
JARDIANCE tabletti, kalvopäällysteinen 10 mg, 25 mg
Vaikuttavat aineet ja niiden määrät
Jardiance 10 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää 10 mg empagliflotsiinia.
Apuaineet, joiden vaikutukset tunnetaan
Yksi tabletti sisältää laktoosimonohydraattia, joka vastaa 154,3 mg vedetöntä laktoosia.
Jardiance 25 mg kalvopäällysteiset tabletit
Yksi tabletti sisältää 25 mg empagliflotsiinia.
Apuaineet, joiden vaikutukset tunnetaan
Yksi tabletti sisältää laktoosimonohydraattia, joka vastaa 107,4 mg vedetöntä laktoosia.
Täydellinen apuaineluettelo, ks. kohta Apuaineet.
Lääkemuoto
Tabletti, kalvopäällysteinen (tabletti).
Kliiniset tiedot
Käyttöaiheet
Tyypin 2 diabetes
Jardiance on tarkoitettu tyypin 2 diabeteksen hoitoon aikuispotilaille ja vähintään 10 vuoden ikäisille lapsipotilaille, joilla sairaus ei ole riittävässä hoitotasapainossa, liikunnan ja ruokavaliohoidon ohella
- monoterapiana, kun metformiinia ei sietokyvyttömyyden takia pidetä sopivana vaihtoehtona
- muiden diabeteksen hoitoon tarkoitettujen lääkevalmisteiden lisänä
Tutkimustulokset eri yhdistelmähoidoista, vaikutuksista glukoositasapainoon, sydän- ja verisuonitapahtumiin ja munuaistapahtumiin sekä tutkimuspopulaatioista on luettavissa kohdissa Varoitukset ja käyttöön liittyvät varotoimet, Yhteisvaikutukset ja Farmakodynamiikka.
Sydämen vajaatoiminta
Jardiance on tarkoitettu oireisen kroonisen sydämen vajaatoiminnan hoitoon aikuisille.
Krooninen munuaistauti
Jardiance on tarkoitettu kroonisen munuaistaudin hoitoon aikuisille.
Annostus ja antotapa
Annostus
Tyypin 2 diabetes
Suositeltu aloitusannos empagliflotsiinia on 10 mg kerran vuorokaudessa monoterapiana ja yhdistelmähoidossa muiden diabeteksen hoitoon tarkoitettujen lääkevalmisteiden kanssa. Annos voidaan nostaa 25 mg:aan kerran vuorokaudessa potilaille, jotka sietävät empagliflotsiinia 10 mg kerran vuorokaudessa ja joiden eGFR on ≥ 60 ml/min/1,73 m2 ja jotka tarvitsevat parempaa glukoositasapainoa. Enimmäisannos on 25 mg vuorokaudessa (ks. alla ja kohta Varoitukset ja käyttöön liittyvät varotoimet).
Sydämen vajaatoiminta
Suositeltu annos empagliflotsiinia on 10 mg kerran vuorokaudessa.
Krooninen munuaistauti
Suositeltu annos empagliflotsiinia on 10 mg kerran vuorokaudessa.
Kaikki käyttöaiheet
Kun empagliflotsiinia käytetään yhdessä sulfonyyliurean tai insuliinin kanssa, voidaan harkita sulfonyyliurean tai insuliinin annoksen pienentämistä hypoglykemian riskin pienentämiseksi (ks. kohdat Yhteisvaikutukset ja Haittavaikutukset).
Jos potilas unohtaa ottaa annoksen, hänen pitää ottaa se heti muistaessaan. Kaksinkertaista annosta ei kuitenkaan saa ottaa saman vuorokauden aikana.
Erityisryhmät
Munuaisten vajaatoiminta
Koska kokemusta on vain vähän, empagliflotsiinihoidon aloittamista ei suositella potilaille, joiden eGFR on < 20 ml/min/1,73 m2.
Empagliflotsiinin annos on 10 mg vuorokaudessa potilaille, joiden eGFR on < 60 ml/min/1,73 m2.
Tyypin 2 diabetesta sairastavilla potilailla empagliflotsiinin glukoosipitoisuutta alentava teho on heikentynyt, jos potilaan eGFR on < 45 ml/min/1,73 m2, ja todennäköisesti olematon, jos potilaan eGFR on < 30 ml/min/1,73 m2. Siksi potilaille, joiden eGFR laskee alle arvon < 45 ml/min/1,73 m2, on tarpeen mukaan harkittava lisäksi jotakin glukoosipitoisuutta alentavaa hoitoa (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet, Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka).
Maksan vajaatoiminta
Annosta ei tarvitse muuttaa maksan vajaatoimintaa sairastaville potilaille. Empagliflotsiinialtistus on suurempaa vaikeaa maksan vajaatoimintaa sairastavilla potilailla. Kokemusta vaikeaa maksan vajaatoimintaa sairastavien potilaiden hoidosta on vähän, eikä sen käyttöä näin ollen suositella tässä potilasryhmässä (ks. kohta Farmakokinetiikka).
Iäkkäät
Annosta ei suositella muutettavan iän perusteella. 75-vuotiailla ja sitä vanhemmilla potilailla on otettava huomioon suurempi nestehukan riski (ks. kohdat Varoitukset ja käyttöön liittyvät varotoimet ja Haittavaikutukset).
Pediatriset potilaat
Suositeltu aloitusannos empagliflotsiinia on 10 mg kerran vuorokaudessa. Annos voidaan nostaa 25 mg:aan kerran vuorokaudessa potilaille, jotka sietävät empagliflotsiinia 10 mg kerran vuorokaudessa ja jotka tarvitsevat parempaa glukoositasapainoa (ks. kohdat kohdat Farmakodynamiikka ja Farmakokinetiikka). Tietoja ei ole saatavilla lapsista, joiden eGFR-arvo on < 60 ml/min/1,73 m2, tai alle 10-vuotiaista lapsista.
Empagliflotsiinin turvallisuutta ja tehoa alle 18 vuoden ikäisten lasten sydämen vajaatoiminnan tai kroonisen munuaistaudin hoidossa ei ole varmistettu. Tietoja ei ole saatavilla.
Antotapa
Tabletit voidaan ottaa joko ruoan kanssa tai tyhjään mahaan, ne niellään kokonaisina veden kanssa.
Vasta-aiheet
Yliherkkyys vaikuttavalle aineelle tai kohdassa Apuaineet mainituille apuaineille.
Varoitukset ja käyttöön liittyvät varotoimet
Yleistä
Empagliflotsiinia ei pidä käyttää potilaille, joilla on tyypin 1 diabetes (ks. ”Ketoasidoosi” kohdassa Varoitukset ja käyttöön liittyvät varotoimet).
Ketoasidoosi
Toisinaan henkeä uhanneita tai kuolemaan johtaneita ketoasidoosin tapauksia on raportoitu diabetespotilailla, joita on hoidettu SGLT2:n estäjillä, mukaan lukien empagliflotsiini. Ketoasidoosi oli monissa näistä tapauksista epätyypillistä siten, että verensokeripitoisuuksien havaittiin olevan vain kohtalaisesti suurentuneet, alle 14 mmol/l. Ei tiedetä, onko ketoasidoosin ilmeneminen todennäköisempää suuremmilla empagliflotsiiniannoksilla. Ketoasidoosin esiintyminen on epätodennäköisempää, jos potilaalla ei ole diabetesta, mutta tapauksia on raportoitu myös tällaisilla potilailla.
Jos potilaalla on epäspesifisiä oireita, kuten pahoinvointia, oksentelua, ruokahaluttomutta, vatsakipua, voimakasta janoa, hengitysvaikeuksia, sekavuutta, epätavallista uupumusta tai uneliaisuutta, ketoasidoosin riski on otettava huomioon. Potilaat tulee tutkia ketoasidoosin varalta välittömästi, jos näitä oireita ilmenee, veren glukoosipitoisuudesta huolimatta.
Jos potilaalla epäillään tai todetaan ketoasidoosi, hoito empagliflotsiinilla pitää heti lopettaa.
Hoito on keskeytettävä potilailla, jotka joutuvat sairaalahoitoon suuren kirurgisen toimenpiteen tai äkillisen vakavan sairauden takia. Näillä potilailla suositellaan ketonien seurantaa. Ketonipitoisuus kannattaa mitata verestä eikä virtsasta. Hoito empagliflotsiinilla voidaan aloittaa uudelleen, kun ketonipitoisuus on normaali ja potilaan tila on jälleen vakaa.
Potilaan ketoasidoosille altistavat aiemmat sairaudet pitää ottaa huomioon ennen kuin hoito empagliflotsiinilla aloitetaan.
Empagliflotsiinin käytön yhteydessä on havaittu pitkittynyttä ketoasidoosia ja pitkittynyttä glukosuriaa. Ketoasidoosi voi jatkua empagliflotsiinihoidon keskeyttämisen jälkeen pidempään kuin puoliintumisaika plasmassa antaisi odottaa (ks. kohta Farmakokinetiikka). Empagliflotsiinista riippumattomat tekijät, kuten insuliinin puute, voivat vaikuttaa ketoasidoosin pitkittymiseen.
Ketoasidoosin riski saattaa olla suurempi potilailla, joilla on pieni beetasolujen toimintareservi (esim. tyypin 2 diabetesta sairastavat potilaat, joilla on pieni C-peptidipitoisuus, aikuisen piilevä autoimmuunidiabetes (LADA) tai haimatulehduksen aiemmin sairastaneet potilaat); potilailla, joilla on ruoan saantia rajoittava sairaus tai vaikea elimistön nestevajaus; potilailla, joiden insuliiniannosta on pienennetty sekä potilailla, joilla on akuutin sairauden, leikkaustoimenpiteen tai alkoholin väärinkäytön vuoksi lisääntynyt insuliinintarve. SGLT2:n estäjien käytössä näiden potilasryhmien hoitoon pitää olla varovainen.
SGLT2:n estäjähoidon aloittamista uudelleen ei suositella, jos potilaalla on aiemmin ollut ketoasidoosi SGLT2:n estäjähoidon aikana, paitsi jos tunnistetaan jokin toinen ketoasidoosia edistävä tekijä ja se on hävinnyt.
Jardiance-valmistetta ei pidä käyttää potilaille, joilla on tyypin 1 diabetes. Tyypin 1 diabetesta sairastavilla potilailla tehdystä kliinisestä tutkimusohjelmasta saatujen tietojen mukaan ketoasidoosin esiintyvyys suureni lumelääkkeeseen verrattuna yleiseksi potilailla, jotka saivat 10 mg ja 25 mg empagliflotsiinia insuliinin lisänä.
Munuaisten vajaatoiminta
Koska kokemusta on vain vähän, empagliflotsiinihoidon aloittamista ei suositella potilaille, joiden eGFR on < 20 ml/min/1,73 m2.
Empagliflotsiinin annos on 10 mg vuorokaudessa potilaille, joiden eGFR on < 60 ml/min/1,73 m2 (ks. kohta Annostus ja antotapa).
Empagliflotsiinin glukoosipitoisuutta alentava teho riippuu munuaisten toiminnasta ja on heikentynyt, jos potilaan eGFR on < 45 ml/min/1,73 m2, ja todennäköisesti olematon, jos potilaan eGFR on < 30 ml/min/1,73 m2 (ks. kohdat Annostus ja antotapa, Farmakodynamiikka ja Farmakokinetiikka).
Munuaisten toiminnan seuraaminen
Munuaisten toimintaa on suositeltavaa arvioida seuraavasti:
- Ennen empagliflotsiinin aloittamista ja säännöllisin väliajoin hoidon aikana, ts. ainakin vuosittain (ks. kohdat Annostus ja antotapa, Haittavaikutukset, Farmakodynamiikka ja Farmakokinetiikka)
- Ennen minkään sellaisen samanaikaisesti käytettävän lääkevalmisteen käytön aloittamista, jolla voi olla haitallinen vaikutus munuaisten toimintaan.
Nestehukan riski
Perustuen SGLT2-estäjien vaikutustapaan glukosuriaan liittyvä osmoottinen diureesi voi johtaa verenpaineen lievään laskuun (ks. kohta Farmakodynamiikka). Varovaisuutta on siksi noudatettava potilailla, joilla empagliflotsiinin aiheuttama verenpaineen aleneminen voi muodostaa riskin, kuten potilailla, joilla tiedetään olevan sydän- ja verisuonitauti, potilailla, joilla on verenpainelääkitys ja anamneesissa hypotensio, tai 75-vuotiailla ja sitä vanhemmilla potilailla.
Kun empagliflotsiinia käyttävillä potilailla on sairauksia, jotka voivat johtaa nesteen menetykseen (esim. ruoansulatuskanavan sairaus), suositellaan nestetasapainon huolellista seurantaa (esim. lääkärintarkastus, verenpainemittaukset, laboratoriotutkimukset mukaan lukien hematokriitti) ja elektrolyyttitasapainon seurantaa. Empagliflotsiinihoidon tilapäistä keskeyttämistä on harkittava, kunnes nesteen menetys on korjattu.
Iäkkäät
Empagliflotsiinin vaikutus, glukoosin erittymiseen virtsaan liitetään osmoottiseen diureesiin, joka saattaa vaikuttaa nesteytykseen. 75-vuotiailla ja sitä vanhemmilla potilailla saattaa olla lisääntynyt riski nestehukkaan. Empagliflotsiinihoitoa saaneilla potilailla oli enemmän nestehukkaan liittyviä haittavaikutuksia kuin lumelääkkeellä hoidetuilla potilailla (ks. kohta Haittavaikutukset). Siksi näiden potilaiden kohdalla on kiinnitettävä erityistä huomiota nesteen nauttimiseen, jos he käyttävät samanaikaisesti lääkkeitä, jotka voivat aiheuttaa nestehukkaa (esim. diureetteja, ACE:n estäjiä).
Komplisoituneet virtsatieinfektiot
Komplisoituneita virtsatieinfektioita mukaan lukien pyelonefriitti ja urosepsis on raportoitu empagliflotsiinia saaneilla potilailla (ks. kohta Haittavaikutukset). Empagliflotsiinin tilapäistä keskeyttämistä on harkittava potilailla, joilla on komplisoituneita virtsatieinfektioita.
Välilihan nekrotisoiva faskiitti (Fournier’n gangreeni)
Välilihan nekrotisoivan faskiitin (tämä tunnetaan myös nimellä Fournier’n gangreeni) tapauksista on ilmoitettu nais- ja miespotilailla, jotka käyttävät SGLT2:n estäjiä, kuten empagliflotsiinia. Tämä on harvinainen, mutta vakava ja mahdollisesti hengenvaarallinen tapahtuma, joka edellyttää kiireellistä leikkausta ja antibioottihoitoa.
Potilaita on kehotettava kääntymään lääkärin puoleen, jos heillä on kipua, aristusta, punoitusta tai turvotusta genitaali- tai perineaalialueella ja tähän liittyy kuumetta tai huonovointisuutta. On syydä huomioida, että nekrotisoivaa faskiittia voi edeltää urogenitaali-infektio tai perineaaliabsessi. Jos Fournier’n gangreenia epäillään, Jardiance-valmisteen käyttö on keskeytettävä ja hoito (mukaan lukien antibioottihoito ja puhdistusleikkaus) on aloitettava.
Alaraajan amputaatiot
Pitkän aikavälin kliinisissä tutkimuksissa toisella SGLT2:n estäjällä on havaittu alaraajan (pääasiallisesti varpaan) amputaatioiden määrän lisääntymistä. Ei tiedetä, onko kyseessä lääkeluokkaan liittyvä vaikutus. Kuten kaikkien diabetespotilaiden kohdalla, on tärkeää antaa potilaille neuvoja säännöllisestä ehkäisevästä jalkahoidosta.
Maksavaurio
Kliinisissä tutkimuksissa on raportoitu maksavaurioita empagliflotsiinia käytettäessä. Syy-yhteyttä empagliflotsiinin ja maksavaurion välillä ei ole osoitettu.
Kohonnut hematokriitti
Hematokriitin kohoamista havaittiin empagliflotsiinihoidon yhteydessä (ks. kohta Haittavaikutukset). Potilaita, joiden hematokriitti on kohonnut huomattavasti, on seurattava, ja heille on tehtävä tutkimuksia taustalla olevan hematologisen sairauden varalta.
Krooninen munuaistauti
Albuminuriapotilailla empagliflotsiinihoidosta voi olla enemmän hyötyä.
Infiltratiivinen sairaus tai takotsubo-kardiomyopatia
Potilaita, joilla on infiltratiivinen sairaus tai takotsubo-kardiomyopatia, ei ole spesifisesti tutkittu. Siksi tehoa näiden potilaiden hoidossa ei ole varmistettu.
Virtsan laboratoriotutkimukset
Jardiance-valmisteen toimintamekanismin vuoksi potilaiden virtsan glukoosimääritys on positiivinen.
Häiriöt 1,5-anhydroglusitolin (1,5-AG) määrityksessä
Glukoositasapainon seurantaa 1,5-AG-määrityksellä ei suositella, sillä SGLT2:n estäjiä käyttäviltä potilailta 1,5-AG-määrityksellä mitatut arvot eivät luotettavasti kuvaa glukoositasapainoa. Vaihtoehtoisten menetelmien käyttöä glukoositasapainon seurantaan suositellaan.
Laktoosi
Tabletit sisältävät laktoosia. Potilaiden, joilla on harvinainen perinnöllinen galaktoosi-intoleranssi, täydellinen laktaasinpuutos tai glukoosi-galaktoosi-imeytymishäiriö, ei pidä käyttää tätä lääkettä.
Natrium
Yksi tabletti sisältää alle 1 mmol natriumia (23 mg) eli sen voidaan sanoa olevan ”natriumiton”.
Yhteisvaikutukset
Farmakodynaamiset yhteisvaikutukset
Diureetit
Empagliflotsiini saattaa lisätä tiatsidien ja loop-diureettien diureettista vaikutusta ja lisätä elimistön kuivumisen ja hypotension riskiä (ks. kohta Varoitukset ja käyttöön liittyvät varotoimet).
Insuliini ja insuliinin eritystä lisäävät lääkeaineet
Insuliini ja insuliinin eritystä lisäävät lääkeaineet, kuten sulfonyyliureat, saattavat lisätä hypoglykemian riskiä. Insuliinin tai insuliinin eritystä lisäävän lääkeaineen pienempi annos saattaa siksi olla tarpeen hypoglykemiariskin pienentämiseksi, kun sitä käytetään empagliflotsiinin kanssa (ks. kohdat Annostus ja antotapa ja Haittavaikutukset).
