
KADCYLA kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos 100 mg, 160 mg
 Lääketurva
Lääketurva
Riskienminimointimateriaalit
Terveydenhuollon ammattilainen
Opas sisältää tärkeää turvallisuuteen liittyvää tietoa terveydenhuollon ammattilaisille Kadcyla-lääkehoidosta lääkitysvirheiden ehkäisemiseksi.
Vaikuttavat aineet ja niiden määrät
Kadcyla 100 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo kuiva-ainetta välikonsentraatiksi infuusionestettä varten, liuosta, sisältää 100 mg trastutsumabiemtansiinia. Käyttökuntoon saattamisen jälkeen yksi 5 ml:n injektiopullo sisältää 20 mg/ml trastutsumabiemtansiinia (ks. kohta Käyttö- ja käsittelyohjeet).
Kadcyla 160 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Yksi injektiopullo kuiva-ainetta välikonsentraatiksi infuusionestettä varten, liuosta, sisältää 160 mg trastutsumabiemtansiinia. Käyttökuntoon saattamisen jälkeen yksi 8 ml:n injektiopullo sisältää 20 mg/ml trastutsumabiemtansiinia (ks. kohta Käyttö- ja käsittelyohjeet).
Apuaineet, joiden vaikutus tunnetaan
Yksi 100 mg injektiopullo sisältää 1,38 mg natriumia ja 1,1 mg polysorbaattia 20.
Yksi 160 mg injektiopullo sisältää 2,24 mg natriumia ja 1,7 mg polysorbaattia 20.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Trastutsumabiemtansiini on trastutsumabia sisältävä vasta‑ainekonjugaatti, jossa trastutsumabi on nisäkkään (kiinanhamsterin munasarja) solususpensioviljelmässä tuotettu humanisoitu monoklonaalinen IgG1-vasta-aine, joka on liitetty stabiilin tioeetterilinkkerin MCC:n (4‑[N‑maleimidometyyli]sykloheksaani‑1‑karboksylaatin) muodostamalla kovalenttisidoksella mikrotubulusten muodostumista estävään DM1:een.
Lääkemuoto
Kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos.
Kliiniset tiedot
Käyttöaiheet
Varhaisvaiheen rintasyöpä
Kadcyla on tarkoitettu käytettäväksi ainoana lääkeaineena aikuisten potilaiden varhaisvaiheen HER2-positiivisen rintasyövän adjuvanttihoitoon, kun potilaalla on invasiivinen jäännöstauti rinnassa ja/tai imusolmukkeissa, potilaan saatua taksaanipohjaista ja HER2-kohdennettua neoadjuvanttihoitoa.
Metastasoitunut rintasyöpä
Kadcyla on tarkoitettu käytettäväksi ainoana lääkeaineena aikuisten potilaiden HER2-positiivisen, leikkaushoitoon soveltumattoman paikallisesti edenneen tai metastasoituneen rintasyövän hoitoon, kun potilas on aiemmin saanut trastutsumabia ja jotakin taksaania erikseen tai yhdistelmänä. Potilaan
-
on pitänyt aiemmin saada hoitoa paikallisesti edenneeseen tai metastasoituneeseen tautiin tai
-
taudin on pitänyt uusiutua adjuvanttihoidon aikana tai kuuden kuukauden kuluessa adjuvanttihoidon päättymisestä.
Ehto
Valmistetta saa määrätä vain syöpäpotilaiden hoitoon perehtynyt lääkäri ja sitä saa antaa vain syöpäpotilaiden hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa.
Annostus ja antotapa
Kadcylaa saa määrätä vain syöpäpotilaiden hoitoon perehtynyt lääkäri ja sitä saa antaa infuusiona laskimoon vain syöpäpotilaiden hoitoon perehtyneen terveydenhuollon ammattilaisen valvonnassa (eli valmius hoitaa allergiset tai anafylaktiset infuusioreaktiot hoitoympäristössä, jossa kaikki elvytysvälineet ovat välittömästi saatavilla (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet)).
Trastutsumabiemtansiinihoitoa saavilla potilailla on oltava immunohistokemiallisella menetelmällä (IHC3+) tai in vitro -diagnostisella (IVD) in situ hybridisaatiomenetelmällä (ISH) tai fluoresenssi in situ ‑hybridisaatiomenetelmällä (FISH) suhdeluku ≥ 2,0 määritelty HER2- positiivinen kasvain. Jos CE-merkittyä IVD-testiä ei ole käytettävissä, HER2-status on määritettävä jollakin toisella validoidulla testimenetelmällä.
Lääkitysvirheiden välttämiseksi on tärkeää varmistaa injektiopullon etiketistä, että valmistettava ja annettava lääkevalmiste on Kadcyla (trastutsumabiemtansiini) eikä toinen trastutsumabia sisältävä valmiste (esim. trastutsumabi tai trastutsumabi-derukstekaani).
Annostus
Suositeltu trastutsumabiemtansiiniannos on 3,6 mg/painokg, joka annetaan infuusiona laskimoon 3 viikon välein (21 vrk:n hoitosykli).
Alkuannos on annettava 90 minuutin kestoisena infuusiona laskimoon. Potilasta on tarkkailtava infuusion annon aikana ja vähintään 90 minuutin ajan ensimmäisen infuusion jälkeen kuumeen, vilunväristysten tai muiden infuusioon liittyvien reaktioiden havaitsemiseksi. Infuusiokohtaa on seurattava tarkoin annon aikana mahdollisen ihonalaisen infiltraation havaitsemiseksi. Valmisteen markkinoille tultua on ekstravasaation seurauksena havaittu viivästyneesti ilmenneitä epidermiksen vaurioita tai epidermaalista nekroosia (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Jos potilas sieti aiemman infuusion hyvin, seuraavat trastutsumabiemtansiiniannokset voidaan antaa 30 minuutin kestoisina infuusioina. Potilasta on seurattava infuusion annon aikana ja vähintään 30 minuutin ajan infuusion jälkeen.
Jos potilaalle kehittyy infuusioon liittyviä oireita, trastutsumabiemtansiini-infuusion antonopeutta pitää hidastaa tai anto keskeyttää (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset). Jos potilaalle ilmaantuu hengenvaarallisia infuusioreaktioita, trastutsumabiemtansiinin anto pitää lopettaa.
Hoidon kesto
Varhaisvaiheen rintasyöpä
Potilaalle annetaan yhteensä 14 hoitosykliä, paitsi jos tauti uusiutuu tai ilmenee toksisuutta, joka ei ole hoidettavissa.
Metastasoitunut rintasyöpä
Potilaalle annetaan hoitoa, kunnes tauti etenee tai ilmenee toksisuutta, joka ei ole hoidettavissa.
Annosmuutokset
Haittavaikutusten oireiden hoito saattaa vaatia hoidon keskeyttämistä tilapäisesti, annoksen pienentämistä tai trastutsumabiemtansiinihoidon lopettamista tekstissä ja taulukoissa 1 ja 2 annettujen ohjeiden mukaisesti.
Trastutsumabiemtansiiniannosta ei saa suurentaa uudelleen, jos annosta on pienennetty.
Taulukko 1Annoksen pienentäminen
Annoksen pienentäminen | Annettava annos |
Ensimmäinen pienennetty annos | 3 mg/kg |
Toinen pienennetty annos | 2,4 mg/kg |
Jos annosta tarvitsee yhä pienentää | Lopeta hoito |
Taulukko 2 Ohjeet annoksen muuttamiseen
Annosmuutokset varhaisvaiheen rintasyövän hoidossa | ||
Haittavaikutus | Vaikeusaste | Hoidon muutos |
Trombosytopenia | Gradus 2–3 sovittuna hoitopäivänä (25 000 – < 75 000/mm3) | Älä anna trastutsumabiemtansiinia ennen kuin trombosyyttiarvo korjautuu gradukseen ≤ 1 (≥ 75 000/mm3); jatka sitten hoitoa samalla annoksella. Jos hoidon antamista on siirrettävä myöhemmäksi 2 kertaa trombosytopenian vuoksi, harkitse annoksen pienentämistä yhdellä annostasolla. |
Gradus 4 milloin tahansa < 25 000/mm3 | Älä anna trastutsumabiemtansiinia ennen kuin trombosyyttimäärä korjautuu gradukseen ≤ 1 (≥ 75 000/mm3); pienennä sitten annosta yhdellä annostasolla. | |
Suurentunut alaniinitransaminaasiarvo (ALAT) | Gradus 2–3 (> 3,0 – ≤ 20 × ULN sovittuna hoitopäivänä) | Älä anna trastutsumabiemtansiinia ennen kuin ALAT-arvo korjautuu gradukseen ≤ 1; pienennä sitten annosta yhdellä annostasolla. |
Gradus 4 (> 20 × ULN milloin tahansa) | Lopeta trastutsumabiemtansiinihoito. | |
Suurentunut aspartaattitransaminaasiarvo (ASAT) | Gradus 2 (> 3,0 – ≤ 5 × ULN sovittuna hoitopäivänä) | Älä anna trastutsumabiemtansiinia ennen kuin ASAT-arvo korjautuu gradukseen ≤ 1; jatka sitten hoitoa samalla annostasolla. |
Gradus 3 (> 5 – ≤ 20 × ULN sovittuna hoitopäivänä) | Älä anna trastutsumabiemtansiinia ennen kuin ASAT-arvo korjautuu gradukseen ≤ 1; pienennä sitten annosta yhdellä annostasolla. | |
Gradus 4 (> 20 × ULN milloin tahansa) | Lopeta trastutsumabiemtansiinihoito. | |
Hyperbilirubinemia | Kokonaisbilirubiinipitoisuus > 1,0 – ≤ 2,0 × ULN sovittuna hoitopäivänä | Älä anna trastutsumabiemtansiinia ennen kuin kokonaisbilirubiinipitoisuus korjautuu arvoon ≤ 1,0 × ULN; pienennä sitten annosta yhdellä annostasolla. |
Kokonaisbilirubiinipitoisuus > 2 × ULN milloin tahansa | Lopeta trastutsumabiemtansiinihoito. | |
Lääkkeestä aiheutunut maksavaurio | Seerumin transaminaasipitoisuus > 3 × ULN ja samanaikaisesti kokonaisbilirubiinipitoisuus > 2 × ULN | Lopeta trastutsumabiemtansiinihoito pysyvästi, jos kohonneisiin maksaentsyymi- ja bilirubiinipitoisuuksiin ei ole muuta todennäköistä syytä, esim. maksametastaasia tai samanaikaista lääkitystä. |
Nodulaarinen regeneratiivinen hyperplasia (NRH) | Kaikki gradukset | Lopeta trastutsumabiemtansiinihoito pysyvästi. |
Perifeerinen neuropatia | Gradus 3–4 | Älä anna trastutsumabiemtansiinia ennen kuin tila korjautuu gradukseen ≤ 2. |
Vasemman kammion toimintahäiriö | LVEF < 45 % | Älä anna trastutsumabiemtansiinia. Arvioi LVEF uudelleen 3 viikon kuluessa. Jos LVEF < 45 % varmistuu, lopeta trastutsumabiemtansiinihoito. |
LVEF 45 % – < 50 % ja pienentynyt ≥ 10 prosenttiyksikköä lähtötilanteesta* | Älä anna trastutsumabiemtansiinia. Arvioi LVEF uudelleen 3 viikon kuluessa. Jos LVEF on edelleen < 50 % eikä ole korjautunut < 10 prosenttiyksikköön lähtötilanteesta, lopeta trastutsumabiemtansiinihoito. | |
LVEF 45 % – < 50 % ja pienentynyt < 10 prosenttiyksikköä lähtötilanteesta* | Jatka trastutsumabiemtansiinihoitoa. Arvioi LVEF uudelleen 3 viikon kuluessa. | |
LVEF ≥ 50 % | Jatka trastutsumabiemtansiinihoitoa. | |
Sydämen vajaatoiminta | Oireinen kongestiivinen sydämen vajaatoiminta, graduksen 3–4 LVSD tai graduksen 3–4 sydämen vajaatoiminta tai graduksen 2 sydämen vajaatoiminta, johon liittyy LVEF < 45 % | Lopeta trastutsumabiemtansiinihoito. |
Keuhkotoksisuus | Interstitiaalinen keuhkosairaus tai pneumoniitti | Lopeta trastutsumabiemtansiinihoito pysyvästi. |
Sädehoitoon liittyvä pneumoniitti | Gradus 2 | Lopeta trastutsumabiemtansiinihoito, jos tilanne ei korjaudu vakiohoidolla. |
Gradus 3–4 | Lopeta trastutsumabiemtansiinihoito. | |
Annosmuutokset metastasoituneen rintasyövän hoidossa | ||
Haittavaikutus | Vaikeusaste | Hoidon muutos |
Trombosytopenia | Gradus 3 (25 000 – < 50 000/mm3) | Älä anna trastutsumabiemtansiinia ennen kuin trombosyyttimäärä korjautuu gradukseen ≤ 1 (≥ 75 000/mm3); jatka sitten hoitoa samalla annostasolla. |
Gradus 4 (< 25 000/mm3) | Älä anna trastutsumabiemtansiinia ennen kuin trombosyyttimäärä korjautuu gradukseen ≤ 1 (≥ 75 000/mm3); pienennä sitten annosta yhdellä annostasolla. | |
Suurentunut transaminaasiarvo (ALAT/ASAT) | Gradus 2 (> 2,5 – ≤ 5 × ULN) | Hoida samalla annostasolla. |
Gradus 3 (> 5 – ≤ 20 × ULN) | Älä anna trastutsumabiemtansiinia ennen kuin ASAT/ALAT-arvo korjautuu gradukseen ≤ 2; pienennä sitten annosta yhdellä annostasolla. | |
Gradus 4 (> 20 × ULN) | Lopeta trastutsumabiemtansiinihoito. | |
Hyperbilirubinemia | Gradus 2 (> 1,5 – ≤ 3 × ULN) | Älä anna trastutsumabiemtansiinia ennen kuin kokonaisbilirubiinipitoisuus korjautuu gradukseen ≤ 1; jatka sitten hoitoa sitten samalla annostasolla. |
Gradus 3 (> 3 – ≤ 10 × ULN) | Älä anna trastutsumabiemtansiinia ennen kuin kokonaisbilirubiinipitoisuus korjautuu gradukseen ≤ 1; pienennä sitten annosta yhdellä annostasolla. | |
Gradus 4 (> 10 × ULN) | Lopeta trastutsumabiemtansiinihoito. | |
Lääkkeestä aiheutunut maksavaurio | Seerumin transaminaasipitoisuus > 3 × ULN ja samanaikaisesti kokonaisbilirubiinipitoisuus > 2 × ULN | Lopeta trastutsumabiemtansiinihoito pysyvästi, jos kohonneisiin maksaentsyymi- ja bilirubiinipitoisuuksiin ei ole muuta todennäköistä syytä, esim. maksametastaasia tai samanaikaista lääkitystä. |
Nodulaarinen regeneratiivinen hyperplasia (NRH) | Kaikki gradukset | Lopeta trastutsumabiemtansiinihoito pysyvästi. |
Vasemman kammion toimintahäiriö | Oireinen kongestiivinen sydämen vajaatoiminta | Lopeta trastutsumabiemtansiinihoito. |
LVEF < 40 % | Älä anna trastutsumabiemtansiinia. Arvioi LVEF uudelleen 3 viikon kuluessa. Jos LVEF < 40 % varmistuu, lopeta trastutsumabiemtansiinihoito. | |
LVEF 40 % – ≤ 45 % ja pienentynyt ≥ 10 prosenttiyksikköä lähtötilanteesta | Älä anna trastutsumabiemtansiinia. Arvioi LVEF uudelleen 3 viikon kuluessa. Jos LVEF ei ole korjautunut 10 prosenttiyksikön sisälle lähtötilanteesta, lopeta trastutsumabiemtansiinihoito. | |
LVEF 40 % – ≤ 45 % ja pienentynyt < 10 prosenttiyksikköä lähtötilanteesta | Jatka trastutsumabiemtansiinihoitoa. Arvioi LVEF uudelleen 3 viikon kuluessa. | |
LVEF > 45 % | Jatka trastutsumabiemtansiinihoitoa. | |
Perifeerinen neuropatia | Gradus 3–4 | Älä anna trastutsumabiemtansiinia ennen kuin tila korjautuu gradukseen ≤ 2. |
Keuhkotoksisuus | Interstitiaalinen keuhkosairaus tai pneumoniitti | Lopeta trastutsumabiemtansiinihoito pysyvästi. |
ALAT = alaniinitransaminaasi; ASAT = aspartaattitransaminaasi, LVEF = vasemman kammion ejektiofraktio (left ventricular ejection fraction), LVSD = vasemman kammion systolinen toimintahäiriö (left ventricular systolic dysfunction), ULN = viitearvojen yläraja (upper limit of normal)
* Ennen trastutsumabiemtansiinihoidon aloittamista.
