
VERZENIOS tabletti, kalvopäällysteinen 50 mg, 100 mg, 150 mg
Vaikuttavat aineet ja niiden määrät
Verzenios 50 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 50 mg abemasiklibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 14 mg laktoosimonohydraattia.
Verzenios 100 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 100 mg abemasiklibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 28 mg laktoosimonohydraattia.
Verzenios 150 mg kalvopäällysteinen tabletti
Yksi kalvopäällysteinen tabletti sisältää 150 mg abemasiklibia.
Apuaineet, joiden vaikutus tunnetaan
Yksi kalvopäällysteinen tabletti sisältää 42 mg laktoosimonohydraattia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Varhaisvaiheen rintasyöpä
Verzenios on tarkoitettu käytettäväksi adjuvanttihoitona hormonaalisen hoidon kanssa aikuispotilaille, joilla on hormonireseptoripositiivinen, HER2-negatiivinen, imusolmukkeisiin levinnyt varhaisvaiheen rintasyöpä, jonka uusiutumisriski on suuri (ks. kohta Farmakodynamiikka).
Pre- tai perimenopausaalisilla naisilla hormonaalinen aromataasin estäjähoito on yhdistettävä LHRH-agonistin kanssa.
Paikallisesti edennyt tai etäpesäkkeinen rintasyöpä
Verzenios on tarkoitettu käytettäväksi naisille, joilla on hormonireseptoripositiivinen, HER2-negatiivinen paikallisesti edennyt tai etäpesäkkeinen rintasyöpä, yhdessä aromataasin estäjän tai fulvestrantin kanssa ensimmäisenä hormonaalisena hoitona tai naisille, jotka ovat saaneet aiemmin hormonaalista hoitoa.
Pre- tai perimenopausaalisilla naisilla hormonaalinen hoito on yhdistettävä LHRH-agonistin kanssa.
Ehto
Hoito tulee aloittaa ja hoitoa tulee jatkaa syöpähoitoihin perehtyneen lääkärin valvonnassa.
Annostus ja antotapa
Verzenios-hoidon aloittaa ja sitä valvoo lääkäri, jolla on kokemusta syöpähoitojen toteuttamisesta.
Annostus
Abemasiklibin suositusannos on 150 mg kahdesti vuorokaudessa yhdistettynä hormonaaliseen hoitoon. Samanaikaisesti käytettävän hormonaalisen valmisteen suositusannostus, ks. kyseisen valmisteen valmisteyhteenveto.
Hoidon kesto
Varhaisvaiheen rintasyöpä
Verzenios-hoitoa käytetään jatkuvasti kahden vuoden ajan tai kunnes sairaus uusiutuu tai ilmenee toksisuutta, joka ei ole hyväksyttävää.
Paikallisesti edennyt taietäpesäkkeinen rintasyöpä
Verzenios-hoitoa käytetään jatkuvasti niin kauan kuin potilas hyötyy siitä kliinisesti tai kunnes ilmenee toksisuutta, joka ei ole hyväksyttävää.
Jos potilas oksentaa tai Verzenios-annos jää väliin, uutta annosta ei pidä ottaa, vaan potilasta kehotetaan ottamaan seuraava annos tavanomaiseen aikaan.
Annosmuutokset
Joidenkin haittavaikutusten hoito voi edellyttää hoidon tauottamista ja/tai annoksen pienentämistä taulukoissa 1–7 kuvattavaan tapaan.
| Taulukko 1. Suositukset annoksen muuttamisesta haittavaikutusten vuoksi | |
Verzenios-annos (yhdistelmähoito) | |
| Suositusannos | 150 mg kahdesti vuorokaudessa |
| 1. annosmuutos | 100 mg kahdesti vuorokaudessa |
| 2. annosmuutos | 50 mg kahdesti vuorokaudessa |
Taulukko 2. Hoitosuositukset hematologisen toksisuuden yhteydessä Täydellistä verenkuvaa seurataan ennen Verzenios-hoidon aloittamista, kahden viikon välein ensimmäisten kahden kuukauden ajan, kerran kuukaudessa seuraavien kahden kuukauden ajan ja kliinisen tarpeen mukaan. Ennen hoidon aloittamista on suositeltavaa, että absoluuttinen neutrofiilimäärä (ANC) on ≥ 1,5 x 109/l, trombosyyttiarvo on ≥ 100 x 109/l ja hemoglobiiniarvo on ≥ 80 g/l. | |
| Toksisuusa, b | Hoitosuositukset |
| Aste 1 tai 2 | Annosta ei tarvitse muuttaa. |
| Aste 3 | Hoito tauotetaan, kunnes toksisuus korjautuu asteen 2 tasolle tai lievemmäksi. Annosta ei tarvitse pienentää. |
| Aste 3, uusiutunut; tai aste 4 | Hoito tauotetaan, kunnes toksisuus korjautuu asteen 2 tasolle tai lievemmäksi. Hoitoa jatketaan yhtä annostasoa pienemmällä annoksella. |
| Potilas tarvitsee verisolujen kasvutekijöitä | Abemasiklibin anto tauotetaan vähintään 48 tunniksi verisolukasvutekijöiden viimeisen annoksen jälkeen ja kunnes toksisuus korjautuu asteen 2 tasolle tai lievemmäksi. Hoitoa jatketaan yhtä annostasoa pienemmällä annoksella, ellei annosta ole jo pienennetty kasvutekijähoitoon johtaneen toksisuuden vuoksi. |
a National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events ‑haittatapahtumaluokitus (CTCAE)
b ANC: Aste 1: ANC < viitearvojen alaraja – 1,5 x 109/l; Aste 2: ANC 1,0 x 109/l – < 1,5 x 109/l; Aste 3: ANC 0,5 x 109/l – < 1,0 x 109/l; Aste 4: ANC < 0,5 x 109/l
Taulukko 3. Hoitosuositukset ripulin yhteydessä Ripulilääkkeiden kuten loperamidin käyttö aloitetaan heti ensimmäisen löysän ulosteen ilmetessä. | |
| Toksisuusa | Hoitosuositukset |
| Aste 1 | Annosta ei tarvitse muuttaa. |
| Aste 2 | Jos toksisuus ei korjaudu 24 tunnissa asteen 1 tasolle tai lievemmäksi, hoito tauotetaan toksisuuden korjautumiseen asti. Annosta ei tarvitse pienentää. |
| Aste 2, joka pitkittyy tai uusiutuu maksimaalisista tukitoimista huolimatta, kun saman annoksen käyttö on aloitettu uudelleen | Hoito tauotetaan, kunnes toksisuus korjautuu asteen 1 tasolle tai lievemmäksi. Hoitoa jatketaan yhtä annostasoa pienemmällä annoksella. |
| Aste 3 tai 4 tai sairaalahoidon tarve | |
a NCI:n CTCAE-luokitus
Taulukko 4. Hoitosuositukset aminotransferaasiarvojen nousun yhteydessä Alaniiniaminotransferaasi (ALAT)- ja aspartaattiaminotransferaasi (ASAT) -arvoja seurataan ennen Verzenios-hoidon aloittamista, kahden viikon välein ensimmäisten kahden kuukauden ajan, kerran kuukaudessa seuraavien kahden kuukauden ajan ja kliinisen tarpeen mukaan. | |
| Toksisuusa | Hoitosuositukset |
Aste 1 (> viitearvojen yläraja [ULN] – 3,0 x ULN) Aste 2 (> 3,0 – 5,0 x ULN) | Annosta ei tarvitse muuttaa. |
| Pitkittynyt tai uusiutunut aste 2, tai aste 3 (> 5,0 – 20,0 x ULN) | Hoito tauotetaan, kunnes toksisuus korjautuu lähtötasolle tai asteen 1 tasolle. Hoitoa jatketaan yhtä annostasoa pienemmällä annoksella. |
| ALAT– tai ASAT –arvojen nousu > 3 x ULN ja lisäksi kokonaisbilirubiini > 2 x ULN, mutta ei kolestaasia | Abemasiklibin käyttö lopetetaan. |
| Aste 4 (> 20,0 x ULN) | Abemasiklibin käyttö lopetetaan. |
a NCI:n CTCAE-luokitus
ULN = upper limit of normal, viitearvojen yläraja
| Taulukko 5. Hoitosuositukset interstitiaalisen keuhkosairauden/keuhkokuumeen yhteydessä | |
| Toksisuusa | Hoitosuositukset |
| Aste 1 tai 2 | Annosta ei tarvitse muuttaa. |
| Pitkittynyt tai uusiutunut asteen 2 toksisuus, joka ei korjaudu maksimaalisilla tukitoimilla 7 päivässä lähtötasolle tai asteen 1 tasolle | Hoito tauotetaan, kunnes toksisuus korjautuu lähtötasolle tai asteen 1 tasolle. Hoitoa jatketaan yhtä annostasoa pienemmällä annoksella. |
| Aste 3 tai 4 | Abemasiklibin käyttö lopetetaan. |
a NCI:n CTCAE-luokitus
| Taulukko 6. Hoitosuositukset tromboembolisten laskimotapahtumien yhteydessä | |
| Toksisuusa | Hoitosuositukset |
| Varhaisvaiheen rintasyöpä | |
| Kaikki asteet (1, 2, 3 tai 4) | Hoito tauotetaan ja potilasta hoidetaan kliinisen tarpeen mukaan. Abemasiklibi voidaan aloittaa uudelleen, kun potilaan tila on kliinisesti vakaa. |
| Paikallisesti edennyt taietäpesäkkeinen rintasyöpä | |
| Aste 1 tai 2 | Annosta ei tarvitse muuttaa. |
| Aste 3 tai 4 | Hoito tauotetaan ja potilasta hoidetaan kliinisen tarpeen mukaan. Abemasiklibi voidaan aloittaa uudelleen, kun potilaan tila on kliinisesti vakaa. |
a NCI:n CTCAE-luokitus
| Taulukko 7. Hoitosuositukset ei-hematologisen toksisuuden yhteydessä (ripulia, aminotransferaasiarvojen nousua, interstitiaalista keuhkosairautta/keuhkokuumetta ja tromboembolisia laskimotapahtumia lukuun ottamatta) | |
| Toksisuusa | Hoitosuositukset |
| Aste 1 tai 2 | Annosta ei tarvitse muuttaa. |
| Pitkittynyt tai uusiutunut asteen 2 toksisuus, joka ei korjaudu maksimaalisilla tukitoimilla 7 päivässä lähtötasolle tai asteen 1 tasolle | Hoito tauotetaan, kunnes toksisuus korjautuu asteen 1 tasolle tai lievemmäksi. Hoitoa jatketaan yhtä annostasoa pienemmällä annoksella. |
| Aste 3 tai 4 | |
a NCI:n CTCAE-luokitus
CYP3A4:n estäjät
Vahvojen CYP3A4:n estäjien samanaikaista käyttöä on vältettävä. Jos vahvoja CYP3A4:n estäjiä ei voida välttää, abemasiklibiannos on pienennettävä 100 mg:aan kahdesti vuorokaudessa.
