adverse Events).
Endometriumkarsinooma (EC)
Suositeltu LENVIMA-annos on 20 mg suun kautta kerran vuorokaudessa yhdistelmähoitona pembrolitsumabin kanssa, jota annetaan joko 200 mg kolmen viikon välein tai 400 mg kuuden viikon välein 30 minuutin kestoisena laskimoinfuusiona. Yhdistelmähoitoa jatketaan, kunnes ei hyväksyttävissä olevaa toksisuutta ilmenee tai kunnes tauti etenee (ks. kohta Farmakodynamiikka).
Ks. lisätietoja annostuksesta pembrolitsumabin valmisteyhteenvedosta.
Annoksen säätäminen ja hoidon lopettaminen endometriumkarsinoomassa
Ks. lenvatinibiin liittyvät toksisuudet taulukosta 4. Kun LENVIMA-valmistetta annetaan yhdistelmähoitona pembrolitsumabin kanssa, keskeytä LENVIMA-hoito, pienennä annosta tai lopeta LENVIMA-hoito tarpeen mukaan (ks. taulukko 3). Keskeytä tai lopeta pembrolitsumabihoito pembrolitsumabin valmisteyhteenvedon ohjeiden mukaisesti. Pembrolitsumabiannoksen pienentämistä ei suositella.
| Taulukko 3 EC-potilaiden annoksen säätäminen suositellusta lenvatinibin vuorokausiannoksestaa | ||
|---|---|---|
Aloitusannos yhdistelmähoitona pembrolitsumabin kanssa | 20 mg suun kautta kerran vuorokaudessa (kaksi 10 mg:n kapselia) | |
Jatkuvat ja ei-siedettävät asteen 2 tai 3 toksisuudet | ||
| Haittavaikutus | Muutos | Säädetty annos |
| Ilmaantuu ensimmäisen kerran | Keskeytä hoito, kunnes lieventynyt asteeseen 0–1 tai lähtötasoon | 14 mg suun kautta kerran vuorokaudessa (yksi 10 mg:n kapseli + yksi 4 mg:n kapseli) |
Ilmaantuu toisen kerran (sama reaktio tai uusi reaktio)
| Keskeytä hoito, kunnes lieventynyt asteeseen 0–1 tai lähtötasoon | 10 mg suun kautta kerran vuorokaudessa (yksi 10 mg:n kapseli) |
Ilmaantuu kolmannen kerran (sama reaktio tai uusi reaktio)
| Keskeytä hoito, kunnes lieventynyt asteeseen 0–1 tai lähtötasoon | 8 mg suun kautta kerran vuorokaudessa (kaksi 4 mg:n kapselia) |
| Hengenvaaralliset toksisuudet (aste 4): lopeta hoitob | ||
a. Vain vähän tietoja on saatavilla alle 8 mg:n annoksista. b. Hoito on lopetettava, jos haittavaikutukset ovat hengenvaarallisia (esim. aste 4), lukuun ottamatta laboratoriokokeiden poikkeamia, joita ei pidetä henkeä uhkaavina, jolloin ne on hoidettava vaikeina (esim. asteen 3) haittavaikutuksina. | ||
| Taulukko 4 Haittavaikutukset, jotka edellyttävät lenvatinibiannoksen säätämistä | |||
|---|---|---|---|
| Haittavaikutus | Vaikeusaste | Toimenpide | Annoksen pienennys ja lenvatinibihoidon uudelleen aloitus |
| Hypertensio | Aste 3 (optimaalisesta verenpaine-lääkityksestä huolimatta) | Hoidon keskeytys | Lieventyessä asteeseen 0, 1 tai 2. Ks. tarkat ohjeet taulukosta 5 kohdassa Varoitukset ja käyttöön liittyvät varotoimet. |
| Aste 4 | Hoidon lopetus | Ei aloiteta uudelleen. | |
| Proteinuria | ≥ 2 g / 24 h | Hoidon keskeytys | Lieventyessä alle 2 g:aan / 24 h. |
| Nefroottinen oireyhtymä | ------- | Hoidon lopetus | Ei aloiteta uudelleen. |
Munuaisten toiminnan heikkeneminen tai vajaatoiminta
| Aste 3 | Hoidon keskeytys | Lieventyessä asteeseen 0–1 tai lähtötilanteen tasolle. |
| Aste 4* | Hoidon lopetus | Ei aloiteta uudelleen. | |
| Sydämen toimintahäiriöt | Aste 3 | Hoidon keskeytys | Lieventyessä asteeseen 0–1 tai lähtötilanteen tasolle. |
| Aste 4 | Hoidon lopetus | Ei aloiteta uudelleen. | |
| Posteriorinen reversiibeli enkefalopatiaoire-yhtymä (PRES) / reversiibeli posteriorinen leukoenkefalopatia-oireyhtymä (RPLS) | Mikä tahansa vaikeusaste | Hoidon keskeytys | Lieventyessä asteeseen 0–1 harkitse uudelleen aloitusta pienennetyllä annoksella. |
| Maksatoksisuus | Aste 3 | Hoidon keskeytys | Lieventyessä asteeseen 0–1 tai lähtötilanteen tasolle. |
| Aste 4* | Hoidon lopetus | Ei aloiteta uudelleen. | |
| Valtimo-tromboemboliat | Mikä tahansa vaikeusaste | Hoidon lopetus | Ei aloiteta uudelleen. |
| Verenvuoto | Aste 3 | Hoidon keskeytys | Lieventyessä asteeseen 0–1. |
| Aste 4 | Hoidon lopetus | Ei aloiteta uudelleen. | |
| Maha-suolikanavan puhkeama tai fisteli | Aste 3 | Hoidon keskeytys | Lieventyessä asteeseen 0–1 tai lähtötilanteen tasolle. |
| Aste 4 | Hoidon lopetus | Ei aloiteta uudelleen. | |
| Muualla kuin maha-suolikanavassa oleva fisteli | Aste 4 | Hoidon lopetus | Ei aloiteta uudelleen. |
| QT-ajan pidentymä | > 500 ms | Hoidon keskeytys | Lieventyessä < 480 ms:iin tai lähtötilanteen tasolle. |
| Ripuli | Aste 3 | Hoidon keskeytys | Lieventyessä asteeseen 0–1 tai lähtötilanteen tasolle. |
| Aste 4 (lääketieteellisestä hoidosta huolimatta) | Hoidon lopetus | Ei aloiteta uudelleen. | |
| *Asteen 4 laboratoriotulosten poikkeamia, joita ei pidetä henkeä uhkaavina, voidaan hoitaa vaikeina (esim. asteen 3) haittavaikutuksina. | |||
Erityisryhmät
DTC
Potilailla, jotka ovat ≥ 75-vuotiaita, rodultaan aasialaisia, joilla on lisäsairauksia (esim. hypertensio ja maksan tai munuaisten vajaatoiminta) tai jotka painavat alle 60 kg, vaikuttaa olevan alentunut sietokyky lenvatinibille (ks. kohta Haittavaikutukset). Kaikilla paitsi vaikeaa maksan tai munuaisten vajaatoimintaa sairastavilla potilailla (ks. alla olevat kohdat) hoito tulee aloittaa 24 mg:n suositusannoksella, minkä jälkeen annosta säädetään yksilöllisen siedettävyyden perusteella.
HCC
Potilailla, jotka ovat ≥ 75-vuotiaita, rodultaan valkoisia tai sukupuoleltaan naisia tai joiden maksan vajaatoiminta on pahempi lähtötilanteessa (Child-Pugh A-pisteet 6 verrattuna pisteisiin 5), vaikuttaa olevan alentunut sietokyky lenvatinibille.
Niiden HCC-potilaiden, joilla ei ole keskivaikeaa tai vaikeaa maksan vajaatoimintaa tai vaikeaa munuaisten vajaatoimintaa, tulee aloittaa hoito suositellulla aloitusannoksella 8 mg (kaksi 4 mg:n kapselia), kun potilaan paino on < 60 kg, ja annoksella 12 mg (kolme 4 mg:n kapselia), kun potilaan paino on ≥ 60 kg, minkä jälkeen annosta säädetään yksilöllisen siedettävyyden perusteella.
Hypertensiota sairastavat potilaat
Verenpaineen on oltava hyvin hallinnassa ennen lenvatinibihoitoa, ja sitä on seurattava säännöllisesti hoidon aikana (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Maksan vajaatoimintaa sairastavat potilaat
DTC
Aloitusannoksen muuttaminen maksan toiminnan perusteella ei ole tarpeen potilailla, joilla on lievä (Child-Pugh A) tai keskivaikea (Child-Pugh B) maksan vajaatoiminta. Vaikeaa maksan vajaatoimintaa (Child-Pugh C) sairastaville potilaille suositeltu aloitusannos on 14 mg kerran päivässä. Annosta voi tämän jälkeen olla tarpeen säätää yksilöllisen sietokyvyn mukaan. Ks. myös kohta Haittavaikutukset).
HCC
HCC-tutkimukseen osallistuneissa potilasryhmissä annosta ei ollut tarpeen muuttaa maksan toiminnan perusteella, kun potilaalla oli lievä maksan vajaatoiminta (Child-Pugh A). Saatavilla olevat hyvin niukat tiedot eivät riitä annossuosituksen muodostamiseen HCC-potilaille, joilla on keskivaikea maksan vajaatoiminta (Child-Pugh B). Näiden potilaiden tarkka turvallisuuden seuranta on suositeltavaa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka). Lenvatinibia ei ole tutkittu potilailla, joilla on vaikea maksan vajaatoiminta (Child-Pugh C), eikä sitä suositella käytettäväksi näille potilaille.
EC
Lenvatinibin käytöstä yhdistelmähoitona pembrolitsumabin kanssa maksan vajaatoimintaa sairastavilla potilailla on rajallisesti tietoa. Yhdistelmähoidon aloitusannoksen muuttaminen maksan toiminnan perusteella ei ole tarpeen potilailla, joilla on lievä (Child-Pugh-asteikolla A) tai keskivaikea (Child-Pugh-asteikolla B) maksan vajaatoiminta. Vaikeaa maksan vajaatoimintaa (Child-Pugh-asteikolla C) sairastaville potilaille suositeltu aloitusannos on 10 mg kerran päivässä. Ks. pembrolitsumabin valmisteyhteenvedosta tietoja annostuksesta maksan vajaatoimintaa sairastavilla potilailla. Annosta voi tämän jälkeen olla tarpeen säätää yksilöllisen sietokyvyn mukaan.
Munuaisten vajaatoimintaa sairastavat potilaat
DTC
Aloitusannoksen muuttaminen munuaisten toiminnan perusteella ei ole tarpeen potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa sairastaville potilaille suositeltu aloitusannos on 14 mg kerran päivässä. Annosta voi tämän jälkeen olla tarpeen säätää yksilöllisen sietokyvyn mukaan. Loppuvaiheen munuaissairautta sairastavia potilaita ei ole tutkittu, minkä vuoksi lenvatinibia ei suositella näille potilaille (ks. kohta Haittavaikutukset).
HCC
Annosta ei ole tarpeen muuttaa munuaisten toiminnan perusteella potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta. Saatavilla olevat tiedot eivät riitä annossuosituksen muodostamiseen HCC-potilaille, joilla on vaikea munuaisten vajaatoiminta.
EC
Aloitusannoksen muuttaminen munuaisten toiminnan perusteella ei ole tarpeen potilailla, joilla on lievä tai keskivaikea munuaisten vajaatoiminta. Vaikeaa munuaisten vajaatoimintaa sairastaville potilaille suositeltu aloitusannos on 10 mg kerran päivässä. Ks. pembrolitsumabin valmisteyhteenvedosta tietoja annostuksesta munuaisten vajaatoimintaa sairastavilla potilailla. Annosta voi tämän jälkeen olla tarpeen säätää yksilöllisen sietokyvyn mukaan. Loppuvaiheen munuaissairautta sairastavia potilaita ei ole tutkittu, minkä vuoksi lenvatinibia ei suositella näille potilaille.
Iäkkäät potilaat
Aloitusannosta ei tarvitse muuttaa iän perusteella. Käytöstä ≥ 75-vuotiailla potilaille on rajallisesti tietoa (ks. kohta Haittavaikutukset).
Pediatriset potilaat
Lenvatinibin turvallisuutta ja tehoa 2 - alle 18 vuoden ikäisten lasten hoidossa ei ole varmistettu. Saatavissa olevan tiedon perusteella, joka on kuvattu kohdissa Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka, ei voida antaa suosituksia annostuksesta. Lenvatinibia ei pidä käyttää alle 2-vuotiaille lapsille eläintutkimuksissa havaittujen turvallisuusongelmien vuoksi (ks. kohta Prekliiniset tiedot turvallisuudesta).
Rotu
Aloitusannosta ei tarvitse muuttaa rodun perusteella (ks. kohta Farmakokinetiikka). Käytöstä rodultaan muille kuin valkoihoisille tai aasialaisille potilaille on rajallisesti tietoa (ks. kohta Haittavaikutukset).
Antotapa
Suun kautta. Kapselit tulee ottaa suurin piirtein samaan aikaan joka päivä ruoan kanssa tai ilman (ks. kohta Farmakokinetiikka). Hoitajien ei pidä avata kapselia, jotta he välttyvät toistuvalta altistukselta kapselin sisällölle.
Lenvatinibikapselit voidaan niellä kokonaisina veden kanssa tai ne voidaan antaa suspensiona liuottamalla kokonainen kapseli tai kokonaiset kapselit veteen, omenamehuun tai maitoon. Suspensio voidaan antaa suun kautta tai ravitsemusletkun kautta. Jos suspensio annetaan ravitsemusletkun kautta, se on valmistettava veteen (ks. kohta Käyttö- ja käsittelyohjeet, Suspension valmistus ja anto).
Jos lenvatinibisuspensiota ei käytetä valmistusajankohtana, sitä voidaan säilyttää peitetyssä astiassa kylmässä 2–8 ºC:ssa enintään 24 tuntia. Kun suspensio otetaan jääkaapista, sitä on ravisteltava noin 30 sekunnin ajan ennen käyttöä. Jos suspensiota ei anneta 24 tunnin kuluessa, se on hävitettävä.
Käyttö yhdistelmähoitona pembrolitsumabin kanssa, ks. pembrolitsumabin valmisteyhteenveto.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Imetys, ks. kohta Raskaus ja imetys.
Varoitukset ja käyttöön liittyvät varotoimet
Hypertensio
Lenvatinibihoitoa saavilla potilailla on raportoitu hypertensiota, jota on yleensä esiintynyt hoidon alkuvaiheessa (ks. kohta Haittavaikutukset). Verenpaineen on oltava hyvin hallinnassa ennen lenvatinibihoitoa, ja jos potilaalla tiedetään olevan verenpainetta, potilasta on hoidettava vakiintuneella verenpainelääkeannoksella vähintään 1 viikon ajan ennen lenvatinibihoitoa. Vakavia huonossa hoitotasapainossa olevan hypertension aiheuttamia komplikaatioita, mukaan lukien aortan dissekaatioita, on raportoitu. Hypertension varhainen havaitseminen ja tehokas hoito ovat tärkeitä, jotta lenvatinibihoidon keskeytysten ja annostuksen pienennysten tarve voidaan minimoida. Verenpainelääkitys on aloitettava niin pian kuin verenpaineen nousu on varmistettu. Verenpaine tulee mitata, kun lenvatinibihoitoa on kulunut 1 viikko, ja sen jälkeen 2 viikon välein ensimmäisten 2 kuukauden ajan, ja tämän jälkeen kerran kuukaudessa. Verenpainelääkityksen valinta on tehtävä yksilöllisesti potilaan kliinisten olosuhteiden mukaan ja tavanomaista lääketieteellistä hoitoa noudattamalla. Aiemmin normotensiivisten potilaiden monoterapia yhdellä antihypertensiolääkkeiden luokalla on aloitettava kun kohonnut verenpaine havaitaan. Antihypertensiovalmistetta jo saavien potilaiden nykyistä lääkettä voidaan lisätä, jos se on asianmukaista, tai hoitoon on lisättävä yksi tai useampi eri luokan hypertensiolääke. Hoida hypertensiota tarvittaessa taulukon 4 suositusten mukaisesti.
| Taulukko 5 Hypertension suositeltu hoito | |
| Verenpaine | Suositellut toimenpiteet |
| Systolinen verenpaine ≥ 140 - < 160 mmHg tai diastolinen verenpaine ≥ 90 - < 100 mmHg | Jatka lenvatinibihoitoa ja aloita hypertensiolääkitys, ellei sitä ole jo aloitettu. TAI Jatka lenvatinibihoitoa ja suurenna nykyisen hypertensiolääkkeen annosta tai aloita ylimääräinen hypertensiolääkitys. |
| Systolinen verenpaine ≥ 160 mmHg tai diastolinen verenpaine ≥ 100 mmHg optimaalisesta hypertensiolääkityksestä huolimatta | 1. Keskeytä lenvatinibihoito. 2. Kun systolinen verenpaine on ≤ 150 mmHg, diastolinen verenpaine ≤ 95 mmHg ja potilas on saanut hypertensiolääkitystä stabiilina annoksena vähintään 48 tunnin ajan, jatka lenvatinibihoitoa pienennetyllä annoksella (ks. kohta Annostus ja antotapa). |
| Hengenvaaralliset seuraukset (pahanlaatuinen hypertensio, neurologiset puutosoireet tai hypertensiivinen kriisi) | Pikainen interventio on tarpeen. Lopeta lenvatinibihoito ja aloita asianmukainen lääketieteellinen hoito. |
Aneurysmat ja valtimon dissekaatiot
VEGF-reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava tarkoin ennen lenvatinibihoidon aloittamista potilaille, joilla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Proteinuria
Lenvatinibihoitoa saavilla potilailla on raportoitu proteinuriaa, jota on yleensä esiintynyt hoidon alkuvaiheessa (ks. kohta Haittavaikutukset). Virtsan proteiinipitoisuus tulee mitata säännöllisesti. Jos proteinuria on virtsan liuskatestillä mitattuna ≥ 2+, hoidon keskeyttäminen, annoksen muuttaminen tai hoidon lopettaminen saattaa olla tarpeen (ks. kohta Annostus ja antotapa). Lenvatinibia käyttävillä potilailla on raportoitu nefroottista oireyhtymää. Lenvatinibihoito on lopetettava nefroottisen oireyhtymän tapauksessa.
Maksatoksisuus
Yleisimmin raportoidut maksaan liittyvät haittavaikutukset lenvatinibihoitoa saavilla DTC-potilailla olivat alaniiniaminotransferaasi (ALT)-, aspartaattiaminotransferaasi (ASAT)- ja veren bilirubiiniarvojen kohoaminen. Lenvatinibihoitoa saavilla DTC-potilailla on raportoitu maksan vajaatoimintaa ja akuuttia maksatulehdusta (< 1 %; ks. kohta Haittavaikutukset). Raportit maksan vajaatoiminnasta koskivat yleensä potilaita, joilla oli etenevä metastaattinen maksasairaus.
REFLECT-tutkimuksessa lenvatinibihoitoa saaneilla HCC-potilailla maksaan liittyviä haittavaikutuksia, kuten maksan enkefalopatiaa ja maksan vajaatoimintaa (mukaan lukien kuolemaan johtaneita reaktioita) ilmoitettiin enemmän (ks. kohta Haittavaikutukset) verrattuna sorafenibillä hoidettuihin potilaisiin. Potilailla, joilla oli vaikeampi maksan vajaatoiminta ja/tai suurempi maksan kasvainkuorma lähtötilanteessa, oli suurempi vaara maksan enkefalopatian ja maksan vajaatoiminnan ilmenemiseen. Maksan enkefalopatiaa ilmeni useammin myös vähintään 75-vuotiailla potilailla. Noin puolet maksan vajaatoiminnan tapauksista ja kolmannes maksan enkefalopatian tapauksista ilmoitettiin potilailla, joiden sairaus oli edennyt.
Tiedot HCC-potilaista, joilla oli keskivaikea maksan vajaatoiminta (Child-Pugh B), ovat hyvin niukkoja, eikä tietoja vaikeaa maksan vajaatoimintaa (Child-Pugh C) sairastavista HCC-potilaista ole. Koska lenvatinibi eliminoituu pääasiassa maksametabolian kautta, keskivaikeaa tai vaikeaa maksan vajaatoimintaa sairastavien potilaiden altistuksen oletetaan olevan suurempi.