Annosten viivästyminen tai antamatta jääminen
Jos suunniteltu annos jää antamatta, se on annettava mahdollisimman pian odottamatta seuraavaan suunniteltuun hoitosykliin. Antoaikataulua on sovitettava siten, että annosvälinä säilyy 3 viikkoa. Seuraava annos on annettava annossuositusten mukaisesti.
Perifeerinen neuropatia
Jos potilaalla on gradus 3 tai 4 perifeerinen neuropatia, trastutsumabiemtansiinihoito on keskeytettävä tilapäisesti, kunnes perifeerinen neuropatia korjautuu gradukseen ≤ 2. Uusintahoidossa saattaa olla tarpeen harkita annoksen pienentämistä annettujen ohjeiden mukaisesti (ks. taulukko 1).
Erityiset potilasryhmät
Iäkkäät potilaat
≥ 65‑vuotiaiden potilaiden annosta ei tarvitse muuttaa. Tietoja ei ole riittävästi hoidon turvallisuuden ja tehon varmistamiseksi ≥ 75‑vuotiailla potilailla, koska tästä potilasryhmästä on vähän tietoja. MO28231-tutkimuksessa 345 potilaasta tehty potilasryhmäanalyysi kuitenkin osoitti, että ≥ 65‑vuotiailla gradusten 3, 4 ja 5 haittavaikutusten, vakavien haittavaikutusten ja hoidon lopettamiseen/keskeyttämiseen johtavien haittavaikutusten ilmaantuvuus on suurempi, mutta graduksen 3 ja vakavampien hoitoon liittyviksi luokiteltujen haittavaikutusten ilmaantuvuus on samankaltainen.
Populaatiofarmakokineettisen analyysin mukaan iällä ei ole kliinisesti merkitsevää vaikutusta trastutsumabiemtansiinin farmakokinetiikkaan (ks. kohdat Farmakodynamiikka ja Farmakokinetiikka).
Munuaisten vajaatoiminta
Lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavien potilaiden aloitusannosta ei tarvitse muuttaa (ks. kohta Farmakokinetiikka). Vaikeaa munuaisten vajaatoimintaa sairastavien potilaiden mahdollista annosmuutostarvetta ei voida määritellä, koska tietoja ei ole riittävästi, joten vaikeaa munuaisten vajaatoimintaa sairastavia potilaita on tarkkailtava huolellisesti.
Maksan vajaatoiminta
Lievää tai keskivaikeaa maksan vajaatoimintaa sairastavien potilaiden aloitusannosta ei tarvitse muuttaa. Trastutsumabiemtansiinia ei ole tutkittu vaikeaa maksan vajaatoimintaa sairastavilla potilailla. Maksan vajaatoimintaa sairastavien potilaiden hoidossa pitää olla varovainen, koska trastutsumabiemtansiinin käytössä tiedetään esiintyvän maksatoksisuutta (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet ja Farmakokinetiikka).
Pediatriset potilaat
Turvallisuutta ja tehoa lasten ja alle 18 vuoden ikäisten nuorten hoidossa ei ole varmistettu, koska ei ole asianmukaista käyttää Kadcyla-valmistetta pediatristen potilaiden rintasyövän hoitoon.
Antotapa
Kadcyla annetaan laskimoon. Terveydenhuollon ammattilaisen on saatettava trastutsumabiemtansiini käyttökuntoon ja annettava se infuusiona laskimoon. Sitä ei saa antaa nopeana infuusiona eikä boluksena laskimoon.
Ks. kohdasta Käyttö- ja käsittelyohjeet ohjeet lääkevalmisteen saattamisesta käyttökuntoon ja laimentamisesta ennen lääkkeen antoa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Jäljitettävyys
Biologisten lääkevalmisteiden jäljitettävyyden parantamiseksi on annetun valmisteen nimi ja eränumero dokumentoitava selkeästi.
Lääkitysvirheiden välttämiseksi on tärkeää varmistaa injektiopullon etiketistä, että valmistettava ja annettava lääkevalmiste on Kadcyla (trastutsumabiemtansiini) eikä toinen trastutsumabia sisältävä valmiste (esim. trastutsumabi tai trastutsumabi-derukstekaani).
Trombosytopenia
Trombosytopeniaa eli verihiutaleiden määrän vähenemistä raportoitiin yleisesti trastutsumabiemtansiinihoidon yhteydessä, ja se oli yleisin hoidon lopettamiseen, annoksen pienentämiseen ja hoidon keskeyttämiseen johtanut haittavaikutus (ks. kohta Haittavaikutukset). Trombosytopenian ilmaantuvuus ja vaikeusaste olivat kliinisissä tutkimuksissa suuremmat aasialaisilla potilailla (ks. kohta Haittavaikutukset).
Trombosyyttimäärää suositellaan seuraamaan ennen kutakin trastutsumabiemtansiiniannosta. Jos potilaalla on trombosytopenia (≤ 100 000/mm3) ja potilas käyttää veren hyytymistä estävää lääkehoitoa (esim. varfariinia, hepariinia, pienimolekyylipainoista hepariinia), potilasta on seurattava huolellisesti trastutsumabiemtansiinihoidon aikana. Trastutsumabiemtansiinia ei ole tutkittu potilailla, joiden hoitoa edeltävä trombosyyttimäärä oli ≤ 100 000/mm3. Jos trombosyyttimäärä vähenee gradukseen 3 tai enemmän (< 50 000/mm3), älä anna trastutsumabiemtansiinia ennen kuin trombosyyttimäärä korjautuu gradukseen 1 (≥ 75 000/mm3) (ks. kohta Annostus ja antotapa).
Verenvuodot
Trastutsumabiemtansiinihoidossa on raportoitu verenvuototapahtumia, mukaan lukien keskushermoston, hengityselimistön ja maha-suolikanavan verenvuotoja. Osa näistä verenvuototapahtumista on johtanut potilaan kuolemaan. Joissakin havaituissa tapauksissa potilaalla oli trombosytopeniaa tai potilas sai myös veren hyytymistä tai verihiutaleiden aggregaatiota estävää hoitoa, mutta muissa tapauksissa ei ollut tunnettuja lisäriskitekijöitä. Näiden lääkeaineiden käytössä on oltava varovainen, ja on harkittava lisäseurantaa, jos samanaikainen käyttö on sairauden hoidon kannalta välttämätöntä.
Maksatoksisuus
Kliinisissä tutkimuksissa annetun trastutsumabiemtansiinihoidon aikana on havaittu maksatoksisuutta, joka on ilmennyt pääasiassa oireettomana seerumin transaminaasipitoisuuksien suurenemisena (gradus 1–4 transaminiitti) (ks. kohta Haittavaikutukset). Transaminaasipitoisuuden suureneminen oli yleensä ohimenevää ja suurimmillaan 8. päivänä hoidon antamisen jälkeen, minkä jälkeen se korjautui ennen seuraavaa hoitosykliä gradukseen 1 tai sen alle. Myös kumulatiivinen vaikutus transaminaasipitoisuuksiin on havaittu (gradus 1–2 ALAT-/ASAT-poikkeavuuksien määrä lisääntyy hoitosyklien määrän lisääntyessä).
Potilaiden suurentuneet transaminaasipitoisuudet korjautuivat useimmiten gradukseen 1 tai normaaleiksi 30 vuorokauden kuluessa viimeisen trastutsumabiemtansiiniannoksen antamisen jälkeen (ks. kohta Haittavaikutukset).
Trastutsumabiemtansiinihoitoa saaneilla potilailla on havaittu vakavia maksan ja sappitiehyiden häiriöitä, kuten maksan nodulaarista regeneratiivista hyperplasiaa ja toisinaan kuolemaan johtaneita lääkkeestä aiheutuneita maksavaurioita. Havaittuihin tapauksiin on saattanut liittyä sekoittavina tekijöinä muita samanaikaisia sairauksia ja/tai tunnetusti maksatoksisten lääkkeiden samanaikainen käyttö.
Maksan toimintaa on seurattava ennen hoidon aloittamista ja ennen kutakin annosta. Potilaat, joiden ALAT-arvo on koholla jo ennen hoidon aloittamista (esim. maksametastaasien vuoksi), saattavat olla alttiita maksavauriolle ja heillä saattaa olla suurempi graduksen 3–5 maksatapahtumien tai suurentuneiden maksan toimintakoetulosten riski. Annoksen pienentäminen tai hoidon lopettaminen suurentuneiden seerumin transaminaasipitoisuuksien ja kokonaisbilirubiinipitoisuuksien vuoksi esitetään kohdassa Annostus ja antotapa.
Trastutsumabiemtansiinihoitoa saaneille potilaille tehtyjen maksabiopsioiden perusteella on todettu maksan nodulaarista regeneratiivista hyperplasiaa. Nodulaarinen regeneratiivinen hyperplasia on harvinainen maksasairaus, jolle on tyypillistä maksan parenkyymin laaja-alainen hyvänlaatuinen muuttuminen pieniksi regeneratiivisiksi kyhmyiksi. Nodulaarinen regeneratiivinen hyperplasia saattaa johtaa ei‑kirroottiseen kohonneeseen porttilaskimopaineeseen. Nodulaarisen regeneratiivisen hyperplasian diagnoosi voidaan varmistaa vain histopatologisesti. Nodulaarisen regeneratiivisen hyperplasian mahdollisuus on huomioitava kaikilla potilailla, joilla on kohonneen porttilaskimopaineen kliinisiä oireita ja/tai joiden maksassa on tietokonetomografiassa (TT) nähtävissä kirroosityyppinen muutos, mutta joiden transaminaasipitoisuus on normaali eikä muita kirroosin merkkejä esiinny. Jos nodulaarinen regeneratiivinen hyperplasia todetaan, trastutsumabiemtansiinihoito on lopetettava pysyvästi.
Trastutsumabiemtansiinia ei ole tutkittu potilailla, joiden seerumin transaminaasipitoisuus on > 2,5 × ULN tai kokonaisbilirubiinipitoisuus on > 1,5 × ULN ennen hoidon aloittamista. Jos potilaan seerumin transaminaasipitoisuus on > 3 × ULN ja kokonaisbilirubiinipitoisuus on samaan aikaan > 2 × ULN, hoito on lopetettava pysyvästi. Maksan vajaatoimintaa sairastavien potilaiden hoidossa pitää olla varovainen (ks. kohdat Annostus ja antotapa ja Farmakokinetiikka).
Neurotoksisuus
Trastutsumabiemtansiinilla tehdyissä kliinisissä tutkimuksissa on raportoitu perifeeristä neuropatiaa, joka on ollut vaikeusasteeltaan pääasiassa gradus 1 ja useimmiten sensorista. Metastasoituneen rintasyövän potilaita, joilla oli lähtötilanteessa gradus ≥ 3 ja varhaisvaiheen rintasyövän potilaita, joilla oli lähtötilanteessa gradus ≥ 2 perifeerinen neuropatia, ei otettu mukaan kliinisiin tutkimuksiin. Jos potilaalla on graduksen 3 tai 4 perifeerinen neuropatia, trastutsumabiemtansiinihoito on keskeytettävä tilapäisesti, kunnes oireet häviävät tai korjautuvat gradukseen ≤ 2. Potilaiden on oltava jatkuvassa kliinisessä seurannassa neurotoksisten oireiden/löydösten havaitsemiseksi.
Sydämen vasemman kammion toimintahäiriö
Trastutsumabiemtansiinihoitoa saaneilla potilailla on suurentunut sydämen vasemman kammion toimintahäiriöiden kehittymisen riski. Trastutsumabiemtansiinihoitoa saaneilla potilailla on todettu < 40 %:n vasemman kammion ejektiofraktiota (LVEF), joten oireinen kongestiivinen sydämen vajaatoiminta on mahdollinen riski (ks. kohta Haittavaikutukset). Sydäntapahtumien yleisiä riskitekijöitä sekä trastutsumabia adjuvanttihoitona rintasyöpätutkimuksissa käytettäessä todettuja riskitekijöitä ovat korkeampi ikä (> 50 vuotta), lähtötilanteen pieni LVEF (< 55 %), pieni LVEF ennen adjuvanttihoitoa paklitakselilla tai sen jälkeen, aiempi tai samanaikainen verenpainelääkehoito, aiempi hoito jollakin antrasykliinillä ja korkea painoindeksi (BMI > 25 kg/m2).
