
OFEV kapseli, pehmeä 25 mg, 100 mg, 150 mg
Vaikuttavat aineet ja niiden määrät
Ofev 25 mg pehmeät kapselit
Yksi pehmeä kapseli sisältää 25 mg nintedanibia (esilaattina)
Apuaine, jonka vaikutus tunnetaan
Yksi 25 mg pehmeä kapseli sisältää 0,3 mg soijalesitiiniä.
Ofev 100 mg pehmeät kapselit
Yksi pehmeä kapseli sisältää 100 mg nintedanibia (esilaattina)
Apuaine, jonka vaikutus tunnetaan
Yksi 100 mg pehmeä kapseli sisältää 1,2 mg soijalesitiiniä.
Ofev 150 mg pehmeät kapselit
Yksi pehmeä kapseli sisältää 150 mg nintedanibia (esilaattina)
Apuaine, jonka vaikutus tunnetaan
Yksi 150 mg pehmeä kapseli sisältää 1,8 mg soijalesitiiniä.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Kapseli, pehmeä (kapseli).
Kliiniset tiedot
Käyttöaiheet
Ofev on tarkoitettu aikuisille idiopaattisen keuhkofibroosin (IPF) hoitoon.
Ofev on tarkoitettu aikuisille myös muiden kroonisten, fenotyypiltään etenevien, fibrotisoivien interstitiaalisten keuhkosairauksien (ILD) hoitoon (ks. kohta Farmakodynamiikka).
Ofev on tarkoitettu 6–17-vuotiaille lapsille ja nuorille kliinisesti merkittävien, etenevien, fibrotisoivien interstitiaalisten keuhkosairauksien (ILD) hoitoon (ks. kohdat Annostus ja antotapa ja Farmakodynamiikka).
Ofev on tarkoitettu aikuisille, sekä vähintään 6-vuotiaille lapsille ja nuorille, systeemiseen skleroosiin liittyvän interstitiaalisen keuhkosairauden (SSc‑ILD) hoitoon.
Ehto
Valmiste on tarkoitettu käytettäväksi käyttöaiheessa mainitun sairauden diagnosointiin ja hoitoon perehtyneen lääkärin ohjauksessa ja seurannassa.
Annostus ja antotapa
Aikuiset: Hoidon saa aloittaa vain lääkäri, jolla on kokemusta niiden sairauksien hoidosta, joihin Ofev on hyväksytty.
Pediatriset potilaat: Hoidon saa aloittaa vasta fibrotisoivien interstitiaalisten keuhkosairauksien (ILD) diagnosointiin ja hoitoon perehtyneen moniammatillisen ryhmän (lääkäri, radiologi, patologi) konsultaation jälkeen.
Annostus
Aikuiset
- idiopaattinen keuhkofibroosi (IPF)
- muut fenotyypiltään etenevät krooniset fibrotisoivat interstitiaaliset keuhkosairaudet (ILD)
- systeeminen skleroosiin liittyvä interstitiaalinen keuhkosairaus (SSc ILD)
Suositeltu annos on 150 mg nintedanibia kahdesti vuorokaudessa. Annokset otetaan noin 12 tunnin välein.
100 mg:n annosta kahdesti vuorokaudessa suositellaan käytettäväksi ainoastaan potilaille, jotka eivät siedä 150 mg:n annosta kahdesti vuorokaudessa.
Jos annos jää väliin, hoitoa jatketaan suositellulla annoksella tavanomaisen aikataulun mukaisesti. Jos yksi annos jää väliin, potilaan ei pidä ottaa ylimääräistä annosta. Suositeltua 300 mg:n enimmäisvuorokausiannosta ei saa ylittää.
Annosmuutokset
Ofev‑valmisteen haittavaikutusten hoidossa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset) oireenmukaisen hoidon lisäksi annosta voidaan pienentää ja hoito voidaan tarvittaessa keskeyttää väliaikaisesti, kunnes kyseinen haittavaikutus on lievittynyt niin, että hoitoa voidaan jatkaa. Ofev-hoitoa voidaan jatkaa täydellä annoksella (150 mg kahdesti vuorokaudessa aikuispotilaille) tai pienemmällä annoksella (100 mg kahdesti vuorokaudessa aikuispotilaille). Jos aikuispotilas ei siedä 100 mg:n annosta kahdesti vuorokaudessa, Ofev-hoito pitää lopettaa.
Jos potilaalla esiintyy pitkittynyttä ripulia, pahoinvointia ja/tai oksentelua asianmukaisesta tukihoidosta (mm. antiemeettisestä hoidosta) huolimatta, annoksen pienentäminen tai hoidon keskeyttäminen saattaa olla tarpeen. Hoitoa voidaan jatkaa pienemmällä annoksella (100 mg kahdesti vuorokaudessa aikuispotilaille) tai täydellä annoksella (150 mg kahdesti vuorokaudessa aikuispotilaille). Jos potilaalla on pitkittynyttä, vaikeaa ripulia, pahoinvointia ja/tai oksentelua oireenmukaisesta hoidosta huolimatta, Ofev-hoito on lopetettava (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Jos hoito joudutaan keskeyttämään aspartaattiaminotransferaasiarvon (ASAT) tai alaniiniaminotransferaasiarvon (ALAT) kohoamisen takia (> 3 × viitealueen yläraja (ULN)), Ofev‑hoitoa voidaan jatkaa pienemmällä annoksella (100 mg kahdesti vuorokaudessa aikuispotilaille) transaminaasiarvojen palattua lähtötasolle. Myöhemmin annosta voidaan nostaa täyteen annokseen (150 mg kahdesti vuorokaudessa aikuispotilaille) (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Erilliset suositukset annoksen pienentämisestä haittavaikutusten hallitsemiseksi pediatrisilla potilailla, ks. taulukko 1.
6–17-vuotiaat lapset ja nuoret
- kliinisesti merkittävien etenevien fibrotisoivien interstitiaalisten keuhkosairauksien (ILD) hoitoon
- systeemiseen skleroosiin liittyvän interstitiaalisen keuhkosairauden (SSc ‑ILD) hoitoon
Kasvua on seurattava säännöllisesti ja kasvulevyjen muutoksien arviointia luiden kuvantamisella suositellaan vuosittain potilaille, joilla on vielä avoimia epifyysejä. Hoidon keskeyttämistä on harkittava potilailla, joille kehittyy kasvun häiriintymisen oireita, tai kasvulevyjen muutoksia (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Säännöllinen suun ja hampaiden tutkimus on tehtävä vähintään 6 kuukauden välein, kunnes hampaisto on täysin kehittynyt (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Suositeltu Ofev-annos 6–17-vuotialle pediatrisille potilaille määritetään potilaan painon mukaan ja se annostellaan kahdesti päivässä noin 12 tunnin välein (ks. taulukko 1). Annosta on muutettava painon mukaan hoidon edetessä.
Taulukko 1: Ofev-annossuositus ja annoksen pienentämissuositus milligrammoina (mg) kilogrammoina (kg) ilmoitetun kehon painon mukaan 6–17-vuotiaille pediatrisille potilaille
| Paino-alue | Ofev-annos | Pienennetty Ofev-annos* |
| 13,5**–22,9 kg | 50 mg (kaksi 25 mg kapselia) kahdesti vuorokaudessa | 25 mg (yksi 25 mg kapseli) kahdesti vuorokaudessa |
| 23,0–33,4 kg | 75 mg (kolme 25 mg kapselia) kahdesti vuorokaudessa | 50 mg (kaksi 25 mg kapselia) kahdesti vuorokaudessa |
| 33,5–57,4 kg | 100 mg (yksi 100 mg kapseli tai neljä 25 mg kapselia) kahdesti vuorokaudessa | 75 mg (kolme 25 mg kapselia) kahdesti vuorokaudessa |
| vähintään 57,5 kg | 150 mg (yksi 150 mg kapseli tai kuusi 25 mg kapselia) kahdesti vuorokaudessa | 100 mg (yksi 100 mg kapseli tai neljä 25 mg kapselia) kahdesti vuorokaudessa |
| * Pienennettyä annosta suositellaan lapsille ja nuorille, joilla on lievä maksan vajaatoiminta (Child Pugh A) ja haittavaikutusten hallitsemiseksi pediatrisilla potilailla. Katso yllä olevasta lisätietoa haittavaikutusten hallitsemisesta. | ||
** Paino alle 13,5 kg: Hoito on keskeytettävä, jos potilaan paino laskee alle 13,5 kg:n. | ||
Erityisryhmät
Iäkkäät potilaat (≥ 65‑vuotiaat)
Iäkkäiden potilaiden kohdalla ei havaittu yleisesti ottaen eroja lääkkeen turvallisuudessa ja tehossa. Annosta ei ennakoivasti tarvitse muuttaa iäkkäille potilaille. 75‑vuotiaiden ja tätä vanhempien potilaiden annosta on todennäköisemmin tarpeen pienentää haittavaikutusten hoitamiseksi (ks. kohta Farmakokinetiikka).
Munuaisten vajaatoiminta
Aikuisten ja pediatristen potilaiden aloitusannosta ei tarvitse muuttaa lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla. Nintedanibin turvallisuutta, tehoa ja farmakokinetiikkaa ei ole tutkittu vaikeaa munuaisten vajaatoimintaa sairastavilla aikuisilla ja pediatrisilla potilailla (kreatiniinipuhdistuma < 30 ml/min).
Maksan vajaatoiminta
Lievää maksan vajaatoimintaa sairastavilla aikuispotilailla (Child Pugh A) suositeltu Ofev-annos on 100 mg kahdesti vuorokaudessa noin 12 tunnin välein. Lievää maksan vajaatoimintaa sairastavilla (Child Pugh A) pediatrisilla potilailla suositellaan pienennettyä aloitusannosta (ks. taulukko 1). Lievää maksan vajaatoimintaa sairastavilla aikuisilla ja pediatrisilla potilailla (Child Pugh A) on hoidon keskeyttämistä, tai lopettamista, harkittava haittavaikutusten hallitsemiseksi.
Nintedanibin turvallisuutta ja tehoa ei ole tutkittu aikuisilla, eikä pediatrisilla potilailla, joilla on Child Pugh ‑luokan B tai C maksan vajaatoiminta. Keskivaikeaa (Child Pugh B) tai vaikeaa (Child Pugh C) maksan vajaatoimintaa sairastavien aikuisten tai pediatristen potilaiden hoitoa Ofev-valmisteella ei suositella (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Nintedanibin tehoa ja turvallisuutta ei ole tutkittu alle 6-vuotiailla lapsipotilailla. Siksi nintedanibihoidon antamista ei suositella alle 6-vuotiaille lapsille. Nintedanibin käyttöä ei ole tutkittu alle 13,5 kg painavilla potilaille, ja siksi sen käyttöä ei suositella tälle potilasryhmälle (ks. kohta Farmakodynamiikka).
Antotapa
Ofev-kapselit otetaan suun kautta. Kapselit otetaan aterian yhteydessä ja ne niellään kokonaisina veden kera. Kapseleita ei saa pureskella. Kapselia ei saa avata tai murskata (ks. kohta Käyttö- ja käsittelyohjeet). Ofev-kapselien kanssa voidaan ottaa pieni määrä (yksi teelusikallinen) kylmää tai huoneenlämpöistä pehmeää ruokaa, kuten omenasosetta tai suklaavanukasta. Kapseli on nieltävä välittömästi pureskelematta, jotta se pysyy ehjänä.
Vasta-aiheet
- Raskaus (ks. kohta Raskaus ja imetys)
- Yliherkkyys nintedanibille, maapähkinälle tai soijalle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Maha-suolikanavan häiriöt
Ripuli
Kliinisissä tutkimuksissa ripuli oli yleisin maha-suolikanavan haittavaikutus (ks. kohta Haittavaikutukset). Useimmilla potilailla haittavaikutus oli vaikeusasteeltaan lievä tai kohtalainen ja ilmeni ensimmäisen kolmen hoitokuukauden aikana.
Myyntiluvan myöntämisen jälkeisessä käytössä on ilmoitettu vakavista, nestehukkaan ja elektrolyyttihäiriöihin johtaneista ripulitapauksista. Potilaita on hoidettava heti ensimerkkien ilmaannuttua riittävällä nesteytyksellä ja ripulilääkkeillä, esim. loperamidilla. Oireet saattavat vaatia annoksen pienentämistä tai hoidon keskeyttämistä. Ofev-hoitoa voidaan jatkaa pienemmällä annoksella tai täydellä annoksella (ks. kohta Annostus ja antotapa). Jos potilaalla on pitkittynyt, vaikea ripuli oireenmukaisesta hoidosta huolimatta, Ofev-hoito on lopetettava.
Pahoinvointi ja oksentelu
Pahoinvointi ja oksentelu olivat usein raportoituja maha-suolikanavan haittavaikutuksia (ks. kohta Haittavaikutukset). Useimmilla potilailla pahoinvointi ja oksentelu oli vaikeusasteeltaan lievää tai kohtalaista. Kliinisissä tutkimuksissa pahoinvoinnin takia Ofev-hoidon lopetti enintään 2,1 % potilaista ja oksentelun takia enintään 1,4 % potilaista.
Jos oireet pitkittyvät asianmukaisesta tukihoidosta (mm. antiemeettisestä hoidosta) huolimatta, annoksen pienentäminen tai hoidon keskeyttäminen saattaa olla tarpeen. Hoitoa voidaan jatkaa pienemmällä annoksella tai täydellä annoksella (ks. kohta Annostus ja antotapa). Jos potilaalla on pitkittyneitä, vaikeita oireita, Ofev-hoito on lopetettava.
Maksan toiminta
Ofev-valmisteen turvallisuutta ja tehoa ei ole tutkittu keskivaikeaa (Child Pugh B) tai vaikeaa (Child Pugh C) maksan vajaatoimintaa sairastavilla potilailla. Siksi Ofev-valmistetta ei suositella näille potilaille (ks. kohta Annostus ja antotapa). Koska altistus kasvaa, haittavaikutusten riski saattaa kasvaa lievää maksan vajaatoimintaa sairastavilla potilailla (Child Pugh A). Lievää maksan vajaatoimintaa sairastavia aikuispotilaita (Child Pugh A) on hoidettava pienemmällä Ofev-annoksella (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Nintedanibihoidon yhteydessä on todettu lääkeaineen aiheuttamia maksavaurioita, mukaan lukien vaikea kuolemaan johtanut maksavaurio. Suurin osa maksatapahtumista ilmenee hoidon ensimmäisten kolmen kuukauden aikana. Siksi maksan transaminaasi- ja bilirubiiniarvot on tutkittava ennen Ofev-hoidon aloittamista ja hoidon ensimmäisen kuukauden aikana. Tämän jälkeen potilaita on seurattava säännöllisesti hoidon seuraavien kahden kuukauden aikana ja tämän jälkeen määräajoin esim. jokaisella potilaskäynnillä tai kliinisen tarpeen mukaan.
Maksaentsyymiarvojen (ALAT, ASAT, veren alkalinen fosfataasi (AFOS), gammaglutamyylitransferaasi (GT), ks. kohta Haittavaikutukset) ja bilirubiinipitoisuuden nousut olivat useimmissa tapauksissa palautuvia annoksen pienentämisen tai hoidon lopettamisen jälkeen. Jos transaminaasipitoisuuksien (ASAT tai ALAT) havaitaan kohonneen > 3 × ULN, suositellaan annoksen pienentämistä tai Ofev-hoidon keskeyttämistä. Potilaan vointia on seurattava tarkasti. Kun transaminaasiarvot ovat palautuneet lähtötasolle, Ofev-hoitoa voidaan jatkaa täydellä annoksella tai se voidaan aloittaa uudelleen pienemmällä annoksella, joka voidaan myöhemmin nostaa täydeksi annokseksi (ks. kohta Annostus ja antotapa Annosmuutokset). Jos maksaentsyymipitoisuuksien kohoamiseen liittyy maksavaurion kliinisiä oireita tai löydöksiä, kuten keltaisuutta, Ofev-hoito on lopetettava pysyvästi. Maksaentsyymipitoisuuksien kohoamisen muut mahdolliset syyt on tutkittava.
Aikuispotilailla, joiden kehonpaino on pieni (< 65 kg), aasialaisilla potilailla ja naispotilailla on suurempi maksaentsyymiarvojen kohoamisen riski. Nintedanibialtistus kasvoi lineaarisesti suhteessa potilaan ikään, mikä saattaa myös lisätä maksaentsyymiarvojen kohoamisen riskiä (ks. kohta Farmakokinetiikka). Tarkka seuranta on suositeltavaa, jos potilaalla on näitä riskitekijöitä.
Munuaisten toiminta
Nintedanibin käytön yhteydessä on raportoitu munuaisten toimintahäiriötä/vajaatoimintaa, joka on joissakin tapauksissa johtanut kuolemaan (ks. kohta Haittavaikutukset).
Potilaita on seurattava nintedanibihoidon aikana, ja erityistä huomiota on kiinnitettävä niihin potilaisiin, joilla on munuaisten toimintahäiriön/vajaatoiminnan riskitekijöitä. Hoidon mukauttamista on harkittava, jos potilaalla ilmenee munuaisten toimintahäiriö/vajaatoiminta (ks. kohta Annostus ja antotapa, Annosmuutokset).
Verenvuoto
Verisuonten endoteelikasvutekijän reseptorin (VEGFR) estoon voi liittyä kohonnut verenvuotoriski.
Kliinisiin tutkimuksiin ei otettu potilaita, joilla tiedettiin olevan verenvuotoriski. Tämä koski myös potilaita, joilla oli perinnöllinen verenvuotoalttius tai jotka saivat täysiannoksista antikoagulaatiohoitoa. Myyntiluvan myöntämisen jälkeen on raportoitu sekä ei-vakavista verenvuototapauksista että vakavista verenvuototapauksista (sekä potilailla, jotka saavat antikoagulaatiohoitoa tai muita mahdollisesti verenvuotoa aiheuttavia lääkevalmisteita, että potilailla, jotka eivät käytä näitä lääkevalmisteita), joista jotkut johtivat kuolemaan. Siksi näitä potilaita saa hoitaa Ofev-valmisteella ainoastaan, jos odotettavissa oleva hyöty ylittää mahdollisen riskin.