Yleisimmin raportoidut maksaan liittyvät haittavaikutukset lenvatinibi- ja pembrolitsumabihoitoa saavilla EC-potilailla olivat alaniiniaminotransferaasi (ALAT)- ja aspartaattiaminotransferaasi (ASAT) -arvojen kohoaminen. Lenvatinibi- ja pembrolitsumabihoitoa saavilla EC-potilailla on raportoitu maksan vajaatoimintaa ja maksatulehdusta (< 1 %; ks. kohta Haittavaikutukset).
Tarkka turvallisuuden seuranta on suositeltavaa, kun potilaalla on lievä tai keskivaikea maksan vajaatoiminta (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka). Maksan toimintakokeet tulee tehdä ennen hoidon aloittamista, ja sen jälkeen 2 viikon välein ensimmäisten 2 kuukauden ajan ja tämän jälkeen kerran kuukaudessa. HCC-potilaita on seurattava maksan toiminnan pahenemisen sekä maksan enkefalopatian varalta. Hoidon keskeyttäminen, annoksen muuttaminen tai hoidon lopettaminen saattaa olla tarpeen maksatoksisuustapauksissa (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Lenvatinibihoitoa saavilla potilailla on raportoitu munuaisten vajaatoimintaa (ks. kohta Haittavaikutukset). Ensisijainen havaittu riskitekijä oli maha-suolikanavan toksisuudesta johtuva kuivuminen ja/tai hypovolemia. Maha-suolikanavan toksisuutta on hoidettava aktiivisesti munuaisten vajaatoiminnan riskin vähentämiseksi. Hoidon keskeyttäminen, annoksen muuttaminen tai hoidon lopettaminen saattaa olla tarpeen (ks. kohta Annostus ja antotapa).
Vaikea-asteista munuaisten vajaatoimintaa sairastavien potilaiden aloitusannosta tulee muuttaa (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Ripuli
Ripulia on raportoitu lenvatinibihoitoa saavilla potilailla usein, etenkin hoidon alkuvaiheessa (ks. kohta Haittavaikutukset). Ripulin lääketieteellinen hoito tulee aloittaa ripeästi elimistön kuivumisen välttämiseksi. Lenvatinibihoito on lopetettava tapauksissa, joissa asteen 4 ripuli jatkuu lääketieteellisestä hoidosta huolimatta.
Sydämen toimintahäiriöt
Lenvatinibihoitoa saavilla potilailla on raportoitu sydämen vajaatoimintaa (< 1 %) ja vasemman kammion ejektiofraktion pienentymistä (ks. kohta Haittavaikutukset). Potilaita on tarkkailtava sydämen dekompensaation kliinisten oireiden tai löydösten varalta, sillä hoidon keskeyttäminen, annoksen muuttaminen tai hoidon lopettaminen saattaa olla tarpeen (ks. kohta Annostus ja antotapa).
Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES)/Reversiibeli posteriorinen leukoenkefalopatiaoireyhtymä (RPLS)
Lenvatinibihoitoa saavilla potilailla on raportoitu posteriorista reversiibeliä enkefalopatiaoireyhtymää (PRES, tunnetaan myös RPLS) (< 1 %; ks. kohta Haittavaikutukset). PRES on neurologinen sairaus, joka voi ilmetä päänsärkynä, kouristuskohtauksina, letargiana, sekavuutena, psyykkisen toiminnan muutoksina, sokeutena tai muina näköön liittyvinä tai neurologisina häiriöinä. Lievää tai keskivaikeaa hypertensiota voi esiintyä. Magneettikuvaus on välttämätöntä PRES-diagnoosin vahvistamiseksi. Verenpainetta tulee hallita asianmukaisin keinoin (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Hoidon keskeyttäminen, annoksen muuttaminen tai hoidon lopettaminen saattaa olla tarpeen potilailla, joilla on PRES-oireyhtymän löydöksiä tai oireita (ks. kohta Annostus ja antotapa).
Valtimotromboemboliat
Lenvatinibihoitoa saavilla potilailla on raportoitu valtimotromboembolioita (aivoverisuonitapahtumia, ohimeneviä aivoverenkiertohäiriöitä ja sydänkohtauksia, ks. kohta Haittavaikutukset). Lenvatinibia ei ole tutkittu potilailla, joilla on ollut valtimotromboembolia hoitoa edeltävän 6 kuukauden aikana, joten sitä on käytettävä varoen tällaisille potilaille. Hoitopäätöksen tulee perustua potilaan yksilöllisen hyöty-riskisuhteen arviointiin. Lenvatinibihoito on lopetettava valtimotromboembolisen tapahtuman jälkeen.
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokasta ehkäisyä lenvatinibihoidon aikana sekä yhden kuukauden ajan sen päättymisestä (ks. kohta Raskaus ja imetys). Tällä hetkellä ei tiedetä lisääkö lenvatinibi tromboembolisten tapahtumien riskiä kun sitä annetaan yhdessä suun kautta otettavien ehkäisyvalmisteiden kanssa.
Verenvuoto
Kliinisissä tutkimuksissa on esiintynyt ja markkinoille tulon jälkeen on raportoitu vakavia kasvaimeen liittyviä verenvuotoja, mukaan lukien kuolemaan johtaneita verenvuototapahtumia (ks. kohta Haittavaikutukset). Markkinoille tulon jälkeisen seurannan aikana vakavia ja kuolemaan johtaneita kaulavaltimon verenvuotoja esiintyi useammin niillä potilailla, joilla oli erilaistumaton kilpirauhaskarsinooma, kuin niillä potilailla, joilla oli erilaistunut kilpirauhaskarsinooma tai muita kasvaintyyppejä. Kasvaimen suuriin verisuoniin (esim. kaulavaltimoon) tunkeutumisen/infiltraation aste tulee ottaa huomioon lenvatinibihoidon aikaansaaman kasvaimen kutistumiseen/nekroosiin liittyvän vaikean verenvuodon riskin vuoksi. Jotkin verenvuototapaukset ovat aiheutuneet sekundaarisesti kasvaimen kutistumisesta ja fistelin muodostumisesta esim. henkitorven ja ruokatorven välille. Kuolemaan johtanutta kallonsisäistä verenvuotoa on raportoitu joillakin potilailla, joista osalla oli ja osalla ei ollut aivometastaaseja. Verenvuotoa muualla kuin aivoissa (esim. henkitorvessa, vatsaontelossa, keuhkoissa) on myös raportoitu. Yksi kuolemaan johtanut maksakasvaimen verenvuototapaus HCC-potilaalla on raportoitu.
Maksakirroosipotilaille on tehtävä ruokatorven suonikohjujen seulonta ja annettava vakiohoitoa ennen lenvatinibihoidon aloittamista.
Hoidon keskeyttäminen, annoksen muuttaminen tai hoidon lopettaminen saattaa olla tarpeen verenvuototapauksissa (ks. kohta Annostus ja antotapa, taulukko 3).
Maha-suolikanavan puhkeama ja fistelin muodostuminen
Lenvatinibihoitoa saavilla potilailla on raportoitu maha-suolikanavan puhkeamia ja fisteleitä (ks. kohta Haittavaikutukset). Maha-suolikanavan puhkeamia ja fisteleitä esiintyi useimmiten potilailla, joilla oli riskitekijöitä, kuten aiempi leikkaus tai aiemmin saatu sädehoito. Hoidon keskeyttäminen, annoksen muuttaminen tai hoidon lopettaminen saattaa olla tarpeen maha-suolikanavan puhkeaman tai fistelin tapauksessa (ks. kohta Annostus ja antotapa).
Muualla kuin maha-suolikanavassa oleva fisteli
Lenvatinibihoitoa saavilla potilailla voi olla tavallista suurempi fistelien kehittymisen riski. Kliinisissä tutkimuksissa ja markkinoille tulon jälkeen on havaittu myös maha-suolikanavan ulkopuolisten fistelien muodostumista tai suurentumista (esim. henkitorvessa, henki- ja ruokatorven välillä, ruokatorvessa, ihossa, naisten sukupuolielimissä). Myös ilmarintaa on raportoitu, ja sen yhteydessä on saattanut joskus esiintyä selkeää näyttöä bronkopleuraalisesta fistelistä. Kasvaimen regression tai nekroosin yhteydessä on joskus raportoitu fisteleitä ja ilmarintaa. Aiempi leikkaus- ja sädehoito saattavat olla myötävaikuttavia riskitekijöitä. Myös keuhkometastaasit saattavat lisätä ilmarinnan riskiä. Pahenemisen välttämiseksi lenvatinibihoitoa ei pidä aloittaa potilaille, joilla on fisteli, ja lenvatinibihoito on lopetettava pysyvästi, jos potilaalla havaitaan fisteli, joka on yhteydessä ruokatorveen, henkitorveen tai keuhkoputkiin, tai mikä tahansa asteen 4 fisteli (ks. kohta Annostus ja antotapa); lenvatinibihoidon keskeyttämisestä tai annoksen pienentämisestä muiden tapahtumien hoidon yhteydessä on rajallisesti tietoa, mutta pahentumista on joissakin tapauksissa havaittu, minkä vuoksi varovaisuutta tulee noudattaa. Lenvatinibi saattaa vaikuttaa negatiivisesti haavojen paranemisprosessiin, kuten muutkin saman luokan lääkeaineet.
QT-ajan pidentyminen
QT/QTc-ajan pidentymistä raportoitiin useammin lenvatinibihoitoa saavilla kuin lumelääkettä saavilla potilailla (ks. kohta Haittavaikutukset). Sydämen sähkökäyrää tulee seurata lähtötilanteessa ja säännöllisesti hoidon aikana kaikilla potilailla kiinnittäen erityistä huomiota niihin potilaisiin, joilla on synnynnäinen pitkä QT -oireyhtymä, kongestiivinen sydämen vajaatoiminta, bradyarytmia tai jotka ottavat lääkevalmisteita, joiden tiedetään pidentävän QT-aikaa, mukaan lukien luokkien Ia ja III rytmihäiriölääkkeitä. Lenvatinibihoito tulee keskeyttää, jos QT-ajan pidentymä on > 500 ms. Lenvatinibihoitoa jatketaan pienennetyllä annoksella, kun QTc-ajan pidentymä on lieventynyt < 480 ms:iin tai lähtötilanteen tasolle.
Elektrolyyttihäiriöt, kuten hypokalemia, hypokalsemia tai hypomagnesemia, lisäävät QT-ajan pidentymisen riskiä, minkä vuoksi mahdolliset elektrolyyttihäiriöt tulee todeta ja korjata kaikilla potilailla ennen hoidon alkamista. Elektrolyyttejä (magnesium, kalium ja kalsium) on tarkkailtava ajoittain hoidon aikana. Veren kalsiumpitoisuutta tulee seurata vähintään kuukausittain ja kalsiumin puutos korvata tarpeen mukaan lenvatinibihoidon aikana. Lenvatinibihoito tulee tarvittaessa keskeyttää tai annosta säätää vaikeusasteen, mahdollisten EKG-muutosten ja hypokalsemian keston mukaan.
Tyreotropiinin suppression heikentyminen / kilpirauhasen toimintahäiriöt
Kilpirauhasen vajaatoimintaa on raportoitu lenvatinibihoitoa saavilla potilailla (ks. kohta Haittavaikutukset). Kilpirauhasen toimintaa tulee seurata ennen lenvatinibihoidon aloittamista ja säännöllisesti koko hoidon aikana. Vajaatoimintaa hoidetaan ylläpitämällä normaalia kilpirauhastoimintaa tavanomaisen lääketieteellisen käytännön mukaisesti.
Lenvatinibi heikentää eksogeenista kilpirauhasen suppressiota (ks. kohta Haittavaikutukset). Tyreotropiiniarvo (TSH) tulee mitata säännöllisesti ja kilpirauhashormonin antoa on säädettävä asianmukaisen tyreotropiiniarvon saavuttamiseksi potilaan hoitotavoitteen mukaisesti.
Haavojen paranemisen komplikaatiot
Tutkimuksia erityisesti lenvatinibin vaikutuksista haavojen paranemiseen ei ole tehty. Lenvatinibia saavilla potilailla on raportoitu heikentynyttä haavojen paranemista. Lenvatinibihoidon väliaikaista keskeyttämistä tulee harkita potilailla, joille tehdään suuria leikkauksia. Lenvatinibihoidon uudelleenaloittamisen ajankohdasta suuren leikkauksen jälkeen on niukasti kliinistä kokemusta. Siksi lenvatinibihoidon uudelleenaloittamispäätöksen tulisi perustua kliiniseen arvioon riittävästä haavan paranemisesta leikkauksen jälkeen.
Leuan luukuolio
Lenvatinibilla hoidetuilla potilailla on raportoitu leuan luukuoliotapauksia. Joitain tapauksia havaittiin potilailla, jotka olivat aiemmin saaneet tai saivat samanaikaista luuston antiresorptiivista hoitoa ja/tai muita angiogeneesin estäjiä, kuten bevasitsumabia, tyrosiinikinaasin estäjiä tai mTOR-estäjiä. Käytettäessä lenvatinibia on siis noudatettava varovaisuutta, kun sitä käytetään samanaikaisesti antiresorptiivisen hoidon ja/tai muiden angiogeneesin estäjien kanssa tai niiden jälkeen.
Invasiiviset hammastoimenpiteet ovat tunnistettu riskitekijä. Ennen lenvatinibihoitoa on harkittava hammastutkimusta ja asianmukaista ennaltaehkäisevää hammashoitoa. Invasiivisia hammastoimenpiteitä on mahdollisuuksien mukaan vältettävä potilailla, jotka ovat aiemmin saaneet tai saavat parhaillaan laskimonsisäisiä bisfosfonaatteja (ks. kohta Haittavaikutukset).
Tuumorilyysioireyhtymä (TLS)
Lenvatinibi voi aiheuttaa tuumorilyysioireyhtymän, joka voi johtaa kuolemaan. Tuumorilyysioireyhtymän riskitekijöitä ovat muun muassa suuri kasvaintaakka, jo olemassa oleva munuaisten vajaatoiminta ja dehydraatio. Näitä potilaita on seurattava tiiviisti ja hoidettava kliinisen tarpeen mukaisesti. Ennaltaehkäisevää nesteytystä on harkittava.
Erityisryhmät
Käytöstä rodultaan muille kuin valkoihoisille tai aasialaisille potilaille sekä vähintään 75-vuotiaille potilaille on rajallisesti tietoa. Aasialaisilla ja iäkkäillä potilailla havaitun huonomman lenvatinibin siedettävyyden vuoksi lenvatinibia on käytettävä varoen tällaisille potilaille (ks. kohta Haittavaikutukset).
Lenvatinibin käytöstä välittömästi sorafenibin tai muiden syöpähoitojen jälkeen ei ole tietoja saatavissa, ja lisääntyvien toksisuuksien mahdollinen riski on olemassa ellei hoitojen välillä pidetä riittävää väliä. Lyhyin tauko hoitojen välillä kliinisissä tutkimuksissa oli 4 viikkoa.
Potilaat, joiden ECOG-toimintakykyluokka oli ≥ 2, suljettiin kliinisten tutkimusten ulkopuolelle (kilpirauhaskarsinoomapotilaita lukuun ottamatta).
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutus lenvatinibiin
Solunsalpaajat
Lenvatinibin, karboplatiinin ja paklitakselin samanaikaisella annolla ei ole merkittävää vaikutusta näiden aineiden farmakokinetiikkaan.
Lenvatinibin vaikutus muihin lääkevalmisteisiin
Syöpäpotilailla tehty, lääkkeiden välisiä yhteisvaikutuksia selvittänyt kliininen tutkimus osoitti, että midatsolaamin (herkkä CYP3A:n ja P-gp:n substraatti) pitoisuudet plasmassa eivät muuttuneet lenvatinibin vaikutuksesta. Lenvatinibin ja muiden CYP3A4:n/P-gp:n substraattien välillä ei siis ole odotettavissa merkittäviä yhteisvaikutuksia.
Suun kautta otettavat ehkäisyvalmisteet
Tällä hetkellä ei tiedetä, voiko lenvatinibi heikentää hormonaalisten ehkäisyvalmisteiden tehoa, ja siksi hormonaalista ehkäisyä käyttävien naisten tulee käyttää ehkäisynä myös estemenetelmää (ks. kohta Raskaus ja imetys).
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi
Naisten, jotka voivat tulla raskaaksi, tulee välttää raskaaksi tulemista ja käyttää erittäin tehokasta ehkäisyä lenvatinibihoidon aikana sekä vähintään kuukauden ajan hoidon päättymisestä. Tällä hetkellä ei tiedetä, voiko lenvatinibi heikentää hormonaalisten ehkäisyvalmisteiden tehoa, ja siksi hormonaalista ehkäisyä käyttävien naisten tulee käyttää ehkäisynä myös estemenetelmää.
Raskaus
Ei ole olemassa tietoja lenvatinibin käytöstä raskaana oleville naisille. Lenvatinibi oli alkiotoksinen ja teratogeeninen, kun sitä annettiin rotille ja kaneille (ks. kohta Prekliiniset tiedot turvallisuudesta).
Lenvatinibia ei pidä käyttää raskauden aikana, ellei se ole selvästi välttämätöntä ja ellei äidin tarpeita ja sikiöön kohdistuvia riskejä ole arvioitu huolellisesti.
Imetys
Ei tiedetä, erittyykö lenvatinibia ihmisen rintamaitoon. Lenvatinibi ja sen metaboliitit erittyvät rottien rintamaitoon (ks. kohta Prekliiniset tiedot turvallisuudesta). Vastasyntyneeseen/imeväiseen kohdistuvia riskejä ei voida poissulkea, ja lenvatinibi on siksi vasta-aiheista imetyksen aikana (ks. kohta Vasta-aiheet).
Hedelmällisyys
Vaikutuksia ihmisellä ei tunneta. Kivesten ja munasarjojen toksisuutta on kuitenkin havaittu rotilla, koirilla ja apinoilla (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Lenvatinibilla on vähäinen vaikutus ajokykyyn ja koneiden käyttökykyyn haittavaikutusten kuten väsymyksen ja huimauksen vuoksi. Potilaiden, joilla näitä oireita esiintyy, on noudettava varovaisuutta ajamisen ja koneiden käytön yhteydessä.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
DTC
Yleisimmin raportoidut haittavaikutukset (esiintyy ≥ 30 %:lla potilaista) ovat hypertensio (68,6 %), ripuli (62,8 %), ruokahalun heikentyminen (51,5 %), painon lasku (49,1 %), väsymys (45,8 %), pahoinvointi (44,5 %), proteinuria (36,9 %), suutulehdus (35,8 %), oksentelu (34,5 %), ääntöhäiriö (34,1 %), päänsärky (34,1 %) ja palmoplantaarinen erytrodysestesia (PPE, eli ns. käsi-jalkaoireyhtymä) (32,7 %). Hypertensiota ja proteinuriaa esiintyy yleensä levatinibihoidon alkuvaiheessa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Suurin osa asteen 3 tai 4 haittavaikutuksista esiintyi 6 ensimmäisen hoitokuukauden aikana. Poikkeuksia olivat ripuli, jota esiintyi koko hoidon aikana, ja painon lasku, joka yleensä kumuloitui ajan myötä.
Tärkeimmät vakavat haittavaikutukset olivat munuaisten vajaatoiminta ja toiminnan heikkeneminen (2,4 %), valtimotromboemboliat (3,9 %), sydämen vajaatoiminta (0,7 %), kallonsisäinen verenvuoto (0,7 %), PRES/RPLS (0,2 %), maksan vajaatoiminta (0,2 %) ja valtimotromboemboliat (aivoverisuonitapahtumat (1,1 %), ohimenevät aivoverenkiertohäiriöt (0,7 %) ja sydänkohtaukset (0,9 %).