Jos potilaan abemasiklibiannos on pienennetty 100 mg:aan kahdesti vuorokaudessa eikä vahvan CYP3A4:n estäjän samanaikaista antoa voida välttää, abemasiklibiannosta pienennetään edelleen 50 mg:aan kahdesti vuorokaudessa.
Jos potilaan abemasiklibiannos on pienennetty 50 mg:aan kahdesti vuorokaudessa eikä vahvan CYP3A4:n estäjän samanaikaista antoa voida välttää, abemasiklibihoitoa voidaan jatkaa samalla annoksella, mutta potilasta on seurattava huolellisesti toksisuuden merkkien varalta. Vaihtoehtoisesti abemasiklibiannos voidaan pienentää 50 mg:aan kerran vuorokaudessa tai hoito voidaan lopettaa.
Jos CYP3A4:n estäjän käyttö lopetetaan, abemasiklibiannos nostetaan ennen kyseisen CYP3A4:n estäjän käyttöönottoa käytetyksi annokseksi, kun on kulunut 3–5 kyseisen CYP3A4:n estäjän puoliintumisaikaa.
Erityisryhmät
Iäkkäät
Annosta ei tarvitse muuttaa iän perusteella (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta. Abemasiklibin antamisesta vaikeaa munuaisten vajaatoimintaa tai loppuvaiheen munuaisten vajaatoimintaa sairastaville tai dialyysihoidossa oleville potilaille ei ole tietoja (ks. kohta Farmakokinetiikka). Abemasiklibia on annettava varoen, jos potilaalla on vaikea munuaisten vajaatoiminta, ja potilasta on seurattava tarkoin toksisuuden merkkien varalta.
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä (Child–Pugh A) tai keskivaikea (Child–Pugh B) maksan vajaatoiminta. Vaikeaa (Child–Pugh C) maksan vajaatoimintaa sairastavilla on suositeltavaa pienentää antotiheys yhteen antokertaan vuorokaudessa (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Abemasiklibin turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten ja nuorten hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Verzenios otetaan suun kautta.
Annos voidaan ottaa ruoan kanssa tai ilman ruokaa. Ei saa ottaa greipin eikä greippimehun kanssa (ks. kohta Yhteisvaikutukset).
Lääkeannokset on otettava suunnilleen samaan aikaan joka päivä.
Tabletti niellään kokonaisena (sitä ei saa pureskella, murskata eikä pilkkoa ennen nielemistä).
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Neutropenia
Abemasiklibia saavilla potilailla on ilmoitettu neutropeniaa. On suositeltavaa muuttaa annosta, jos potilaalle kehittyy asteen 3 tai 4 neutropenia (ks. kohta Annostus ja antotapa). Kuolemaan johtaneita neutropeenisia sepsistapahtumia esiintyi < 1 %:lla etäpesäkkeistä rintasyöpää sairastavista potilaista. Potilaita on neuvottava ilmoittamaan kaikista kuumejaksoista terveydenhuollon ammattilaiselle.
Infektiot
Infektioita ilmoitettiin useammin abemasiklibia ja endokriinistä hoitoa saaneilla potilailla kuin endokriinistä hoitoa saaneilla potilailla. Keuhkoinfektiota ilmoitettiin abemasiklibia saavilla potilailla ilman samanaikaista neutropeniaa. Kuolemaan johtaneita tapahtumia esiintyi < 1 %:lla etäpesäkkeistä rintasyöpää sairastavista potilaista. Potilaita on seurattava infektioiden merkkien ja oireiden varalta ja hoidettava lääketieteellisesti asianmukaiseen tapaan.
Tromboemboliset laskimotapahtumat
Tromboembolisia laskimotapahtumia ilmoitettiin abemasiklibia ja endokriinistä hoitoa saaneilla potilailla. Potilaita on seurattava syvän laskimotukoksen ja keuhkoembolian merkkien ja oireiden varalta ja hoidettava lääketieteellisesti asianmukaiseen tapaan. Tromboembolisen laskimotapahtuman asteesta riippuen abemasiklibin annosta voi olla tarpeen muuttaa (ks. kohta Annostus ja antotapa).
Tromboemboliset valtimotapahtumat
Metastaattista rintasyöpää koskeneissa tutkimuksissa on havaittu mahdollisesti suurentunut riski vakaville tromboembolisille valtimotapahtumille, mukaan lukien iskeeminen aivohalvaus ja sydäninfarkti, kun abemasiklibia annettiin yhdessä endokriinisten hoitojen kanssa. Abemasiklibihoidon jatkamisen hyödyt ja riskit on otettava huomioon potilailla, joilla on vakava tromboembolinen valtimotapahtuma.
Aminotransferaasiarvojen nousu
Abemasiklibia saavilla potilailla on ilmoitettu ALAT- ja ASAT-arvojen nousua. ALAT- ja ASAT -arvojen nousun voimakkuudesta riippuen abemasiklibin annosta on mahdollisesti muutettava (ks. kohta Annostus ja antotapa).
Ripuli
Ripuli on yleisin haittavaikutus. Kliinisissä tutkimuksissa mediaaniaika ensimmäisen ripulitapahtuman alkuun oli noin 6–8 päivää ja ripulin mediaanikesto 7–12 päivää (aste 2) ja 5–8 päivää (aste 3). Ripuliin voi liittyä nestehukkaa. Potilaiden on aloitettava ripulilääkkeen kuten loperamidin käyttö heti ensimmäisen löysän ulosteen ilmetessä. Potilaiden on lisättävä nesteiden nauttimista suun kautta ja ilmoitettava terveydenhuollon ammattilaiselle. On suositeltavaa muuttaa annosta, jos potilaalle kehittyy asteen ≥ 2 ripuli (ks. kohta Annostus ja antotapa).
Interstitiaalinen keuhkosairaus/keuhkokuume
Interstitiaalista keuhkosairautta/keuhkokuumetta ilmoitettiin abemasiklibia saavilla potilailla. Seuraa potilaita interstitiaaliseen keuhkosairauteen/keuhkokuumeeseen viittaavien keuhko-oireiden varalta, ja hoida lääketieteellisesti asianmukaisesti. Abemasiklibin annosta saatetaan joutua muuttamaan interstitiaalisen keuhkosairauden/keuhkokuumeen asteen perusteella (ks. kohta Annostus ja antotapa). Lopeta abemasiklibi pysyvästi, mikäli potilaalla todetaan asteen 3 tai 4 interstitiaalinen keuhkosairaus/keuhkokuume.
CYP3A4:n induktorien samanaikainen käyttö
CYP3A4:n induktorien samanaikaista käyttöä on vältettävä, sillä yhdistelmän käyttöön liittyy abemasiklibin tehon heikkenemisen riski (ks. kohta Yhteisvaikutukset).
Viskeraalinen kriisi
Abemasiklibin tehosta ja turvallisuudesta ei ole tietoa potilailla, joilla on viskeraalinen kriisi.
Laktoosi
Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Natrium
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per tabletti eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Muiden lääkevalmisteiden vaikutus abemasiklibin farmakokinetiikkaan
Abemasiklibi metaboloituu lähinnä CYP3A4-välitteisesti.
CYP3A4:n estäjät
Abemasiklibin käyttö yhdessä CYP3A4:n estäjien kanssa voi suurentaa abemasiklibin pitoisuuksia plasmassa. Pitkälle edennyttä ja/tai etäpesäkkeistä syöpää sairastavilla CYP3A4:n estäjä klaritromysiinin samanaikainen käyttö johti plasman abemasiklibialtistuksen suurenemiseen 3,4‑kertaiseksi ja sitoutumattoman abemasiklibin ja sen aktiivisten metaboliittien yhteenlasketun, vaikutuskyvyn suhteen korjatun plasman altistuksen suurenemiseen 2,5‑kertaiseksi.
Vahvojen CYP3A4:n estäjien ja abemasiklibin samanaikaista käyttöä on vältettävä. Abemasiklibiannosta on pienennettävä, jos vahvan CYP3A4:n estäjän samanaikainen anto on tarpeen (ks. kohta Annostus ja antotapa). Tämän jälkeen potilasta on seurattava tarkasti haittojen varalta. Esimerkkejä vahvoista CYP3A4:n estäjistä ovat mm. klaritromysiini, itrakonatsoli, ketokonatsoli, lopinaviiri/ritonaviiri, posakontsoli tai vorikonatsoli. Greippiä ja greippimehua on vältettävä.
Annosmuutos ei ole tarpeen potilailla, jotka käyttävät keskivahvoja tai heikkoja CYP3A4 estäjiä. Potilaita tulee kuitenkin seurata tarkasti toksisuuden merkkien varalta.
CYP3A4:n induktorit
Abemasiklibin anto vahvan CYP3A4:n induktori rifampisiinin kanssa pienensi plasman abemasiklibipitoisuutta 95 % ja sitoutumattoman abemasiklibin ja aktiivisten metaboliittien vaikutuskyvyn suhteen korjattua pitoisuutta plasmassa 77 % AUC0-∞-arvon perusteella. Vahvojen CYP3A4:n induktorien (esimerkiksi karbamatsepiinin, fenytoiinin, rifampisiinin ja mäkikuisman) samanaikaista käyttöä on vältettävä, sillä yhdistelmän käyttöön liittyy abemasiklibin tehon heikkenemisen riski.
Abemasiklibin vaikutus muiden lääkevalmisteiden farmakokinetiikkaan
Lääkevalmisteet, jotka ovat kuljettajaproteiinien substraatteja
Abemasiklibi ja sen tärkeimmät aktiiviset metaboliitit estävät munuaisten kuljettajaproteiinien OCT2 (orgaanisten kationien kuljettajaproteiini 2), MATE1 (multidrug and extrusion toxin protein 1) ja MATE2‑K toimintaa. Abemasiklibilla voi olla in vivo ‑yhteisvaikutuksia näiden kuljettajaproteiinien kliinisesti merkittävien substraattien kuten dofetilidin tai kreatiniinin kanssa (ks. kohta Haittavaikutukset). Kliinisessä lääkeaineinteraktiotutkimuksessa, jossa metformiinia (OCT2:n, MATE1:n ja MATE2:n substraatti) annettiin yhdessä abemasiklibin (400 mg) kanssa, todettiin plasman metformiinialtistuksen suurenevan hieman, mutta ei kliinisesti merkittävästi (37 %). Ilmiön todettiin johtuvan munuaisissa tapahtuvan erittymisen vähenemisestä; glomerulussuodatus ei muuttunut.
Terveillä henkilöillä abemasiklibin ja P‑glykoproteiinin (P-gp) substraatti loperamidin samanaikainen anto johti plasman loperamidialtistuksen suurenemiseen 9 % AUC0-∞-arvon perusteella ja 35 % Cmax-arvon perusteella. Tätä ei pidetty kliinisesti merkittävänä. Abemasiklibin on kuitenkin todettu estävän P-gp-toimintaa ja rintasyöpäresistenssiproteiini BCRP:n toimintaa in vitro. Abemasiklibilla saattaa siis olla in vivo ‑yhteisvaikutuksia sellaisten näiden kuljettajaproteiinien substraattien kanssa, joiden terapeuttinen leveys on kapea, esim. digoksiinin tai dabigatraanieteksilaatin kanssa.