Farmakokineettiset yhteisvaikutukset
Muiden lääkevalmisteiden vaikutukset empagliflotsiiniin
In vitro -tulokset viittaavat siihen, että empagliflotsiini metaboloituu ihmisillä ensisijaisesti glukuronidaatiolla uridiini-5’‑difosfoglukuronosyylitransferaasien UGT1A3:n, UGT1A8:n, UGT1A9:n ja UGT2B7:n vaikutuksesta. Empagliflotsiini on ihmisen sisäänotto-kuljettajaproteiinien OAT3:n, OATP1B1:n ja OATP1B3:n substraatti, mutta ei OAT1:n tai OCT2:n substraatti. Empagliflotsiini on P-glykoproteiinin (P-gp) ja rintasyöpäresistenssiproteiinin (BCRP) substraatti.
Kun empagliflotsiinia annettiin UGT-entsyymien ja OAT3:n estäjän probenesidin kanssa, empagliflotsiinin huippupitoisuudet plasmassa (Cmax) nousivat 26 % ja pitoisuus–aikakäyrän alle jäävä pinta-ala (AUC) suureni 53 %. Näitä muutoksia ei pidetty kliinisesti merkittävinä.
UGT:n induktion (esim. rifampisiinin tai fenytoiinin aiheuttaman induktion) vaikutusta empagliflotsiiniin ei ole tutkittu. Samanaikaista hoitoa tunnettujen UGT-entsyymien indusorien kanssa ei suositellamahdollisen tehoa alentavan vaikutuksen vuoksi. Jos UGT‑entsyymien indusoreja joudutaan antamaan samanaikaisesti, glukoositasapainoa on syytä seurata Jardiance‑valmisteen vasteen arvioimiseksi.
Yhteisvaikutustutkimus gemfibrotsiilin kanssa, joka on OAT3- ja OATP1B1/1B3-kuljettajaproteiinien in vitro -estäjä, osoitti että empagliflotsiinin Cmax nousi 15 % ja AUC suureni 59 % yhtäaikaisen käytön jälkeen. Näitä muutoksia ei pidetty kliinisesti merkittävinä.
OATP1B1/1B3-kuljettajaproteiinien estäminen samanaikaisesti annetun rifampisiinin avulla nosti empagliflotsiinin Cmax-arvoa 75 % ja AUC-arvoa 35 %. Näitä muutoksia ei pidetty kliinisesti merkittävinä.
Empagliflotsiinialtistus oli samankaltaista annettaessa yhdessä P-gp:n estäjän verapamiilin kanssa ja ilman verapamiilia. Tämä viittaa siihen, että P-gp:n estämisellä ei ole kliinisesti merkittävää vaikutusta empagliflotsiiniin.
Yhteisvaikutustutkimusten tulokset viittaavat siihen, että samanaikaisesti annetut metformiini, glimepiridi, pioglitatsoni, sitagliptiini, linagliptiini, varfariini, verapamiili, ramipriili, simvastatiini, torasemidi tai hydroklooritiatsidi eivät vaikuttaneet empagliflotsiinin farmakokinetiikkaan.
Empagliflotsiinin vaikutukset muihin lääkevalmisteisiin
Empagliflotsiini saattaa lisätä litiumin erittymistä munuaisten kautta, ja veren litiumpitoisuudet saattavat pienentyä. Seerumin litiumpitoisuutta on seurattava useammin empagliflotsiinihoidon aloittamisen ja annosmuutosten jälkeen. Potilas tulee ohjata litiumia määränneen lääkärin vastaanotolle seerumin litiumpitoisuuden seurantaa varten.
In vitro -tutkimusten perusteella empagliflotsiini ei estä, inaktivoi tai indusoi CYP450-isoformeja. Empagliflotsiini ei estä UGT1A1:tä, UGT1A3:a, UGT1A8:aa, UGT1A9:ää eikä UGT2B7:ää. Siksi yhteisvaikutukset merkittävimpien CYP450- ja UGT-isoformien kanssa ovat epätodennäköisiä, kun empagliflotsiinia annetaan samanaikaisesti näiden entsyymien substraattien kanssa.
Empagliflotsiini ei estä P-gp:tä terapeuttisilla annoksilla. In vitro -tutkimusten perusteella on epätodennäköistä, että empagliflotsiinilla olisi yhteisvaikutuksia sellaisten vaikuttavien aineiden kanssa, jotka ovat P-gp:n substraatteja. Kun digoksiinia, P-gp:n substraattia, annettiin empagliflotsiinin kanssa, digoksiinin AUC suureni 6 % ja Cmax 14 %. Näitä muutoksia ei pidetty kliinisesti merkittävinä.
Empagliflotsiini ei estä ihmisen sisäänotto-kuljettajaproteiineja kuten OAT3:a, OATP1B1:tä ja OATP1B3:a in vitro kliinisesti merkittävillä pitoisuuksilla plasmassa, ja näin ollen lääkeaineiden väliset yhteisvaikutukset näiden sisäänotto-kuljettajaproteiinien substraattien kanssa ovat epätodennäköisiä.
Terveillä vapaaehtoisilla tehtyjen yhteisvaikutustutkimusten tiedot viittaavat siihen, että empagliflotsiinilla ei ollut kliinisesti merkittävää vaikutusta metformiinin, glimepiridin, pioglitatsonin, sitagliptiinin, linagliptiinin, simvastatiinin, varfariinin, ramipriilin, digoksiinin, diureettien ja suun kautta otettavien ehkäisyvalmisteiden farmakokinetiikkaan.
Pediatriset potilaat
Yhteisvaikutuksia on tutkittu vain aikuisille tehdyissä tutkimuksissa.
Raskaus ja imetys
Raskaus
Empagliflotsiinin käytöstä raskaana oleville naisille ei ole olemassa tietoja. Eläimillä tehtyjen tutkimusten mukaan empagliflotsiini läpäisee istukan raskauden myöhäisvaiheessa hyvin rajallisessa määrin, mutta sillä ei vaikuta olevan suoria tai epäsuoria haitallisia vaikutuksia varhaiseen alkion kehitykseen. Eläimillä tehdyissä tutkimuksissa on kuitenkin havaittu haittavaikutuksia postnataaliseen yksilönkehitykseen (ks. kohta Prekliiniset tiedot turvallisuudesta). Varmuuden vuoksi Jardiance-valmisteen käyttöä on suositeltavaa välttää raskauden aikana.
Imetys
Ei tiedetä, erittyykö empagliflotsiini ihmisillä äidinmaitoon. Olemassa olevat toksikologiset tiedot koe-eläimistä ovat osoittaneet empagliflotsiinin erittyvän maitoon. Imetettävään vauvaan kohdistuvia riskejä ei voida sulkea pois. Jardiance-valmistetta ei pidä käyttää imetyksen aikana.
Hedelmällisyys
Jardiance-valmisteen vaikutusta ihmisen hedelmällisyyteen ei ole tutkittu. Eläimillä tehdyissä tutkimuksissa ei ole havaittu suoria tai epäsuoria hedelmällisyydelle haitallisia vaikutuksia (ks. kohta Prekliiniset tiedot turvallisuudesta).
Vaikutus ajokykyyn ja koneiden käyttökykyyn
Jardiance-valmisteella on vähäinen vaikutus ajokykyyn ja koneidenkäyttökykyyn. Potilaita on neuvottava noudattamaan varovaisuutta hypoglykemian välttämiseksi ajamisen ja koneiden käytön aikana, erityisesti kun Jardiance-valmistetta käytetään sulfonyyliurean ja/tai insuliinin kanssa.
Haittavaikutukset
Turvallisuusprofiilin yhteenveto
Tyypin 2 diabetes
Kaikkiaan 15 582 tyypin 2 diabetesta sairastavaa potilasta osallistui kliinisiin tutkimuksiin, joissa arvioitiin empagliflotsiinin turvallisuutta. 10 004 potilasta sai empagliflotsiinia joko ainoana lääkkeenä tai metformiinin, sulfonyyliurean, pioglitatsonin, DPP-4:n estäjien tai insuliinin kanssa.
Kuuteen lumekontrolloituun 18–24 viikon pituiseen tutkimukseen osallistui 3 534 potilasta, joista 1 183 hoidettiin lumelääkkeellä ja 2 351 empagliflotsiinilla. Empagliflotsiinilla hoidettujen potilaiden haittatapahtumien kokonaisesiintyvyys oli samankaltainen kuin lumelääkkeellä. Useimmin raportoitu haittavaikutus oli hypoglykemia yhdistelmähoitona sulfonyyliurean tai insuliinin kanssa käytettynä (katso valikoitujen haittavaikutusten kuvaus).
Sydämen vajaatoiminta
EMPEROR-tutkimuksiin osallistui vajaatoimintapotilaita, joilla oli joko alentunut ejektiofraktio (N = 3 726) tai säilynyt ejektiofraktio (N = 5 985) ja jotka saivat 10 mg empagliflotsiinia tai lumelääkettä. Noin puolella potilaista oli tyypin 2 diabetes. Yleisin haittavaikutus yhdistetyissä EMPEROR-Reduced- ja EMPEROR-Preserved-tutkimuksissa oli nestehukka (empagliflotsiini 10 mg: 11,4 %; lumelääke: 9,7 %).
Krooninen munuaistauti
EMPA-KIDNEY-tutkimukseen osallistui kroonista munuaistautia sairastavia potilaita (N = 6 609), jotka saivat 10 mg empagliflotsiinia tai lumelääkettä. Noin 44 %:lla potilaista oli tyypin 2 diabetes. Yleisimpiä haittatapahtumia EMPA-KIDNEY-tutkimuksessa olivat kihti (empagliflotsiini 7,0 % vs. lumelääke 8,0 %) ja akuutti munuaisvaurio (empagliflotsiini 2,8 % vs. lumelääke 3,5 %), joita raportoitiin useammin lumelääkettä saaneilla potilailla.
Empagliflotsiinin yleinen turvallisuusprofiili oli yleisesti ottaen johdonmukainen tutkituissa käyttöaiheissa.
Taulukoitu luettelo haittavaikutuksista
Elinjärjestelmäluokan ja MedDRA-termien perusteella luokitellut haittavaikutukset, jotka ilmoitettiin empagliflotsiinia lumekontrolloiduissa tutkimuksissa saaneilla potilailla, esitetään alla olevassa taulukossa (taulukko 1).
Haittavaikutukset on lueteltu absoluuttisen esiintyvyyden mukaisesti. Esiintyvyydet on määritelty seuraavasti: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100, < 1/10), melko harvinainen (≥ 1/1 000, < 1/100), harvinainen (≥ 1/10 000, < 1/1 000) tai hyvin harvinainen (< 1/10 000), tuntematon (koska saatavissa oleva tieto ei riitä esiintyvyyden arviointiin).
Taulukko 1: Taulukoitu luettelo haittavaikutuksista (MedDRA) lumekontrolloiduissa tutkimuksissa ja myyntiluvan myöntämisen jälkeisessä käytössä
| Elinjärjestelmä | Hyvin yleinen | Yleinen | Melko harvinainen | Harvinainen | Hyvin harvinainen |
| Infektiot | Vaginan kandidiaasi, vulvovaginiitti, balaniitti ja muu genitaali-infektioa Virtsatieinfektio (mukaan lukien pyelonefriitti ja urosepsis)a | Välilihan nekrotisoiva faskiitti (Fournier’n gangreeni)* | |||
| Aineenvaihdunta ja ravitsemus | Hypoglykemia (käytettäessä sulfonyyliurean tai insuliinin kanssa)a | Jano | Ketoasidoosi* | ||
| Ruoansulatuskanava | Ummetus | ||||
| Iho ja ihonalainen kudos | Kutina (yleinen) Ihottuma | Urtikaria Angioedeema | |||
| Verisuonisto | Nestehukkaa | ||||
| Munuaiset ja virtsatiet | Lisääntynyt virtsaaminena | Dysuria | Tubulo-interstitiaalinen nefriitti | ||
| Tutkimukset | Kohonneet seerumin lipidiarvota | Kohonnut veren kreatiniinipitoisuus / Alentunut glomerulusten suodatusnopeusa Kohonnut hematokriittia |
a katso lisätietoja alla olevista alakohdista
* ks. kohta Varoitukset ja käyttöön liittyvät varotoimet
Valikoitujen haittavaikutusten kuvaus
Hypoglykemia
Hypoglykemian esiintyvyys tutkimuksissa riippui potilaalla käytöstä olleesta taustalääkityksestä ja se oli samanlainen empagliflotsiinia ja lumelääkettä monoterapiana saaneiden potilaiden ryhmässä, yhdistettynä metformiiniin, yhdistettynä pioglitatsoniin metformiinin kanssa tai ilman, linagliptiinin ja metformiinin kanssa ja yhdistettynä tavanomaiseen hoitoon ja empagliflotsiinin ja metformiinin yhdistelmähoidossa potilailla, jotka eivät aiemmin olleet saaneet lääkehoitoa, verrattuna potilaisiin, joita hoidettiin empagliflotsiinilla ja metformiinilla yksittäisinä ainesosina. Esiintymistiheyden havaittiin lisääntyvän yhdistettynä metformiinin ja sulfonyyliurean kanssa (empagliflotsiini 10 mg: 16,1 %, empagliflotsiini 25 mg: 11,5 %, lumelääke: 8,4 %), yhdistettynä perusinsuliiniin metformiinin kanssa tai ilman ja sulfonyyliurean kanssa tai ilman (empagliflotsiini 10 mg: 19,5 %, empagliflotsiini 25 mg: 28,4 %, lumelääke: 20,6 % 18 viikon pituisen aloitushoidon aikana, kun insuliiniannosta ei voitu säätää; empagliflotsiini 10 mg ja 25 mg: 36,1 %, lumelääke 35,3 % 78 viikon pituisen tutkimuksen aikana) ja yhdistettynä monipistosinsuliinihoitoon metformiinin kanssa tai ilman (empagliflotsiini 10 mg: 39,8 %, empagliflotsiini 25 mg: 41,3 %, lumelääke: 37,2 % ensimmäisten 18 hoitoviikon aikana, kun insuliiniannosta ei voitu säätää; empagliflotsiini 10 mg: 51,1 %, empagliflotsiini 25 mg: 57,7 %, lumelääke: 58 % 52 viikon pituisen tutkimuksen aikana).
Sydämen vajaatoimintaa koskeneissa EMPEROR-tutkimuksissa hypoglykemian esiintyvyys oli samankaltainen, kun hoitoa annettiin yhdistettynä sulfonyyliureaan tai insuliiniin (empagliflotsiini 10 mg: 6,5 %; lumelääke: 6,7 %).
Merkittävä hypoglykemia (hoitoa vaativat tapahtumat)
Merkittävän hypoglykemian ei havaittu lisääntyvän empagliflotsiinia monoterapiana saaneilla potilailla verrattuna lumelääkettä monoterapiana saaneisiin potilaisiin, metformiiniin yhdistettynä, metformiiniin ja sulfonyyliureaan yhdistettynä, pioglitatsoniin yhdistettynä metformiinin kanssa tai ilman, linagliptiinin ja metformiinin kanssa, yhdistettynä tavanomaiseen hoitoon ja empagliflotsiinin ja metformiinin yhdistelmähoidossa potilailla, jotka eivät aiemmin olleet saaneet lääkehoitoa, verrattuna potilaisiin, joita hoidettiin empagliflotsiinilla ja metformiinilla yksittäisinä ainesosina. Esiintyvyys lisääntyi yhdistettäessä perusinsuliiniin metformiinin kanssa tai ilman ja sulfonyyliurean kanssa tai ilman (empagliflotsiini 10 mg: 0 %, empagliflotsiini 25 mg: 1,3 %, lumelääke: 0 % 18 viikon pituisen aloitushoidon aikana kun insuliiniannosta ei voitu säätää; empagliflotsiini 10 mg: 0 %, empagliflotsiini 25 mg: 1,3 %, lumelääke 0 % 78 viikon pituisen tutkimuksen aikana) ja yhdistettynä monipistosinsuliinihoitoon metformiinin kanssa tai ilman (empagliflotsiini 10 mg: 0,5 %, empagliflotsiini 25 mg: 0,5 %, lumelääke: 0,5 % ensimmäisten 18 hoitoviikon aikana, kun insuliiniannosta ei voitu säätää; empagliflotsiini 10 mg: 1,6 %, empagliflotsiini 25 mg: 0,5 %, lumelääke: 1,6 % 52 viikon pituisen tutkimuksen aikana).
Sydämen vajaatoimintaa koskeneissa EMPEROR-tutkimuksissa merkittävän hypoglykemian esiintyvyys diabetespotilailla oli samankaltainen riippumatta siitä, annettiinko potilaille empagliflotsiinia vai lumelääkettä yhdistettynä sulfonyyliureaan tai insuliiniin (empagliflotsiini 10 mg: 2,2 %; lumelääke: 1,9 %).
Vaginan kandidiaasi, vulvovaginiitti, balaniitti ja muu genitaali-infektio
Vaginan kandidiaasia, vulvovaginiittia, balaniittia ja muita genitaali-infektioita raportoitiin yleisemmin empagliflotsiinihoitoa saaneilla potilailla (empagliflotsiini 10 mg: 4,0 %, empagliflotsiini 25 mg: 3,9 %) kuin lumelääkettä saaneilla (1,0 %). Näitä infektioita raportoitiin yleisemmin empagliflotsiinia saaneilla naisilla kuin lumelääkettä saaneilla, ja esiintyvyyden ero oli pienempi miehillä. Sukupuolielinten infektiot olivat voimakkuudeltaan lieviä tai keskivaikeita.
Sydämen vajaatoimintaa koskeneissa EMPEROR-tutkimuksissa näitä infektioita esiintyi lumelääkkeeseen verrattuna empagliflotsiinihoidon yhteydessä enemmän diabetespotilailla (empagliflotsiini 10 mg: 2,3 %; lumelääke: 0,8 %) kuin potilailla, joilla ei ollut diabetesta (empagliflotsiini 10 mg: 1,7 %; lumelääke: 0,7 %).
Lisääntynyt virtsaaminen
Lisääntynyttä virtsaamista (mukaan lukien ennaltamääritellyt termit pollakisuria, polyuria ja nokturia) havaittiin esiintyvän enemmän empagliflotsiinihoitoa saaneilla potilailla (empagliflotsiini 10 mg: 3,5 %, empagliflotsiini 25 mg: 3,3 %) kuin lumelääkettä saaneilla (1,4 %). Lisääntynyt virtsaaminen oli useimmiten voimakkuudeltaan lievää tai kohtalaista. Raportoidun nokturian esiintyvyys oli samanlainen lumelääkkeellä ja empagliflotsiinilla (< 1 %).
Sydämen vajaatoimintaa koskeneissa EMPEROR-tutkimuksissa lisääntyneen virtsaamisen esiintyvyys oli samankaltainen empagliflotsiinia ja lumelääkettä saaneilla potilailla (empagliflotsiini 10 mg: 0,9 %; lumelääke: 0,5 %).