Tromboemboliset valtimotapahtumat
Kliinisistä tutkimuksista suljettiin pois potilaat, joilla oli ollut äskettäin sydäninfarkti tai aivohalvaus. Kliinisissä tutkimuksissa aikuispotilailla raportoitiin tromboembolisia valtimotapahtumia harvoin (INPULSIS-tutkimuksessa Ofev 2,5 % vs. lumelääke 0,7 %; INBUILD-tutkimuksessa Ofev 0,9 % vs. lumelääke 0,9 %; SENSCIS-tutkimuksessa Ofev 0,7 % vs. lumelääke 0,7 %). INPULSIS-tutkimuksessa sydäninfarktin saaneiden potilaiden prosentuaalinen osuus oli suurempi Ofev-ryhmässä (1,6 %) kuin lumeryhmässä (0,5 %), kun taas iskeemiseen sydänsairauteen liittyvät haittatapahtumat jakautuivat tasaisesti Ofev- ja lumeryhmän välillä. INBUILD-tutkimuksessa sydäninfarktien yleisyys oli vähäistä: Ofev 0,9 % vs. lumelääke 0,9 %. SENSCIS-tutkimuksessa sydäninfarktien yleisyys oli vähäistä lumeryhmässä (0,7 %), eikä niitä havaittu Ofev-ryhmässä.
Varovaisuutta on noudatettava, jos potilaalla on suurentunut sydän- ja verisuonitapahtumien riski, mukaan lukien tiedossa oleva sepelvaltimotauti. Hoidon keskeyttämistä on harkittava, jos potilaalle kehittyy akuutin sydänlihasiskemian merkkejä tai oireita.
Aneurysmat ja valtimon dissekaatiot
VEGF‑reitin estäjien käyttö potilailla, joilla on kohonnut verenpaine tai joilla ei ole kohonnutta verenpainetta, saattaa edistää aneurysmien ja/tai valtimon dissekaatioiden muodostumista. Tämä riski on arvioitava tarkoin ennen Ofev-hoidon aloittamista potilaille, joilla on riskitekijöitä, kuten kohonnut verenpaine tai aikaisempi aneurysma.
Laskimotromboembolia
Kliinisissä tutkimuksissa nintedanibihoitoa saaneilla potilailla ei havaittu kohonnutta laskimotromboembolian riskiä. Nintedanibin vaikutusmekanismin takia potilailla saattaa olla kohonnut tromboembolisten tapahtumien riski.
Maha-suolikanavan perforaatiot ja iskeeminen koliitti
Kliinisissä tutkimuksissa aikuispotilailla perforaation yleisyys oli molemmissa hoitoryhmissä enintään 0,3 %. Nintedanibin vaikutusmekanismin takia potilailla saattaa olla kohonnut maha-suolikanavan perforaatioiden riski. Myyntiluvan myöntämisen jälkeen on raportoitu maha-suolikanavan perforaatioita ja iskeemisiä koliitteja, joista jotkut johtivat kuolemaan. Erityistä varovaisuutta on noudatettava, jos potilaalle on tehty aiemmin vatsan alueen leikkaus, jos hän on sairastanut peptisen haavauman tai divertikkelitaudin tai jos hän saa samanaikaisesti kortikosteroidi- tai tulehduskipulääkehoitoa. Ofev-hoidon saa aloittaa aikaisintaan 4 viikon kuluttua vatsan alueen leikkauksesta. Ofev-hoito on lopetettava pysyvästi, jos potilaalle kehittyy maha-suolikanavan perforaatio tai iskeeminen koliitti. Poikkeustapauksissa Ofev-hoito voidaan aloittaa uudestaan, kun iskeeminen koliitti on täysin parantunut ja kun potilaan tila ja muut riskitekijät on arvioitu huolellisesti.
Nefroottinen proteinuria ja tromboottinen mikroangiopatia
Hyvin harvoja nefroottisen proteinurian tapauksia, joihin liittyi tai ei liittynyt munuaisten vajaatoimintaa, on raportoitu valmisteen markkinoille tulon jälkeen. Yksittäisissä tapauksissa histologiset löydökset sopivat glomerulaariseen mikroangiopatiaan, johon liittyi tai ei liittynyt munuaisten veritulppia. Oireiden korjaantumista on todettu Ofev-hoidon lopettamisen jälkeen, mutta joissakin tapauksissa proteinuria ei korjaantunut täysin. Hoidon keskeyttämistä on harkittava, jos potilaalle kehittyy nefroottisen oireyhtymän merkkejä tai oireita.
Tromboottista mikroangiopatiaa (TMA) on liittynyt VEGF‑reitin estäjien, ja hyvin harvoissa tapauksissa myös nintedanibin, käyttöön. Jos nintedanibia saavalla potilaalla havaitaan TMA:han liittyviä laboratoriolöydöksiä tai kliinisiä löydöksiä, on nintedanibihoito lopetettava ja potilas tutkittava huolellisesti TMA:n varalta.
Posteriorinen reversiibeli enkefalopatiaoireyhtymä (PRES)
Markkinoille tulon jälkeisessä käytössä on raportoitu joitakin posteriorisen reversiibelin enkefalopatiaoireyhtymän (PRES) tapauksia.
PRES on neurologinen häiriö (vahvistetaan magneettikuvauksella), joka voi ilmetä päänsärkynä, hypertensiona, näköhäiriöinä, kouristuskohtauksina, letargiana, sekavuutena ja muina visuaalisina ja neurologisina häiriöinä, ja se voi johtaa kuolemaan. PRES-oireyhtymää on raportoitu muiden VEGF-estäjien käytön yhteydessä.
Nintedanibihoito on keskeytettävä, jos epäillään PRESiä. Ei tiedetä, voiko nintedanibihoidon aloittaa uudelleen potilaille, joilla on aiemmin ollut PRES, ja tämä on jätettävä lääkärin harkintaan.
Hypertensio
Ofev-hoito saattaa nostaa verenpainetta. Systeeminen verenpaine on mitattava määräajoin ja kliinisen tarpeen mukaan.
Keuhkoverenpainetauti
Ofev-valmisteen käytöstä keuhkoverenpainetautia sairastaville potilaille on vain vähän tietoja.
Potilaat, joilla on merkittävä keuhkoverenpainetauti (sydämen minuutti-indeksi ≤ 2 l/min/m² tai parenteraalinen epoprostenoli/treprostiniili tai merkittävä oikeanpuoleinen sydämen vajaatoiminta), suljettiin pois INBUILD‑ ja SENSCIS-tutkimuksista.
Ofev-valmistetta ei pidä käyttää potilaille, joilla on vaikea keuhkoverenpainetauti. Tarkkaa seurantaa suositellaan potilaille, joilla on lievä tai keskivaikea keuhkoverenpainetauti.
Haavan paranemiseen liittyvät komplikaatiot
Kliinisissä tutkimuksissa haavojen paranemisen heikentymisen ei havaittu lisääntyneen. Nintedanibi saattaa vaikutusmekanisminsa takia heikentää haavojen paranemista. Erityisiä tutkimuksia nintedanibin vaikutuksesta haavan paranemiseen ei tehty. Ofev-hoito voidaan siksi aloittaa tai hoitoa voidaan jatkaa (jos se on keskeytetty leikkaustoimenpiteen vuoksi) ainoastaan, kun haavojen on kliinisen arvion perusteella todettu parantuneen riittävällä tavalla.
Samanaikainen anto pirfenidonin kanssa
Nintedanibin ja pirfenidonin samanaikaista käyttöä tutkittiin asiaa nimenomaisesti koskeneessa farmakokineettisessä tutkimuksessa idiopaattista keuhkofibroosia sairastavilla potilailla. Näistä tuloksista ei saatu näyttöä siitä, että nintedanibin ja pirfenidonin välillä olisi mitään oleellisia farmakokineettisiä lääkeyhteisvaikutuksia, kun näitä lääkeaineita käytetään samanaikaisesti (ks. kohta Farmakokinetiikka). Molempien lääkevalmisteiden turvallisuusprofiilit ovat samankaltaiset, joten additiivisia haittavaikutuksia, maha-suolikanavan haittatapahtumat ja maksaan kohdistuvat haittatapahtumat mukaan lukien, voi olla odotettavissa. Pirfenidonin samanaikaisen käytön hyöty-riskisuhdetta ei ole vahvistettu.
Vaikutus QT‑aikaan
Nintedanibin kliinisessä tutkimusohjelmassa ei havaittu näyttöä QT‑ajan pitenemisestä (kohta Farmakodynamiikka). Koska joidenkin tyrosiinikinaasin estäjien tiedetään vaikuttavan QT‑aikaan, varovaisuutta on noudatettava annettaessa nintedanibia potilaille, joilla QTc‑aika saattaa pidentyä.
Allerginen reaktio
Soijaa sisältävien ravintovalmisteiden tiedetään aiheuttavan soija-allergisille henkilöille allergisia reaktioita, mukaan lukien vakavaa anafylaksiaa (ks. kohta Vasta-aiheet). Maapähkinäproteiinille allergisilla henkilöillä soijavalmisteiden aiheuttamien vaikeiden reaktioiden riski on suurentunut.
Pediatriset potilaat
Nintedanibin käytöstä pediatrisilla potilailla on vain rajatusti tietoa fibrotisoivia interstitiaalisia keuhkosairauksia sairastavien alajoukossa (ks. kohta Farmakodynamiikka). Tämä alajoukko ei kata kaikkia pediatrisilla potilailla fibrotisoivia intestitiaalisia keuhkosairauksia aiheuttavia tekijöitä.
Hoidon hyötyihin liittyy pediatrisilla potilailla suurempi epävarmuus kuin aikuisilla potilailla.
Aikuispotilaille yllä ilmoitettuja varotoimia on noudatettava myös pediatristen potilaiden hoidossa.
Katso erilliset pediatristen potilaiden annoksen pienentämissuositukset taulukosta 1.
Pediatristen potilaiden erityisominaisuudet on selitetty alla.
Luun kehitys ja kasvu
Prekliinisissä tutkimuksissa (ks. kohta Prekliiniset tiedot turvallisuudesta) havaittiin palautuvia kasvulevyn muutoksia. Pediatrisessa kliinisessä tutkimuksessa nintedanibihoidon aikana ei todettu merkittävää kasvun hidastumista. Pediatrisista potilaista ei kuitenkaan ole saatavilla pitkäaikaisturvallisuutta koskevia tietoja.
Kasvua on seurattava säännöllisesti ja kasvulevyjen muutoksien arviointia luiden kuvantamisella suositellaan vuosittain potilaille, joilla on vielä avoimia epifyysejä. Hoidon keskeyttämistä on harkittava, jos potilaalle kehittyy merkkejä kasvun heikentymisestä tai kasvulevyjen muutoksista.
Hampaiden kehityshäiriöt
Prekliinisissä tutkimuksissa (ks. kohta Prekliiniset tiedot turvallisuudesta) havaittiin hampaiden kehityshäiriöitä. Pediatrisessa kliinisessä tutkimuksessa hampaiden kehityshäiriöiden riskiä ei vahvistettu.
Varotoimena säännöllinen suun ja hampaiden tutkimus on tehtävä vähintään 6 kuukauden välein, kunnes hampaisto on täysin kehittynyt.
Yhteisvaikutukset
P‑glykoproteiini (P‑gp)
Nintedanibi on P‑gp:n substraatti (ks. kohta Farmakokinetiikka). Anto yhdessä voimakkaan P‑gp:n estäjän ketokonatsolin kanssa lisäsi nintedanibialtistuksen AUC‑arvon perusteella 1,61‑kertaiseksi ja Cmax-arvon perusteella 1,83‑kertaiseksi erityisessä lääkkeiden yhteisvaikutuksia arvioineessa tutkimuksessa. Lääkkeiden yhteisvaikutustutkimuksessa voimakkaan P‑gp:n indusoijan rifampisiinin samanaikainen käyttö vähensi nintedanibialtistuksen AUC‑arvon perusteella 50,3 %:iin ja Cmax-arvon perusteella 60,3 %:iin pelkkään nintedanibiin verrattuna. Jos voimakkaita P‑gp:n estäjiä (esim. ketokonatsolia, erytromysiiniä tai siklosporiinia) annetaan samanaikaisesti Ofev‑valmisteen kanssa, nintedanibialtistus saattaa kasvaa. Tällaisissa tapauksissa potilasta on seurattava tarkasti nintedanibin siedettävyyden suhteen. Ofev-hoidon keskeyttäminen, annoksen pienentäminen tai hoidon lopettaminen voi olla välttämätöntä haittavaikutusten hoitamiseksi (ks. kohta Annostus ja antotapa).
Voimakkaat P‑gp:n indusoijat (esim. rifampisiini, karbamatsepiini, fenytoiini ja mäkikuisma) saattavat vähentää nintedanibialtistusta. Samanaikaisessa käytössä on harkittava jonkun muun lääkevalmisteen käyttöä, jonka mahdollinen P‑gp:tä indusoiva vaikutus on hyvin pieni tai sitä ei ole lainkaan.
CYP‑entsyymit
Vain pieni osuus nintedanibin biotransformaatiosta tapahtuu CYP‑välitteisesti. Nintedanibi ja sen metaboliitit, vapaa happo-osa BIBF 1202 ja sen glukuronidimuoto BIBF 1202 -glukuronidi, eivät estäneet eivätkä indusoineet CYP‑entsyymejä prekliinisissä tutkimuksissa (ks. kohta Farmakokinetiikka). Näin ollen nintedanibin ja muiden lääkkeiden CYP-metaboliaan perustuvien yhteisvaikutusten todennäköisyys on pieni.
Samanaikainen anto muiden lääkevalmisteiden kanssa
Nintedanibin ja suun kautta otettavien hormonaalisten ehkäisyvalmisteiden samanaikainen käyttö ei muuttanut suun kautta otettavien hormonaalisten ehkäisyvalmisteiden farmakokinetiikkaa oleellisesti (ks. kohta Farmakokinetiikka).
Nintedanibin samanaikainen anto bosentaanin kanssa ei muuttanut nintedanibin farmakokinetiikkaa (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Naiset, jotka voivat tulla raskaaksi / Raskauden ehkäisy
Nintedanibi voi aiheuttaa haittaa ihmisen sikiölle (ks. kohta Prekliiniset tiedot turvallisuudesta). Naisia, jotka voivat tulla raskaaksi, on neuvottava välttämään raskautta Ofev‑hoidon aikana ja käyttämään erittäin tehokkaita ehkäisymenetelmiä hoitoa aloitettaessa, hoidon aikana sekä vähintään kolme kuukautta viimeisen Ofev-annoksen jälkeen. Nintedanibi ei vaikuta oleellisesti etinyyliestradiolin ja levonorgestreelin pitoisuuksiin plasmassa (ks. kohta Farmakokinetiikka). Oksentelu ja/tai ripuli tai muut imeytymiseen mahdollisesti vaikuttavat sairaudet saattavat heikentää suun kautta otettavien hormonaalisten ehkäisyvalmisteiden tehoa. Jos tällaisia sairaustiloja esiintyy suun kautta otettavia hormonaalisia ehkäisyvalmisteita käyttävillä naisilla, heidän tulisi käyttää jotakin muuta erittäin tehokasta ehkäisymenetelmää.
Raskaus
Ofev-valmisteen käytöstä raskaana oleville naisille ei ole tietoa, mutta prekliinisissä eläimillä tehdyissä tutkimuksissa vaikuttavalla aineella on havaittu lisääntymistoksisuutta (ks. kohta Prekliiniset tiedot turvallisuudesta). Koska nintedanibi voi aiheuttaa haittaa myös ihmissikiölle, sitä ei pidä käyttää raskauden aikana (ks. kohta Vasta-aiheet), ja naiselle on tehtävä raskaustesti ennen Ofev-hoitoa ja hoidon aikana, jos tarpeen.
Naispotilaita on neuvottava kertomaan lääkärille tai apteekkihenkilökunnalle, jos he tulevat raskaaksi Ofev‑hoidon aikana.
Jos potilas tulee raskaaksi Ofev‑hoidon aikana, hoito on lopetettava ja potilaalle on kerrottava sikiöön mahdollisesti kohdistuvista riskeistä.
Imetys
Ei tiedetä, erittyvätkö nintedanibi ja sen metaboliitit ihmisillä äidinmaitoon.
Prekliiniset tutkimukset osoittivat, että pieni määrä nintedanibia ja sen metaboliitteja (≤ 0,5 % annetusta annoksesta) erittyi maitoon imettävillä rotilla. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. Imetys on lopetettava Ofev‑hoidon ajaksi.
Hedelmällisyys
Prekliinisten tutkimusten perusteella ei ole näyttöä miesten hedelmällisyyden heikentymisestä (ks. kohta Prekliiniset tiedot turvallisuudesta). Subkroonista ja kroonista toksisuutta koskevien tutkimusten perusteella naarasrottien hedelmällisyys ei heikentynyt, kun systeeminen altistus oli verrattavissa ihmisen suurimmalla suositusannoksella (150 mg kahdesti vuorokaudessa) aiheutuvaan altistukseen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Ofev‑valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaita on neuvottava olemaan varovaisia ajaessaan autoa tai käyttäessään koneita Ofev‑hoidon aikana.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Kliinisissä tutkimuksissa ja myyntiluvan myöntämisen jälkeen saadun kokemuksen perusteella nintedanibihoidon yhteydessä yleisimmin raportoituja haittavaikutuksia olivat ripuli, pahoinvointi ja oksentelu, vatsakipu, ruokahalun heikentyminen, painon lasku ja maksaentsyymiarvojen nousu.
Tietoa valikoitujen haittavaikutusten hoidosta, ks. kohta Varoitukset ja käyttöön liittyvät varotoimet.