Radiojodihoidolle resistenttiä erilaistunutta kilpirauhaskarsinoomaa sairastavasta 452 potilaasta 63,1 %:lla annosta pienennettiin haittavaikutuksen vuoksi ja 19,5 %:lla hoito lopetettiin samasta syystä. Yleisimmät annoksen pienentämiseen johtaneet haittavaikutukset (≥ 5 % potilaista) olivat hypertensio, proteinuria, ripuli, väsymys, käsi-jalkaoireyhtymä (PPE), painon lasku ja ruokahalun heikentyminen. Yleisimmät lenvatinibihoidon lopettamiseen johtaneet haittavaikutukset olivat proteinuria, voimattomuus, hypertensio, aivoverisuonitapahtuma, ripuli ja keuhkoembolia.
HCC
Tavallisimmin ilmoitettuja haittavaikutuksia (joita ilmenee ≥ 30 %:lla potilaista) ovat hypertensio (44,0 %), ripuli (38,1 %), heikentynyt ruokahalu (34,9 %), väsymys (30,6 %) ja painon lasku (30,4 %).
Tärkeimmät vakavat haittavaikutukset olivat maksan vajaatoiminta (2,8 %), maksan enkefalopatia (4,6 %), ruokatorven suonikohjujen verenvuoto (1,4 %), aivoverenvuoto (0,6 %), valtimon tromboembolia (2,0 %), mukaan lukien sydäninfarkti (0,8 %), aivoinfarkti (0,4 %) ja aivoverisuonitapahtuma (0,4 %) sekä munuaisten vajaatoiminta (1,4 %). HCC-potilailla oli useammin alentunut neutrofiilimäärä (8,7 % lenvatinibihoitoa saaneista potilaista verrattuna muihin ei-HCC-kasvaintyypin potilaisiin [1,4 %]), mihin ei liittynyt infektiota, sepsistä tai bakteeriperäistä peritoniittia.
496 HCC-potilaasta 62,3 %:n annosta säädettiin (annostus keskeytettiin tai annosta pienennettiin) ja hoito lopetettiin 20,2 %:lla potilaista haittavaikutuksen vuoksi. Haittavaikutukset, jotka yleisimmin aiheuttivat annoksen muuttamisen (≥ 5 %:lla potilaista), olivat heikentynyt ruokahalu, ripuli, proteinuria, hypertensio, väsymys, käsi-jalkaoireyhtymä (PPE) ja alentunut verihiutalemäärä. Haittavaikutukset, jotka yleisimmin aiheuttivat lenvatinibihoidon lopetuksen, olivat maksan enkefalopatia, väsymys, kohonnut veren bilirubiiniarvo, proteinuria ja maksan vajaatoiminta.
EC
Lenvatinibin ja pembrolitsumabin yhdistelmähoidon turvallisuutta arvioitiin 530:llä edennyttä EC:tä sairastaneella potilaalla, jotka saivat 20 mg lenvatinibia kerran vuorokaudessa ja 200 mg pembrolitsumabia kolmen viikon välein. Yleisimmät haittavaikutukset (joita ilmeni ≥ 20 %:lla potilaista) olivat hypertensio (63 %), ripuli (57 %), kilpirauhasen vajaatoiminta (56 %), pahoinvointi (51 %), heikentynyt ruokahalu (47 %), oksentelu (39 %), väsymys (38 %), painon lasku (35 %), nivelsärky (33 %), proteinuria (29 %), ummetus (27 %), päänsärky (27 %), virtsatieinfektio (27 %), ääntöhäiriö (25 %), vatsakipu (23 %), voimattomuus (23 %), käsi-jalkaoireyhtymä (23 %), suutulehdus (23 %), anemia (22 %) ja hypomagnesemia (20 %).
Yleisimmät (joita ilmeni ≥ 5 %:lla potilaista) vaikeat (aste ≥ 3) haittavaikutukset olivat hypertensio (37,2 %), painon lasku (9,1 %), ripuli (8,1 %), lipaasipitoisuuden suureneminen (7,7 %), heikentynyt ruokahalu (6,4 %), voimattomuus (6 %), väsymys (6 %), hypokalemia (5,7 %), anemia (5,3 %) ja proteinuria (5,1 %).
Haittavaikutuksen takia lenvatinibihoidon lopettaneiden osuus oli 30,6 % potilaista ja sekä lenvatinibi- että pembrolitsumabihoidon lopettaneiden osuus oli 15,3 % potilaista. Yleisimmin (ilmeni ≥ 1 %:lla potilaista) lenvatinibihoidon lopettamiseen johtaneet haittavaikutukset olivat hypertensio (1,9 %), ripuli (1,3 %), voimattomuus (1,3 %), heikentynyt ruokahalu (1,3 %), proteinuria (1,3 %) ja painon lasku (1,1 %).
Haittavaikutuksen takia lenvatinibihoidon keskeyttäneiden osuus oli 63,2 % potilaista. Haittavaikutuksen takia lenvatinibi- ja pembrolitsumabihoidon keskeyttäneiden osuus oli 34,3 % potilaista. Yleisimmin (ilmeni ≥ 5 %:lla potilaista) lenvatinibihoidon keskeyttämiseen johtaneet haittavaikutukset olivat hypertensio (12,6 %), ripuli (11,5 %), proteinuria (7,2 %), oksentelu (7 %), väsymys (5,7 %) ja heikentynyt ruokahalu (5,7 %).
Lenvatinibiannosta pienennettiin haittavaikutusten takia 67,0 %:lla potilaista. Yleisimmin (ilmeni ≥ 5 %:lla potilaista) lenvatinibiannoksen pienentämiseen johtaneet haittavaikutukset olivat hypertensio (16,2 %), ripuli (12,5 %), käsi-jalkaoireyhtymä (9,1 %), väsymys (8,7 %), proteinuria (7,7 %), heikentynyt ruokahalu (6,6 %), pahoinvointi (5,5 %), voimattomuus (5,1 %) ja painon lasku (5,1 %).
Haittavaikutusten luettelo
Monoterapiana annetun lenvatinibin turvallisuusprofiili perustuu tietoihin 452 DTC-potilaasta ja 496 HCC-potilaasta, mikä mahdollistaa vain yleisten haittavaikutusten luokittelun DTC- ja HCC-potilailla. Tässä osassa esitetyt haittavaikutukset perustuvat sekä DTC- että HCC-potilaiden turvallisuustietoihin (ks. kohta Farmakodynamiikka).
Yhdistelmähoitona annetun lenvatinibin turvallisuusprofiili perustuu tietoihin 530 EC-potilaasta, jotka saivat lenvatinibia yhdistelmähoitona pembrolitsumabin kanssa (ks. kohta Farmakodynamiikka).
DTC-, HCC- ja EC-potilailla tehdyissä kliinisissä tutkimuksissa havaitut ja lenvatinibin markkinoilletulon jälkeisessä käytössä raportoidut haittavaikutukset on lueteltu taulukossa 6. Haittavaikutuksen esiintymistiheysluokka edustaa konservatiivisinta arviota esiintymistiheydestä näissä ryhmissä. Lenvatinibiin tai erikseen annettuihin yhdistelmähoidon osiin liittyviä tunnettuja haittavaikutuksia saattaa esiintyä hoidon aikana, kun näitä lääkevalmisteita käytetään yhdistelmähoitona, vaikka kyseisiä vaikutuksia ei olisi ilmoitettu yhdistelmähoidon kliinisissä tutkimuksissa.
Turvallisuuteen liittyviä lisätietoja lenvatinibin käytöstä yhdistelmähoidossa on kyseisen yhdistelmähoidon toisen osan valmisteyhteenvedossa (pembrolitsumabi).
Esiintymistiheydet:
- Hyvin yleinen (≥ 1/10)
- Yleinen (≥ 1/100, < 1/10)
- Melko harvinainen (≥ 1/1000, < 1/100)
- Harvinainen (≥ 1/10 000, < 1/1000)
- Hyvin harvinainen (< 1/10 000)
- Tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin)
Haittavaikutukset on kussakin esiintymistiheysryhmässä esitetty vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 6 Lenvatinibihoitoa saaneilla potilailla raportoidut haittavaikutukset § | ||
| Elinjärjestelmä | Lenvatinibi monoterapiana | Yhdistelmähoitona pembrolitsumabin kanssa |
| (MedDRA-sanasto) | ||
| Infektiot | ||
| Hyvin yleinen | Virtsatieinfektio | Virtsatieinfektio |
| Melko harvinainen | Välilihan absessi | Välilihan absessi |
| Veri ja imukudos | ||
| Hyvin yleinen | Trombosytopeniaa,‡ Lymfopenia a,‡ Leukopeniaa,‡ Neutropeniaa ‡ | Trombosytopeniaa,‡ Lymfopeniaa,‡ Leukopeniaa,‡ Neutropeniaa,‡ Anemia |
| Melko harvinainen | Pernainfarkti | |
| Umpieritys | ||
| Hyvin yleinen | Kilpirauhasen vajaatoiminta Veren tyreotropiiniarvon kohoaminen*,‡ | Kilpirauhasen vajaatoiminta |
Veren tyreotropiiniarvon kohoaminen * Kilpirauhasen liikatoiminta | ||
| Yleinen | Lisämunuaisten vajaatoiminta | |
| Melko harvinainen | Lisämunuaisen vajaatoiminta | |
| Aineenvaihdunta ja ravitsemus | ||
| Hyvin yleinen | Hypokalsemia*‡ | Hypokalsemia*,‡ Hypokalemia‡ |
| Hypokalemia‡ | Hyperkolesterolemiab,‡ | |
Hyperkolesterolemia b,‡ Hypomagnesemia b,‡ Painon lasku | Hypomagnesemiab,‡ | |
| Ruokahalun heikentyminen | Painon lasku Ruokahalun heikentyminen | |
| Yleinen | Kuivuminen | Kuivuminen |
| Harvinainen | Tuumorilyysioireyhtymä† | Tuumorilyysioireyhtymä† |
| Psyykkiset häiriöt | ||
| Hyvin yleinen | Unettomuus | |
| Yleinen | Unettomuus | |
| Hermosto | ||
| Hyvin yleinen | Huimaus | Huimaus |
| Päänsärky | Päänsärky | |
| Makuhäiriö | Makuhäiriö | |
| Yleinen | Aivoverisuonitapahtuma† | |
| Melko harvinainen | Posteriorinen reversiibeli enkefalopatiaoireyhtymä | Posteriorinen reversiibeli enkefalopatiaoireyhtymä |
| Monopareesi | Aivoverisuonitapahtuma† | |
| Ohimenevä aivoverenkiertohäiriö | Monopareesi | |
| Ohimenevä aivoverenkiertohäiriö | ||
| Sydän | ||
| Yleinen | Sydäninfarktic,† | QT-ajan pidentymä |
| Sydämen vajaatoiminta | ||
| QT-ajan pidentymä | ||
| Ejektiofraktion pienentyminen | ||
| Melko harvinainen | Sydäninfarktic,† | |
| Sydämen vajaatoiminta | ||
| Ejektiofraktion pienentyminen | ||
| Verisuonisto | ||
| Hyvin yleinen | Verenvuotod, *,† | Verenvuotod, *,† |
Hypertensioe,* Hypotensio | Hypertensioe,* | |
| Yleinen | Hypotensio | |
| Tuntematon | Aneurysmat ja valtimon dissekaatiot | |
| Hengityselimet, rintakehä ja välikarsina | ||
| Hyvin yleinen | Ääntöhäiriö | Ääntöhäiriö |
| Yleinen | Keuhkoembolia† | Keuhkoembolia† |
| Melko harvinainen | Ilmarinta | Ilmarinta |
| Ruoansulatuselimistö | ||
| Hyvin yleinen | Ripuli | Ripuli |
| Maha-suolikanavan kipu ja vatsakipuf | Maha-suolikanavan kipu ja vatsakipuf | |
| Oksentelu | Oksentelu | |
| Pahoinvointi | Pahoinvointi | |
| Suutulehdusg | Suutulehdusg | |
| Suukipuh | Suukipuh | |
| Ummetus | Ummetus | |
| Ylävatsavaivat | Suun kuivuminen | |
Suun kuivuminen Lipaasipitoisuuden suureneminen‡ Amylaasipitoisuuden suureneminen‡ | Lipaasipitoisuuden suureneminen Amylaasipitoisuuden suureneminen ‡ | |
| Yleinen | Peräaukon fisteli | Haimatulehdusi |
| Ilmavaivat | Ilmavaivat | |
| Mahasuolikanavan puhkeama | Ylävatsavaivat | |
| Paksusuolentulehdus | ||
| Mahasuolikanavan puhkeama | ||
| Melko harvinainen | Haimatulehdusi Paksusuolentulehdus | Peräaukon fisteli |
| Maksa ja sappi | ||
| Hyvin yleinen | Veren bilirubiinipitoisuuden suureneminen j,*,‡ Hypoalbuminemiaj,*,‡ | Veren bilirubiinipitoisuuden suureneminen j,*,‡ Hypoalbuminemiaj,*,‡ Alaniiniaminotransferaasiarvon nousu*, ‡ |
Alaniiniaminotransferaasiarvon nousu*,‡ Aspartaattiaminotransferaasiarvon nousu*,‡ Veren alkalisen fosfataasin kohoaminen‡ Gammaglutamyylitransferaasin kohoaminen‡ | Aspartaattiaminotransferaasiarvon nousu*, ‡ Veren alkalisen fosfataasin kohoaminen ‡ | |
| Yleinen | Maksan vajaatoimintak,*,† | Sappirakkotulehdus |
| Maksan enkefalopatial,*,† | Maksan toiminnan poikkeavuudet | |
| Maksan toiminnan poikkeavuudet | ||
| Sappirakkotulehdus | Gammaglutamyylitransferaasin kohoaminen | |
| Melko harvinainen | Maksasoluvauriot/maksatulehdusm | Maksan vajaatoimintak,*† Maksan enkefalopatial,† |
| Maksasoluvauriot/maksatulehdusm | ||
| Iho ja ihonalainen kudos | ||
| Hyvin yleinen | Palmoplantaarinen erytrodysestesia (käsi-jalkaoireyhtymä) | Palmoplantaarinen erytrodysestesia (käsi-jalkaoireyhtymä) |
| Ihottuma | Ihottuma | |
| Hiustenlähtö | ||
| Yleinen | Hyperkeratoosi | Hiustenlähtö |
| Melko harvinainen | Hyperkeratoosi | |
| Luusto, lihakset ja sidekudos | ||
| Hyvin yleinen | Selkäkipu | Selkäkipu |
| Nivelsärky | Nivelsärky | |
| Lihassärky | Lihassärky | |
| Raajakipu | Raajakipu | |
| Luusto- ja lihaskipu | ||
| Yleinen | Luusto- ja lihaskipu | |
| Melko harvinainen | Leuan luukuolio | |
| Munuaiset ja virtsatiet | ||
| Hyvin yleinen | Proteinuria* | Proteinuria* |
| Veren kreatiniinin kohoaminen‡ | Veren kreatiniinin kohoaminen ‡ | |
| Yleinen | Munuaisten vajaatoimintan, *,† | Munuaisten vajaatoimintan, *,† |
| Munuaistoiminnan heikentyminen* | ||
| Veren urean kohoaminen | ||
| Melko harvinainen | Nefroottinen oireyhtymä | Munuaistoiminnan heikentyminen* Veren urean kohoaminen |
| Yleisoireet ja antopaikassa todettavat haitat | ||
| Hyvin yleinen | Väsymys | Väsymys |
| Voimattomuus | Voimattomuus | |
| Perifeerinen turvotus | Perifeerinen turvotus | |
| Yleinen | Huonovointisuus | Huonovointisuus |
| Melko harvinainen | Heikentynyt haavan paraneminen | Heikentynyt haavan paraneminen |
| Tuntematon | Muualla kuin maha-suolikanavassa oleva fistelio | |
§: Taulukossa 6 esitetyt haittavaikutusten esiintyvyydet eivät välttämättä johdu pelkästään lenvatinibista, vaan niihin voivat vaikuttaa taustasairaus tai muut yhdistelmänä käytetyt lääkevalmisteet.
*: Ks. lisätietoja kohdasta Haittavaikutukset Valikoitujen haittavaikutusten kuvaus.
†: Käsittää myös kuolemaan johtaneita tapauksia.
‡: Esiintyvyys perustuu laboratoriotietoihin.
Seuraavilla termeillä on yhdistetty merkitys:
a: Trombosytopenia käsittää trombosytopenian ja verihiutalemäärän pienentymisen. Neutropenia käsittää neutropenian ja neutrofiilimäärän pienentymisen. Leukopenia käsittää leukopenian ja valkosolumäärän pienentymisen. Lymfopenia käsittää lymfopenian ja lymfosyyttimäärän pienentymisen.
b: Hypomagnesemia käsittää hypomagnesemian ja veren magnesiumpitoisuuden pienenemisen. Hyperkolesterolemia käsittää hyperkolesterolemian ja veren kolesterolipitoisuuden suurentumisen.
c: Sydäninfarkti käsittää sydäninfarktin ja akuutin sydäninfarktin.
d: Käsittää kaikki verenvuototermit.
Verenvuototermit, joita ilmeni vähintään viidellä DTC-tutkittavalla, olivat: nenäverenvuoto, veriyskä, verivirtsaisuus, kontuusio, ulosteen verisyys, ienten verenvuoto, hiussuonipurkaumat, keuhkoverenvuoto, peräsuolen verenvuoto, verivirtsaisuus, mustelmat ja emättimen verenvuoto.
Verenvuototermit, joita ilmeni vähintään viidellä HCC-tutkittavalla, olivat: nenäverenvuoto, verivirtsaisuus, ienten verenvuoto, veriyskä, ruokatorven suonikohjujen verenvuoto, peräpukamien verenvuoto, suun verenvuoto, peräsuolen verenvuoto ja maha-suolikanavan yläosan verenvuoto.
Verenvuototermi, jota ilmeni vähintään viidellä EC-tutkittavalla, oli: emättimen verenvuoto.
e: Hypertensio käsittää hypertension, hypertensiivisen kriisin, diastolisen verenpaineen nousun, ortostaattisen hypertension ja verenpaineen nousun.
f: Maha-suolikanavan kipu ja vatsakipu käsittää seuraavat: vatsavaivat, vatsakivun, alavatsakivun, ylävatsakivun, vatsan arkuuden, keskiylävatsavaivat ja maha-suolikanavan kivun.
g: Suutulehdus käsittää seuraavat: aftaisen suutulehduksen, aftaisen mahahaavan, ienten eroosion, ienten haavaumat, suun limakalvon rakkulat, suutulehduksen, kielitulehduksen, suun haavaumat ja limakalvotulehduksen.
h: Suukipu käsittää seuraavat: suukivun, kielikivun, ienkivun, suunielun epämukavuuden, suunielun kivun ja kielen epämukavuuden.
i: Haimatulehdus käsittää haimatulehduksen ja akuutin haimatulehduksen.
j: Hyperbilirubinemia käsittää hyperbilirubinemian, veren bilirubiinitason nousun, keltaisuuden ja konjugoituneen bilirubiinin määrän suurenemisen. Hypoalbuminemia käsittää hypoalbuminemian ja veren albumiinitason laskun.
k: Maksan vajaatoiminta käsittää maksan vajaatoiminnan, akuutin maksan vajaatoiminnan ja kroonisen maksan vajaatoiminnan.
l: Maksan enkefalopatia käsittää maksan enkefalopatian, maksakooman, metabolisen enkefalopatian ja enkefalopatian.
m: Maksasoluvauriot ja maksatulehdus käsittää seuraavat: lääkeaineperäisen maksavaurion, maksan rasvoittumisen ja kolestaattisen maksavaurion.
n: Munuaisten vajaatoiminta käsittää seuraavat: akuutin prerenaalisen vajaatoiminnan, munuaisten vajaatoiminnan, munuaisten akuutin vajaatoiminnan, akuutin munuaisvaurion ja munuaistiehyiden kuolion.
o: Muualla kuin maha-suolikanavassa oleva fisteli käsittää tapaukset, joissa fisteli sijaitsi mahalaukun ja suoliston ulkopuolella, kuten henkitorvessa, henki- ja ruokatorven välillä, ruokatorvessa, naisten sukupuolielimissä tai ihossa.