Virtsatieinfektio
Haittatapahtumana ilmoitetun virtsatieinfektion kokonaisesiintyvyys oli samanlainen empagliflotsiinihoitoa (25 mg) ja lumelääkettä saaneilla potilailla (7,0 % ja 7,2 %), ja korkeampi empagliflotsiinihoitoa (10 mg) saaneilla potilailla (8,8 %). Samoin kuin lumelääkettä saaneilla potilailla virtsatieinfektio raportoitiin yleisemmin sellaisilla empagliflotsiinilla hoidetuilla potilailla, joilla oli ollut aikaisemmin kroonisia tai uusiutuvia virtsatieinfektioita. Virtsatieinfektion voimakkuus (lievä, keskivaikea, vaikea) oli samankaltainen empagliflotsiinia ja lumelääkettä saaneilla potilailla. Virtsatieinfektio raportoitiin yleisemmin empagliflotsiinihoitoa saaneilla naisilla kuin lumelääkettä saaneilla; eroa ei ollut miehillä.
Nestehukka
Nestehukan kokonaisesiintyvyys (mukaan lukien ennaltamääritellyt termit alentunut verenpaine (ambulatorinen), alentunut systolinen verenpaine, dehydraatio, hypotensio, hypovolemia, ortostaattinen hypotensio ja pyörtyminen) oli samankaltainen empagliflotsiinihoitoa saaneilla potilailla (empagliflotsiini 10 mg: 0,6 %, empagliflotsiini 25 mg: 0,4 %) ja lumelääkettä saaneilla potilailla (0,3 %). Nestehukan esiintyvyys lisääntyi 75-vuotiailla ja sitä vanhemmilla potilailla, jotka saivat 10 mg:n empagliflotsiinihoitoa (2,3 %) tai 25 mg:n empagliflotsiinihoitoa (4,3 %), verrattuna lumelääkettä saaneisiin (2,1 %).
Kohonnut veren kreatiniinipitoisuus / Alentunut glomerulusten suodatusnopeus
Niiden potilaiden, joilla veren kreatiniinipitoisuus oli kohonnut ja glomerulusten suodatusnopeus alentunut, kokonaisesiintyvyys oli yhtä suuri empagliflotsiini- ja lumelääkeryhmissä (veren kreatiniinipitoisuus kohonnut: empagliflotsiini 10 mg 0,6 %, empagliflotsiini 25 mg 0,1 %, lumelääke 0,5 %; glomerulusten suodatusnopeus alentunut: empagliflotsiini 10 mg 0,1 %, empagliflotsiini 25 mg 0 %, lumelääke 0,3 %).
Empagliflotsiinihoitoa saaneilla potilailla hoidon alussa havaittu kreatiniinipitoisuuden nousu ja hoidon alussa arvioidussa glomerulusten suodatusnopeudessa havaittu alenema oli hoidon jatkuessa yleensä ohimenevää tai lääkehoidon keskeytyessä palautuvaa.
Vastaavasti EMPA-REG OUTCOME -tutkimuksessa empagliflotsiinia saaneiden potilaiden laskennallinen glomerulusten suodatusnopeus (eGFR) pieneni aluksi (keskiarvo: 3 ml/min/1,73 m2). Sen jälkeen eGFR pysyi samalla tasolla, kun hoitoa jatkettiin. Keskimääräinen eGFR palasi lähtöarvoihin hoidon lopettamisen jälkeen, mikä viittaa siihen, että äkillisillä hemodynaamisilla muutoksilla voi olla osuutensa näissä munuaistoiminnan muutoksissa. Tämä ilmiö todettiin myös sydämen vajaatoimintaa koskeneissa EMPEROR-tutkimuksissa ja EMPA-KIDNEY-tutkimuksessa.
Seerumin lipidiarvojen nousu
Keskimääräiset prosentuaaliset nousut lähtöarvoista empagliflotsiinin 10 mg:n ja 25 mg:n annoksilla verrattuna lumelääkkeeseen olivat: kokonaiskolesteroli 4,9 % ja 5,7 % vs. 3,5 %; HDL-kolesteroli 3,3 % ja 3,6 % vs. 0,4 %; LDL-kolesteroli 9,5 % ja 10,0 % vs. 7,5 %; triglyseridit 9,2 % ja 9,9 % vs. 10,5 %.
Hematokriittiarvon nousu
Hematokriittiarvon keskimääräinen muutos lähtöarvosta oli 3,4 % empagliflotsiinia 10 mg saaneilla ja 3,6 % empagliflotsiinia 25 mg saaneilla sekä 0,1 % lumelääkettä saaneilla. EMPA-REG Outcome -tutkimuksessa hematokriittiarvot palasivat lähtöarvoja kohti hoidon lopettamista seuranneen 30 vuorokauden seurantavaiheen jälkeen.
Pediatriset potilaat
DINAMO-tutkimuksessa hoidettiin 157 lasta, jotka olivat iältään vähintään 10 vuotta ja joilla oli tyypin 2 diabetes. Tutkimuksessa 52 potilasta sai empagliflotsiinia, 52 linagliptiinia ja 53 lumelääkettä (ks. kohta Farmakodynamiikka).
Lumelääkekontrolloidun vaiheen aikana yleisin haittavaikutus oli hypoglykemia, jota esiintyi yhdistetyn empagliflotsiiniryhmän potilailla enemmän kuin lumelääkettä saaneilla (empagliflotsiini 10 mg ja 25 mg, yhdistettynä: 23,1 %, lumelääke: 9,4 %). Mikään haittatapahtumista ei ollut vaikea tai edellyttänyt hoitoa.
Yleisesti ottaen turvallisuusprofiili lapsilla oli samanlainen kuin turvallisuusprofiili aikuisilla, joilla on tyypin 2 diabetes.
Epäillyistä haittavaikutuksista ilmoittaminen
On tärkeää ilmoittaa myyntiluvan myöntämisen jälkeisistä lääkevalmisteen epäillyistä haittavaikutuksista. Se mahdollistaa lääkevalmisteen hyöty–haittatasapainon jatkuvan arvioinnin. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista seuraavalle taholle:
www-sivusto: www.fimea.fi
Lääkealan turvallisuus- ja kehittämiskeskus Fimea
Lääkkeiden haittavaikutusrekisteri
PL 55
00034 FIMEA
Yliannostus
Oireet
Kontrolloiduissa kliinisissä tutkimuksissa, joissa terveille vapaaehtoisille annettiin enintään 800 mg:n kerta-annoksia empagliflotsiinia ja tyypin 2 diabetesta sairastaville potilaille toistuvina annoksina enintään 100 mg empagliflotsiinia vuorokaudessa, ei ilmennyt mitään toksisuutta. Empagliflotsiini lisäsi glukoosin erittymistä virtsaan, mikä johti virtsamäärän nousuun. Havaittu virtsamäärän nousu ei ollut annosriippuvaista eikä ole kliinisesti merkittävää. Yli 800 mg:n suuruisista annoksista ihmisillä ei ole kokemuksia.
Hoito
Yliannostustapauksessa on aloitettava hoito potilaan kliinisen tilan mukaisesti. Empagliflotsiinin poistamista hemodialyysin avulla ei ole tutkittu.
Farmakologiset ominaisuudet
Farmakodynamiikka
Farmakoterapeuttinen ryhmä: Diabeteslääkkeet, Natrium-glukoosi-kuljettajaproteiini 2:n (SGLT2) estäjät, ATC-koodi: A10BK03
Vaikutusmekanismi
Empagliflotsiini on reversiibeli, erittäin voimakas (IC50 1,3 nmol) ja selektiivinen kilpaileva natriumin- ja glukoosinkuljettajaproteiini 2:n (SGLT2) estäjä. Empagliflotsiini ei estä muita glukoosinkuljettajaproteiineja, jotka ovat tärkeitä glukoosin kuljetuksessa ääreiskudoksiin, ja se on 5 000 kertaa selektiivisempi SGLT2:n kuin SGLT1:n suhteen. SGLT1 on kuljettajaproteiini, joka pääasiallisesti vastaa glukoosin imeytymisestä suolessa. SGLT2:ta esiintyy pääosin munuaisessa, kun taas muissa kudoksissa sitä ei ole lainkaan tai sen määrä on hyvin alhainen. Se vastaa pääasiallisesti glukoosin takaisinimeytymisestä glomerulussuodoksesta takaisin verenkiertoon. Tyypin 2 diabetesta ja hyperglykemiaa sairastavilla potilailla suurempi määrä glukoosia suodattuu ja imeytyy takaisin.
Empagliflotsiini parantaa tyypin 2 diabetesta sairastavien potilaiden glukoositasapainoa vähentämällä glukoosin takaisinimeytymistä munuaisissa. Munuaisen poistaman glukoosin määrä tällä glukosuurisella mekanismilla riippuu veren glukoosipitoisuudesta ja GFR-arvosta. SGLT2:n esto tyypin 2 diabetesta ja hyperglykemiaa sairastavilla potilailla johtaa liiallisen glukoosin erittymiseen virtsaan. Lisäksi empagliflotsiinihoidon aloittaminen lisää natriumin eritystä, mikä johtaa osmoottiseen diureesiin ja suontensisäisen tilavuuden laskuun.
Tyypin 2 diabetesta sairastavilla potilailla glukoosin erittyminen virtsaan lisääntyi välittömästi empagliflotsiinin ensimmäisen annoksen jälkeen ja jatkuu 24 tunnin annosvälin ajan. Glukoosin lisääntynyt erittyminen virtsaan jatkui 4 viikon hoitojakson loppuun asti, ja sitä erittyi keskimäärin 78 g/vrk. Glukoosin lisääntynyt erittyminen virtsaan alensi välittömästi plasman glukoosiarvoja tyypin 2 diabetesta sairastavilla potilailla.
Empagliflotsiini parantaa plasman glukoosiarvoja sekä paaston että aterian jälkeen. Empagliflotsiinin vaikutusmekanismi on riippumaton beetasolujen toiminnasta ja insuliinierityksestä, ja tämä vaikuttaa osaltaan hypoglykemian vähäiseen riskiin. Beetasolujen toimintaa kuvaavissa markkereissa, mukaan lukien homeostaasimallimääritys (HOMA-β), havaittiin paranemista. Lisäksi glukoosin erittyminen virtsaan aiheuttaa kalorien hävikkiä, mihin liittyy kehon rasvakudoksen vähenemistä ja painon laskua. Empagliflotsiinista aiheutuvaan glukosuriaan liittyy diureesi, joka voi osaltaan johtaa pitkäaikaiseen kohtalaiseen verenpaineen laskuun.
Empagliflotsiini myös vähentää natriumin takaisinimeytymistä ja lisää natriumin kuljetusta distaalisiin tubuluksiin. Tämä voi vaikuttaa moniin fysiologisiin toimintoihin ja esimerkiksi lisätä tubuloglomerulaarista palautetta ja vähentää glomerulusten sisäistä painetta, vähentää sydämen esi- ja jälkikuormitusta, vaimentaa sympaattisen hermoston toimintaa ja vähentää vasemman kammion seinämän kuormitusta, mikä näkyy matalampina NT-proBNP-arvoina, millä voi olla suotuisia vaikutuksia sydämen uudelleen muotoutumiseen, täyttöpaineisiin ja diastoliseen toimintaan sekä munuaisten rakenteen ja toiminnan säilymiseen. Muut vaikutukset, kuten hematokriittiarvon nousu sekä painon ja verenpaineen lasku, saattavat osaltaan myötävaikuttaa suotuisiin sydän- ja munuaisvaikutuksiin.
Kliininen teho ja turvallisuus
Tyypin 2 diabetes
Sekä glukoositasapainon parantaminen että sydän- ja verisuonisairauksien ja -kuolemien vähentäminen ovat olennainen osa tyypin 2 diabeteksen hoitoa.
Glukoositasapainoa parantavaa tehoa ja sydän- ja verisuonisairauksia koskevia tuloksia arvoitiin yhteensä 14 663:lla tyypin 2 diabetesta sairastavalla potilaalla, joita hoidettiin 12 kaksoissokkoutetussa, lume- ja aktiiviainekontrolloidussa kliinisessä tutkimuksessa, joissa 9 295 potilasta sai empagliflotsiinia (empagliflotsiini 10 mg: 4 165 potilasta; empagliflotsiini 25 mg: 5 130 potilasta). Viidessä tutkimuksessa hoito kesti 24 viikkoa; näiden tutkimusten jatkotutkimuksissa sekä muissa tutkimuksissa potilaat altistuivat empagliflotsiinille korkeintaan 102 viikon ajan.
Empagliflotsiinihoito monoterapiana sekä metformiinin, pioglitatsonin, sulfonyyliurean, DPP-4:n estäjien ja insuliinin kanssa paransi kliinisesti merkittävästi HbA1c-arvoa, paastoglukoosiarvoa , painoa sekä systolista ja diastolista verenpainetta. Suurempi osa 25 mg:n empagliflotsiiniannoksella hoidetuista potilaista saavutti tavoitteen HbA1c alle 53 mmol/mol (7 %) ja pienempi osa tarvitsi lisälääkitystä glukoositasapainon korjaamiseksi kuin 10 mg:n empagliflotsiiniannoksella tai lumelääkkeellä hoidetuista potilaista. Potilailla, joilla HbA1c-arvo tutkimuksen alussa oli korkeampi, havaittiin myös suurempi HbA1c-arvon aleneminen. Lisäksi empagliflotsiini yhdistettynä tavanomaiseen hoitoon vähensi sydän- ja verisuonikuolemia tyypin 2 diabetespotilailla, joilla oli sydän- ja verisuonisairaus.
Monoterapia
Empagliflotsiinin tehoa ja turvallisuutta monoterapiana arvioitiin kaksoissokkoutetussa, lume- ja aktiivikontrolloidussa 24 viikon pituisessa tutkimuksessa aiemmin hoitamattomilla potilailla. Empagliflotsiinihoito alensi tilastollisesti merkitsevästi (p < 0,0001) HbA1c-arvoa lumelääkkeeseen verrattuna (taulukko 2) ja kliinisesti merkittävästi paastoglukoosiarvoa.
Ennalta määritellyssä potilasanalyysissä (N = 201), jossa lähtötilanteen HbA1c ≥ 69 mmol/mol (≥ 8,5 %), hoito alensi lähtötilanteen HbA1c-arvoa ‑15,69 mmol/mol (-1,44 %) 10 mg empagliflotsiinia saaneilla, ‑15,64 mmol/mol (-1,43 %) 25 mg empagliflotsiinia saaneilla ja ‑11,34 mmol/mol (-1,04 %) sitagliptiinia saaneilla, ja nosti arvoa 0,07 mmol/mol (0,01 %) lumelääkettä saaneilla.
Tämän tutkimuksen kaksoissokkoutetussa, lumekontrolloidussa jatkovaiheessa HbA1c-arvon, painon ja verenpaineen aleneminen säilyi viikolle 76 asti.
Taulukko 2: 24 viikon lumekontrolloidun empagliflotsiini-monoterapiatutkimuksen tehon tulokseta
| Lumelääke | Jardiance | Sitagliptiini | ||
| 10 mg | 25 mg | 100 mg | ||
| N | 228 | 224 | 224 | 223 |
| HbA1c mmol/mol (%) | ||||
| Lähtötilanne (keskiarvo) | 62,99 (7,91) | 62,55 (7,87) | 62,42 (7,86) | 62,31 (7,85) |
| Muutos lähtötilanteesta1 | 0,82 (0,08) | -7,22 (-0,66) | -8,48 (-0,78) | -7,19 (-0,66) |
| Ero lumelääkkeeseen1 (97,5 % lv) | -8,04 [‑9,82, ‑6,26] (-0,74* [-0,90, -0,57]) | -9,30 [‑11,08, ‑7,52] (‑0,85* [‑1,01, ‑0,69]) | -8,01 [‑9,57, ‑6,44] (‑0,73 [‑0,88, ‑0,59])3 | |
| N | 208 | 204 | 202 | 200 |
| HbA1c-arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c-arvon ollessa ≥ 53 mmol/mol (7 %2) | 12,0 | 35,3 | 43,6 | 37,5 |
| N | 228 | 224 | 224 | 223 |
| Paino (kg) | ||||
| Lähtötilanne (keskiarvo) | 78,23 | 78,35 | 77,80 | 79,31 |
| Muutos lähtötilanteesta1 | -0,33 | -2,26 | -2,48 | 0,18 |
| Ero lumelääkkeeseen1 (97,5 % lv) | -1,93* (-2,48, -1,38) | -2,15* (-2,70,-1,60) | 0,52 (-0,04, 1,00)3 | |
| N | 228 | 224 | 224 | 223 |
| Systolinen verenpaine (SVP, mmHg)4 | ||||
| Lähtötilanne (keskiarvo) | 130,4 | 133,0 | 129,9 | 132,5 |
| Muutos lähtötilanteesta1 | -0,3 | -2,9 | -3,7 | 0,5 |
| Ero lumelääkkeeseen1 (97,5 % lv) | -2,6* (-5,2, -0,0) | -3,4* (‑6,0, ‑0,9) | 0,8 (-1,4, 3,1)3 | |
a Koko analyysijoukko (FAS) käyttäen viimeisimmästä havainnosta laskettua arviota (last observation carried forward, LOCF), ennen lisälääkityksen ottamista glukoositasapainon korjaamiseksi
1 Korjattu keskiarvo lähtötilanteeseen nähden
2 Tilastollista merkitsevyyttä ei arvioitu perättäisen vahvistavan testimenetelmän vuoksi
3 95 % lv
4 LOCF, verenpainetta alentavan lisälääkkeen ottamisen jälkeiset arvot on poistettu
*p-arvo < 0,0001
Yhdistelmähoito
Empagliflotsiini yhdistettynä metformiiniin, sulfonyyliureaan, pioglitatsoniin
Empagliflotsiini yhdistettynä metformiiniin, metformiiniin ja sulfonyyliureaan tai pioglitatsoniin metformiinin kanssa tai ilman, alensi tilastollisesti merkitsevästi (p < 0,0001) HbA1c-arvoa ja painoa lumelääkkeeseen verrattuna (taulukko 3). Lisäksi se alensi kliinisesti merkittävästi paastoglukoosiarvoa sekä systolista ja diastolista verenpainetta lumelääkkeeseen verrattuna.
Näiden tutkimusten kaksoissokkoutetussa, lumekontrolloidussa jatkovaiheessa HbA1c-arvon, painon ja verenpaineen aleneminen säilyi viikolle 76 asti.