Tavanomaiset sydämen toimintakokeet (kaikukardiogrammi tai tasapainotila-angiografia) on tehtävä ennen hoidon aloittamista sekä säännöllisesti (esim. kolmen kuukauden välein) hoidon aikana. Jos potilaalla on vasemman kammion toimintahäiriö, annoksen antamista on lykättävä myöhemmäksi tai hoito on tarvittaessa keskeytettävä (ks. kohta Annostus ja antotapa). Kliinisissä tutkimuksissa potilaiden LVEF oli lähtötilanteessa ≥ 50 %. Kliinisiin tutkimuksiin ei otettu mukaan potilaita, joilla oli aiemmin ollut kongestiivista sydämen vajaatoimintaa, hoitoa vaatineita vakavia sydämen rytmihäiriöitä, aiemmin sairastettu sydäninfarkti tai epästabiili angina pectoris 6 kuukauden sisällä satunnaistamisesta tai parhaillaan pitkälle edenneestä syöpäsairaudesta aiheutuvaa levossa esiintyvää hengenahdistusta. Havainnointitutkimus (BO39807) tehtiin metastasoitunutta rintasyöpää sairastavilla potilailla, joiden LVEF oli reaalimaailman (real world setting) lähtötilanteessa 40–49 %, ja tutkimuksessa havaittiin LVEF:n pienenemistä > 10 % lähtötilanteesta ja/tai kongestiivista sydämen vajaatoimintaa. Päätöksen trastutsumabiemtansiinin antamisesta metastasoitunutta rintasyöpää sairastaville potilaille, joiden LVEF on pieni, pitää perustua huolelliseen hyöty-riskiarvioon, ja näiden potilaiden sydämen toimintaa pitää seurata tarkoin (ks. kohta Haittavaikutukset).
Keuhkotoksisuus
Trastutsumabiemtansiinilla tehdyissä kliinisissä tutkimuksissa on raportoitu interstitiaalista keuhkosairautta, pneumoniitti mukaan lukien, joka on toisinaan johtanut aikuisen hengitysvaikeusoireyhtymän kehittymiseen tai potilaan kuolemaan (ks. kohta Haittavaikutukset). Oireita ja löydöksiä ovat hengenahdistus, yskä, uupumus ja keuhkoinfiltraatit.
Jos potilaalla todetaan interstitiaalinen keuhkosairaus tai pneumoniitti, trastutsumabiemtansiinihoito suositellaan lopettamaan pysyvästi, paitsi jos on kyse sädepneumoniitista adjuvanttihoidon yhteydessä, jolloin trastutsumabiemtansiinihoito pitää lopettaa pysyvästi, kun graduksen ≥ 3 tai graduksen 2 sädepneumoniittiin ei saada vastetta vakiohoidolla (ks. kohta Annostus ja antotapa).
Keuhkotapahtumien riski saattaa olla suurentunut, jos sädehoitoa keuhkoihin samanaikaisesti saavalla potilaalla on pitkälle edenneen syöpäsairauden ja muiden samanaikaisten sairauksien komplikaationa levossa esiintyvää hengenahdistusta.
Infuusioon liittyvät reaktiot
Trastutsumabiemtansiinihoitoa ei ole tutkittu potilailla, joiden trastutsumabihoito lopetettiin pysyvästi infuusioon liittyneen reaktion vuoksi. Hoitoa ei suositella tälle potilasryhmälle. Potilaita pitää tarkkailla tiiviisti infuusioon liittyvien reaktioiden havaitsemiseksi, etenkin ensimmäisen infuusion aikana.
Infuusioon liittyviä reaktioita (sytokiinien vapautumisen seurauksena), joille on tyypillistä yksi tai useampi seuraavista oireista, on raportoitu: kasvojen ja kaulan punoitus, vilunväristykset, kuume, hengenahdistus, matala verenpaine, hengityksen vinkuminen, bronkospasmi ja takykardia. Nämä oireet eivät yleensä olleet vaikea-asteisia (ks. kohta Haittavaikutukset). Nämä reaktiot hävisivät useimmilla potilailla muutamien tuntien tai vuorokauden kuluessa infuusion päättymisen jälkeen. Jos potilaalle ilmaantuu vaikea-asteinen infuusioon liittyvä reaktio, hoito on keskeytettävä, kunnes oireet ja löydökset häviävät. Uutta hoitokertaa on harkittava reaktion vaikeusasteen kliinisen arvion perusteella. Jos potilaalle ilmaantuu hengenvaarallinen infuusioon liittyvä reaktio, hoito on lopetettava pysyvästi (ks. kohta Annostus ja antotapa).
Yliherkkyysreaktiot
Trastutsumabiemtansiinihoitoa ei ole tutkittu potilailla, joiden trastutsumabihoito lopetettiin pysyvästi yliherkkyyden vuoksi. Trastutsumabiemtansiinihoitoa ei suositella tälle potilasryhmälle.
Potilasta on tarkkailtava huolellisesti sellaisten yliherkkyys-/allergisten reaktioiden havaitsemiseksi, joiden kliininen ilmenemismuoto saattaa olla sama kuin infuusioon liittyvillä reaktioilla. Trastutsumabiemtansiinilla tehdyissä kliinisissä tutkimuksissa on havaittu vakavia anafylaktisia reaktioita. Tällaisten reaktioiden hoitoon käytettävät lääkevalmisteet sekä ensiapuvälineet on oltava heti saatavilla. Jos todellinen yliherkkyysreaktio (jossa vaikeusaste pahenee seuraavien infuusioiden yhteydessä) ilmaantuu, trastutsumabiemtansiinihoito on lopetettava pysyvästi.
Infuusiokohdan reaktiot
Infuusiona laskimoon annettavan trastutsumabiemtansiinin ekstravasaatiosta voi aiheutua paikallista kipua. Poikkeustapauksissa voi ilmetä vaikea-asteisia kudosvaurioita ja epidermaalista nekroosia. Jos ekstravasaatio tapahtuu, infuusion anto on lopetettava heti ja potilas on tutkittava säännöllisin väliajoin, sillä nekroosi voi ilmetä päivien tai viikkojen kuluttua infuusiosta.
Apuaineet, joiden vaikutus tunnetaan
Tämä lääkevalmiste sisältää 1,1 mg polysorbaattia 20 yhdessä 100 mg injektiopullossa ja 1,7 mg polysorbaattia 20 yhdessä 160 mg injektiopullossa. Polysorbaatit voivat aiheuttaa allergisia reaktioita.
Tämä lääkevalmiste sisältää alle 1 mmol natriumia (23 mg) per annos eli se on olennaisesti natriumiton.
Yhteisvaikutukset
Varsinaisia yhteisvaikutustutkimuksia ei ole tehty.
Ihmisen maksan mikrosomeilla tehdyt in vitro ‑metaboliatutkimukset viittaavat siihen, että trastutsumabiemtansiinin komponentti DM1 metaboloituu pääasiassa CYP3A4:n välityksellä ja vähemmässä määrin CYP3A5:n välityksellä. Voimakkaiden CYP3A4:n estäjien (esim. ketokonatsoli, itrakonatsoli, klaritromysiini, atatsanaviiri, indinaviiri, nefatsodoni, nelfinaviiri, ritonaviiri, sakinaviiri, telitromysiini ja vorikonatsoli) samanaikaista käyttöä trastutsumabiemtansiinin kanssa on vältettävä, koska DM1-altistus ja toksisuus saattavat lisääntyä. Harkitse vaihtoehtoista lääkevalmistetta, joka ei estä CYP3A4:ää tai estää sitä vain minimaalisesti. Jos voimakkaiden CYP3A4:n estäjien samanaikaista käyttöä ei voi välttää, harkitse trastutsumabiemtansiinihoidon siirtämistä siihen saakka, kunnes voimakkaat CYP3A4:n estäjät ovat poistuneet verenkierrosta (noin kolme estäjien eliminaation puoliintumisaikaa), jos mahdollista. Jos voimakasta CYP3A4:n estäjää käytetään samanaikaisesti eikä trastutsumabiemtansiinihoitoa voi siirtää myöhemmäksi, potilasta on seurattava tarkoin haittavaikutusten havaitsemiseksi.
Raskaus ja imetys
Ehkäisy miehille ja naisille
Naisten, jotka voivat tulla raskaaksi, on käytettävä tehokasta raskauden ehkäisyä trastutsumabiemtansiinin käytön aikana sekä vähintään 7 kuukauden ajan viimeisen trastutsumabiemtansiiniannoksen jälkeen. Miespotilaiden, joiden naispuolinen kumppani voi tulla raskaaksi, on käytettävä tehokasta raskauden ehkäisyä trastutsumabiemtansiinihoidon aikana ja vähintään 4 kuukauden ajan viimeisen trastutsumabiemtansiiniannoksen jälkeen.
Raskaus
Ei ole olemassa tietoja trastutsumabiemtansiinin käytöstä raskaana oleville naisille. Raskaana olevalle naiselle annetun trastutsumabiemtansiinin trastutsumabikomponentti voi vahingoittaa sikiötä tai aiheuttaa sikiön kuoleman. Trastutsumabia raskauden aikana saaneilla naisilla on raportoitu valmisteen markkinoille tulon jälkeen lapsiveden niukkuutta, johon liittyi toisinaan kuolemaan johtanut keuhkojen vajaakehitys. DM1:n kanssa samaan maytansinoidien luokkaan kuuluvalla kemialliselta rakenteeltaan hyvin samankaltaisella maytansiinilla tehdyt eläinkokeet viittaavat siihen, että trastutsumabiemtansiinin mikrotubulusten muodostumista estävä solunsalpaajakomponentti DM1 on oletettavasti teratogeeninen ja mahdollisesti alkiotoksinen (ks. kohta Prekliiniset tiedot turvallisuudesta).
Trastutsumabiemtansiinin antamista raskaana olevalle naiselle ei suositella, ja naiselle on kerrottava sikiölle aiheutuvan haitan mahdollisuudesta ennen kuin hän tulee raskaaksi. Jos nainen tulee raskaaksi, hänen on otettava viipymättä yhteyttä lääkäriin. Jos raskaana olevaa naista hoidetaan trastutsumabiemtansiinilla, potilas suositellaan ottamaan moniammatillisen hoitotiimin tarkkaan seurantaan.
Imetys
Ei tiedetä, erittyykö trastutsumabiemtansiini ihmisen rintamaitoon. Koska monet lääkevalmisteet erittyvät rintamaitoon ja koska vakavat haittavaikutukset imetettävälle lapselle ovat mahdollisia, naisen pitää lopettaa imetys ennen trastutsumabiemtansiinihoidon aloittamista. Imetys voidaan aloittaa 7 kuukauden kuluttua hoidon päättymisen jälkeen.
Hedelmällisyys
Trastutsumabiemtansiinilla ei ole tehty lisääntymistutkimuksia eikä kehitystoksikologisia tutkimuksia.
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Trastutsumabiemtansiinilla on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Raportoitujen haittavaikutusten, kuten uupumuksen, päänsäryn, heitehuimauksen ja näön sumenemisen, merkitystä ajokyvyn tai koneidenkäyttökyvyn kannalta ei tiedetä. Jos potilaalle ilmaantuu infuusioon liittyvä reaktio (kasvojen ja kaulan punoitusta, vilunväristyksiä, kuumetta, hengitysvaikeuksia, matala verenpaine, hengityksen vinkumista, bronkospasmi ja takykardia), häntä on kehotettava olemaan ajamatta autoa ja käyttämättä koneita, kunnes oireet häviävät.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Trastutsumabiemtansiinin turvallisuutta on tutkittu kliinisissä tutkimuksissa 2 611 rintasyöpäpotilaalla. Tässä potilasryhmässä
-
yleisimmät vakavat haittavaikutukset (> 0,5 %:lla potilaista) olivat verenvuoto, kuume, trombosytopenia, hengenahdistus, vatsakipu, tuki- ja liikuntaelimistön kipu ja oksentelu.
-
yleisimmät trastutsumabiemtansiinin käytön yhteydessä esiintyneet haittavaikutukset (≥ 25 %) olivat pahoinvointi, uupumus, tuki- ja liikuntaelimistön kipu, verenvuoto, päänsärky, suurentunut transaminaasipitoisuus, trombosytopenia ja perifeerinen neuropatia. Suurin osa haittavaikutuksista oli vaikeusasteeltaan gradus 1 tai 2.
-
yleisimmät Yhdysvaltain kansallisen syöpäinstituutin yleisten haittavaikutusten luokituksen (National Cancer Institute - Common Terminology Criteria for Adverse Events [NCI-CTCAE]) mukaiset gradus ≥ 3 haittavaikutukset (> 2 %) olivat trombosytopenia, suurentuneet transaminaasipitoisuudet, anemia, neutropenia, uupumus ja hypokalemia.
Haittavaikutustaulukko
2 611:llä trastutsumabiemtansiinihoitoa saaneella potilaalla esiintyneet haittavaikutukset esitetään taulukossa 3. Haittavaikutukset luetellaan seuraavassa MedDRA-elinjärjestelmän ja yleisyysluokan mukaisesti. Tässä osiossa käytetään seuraavia yleisyysluokkia: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000), hyvin harvinainen (< 1/10 000) ja tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin). Haittavaikutukset on esitetty kussakin yleisyysluokassa ja elinjärjestelmässä haittavaikutuksen vakavuuden mukaan alenevassa järjestyksessä. Haittavaikutukset on raportoitu NCI-CTCAE-luokituksen toksisuusarvion mukaisesti.