Rintasyöpäpotilailla toteutetussa kliinisessä tutkimuksessa abemasiklibilla ei todettu olevan kliinisesti merkittäviä farmakokineettisiä lääkeaineinteraktioita anastrotsolin, fulvestrantin, eksemestaanin, letrotsolin eikä tamoksifeenin kanssa.
Toistaiseksi ei tiedetä, huonontaako abemasiklibi systeemisesti vaikuttavien hormonaalisten ehkäisyvalmisteiden vaikuttavuutta.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / raskauden ehkäisy naisilla
Naisten, jotka voivat tulla raskaaksi, on käytettävä erittäin tehokkaita ehkäisymenetelmiä (esim. kahta estemenetelmää) hoidon aikana ja vähintään 3 viikon ajan hoidon päättymisen jälkeen (ks. kohta Yhteisvaikutukset).
Raskaus
Ei ole olemassa tietoja abemasiklibin käytöstä raskaana oleville naisille. Eläinkokeissa on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Verzenios-valmisteen käyttöä ei suositella raskauden aikana eikä hedelmällisessä iässä oleville naisille, jotka eivät käytä ehkäisyä.
Imetys
Ei tiedetä, erittyykö abemasiklibi ihmisen rintamaitoon. Imeväiseen kohdistuvia riskejä ei voida poissulkea. Abemasiklibia saavien potilaiden ei pidä imettää.
Hedelmällisyys
Abemasiklibin vaikutusta ihmisen hedelmällisyyteen ei tunneta. Vaikutuksia hedelmällisyyteen ei havaittu urosrotilla, mutta sukuelimiin kohdistuvat sytotoksiset vaikutukset uroshiirillä, -rotilla ja ‑koirilla viittaavat siihen, että abemasiklibi saattaa heikentää miesten hedelmällisyyttä. Sukuelimiin kohdistuvia vaikutuksia ei havaittu naarashiirillä, -rotilla eikä -koirilla, eikä naarasrotillakaan havaittu vaikutuksia hedelmällisyyteen eikä alkion varhaiskehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Verzenios-valmisteella on vähäinen vaikutus ajokykyyn ja koneiden käyttökykyyn. Potilaita on kehotettava olemaan varovaisia ajaessaan autoa tai käyttäessään koneita, jos heillä on uupumusta tai huimausta Verzenios-hoidon aikana (ks. kohta Haittavaikutukset).
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yleisimmin esiintyviä haittavaikutuksia ovat ripuli, infektiot, neutropenia, leukopenia, anemia, uupumus, pahoinvointi, oksentelu, hiustenlähtö ja ruokahalun heikentyminen.
Yleisimpien haittavaikutusten osalta asteen ≥ 3 tapahtumia oli < 5 % (pois lukien neutropenia, leukopenia ja ripuli).
Haittavaikutustaulukko
Seuraavassa taulukossa luetellaan haittavaikutukset MedDRA-elinjärjestelmäluokan ja yleisyyden mukaan järjestettyinä. Yleisyysluokat ovat seuraavat: hyvin yleinen (≥ 1/10); yleinen (≥ 1/100, < 1/10); melko harvinainen (≥ 1/1 000, < 1/100); harvinainen (≥ 1/10 000, < 1/1 000); hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa vakavuuden mukaan alenevassa järjestyksessä.
| Taulukko 8. Ilmoitetut haittavaikutukset vaiheen 3 tutkimuksissa, joissa abemasiklibia annettiin hormonaalisen hoidon kanssaa(N = 3 559), ja markkinoille tulon jälkeen | ||||
| Elinjärjestelmäluokka | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen |
| Infektiot | Infektiotb | |||
Veri ja imukudos
| Neutropenia Leukopenia Anemia Trombosytopenia Lymfopeniah |
| Kuumeinen neutropeniae
| |
| Aineenvaihdunta ja ravitsemus | Ruokahalun heikkeneminen | |||
| Hermosto | Päänsärkyf Makuaistin häiriötg Huimausg |
|
| |
| Silmät | Lisääntynyt kyynelnesteen eritys | Fotopsia Keratiitti | ||
Verisuonisto
|
| Laskimotromboemboliac | ||
Hengityselimet, rintakehä ja välikarsina
| Interstitiaalinen keuhkosairaus / pneumoniittid |
| ||
Ruoansulatuselimistö
| Ripuli Oksentelu Pahoinvointi Suutulehdusf | Dyspepsiaf
|
| |
| Iho ja ihonalainen kudos | Hiustenlähtög Kutinag Ihottumag | Kynsimuutoksetf Kuiva ihoe
|
| Erythema multiforme |
| Luusto, lihakset ja sidekudos | Lihasheikkouse | |||
| Yleisoireet ja antopaikassa todettavat haitat | Kuumee Uupumus | |||
| Tutkimukset | Alaniiniaminotransferaasiarvojen nousug Aspartaattiaminotransferaasiarvojen nousug |
|
| |
a Abemasiklibi yhdessä anastrotsolin, letrotsolin, eksemestaanin, tamoksifeenin tai fulvestrantin kanssa.
b Infektiot-termi kattaa kaikki infektiot-elinjärjestelmäluokkaan kuuluvat haittavaikutustermit.
c Tromboembolisia laskimotapahtumia ovat syvä laskimotukos, keuhkoembolia, aivojen laskimosinuksen tromboosi, solislaskimon, kainalolaskimon tromboosi, alaonttolaskimon syvä laskimotukos ja lantion alueen laskimotromboosi.
d Interstitiaalinen keuhkosairaus / pneumoniitti varhaisvaiheen rintasyövässä kattaa kaikki MedDRA-hakulausekkeen ”interstitiaalinen keuhkosairaus” haittavaikutustermit. Etäpesäkkeisen rintasyövän osalta haittavaikutustermejä ovat interstitiaalinen keuhkosairaus, pneumoniitti, organisoituva pneumonia, keuhkofibroosi ja obliteroiva bronkioliitti.
e Haittavaikutukset huomioitu vain etäpesäkkeisen rintasyövän osalta (tutkimukset MONARCH 2 ja MONARCH 3).
f Haittavaikutukset huomioitu vain varhaisvaiheen rintasyövän osalta (tutkimus monarchE).
g Esiintymistiheys yleinen varhaisvaiheen rintasyövässä (monarchE) ja hyvin yleinen etäpesäkkeisessä rintasyövässä (MONARCH 2 ja MONARCH 3).
h Esiintymistiheys yleinen etäpesäkkeisessä rintasyövässä (MONARCH 2 ja MONARCH 3) ja hyvin yleinen varhaisvaiheen rintasyövässä (monarchE).
Valikoitujen haittavaikutusten kuvaus
Neutropenia
Neutropeniaa ilmoitettiin yleisesti kaikissa tutkimuksissa. MonarchE-tutkimuksessa neutropeniaa ilmoitettiin 45,8 %:lla potilaista. Laboratoriokokeisiin perustuvaa asteen 3 tai 4 neutrofiiliarvojen laskua ilmoitettiin 19,1 %:lla potilaista, jotka saivat abemasiklibia hormonaalisen hoidon kanssa; mediaaniaika neutropenian alkamiseen oli 30 päivää ja mediaaniaika sen korjautumiseen 16 päivää. Kuumeista neutropeniaa ilmoitettiin 0,3 %:lla potilaista. MONARCH 2- ja MONARCH 3 ‑tutkimuksissa neutropeniaa ilmoitettiin 45,1 %:lla potilaista. Asteen 3 tai 4 neutrofiiliarvojen laskua (laboratoriokokeiden perusteella) ilmoitettiin 28,2 %:lla potilaista, jotka saivat abemasiklibia yhdessä aromataasin estäjän tai fulvestrantin kanssa. Mediaaniaika asteen 3 tai 4 neutropenian alkamiseen oli 29–33 päivää ja mediaaniaika sen korjautumiseen 11–15 päivää. Kuumeista neutropeniaa ilmoitettiin 0,9 %:lla potilaista. On suositeltavaa muuttaa annosta, jos potilaalle kehittyy asteen 3 tai 4 neutropenia (ks. kohta Annostus ja antotapa).
Ripuli
Ripuli oli yleisimmin ilmoitettu haittavaikutus (ks. taulukko 8). Sen ilmaantuvuus oli suurimmillaan ensimmäisen abemasiklibihoitokuukauden aikana ja pieneni myöhemmin. MonarchE-tutkimuksessa mediaaniaika ensimmäisen, minkä tahansa asteen ripulitapahtuman alkuun oli 8 päivää. Asteen 2 ripulin mediaanikesto oli 7 päivää ja asteen 3 ripulin mediaanikesto 5 päivää. MONARCH 2- ja MONARCH 3 -tutkimuksissa mediaaniaika ensimmäisen, minkä tahansa asteen ripulitapahtuman alkuun oli noin 6–8 päivää. Ripulin mediaanikesto oli 9–12 päivää asteen 2 osalta ja 6–8 päivää asteen 3 osalta. Ripuli korjautui lähtötasolle tai tätä lievemmäksi tukihoidolla kuten loperamidilla ja/tai annosmuutoksilla (ks. kohta Annostus ja antotapa).
Aminotransferaasiarvojen suureneminen
MonarchE-tutkimuksessa ALAT- ja ASAT-arvojen nousua ilmoitettiin usein potilailla, jotka saivat abemasiklibia hormonaalisen hoidon kanssa (ALAT-arvon nousua 12,3 %:lla ja ASAT-arvon nousua 11,8 %:lla). Laboratoriokokeisiin perustuvaa asteen 3 tai 4 ALAT-arvon nousua ilmoitettiin 2,6 %:lla potilaista ja asteen 3 tai 4 ASAT-arvon nousua 1,6 %:lla potilaista. Mediaaniaika asteen 3 tai 4 ALAT-nousun alkamiseen oli 118 päivää ja mediaaniaika sen korjautumiseen 14,5 päivää. Mediaaniaika asteen 3 tai 4 ASAT-nousun alkamiseen oli 90,5 päivää ja mediaaniaika sen korjautumiseen 11 päivää. MONARCH 2- ja MONARCH 3 -tutkimuksissa ilmoitettiin usein ALAT-arvon nousua (15,1 %) ja ASAT-arvon nousua (14,2 %) potilailla, jotka saivat abemasiklibia yhdessä aromataasin estäjän tai fulvestrantin kanssa. Asteen 3 tai 4 nousua (laboratoriokokeiden perusteella) ilmoitettiin ALAT-arvoissa 6,1 %:lla ja ASAT-arvoissa 4,2 %:lla. Mediaaniaika asteen 3 tai 4 ALAT-arvon nousun alkamiseen oli 57–61 päivää ja mediaaniaika sen korjautumiseen 14 päivää. Mediaaniaika asteen 3 tai 4 ASAT-arvon nousun alkamiseen oli 71–185 päivää ja mediaaniaika sen korjautumiseen 13–15 päivää. On suositeltavaa muuttaa annosta, jos potilaalle kehittyy asteen 3 tai 4 ALAT- tai ASAT-arvon nousua (ks. kohta Annostus ja antotapa).