Taulukko 3: 24 viikon lumekontrolloitujen tutkimusten tehon tulokseta
| Yhdistettynä metformiinihoitoon | |||
| Lumelääke | Jardiance | ||
| 10 mg | 25 mg | ||
| N | 207 | 217 | 213 |
| HbA1c mmol/mol (%) | |||
| Lähtötilanne (keskiarvo) | 62,87 (7,90) | 63,28 (7,94) | 62,35 (7,86) |
| Muutos lähtötilanteesta1 | -1,45 (-0,13) | -7,64 (-0,70) | -8,40 (-0,77) |
| Ero lumelääkkeeseen1 (97,5 % lv) | 6,20 [‑7,85, ‑4,54] (-0,57* [-0,72, -0,42]) | -6,95 [-8,62, ‑5,29] (-0,64* [-0,79, -0,48]) | |
| N | 184 | 199 | 191 |
| HbA1c-arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c-arvon ollessa ≥ 53 mmol/mol (7 %2) | 12,5 | 37,7 | 38,7 |
| N | 207 | 217 | 213 |
| Paino (kg) | |||
| Lähtötilanne (keskiarvo) | 79,73 | 81,59 | 82,21 |
| Muutos lähtötilanteesta1 | -0,45 | -2,08 | -2,46 |
| Ero lumelääkkeeseen1 (97,5 % lv) | -1,63* (-2,17, -1,08) | -2,01* (-2,56, -1,46) | |
| N | 207 | 217 | 213 |
| SVP (mmHg)2 | |||
| Lähtötilanne (keskiarvo) | 128,6 | 129,6 | 130,0 |
| Muutos lähtötilanteesta1 | -0,4 | -4,5 | -5,2 |
| Ero lumelääkkeeseen1 (95 % lv) | -4,1* (-6,2, -2,1) | -4,8* (-6,9, -2,7) | |
| Yhdistettynä metformiini- ja sulfonyyliureahoitoon | |||
| Lumelääke | Jardiance | ||
| 10 mg | 25 mg | ||
| N | 225 | 225 | 216 |
| HbA1c mmol/mol (%) | |||
| Lähtötilanne (keskiarvo) | 65,57 (8,15) | 64,66 (8,07) | 64,98 (8,10) |
| Muutos lähtötilanteesta1 | -1,90 (-0,17) | -8,92 (-0,82) | -8,38 (-0,77) |
| Ero lumelääkkeeseen1 (97,5 % lv) | ‑7,02 [‑8,67, ‑5,38] (-0,64* [-0,79, -0,49]) | ‑6,48 [‑8,14, ‑4,81] (-0,59* [-0,74, -0,44]) | |
| N | 216 | 209 | 202 |
| HbA1c-arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c-arvon ollessa ≥ 53 mmol/mol (7 %2) | 9,3 | 26,3 | 32,2 |
| N | 225 | 225 | 216 |
| Paino (kg) | |||
| Lähtötilanne (keskiarvo) | 76,23 | 77,08 | 77,50 |
| Muutos lähtötilanteesta1 | -0,39 | -2,16 | -2,39 |
| Ero lumelääkkeeseen1 (97,5 % lv) | -1,76* (-2,25, -1,28) | -1,99* (-2,48, -1,50) | |
| N | 225 | 225 | 216 |
| SVP (mmHg)2 | |||
| Lähtötilanne (keskiarvo) | 128,8 | 128,7 | 129,3 |
| Muutos lähtötilanteesta1 | -1,4 | -4,1 | -3,5 |
| Ero lumelääkkeeseen1 (95 % lv) | -2,7 (-4,6, -0,8) | -2,1 (-4,0, -0,2) | |
| Yhdistettynä pioglitatsoni +/- metformiinihoitoon | |||
| Lumelääke | Jardiance | ||
| 10 mg | 25 mg | ||
| N | 165 | 165 | 168 |
| HbA1c mmol/mol (%) | |||
| Lähtötilanne (keskiarvo) | 65,63 (8,16) | 64,70 (8,07) | 64,56 (8,06) |
| Muutos lähtötilanteesta1 | -1,18 (-0,11) | -6,41 (-0,59) | -7,82 (-0,72) |
| Ero lumelääkkeeseen1 (97,5 % lv) | ‑5,23 [‑7,55, ‑2,92] (-0,48* [-0,69, -0,27]) | ‑6,64 [‑8,95, ‑4,33] (-0,61* [-0,82, -0,40]) | |
| N | 155 | 151 | 160 |
| HbA1c-arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c-arvon ollessa ≥ 53 mmol/mol (7 %2) | 7,7 | 24 | 30 |
| N | 165 | 165 | 168 |
| Paino (kg) | |||
| Lähtötilanne (keskiarvo) | 78,1 | 77,97 | 78,93 |
| Muutos lähtötilanteesta1 | 0,34 | -1,62 | -1,47 |
| Ero lumelääkkeeseen1 (97,5 % lv) | -1,95* (-2,64, -1,27) | -1,81* (-2,49, -1,13) | |
| N | 165 | 165 | 168 |
| SVP (mmHg)3 | |||
| Lähtötilanne (keskiarvo) | 125,7 | 126,5 | 126 |
| Muutos lähtötilanteesta1 | 0,7 | -3,1 | -4,0 |
| Ero lumelääkkeeseen1 (95 % lv) | -3,9 (-6,23, -1,50) | -4,7 (-7,08, -2,37) | |
a Koko analyysijoukko (FAS) käyttäen viimeisimmästä havainnosta laskettua arviota (last observation carried forward, LOCF), ennen lisälääkityksen ottamista glukoositasapainon korjaamiseksi
1 Korjattu keskiarvo lähtötilanteeseen nähden
2 Tilastollista merkitsevyyttä ei arvioitu perättäisen vahvistavan testimenetelmän vuoksi
3 LOCF, verenpainetta alentavan lisälääkkeen ottamisen jälkeiset arvot on poistettu
* p-arvo < 0,0001
Yhdessä metformiinin kanssa potilailla, jotka eivät aiemmin olleet saaneet lääkehoitoa
Empagliflotsiinin tehoa ja turvallisuutta arvioitiin 24 viikon pituisessa faktorikoeasetelmatutkimuksessa potilailla, jotka eivät aiemmin olleet saaneet lääkehoitoa. Empagliflotsiinin ja metformiinin yhdistelmähoito (5 mg ja 500 mg; 5 mg ja 1 000 mg; 12,5 mg ja 500 mg ja 12,5 mg ja 1 000 mg kaksi kertaa vuorokaudessa annettuna) paransi HbA1c‑arvoja tilastollisesti merkitsevästi (taulukko 4) ja johti suurempiin paastoglukoosiarvojen (yksittäisiin ainesosiin verrattuna) ja painon alenemiseen (metformiiniin verrattuna).
Taulukko 4: Tehon tulokset 24 viikon kohdalla, kun empagliflotsiinin ja metformiinin yhdistelmähoitoa verrattiin yksittäisiin ainesosiina
| Empagliflotsiini 10 mgb | Empagliflotsiini 25 mgb | Metformiinic | ||||||
| + Met 1 000 mgc | + Met 2 000 mgc | Ei metformiinia | + Met 1 000 mgc | + Met 2 000 mgc | Ei metformiinia | 1 000 mg | 2 000 mg | |
| N | 161 | 167 | 169 | 165 | 169 | 163 | 167 | 162 |
| HbA1c mmol/mol (%) | ||||||||
| Lähtö-tilanne (keski-arvo) | 71,40 (8,68) | 71,04 (8,65) | 70,71 (8,62) | 73,07 (8,84) | 71,18 (8,66) | 73,27 (8,86) | 71,46 (8,69) | 69,97 (8,55) |
| Muutos lähtötilanteeseen nähden1 | -21,60 (‑1,98) | ‑22,66 (‑2,07) | ‑14,75 (‑1,35) | ‑21,11 (‑1,93) | ‑22,69 (‑2,08) | ‑14,85 (‑1,36) | ‑12,93 (‑1,18) | ‑19,09 (‑1,75) |
| Vertailu empagliflotsiiniin nähden [95 % lv]1 | -6,85 [‑9,38, ‑4,32] | ‑‑7,92 [‑10,43, ‑5,40](-0,72*[-0,96, -0,49]) | ‑6,26 | ‑7,84 | ||||
| Vertailu metformiiniin nähden [95 % lv]1 | ‑‑8,67 [‑11,23, ‑6,11](-0,79*[-1,03, -0,56]) | ‑3,57 | ‑8,18 | ‑3,60 | ||||
Met = metformiini
1 Korjattu keskiarvo lähtötilanteeseen nähden
a Analyysit suoritettiin koko analyysiaineiston (FAS) perusteella havaittujen tapausten (OC) lähestymistapaa noudattaen
b Annettiin jaettuna kahteen yhtä suureen annokseen vuorokaudessa annettaessa yhdessä metformiinin kanssa
c Annettiin jaettuna kahteen yhtä suureen annokseen vuorokaudessa
* HbA1c‑arvon osalta p ≤ 0,0062
Empagliflotsiini potilailla, joiden hoitotasapaino on riittämätön, kun käytössä on metformiini ja linagliptiini
Potilailla, joiden hoitotasapaino oli riittämätön metformiinilla ja 5 mg linagliptiinilla, empagliflotsiinihoito 10 mg:n tai 25 mg:n annoksella alensi tilastollisesti merkitsevästi (p<0,0001) HbA1c-arvoa ja painoa lumelääkkeeseen verrattuna (taulukko 5). Lisäksi se alensi kliinisesti merkittävästi paastoglukoosiarvoa sekä systolista ja diastolista verenpainetta lumelääkkeeseen verrattuna.
Taulukko 5: Tehon tulokset 24 viikon lumelääkekontrolloidussa tutkimuksessa potilailla, joiden hoitotasapaino oli riittämätön metformiinilla ja 5 mg linagliptiinilla
| Metformiinin ja 5 mg linagliptiinin kanssa | |||
| Lumehoito5 | Empagliflotsiini6 | ||
| 10 mg | 25 mg | ||
| N | 106 | 109 | 110 |
| HbA1c mmol/mol (%)3 | |||
| Lähtötilanne (keskiarvo) | 63,45 (7,96) | 63,63 (7,97) | 63,64 (7,97) |
| Muutos lähtötilanteesta1 | 1,49 (0,14) | ‑7,14 (-0,65) | ‑6,12 (-0,56) |
| Ero lumelääkkeeseen [95 % lv] | ‑8,63 [‑11,19, ‑6,06](-0,79* [-1,02, -0,55]) | ‑7,61 [‑10,18, ‑5,04](-0,70* [-0,93, -0,46]) | |
| N | 100 | 100 | 107 |
| HbA1c-arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c-arvon ollessa ≥ 53 mmol/mol (7 %2) | 17,0 | 37,0 | 32,7 |
| N | 106 | 109 | 110 |
| Paino (kg)3 | |||
| Lähtötilanne (keskiarvo) | 82,3 | 88,4 | 84,4 |
| Muutos lähtötilanteesta1 | -0,3 | -3,1 | -2,5 |
| Ero lumelääkkeeseen (95 % lv) | -2,8* (-3,5, -2,1) | -2,2* (-2,9, -1,5) | |
| N | 106 | 109 | 110 |
| SVP (mmHg)4 | |||
| Lähtötilanne (keskiarvo) | 130,1 | 130,4 | 131,0 |
| Muutos lähtötilanteesta1 | -1,7 | -3,0 | -4,3 |
| Ero lumelääkkeeseen (95% CI) | -1,3 (-4,2, 1,7) | -2,6 (-5,5, 0,4) | |
1 Korjattu keskiarvo lähtötilanteeseen nähden
2 Tilastollista merkitsevyyttä ei ole arvioitu; ei mukana toissijaisten päätetapahtumien sekventiaalisessa testimenetelmässä.
3 FAS:n (OC) MMRM-malli; mukana lähtötilanteen HbA1c-arvo, lähtötilanteen eGFR-arvo (MDRD), maantieteellinen alue, käynti ja hoito sekä käynnin ja hoidon vuorovaikutus. Painon osalta huomioitiin lähtötilanteen paino.
4 MMRM-malli; mukana lineaarisina kovariaatteina lähtötilanteen systolinen verenpaine ja lähtötilanteen HbA1c-arvo ja kiinteinä vaikutuksina lähtötilanteen eGFR-arvo, maantieteellinen alue, hoito, käynti sekä käynnin ja hoidon vuorovaikutus.
5 Lumeryhmään satunnaistetut potilaat saivat lumelääkettä ja 5 mg linagliptiinia sekä lisäksi metformiinia
6 Empagliflotsiini 10 mg tai empagliflotsiini 25 mg -ryhmään satunnaistetut potilaat saivat 10 mg tai 25 mg empagliflotsiinia ja 5 mg linagliptiinia sekä lisäksi metformiinia
*p-arvo <0,0001
Ennalta määrätyssä alaryhmässä, jossa potilaiden lähtötilanteen HbA1c-arvo oli suurempi tai yhtä suuri kuin 69 mmol/mol (8,5 %), HbA1c-arvon alenema oli lähtötilanteeseen nähden ‑13,8 mmol/mol (-1,3 %) empagliflotsiini10 mg tai empagliflotsiini 25 mg -hoidossa viikolla 24 (p<0,0001) lumelääkkeeseen verrattuna.
Empagliflotsiinin 24 kuukauden tulokset, yhdistettynä metformiiniin ja verrattuna glimepiridiin
Tutkimuksessa, jossa verrattiin empagliflotsiinin 25 mg:n tehoa ja turvallisuutta glimepiridiin (korkeintaan 4 mg vuorokaudessa) potilailla, joilla oli riittämätön glukoositasapaino pelkkää metformiinia käytettäessä, päivittäinen empagliflotsiinihoito alensi HbA1c-arvoa enemmän kuin glimepiridi (taulukko 6). Lisäksi päivittäinen empagliflotsiinihoito alensi paastoglukoosiarvoa kliinisesti merkittävästi glimepiridiin verrattuna. Päivittäin käytetty empagliflotsiini alensi glimepiridiin verrattuna tilastollisesti merkitsevästi painoa, systolista ja diastolista verenpainetta ja pienensi tilastollisesti merkitsevästi niiden potilaiden osuutta, joilla oli hypoglykeemisiä tapahtumia (2,5 % empagliflotsiinihoitoa ja 24,2 % glimepiridihoitoa saaneista, p < 0,0001).
Taulukko 6: Tehon tulokset viikolla 104 vertailututkimuksessa, jossa verrattiin empagliflotsiinia glimepiridiin metformiiniin lisähoitonaa
| Empagliflotsiini 25 mg | Glimepiridib | |
| N | 765 | 780 |
| HbA1c mmol/mol (%) | ||
| Lähtötilanne (keskiarvo) | 63,02 (7,92) | 63,01 (7,92) |
| Muutos lähtötilanteesta1 | ‑7,18 (-0,66) | ‑6,02 (-0,55) |
| Ero glimepiridiin1 (97,5 % lv) | ‑1,16 [‑2,24, ‑0,09] | |
| N | 690 | 715 |
| HbA1c-arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c-arvon ollessa ≥ 53 mmol/mol (7 %2) | 33,6 | 30,9 |
| N | 765 | 780 |
| Paino (kg) | ||
| Lähtötilanne (keskiarvo) | 82,52 | 83,03 |
| Muutos lähtötilanteesta1 | -3,12 | 1,34 |
| Ero glimepiridiin1 (97,5 % lv) | -4,46** (-4,87, -4,05) | |
| N | 765 | 780 |
| SVP (mmHg)2 | ||
| Lähtötilanne (keskiarvo) | 133,4 | 133,5 |
| Muutos lähtötilanteesta1 | -3,1 | 2,5 |
| Ero glimepiridiin1 (97,5 % lv) | -5,6** (-7,0, -4,2) | |
a Koko analyysijoukko (FAS) käyttäen viimeisimmästä havainnosta laskettua arviota (last observation carried forward, LOCF), ennen lisälääkityksen ottamista glukoositasapainon korjaamiseksi
b Korkeintaan 4 mg glimepiridia
1 Korjattu keskiarvo lähtötilanteeseen nähden
2 LOCF, verenpainetta alentavan lisälääkkeen ottamisen jälkeiset arvot on poistettu
* p-arvo < 0,0001 samanveroisuudelle, ja p-arvo = 0,0153 paremmuudelle
** p-arvo < 0,0001
Yhdistettynä insuliinihoitoon
Empagliflotsiini yhdistettynä päivittäiseen moniannoksiseen insuliiniin
Empagliflotsiinin tehoa ja turvallisuutta yhdistettynä päivittäiseen moniannoksiseen insuliiniin joko samanaikaisesti käytetyn metformiinihoidon kanssa tai ilman arvioitiin kaksoissokkoutetussa, lumekontrolloidussa, 52 viikon pituisessa tutkimuksessa. Ensimmäisten 18 viikon ja viimeisten 12 viikon aikana insuliiniannos pidettiin vakaana, mutta viikkojen 19 ja 40 välillä sitä säädettiin, jotta saavutettiin ateriaa edeltävät glukoosiarvot < 5,5 mmol/l ja aterianjälkeiset glukoosiarvot < 7,8 mmol/l.
Viikolla 18 empagliflotsiini paransi tilastollisesti merkitsevästi HbA1c-arvoa lumelääkkeeseen verrattuna (taulukko 7).
Viikolla 52 empagliflotsiinihoito alensi tilastollisesti merkitsevästi HbA1c-arvoa ja vähensi insuliinin tarvetta lumelääkkeeseen verrattuna sekä alensi paastoglukoosiarvoa ja painoa.