Taulukko 3Trastutsumabiemtansiinihoitoa kliinisissä tutkimuksissa saaneiden potilaiden haittavaikutustaulukko
Elinjärjestelmä | Yleisyys | Haittavaikutukset |
Infektiot ja infestaatiot | Hyvin yleinen | Virtsatieinfektio |
Veri ja imukudos | Hyvin yleinen | Trombosytopenia, anemia |
Yleinen | Neutropenia, leukopenia | |
Immuunijärjestelmä | Yleinen | Lääkeaineyliherkkyys |
Aineenvaihdunta ja ravitsemus | Yleinen | Hypokalemia |
Psyykkiset häiriöt | Hyvin yleinen | Unettomuus |
Hermosto | Hyvin yleinen | Perifeerinen neuropatia, päänsärky |
Yleinen | Heitehuimaus, makuaistin häiriö, muistin heikkeneminen | |
Silmät | Yleinen | Kuivat silmät, konjunktiviitti, näön sumeneminen, lisääntynyt kyynelnesteen eritys |
Sydän | Yleinen | Sydämen vasemman kammion toimintahäiriö |
Verisuonisto | Hyvin yleinen | Verenvuoto |
Yleinen | Hypertensio | |
Hengityselimet, rintakehä ja välikarsina | Hyvin yleinen | Nenäverenvuoto, yskä, hengenahdistus |
Melko harvinainen | Pneumoniitti (interstitiaalinen keuhkosairaus) | |
Ruoansulatuselimistö | Hyvin yleinen | Stomatiitti, ripuli, oksentelu, pahoinvointi, ummetus, suun kuivuminen, vatsakipu |
Yleinen | Dyspepsia, verenvuoto ikenistä | |
Maksa ja sappi | Hyvin yleinen | Suurentuneet transaminaasi-pitoisuudet |
Yleinen | Suurentunut veren alkalisen fosfataasin pitoisuus, suurentunut veren bilirubiinipitoisuus | |
Melko harvinainen | Maksatoksisuus, nodulaarinen regeneratiivinen hyperplasia, kohonnut porttilaskimopaine | |
Harvinainen | Maksan vajaatoiminta | |
Iho ja ihonalainen kudos | Yleinen | Ihottuma, kutina, alopesia, kynsihäiriöt, käsi-jalkaoireyhtymä (kämmenien ja jalkapohjien erytrodysestesia), urtikaria |
Luusto, lihakset ja sidekudos | Hyvin yleinen | Tuki- ja liikuntaelimistön kipu, nivelkipu, lihaskipu |
Yleisoireet ja antopaikassa todettavat haitat | Hyvin yleinen | Uupumus, kuume, astenia |
Yleinen | Perifeerinen edeema, vilunväristykset | |
Melko harvinainen | Injektiokohdan ekstravasaatio | |
Vammat, myrkytykset ja hoitokomplikaatiot | Yleinen | Infuusioon liittyvät reaktiot |
Melko harvinainen | Sädepneumoniitti |
Taulukossa 3 esitetään metastasoitunutta rintasyöpää koskevien tutkimusten koko hoitojakson (N = 1 871; trastutsumabiemtansiinihoitosyklien lukumäärän mediaani oli 10) ja tutkimuksen KATHERINE (N = 740; hoitosyklien lukumäärän mediaani oli 14) yhdistetyt tiedot.
Valikoitujen haittavaikutusten kuvaukset
Trombosytopenia
Trombosytopeniaa eli verihiutalemäärän vähenemistä raportoitiin trastutsumabiemtansiinilla tehdyissä metastasoitunutta rintasyöpää koskeneissa kliinisissä tutkimuksissa 24,9 %:lla potilaista, ja se oli yleisin (2,6 %) hoidon lopettamiseen johtanut haittavaikutus. Trombosytopeniaa raportoitiin trastutsumabiemtansiinilla tehdyissä varhaisvaiheen rintasyöpää koskeneissa kliinisissä tutkimuksissa 28,6 %:lla potilaista, ja se oli yleisimmin kaikkina graduksina ja graduksena ≥ 3 raportoitu haittavaikutus sekä yleisimmin hoidon lopettamiseen (4,2 %), hoidon keskeyttämiseen ja annoksen pienentämiseen johtanut haittavaikutus. Suurimmalla osalla potilaista tapahtumat olivat gradus 1 tai 2 (≥ 50 000/mm3), ja trombosyyttimäärä oli pienimmillään päivänä 8, mistä se yleensä koheni gradukseen 0 tai 1 (≥ 75 000/mm3) seuraavaan hoitosuunnitelman mukaiseen annokseen mennessä. Trombosytopenian ilmaantuvuus ja vaikeusaste olivat kliinisissä tutkimuksissa suurempia aasialaisilla potilailla. Graduksen 3 tai 4 tapahtumien (< 50 000/mm3) ilmaantuvuus oli etnisestä taustasta riippumatta trastutsumabiemtansiinihoitoa saaneilla metastasoitunutta rintasyöpää sairastavilla potilailla 8,7 % ja varhaisvaiheen rintasyöpää sairastavilla potilailla 5,7 %. Annosmuutokset trombosytopenian yhteydessä, ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
Verenvuodot
Verenvuototapahtumia raportoitiin trastutsumabiemtansiinilla tehdyissä metastasoitunutta rintasyöpää koskeneissa kliinisissä tutkimuksissa 34,8 %:lla potilaista, ja vaikea-asteisia verenvuototapahtumia (gradus ≥ 3) esiintyi 2,2 %:lla potilaista. Verenvuototapahtumia raportoitiin 29,2 %:lla varhaisvaiheen rintasyöpää sairastavista potilaista, ja vaikea-asteisten verenvuototapahtumien (gradus ≥ 3) ilmaantuvuus oli 0,4 %, mihin lukeutui yksi graduksen 5 tapahtuma. Joissakin havaituissa tapauksissa potilaalla oli trombosytopenia tai potilas sai myös veren hyytymistä tai verihiutaleiden aggregaatiota estävää hoitoa, mutta muissa tapauksissa ei ollut tunnettuja lisäriskitekijöitä. Kuolemaan johtaneita verenvuototapahtumia on havaittu sekä metastasoitunutta rintasyöpää että varhaisvaiheen rintasyöpää sairastavilla potilailla.
Suurentuneet transaminaasipitoisuudet (ASAT tai ALAT)
Kliinisissä tutkimuksissa on havaittu trastutsumabiemtansiinihoidon aikana suurentuneita seerumin transaminaasipitoisuuksia (gradus 1–4) (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet). Transaminaasipitoisuuden suureneminen oli yleensä ohimenevää. Trastutsumabiemtansiinin on havaittu vaikuttavan kumulatiivisesti transaminaaseihin, vaikutus yleensä hävisi, kun hoito lopetettiin. Suurentuneita transaminaasipitoisuuksia raportoitiin metastasoitunutta rintasyöpää koskeneissa kliinisissä tutkimuksissa 24,2 %:lla potilaista. ASAT-arvojen gradus 3 tai 4 suurenemista raportoitiin 4,2 %:lla potilaista, ja ALAT-arvojen gradus 3 tai 4 suurenemista raportoitiin 2,7 %:lla metastasoitunutta rintasyöpää sairastavista potilaista. Tätä esiintyi tavallisesti ensimmäisten hoitosyklien (1–6) aikana. Suurentuneita transaminaasipitoisuuksia raportoitiin 32,6 %:lla varhaisvaiheen rintasyöpää sairastavista potilaista. Graduksen 3 ja 4 suurentuneita transaminaasipitoisuuksia raportoitiin 1,6 %:lla varhaisvaiheen rintasyöpää sairastavista potilaista. Graduksen ≥ 3 maksatapahtumiin ei yleensä liittynyt huonoa kliinistä hoitotulosta. Myöhemmät seuranta‑arvot kohenivat yleensä siinä määrin, että potilaiden oli mahdollista jatkaa mukana tutkimuksessa ja saada tutkimushoitoa edelleen samalla tai pienemmällä annoksella. Trastutsumabiemtansiinialtistuksen (AUC), trastutsumabiemtansiinin maksimipitoisuuden seerumissa (Cmax), trastutsumabin kokonaisaltistuksen (AUC) tai DM1:n Cmax-arvon ja transaminaasipitoisuuksien suurenemisen välillä ei havaittu yhteyttä. Annosmuutokset suurentuneiden transaminaasipitoisuuksien yhteydessä, ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
Sydämen vasemman kammion toimintahäiriö
Sydämen vasemman kammion toimintahäiriöitä raportoitiin trastutsumabiemtansiinilla tehdyissä metastasoitunutta rintasyöpää koskeneissa kliinisissä tutkimuksissa 2,2 %:lla potilaista. Suurin osa tapahtumista oli oireettomia graduksen 1 tai 2 vasemman kammion ejektiofraktion (LVEF) pienenemisiä. Graduksen 3 tai 4 tapahtumia raportoitiin 0,4 %:lla metastasoitunutta rintasyöpää sairastavista potilaista. Havainnointitutkimuksessa (BO39807) noin 22 %:lla (7/32) metastasoitunutta rintasyöpää sairastavista potilaista, joille trastutsumabiemtansiinihoito aloitettiin ja joiden LVEF oli lähtötilanteessa 40–49 %, LVEF pieneni > 10 % lähtötilanteesta ja/tai heillä oli kongestiivista sydämen vajaatoimintaa. Valtaosalla näistä potilaista oli muita sydämen ja verisuonten riskitekijöitä. Sydämen vasemman kammion toimintahäiriöitä raportoitiin 3,0 %:lla varhaisvaiheen rintasyöpää sairastavista potilaista. Graduksen 3 toimintahäiriöitä raportoitiin 0,5 %:lla potilaista, ja vaikeampiasteisia tapahtumia ei raportoitu. Annosmuutokset LVEF:n pienenemisen yhteydessä, ks. taulukko 2 kohdassa Annostus ja antotapa sekä kohta Varoitukset ja käyttöön liittyvät varotoimet.
Perifeerinen neuropatia
Perifeeristä neuropatiaa, joka oli pääasiassa gradus 1 ja lähinnä sensorista, raportoitiin kliinisissä trastutsumabiemtansiinitutkimuksissa. Metastasoitunutta rintasyöpää sairastavilla potilailla perifeerisen neuropatian kokonaisilmaantuvuus oli 29,0 % ja graduksen ≥ 2 perifeerisen neuropatian ilmaantuvuus oli 8,6 %. Varhaisvaiheen rintasyöpää sairastavilla potilailla kokonaisilmaantuvuus oli 32,0 % ja graduksen ≥ 2 perifeerisen neuropatian ilmaantuvuus oli 10,1 %.
Infuusioon liittyvät reaktiot
Infuusioon liittyville reaktioille on tyypillistä yksi tai useampi seuraavista oireista: kasvojen ja kaulan punoitus, vilunväristykset, kuume, hengenahdistus, matala verenpaine, hengityksen vinkuminen, bronkospasmi ja takykardia. Infuusioon liittyviä reaktioita raportoitiin trastutsumabiemtansiinilla tehdyissä metastasoitunutta rintasyöpää koskeneissa kliinisissä tutkimuksissa 4,0 %:lla potilaista. Raportoiduista tapauksista kuusi oli gradus 3, ja yhtään gradus 4 tapausta ei raportoitu. Infuusioon liittyviä reaktioita raportoitiin 1,6 %:lla varhaisvaiheen rintasyöpää sairastavista potilaista. Graduksen 3 tai 4 tapahtumia ei raportoitu. Infuusioon liittyvät reaktiot hävisivät muutamien tuntien tai vuorokauden kuluessa infuusion päättymisen jälkeen. Kliinisissä tutkimuksissa ei havaittu suhdetta annokseen. Annosmuutokset infuusioon liittyvien reaktioiden yhteydessä, ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
Yliherkkyysreaktiot
Yliherkkyyttä raportoitiin trastutsumabiemtansiinilla tehdyissä metastasoitunutta rintasyöpää koskeneissa kliinisissä tutkimuksissa 2,6 %:lla potilaista, ja raportoiduista tapauksista yksi oli gradus 3 ja yksi oli gradus 4. Yliherkkyyttä raportoitiin 2,7 %:lla varhaisvaiheen rintasyöpää sairastavista potilaista. Graduksen 3 yliherkkyyttä raportoitiin 0,4 %:lla potilaista, ja vaikeampiasteisia tapahtumia ei raportoitu. Suurin osa yliherkkyysreaktioista oli vaikeusasteeltaan lieviä tai keskivaikeita, ja ne hävisivät hoidon avulla. Annosmuutokset yliherkkyysreaktioiden yhteydessä, ks. kohdat Annostus ja antotapa ja Varoitukset ja käyttöön liittyvät varotoimet.
Immunogeenisuus
Trastutsumabiemtansiinin käytössä, kuten kaikkien terapeuttisten proteiinien käytössä, on immuunivasteen mahdollisuus. Seitsemässä kliinisessä tutkimuksessa yhteensä 1 243 potilaalta testattiin useana eri ajankohtana lääkevasta-ainevaste trastutsumabiemtansiinille. Potilaista 5,1 % (64/1 243) sai trastutsumabiemtansiinin annon jälkeen yhtenä tai useana ajankohtana positiivisen testituloksen trastutsumabiemtansiinin vasta-ainetestissä. Vaiheen I ja vaiheen II tutkimuksissa 6,4 % (24/376) potilaista sai positiivisen tuloksen trastutsumabiemtansiinin vasta-ainetestissä. EMILIA-tutkimuksessa (TDM4370g/BO21977) 5,2 % (24/466) potilaista sai positiivisen tuloksen trastutsumabiemtansiinin vasta-ainetestissä, ja näistä 13 potilaan tulos oli positiivinen myös neutraloivien vasta-aineiden osalta. KATHERINE-tutkimuksessa (BO27938) 4,0 % (16/401) potilaista sai positiivisen tuloksen trastutsumabiemtansiinin vasta-ainetestissä, ja näistä 5 potilaan tulos oli positiivinen myös neutraloivien vasta-aineiden osalta. Lääkevasta-aineiden esiintyvyys oli vähäinen, joten näiden vasta-aineiden vaikutusta trastutsumabiemtansiinin farmakokinetiikkaan, farmakodynamiikkaan, turvallisuuteen ja/tai tehoon ei tunneta.
Ekstravasaatio
Trastutsumabiemtansiinilla tehdyissä kliinisissä tutkimuksissa on havaittu ekstravasaatiosta aiheutuneita reaktioita. Nämä reaktiot olivat tavallisesti lieviä tai keskivaikeita ja ilmenivät eryteemana, arkuutena, ihoärsytyksenä, kipuna tai infuusiokohdan turpoamisena. Tällaisia reaktioita on havaittu useimmiten 24 tunnin kuluessa infuusion antamisesta. Valmisteen markkinoille tultua on havaittu ekstravasaation seurauksena esiintyneitä epidermiksen vaurioita tai epidermaalista nekroosia, jotka ovat poikkeuksellisesti ilmenneet päivien tai viikkojen kuluessa infuusiosta. Trastutsumabiemtansiinin ekstravasaation spesifisestä hoidosta ei ole tällä hetkellä tietoja (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Laboratorioarvojen poikkeavuudet
Taulukoissa 4 ja 5 esitetään trastutsumabiemtansiinia kliinisessä TDM4370g/BO21977/EMILIA-tutkimuksessa ja BO27938/KATHERINE-tutkimuksessa saaneilla potilailla havaitut laboratorioarvojen poikkeavuudet.