Valikoitujen haittavaikutusten kuvaus
Hypertensio (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) hypertensiota (käsittää hypertension, hypertensiivisen kriisin, diastolisen verenpaineen nousun ja verenpaineen nousun) raportoitiin 72,8 %:lla lenvatinibihoitoa saaneista ja 16,0 %:lla lumelääkettä saaneista potilaista. Hypertension alkamista edeltävän ajan mediaani lenvatinibihoitoa saaneilla potilailla oli 16 päivää. Vähintään asteen 3 haittavaikutuksia (käsitti yhden asteen 4 haittavaikutuksen) esiintyi 44,4 %:lla lenvatinibihoitoa saaneista ja 3,8 %:lla lumelääkettä saaneista potilaista. Suurin osa näistä haittavaikutuksista hävisi tai lieveni, kun hoito keskeytettiin tai annosta pienennettiin. Hoito keskeytettiin 13,0 %:lla potilaista ja annosta pienennettiin 13,4 %:lla potilaista. Hypertensio johti hoidon pysyvään lopettamiseen 1,1 %:lla potilaista.
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) hypertensiota (mukaan lukien hypertensio, verenpaineen nousu, diastolisen verenpaineen nousu ja ortostaattinen hypertensio) raportoitiin 44,5 %:lla lenvatinibihoitoa saaneista potilaista. Asteen 3 hypertensiota esiintyi 23,5 %:lla. Mediaaniaika oireiden alkuun oli 26 päivää. Suurin osa tapauksista oli palautuvia annoksen keskeytyksen (3,6 %) tai pienennyksen (3,4 %) jälkeen. Yksi tutkittava (0,2 %) lopetti lenvatinibihoidon hypertension takia.
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) hypertensiota raportoitiin 65 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista. Vähintään asteen 3 haittavaikutuksia esiintyi 38,4 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista. Mediaaniaika oireiden alkuun oli 15 päivää lenvatinibi- ja pembrolitsumabihoitoa saaneiden potilaiden ryhmässä. Lenvatinibihoidon keskeytti 11,6 % potilaista, annosta pienennettiin 17,7 %:lla potilaista ja hoidon lopetti 2,0 % potilaista.
Proteinuria (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) proteinuriaa raportoitiin 33,7 %:lla lenvatinibihoitoa saaneista ja 3,1 %:lla lumelääkettä saaneista potilaista. Proteinurian alkamista edeltävän ajan mediaani oli 6,7 viikkoa. Asteen 3 haittavaikutuksia esiintyi 10,7 %:lla lenvatinibihoitoa saaneista ja 0 %:lla lumelääkettä saaneista potilaista. Suurin osa näistä haittavaikutuksista hävisi tai lieveni, kun hoito keskeytettiin tai annosta pienennettiin. Hoito keskeytettiin 16,9 %:lla potilaista ja annosta pienennettiin 10,7 %:lla potilaista. Proteinuria johti hoidon pysyvään lopettamiseen 0,8 %:lla potilaista.
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) proteinuriaa raportoitiin 26,3 %:lla lenvatinibihoitoa saaneista potilaista, ja asteen 3 reaktioita esiintyi 5,9 %:lla. Mediaaniaika oireiden alkamiseen oli 6,1 viikkoa. Suurin osahaittavaikutuksista hävisi annoksen keskeytyksen (6,9 %) tai annoksen pienennyksen (2,5 %) jälkeen. Proteinuria aiheutti pysyvän hoidon lopetuksen 0,6 %:lle potilaista.
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) proteinuriaa raportoitiin 29,6 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista ja asteen ≥ 3 reaktioita esiintyi 5,4 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 34,5 päivää. Lenvatinibihoidon keskeytti 6,2 % potilaista, annosta pienennettiin 7,9 %:lla potilaista ja hoidon lopetti 1,2 % potilaista.
Munuaisten vajaatoiminta ja toiminnan heikkeneminen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) 5,0 %:lle potilaista kehittyi munuaisten vajaatoiminta ja 1,9 %:lla munuaisten toiminta heikentyi (3,1 %:lla potilaista havaittiin asteen ≥ 3 munuaisten vajaatoiminta tai toiminnan heikkeneminen). Lumelääkeryhmässä munuaisten vajaatoimintaa tai toiminnan heikkenemistä havaittiin 0,8 %:lla potilaista (kaikilla aste ≥ 3).
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) 7,1 %:lle lenvatinibihoitoa saaneista potilaista kehittyi munuaisten vajaatoiminta/toimintahäiriö. Asteen 3 tai vaikeampia reaktioita esiintyi 1,9 %:lla lenvatinibilla hoidetuista potilaista.
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) 18,2 %:lle lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista kehittyi munuaisten vajaatoiminta/toimintahäiriö. Asteen ≥ 3 reaktioita esiintyi 4,2 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 86,0 päivää. Lenvatinibihoidon keskeytti 3,0 % potilaista, annosta pienennettiin 1,7 %:lla potilaista ja hoidon lopetti 1,2 % potilaista.
Sydämen toimintahäiriöt (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka), ejektiofraktion pienentymistä / sydämen vajaatoimintaa raportoitiin 6,5 %:lla lenvatinibihoitoa saaneista potilaista (1,5 %:lla aste ≥ 3) ja 2,3 %:lla lumelääkeryhmän potilaista (ei asteen ≥ 3 tapahtumia).
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) sydämen toimintahäiriöitä (mukaan lukien kongestiivista sydämen vajaatoimintaa, kardiogeenistä sokkia ja kardiopulmonaarista vajaatoimintaa) raportoitiin 0,6 %:lla lenvatinibihoitoa saaneista potilaista (0,4 % oli astetta ≥ 3).
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) sydämen toimintahäiriöitä raportoitiin 1,0 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista ja asteen ≥ 3 reaktioita esiintyi 0,5 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 112,0 päivää. Lenvatinibiannosta pienennettiin 0,2 %:lla potilaista ja hoidon lopetti 0,2 % potilaista.
Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES) / reversiibeli posteriorinen leukoenkefalopatiaoireyhtymä (RPLS) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) lenvatinibihoitoa saavilla potilailla raportoitiin yksi PRES-tapahtuma (aste 2) ja lumelääkeryhmässä ei yhtäkään.
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) esiintyi yksi PRES-tapahtuma (aste 2) lenvatinibihoitoa saaneessa ryhmässä.
Yhteensä 1 823:n lenvatinibia monoterapiana kliinisissä tutkimuksissa saaneen potilaan joukossa oli viisi PRES-tapausta (0,2 %, kaikissa aste 3 tai 4), jotka kaikki paranivat hoidon ja/tai lenvatinibihoidon keskeytyksen tai pysyvän lopettamisen jälkeen.
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) esiintyi yksi PRES-tapahtuma (aste 1) lenvatinibi- ja pembrolitsumabihoitoa saaneessa ryhmässä, minkä takia lenvatinibihoito keskeytettiin.
Maksatoksisuus (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) yleisimmin raportoidut maksaan liittyvät haittavaikutukset olivat hypoalbuminemia (9,6 % lenvatinibilla vs. 1,5 % lumelääkkeellä) ja maksaentsyymiarvojen kohoaminen mukaan lukien alaniiniaminotransferaasi (7,7 % lenvatinibilla vs. 0 % lumelääkkeellä), aspartaattiaminotransferaasi (6,9 % lenvatinibilla vs. 1,5 % lumelääkkeellä) ja veren bilirubiini (1,9 % lenvatinibilla vs. 0 % lumelääkkeellä). Maksavaikutusten alkamista edeltävän ajan mediaani lenvatinibihoitoa saaneilla potilailla oli 12,1 viikkoa. Maksaan liittyviä vähintään asteen 3 haittavaikutuksia (käsitti yhden asteen 5 maksan toiminnan pettämisen) esiintyi 5,4 %:lla lenvatinibihoitoa saaneista ja 0,8 %:lla lumelääkettä saaneista potilaista. Maksaan liittyvät haittavaikutukset johtivat hoidon keskeyttämiseen 4,6 %:lla, annoksen pienentämiseen 2,7 %:lla ja hoidon pysyvään lopettamiseen 0,4 %:lla potilaista.
Yhteensä 1 166:sta lenvatinibihoitoa saaneesta potilaasta maksan toiminta petti kolmella henkilöllä (0,3 %), mikä johti kaikissa kolmessa tapauksessa kuolemaan. Yksi tapauksista oli potilaalla, jolla ei ollut maksametastaaseja. Myös yksi akuutti maksatulehdustapaus raportoitiin potilaalla, jolla ei ollut maksametastaaseja.
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) yleisimmin raportoituja hepatotoksisia haittavaikutuksia olivat veren suurentunut bilirubiinipitoisuus (14,9 %), suurentunut aspartaattiaminotransferaasipitoisuus (13,7 %), suurentunut alaniiniaminotransferaasipitoisuus (11,1 %), hypoalbuminemia (9,2 %), maksan enkefalopatia (8,0 %), suurentunut gammaglutamyylitransferaasipitoisuus (7,8 %) ja suurentunut veren alkalifosfataasipitoisuus (6,7 %). Mediaaniaika hepatotoksisten haittavaikutusten alkamiseen oli 6,4 viikkoa. Asteen ≥ 3 hepatotoksisia reaktioita esiintyi 26,1 %:lla lenvatinibihoitoa saaneista potilaista. Maksan vajaatoimintaa (mukaan lukien 12 potilaan kuolemaan johtaneet tapahtumat) esiintyi 3,6 %:lla potilaista (kaikki tapaukset olivat astetta ≥ 3). Maksan enkefalopatiaa (mukaan lukien 4 potilaan kuolemaan johtaneet tapahtumat) esiintyi 8,4 %:lla potilaista (5,5 % oli astetta ≥ 3). Lenvatinibiryhmässä hepatotoksiset tapahtumat aiheuttivat 17 kuolemaa (3,6 %) ja sorafenibiryhmässä puolestaan 4 kuolemaa (0,8 %). Hepatotoksiset haittavaikutukset aiheuttivat annostuksen keskeytyksen 12,2 %:lla lenvatinibia saaneista potilaista ja annoksen pienennyksen 7,4 %:lla lenvatinibia saaneista potilaista sekä pysyvän hoidon lopetuksen 5,5 %:ssa tapauksista.
Kliinisissä kokeissa, joissa 1327 potilasta sai lenvatinibia monoterapiana muihin indikaatioihin kuin HCC:n hoitoon, maksan vajaatoimintaa (mukaan lukien kuolemaan johtaneita tapauksia) raportoitiin 4 potilaalla (0,3 %), maksavaurioita 2 potilaalla (0,2 %), akuuttia hepatiittia 2 potilaalla (0,2 %) ja hepatosellulaarisia vaurioita yhdellä potilaalla (0,1 %).
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) maksatoksisuutta raportoitiin 33,7 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista ja asteen ≥ 3 reaktioita esiintyi 12,1 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 56,0 päivää. Lenvatinibihoidon keskeytti 5,2 % potilaista, annosta pienennettiin 3,0 %:lla potilaista ja hoidon lopetti 1,2 % potilaista.
Valtimotromboemboliat (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) valtimotromboemboliatapahtumia raportoitiin 5,4 %:lla lenvatinibihoitoa saaneista ja 2,3 %:lla lumelääkeryhmän potilaista.
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) valtimotromboemboliatapahtumia raportoitiin 2,3 %:lla lenvatinibihoitoa saaneista potilaista.
Yhteensä 1 823:n lenvatinibia monoterapiana kliinisissä kokeissa saaneen potilaan joukossa oli kymmenen kuolemaan johtavanutta valtimotromboemboliatapausta (0,5 %) (viisi sydäninfarktia ja viisi aivoverisuonitapahtumaa).
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) valtimotromboembolioita raportoitiin 3,7 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista ja asteen ≥ 3 reaktioita esiintyi 2,2 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 59,0 päivää. Lenvatinibihoidon keskeytti 0,2 % potilaista ja hoidon lopetti 2,0 % potilaista.
Verenvuoto (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) verenvuotoa raportoitiin 34,9 %:lla lenvatinibihoitoa saaneista potilaista (1,9 %:lla aste ≥ 3) ja 18,3 %:lla lumelääkettä saaneista potilaista (3,1 %:lla aste ≥ 3). Haittavaikutuksia, joiden esiintyvyys oli ≥ 0,75 % suurempi kuin lumelääkettä saavilla, olivat seuraavat: nenäverenvuoto (11,9 %), verivirtsaisuus (6,5 %), kontuusio (4,6 %), ienten verenvuoto (2,3 %), ulosteen verisyys (2,3 %), peräsuolen verenvuoto (1,5 %), verenpurkauma (1,1 %), peräpukamien verenvuoto (1,1 %), kurkunpään verenvuoto (1,1 %), hiussuonipurkauma (1,1 %) ja kallonsisäisen kasvaimen verenvuoto (0,8 %). Niistä tässä tutkimuksessa lenvatinibia saaneesta 16 potilaasta, joilla oli lähtötilanteessa keskushermostometastaaseja, yksi sai kuolemaan johtaneen kallonsisäisen verenvuodon.
Ensimmäisen verenvuotoepisodin alkamista edeltävän ajan mediaani lenvatinibihoitoa saaneilla potilailla oli 10,1 viikkoa. Lenvatinibihoitoa ja lumelääkettä saavien potilaiden välillä ei havaittu eroa vakavien haittavaikutusten esiintyvyydessä (3,4 % vs. 3,8 %), hoidon ennenaikaiseen lopettamiseen johtaneiden haittavaikutusten esiintyvyydessä (1,1 % vs. 1,5 %), hoidon keskeyttämiseen johtaneiden haittavaikutusten esiintyvyydessä (3,4 % vs. 3,8 %) tai annoksen pienentämiseen johtaneiden haittavaikutusten esiintyvyydessä (0,4 % vs. 0 %).
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) verenvuotoa raportoitiin 24,6 %:lla potilaista ja 5,0 % näistä oli astetta ≥ 3. Asteen 3 reaktioita esiintyi 3,4 %:lla, asteen 4 reaktioita 0,2 %:lla ja 7 potilaalla (1,5 %) ilmeni asteen 5 reaktio, joita olivat aivoverenvuoto, maha-suolikanavan yläosan verenvuoto, suolistoverenvuoto ja kasvaimen verenvuoto. Mediaaniaika oireiden alkamiseen oli 11,9 viikkoa. Verenvuototapahtuma aiheutti annostuksen keskeytyksen 3,2 %:lla potilaista ja annoksen pienennyksen 0,8 %:lla potilaista sekä hoidon lopetuksen 1,7 %:lla potilaista.
Yhteensä 1 327:sta lenvatinibia monoterapiana kliinisissä kokeissa muuhun kuin HCC:n hoitoon saaneesta potilaasta 2 %:lla raportoitiin vähintään asteen 3 verenvuotoa; kolmella potilaalla (0,2 %) esiintyi asteen 4 verenvuotoa ja kahdeksalla potilaalla (0,6 %) asteen 5 verenvuotoa, mukaan lukien valtimoverenvuotoa, aivoverenvuotoa, kallonsisäistä verenvuotoa, kallonsisäisen kasvaimen verenvuotoa, verioksennusta, mustaa veriulostetta veriyskää ja kasvaimen verenvuotoa.
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) verenvuotoa raportoitiin 24,4 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista ja asteen ≥ 3 reaktioita esiintyi 3,0 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 65,0 päivää. Lenvatinibihoidon keskeytti 1,7 % potilaista, annosta pienennettiin 1,2 %:lla potilaista ja hoidon lopetti 1,7 % potilaista.
Hypokalsemia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) hypokalsemiaa raportoitiin 12,6 %:lla lenvatinibihoitoa saaneista ja 0 %:lla lumelääkettä saaneista potilaista. Hypokalsemian alkamista edeltävän ajan mediaani lenvatinibihoitoa saaneilla potilailla oli 11,1 viikkoa. Asteen 3 tai 4 haittavaikutuksia esiintyi 5,0 %:lla lenvatinibihoitoa saaneista ja 0 %:lla lumelääkettä saaneista potilaista. Suurin osa näistä haittavaikutuksista hävisi tai lieveni, kun hoito keskeytettiin tai annosta pienennettiin. Hoito keskeytettiin 1,5 %:lla potilaista ja annosta pienennettiin 1,1 %:lla potilaista. Hoito lopetettiin pysyvästi yhdellä potilaalla, jolla oli asteen 4 hypokalsemia.
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) hypokalsemiaa raportoitiin 1,1 %:lla potilaista, ja asteen 3 reaktioita esiintyi 0,4 %:lla. Lenvatinibiannostus keskeytettiin hypokalsemian vuoksi yhdellä tutkittavalla (0,2 %), eikä annoksen pienennyksiä tai hoidon lopetuksia tapahtunut.
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) hypokalsemiaa raportoitiin 3,9 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista ja asteen ≥ 3 reaktioita esiintyi 1,0 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 148,0 päivää. Lenvatinibiannoksen muutoksia ei raportoitu.
Maha-suolikanavan puhkeama ja fistelin muodostuminen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) maha-suolikanavan puhkeama tai fisteli raportoitiin 1,9 %:lla lenvatinibihoitoa saaneista potilaista ja 0,8 %:lla lumelääkeryhmän potilaista.
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) maha-suolikanavan puhkeama tai fisteli raportoitiin 1,9 %:lla lenvatinibihoitoa saaneista potilaista.
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) fistelin muodostumisia raportoitiin 2,5 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista ja asteen ≥ 3 reaktioita esiintyi 2,5 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 117,0 päivää. Lenvatinibihoidon lopetti 1,0 % potilaista. Maha-suolikanavan puhkeamia raportoitiin 3,9 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista ja asteen ≥ 3 reaktioita esiintyi 3,0 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 42 päivää. Lenvatinibihoidon keskeytti 0,5 % potilaista ja hoidon lopetti 3,0 % potilaista.
Muualla kuin maha-suolikanavassa olevat fistelit (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
Lenvatinibin käyttöön on yhdistetty fistelitapauksia, joista osa on johtanut kuolemaan. Mahan ja suoliston ulkopuolella sijaitsevia fisteleitä on raportoitu useassa eri käyttöaiheessa. Niiden ilmaantuminen on ajoittunut kahdesta viikosta yhteen vuoteen lenvatinibihoidon aloittamisen jälkeen; ilmaantumista edeltävän ajan mediaani on noin 3 kuukautta.
QT-ajan pidentyminen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) QT/QTc-ajan pidentymistä raportoitiin 8,8 %:lla lenvatinibihoitoa saaneista potilaista ja 1,5 %:lla lumelääkeryhmän potilaista. QT-ajan pidentymä ylitti 500 ms 2 %:lla lenvatinibihoitoa saaneista potilaista; lumelääkeryhmässä vastaavia pidentymiä ei raportoitu.
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) QT/QTc-ajan pidentymistä raportoitiin 6,9 %:lla lenvatinibihoitoa saaneista potilaista. Yli 500 ms:n QTcF-ajan pidentymä esiintyi 2,4 %:lla potilaista.
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) QT-ajan pidentymistä raportoitiin 3,9 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista ja asteen ≥ 3 reaktioita esiintyi 0,5 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 115,5 päivää. Lenvatinibihoidon keskeytti 0,2 % potilaista ja lenvatinibiannosta pienennettiin 0,5 %:lla potilaista.
Veren tyreotropiinin kohoaminen (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) TSH-arvo oli 88 %:lla potilaista lähtötilanteessa korkeintaan 0,5 mU/l. Potilaista, joiden TSH-arvo oli lähtötilanteessa normaali, TSH-arvo kohosi lähtötilanteen jälkeen suuremmaksi kuin 0,5 mU/l 57 %:lla lenvatinibihoitoa saaneista ja 14 %:lla lumelääkettä saaneista potilaista.
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) 89,6 %:lla potilaista oli lähtötilanteessa TSH-taso alle normaalin ylärajan. TSH-arvon nousu normaalin ylärajan yli havaittiin lähtötilanteen jälkeen 69,6 %:lla lenvatinibihoitoa saaneista potilaista.
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) kilpirauhasen vajaatoimintaa raportoitiin 68,2 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista ja asteen ≥ 3 reaktioita esiintyi 1,2 %:lla potilaista. Mediaaniaika oireiden alkamiseen oli 62,0 päivää. Lenvatinibihoidon keskeytti 2,2 % potilaista ja lenvatinibiannosta pienennettiin 0,7 %:lla potilaista.
Veren tyreotropiinipitoisuuden kohoamista raportoitiin 12,8 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista eikä yhdelläkään potilaalla raportoitu asteen ≥ 3 reaktioita. Hoidon keskeytti 0,2 % potilaista.