Taulukko 7: Tehon tulokset viikoilla 18 ja 52 lumekontrolloidussa tutkimuksessa, jossa empagliflotsiini yhdistettiin päivittäiseen moniannoksiseen insuliiniin metformiinin kanssa tai ilman
| Lumelääke | Jardiance | ||
| 10 mg | 25 mg | ||
| N | 188 | 186 | 189 |
| HbA1c mmol/mol (%) viikolla 18 | |||
| Lähtötilanne (keskiarvo) | 67,53 (8,33) | 68,21 (8,39) | 67,13 (8,29) |
| Muutos lähtötilanteesta1 | ‑5,43 (-0,50) | ‑10,26 (-0,94) | ‑11,11 (-1,02) |
| Ero lumelääkkeeseen1 (97,5 % lv) | ‑4,83 [‑6,68, ‑2,98](-0,44* [-0,61, -0,27]) | ‑5,68 [‑7,52, ‑3,85](-0,52* [-0,69, -0,35]) | |
| N | 115 | 119 | 118 |
| HbA1c mmol/mol (%) viikolla 522 | |||
| Lähtötilanne (keskiarvo) | 66,62 (8,25) | 68,31 (8,40) | 67,93 (8,37) |
| Muutos lähtötilanteesta1 | ‑8,83 (-0,81) | ‑12,94 (-1,18) | ‑13,87 (-1,27) |
| Ero lumelääkkeeseen1 (97,5 % lv) | ‑4,11 [‑6,75, ‑1,46](-0,38*** [-0,62, -0,13]) | ‑5,04 [‑7,68, ‑2,39](-0,46* [-0,70, -0,22]) | |
| N | 113 | 118 | 118 |
| HbA1c-arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c-arvon ollessa ≥ 53 mmol/mol (7 %2) viikolla 52 | 26,5 | 39,8 | 45,8 |
| N | 115 | 118 | 117 |
| Insuliiniannos (IU/vrk) viikolla 522 | |||
| Lähtötilanne (keskiarvo) | 89,94 | 88,57 | 90,38 |
| Muutos lähtötilanteesta1 | 10,16 | 1,33 | -1,06 |
| Ero lumelääkkeeseen1 (97,5 % lv) | -8,83# (-15,69, -1,97) | -11,22** (-18,09, -4,36) | |
| N | 115 | 119 | 118 |
| Paino (kg) viikko 522 | |||
| Lähtötilanne (keskiarvo) | 96,34 | 96,47 | 95,37 |
| Muutos lähtötilanteesta1 | 0,44 | -1,95 | -2,04 |
| Ero lumelääkkeeseen1 (97,5 % lv) | -2,39* (-3,54, -1,24) | -2,48* (-3,63, -1,33) | |
1 Keskiarvo sovitettu lähtöarvon suhteen
2 Viikko 19–40: hoito-ohjelma tavoitteen saavuttamiseksi: insuliiniannosta muutettiin siten, että saavutettiin ennalta määritellyt verensokerin tavoitetasot (ateriaa edeltävä < 5,5 mmol/l, aterianjälkeinen < 7,8 mmol/l
* p-arvo < 0,0001
** p-arvo = 0,0003
*** p-arvo = 0,0005
# p-arvo = 0,0040
Empagliflotsiini yhdistettynä perusinsuliiniin
Empagliflotsiinin tehoa ja turvallisuutta yhdistettynä perusinsuliiniin metformiinin kanssa tai ilman ja/tai sulfonyyliurean kanssa tai ilman arvioitiin kaksoissokkoutetussa, lumekontrolloidussa, 78 viikon pituisessa tutkimuksessa. Ensimmäisten 18 viikon aikana insuliiniannos pidettiin vakaana, mutta seuraavien 60 viikon aikana sitä säädettiin, jotta saavutettiin paastoglukoosiarvo < 6,1 mmol/l.
Viikolla 18 empagliflotsiini paransi tilastollisesti merkitsevästi HbA1c-arvoa (taulukko 8).
Viikolla 78 empagliflotsiini alensi HbA1c-arvoa tilastollisesti merkitsevästi ja vähensi insuliinin tarvetta lumelääkkeeseen verrattuna. Lisäksi empagliflotsiini alensi paastoglukoosiarvoa, painoa ja verenpainetta.
Taulukko 8: Tehon tulokset viikoilla 18 ja 78 lumekontrolloidussa tutkimuksessa, jossa empagliflotsiini yhdistettiin perusinsuliiniin metformiinin kanssa tai ilman tai sulfonyyliureaana
| Lumelääke | Empagliflotsiini 10 mg | Empagliflotsiini 25 mg | |
| N | 125 | 132 | 117 |
| HbA1c mmol/mol (%) viikolla 18 | |||
| Lähtötilanne (keskiarvo) | 64,98 (8,10) | 66,72 (8,26) | 67,68 (8,34) |
| Muutos lähtötilanteesta1 | ‑0,14 (-0,01) | ‑6,22 (-0,57) | ‑7,80 (-0,71) |
Ero lumelääkkeeseen1 (97,5% lv) | ‑6,08 [‑8,51, ‑3,64](-0,56* [-0,78, -0,33]) | ‑7,66 [‑10,18, ‑5,14](-0,70* [-0,93, -0,47]) | |
| N | 112 | 127 | 110 |
| HbA1c mmol/mol (%) viikolla 78 | |||
| Lähtötilanne (keskiarvo) | 64,89 (8,09) | 66,89 (8,27) | 67,12 (8,29) |
| Muutos lähtötilanteesta1 | ‑0,18 (-0,02) | ‑5,24 (-0,48) | ‑7,00 (-0,64) |
| Ero lumelääkkeeseen1 [97,5% lv] | ‑5,06 [‑8,02, ‑2,11](-0,46* [-0,73, -0,19]) | ‑6,82 [‑9,88, ‑3,76](-0,62* [-0,90, -0,34]) | |
| N | 112 | 127 | 110 |
| Perusinsuliinin annos (IU/vrk) viikolla 78 | |||
| Lähtötilanne (keskiarvo) | 47,84 | 45,13 | 48,43 |
| Muutos lähtötilanteesta1 | 5,45 | -1,21 | -0,47 |
Ero lumelääkkeeseen1 (97,5% lv) | -6,66** (-11,56, -1,77) | -5,92** (-11,00, -0,85) |
a Koko analyysijoukko (FAS) käyttäen viimeisimmästä havainnosta laskettua arviota (last observation carried forward, LOCF), ennen lisälääkityksen ottamista glukoositasapainon korjaamiseksi
1 Korjattu keskiarvo lähtötilanteeseen nähden
* p-arvo < 0,0001
** p-arvo < 0,025
Munuaisten vajaatoimintaa sairastavat potilaat, 52 viikon lumekontrolloidut tulokset
Empagliflotsiinin tehoa ja turvallisuutta yhdistettynä muuhun diabeteshoitoon arvioitiin munuaisten vajaatoimintaa sairastavilla potilailla kaksoissokkoutetussa, lumekontrolloidussa 52 viikon pituisessa tutkimuksessa. Empagliflotsiinihoito alensi HbA1c-arvoa tilastollisesti merkitsevästi (taulukko 9) ja paransi paastoglukoosiarvoa kliinisesti merkittävästi lumelääkkeeseen verrattuna viikolla 24. HbA1c-arvon, painon ja verenpaineen lasku säilyi viikolle 52 asti.
Taulukko 9: Tulokset viikolla 24 lumekontrolloidussa tutkimuksessa, jossa empagliflotsiinia käytettiin munuaisten vajaatoimintaa sairastavilla tyypin 2 diabetespotilaillaa
| Lumelääke | Empagliflotsiini 10 mg | Empagliflotsiini 25 mg | Lumelääke | Empagliflotsiini 25 mg | |
| eGFR ≥ 60 – < 90 ml/min/1,73 m² | eGFR ≥ 30 – < 60 ml/min/1,73 m² | ||||
| N | 95 | 98 | 97 | 187 | 187 |
| HbA1c mmol/mol (%) | |||||
| Lähtötilanne (keskiarvo) | 64,93 (8,09) | 64,18 (8,02) | 63,51 (7,96) | 64,37 (8,04) | 64,28 (8,03) |
| Muutos lähtötilanteesta1 | 0,65 (0,06) | ‑5,02 (-0,46) | ‑6,83 (-0,63) | 0,51 (0,05) | ‑4,05 (-0,37) |
| Ero lumelääkkeeseen1[95 % lv] | ‑5,68 [‑7,83, ‑3,52] (-0,52* | ‑7,48 [‑9,65, ‑5,32] (-0,68*[-0,88, -0,49]) | ‑4,56 [‑6,07, ‑3,05] (-0,42*[-0,56, -0,28]) | ||
| N | 89 | 94 | 91 | 178 | 175 |
| HbA1c-arvon < 53 mmol/mol (7 %) saavuttaneet potilaat (%) lähtötilanteen HbA1c-arvon ollessa ≥ 53 mmol/mol (7 %2) | 6,7 | 17,0 | 24,2 | 7,9 | 12,0 |
| N | 95 | 98 | 97 | 187 | 187 |
| Paino (kg)2 | |||||
| Lähtötilanne (keskiarvo) | 86,00 | 92,05 | 88,06 | 82,49 | 83,22 |
| Muutos lähtötilanteesta1 | -0,33 | -1,76 | -2,33 | -0,08 | -0,98 |
| Ero lumelääkkeeseen1 (95 % lv) | -1,43 (-2,09, -0,77) | -2,00 (-2,66, -1,34) | -0,91 (-1,41, -0,41) | ||
| N | 95 | 98 | 97 | 187 | 187 |
| SVP (mmHg)2 | |||||
| Lähtötilanne (keskiarvo) | 134,69 | 137,37 | 133,68 | 136,38 | 136,64 |
| Muutos lähtötilanteesta1 | 0,65 | -2,92 | -4,47 | 0,40 | -3,88 |
| Ero lumelääkkeeseen1 (95 % lv) | -3,57 (-6,86, -0,29) | -5,12 (-8,41, -1,82) | -4,28 (-6,88, -1,68) | ||
a Koko analyysijoukko (FAS) käyttäen viimeisimmästä havainnosta laskettua arviota (last observation carried forward, LOCF), ennen lisälääkityksen ottamista glukoositasapainon korjaamiseksi
1 Korjattu keskiarvo lähtötilanteeseen nähden
2 Tilastollista merkitsevyyttä ei arvioitu perättäisen vahvistavan testimenetelmän vuoksi
* p < 0,0001
Sydän- ja verisuonisairauksiin liittyvät tulokset
Lumekontrolloitu EMPA-REG OUTCOME -kaksoissokkotutkimus vertasi yhdistetyssä analyysissä 10 mg:n ja 25 mg:n empagliflotsiiniannoksia lumelääkkeeseen; lisättynä tavanomaisen hoidon rinnalle tyypin 2 diabetespotilailla, joilla oli sydän- ja verisuonisairaus. Kaikkiaan 7 020 potilasta sai hoitoa (empagliflotsiini 10 mg: 2 345, empagliflotsiini 25 mg: 2 342, lumelääke: 2 333), ja seurantajakson mediaanipituus oli 3,1 vuotta. Keski-ikä oli 63 vuotta, keskimääräinen HbA1c-arvo oli 64,7 mmol/mol (8,1 %) ja 71,5 % potilaista oli miehiä. Lähtötilanteessa 74 % potilaista sai metformiini-, 48 % insuliini- ja 43 % sulfonyyliureahoitoa. Noin puolella potilaista (52,2 %) eGFR oli 60‒90 ml/min/1,73 m2, 17,8 %:lla 45‒60 ml/min/1,73 m2 ja 7,7 %:lla 30‒45 ml/min/1,73 m2.
Viikolla 12 korjattu keskimääräinen parannus (keskivirhe) HbA1c-arvossa lähtötilanteeseen nähden oli lumeryhmässä 1,20 mmol/mol (0,16) [0,11 % (0,02)], empagliflotsiini 10 mg -ryhmässä ‑7,06 mmol/mol (0,16) [0,65 % (0,02)] ja empagliflotsiini 25 mg -ryhmässä 0,71 % (0,02). Ensimmäisten 12 viikon jälkeen glukoositasapainoa optimoitiin tutkimushoidosta riippumatta. Siksi vaikutus oli viikolla 94 heikentynyt; korjattu keskimääräinen parannus (keskivirhe) HbA1c-arvossa oli lumeryhmässä ‑0,89 mmol/mol (0,25) [0,08 % (0,02)], empagliflotsiini 10 mg -ryhmässä ‑5,49 mmol/mol (0,25) [0,50 % (0,02)] ja empagliflotsiini 25 mg -ryhmässä ‑6,05 mmol/mol (0,25) [0,55 % (0,02)].
Empagliflotsiini oli parempi ehkäisemään ensisijaista yhdistelmäpäätetapahtumaa eli sydän- ja verisuonikuolemia, ei-kuolemaan johtaneita sydäninfarkteja tai ei-kuolemaan johtaneita aivoinfarkteja lumehoitoon verrattuna. Hoidon vaikutus johtui pääasiassa sydän- ja verisuonikuolemien merkitsevästä vähenemisestä. Ei-kuolemaan johtaneissa sydäninfarkteissa tai ei-kuolemaan johtaneissa aivoinfarkteissa ei ollut merkitsevää muutosta. Sydän- ja verisuonikuolemat vähenivät empagliflotsiini 10 mg- ja 25 mg -hoidossa yhtä paljon (kuva 1), ja se näkyi myös kokonaiseloonjäännin paranemisena (taulukko 10). EMPA-REG OUTCOME ‑tutkimuksessa empagliflotsiinin vaikutus ensisijaiseen yhdistelmäpäätetapahtumaan eli sydän- ja verisuonikuolemiin, ei-kuolemaan johtaneisiin sydäninfarkteihin tai ei-kuolemaan johtaneisiin aivoinfarkteihin oli yleisesti riippumaton glukoositasapainosta tai munuaistoiminnasta (eGFR). Empagliflotsiinin vaikutus oli yleisesti yhtäläinen eri eGFR-luokissa aina eGFR-tasoon 30 ml/min/1,73 m2 saakka.
EMPA-REG OUTCOME –tutkimuksessa DPP-4:n estäjien käyttäjät ja tummaihoiset potilaat olivat rajallisesti edustettuina. Tehoa sydän- ja verisuonikuolemien ehkäisemisessä ei ole vakuuttavasti osoitettu tällaisilla potilailla, jotka käyttävät samanaikaisesti empagliflotsiinia.
Taulukko 10: Hoidon vaikutus ensisijaiseen yhdistelmäpäätetapahtumaan, sen osatekijöihin ja kuolleisuuteena
| Lumelääke | Empagliflotsiinib | |
| N | 2 333 | 4 687 |
Ensimmäiseen sydän- ja verisuonikuolemaan, ei-kuolemaan johtaneeseen sydäninfarktiin tai ei-kuolemaan johtaneeseen aivoinfarktiin kulunut aika N (%) | 282 (12,1) | 490 (10,5) |
| Riskisuhde vs. lume (95,02 % lv)* | 0,86 (0,74; 0,99) | |
| Paremmuuden p-arvo | 0,0382 | |
Sydän- ja verisuonikuolemat N (%) | 137 (5,9) | 172 (3,7) |
| Riskisuhde vs. lume (95 % lv) | 0,62 (0,49; 0,77) | |
| p-arvo | <0,0001 | |
Ei-kuolemaan johtanut sydäninfarkti N (%) | 121 (5,2) | 213 (4,5) |
| Riskisuhde vs. lume (95 % lv) | 0,87 (0,70; 1,09) | |
| p-arvo | 0,2189 | |
Ei-kuolemaan johtanut aivoinfarkti N (%) | 60 (2,6) | 150 (3,2) |
| Riskisuhde vs. lume (95 % lv) | 1,24 (0,92; 1,67) | |
| p-arvo | 0,1638 | |
Kokonaiskuolleisuus N (%) | 194 (8,3) | 269 (5,7) |
| Riskisuhde vs. lume (95 % lv) | 0,68 (0,57; 0,82) | |
| p-arvo | <0,0001 | |
| Muu kuin sydän- ja verisuonikuolleisuus N (%) | 57 (2,4) | 97 (2,1) |
| Riskisuhde vs. lume (95 % lv) | 0,84 (0,60; 1,16) |
a Hoidetut potilaat eli potilaat, jotka olivat saaneet vähintään yhden annoksen tutkittavaa lääkettä
b 10 mg:n ja 25 mg:n empagliflotsiiniannosten yhdistetty analyysi
* Koska tutkimustulokset otettiin mukaan välianalyysiin, sovellettiin kaksipuolista 95,02 %:n luottamusväliä, joka vastaa merkitsevyyttä kuvaavaa p-arvoa < 0,0498.
Kuva 1. Sydän- ja verisuonikuolemaan kulunut aika EMPA-REG OUTCOME -tutkimuksessa
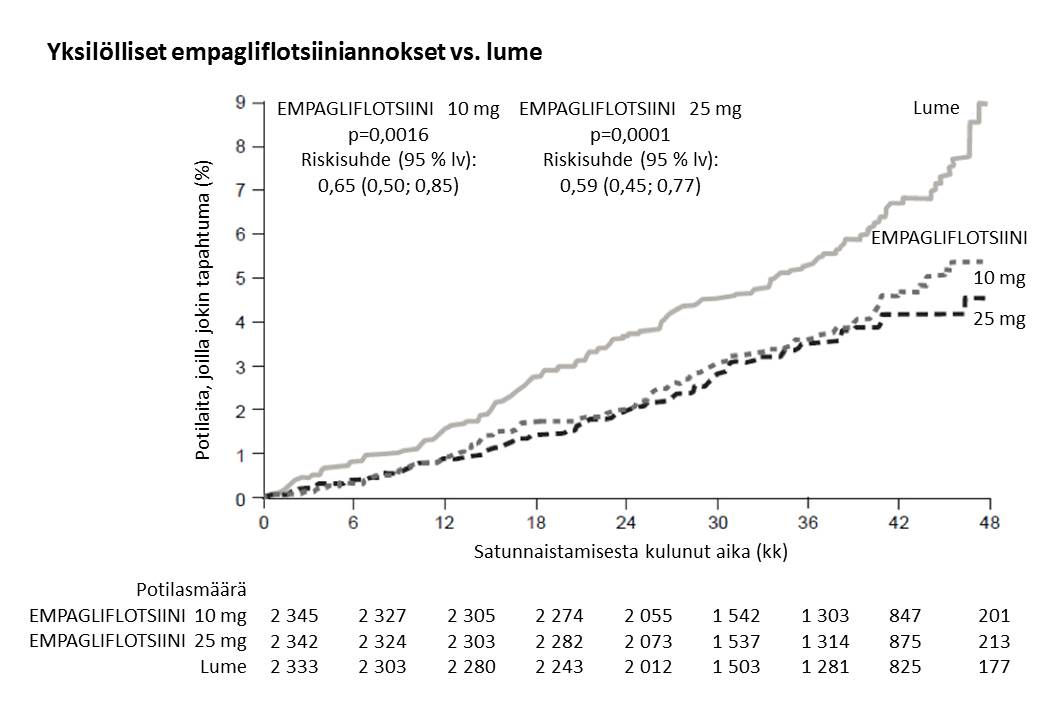
Sairaalahoitoa vaativa sydämen vajaatoiminta
EMPA-REG OUTCOME -tutkimuksessa empagliflotsiini vähensi sydämen vajaatoiminnasta johtuvien sairaalahoitojen riskiä lumelääkkeeseen verrattuna (empagliflotsiini 2,7 %; lumelääke 4,1 %; riskisuhde 0,65; 95 %:n luottamusväli 0,50; 0,85).
Nefropatia
EMPA-REG OUTCOME -tutkimuksessa ensimmäiseen nefropatiatapahtumaan kuluneen ajan riskisuhde oli 0,61 (95 %:n luottamusväli 0,53; 0,70) empagliflotsiinille (12,7 %) lumelääkkeeseen verrattuna (18,8 %).
Lisäksi empagliflotsiinihoitoa saaneilla potilailla, joilla oli lähtötilanteessa makroalbuminuria, esiintyi enemmän (riskisuhde 1,82; 95 %:n luottamusväli 1,40; 2,37) pitkäkestoista normo- tai mikroalbuminuriaa (49,7 %) lumelääkkeeseen verrattuna (28,8 %).
