
AIMOVIG injektioneste, liuos, esitäytetty kynä 70 mg, 140 mg
Vaikuttavat aineet ja niiden määrät
Aimovig 70 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 70 mg erenumabia.
Aimovig 140 mg injektioneste, liuos, esitäytetty ruisku
Yksi esitäytetty ruisku sisältää 140 mg erenumabia.
Aimovig 70 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kynä sisältää 70 mg erenumabia.
Aimovig 140 mg injektioneste, liuos, esitäytetty kynä
Yksi esitäytetty kynä sisältää 140 mg erenumabia.
Erenumabi on täysin humaani monoklonaalinen IgG2-vasta-aine, joka valmistetaan yhdistelmä-DNA-tekniikalla kiinanhamsterin munasarjasolulinjassa (CHO).
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Injektioneste, liuos (injektioneste)
Kliiniset tiedot
Käyttöaiheet
Aimovig on tarkoitettu migreenin estohoitoon aikuisilla, joilla on vähintään 4 migreenipäivää kuukaudessa.
Ehto
Hoidon saa aloittaa migreenin toteamiseen ja hoitoon perehtynyt lääkäri.
Annostus ja antotapa
Hoidon aloittaa migreenin toteamiseen ja hoitoon perehtynyt lääkäri.
Annostus
Hoito on tarkoitettu potilaille, joilla on vähintään 4 migreenipäivää kuukaudessa erenumabi-hoitoa aloitettaessa.
Suositusannos aikuisilla on 70 mg erenumabia 4 viikon välein. Jotkut potilaat voivat hyötyä 140 mg annoksesta 4 viikon välein (ks. kohta Farmakodynamiikka).
Jokainen 140 mg:n annos annetaan yhtenä 140 mg:n injektiona ihon alle tai kahtena 70 mg:n injektiona ihon alle.
Kliinisten tutkimusten mukaan valtaosalla niistä potilaista, jotka saivat hoitovasteen, havaittiin kliinistä hyötyä 3 kk kuluessa. Jos potilaalla ei ole todettu vastetta 3 hoitokuukauden jälkeen, on harkittava hoidon lopettamista. Hoidon jatkamistarve on suositeltavaa arvioida säännöllisin välein tämän jälkeen.
Erityisryhmät
Iäkkäät (65 vuotta täyttäneet)
Aimovigia ei ole tutkittu iäkkäillä potilailla. Annosta ei tarvitse muuttaa, sillä ikä ei vaikuta erenumabin farmakokinetiikkaan.
Munuaisten/maksan vajaatoiminta
Annosta ei tarvitse muuttaa, jos potilaalla on lievä tai keskivaikea munuaisten vajaatoiminta tai maksan vajaatoiminta (ks. kohta Farmakokinetiikka).
Pediatriset potilaat
Aimovig-valmisteen turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten hoidossa ei ole vielä varmistettu. Tietoja ei ole saatavilla.
Antotapa
Aimovig annetaan ihon alle.
Aimovig on tarkoitettu potilaan itse pistettäväksi asianmukaisen opastuksen jälkeen. Injektiot voi myös antaa toinen henkilö, joka on saanut asianmukaisen opastuksen. Injektio voidaan antaa vatsan alueelle, reiteen tai olkavarren ulkosyrjään (olkavartta voidaan käyttää vain, jos injektion antaa joku muu kuin potilas itse; ks. kohta Farmakokinetiikka). Injektiokohtia tulee vaihdella, eikä valmistetta saa injisoida alueelle, jossa iho aristaa tai on mustelmilla, punoittava tai kovettunut.
Esitäytetty ruisku
Esitäytetyn Aimovig-ruiskun koko sisältö on injisoitava. Jokainen esitäytetty ruisku on tarkoitettu vain yhtä käyttökertaa varten. Ruiskut on suunniteltu siten, että niiden koko sisältö annetaan kerralla eikä niihin jää lainkaan jäämiä.
Kattavat käyttöohjeet löytyvät pakkausselosteesta.
Esitäytetty kynä
Esitäytetyn Aimovig-kynän koko sisältö on injisoitava. Jokainen esitäytetty kynä on tarkoitettu vain yhtä käyttökertaa varten. Kynät on suunniteltu siten, että niiden koko sisältö annetaan kerralla eikä niihin jää lainkaan jäämiä.
Kattavat käyttöohjeet löytyvät pakkausselosteesta.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Sydän- ja verisuonivaikutukset
Potilaat, joilla oli tiettyjä merkittäviä sydän- ja verisuonisairauksia, suljettiin pois kliinisistä tutkimuksista (ks. kohta Farmakodynamiikka). Turvallisuustiedot näiden potilaiden osalta puuttuvat.
Yliherkkyysreaktiot
Vakavia yliherkkyysreaktioita, kuten ihottumaa, angioedeemaa ja anafylaktisia reaktioita, on ilmoitettu erenumabihoidon yhteydessä myyntiluvan saamisen jälkeen. Nämä reaktiot voivat ilmetä minuuttien sisällä hoidosta, mutta jotkut reaktiot voivat ilmetä vasta yli viikon kuluttua hoidon jälkeen. Tästä syystä, potilaita tulee varoittaa yliherkkyysreaktioihin liittyvistä oireista. Jos potilaalla ilmenee vakava tai vaikea yliherkkyysreaktio, on asianmukainen hoito aloitettava ja erenumabihoito tulisi lopettaa (ks. kohta Vasta-aiheet).
Ummetus
Ummetus on erenumabin yleinen haittavaikutus, ja on vakavuusasteeltaan yleensä lievä tai keskivaikea. Useimmissa tapauksissa ummetuksen puhkeaminen ilmoitettiin ensimmäisen erenumab-annoksen jälkeen; potilailla on kuitenkin ollut ummetusta myös hoidon myöhemmässä vaiheessa. Suurimmassa osassa tapauksista ummetus helpottui kolmessa kuukaudessa. Markkinoille tulon jälkeen ummetukseen on raportoitu liittyneen vakavia komplikaatioita. Muutamissa vakavissa tapauksissa tarvittiin sairaalahoitoa, osassa tapauksista tarvittiin leikkaushoitoa. Aikaisemmin koettu ummetus, tai samanaikainen suolen liikkeitä vähentävä lääkitys, saattaa lisätä vakavamman ummetuksen riskiä, sekä mahdollisuutta ummetukseen liittyville komplikaatioille. Potilaita on varoitettava ummetuksen riskistä, ja heitä on kehotettava hakeutumaan hoitoon, jos ummetus ei helpotu tai se pahenee. Potilaiden on hakeuduttava välittömästi hoitoon, mikäli heille kehittyy vakava ummetus. Ummetusta on hoidettava pikaisesti kliinisesti tarkoituksenmukaisella tavalla. Lääkityksen lopettamista on harkittava vakavan ummetuksen yhteydessä.