Taulukko 4Trastutsumabiemtansiinilla tehdyssä TDM4370g/BO21977/EMILIA-tutkimuksessa hoitoa saaneilla potilailla havaitut laboratorioarvojen poikkeavuudet
Parametri | Trastutsumabiemtansiini (N = 490) | ||
Kaikki vaikeusasteet (%) | Gradus 3 (%) | Gradus 4 (%) | |
Maksa | |||
suurentunut bilirubiinipitoisuus | 21 | < 1 | 0 |
suurentunut ASAT-arvo | 98 | 8 | < 1 |
suurentunut ALAT-arvo | 82 | 5 | < 1 |
Hematologia | |||
vähentynyt trombosyyttimäärä | 85 | 14 | 3 |
pienentynyt hemoglobiinipitoisuus | 63 | 5 | 1 |
vähentynyt neutrofiilimäärä | 41 | 4 | < 1 |
Kalium | |||
pienentynyt kaliumpitoisuus | 35 | 3 | < 1 |
Taulukko 5 Trastutsumabiemtansiinilla tehdyssä BO27938/KATHERINE-tutkimuksessa hoitoa saaneilla potilailla havaitut laboratorioarvojen poikkeavuudet
Parametri | Trastutsumabiemtansiini (N = 740) | ||
Kaikki vaikeusasteet (%) | Gradus 3 (%) | Gradus 4 (%) | |
Maksa | |||
suurentunut bilirubiinipitoisuus | 11 | 0 | 0 |
suurentunut ASAT-arvo | 79 | < 1 | 0 |
suurentunut ALAT-arvo | 55 | < 1 | 0 |
Hematologia | |||
vähentynyt trombosyyttimäärä | 51 | 4 | 2 |
pienentynyt hemoglobiinipitoisuus | 31 | 1 | 0 |
vähentynyt neutrofiilimäärä | 24 | 1 | 0 |
Kalium | |||
pienentynyt kaliumpitoisuus | 26 | 2 | < 1 |
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty-haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Trastutsumabiemtansiinin yliannostukseen ei tunneta vastalääkettä. Yliannoksen yhteydessä potilasta on seurattava tarkoin haittavaikutusten oireiden ja löydösten havaitsemiseksi ja sopiva oireenmukainen hoito on aloitettava. Trastutsumabiemtansiinihoidon yhteydessä on raportoitu yliannostapauksia, joista useimpiin liittyi trombosytopeniaa. Yhdessä tapauksessa potilas kuoli. Kuolemaan johtaneessa tapauksessa potilas sai virheellisesti trastutsumabiemtansiinia 6 mg/kg ja kuoli noin 3 viikon kuluttua yliannoksesta. Syy-yhteyttä trastutsumabiemtansiiniin ei varmistettu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Syöpälääkkeet ja immuunivasteen muuntajat, monoklonaaliset vasta-aineet ja vasta-ainekonjugoidut lääkkeet, ihmisen epidermaalisen kasvutekijän reseptori 2:n (HER2) estäjät. ATC-koodi: L01FD03.
Vaikutusmekanismi
Kadcyla, trastutsumabiemtansiini, on HER‑2-reseptoriin täsmäkohdennettu vasta‑ainekonjugaatti, joka sisältää humanisoitua anti-HER‑2 IgG1:tä, trastutsumabia, joka on liitetty kovalenttisidoksella mikrotubulusten muodostumista estävään DM1:een (maytansiinijohdos) stabiilin tioeetterilinkkerin MCC:n (4‑[N‑maleimidometyyli]sykloheksaani‑1‑karboksylaatin) välityksellä. Emtansiini viittaa MCC‑DM1-kompleksiin. Kuhunkin trastutsumabimolekyyliin on konjugoitu keskimäärin 3,5 DM1-molekyyliä.
DM1:n konjugaatio trastutsumabiin tekee solunsalpaajan HER2:ta yli‑ilmentäville kasvainsoluille selektiiviseksi, jolloin DM1:tä kulkeutuu enemmän suoraan syöpäsolujen sisään. HER2-reseptoriin sitouduttuaan trastutsumabiemtansiini käy läpi reseptorivälitteisen internalisaation ja sitä seuraavan lysosomaalisen degradaation, jolloin DM1:tä sisältävät sytotoksiset kataboliitit (pääasiassa lysiini‑MCC-DM1) vapautuvat.
Trastutsumabiemtansiinilla on sekä trastutsumabin että DM1:n vaikutusmekanismit:
-
trastutsumabiemtansiini, kuten trastutsumabikin, sitoutuu HER2:n ekstrasellulaarisen domeenin (ECD) aladomeeniin IV sekä Fcγ-reseptoreihin ja komplementtiin C1q. Trastutsumabiemtansiini, kuten trastutsumabikin, estää lisäksi HER2:n ekstrasellulaarisen domeenin vapautumista, estää fosfatidyylinositoli-3‑kinaasi (PI3‑K) ‑viestintäreitin kautta kulkevaa signaalinvälitystä ja välittää HER2:ta yli-ilmentävissä ihmisen rintasyöpäsoluissa vasta‑aineriippuvaista soluvälitteistä sytotoksisuutta (ADCC, antibody‑dependent cell‑mediated cytotoxicity).
-
trastutsumabiemtansiinin solunsalpaajakomponentti DM1 sitoutuu tubuliiniin. Sekä DM1 että trastutsumabiemtansiini estävät tubuliinin polymerisaatiota, jolloin solusykli pysähtyy vaiheeseen G2/M, mikä johtaa lopulta apoptoottiseen solukuolemaan. Sytotoksisuusmääritysten in vitro tulokset osoittavat, että DM1 on 20–200 kertaa voimakkaampi kuin taksaanit ja vinka-alkaloidit.
-
MCC-linkkerin on tarkoitus vähentää DM1:n systeemistä vapautumista ja tehostaa sen täsmäkuljetusta, minkä osoittavat hyvin pienet plasmassa todetut vapaan DM1:n pitoisuudet.
Kliininen teho
Varhaisvaiheen rintasyöpä
BO27938 (KATHERINE)
BO27938 (KATHERINE) oli satunnaistettu 1 486 HER2-positiivista varhaisvaiheen rintasyöpää sairastavan potilaan avoin monikeskustutkimus. Potilailla oli kemoterapiaa ja HER2-kohdennettua hoitoa sisältäneen preoperatiivisen systeemisen lääkehoidon jälkeen invasiivinen jäännöskasvain rinnassa ja/tai kainaloiden imusolmukkeissa (potilaat eivät olleet saaneet täydellistä patologista vastetta [pathological complete response, pCR]). Potilaat olivat saattaneet saada useampaa kuin yhtä HER2-kohdennettua hoitoa. Potilaat saivat sädehoitoa ja/tai hormonihoitoa samaan aikaan tutkimushoidon kanssa paikallisten hoitosuositusten mukaisesti. Rintarauhasen kasvaimesta otetuista näytteistä osoitettiin HER2:n yli-ilmentyminen, joksi määriteltiin keskuslaboratoriossa immunohistokemiallisella menetelmällä (IHC) 3+ tai in situ ‑hybridisaatiomenetelmällä (ISH) määritetty suhdeluku ≥ 2,0. Potilaat satunnaistettiin (1:1) saamaan trastutsumabia tai trastutsumabiemtansiinia. Satunnaistaminen ositettiin preoperatiivisen hoidon jälkeen arvioidun kliinisen levinneisyysasteen perusteella potilaan tutkimukseen tullessa (leikattavissa vs ei leikattavissa), hormonireseptoristatuksen, preoperatiivisen HER2-kohdennetun hoidon (trastutsumabi, trastutsumabin ja toisen HER2-kohdennetun lääkeaineen yhdistelmähoidon) ja patologisen imusolmukestatuksen perusteella.
Trastutsumabiemtansiinia annettiin laskimoon annoksina 3,6 mg/kg 21 vuorokauden pituisen hoitosyklin päivänä 1. Trastutsumabia annettiin laskimoon annoksina 6 mg/kg 21 vuorokauden pituisen hoitosyklin päivänä 1. Potilaat saivat trastutsumabiemtansiini- tai trastutsumabihoitoa yhteensä 14 hoitosykliä, paitsi jonkin seuraavista ilmaantuessa: tauti uusiutui, potilas perui suostumuksensa tutkimukseen osallistumisesta tai ilmaantui toksisuutta, joka ei ollut hyväksyttävissä. Trastutsumabiemtansiinihoidon lopettaneiden potilaiden oli mahdollista jatkaa HER2-kohdennettua trastutsumabihoitoa tutkimuksen mukaisesti enintään 14 hoitosykliä, jos se oli toksisuuden kannalta ja tutkijan arvion mukaan tarkoituksenmukaista.
Tutkimuksen ensisijainen tehon päätetapahtuma oli elossaolo ilman invasiivista tautia (Invasive Disease-Free Survival, IDFS). Elossaolo ilman invasiivista tautia määriteltiin seuraavasti: aika satunnaistamispäivämäärästä invasiivisen rintarauhaskasvaimen ensimmäiseen uusiutumiseen samassa rinnassa, paikallisen tai alueellisen invasiivisen rintasyövän uusiutumiseen samassa rinnassa, taudin uusiutumiseen muualla elimistössä, invasiiviseen rintasyöpään toisessa rinnassa tai kuolemaan mistä tahansa syystä. Muita päätetapahtumia olivat elossaolo ilman invasiivista tautia, mukaan lukien toinen muu primaarisyöpä kuin rintasyöpä, elossaolo ilman tautia (disease-free survival, DFS), kokonaiselossaolo (overall survival, OS) ja muualla elimistössä olevan syövän uusiutumattomuusaika (distant recurrence-free interval, DRFI).
Potilaiden demografiset ominaisuudet ja kasvaimen ominaisuudet lähtötilanteessa olivat tasapainossa hoitohaarojen kesken. Iän mediaani oli noin 49 vuotta (vaihteluväli 23–80 vuotta), 72,8 % oli valkoihoisia, 8,7 % oli aasialaisia ja 2,7 % oli mustaihoisia tai afroamerikkalaisia. Viittä potilasta lukuun ottamatta kaikki olivat naisia; trastutsumabihaarassa oli mukana 3 miestä ja trastutsumabiemtansiinihaarassa oli mukana 2 miestä. Potilaista 22,5 % tuli tutkimukseen mukaan Pohjois-Amerikassa, 54,2 % Euroopassa ja 23,3 % muualla maailmassa. Kasvaimen ennustetta koskevat ominaisuudet, mukaan lukien hormonireseptoristatus (positiivinen: 72,3 %; negatiivinen: 27,7 %), kliininen levinneisyysaste tutkimukseen tullessa (ei leikattavissa: 25,3 %, leikattavissa: 74,8 %) ja patologisten imusolmukkeiden status leikkausta edeltävän hoidon jälkeen (levinnyt imusolmukkeisiin: 46,4 %, ei levinnyt imusolmukkeisiin tai imusolmukkeiden statusta ei arvioitu: 53,6 %) olivat tutkimushaaroissa samankaltaiset.
Valtaosa potilaista (76,9 %) oli saanut antrasykliiniä sisältävää solunsalpaajahoitoa ennen leikkausta. Potilaista 19,5 % sai neoadjuvanttihoidon osana trastutsumabin lisäksi toista HER2-kohdennettua lääkeainetta, joka 93,8 %:lla näistä potilaista oli pertutsumabi. Kaikki potilaat olivat saaneet taksaaneja sisältävää solunsalpaajahoitoa ennen leikkausta.
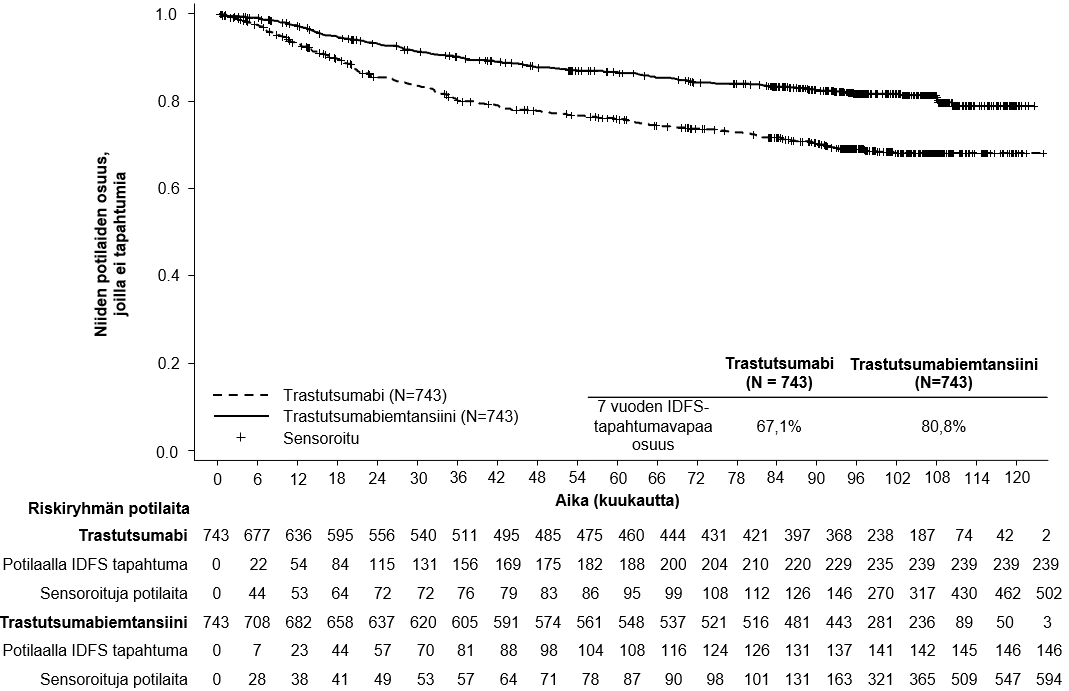
Elossaolossa ilman invasiivista tautia havaittiin primaarianalyysin ajankohtana tilastollisesti merkitsevä paraneminen trastutsumabiemtansiinia saaneilla potilailla verrattuna trastutsumabia saaneisiin potilaisiin, ks. taulukko 6.