Plasman paastoglukoosi
Neljässä lumekontrolloidussa tutkimuksessa empagliflotsiinihoito monoterapiana tai yhdistelmähoitona metformiinin, pioglitatsonin tai metformiinin ja sulfonyyliurean kanssa, laski plasman paastoglukoosiarvoa keskimäärin 1,14 mmol/l empagliflotsiini 10 mg:aa saaneilla ja 1,29 mmol/l empagliflotsiini 25 mg:aa saaneilla lähtötilanteesta viikkoon 24 mennessä. Lumelääkkeellä vastaava muutos oli 0,41 mmol/l. Vaikutus säilyi viikolle 76 asti.
Glukoosipitoisuus 2 tuntia aterian jälkeen
Empagliflotsiinihoito yhdistettynä metformiiniin tai metformiiniin ja sulfonyyliureaan alensi kliinisesti merkittävästi 2 tuntia aterian jälkeen mitattua glukoosipitoisuutta (ateriarasituskoe) viikolla 24 (yhdistettynä metformiiniin: lumelääke +0,33 mmol/l, empagliflotsiini 10 mg: ‑2,56 mmol/l, empagliflotsiini 25 mg: ‑2,48 mmol/l, yhdistettynä metformiiniin ja sulfonyyliureaan: lumelääke ‑0,13 mmol/l, empagliflotsiini 10 mg: ‑1,98 mmol/l, empagliflotsiini 25 mg: ‑2,03 mmol/l).
Potilaat, joiden HbA1c-arvo lähtötilanteessa oli > 10 %
Kolmen faasi 3 -tutkimuksen ennalta määritellyssä yhteisanalyysissä avoin hoito empagliflotsiini 25 mg:n annoksella vaikeaa hyperglykemiaa sairastavilla potilailla (N = 184, keskimääräinen lähtötilanteen HbA1c 98,33 mmol/mol [11,15 %]) alensi kliinisesti merkittävästi lähtötilanteen HbA1c-arvoa ‑35,74 mmol/mol (3,27 %) viikolla 24. Näissä tutkimuksissa ei ollut lumelääkeryhmää tai empagliflotsiini 10 mg -ryhmää.
Paino
Neljän lumekontrolloidun tutkimuksen ennalta määritellyssä yhteisanalyysissä empagliflotsiinihoito alensi painoa (‑0,24 kg lumelääkettä saaneilla, ‑2,04 kg empagliflotsiini 10 mg:aa saaneilla ja ‑2,26 kg empagliflotsiini 25 mg:aa saaneilla) viikolla 24. Painon lasku säilyi viikolle 52 asti (‑0,16 kg lumelääkettä saaneilla, ‑1,96 kg empagliflotsiini 10 mg:aa saaneilla ja ‑2,25 kg empagliflotsiini 25 mg:aa saaneilla).
Verenpaine
Empagliflotsiinin tehoa ja turvallisuutta arvioitiin kaksoissokkoutetussa, lumekontrolloidussa 12 viikon pituisessa tutkimuksessa, johon osallistui tyypin 2 diabetesta ja korkeaa verenpainetta sairastavia potilaita, joilla oli käytössään eri diabeteslääkkeitä ja korkeintaan kaksi verenpainetta alentavaa lääkitystä. Empagliflotsiinihoito kerran vuorokaudessa paransi tilastollisesti merkitsevästi HbA1c-arvoa sekä 24 tunnin keskimääräistä systolista ja diastolista verenpainetta ambulatorisessa verenpaineen rekisteröinnissä (taulukko 11). Empagliflotsiinihoito alensi istuvassa asennossa mitattua systolista ja diastolista verenpainetta.
Taulukko 11: Tehon tulokset viikolla 12 lumekontrolloidussa empagliflotsiinitutkimuksessa, johon osallistui tyypin 2 diabetesta ja kontrolloimatonta verenpainetta sairastavia potilaitaa
| Lumelääke | Jardiance | ||
| 10 mg | 25 mg | ||
| N | 271 | 276 | 276 |
| HbA1c mmol/mol (%) viikolla 121 | |||
| Lähtötilanne (keskiarvo) | 62,88 (7,90) | 62,55 (7,87) | 63,03 (7,92) |
| Muutos lähtötilanteesta2 | 0,31 (0,03) | ‑6,49 (-0,59) | ‑6,82 (-0,62) |
| Ero lumelääkkeeseen2 (95 % lv) | ‑6,80 [‑7,88, ‑5,73](-0,62* [-0,72, -0,52]) | ‑7,12 [‑8,19, ‑6,06](-0,65* [-0,75, -0,55]) | |
| Systolinen verenpaine 24 tunnin kohdalla viikolla 123 | |||
| Lähtötilanne (keskiarvo) | 131,72 | 131,34 | 131,18 |
| Muutos lähtötilanteesta4 | 0,48 | -2,95 | -3,68 |
| Ero lumelääkkeeseen4 (95 % lv) | -3,44* (-4,78, -2,09) | -4,16* (-5,50, -2,83) | |
| Diastolinen verenpaine 24 tunnin kohdalla viikolla 123 | |||
| Lähtötilanne (keskiarvo) | 75,16 | 75,13 | 74,64 |
| Muutos lähtötilanteesta5 | 0,32 | -1,04 | -1,40 |
| Ero lumelääkkeeseen5 (95 % lv) | -1,36** (-2,15, -0,56) | -1,72* (-2,51, -0,93) | |
a Koko analyysijoukko (FAS)
1 LOCF, glukoositasapainoa korjaavan lisälääkityksen ottamisen jälkeiset arvot on poistettu
2 Korjattu keskiarvo lähtötilanteen HbA1c-arvoon, lähtötilanteen eGFR-arvoon, maantieteelliseen alueeseen ja verenpainetta alentavien lääkevalmisteiden lukumäärään nähden
3 LOCF, glukoositasapainoa parantavan lisähoidon ottamisen jälkeiset arvot ja verenpainetta alentavan lisälääkkeen vaihtamisen jälkeiset arvot on poistettu
4 Korjattu keskiarvo lähtötilanteen systoliseen verenpaineeseen, lähtötilanteen HbA1c-arvoon, lähtötilanteen eGFR-arvoon, maantieteelliseen alueeseen ja verenpainetta alentavien lääkevalmisteiden lukumäärään nähden
5 Korjattu keskiarvo lähtötilanteen diastoliseen verenpaineeseen, lähtötilanteen HbA1c-arvoon, lähtötilanteen eGFR-arvoon, maantieteelliseen alueeseen ja verenpainetta alentavien lääkevalmisteiden lukumäärään nähden
* p-arvo < 0,0001
** p-arvo < 0,001
Neljän lumekontrolloidun tutkimuksen ennalta määritellyssä yhteisanalyysissä empagliflotsiinihoito alensi systolista verenpainetta (empagliflotsiini 10 mg:aa saaneilla ‑3,9 mmHg; empagliflotsiini 25 mg:aa saaneilla ‑4,3 mmHg) verrattuna lumelääkettä saaneisiin (‑0,5 mmHg) ja diastolista verenpainetta (empagliflotsiini 10 mg:aa saaneilla ‑1,8 mmHg; empagliflotsiini 25 mg:aa saaneilla ‑2,0 mmHg) verrattuna lumelääkettä saaneisiin (‑0,5 mmHg). Verenpaineen aleneminen havaittiin viikolla 24 ja se säilyi viikolle 52 asti.
Sydämen vajaatoiminta
Empagliflotsiini potilailla, joilla oli sydämen vajaatoiminta ja alentunut ejektiofraktio
Satunnaistettuun, kaksoissokkoutettuun, lumekontrolloituun tutkimukseen (EMPEROR-Reduced) osallistui 3 730 potilasta, joilla oli krooninen sydämen vajaatoiminta (New York Heart Association [NYHA] II–IV) ja alentunut ejektiofraktio (LVEF ≤ 40 %). Tässä tutkimuksessa arvioitiin tavanomaisen sydämen vajaatoimintahoidon lisänä kerran vuorokaudessa annetun empagliflotsiinin (10 mg) tehoa ja turvallisuutta. Ensisijainen päätetapahtuma oli ensimmäiseen vahvistettuun tapahtumaan eli joko sydän- ja verisuoniperäiseen (CV) kuolemaan tai sydämen vajaatoiminnasta johtuvaan sairaalahoitoon (HHF) kulunut aika. Varmistustesteihin sisältyivät vahvistetun sydämen vajaatoiminnasta johtuvan sairaalahoidon (ensimmäisen kerran ja toistumisen) esiintyminen sekä eGFR (CKD‑EPI)cr ‑muutoksen voimakkuus lähtötilanteeseen nähden. Sydämen vajaatoiminnan hoitoja lähtötilanteessa olivat ACE:n estäjät / angiotensiinireseptorin salpaajat / angiotensiinireseptorin ja neprilysiinin estäjät (88,3 %), beetasalpaajat (94,7 %), mineralokortikoidireseptorin salpaajat (71,3 %) ja diureetit (95,0 %).
Yhteensä 1 863 potilasta satunnaistettiin saamaan empagliflotsiinia 10 mg (lumelääkettä: 1 867), ja seuranta-ajan mediaani oli 15,7 kuukautta. Tutkimuspopulaatiosta 76,1 % oli miehiä ja 23,9 % naisia. Tutkittavien keskimääräinen ikä oli 66,8 vuotta (vaihteluväli: 25–94 vuotta), ja 26,8 % oli vähintään 75‑vuotiaita. Tutkimuspopulaatiosta 70,5 % oli valkoihoisia, 18,0 % aasialaisia ja 6,9 % tummaihoisia/afroamerikkalaisia. Satunnaistamisvaiheessa 75,1 % potilaista kuului NYHA-luokkaan II, 24,4 % luokkaan III ja 0,5 % luokkaan IV. Keskimääräinen LVEF oli 27,5 %. Lähtötilanteessa keskimääräinen eGFR oli 62,0 ml/min/1,73 m2 ja virtsan albumiinin ja kreatiniinin suhteen (UACR) mediaani oli 22 mg/g. Noin puolella potilaista (51,7 %) eGFR oli ≥ 60 ml/min/1,73 m2, 24,1 %:lla se oli 45 – < 60 ml/min/1,73 m2, 18,6 %:lla 30 – < 45 ml/min/1,73 m2 ja 5,3 %:lla 20 – < 30 ml/min/1,73 m2.
Empagliflotsiini pienensi ensisijaisen yhdistelmäpäätetapahtuman eli sydän- ja verisuoniperäisen kuoleman tai sydämen vajaatoiminnasta johtuvan sairaalahoidon riskiä enemmän kuin lumelääke. Lisäksi empagliflotsiini pienensi sydämen vajaatoiminnasta johtuvan sairaalahoidon (HHF) (ensimmäisen kerran ja toistumisen) riskiä merkitsevästi ja hidasti eGFR-arvon laskua merkitsevästi (taulukko 12; kuva 2).
Taulukko 12: Hoidon vaikutus ensisijaiseen yhdistelmäpäätetapahtumaan, sen komponentteihin ja kahteen tärkeimpään toissijaiseen päätetapahtumaan, jotka sisältyivät ennalta määritettyihin varmistustesteihin
| Lume | Empagliflotsiini 10 mg | |
| N | 1 867 | 1 863 |
| Ensimmäiseen CV-kuolemaan tai HHF-tapahtumaan kulunut aika, N (%) | 462 (24,7) | 361 (19,4) |
| Riskisuhde vs. lume (95 % lv)* | 0,75 (0,65; 0,86) | |
| paremmuuden p-arvo | < 0,0001 | |
| CV-kuolema, N (%) | 202 (10,8) | 187 (10,0) |
| Riskisuhde vs. lume (95 % lv)* | 0,92 (0,75; 1,12) | |
| HHF (ensimmäinen kerta), N (%) | 342 (18,3) | 246 (13,2) |
| Riskisuhde vs. lume (95 % lv)* | 0,69 (0,59; 0,81) | |
| HHF (ensimmäinen kerta ja toistuminen), tapahtumien N | 553 | 388 |
| Riskisuhde vs. lume (95 % lv)* | 0,70 (0,58; 0,85) | |
| p−arvo | 0,0003 | |
| eGFR (CKD-EPI)cr -kulmakerroin**, laskunopeus (ml/min/1,73 m2/vuosi) | -2,28 | -0,55 |
| Hoitoero vs. lume (95 % lv) | 1,73 (1,10; 2,37) | |
| p-arvo | < 0,0001 |
CV = sydän- ja verisuoniperäinen, HHF = sydämen vajaatoiminnasta johtuva sairaalahoito, eGFR = arvioitu glomerulusten suodatusnopeus, CKD EPI = Chronic Kidney Disease Epidemiology Collaboration -laskentakaava
* Sydän- ja verisuoniperäiset kuolemat ja sydämen vajaatoiminnasta johtuvat sairaalahoitotapahtumat vahvisti riippumaton kliininen arviointilautakunta, ja niiden analysointiin käytettiin satunnaistettua potilasjoukkoa.
**eGFR-kulmakertoimen analysointiin käytettiin hoidettua potilasjoukkoa. Leikkauspiste on lumelääkkeelle -0,95 ml/min/1,73 m2 ja empagliflotsiinille -3,02 ml/min/1,73 m2. Leikkauspiste kuvaa eGFR-arvoon kohdistuvaa akuuttia vaikutusta ja kulmakerroin puolestaan pitkäaikaisvaikutusta.
Kuva 2 Ensimmäiseen vahvistettuun sydän- ja verisuoniperäiseen kuolemaan tai sydämen vajaatoiminnasta johtuvaan sairaalahoitoon kulunut aika
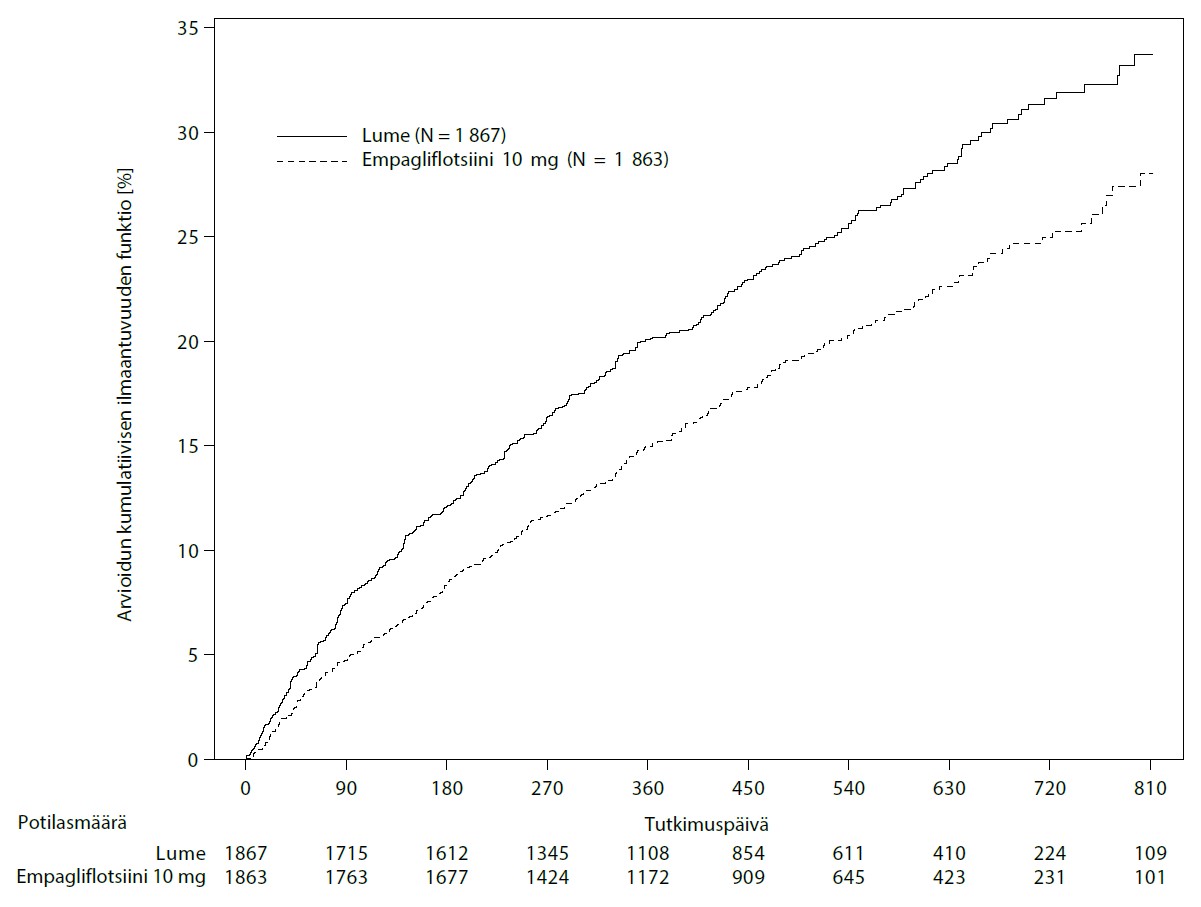
Ensisijaisen yhdistelmäpäätetapahtuman tuloksissa riskisuhde (HR) oli yleensä johdonmukaisesti alle 1 ennalta määritetyissä alaryhmissä, mukaan lukien sydämen vajaatoimintapotilaat, jotka sairastivat tai eivät sairastaneet tyypin 2 diabetesta ja joilla oli tai ei ollut munuaisten vajaatoimintaa (eGFR vähintään 20 ml/min/1,73 m2).
Empagliflotsiini potilailla, joilla oli sydämen vajaatoiminta ja säilynyt ejektiofraktio
Satunnaistettuun, kaksoissokkoutettuun, lumekontrolloituun tutkimukseen (EMPEROR-Preserved) osallistui 5 988 potilasta, joilla oli krooninen sydämen vajaatoiminta (NYHA II–IV) ja säilynyt ejektiofraktio (LVEF > 40 %). Tässä tutkimuksessa arvioitiin tavanomaisen hoidon lisänä kerran vuorokaudessa annetun empagliflotsiinin (10 mg) tehoa ja turvallisuutta. Ensisijainen päätetapahtuma oli ensimmäiseen vahvistettuun tapahtumaan eli joko sydän- ja verisuoniperäiseen (CV) kuolemaan tai sydämen vajaatoiminnasta johtuvaan sairaalahoitoon (HHF) kulunut aika. Varmistustesteihin sisältyivät vahvistetun sydämen vajaatoiminnasta johtuvan sairaalahoidon (ensimmäisen kerran ja toistumisen) esiintyminen sekä eGFR (CKD‑EPI)cr ‑muutoksen voimakkuus lähtötilanteeseen nähden. Lähtötilanteen hoitoja olivat ACE:n estäjät / angiotensiinireseptorin salpaajat / angiotensiinireseptorin ja neprilysiinin estäjät (80,7 %), beetasalpaajat (86,3 %), mineralokortikoidireseptorin salpaajat (37,5 %) ja diureetit (86,2 %).