Kreatiniini
Kyseessä ei ole haittavaikutus, mutta abemasiklibin on todettu suurentavan seerumin kreatiniinipitoisuutta. MonarchE-tutkimuksessa 99,3 %:lla potilaista esiintyi seerumin kreatiniinipitoisuuden suurenemista (laboratoriokokeiden perusteella); heistä 0,5 %:lla tämä oli astetta 3 tai 4. Pelkkää hormonaalista hoitoa saaneista seerumin kreatiniinipitoisuuden suurenemista (kaikki laboratoriokoetulosten asteet) ilmoitettiin 91,0 %:lla. MONARCH 2- ja MONARCH 3 -tutkimuksissa 98,3 %:lla potilaista esiintyi seerumin kreatiniinipitoisuuden suurenemista (laboratoriokokeiden perusteella); heistä 1,9 %:lla tämä oli astetta 3 tai 4. Seerumin kreatiniinipitoisuuden nousua (kaikki laboratoriokoetulosten asteet) ilmoitettiin 78,4 %:lla pelkkää aromataasin estäjää tai fulvestranttia saaneista. Abemasiklibin on todettu suurentavan seerumin kreatiniinipitoisuutta estämällä munuaistubuluksissa tapahtuvaan erittymiseen osallistuvien kuljettajaproteiinien toimintaa mutta vaikuttamatta glomerulusten toimintaan (joheksolin puhdistumalla mitattuna) (ks. kohta Yhteisvaikutukset). Kliinisissä tutkimuksissa seerumin kreatiniinipitoisuuden suureneminen tapahtui ensimmäisen abemasiklibihoitokuukauden aikana, arvot pysyivät koholla mutta vakaina koko hoitojakson ajan, muutos korjautui hoidon päättyessä eikä samaan aikaan esiintynyt muutoksia munuaistoiminnan markkereissa kuten veren ureatyppiarvoissa, kystatiini C ‑arvoissa eikä kystatiini C ‑pitoisuuksien perusteella lasketussa glomerulusten suodatusnopeudessa.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haitta-tasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Abemasiklibiyliannostuksen yhteydessä voi esiintyä uupumusta ja ripulia. Potilaalle on järjestettävä yleistä tukihoitoa.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut syöpälääkkeet, proteiinikinaasin estäjät, ATC-koodi: L01EF03
Vaikutusmekanismi
Abemasiklibi on vahva, selektiivinen sykliineistä riippuvaisten kinaasien 4 ja 6 (CDK4 ja CDK6) estäjä ja vaikuttaa entsymaattisissa kokeissa tehokkaimmin sykliini D1/CDK4-toimintaan. Abemasiklibi estää retinoblastoomaproteiinin (Rb) fosforylaatiota ja salpaa solusyklin etenemisen G1-vaiheesta solunjakautumisen S-vaiheeseen, mikä estää kasvaimen kasvua. Estrogeenireseptoripositiivisissa rintasyöpäsolulinjoissa abemasiklibilla saavutettu pitkäkestoinen kohdemolekyylin esto esti Rb-fosforylaation rebound-lisääntymisen, mikä johti solujen vanhenemiseen ja apoptoosiin. Rb-negatiiviset ja Rb-vajeiset syöpäsolulinjat ovat yleensä vähemmän herkkiä abemasiklibille in vitro. Rintasyövän ksenograftimalleissa abemasiklibin käyttö päivittäin ilman taukoja kliinisesti merkittävillä pitoisuuksilla joko yksinään tai yhdessä antiestrogeenien kanssa johti kasvaimen koon pienenemiseen.
Farmakodynaamiset vaikutukset
Syöpäpotilailla abemasiklibi estää CDK4- ja CDK6-kinaasien toimintaa, mikäli ilmenee Rb-proteiinin ja topoisomeraasi II alfan fosforylaation estona. Tämä johtaa solusyklin estymiseen G1-tarkastuspisteestä ylävirtaan.
Sydämen elektrofysiologia
Abemasiklibin vaikutusta QTcF-aikaan arvioitiin 144 potilaalla, joilla oli pitkälle edennyt syöpä. QTcF-ajassa ei todettu suurta (eli > 20 ms) muutosta vakaan tilan suurimpien havaittujen abemasiklibipitoisuuksien keskiarvopitoisuuden yhteydessä hoitoannosten ja ‑antoaikataulun jälkeen.
Terveillä henkilöillä toteutetussa altistus‑vasteanalyysissä, jossa altistukset vastasivat annosta 200 mg kahdesti vuorokaudessa, abemasiklibi ei pidentänyt QTcF‑aikaa kliinisesti merkittävästi.
Kliininen teho ja turvallisuus
Varhaisvaiheen rintasyöpä
Satunnaistettu vaiheen 3 tutkimus monarchE: Verzenios yhdistettynä hormonaaliseen hoitoon
Verzenios-valmisteen tehoa ja turvallisuutta arvioitiin yhdistelmähoidossa hormonaalisen adjuvanttihoidon kanssa monarchE-tutkimuksessa. Kyseessä oli avoin, satunnaistettu, vaiheen 3 kaksi kohorttinen tutkimus naisilla ja miehillä, joilla on hormonireseptoripositiivinen, HER2-negatiivinen, imusolmukkeisiin levinnyt varhaisvaiheen rintasyöpä, jonka uusiutumisriski on suuri. Suuri uusiutumisriski kohortissa 1 määriteltiin kliinisten ja patologisten piirteiden perusteella: positiivisten kainaloimusolmukkeiden määrä joko ≥ 4 tai positiivisten kainaloimusolmukkeiden määrä 1–3 ja vähintään yksi seuraavista: kasvaimen koko ≥ 5 cm tai histologinen luokka 3.
Kaikkiaan 5 637 potilasta satunnaistettiin suhteessa 1:1 saamaan joko pelkkää tavanomaista hormonaalista hoitoa tai Verzenios 150 mg x 2 -hoitoa yhdistettynä lääkärin määräämään tavanomaiseen hormonaaliseen hoitoon kahden vuoden ajan. Satunnaistaminen stratifioitiin aiemman solunsalpaajahoidon, menopausaalisen statuksen ja alueen mukaan. Miehet stratifioitiin postmenopausaalisiksi. Potilaat olivat saaneet aiemmin definitiivistä lokoregionaalista hoitoa (johon saattoi kuulua solunsalpaajahoitoa neoadjuvantti- tai adjuvanttihoitona). Potilaiden oli täytynyt toipua aiemman solunsalpaajahoidon tai sädehoidon akuuteista haittavaikutuksista. Ennen satunnaistamista vaadittiin 21 päivän lääkkeetön jakso solunsalpaajahoidon jälkeen ja 14 päivän jakso sädehoidon jälkeen. Ennen satunnaistamista potilailla oli ollut mahdollisuus saada hormonaalista adjuvanttihoitoa enintään 12 viikon ajan viimeisimmän muun hoidon (kirurgia, kemoterapia tai sädehoito) jälkeen. Adjuvanttihoitoa fulvestrantilla ei sallittu tavanomaisena hormonaalisena hoitona. Tutkimukseen saivat osallistua potilaat, joiden ECOG (eastern cooperative oncology group) -toimintakykyluokka oli 0 tai 1. Tutkimuksesta suljettiin pois potilaat, joilla oli anamneesissa tromboembolisia laskimotapahtumia. Tutkimushoitovaiheen päätyttyä hormonaalista adjuvanttihoitoa jatkettiin kummassakin hoitoryhmässä; kumulatiivinen kesto oli 5–10 vuotta lääketieteellisen tarpeen mukaan. LHRH-agonisteja annettiin pre- tai perimenopausaalisille naisille ja miehille kliinisen tarpeen mukaan.
Satunnaistetuista 5 637 potilaasta kohorttiin 1 otettiin mukaan 5 120 potilasta, jotka edustivat 91 % ITT-populaatiosta. Kohortin 1 potilaiden demografiset tiedot ja kasvainta koskevat lähtötilanteen ominaisuudet olivat samankaltaiset ITT-populaatiossa. Tutkimukseen otettujen potilaiden mediaani-ikä oli noin 51 vuotta (vaihteluväli 22–89 vuotta). 15 % potilaista oli vähintään 65-vuotiaita; 99 % oli naisia, 71 % oli valkoihoisia, 24 % aasialaisia ja 5 % muuta alkuperää. 43 % potilaista oli pre- tai perimenopausaalisia. Suurin osa oli saanut aiempaa solunsalpaajahoitoa (36 % neoadjuvanttihoitoa, 62 % adjuvanttihoitoa) ja aiempaa sädehoitoa (96 %). Ensimmäisenä hormonaalisena hoitona oli esimerkiksi letrotsoli (39 %), tamoksifeeni (31 %), anastrotsoli (22 %) tai eksemestaani (8 %).
65 %:lla potilaista oli vähintään 4 positiivista imusolmuketta, 41 %:lla oli luokan 3 kasvain, 24 %:lla kasvaimen patologinen koko oli leikkauksessa ≥ 5 cm.
Ensisijainen päätetapahtuma oli IDFS (invasive disease free survival, tautivapaa elossaolo invasiivisen syövän suhteen) ITT-populaatiossa. Se määriteltiin aikana, joka kului satunnaistamisesta johonkin seuraavista: invasiivisen rintasyövän ensimmäinen uusiutuminen ipsilateraalisesti, paikallisen invasiivisen rintasyövän uusiutuminen, etäpesäkkeinen uusiutuminen, kontralateraalinen invasiivinen rintasyöpä, toinen primaarinen invasiivinen syöpä muualla kuin rinnassa, tai mistä tahansa syystä johtuva kuolema.
Tärkeä toissijainen päätetapahtuma kohortissa 1 oli DRFS (distant relapse free survival, tautivapaa elossaolo etäpesäkkeisen syövän suhteen). Se määriteltiin aikana, joka kului satunnaistamisesta etäpesäkkeisen syövän ensimmäiseen uusiutumiseen tai mistä tahansa syystä johtuvaan kuolemaan.
Tutkimuksen ensisijainen tavoite saavutettiin etukäteen suunnitellussa välianalyysissa (tiedonkeruun katkaisupäivä 16.3.2020). Tilastollisesti merkitsevä IDFS-hyöty havaittiin potilailla, jotka saivat Verzenios-valmistetta yhdessä hormonaalisen hoidon kanssa, verrattuna pelkkää hormonaalista hoitoa saaneisiin ITT-populaatiossa. Lopullisessa kokonaiselossaoloajan (OS, overall survival) analyysissä (tiedonkeruun katkaisupäivä 15.7.2025) havaittiin tilastollisesti merkitsevä kokonaiselossaoloajan pidentyminen potilailla, jotka saivat Verzenios-valmistetta yhdessä hormonaalisen hoidon kanssa verrattuna pelkkää hormonaalista hoitoa saaneisiin ITT-populaatiossa. Hyväksyntä myönnettiin suuremmalle alaryhmälle, kohortille 1.
Lopullisessa kokonaiselossaoloajan analyysissä kaikki kohortin 1 potilaat olivat saaneet päätökseen 2 vuotta jatkuneen tutkimushoitovaiheen; seurannan mediaanikesto oli 76 kk (6,3 vuotta).