Ripuli (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)
DTC
Keskeisessä faasi III SELECT-tutkimuksessa (ks. kohta Farmakodynamiikka) ripulia raportoitiin 67,4 %:lla lenvatinibihoitoa saaneista potilaista (9,2 %:lla aste ≥ 3) ja 16,8 %:lla lumelääkeryhmän potilaista (ei asteen ≥ 3 tapahtumia).
HCC
Vaiheen 3 REFLECT-tutkimuksessa (ks. kohta Farmakodynamiikka) ripulia raportoitiin 38,7 %:lla lenvatinibihoitoa saaneista potilaista (4,2 %:lla aste ≥ 3).
EC
Vaiheen 3 tutkimuksessa 309 (ks. kohta Farmakodynamiikka) ripulia raportoitiin 54,2 %:lla lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista (7,6 % oli asteen ≥ 3 reaktioita). Lenvatinibihoidon keskeytti 10,6 % potilaista, annosta pienennettiin 11,1 %:lla potilaista ja hoidon lopetti 1,2 % potilaista.
Pediatriset potilaat
Pediatrisissa tutkimuksissa 207, 216, 230 ja 231 (ks. kohta Farmakodynamiikka) lenvatinibin yleinen turvallisuusprofiili monoterapiana tai yhdistelmähoitona joko ifosfamidin ja etoposidin tai everolimuusin kanssa oli yhdenmukainen lenvatinibihoitoa saaneilla aikuisilla todetun turvallisuusprofiilin kanssa.
Potilailla, joilla oli uusiutunut/refraktorinen osteosarkooma, raportoitiin ilmarintaa yleisemmin kuin on havaittu aikuisilla DTC-, HCC-, RCC- ja EC-potilailla. Tutkimuksessa 207 ilmarinta todettiin 6 potilaalla (10,9 %), jotka saivat lenvatinibia monoterapiana, ja 7 potilaalla (16,7 %), jotka saivat lenvatinibia yhdistelmähoitona ifosfamidin ja etoposidin kanssa. Yhteensä 2 potilasta lopetti tutkimushoidon ilmarinnan vuoksi. Tutkimuksessa 230 ilmarintaa raportoitiin yhteensä 14 potilaalla (11 potilaalla [28,2 %], jotka saivat lenvatinibia yhdistelmähoitona ifosfamidin ja etoposidin kanssa, ja 3 potilaalla [7,7 %], joka sai ifosfamidia ja etoposidia). Tutkimuksessa 216 ilmarinta todettiin kolmella potilaalla (4,7 %), joilla oli Ewingin sarkooma, rabdomyosarkooma (RMS) ja Wilmsin kasvain; kaikilla kolmella potilaalla oli lähtötilanteessa keuhkometastaaseja. Tutkimuksessa 231 ilmarinta todettiin kahdeksalla potilaalla (6,3 %), joilla oli sukkulasolusarkooma, erilaistumaton sarkooma, RMS, pahanlaatuinen perifeerinen hermotuppikasvain, synoviaalisarkooma, sukkulasolukarsinooma ja pahanlaatuinen fibromyksoidi luutuva kasvain; kaikilla kahdeksalla potilaalla oli lähtötilanteessa keuhkometastaaseja tai rintakehän seinämän tai keuhkopussinontelon primaaritauti. Yksikään tutkimuksiin 216 ja 230 osallistuneista potilaista ei lopettanut tutkimushoitoa ilmarinnan vuoksi, yksi tutkimukseen 231 osallistuneista potilaista lopetti tutkimushoidon ilmarinnan vuoksi. Ilmarinnan ilmeneminen vaikutti liittyvän lähinnä keuhkoetäpesäkkeisiin ja taustasairauteen.
Tutkimuksen 207 monoterapian annoshakukohortissa yleisimmin (≥ 40 %) raportoidut haittavaikutukset olivat ruokahalun heikentyminen, ripuli, kilpirauhasen vajaatoiminta, oksentelu, vatsakipu, kuume, hypertensio ja painon lasku. Laajennetussa monoterapiakohortissa, johon kuuluneilla potilailla oli uusiutunut tai refraktorinen osteosarkooma, yleisimmin (≥ 40 %) raportoidut haittavaikutukset olivat ruokahalun heikentyminen, päänsärky, oksentelu, kilpirauhasen vajaatoiminta ja proteinuria.
Tutkimuksen 207 yhdistelmähoidon annoshakukohortissa yleisimmin (≥ 50 %) raportoidut haittavaikutukset olivat oksentelu, anemia, pahoinvointi, ripuli, kilpirauhasen vajaatoiminta, vatsakipu, nivelsärky, nenäverenvuoto, neutropenia, ummetus, päänsärky ja raajakipu. Laajennetussa yhdistelmähoitokohortissa yleisimmin (≥ 50 %) raportoidut haittavaikutukset olivat anemia, pahoinvointi, valkosolumäärän pieneneminen, ripuli, oksentelu ja verihiutalemäärän pieneneminen.
Tutkimuksen 216 vaiheessa 1 (yhdistelmähoidon annoshakukohortti) yleisimmin (≥ 40 %) raportoidut haittavaikutukset olivat hypertensio, kilpirauhasen vajaatoiminta, hypertriglyseridemia, vatsakipu ja ripuli, ja vaiheessa 2 (laajennettu yhdistelmähoitokohortti) yleisimmin (≥ 35 %) raportoidut haittavaikutukset olivat hypertriglyseridemia, proteinuria, ripuli, lymfosyyttimäärän pieneneminen, valkosolumäärän pieneneminen, veren kolesterolipitoisuuden nousu, väsymys ja verihiutalemäärän pieneneminen.
OLIE-tutkimuksessa (tutkimus 230) yleisimmin (≥ 35 %) raportoidut haittavaikutukset olivat kilpirauhasen vajaatoiminta, anemia, pahoinvointi, verihiutalemäärän pieneneminen, proteinuria, oksentelu, selkäkipu, kuumeinen neutropenia, hypertensio, ummetus, ripuli, neutrofiilimäärän pieneneminen ja kuume.
Tutkimuksessa 231 yleisimmin raportoidut (≥ 15 %) haittavaikutukset olivat kilpirauhasen vajaatoiminta, hypertensio, proteinuria, ruokahalun heikentyminen, ripuli ja verihiutalemäärän pieneneminen.
Muut erityisryhmät
Iäkkäät potilaat
DTC
Vähintään 75-vuotiailla potilailla esiintyi nuorempia potilaita enemmän asteen 3 tai 4 hypertensiota, proteinuriaa, ruokahalun heikentymistä ja kuivumista.
HCC
Vähintään 75-vuotiailla potilailla esiintyi nuorempia potilaita enemmän hypertensiota, proteinuriaa, ruokahalun heikentymistä, heikkoutta, kuivumista, pyörrytystä, pahoinvointia, perifeeristä turvotusta, kutinaa ja maksan enkefalopatiaa. Maksan enkefalopatiaa esiintyi yli kaksi kertaa enemmän vähintään 75-vuotiailla potilailla (17,2 %) kuin sitä nuoremmilla (7,1 %). Maksan enkefalopatiaan liittyi usein sairauden haitallisia piirteitä lähtötilanteessa tai muiden lääkevalmisteiden käyttöä samanaikaisesti. Myös valtimotromboemboliatapahtumia esiintyi enemmän tässä ikäryhmässä.
EC
Vähintään 75-vuotiailla potilailla esiintyi nuorempia potilaita enemmän virtsatieinfektioita ja asteen ≥ 3 hypertensiota (lisäys ≥ 10 % verrattuna alle 65-vuotiaisiin potilaisiin).
Sukupuoli
DTC
Naisilla esiintyi miehiä enemmän hypertensiota (mukaan lukien asteen 3 tai 4 hypertensiota), proteinuriaa ja käsi-jalkaoireyhtymää, kun taas miehillä esiintyi naisia useammin ejektiofraktion pienentymistä, maha-suolikanavan puhkeamia ja fisteleitä.
HCC
Naisilla esiintyi miehiä enemmän hypertensiota, väsymystä, EKG:ssä näkyvää QT-ajan pidentymistä ja hiustenlähtöä. Miehillä dysfoniaa esiintyi enemmän (26,5 %) kuin naisilla (12,3 %). Samoin painonlasku ja verihiutalemäärän pieneneminen oli miehillä yleisempää. Maksan vajaatoimintaa havaittiin vain miehillä.
Etninen alkuperä
DTC
Aasialaisilla potilailla esiintyi valkoihoisia potilaita enemmän (ero ≥ 10 %) perifeeristä turvotusta, hypertensiota, väsymystä, käsi-jalkaoireyhtymää, proteinuriaa, suutulehdusta, trombosytopeniaa ja lihassärkyä. Valkoihoisilla potilailla esiintyi vastaavasti enemmän ripulia, painon laskua, pahoinvointia, oksentelua, ummetusta, voimattomuutta, vatsakipua, kipua raajoissa ja suun kuivumista. Lenvatinibiannosta pienennettiin suuremmalla osalla aasialaispotilaista verrattuna valkoihoisiin potilaisiin. Mediaaniaika annoksen ensimmäiseen pienennyskertaan oli aasialaisilla potilailla valkoihoisia potilaita lyhyempi ja päivittäin otettava keskimääräinen annos pienempi.
HCC
Aasialaisilla potilailla esiintyi valkoihoisia potilaita enemmän proteinuriaa, neutrofiilimäärän pienentymistä, verihiutalemäärän pienentymistä, valkosolumäärän pienentymistä ja PPE:tä, kun taas valkoihoisilla potilailla esiintyi enemmän väsymystä, maksan enkefalopatiaa, akuutteja munuaisvaurioita, ahdistusta, heikkoutta, pahoinvointia, trombosytopeniaa ja oksentelua.
EC
Aasialaisilla potilailla esiintyi valkoihoisia potilaita enemmän (ero ≥ 10 %) anemiaa, huonovointisuutta, neutrofiilimäärän pienentymistä, suutulehdusta, verihiutalemäärän pienentymistä, proteinuriaa ja PPE:tä, kun taas valkoihoisilla potilailla esiintyi enemmän limakalvotulehdusta, vatsakipua, ripulia, virtsatieinfektioita, painon laskua, hypomagnesemiaa, huimausta, voimattomuutta ja väsymystä.
Hypertensio lähtötilanteessa
DTC
Potilailla, joilla oli lähtötilanteessa hypertensio, esiintyi enemmän asteen 3 tai 4 hypertensiota, proteinuriaa, ripulia ja kuivumista sekä myös seuraavien haittavaikutusten vakavia tapauksia: kuivuminen, hypotensio, keuhkoembolia, pahanlaatuinen pleuraeffuusio, eteisvärinä ja maha-suolikanavan oireet (vatsakipu, ripuli, oksentelu).
Maksan vajaatoiminta
DTC
Potilailla, joilla oli lähtötilanteessa maksan vajaatoiminta, esiintyi enemmän hypertensiota, käsi-jalkaoireyhtymää sekä asteen 3 tai 4 hypertensiota, voimattomuutta, väsymystä ja hypokalsemiaa kuin potilailla, joiden maksan toiminta oli normaali.
HCC
Potilailla, joilla oli lähtötilanteessa Child-Pugh (CP) ‑pisteet 6 (noin 20 % REFLECT-tutkimuksen potilaista), esiintyi enemmän ruokahalun heikkenemistä, väsymystä, proteinuriaa, maksan enkefalopatiaa ja maksan vajaatoimintaa verrattuna potilaisiin, joiden CP-pisteet lähtötilanteessa olivat 5. Hepatotoksisia tapahtumia ja verenvuototapahtumia esiintyi myös enemmän CP-pisteet 6 saaneilla potilailla kuin CP-pisteet 5 saaneilla.
Munuaisten vajaatoiminta
DTC
Potilailla, joilla oli lähtötilanteessa munuaisten vajaatoiminta, esiintyi enemmän asteen 3 tai 4 hypertensiota, proteinuriaa, väsymystä, suutulehduksia, perifeeristä turvotusta, trombosytopeniaa, kuivumista, QT-ajan pidentymää, kilpirauhasen vajaatoimintaa, hyponatremiaa, veren tyreotropiinin kohoamista ja keuhkokuumetta kuin potilailla, joiden munuaisten toiminta oli normaali. Näillä potilailla esiintyi myös enemmän munuaisiin ja maksaan liittyviä haittavaikutuksia.
HCC
Potilailla, joilla oli lähtötilanteessa munuaisten vajaatoiminta, esiintyi enemmän väsymystä, hypotyroidismia, kuivumista, ripulia, ruokahalun heikkenemistä, proteinuriaa ja maksan enkefalopatiaa. Näillä potilailla esiintyi myös enemmän munuaisreaktioita ja valtimotromboembolisia tapahtumia.
Potilaat, joiden kehonpaino on < 60 kg
DTC
Alle 60 kg painavilla potilailla esiintyi enemmän käsi-jalkaoireyhtymää, proteinuriaa, asteen 3 tai 4 hypokalsemiaa ja hyponatremiaa sekä taipumus yleisempään asteen 3 tai 4 vähentyneeseen ruokahaluun.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
FI-00034 FIMEA
Yliannostus
Suurimmat kliinisesti tutkitut lenvatinibiannokset olivat 32 mg ja 40 mg päivässä. Kliinisissä tutkimuksissa on vahingossa tapahtuneiden annostusvirheiden takia otettu 40–48 mg:n kerta-annoksia. Useimmin havaitut haittavaikutukset näillä annoksilla olivat hypertensio, pahoinvointi, ripuli, väsymys, suutulehdus, proteinuria, päänsärky ja käsi-jalkaoireyhtymän paheneminen. Myös sellaisista yliannostustapauksista on raportoitu, joissa lenvatinibia on otettu 6–10 kertaa suositeltua vuorokausiannosta suurempana kerta-annoksena. Näissä tapauksissa haittavaikutukset (munuaisten ja sydämen vajaatoiminta) olivat yhdenmukaisia lenvatinibin tunnetun turvallisuusprofiilin kanssa, tai haittavaikutuksia ei esiintynyt ollenkaan.
Oireet ja hoito
Lenvatinibin yliannostukseen ei ole saatavilla erityistä vastalääkettä. Jos yliannostusta epäillään, lenvatinibihoito tulee keskeyttää ja potilaalle tulee tarvittaessa antaa sopivaa elintoimintoja tukevaa hoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Antineoplastiset aineet, proteiinikinaasin estäjät, ATC-koodi: L01EX08
Lenvatinibi on multikinaasiestäjä, jolla on osoitettu olevan pääasiassa antiangiogeenisiä ominaisuuksia in vitro ja in vivo, ja in vitro –malleissa havaittiin myös suoraa tuumorin kasvua estävää vaikutusta.
Vaikutusmekanismi
Lenvatinibi on reseptorityrosiinikinaasin (RTK) estäjä, joka estää valikoivasti endoteelikasvutekijän (VEGF) reseptoreiden VEGFR1 (FLT1), VEGFR2 (KDR) ja VEGFR3 (FLT4) kinaasin aktiivisuutta ja lisäksi muita angiogeneesiä edistäviin ja onkogeenisiin reitteihin liittyviä reseptorityrosiinikinaaseja, mukaan lukien fibroblastikasvutekijän (FGF) reseptorit FGFR1, 2, 3 ja 4 sekä verihiutalekasvutekijän (PDGF) reseptorit PDGFRα, KIT ja RET.
Lisäksi lenvatinibilla oli selektiivinen, suora antiproliferatiivinen aktiivisuus hepatosellulaarisissa solulinjoissa, mikä oli riippuvainen aktivoidusta FGFR-signaloinnista, mikä puolestaan liittyy lenvatinibin inhiboimaan FGFR-signalointiin.
Syngeenisissä hiiren kasvainmalleissa lenvatinibi vähensi kasvaimeen liittyviä makrofageja, lisäsi aktivoituneiden sytotoksisten T-solujen määrää ja sillä osoitettiin olevan suurempi kasvaimen kasvua ehkäisevä vaikutus yhdessä PD-1:n monoklonaalisen vasta-aineen kanssa verrattuna jompaankumpaan hoitoon yksinään.
Vaikka lenvatinibilla ei ole tehty suoranaisia tutkimuksia, hypertension vaikutusmekanismin oletetaan välittyvän VEGFR2:n eston avulla verisuonten endoteelisoluissa. Samalla tavalla ilman suoranaisia tutkimuksia oletetaan myös proteinurian vaikutusmekanismin välittyvän VEGFR1:n ja VEGFR2:n suppression avulla glomerulusten epiteelisoluissa.
Kilpirauhasen vajaatoiminnan toimintamekanismia ei ole täysin selvitetty.
Kliininen teho
Radiojodille resistentti erilaistunut kilpirauhassyöpä
SELECT-tutkimus oli satunnaistettu, kaksoissokkoutettu, lumelääkekontrolloitu monikeskustutkimus, johon osallistuneilla 392 potilaalla oli radiojodille resistentti erilaistunut kilpirauhassyöpä, jonka etenemisestä oli näyttöä riippumattoman, keskitetysti tarkistetun röntgenologisen tarkastelun perusteella tutkimukseen osallistumista edeltävien 12 kuukauden aikana (+ 1 kuukauden ikkuna). Radiojodille resistenssi määriteltiin yhtenä tai useampana mitattavissa olevana leesiona, johon ei kertynyt jodia tai joka eteni radiojodihoidosta huolimatta tai jonka kumulatiivinen radiojodiaktiivisuus oli > 600 mCi tai 22 GBq viimeisen annoksen yhteydessä vähintään 6 kuukautta ennen tutkimukseen osallistumista. Satunnaistaminen ositettiin maantieteellisen alueen (Eurooppa, Pohjois-Amerikka tai muu), aiemman VEGF/VEGFR-täsmähoidon (0 tai 1 aiemmin saatua VEGF/VEGFR-täsmähoitoa) ja iän (≤ 65 vuotta tai > 65 vuotta) perusteella. Ensisijainen tehoa koskevan lopputuloksen vertailuperuste oli etenemisvapaa elossaoloaika, joka määritettiin sokkoutetun riippumattoman röntgenologisen arvion ja RECIST 1.1. -kriteerien (Response Evaluation Criteria in Solid Tumours 1.1) avulla. Toissijaiset tehoa koskevan lopputuloksen vertailuperusteet olivat kokonaisvasteprosentti ja kokonaiselossaoloaika. Lumelääkehaaran potilailla oli mahdollisuus lenvatinibihoidon saamiseen heti, kun taudin eteneminen vahvistettiin.
Tutkimukseen sopivat potilaat, joiden sairaus oli mitattavissa RECIST 1.1 -kriteerein, satunnaistettiin suhteessa 2:1 saamaan joko lenvatinibia 24 mg kerran päivässä (n = 261) tai lumelääkettä (n = 131). Lähtötilanteen demografiset ja sairautta koskevat ominaisuudet olivat hyvin tasapainotettuja kummassakin hoitoryhmässä. Satunnaistetusta 392 potilaasta 76,3 % ei ollut aiemmin saanut VEGF/VEGFR-täsmähoitoa, 49,0 % oli naisia ja 49,7 % oli eurooppalaisia. Potilaiden iän mediaani oli 63 vuotta. Histologisesti 66,1 %:lla oli vahvistetusti diagnosoitu papillaarinen kilpirauhassyöpä ja 33,9 %:lla follikulaarinen kilpirauhassyöpä, mukaan lukien 14,8 %:lla hürthle-solukarsinooma ja 3,8 %:lla kirkassolukarsinooma. Metastaaseja oli 99 %:lla potilaista: keuhkoissa 89,3 %:lla, imusolmukkeissa 51,5 %:lla, luustossa 38,8 %, maksassa 18,1 %:lla, keuhkopussissa 16,3 %:lla ja aivoissa 4,1 %:lla. ECOG-toimintakykyluokka oli suurimmalla osalla potilaista 0, 42,1 %:lla 1 ja 3,9 %:lla yli 1. Ennen tutkimukseen osallistumista annetun radiojodin kumuloituneen aktiivisuuden mediaani oli 350 mCi (12,95 GBq).