Haittavaikutustaulukko
Taulukossa 2 on yhteenveto lääkehaittavaikutuksista MedDRA:n elinjärjestelmäluokittain ja esiintymistiheyden mukaan seuraavasti:
hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 2: Yhteenveto haittavaikutuksista esiintymistiheyden mukaan
| Esiintyvyys | |||
Elinjärjestelmäluokka suositeltu termi | Idiopaattinen keuhkofibroosi | Muut krooniset, fenotyypiltään etenevät fibrotisoivat interstitiaaliset keuhkosairaudet | Systeemiseen skleroosiiin liittyvä interstitiaalinen keuhkosairaus |
| Veri ja imukudos | |||
| Trombosytopenia | Melko harvinainen | Melko harvinainen | Melko harvinainen |
| Aineenvaihdunta ja ravitsemus | |||
| Painon lasku | Yleinen | Yleinen | Yleinen |
| Ruokahalun heikentyminen | Yleinen | Hyvin yleinen | Yleinen |
| Nestehukka | Melko harvinainen | Melko harvinainen | Tuntematon |
| Hermosto | |||
| Päänsärky | Yleinen | Yleinen | Yleinen |
| Posteriorinen reversiibeli enkefalopatiaoireyhtymä | Tuntematon | Tuntematon | Tuntematon |
| Sydän | |||
| Sydäninfarkti | Melko harvinainen | Melko harvinainen | Tuntematon |
| Verisuonisto | |||
| Verenvuoto (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Yleinen | Yleinen | Yleinen |
| Hypertensio | Melko harvinainen | Yleinen | Yleinen |
| Aneurysmat ja valtimon dissekaatiot | Tuntematon | Tuntematon | Tuntematon |
| Ruoansulatuselimistö | |||
| Ripuli | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| Pahoinvointi | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| Vatsakipu | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| Oksentelu | Yleinen | Hyvin yleinen | Hyvin yleinen |
| Haimatulehdus | Melko harvinainen | Melko harvinainen | Tuntematon |
| Koliitti | Melko harvinainen | Melko harvinainen | Melko harvinainen |
| Maksa ja sappi | |||
| Lääkeaineen aiheuttama maksavaurio | Melko harvinainen | Yleinen | Melko harvinainen |
| Maksaentsyymipitoisuuksien kohoaminen | Hyvin yleinen | Hyvin yleinen | Hyvin yleinen |
| Kohonnut alaniiniaminotransferaasi (ALAT) | Yleinen | Hyvin yleinen | Yleinen |
| Kohonnut aspartaattiaminotransferaasi (ASAT) | Yleinen | Yleinen | Yleinen |
| Kohonnut gammaglutamyylitransferaasi (GT) | Yleinen | Yleinen | Yleinen |
| Hyperbilirubinemia | Melko harvinainen | Melko harvinainen | Tuntematon |
| Kohonnut veren alkalinen fosfataasi (AFOS) | Melko harvinainen | Yleinen | Yleinen |
| Iho ja ihonalainen kudos | |||
| Ihottuma | Yleinen | Yleinen | Melko harvinainen |
| Kutina | Melko harvinainen | Melko harvinainen | Melko harvinainen |
| Alopesia | Melko harvinainen | Melko harvinainen | Tuntematon |
| Munuaiset ja virtsatiet | |||
| Munuaisten vajaatoiminta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) | Tuntematon | Tuntematon | Melko harvinainen |
| Proteinuria | Melko harvinainen | Melko harvinainen | Tuntematon |
Valikoitujen haittavaikutusten kuvaus
Ripuli
Kliinisissä tutkimuksissa (ks. kohta Farmakodynamiikka) ripuli oli yleisin raportoitu maha-suolikanavan tapahtuma. Useimmilla potilailla se oli voimakkuudeltaan lievä tai kohtalainen. Useampi kuin kaksi kolmesta ripulioireita saaneesta potilaasta kertoi oireiden alkaneen jo kolmen ensimmäisen hoitokuukauden aikana. Useimmilla potilailla ripuli hoidettiin ripulilääkkeillä, pienentämällä annosta tai keskeyttämällä hoito (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Taulukossa 3 esitetään yhteenveto kliinisissä tutkimuksissa raportoiduista ripulitapahtumista:
Taulukko 3: Ripulin esiintyminen kliinisissä tutkimuksissa 52 viikon aikana
| INPULSIS | INBUILD | SENSCIS | ||||
| Lumelääke | Ofev | Lumelääke | Ofev | Lumelääke | Ofev | |
| Ripuli | 18,4 % | 62,4 % | 23,9 % | 66,9 % | 31,6 % | 75,7 % |
| Vaikea ripuli | 0,5 % | 3,3 % | 0,9 % | 2,4 % | 1,0 % | 4,2 % |
| Ofev-annoksen pienentämiseen johtanut ripuli | 0 % | 10,7 % | 0,9 % | 16,0 % | 1,0 % | 22,2 % |
| Ofev-hoidon lopettamiseen johtanut ripuli | 0,2 % | 4,4 % | 0,3 % | 5,7 % | 0,3 % | 6,9 % |
Maksaentsyymipitoisuuksienkohoaminen
INPULSIS-tutkimuksissa maksaentsyymipitoisuuksien kohoamista (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet) raportoitiin 13,6 %:lla Ofev-hoitoa saaneista potilaista ja 2,6 %:lla lumehoitoa saaneista potilaista. INBUILD-tutkimuksessa maksaentsyymipitoisuuksien kohoamista raportoitiin 22,6 %:lla Ofev-hoitoa saaneista potilaista ja 5,7 %:lla lumehoitoa saaneista potilaista. SENSCIS-tutkimuksessa maksaentsyymipitoisuuksien kohoamista raportoitiin 13,2 %:lla Ofev-hoitoa saaneista potilaista ja 3,1 %:lla lumehoitoa saaneista potilaista. Maksaentsyymipitoisuuksien kohoaminen oli palautuvaa, eikä siihen liittynyt kliinisesti ilmeistä maksasairautta.
Lisätietoa erityisryhmistä, ripulia ja maksaentsyymipitoisuuksien kohoamista koskevista suositelluista toimenpiteistä ja annosmuutoksista, ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Annostus ja antotapa.
Verenvuoto
Kliinisissä tutkimuksissa verenvuodon yleisyys oli joko hieman suurempi Ofev-hoitoa saaneilla potilailla tai samaa luokkaa molemmissa hoitoryhmissä (INPULSIS-tutkimuksessa Ofev 10,3 % vs. lumelääke 7,8 %; INBUILD-tutkimuksessa Ofev 11,1 % vs. lumelääke 12,7 %; SENSCIS-tutkimuksessa Ofev 11,1 % vs. lumelääke 8,3 %). Ei-vakava nenäverenvuoto oli yleisin raportoitu verenvuototapahtuma. Vakavien verenvuototapahtumien yleisyys oli vähäistä molemmissa hoitoryhmissä (INPULSIS-tutkimuksessa Ofev 1,3 % vs. lumelääke 1,4 %; INBUILD-tutkimuksessa Ofev 0,9 % vs. lumelääke 1,5 %; SENSCIS-tutkimuksessa Ofev 1,4 % vs. lumelääke 0,7 %).
Myyntiluvan myöntämisen jälkeen raportoituja verenvuototapahtumia ovat olleet esimerkiksi maha-suolikanavan, hengityselinten ja keskushermoston verenvuodot, joista maha-suolikanavan verenvuodot ovat olleet yleisimpiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Proteinuria
Kliinisissä tutkimuksissa proteinurian yleisyys oli vähäistä ja samaa luokkaa molemmissa hoitoryhmissä (INPULSIS-tutkimuksessa Ofev 0,8 % vs. lumelääke 0,5 %; INBUILD-tutkimuksessa Ofev 1,5 % vs. lumelääke 1,8 %; SENSCIS-tutkimuksessa Ofev 1,0 % vs. lumelääke 0,0 %). Kliinisissä tutkimuksissa ei ole raportoitu nefroottista oireyhtymää. Hyvin harvoja nefroottisen proteinurian tapauksia, joihin liittyi tai ei liittynyt munuaisten vajaatoimintaa, on raportoitu valmisteen markkinoille tulon jälkeen. Yksittäisissä tapauksissa histologiset löydökset sopivat glomerulaariseen mikroangiopatiaan, johon liittyi tai ei liittynyt munuaisten veritulppia. Oireiden korjaantumista on todettu Ofev-hoidon lopettamisen jälkeen, mutta joissakin tapauksissa proteinuria ei korjaantunut täysin. Hoidon keskeyttämistä on harkittava, jos potilaalle kehittyy nefroottisen oireyhtymän merkkejä tai oireita (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Pediatriset potilaat
Nintedanibin turvallisuudesta pediatrisilla potilailla on vain vähän tietoa.
Yhteensä 39 iältään 6–17‑vuotiasta potilasta sai hoitoa satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa, 24 viikon pituisessa tutkimuksessa, jonka jälkeen nintedanibihoitoa annettiin avoimesti vaihtelevan pituisia aikoja (ks. kohta Farmakodynamiikka). Idiopaattista keuhkofibroosia, muita kroonisia, fenotyypiltään eteneviä fibrotisoivia interstitiaalisia keuhkosairauksia ja systeemiseen skleroosiin liittyvää interstitiaalista keuhkosairautta sairastavilla aikuispotilailla todettua turvallisuusprofiilia vastaavasti yleisimpiä nintedanibihoidon yhteydessä raportoituja haittavaikutuksia lumekontrolloidun jakson aikana olivat ripuli (38,5 %), oksentelu (26,9 %), pahoinvointi (19,2 %), vatsakipu (19,2 %) ja päänsärky (11,5 %).
Nintedanibihoidon yhteydessä raportoituja maksan ja sapen häiriöitä lumekontrolloidun jakson aikana olivat maksavaurio (3,8 %) ja maksan toimintakoearvojen nousu (3,8 %). Vähäisten tietojen vuoksi ei tiedetä varmasti, onko lääkkeen aiheuttaman maksavaurion riski samaa luokkaa lapsilla ja aikuisilla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Prekliinisten löydösten perusteella pediatrisessa kliinisessä tutkimuksessa seurattiin luustoa, kasvua ja hampaiden kehitystä (ks. kohdat Annostus ja antotapa, Varoitukset ja käyttöön liittyvät varotoimet ja Prekliiniset tiedot turvallisuudesta) mahdollisina riskeinä.
Niiden potilaiden prosenttiosuus, joilla esiintyi hoidon aikana ilmenneitä epifyysilevyjen patologisia löydöksiä. Esiintyvyydet olivat hoitoryhmissä samaa luokkaa viikolla 24 (7,7 % molemmissa hoitoryhmissä). Viikkoon 52 mennessä niiden potilaiden prosenttiosuudet, joilla patologisia löydöksiä esiintyi, olivat nintedanibi/nintedanibi-ryhmässä 11,5 % ja lumelääke/nintedanibi-ryhmässä 15,4 %.
Niiden potilaiden prosenttiosuus, joilla esiintyi hoidon aikana ilmenneitä patologisia löydöksiä hampaiden tutkimuksessa tai kuvantamisessa. Esiintyvyydet olivat 46,2 % nintedanibiryhmässä ja 38,5 % lumeryhmässä viikkoon 24 mennessä. Viikkoon 52 mennessä niiden potilaiden prosenttiosuudet, joilla patologisia löydöksiä esiintyi, olivat nintedanibi/nintedanibi-ryhmässä 50,0 % ja lumelääke/nintedanibi-ryhmässä 46,2 %.
Pediatrisista potilaista ei ole saatavilla pitkäaikaisturvallisuutta koskevia tietoja. Kasvuun, hampaiden kehitykseen ja murrosikään kohdistuvaan vaikutukseen ja maksavaurion riskiin liittyy epävarmuustekijöitä.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty‑haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www‐sivusto: www.fimea.fi
Lääkealan turvallisuus‐ ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Ofev‑valmisteen yliannostukseen ei ole olemassa erityistä vastalääkettä eikä hoitoa. Onkologisessa tutkimusohjelmassa kaksi potilasta sai yliannostuksen, joka oli korkeintaan 600 mg kahdesti vuorokaudessa enintään kahdeksan vuorokauden ajan. Havaitut haittavaikutukset (maksaentsyymiarvojen kohoaminen ja maha-suolikanavan oireet) olivat yhdenmukaisia nintedanibin tunnetun turvallisuusprofiilin kanssa. Kumpikin potilas toipui näistä haittavaikutuksista. INPULSIS‑tutkimuksissa yksi potilas altistui tahattomasti 600 mg/vrk annokselle yhteensä 21 vuorokauden ajan. Yksi ei-vakava haittatapahtuma (nenänielutulehdus) ilmaantui ja hävisi väärän annostuksen käytön aikana. Muita tapahtumia ei raportoitu. Yliannostustilanteessa hoito on keskeytettävä ja yleisiin tukitoimenpiteisiin on ryhdyttävä tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Muut syöpälääkkeet, proteiinikinaasin estäjät, ATC‑koodi: L01EX09
Vaikutusmekanismi
Nintedanibi on pienimolekyylinen tyrosiinikinaasin estäjä, mukaan lukien seuraavat reseptorit: verihiutalekasvutekijän reseptori (PDGFR) α ja ß, fibroblastikasvutekijän reseptori (FGFR) 1–3 ja VEGFR 1–3. Lisäksi nintedanibi estää imusoluspesifistä tyrosiiniproteiinikinaasia (Lck), tyrosiiniproteiinikinaasia (Lyn), esisyöpägeenin tyrosiiniproteiinikinaasia (Src) ja kantasoluryhmiä stimuloivan kasvutekijän 1 reseptorin (CSF1R) kinaasia. Nintedanibi sitoutuu kilpailevasti näiden kinaasien adenosiinitrifosfaattia (ATP) sitovaan taskuun ja estää solunsisäisiä signaalikaskadeja, joiden on osoitettu olevan osallisina interstitiaalisissa keuhkosairauksissa fibroottisen kudoksen uudelleenmuotoutumisen patogeneesissä.
Farmakodynaamiset vaikutukset
In vitro -tutkimuksissa, joissa on käytetty ihmissoluja, nintedanibin on osoitettu estävän prosesseja, joiden oletetaan osallistuvan fibroottisen patogeneesin käynnistämiseen, pro-fibroottisten välittäjäaineiden vapautumiseen ääreisverenkierron monosyyttisoluista ja makrofagien polarisaatioon vaihtoehtoisesti aktivoituneiksi makrofageiksi. Nintedanibin on osoitettu estävän elinten fibrooseissa keskeisiä prosesseja, fibroblastien proliferaatiota, migraatiota ja muuntumista aktiiviseksi myofibroblastifenotyypiksi sekä soluväliaineen sekreetiota. Eläimillä tehdyistä tutkimuksista saaduissa idiopaattisen keuhkofibroosin (IPF), systeemisen skleroosin ja systeemiseen skleroosiin liittyvän interstitiaalisen keuhkosairauden (SSc/SSc‑ILD), nivelreumaan liittyvän interstitiaalisen keuhkosairauden (RA-ILD) ja muiden elinten fibroosien malleissa nintedanibi on osoittanut anti-inflammatorisia ja antifibroottisia vaikutuksia keuhkoissa, iholla, sydämessä, munuaisissa ja maksassa. Nintedanibi osoitti myös verisuoniaktiivisuutta. Se vähensi dermaalisten pienten verisuonten endoteelisolujen apoptoosia ja heikensi keuhkoverisuonten uudelleenmuotoutumista vähentämällä verisuonten sileiden lihasten solujen proliferaatiota, keuhkoverisuonten seinämien paksuutta ja tukkeutuneiden keuhkoverisuonten prosentuaalista osuutta.
Kliininen teho ja turvallisuus
Idiopaattinen keuhkofibroosi (IPF)
Nintedanibin kliinistä tehoa on tutkittu idiopaattista keuhkofibroosia sairastavilla potilailla kahdessa vaiheen III satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa tutkimuksessa, joiden tutkimusasetelmat olivat samanlaiset (INPULSIS‑1 (1199.32) ja INPULSIS‑2 (1199.34)). Potilaat, joilla nopea vitaalikapasiteetti (FVC) oli lähtötilanteessa < 50 % odotusarvosta tai hiilimonoksidin diffuusiokapasiteetti (DLCO, hemoglobiinikorjattu) oli lähtötilanteessa < 30 % odotusarvosta, suljettiin pois tutkimuksesta. Potilaat satunnaistettiin suhteessa 3:2 saamaan Ofev 150 mg -valmistetta tai lumetta kahdesti vuorokaudessa 52 viikon ajan.
Ensisijainen päätetapahtuma oli FVC:n vuosittainen alenema. Keskeisiä toissijaisia päätetapahtumia olivat Saint George's Respiratory Questionnaire (SGRQ) ‑kokonaispistemäärän muutos lähtötilanteesta 52 viikon kohdalla ja idiopaattisen keuhkofibroosin ensimmäiseen äkilliseen pahenemisvaiheeseen kulunut aika.
FVC:n vuosittainen alenema
FVC:n vuosittainen alenema (ml) oli nintedanibia saaneilla potilailla merkitsevästi pienempi verrattuna lumetta saaneisiin potilaisiin. Hoidon vaikutus oli yhdenmukainen molemmissa tutkimuksissa. Tutkimuskohtaiset ja yhdistetyt tulokset, ks. taulukko 4.
Taulukko 4: FVC:n vuosittainen alenema (ml) INPULSIS‑1- ja INPULSIS‑2 -tutkimuksissa sekä tutkimusten yhdistetyt tulokset – hoidetut potilaat
| INPULSIS‑1 | INPULSIS‑2 | INPULSIS‑1 ja INPULSIS‑2 yhdistetty | ||||
| Lume | Ofev 150 mg kahdesti vuorokaudessa | Lume | Ofev 150 mg kahdesti vuorokaudessa | Lume | Ofev 150 mg kahdesti vuorokaudessa | |
| Analysoitujen potilaiden lukumäärä | 204 | 309 | 219 | 329 | 423 | 638 |
| Alenema1 (keskivirhe) 52 viikon aikana | ‑239,9 (18,71) | ‑114,7 (15,33) | ‑207,3 (19,31) | ‑113,6 (15,73) | ‑223,5 (13,45) | ‑113,6 (10,98) |
| Vertailu lumeeseen | ||||||
| Ero1 | 125,3 | 93,7 | 109,9 | |||
| 95 % lv | (77,7; 172,8) | (44,8; 142,7) | (75,9; 144,0) | |||
| p‑arvo | < 0,0001 | 0,0002 | < 0,0001 | |||
1 Arvio perustuu satunnaiskerroinregressiomalliin. lv: luottamusväli | ||||||
Herkkyysanalyysissä, jossa oletettiin, että FVC:n alenema viimeisen havaitun arvon jälkeen potilailla, joilta puuttui tietoja viikolla 52, olisi samankaltainen kuin kaikilla lumeryhmän potilailla, korjattu ero vuosittaisessa alenemassa oli nintedanibi- ja lumeryhmän välillä 113,9 ml/vuosi (95 % lv 69,2; 158,5) INPULSIS‑1‑tutkimuksessa ja 83,3 ml/vuosi (95 % lv 37,6; 129,0) INPULSIS‑2‑tutkimuksessa.