Tehotulokset kohortissa 1 esitetään yhteenvetona taulukossa 9, kuvassa 1 ja kuvassa 2.
| Taulukko 9. monarchE: tehotietojen yhteenveto (kohortin 1 populaatio) | ||
| Verzenios + hormonaalinen hoito N = 2 555 | Pelkkä hormonaalinen hoito N = 2 565 |
Invasiivisen syövän uusiutumisvapaa elossaoloaika (IDFS) |
|
|
Tapahtuman kokeneiden potilaiden osuus (n, %) | 512 (20,0) | 678 (26,4) |
Riskisuhde (95 % lv) | 0,726 (0,648–0,815) | |
IDFS 84 kk kohdalla (%, 95 % lv) | 77,0 (75,0–78,8) | 70,1 (68,0–72,1) |
Etäpesäkkeisen syövän uusiutumisvapaa elossaoloaika (DRFS) |
|
|
Tapahtuman kokeneiden potilaiden osuus (n, %) | 448 (17,5) | 589 (23,0) |
Riskisuhde (95 % lv) | 0,736 (0,651–0,832) | |
DRFS 84 kk kohdalla (%, 95 % lv) | 79,5 (77,6–81,2) | 74,0 (72,0–75,9) |
Kokonaiselossaoloaika (OS) |
|
|
Tapahtumien määrä, n (%) | 286 (11,2) | 344 (13,4) |
Riskisuhde (95 % lv) | 0,835 (0,713–0,977) | |
Lyhenne: lv = luottamusväli.
Tiedonkeruun katkaisupäivä 15. heinäkuuta 2025
Kuva 1. monarchE: Invasiivisen syövän uusiutumisvapaan elossaoloajan Kaplan–Meier -käyrä (tutkijan arvio, kohortin 1 populaatio)
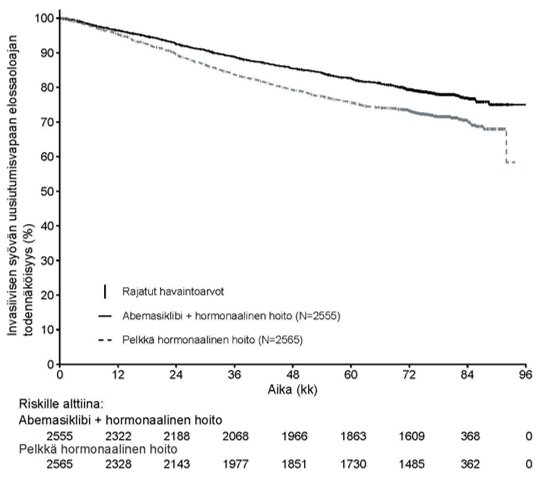
Lyhenteet: N = potilaiden lukumäärä populaatiossa.
Tiedonkeruun katkaisupäivä 15. heinäkuuta 2025.
Kuva 2. monarchE: Kokonaiselossaoloajan Kaplan-Meier -käyrä (kohortin 1 populaatio)
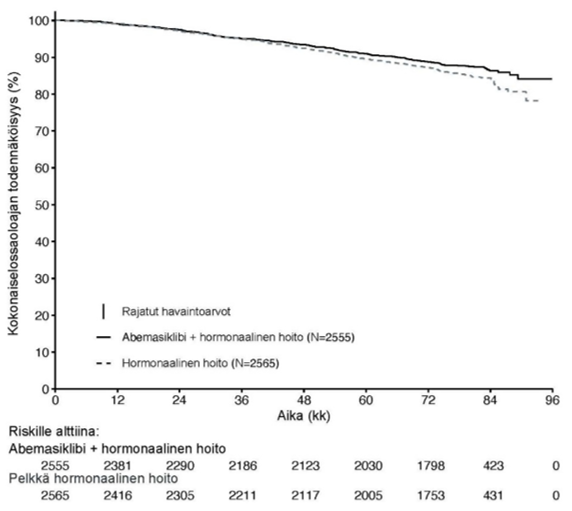
Lyhenteet: N = potilaiden lukumäärä populaatiossa.
Tiedonkeruun katkaisupäivä 15. heinäkuuta 2025
Hyöty havaittiin potilaiden kaikissa kohortin 1 alaryhmissä, jotka oli määritelty potilaiden maantieteellisen alueen, menopausaalisen statuksen ja aiemman solunsalpaajahoidon perusteella.
Paikallisesti edennyt taietäpesäkkeinen rintasyöpä
Satunnaistettu vaiheen 3 tutkimus MONARCH 3: Verzenios yhdistettynä aromataasin estäjiin
Verzenios-hoidon tehoa ja turvallisuutta aromataasin estäjään (anastrotsoli tai letrotsoli) yhdistettynä arvioitiin MONARCH 3 ‑tutkimuksessa. Kyseessä oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen 3 tutkimus hormonireseptoripositiivista, HER2‑negatiivista paikallisesti edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla naisilla, jotka eivät olleet saaneet aiempaa systeemistä hoitoa tässä tautitilanteessa. Potilaat satunnaistettiin suhteessa 2:1 saamaan Verzenios-valmistetta 150 mg kahdesti vuorokaudessa ja ei-steroidaalista aromataasin estäjää päivittäin suositusannoksina tai lumelääkettä ja ei-steroidaalista aromataasin estäjää saman aikataulun mukaan annettuna. Ensisijainen päätetapahtuma oli tutkijan arvioima etenemisvapaa elossaoloaika (PFS) RECIST 1.1 ‑kriteereillä arvioituna; tärkeimpiä toissijaisia tehon päätetapahtumia olivat objektiivinen kokonaisvaste (ORR), kliininen hyötyprosentti (CBR) ja kokonaiselossaoloaika (OS).
Mukaan otettujen potilaiden mediaani-ikä oli 63 vuotta (vaihteluväli 32–88). Noin 39 % potilaista oli saanut solunsalpaajahoitoa ja 44 % oli saanut hormonihoitoa (neo)adjuvanttihoitona. Jos potilas oli saanut aiemmin endokriinistä (neo)adjuvanttihoitoa, oli hoidon päättymisestä pitänyt kulua vähintään 12 kk ennen tutkimukseen satunnaistamista. Useimmilla potilailla (96 %) oli lähtötilanteessa etäpesäkkeinen tauti. Noin 22 %:lla potilaista oli tautimuutoksia vain luustossa, ja 53 %:lla oli viskeraalisia etäpesäkkeitä.
Tutkimuksen ensisijainen päätetapahtuma saavutettiin eli etenemisvapaa elossaoloaika piteni. Ensisijaisten tehotulosten yhteenveto esitetään taulukossa 10 ja kuvassa 3.
| Taulukko 10. MONARCH 3: Tehotietojen yhteenveto (tutkijan arvio, lähtöryhmien mukainen analyysi [ITT]) | |||
| Verzenios + aromataasin estäjä | Lumelääke + aromataasin estäjä | ||
| Etenemisvapaa elossaoloaika (PFS) | N = 328 | N = 165 | |
| Tutkijan arvio, tapahtumamäärä (%) | 138 (42,1) | 108 (65,5) | |
| Mediaani [kk] (95 %:n lv) | 28,18 (23,51; ei saav.) | 14,76 (11,24–19,20) | |
| Riskisuhde (95 %:n lv) ja p-arvo | 0,540 (0,418–0,698), p = 0,000002 | ||
| Riippumaton radiologinen arviointi, tapahtumamäärä (%) | 91 (27,7) | 73 (44,2) | |
| Mediaani [kk] (95 %:n lv) | Ei saav. (ei saav.; ei saav.) | 19,36 (16,37–27,91) | |
| Riskisuhde (95 %:n lv) ja p-arvo | 0,465 (0,339–0,636); p < 0,000001 | ||
| Objektiivinen kokonaisvasteb [%] (95 %:n lv) | 49,7 (44,3–55,1) | 37,0 (29,6–44,3) | |
| Vasteen kesto [kk] (95 %:n lv) | 27,39 (25,74; ei saav.) | 17,46 (11,21–22,19) | |
| Objektiivinen vaste potilailla, joilla mitattavissa oleva tautia | N = 267 | N = 132 | |
| Objektiivinen kokonaisvasteb [%] (95 %:n lv) | 61,0 (55,2–66,9) | 45,5 (37,0–53,9) | |
| Täydellinen vaste (%) | 3,4 | 0 | |
| Osittainen vaste (%) | 57,7 | 45,5 | |
| Kliininen hyötyprosenttic(mitattavissa oleva tauti) [%] (95 %:n lv) | 79,0 (74,1–83,9) | 69,7 (61,9–77,5) | |
a Mitattavissa oleva tauti RECIST‑kriteerien version 1.1 perusteella määriteltynä
b Täydellinen vaste + osittainen vaste
c Täydellinen vaste + osittainen vaste + taudin etenemisen pysähtyminen ≥ 6 kk ajaksi
N = potilasmäärä; lv = luottamusväli; ei saav. = ei saavutettu.
Kuva 3. MONARCH 3: Etenemisvapaan elossaoloajan Kaplan–Meier -käyrä (tutkijan arvio, lähtöryhmien mukainen populaatio)
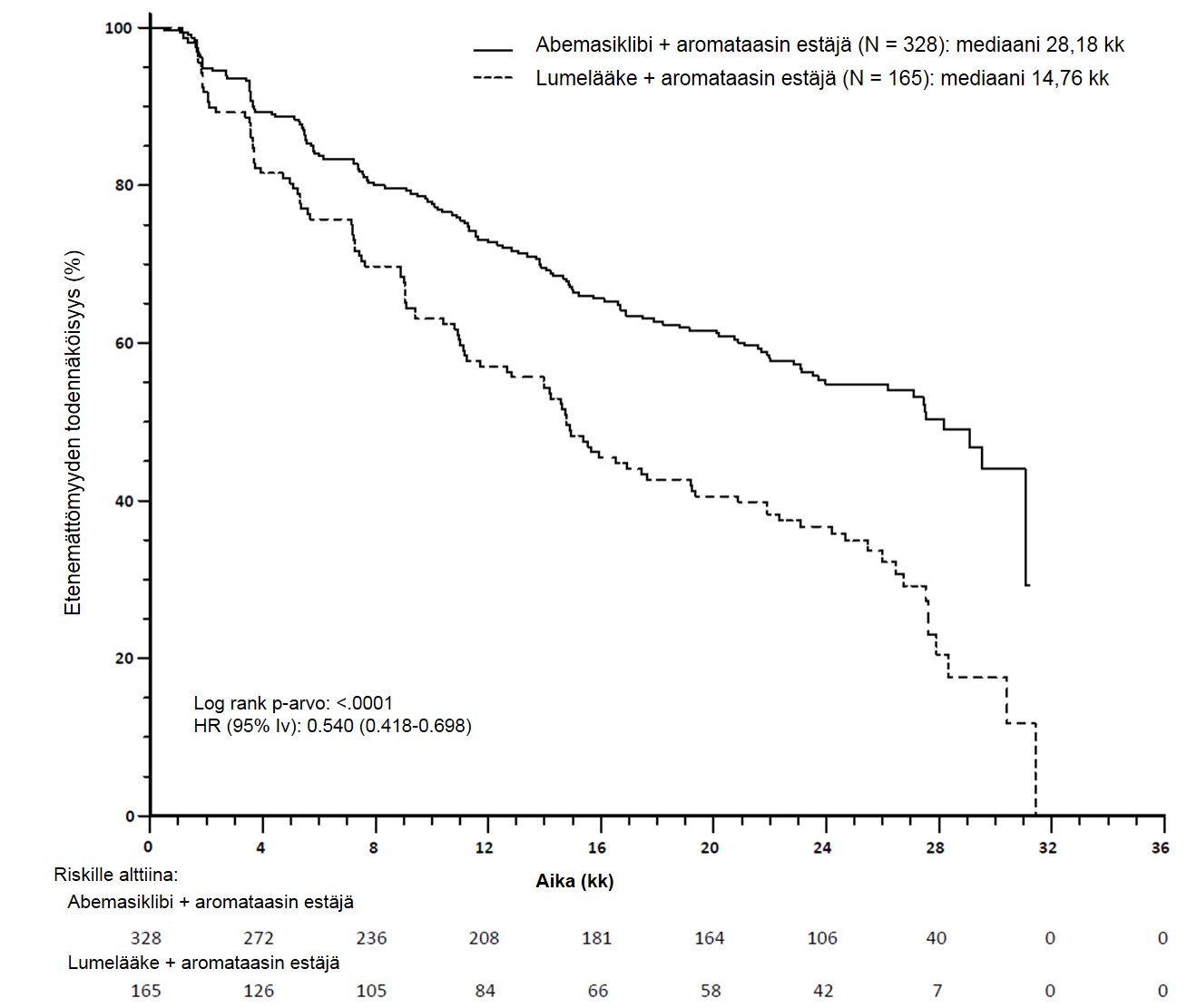
Nämä tulokset vastaavat taudin etenemisen tai kuoleman riskin kliinisesti merkittävää pienenemistä 46 % abemasiklibia ja aromataasin estäjää saaneilla potilailla.