Elossaoloajan ilman taudin etenemistä osoitettiin olevan tilastollisesti merkitsevästi pidempi lenvatinibihoitoa saaneilla kuin lumelääkettä saaneilla potilailla (p < 0,0001) (ks. kuva 1). Myönteinen vaikutus etenemisvapaaseen elossaoloaikaan nähtiin iän (yli tai alle 65 vuotta), sukupuolen, rodun, histologisen alatyypin, maantieteellisen alueen ja aiemmin saadun VEGF/VEGFR-täsmähoitojen (0 tai 1 kertaa) mukaisissa alaryhmissä. Ensisijaisen tehoanalyysin tekohetkellä lumelääkettä saamaan satunnaistetuista potilaista 109 (83,2 %) oli vaihtanut avoimeen lenvatinibihaaraan riippumattomaan arvioon perustuvan sairauden etenemisen vahvistuksen jälkeen.
Objektiivinen vasteprosentti (täydellinen vaste [CR] + osittainen vaste [PR]) oli riippumattoman röntgenologisen arvion perusteella merkitsevästi (p < 0,0001) suurempi lenvatinibiryhmässä (64,8 %) kuin lumelääkeryhmässä (1,5 %). Lenvatinibihoitoa saaneista potilaista 4 (1,5 %) saavutti täydellisen vasteen ja 165 (63,2 %) osittaisen vasteen. Lumelääkettä saaneista potilaista kukaan ei saavuttanut täydellistä vastetta ja 2 (1,5 %) saavutti osittaisen vasteen.
Ensimmäistä annoksen pienennystä edeltävän ajan mediaani oli 2,8 kuukautta. Objektiivista vastetta edeltävän ajan mediaani oli 2,0 kuukautta (95 %:n luottamusväli: 1,9; 3,5). Potilaista, joilla lenvatinibi sai aikaan täydellisen tai osittaisen vasteen, 70,4 %:lla vaste havaittiin kuitenkin 30 vuorokauden kuluessa siitä, kun hoito 24 mg:n annoksilla oli aloitettu.
Kokonaiselossaolon analyysia sekoitti se, että lumelääkettä saavilla potilailla, joiden tauti vahvistetusti eteni, oli mahdollisuus vaihtaa avoimeen lenvatinibihaaraan. Hoitoryhmien kokonaiseloonjäännissä ei ollut tilastollisesti merkitsevää eroa ensisijaisen tehoanalyysin kohdalla (riskisuhde = 0,73; 95 %:n luottamusväli: 0,50; 1,07, p = 0,1032). Tällöin kokonaiseloonjäännin mediaania ei ollut saavutettu lenvatinibiryhmässä tai lumelääkkeestä vaihtaneiden ryhmässä.
Taulukko 7 Tehoa koskevat tulokset DTC-potilaista
Lenvatinibi N = 261 | Lumelääke N = 131 | |
| Elossaoloaika ilman taudin etenemistä (PFS)a | ||
| Etenemisten tai kuolemien lukumäärä (%) | 107 (41,0) | 113 (86,3) |
| PFS:n mediaani kuukausina (95 %:n CI) | 18,3 (15,1; EA) | 3,6 (2,2; 3,7) |
| Riskisuhde (99 %:n CI)b,c | 0,21 (0,14; 0,31) | |
| P-arvob | < 0,0001 | |
| Potilaat, jotka olivat saaneet 0 aiempaa VEGF/VEGFR-täsmähoitoa (%) | 195 (74,7) | 104 (79,4) |
| Etenemisten tai kuolemien lukumäärä | 76 | 88 |
| PFS:n mediaani kuukausina (95 %:n CI) | 18,7 (16,4; EA) | 3,6 (2,1; 5,3) |
| Riskisuhde (95 %:n CI)b,c | 0,20 (0,14; 0,27) | |
| Potilaat, jotka olivat saaneet 1 aiempaa VEGF/VEGFR-täsmähoitoa (%) | 66 (25,3) | 27 (20,6) |
| Etenemisten tai kuolemien lukumäärä | 31 | 25 |
| PFS:n mediaani kuukausina (95 %:n CI) | 15,1 (8,8; EA) | 3,6 (1,9; 3,7) |
| Riskisuhde (95 %:n CI)b,c | 0,22 (0,12; 0,41) | |
| Objektiivinen vasteprosenttia | ||
| Objektiivisten vasteiden lukumäärä (%) | 169 (64,8) | 2 (1,5) |
| (95 %:n CI) | (59,0; 70,5) | (0,0; 3,6) |
| P-arvob | < 0,0001 | |
| Täydellisten vasteiden lukumäärä | 4 | 0 |
| Osittaisten vasteiden lukumäärä | 165 | 2 |
| Objektiivista vastetta edeltävän ajan mediaanid kuukausina (95 %:n CI) | 2,0 (1,9; 3,5) | 5,6 (1,8; 9,4) |
| Vasteen keston mediaanid kuukausina (95 %:n CI) | EA (16,8; EA) | EA (EA, EA) |
| Kokonaiseloonjäänti | ||
| Kuolemien lukumäärä (%) | 71 (27,2) | 47 (35,9) |
| Kokonaiseloonjäännin mediaani kuukausina (95 %:n CI) | EA (22,0; EA) | EA(20,3; EA) |
| Riskisuhde (95 %:n CI) b, e | 0,73 (0,50; 1,07) | |
| P-arvob, e | 0,1032 | |
CI = luottamusväli, EA = ei arvioitavissa, PFS = eloonjäänti ilman taudin etenemistä, RPSFT = järjestysluvun säilyttävä rakenteellinen vikaantumisaikamalli (Rank Preserving Structural Failure Time -malli), VEGF/VEGFR = endoteelikasvutekijä / endoteelikasvutekijän reseptori. a: Riippumaton radiologinen arvio. b: Ositettu maantieteellisen alueen (Eurooppa vs. Pohjois-Amerikka vs. muu), ikäryhmän (≤ 65 vuotta vs. > 65 vuotta) ja aiemman VEGF/VEGFR-täsmähoidon (0 vs. 1) mukaan. c: Arvioitu Coxin verrannollisten riskitiheyksien mallilla. d: Arvioitu Kaplan-Meierin menetelmällä; 95 %:n luottamusväli luotiin yleistetyn Brookmeyerin ja Crowleyn menetelmän avulla potilailla, joilla oli paras kokonaisvaste (täydellinen tai osittainen vaste). e: Vaihdon vaikutusta ei korjattu. | ||
Kuva 1 Kaplan-Meierin käyrä etenemisvapaasta elossaoloajasta – DTC
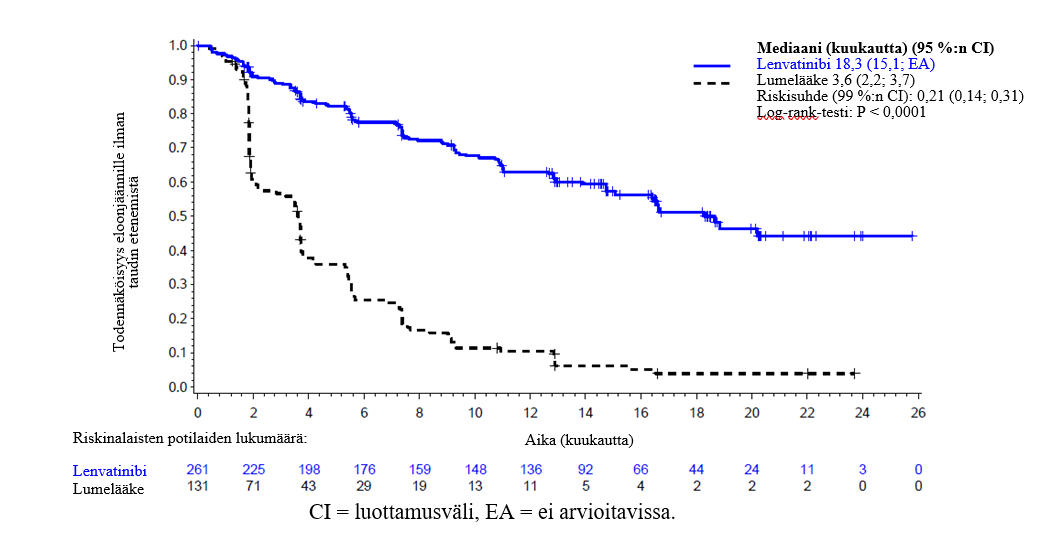
Hepatosellulaarinen karsinooma
Lenvatinibin kliininen tehokkuus ja turvallisuus on arvioitu kansainvälisessä, avoimessa, satunnaistetussa vaiheen 3 monikeskustutkimuksessa (REFLECT) potilailla, joilla on ei-resektoitavissa oleva hepatosellulaarinen karsinooma (HCC).
Yhteensä 954 potilasta satunnaistettiin 1:1 saamaan joko lenvatinibia (12 mg [jos paino lähtötilanteessa oli vähintään 60 kg] tai 8 mg [jos paino lähtötilanteessa oli alle 60 kg]) suun kautta kerran vuorokaudessa tai sorafenibia 400 mg suun kautta kaksi kertaa vuorokaudessa.
Potilaat voivat osallistua tutkimukseen, jos heidän maksan toimintakyky oli Child-Pugh-luokkaa A ja toimintakyky Eastern Cooperative Oncology Group Performance Status (ECOG PS) -luokittelun mukaan 0 tai 1. Potilaat, jotka olivat aiemmin saaneet systeemistä syöpähoitoa edenneeseen/ei-resektoitavaan hepatosellulaariseen karsinoomaan tai saaneet anti-VEGF-hoitoa, suljettiin tutkimuksen ulkopuolelle. Aiemmin sädehoidolla tai lokoregionaalisella hoidolla hoidetuissa kohdeleesioissa piti näkyä radiografisia merkkejä sairauden etenemisestä. Potilaat, joilla näkyi kuvauksessa ≥ 50 %:n taudin maksa-affiisio, selkeä invaasio sappitiehyeeseen tai porttilaskimon päähaaraan (Vp4), suljettiin myös tutkimuksen ulkopuolelle.
- Lenvatinibi- ja sorafenibiryhmien väestöominaisuudet ja lähtötilanteen sairauden ominaisuudet olivat samanlaiset, ja ne on esitetty jäljempänä kaikista 954 satunnaistetusta potilaasta:
- mediaani-ikä: 62 vuotta
- miehiä: 84 %
- valkoihoisia: 29 %, aasialaisia: 69 %, mustia tai afrikkalaisamerikkalaisia: 1,4 %
- paino: < 60 kg – 31 %, 60–80 kg – 50 %, > 80 kg – 19 %
- Eastern Cooperative Oncology Group Performance Status (ECOG PS) 0: 63 %, ECOG PS 1: 37 %
- Child-Pugh A: 99 %, Child-Pugh B: 1 %
- etiologia: hepatiitti B (50 %), hepatiitti C (23 %), alkoholi (6 %)
- makroskooppisen porttilaskimoinvaasion puuttuminen (MPVI): 79 %
- MPVI:n puuttuminen, kasvaimen leviäminen maksan ulkopuolelle (EHS) tai molemmat: 30 %
- piilevä kirroosi (erillisessä kuvantamisessa): 75 %
- Barcelona Clinic Liver Cancer (BCLC) ‑aste B: 20 %; BCLC-aste C: 80 %
- aiemmat hoidot: maksaresektio (28 %), sädehoito (11 %), lokoregionaalinen hoito, kuten transarteriaalinen (kemo-)embolisaatio (52 %), radiotaajuusablaatio (21 %) ja perkutaaninen etanoli-injektio (4 %)
Ensisijainen hoidon tehon päätemuuttuja oli kokonaiselinaika (OS). Lenvatinibi oli samanveroinen elinajan suhteen sorafenibiin verrattuna, HR = 0,92 [95 %:n CI (0,79; 1,06)] ja mediaanielinaika oli 13,6 kuukautta vs. 12,3 kuukautta (ks. taulukko 8 ja kuva 2). Sijaispäätemuuttujien (surrogate endpoints) (PFS ja ORR) tulokset on esitetty alla taulukossa 8.
| Taulukko 8: REFLECT-tutkimuksen tehokkuustulokset HCC-potilaista | ||||
| Tehoparametri | Hasardisuhdea, b (95 %:n CI) | P-arvo d | Mediaani (95 %:n CI) e | |
Lenvatinibi (N = 478) | Sorafenibi (N = 476) | |||
| OS | 0,92 (0,79; 1,06) | – | 13,6 (12,1; 14,9) | 12,3 (10,4; 13,9) |
| PFSg (mRECIST) | 0,64 (0,55; 0,75) | < 0,00001 | 7,3 (5,6; 7,5) | 3,6 (3,6; 3,7) |
| Prosenttiosuudet (95 %:n CI) | ||||
| ORRc, f, g (mRECIST) | – | < 0,00001 | 41 % (36 %; 45 %) | 12 % (9 %; 15 %) |
| Tietojen katkaisupäivä: 13. marraskuuta 2016. |
- Hasardisuhde (HR) on esitetty lenvatinibille verrattuna sorafenibiin, ja se perustuu Cox-malliin, joka sisältää hoitoryhmän yhtenä tekijänä.
- Ositettu (stratifioitu) alueittain (alue 1: Aasian–Tyynenmeren alue; alue 2: länsimaat), makroskooppinen porttilaskimoinvaasio tai leviäminen maksan ulkopuolelle tai molemmat (kyllä, ei), ECOG PS (0; 1) ja kehon paino (< 60 kg, ≥ 60 kg).
- Tulokset perustuvat vahvistettuihin ja vahvistamattomiin vasteisiin.
- P-arvo on lenvatinibin versus sorafenibin paremmuustestistä.
- Kvartiilit on arvioitu Kaplan–Meierin menetelmällä, ja 95 %:n luottamusvälit (CI) on arvioitu yleistetyllä Brookmeyerin ja Crowleyn menetelmällä.
- Kokonaisvaste (response rate) = (täydellinen tai osittainen vaste)
-
Riippumattoman radiologisen tarkastuksen retrospektiivisen analyysin mukaan. Objektiivisen vasteen mediaanikesto oli 7,3 (95 %:n CI 5,6; 7,4) kuukautta lenvatinibiryhmässä ja 6,2 (95 %:n CI 3,7; 11,2) kuukautta sorafenibiryhmässä.
Kuva 2 Kaplan–Meierin käyrä elinajan suhteen – HCC
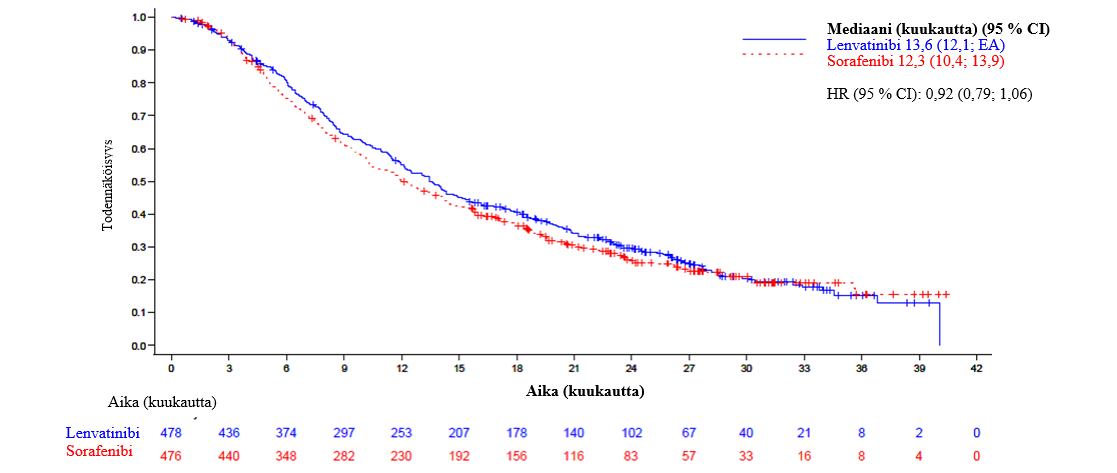
1. Tietojen katkaisupäivä: 13. marraskuuta 2016.
2. Hasardisuhteen ei-huonommuusmarginaali (HR: lenvatinibi vs. sorafenibi = 1,08).
3. Mediaani arvioitiin Kaplan–Meierin menetelmällä ja 95 %:n luottamusväli rakennettiin yleistetyllä Brookmeyerin ja Crowleyn menetelmällä.
4. HR arvioitiin Coxin suhteellisesta hasardimallista niin, että hoito oli erillinen muuttuja, ja ryhmiteltiin IxRS-ryhmittelykertoimilla. Efron-menetelmää käytettiin tasapistetilanteessa.
5. + = sensuroidut havainnot.
Aliryhmien stratifiointitekijöiden perusteella tehdyissä analyyseissä (MPVI:n tai EHS:n esiintyminen tai puuttuminen tai molemmat, ECOG PS 0 tai 1, kehonpaino < 60 kg tai ≥ 60 kg ja alue) HR yhdenmukaisesti suosi lenvatinibia sorafeinibin sijaan, lukuun ottamatta länsimaiden aluetta [HR 1,08 (95 %:n CI 0,82; 1,42], potilaita, joilla ei ollut EHS:ä [HR 1,01 (95 %:n CI 0,78; 1,30)], ja potilaita, joilla ei ollut MPVI:tä, EHS:ää tai kumpaakaan [HR 1,05 (0,79; 1,40)]. Aliryhmäanalyysien tuloksia on tulkittava varoen.
Hoidon mediaanikesto oli 5,7 kuukautta (Q1: 2,9; Q3: 11,1) lenvatinibiryhmässä ja 3,7 kuukautta (Q1: 1,8; Q3: 7,4) sorafenibiryhmässä.
REFLECT-tutkimuksen molemmissa hoitoryhmissä mediaani kokonaiselinaika oli noin 9 kuukautta pidempi tutkittavilla, jotka saivat hoidon jälkeen syöpähoitoa, verrattuna niihin, jotka eivät saaneet. Lenvatinibiryhmässä mediaani kokonaiselinaika oli 19,5 kuukautta (95 %:n CI: 15,7; 23,0) tutkittavilla, jotka saivat hoidon jälkeen syöpähoitoa (43 %), ja 10,5 kuukautta (95 %:n CI: 8,6; 12,2) tutkittavilla, jotka eivät sitä saaneet. Sorafenibiryhmässä mediaani kokonaiselinaika oli 17,0 kuukautta (95 %:n CI: 14,2; 18,8) tutkittavilla, jotka saivat hoidon jälkeen syöpähoitoa (51 %), ja 7,9 kuukautta (95 %:n CI: 6,6; 9,7) tutkittavilla, jotka eivät sitä saaneet. Mediaani kokonaiselinaika oli noin 2,5 kuukautta pidempi lenvatinibiryhmässä verrattuna sorafenibiryhmään molemmissa tutkittavien alaryhmissä (hoidon jälkeisestä syöpähoidosta riippumatta).
Endometriumkarsinooma
Lenvatinibin ja pembrolitsumabin yhdistelmähoidon tehoa tutkittiin tutkimuksessa 309. Tämä oli satunnaistettu, avoin, aktiivikontrolloitu monikeskustutkimus, joka tehtiin edennyttä EC:tä sairastavilla potilailla, jotka olivat aiemmin saaneet vähintään yhtä platinapohjaista solunsalpaajahoitoa minä tahansa hoitomuotona, mukaan lukien neoadjuvanttihoitona tai adjuvanttihoitona. Tutkittavat olivat saattaneet saada yhteensä kahtakin platinapohjaista hoitoa, kunhan toinen oli annettu neoadjuvanttihoitona ja toinen adjuvanttihoitona. Tutkimukseen ei otettu mukaan potilaita, joilla oli endometriaalinen stroomasarkooma (mukaan lukien karsinosarkooma), eikä potilaita, joilla oli aktiivinen autoimmuunisairaus tai jokin sairaus, joka edellytti immuunivasteen heikentämistä. Satunnaistaminen ositettiin MMR-statuksen (mismatch repair, kahdentumisvirheiden korjaus) (dMMR tai pMMR [ei dMMR]) mukaan validoidun immunohistokemiallisen määrityksen avulla. pMMR-osite ositettiin edelleen ECOG-toimintakykyluokan, maantieteellisen alueen ja aiemman lantion alueen sädehoidon mukaan. Potilaat satunnaistettiin (1:1) yhteen seuraavista hoitoryhmistä:
- lenvatinibi 20 mg suun kautta kerran vuorokaudessa yhdistelmähoitona pembrolitsumabin kanssa, jota annettiin laskimoon 200 mg kolmen viikon välein
- tutkijan valinnan mukaan joko doksorubisiini 60 mg/m2 kolmen viikon välein tai paklitakseli 80 mg/m2 viikoittain, 3 viikkoa hoitoa / 1 viikon tauko.