Yhteensä 2 997 potilasta satunnaistettiin saamaan empagliflotsiinia 10 mg (lumelääkettä: 2 991), ja seuranta-ajan mediaani oli 26,2 kuukautta. Tutkimuspopulaatiosta 55,3 % oli miehiä ja 44,7 % naisia. Tutkittavien keskimääräinen ikä oli 71,9 vuotta (vaihteluväli: 22–100 vuotta), ja 43,0 % oli vähintään 75‑vuotiaita. Tutkimuspopulaatiosta 75,9 % oli valkoihoisia, 13,8 % aasialaisia ja 4,3 % tummaihoisia/afroamerikkalaisia. Satunnaistamisvaiheessa 81,5 % potilaista kuului NYHA-luokkaan II, 18,1 % luokkaan III ja 0,3 % luokkaan IV. EMPEROR-Preserved-tutkimuksen tutkimuspopulaatioon kuului potilaita, joiden LVEF oli < 50 % (33,1 %), potilaita, joiden LVEF oli 50 – < 60 % (34,4 %) ja potilaita, joiden LVEF oli ≥ 60 % (32,5 %). Lähtötilanteessa keskimääräinen eGFR oli 60,6 ml/min/1,73 m2 ja virtsan albumiinin ja kreatiniinin suhteen (UACR) mediaani oli 21 mg/g. Noin puolella potilaista (50,1 %) eGFR oli ≥ 60 ml/min/1,73 m2, 26,1 %:lla se oli 45 – < 60 ml/min/1,73 m2, 18,6 %:lla 30 – < 45 ml/min/1,73 m2 ja 4,9 %:lla 20 – < 30 ml/min/1,73 m2.
Empagliflotsiini pienensi ensisijaisen yhdistelmäpäätetapahtuman eli sydän- ja verisuoniperäisen kuoleman tai sydämen vajaatoiminnasta johtuvan sairaalahoidon riskiä enemmän kuin lumelääke. Lisäksi empagliflotsiini pienensi sydämen vajaatoiminnasta johtuvan sairaalahoidon (HHF) (ensimmäisen kerran ja toistumisen) riskiä merkitsevästi ja hidasti eGFR-arvon laskua merkitsevästi (taulukko 13; kuva 3).
Taulukko 13: Hoidon vaikutus ensisijaiseen yhdistelmäpäätetapahtumaan, sen komponentteihin ja kahteen tärkeimpään toissijaiseen päätetapahtumaan, jotka sisältyivät ennalta määritettyihin varmistustesteihin
| Lume | Empagliflotsiini 10 mg | |
| N | 2 991 | 2 997 |
| Ensimmäiseen CV-kuolemaan tai HHF-tapahtumaan kulunut aika, N (%) | 511 (17,1) | 415 (13,8) |
| Riskisuhde vs. lume (95 % lv)* | 0,79 (0,69; 0,90) | |
| paremmuuden p-arvo | 0,0003 | |
| CV-kuolema, N (%) | 244 (8,2) | 219 (7,3) |
| Riskisuhde vs. lume (95 % lv) | 0,91 (0,76; 1,09) | |
| HHF (ensimmäinen kerta), N (%) | 352 (11,8) | 259 (8,6) |
| Riskisuhde vs. lume (95 % lv) | 0,71 (0,60; 0,83) | |
| HHF (ensimmäinen kerta ja toistuminen), tapahtumien N | 541 | 407 |
| Riskisuhde vs. lume (95 % lv)* | 0,73 (0,61; 0,88) | |
| p−arvo | 0,0009 | |
| eGFR (CKD-EPI)cr -kulmakerroin**, laskunopeus (ml/min/1,73 m2/vuosi) | -2,62 | -1,25 |
| Hoitoero vs. lume (95 % lv) | 1,36 (1,06; 1,66) | |
| p-arvo | < 0,0001 |
CV = sydän- ja verisuoniperäinen, HHF = sydämen vajaatoiminnasta johtuva sairaalahoito, eGFR = arvioitu glomerulusten suodatusnopeus, CKD EPI = Chronic Kidney Disease Epidemiology Collaboration -laskentakaava
* Sydän- ja verisuoniperäiset kuolemat ja sydämen vajaatoiminnasta johtuvat sairaalahoitotapahtumat vahvisti riippumaton kliininen arviointilautakunta, ja niiden analysointiin käytettiin satunnaistettua potilasjoukkoa.
**eGFR-kulmakertoimen analysointiin käytettiin hoidettua potilasjoukkoa. Leikkauspiste on lumelääkkeelle -0,18 ml/min/1,73 m2 ja empagliflotsiinille -3,02 ml/min/1,73 m2. Leikkauspiste kuvaa eGFR-arvoon kohdistuvaa akuuttia vaikutusta ja kulmakerroin puolestaan pitkäaikaisvaikutusta.
Kuva 3 Ensimmäiseen vahvistettuun sydän- ja verisuoniperäiseen kuolemaan tai sydämen vajaatoiminnasta johtuvaan sairaalahoitoon kulunut aika
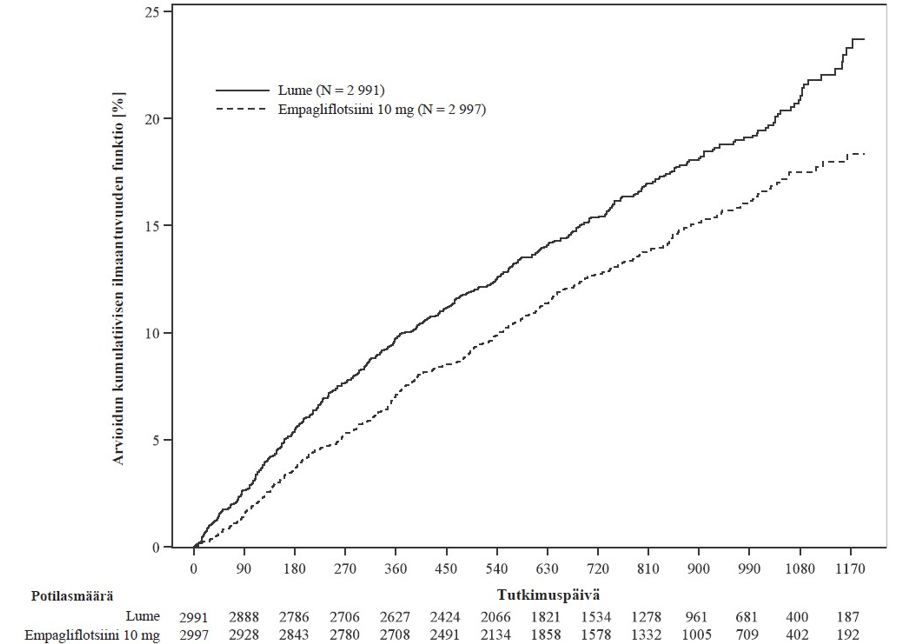
Ensisijaisen yhdistelmäpäätetapahtuman tulokset olivat johdonmukaisia kaikissa ennalta määritetyissä alaryhmissä, jotka luokiteltiin esim. LVEF-arvon, diabeteksen tai munuaistoiminnan (eGFR vähintään 20 ml/min/1,73 m2) mukaan.
Krooninen munuaistauti
Satunnaistettu, kaksoissokkoutettu, lumekontrolloitu tutkimus, jossa empagliflotsiinia käytettiin annoksena 10 mg kerran vuorokaudessa (EMPA-KIDNEY), tehtiin 6 609 potilaalla, joilla oli krooninen munuaistauti (eGFR ≥ 20 – < 45 ml/min/1,73 m2; tai eGFR ≥ 45 – < 90 ml/min/1,73 m2 ja virtsan albumiinin ja kreatiniinin suhde (UACR) ≥ 20 mg/mmol (200 mg/g). Tutkimuksessa arvioitiin sydän- ja munuaistuloksia, kun empagliflotsiinia käytettiin tavanomaisen hoidon lisänä. Ensisijainen päätetapahtuma oli munuaistaudin ensimmäiseen etenemiseen kulunut aika (pysyvä ≥ 40 %:n eGFR:n lasku satunnaistamisesta, eGFR pysyvästi < 10 ml/min/1,73 m², loppuvaiheen munuaisten vajaatoiminta tai munuaisperäinen kuolema) tai sydän- ja verisuoniperäinen kuolema. Varmistustesteihin sisältyivät sairaalaan joutuminen ensimmäistä kertaa sydämen vajaatoiminnan takia tai sydän- ja verisuoniperäinen kuolema, mistä tahansa syystä johtuva sairaalahoito (ensimmäinen kerta ja toistuminen), sekä mistä tahansa syystä johtuva kuolema. Lähtötilanteessa 85,2 %:lla oli käytössä ACE:n estäjä tai angiotensiinireseptorin salpaaja.
Yhteensä 3 304 potilasta satunnaistettiin saamaan 10 mg empagliflotsiinia (lumelääke: 3 305), ja heitä seurattiin 24,3 kuukauden ajan (mediaani). Tutkimuspopulaatiosta 66,8 % oli miehiä ja 33,2 % naisia. Tutkittavien keskimääräinen ikä oli 63,3 vuotta (vaihteluväli: 18–94 vuotta) ja 23,0 % oli 75‑vuotiaita tai vanhempia. Tutkimuspopulaatiosta 58,4 % oli valkoihoisia, 36,2 % aasialaisia ja 4,0 % tummaihoisia/afroamerikkalaisia.
Lähtötilanteessa keskimääräinen eGFR oli 37,3 ml/min/1,73 m2. Potilaista 21,2 %:lla eGFR oli ≥ 45 ml/min/1,73 m2, 44,3 %:lla 30 – < 45 ml/min/1,73 m2 ja 34,5 %:lla < 30 ml/min/1,73 m2, mukaan lukien 254 potilaista, joiden eGFR oli < 20 ml/min/1,73 m2. UACR-arvon mediaani oli 37,18 mg/mmol (329 mg/g). Potilaista 20,1 %:lla UACR oli < 3 mg/mmol (30 mg/g), 28,2 %:lla UACR oli 3– ≤ 30 mg/mmol (30 – ≤ 300 mg/g) ja 51,7 %:lla UACR oli > 30 mg/mmol (300 mg/g); potilaista 41,1 %:lla UACR oli < 200 mg/g. Kroonisen munuaistaudin ensisijaisia syitä olivat diabeettinen nefropatia / diabeettinen munuaistauti (31 %), glomerulussairaus (25 %), hypertensiivinen/renovaskulaarinen sairaus (22 %) ja muu/tuntematon (22 %).
Empagliflotsiini pienensi ensisijaisen yhdistelmäpäätetapahtuman eli munuaistaudin etenemisen tai sydän- ja verisuoniperäisen kuoleman riskiä verrattuna lumelääkkeeseen (ks. taulukko 14). Lisäksi empagliflotsiini pienensi mistä tahansa syystä johtuvan sairaalahoidon (ensimmäinen kerta ja toistuminen) riskiä merkitsevästi.
Taulukko 14: Hoidon vaikutus ensisijaiseen yhdistelmäpäätetapahtumaan, sen komponentteihin ja tärkeimpiin toissijaisiin päätetapahtumiin, jotka sisältyivät ennalta määritettyihin varmistustesteihin
| Lume | Empagliflotsiini 10 mg | |
| N | 3 305 | 3 304 |
| Munuaistaudin ensimmäiseen etenemiseen kulunut aika (pysyvä ≥ 40 %:n eGFR:n lasku satunnaistamisesta, eGFR pysyvästi < 10 ml/min/1,73 m², loppuvaiheen munuaisten vajaatoiminta* (ESKD) tai munuaisperäinen kuolema) tai CV-kuolema, N (%) | 558 (16,9) | 432 (13,1) |
| Riskisuhde vs. lume (99,83 % lv) | 0,72 (0,59; 0,89) | |
| paremmuuden p‑arvo | < 0,0001 | |
| Pysyvä ≥ 40 %:n eGFR:n lasku satunnaistamisesta, N (%) | 474 (14,3) | 359 (10,9) |
| Riskisuhde vs. lume (95 % lv) | 0,70 (0,61; 0,81) | |
| p-arvo | < 0,0001 | |
| ESKD* tai eGFR pysyvästi < 10 ml/min/1,73 m², N (%) | 221 (6,7) | 157 (4,8) |
| Riskisuhde vs. lume (95 % lv) | 0,69 (0,56; 0,84) | |
| p-arvo | 0,0003 | |
| Munuaisperäinen kuolema, N (%)** | 4 (0,1) | 4 (0,1) |
| Riskisuhde vs. lume (95 % lv) | ||
| p-arvo | ||
| CV-kuolema, N (%) | 69 (2,1) | 59 (1,8) |
| Riskisuhde vs. lume (95 % lv) | 0,84 (0,60; 1,19) | |
| p-arvo | 0,3366 | |
| ESKD tai CV-kuolema, N (%)# | 217 (6,6) | 163 (4,9) |
| Riskisuhde vs. lume (95 % lv) | 0,73 (0,59; 0,89) | |
| p-arvo | 0,0023 | |
| Mistä tahansa syystä johtuva sairaalahoito (ensimmäinen kerta ja toistuminen), tapahtumien N | 1 895 | 1 611 |
| Riskisuhde vs. lume (99,03 % lv) | 0,86 (0,75; 0,98) | |
| p-arvo | 0,0025 |
CV = sydän- ja verisuoniperäinen, eGFR = arvioitu glomerulusten suodatusnopeus
* Loppuvaiheen munuaisten vajaatoiminta (ESKD) määritellään ylläpitodialyysin aloittamisena tai munuaisensiirtona.
** Munuaisperäisiä kuolemia oli liian vähän luotettavan riskisuhteen laskemiseksi.
# Etukäteen määritelty toiseksi kahdesta keskeytyskriteeristä ennalta suunnitellussa välianalyysissa.
Kuva 4 Munuaistaudin ensimmäiseen etenemiseen tai vahvistettuun sydän- ja verisuoniperäiseen kuolemaan kulunut aika, arvioidun kumulatiivisen ilmaantuvuuden funktio
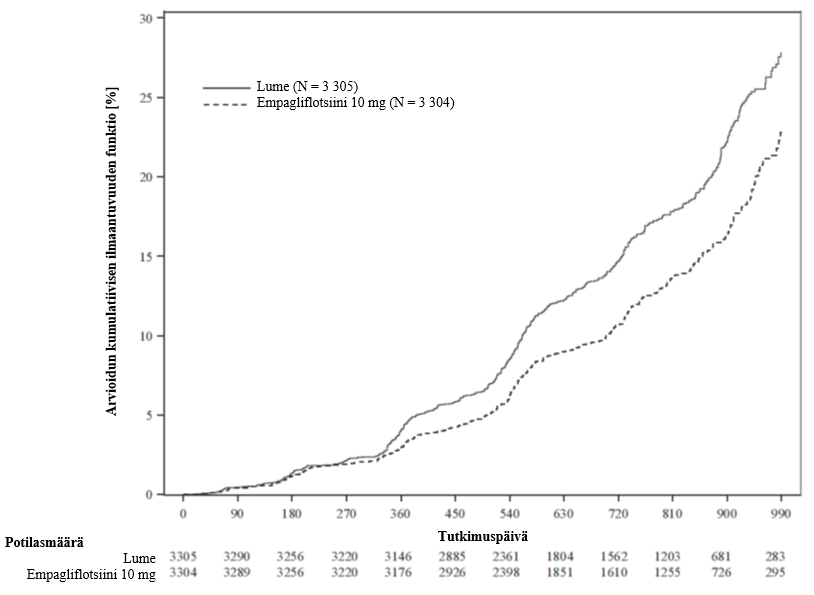
Ensisijaisen yhdistelmäpäätetapahtuman tulokset olivat yleisesti ottaen johdonmukaisia kaikissa ennalta määritellyissä alaryhmissä, joita olivat muun muassa eGFR-luokat, munuaistaudin perussyy, diabetesstatus tai RAS-estäjien käyttö. Hoidon hyödyt olivat selvemmin nähtävissä potilailla, joilla oli selvästi lisääntynyt albuminuria.
Hoidon aikana eGFR-arvo laski ajan kuluessa hitaammin empagliflotsiiniryhmässä kuin lumeryhmässä (kuva 5). Empagliflotsiini hidasti eGFR-arvon vuosittaista laskua 1,37 ml/min/1,73 m2/vuosi (95 % lv 1,16; 1,59) lumelääkkeeseen verrattuna kaikkien kuukauden 2 käynnin ja viimeisen seurantakäynnin välisenä aikana tehtyjen eGFR-mittausten ennalta määritellyn analyysin perusteella. Useissa empagliflotsiinitutkimuksissa empagliflotsiinihoitoa saaneiden potilaiden eGFR laski aluksi mutta palasi kohti lähtöarvoja hoidon lopettamisen jälkeen, mikä tukee oletusta, että hemodynaamisilla muutoksilla on osuutta empagliflotsiinin eGFR-arvoon kohdistuviin akuutteihin vaikutuksiin.
Kuva 5 eGFR-arvon muutos ajan kuluessa*
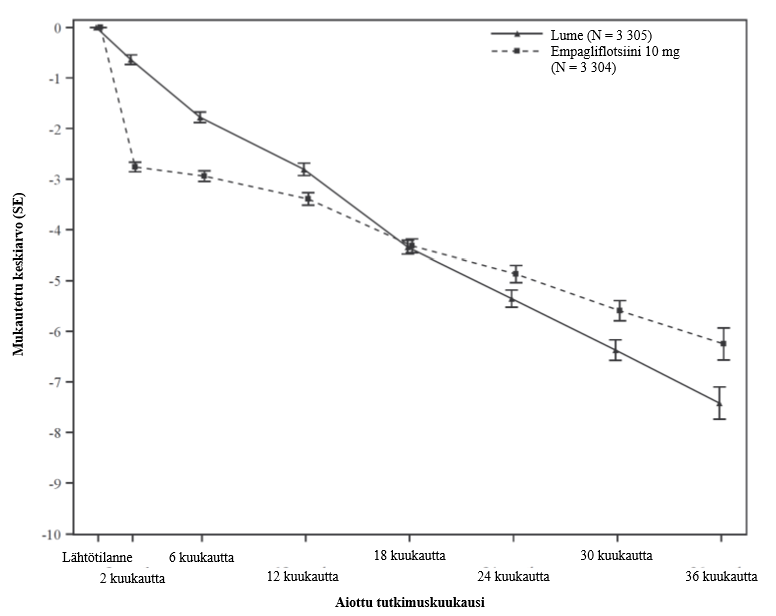
*eGFR (CKD-EPI) (ml/min/1,73 m2) MMRM-tulokset ajan kuluessa – satunnaistettu potilasjoukko.