Kokonaiselossaoloajan tiedot eivät olleet valmiit PFS-loppuanalyysin ajankohtana (jolloin hoitoryhmissä oli todettu 93 tapahtumaa). HR oli 1,057 (95 %:n lv 0,683–1,633), p = 0,8017.
Useissa etukäteen määritellyissä etenemisvapaan elossaoloajan alaryhmäanalyyseissä saatiin johdonmukaiset tulokset eri alaryhmissä, joiden muodostamisperusteina olivat mm. ikä (< 65 v tai ≥ 65 v), taudin sijainti, tautitilanne (de novo etäpesäkkeinen vs. uusiutunut etäpesäkkeinen vs. uusiutunut paikallisesti edennyt tauti), taudin mitattavuus, progesteronireseptoristatus ja lähtötilanteen ECOG-toimintakykyluokka. Taudin etenemisen tai kuoleman riskin todettiin pienentyneen potilailla, joilla oli viskeraalinen tauti (HR 0,567 [95 %:n lv 0,407–0,789], etenemisvapaan elossaoloajan mediaani 21,6 kk vs. 14,0 kk); potilailla, joilla oli vain luustomuutoksia (HR 0,565 [95 %:n lv 0,306–1,044]); ja potilailla, joiden tauti oli mitattavissa (HR 0,517 (95 %:n lv 0,392–0,681]).
Ensimmäisessä kokonaiselossaoloajan välianalyysissä yhteensä 197 tapahtumaa havaittiin kummassakin ryhmässä ja HR oli 0,786 (95 %:n lv 0,589–1,049).
Toisessa kokonaiselossaoloajan välianalyysissä yhteensä 255 tapahtumaa havaittiin kummassakin ryhmässä ja HR oli 0,754 (95 %:n lv 0,584–0,974).
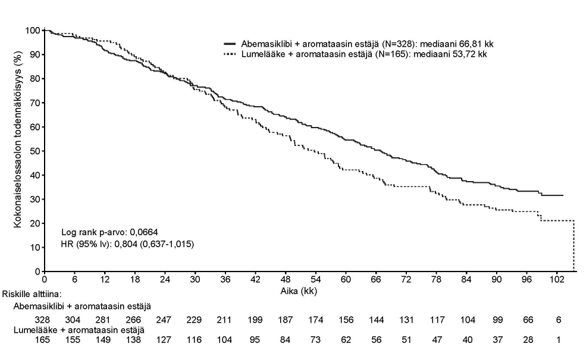
Tulokset lopullisesta kokonaiselossaoloajan analyysistä eivät olleet tilastollisesti merkitseviä (yhteenveto taulukossa 11 ja kuvassa 4).
| Taulukko 11. MONARCH 3: Kokonaiselossaoloajan tietojen yhteenveto (lähtöryhmien mukainen populaatio, ITT) | ||
|---|---|---|
| Verzenios + anastrotsoli tai letrotsoli | Lumelääke + anastrotsoli tai letrotsoli | |
| Kokonaiselossaoloaika | N = 328 | N = 165 |
| Tapahtumien lukumäärä (n, %) | 198 (60,4) | 116 (70,3) |
| Mediaani [kk] (95 %:n lv) | 66,81 (59,21–74,83) | 53,72 (44,75–59,34) |
| Riskisuhde (95 %:n lv) | 0,804 (0,637–1,015) | |
N = potilasmäärä; lv = luottamusväli
Kokonaiselossaoloajan analyyseissä potilailla, joilla oli viskeraalinen tauti, osoitettiin kokonaiselossaoloajan riskisuhteeksi 0,758 (95 %:n lv: 0,558–1,030). Kokonaiselossaoloajan mediaani oli 63,72 kk abemasiklibi + aromataasin estäjä -ryhmässä ja 48,82 kk lumelääke + aromataasin estäjä -ryhmässä. Tulokset eivät olleet tilastollisesti merkitseviä, kuten eivät olleet lähtöryhmien mukaisessa populaatiossakaan.
Kuva 4. MONARCH 3: Kokonaiselossaoloajan Kaplan-Meier -käyrä (lähtöryhmien mukainen populaatio, ITT)
Satunnaistettu vaiheen 3 tutkimus MONARCH 2: Verzenios yhdessä fulvestrantin kanssa
Verzenios-hoidon tehoa ja turvallisuutta yhdessä fulvestrantin kanssa arvioitiin MONARCH 2 ‑tutkimuksessa. Kyseessä oli satunnaistettu, kaksoissokkoutettu, lumekontrolloitu vaiheen 3 tutkimus hormonireseptoripositiivista, HER2‑negatiivista paikallisesti edennyttä tai etäpesäkkeistä rintasyöpää sairastavilla naisilla. Potilaat satunnaistettiin suhteessa 2:1 saamaan Verzenios-valmistetta (150 mg kahdesti vuorokaudessa) + fulvestranttia (500 mg yhden kuukauden välein ja lisäksi 500 mg:n lisäannos 2 viikon kuluttua aloitusannoksesta) vs. lumetta ja fulvestranttia saman aikataulun mukaan annettuna. Ensisijainen päätetapahtuma oli tutkijan arvioima etenemisvapaa elossaoloaika RECIST 1.1 ‑kriteereillä arvioituna; tärkeimpiä toissijaisia tehon päätetapahtumia olivat objektiivinen kokonaisvaste (ORR), kliininen hyötyprosentti (CBR) ja kokonaiselossaoloaika (OS).
Mukaan otettujen potilaiden mediaani-ikä oli 60 vuotta (vaihteluväli 32–91 v). Kussakin hoitoryhmässä valtaosa potilaista oli valkoihoisia, eikä ollut saanut solunsalpaajahoitoa etäpesäkkeisen taudin hoitoon. Potilaista 17 % oli pre- tai perimenopausaalisia (GnRH-agonistilla toteutettu munasarjasuppressio). Noin 56 %:lla potilaista oli viskeraalisia etäpesäkkeitä. Noin 25 %:lla potilaista oli primaarinen resistenssi endokriiniselle hoidolle (metastaattisen rintasyövän eteneminen 2 ensimmäisen vuoden endokriinisen adjuvanttihoidon aikana tai ensilinjan endokriinisen hoidon ensimmäisen 6 kk aikana), ja valtaosalle resistenssi endokriiniselle hoidolle kehittyi myöhemmin. Potilaista 59 %:lla viimeisin endokriininen hoito oli (neo)adjuvanttihoitoa ja 38 %:lla potilaista etäpesäkkeisen taudin hoitoa.
Tutkimuksen ensisijainen päätetapahtuma saavutettiin eli etenemisvapaa elossaoloaika piteni. Ensisijaisten tehotulosten yhteenveto esitetään taulukossa 12 ja kuvassa 5.