Lateksiyliherkkyys
Tämän lääkevalmisteen irrotettava suojus sisältää lateksikumia. Saattaa aiheuttaa vakavia allergisia reaktioita.
Natriumpitoisuus
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Samanaikaisesti käytettävien lääkevalmisteiden ei odoteta vaikuttavan altistukseen monoklonaalisten vasta-aineiden metaboliareittien perusteella. Terveillä koehenkilöillä toteutetuissa tutkimuksissa ei havaittu yhteisvaikutuksia ehkäisytablettien (etinyyliestradioli/norgestimaatti) eikä sumatriptaanin kanssa.
Raskaus ja imetys
Raskaus
On vain vähän tietoja erenumabin käytöstä raskaana oleville naisille. Eläinkokeissa ei ole havaittu suoria tai epäsuoria lisääntymistoksisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi Aimovig-valmisteen käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä erittyykö erenumabi rintamaitoon. Ihmisen IgG:n tiedetään erittyvän rintamaitoon ensimmäisten päivien aikana synnytyksestä ja pitoisuudet vähenevät pian tämän jälkeen. Täten riskiä imeväiselle ei voida sulkea pois tämän lyhyen ajanjakson aikana. Tämän jälkeen Aimovigin käyttöä voidaan harkita imetyksen aikana vain jos se on kliinisesti tarpeen.
Hedelmällisyys
Eläintutkimuksissa ei havaittu vaikutuksia naaraiden eikä urosten hedelmällisyyteen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Aimovig-valmisteella ei oletettavasti ole haitallista vaikutusta ajokykyyn ja koneidenkäyttökykyyn.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Yhteensä yli 2 500 potilasta (yli 2 600 potilasvuotta) on saanut Aimovig-hoitoa myyntilupaa varten suoritetuissa tutkimuksissa. Näistä potilaista yli 1 300 altistui vähintään 12 kk ajan ja 218 potilasta altistui 5 vuotta. Aimovigin kokonaisturvallisuusprofiili oli yhdenmukainen 5 vuoden pitkäaikaisen avoimen hoidon vaiheessa.
70 mg:n ja 140 mg:n yhteydessä ilmoitettuja haittavaikutuksia olivat injektiokohdan reaktiot (5,6 %/4,5 %), ummetus (1,3 %/3,2 %), lihasspasmit (0,1 %/2,0 %) ja kutina (0,7 %/1,8 %). Useimmat vaikutukset olivat vaikeusasteeltaan lieviä tai keskivaikeita. Alle 2 % näiden tutkimusten potilaista lopetti tutkimuksen haittavaikutusten vuoksi.
Haittavaikutustaulukko
Taulukossa 1 luetellaan kaikki haittavaikutukset, joita Aimovig-hoitoa saaneilla potilailla esiintyi tutkimusten 12 viikon pituisten lumekontrolloitujen vaiheiden aikana, sekä myyntiluvan myöntämisen jälkeen. Haittavaikutukset on luokiteltu kussakin elinjärjestelmäluokassa yleisyysjärjestyksessä yleisimmistä alkaen. Kunkin yleisyysluokan haittavaikutukset on esitetty vakavuusjärjestyksessä vakavimmasta alkaen. Kunkin haittavaikutuksen kohdalla mainittava yleisyysluokka perustuu seuraavaan käytäntöön: hyvin yleinen (≥1/10), yleinen (≥1/100, <1/10), melko harvinainen (≥1/1 000, <1/100), harvinainen (≥1/10 000, <1/1 000), hyvin harvinainen (<1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1 Luettelo haittavaikutuksista
| Elinjärjestelmä | Haittavaikutus | Yleisyysluokka |
| Immuunijärjestelmä | Yliherkkyysreaktiota mukaan lukien anafylaksia, angioedeema, ihottuma, turvotus/edeema ja urtikaria | Yleinen |
| Ruoansulatuselimistö | Ummetus | Yleinen |
| Suun haavaumatb | Tuntematon | |
| Iho ja ihonalainen kudos | Kutinac | Yleinen |
Alopesia Ihottumad | Tuntematon | |
| Luusto, lihakset ja sidekudos | Lihasnykäykset | Yleinen |
| Yleisoireet ja antopaikassa todettavat haitat | Pistoskohdan reaktiota | Yleinen |
a Ks. kohta ”Valikoitujen haittavaikutusten kuvaus” b Suun haavaumat sisältää seuraavat MedDRA-termit: stomatiitti, suun ulseraatio, suun limakalvon rakkulointia. c Kutina sisältää seuraavat MedDRA-termit: yleistynyt kutina, kutina ja kutiava ihottuma. d Ihottuma sisältää seuraavat MedDRA-termit: papulaarinen ihottuma, kesivä ihottuma, punoittava ihottuma, urtikaria, rakkula. | ||
Valikoitujen haittavaikutusten kuvaus
Injektiokohdan reaktiot
Tutkimusten yhdistetyssä, 12-viikkoisessa lumekontrolloidussa vaiheessa injektiokohdan reaktiot olivat lieviä ja useimmiten ohimeneviä. Yhdessä tapauksessa, potilaan saadessa 70 mg:n annoksen, hoito keskeytettiin injektiokohdan ihottuman vuoksi. Yleisimmät injektiokohdan reaktiot olivat paikallinen kipu, punoitus ja kutina. Injektiokohdan kipu lievittyi yleensä 1 tunnin kuluessa lääkkeenannosta.
Iho- ja yliherkkyysreaktiot
Tutkimusten yhdistetyssä, 12-viikkoisessa lumekontrolloidussa vaiheessa havaittiin ei-vakavia ihottuma-, kutina- ja turvotus-/edeematapauksia. Valtaosassa tapauksista reaktiot olivat lieviä, eivätkä johtaneet hoidon keskeyttämiseen.