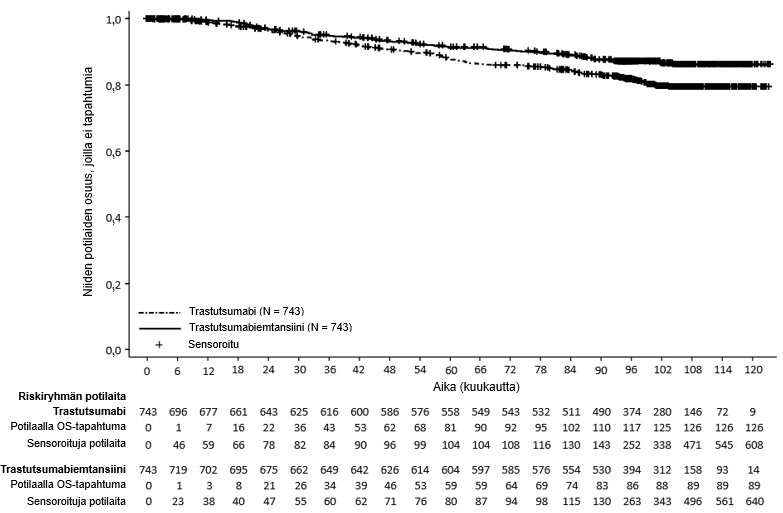
Lopullinen deskriptiivinen IDFS-analyysi suoritettiin, kun 385 IDFS-tapahtumaa oli havaittu, ja tulokset olivat yhdenmukaisia ensisijaisen analyysin kanssa (riskisuhde = 0,54, 95 %:n luottamusväli: 0,44–0,66), ks. kuva 1. Toinen kokonaiselinajan välianalyysi suoritettiin keskimääräisen 101 kuukauden seuranta-ajan jälkeen ja se osoitti tilastollisesti merkitsevän parannuksen kokonaiselinajassa potilailla, jotka saivat trastutsumabiemtansiinia verrattuna trastutsumabiin (stratifioimaton riskisuhde = 0,66, 95 %:n luottamusväli: 0,51–0,87, p = 0,0027). Ks. taulukko 6 ja kuva 2.
Taulukko 6 Yhteenveto hoidon tehosta BO27938 (KATHERINE) -tutkimuksessa
| Trastutsumabi N = 743 | Trastutsumabiemtansiini N = 743 |
Ensisijainen päätetapahtuma |
| |
Elossaolo ilman invasiivista tautia1,3 |
| |
Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 165 (22,2 %) | 91 (12,2 %) |
Riskisuhde (95 %:n luottamusväli) | 0,50 (0,39; 0,64) | |
p-arvo (log-rank-testi, osittamaton) | < 0,0001 | |
3 vuoden tapahtumavapaa osuus2 (%) (95 %:n luottamusväli) | 77,02 (73,78; 80,26) | 88,27 (85,81; 90,72) |
Toissijaiset päätetapahtumat3 |
| |
Kokonaiselossaolo4 |
| |
Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 126 (17,0 %) | 89 (12,0 %) |
Riskisuhde (95 %:n luottamusväli) | 0,66 (0,51; 0,87) | |
p-arvo (log-rank-testi, osittamaton) | 0,0027 | |
7 vuoden elossaolo-osuus2, % (95 %:n luottamusväli) | 84,4 (81,58; 87,16) | 89,1 (86,71; 91,42) |
Elossaolo ilman invasiivista tautia, mukaan lukien toinen primaarisyöpä muualla kuin rintarauhasessa1,5 |
| |
Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 167 (22,5 %) | 95 (12,8 %) |
Riskisuhde (95 %:n luottamusväli) | 0,51 (0,40; 0,66) | |
p-arvo (log-rank-testi, osittamaton) | < 0,0001 | |
3 vuoden tapahtumavapaa osuus2, % [95 %:n luottamusväli] | 76,9 (73,65; 80,14) | 87,7 (85,18; 90,18) |
Elossaolo ilman tautia1,5 |
| |
Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 167 (22,5 %) | 98 (13,2 %) |
Riskisuhde (95 %:n luottamusväli) | 0,53 (0,41; 0,68) | |
p-arvo (log-rank-testi, osittamaton) | < 0,0001 | |
3 vuoden tapahtumavapaa osuus2 (%) (95 %:n luottamusväli) | 76,9 (73,65; 80,14) | 87,41 (84,88; 89,93) |
Muualla elimistössä olevan syövän uusiutumattomuusaika1,5 |
| |
Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 121 (16,3 %) | 78 (10,5 %) |
Riskisuhde (95 %:n luottamusväli) | 0,60 (0,45; 0,79) | |
p-arvo (log-rank-testi, osittamaton) | 0,0003 | |
3 vuoden tapahtumavapaa osuus2 (%) (95 %:n luottamusväli) | 83,0 (80,10; 85,92) | 89,7 (87,37; 92,01) |
1. Primaarianalyysin tiedot
2. 3 vuoden tapahtumavapaa osuus ja 7 vuoden elossaolo saatu Kaplan-Meierin estimaateista
3. Elossaoloa ilman invasiivista tautia ja kokonaiselossaoloa koskeva hierarkkinen testaus
4. Kokonaiselossaolon toisen välianalyysin tiedot
5. Näitä toissijaisia päätetapahtumia ei korjattu toistuvuuden suhteen
Kuva 1 Invasiivisesta taudista vapaan elossaolon Kaplan–Meier-käyrät KATHERINE-tutkimuksessa (päivitetty analyysi)
Kuva 2 Kokonaiselossaolon Kaplan–Meier-käyrät KATHERINE-tutkimuksessa (päivitetty analyysi)
KATHERINE-tutkimuksessa trastutsumabiemtansiinihoidosta todettiin invasiivisesta taudista vapaan elossaolon osalta yhdenmukainen hyöty kaikissa ennalta määritellyissä arvioiduissa potilasryhmissä, mikä tukee kokonaistulosta.
Metastasoitunut rintasyöpä
TDM4370g/BO21977/(EMILIA)
Vaiheen III, satunnaistettu, kansainvälinen, avoin, kliininen monikeskustutkimus toteutettiin HER2‑positiivista, leikkaushoitoon soveltumatonta paikallisesti edennyttä tai metastasoitunutta rintasyöpää sairastavilla potilailla, jotka olivat aiemmin saaneet taksaani‑ ja trastutsumabi-pohjaista hoitoa, mukaan lukien potilaat, jotka olivat saaneet aiemmin trastutsumabihoitoa ja jotakin taksaania adjuvanttihoitona ja joiden tauti oli edennyt adjuvanttihoidon aikana tai kuuden kuukauden kuluessa adjuvanttihoidon päättymisestä. Tutkimukseen otettiin mukaan vain potilaat, joiden Eastern Cooperative Oncology Group (ECOG) ‑toimintakykyluokka oli 0 tai 1. Potilaiden rintakasvainnäyte piti ennen tutkimukseen mukaan tuloa varmistaa keskuslaboratoriossa HER2‑positiiviseksi, joksi oli määritelty pisteet 3+ IHC-menetelmällä tai in situ ‑hybridisaatiolla todettu geenimonistuma. Potilaiden ja kasvainten lähtötilanteen ominaisuudet olivat hyvin tasapainossa hoitoryhmien välillä. Potilaat, joilla oli etäpesäkkeitä aivoissa, soveltuivat mukaan tutkimukseen, jos he eivät tarvinneet hoitoa oireiden hallinnassa pitämiseen. Trastutsumabiemtansiinihoitoon satunnaistettujen potilaiden iän mediaani oli 53 vuotta, suurin osa potilaista (99,8 %) oli naisia, suurin osa (72 %) oli valkoihoisia ja 57 %:lla oli estrogeenireseptori‑ ja/tai progesteronireseptoripositiivinen tauti. Tutkimuksessa verrattiin trastutsumabiemtansiinin turvallisuutta ja tehoa lapatinibista ja kapesitabiinista koostuvan yhdistelmähoidon turvallisuuteen ja tehoon. Yhteensä 991 potilasta satunnaistettiin saamaan trastutsumabiemtansiinia tai lapatinibin ja kapesitabiinin yhdistelmää seuraavasti:
-
trastutsumabiemtansiiniryhmä: 3,6 mg/kg trastutsumabiemtansiinia laskimoon 30–90 minuutin kestoisena infuusiona 21 vuorokauden pituisen hoitosyklin päivänä 1
-
vertailuryhmä (lapatinibin ja kapesitabiinin yhdistelmähoito): lapatinibia 1250 mg/vrk suun kautta kerran päivässä 21 vuorokauden pituisen hoitosyklin jokaisena päivänä sekä kapesitabiinia 1000 mg/m2 suun kautta kaksi kertaa päivässä 21 vuorokauden pituisen hoitosyklin päivinä 1–14.
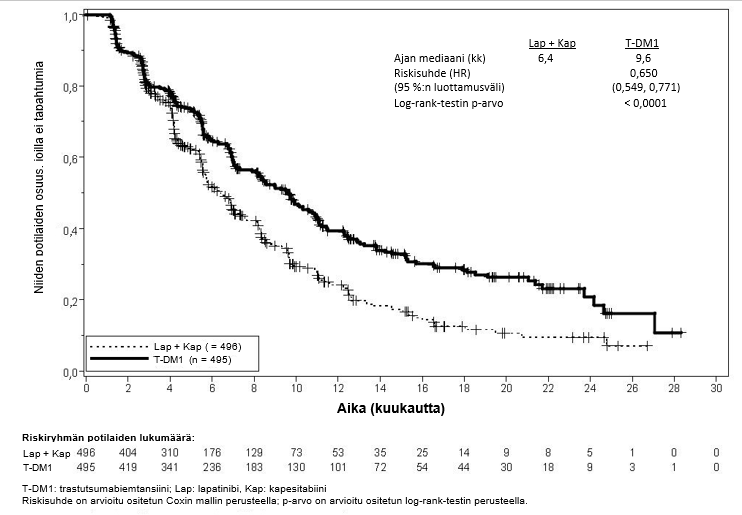
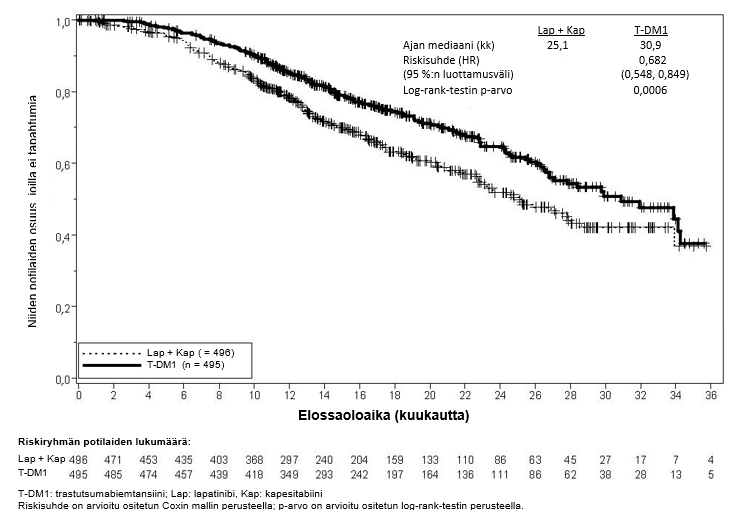
Tutkimuksen ensisijaiset tehon päätetapahtumat olivat riippumattoman arviointilautakunnan arvioima taudin etenemisvapaa aika (progression‑free survival, PFS) ja kokonaiselossaolo (overall survival, OS) (ks. taulukko 7 sekä kuvat 3 ja 4).
Kliinisen tutkimuksen aikana arvioitiin myös aikaa oireiden etenemiseen, mikä määriteltiin 5 pisteen vähenemisenä syövän hoitotuloksen selvittämiseen tutkimuksessa käytetyn Trials Outcome Index‑Breast (TOI‑B) ‑kyselyn toiminnallista elämänlaatua koskevan the Functional Assessment of Cancer Therapy‑Breast Quality of Life (FACT‑B QoL) ‑osiossa. TOI‑B-kyselytuloksen 5 pisteen muutos katsotaan kliinisesti merkitseväksi. Kadcyla pidensi potilaiden raportoimaa aikaa oireiden etenemiseen 7,1 kuukauteen verrattuna 4,6 kuukauteen vertailuryhmässä (riskisuhde 0,796 (0,667, 0,951); p-arvo 0,0121). Tiedot on saatu avoimesta tutkimuksesta eikä varmoja johtopäätöksiä voida tehdä.
Taulukko 7Tutkimuksen TDM4370g/BO21977 (EMILIA) tehon yhteenveto
| Lapatinibi + kapesitabiini n = 496 | Trastutsumabi-emtansiini n = 495 |
Ensisijaiset päätetapahtumat | ||
Riippumattoman arviointilautakunnan arvio taudin etenemisvapaasta ajasta |
| |
Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 304 (61,3 %) | 265 (53,5 %) |
Taudin etenemisvapaan elossaoloajan mediaani (kuukautta) | 6,4 | 9,6 |
Riskisuhde (ositettu*) | 0,650 | |
Riskisuhteen 95 %:n luottamusväli | (0,549, 0,771) | |
p-arvo (Log-rank-testi, ositettu*) | < 0,0001 | |
Kokonaiselossaolo** |
| |
Kuolleiden potilaiden lukumäärä (%) | 182 (36,7 %) | 149 (30,1 %) |
Elossaoloajan mediaani (kuukautta) | 25,1 | 30,9 |
Riskisuhde (ositettu*) | 0,682 | |
Riskisuhteen 95 %:n luottamusväli | (0,548, 0,849) | |
p-arvo (Log-rank-testi*) | 0,0006 | |
Keskeiset toissijaiset päätetapahtumat | ||
Tutkijan arvio taudin etenemisvapaasta ajasta |
| |
Niiden potilaiden lukumäärä (%), joilla tapahtuma esiintyi | 335 (67,5 %) | 287 (58,0 %) |
Taudin etenemisvapaan ajan mediaani (kuukautta) | 5,8 | 9,4 |
Riskisuhde (95 %:n luottamusväli) | 0,658 (0,560, 0,774) | |
p-arvo (Log-rank-testi*) | < 0,0001 | |
Objektiivinen vasteluku |
| |
Potilaat, joilla mitattavissa oleva tauti | 389 | 397 |
Niiden potilaiden lukumäärä, joilla todettiin objektiivinen vaste (%) | 120 (30,8 %) | 173 (43,6 %) |
Ero (95 %:n luottamusväli) | 12,7 % (6,0, 19,4) | |
p-arvo (Mantel-Haenszelin khiin neliö ‑testi*) | 0,0002 | |
Objektiivisen vasteen kestoaika (kuukautta) |
| |
Niiden potilaiden lukumäärä, joilla todettiin objektiivinen vaste | 120 | 173 |
95 %:n luottamusvälin mediaani | 6,5 (5,5, 7,2) | 12,6 (8,4, 20,8) |
Kokonaiselossaolo: overall survival, OS; taudin etenemisvapaa aika: progression-free survival, PFS; objektiivinen vasteluku: objective response rate, ORR; objektiivinen vaste: objective response, OR; riippumaton arviointilautakunta: independent review committee, IRC; riskisuhde: hazard ratios, HR; luottamusväli: confidence interval, CI
* Ositus: maantieteellinen alue (Yhdysvallat, Länsi-Eurooppa, muu), paikallisesti edenneen tai metastasoituneen taudin hoitoon aikaisemmin annettujen solunsalpaajahoitojen lukumäärä (0–1 vs. > 1), ja viskeraalinen vs. ei-viskeraalinen tauti.