| Taulukko 12. MONARCH 2: Tehotietojen yhteenveto (tutkijan arvio, lähtöryhmien mukainen analyysi [ITT]) | ||
| Verzenios + fulvestrantti | Lumelääke + fulvestrantti | |
| Etenemisvapaa elossaoloaika (PFS) | N = 446 | N = 223 |
| Tutkijan arvio, tapahtumamäärä (%) | 222 (49,8) | 157 (70,4) |
| Mediaani [kk] (95 %:n lv) | 16,4 (14,4–19,3) | 9,3 (7,4–12,7) |
| Riskisuhde (95 %:n lv) ja p-arvo | 0,553 (0,449–0,681), p = 0,0000001 | |
| Riippumaton radiologinen arviointi, tapahtumamäärä (%) | 164 (36,8) | 124 (55,6) |
| Mediaani [kk] (95 %:n lv) | 22,4 (18,3; ei saav.) | 10,2 (5,8–14,0) |
| Riskisuhde (95 %:n lv) ja p-arvo | 0,460 (0,363–0,584); p < 0,000001 | |
| Objektiivinen vasteprosenttib [%] (95 %:n lv) | 35,2 (30,8–39,6) | 16,1 (11,3–21,0) |
| Vasteen kesto [kk] (95 %:n lv) | ei saav. (18,05; ei saav.) | 25,6 (11,9–25,6) |
| Objektiivinen vaste potilailla, joilla mitattavissa oleva tautia | N = 318 | N = 164 |
| Objektiivinen vasteprosenttib [%] (95 %:n lv) | 48,1 (42,6–53,6) | 21,3 (15,1–27,6) |
| Täydellinen vaste (%) | 3,5 | 0 |
| Osittainen vaste (%) | 44,7 | 21,3 |
| Kliininen hyötyprosenttic(mitattavissa oleva tauti) [%] (95 %:n lv) | 73,3 (68,4–78,1) | 51,8 (44,2–59,5) |
a Mitattavissa oleva tauti RECIST‑kriteerien version 1.1 perusteella määriteltynä
b Täydellinen vaste + osittainen vaste
c Täydellinen vaste + osittainen vaste + taudin etenemisen pysähtyminen ≥ 6 kk ajaksi
N = potilasmäärä; lv = luottamusväli
Kuva 5. MONARCH 2: Etenemisvapaan elossaoloajan Kaplan–Meier -käyrä (tutkijan arvio, lähtöryhmien mukainen populaatio)
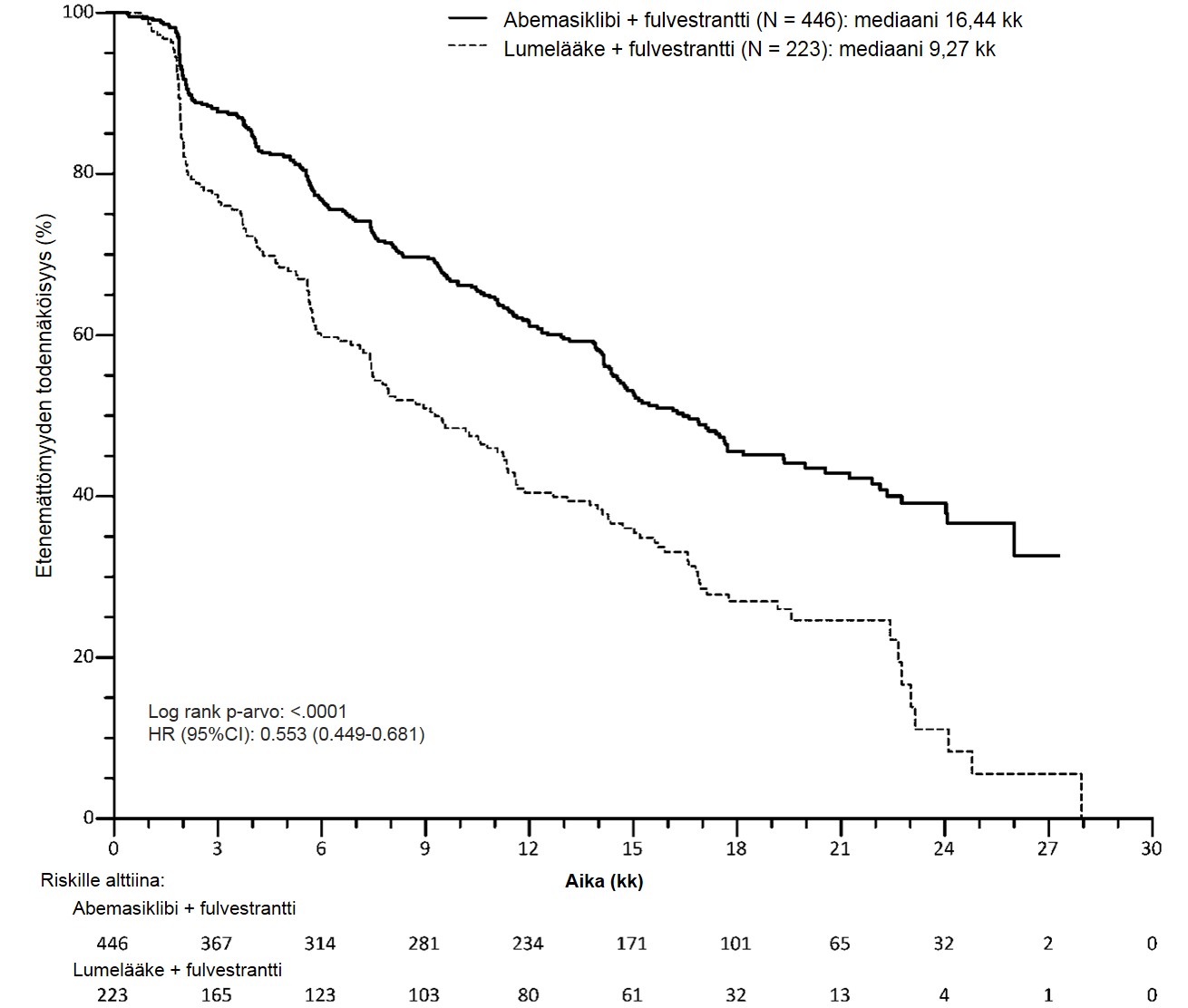
Nämä tulokset vastaavat taudin etenemisen tai kuoleman riskin kliinisesti merkittävää pienenemistä 44,7 % Verzeniosta + fulvestranttia saaneilla potilailla. Verzenios + fulvestrantti ‑yhdistelmähoito pidensi etenemisvapaata elossaoloaikaa eikä huonontanut terveyteen liittyvää elämänlaatua merkittävästi eikä kliinisesti merkitsevästi.
Useissa etukäteen määritellyissä etenemisvapaan elossaoloajan alaryhmäanalyyseissä saatiin johdonmukaiset tulokset eri alaryhmissä, joiden muodostamisperusteina olivat mm. ikä (< 65 v tai ≥ 65 v), rotu, maantieteellinen alue, taudin sijainti, resistenssi endokriiniselle hoidolle, taudin mitattavuus, progesteronireseptoristatus ja vaihdevuosistatus. Taudin etenemisen tai kuoleman riskin todettiin pienentyneen potilailla, joilla oli viskeraalinen tauti (HR 0,481 [95 %:n lv 0,369–0,627], etenemisvapaan elossaoloajan mediaani 14,7 kk vs. 6,5 kk); potilailla, joilla oli vain luustomuutoksia (HR 0,543 [95 %:n lv 0,355–0,833]); ja potilailla, joiden tauti oli mitattavissa (HR 0,523 [95 %:n lv 0,412–0,644]). Pre- tai perimenopausaalisilla potilailla riskisuhde oli 0,415 (95 %:n lv 0,246–0,698); progesteronireseptorinegatiivisilla potilailla HR oli 0,509 [95 %:n lv 0,325–0,797]).
Etenemisvapaa elossaoloaika (PFS) oli johdonmukainen myös alaryhmässä, jonka tutkittavilla oli paikallisesti edennyt tai etäpesäkkeinen tauti ja jonka tutkittavat eivät olleet saaneet aiempaa endokriinistä hoitoa.
Ennalta määritellyn välivaiheen kokonaiselossaoloajan analyysi (tiedonkeruun katkaisupäivä 20. kesäkuuta 2019) lähtöryhmien mukaisessa populaatiossa osoitti tilastollisesti merkitsevän pitenemisen potilailla, jotka saivat Verzenios-lääkevalmistetta + fulvestranttia verrattuna potilaisiin, jotka saivat lumelääkettä + fulvestranttia. Kokonaiselossaoloajan tulosten yhteenveto esitetään taulukossa 13.
| Taulukko 13. MONARCH 2: Kokonaiselossaoloajan tietojen yhteenveto (lähtöryhmien mukainen populaatio) | ||
| Verzenios + fulvestrantti | Lumelääke + fulvestrantti | |
| Kokonaiselossaoloaika | N = 446 | N = 223 |
| Tapahtumien lukumäärä (n, %) | 211 (47,3) | 127 (57,0) |
| Mediaani [kk] (95 %:n lv) | 46,7 (39,2–52,2) | 37,3 (34,4–43,2) |
| Riskisuhde (95 %:n lv) | 0,757 (0,606–0,945) | |
| p-arvo | 0,0137 | |
N = potilasmäärä; lv = luottamusväli
Stratifiointitekijöiden mukaan tehdyissä kokonaiselossaoloaikojen analyyseissä osoitettiin kokonaiselossaoloajan riskisuhteeksi 0,675 (95 %:n lv: 0,511–0,891) potilailla, joilla oli viskeraalinen tauti, ja 0,686 (95 %:n lv: 0,451–1,043) potilailla, joilla oli primaarinen resistenssi endokriiniselle hoidolle.
Ennalta määritellyssä lopullisessa kokonaiselossaoloajan analyysissä (tiedonkeruun katkaisupäivä 18. maaliskuuta 2022) havaittiin yhteensä 440 tapahtumaa kahdessa tutkimushaarassa. Aiemmin välivaiheen kokonaiselossaoloajan analyysissä (tiedonkeruun katkaisupäivä 20. kesäkuuta 2019) havaittu kokonaiselossaoloajan paraneminen säilyi abemasiklibia ja fulvestranttia saaneessa ryhmässä verrattuna lumelääkettä ja fulvestranttia saaneeseen ryhmään, ja sen HR oli 0,784 (95 %:n lv: 0,644–0,955). Kokonaiselossaoloajan mediaani oli 45,8 kuukautta abemasiklibia ja fulvestranttia saaneessa ryhmässä ja 37,25 kuukautta lumelääkettä ja fulvestranttia saaneessa ryhmässä. Kokonaiselossaoloajan tulokset on esitetty kuvassa 6.
Kuva 6. MONARCH 2: Kokonaiselossaoloajan Kaplan–Meier -käyrä (lähtöryhmien mukainen populaatio)
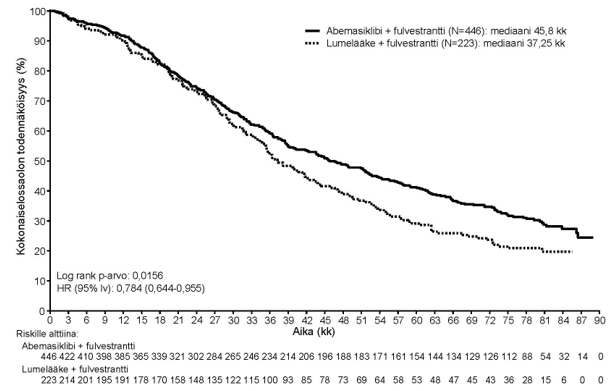
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Verzenios-valmisteen käytöstä kaikkien pediatristen potilasryhmien hoidossa rintasyövässä (ks. kohta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Verzenios-hoidon tehoa ja turvallisuutta yhdessä irinotekaanin ja temotsolomidin kanssa arvioitiin tutkimuksessa J1S-MC-JP04. Kyseessä oli satunnaistettu, avoin vaiheen 2 -monikeskustutkimus potilailla, joilla oli uusiutunut tai hoitoresistentti Ewingin sarkooma. Ensisijainen päätetapahtuma oli etenemisvapaa elossaoloaika (PFS), jonka arvioi sokkoutettu riippumaton arviointikomitea. Tutkimukseen osallistui 46 potilasta, iältään 3–35 vuotta, jotka satunnaistettiin suhteessa 2:1 saamaan Verzenios-valmistetta + irinotekaania + temotsolomidia tai irinotekaania + temotsolomidia. Potilaista 58,7 % (27 henkilöä) oli alle 18-vuotiaita. Potilaista 45 sai hoitoa 21 päivän sykleissä, kunnes tauti eteni tai he saavuttivat muun keskeyttämiskriteerin. Verzenios-valmisteen lisäämisellä ei havaittu eroa PFS-arvossa. PFS-mediaani oli 2,8 kuukautta Verzenios + irinotekaani + temotsolomidi -hoitoa saaneilla potilailla ja 2,9 kuukautta irinotekaani + temotsolomidi -hoitoa saaneilla potilailla (riskisuhde 0,64 [95 % lv: 0,28–1,45]).