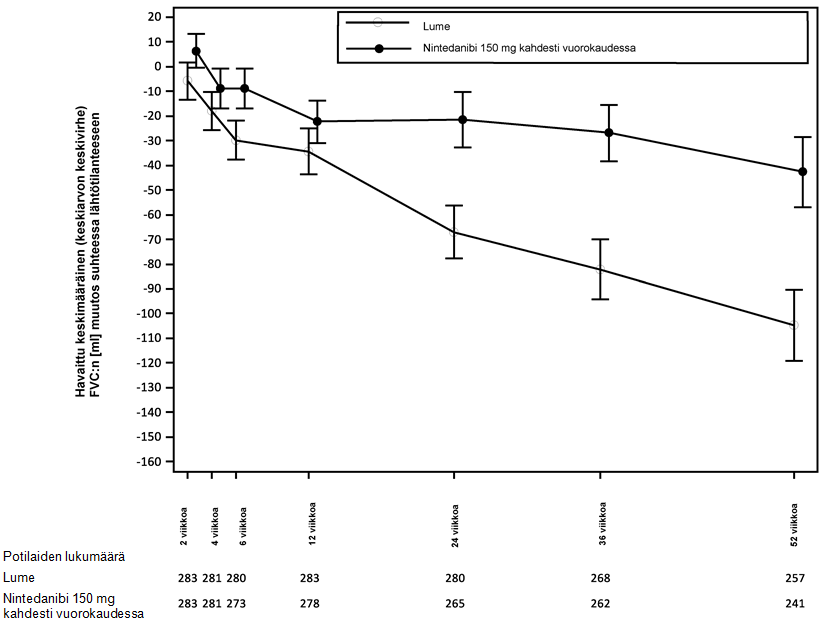
Muutoksen kehittyminen suhteessa lähtötilanteeseen ja aikaan kummassakin hoitoryhmässä (INPULSIS‑1- ja INPULSIS‑2-tutkimusten yhdistetty analyysi), ks. kuva 1.
Kuva 1: Havaitut keskimääräiset (keskiarvon keskivirhe) FVC‑muutokset suhteessa lähtötilanteeseen (ml) eri ajankohtina, yhdistetyt INPULSIS‑1‑ ja INPULSIS‑2-tutkimukset
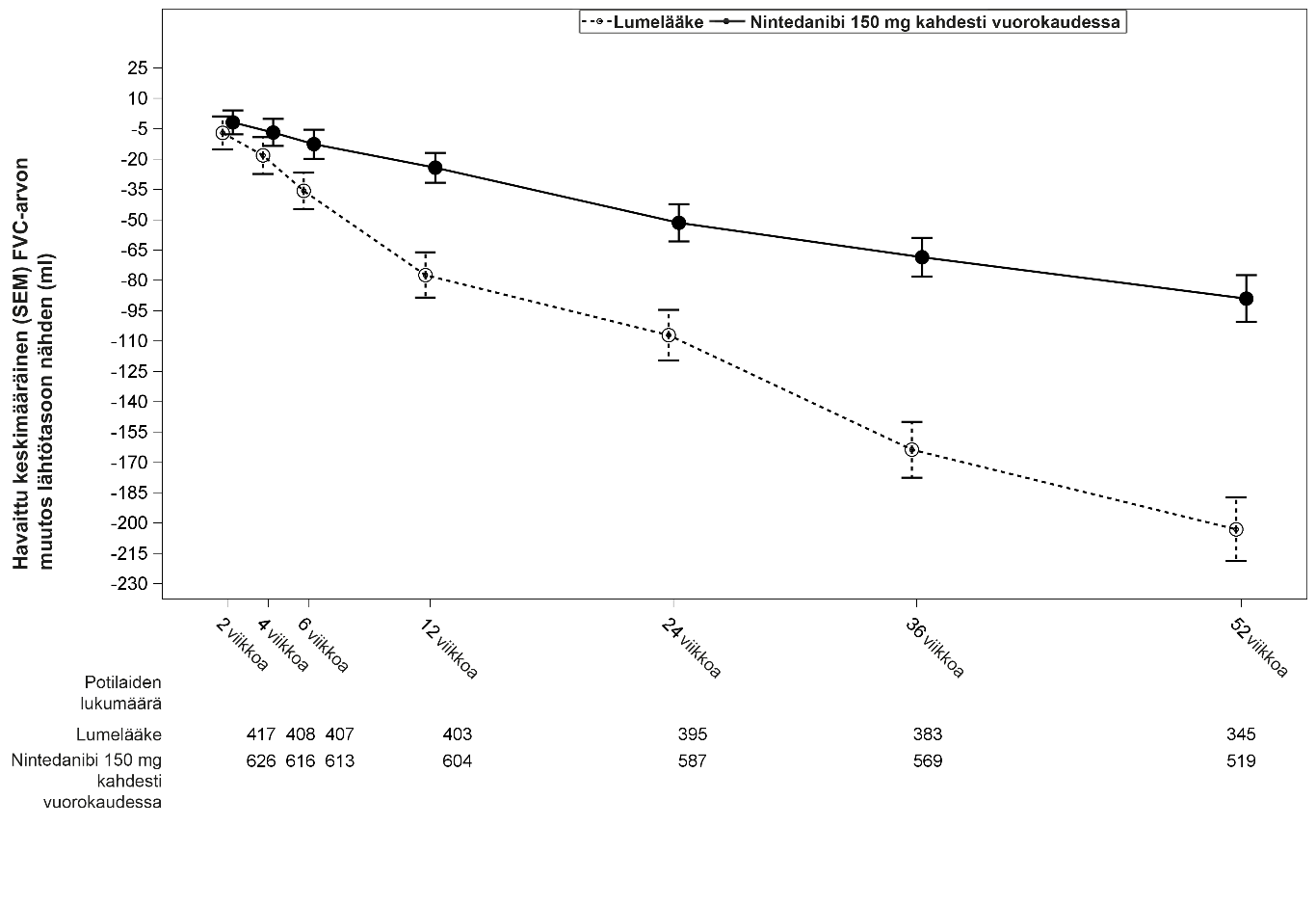
Potilaiden lukumäärä
Lume
Nintedanibi 150 mg kahdesti vuorokaudessa
Havaittu keskimääräinen (keskiarvon keskivirhe) FVC-muutos suhteessa lähtötilanteeseen (ml)
FVC ‑ vasteanalyysi
Molemmissa INPULSIS‑tutkimuksissa FVC‑vasteen saavuttaneiden osuus oli nintedanibiryhmässä merkitsevästi suurempi kuin lumeryhmässä. Vasteen saavuttaneiksi määriteltiin potilaat, joiden FVC:n prosenttiosuus odotusarvosta pieneni absoluuttisesti enintään 5 % (raja-arvo, joka merkitsee kuolleisuusriskin suurenemista idiopaattisessa keuhkofibroosissa). Tulokset olivat samaa luokkaa analyyseissä, joissa käytettiin varovaisempaa raja-arvoa (10 %). Tutkimuskohtaiset ja yhdistetyt tulokset, ks. taulukko 5.
Taulukko 5: FVC‑vasteen saavuttaneiden potilaiden osuus 52 viikon kohdalla INPULSIS‑1- ja INPULSIS‑2-tutkimuksissa sekä tutkimusten yhdistetyt tulokset – hoidetut potilaat
| INPULSIS‑1 | INPULSIS‑2 | INPULSIS‑1 ja INPULSIS‑2 yhdistetty | ||||
| Lume | Ofev 150 mg kahdesti vuorokaudessa | Lume | Ofev 150 mg kahdesti vuorokaudessa | Lume | Ofev 150 mg kahdesti vuorokaudessa | |
| Analysoitujen potilaiden lukumäärä | 204 | 309 | 219 | 329 | 423 | 638 |
| 5 %:n raja-arvo | ||||||
| FVC‑vasteen saavuttaneiden lukumäärä ja osuus (%)1 | 78 (38,2) | 163 (52,8) | 86 (39,3) | 175 (53,2) | 164 (38,8) | 338 (53,0) |
| Vertailu lumeeseen | ||||||
| Ristitulosuhde | 1,85 | 1,79 | 1,84 | |||
| 95 % lv | (1,28; 2,66) | (1,26; 2,55) | (1,43; 2,36) | |||
| p‑arvo2 | 0,0010 | 0,0011 | < 0,0001 | |||
| 10 %:n raja-arvo | ||||||
| FVC‑vasteen saavuttaneiden lukumäärä ja osuus (%)1 | 116 (56,9) | 218 (70,6) | 140 (63,9) | 229 (69,6) | 256 (60,5) | 447 (70,1) |
| Vertailu lumeeseen | ||||||
| Ristitulosuhde | 1,91 | 1,29 | 1,58 | |||
| 95 % lv | (1,32; 2,79) | (0,89; 1,86) | (1,21; 2,05) | |||
| p‑arvo2 | 0,0007 | 0,1833 | 0,0007 | |||
1 Vasteen saavuttaneita ovat potilaat, joilla absoluuttinen alenema FVC‑arvon prosenttiosuudessa odotusarvosta oli raja-arvosta riippuen enintään 5 % tai enintään 10 % ja joilla FVC arvioitiin 52 viikon kohdalla.
2 Perustuu logistiseen regressioon.
Etenemiseen kulunut aika (≥ 10 %:n absoluuttinen pieneneminen FVC:n prosenttiosuudessa odotusarvosta tai kuolema)
Etenemisen riski oli kummassakin INPULSIS‑tutkimuksessa tilastollisesti merkitsevästi pienempi nintedanibilla hoidetuilla potilailla verrattuna lumeella hoidettuihin potilaisiin. Yhdistetyssä analyysissä riskisuhde oli 0,60 viitaten 40 % pienempään etenemisriskiin nintedanibilla hoidetuilla potilailla verrattuna lumeella hoidettuihin potilaisiin.
Taulukko 6: Niiden potilaiden määrä, joilla absoluuttinen pieneneminen FVC:n prosenttiosuudessa odotusarvosta oli ≥ 10 % tai jotka menehtyivät 52 viikon aikana, ja aika etenemiseen INPULSIS‑1‑ ja INPULSIS‑2‑tutkimuksissa sekä tutkimusten yhdistetyt tulokset – hoidetut potilaat
| INPULSIS‑1 | INPULSIS‑2 | INPULSIS‑1 ja INPULSIS‑2 yhdistetty | ||||
| Lume | Ofev 150 mg kahdesti vuorokaudessa | Lume | Ofev 150 mg kahdesti vuorokaudessa | Lume | Ofev 150 mg kahdesti vuorokaudessa | |
| Riskiryhmään kuuluvien potilaiden määrä | 204 | 309 | 219 | 329 | 423 | 638 |
| Tapahtuman kokeneita potilaita, n (%) | 83 (40,7) | 75 (24,3) | 92 (42,0) | 98 (29,8) | 175 (41,4) | 173 (27,1) |
| Vertailu lumeeseen1 | ||||||
| p‑arvo2 | 0,0001 | 0,0054 | < 0,0001 | |||
| Riskisuhde3 | 0,53 | 0,67 | 0,60 | |||
| 95 % lv | (0,39; 0,72) | (0,51; 0,89) | (0,49; 0,74) | |||
1 Perustuu enintään 372 päivän aikana kerättyihin tietoihin (52 viikkoa + 7 päivän marginaali).
2 Perustuu log-rank -testiin.
3 Perustuu Coxin regressiomalliin.
SGRQ-kokonaispisteiden muutos lähtötilanteesta 52 viikon kohdalla
INPULSIS-tutkimusten yhdistetyssä analyysissä lähtötason SGRQ-pisteet olivat nintedanibiryhmässä 39,51 ja lumeryhmässä 39,58. SGRQ-kokonaispistemäärän arvioitu keskimuutos lähtötilanteesta 52 viikon kohdalla oli pienempi nintedanibiryhmässä (3,53) kuin lumeryhmässä (4,96). Hoitoryhmien välinen ero oli ‑1,43 (95 % lv: ‑3,09; 0,23; p = 0,0923). Kaikkiaan nintedanibin vaikutus terveyteen liittyvään elämänlaatuun oli SGRQ-kokonaispistein mitattuna kohtalainen. Elämänlaatu heikentyi vähemmän kuin lumehoidossa.
Aika ensimmäiseen äkilliseen idiopaattisen keuhkofibroosin pahenemisvaiheeseen
INPULSIS-tutkimusten yhdistetyssä analyysissä ensimmäisen äkillisen pahenemisvaiheen riski oli numeerisesti pienempi nintedanibia saaneilla potilailla kuin lumetta saaneilla potilailla. Tutkimuskohtaiset tulokset ja yhdistetyt tulokset, ks. taulukko 7.
Taulukko 7: Idiopaattisen keuhkofibroosin äkillisen pahenemisvaiheen kokeneiden potilaiden määrä 52 viikon aikana ja aika ensimmäiseen pahenemisvaiheeseen. Analyysi, joka perustuu tutkijan raportoimiin tapahtumiin INPULSIS‑1- ja INPULSIS‑2-tutkimuksissa sekä tutkimusten yhdistetyt tulokset – hoidetut potilaat
| INPULSIS‑1 | INPULSIS‑2 | INPULSIS‑1 ja INPULSIS‑2 yhdistetty | ||||
| Lume | Ofev 150 mg kahdesti vuorokaudessa | Lume | Ofev 150 mg kahdesti vuorokaudessa | Lume | Ofev 150 mg kahdesti vuorokaudessa | |
| Riskiryhmään kuuluvien potilaiden määrä | 204 | 309 | 219 | 329 | 423 | 638 |
| Tapahtuman kokeneita potilaita, n (%) | 11 (5,4) | 19 (6,1) | 21 (9,6) | 12 (3,6) | 32 (7,6) | 31 (4,9) |
| Vertailu lumeeseen1 | ||||||
| p‑arvo2 | 0,6728 | 0,0050 | 0,0823 | |||
| Riskisuhde3 | 1,15 | 0,38 | 0,64 | |||
| 95 % lv | (0,54; 2,42) | (0,19; 0,77) | (0,39; 1,05) | |||
1 Perustuu enintään 372 päivän aikana kerättyihin tietoihin (52 viikkoa + 7 päivän marginaali).
2 Perustuu log-rank -testiin.
3 Perustuu Coxin regressiomalliin.
Ennalta määritellyssä herkkyysanalyysissä vähintään yhden arvioidun pahenemisvaiheen 52 viikon aikana kokeneiden potilaiden määrä oli pienempi nintedanibiryhmässä (1,9 % potilaista) kuin lumeryhmässä (5,7 % potilaista). Kun arvioituun pahenemisvaihetapahtumaan kulunutta aikaa analysoitiin yhdistettyjen tietojen perusteella, riskisuhteeksi saatiin 0,32 (95 % lv 0,16; 0,65; p = 0,0010).
Elossaoloanalyysi
INPULSIS-tutkimusten elossaolotietojen ennalta määritellyssä yhdistetyssä analyysissä kokonaiskuolleisuus 52 viikon aikana oli nintedanibiryhmässä pienempi (5,5 %) kuin lumeryhmässä (7,8 %). Kuolemaan kuluneen ajan analyysissä riskisuhteeksi saatiin 0,70 (95 % lv 0,43, 1,12; p = 0,1399). Kaikkien elossaolopäätetapahtumien (kuten hoidonaikainen kuolleisuus ja hengityselimiin liittyvä kuolleisuus) tuloksissa oli yhdenmukainen numeerinen ero nintedanibin eduksi.
Taulukko 8: Mistä tahansa syystä johtuva kuolleisuus 52 viikon aikana INPULSIS‑1- ja INPULSIS‑2 -tutkimuksissa sekä tutkimusten yhdistetyt tulokset – hoidetut potilaat
| INPULSIS‑1 | INPULSIS‑2 | INPULSIS‑1 ja INPULSIS‑2 yhdistetty | ||||
| Lume | Ofev 150 mg kahdesti vuorokaudessa | Lume | Ofev 150 mg kahdesti vuorokaudessa | Lume | Ofev 150 mg kahdesti vuorokaudessa | |
| Riskiryhmään kuuluvien potilaiden määrä | 204 | 309 | 219 | 329 | 423 | 638 |
| Tapahtuman kokeneita potilaita, n (%) | 13 (6,4) | 13 (4,2) | 20 (9,1) | 22 (6,7) | 33 (7,8) | 35 (5,5) |
| Vertailu lumeeseen1 | ||||||
| p‑arvo2 | 0,2880 | 0,2995 | 0,1399 | |||
| Riskisuhde3 | 0,63 | 0,74 | 0,70 | |||
| 95 % lv | (0,29; 1,36) | (0,40; 1,35) | (0,43; 1,12) | |||
1 Perustuu enintään 372 päivän aikana kerättyihin tietoihin (52 viikkoa + 7 päivän marginaali).
2 Perustuu log-rank -testiin.
3 Perustuu Coxin regressiomalliin.
Pitkäkestoinen Ofev-hoito idiopaattista keuhkofibroosia sairastaville potilaille (INPULSIS-ON)
Ofev-valmisteella tehtyyn avoimeen jatkotutkimukseen osallistui 734 potilasta, joilla oli idiopaattinen keuhkofibroosi. Potilaat, jotka suorittivat INPULSIS-tutkimuksessa 52 viikon pituisen hoitojakson loppuun, saivat INPULSIS-ON-jatkotutkimuksessa avoimesti Ofev-hoitoa. Sekä INPULSIS- että INPULSIS-ON-tutkimuksissa mukana olleiden Ofev-hoitoa saaneiden potilaiden altistuksen keston mediaani oli 44,7 kuukautta (vaihteluväli 11,9–68,3). Eksploratiivisiin tehon päätetapahtumiin sisältyi FVC:n vuosittainen alenema 192 viikon aikana, joka oli kaikilla hoitoa saaneilla potilailla ‑135,1 (5,8) ml/vuosi ja yhdenmukainen vaiheen III INPULSIS-tutkimuksissa Ofev-hoitoa saaneilla potilailla todetun FVC:n vuosittaisen aleneman (‑113,6 ml/vuosi) kanssa. Ofev-valmisteen haittatapahtumaprofiili INPULSIS-ON-tutkimuksessa oli yhdenmukainen vaiheen III INPULSIS-tutkimuksissa todetun haittatapahtumaprofiilin kanssa.