Myyntiluvan myöntämisen jälkeen havaittiin anafylaksia- ja angioedeematapauksia.
Immunogeenisuus
Kliinisten tutkimusten kaksoissokkoutetun vaiheen aikana erenumabivasta-aineiden ilmaantuvuus oli 70 mg erenumabiannoksia saaneilla 6,3 % (56/884) (näistä 3:lla oli in vitro neutraloiva vaikutus) ja 140 mg erenumabiannoksia saaneilla 2,6 % (13/504) (yhdelläkään ei ollut in vitro neutraloivaa vaikutusta). 256 viikon avoimen vaiheen tutkimuksessa, erenumabivasta-aineiden ilmaantuvuus oli ainoastaan 70 mg tai 140 mg Aimovigia, koko tutkimuksen aikana, saaneilla potilailla 11,0 % (25/225) (näistä 2:lla oli in vitro neutraloiva vaikutus). Erenumabivasta-aineiden kehittyminen ei vaikuttanut erenumabin tehoon eikä turvallisuuteen.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta.
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Kliinisissä tutkimuksissa ei ole ilmoitettu yhtään yliannostustapausta.
Kliinisissä tutkimuksissa ihon alle on annettu enintään 280 mg annoksia ilman näyttöä annosta rajoittavasta toksisuudesta.
Yliannostustapauksessa potilasta tulee hoitaa oireenmukaisesti, ja tukitoimiin tulee ryhtyä tarpeen mukaan.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Analgeetit, migreenilääkkeet, ATC-koodi: N02CD01
Vaikutusmekanismi
Erenumabi on humaani monoklonaalinen vasta-aine, joka sitoutuu kalsitoniinigeeniin liittyvän peptidin (CGRP) reseptoriin. CGRP-reseptoreja on migreenin patofysiologian kannalta oleellisilla alueilla kuten ganglion trigeminalessa (puolikuuhermosolmussa). Erenumabi kilpailee CGRP-reseptoriin sitoutumisesta voimakkaasti ja spesifisesti CGRP:n kanssa ja estää sen toimintaa reseptorissa, eikä sillä ole merkittävää vaikutusta muihin kalsitoniiniperheen reseptoreihin.
CGRP on nosiseptiivistä signalointia säätelevä neuropeptidi ja migreenin patofysiologiaan yhdistetty vasodilataattori. Toisin kuin muiden neuropeptidien, CGRP-pitoisuuksien on osoitettu suurenevan merkitsevästi migreenin aikana ja palautuvan normaaleiksi päänsäryn lievittyessä. CGRP:n infusoiminen laskimoon indusoi potilailla migreenityyppisen päänsäryn.
CGRP:n vaikutusten estäminen voisi teoriassa vaimentaa kompensatorista vasodilataatiota iskemiaan liittyvissä tiloissa. Yhdessä tutkimuksessa arvioitiin laskimoon annetun 140 mg Aimovig-kerta-annoksen vaikutusta kontrolloiduissa liikuntaolosuhteissa tutkittavilla, joilla oli stabiili angina pectoris. Aimovig-potilailla liikuntaharjoittelun kesto oli samankaltainen kuin lumelääkettä saaneilla, eikä valmiste pahentanut sydänlihasiskemiaa näillä potilailla.
Kliininen teho ja turvallisuus
Erenumabi arvioitiin migreenin estohoidossa kahdessa avaintutkimuksessa, jotka kattoivat kroonisen ja episodisen migreenin kirjon. Kumpaankin tutkimukseen otetuilla potilailla oli vähintään 12 kk:n migreenianamneesi (auraoirein tai ilman) ICHD-III-luokituksen (International Classification of Headache Disorders) diagnoosikriteerien mukaisesti. Iäkkäät potilaat (> 65-vuotiaat), potilaat, joilla oli opioidien liikakäyttöä (kroonisen migreenin tutkimuksessa), potilaat, joilla oli lääkityksen liikakäyttöä (episodisen migreenin tutkimuksessa), sekä potilaat, joilla oli ollut sydäninfarkti, aivohalvaus, TIA-kohtauksia, epästabiili angina pectoris, sepelvaltimon ohitusleikkaus tai muu revaskularisaatiotoimenpide seulontaa edeltävien 12 kk aikana, suljettiin pois tutkimuksesta. Potilaat, joilla oli huonossa hoitotasapainossa oleva verenpainetauti tai joiden BMI oli > 40, suljettiin pois tutkimuksesta 1.
Krooninen migreeni
Tutkimus 1
Erenumabi arvioitiin ainoana lääkkeenä kroonisen migreenin estohoitoon satunnaistetussa, 12 viikon pituisessa, lumekontrolloidussa, kaksoissokkoutetussa monikeskustutkimuksessa potilailla, joilla oli migreeni auraoirein tai ilman (≥ 15 päänsärkypäivää kuukaudessa, ≥ 8 migreenipäivää kuukaudessa).
667 potilasta satunnaistettiin suhteessa 3:2:2 saamaan lumelääkettä (n = 286) tai 70 mg (n = 191) tai 140 mg (n = 190) erenumabia, ositettuna akuuttilääkityksen liikakäytön mukaan (esiintyi 41 %:lla kaikista potilaista). Akuuttihoito päänsäryn lievittämiseen oli tutkimuksen aikana sallittua.
Tutkimusryhmien demografiset tiedot ja taudin lähtötilannetiedot olivat tasapainossa ja toisiinsa verrattavissa. Potilaiden iän mediaani oli 43 vuotta, 83 % oli naisia ja 94 % valkoihoisia. Migreenikohtausten esiintymistiheyden keskiarvo oli lähtötilanteessa noin 18 migreenipäivää kuukaudessa. Yhteensä 68 %:lla yksi tai useampi estolääkehoito oli epäonnistunut riittämättömän tehon tai huonon siedettävyyden vuoksi, ja 49 %:lla oli epäonnistunut kaksi tai useampi aiempaa estolääkehoitoa riittämättömän tehon tai huonon siedettävyyden vuoksi. Erenumabiryhmissä yhteensä 366 potilasta (96 %) ja lumeryhmässä 265 potilasta (93 %) suoritti tutkimuksen loppuun (eli suoritti viikon 12 arvioinnin).