Farmakokinetiikka
Imeytyminen
Abemasiklibi imeytyy hitaasti; sen Tmax-aika on 8 tuntia ja absoluuttisen biologisen hyötyosuuden keskiarvo noin 45 %. Terapeuttisella 50–200 mg:n annosalueella plasman altistus (AUC) ja Cmax ovat suunnilleen suhteessa annokseen. Vakaa tila saavutettiin 5 päivässä, kun valmistetta otettiin toistuvasti kahdesti vuorokaudessa. Abemasiklibi kumuloitui elimistöön, ja sen kumulaatiokertoimen geometrinen keskiarvo oli Cmax-arvon perusteella laskettuna 3,7 (CV 58 %) ja AUC-arvon perusteella laskettuna 5,8 (CV 65 %). Runsasrasvainen ateria suurensi abemasiklibin ja sen aktiivisten metaboliittien yhteenlaskettua AUC‑arvoa 9 % ja Cmax-arvoa 26 %. Muutoksia ei pidetty kliinisesti merkittävinä. Abemasiklibi voidaan siis ottaa ruoan kanssa tai ilman ruokaa.
Jakautuminen
Abemasiklibi sitoutuu voimakkaasti ihmisen plasman proteiineihin (sitoutunut fraktio keskimäärin 96–98 %). Systeemisen jakautumistilavuuden geometrinen keskiarvo on noin 750 l (CV 69 %), mikä viittaa siihen, että abemasiklibi jakautuu kudoksiin.
Abemasiklibin ja sen aktiivisten metaboliittien pitoisuudet aivo-selkäydinnesteessä ovat verrattavissa sitoutumattoman lääkeaineen pitoisuuksiin plasmassa.
Biotransformaatio
Maksametabolia on abemasiklibin tärkein puhdistumareitti. Abemasiklibi metaboloituu useiksi eri metaboliiteiksi lähinnä sytokromi P450 (CYP) 3A4:n vaikutuksesta. Ensisijainen biotransformaatio on hydroksylaatio kiertäväksi metaboliitiksi, jonka AUC on 77 % kanta-aineesta. Lisäksi kiertävän N-desetyylimetaboliitin AUC on 39 % kanta-aineesta ja kiertävän N-desetyylihydoksimetaboliitin 15 % kanta-aineesta. Kiertävät metaboliitit ovat aktiivisia, ja niiden vaikutuskyky on samaa luokkaa kuin abemasiklibin.
Eliminaatio
Abemasiklibin maksapuhdistuman (CL) geometrinen keskiarvo oli 21,8 l/h (CV 39,8 %) ja sen eliminaation puoliintumisaika potilaiden plasmasta keskimäärin 24,8 tuntia (CV 52,1 %). Peroraalisen [14C]‑abemasiklibikerta-annoksen jälkeen noin 81 % annoksesta erittyi ulosteeseen ja 3,4 % virtsaan. Valtaosa ulosteeseen erittyneestä määrästä oli metaboliittien muodossa.
Erityisryhmät
Ikä, sukupuoli ja paino
Ikä, sukupuoli ja paino eivät vaikuttaneet abemasiklibialtistukseen syöpäpotilailla tehdyssä populaatiofarmakokinetiikan analyysissä (135 miestä ja 859 naista; iän vaihteluväli 24–91 v; painon vaihteluväli 36–175 kg).
Maksan vajaatoiminta
Abemasiklibi metaboloituu maksassa. Lievä (Child–Pugh A) ja keskivaikea (Child–Pugh B) maksan vajaatoiminta eivät vaikuttaneet abemasiklibialtistukseen. Vaikeaa maksan vajaatoimintaa (Child–Pugh C) sairastavilla abemasiklibin AUC0-∞-arvo suureni 2,1-kertaiseksi ja vaikutuskyvyn suhteen korjatun, sitoutumattoman abemasiklibin ja sen aktiivisten metaboliittien AUC0-∞-arvo 2,4-kertaiseksi. Abemasiklibin puoliintumisaika piteni 24 tunnista 55 tuntiin (ks. kohta Annostus ja antotapa).
Munuaisten vajaatoiminta
Abemasiklibin ja sen metaboliittien puhdistuminen munuaisteitse on vähäistä. Lievä ja keskivaikea munuaisten vajaatoiminta eivät vaikuttaneet abemasiklibialtistukseen. Vaikeaa munuaisten vajaatoimintaa tai loppuvaiheen munuaisten vajaatoimintaa sairastavien tai dialyysihoidossa olevien potilaiden hoidosta ei ole tietoja.
Prekliiniset tiedot turvallisuudesta
Tärkeimpiä kohde-elinlöydöksiä, joilla on mahdollisesti merkitystä ihmiselle, olivat ruoansulatuskanavaan, hematolymfopoieettisiin elimiin ja urosten sukuelimiin kohdistuvat vaikutukset, joita todettiin enintään 13 viikon pituisissa tutkimuksissa hiirillä, rotilla ja koirilla. Silmiin ja sydänläppiin kohdistuvia vaikutuksia esiintyi kliinisesti merkityksellisillä altistustasoilla vain jyrsijöillä. Keuhkoihin ja luustolihaksiin kohdistuvia vaikutuksia esiintyi vain jyrsijöillä, joiden altistustaso oli vähintään 2-kertaisesti suurempi kuin ihmisen altistustaso. Munuaisiin kohdistuvia vaikutuksia esiintyi vain jyrsijöillä, joiden altistustaso oli vähintään 6-kertaisesti suurempi kuin ihmisen altistustaso. 28-päiväisen toipumisjakson lopussa todettiin, että kaikkien kohde-elinten löydökset korjautuivat täysin tai osittain lukuun ottamatta urosten sukuelimiin kohdistuvia vaikutuksia.
Genotoksisuus
Abemasiklibi ei ollut mutageeninen bakteereilla tehdyssä käänteismutaatiokokeessa (Amesin testi). Se ei ollut klastogeeninen ihmisen ääreisveren lymfosyyttien kromosomipoikkeavuuskokeessa in vitro eikä rotan luuytimen mikrotumakokeessa in vivo.
Karsinogeenisuus
Abemasiklibin karsinogeenisuutta arvioitiin 2 vuotta kestäneissä tutkimuksissa rotilla ja hiirillä. Kun abemasiklibia annettiin urosrotille suun kautta päivittäin, esiintyi hyvänlaatuisia kivesten interstitiaaliadenoomia altistuksen ollessa noin 1,5 kertaa ihmiselle koituva kliininen altistus. Lisäksi havaittiin interstitiaalisolujen hyperplasiaa altistuksen ollessa noin 0,1 kertaa ihmiselle koituva kliininen altistus. Näiden vaikutusten esiintymistä ihmisillä ei tunneta. Hiirillä ja naarasrotilla abemasiklibin antoon ei liittynyt neoplastisia löydöksiä.
Hedelmällisyyden heikentyminen
Abemasiklibi saattaa heikentää lisääntymiskykyisten miesten hedelmällisyyttä. Toistuvaisannosten toksisuutta koskeneissa, enintään 3 kuukauden pituisissa tutkimuksissa abemasiklibiin liittyneitä löydöksiä havaittiin kiveksissä, lisäkiveksissä, eturauhasessa ja rakkularauhasissa. Löydöksiä olivat elinten painon pieneneminen, intratubulaarinen solujäte, hypospermia sekä siementiehyiden laajentuminen, atrofia ja degeneraatio/nekroosi. Näitä vaikutuksia esiintyi rotilla ja koirilla (altistus rotilla noin 2 kertaa ja koirilla noin 0,02 kertaa ihmiselle koituva kliininen altistus). Urosrottien hedelmällisyystutkimuksessa abemasiklibi ei vaikuttanut lisääntymiskykyyn.
Naarasrottien hedelmällisyyttä ja alkion varhaiskehitystä kartoittaneessa tutkimuksessa ja toistuvaisannosten toksisuustutkimuksissa abemasiklibi ei vaikuttanut millään tavoin lisääntymiskykyyn eikä aiheuttanut sellaisia olennaisia vaikutuksia naaraiden sukuelimiin, jotka viittaisivat naaraiden hedelmällisyyden heikkenemiseen.
Kehitystoksisuus
Abemasiklibi oli teratogeeninen ja aiheutti sikiöiden painon laskua, kun emon altistus oli ihmisen suositusannosten luokkaa.
Farmaseuttiset tiedot
Apuaineet
Tabletin ydin
kroskarmelloosinatrium
laktoosimonohydraatti
mikrokiteinen selluloosa
hydratoitu kolloidinen piidioksidi
natriumstearyylifumaraatti
Kalvopäällyste
Verzenios 50 mg kalvopäällysteinen tabletti
polyvinyylialkoholi (E1203)
titaanidioksidi (E171)
makrogoli (E1521)
talkki (E553b)
keltainen rautaoksidi (E172)
punainen rautaoksidi (E172)
Verzenios 100 mg kalvopäällysteinen tabletti
polyvinyylialkoholi (E1203)
titaanidioksidi (E171)
makrogoli (E1521)
talkki (E553b)
Verzenios 150 mg kalvopäällysteinen tabletti
polyvinyylialkoholi (E1203)
titaanidioksidi (E171)
makrogoli (E1521)
talkki (E553b)
keltainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta.
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
VERZENIOS tabletti, kalvopäällysteinen
50 mg (L:kyllä) 56 fol (2402,33 €)
100 mg (L:kyllä) 56 fol (2402,33 €)
150 mg (L:kyllä) 56 fol (2402,33 €)
PF-selosteen tieto
14, 28, 42, 56, 70 tai 168 kalvopäällysteistä tablettia alumiinifoliolla sinetöidyissä PCTFE/PE/PVC-läpipainopakkauksissa.
28 x 1 kalvopäällysteistä tablettia yksittäispakatuissa alumiini/alumiiniläpipainopakkauksissa.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Verzenios 50 mg kalvopäällysteinen tabletti
Beige, soikea tabletti, jonka koko on 5,2 x 9,5 mm ja jonka toisella puolella on kaiverrus ”Lilly” ja toisella puolella kaiverrus ”50”.
Verzenios 100 mg kalvopäällysteinen tabletti
Valkoinen, soikea tabletti, jonka koko on 6,6 x 12,0 mm ja jonka toisella puolella on kaiverrus ”Lilly” ja toisella puolella kaiverrus ”100”.
Verzenios 150 mg kalvopäällysteinen tabletti
Keltainen, soikea tabletti, jonka koko on 7,5 x 13,7 mm ja jonka toisella puolella on kaiverrus ”Lilly” ja toisella puolella kaiverrus ”150”.
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
VERZENIOS tabletti, kalvopäällysteinen
50 mg 56 fol
100 mg 56 fol
150 mg 56 fol
- Ylempi erityiskorvaus (100 %). Abemasiklibi: Hormonireseptoripositiivisen ja HER2-negatiivisen rintasyövän hoito erityisin edellytyksin (1515).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Abemasiklibi: Hormonireseptoripositiivisen ja HER2-negatiivisen paikallisesti edenneen tai etäpesäkkeisen rintasyövän hoito erityisin edellytyksin (3030).
ATC-koodi
L01EF03
Valmisteyhteenvedon muuttamispäivämäärä
29.01.2026
Yhteystiedot
 OY ELI LILLY FINLAND AB
OY ELI LILLY FINLAND AB Mannerheimintie 117
00280 Helsinki
09 854 5250
www.lilly.com/fi
medinfo_finland@lilly.com
Lääketietopalvelu puh. 0800-140 240