Idiopaattista keuhkofibroosia sairastavat potilaat, joilla on pitkälle edennyt keuhkojen toiminnanvajaus (INSTAGE)
INSTAGE-tutkimus oli monikansallinen, prospektiivinen, satunnaistettu, kaksoissokkoutettu, rinnakkaisryhmillä toteutettu kliininen monikeskustutkimus, johon osallistui 24 viikon ajan idiopaattista keuhkofibroosia sairastavia potilaita, joilla oli pitkälle edennyt keuhkojen toiminnanvajaus (DLCO ≤ 35 % odotusarvosta). 136 potilasta sai Ofev-valmistetta monoterapiana. Ensisijaisen päätetapahtuman tulokset osoittivat St. George’s Respiratory Questionnaire (SGRQ) ‑kokonaispisteiden laskeneen ‑0,77 yksiköllä viikolla 12 perustuen mukautettuun keskimääräiseen muutokseen lähtötilanteesta. Post hoc -vertailu osoitti, että näillä potilailla todettu FVC:n lasku vastasi FVC:n laskua niillä potilailla, joiden tauti ei ollut edennyt yhtä pitkälle ja jotka saivat Ofev-hoitoa vaiheen III INPULSIS-tutkimuksissa.
Ofev-valmisteen turvallisuus- ja siedettävyysprofiili idiopaattista keuhkofibroosia sairastavilla potilailla, joilla on pitkälle edennyt keuhkojen toiminnanvajaus, oli yhdenmukainen vaiheen III INPULSIS-tutkimuksissa tehtyjen havaintojen kanssa.
Lisätiedot vaiheen IV INJOURNEY-tutkimuksesta, jossa tutkittaville annettiin Ofev-valmistetta 150 mg kahdesti vuorokaudessa ja lisäksi pirfenidonia
Nintedanibin ja pirfenidonin samanaikaista käyttöä on tutkittu eksploratiivisessa, avoimessa, satunnaistetussa tutkimuksessa, jossa nintedanibia annoksena 150 mg kahdesti vuorokaudessa ja sen lisäksi annettua pirfenidonia (jonka annos titrattiin tasolle 801 mg kolme kertaa vuorokaudessa) verrattiin pelkkään nintedanibiin annoksena 150 mg kahdesti vuorokaudessa 105 satunnaistetulla potilaalla 12 viikon ajan. Ensisijainen päätetapahtuma oli niiden potilaiden prosenttiosuus, joille kehittyi maha-suolikanavan haittatapahtumia lähtötilanteen ja viikon 12 välisenä aikana. Maha-suolikanavan haittatapahtumat olivat yleisiä ja molempien lääkeaineiden vakiintuneiden turvallisuusprofiilien mukaisia. Yleisimpiä haittatapahtumia olivat ripuli, pahoinvointi ja oksentelu, joita ilmoitettiin nintedanibin ja pirfenidonin yhdistelmää saaneilla potilailla ja pelkkää nintedanibia saaneilla potilailla.
Lähtötilanteen jälkeen tapahtuneen FVC:n absoluuttisen muutoksen keskiarvo (keskivirhe) viikolla 12 oli ‑13,3 (17,4) ml potilailla, jotka saivat nintedanibin ja pirfenidonin yhdistelmää (n = 48) ja ‑40,9 (31,4) ml potilailla, jotka saivat pelkkää nintedanibia (n = 44).
Muut krooniset, fenotyypiltään etenevät fibrotisoivat interstitiaaliset keuhkosairaudet (ILD)
Ofev-valmisteen kliinistä tehoa on tutkittu kaksoissokkoutetussa satunnaistetussa lumekontrolloidussa vaiheen III tutkimuksessa (INBUILD) potilailla, jotka sairastivat muita kroonisia, fenotyypiltään eteneviä fibrotisoivia interstitiaalisia keuhkosairauksia. Idiopaattista keuhkofibroosia sairastavat potilaat suljettiin pois tutkimuksesta. Potilaat, joiden kliininen diagnoosi oli krooninen fibrotisoiva interstitiaalinen keuhkosairaus, valittiin, jos heillä oli rintakehän ohutleikekuvauksessa (HRCT) selkeää fibroosia (yli 10 % fibroottisia piirteitä) ja kliinisiä merkkejä etenemisestä (määritelmän mukaan FVC:n alenema ≥ 10 %, FVC:n alenema ≥ 5 % ja < 10 % sekä oireiden pahenemista tai löydöksiä kuvantamistutkimuksessa, tai oireiden pahenemista ja kuvantamistutkimuksen löydösten pahenemista seulontaa edeltävien 24 kuukauden aikana). Potilaiden FVC‑arvon piti olla vähintään 45 % odotusarvosta ja DLCO‑arvon vähintään 30 % ja alle 80 % odotusarvosta. Potilaiden sairauden piti olla edennyt huolimatta siitä, että he olivat saaneet hoitoa, joka katsottiin asianmukaiseksi potilaan interstitiaalisen keuhkosairauden tyypissä.
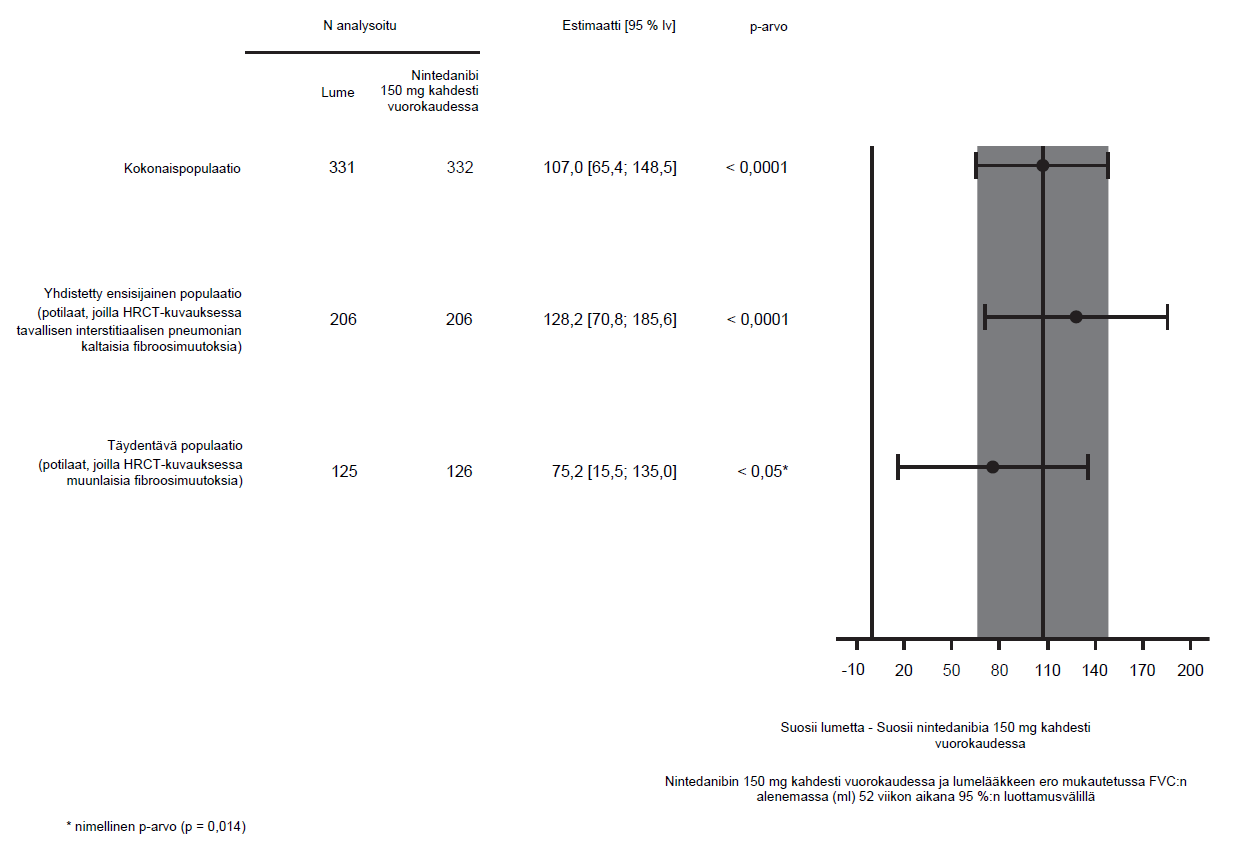
Kaikkiaan 663 potilasta satunnaistettiin suhteessa 1:1 saamaan joko Ofev-valmistetta 150 mg kahdesti vuorokaudessa tai vastaavaa lumetta vähintään 52 viikon ajan. Mediaani Ofev-altistus oli 17,4 kuukautta ja keskimääräinen Ofev-altistus 15,6 kuukautta koko tutkimuksen aikana. Satunnaistaminen stratifioitiin HRCT-kuvauksessa todettujen, keskitetysti arvioitujen fibroosimuutosten perusteella. Tutkimukseen satunnaistettiin 412 potilasta, joilla todettiin HRCT-kuvauksessa tavallisen interstitiaalisen pneumonian (UIP) kaltaisia fibroosimuutoksia, ja 251 potilasta, joilla todettiin HRCT-kuvauksessa muunlaisia fibroosimuutoksia. Tässä tutkimuksessa analyysejä varten määritettiin kaksi yhtäläisesti ensisijaista populaatiota: kaikki potilaat (kokonaispopulaatio) ja potilaat, joilla todettiin HRCT-kuvauksessa tavallisen interstitiaalisen pneumonian kaltaisia fibroosimuutoksia. Potilaista, joilla todettiin HRCT-kuvauksessa muunlaisia fibroosimuutoksia, muodostettiin ”täydentävä” populaatio.
Ensisijainen päätetapahtuma oli nopean vitaalikapasiteetin (FVC) vuosittainen alenema (ml) 52 viikon aikana. Tärkeimpiä toissijaisia päätetapahtumia olivat King’s Brief Interstitial Lung Disease (K-BILD) -kyselyn kokonaispisteiden absoluuttinen muutos lähtötilanteesta viikolla 52, interstitiaalisen keuhkosairauden ensimmäiseen äkilliseen pahenemisvaiheeseen tai kuolemaan kulunut aika 52 viikon ajanjaksolla sekä kuolemaan kulunut aika 52 viikon ajanjaksolla.
Potilaiden keskimääräinen (keskihajonta [SD], pienin-suurin) ikä oli 65,8 (9,8; 27–87) vuotta, ja heidän keskimääräinen FVC‑prosenttiosuutensa odotusarvosta oli 69,0 % (15,6; 42–137). Tutkimusryhmissä taustalla olevia kliinisiä interstitiaalisen keuhkosairauden diagnooseja olivat allerginen alveoliitti (26,1 %), autoimmuunit interstitiaaliset keuhkosairaudet (25,6 %), idiopaattinen epäspesifinen interstitiaalinen pneumonia (18,9 %), luokittelematon idiopaattinen interstitiaalinen pneumonia (17,2 %) ja muut interstitiaaliset keuhkosairaudet (12,2 %).
INBUILD-tutkimusta ei suunniteltu osoittamaan eikä sillä ollut voimaa osoittaa nintedanibin hyötyjä spesifisissä diagnostisissa alaryhmissä. Interstitiaalisten keuhkosairauksien diagnooseihin perustuvissa alaryhmissä osoitettiin johdonmukaisia vaikutuksia. Nintedanibin käytöstä hyvin harvinaisia eteneviä fibrotisoivia interstitiaalisia keuhkosairauksia sairastaville potilaille on vain vähän tietoa.
FVC:n vuosittainen alenema
FVC:n vuosittainen alenema (ml) 52 viikon aikana väheni merkitsevästi 107,0 ml Ofev-valmistetta saaneilla potilailla verrattuna lumetta saaneisiin potilaisiin (taulukko 9), mikä vastaa 57,0 %:n suhteellista hoitovaikutusta.
Taulukko 9: FVC:n vuosittainen alenema (ml) 52 viikon aikana
| Lume | Ofev 150 mg kahdesti vuorokaudessa | |
| Analysoitujen potilaiden lukumäärä | 331 | 332 |
| Alenema1 (keskivirhe) 52 viikon aikana | ‑187,8 (14,8) | ‑80,8 (15,1) |
| Vertailu lumeeseen | ||
| Ero1 | 107,0 | |
| 95 % lv | (65,4; 148,5) | |
| p‑arvo | < 0,0001 |
- Perustuu seuraaviin: satunnaiskertoimien regressio ja kiinteät kategoriset hoidon vaikutukset, HRCT-kuvauksessa todetut muutokset, kiinteät jatkuvat ajan vaikutukset, lähtötilanteen FVC [ml], mukaan lukien hoidon ja ajan sekä lähtötilanteen ja ajan yhteisvaikutukset.
Samankaltaisia tuloksia saatiin siitä ensisijaisesta populaatiosta, jonka potilailla oli HRCT-kuvauksessa todettu tavallisen interstitiaalisen pneumonian kaltaisia fibroosimuutoksia. Hoitovaikutus oli yhdenmukainen myös täydentävässä populaatiossa, jonka potilailla oli HRCT-kuvauksessa todettu muunlaisia fibroosimuutoksia (yhteisvaikutuksen p‑arvo 0,2268) (kuva 2).
Kuva 2: Forest plot -kaavio FVC:n vuosittaisesta alenemasta (ml) 52 viikon aikana potilaspopulaatioissa
Ofev-valmisteen vaikutus FVC:n vuosittaisen aleneman vähenemiseen vahvistettiin kaikissa ennalta määritetyissä herkkyysanalyyseissä, ja tulokset olivat yhdenmukaisia ennalta määritetyissä tehon alaryhmissä, joita olivat sukupuoli, ikäryhmä, rotu, FVC:n prosenttiosuus odotusarvosta lähtötilanteessa ja alkuperäisen taustalla olevan interstitiaalisen keuhkosairauden diagnoosi ryhmissä.
Kuvassa 3 esitetään FVC:n muutoksen kehitys hoitoryhmissä lähtötilanteen jälkeen ajan mittaan.
Kuva 3: Havaittu keskimääräinen (keskiarvon keskivirhe) FVC‑muutos suhteessa lähtötilanteeseen (ml) 52 viikon aikana
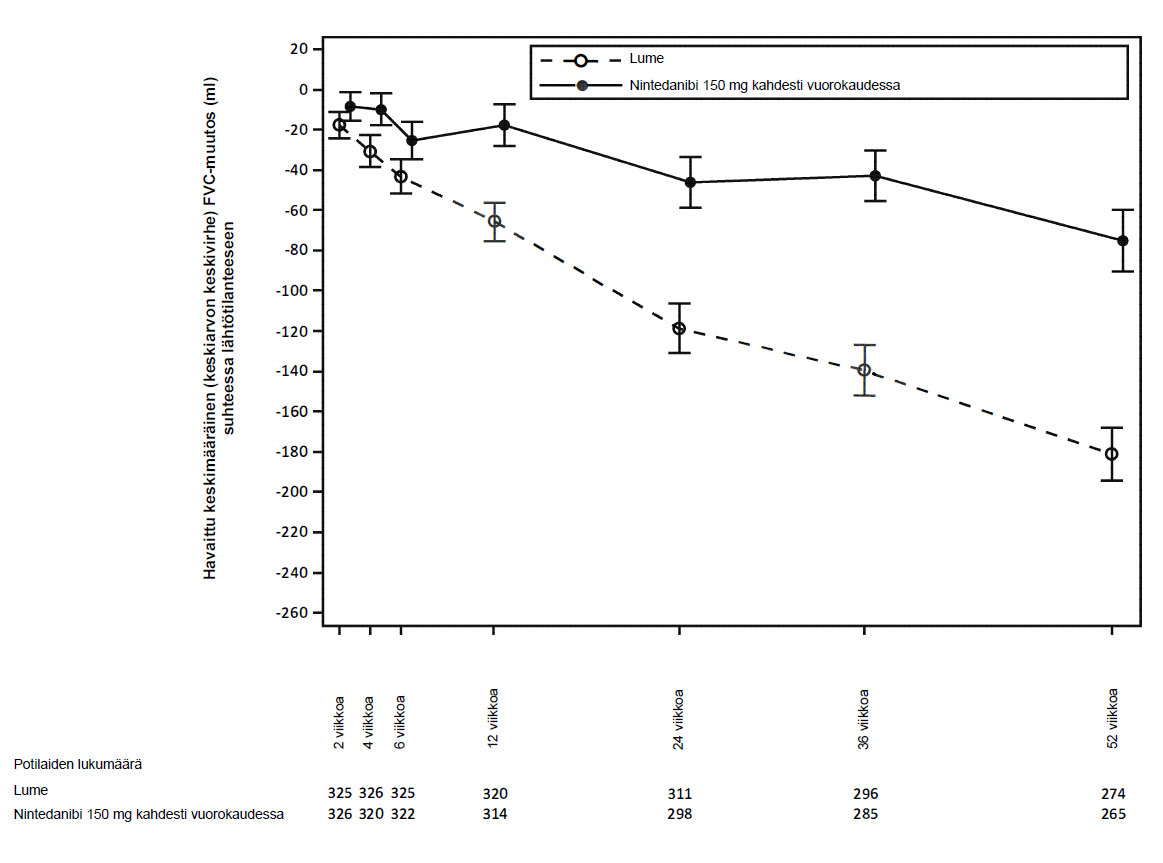
Lisäksi Ofev-valmisteella todettiin olevan suotuisia vaikutuksia lähtötilanteen FVC:n prosenttiosuuden (odotusarvosta) absoluuttisen muutoksen mukautettuun keskiarvoon viikolla 52. Lähtötilanteen FVC:n prosenttiosuuden (odotusarvosta) absoluuttisen muutoksen mukautettu keskiarvo oli pienempi nintedanibiryhmässä (‑2,62 %) kuin lumeryhmässä (‑5,86 %). Mukautettu keskiero hoitoryhmien välillä oli 3,24 (95 % lv: 2,09; 4,40, nimellinen p < 0,0001).
FVC ‑ vasteen saavuttaneiden analyysi
FVC‑vasteen saavuttaneiden osuus eli niiden potilaiden osuus, joilla FVC:n prosenttiosuus odotusarvosta aleni suhteellisesti enintään 5 %, oli suurempi Ofev-ryhmässä kuin lumeryhmässä. Samankaltaisia tuloksia saatiin analyyseistä, joissa raja-arvo oli 10 % (taulukko 10).