Kuukausittaisten migreenipäivien (MMD) keskiarvon pienenemistä lumelääkkeeseen nähden havaittiin kuukausianalyysissä kuukaudesta 1 alkaen, ja seurantaviikkoanalyysissä erenumabin vaikutuksen havaittiin alkavan ensimmäisestä antoviikosta alkaen.
Kuva 1 Muutos lähtötilanteesta: kuukausittaiset migreenipäivät ajan myötä tutkimuksessa 1 (sisältäen ensisijaisen päätetapahtuman kk 3 kohdalla)
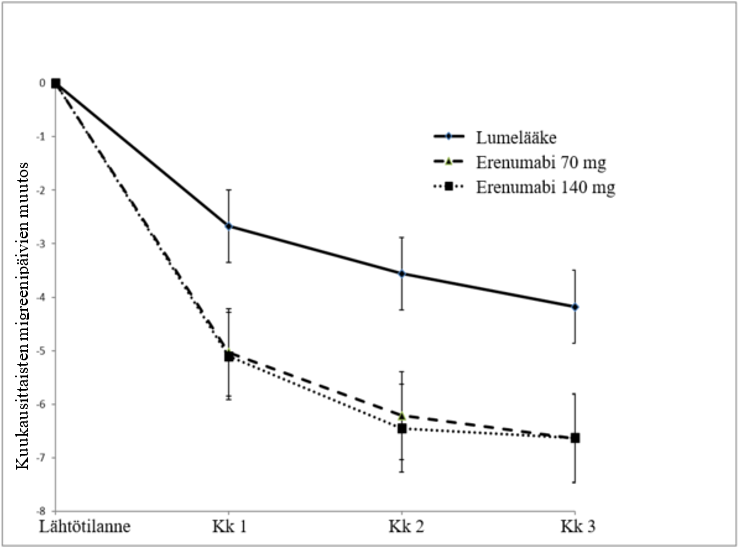
Taulukko 2 Tehon ja potilaan raportoimien tulosten muutos lähtötilanteesta viikon 12 kohdalla tutkimuksessa 1
Aimovig (erenumabi) 140 mg (n = 187) | Aimovig (erenumabi) 70 mg (n = 188) | Lume (n = 281) | Hoitojen ero (95 % lv) | p-arvo | |
| Tehotulokset | |||||
MMD Keskimuutos (95 % lv) Lähtötilanne (SD) | ‑6,6 (‑7,5, ‑5,8) 17,8 (4,7) | ‑6,6 (‑7,5, ‑5,8) 17,9 (4,4) | ‑4,2 (‑4,9, ‑3,5) 18,2 (4,7) | Molemmat ‑2,5 (‑3,5, ‑1,4) | Molemmat < 0,001 |
≥ 50 % MMD –vasteen saaneet Prosenttiosuus [%] | 41,2 % | 39,9 % | 23,5 % | n/a | Molemmat: < 0,001a,d |
≥ 75 % MMD –vasteen saaneet Prosenttiosuus [%] | 20,9 % | 17,0 % | 7,8 % | n/a | n/ab |
Migreenispesifisen akuutti- lääkityksen päiviä/kk Keskimuutos (95 % lv) | ‑4,1 (‑4,7, ‑3,6) | ‑3,5 (‑4,0, ‑2,9) | ‑1,6 (‑2,1, ‑1,1) | 70 mg: ‑1,9 (‑2,6, ‑1,1) 140 mg: ‑2,6 (‑3,3, ‑1,8) | Molemmat: < 0,001a |
| Lähtötilanne (keskihajonta) | 9,7 (7,0) | 8,8 (7,2) | 9,5 (7,6) | ||
| Potilaan raportoimat tulokset | |||||
HIT‑6 Keskimuutosc (95 % lv) | ‑5,6 (‑6,5, ‑4,6) | ‑5,6 (‑6,5, ‑4,6) | ‑3,1 (‑3,9, ‑2,3) | 70 mg: ‑2,5 (‑3,7, ‑1,2) 140 mg: ‑2,5 (‑3,7, ‑1,2) | n/ab |
MIDAS-kokonaispisteet Keskimuutosc (95 % lv) | ‑19,8 (‑25,6, ‑14,0) | ‑19,4 (‑25,2, ‑13,6) | ‑7,5 (‑12,4, ‑2,7) | 70 mg: ‑11,9 (‑19,3, ‑4,4) 140 mg: ‑12,2 (‑19,7, ‑4,8) | n/ab |
lv = luottamusväli; MMD = kuukausittaiset migreenipäivät; HIT-6 = Headache Impact Test; MIDAS = Migraine Disability Assessment; n/a = not applicable. a Toissijaisten päätetapahtumien osalta kaikki p-arvot ilmoitetaan korjaamattomina, ja ne ovat tilastollisesti merkitseviä monivertailukorjauksen jälkeen. b Eksploratiivisten päätetapahtumien p-arvoa ei esitetä. c HIT-6: Muutos ja väheneminen lähtötilanteesta arvioitiin 12-viikkoisen kaksoissokkoutetun hoitovaiheen viimeisten 4 viikon aikana. MIDAS: Muutos ja väheneminen lähtötilanteesta arvioitiin 12 viikon aikana. Tiedot kerättiin 3 kuukautta takautuvasti. d p-arvo laskettiin vetosuhteiden (OR, odds ratio) perusteella. | |||||
Potilailla, joilla vähintään yksi estolääkehoito oli epäonnistunut, hoitojen ero kuukausittaisten migreenipäivien (MMD) vähenemisessä oli erenumabi 140 mg -ryhmän ja lumeryhmän välillä ‑3,3 vrk (95 % lv: ‑4,6, ‑2,1) ja erenumabi 70 mg -ryhmän ja lumeryhmän välillä ‑2,5 vrk (95 % lv: ‑3,8, ‑1,2). Potilailla, joilla vähintään kaksi estolääkehoitoa oli epäonnistunut, hoitojen ero oli erenumabi 140 mg ‑ryhmän ja lumeryhmän välillä ‑4,3 vrk (95 % lv: ‑5,8; ‑2,8) ja erenumabi 70 mg ‑ryhmän ja lumeryhmän välillä ‑2,7 vrk (95 % lv: ‑4,2, ‑1,2). Myös tarkasteltaessa potilaita, joilla vähintään yksi estolääkehoito oli epäonnistunut, MMD-päivät vähenivät vähintään 50 % suuremmalla osuudella erenumabi-ryhmän potilaista kuin lumeryhmän potilaista (140 mg -hoidolla 40,8 % ja 70 mg -hoidolla 34,7 % vs. lumehoidolla 17,3 %). 140 mg -hoidolla vetosuhde (odds ratio) oli 3,3 (95 % lv: 2,0, 5,5) ja 70 mg -hoidolla 2,6 (95 % lv: 1,6, 4,5). Potilailla, joilla vähintään kaksi estolääkehoitoa oli epäonnistunut, osuus oli 140 mg -hoidolla 41,3 %, vetosuhde 4,2 (95 % lv: 2,2, 7,9); 70 mg -hoidolla osuus oli 35,6 %, vetosuhde 3,5 (95 % lv: 1,8, 6,6), vs. lumehoidon osuus 14,2 %.