** Kokonaiselossaolon (OS) välianalyysi tehtiin, kun oli havaittu 331 tapahtumaa. Koska tässä analyysissä ylitettiin tehon raja, se katsottiin lopulliseksi analyysiksi.
Hoidosta todettiin olevan hyötyä niille potilaille, joiden tauti oli relapsoitunut 6 kuukauden kuluessa adjuvanttihoidon päättymisestä ja jotka eivät olleet aiemmin saaneet mitään systeemistä syöpähoitoa metastasoituneeseen tautiin (n = 118); taudin etenemisvapaan ajan riskisuhde oli 0,51 (95 %:n luottamusväli: 0,30, 0,85) ja kokonaiselossaolon riskisuhde oli 0,61 (95 %:n luottamusväli: 0,32, 1,16). Taudin etenemisvapaan ajan mediaani oli trastutsumabiemtansiinia saaneessa ryhmässä 10,8 kuukautta, mutta kokonaiselossaolon mediaaniaikaa ei saavutettu, ja vastaavat ajat lapatinibin ja kapesitabiinin yhdistelmää saaneessa ryhmässä olivat 5,7 kuukautta ja 27,9 kuukautta.
Kuva 3Riippumattoman arviointilautakunnan (IRC) arvioiman taudin etenemisvapaan elossaoloajan Kaplan-Meier-käyrä
Kuva 4Kokonaiselossaoloajan (OS) Kaplan-Meier-käyrä
Tutkimuksessa TDM4370g/BO21977 todettiin trastutsumabiemtansiinihoidosta yhdenmukainen hyöty suurimmassa osassa ennalta määritellyistä arvioiduista potilasryhmistä, mikä tukee kokonaistuloksen painoarvoa. Potilasryhmässä, joiden tauti oli hormonireseptorinegatiivinen (n = 426), taudin etenemisvapaan ajan riskisuhde oli 0,56 (95 %:n luottamusväli: 0,44, 0,72) ja kokonaiselossaolon riskisuhde oli 0,75 (95 %:n luottamusväli: 0,54, 1,03). Potilasryhmässä, joiden tauti oli hormonireseptoripositiivinen (n = 545), taudin etenemisvapaan ajan riskisuhde oli 0,72 (95 %:n luottamusväli: 0,58, 0,91) ja kokonaiselossaolon riskisuhde oli 0,62 (95 %:n luottamusväli: 0,46, 0,85).
Potilasryhmässä, joiden tauti ei ollut mitattavissa (n = 205), taudin etenemisvapaan ajan riskisuhde oli riippumattoman arviointilautakunnan arvion perusteella 0,91 (95 %:n luottamusväli: 0,59, 1,42) ja kokonaiselossaolon riskisuhde oli 0,96 (95 %:n luottamusväli: 0,54, 1,68). Vähintään 65-vuotiaiden potilaiden (n = 138 yhteensä kummassakin hoitoryhmässä) taudin etenemisvapaan ajan riskisuhde oli 1,06 (95 %:n luottamusväli: 0,68, 1,66) ja kokonaiselossaolon riskisuhde oli 1,05 (95 %:n luottamusväli: 0,58; 1,91). 65–74-vuotiaiden potilaiden (n = 113), taudin etenemisvapaan ajan riskisuhde oli riippumattoman arviointilautakunnan arvion perusteella 0,88 (95 %:n CI: 0,53, 1,45) ja kokonaiselossaolon riskisuhde oli 0,74 (95 %:n luottamusväli: 0,37, 1,47). Potilasryhmässä, jossa potilaat olivat vähintään 75-vuotiaita, taudin etenemisvapaan ajan riskisuhde oli riippumattoman arviointilautakunnan arvion perusteella 3,51 (95 %:n luottamusväli: 1,22, 10,13) ja kokonaiselossaolon riskisuhde oli 3,45 (95 %:n luottamusväli: 0,94, 12,65). Vähintään 75-vuotiaiden potilaiden ryhmässä ei hoidosta osoitettu hyötyä taudin etenemisvapaan ajan eikä kokonaiselossaolon suhteen, mutta potilasryhmä oli liian pieni (n = 25), jotta selkeitä johtopäätöksiä olisi voitu tehdä.
Seuranta-ajan deskriptiivisessä kokonaiselossaoloanalyysissä riskisuhde oli 0,75 (95 %:n luottamusväli: 0,64, 0,88). Kokonaiselossaoloajan mediaani oli trastutsumabiemtansiiniryhmässä 29,9 kuukautta verrattuna 25,9 kuukauteen lapatinibin ja kapesitabiinin yhdistelmää saaneessa ryhmässä. Seuranta-ajan deskriptiivisen kokonaiselossaoloanalyysin ajankohtana yhteensä 27,4 % lapatinibin ja kapesitabiinin yhdistelmää saaneen ryhmän potilaista oli siirtynyt trastutsumabiemtansiiniryhmään. Herkkyysanalyysissä, jossa hoitoryhmästä toiseen siirtyneet potilaat sensuroitiin siirtymisajankohtana, riskisuhde oli 0,69 (95 %:n luottamusväli 0,59, 0,82). Seuranta-ajan deskriptiivisen analyysin tulokset ovat yhdenmukaiset kokonaiselossaolon varmistusanalyysin kanssa.
TDM4450g
Satunnaistetussa, avoimessa, vaiheen II monikeskustutkimuksessa verrattiin trastutsumabiemtansiinin sekä trastutsumabin ja dosetakselin yhdistelmän vaikutusta HER‑2‑positiivista metastasoitunutta rintasyöpää sairastavilla potilailla, jotka eivät olleet aiemmin saaneet solunsalpaajahoitoa metastasoituneen taudin hoitoon. Potilaat satunnaistettiin saamaan 3,6 mg/kg trastutsumabiemtansiinia laskimoon 3 viikon välein (n = 67) tai aloitusannoksen 8 mg/kg trastutsumabia laskimoon ja sen jälkeen 6 mg/kg laskimoon 3 viikon välein yhdistelmänä laskimoon 3 viikon välein annetun dosetakseliannoksen 75–100 mg/m2 kanssa (n = 70).
Ensisijainen päätetapahtuma oli tutkijan arvioima taudin etenemisvapaa aika. Taudin etenemisvapaan ajan mediaani oli trastutsumabin ja dosetakselin yhdistelmää saaneessa ryhmässä 9,2 kuukautta ja trastutsumabiemtansiinia saaneessa ryhmässä 14,2 kuukautta (riskisuhde 0,59, p = 0,035), ja seuranta‑ajan mediaani oli kummassakin ryhmässä noin 14 kuukautta. Objektiivinen vasteluku (ORR) oli trastutsumabin ja dosetakselin yhdistelmää saaneessa ryhmässä 58,0 % ja trastutsumabiemtansiinia saaneessa ryhmässä 64,2 %. Vasteen kestoajan mediaania ei saavutettu trastutsumabiemtansiiniryhmässä vs. 9.5 kuukauteen vertailuryhmässä.
TDM4374g
Vaiheen II yhden ryhmän avoimessa tutkimuksessa selvitettiin trastutsumabiemtansiinin vaikutuksia HER2‑positiivista parantumatonta, paikallisesti edennyttä tai metastasoitunutta rintasyöpää sairastavilla potilailla. Kaikki potilaat olivat saaneet aiemmin HER2‑kohdennettua hoitoa (trastutsumabia ja lapatinibia) ja solunsalpaajaa (antrasykliiniä, taksaania ja kapesitabiinia) neoadjuvanttihoidon, adjuvanttihoidon, paikallisesti edenneen tai metastasoituneen taudin yhteydessä. Potilaan missä tahansa tilanteessa saamien syöpälääkkeiden lukumäärän mediaani oli 8,5 (vaihteluväli 5–19) ja metastasoituneen taudin yhteydessä 7,0 (vaihteluväli 3–17), mukaan lukien kaikki rintasyövän hoitoon tarkoitetut lääkeaineet.
Potilaat (n = 110) saivat 3,6 mg/kg trastutsumabiemtansiinia laskimoon 3 viikon välein, kunnes tauti eteni tai potilaalle ilmaantui haittaavaa toksisuutta.
Keskeiset tehon analyysit olivat riippumattomaan radiologiseen arvioon perustuva objektiivinen vasteluku (ORR) ja objektiivisen vasteen kestoaika. Objektiivinen vasteluku oli sekä riippumattoman arviointilautakunnan että tutkijan arvion perusteella 32,7 % (95 %:n luottamusväli: 24,1, 42,1), n = 36 vasteen saanutta. Vasteen kestoajan mediaania ei riippumattoman arviointilautakunnan mukaan saavutettu (95 %:n luottamusväli, 4,6 kuukautta – ei arvioitavissa).
Pediatriset potilaat
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset trastutsumabiemtansiinin käytöstä rintasyövän hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Populaatiofarmakokineettinen analyysi viittasi siihen, että altistuksessa trastutsumabiemtansiinille ei ollut eroja taudin statuksen perusteella (adjuvantti vs. metastasoitunut).
Imeytyminen
Trastutsumabiemtansiini annetaan laskimoon. Muita antoreittejä ei ole tutkittu.
Jakautuminen
TDM4370g/BO21977-tutkimuksessa ja BO29738-tutkimuksessa trastutsumabiemtansiinia 3,6 mg/kg laskimoon 3 viikon välein saaneiden potilaiden seerumissa trastutsumabiemtansiinin keskimääräinen maksimipitoisuus (Cmax) hoitosyklissä 1 oli 83,4 (± 16,5) μg/ml (tutkimus TDM4370g/BO21977) ja 72,6 (± 24,3) μg/ml (tutkimus BO29738). Laskimoon annetun trastutsumabiemtansiinin sentraalinen jakautumistilavuus oli populaatiofarmakokineettisen analyysin perusteella 3,13 l ja likimäärin plasman tilavuus.
Biotransformaatio (trastutsumabiemtansiini ja DM1)
Trastutsumabiemtansiini käy oletettavasti solujen lysosomeissa läpi proteolyyttisen dekonjugaation ja katabolian.
Ihmisen maksan mikrosomeilla tehdyt in vitro ‑metaboliatutkimukset viittaavat siihen, että trastutsumabiemtansiinin pienimolekyylinen komponentti DM1 metaboloituu pääasiassa CYP3A4:n välityksellä ja vähemmässä määrin CYP3A5:n välityksellä. DM1 ei estänyt pääasiallisia CYP450-entsyymejä in vitro. Trastutsumabiemtansiinin kataboliitteja MCC-DM1, Lys-MCC-DM1 ja DM1 havaittiin pieninä pitoisuuksina ihmisen plasmassa. DM1 oli P-glykoproteiinin (P-gp) substraatti in vitro.
Eliminaatio
Trastutsumabiemtansiinin puhdistuma HER2‑positiivista metastasoitunutta rintasyöpää sairastaville potilaille laskimoon annettuna oli populaatiofarmakokineettisen analyysin perusteella 0,68 l/vrk ja eliminaation puoliintumisaika (t1/2) oli noin 4 vuorokautta. Trastutsumabiemtansiinin kumuloitumista ei havaittu 3 viikon välein toistettujen laskimoon annettujen infuusioiden jälkeen.
Trastutsumabiemtansiinin tilastollisesti merkitseviksi farmakokineettisten parametrien kovariaateiksi tunnistettiin populaatiofarmakokineettisen analyysin perusteella kehon paino, albumiini, kohdekasvainten pisimmän läpimitan summa kiinteiden kasvainten hoitovasteen arviointiin käytettävien RECIST (Response Evaluation Criteria In Solid Tumors) ‑kriteerien perusteella, HER2:n ekstrasellulaarisen domeenin vapautuminen, lähtötilanteen trastutsumabipitoisuudet ja aspartaattiaminotransferaasi (ASAT). Näiden kovariaattien vaikutuksen voimakkuus trastutsumabiemtansiinialtistukseen viittaa kuitenkin siihen, ettei näillä kovariaateilla todennäköisesti ole kliinisesti merkityksellistä vaikutusta trastutsumabiemtansiinialtistukseen. Eksploratiivinen analyysi osoitti lisäksi, että kovariaattien (eli munuaisten toiminta, rotu ja ikä) vaikutus kokonaistrastutsumabin ja DM1:n farmakokinetiikkaan oli vähäistä eikä sillä ollut kliinistä merkitystä. Trastutsumabiemtansiinin kataboliitit, DM1, Lys‑MCC‑DM1, ja MCC‑DM1 mukaan lukien, erittyivät nonkliinisissä tutkimuksissa pääasiassa sappinesteeseen ja eliminoituivat minimaalisesti virtsaan.
Lineaarisuus/ei‑lineaarisuus
Laskimoon 3 viikon välein annetun trastutsumabiemtansiinin farmakokinetiikka oli lineaarinen annosalueella 2,4–4,8 mg/kg. Puhdistuma oli nopeampi, jos potilaan saama annos oli 1,2 mg/kg tai pienempi.