Taulukko 10: FVC‑vasteen saavuttaneiden potilaiden osuus 52 viikon kohdalla INBUILD-tutkimuksessa
| Lume | Ofev 150 mg kahdesti vuorokaudessa | |
| Analysoitujen potilaiden lukumäärä | 331 | 332 |
| 5 %:n raja-arvo | ||
| FVC‑vasteen saavuttaneiden lukumäärä (%)1 | 104 (31,4) | 158 (47,6) |
| Vertailu lumeeseen | ||
| Ristitulosuhde² | 2,01 | |
| 95 % lv | (1,46; 2,76) | |
| Nimellinen p‑arvo | < 0,0001 | |
| 10 %:n raja-arvo | ||
| FVC‑vasteen saavuttaneiden lukumäärä (%)1 | 169 (51,1) | 197 (59,3) |
| Vertailu lumeeseen | ||
| Ristitulosuhde² | 1,42 | |
| 95 % lv | (1,04; 1,94) | |
| Nimellinen p‑arvo | 0,0268 | |
- Vasteen saavuttaneita ovat potilaat, joilla suhteellinen alenema FVC‑arvon prosenttiosuudessa odotusarvosta oli raja-arvosta riippuen enintään 5 % tai enintään 10 % ja joilla FVC arvioitiin 52 viikon kohdalla (potilaat, joista puuttui tietoja viikon 52 kohdalla, katsottiin hoitoon vastaamattomiksi).
- Perustuu logistiseen regressiomalliin, jossa jatkuvana kovariaattina on FVC:n prosenttiosuus odotusarvosta lähtötilanteessa ja binäärisenä kovariaattina HRCT‑kuvauksessa todetut muutokset.
Interstitiaalisen keuhkosairauden ensimmäiseen äkilliseen pahenemisvaiheeseen tai kuolemaan kulunut aika
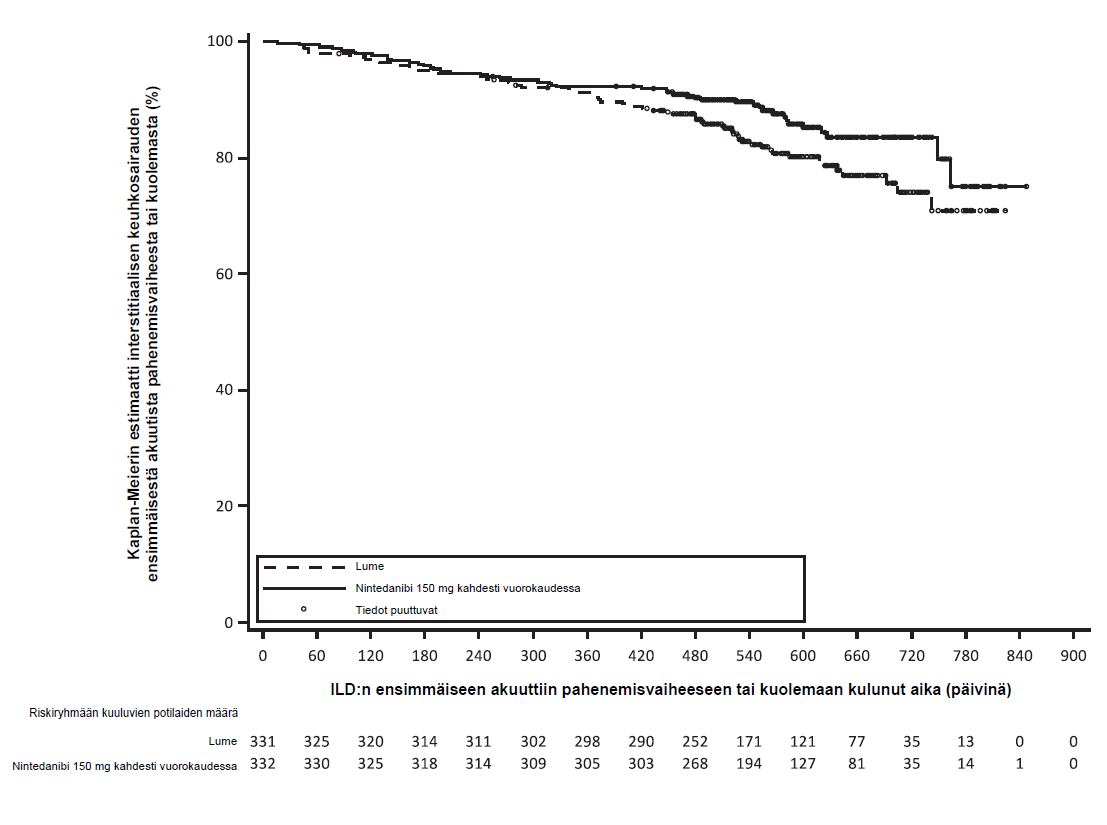
Koko tutkimuksen aikana niiden potilaiden osuus, joilla esiintyi interstitiaalisen keuhkosairauden ensimmäinen äkillinen pahenemisvaihe, kuolema tai molemmat, oli Ofev-ryhmässä 13,9 % ja lumeryhmässä 19,6 %. Riskisuhde oli 0,67 (95 % lv: 0,46; 0,98; nimellinen p = 0,0387), mikä tarkoittaa interstitiaalisen keuhkosairauden ensimmäisen äkillisen pahenemisvaiheen tai kuoleman riskin pienenemistä 33 %:lla Ofev-hoitoa saaneilla potilailla lumehoitoa saaneisiin verrattuna (kuva 4).
Kuva 4: Kaplan‑Meierin kaavio interstitiaalisen keuhkosairauden ensimmäiseen äkilliseen pahenemisvaiheeseen tai kuolemaan kuluneesta ajasta koko tutkimuksen aikana
Elossaoloanalyysi
Kuoleman riski oli pienempi Ofev-ryhmässä kuin lumeryhmässä. Riskisuhde oli 0,78 (95 % lv: 0,50; 1,21; nimellinen p = 0,2594), mikä tarkoittaa kuoleman riskin pienenemistä 22 %:lla Ofev-hoitoa saaneilla potilailla lumehoitoa saaneisiin verrattuna.
Etenemiseen (≥ 10 %:n absoluuttinen alenema FVC:n prosenttiosuudessa odotusarvosta) tai kuolemaan kulunut aika
INBUILD-tutkimuksessa etenemisen (≥ 10 %:n absoluuttinen alenema FVC:n prosenttiosuudessa odotusarvosta) tai kuoleman riski oli pienempi Ofev-hoitoa saaneilla potilailla. Niiden potilaiden osuus, joilla esiintyi jokin tapahtuma, oli Ofev-ryhmässä 40,4 % ja lumeryhmässä 54,7 %. Riskisuhde oli 0,66 (95 % lv: 0,53; 0,83; p = 0,0003), mikä tarkoittaa sairauden etenemisen (≥ 10 %:n absoluuttinen alenema FVC:n prosenttiosuudessa odotusarvosta) tai kuoleman riskin pienenemistä 34 %:lla Ofev-hoitoa saaneilla potilailla lumehoitoa saaneisiin verrattuna.
Elämänlaatu
K-BILD-kokonaispisteiden mukautettu keskimääräinen muutos lähtötilanteesta 52 viikon kohdalla oli lumeryhmässä ‑0,79 yksikköä ja Ofev-ryhmässä 0,55 yksikköä. Hoitoryhmien välinen ero oli 1,34 (95 % lv: ‑0,31; 2,98; nimellinen p = 0,1115).
Living with Pulmonary Fibrosis (L‑PF) -oirekyselyn hengenahdistusosion pisteiden mukautettu keskimääräinen absoluuttinen muutos lähtötilanteesta 52 viikon kohdalla oli Ofev-ryhmässä 4,28 ja lumeryhmässä 7,81. Mukautettu keskiero hoitoryhmien välillä oli ‑3,53 (95 % lv: ‑6,14; ‑0,92; nimellinen p = 0,0081) Ofev-hoidon eduksi. L‑PF‑oirekyselyn yskäosion pisteiden mukautettu keskimääräinen absoluuttinen muutos lähtötilanteesta 52 viikon kohdalla oli Ofev-ryhmässä ‑1,84 ja lumeryhmässä 4,25. Mukautettu keskiero hoitoryhmien välillä oli ‑6,09 (95 % lv: ‑9,65; ‑2,53; nimellinen p = 0,0008) Ofev-hoidon eduksi.
Systeemiseen skleroosiin liittyvä interstitiaalinen keuhkosairaus (SSc‑ILD)
Ofev-valmisteen kliinistä tehoa on tutkittu kaksoissokkoutetussa satunnaistetussa lumekontrolloidussa vaiheen III tutkimuksessa (SENSCIS) potilailla, jotka sairastivat systeemiseen skleroosiin liittyvää interstitiaalista keuhkosairautta (SSc‑ILD). Potilaiden SSc‑ILD diagnoosi perustui systeemisen skleroosin vuoden 2013 ACR/EULAR (American College of Rheumatology / European League Against Rheumatism) -luokittelukriteereihin ja edellisten 12 kuukauden aikana tehtyyn rintakehän ohutleikekuvaukseen (HRCT). Kaikkiaan 580 potilasta satunnaistettiin suhteessa 1:1 saamaan joko Ofev 150 mg -valmistetta kahdesti vuorokaudessa tai vastaavaa lumetta vähintään 52 viikon ajan. Näistä 576 potilasta hoidettiin. Satunnaistaminen stratifioitiin anti-topoisomeraasin vasta-ainepitoisuuden (ATA) avulla. Potilaat olivat mukana sokkoutetussa tutkimushoidossa enintään 100 viikkoa (mediaani Ofev-altistus 15,4 kuukautta; keskimääräinen Ofev-altistus 14,5 kuukautta).
Ensisijainen päätetapahtuma oli nopean vitaalikapasiteetin (FVC) vuosittainen alenema 52 viikon aikana. Keskeisiä toissijaisia päätetapahtumia olivat muunnetun Rodnan Skin Score (mRSS) ‑pistemäärän absoluuttinen muutos lähtötilanteesta 52 viikon kohdalla ja Saint George’s Respiratory Questionnaire (SGRQ) ‑kokonaispistemäärän absoluuttinen muutos lähtötilanteesta 52 viikon kohdalla.
Kokonaispopulaatiossa 75,2 % potilaista oli naisia. Keskimääräinen (keskihajonta [SD], pienin-suurin) ikä oli 54,0 (12,2; 20–79) vuotta. Kaikkiaan 51,9 %:lla potilaista oli yleistynyt kutaaninen systeeminen skleroosi ja 48,1 %:lla oli rajoittunut kutaaninen systeeminen skleroosi. Keskimääräinen (SD) aika jonkin muun oireen kuin Raynaudʼn ilmiön alkamisesta oli 3,49 (1,7) vuotta. Lähtötilanteessa 49,0 % potilaista sai vakiintunutta mykofenolaattihoitoa (46,5 % mykofenolaattimofetiilia, 1,9 % mykofenolaattinatriumia ja 0,5 % mykofenolihappoa). Potilaiden turvallisuusprofiili oli vertailukelpoinen riippumatta siitä, saivatko potilaat mykofenolaattia lähtötilanteessa vai eivät.
FVC:n vuosittainen alenema
FVC:n vuosittainen alenema (ml) 52 viikon aikana väheni merkitsevästi 41,0 ml Ofev-valmistetta saavilla potilailla verrattuna lumetta saaneisiin potilaisiin (taulukko 11), mikä vastaa 43,8 %:n suhteellista hoitovaikutusta.
Taulukko 11: FVC:n vuosittainen alenema (ml) 52 viikon aikana
| Lume | Ofev 150 mg kahdesti vuorokaudessa | |
| Analysoitujen potilaiden lukumäärä | 288 | 287 |
| Alenema1 (keskivirhe) 52 viikon aikana | ‑93,3 (13,5) | ‑52,4 (13,8) |
| Vertailu lumeeseen | ||
| Ero1 | 41,0 | |
| 95 % lv | (2,9; 79,0) | |
| p‑arvo | < 0,05 |
1Perustuu seuraaviin: satunnaiskertoimien regressio ja kiinteät kategoriset hoidon vaikutukset, ATA-pitoisuus, sukupuoli, kiinteät jatkuvat ajan vaikutukset, lähtötilanteen FVC [ml], ikä, pituus, mukaan lukien hoidon ja ajan sekä lähtötilanteen ja ajan yhteisvaikutukset. Satunnaisvaikutusta käytettiin potilaskohtaisen vakiotermin ja ajan kohdalla. Potilaskohtaiset virheet mallinnettiin strukturoimattomalla varianssi-kovarianssimatriisilla. Yksilöiden välinen vaihtelu mallinnettiin varianssikomponenttien varianssi-kovarianssimatriisilla.
Ofev-valmisteen vaikutus FVC:n vuosittaisen aleneman vähenemisessä oli samanlainen kaikissa ennalta määritetyissä herkkyysanalyyseissä, eikä ennalta määritetyissä alaryhmissä (esim. ikä, sukupuoli ja mykofenolaatin käyttö) havaittu mitään heterogeenisyyttä.
Samankaltaisia vaikutuksia havaittiin myös muissa keuhkotoimintaa mittaavissa päätetapahtumissa, esim. FVC:n (ml) absoluuttinen muutos lähtötilanteesta 52 viikon kohdalla (kuva 5 ja taulukko 12) ja FVC:n odotettu alenema (%) 52 viikon aikana (taulukko 13). Näistä saatiin lisänäyttöä Ofev-valmisteen systeemiseen skleroosiin liittyvän interstitiaalisen keuhkosairauden etenemistä hidastavista vaikutuksista. Lisäksi Ofev-ryhmässä pienemmällä määrällä potilaita absoluuttinen FVC:n alenema oli > 5 % odotusarvosta (20,6 % Ofev-ryhmässä vs. 28,5 % lumeryhmässä OR = 0,65, p = 0,0287). FVC:n suhteellinen alenema (ml) > 10 % oli vertailukelpoinen molempien ryhmien välillä (16,7 % Ofev-ryhmässä vs. 18,1 % lumeryhmässä, OR = 0,91, p = 0,6842). Viikon 52 kohdalla puuttuneille FVC‑arvoille annettiin näissä analyyseissa potilaan huonointa hoidonaikaista arvoa vastaava arvo.
Enintään 100 viikolta kerättyjen tietojen (hoidon pisin mahdollinen kesto SENSCIS-tutkimuksessa) eksploratiivinen analyysi viittasi siihen, että Ofev-valmisteen hoidonaikainen vaikutus systeemiseen skleroosiin liittyvän interstitiaalisen keuhkosairauden etenemisen hidastamiseen jatkui yli 52 viikon ajan.
Kuva 5: Havaittu keskimääräinen (keskiarvon keskivirhe) FVC‑muutos suhteessa lähtötilanteeseen (ml) 52 viikon aikana
Taulukko 12: FVC:n (ml) absoluuttinen muutos lähtötilanteesta 52 viikon kohdalla
| Lume | Ofev 150 mg kahdesti vuorokaudessa | |
| Analysoitujen potilaiden lukumäärä | 288 | 288 |
| Keskiarvo (SD) lähtötilanteessa | 2 541,0 (815,5) | 2 458,5 (735,9) |
| Keskimääräinen1 (SE) muutos lähtötilanteesta viikon 52 kohdalla | ‑101,0 (13,6) | ‑54,6 (13,9) |
| Vertailu lumeeseen | ||
| Keskiarvo1 | 46,4 | |
| 95 % lv | (8,1; 84,7) | |
| p‑arvo | < 0,05 |
1Perustuu toistomittausten sekamalliin (MMRM) ja seuraaviin kiinteisiin kategorisiin vaikutuksiin: ATA-pitoisuus, käynti, hoidon ja käynnin yhteisvaikutus, lähtötilanteen ja käynnin yhteisvaikutus, ikä, sukupuoli ja pituus. Käynti oli toistomittausmuuttuja. Potilaskohtaiset virheet mallinnettiin strukturoimattomalla varianssi-kovarianssirakenteella. Mukautettu keskiarvo perustui mallin kaikkiin analysoituihin potilaisiin (ei pelkästään potilaisiin, joilla oli lähtötilanne ja mittaustulos 52 viikon kohdalla).
Taulukko 13: FVC:n vuotuinen alenema (% odotusarvosta) 52 viikon aikana
| Lume | Ofev 150 mg kahdesti vuorokaudessa | |
| Analysoitujen potilaiden lukumäärä | 288 | 287 |
| Alenema1 (SE) 52 viikon aikana | ‑2,6 (0,4) | ‑1,4 (0,4) |
| Vertailu lumeeseen | ||
| Ero1 | 1,15 | |
| 95 % lv | (0,09; 2,21) | |
| p‑arvo | < 0,05 |
- Perustuu seuraaviin: satunnaiskertoimien regressio ja kiinteät kategoriset hoidon vaikutukset, ATA-pitoisuus, ajan kiinteät jatkuvat vaikutukset, lähtötilanteen FVC [% oletusarvosta], mukaan lukien hoidon ja ajan sekä lähtötilanteen ja ajan yhteisvaikutukset. Satunnaisvaikutusta käytettiin potilaskohtaisen vakiotermin ja ajan kohdalla. Potilaskohtaiset virheet mallinnettiin strukturoimattomalla varianssi-kovarianssimatriisilla. Yksilöidenvälinen vaihtelu mallinnettiin varianssikomponenttien varianssi-kovarianssimatriisilla
mRSS-pistemäärän (muunnettu Rodnan Skin Score) muutos lähtötilanteesta 52 viikon kohdalla
mRSS:n mukautettu keskimääräinen absoluuttinen muutos lähtötilanteesta 52 viikon kohdalla oli vertailukelpoinen Ofev-ryhmän [‑2,17 (95 % lv ‑2,69; ‑1,65)] ja lumeryhmän [‑1,96 (95 % lv ‑2,48, ‑1,45)] välillä. Mukautettu keskiero hoitoryhmien välillä oli ‑0,21 (95 % lv ‑0,94; 0,53; p = 0,5785).
SGRQ (St. George's Respiratory Questionnaire) -kokonaispisteiden muutos lähtötilanteesta 52 viikon kohdalla
SGRQ-kokonaispisteiden mukautettu keskimääräinen absoluuttinen muutos lähtötilanteesta 52 viikon kohdalla oli vertailukelpoinen Ofev-ryhmän [0,81 (95 % lv ‑0,92; 2,55)] ja lumeryhmän [‑0,88 (95 % lv ‑2,58; 0,82)] välillä. Mukautettu keskiero hoitoryhmien välillä oli 1,69 (95 % lv ‑0,73; 4,12; p = 0,1711).
Elossaoloanalyysi
Kuolleisuus Ofev-ryhmän (N = 10; 3,5 %) ja lumeryhmän (N = 9; 3,1 %) välillä oli vertailukelpoinen koko tutkimuksen ajan. Kuolemaan kuluneen ajan analyysissä riskisuhteeksi saatiin 1,16 (95 % lv 0,47; 2,84; p = 0,7535).