Noin 41 %:lla tutkimuksen potilaista oli lääkityksen liikakäyttöä. Erenumabi 140 mg- ja lumehoidon välinen sekä erenumabi 70 mg- ja lumehoidon välinen ero MMD-päivien vähentämisessä oli molemmissa tapauksissa ‑3,1 päivää (95 % lv: ‑4,8, ‑1,4) ja migreenispesifisen akuuttilääkityksen päivien vähentämisessä 140 mg -hoidolla ‑2,8 (95 % lv: ‑4,2, ‑1,4) ja 70 mg hoidolla ‑3,3 (95 % lv: ‑4,7, ‑1,9). MMD-päivien vähenemistä vähintään 50 % todettiin suuremmalla osuudella erenumabi-ryhmän potilaista kuin lumeryhmän potilaista (140 mg ‑hoidolla 34,6 %, 70 mg -hoidolla 36,4 % vs. lumehoidolla 17,7 %), 140 mg ‑hoidon vetosuhde 2,5 (95 % lv: 1,3, 4,9) ja 70 mg ‑hoidon vetosuhde 2,7 (95 % lv: 1,4, 5,2).
Teho säilyi 1 vuoteen asti tutkimuksen 1 avoimessa jatkotutkimuksessa, jossa potilaat saivat 70 mg ja/tai 140 mg erenumabia. 74,1 % potilaista osallistui koko 52 viikon jatkotutkimukseen. Kun molemmilla annoksilla saadut tulokset yhdistettiin, havaittiin 52 viikon jälkeen -9,3 MMD:n väheneminen suhteessa päätutkimuksen lähtötilanteeseen. 59 % koko tutkimukseen osallistuneista potilaista saavutti 50 % vasteen tutkimuksen viimeisen kuukauden aikana.
Episodinen migreeni
Tutkimus 2
Erenumabi arvioitiin episodisen migreenin estohoitoon satunnaistetussa, 24 viikon pituisessa, lumekontrolloidussa, kaksoissokkoutetussa monikeskustutkimuksessa potilailla, joilla oli migreeni auraoirein tai ilman (4–14 migreenipäivää kuukaudessa).
955 potilasta satunnaistettiin suhteessa 1:1:1 saamaan 140 mg (n = 319) tai 70 mg erenumabia (n = 317) tai lumelääkettä (n = 319). Akuuttihoito päänsäryn lievittämiseen oli tutkimuksen aikana sallittua.
Tutkimusryhmien demografiset tiedot ja taudin lähtötilannetiedot olivat tasapainossa ja toisiinsa verrattavissa. Potilaiden iän mediaani oli 42 vuotta, 85 % oli naisia ja 89 % valkoihoisia. Migreenikohtausten esiintymistiheyden keskiarvo oli lähtötilanteessa noin 8 migreenipäivää kuukaudessa. Yhteensä 39 %:lla potilaista vähintään yksi aiempi estolääkehoito oli epäonnistunut tehon puutteen tai huonon siedettävyyden vuoksi. 140 mg -ryhmässä yhteensä 294 potilasta (92 %), 70 mg -ryhmässä 287 (91 %) ja lumeryhmässä 284 potilasta (89 %) suoritti kaksoissokkoutetun vaiheen loppuun.
Migreenipäivien tiheys lähtötilanteeseen nähden pieneni erenumabia saaneilla kliinisesti merkittävästi ja tilastollisesti merkitsevästi enemmän kuukausina 4–6 (kuva 2) verrattuna lumehoitoa saaneisiin potilaisiin. Ero lumeeseen nähden havaittiin kuukaudesta 1 alkaen.