Iäkkäät potilaat
Populaatiofarmakokineettinen analyysi osoitti, ettei ikä vaikuttanut trastutsumabiemtansiinin farmakokinetiikkaan. Trastutsumabiemtansiinin farmakokinetiikassa ei havaittu merkittäviä eroja < 65‑vuotiaiden (n = 577), 65–75-vuotiaiden (n = 78) ja > 75-vuotiaiden (n = 16) potilaiden välillä.
Munuaisten vajaatoiminta
Munuaisten vajaatoimintaa sairastavilla potilailla ei ole tehty varsinaisia farmakokineettisiä tutkimuksia. Populaatiofarmakokineettinen analyysi osoitti, ettei kreatiniinipuhdistuma vaikuta trastutsumabiemtansiinin farmakokinetiikkaan. Trastutsumabiemtansiinin farmakokinetiikka oli lievää tai keskivaikeaa munuaisten vajaatoimintaa sairastavilla potilailla (kreatiniinipuhdistuma CLcr 60−89 ml/min, n = 254 ja vastaavasti CLcr 30–59 ml/min, n = 53) samankaltainen kuin potilailla, joiden munuaisten toiminta oli normaali (CLcr ≥ 90 ml/min, n = 361). Vain vähän farmakokineettisiä tietoja on saatavilla vaikeaa munuaisten vajaatoimintaa (CLcr 15–29 ml/min) sairastavista potilaista (n = 1), jonka vuoksi annostussuosituksia ei voida antaa.
Maksan vajaatoiminta
Maksa on ensisijainen DM1:tä ja DM1:tä sisältäviä kataboliitteja elimistöstä poistava elin. Trastutsumabiemtansiinin ja DM1:tä sisältävien kataboliittien farmakokinetiikkaa tutkittiin antamalla trastutsumabiemtansiinia annoksina 3,6 mg/kg metastasoitunutta HER2-positiivista rintasyöpää sairastaville potilaille, joiden maksan toiminta oli normaali (n = 10) tai jotka sairastivat lievää (Child-Pugh A; n = 10) tai keskivaikeaa (Child-Pugh B; n = 8) maksan vajaatoimintaa.
-
DM1:n ja DM1:tä sisältävien kataboliittien (Lys-MCC-DM1 ja MCC-DM1) pitoisuudet plasmassa olivat pienet ja verrannolliset maksan vajaatoimintaa sairastavien ja sairastamattomien välillä.
-
Systeeminen altistus (AUC) trastutsumabiemtansiinille oli hoitosyklissä 1 lievää maksan vajaatoimintaa sairastavilla potilailla noin 38 % ja keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla noin 67 % pienempi kuin potilailla, joiden maksan toiminta oli normaali. Toistettujen annosten jälkeen hoitosyklissä 3 altistus trastutsumabiemtansiinille (AUC) oli lievää tai keskivaikeaa maksan vajaatoimintaa sairastavilla potilailla samansuuruinen kuin potilailla, joiden maksan toiminta oli normaali.
Vaikeaa maksan vajaatoimintaa (Child-Pugh-luokka C) sairastavista potilaista ei ole tehty varsinaista farmakokineettistä tutkimusta, eikä tästä potilasryhmästä ole kerätty populaatiofarmakokineettisiä tietoja.
Muut erityisryhmät
Populaatiofarmakokineettinen analyysi osoitti, ettei rotu oletettavasti vaikuta trastutsumabiemtansiinin farmakokinetiikkaan. Koska suurin osa trastutsumabiemtansiinilla tehtyihin kliinisiin tutkimuksiin osallistuneista potilaista oli naisia, sukupuolen vaikutusta trastutsumabiemtansiinin farmakokinetiikkaan ei ole varsinaisesti tutkittu.
Prekliiniset tiedot turvallisuudesta
Eläintoksisuus ja/tai -farmakologia
Rotat ja apinat sietivät hyvin annetut trastutsumabiemtansiiniannokset, joka olivat enimmillään 20 mg/kg (rotille) ja 10 mg/kg (apinoille), mikä vastaa kummallakin lajilla annosta 2040 μg DM1/m2, joka puolestaan vastaa suunnilleen potilaille annettavaa trastutsumabiemtansiinin kliinistä annosta. GLP-toksisuustutkimuksissa todettiin kummassakin eläinmallissa osittain tai täysin korjautuvaa annosriippuvaista toksisuutta, mistä poikkeuksen muodostivat korjautumaton perifeerinen aksonaalinen toksisuus (jota havaittiin vain apinalla annoksilla ≥ 10 mg/kg) sekä lisääntymiselintoksisuus (havaittiin vain rotalla annoksilla 60 mg/kg). Toksisuus kohdistui pääasiassa maksaan (kohonneet maksaentsyymipitoisuudet) annoksilla ≥ 20 mg/kg (rotalla) ja ≥ 10 mg/kg (apinalla), luuytimeen (vähentynyt trombosyytti- ja veren valkosolumäärä)/veriarvoihin annoksilla ≥ 20 mg/kg (rotalla) ja ≥ 10 mg/kg (apinalla) ja imukudoselimiin annoksilla ≥ 20 mg/kg (rotalla) ja ≥ 3 mg/kg (apinalla).
Mutageenisuus
DM1 oli kerta-annoksilla in vivo tehdyssä rotan luuytimen mikrotumamäärityksessä aneugeeninen ja klastogeeninen altistuksilla, jotka olivat verrannolliset trastutsumabiemtansiinia saaneilla ihmisillä mitattuihin DM1:n keskimääräisiin maksimipitoisuuksiin. DM1 ei ollut mutageeninen in vitro bakteerien käänteismutaatiomäärityksessä (Ames).
Hedelmällisyyden heikkeneminen ja teratogeenisuus
Trastutsumabiemtansiinin vaikutuksen arvioimiseksi ei ole tehty hedelmällisyyttä koskevia eläinkokeita. Hedelmällisyyteen kohdistuvia haittavaikutuksia voidaan yleisten eläintoksisuustutkimusten tulosten perusteella kuitenkin olettaa esiintyvän.
Trastutsumabiemtansiinilla ei ole tehty erityisesti alkion ja sikiön kehitystä koskevia eläintutkimuksia. Trastutsumabilla on todettu kliinisessä käytössä kehitystoksisuutta, vaikka sitä ei ennakoitu nonkliinisessä tutkimusohjelmassa. Maytansiinilla on lisäksi todettu nonkliinisissä tutkimuksissa kehitystoksisuutta, mikä viittaa siihen, että trastutsumabiemtansiinin mikrotubulusten muodostumista estävä maytansiinin sukuinen solunsalpaajakomponentti DM1 on samalla tavoin teratogeeninen ja mahdollisesti alkiotoksinen.
Farmaseuttiset tiedot
Apuaineet
Sukkiinihappo
Natriumhydroksidi
Sakkaroosi
Polysorbaatti 20
Yhteensopimattomuudet
Lääkevalmistetta ei saa sekoittaa muiden lääkevalmisteiden kanssa eikä laimentaa muilla lääkevalmisteilla, lukuun ottamatta niitä, jotka mainitaan kohdassa Käyttö- ja käsittelyohjeet.
Kuiva-aineen liuottamisessa tai laimentamisessa ei saa käyttää glukoosiliuosta (5 %), koska se aiheuttaa proteiinien aggregoitumista.
Kestoaika
Avaamaton injektiopullo
4 vuotta.
Liuotettu kuiva-aine eli välikonsentraatti
Välikonsentraatin on osoitettu olevan kemiallisesti ja fysikaalisesti stabiili enintään 120 tuntia (5 vuorokautta) 2 °C – 8 °C:ssa, kun se on saatettu käyttökuntoon steriilillä injektionesteisiin käytettävällä vedellä tai 4,5 mg/ml (0,45 %) natriumkloridiliuoksella. Mikrobiologiselta kannalta valmiste tulisi käyttää heti. Jos valmistetta ei käytetä heti, käyttäjä vastaa välikonsentraatin säilytysajoista ja -olosuhteista ennen käyttöä. Ne eivät saa tavallisesti ylittää 24:ää tuntia 2 °C – 8 °C:ssa, jollei käyttökuntoon saattaminen ole tapahtunut kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa.
Laimennettu liuos
9 mg/ml (0,9 %) natriumkloridi-infuusioliuosta tai 4,5 mg/ml (0,45 %) natriumkloridi-infuusioliuosta sisältävään infuusiopussiin laimennettu Kadcyla on stabiili enintään 24 tunnin ajan 2 °C – 8 °C:ssa, edellyttäen että liuos on valmistettu kontrolloiduissa ja validoiduissa aseptisissa olosuhteissa. Liuoksessa saattaa olla säilytyksen aikana havaittavissa hiukkasia, jos se on laimennettu 0,9-prosenttiseen natriumkloridiin (ks. kohta Käyttö- ja käsittelyohjeet).
Säilytys
Säilytä jääkaapissa (2 °C – 8 °C).
Välikonsentraatin ja laimennetun lääkevalmisteen säilytys, ks. kohta Kestoaika.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
KADCYLA kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
100 mg (L:ei) 1 kpl (15 ml pullo) (2243,41 €)
160 mg (L:ei) 1 kpl (20 ml pullo) (3481,09 €)
PF-selosteen tieto
Kadcyla 100 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Kadcyla on pakattu 15 ml:n (100 mg) tyypin 1 lasiseen injektiopulloon, joka on suljettu fluoriresiinilaminaattipäällysteisellä harmaalla butyylikumitulpalla ja sinetöity alumiinisinetillä, jossa on valkoinen irti napsautettava muovilevy.
Pakkauksessa on 1 injektiopullo.
Kadcyla 160 mg kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
Kadcyla on pakattu 20 ml:n (160 mg) tyypin 1 lasiseen injektiopulloon, joka on suljettu fluoriresiinilaminaattipäällysteisellä harmaalla butyylikumitulpalla ja sinetöity alumiinisinetillä, jossa on purppuranvärinen irti napsautettava muovilevy.
Pakkauksessa on 1 injektiopullo.
Valmisteen kuvaus:
Valkoinen tai luonnonvalkoinen kylmäkuivattu kuiva-aine.
Käyttö- ja käsittelyohjeet
Asianmukaista aseptista tekniikkaa on noudatettava. Asianmukaisia solunsalpaajalääkevalmisteiden valmistusmenetelmiä on noudatettava.
Välikonsentraatti on laimennettava edelleen polyvinyylikloridi (PVC) infuusiopussissa tai lateksia ja PVC:tä sisältämättömässä polyolefiini-infuusiopussissa.
Kun välikonsentraatti on laimennettu 9 mg/ml (0,9 %) natriumkloridi-infuusioliuokseen, infuusioletkussa on käytettävä 0,20 tai 0,22 mikronin polyeetterisulfonisuodatinta (PES-suodatinta).
Lääkitysvirheiden välttämiseksi on tärkeää varmistaa injektiopullon etiketistä, että valmistettava ja annettava lääkevalmiste on Kadcyla (trastutsumabiemtansiini) eikä toinen trastutsumabia sisältävä valmiste (esim. trastutsumabi tai trastutsumabi-derukstekaani).
Ohjeet kuiva-aineen liuottamiseen eli välikonsentraatin valmistamiseen
-
100 mg trastutsumabiemtansiinia sisältävä injektiopullo: Injisoi steriilin ruiskun avulla 5 ml steriiliä injektionesteisiin käytettävää vettä tai 4,5 mg/ml (0,45 %) natriumkloridiliuosta hitaasti injektiopulloon.
-
160 mg trastutsumabiemtansiinia sisältävä injektiopullo: Injisoi steriilin ruiskun avulla 8 ml steriiliä injektionesteisiin käytettävää vettä tai 4,5 mg/ml (0,45 %) natriumkloridiliuosta hitaasti injektiopulloon.
-
Pyörittele injektiopulloa varovasti, kunnes kuiva-aine on liuennut täysin. Älä ravista.
Välikonsentraatti on tarkistettava silmämääräisesti ennen antoa, ettei siinä ole hiukkasia ja värimuutoksia havaittavissa. Välikonsentraatissa ei saa olla hiukkasia silmin havaittavissa, ja sen on oltava kirkasta tai hieman opaalinhohtoista (väriltään väritöntä tai vaaleanruskeaa). Älä käytä liuosta, jos siinä on hiukkasia näkyvissä tai se on sameaa tai sen väri on muuttunut.
Laimennusohjeet
Laske tarvittava välikonsentraatin liuostilavuus annoksen 3,6 mg trastutsumabiemtansiinia/painokg perusteella (ks. kohta Annostus ja antotapa):
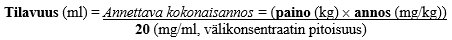
Vedä injektiopullosta asianmukainen liuostilavuus ja lisää se 250 ml 4,5 mg/ml (0,45 %) natriumkloridi-infuusioliuosta tai 9 mg/ml (0,9 %) natriumkloridi-infuusioliuosta sisältävään infuusiopussiin. Glukoosiliuosta (5 %) ei saa käyttää (ks. kohta Yhteensopimattomuudet). 4,5 mg/ml (0,45 %) natriumkloridi-infuusioliuosta käytettäessä infuusioletkussa ei tarvitse olla 0,20 tai 0,22 μm:n polyeetterisulfonisuodatinta (PES-suodatinta). Jos infuusioon käytetään 9 mg/ml (0,9 %) natriumkloridi-infuusioliuosta, infuusioletkussa on käytettävä 0,20 tai 0,22 mikronin polyeetterisulfonisuodatinta (PES-suodatinta). Infuusio on annettava heti liuoksen käyttökuntoon saattamisen jälkeen. Infuusioliuos ei saa jäätyä eikä sitä saa ravistaa säilytyksen aikana.
Hävittäminen
Käyttökuntoon saatettu valmiste ei sisällä säilytysainetta ja se on tarkoitettu vain yhtä käyttökertaa varten. Hävitä käyttämättä jäänyt liuos.
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
KADCYLA kuiva-aine välikonsentraatiksi infuusionestettä varten, liuos
100 mg 1 kpl
160 mg 1 kpl
- Ei korvausta.
ATC-koodi
L01FD03
Valmisteyhteenvedon muuttamispäivämäärä
16.06.2026
Yhteystiedot
 ROCHE OY
ROCHE OY Revontulenpuisto 2 C, P.O. Box 112
02101 Espoo
010 554 500
www.roche.fi
etunimi.sukunimi@roche.com