QT-aika
QT/QTc‑tulokset kirjattiin munuaissyöpäpotilailla toteutetussa tutkimuksessa. Tässä tutkimuksessa QTcF‑aika ei pidentynyt, kun potilaille annettiin nintedanibia suun kautta 200 mg:n kerta-annoksena tai toistuvina 200 mg:n annoksina kahdesti vuorokaudessa 15 vuorokauden ajan.
Pediatriset potilaat
Kliinisesti merkittävät, etenevät fibrotisoivat interstitiaaliset keuhkosairaudet (ILD) ja interstitiaaliseen keuhkosairauteen liittyvä systeeminen skleroosi (SSc-ILD) 6–17-vuotiailla lapsilla ja nuorilla
Ofev-valmisteen kliinistä turvallisuutta ja tehoa 6–17 vuoden ikäisillä, kliinisesti merkittäviä fibrotisoivia interstitiaalisia keuhkosairauksia (ILD) sairastavilla lapsilla ja nuorilla on tutkittu eksploratorisessa, satunnaistetussa, kaksoissokkoutetussa, lumekontrolloidussa vaiheen III tutkimuksessa (InPedILD 1199.337).
Potilaat satunnaistettiin suhteessa 2:1 saamaan joko Ofev-valmistetta kahdesti vuorokaudessa (annokset mukautettiin painon mukaan käyttäen myös 25 mg:n kapseleita) tai vastaavaa lumetta 24 viikon ajan, mitä seurasi pituudeltaan vaihteleva avoin nintedanibihoitojakso. Standardihoidon käyttö sallittiin, mikäli hoitava lääkäri katsoi sen kliinisesti aiheelliseksi.
InPedILD-tutkimuksen ensisijainen tavoite oli arvioida annosaltistusta ja nintedanibin turvallisuutta lapsilla ja nuorilla, joilla oli kliinisesti merkittäviä fibrotisoivia intestitiaalisia keuhkosairauksia (ILD). Tehon arviointi oli vain toissijainen tavoite.
InPedILD-tutkimukseen otettiin 6–17‑vuotiaita lapsia ja nuoria, joilla oli kliinisesti merkittävä fibrotisoiva interstitiaalinen keuhkosairaus ja joiden FVC oli vähintään 25 % odotusarvosta. Potilaiden sairaus luokiteltiin fibrotisoivaksi interstitiaaliseksi keuhkosairaudeksi kahdessa rintakehän ohutleikekuvauksessa (joista toinen oli tehty edellisten 12 kuukauden aikana) todetun fibroosin perusteella tai keuhkobiopsiassa ja yhdessä edellisten 12 kuukauden aikana tehdyssä rintakehän ohutleikekuvauksessa todetun fibroosin perusteella.
Kliinisesti merkittävä tauti määriteltiin Fan-pistearvoksi ≥ 3 tai dokumentoiduksi näytöksi taudin kliinisestä etenemisestä minkä tahansa ajanjakson aikana. Näyttö taudin kliinisestä etenemisestä perustui FVC‑arvon suhteelliseen laskuun tasolta ≥ 10 % odotusarvosta, FVC‑arvon suhteelliseen laskuun 5–10 %:lla odotusarvosta ja oireiden pahenemiseen, pahenevaan fibroosiin rintakehän ohutleikekuvauksessa tai muihin etenevästä keuhkofibroosista johtuvan kliinisen pahenemisen mittareihin (esim. lisääntynyt hapen tarve, vähentynyt diffuusiokapasiteetti), vaikka tämä ei ollutkaan vaatimus tutkimukseenotolle potilaille, joiden Fan-pistearvo oli ≥ 3.
Kaikkiaan 39 potilasta satunnaistettiin (61,5 % tyttöjä). Lähtötilanteen ominaisuudet:
- ikä 6–11 vuotta: 12 potilasta, ikä 12–17 vuotta: 27 potilasta. Keskimääräinen ikä [keskihajonta (SD)] oli 12,6 (3,3) vuotta.
- Keskimääräinen (SD) paino oli 42,2 kg (17,8 kg); ikä 6–11 vuotta: 26,6 kg (10,4 kg), ikä 12–17 vuotta: 49,1 kg (16,0 kg).
- Lähtötilanteessa yleinen iänmukainen BMI-Z -arvo (standardoitu pistemäärä) (SD) oli keskimäärin ‑0,6 (1,8).
- Yleinen FVC-Z -arvon (SD) keskiarvo ‑3,5 (1,9).
Tutkimukseen otettujen potilaiden yleisimpiä taustalla olevia yksittäisiä interstitiaalisen keuhkosairauden diagnooseja olivat:
- surfaktanttiproteiinien puutos (nintedanibi: 26,9 %, lumelääke: 38,5 %)
- systeeminen skleroosi (nintedanibi: 15,4 %, lumelääke: 23,1 %)
- toksinen / säteilyn aiheuttama / lääkkeen aiheuttama pneumoniitti (nintedanibi: 11,5 %, lumelääke: 7,7 %)
- yliherkkyydestä johtuvaa kroonista pneumoniittia ilmoitettiin 2 tutkittavalla (nintedanibi: 7,7 %).
-
muita taustalla olevia interstitiaalisen keuhkosairauden diagnooseja, joita kutakin raportoitiin yhdellä potilaalla, olivat:
- hematopoieettisen kantasolusiirron (HSCT) jälkeinen fibroosi
- lastenreuma
- juveniili idiopaattinen artriitti
- dermatomyosiitti (DM)
- deskvamatiivinen interstitiaalinen pneumoniitti
- H1N1‑influenssa
- epäselvä (krooninen diffuusi keuhkosairaus)
- COPA-oireyhtymä
- COPA-geenin mutaatio
- määrittelemätön sidekudossairaus
- infektion jälkeinen ahtauttava bronkioliitti
- määrittelemätön interstitiaalinen keuhkosairaus
- idiopaattinen
- pistoon liittyvä vaskulopatia.
Ensisijaisen päätetapahtuman tuloksia olivat:
-
Nintedanibialtistus:
- AUCτ,ss-arvona kuvattu nintedanibialtistus vakaassa tilassa otettujen näytteiden perusteella oli suurin piirtein samaa luokkaa lapsilla ja nuorilla ja verrattavissa aikuisilla todettuun AUCτ,ss-arvoon (ks. kohta Farmakokinetiikka).
-
Hoidon aikana ilmenneet haittatapahtumat (viikko 24):
- Nintedanibiryhmä: 84,6 % potilaista (ikä 6–11 vuotta: 75,0 %, ikä 12–17 vuotta: 88,9 %)
- Lumeryhmä: 84,6 % potilaista (ikä 6–11 vuotta: 100,0 %, ikä 12–17 vuotta: 77,8 %)
Nopean vitaalikapasiteetin (FVC) (prosenttiosuus odotusarvosta) muutos lähtötilanteesta arvioitiin toissijaisena tehon päätetapahtumana. Tulokset (kuva 6):
-
Viikko 24:
- Nintedanibiryhmä: mukautettu keskimääräinen muutos = 0,31 (95 % lv: ‑2,36; 2,98)
- Lumeryhmä: mukautettu keskimääräinen muutos = ‑0,89 (95 % lv: ‑4,61; 2,82)
- Ero FVC % -arvon odotusarvosta 1,21 (95 % lv: ‑3,40; 5,81) nintedanibin eduksi
-
Viikko 52:
- Satunnaistettu nintedanibiryhmä: mukautettu keskimääräinen muutos = 0,79 (95 % lv: ‑2,95; 4,53)
- Satunnaistettu lumeryhmä: mukautettu keskimääräinen muutos = ‑0,98 (95 % lv: ‑6,26; 4,30)
Pediatrisilla potilailla todettiin suurta vaihtelua nintedanibihoidolla saavutetuissa vasteissa FVC:n (prosenttiosuus odotusarvosta) päätetapahtumassa ja useissa muissa eksploratorisissa tehon päätetapahtumissa.
Kuva 6: Absoluuttisen muutoksen mukautettu keskiarvo (SE) FVC:n odotusarvosta lasketun prosenttiosuuden muutoksesta lähtötilanteen ja viikon 52 välillä – hoidetut potilaat*
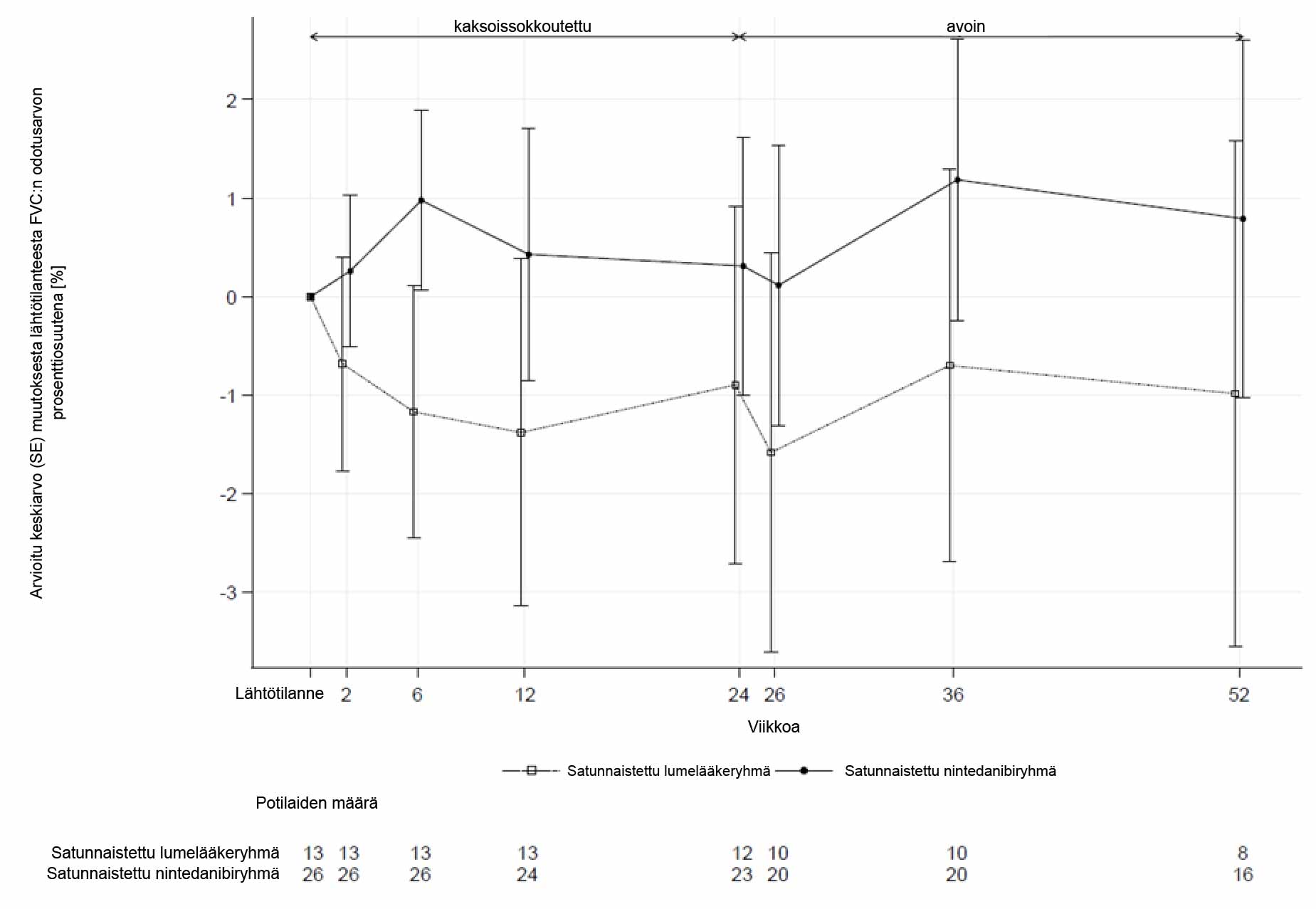
*24 hoitoviikon jälkeen kaikki potilaat saivat avoimesti nintedanibia.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Ofev-valmisteen käytöstä idiopaattisen keuhkofibroosin hoidossa kaikissa pediatrisissa potilasryhmissä.
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Ofev-valmisteen käytöstä fibrotisoivien interstitiaalisten keuhkosairauksien hoidossa alle 6 vuoden ikäisillä pediatrisilla potilailla (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Nintedanibin enimmäispitoisuus plasmassa saavutettiin noin 2–4 tuntia suun kautta tapahtuneen pehmeän liivatekapselin annon jälkeen, kun potilas oli aterioinut (vaihteluväli 0,5–8 tuntia). 100 mg:n annoksen absoluuttinen biologinen hyötyosuus oli terveillä vapaaehtoisilla 4,69 % (90 % lv: 3,615–6,078). Imeytymistä ja biologista hyötyosuutta heikentävät kuljettajaproteiinien vaikutukset ja merkittävä ensikierron metabolia. Nintedanibialtistus suureni suhteessa annokseen annosalueella 50–450 mg kerran vuorokaudessa ja 150–300 mg kahdesti vuorokaudessa. Vakaan tilan pitoisuudet plasmassa saavutettiin viimeistään viikon kuluessa annostelusta.
Aterian jälkeen nintedanibialtistus lisääntyi noin 20 % paasto-olosuhteissa suoritettuun annosteluun verrattuna (lv: 95,3–152,5 %) ja imeytyminen hidastui (tmax-ajan mediaani paasto-olosuhteissa: 2,00 h; aterian jälkeen: 3,98 h).
In vitro -tutkimuksessa nintedanibikapseleiden sekoittaminen pieneen määrään omenasosetta tai suklaavanukasta enintään 15 minuutin ajaksi ei vaikuttanut farmaseuttiseen laatuun. Pidempiaikaisen pehmeälle ruoalle altistumisen todettiin aiheuttavan kapselien turpoamista ja epämuotoisuutta veden imeytyessä kapselin liivatekuoreen. Siksi kapseleiden ottaminen pehmeän ruoan kanssa ei oletettavasti muuta kliinistä vaikutusta, kun ne otetaan välittömästi.
Nintedanibin biologisen hyötyosuuden kerta-annostutkimuksessa terveille aikuisille miestutkittaville annettiin joko yksi 100 mg:n pehmeä liivatekapseli tai neljä 25 mg:n pehmeää liivatekapselia ja molemmissa hoidoissa biologinen hyötyosuus oli samanlainen.
Jakautuminen
Nintedanibin jakautumiskinetiikka on vähintään kaksivaiheinen. Laskimoon annetun infuusion jälkeen havaittiin suuri jakautumistilavuus (Vss: 1 050 l, 45,0 % gCV).
Nintedanibi sitoutui voimakkaasti ihmisen plasman proteiineihin in vitro; sitoutunut fraktio oli 97,8 %. Seerumin albumiinia pidetään merkittävimpänä sitoutumisproteiinina. Nintedanibi jakautuu ensisijaisesti plasmaan. Suhdeluku veren ja plasman välillä on 0,869.
Biotransformaatio
Nintedanibin pääasiallinen metabolinen reaktio on esteraasin aiheuttama hydrolyysi, jonka seurauksena muodostuu vapaa happo-osa BIBF 1202. BIBF 1202 glukuronidoituu edelleen uridiini‑5’-difosfoglukuronyylitransferaasi (UGT) -entsyymien UGT 1A1, UGT 1A7, UGT 1A8 ja UGT 1A10 avulla BIBF 1202 -glukuronidiksi.
Vain pieni osuus nintedanibin biotransformaatiosta tapahtuu CYP‑välitteisesti, ja CYP 3A4 on tärkein vaikuttava entsyymi. Tärkeintä CYP‑riippuvaista metaboliittia ei havaittu ihmisen plasmassa ADME-tutkimuksessa. In vitro CYP‑riippuvaisen metabolian osuus oli noin 5 % ja esteraasin noin 25 %. Nintedanibi, BIBF 1202 ja BIBF 1202 -glukuronidi eivät myöskään prekliinisissä tutkimuksissa estäneet tai indusoineet CYP‑entsyymejä. Yhteisvaikutukset nintedanibin ja CYP:n substraattien, CYP:n estäjien ja CYP:n indusoijien välillä ovat siksi epätodennäköisiä.
Eliminaatio
Plasman kokonaispuhdistuma oli laskimoon annetun infuusion jälkeen korkea (puhdistuma: 1 390 ml/min, 28,8 % gCV). Muuttumatonta vaikuttavaa ainetta erittyi virtsaan 48 tunnin aikana noin 0,05 % annoksesta (31,5 % gCV) suun kautta tapahtuneen annon ja noin 1,4 % annoksesta (24,2 % gCV) laskimoon tapahtuneen annon jälkeen. Munuaispuhdistuma oli 20 ml/min (32,6 % gCV). Pääosa suun kautta annetun [14C]-nintedanibin radioaktiivisuudesta eliminoitui ulosteeseen/sappeen (93,4 % annoksesta, 2,61 % gCV). Munuaisten kautta tapahtuvan erittymisen osuus kokonaispuhdistumasta oli vähäinen (0,649 % annoksesta, 26,3 % gCV). Lääkeaineen katsottiin erittyneen täydellisesti (yli 90 %) neljän päivän kuluttua annostelusta. Nintedanibin terminaalinen puoliintumisaika oli 10–15 tuntia (gCV-% noin 50 %).
Lineaarisuus/ei-lineaarisuus
Nintedanibin farmakokinetiikan voidaan katsoa olevan lineaarinen ajan suhteen (eli kerta-annoksen tiedot voidaan ekstrapoloida toistuvia annoksia koskeviksi). Kumulaatio toistuvien antokertojen yhteydessä oli 1,04‑kertainen (Cmax) ja 1,38‑kertainen (AUCτ). Nintedanibin jäännöspitoisuudet pysyivät vakaina yli vuoden ajan.
Kuljetus
Nintedanibi on P‑gp:n substraatti. Tiedot nintedanibin mahdollisista yhteisvaikutuksista tämän kuljettajaproteiinin kanssa, ks. kohta Yhteisvaikutukset. On osoitettu, että nintedanibi ei ole OATP‑1B1:n, OATP‑1B3:n, OATP‑2B1:n, OCT‑2:n tai MRP‑2:n substraatti tai estäjä in vitro. Nintedanibi ei myöskään ole BCRP:n substraatti. Lääkkeellä oli vain heikko OCT‑1:tä, BCRP:tä ja P‑gp:tä estävä vaikutus in vitro, ja tällä katsotaan olevan vain vähäistä kliinistä merkitystä. Sama koskee nintedanibin toimintaa OCT‑1:n substraattina.