Kuva 2 Muutos lähtötilanteesta: kuukausittaiset migreenipäivät ajan myötä tutkimuksessa 2 (sisältäen ensisijaisen päätetapahtuman kuukausina 4, 5 ja 6)
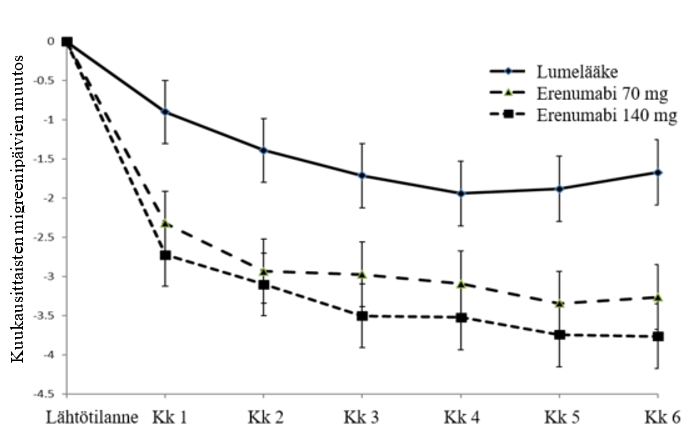
Taulukko 3 Muutos lähtötilanteesta tehossa ja potilaan raportoimissa tuloksissa viikkojen 13–24 kohdalla tutkimuksessa 2
Aimovig (erenumabi) 140 mg (n = 318) | Aimovig (erenumabi) 70 mg (n = 312) | Lume (n = 316) | Hoitojen ero (95 % lv) | p-arvo | |
| Tehotulokset | |||||
MMD Keskimuutos (95 % lv) Lähtötilanne (SD) | ‑3,7 (‑4,0, ‑3,3) 8,3 (2,5) | ‑3,2 (‑3,6, ‑2,9) 8,3 (2,5) | ‑1,8 (‑2,2, ‑1,5) 8,2 (2,5) | 70 mg: ‑1,4 (‑1,9, ‑0,9) 140 mg: ‑1,9 (‑2,3, ‑1,4) | Molemmat < 0,001a |
≥ 50 % MMD –vasteen saaneet Prosenttiosuus [%] | 50,0 % | 43,3 % | 26,6 % | n/a | Molemmat: < 0,001a,d |
≥ 75 % MMD –vasteen saaneet Prosenttiosuus [%] | 22,0 % | 20,8 % | 7,9 % | n/a | n/ab |
Migreenispesifisen akuuttilääkityksen päiviä/kk Keskimuutos (95 % lv) | ‑1,6 (‑1,8, ‑1,4) | ‑1,1 (‑1,3, ‑0,9) | ‑0,2 (‑0,4, 0,0) | 70 mg: ‑0,9 (‑1,2, ‑0,6) 140 mg: ‑1,4 (‑1,7, ‑1,1) | Molemmat: < 0,001a |
| Lähtötilanne (SD) | 3,4 (3,5) | 3,2 (3,4) | 3,4 (3,4) | ||
| Potilaan raportoimat tulokset | |||||
HIT‑6 Keskimuutosc (95 % lv) | ‑6,9 (‑7,6, ‑6,3) | ‑6,7 (‑7,4, ‑6,0) | ‑4,6 (‑5,3, ‑4,0) | 70 mg: ‑2,1 (‑3,0, ‑1,1) 140 mg: ‑2,3 (‑3,2, ‑1,3) | n/ab |
MIDAS-kokonaispisteet (muokattu) Keskimuutosc (95 % lv) | ‑7,5 (‑8,3, ‑6,6) | ‑6,7 (‑7,6, ‑5,9) | ‑4,6 (‑5,5, ‑3,8) | 70 mg: ‑2,1 (‑3,3, ‑0,9) 140 mg: ‑2,8 (‑4,0, ‑1,7) | n/ab |
lv = luottamusväli; MMD = kuukausittaiset migreenipäivät; HIT-6 = Headache Impact Test; MIDAS = Migraine Disability Assessment; n/a = not applicable. a Toissijaisten päätetapahtumien osalta kaikki p-arvot ilmoitetaan korjaamattomina, ja ne ovat tilastollisesti merkitseviä monivertailukorjauksen jälkeen. b Eksploratiivisten päätetapahtumien p-arvoa ei esitetty. c HIT-6: Muutos ja väheneminen lähtötilanteesta arvioitiin 12-viikkoisen kaksoissokkoutetun hoitovaiheen viimeisten 4 viikon aikana. MIDAS: Muutos ja väheneminen lähtötilanteesta arvioitiin 24 viikon aikana. Tiedot kerättiin 1 kk takautuvasti. d p-arvo laskettiin vetosuhteiden (OR, odds ratio) perusteella. | |||||
Potilailla, joilla vähintään yksi estolääkehoito oli epäonnistunut, hoitojen ero MMD-päivien vähenemisessä oli erenumabi 140 mg -ryhmän ja lumeryhmän välillä ‑2,5 (95 % lv: ‑3,4, ‑1,7) ja erenumabi 70 mg -ryhmän ja lumeryhmän välillä ‑2,0 (95 % lv: ‑2,8, ‑1,2). MMD-päivien vähenemistä vähintään 50 % todettiin suuremmalla osuudella erenumabi-ryhmän potilaista kuin lumeryhmän potilaista (140 mg -hoidolla 39,7 % potilaista ja 70 mg -hoidolla 38,6 %). 140 mg -hoidolla vetosuhde (odds ratio) oli 3,1 [95 % lv: 1,7, 5,5] ja 2,9 [95 %:n lv: 1,6, 5,3]).
Teho säilyi 1 vuoteen asti tutkimuksen 2 aktiivisessa uudelleensatunnaistamisvaiheessa. Aktiivista hoitovaihetta (ATP, active treatment phase) varten potilaat satunnaistettiin uudestaan saamaan joko 70 mg tai 140 mg erenumabia. 79,8 % potilaista oli tutkimuksessa mukana viikon 52 loppuun asti. Kuukausittaisten migreenipäivien väheneminen lähtötilanteesta viikkoon 52 saakka oli -4,22 päivää 70 mg saaneiden ATP-ryhmässä ja -4,64 päivää 140 mg saaneiden ATP-ryhmässä. Viikolla 52 vähintään 50 %:in vähenemisen migreenipäivien lukumäärässä lähtötilanteeseen nähden saavuttaneiden potilaiden osuudet olivat 61,0 % ATP-ryhmässä, joka sai 70 mg, ja 64,9 % ATP-ryhmässä, joka sai 140 mg.
Pitkäaikaisseurannantutkimus
Lumekontrolloidun tutkimuksen jälkeen, 383 potilasta jatkoivat 5 vuoden avoimen vaiheen jatkotutkimukseen saamaan aluksi 70 mg erenumabia (keskimääräinen altistus: 2,0 vuotta), joista 250 potilaan annos nostettiin 140 mg:aan erenumabia (keskimääräinen altistus: 2,7 vuotta). 214 potilasta suoritti 5 vuoden avoimen vaiheen jatkotutkimuksen loppuun. 383 potilaasta, 168 potilasta (43,9 %) keskeyttivät tutkimuksen yleisimmällä syyllä potilaan pyynnöstä (84 potilasta; 21,9 %), haittatapahtumat (19 potilasta; 5,0 %), kadonnut seurannasta (14 potilasta; 3,7 %) ja tehonpuute (12 potilasta; 3,1 %). Nämä tulokset osoittavat, että teho säilyi 5 vuotta avoimen vaiheen tutkimuksessa.
Tutkimus 3: Tutkimus potilailla, joilla oli 2-4 aiemmin epäonnistunutta tai soveltumatonta migreenin estolääkehoitoa.