Lenvatinibi- ja pembrolitsumabihoitoa jatkettiin, kunnes tauti eteni RECIST v1.1. -kriteerien mukaisesti sokkoutetun riippumattoman keskitetyn arvioijatahon (Blinded Independent Central Review, BICR) arvioimana, kunnes ilmaantui toksisuutta, joka ei ollut hyväksyttävissä, tai pembrolitsumabihoitoa enintään 24 kuukauden ajan. Tutkimushoitoa voitiin jatkaa RECIST-kriteerien mukaisen taudin etenemisen jälkeenkin, jos tutkijalääkäri katsoi, että hoidosta oli kliinistä hyötyä potilaalle ja potilas sieti hoitoa. Kaikkiaan 121/411 (29 %) lenvatinibi- ja pembrolitsumabihoitoa saaneista potilaista jatkoi tutkimushoitoa RECIST-kriteerien mukaisen taudin etenemisen jälkeen. Etenemisen jälkeisen hoidon mediaanikesto oli 2,8 kuukautta. Kasvaimen tila arvioitiin 8 viikon välein.
Yhteensä 827 potilasta otettiin mukaan tutkimukseen ja satunnaistettiin saamaan joko lenvatinibin japembrolitsumabin yhdistelmähoitoa (n = 411) tai tutkijan valitsemana joko doksorubisiinia (n = 306) tai paklitakselia (n = 110). Näiden potilaiden lähtötilanteen ominaisuudet olivat seuraavat: iän mediaani 65 vuotta (vaihteluväli 30–86), 50 % vähintään 65 vuotta; 61 % valkoihoisia, 21 % aasialaisia ja 4 % mustaihoisia; ECOG-toimintakykyluokka 0 (59 %) tai 1 (41 %); 84 %:lla pMMR-kasvainstatus ja 16 %:lla dMMR-kasvainstatus. Histologiset alatyypit olivat endometrioidi karsinooma (60 %), seroosinen karsinooma (26 %), kirkassolukarsinooma (6 %), sekamuotoinen (5 %) ja muu (3 %). Kaikki 827 potilasta olivat saaneet aiempaa systeemistä hoitoa EC:hen: 69 % oli saanut yhtä, 28 % kahta ja 3 % vähintään kolmea aiempaa systeemistä hoitoa. 37 % potilaista oli saanut aiemmin vain neodjuvanttihoitoa tai adjuvanttihoitoa.
Tutkimushoidon keston mediaani oli 7,6 kuukautta (vaihteluväli: 1 päivästä 26,8 kuukauteen). Lenvatinibialtistuksen keston mediaani oli 6,9 kuukautta (vaihteluväli: 1 päivästä 26,8 kuukauteen).
Ensisijaiset tehoa mittaavat lopputulosmuuttujat olivat kokonaiselinaika (OS) ja elinaika ilman taudin etenemistä (PFS) (jotka arvioi BICR käyttämällä RECIST 1.1 -kriteerejä). Toissijaisia tehoa mittaavia lopputulosmuuttujia olivat objektiivisten vasteiden osuus (ORR), jonka arvioi BICR käyttämällä RECIST 1.1 -kriteerejä. Etukäteen määritetyn välianalyysin ajankohtana, jolloin seuranta-ajan mediaani oli 11,4 kuukautta (vaihteluväli: 0,3–26,9 kuukautta), tutkimus osoitti kokonaiselinajan ja elinajan ilman taudin etenemistä parantuneen tilastollisesti merkitsevästi kaikki potilaat kattavassa populaatiossa.
Tehon tulokset MMR-aliryhmittäin olivat yhdenmukaisia tutkimuksen kokonaistulosten kanssa.
Etukäteen määritetty lopullinen kokonaiselinajan analyysi tehtiin ilman korjausta multiplisiteetin suhteen. Tällöin seurantaa oli jatkettu välianalyysin jälkeen vielä noin 16 kuukautta (seuranta-ajan mediaani yhteensä 14,7 kuukautta [vaihteluväli: 0,3–43,0 kuukautta]). Tehon tuloksista kaikki potilaat kattavassa populaatiossa on yhteenveto taulukossa 9 ja Kaplan–Meierin käyrä lopullisesta kokonaiselinajan analyysista on esitetty kuvassa 3 ja elinaikaa ilman taudin etenemistä koskevasta välianalyysista kuvassa 4.
Taulukko 9 Tutkimuksen 309 tehoa koskevat tulokset endometriumkarsinoomassa
| |||
| Päätetapahtuma | LENVIMA ja pembrolitsumabi N = 411 | Doksorubisiini tai paklitakseli N = 416 | |
| OS | |||
| Niiden potilaiden määrä (%), joilla tapahtuma | 276 (67 %) | 329 (79 %) | |
| Mediaani kuukausina (95 %:n CI) | 18,7 (15,6, 21,3) | 11,9 (10,7, 13,3) | |
| Riskisuhdea (95 %:n CI) | 0,65 (0,55, 0,77) | ||
| P-arvob | < 0,0001 | ||
| PFSd | |||
| Niiden potilaiden määrä (%), joilla tapahtuma | 281 (68 %) | 286 (69 %) | |
| Mediaani kuukausina (95 %:n CI) | 7,2 (5,7, 7,6) | 3,8 (3,6, 4,2) | |
| Riskisuhdea (95 %:n CI) | 0,56 (0,47, 0,66) | ||
| P-arvoc | < 0,0001 | ||
| ORRd | |||
| ORRe (95 %:n CI) | 32 % (27, 37) | 15 % (11,18) | |
| Täydellinen vaste | 7 % | 3 % | |
| Osittainen vaste | 25 % | 12 % | |
| P-arvof | < 0,0001 | ||
| Vasteen kestod | |||
| Mediaani kuukausinag (vaihteluväli) | 14,4 (yli 1,6; yli 23,7) | 5,7 (yli 0,0; yli 24,2) | |
a Perustuu ositettuun Coxin regressiomalliin b Yksipuolinen nimellinen P-arvo perustuu ositettuun log-rank-testiin (lopullinen analyysi). Etukäteen määritetyn kokonaiselinaikaa koskevan välianalyysin kohdalla, jolloin seuranta-ajan mediaani oli 11,4 kuukautta (vaihteluväli: 0,3–26,9 kuukautta) lenvatinibin ja pembrolitsumabin yhdistelmän todettiin olevan tilastollisesti merkitsevästi parempi kokonaiselinajan suhteen verrattuna doksorubisiiniin tai paklitakseliin (HR: 0,62 [95 %:n CI: 0,51, 0,75] P-arvo < 0,0001). c Yksipuolinen P-arvo perustuu ositettuun log-rank-testiin d Etukäteen määritetyssä välianalyysissa e Vaste: paras objektiivinen vaste vahvistettuna täydellisenä vasteena tai osittaisena vasteena f Perustuu Miettisen ja Nurmisen menetelmään ositettuna ECOG-toimintakykyluokan, maantieteellisen alueen ja aiemman lantion alueen sädehoidon mukaan. g Perustuu Kaplan–Meierin estimaattiin | |||
Kuva 3 Kaplan–Meierin käyrä kokonaiselinajasta tutkimuksessa 309*
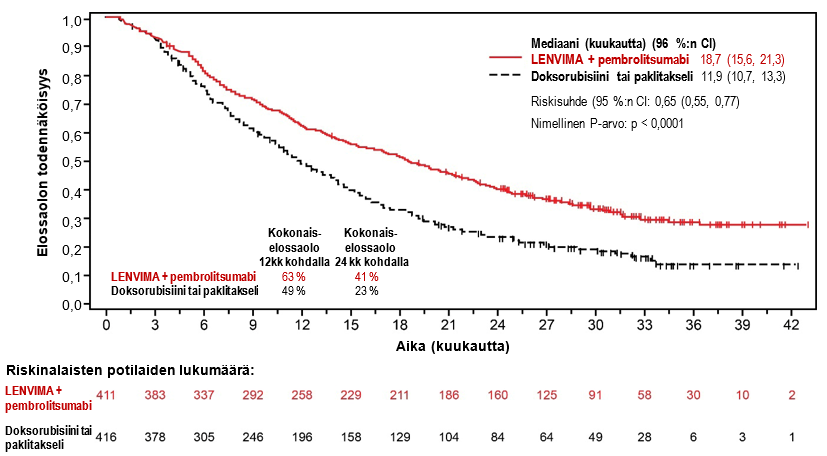
*Tutkimussuunnitelmassa määritetyn lopullisen analyysin perusteella
Kuva 4 Kaplan–Meierin käyrä elinajasta ilman taudin etenemistä tutkimuksessa 309
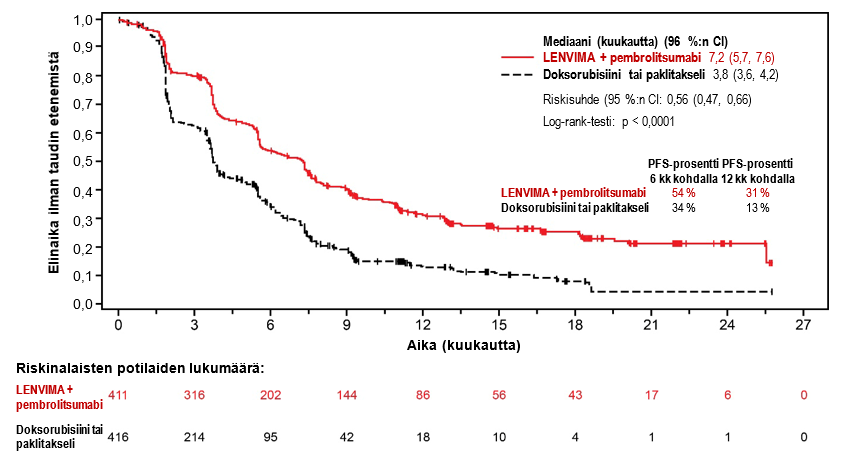
QT-ajan pidentyminen
Terveillä koehenkilöillä tehdyssä perusteellisessa QT-tutkimuksessa 32 mg:n kerta-annos lenvatinibia ei pidentänyt QT/QTc-aikaa. QT/QTc-ajan pidentymistä on kuitenkin raportoitu useammin lenvatinibihoitoa saavilla kuin lumelääkettä saavilla potilailla (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset lenvatinibin käytöstä hepatosellulaarisen karsinooman (HCC) ja endometriumkarsinooman (EC) hoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Pediatriset tutkimukset
Lenvatinibin tehoa arvioitiin, mutta ei varmistettu, kahdessa avoimessa tutkimuksessa:
Tutkimus 207 oli vaiheen 1/2 avoin, monikeskustutkimuksena toteutettu annoshaku- ja aktiivisuusarviointitutkimus, jossa lenvatinibia annettiin monoterapiana ja yhdistelmähoitona ifosfamidin ja etoposidin kanssa pediatrisille potilaille (ikä 2 – < 18 vuotta; osteosarkooman osalta 2 – ≤ 25 vuotta), joilla oli uusiutuneita tai refraktorisia kiinteitä kasvaimia. Tutkimukseen osallistui yhteensä 97 potilasta. Lenvatinibimonoterapian annoshakukohorttiin osallistui 23 potilasta, jotka saivat lenvatinibia suun kautta kerran vuorokaudessa kolmella annostasolla (11, 14 tai 17 mg/m2). Lenvatinibin, ifosfamidin ja etoposidin yhdistelmähoidon annoshakukohorttiin osallistui yhteensä 22 potilasta, jotka saivat lenvatinibia kahdella annostasolla (11 tai 14 mg/m2). Lenvatinibin suositusannokseksi monoterapiana ja yhdistelmähoitona ifosfamidin ja etoposidin kanssa määriteltiin 14 mg/m2 suun kautta kerran vuorokaudessa.
Laajennetussa lenvatinibimonoterapiakohortissa, johon kuuluneilla potilailla oli uusiutunut tai refraktorinen DTC, ensisijainen tehoa koskevan lopputuloksen vertailuperuste oli objektiivinen vasteprosentti (ORR: täydellinen vaste [CR] + osittainen vaste [PR]). Kohorttiin osallistui yksi potilas, ja tämä potilas saavutti osittaisen vasteen. Laajennetussa lenvatinibimonoterapiakohortissa sekä lenvatinibin, ifosfamidin ja etoposidin laajennetussa yhdistelmähoitokohortissa, joihin kuuluneilla potilailla oli uusiutunut tai refraktorinen osteosarkooma, ensisijainen tehoa koskevan lopputuloksen vertailuperuste oli etenemiseltä säästyneiden osuus 4 kuukauden kohdalla (PFS 4). Binomiarvion mukainen PFS 4, johon sisältyivät kaikki 31 lenvatinibimonoterapiaa saanutta potilasta, oli 29 % (95 %:n luottamusväli: 14,2; 48,0). Binomiarvion mukainen PFS 4, johon sisältyivät kaikki 20 lenvatinibin, ifosfamidin ja etoposidin laajennettuun yhdistelmähoitokohorttiin kuulunutta potilasta, oli 50 % (95 %:n luottamusväli: 27,2; 72,8).
Tutkimus 261 oli avoin, yhdellä hoitoryhmällä toteutettu vaiheen 1/2 monikeskustutkimus, jossa yhdistelmähoitona everolimuusin kanssa annetun lenvatinibin turvallisuutta, siedettävyyttä ja kasvaimia tuhoavaa vaikutusta selvitettiin pediatrisilla potilailla (ja ≤ 21 vuoden ikäisillä nuorilla aikuisilla), joilla oli uusiutuneita tai refraktorisia kiinteitä maligniteetteja, mukaan lukien keskushermoston kasvaimet. Tutkimukseen otettiin ja siinä hoidettiin kaikkiaan 64 potilasta. Vaiheeseen 1 (yhdistelmähoidon annoshakuvaihe) otettiin 23 potilasta, jotka saivat hoitoa seuraavasti: 5 potilasta sai hoitoa annostasolla 1 (lenvatinibi 8 mg/m2 ja everolimuusi 3 mg/m2) ja 18 potilasta annostasolla 1 (lenvatinibi 11 mg/m2 ja everolimuusi 3 mg/m2). Yhdistelmähoidon suositeltu annos oli lenvatinibi 11 mg/m2 ja everolimuusi 3 mg/m2 kerran vuorokaudessa. Vaiheeseen 2 (laajennettu yhdistelmähoito) otettiin 41 potilasta, jotka saivat hoitoa suositellulla annoksella seuraavissa kohorteissa: Ewingin sarkooma (EWS, n = 10), rabdomyosarkooma (RMS, n = 20) ja korkea-asteinen gliooma (HGG, n = 11). Ensisijainen tehoa koskevan lopputuloksen vertailuperuste oli arviointikelpoisten potilaiden objektiivinen vasteprosentti (ORR) viikolla 16, jonka tutkija arvioi RECIST v.1.1- tai RANO-kriteerien (HGG-potilaiden kohdalla) perusteella. EWS- ja HGG-kohorteissa ei todettu yhtään objektiivista vastetta, ja RMS-kohortissa todettiin kaksi osittaista ORR-vastetta viikolla 16: 10 % (95 %:n luottamusväli: 1,2; 31,7).
OLIE-tutkimus (tutkimus 230) oli vaiheen 2 avoin, satunnaistettu, kontrolloitu monikeskustutkimus, johon osallistuneilla potilailla (ikä 2 – ≤ 25 vuotta) oli uusiutunut tai refraktorinen osteosarkooma. Yhteensä 81 potilasta satunnaistettiin suhteessa 1:1 (78 hoidettavaa; 39 kummassakin haarassa) saamaan lenvatinibia 14 mg/m2 yhdistelmähoitona ifosfamidin 3 000 mg/m2 ja etoposidin 100 mg/m2 kanssa (ryhmä A) tai ifosfamidia 3 000 mg/m2 ja etoposidia 100 mg/m2 (ryhmä B). Ifosfamidi ja etoposidi annettiin laskimoon kunkin 21 päiväisen syklin päivinä 1–3 enintään 5 syklin ajan. Lenvatinibihoito sallittiin, kunnes tauti eteni RECIST v1.1. -kriteerien mukaisesti sokkoutetun riippumattoman keskitetyn arvioijatahon (Blinded Independent Central Review, BICR) arvioimana tai kunnes ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Ensisijainen tehoa koskevan lopputuloksen vertailuperuste oli RECIST v1.1. -kriteerien mukainen etenemiseltä säästyneiden osuus (PFS) BICR:n arvioimana. Tutkimuksessa ei osoitettu tilastollisesti merkitsevää eroa PFS:n mediaanissa: se oli 6,5 kuukautta (95 %:n luottamusväli: 5.7; 8.2) lenvatinibin, ifosfamidin ja etoposidin yhdistelmähoidossa ja 5,5 kuukautta (95 %:n luottamusväli: 2,9; 6,5) ifosfamidin ja etoposidin yhdistelmähoidossa (HR = 0,54 [95 %:n luottamusväli: 0,27; 1,08]). Tutkimuksen 230 tilastollinen voima ei ollut riittävä tilastollisesti merkittävän eron havaitsemiseen kokonaiselinaikojen suhteen. Tutkimuksen päätösanalyysissa hasardisuhde oli 0,93 (95 %:n luottamusväli: 0,53; 1,62) lenvantinibin, ifosfamidin ja etoposidin yhdistelmähoidon sekä ifosfamidin ja etoposidin vertailussa. Kokonaiselinajan mediaani oli 12,4 kuukautta (95 %:n luottamusväli: 10,4; 19,8) ja 17,2 kuukautta (95 %:n luottamusväli: 11,1; 22,3), ja seuranta-ajan mediaani oli 24,1 kuukautta ja 29,5 kuukautta.
Tutkimus 231 on avoin, vaiheen 2 monikeskus- ja koritutkimus, jossa lenvatinibin kasvaimia tuhoavaa vaikutusta ja turvallisuutta arvioidaan 2 – ≤ 21 vuoden ikäisillä lapsilla, nuorilla ja nuorilla aikuisilla, joilla on uusiutuneita tai refraktorisia kiinteitä maligniteetteja, mukaan lukien EWS, RMS ja HGG. Tutkimukseen otettiin kaikkiaan 127 potilasta, jotka saivat hoitoa lenvatinibin suositellulla annoksella (14 mg/m2) seuraavissa kohorteissa: EWS (n = 9), RMS (n = 17), HGG (n = 8) ja muut kiinteät kasvaimet (n = 9 kaikissa seuraavissa: diffuusi keskilinjan gliooma, medulloblastooma ja ependymooma; kaikki muut kiinteät kasvaimet n = 66). Ensisijainen tehoa koskevan lopputuloksen vertailuperuste oli arviointikelpoisten potilaiden ORR viikolla 16, jonka tutkija arvioi RECIST v.1.1- tai RANO-kriteerien (HGG-potilaiden kohdalla) perusteella. Yhtään objektiivista vastetta ei todettu potilailla, joilla oli HGG, diffuusi keskilinjan gliooma, medulloblastooma tai ependymooma. Sekä EWS- että RMS-kohorteissa todettiin kaksi osittaista ORR-vastetta viikolla 16: 22,2 % (95 %:n luottamusväli: 2,8; 60,0) ja 11,8 % (95 %:n luottamusväli: 1,5; 36,4). Kaikkien muiden kiinteiden kasvainten joukossa todettiin viisi osittaista ORR-vastetta (synoviaalisarkooma [n = 2], kaposiforminen hemangioendoteliooma [n = 1], Wilmsin kasvain eli nefroblastooma [n = 1] ja kirkassolukarsinooma [n = 1]) viikolla 16: 7,7 % (95 %:n luottamusväli: 2,5; 17,0).
Farmakokinetiikka
Lenvatinibin farmakokineettisiä parametreja on tutkittu terveillä koehenkilöillä sekä koehenkilöillä, joilla on maksan vajaatoiminta, munuaisten vajaatoiminta ja kiinteitä kasvaimia.