Populaatiofarmakokineettinen analyysi erityisryhmissä
Nintedanibin farmakokineettiset ominaisuudet olivat samankaltaisia terveillä vapaaehtoisilla, idiopaattista keuhkofibroosia sairastavilla potilailla, muita kroonisia, fenotyypiltään eteneviä fibrotisoivia interstitiaalisia keuhkosairauksia sairastavilla potilailla, systeemiseen skleroosiin liittyvää interstitiaalista keuhkosairautta sairastavilla potilailla ja syöpäpotilailla. Idiopaattista keuhkofibroosia sairastavilla potilailla ja ei-pienisoluista keuhkosyöpää sairastavilla potilailla (n = 1 191) toteutetun populaatiofarmakokineettisen analyysin ja deskriptiivisten tutkimusten tulosten perusteella sukupuoli (kehonpainon mukaan korjattuna), lievä tai keskivaikea munuaisten vajaatoiminta (kreatiniinipuhdistuman mukaan arvioituna), alkoholin kulutus tai P‑gp-genotyyppi eivät vaikuttaneet nintedanibialtistukseen. Populaatiofarmakokineettiset analyysit antoivat viitteitä iän, kehonpainon ja etnisen taustan kohtalaisista vaikutuksista nintedanibialtistukseen (ks. alla). Altistuksen suuren potilaskohtaisen vaihtelun perusteella kohtalaisia vaikutuksia ei pidetä kliinisesti merkityksellisinä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Ikä
Nintedanibialtistus kasvoi lineaarisesti suhteessa ikään. AUCτ,ss pieneni 45‑vuotiaalla potilaalla 16 % ja kasvoi 76‑vuotiaalla potilaalla 13 % verrattuna mediaani-ikäiseen, 62‑vuotiaaseen potilaaseen. Ikähaarukka oli analyysissä 29–85 vuotta; noin 5 % populaatiosta oli yli 75‑vuotiaita. Populaatiofarmakokineettisen mallin perusteella 75‑vuotiailla ja tätä vanhemmilla potilailla havaittiin noin 20–25 % korkeampi nintedanibialtistus kuin alle 65‑vuotiailla potilailla.
Pediatriset potilaat
InPedILD-tutkimuksesta (1199.337) saatujen farmakokineettisten tietojen analyysin perusteella nintedanibin ottaminen suun kautta painoon perustuvan annosalgoritmin mukaan johti altistukseen, joka oli aikuispotilailla todettujen raja-arvojen sisällä. Todetut altistuksen geometriset AUCτ,ss-keskiarvot (geometrinen vaihtelukerroin) olivat 175 ng/ml·h (85,1 %) 10:llä 6–11‑vuotiaalla potilaalla ja 167 ng/ml·h (83,6 %) 23:lla 12–17‑vuotiaalla potilaalla.
InPedILD-tutkimuksen tietojen altistus-vasteanalyysissa ilmeni altistuksen ja FVC:n prosenttiosuuden (odotusarvosta) sekä FVC:n Z-arvon välinen Emax-tyyppinen suhde, jota aikuistutkimuksissa saatu tieto tuki. FVC:n prosenttiosuudelle (odotusarvosta) EC50 oli 4,4 ng/ml (suhteellinen keskivirhe: 28,6 %) ja FVC:n Z-arvolle EC50 oli 5,0 ng/ml (suhteellinen keskivirhe: 75,3 %).
Nintedanibin käyttöä ei ole tutkittu lapsilla ja nuorilla, joilla on maksan vajaatoiminta.
Lapsilla ja nuorilla, joilla on fibrotisoiva ILD ja lievä maksan vajaatoiminta (Child Pugh ‑luokka A), populaatiofarmakokineettinen mallinnus osoittaa, että suositellut annospienennykset (ks. kohta Annostus ja antotapa) johtaisivat altistukseen, joka vastaa lievää maksan vajaatoimintaa (Child Pugh ‑luokka A) sairastavien aikuispotilaiden nintedanibialtistusta, kun heille annetaan vastaava suositellusti pienennetty annos.
Kehonpaino
Kehonpainon ja nintedanibialtistuksen välillä havaittiin käänteinen korrelaatio. AUCT,SS kasvoi 50 kg:n painoisella potilaalla 25 % (5. persentiili) ja pieneni 100 kg:n painoisella potilaalla 19 % (95. persentiili) verrattuna mediaania edustavaan, 71,5 kg:n painoiseen potilaaseen.
Etninen tausta
Nintedanibin keskialtistus väestössä oli 33–50 % korkeampi kiinalaisilla, taiwanilaisilla ja intialaisilla potilailla, 16 % korkeampi japanilaisilla potilailla ja 16–22 % matalampi korealaisilla potilailla suhteessa valkoihoisiin (kehonpainon mukaan korjattuna). Mustaihoisten potilaiden osalta tutkimustietoa oli hyvin rajoitetusti, mutta tulokset olivat samaa luokkaa kuin valkoihoisilla.
Maksan vajaatoiminta
Erityisessä vaiheen I kerta-annostutkimuksessa nintedanibialtistus oli lievää maksan vajaatoimintaa sairastavilla vapaaehtoisilla tutkittavilla Cmax‑ ja AUC‑arvon perusteella 2,2‑kertainen terveisiin henkilöihin verrattuna (Child Pugh A; 90 % lv Cmax‑arvolle 1,3–3,7 ja AUC‑arvolle 1,2–3,8). Keskivaikeaa maksan vajaatoimintaa sairastavilla vapaaehtoisilla tutkittavilla (Child Pugh B) altistus oli Cmax-arvon perusteella 7,6‑kertainen (90 % lv 4,4–13,2) ja AUC‑arvon perusteella 8,7‑kertainen (90 % lv 5,7–13,1) terveisiin vapaaehtoisiin nähden. Vaikeaa maksan vajaatoimintaa sairastavia potilaita (Child Pugh C) ei ole tutkittu.
Samanaikainen pirfenidonihoito
Nintedanibin ja pirfenidonin samanaikaista käyttöä tutkittiin asiaa nimenomaisesti koskeneessa farmakokineettisessä tutkimuksessa idiopaattista keuhkofibroosia sairastavilla potilailla. Ryhmän 1 potilaat saivat 150 mg:n kerta-annoksen nintedanibia ennen pirfenidoniannoksen titraamista vakaassa tilassa tasolle 801 mg kolme kertaa vuorokaudessa ja myös sen jälkeen (hoitoa saaneiden potilaiden n = 20). Ryhmän 2 potilaat saivat vakaassa tilassa 801 mg pirfenidonia kolme kertaa vuorokaudessa, ja heille tehtiin farmakokineettinen profilointi ennen vähintään 7 vuorokautta kestänyttä samanaikaista hoitoa nintedanibilla annoksena 150 mg kahdesti vuorokaudessa sekä myös sen jälkeen (hoitoa saaneiden potilaiden n = 17). Ryhmässä 1 mukautettujen geometristen keskiarvojen suhteet (90 % lv) olivat nintedanibin Cmax‑arvolle 93 % (57–151 %) ja AUC0‑tz‑arvolle 96 % (70–131 %) (yksilöiden välisen vertailun n = 12). Ryhmässä 2 mukautettujen geometristen keskiarvojen suhteet (90 % lv) olivat pirfenidonin Cmax,ss‑arvolle 97 % (86–110 %) ja AUCτ,ss‑arvolle 95 % (86–106 %) (yksilöiden välisen vertailun n = 12).
Näistä tuloksista ei saatu näyttöä siitä, että nintedanibin ja pirfenidonin välillä olisi mitään oleellisia farmakokineettisiä lääkeyhteisvaikutuksia, kun näitä lääkeaineita käytetään samanaikaisesti (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Samanaikainen bosentaanihoito
Ofev-valmisteen ja bosentaanin samanaikaista käyttöä tutkittiin asiaa nimenomaisesti koskeneessa farmakokineettisessä tutkimuksessa terveillä vapaaehtoisilla. Tutkittavat saivat 150 mg:n Ofev-kerta-annoksen ennen bosentaanin 125 mg toistuvan annoksen saamista ja sen jälkeen. Bosentaani annettiin kahdesti vuorokaudessa vakaassa tilassa. Mukautettujen geometristen keskiarvojen suhteet (90 % lv) olivat nintedanibin Cmax‑arvolle 103 % (86–124 %) ja AUC0‑tz‑arvolle 99 % (91–107 %) (n = 13), mikä osoittaa, että nintedanibin ja bosentaanin samanaikainen käyttö ei vaikuttanut nintedanibin farmakokinetiikkaan.
Samanaikainen suun kautta otettavien hormonaalisten ehkäisyvalmisteiden käyttö
Asiaa nimenomaisesti koskeneessa farmakokineettisessä tutkimuksessa systeemiseen skleroosiin liittyvää interstitiaalista keuhkosairautta sairastaville naispotilaille annettiin yhtenä kerta-annoksena 30 mikrog etinyyliestradiolia ja 150 mikrog levonorgestreeliä ennen kahdesti vuorokaudessa annettavaa 150 mg:n nintedanibiannosta ja sen jälkeen vähintään 10 vuorokauden ajan. Mukautettujen geometristen keskiarvojen suhteet (90 % lv) olivat etinyyliestradiolille 117 % (108–127 %; Cmax) ja 101 % (93–111 %; AUC0‑tz) sekä levonorgestreelille 101 % (90–113 %; Cmax) ja 96 % (91–102 %; AUC0‑tz) (n = 15), mikä osoittaa, että nintedanibin samanaikainen käyttö ei vaikuta oleellisesti etinyyliestradiolin ja levonorgestreelin pitoisuuksiin plasmassa.
Altistus-vastesuhde
Altistus-vasteanalyysit potilailla, jotka sairastivat idiopaattista keuhkofibroosia tai muita kroonisia, fenotyypiltään eteneviä fibrotisoivia interstitiaalisia keuhkosairauksia, viittasivat heikkoon suhteeseen plasman nintedanibialtistuksen ja ALAT- ja/tai ASAT-arvojen kohoamisen välillä. Todellinen annettu annos saattaa ennustaa minkä tahansa asteisen ripulin kehittymisriskiä paremmin, vaikka plasman lääkeainealtistusta riskiä määrittävänä tekijänä ei voitukaan sulkea pois (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Tiedot pediatristen potilaiden altistus-vasteanalyysistä, ks. pediatristen potilaiden alakohta.
Prekliiniset tiedot turvallisuudesta
Yleinen toksikologia
Kerta-annosten toksisuustutkimuksissa rotilla ja hiirillä nintedanibin havaittiin aiheuttavan vähäistä akuuttia toksisuutta. Toistuvien annosten toksikologiaa koskevissa tutkimuksissa nuorilla rotilla havaittiin jatkuvasti ja nopeasti kasvavien etuhampaiden (mutta ei väli- eikä poskihampaiden) kiilteen ja hammasluun pysyviä muutoksia. Lisäksi todettiin epifyysilevyjen paksuuntumista luiden kasvuvaiheissa, ja nämä muutokset korjaantuivat hoidon lopettamisen jälkeen. Nämä muutokset tunnetaan muiden VEGFR‑2:n estäjien perusteella, ja niiden voidaan katsoa olevan luokkavaikutuksia.
Muilla kuin jyrsijöillä tehdyissä toksisuustutkimuksissa havaittiin ripulia ja oksentelua yhdessä vähentyneen ruoankulutuksen ja kehonpainon laskun kanssa.
Maksaentsyymiarvojen kohoamisen merkkejä ei havaittu rotilla, koirilla eikä makakiapinoilla. Lievää maksaentsyymiarvojen kohoamista, joka ei johtunut vakavista haittavaikutuksista kuten ripulista, havaittiin ainoastaan reesusapinoilla.
Lisääntymistoksisuus
Rotilla havaittiin alkio- ja sikiökuolemia ja teratogeenisuutta, kun altistus oli pienempi kuin ihmisen altistus suurimmalla suositusannoksella (150 mg × 2). Aksiaalisen luuston ja suurten valtimoiden kehitykseen kohdistuvia vaikutuksia havaittiin myös subterapeuttisilla altistustasoilla.
Kaniineilla havaittiin alkio- ja sikiökuolemia ja teratogeenisuutta, kun altistus oli noin kolme kertaa suurempi kuin ihmisen suurimmalla suositusannoksella aiheutuva altistus. Alkio- ja sikiöaikaisessa aksiaalisen luuston ja sydämen kehityksessä havaittiin epäselviä vaikutuksia jo silloin, kun altistus oli pienempi kuin ihmisen suurimmalla suositusannoksella (150 mg × 2) aiheutuva altistus.
Pre- ja postnataalista kehitystä koskeneessa tutkimuksessa rotilla havaittiin pre- ja postnataaliseen kehitykseen kohdistuvia vaikutuksia, kun altistus oli pienempi kuin ihmisen suurimmalla suositusannoksella aiheutuva altistus.
Rotilla tehdyssä tutkimuksessa, jossa arvioitiin urosten hedelmällisyyttä ja alkionkehitystä varhaisvaiheesta implantaatioon, ei havaittu urosten sukuelimiin eikä urosten hedelmällisyyteen kohdistuvia vaikutuksia.
Rotilla pieniä määriä radioleimattua nintedanibia ja/tai sen metaboliitteja erittyi maitoon (≤ 0,5 % annetusta annoksesta).
Kaksivuotisessa hiirillä ja rotilla tehdyssä karsinogeenisuustutkimuksessa ei saatu näyttöä nintedanibin karsinogeenisuudesta.
Genotoksisuustutkimuksissa nintedanibin ei havaittu olevan mutageeninen.
Farmaseuttiset tiedot
Apuaineet
Kapselin sisältö
keskipitkäketjuiset triglyseridit
kova rasva
lesitiini (soija) (E322)
Kapselin kuori
liivate
glyseroli (85 %)
titaanidioksidi (E171)
punainen rautaoksidi (E172)
keltainen rautaoksidi (E172)
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
Ofev 25 mg pehmeät kapselit
4 vuotta
Ofev 100 mg ja 150 mg pehmeät kapselit
3 vuotta
Säilytys
Läpipainopakkaus
Säilytä alle 25 °C. Säilytä alkuperäispakkauksessa. Herkkä kosteudelle.
Purkki
Säilytä alle 25 °C. Pidä purkki tiiviisti suljettuna. Herkkä kosteudelle.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
OFEV kapseli, pehmeä
100 mg (L:ei) 60 x 1 fol (1280,22 €)
150 mg (L:ei) 60 x 1 fol (2262,09 €)
PF-selosteen tieto
Ofev 25 mg pehmeät kapselit
Ofev 25 mg pehmeistä kapseleista on saatavana seuraavat pakkauskoot:
- 60 × 1 pehmeä kapseli yksittäispakattuina perforoiduissa alumiini/alumiini-läpipainopakkauksissa
- 60 pehmeää kapselia HDPE-(muovi)purkissa, jossa kierrettävä korkki
- 120 pehmeää kapselia HDPE-(muovi)purkissa, jossa kierrettävä korkki
- 180 pehmeää kapselia HDPE-(muovi)purkissa, jossa kierrettävä korkki
Ofev 100 mg pehmeät kapselit
Ofev 100 mg pehmeistä kapseleista on saatavana seuraavat pakkauskoot:
- 30 × 1 pehmeä kapseli yksittäispakattuina perforoiduissa alumiini/alumiini-läpipainopakkauksissa
- 60 × 1 pehmeä kapseli yksittäispakattuina perforoiduissa alumiini/alumiini-läpipainopakkauksissa
Ofev 150 mg pehmeät kapselit
Ofev 150 mg pehmeistä kapseleista on saatavana seuraavat pakkauskoot:
- 30 × 1 pehmeä kapseli yksittäispakattuina perforoiduissa alumiini/alumiini-läpipainopakkauksissa
- 60 × 1 pehmeä kapseli yksittäispakattuina perforoiduissa alumiini/alumiini-läpipainopakkauksissa
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Ofev 25 mg pehmeät kapselit
Ofev 25 mg pehmeät kapselit ovat oransseja, läpinäkymättömiä, soikeita pehmeitä liivatekapseleita (noin 8 × 5 mm), joiden toiselle puolelle on merkitty ”25”.
Ofev 100 mg pehmeät kapselit
Ofev 100 mg pehmeät kapselit ovat persikanvärisiä, läpinäkymättömiä, pitkänomaisia pehmeitä liivatekapseleita (noin 16 × 6 mm), joiden toiselle puolelle on merkitty Boehringer Ingelheimin logo ja numero ”100”.
Ofev 150 mg pehmeät kapselit
Ofev 150 mg pehmeät kapselit ovat ruskeita, läpinäkymättömiä, pitkänomaisia pehmeitä liivatekapseleita (noin 18 × 7 mm), joiden toiselle puolelle on merkitty Boehringer Ingelheimin logo ja numero ”150”.
Käyttö- ja käsittelyohjeet
Jos kädet joutuvat kosketuksiin kapselin sisällön kanssa, ne on pestävä välittömästi runsaalla vedellä (ks. kohta Annostus ja antotapa).
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
OFEV kapseli, pehmeä
100 mg 60 x 1 fol
150 mg 60 x 1 fol
- Alempi erityiskorvaus (65 %). Nintedanibi ja pirfenidoni: Idiopaattisen keuhkofibroosin hoito erityisin edellytyksin (284).
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Nintedanibi ja pirfenidoni: Idiopaattisen keuhkofibroosin hoito aikuisilla potilailla erityisin edellytyksin / Nintedanibi: Muiden kroonisten, etenevien fibrotisoivien interstitiaalisten keuhkosairauksien hoito aikuisilla ja vähintään 6-vuotiailla lapsilla ja nuorilla erityisin edellytyksin (356).
OFEV kapseli, pehmeä
25 mg 60 x 1 fol
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Nintedanibi ja pirfenidoni: Idiopaattisen keuhkofibroosin hoito aikuisilla potilailla erityisin edellytyksin / Nintedanibi: Muiden kroonisten, etenevien fibrotisoivien interstitiaalisten keuhkosairauksien hoito aikuisilla ja vähintään 6-vuotiailla lapsilla ja nuorilla erityisin edellytyksin (356).
ATC-koodi
L01EX09
Valmisteyhteenvedon muuttamispäivämäärä
05.01.2026
Yhteystiedot
 BOEHRINGER INGELHEIM FINLAND KY
BOEHRINGER INGELHEIM FINLAND KY Tammasaarenkatu 5
00180 Helsinki
010 310 2800
www.boehringer-ingelheim.fi
medinfo.finland@boehringer-ingelheim.com