246 aikuispotilasta, joilla oli episodinen migreeni, satunnaistettiin suhteessa 1:1 saamaan joko 140 mg erenumabia (n = 121) tai lumelääkettä (n = 125) 12 viikon ajan. Kolme potilasta (erenumabi: 2, lumelääke: 1) jätettiin ensisijaisen analyysin ulkopuolelle, koska he eivät saaneet tutkimushoitoa. Kaksoissokkoutetun hoidon neljän viimeisen viikon aikana 30,3 % (36/119) erenumabiryhmän potilaista saavutti vähintään 50 % MMD-päivien vähenemisen lähtötasosta verrattuna lumelääkeryhmän 13,7 % (17/124) (p = 0,002).
Tutkimus 4: Tutkimus siedettävyyden (ensisijainen päätetapahtuma) ja tehon arvioimiseksi topiramaattiin verrattuna
777 aikuispotilasta, joilla oli episodinen tai krooninen migreeni, satunnaistettiin suhteessa 1:1 saamaan joko erenumabia (70 mg tai 140 mg, n = 389) tai topiramaattia 50 - 100 mg (n = 388) 24 viikon ajan (kaksoissokkoutettu hoitovaihe). Turvallisuus- ja tehotiedot yhdistettiin potilaista, jotka saivat 70 mg ja 140 mg: n erenumabiannoksia, ja niitä verrattiin topiramaattia saavien potilaiden tietoihin.
Erenumabin siedettävyys oli parempi haittatapahtumien aiheuttaman hoidon keskeyttämisen määrän suhteen verrattuna topiramaattiin (erenumabi: 10,5 %, topiramaatti: 38,9 %; p < 0,001; ensisijainen päätetapahtuma). Lisäksi 55,4 %:lla erenumabiryhmän potilaista MMD-päivien määrä väheni vähintään 50 % lähtötasosta tutkimuksen kolmen viimeisen kuukauden aikana, kun taas topiramaattiryhmässä vastaava luku oli 31,2 % (p < 0,001).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt lykkäyksen velvoitteelle toimittaa tutkimustulokset Aimovig-valmisteen käytöstä migreenipäänsäryn estohoidossa yhdessä tai useammassa pediatrisessa potilasryhmässä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Erenumabin kinetiikka on ei-lineaarinen CGRP-reseptoriin sitoutumisen vuoksi. Terapeuttisesti relevanteilla annoksilla ihon alle 4 viikon välein annetun erenumabin farmakokinetiikka on kuitenkin vallitsevasti lineaarinen, sillä sitoutuminen CGRP-reseptoriin saturoituu. Kun terveille koehenkilöille annettiin ihon alle 140 mg annos kerran kuukaudessa, Cmax-keskiarvo (keskihajonta [SD]) oli 15,8 (4,8) mikrog/ml ja AUClast-keskiarvo (SD) oli 505 (139) vrk*mikrog/ml. Annoksella 70 mg kerran kuukaudessa Cmax-keskiarvo (SD) oli 6,1 (2,1) µg/ml ja AUClast-keskiarvo (SD) oli 159 (58) vrk*mikrog/ml.
Ihon alle 4 viikon välein annettujen 140 mg annosten havaittiin johtavan minimipitoisuuksien alle 2-kertaiseen kertymiseen seerumissa, ja seerumin minimipitoisuudet lähestyivät vakaata tilaa 12 viikon annostelun jälkeen.
Imeytyminen
Kun terveille aikuisille annettiin ihon alle 140 mg tai 70 mg kerta-annos erenumabia, seerumin huippupitoisuuksien mediaani saavutettiin 4–6 vuorokaudessa, ja absoluuttinen biologinen hyötyosuus oli arviolta 82 %.
Jakautuminen
Kun valmistetta annettiin 140 mg kerta-annoksena laskimoon, terminaalivaiheen (Vz) jakautumistilavuuden keskiarvo (SD) oli arviolta 3,86 (0,77) l.
Biotransformaatio/eliminaatio
Erenumabilla havaittiin kaksi eliminaatiovaihetta. Pieninä pitoisuuksina eliminaatio tapahtuu lähinnä saturoituvan kohteeseen (CGRP-R) sitoutumisen kautta, kun taas suurempina pitoisuuksina erenumabi eliminoituu suuressa määrin epäspesifisen proteolyysireitin kautta. Koko annostelujakson ajan erenumabi eliminoituu lähinnä epäspesifisen proteolyysireitin kautta, ja efektiivinen puoliintumisaika on 28 vrk.
Erityisryhmät
Munuaisten vajaatoimintapotilaat
Potilaita, joilla on vaikea munuaisten vajaatoiminta (eGFR < 30 ml/min/1,73 m2) ei ole tutkittu. Kliinisten Aimovig-tutkimusten integroiduista tiedoista tehtiin populaatiofarmakokinetiikan analyysi, jossa erenumabin farmakokinetiikassa ei havaittu eroa lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien ja normaalin munuaistoiminnan omaavien välillä (ks. kohta Annostus ja antotapa).
Maksan vajaatoimintapotilaat
Maksan vajaatoimintapotilaita koskevia tutkimuksia ei ole tehty. Erenumabi on humaani monoklonaalinen vasta-aine, eikä se siksi metaboloidu sytokromi P450 -entsyymien vaikutuksesta. Maksapuhdistuma ei ole erenumabille merkittävä eliminaatioreitti (ks. kohta Annostus ja antotapa).
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, toistuvan altistuksen aiheuttamaa toksisuutta sekä lisääntymis- ja kehitystoksisuutta koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Erenumabilla ei ole tehty karsinogeenisuustutkimuksia. Erenumabi ei ole farmakologisesti aktiivinen jyrsijöillä. Sillä on biologista vaikutusta jaavanmakakiapinoilla, mutta tämä laji ei sovellu malliksi tuumorigeenisuuden riskin arviointiin. Erenumabin mutageenisuutta ei ole arvioitu. Monoklonaalisten vasta-aineiden ei kuitenkaan oleteta muuttavan DNA:ta eikä kromosomeja.