Pediatriset potilaat
Tyypin 2 diabetes
Kerran vuorokaudessa otettavan empagliflotsiinin (10 mg ja mahdollinen annoksen suurennus 25 mg:aan) ja linagliptiinin (5 mg) kliinistä tehoa ja turvallisuutta on tutkittu 10–17-vuotiailla lapsilla ja nuorilla, joilla oli tyypin 2 diabetes, lumelääkekontrolloidussa tutkimuksessa (DINAMO) 26 viikon ajan ja turvallisuusjatkovaiheessa enintään 52 viikon ajan. Taustahoidot ruokavalion ja liikunnan lisänä sisälsivät metformiinia (51 %), metformiinin ja insuliinin yhdistelmää (40,1 %), insuliinia (3,2 %) tai ei mitään (5,7 %).
HbA1c-arvon mukautettu keskimääräinen muutos empagliflotsiinin (N = 52) ja lumelääkkeen (N = 53) välillä oli ‑9,23 mmol/mol (-0,84 %) viikolla 26; muutos oli kliinisesti merkittävä ja tilastollisesti merkitsevä (95 %:n lv ‑16,39; ‑2,07 [-1,50; -0,19]); p = 0,0116). Lisäksi empagliflotsiinihoidolla saavutettiin lumelääkkeeseen verrattuna kliinisesti merkittävä mukautettu keskimääräinen muutos plasman paastoglukoosiarvossa, -1,95 mmol/l (95 %:n lv (-3,25; -0,65).
Sydämen vajaatoiminta ja krooninen munuaistauti
Euroopan lääkevirasto on myöntänyt vapautuksen velvoitteesta toimittaa tutkimustulokset Jardiance-valmisteen käytöstä sydämen vajaatoiminnan hoidossa ja kroonisen munuaistaudin hoidossa kaikissa pediatrisissa potilasryhmissä (ks. kohdasta Annostus ja antotapa ohjeet käytöstä pediatristen potilaiden hoidossa).
Farmakokinetiikka
Imeytyminen
Empagliflotsiinin farmakokinetiikkaa on kuvattu laajasti terveillä vapaaehtoisilla ja tyypin 2 diabetesta sairastavilla potilailla. Suun kautta annosteltuna empagliflotsiini imeytyi nopeasti, ja huippupitoisuus plasmassa (tmax-arvon mediaani) saavutettiin keskimäärin 1,5 tuntia annoksen jälkeen. Tämän jälkeen pitoisuudet plasmassa alenivat kaksivaiheisesti (nopea jakautumisvaihe ja suhteellisen hidas terminaalinen vaihe). Vakaan tilan keskimääräinen plasman AUC oli 1 870 nmol.h/l ja Cmax 259 nmol/l empagliflotsiinin 10 mg:n vuorokausiannoksella ja 4 740 nmol.h/l ja 687 nmol/l empagliflotsiinin 25 mg:n vuorokausiannoksella. Systeeminen empagliflotsiinialtistus lisääntyi suhteessa annokseen. Farmakokineettiset parametrit olivat samanlaisia empagliflotsiinin kerta–annoksen jälkeen ja vakaassa tilassa, mikä viittaa farmakokinetiikan olevan lineaarinen ajan suhteen. Empagliflotsiinin farmakokinetiikassa ei ollut kliinisesti merkittäviä eroja terveiden vapaaehtoisten ja tyypin 2 diabetesta sairastavien potilaiden välillä.
Kun empagliflotsiinia annettiin 25 mg runsasrasvaisen ja kaloripitoisen aterian jälkeen, altistus oli hieman vähäisempää; AUC pieneni noin 16 % ja Cmax noin 37 % paastotilanteeseen verrattuna. Havaittua ruoan vaikutusta empagliflotsiinin farmakokinetiikkaan ei pidetty kliinisesti merkittävänä ja empagliflotsiinia voidaan ottaa ruoan kanssa tai ilman.
Jakautuminen
Näennäisen vakaan tilan jakautumistilavuuden arvioitiin olevan 73,8 litraa perustuen populaatiofarmakokineettiseen analyysiin. Kun terveille vapaaehtoisille annettiin suun kautta [14C]‑empagliflotsiiniliuosta, punasoluihin jakautui noin 37 % ja plasman proteiineihin sitoutui 86 %.
Biotransformaatio
Empagliflotsiinin merkittäviä metaboliitteja ei havaittu ihmisen plasmassa, ja yleisimmät metaboliitit olivat kolme glukuronidikonjugaattia (2‑, 3‑ ja 6‑O-glukuronidi). Systeeminen altistuminen jokaiselle metaboliitille oli alle 10 % lääkeaineen kokonaismäärästä. In vitro -tutkimustulokset viittaavat siihen, että empagliflotsiinin ensisijainen metaboliareitti ihmisillä on glukuronidaatio uridiini-5’-difosfoglukuronosyylitransferaasien UGT2B7, UGT1A3, UGT1A8 ja UGT1A9 vaikutuksesta.
Eliminaatio
Populaatiofarmakokineettisen analyysin perusteella empagliflotsiinin näennäisen terminaalisen eliminaation puoliintumisajan arvioitiin olevan 12,4 tuntia ja näennäinen oraalinen puhdistuma oli 10,6 litraa/tunti. Suun kautta otetun empagliflotsiinin puhdistuma vaihteli tutkittavien välillä 39,1 % ja jäännöspitoisuus 35,8 %. Kun empagliflotsiinia otettiin kerran vuorokaudessa, vakaan tilan pitoisuus plasmassa saavutettiin viidennellä annoksella. Vakaassa tilassa havaittiin korkeintaan 22 % kertyminen plasman AUC:n suhteen, mikä oli yhdenmukaista puoliintumisajan kanssa. Kun terveille vapaaehtoisille annetiin suun kautta [14C]‑empagliflotsiiniliuosta, noin 96 % lääkeaineeseen liittyvästä radioaktiivisuudesta poistui ulosteen mukana (41 %) tai virtsan mukana (54 %). Ulosteessa suurin osa lääkeaineeseen liittyvästä radioaktiivisuudesta oli muuttumatonta kantalääkettä. Virtsaan erittyneestä lääkeaineeseen liittyvästä radioaktiivisuudesta noin puolet oli muuttumatonta kantalääkettä.
Erityisryhmät
Munuaisten vajaatoiminta
Lievää, kohtalaista tai vaikeaa munuaisten vajaatoimintaa sairastavilla (eGFR < 30‑< 90 ml/min/1,73 m2) ja loppuvaiheen munuaisten vajaatoimintaa (ESKD) sairastavilla empagliflotsiinin AUC suureni vastaavasti noin 18 %, 20 %, 66 % ja 48 %, verrattuna tutkittaviin, joilla oli normaali munuaistoiminta. Empagliflotsiinin huippupitoisuudet plasmassa olivat kohtalaista munuaisten vajaatoimintaa ja munuaisten vajaatoimintaa/loppuvaiheen munuaisten vajaatoimintaa (ESKD) sairastavilla samanlaiset kuin potilailla, joiden munuaistoiminta oli normaali. Empagliflotsiinin huippupitoisuudet plasmassa lievää ja vaikeaa munuaisten vajaatoimintaa sairastavilla olivat noin 20 % korkeampia kuin potilailla, joiden munuaistoiminta oli normaali. Populaatiofarmakokineettinen analyysi osoitti, että suun kautta otetun empagliflotsiinin näennäinen puhdistuma aleni yhdessä eGFR:n alenemisen kanssa, mikä lisäsi lääkeainealtistusta.
Maksan vajaatoiminta
Child-Pugh-luokituksen mukaisesti lievää, kohtalaista tai vaikeaa maksan vajaatoimintaa sairastavilla potilailla empagliflotsiinin AUC suureni vastaavasti noin 23 %, 47 % ja 75 %, ja Cmax vastaavasti 4 %, 23 % ja 48 % verrattuna tutkittaviin, joiden maksantoiminta oli normaali.
Painoindeksi
Painoindeksillä ei ollut kliinisesti merkittävää vaikutusta empagliflotsiinin farmakokinetiikkaan populaatiofarmakokineettisen analyysin perusteella. Tässä analyysissä AUC-arvon arvioitiin olevan 5,82 %, 10,4 % ja 17,3 % alempi tutkittavilla, joiden BMI oli vastaavasti 30, 35 ja 45 kg/m2, verrattuna tutkittaviin, joiden painoindeksi oli 25 kg/m2.
Sukupuoli
Sukupuolella ei ollut kliinisesti merkittävää vaikutusta empagliflotsiinin farmakokinetiikkaan populaatiofarmakokineettisen analyysin perusteella.
Etninen tausta
Populaatiofarmakokineettisesä analyysissä AUC-arvon arvioitiin olevan 13,5 % korkeampi aasialaisilla, joiden painoindeksi oli 25 kg/m2, verrattuna ei-aasialaisiin, joiden painoindeksi oli 25 kg/m2.
Iäkkäät
Iällä ei ollut kliinisesti merkittävää vaikutusta empagliflotsiinin farmakokinetiikkaan populaatiofarmakokineettisen analyysin perusteella.
Pediatriset potilaat
Pediatrisessa vaiheen 1 tutkimuksessa tutkittiin empagliflotsiinin (5 mg, 10 mg ja 25 mg) farmakokinetiikkaa ja farmakodynamiikkaa ≥ 10 - < 18 vuoden ikäisillä tyypin 2 diabetesta sairastavilla lapsilla ja nuorilla. Havaitut farmakokineettiset ja farmakodynaamiset vasteet vastasivat aikuispotilaiden vasteita.
Pediatrisessa vaiheen 3 tutkimuksessa tutkittiin empagliflotsiinin 10 mg:n annoksen sekä mahdollisen 25 mg:aan suurennetun annoksen farmakokinetiikkaa ja farmakodynamiikkaa (HbA1c-arvon muutosta lähtötilanteesta) 10–17 vuoden ikäisillä tyypin 2 diabetesta sairastavilla lapsilla ja nuorilla. Havaittu altistus-vastesuhde oli yleisesti ottaen verrattavissa aikuisten, lasten ja nuorten kesken. Empagliflotsiinin oraalinen annostelu tuotti altistuksen, joka oli aikuispotilailla havaitulla alueella.
Havaitut geometriset keskimääräiset pohjapitoisuudet ja geometriset keskimääräiset pitoisuudet 1,5 tuntia lääkkeen annon jälkeen vakaassa tilassa olivat 26,6 nmol/l ja 308 nmol/l empagliflotsiinin 10 mg:n vuorokausiannoksella ja 67,0 nmol/l ja 525 nmol/l empagliflotsiinin 25 mg:n vuorokausiannoksella.
Prekliiniset tiedot turvallisuudesta
Farmakologista turvallisuutta, genotoksisuutta sekä hedelmällisyyttä ja varhaista alkionkehitystä koskevien konventionaalisten tutkimusten tulokset eivät viittaa erityiseen vaaraan ihmisille.
Jyrsijöillä ja koirilla tehdyissä pitkäaikaisissa toksisuustutkimuksissa toksisuuden merkkejä havaittiin, kun altistukset olivat vähintään kymmenkertaisia empagliflotsiinin kliiniseen annokseen verrattuna.
Suurin osa toksisuudesta oli yhdenmukaista glukoosin virtsaan erittymiseen ja elektrolyyttien epätasapainoon liittyvän sekundaarisen farmakologian kanssa, kuten painon ja kehon rasvakudoksen määrän aleneminen, lisääntynyt ravinnon nauttiminen, ripuli, dehydraatio, seerumin glukoosin aleneminen ja muiden lisääntyneeseen proteiinimetaboliaan ja glukoneogeneesiin viittaavien seerumin parametrien lisääntyminen, muutokset virtsassa kuten polyuria ja glukosuria, ja mikroskooppiset muutokset, mukaan lukien mineralisaatio munuaisissa, joissain pehmytkudoksissa ja verisuonikudoksissa. Joillain lajeilla havaittiin mikroskooppisesti liiallisia farmakologisia vaikutuksia munuaisissa kuten munuaistiehyiden laajenemista ja munuaistiehyiden ja -altaan mineralisaatiota. Näitä havaittiin, kun altistus oli noin nelinkertainen verrattuna kliiniseen AUC-altistukseen empagliflotsiinin 25 mg:n annoksella.
Empagliflotsiini ei ole genotoksinen.
Kahden vuoden mittaisessa karsinogeenisuustutkimuksessa empagliflotsiini ei lisännyt kasvainten esiintyvyyttä naarasrotilla, jotka saivat korkeintaan 700 mg/kg:n vuorokausiannoksia vastaten keskimäärin 72‑kertaista maksimaalista empagliflotsiinin kliinistä AUC-altistusta. Urosrotilla hoitoon liittyviä hyvänlaatuisia vaskulaarisia proliferatiivisia leesioita (hemangioomia) havaittiin mesenteerisessä imusolmukkeessa suurimmilla annoksilla, mutta ei annoksella 300 mg/kg/vrk, joka vastaa keskimäärin 26-kertaista maksimaalista kliinistä empagliflotsiinialtistusta. Kiveksissä olevien interstitiaalisten kasvainten esiintyvyys rotilla oli suurempi 300 mg/kg/vrk ja tätä suuremmilla annoksilla, mutta ei 100 mg/kg/vrk annoksella, joka vastaa keskimäärin 18‑kertaista maksimaalista kliinistä empagliflotsiinialtistusta. Molemmat kasvaimet ovat yleisiä rotilla eivätkä ne todennäköisesti ole merkityksellisiä ihmisille.
Empagliflotsiini ei lisännyt kasvainten esiintyvyyttä naarashiirillä, jotka saivat korkeintaan 1 000 mg/kg:n vuorokausiannoksia, joka vastaa keskimäärin 62-kertaista maksimaalista kliinistä empagliflotsiinialtistusta. Empagliflotsiini aiheutti munuaisen kasvaimia uroshiirillä 1 000 mg/kg:n vuorokausiannoksilla, mutta ei 300 mg/kg:n vuorokausiannoksilla, joka vastaa keskimäärin 11-kertaista maksimaalista kliinistä empagliflotsiinialtistusta. Näiden kasvainten ilmaantuvuus on riippuvainen uroshiirten luontaisesta munuaispatologiasta ja metaboliareitistä, jotka eivät heijasta munuaispatologiaa ja metaboliareittiä ihmisessä. Uroshiirten munuaiskasvaimia ei pidetä merkityksellisinä ihmisille.
Ihmisen terapeuttista annosta riittävästi suuremmilla altistustasoilla empagliflotsiinilla ei ollut haitallisia vaikutuksia hedelmällisyyteen tai varhaiseen alkionkehitykseen. Organogeneesivaiheen aikana annettu empagliflotsiini ei ollut teratogeeninen. Ainoastaan emolle toksisina annoksina empagliflotsiini aiheutti myös taipuneita raajan luita rotalla ja lisäsi alkioiden menetystä kanilla.
Rotilla tehdyissä pre- ja postnataalitoksisuustutkimuksissa havaittiin poikasten painonnousun vähenevän, kun emo altistettiin annoksille, jotka vastasivat keskimäärin 4-kertaista maksimaalista kliinistä empagliflotsiinialtistusta. Vastaavaa vaikutusta ei havaittu systeemisellä altistuksella, joka vastasi maksimaalista kliinistä empagliflotsiinialtistusta. Tämän löydöksen merkitys ihmisille on epäselvä.
Nuorilla rotilla tehdyssä toksisuustutkimuksessa, jossa empagliflotsiinia annosteltiin 21 päivän iästä 90 päivän ikään, nuorilla rotilla havaittiin hyvin vähäistä tai lievää, ei haittavaikutukseksi luokiteltavaa munuaistiehyiden ja munuaisaltaan laajentumaa ainoastaan annoksella 100 mg/kg/vrk, joka on suunnilleen 11-kertainen annos kliiniseen 25 mg:n enimmäisannokseen nähden. Näitä löydöksiä ei enää 13 viikon lääkkeettömän toipumisjakson jälkeen havaittu.
Farmaseuttiset tiedot
Apuaineet
Tablettiydin: laktoosimonohydraatti, mikrokiteinen selluloosa, hydroksipropyyliselluloosa, kroskarmelloosinatrium, kolloidinen vedetön piidioksidi, magnesiumstearaatti.
Kalvopäällyste: hypromelloosi, titaanidioksidi (E171), talkki, makrogoli (400), keltainen rautaoksidi (E172).
Yhteensopimattomuudet
Ei oleellinen.
Kestoaika
3 vuotta
Säilytys
Tämä lääkevalmiste ei vaadi erityisiä säilytysolosuhteita.
Pakkaukset ja valmisteen kuvaus
Markkinoilla olevat pakkaukset
Resepti
JARDIANCE tabletti, kalvopäällysteinen
10 mg (L:kyllä) 30 x 1 fol (55,39 €), 90 x 1 fol (140,80 €)
25 mg (L:kyllä) 30 x 1 fol (55,60 €), 90 x 1 fol (141,74 €)
PF-selosteen tieto
Perforoidut yksittäispakatut PVC/alumiiniläpipainopakkaukset.
Pakkauskoot 7 x 1, 10 x 1, 14 x 1, 28 x 1, 30 x 1, 60 x 1, 70 x 1, 90 x 1 ja 100 x 1 kalvopäällysteistä tablettia.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä.
Valmisteen kuvaus:
Jardiance 10 mg kalvopäällysteiset tabletit
Pyöreä, vaaleankeltainen, kaksoiskupera, viistoreunainen kalvopäällysteinen tabletti, jossa on toisella puolella merkintä "S10" ja toisella puolella Boehringer Ingelheimin logo (tabletin halkaisija: 9,1 mm).
Jardiance 25 mg kalvopäällysteiset tabletit
Soikea, vaaleankeltainen, kaksoiskupera kalvopäällysteinen tabletti, jossa on toisella puolella merkintä "S25" ja toisella puolella Boehringer Ingelheimin logo (tabletin pituus: 11,1 mm, tabletin leveys: 5,6 mm).
Käyttö- ja käsittelyohjeet
Käyttämätön lääkevalmiste tai jäte on hävitettävä paikallisten vaatimusten mukaisesti.
Korvattavuus
JARDIANCE tabletti, kalvopäällysteinen
25 mg 30 x 1 fol, 90 x 1 fol
- Alempi erityiskorvaus (65 %). Diabetes, muu kuin insuliinihoito (215).
- Peruskorvaus (40 %).
JARDIANCE tabletti, kalvopäällysteinen
10 mg 30 x 1 fol, 90 x 1 fol
- Alempi erityiskorvaus (65 %). Diabetes, muu kuin insuliinihoito (215), Dapagliflotsiini ja empagliflotsiini: Kroonisen sydämen vajaatoiminnan hoito erityisin edellytyksin (250).
- Peruskorvaus (40 %).
ATC-koodi
A10BK03
Valmisteyhteenvedon muuttamispäivämäärä
24.03.2026
Yhteystiedot
 BOEHRINGER INGELHEIM FINLAND KY
BOEHRINGER INGELHEIM FINLAND KY Tammasaarenkatu 5
00180 Helsinki
010 310 2800
www.boehringer-ingelheim.fi
medinfo.finland@boehringer-ingelheim.com