Imeytyminen
Lenvatinibi imeytyy nopeasti suun kautta annon jälkeen; tyypillisesti havaittu tmaxarvo on 1–4 tuntia annoksen jälkeen. Ruoka ei vaikuta imeytymisen määrään, mutta se hidastaa imeytymistä. Terveille koehenkilöille ruoan kanssa annettaessa huippupitoisuus plasmassa viivästyy 2 tunnilla. Absoluuttista biologista hyötyosuutta ihmisellä ei ole määritetty, mutta tiedot massatasetutkimuksesta viittaavat noin 85 %:iin. Suun kautta annetun annoksen absoluuttinen biologinen hyötyosuus oli hyvä koirilla (70,4 %) ja apinoilla (78,4 %).
Jakautuminen
Lenvatinibin sitoutuminen ihmisen plasman proteiineihin on runsasta in vitro: 98–99 % (0,3–30 μg/ml, mesilaatti). Lenvatinibi sitoutui pääasiassa albumiiniin sekä vähäisemmässä määrin happamaan alfa-1-glykoproteiiniin ja γ‑globuliiniin.
Veren ja plasman lenvatinibipitoisuuksien suhde in vitro vaihteli välillä 0,589–0,608 (0,1–10 μg/ml, mesilaatti).
Lenvatinibi on P-gp:n ja BCRP:n substraatti. Lenvatinibi ei ole OAT1:n, OAT3:n, OATP1B1:n, OATP1B3:n, OCT1:n, OCT2:n, MATE1:n, MATE2-K:n tai sappisuolan vientipumpun BSEP:n substraatti.
Potilailla ensimmäisen annoksen näennäisen jakaantumistilavuuden mediaani (Vz/F) vaihteli välillä 50,5–92 l, ja se oli yleisesti ottaen yhdenmukainen annosryhmien (3,2–32 mg) välillä. Vastaava vakaan tilan näennäisen jakaantumistilavuuden mediaani vaihteli välillä 43,2–121 l, ja se oli myös yleisesti ottaen yhdenmukainen annosryhmien välillä.
Biotransformaatio
Sytokromi P450 3A4:n osoitettiin in vitro olevan pääasiallinen (> 80 %) lenvatinibin P450-välitteiseen metaboliaan osallistuva isoformi. In vivo -tiedot kuitenkin osoittivat ei-P450-välitteisten reittien osallistuvan merkittävissä määrin lenvatinibin kokonaismetaboliaan. Näin ollen CYP 3A4:n indusoijilla ja estäjillä oli minimaalinen vaikutus lenvatinibille altistukseen in vivo (ks. kohta Yhteisvaikutukset).
Lenvatinibin demetyloituneen muodon (M2) havaittiin olevan pääasiallinen metaboliitti ihmisen maksan mikrosomeissa. Pääasialliset metaboliitit ihmisen ulosteessa, M2’ ja M3’, muodostuivat M2-metaboliitista ja lenvatinibista aldehydioksidaasin avulla.
Plasmanäytteissä, jotka kerättiin 24 tunnin sisällä annosta, lenvatinibin osuus radioaktiivisuudesta plasman radiokromatogrammeissa oli 97 %, kun taas M2-metaboliitin osuus oli 2,5 %. AUC(0–inf)-arvon perusteella lenvatinibin osuus kokonaisradioaktiivisuudesta plasmassa oli 60 % ja osuus kokonaisradioaktiivisuudesta veressä 64 %.
Ihmisillä tehdyn massatasapaino/eritys-tutkimuksen tiedot osoittavat lenvatinibin metaboloituvan ihmisillä voimakkaasti. Pääasiallisten metaboliareittien havaittiin ihmisillä olevan hapettuminen aldehydioksidaasin avulla, demetylaatio CYP3A4:n avulla, glutationin konjugaatio, jossa O-aryyliryhmä (kloorifenyyliosa) eliminoituu, sekä näiden reittien yhdistelmät ja niitä seuraavat biotransformaatiot (esim. glukuronidaatio, glutationiosan hydrolyysi, kysteiiniosan pilkkoutuminen sekä kysteinyyliglysiinin ja kysteiinin konjugaattien molekyylinsisäinen uudelleen ryhmittyminen ja sitä seuraava dimerisaatio). Nämä metaboliset reitit in vivo ovat yhdenmukaisia ihmisten biomateriaaleilla tehtyjen in vitro -kokeiden tulosten kanssa.
Kuljettajaproteiineilla tehdyt in vitro -kokeet
Seuraavien kuljettajaproteiinien kliinisesti merkittävä esto suljettiin pois, kun katkaisukohtana oli
IC50> 50 × Cmax,sitoutumaton: OAT1, OAT3, OATP1B1, OCT1, OCT2 ja BSEP.
Lenvatinibin P-gp- ja rintasyövän resistenssiproteiini (BCRP) ‑välitteistä kuljettajaproteiinitoimintaa estävä vaikutus oli minimaalinen tai olematon. Myöskään P-gp mRNA:n ekspressiota indusoivaa vaikutusta ei havaittu.
Lenvatinibin OATP1B3:a ja MATE2-K:ta estävä vaikutus oli minimaalinen tai olematon. Lenvatinibi estää heikosti MATE1:tä. Lenvatinibi ei estänyt aldehydioksidaasitoimintaa ihmisen maksasolujen sytosolissa.
Eliminaatio
Pitoisuus plasmassa laskee bieksponentiaalisesti huippupitoisuuden (Cmax) jälkeen. Lenvatinibin keskimääräinen terminaalinen eksponentiaalinen puoliintumisaika on noin 28 tuntia.
Kun radioaktiivisesti merkittyä lenvatinibia annettiin 6 potilaalle, joilla oli kiinteitä kasvaimia, noin kaksi kolmannesta radioaktiivisesta merkkiaineesta poistui ulosteen mukana ja yksi neljännes virtsan mukana. M3-metaboliitti oli tärkein analyytti ulosteessa (noin 17 % annoksesta) ja sen jälkeen M2’ (noin 11 % annoksesta) ja M2 (noin 4,4 % annoksesta).
Lineaarisuus/ei-lineaarisuus
Annosten suhteellisuus ja kumuloituminen
Potilailla, joilla oli kiinteitä kasvaimia ja joille annettiin yksittäisiä tai useita lenvatinibiannoksia kerran päivässä, lenvatinibille altistus (Cmax ja AUC) kasvoi suorassa suhteessa annettuun annokseen annosvälillä 3,2–32 mg kerran päivässä.
Lenvatinibin kumuloituminen vakaassa tilassa on minimaalista. Tällä alueella kumuloitumisindeksi (Rac) vaihteli välillä 0,96 (20 mg) - 1,54 (6,4 mg). HCC-tutkittavien, joilla oli lievä tai keskivaikea maksan vajaatoiminta, Rac oli samanlainen kuin muiden kiinteiden kasvainten osalta on ilmoitettu.
Erityisryhmät
Maksan vajaatoiminta
Lenvatinibin farmakokinetiikkaa 10 mg:n kerta-annoksen jälkeen arvioitiin 6 koehenkilöllä, joilla oli lievä (Child-Pugh A) tai keskivaikea (Child-Pugh B) maksan vajaatoiminta. Lisäksi arvioitiin 5 mg:n annosta 6 koehenkilöllä, joilla oli vaikea (Child-Pugh C) maksan vajaatoiminta. Verrokkeina käytettiin kahdeksaa tervettä, demografisesti kaltaistettua henkilöä, jotka saivat 10 mg:n annoksen. Annoskorjattujen AUC0-t - ja AUC0-inf -arvojen perusteella lenvatinibille altistus oli lievää maksan vajaatoimintaa sairastavilla koehenkilöillä 119 %, keskivaikeaa maksan vajaatoimintaa sairastavilla 107 % ja vaikeaa maksan vajaatoimintaa sairastavilla 180 % normaalista. Ei tiedetä, muuttuuko plasman proteiiniin sitoutuminen maksan vajaatoimintaa sairastavilla henkilöillä. Annostussuositukset, ks. kohta Annostus ja antotapa.
HCC-potilaista, joilla on Child-Pugh B (keskivaikea maksan vajaatoiminta, 3 lenvatinibilla hoidettua potilasta keskeisessä tutkimuksessa), ei ole riittäviä tietoja, ja HCC-potilaista, joilla on Child-Pugh C (vaikea maksan vajaatoiminta), ei ole tietoja. Lenvatinibi eliminoituu pääasiassa maksan kautta ja altistus voi olla suurempi näillä potilasryhmillä.
Puoliintumisajan mediaani oli verrannollinen koehenkilöillä, joilla oli lievä, keskivaikea ja vaikea maksan vajaatoimintaa sekä myös normaali maksan toiminta, ja se vaihteli välillä 26–31 tuntia. Lenvatinibiannoksen virtsaan erittyvä osuus oli pieni kaikissa kohorteissa (< 2,16 % hoitokohortista riippumatta).
Munuaisten vajaatoiminta
Lenvatinibin farmakokinetiikkaa 24 mg:n kerta-annoksen jälkeen arvioitiin 6 koehenkilöllä, joilla oli lievä, keskivaikea, tai vaikea munuaisten vajaatoiminta, sekä 8 terveellä, demografisesti kaltaistetulla verrokilla. Loppuvaiheen munuaissairautta sairastavia potilaita ei tutkittu.
Lenvatinibille altistuminen, AUC0-inf tietoihin perustuen, oli lievää munuaisten vajaatoimintaa sairastavilla koehenkilöillä 101 %, keskivaikeaa munuaisten vajaatoimintaa sairastavilla koehenkilöillä 90 % ja vaikeaa munuaisten vajaatoimintaa sairastavilla koehenkilöillä 122 % normaalien koehenkilöiden arvosta. Ei tiedetä, muuttuuko plasman proteiinisidonnaisuus munuaisten vajaatoimintaa sairastavilla henkilöillä. Annostussuositukset, ks. kohta Annostus ja antotapa.
Ikä, sukupuoli, paino, rotu
Populaatiofarmakokineettisessä analyysissa, joka käsitti enintään 24 mg lenvatinibia kerran päivässä saavia potilaita, iällä, sukupuolella, painolla ja rodulla (japanilainen vs. muu, valkoihoinen vs. muu) ei ollut kliinisesti merkittävää vaikutusta puhdistumaan (ks. kohta Annostus ja antotapa).
Pediatriset potilaat
Populaatiofarmakokineettisessä analyysissa, joka suoritettiin 2–12 vuoden ikäisillä pediatrisilla potilailla ja joka käsitti tiedot pediatriseen lenvatinibiohjelmaan kuuluneista 3 pediatrista potilasta, joiden ikä oli 2 – < 3 vuotta, 28 pediatrista potilasta, joiden ikä oli ≥ 3 – < 6 vuotta, ja 89 pediatrista potilasta, joiden ikä oli 6 – ≤ 12 vuotta), lenvatinibin puhdistumaan suun kautta (CL/F) vaikutti tutkittavan paino, ei kuitenkaan ikä. Ennustetut altistustasot käyrän alle jäävän pinta-alan mukaan vakaassa tilassa (AUCss) pediatrisilla potilailla, jotka saivat 14 mg/m2, olivat verrannolliset kiinteän 24 mg:n annoksen saaneiden aikuisten potilaiden altistustasoihin. Näissä tutkimuksissa ei havaittu ilmeisiä eroja vaikuttavan aineen, lenvatinibin, farmakokinetiikassa lapsilla (ikä 2–12 vuotta), nuorilla ja nuorilla aikuisilla, joilla oli tutkittuja kasvaintyyppejä, mutta lapsia koskevat tiedot ovat suhteellisen rajalliset, jotta varmoja päätelmiä ei voida tehdä (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Toistuvien annosten toksisuutta arvioivissa tutkimuksissa (kesto enintään 39 viikkoa) lenvatinibi aiheutti toksikologisia muutoksia eri elimissä ja kudoksissa. Muutokset liittyivät lenvatinibin odotettuihin farmakologisiin vaikutuksiin, ja niitä olivat glomerulupatia, kivesten solukato, munasarjojen follikulaarinen atresia, maha-suolikanavan muutokset, luuston muutokset, lisämunuaisten muutokset (rotilla ja koirilla) ja valtimoleesiot (arteriaalinen fibrinoidi nekroosi, mediaalinen rappeuma tai verenvuoto) rotilla, koirilla ja cynomolgus-apinoilla. Maksatoksisuuteen liittyvää transaminaasipitoisuuksien kohoamista havaittiin myös rotilla, koirilla ja apinoilla. Toksikologisten muutosten havaittiin hävinneen 4 viikkoa kestäneen palautumisjakson lopussa kaikilla tutkituilla eläinlajeilla.
Genotoksisuus
Lenvatinibi ei ollut genotoksinen.
Lenvatinibilla ei ole tehty karsinogeenisuustutkimuksia.
Lisääntymis- ja kehitystoksisuus
Lenvatinibin vaikutusta hedelmällisyyteen arvioivia erityisiä eläintutkimuksia ei ole tehty. Toistuvien annosten toksisuutta arvioivissa eläintutkimuksissa havaittiin kuitenkin muutoksia kiveksissä (siementiehyiden epiteelin solukatoa)ja munasarjoissa (follikulaarista atresiaa) altistuksilla, jotka vastasivat 11–15-kertaista (rotta) tai 0,6–7-kertaista (apina) odotettua kliinistä altistusta (perustuu AUC-arvoon) ihmisen suurimmalla siedetyllä annoksella. Näiden löydösten havaittiin korjautuneen 4 viikon pituisen palautumisjakson lopussa.
Lenvatinibin anto organogeneesin aikana johti rotilla alkiokuolemiin ja teratogeenisuuteen (sikiöiden ulkoisiin ja luuston poikkeavuuksiin) altistuksilla, jotka olivat pienempiä kuin kliininen altistus (perustuu AUC-arvoon) ihmisen suurimmalla siedetyllä annoksella, ja kaneilla alkiokuolemiin ja teratogeenisuuteen (sikiöiden ulkoisiin, sisäelinten ja luuston poikkeavuuksiin) kehon pinta-alan (mg/m2) perusteella ihmisen suurinta siedettyä annosta vastaavalla altistuksella. Nämä löydökset viittaavat mahdolliseen teratogeenisuuteen, joka todennäköisesti liittyy lenvatinibin farmakologiseen aktiivisuuteen verisuonien kasvua estävänä aineena.
Lenvatinibi ja sen metaboliitit erittyvät rottien rintamaitoon.
Toksisuustutkimukset nuorilla eläimillä
Kuolleisuus oli annosta rajoittava toksisuus nuorilla rotilla, joille anto aloitettiin 7:nä tai 21:nä postnataalisena päivänä, ja se havaittiin altistuksilla, jotka olivat 125 tai 12 kertaa pienempiä kuin altistustasot, joilla aikuisilla rotilla havaittaiin kuolleisuutta, mikä viittaa siihen, että herkkyys toksisuudelle lisääntyy nuorenevan iän myötä. Kuolleisuus voidaan siten yhdistää pohjukaissuolen primaarileesioihin liittyviin komplikaatioihin, ja kuolleisuutta voi mahdollisesti lisätä kehittymättömiin elimiiin kohdistunut toksisuus.
Lenvatinibin toksisuus oli voimakkaampaa rotilla, joille anto aloitettiin 7:nä postnataalisena päivänä, kuin rotilla, joille anto aloitettiin 21:nä postnataalisena päivänä ja kuolleisuutta ja tiettyjä toksisuuksia havaittiin aikaisemmin nuorilla rotilla, joiden annos oli 10 mg/kg, kuin aikuisilla rotilla, jotka saivat samansuuruisen annoksen. Nuorilla rotilla havaittiin lisäksi kasvun hidastumista, fyysisen kehityksen sekundaarista viivästymistä ja farmakologisiin vaikutuksiin yhdistettyjä leesioita (etuhampaissa, reisiluussa [kasvurustossa], munuaisissa, lisämunuaisissa ja pohjukkaissuolessa).
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
Kalsiumkarbonaatti, mannitoli, mikrokiteinen selluloosa, hydroksipropyyliselluloosa, matalasubstituoitu hydroksipropyyliselluloosa, talkki.
Kapselin kuori
Hypromelloosi, titaanidioksidi (E171), keltainen rautaoksidi (E172), punainen rautaoksidi (E172).
Painomuste
Sellakka, musta rautaoksidi (E172), kaliumhydroksidi, propyleeniglykoli.
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
4 vuotta.
Säilytys
Säilytä alle 25°C. Säilytä alkuperäisessä läpipainopakkauksessa. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
LENVIMA kapseli, kova
4 mg (L:ei) 30 fol (1360,40 €)
10 mg (L:ei) 30 fol (1579,77 €)
PF-selosteen tieto
Polyamidi/alumiini/PVC/alumiini -läpipainopakkaukset, jotka sisältävät 10 kapselia. Yksi pakkaus sisältää 30, 60 tai 90 kovaa kapselia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
4 mg: Kellertävän punainen runko ja kellertävän punainen hattu. Pituus on noin 14,3 mm. Hattuun on merkitty mustalla musteella ”Є” ja runkoon ”LENV 4 mg”.
10 mg: Keltainen runko ja kellertävän punainen hattu. Pituus on noin 14,3 mm. Hattuun on merkitty mustalla musteella ”Є” ja runkoon ”LENV 10 mg”.
Käyttö- ja käsittelyohjeet
Hoitajien ei pidä avata kapselia, jotta he välttyvät toistuvalta altistukselta kapselin sisällölle.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Suspension valmistus ja anto:
- Suspensio voidaan valmistaa veteen, omenamehuun tai maitoon. Jos suspensio annetaan ravitsemusletkun kautta, se on valmistettava veteen.
- Pane määrättyä annosta vastaava määrä kapseleita (enintään 5 kapselia) pieneen astiaan (tilavuudeltaan noin 20 ml tai 4 teelusikallista) tai mittaruiskuun (20 ml). Älä riko tai murskaa kapseleita.
-
Lisää astiaan tai mittaruiskuun 3 ml nestettä. Odota 10 minuuttia, jotta kapselin kuori (ulkopinta) liukenee, ja sitten sekoita tai ravista seosta 3 minuutin ajan, kunnes kapselit ovat täysin liuenneet.
- Jos käytät mittaruiskua, sulje se korkilla, irrota mäntä ja lisää neste ruiskuun toisella ruiskulla tai kalibroidulla tiputtimella. Kiinnitä mäntä takaisin paikalleen ennen sekoittamista.
- Anna astian tai mittaruiskun koko sisältö. Suspensio voidaan antaa astiasta suoraan suuhun tai mittaruiskusta suoraan suuhun tai ravitsemusletkun kautta.
- Lisää seuraavaksi astiaan tai mittaruiskuun vielä 2 ml nestettä toisella ruiskulla tai tiputtimella. Pyöritä tai ravista nesteseosta ja anna potilaalle. Varmista, että koko annos on otettu, toistamalla tämä vaihe vähintään kahdesti, kunnes jäämiä ei enää näy.
Huomaa: Yhteensopivuus on vahvistettu seuraavien suhteen: polypropeeniruiskut ja läpimitaltaan vähintään 5 F:n ravitsemusletkut (polyvinyylikloridi- tai polyuretaaniletku), läpimitaltaan vähintään 6 F:n (silikoniletku) ja läpimitaltaan enintään 16 F:n polyvinyylikloridi-, polyuretaani- ja silikoniletkut.
Korvattavuus
LENVIMA kapseli, kova
4 mg 30 fol
10 mg 30 fol
- Ylempi erityiskorvaus (100 %). Lenvatinibi: Kilpirauhassyövän, maksasolukarsinooman, munuaissyövän ja kohdunrungon syövän hoito erityisin edellytyksin (1508).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Lenvatinibi: Kilpirauhassyövän, maksasolukarsinooman, munuaissyövän ja kohdunrungon syövän hoito erityisin edellytyksin (3019).
ATC-koodi
L01EX08
Valmisteyhteenvedon muuttamispäivämäärä
26.03.2025
Yhteystiedot
Svärdvägen 15
18233 Danderyd
Ruotsi
+46 8 501 01 60
www.eisai.eu
nordic_medinfo@eisai.net
Edustaja Suomessa: EISAI AB