Toistuvan annostelun toksisuustutkimuksissa ei havaittu haittavaikutuksia sukukypsillä apinoilla, joille annettiin enintään 150 mg/kg valmistetta ihon alle kahdesti viikossa enimmillään 6 kk ajan, jolloin systeeminen altistus oli seerumin AUC-arvon perusteella enintään 123 kertaa suurempi kuin kliininen annos 140 mg neljän viikon välein tai 246 kertaa suurempi kuin kliininen annos 70 mg neljän viikon välein. Hedelmällisyyden korvaaviin markkereihin (sukuelinten anatomiaan tai histologiaan kohdistuvia patologisia muutoksia) kohdistuvia haittavaikutuksia ei myöskään havaittu.
Jaavanmakakiapinoilla toteutetussa lisääntymistutkimuksessa ei myöskään havaittu raskauteen, alkion-/sikiönkehitykseen eikä postnataaliseen kehitykseen (6 kk ikään asti havainnoituna) kohdistuneita vaikutuksia, kun erenumabia annettiin koko tiineyden ajan altistustasoilla, jotka AUC-arvon perusteella olivat noin 17 kertaa suuremmat kuin erenumabia 140 mg neljän viikon välein saavien ja noin 34 kertaa suuremmat kuin 70 mg neljän viikon välein saavien potilaiden altistustasot. Vastasyntyneillä apinoilla havaittiin syntymän aikaan mitattavissa olevia erenumabipitoisuuksia seerumissa. Tämä vahvistaa, että erenumabi läpäisee istukan, kuten muutkin IgG-vasta-aineet.
Farmaseuttiset tiedot
Apuaineet
Sakkaroosi
Polysorbaatti 80
Natriumhydroksidi (pH:n säätelyyn)
Etikkahappo, väkevä
Injektionesteisiin käytettävä vesi
Yhteensopimattomuudet
Koska yhteensopivuustutkimuksia ei ole tehty, tätä lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa.
Kestoaika
3 vuotta
Säilytys
Esitäytetty ruisku
Säilytä jääkaapissa (2 °C – 8 °C). Ei saa jäätyä.
Pidä esitäytetty ruisku ulkopakkauksessa. Herkkä valolle.
Kun Aimovig on otettu pois jääkaapista, se on käytettävä 7 vrk kuluessa, kun se on säilytetty huoneenlämmössä (alle 25 °C) tai hävitettävä. Jos sitä on säilytetty korkeammassa lämpötilassa tai pidemmän aikaa, se on hävitettävä.
Esitäytetty kynä
Säilytä jääkaapissa (2 °C – 8 °C). Ei saa jäätyä.
Pidä esitäytetty kynä ulkopakkauksessa. Herkkä valolle.
Kun Aimovig on otettu pois jääkaapista, se on käytettävä 7 vrk kuluessa, kun se on säilytetty huoneenlämmössä (alle 25 °C) tai hävitettävä. Jos sitä on säilytetty korkeammassa lämpötilassa tai pidemmän aikaa, se on hävitettävä.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
AIMOVIG injektioneste, liuos, esitäytetty kynä
70 mg (L:ei) 1 kpl (1 ml (70 mg/ml)) (323,84 €)
140 mg (L:ei) 1 kpl (1 ml (140 mg/ml)) (323,84 €)
PF-selosteen tieto
Esitäytetty ruisku
Aimovig toimitetaan esitäytetyssä ruiskussa (1 ml, tyypin 1 lasia), jossa on ruostumattomasta teräksestä valmistettu neula ja neulansuojus (lateksia sisältävää kumia).
Aimovig on saatavana pakkauksissa, joissa on 1 esitäytetty ruisku.
Esitäytetty kynä
Aimovig toimitetaan esitäytetyssä kynässä (1 ml, tyypin 1 lasia), jossa on ruostumattomasta teräksestä valmistettu neula ja neulansuojus (lateksia sisältävää kumia).
Aimovig on saatavana pakkauksissa, joissa on 1 esitäytetty kynä, tai monipakkauksissa, joissa on 3 (3 x 1) esitäytettyä kynää.
Kaikkia pakkauskokoja ei välttämättä ole markkinoilla.
Valmisteen kuvaus:
Liuos on kirkas tai opalisoiva, väritön tai hiukan kellertävä.
Käyttö- ja käsittelyohjeet
Ennen lääkkeenantoa liuos on tarkastettava silmämääräisesti. Liuosta ei saa pistää, jos se on sameaa tai selvästi keltaista tai jos siinä on hiutaleita tai hiukkasia.
Esitäytetty ruisku
Injektiokohdan epämukavuuden välttämiseksi esitäytetyn ruiskun / esitäytettyjen ruiskujen on annettava olla huoneenlämmössä (alle 25 °C) vähintään 30 minuutin ajan ennen pistämistä. Se on myös suojattava suoralta auringonvalolta. Esitäytetyn ruiskun / esitäytettyjen ruiskujen koko sisältö / sisällöt on pistettävä. Ruiskua / ruiskuja ei saa ravistaa eikä lämmittää esim. kuumassa vedessä tai mikroaaltouunissa.
Esitäytetty kynä
Injektiokohdan epämukavuuden välttämiseksi esitäytetyn kynän / esitäytettyjen kynien on annettava olla huoneenlämmössä (alle 25 °C) vähintään 30 minuutin ajan ennen pistämistä. Se on myös suojattava suoralta auringonvalolta. Esitäytetyn kynän /esitäytettyjen kynien koko sisältö / sisällöt on pistettävä. Kynää / kyniä ei saa ravistaa eikä lämmittää esim. kuumassa vedessä tai mikroaaltouunissa.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
AIMOVIG injektioneste, liuos, esitäytetty kynä
70 mg 1 kpl
140 mg 1 kpl
- Rajoitettu peruskorvaus lääkärin lausunnolla (40 %). Atogepantti, eptinetsumabi, erenumabi, fremanetsumabi, galkanetsumabi ja rimegepantti (migreenin estohoito): Aikuisten migreenin estohoito erityisin edellytyksin (3007).
ATC-koodi
N02CD01
Valmisteyhteenvedon muuttamispäivämäärä
19.12.2024
Yhteystiedot
 NOVARTIS FINLAND OY
NOVARTIS FINLAND OY Revontulenkuja 1
02100 Espoo
010 613 3200
www.novartis.fi
Lääkeinformaatiopalvelu 010 6133 210,
medinfo.nordics@novartis.com